Embed Size (px)
Citation preview

ONCOGENIC RAS INDUCES ACCELERATED TRANSITION THROUGH G2/M AND PROMOTES DEFECTS IN THE G2 DNA DAMAGE AND MITOTIC SPINDLE CHECKPOINTS Jeffrey A. Knauf*1, Bin Ouyang*1, Erik S. Knudsen2, Kenji Fukasawa2, George Babcock3 James A.
Fagin1,2. From the 1Division of Endocrinology and Metabolism, 2Department of Cell Biology, Neurobiology
and Anatomy, and the 3Division of Burn Surgery/Shriners Burns Institute, University of Cincinnati College of Medicine, Cincinnati, Ohio, 45267 USA
Running title: Oncogenic RAS Induces Defects in DNA Damage and Mitotic Checkpoints *Equal contributors to this work. Address correspondence to: James A. Fagin MD, Division of Endocrinology and Metabolism, University of Cincinnati College of Medicine, 231 Bethesda Ave, Rm 5564, Cincinnati, OH 45267-0547, E-mail: [email protected]
Activating mutations of RAS are prevalent in thyroid follicular neoplasms, which commonly have chromosomal losses and gains. In thyroid cells, acute expression of HRASV12 increases the frequency of chromosomal abnormalities within one or two cell cycles, suggesting that RAS oncoproteins may interfere with cell cycle checkpoints required for maintenance of a stable genome. To explore this, PCCL3 thyroid cells with conditional expression of HRASV12 or HRASV12 effector mutants were presynchronized at the G1/S boundary, followed by activation of expression of RAS mutants and release from the cell cycle block. Expression of HRASV12 accelerated the G2/M phase by ~4 hours, and promoted bypass of the G2 DNA damage and mitotic spindle checkpoints. Accelerated passage through G2/M and bypass of the G2 DNA damage checkpoint, but not bypass of the mitotic spindle checkpoint, required activation of MAPK. However, selective activation of the MAPK pathway was not sufficient to disrupt the G2 DNA damage checkpoint, as cells arrested appropriately in G2 despite conditional expression of HRASV12, S35 or BRAFV600E. By contrast to the MAPK requirement for radiation-induced G2 arrest, RAS-induced bypass of the mitotic spindle checkpoint was not prevented by pretreatment with MEK inhibitors. These data support a direct role for MAPK pathway in control of G2 progression and regulation of the G2 DNA damage checkpoint. We propose that oncogenic RAS activation may predispose cells to genomic instability through both MAPK-
dependent and independent pathways that affect critical checkpoints in G2/M. Human tumors, including those of the thyroid (1-3), arise from a single transformed cell. Despite their clonal origin cells from advanced carcinomas are often genetically heterogeneous. Tumor cell variability is thought to result from genomic instability. Clonal heterogeneity in turn tends to predict a poor outcome, and resistance to therapy. This is also the case in thyroid cancer. In thyroid tumors, the nature of the oncogenic events involved in the initiation of the neoplastic clone may determine the likelihood of genomic instability occurring at a later stage. Among the various forms of thyroid neoplasia, follicular adenomas and carcinomas are commonly aneuploid, whereas abnormalities in chromosome number are comparatively less frequent in papillary thyroid carcinomas (PTC). Mutations of all three RAS genes are found in benign and malignant follicular neoplasms and in follicular variant PTCs, and believed to be one of the early steps in thyroid tumor formation (4-9). By contrast rearrangements of the tyrosine kinase receptor gene RET (RET/PTC) are only found in PTC, which are commonly diploid and less aggressive. A possible explanation for this is that activating mutations of RAS, but not of RET, promote tumor progression in part by decreasing genomic stability. Consistent with this is the observation that acute expression of HRASV12 increases the frequency of micronuclei, an accepted indicator of chromosomal instability, whereas expression RET/PTC does not (10). The rate of spontaneous mutations acquired during the natural life span of a cell is
1
http://www.jbc.org/cgi/doi/10.1074/jbc.M511690200The latest version is at JBC Papers in Press. Published on November 29, 2005 as Manuscript M511690200
Copyright 2005 by The American Society for Biochemistry and Molecular Biology, Inc.
by guest on July 2, 2016http://w
ww
.jbc.org/D
ownloaded from

exceedingly low (11,12). This suggests that one of the early genetic disruptions involved in tumor development may confer cells with a “mutator” phenotype (13,14), hence predisposing to the accumulation of additional abnormalities. Indeed, germline mutations in genes such at p53, ATM (ataxia telangiectasia mutated), and BRCA1/2 that are involved in DNA damage repair and regulation of cell cycle checkpoints are found in cancer susceptibility syndromes. Although proteins encoded by p53, ATM and BRCA1/2 have numerous functions, progression to the malignant state in these cancer syndromes is likely to be at least in part due to genomic instability. Although not as widely appreciated, oncoproteins such as RAS have also been proposed to promote tumor progression through induction of genomic instability. For example, Finney and Bishop reported that replacement of a normal Hras gene with an activated mutant Hras by homologous recombination in rat1 fibroblasts is not in itself sufficient to induce transformation, but rather requires secondary changes such as gene amplification events, including amplification of the mutant Ras allele (15). This study supports the concept that RAS may serve as a mutator gene under physiological conditions, because the mutant HRAS protein in these experiments was expressed under the control of its own promoter. The ability of activated RAS to promote chromosomal instability is also supported by studies that demonstrate that expression of the human HRAS oncogene in p53-null cells leads to premature entry of cells into S phase, increased permissivity for gene amplification, and generation of aberrant chromosomes within a single cell cycle (16-20). Oncogenic RAS has also been shown to produce chromosome aberrations in rat mammary carcinoma cells (21), rat prostatic tumor cells (22), and in a human colon carcinoma cell line (23). The demonstration by Agapova et al (20) that expression of activated HRAS promotes bypass of G2 DNA damage checkpoint in p53 mutant cells suggests that oncogenic RAS-induced genomic instability may potentially be due to a relaxation of this checkpoint. The effectors downstream of RAS that are required for this effect have not been fully elucidated. Activation by RAS of RAL-GEF and RAL is responsible for dampening the G2 arrest induced by ethyl methanesulfonate in p53 deficient MDAH041
fibroblasts (24). On the other hand, activation of the RAS downstream effectors MEK2 and ERK are required for exit from DNA damage-induced G2 cell cycle arrest (25) and the transition from G2 into M (26,27), respectively. A role for the MEK/ERK pathway in G2/M is further supported by the observation that activated ERK associates with the mitotic apparatus of somatic mammalian cells (28,29). ERK was also reported to associate with kinetochores during early prophase, but this association was not apparent at later stages of mitosis. Both ERK and its activator MEK localize to the mitotic spindle from prophase through anaphase, and to the midbody during cytokinesis. Furthermore, activated ERK was found to associate with the spindle–microtubule motor CENP-E during mitosis (29) and is capable of regulating microtubule dynamics during mitosis (30). These results strongly support a role for MEK and ERK in regulating the progression of cells through G2 and mitosis, suggesting that inappropriate activation of these RAS effectors in cells expressing oncogenic RAS could potentially disrupt the orderly transition of these cells through these latter stages of the cells cycle, which are critical for maintaining genomic integrity. We previously showed that acute expression of HRASV12 increases the frequency of chromosome misalignment, multiple spindle formation, centrosome amplification and generation of micronuclei within the first few cell cycles after activation in rat thyroid PCCL3 cells (31). These cells are not transformed, and have wild type p53 genes. Rapid induction of these chromosomal abnormalities by RAS is consistent with a disruption of progression of cells through G2/M and/or alteration of the integrity of critical checkpoints needed to ensure genomic stability. Here we investigated this possibility by presynchronizing PCCL3 thyroid cells with conditional expression of HRASV12 or HRASV12 effector mutants at the G1/S boundary, followed by activation of expression of RAS mutants, and release from the G1 block.into a radiation induced G2 arrest, or a nocodazole activated mitotic checkpoint. This allowed us to follow the progression of cells through G2/M as well as the G2 DNA damage and mitotic spindle checkpoints, and explore the contribution of RAS effectors to this effect.
2
by guest on July 2, 2016http://w
ww
.jbc.org/D
ownloaded from

Material and Methods Cell lines-The well-differentiated rat thyroid cell line PCCL3 was propagated in H4 complete medium, which consisted of Coon’s modification of Ham’s F12 media containing 5% fetal bovine serum, glutamine (286 µg/ml), apo-transferrin (5 µg/ml), hydrocortisone (10 nM), insulin (10 µg/ml), TSH (10 mIU/ml), penicillin and streptomycin. The following cell lines have been previously described: rtTA, PCCL3 cells stably expressing the reverse tetracycline trans-activator rtTA (32); Ras-25, PCCL3 cells with doxycycline (dox)-induced expression of HRASV12 (31,33); PC-BRAFV600E-6 PCCL3 cells with dox-induced expression of constitutively active BRAF mutant, BRAFV600E (34), and MEK1-65 PCCL3 cells with doxycycline (dox)-induced expression of the constitutively active MEK1 mutant, MEK1S217E/S221E (31,33). Using the same approach, we created PCCL3 cells with dox-inducible expression of the previously described RAS effector mutants (35). Briefly, we subcloned the HRASV12,S35, HRASV12,G37, or HRASV12,C40 cDNAs (gifts from Kenji Fukasawa, University of Cincinnati) into pUHG10-3, downstream of seven repeats of a tet operator sequence and a minimal cytomegalovirus promoter. These constructs were co-transfected into rtTA with pTK-hygro using Lipofectamine 2000 (Invitrogen, Carlsbad, CA), and clones selected based on absence of expression under basal conditions and strong induction by dox. Reagents-FITC conjugated anti-BrdU IgG was purchased from Pharmigen (San Diego, CA). Antibodies to phospho-MEK1/2 (sc-7995), MEK2 (sc-524), ERK1 (sc-94), phospho-ERK1/2 (sc-7383), HRAS (SC-520), cyclin B1 (SC-245), HDAC1 (SC-6298), and GAPDH (SC-20357) were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA) and anti phospho-MEK1/2 (9121S) goat polyclonal IgG from Cell Signaling Technology (Beverly, MA). Thymidine, nocodazole, 4’, 6-diamidino-2-phenylindole (DAPI), propidium iodide, TSH, insulin, apo-transferrin, and hydrocortisone were purchased from Sigma (St. Louis, MO), PD98059 and wortmannin were from Calbiochem (San Diego, CA). Coon’s modification of Ham’s F12 medium was from Irvine Scientific (Irvine, CA). Fetal bovine serum, penicillin-streptomycin, and
glutamine were purchased from Life Technologies, Inc. (Gaithersburg, MD). Monitoring cell cycle progression by FACS Cell synchronization-To synchronize cells in G1/S, cells were plated into 60 mm tissue culture dishes at approximately 50% confluence in H4 medium and incubated at 37°C in 5% CO2 for 24 hours. The medium was then replaced with fresh H4 medium containing 4 mM thymidine and cells incubated for 14 h. Cells were then washed twice with PBS, H4 medium added, and cells incubated for 9 h. The medium was then replaced with fresh H4 medium containing 4 mM thymidine and cells incubated in the absence or presence of 1 µg/ml dox for 14 h. Cells were released by washing twice with PBS and then adding fresh H4 medium containing 10 µM BrdU with or without 1 µg/ml dox. After 2 h cells were washed with PBS to remove BrdU, and fresh H4 medium with or without 1 µg/ml dox added. To induce the G2 DNA damage checkpoint cells were irradiated 4 h after release cells with 10 Gy of X-rays or 15 Gy of γ-rays (Faxitron cabinet X-ray irradiator or Cesium irradiator, respectively). To induce the mitotic spindle checkpoint, nocodazole was added 5 h after release from the G1/S block to a final concentration of 0.4 µg/ml. At the indicated times after release, cells were washed with PBS, harvested by trypsinization, fixed in 5 ml of cold (-20°C) 70% ethanol and incubated overnight at 4°C. Cell cycle analysis-The fixed cells were pelleted by centrifugation and resuspended in 50 µl 0.85% NaCl. To denature DNA, 2 ml of 2M HCl was added and cells incubated for 20 minutes at room temperature. Cells were then pelleted by centrifugation, resuspended in 1ml of 0.1 M sodium borate (pH 8.6) and washed 1 time with PBS. Cells were then resuspended in 10 µl of FITC conjugated anti-BrdU IgG (Pharmigen, San Diego, CA) and incubated for 1 h at room temperature with mixing. Five hundred microliters of propidium iodide (PI) staining solution (50 µg/ml PI, 50 µg/ml RNAse A) was added and the number of BrdU positive cells in S, G2/M, and G1 was determined by FACS analysis using a Coulter EPICS XL flow cytometer (Miami, FL) at an excitation range of 488 nm (argon laser), and a 525 BP filter for FITC and 620 BP for propidium iodide.
3
by guest on July 2, 2016http://w
ww
.jbc.org/D
ownloaded from

Monitoring cell cycle progression by manually counting mitotic cells-Cells were synchronized in G1/S with thymidine as described above, except BrdU was not added to the releasing medium. Where indicated nocodazole was added 5 h after release to a final concentration of 0.4 µg/ml to induce the mitotic spindle checkpoint. At the indicated times cells were harvested by trypsinization, washed with PBS, spotted onto a microscope slide, and fixed by incubating with ethanol/acetic acid (19:1) for 20 minutes at room temperature. Cells were then incubated for 5 minutes with PBS containing 0.2% Triton X-100, PBS, and then PBS containing 2 µg/ml 4’, 6-diamidino-2-phenylindole (DAPI). Slides were washed and mounted in Vectorshield (Vector Laboratories, Burlingame, CA). The number of mitotic cells (characterized by condensed chromosome structures observed in prophase through telophase) and non-mitotic cells was determined by manually counting using a fluorescent microscope from a sufficient number of randomly selected fields to obtain at least 5,000 cells. Western blotting-Cells were synchronized in G1/S with thymidine as described above. At the indicated times cells were harvested, washed with cold PBS, resuspended in buffer A (20 mM Tris-HCl pH 7.4, 135 mM NaCl, 2 mM EDTA, 1% Triton X-100, 25 mM β-glycerophosphate, 10% glycerol, 1 mM sodium orthovanadate, sodium fluoride, 1 mM phenylmethylsulfonyl fluoride, 10 µg/ml pepstain, 10 µg/ml aprotinin, 10 µg/ml E-64) and incubated for 20 minutes on ice. Cells were lysed by repeatedly passing through a G16 needle. Lysates were centrifuged, the supernatant collected, and protein concentrations determined using Coomassie Protein Assay as directed by manufacturer (Pierce Biotechnology, Inc., Rockford, IL). Nuclear fractionation-Cells were collected, washed twice with PBS and resuspended in buffer B (10 mM Tris pH7.4, 2 mM MgCl2, 1 mM EDTA, 1 mM sodium orthovanadate, 10 mM sodium fluoride, 1 mM phenylmethylsulfonyl fluoride, 10 µg/ml pepstatin, 10 µg/ml aprotinin, 10 µg/ml E-64). Cells were incubated for 15 minutes on ice, passed through a G16 needle, and centrifuged at 5,000 x g for 8 minutes at 4°C. The supernatant was collected (cytosolic fraction) and
protein concentrations determined using Coomassie. Pellets were washed twice with buffer B, suspended in SDS-PAGE gel loading buffer (10% SDS, 25% glycerol, 0.1% bromophenol blue, 0.75 M Tris-HCl pH 8.8), incubated for 5 minutes at 95°C and loaded on to an SDS polyacrylamide gel. Immunoblotting was performed as previously described (36). Cyclin B1 kinase assay-Protein A/G plus agarose conjugate was incubated with anti-cyclin B1 for 2 hours at 4°C. The anti-cyclin B1/ A/G plus agarose solution was incubated overnight at 4°C with 400 µg of total cell lysate prepared in buffer A. The immunoprecipitate was washed 3 times with buffer A and then once with kinase buffer (25 mM HEPES pH 7.2, 25 mM β-glycerophosphate, 5 mM EGTA, 1 mM NaVO3, 1mM DTT, 10 mM NaF, 2 µg/µl of histone H1 (Calbiochem). The immunoprecipitate was resuspended in kinase buffer and 32PγATP (0.25 µCi) was added. The reaction mixture was incubated at 30°C for 30 minutes and the reaction stopped by addition of SDS-PAGE loading buffer at 98°C for 5 minutes. The agarose conjugate was removed by centrifugation and the supernatant loaded on to a SDS-PAGE gel. The gel was exposed to x-ray film and band intensity determined by densitometry using a Kodak image station.
Results HRASV12 expression accelerates transition through G2/M To examine the effects of activated RAS on progression through G2/M we used Ras-25 cells, PCCL3 cells with dox inducible-expression of HRASV12. Effects of HRASV12 on G2/M were determined by first synchronizing cells at the G1/S boundary by double thymidine treatment, and then inducing expression of HRASV12 by the addition of dox for 14 h prior to release. Cell cycle progression was then monitored as described in Material and Methods. PCCL3 cells expressing HRASV12 had an approximately 2 h delay in progression through S phase (Figure 1), but had an accelerated transition through G2/M as they entered the next G1 approximately 2 h sooner. Thus, expression of HRASV12 resulted in a net acceleration of
4
by guest on July 2, 2016http://w
ww
.jbc.org/D
ownloaded from

G2/M of approximately 4 h. The abnormal duration of G2/M is illustrated by the flattened G2/M peak seen between 8-12 h after release (Figure 1A, upper panels). Treatment of the parental line, rtTA (PCCL3 cells that only express the reverse tetracycline transactivator), with dox did not significantly affect the progression of cells through S or G2/M (Figure 1A, lower panels). Acute expression of HRASV12 did not affect the ability of cells to synchronize at the G1/S border or to release from the double thymidine block (data not shown), indicating that the differences were not due to effects of HRASV12 on cell synchronization. To confirm the accelerated transition through G2/M, cells were collected and mitotic cells quantified after release from the double thymidine block by staining with DAPI and visual counting of cells with condensed chromosomes (Figure 1C). The decreased number of cells in mitosis after RAS activation points to an asynchronous passage of cells through mitosis, consistent with an abbreviated G2 and/or less likely, of the mitotic phase (Figure 1C). A time course of cyclin B1 kinase activity in cells treated as described in Figure 1C demonstrates lower levels of kinase activity between 8-12 h after release in cells expressing HRASV12 (Figure 1D). The decrease in kinase activity corresponded closely with cyclin B1 immunoreactivity indicating no intrinsic effect of HRASV12 on kinase activity (Figure 1E). These results are consistent with a reduced number of cells in G2/M at any of the sampled time points, presumably due to a more rapid transit through these stages in cells expressing HRASV12. Acute HRASV12 expression promotes exit from the DNA damage and mitotic spindle checkpoints Next we determined whether inappropriate activation of HRAS resulted in abnormalities of either the DNA damage or mitotic spindle checkpoints, which could also contribute to the chromosomal abnormalities we observed after
acute expression of HRASV12. To determine the effects of HRASV12 expression on the G2 DNA damage checkpoint Ras-25 cells were synchronized and released as in Figure 1A, except that 4 h after release they were exposed to 15 Gy of ionizing radiation. Cells were harvested at the indicated times, and the cell cycle stage of the BrdU-positive cells determined by FACS analysis. As shown in Figure 2, irradiated cells that did not express HRASV12 remained in G2/M for approximately 4 h longer than unirradiated cells. In the presence of dox there was a more rapid entry of BrdU positive cells into the next G1 indicating that the irradiation-induced G2 arrest was partially overcome by expression of HRASV12 (Figure 2). We next explored the effects of HRASV12 expression on the mitotic spindle checkpoint. Nocodazole, a microtubule disruptor that arrests cells in metaphase, was added 5 h after release from the double thymidine block (a time point when most cells were in S phase) to activate the mitotic checkpoint. Cells were harvested at indicated times, smeared on glass slides, stained with DAPI, and mitotic cells counted manually. In the absence of dox, nocodazole induced an accumulation of Ras-25 cells in mitosis that peaked 15 hours after release and then declined slightly over the next 15 h. In cells expressing HRASV12, nocodazole induced a peak of mitotic cells at 13 hours, which rapidly declined over the next 10 hours (Figure 3A). Cyclin B1, which is rapidly degraded as cells exit mitosis, had reduced activity (not shown), and was expressed at lower levels and declined earlier in HRASV12 expressing cells in spite of the continued presence of nocodazole (Figure 3B). In parental rtTA cells, nocodazole produced an accumulation of cells in mitosis that peaked at 15 hours and did not decline for more than 30 h (data not shown). Compared to parental cells, Ras-25 cells without dox exited the nocodazole induced mitotic arrest slightly faster. This is possibly due to slight leakiness of HRASV12 in the absence of dox. The decline in the number of mitotic cells, and the decrease in cyclin B1 levels and kinase activity indicate that HRASV12 expression promoted bypass of the nocodazole block. However, cells did not undergo cytokinesis, but moved into the next G1 with a 4N DNA content (data not shown). To confirm that differences in cell viability were not responsible
5
by guest on July 2, 2016http://w
ww
.jbc.org/D
ownloaded from

for the HRASV12-induced changes, we confirmed that mitosis-arrested cells underwent cytokenesis after removal of nocodazole. Thus, cells expressing HRASV12 underwent cytokenesis and returned to a 2N DNA content within 2 h after removal of nocodazole, whereas in the absence of dox cells required approximately 3.5 hours to recover from mitotic arrest (Figure 3C). The ability of cells to undergo cytokenesis and return to a 2N DNA content confirms their viability and the faster recovery from the microtubule disruption seen in the HRASV12 expressing cells, which is consistent with decreased sensitivity to the nocodazole-induced checkpoint. Activated MEK and ERK increase in the nucleus in late S and G2/M Recent reports demonstrating that MEK and ERK are involved in the normal progression of cells through G2/M (26,27) suggest that inappropriate activation of this pathway may at least in part be responsible for the acceleration observed in cells expressing HRASV12. To further investigate the role of MEK/ERK in G2/M transition we monitored the phosphorylation status of MEK and ERK through the cell cycle. To do this Ras-25 cells were synchronized by double thymidine as described in Figure 1C,D and cells harvested at the indicated times after release. Cell lysates were prepared from nuclear and cytosolic fractions, and Western blotted for total and phosphorylated MEK and ERK. In the presence of dox there was a marked increase in nuclear phospho-ERK and phospho-MEK levels beginning 6 h and peaking at 8 h after release (corresponding to late S and early G2). In the absence of dox there was a modest increase in nuclear phospho-ERK and phospho-MEK levels which began at 6 h (Figure 4A,B). The increase in nuclear phospho-ERK and phospho-MEK levels in both the absence and presence of dox was associated with higher total MEK and ERK. This suggests that the increase in phospho-ERK and phospho-MEK in late S and early G2 is likely at least in part due to increased nuclear import or retention of MEK and ERK. Cytosolic phospho-ERK and
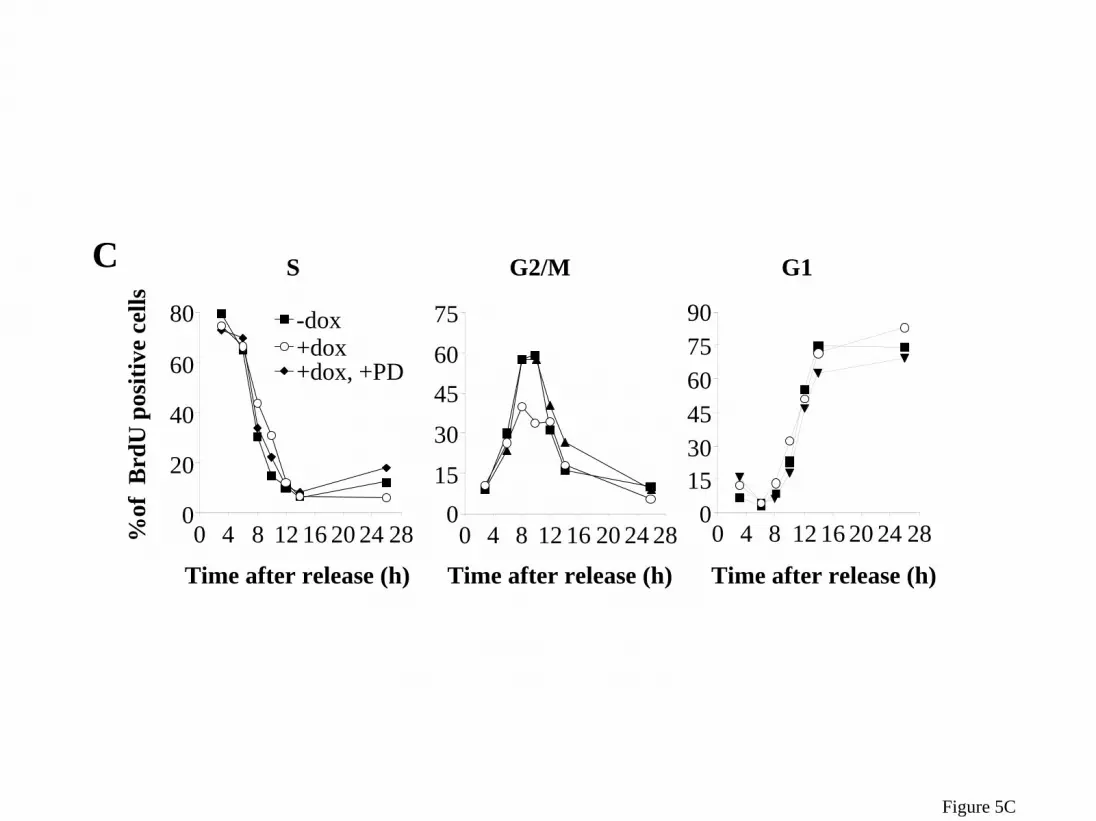
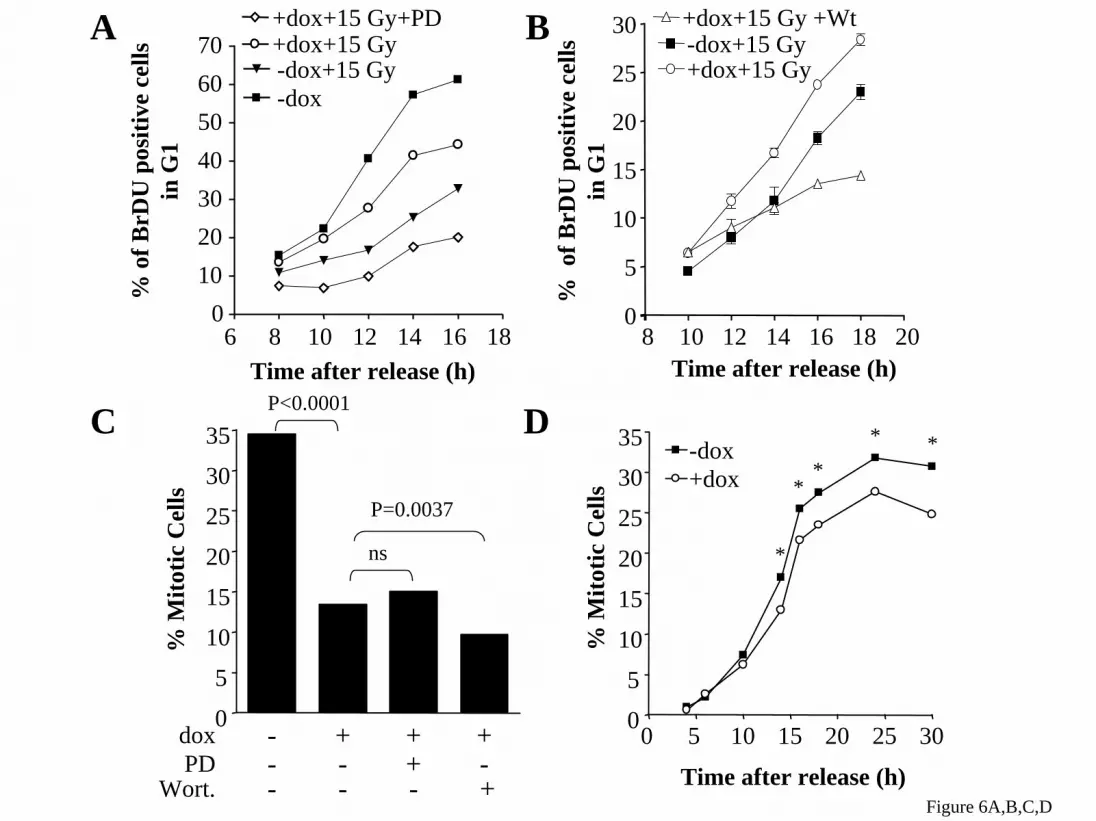
phospho-MEK were also markedly increased in HRASV12 expressing cells, which can not be accounted for by changes in total cytosolic MEK or ERK (Figure 4C,D). Inhibition of the MAPK pathway retards G2/M progression and prevents G2/M acceleration by acute expression of HRASV12 in PCCL3 cells To explore the effects of MAPK on cell cycle progression, we treated rtTA cells with 35 or 70 µM PD98059 beginning at release from a double thymidine block. Treatment with the MEK inhibitor resulted in a modest dose-dependent delay in exit from S phase, and a significant prolongation of G2/M, consistent with a requirement for MEK/ERK in the normal transit through the latter stage of the cell cycle (Figure 5A,B), as reported by others in fibroblasts (25,27). The effects of PD98059 on G2/M are reciprocal to those seen after acute expression of HRASV12. Indeed, pretreatment of Ras-25 cells with PD98059 reversed entirely the HRASV12-mediated acceleration of G2/M (Figure 5C,D). Activation of MEK and PI3K are required for HRASV12-induced bypass of the G2 DNA damage, but not the mitotic spindle checkpoint Treatment of cells with PD98059 (added with dox 14 h prior to release) slowed the entry of irradiated cells expressing HRASV12 into the next G1 (Figure 6A). Wortmannin had similar effects (Figure 6B). We confirmed that the concentration of wortmannin used (300 nM) blunted RAS-induced activation of PI3K without affecting radiation-induced phosphorylation of p53 at Ser-15 by ATM (not shown). These data suggest that activation of MEK and PI3K are required for HRASV12-induced bypass of the G2 DNA damage checkpoint. To determine if the HRASV12-induced bypass of the nocodazole-induced mitotic phase arrest was also dependent on activation of MEK and/or PI3K, Ras-25 cells were treated as in Figure 3A except that PD98059 or wortmannin were added 14 h prior to release, and the number of mitotic cells counted at the indicated times. Addition of PD98059 or wortmannin did not restore the mitotic spindle checkpoint in the HRASV12 expressing cells (Figure 6B,C), suggesting that neither of these pathways alone is required for the effect. However, activation of MEK/ERK pathways by doxycycline-induced expression of BRAFV600E allowed cells to partially
6
by guest on July 2, 2016http://w
ww
.jbc.org/D
ownloaded from

escape the nocodazole-induced mitotic phase arrest (Figure 6D). Activation of single RAS effector pathways is not sufficient to promote bypass of the G2 DNA damage checkpoint To determine if HRASV12-induced activation of the MEK/ERK pathway was sufficient to promote bypass of the G2 DNA damage check, we developed PCCL3 cells with dox-inducible expression of HRASV12,S35, an effector mutant that preferentially activates the MEK/ERK pathway. We also tested the role of the RAL-GDS and the PI3K pathways using PCCL3 cells with dox-inducible expression of HRASV12,G37 and HRASV12,C40, respectively. Western blots probed for phospho-AKT, phospho-ERK, or HRAS demonstrated similar expression of HRAS and selective activation of MEK/ERK pathway by HRASV12,S35, and of the PI3K-AKT pathway by HRASV12,C40 (Figure 7A). As shown in Figure 7B expression of HRASV12,S35, HRASV12,G37, or HRASV12,C40 was not sufficient to promote bypass of the G2 DNA damage check point. This suggests that activation of MEK/ERK in combination with an additional RAS effector pathway may be responsible for bypass of the G2 DNA damage checkpoint seen in HRASV12 expressing cells. Alternatively, the slightly lower activation of MEK/ERK seen after HRASV12,S35-expression as compared to HRASV12 may not have been sufficient to disrupt the checkpoint. To address this possibility we used the previously described PC-BRAFV600E-6 cell line (PCCL3 cells with dox-inducible expression of BRAFV600E (34), a constitutively active mutant of BRAF) that have a slightly greater dox-induced activation of MEK/ERK than Ras-25 cells (data not shown). Expression of BRAFV600E did not promote bypass of the G2 DNA damage checkpoint confirming that activation of the MEK/ERK pathway is not sufficient to disrupt this checkpoint.
Discussion Mutations that constitutively activate RAS proteins are characteristic of many human cancers (reviewed in (37-39)), including those arising from thyroid follicular cells (4-9). The aberrant activation of RAS proteins has been implicated in facilitating virtually all aspects of the malignant phenotype of the cancer cell, including cellular proliferation, invasion and metastasis, inhibition of
apoptosis, and angiogenesis (reviewed in (40-43)). Additionally, RAS oncoproteins may also contribute to oncogenesis through a decrease in genetic stability (16-23). While much is known regarding the mechanisms by which aberrant RAS activation promotes uncontrolled proliferation by promoting entry in S-phase and increasing cell survival, less is known regarding how RAS promotes genetic instability. We previously demonstrated that acute expression of HRASV12 increases the frequency of chromosome misalignment, multiple spindle formation, centrosome amplification, and generation of micronuclei within the first few cell cycles after activation in rat thyroid PCCL3 cells (31). As these changes suggest problems in the G2/M phase of the cell cycle, here we manipulated the timing of expression of HRASV12 at defined points in the cell cycle to explore the effects of activated RAS on progression through G2/M. This demonstrated that expression of activated HRAS accelerates the G2/M phase of the cell cycle by approximately 4 hours. Furthermore, the HRASV12-induced acceleration was blunted by inhibition of MEK, indicating that MEK activity is required for this effect. A role for the MAPK pathway in regulating cell cycle progression during G2 and M is supported by a number of recent studies (25,27-29,44-47). Proteins in the MAPK pathway may regulate organelle disassembly and mitotic structures during G2/M phase transitions. For example, active ERK localization to the mitotic kinetochore may regulate proteins involved in chromosome segregation during metaphase to anaphase transition (28,29). A predisposition to gain or loose whole chromosomes in colorectal cell lines has been linked to abnormalities in the mitotic checkpoint (48), which is activated by the presence of unattached kinetochores (49). Given the key role for RAS in mitotic spindle assembly in fission yeast (50) and the fact that the majority of spindle checkpoint genes are highly conserved between higher and lower eukaryotes it not surprising that expression of oncogenic RAS relaxes the mitotic spindle checkpoint. To our knowledge this is the first report demonstrating that expression of HRASV12 promotes bypass of this important checkpoint. In Schizosaccharomyces pombe,
7
by guest on July 2, 2016http://w
ww
.jbc.org/D
ownloaded from

RAS1, a RAS homolog, signals through two major pathways that appear to regulate distinct functions: mating (through Byr2, functionally analogous to RAF) and cytoskeletal organization (through SCD1, a putative guanine nucleotide exchange factor activating CDC42, functionally analogous to a RHO-like GTPase). SCD1 in turn forms a complex with MOE1 and localizes to the spindle during mitosis (51). Double mutants (MOE1 with RAS1 or SCD1) accumulate in early mitosis and have severe spindle assembly defects, suggesting that the RAS1-SCD1 pathway is required for mitotic functions. Interestingly, SCD1 overexpression also results in spindle damage (52). These data point to potential mechanisms by which constitutively activated RAS could disrupt chromosomal stability. The response to DNA damage is an essential surveillance system to maintain genomic integrity. The ATM kinase is a central transducer of this response. Lack of a functional ATM kinase, such as occurs in the ataxia telangiectasia syndrome, results in chromosomal instability and a predisposition to cancer. Recently, DNA damage has been shown to activate ERK through events downstream of ATM (53,54). Moreover, ERK appears to be required for appropriate function of the DNA damage checkpoint, as cells are unable to recover in a timely fashion from ionizing radiation-induced G2 arrest when MEK2 activation is blocked (25). The apparent role of MEKs and ERK in the G2 checkpoint suggest that this checkpoint could be affected by expression of constitutively activate RAS. Indeed, the irradiation-induced arrest of PCCL3 thyroid cells in G2 was relaxed by expression HRASV12. In contrast to HRASV12 induced bypass of the mitotic spindle checkpoint, bypass of the G2 DNA damage checkpoint was dependent on activation of MEK/ERK pathway, as the checkpoint was restored with addition of the MEK inhibitor PD98059. However, we cannot fully rule out the possibility that in the presence of nocodazole the MEK inhibitor blocks cells in G2, and that this is responsible for the fewer mitotic cells, rather a bypass of the checkpoint. However, activation of the MEK/ERK pathway was not sufficient for this effect. Thus G2 arrest was not dampened by dox-inducible expression of HRASV12,C35, BRAFV600E, or MEK1Glu-217/Glu221. These results, together with the lack of G2 checkpoint bypass following
conditional expression of the RAS effector mutants HRASV12,C37 and HRASV12,C40, which selectively activate RAL-GDS and PI3K, respectively, indicates that signaling through any single RAS effector is not sufficient to compromise the G2 DNA damage checkpoint. In this respect, our data differs from that of Agapova et al (24). Whereas they also noted that expression of mutant HRAS in human MDAH041 fibroblasts and Saos-2 osteosarcoma cells attenuated G2 arrest following DNA damage, they observed that signaling via RAL-GDS could account for this effect. The explanation for the discrepancy is not clear. However, there are experimental differences that could be responsible: 1) The cell lines used by Agapova et al are p53-deficient, whereas PCCL3 cells are not (31). 2) The two studies used different DNA damaging stimuli (radiation vs an alkylating agent or a topoisomerase inhibitor), which could signal G2 arrest through different pathways. Indeed, ATM and ATR, two PI3K-like kinases that initiate the signaling process in response to DNA damage, are differentially activated by different types of DNA damaging agents (55,56). 3) Finally, there may be tissue and/or species difference in the effector pathways required. Since an effector mutant that selectively activates more than one RAS effector pathway is not available it was not possible to investigate further which RAS effectors are sufficient to promote bypass of the G2 DNA damage checkpoint. However, the observation that addition of either wortmannin or PD98059 prevented the HRASV12-induced bypass of the G2 DNA damage checkpoint suggest that both PI3K and the MAPK are required. A role for PI3K pathway is supported by the observation that expression of constitutively active AKT in RAT1a cells alleviates the G2 arrest induced by gamma irradiation (57). The observation that AKT can phosphorylate CHK1 at S280 (a downstream effector of ATM and ATR required for the activation of the G2 DNA damage checkpoint) and inactivate it, provides a plausible mechanism by which RAS-PI3K-induced AKT activation may promote bypass of the DNA damage checkpoint by inactivating CHK1. In summary, we demonstrate that acute expression of HRASV12 promotes an acceleration of G2/M phase that is associated with a bypass of
8
by guest on July 2, 2016http://w
ww
.jbc.org/D
ownloaded from

the G2 DNA damage and mitotic spindle checkpoints. These RAS-induced defects could promote genetic instability through both MEK/ERK-dependent and independent pathways, by at least in part compromising critical checkpoints required to maintain genomic integrity. Recently, Bartkova et al (58) and Gorgoulis et al (59) showed activation of DNA damage checkpoint genes in early precursor lesions of cancer, but not in normal tissue.
Activation of the checkpoint genes would be predicted to block tumor progression and prevent genetic instability, whereas inactivation of DNA damage checkpoint genes (i.e. p53, CHK2, ATM) would disable this protective mechanism. Conceivably early oncogenic events, such as RAS mutations, may under some conditions also promote functional defects in the DNA damage and mitotic checkpoints and thus favor tumor progression.
REFERENCES
1. Namba, H., Matsuo, K., and Fagin, J. A. (1990) J. Clin. Invest. 86, 120-125
2. Aeschimann, S., Kopp, P. A., Kimura, E. T., Zbaeren, J., Tobler, A., Fey, M. F., and Studer, H. (1993) J. Clin. Endocrinol. Metab. 77, 846-851
3. Fey, M. F., Peter, H. J., Hinds, H. L., Zimmermann, A., Liechti-Gallati, S., Gerber, H., Studer, H., and Tobler, A. (1992) Journal of Clinical Investigation 89, 1438-1444
4. Lemoine, N. R., Mayall, E. S., Wyllie, F. S., Farr, C. J., Hughes, D., Padua, R. A., Thurston, V., Williams, E. D., and Wynford-Thomas, D. (1988) Cancer Res. 48, 4459-4463
5. Lemoine, N. R., Mayall, E. S., Wyllie, F. S., Williams, E. D., Goyns, M., Stringer, B., and Wynford-Thomas, D. (1989) Oncogene 4, 159-164
6. Namba, H., Rubin, S. A., and Fagin, J. A. (1990) Mol. Endocrinol. 4, 1474-1479
7. Esapa, C. T., Johnson, S. J., Kendall-Taylor, P., Lennard, T. W., and Harris, P. E. (1999) Clin. Endocrinol. 50, 529-535
8. Suarez, H. G., du Villard, J. A., Severino, M., Caillou, B., Schlumberger, M., Tubiana, M., Parmentier, C., and Monier, R. (1990) Oncogene 5, 565-570
9. Karga, H., Lee, J. K., Vickery, A. L., Jr., Thor, A., Gaz, R. D., and Jameson, J. L. (1991) J. Clin. Endocrinol. Metab 73, 832-836
10. Norppa, H. and Falck, G. C. (2003) Mutagenesis 18, 221-233
11. Oller, A. R., Rastogi, P., Morgenthaler, S., and Thilly, W. G. (1989) Mutat. Res. 216, 149-161
12. Thacker, J. (1985) Mutat. Res. 150, 431-442
13. Loeb, L. A. (1998) Adv. Cancer Res. 72, 25-56
14. Loeb, L. A. (1991) Cancer Res. 51, 3075-3079
15. Finney, R. E. and Bishop, J. M. (1993) Science 260, 1524-1527
16. Denko, N., Stringer, J., Wani, M., and Stambrook, P. (1995) Somat. Cell Mol. Genet. 21, 241-253
9
by guest on July 2, 2016http://w
ww
.jbc.org/D
ownloaded from

17. Denko, N. C., Giaccia, A. J., Stringer, J. R., and Stambrook, P. J. (1994) Proc. Natl. Acad. Sci. U. S. A. 91, 5124-5128
18. Saavedra, H. I., Fukasawa, K., Conn, C. W., and Stambrook, P. J. (1999) J. Biol. Chem. 274, 38083-38090
19. Wani, M. A., Xu, X., and Stambrook, P. J. (1994) Cancer Res. 54, 2504-2508
20. Agapova, L. S., Ivanov, A. V., Sablina, A. A., Kopnin, P. B., Sokova, O. I., Chumakov, P. M., and Kopnin, B. P. (1999) Oncogene 18, 3135-3142
21. Ichikawa, T., Kyprianou, N., and Isaacs, J. T. (1990) Cancer Res. 50, 6349-6357
22. Ichikawa, T., Schalken, J. A., Ichikawa, Y., Steinberg, G. D., and Isaacs, J. T. (1991) Prostate 18, 163-172
23. de Vries, J. E., Kornips, F. H., Marx, P., Bosman, F. T., Geraedts, J. P., and ten Kate, J. (1993) Cancer Genet. Cytogenet. 67, 35-43
24. Agapova, L. S., Volodina, J. L., Chumakov, P. M., and Kopnin, B. P. (2004) J. Biol. Chem. 279, 36382-36389
25. Abbott, D. W. and Holt, J. T. (1999) J. Biol. Chem. 274, 2732-2742
26. Lavoie, J. N., L'Allemain, G., Brunet, A., Muller, R., and Pouyssegur, J. (1996) J. Biol. Chem. 271, 20608-20616
27. Wright, J. H., Munar, E., Jameson, D. R., Andreassen, P. R., Margolis, R. L., Seger, R., and Krebs, E. G. (1999) Proc. Natl. Acad. Sci. U. S. A 96, 11335-11340
28. Shapiro, P. S., Vaisberg, E., Hunt, A. J., Tolwinski, N. S., Whalen, A. M., McIntosh, J. R., and Ahn, N. G. (1998) J. Cell Biol. 142, 1533-1545
29. Zecevic, M., Catling, A. D., Eblen, S. T., Renzi, L., Hittle, J. C., Yen, T. J., Gorbsky, G. J., and Weber, M. J. (1998) J. Cell Biol. 142, 1547-1558
30. Gotoh, Y., Nishida, E., Matsuda, S., Shiina, N., Kosako, H., Shiokawa, K., Akiyama, T., Ohta, K., and Sakai, H. (1991) Nature 349, 251-254
31. Saavedra, H. I., Knauf, J. A., Shirokawa, J. M., Wang, J., Ouyang, B., Elisei, R., Stambrook, P. J., and Fagin, J. A. (2000) Oncogene 19, 3948-3954
32. Wang, J., Knauf, J. A., Basu, S., Puxeddu, E., Kuroda, H., Santoro, M., Fusco, A., and Fagin, J. A. (2003) Mol. Endocrinol. 17, 1425-1436
33. Shirokawa, J. M., Elisei, R., Knauf, J. A., Hara, T., Wang, J., Saavedra, H. I., and Fagin, J. A. (2000) Mol. Endocrinol. 14, 1725-1738
34. Mitsutake, N., Knauf, J. A., Mitsutake, S., Mesa, C., Jr., Zhang, L., and Fagin, J. A. (2005) Cancer Res. 65, 2465-2473
10
by guest on July 2, 2016http://w
ww
.jbc.org/D
ownloaded from

35. Rodriguez-Viciana, P., Warne, P. H., Khwaja, A., Marte, B. M., Pappin, D., Das, P., Waterfield, M. D., Ridley, A., and Downward, J. (1997) Cell 89, 457-467
36. Knauf, J. A., Elisei, R., Mochly-Rosen, D., Liron, T., Chen, X. N., Gonsky, R., Korenberg, J. R., and Fagin, J. A. (1999) J. Biol. Chem. 274, 23414-23425
37. Barbacid, M. (1990) Eur. J. Clin. Invest 20, 225-235
38. Sebolt-Leopold, J. S. (2004) Curr. Pharm. Des 10, 1907-1914
39. Bos, J. L. (1989) Cancer Res. 49, 4682-4689
40. Campbell, P. M. and Der, C. J. (2004) Semin. Cancer Biol. 14, 105-114
41. Malumbres, M. and Barbacid, M. (2001) Nat. Rev. Cancer 1, 222-231
42. Kranenburg, O., Gebbink, M. F., and Voest, E. E. (2004) Biochim. Biophys. Acta 1654, 23-37
43. Downward, J. (1998) Curr. Opin. Genet. Dev. 8, 49-54
44. Cha, H. and Shapiro, P. (2001) J. Cell Biol. 153, 1355-1367
45. Roberts, E. C., Shapiro, P. S., Nahreini, T. S., Pages, G., Pouyssegur, J., and Ahn, N. G. (2002) Mol. Cell Biol. 22, 7226-7241
46. Jesch, S. A., Lewis, T. S., Ahn, N. G., and Linstedt, A. D. (2001) Mol. Biol. Cell 12, 1811-1817
47. Liu, X., Yan, S., Zhou, T., Terada, Y., and Erikson, R. L. (2004) Oncogene 23, 763-776
48. Cahill, D. P., Lengauer, C., Yu, J., Riggins, G. J., Willson, J. K., Markowitz, S. D., Kinzler, K. W., and Vogelstein, B. (1998) Nature 392, 300-303
49. Taylor, S. S. and McKeon, F. (1997) Cell 89, 727-735
50. Segal, M. and Clarke, D. J. (2001) Bioessays 23, 307-310
51. Chen, C. R., Li, Y. C., Chen, J., Hou, M. C., Papadaki, P., and Chang, E. C. (1999) Proc. Natl. Acad. Sci. U. S. A 96, 517-522
52. Li, Y. C., Chen, C. R., and Chang, E. C. (2000) Genetics 156, 995-1004
53. Panta, G. R., Kaur, S., Cavin, L. G., Cortes, M. L., Mercurio, F., Lothstein, L., Sweatman, T. W., Israel, M., and Arsura, M. (2004) Mol. Cell Biol. 24, 1823-1835
54. Tang, D., Wu, D., Hirao, A., Lahti, J. M., Liu, L., Mazza, B., Kidd, V. J., Mak, T. W., and Ingram, A. J. (2002) J. Biol. Chem. 277, 12710-12717
55. Shiloh, Y. (2003) Nat. Rev. Cancer 3, 155-168
56. Shiloh, Y. and Kastan, M. B. (2001) Adv. Cancer Res. 83:209-54., 209-254
11
by guest on July 2, 2016http://w
ww
.jbc.org/D
ownloaded from

57. Kandel, E. S., Skeen, J., Majewski, N., Di, C. A., Pandolfi, P. P., Feliciano, C. S., Gartel, A., and Hay, N. (2002) Mol. Cell Biol. 22, 7831-7841
58. Bartkova, J., Horejsi, Z., Koed, K., Kramer, A., Tort, F., Zieger, K., Guldberg, P., Sehested, M., Nesland, J. M., Lukas, C., Orntoft, T., Lukas, J., and Bartek, J. (2005) Nature 434, 864-870
59. Gorgoulis, V. G., Vassiliou, L. V., Karakaidos, P., Zacharatos, P., Kotsinas, A., Liloglou, T., Venere, M., Ditullio, R. A., Jr., Kastrinakis, N. G., Levy, B., Kletsas, D., Yoneta, A., Herlyn, M., Kittas, C., and Halazonetis, T. D. (2005) Nature 434, 907-913
FOOTNOTES
This work was supported in part by NIH Grant CA72597. The abbreviations used are: MAPK, mitogen-activated protein kinase; MEK, mitogen-activated protein/extracellular signal-regulated kinase kinase; GEF, guanine-nucletide exchange factor; RET, rearranged by transfection; ERK, extracellular signal-regulated kinase; PI3K, phosphatidylinositol 3-kinase; BrdU, bromodeoxyuridine; DAPI, 4’, 6-diamidino-2-phenylindole; TSH, thyrotropin; dox, doxycycline; PI, propidium iodide; ATM (ataxia telangiectasia mutated); ATR (ATM- and RAD3-related).
FIGURE LEGENDS Figure 1: RAS accelerates traversion of PCCL3 cells through G2/M. A) Ras-25 and rtTA cells were synchronized in G1/S with a double thymidine block. Cells were treated with (full squares) or without (open circles) 1 µg/ml dox 14 h prior to release in the continued presence of thymidine. Following release cells were pulsed with BrdU for 2 h. At the indicated times cells were harvested, fixed, and stained with FITC-conjugated anti-BrdU IgG and propidium iodide (PI). FACS analysis was used to determine the number of BrdU positive cells at each stage of the cell cycle. B) Representative FACS histogram of BrdU positive cells. Similar results were obtained in 4 additional independent experiments. C) Cells were synchronized as above except BrdU was not added to releasing medium. At the indicated times after release cells were harvested, spotted onto a microscope slide, fixed, and stained with DAPI. Cover slips were mounted in vectorshield antifade and mitotic and non-mitotic cells counted manually using fluorescence microscope. Data represent the mean of a single experiment performed in duplicate. Similar results were obtained in 2 additional experiments. D) Cells were harvested at the indicated times after release from the double thymidine block, whole cell lysates prepared and immunoprecipitated cyclin B1 used in in vitro kinase assays using MBP as a substrate. E) At the indicated times after release from double thymidine block cells were harvested, whole cell lysates prepared and used for Western blotting with anti-cyclin B1 IgG. Similar results were obtained in 2 additional experiments. Figure 2: Acute RAS activation promotes exit from the DNA damage checkpoint. Ras-25 cells were synchronized in G1/S and released as described in Figure 1A. Four hours after release, cells were exposed to 15 Gy of X-ray radiation (Faxitron cabinet irradiator) and maintained in culture until harvested for FACS analysis of BRDU positive cells at the indicated times. A) In the absence of dox, radiated cells were delayed in G2. Expression of HRASV12 (+dox) resulted in more rapid exit from the G2 block (see 14 h FACS profile). B) Impact of oncogenic RAS expression on the percentage of BRDU positive cells entering the next G1 following radiation-induced DNA damage. A greater number of irradiated cells entered G1 compared to those not expressing the oncoprotein. Data points represent the mean of a single experiment performed in duplicate. Similar results were obtained in 2 additional experiments. Figure 3: RAS accelerates passage of PCCL3 cells through the mitotic spindle checkpoint. Ras-25 cells were synchronized in G1/S and released as described in Figure 1C. Five hours after release nocodazole was added to a final concentration 0.4 µg/ml. A) At the indicated times after release, cells were harvested, spotted onto a microscope slide, fixed, and stained with DAPI. Cover slips were mounted in vectorshield antifade and mitotic cells were counted manually by fluorescence microscopy. Data
12
by guest on July 2, 2016http://w
ww
.jbc.org/D
ownloaded from

represent the mean of a single experiment performed in duplicate. Similar results were obtained in 2 additional independent experiments. B) Cyclin B1 Western blot of total cell lysates prepared from cells harvested at the indicated times after release. C) Ras-25 cells were treated as above, except that 5 h after release nocodazole was added to a final concentration of 0.4 µg/ml. Then 14 h after release nocodazole was removed by washing cells twice with PBS, followed by addition of fresh H4 medium with or without 1.0 µg/ml dox until harvested for FACS analysis. Data shows that the cells are viable as they are able to undergo cytokenesis (4N to 2N DNA content). Moreover, expression of HRASV12 promoted a more rapid recovery from the nocodazole block, as demonstrated by their accelerated entry into the next G1. Figure 4: MEK and ERK phosphorylation during late S and G2/M. Ras-25 cells were synchronized in G1/S as described in Figure 1C. Cells were harvested at the indicated times after release and fractionated into nuclear (A) and cytosolic fractions (B). The corresponding extracts were subjected to SDS-PAGE, transferred to nitrocellulose and membranes probed with the indicated antibodies. Similar results were seen in 3 independent experiments. C&D) Data points represent the average percent change compared to 8 h +dox in the indicated protein in the nuclear (C) or cytosolic (D) fractions after normalization for HDAC1 or GAPDH, respectively. Data correspond to 3 independent experiments. Asterisks above or below the line correspond to +dox and –dox conditions, respectively. * p<0.05 vs the corresponding time 0. Figure 5: PD98059 slows progress of PCCL3 cells through G2/M. Ras-25 and rtTA cells were synchronized in G1/S and released as described in Figure 1A, except that where indicated 35 or 70 µM PD98059 was added to releasing medium. At the indicated times cells were harvested, fixed, and stained with FITC-conjugated anti-BrdU IgG and PI. FACS analysis was used to determine the number of BrdU positive cells at each stage of the cell cycle. A) FACS analysis of BrdU-labeled rtTA cells shows that PD98059 evoked a concentration-dependent prolongation of G2/M and delayed exit from mitosis. B) Corresponding FACS histograms from experiment shown in 4A. C) FACS analysis of BrdU-labeled Ras-25 cells shows 70 µM PD98059 inhibition of RAS-induced acceleration through G2/M. D) Corresponding FACS histogram from experiment shown in 4C. Similar results were obtained in an additional independent experiment. Figure 6: Inhibition of MEK or PI3K blocks HRASV12-induced bypass of the G2 DNA damage, but not of the mitotic spindle checkpoint. A) Ras-25 cells were synchronized in G1/S and released as described in Figure 1A, except that where indicated 70 µM PD98059 was added 14 h prior to release. Four hours after release cells were exposed to 15 Gy of γ-irradiation. At the indicated times cells were harvested, fixed, and stained with FITC-conjugated anti-BrdU IgG and propidium iodide (PI). FACS analysis was used to determine the number of BrdU positive cells in G1. Data represent the mean of a single experiment performed in triplicate. Similar results were obtained in a second experiment. B) Ras-25 cells were synchronized in G1/S and released as described in Figure 1A, except that where indicated 300 nM wortmannin was added 14 h prior to release. Four hours after release cells were exposed to 10 Gy of γ-irradiation. At the indicated times cells were harvested, fixed, and stained with FITC-conjugated anti-BrdU IgG and propidium iodide (PI). FACS analysis was used to determine the number of BrdU positive cells in G1. Data represent the mean of a single experiment performed in triplicate. Similar results were obtained in a second experiment. C&D) Ras-25 cells were synchronized in G1/S and released as described in Figure 1C, except that where indicated 70 µM PD98059 (C) or 300 nM wortmannin (D) was added. Five hours after release nocodazole was added and cells were harvested 16 hours after release. Cells were fixed, stained with DAPI, and mitotic cells counted manually as described in Figure 1C. PD98059 did not affect the ability of HRASV12 to overcome the nocodazole-induced block. . Similar results were obtained in a second independent experiment.
13
by guest on July 2, 2016http://w
ww
.jbc.org/D
ownloaded from

Figure 7: Activation of a single RAS effector pathway is not sufficient to promote bypass of the G2 DNA damage checkpoint. A) Western blots of protein lysates from the indicated cells incubated with 1.0 µg/ml dox for 17 or 25 h. Similar results were obtained in a second independent experiment. B) The indicated cell lines were synchronized in G1/S and released as described in Figure 1A. Four hours after release cells were exposed to 10 Gy of γ- irradiation. At the indicated times cells were harvested, fixed, and stained with FITC-conjugated anti-BrdU IgG and PI. FACS analysis was used to determine the number of BrdU positive cells in G1. Data represent the mean of a single experiment performed in triplicate. Similar results were obtained in at least 2 additional independent experiments.
14
by guest on July 2, 2016http://w
ww
.jbc.org/D
ownloaded from

AG2/M G1S
0 4 8 12 16 20 240
20
40
60
80-dox+dox
0 4 8 12 16 20 240
20
40
60
80
0 4 8 12 16 20 240
20
40
60
80
% o
fBrd
Upo
sitiv
eR
as-2
5 ce
lls
0 4 8 12 16 20 240
153045607590
0 4 8 12 16 20 240
20
40
60
80
100
0 4 8 12 16 20 240
15
30
45
60
75-dox+dox
% o
f B
rdU
posi
tive
rtT
A c
ells
Time after release (h) Time after release (h) Time after release (h)
Figure 1A
by guest on July 2, 2016 http://www.jbc.org/ Downloaded from

3 h
6 h
8 h
10 h
12 h
14 h
Tim
e af
ter
rele
ase
(h)
-dox +dox B
Figure 1B
by guest on July 2, 2016 http://www.jbc.org/ Downloaded from

C
6 8 10 12 140.0
0.5
1.0
1.5
2.0
2.5 -dox+dox
Time after release (h)
% M
itotic
cel
ls
0 2 4 5 6 7 9 12 14 16Time after release (h) -dox cyclin B1
0 2 4 6 8 10 12 140
100000
200000
300000
400000
500000 -dox+dox
Time after release (h)
Cyc
linB
1 ki
nase
act
ivity
D
E
cyclin B1+dox
Figure 1C,D,E
by guest on July 2, 2016 http://www.jbc.org/ Downloaded from

A Time after release (h)
-dox
-dox
+dox
Irra
diat
ed w
ith 1
5 G
y
Uni
rrad
iate
d8 10 12 14 16
Figure 2A
by guest on July 2, 2016 http://www.jbc.org/ Downloaded from

B
6 8 10 12 14 16 180
25
50
75-dox-dox+15 Gy+dox+15 Gy
Time after release (h)
% o
fBrd
Upo
sitiv
e ce
lls in
G1
Figure 2B
by guest on July 2, 2016 http://www.jbc.org/ Downloaded from

A
Time after release (h) 14 16 18 24
-dox
B
0 5 10 15 20 25 300
10
20
30
40
50
60 dox-dox+
Time after release (h)
% M
itotic
cel
ls
cyclin B1
cyclin B1+dox
Figure 3A,B
by guest on July 2, 2016 http://www.jbc.org/ Downloaded from

C
Time after removal of nocodazole (h) 0 1.0 1.5 2.0 2.5
-dox
+dox
Figure 3C
by guest on July 2, 2016 http://www.jbc.org/ Downloaded from

A+dox -dox
Time afterrelease (h) 0 6 8 10 12 14 16 0 6 8 10 12 14 16
pMEK
MEK1
MEK2
pERK
tERK
HDAC1
GAPDH
Figure 4A
by guest on July 2, 2016 http://www.jbc.org/ Downloaded from

B+dox -dox
Time afterrelease (h) 0 6 8 10 12 14 16 0 6 8 10 12 14 16
pMEK
tMEK1
tMEK 2
pERK
tERK
GAPDH
HDAC1
Figure 4B
by guest on July 2, 2016 http://www.jbc.org/ Downloaded from

C
0 2 4 6 8 10 12 140
20
40
60
80
100%
cha
nge
in p
ME
K*
*
**
*
0 2 4 6 8 10 12 140
20
40
60
80
100
% c
hang
e in
tME
K
0 2 4 6 8 1012140
20
40
60
80
100
% c
hang
e in
pE
RK
0 2 4 6 8 10 12 140
20406080
100120
%ch
ange
in tE
RK
Time after release (h)
*
**
*
*
* *
* ** *
*
-dox+dox
Time after release (h)
**
*
*
Figure 4C
by guest on July 2, 2016 http://www.jbc.org/ Downloaded from

D
0 2 4 6 8 10 12 140
20406080
100120140160
% c
hang
e in
tME
K
0 2 4 6 8 10 12 140
20
40
60
80
100
120%
cha
nge
inpM
EK
-dox+dox
0 2 4 6 8 10 12 140
25
50
75
100
125
150
Time after release (h)
% c
hang
e in
tER
K
0 2 4 6 8 10 12 140
30
60
90
120
150
180
Time after release (h)
% c
hang
e in
pER
K
Figure 4D
by guest on July 2, 2016 http://www.jbc.org/ Downloaded from

A S G2/M G1
% o
fBrd
Upo
sitiv
e ce
lls
0 4 8 12 16 20 24 280
102030405060 0 µM
35µM70µM
0 4 8 12 16 20 24 280
15
30
45
60
75
Time after release (h)0 4 8 12 16 20 24 28
0
20
40
60
80
Time after release (h) Time after release (h)
Figure 5A
by guest on July 2, 2016 http://www.jbc.org/ Downloaded from

B PD98059 (µM) 0 35 70
3
6
Tim
e af
ter
rele
ase
(h)
8
10
12
14
Figure 5B
by guest on July 2, 2016 http://www.jbc.org/ Downloaded from
C
%of
Brd
Upo
sitiv
e ce
lls
S G2/M G1
0 4 8 12 16 20 24 280
153045607590
0 4 8 12 16 20 24 280
20
40
60
80 -dox+dox+dox, +PD
0 4 8 12 16 20 24 280
15
30
4560
75
Time after release (h) Time after release (h) Time after release (h)
Figure 5C
by guest on July 2, 2016 http://www.jbc.org/ Downloaded from

D -dox +dox +dox, +PD
3
6T
ime
afte
r re
leas
e (h
)
8
10
12
14
Figure 5D
by guest on July 2, 2016 http://www.jbc.org/ Downloaded from
A
6 8 10 12 14 16 180
10203040506070
-dox-dox+15 Gy+dox+15 Gy+dox+15 Gy+PD
Time after release (h)
% o
fBrD
Upo
sitiv
e ce
lls
in G
1
8 10 12 14 16 18 200
5
10
15
20
25
30-dox+15 Gy+dox+15 Gy
+dox+15 Gy +Wt
Time after release (h)
% o
fBrD
Upo
sitiv
e ce
lls
in G
1
B
C
Figure 6A,B,C,D
0 5 10 15 20 25 300
5
10
15
20
25
30
35 -dox+dox
Time after release (h)
% M
itotic
Cel
ls
0
5
10
15
20
25
30
35
% M
itotic
Cel
ls
doxPD
Wort.
+--
++-
+-+
D
---
P=0.0037
ns
P<0.0001
*
**
**
by guest on July 2, 2016 http://www.jbc.org/ Downloaded from

A H-RASV12,G37 H-RASV12,C40 H-RASV12,S35
Time with dox (h) 0 17 25 0 17 25 0 17 25
pAKT
pMEK
H-RAS
tAKT
Figure 7A
by guest on July 2, 2016 http://www.jbc.org/ Downloaded from

-dox+dox
Ras-25
9 11 13 15 1705
1015202530
% o
f BrD
Upo
sitiv
e ce
lls in
G1
B PC-BRAFV600E-6
9 11 13 15 170
4
8
12
16
20MEK1-65
9 11 13 15 170
10
20
30
40
H-RASV12,G37
9 11 13 15 170
10
20
30
40
50
9 11 13 15 170
10
20
30
40
9 11 13 15 1705
101520253035
Time after release (h)Time after release (h)
H-RASV12,S35 H-RASV12,C40
Time after release (h)
% o
f BrD
Upo
sitiv
e ce
lls in
G1
-dox+dox
Figure 7B
by guest on July 2, 2016 http://www.jbc.org/ Downloaded from

James A. FaginJeffrey A. Knauf, Bin Ouyang, Erik S. Knudsen, Kenji Fukasawa, George Babcock and
the G2 DNA damage and mitotic spindle checkpointsOncogenic ras induces accelerated transition through G2/M and promotes defects in
published online November 29, 2005J. Biol. Chem.
10.1074/jbc.M511690200Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
http://www.jbc.org/content/early/2005/11/29/jbc.M511690200.citation.full.html#ref-list-1
This article cites 0 references, 0 of which can be accessed free at
by guest on July 2, 2016http://w
ww
.jbc.org/D
ownloaded from




























