Embed Size (px)
Citation preview

A
pAudr©
K
1
a[ptTtcasgailos
0d
Electrochimica Acta 52 (2007) 4581–4588
Oxygen reduction reaction on spontaneously and potentiodynamicallyformed Au/TiO2 composite surfaces
Danijela Jasin a, Andjela Abu-Rabi b, Slavko Mentus c,∗,1, Dusan Jovanovic b
a Technical School Zrenjanin, Zrenjanin, Serbiab Institute of Chemistry, Metallurgy and Technology, Center of Catalysis, Njegoseva 12, Belgrade, Serbia
c Belgrade University, Faculty of Physical Chemistry, Studentski trg 12, 11000 Belgrade, Serbia
Received 20 September 2006; received in revised form 20 December 2006; accepted 24 December 2006Available online 21 January 2007
bstract
From aqueous solutions, gold was deposited on freshly polished titanium surface either by displacement reaction or by potentiodynamicolarization. Both of these procedures resulted in the formation of Au/TiO2 composite layer over the titanium surface, although with quite differentu content. The electrochemical behaviour of these composite layers was examined in 0.1 M NaOH solution, both oxygen free and oxygen saturated,
sing rotating disc technique. In the potential region of oxygen reduction reaction, the Au/TiO2 layer obtained by potentiodynamic polarization,isplayed the voltammetric curve similar to that observed on (1 0 0) plane of Au monocrystal. The Koutecky–Levich analysis evidenced that 4e−oute of oxygen reduction dominates on both type of composite layers.2007 Elsevier Ltd. All rights reserved.
ectrod
uo[aosrneso[ncd
eywords: Electrocatalysis; Gold; Oxygen reduction reaction; Rotating disk el
. Introduction
Being very weak chemisorber, gold was considered tradition-lly as a poor catalyst, in terms of activated chemisorption theory1]. However, Haruta et al. published that Au nanoclusters dis-ersed on alumina support displays unexpectedly high activityoward CO oxidation [2,3] and reduction of nitrogen oxides [4].he discovery that nanodisperzed gold enables low tempera-
ure oxidation of CO is very intriguing from the aspect of fuelells. Namely, platinum catalysts used currently in low temper-ture fuel cells, suffer of continuous loss of activity, caused bytrong adsorption of CO present at impurity level in the fuelas. The mutual enhancement of catalytic activity of metal cat-lyst and supporting material was termed strong metal supportnteraction (SMSI) [2–4]. This effect was observed much ear-
ier in titania/platinum system [5], and thus, from the aspectf SMSI, titania was long ago considered as very promisingupporting material for catalysis and electrocatalysis. That stim-∗ Corresponding author. Tel.: +381 11 187 133; fax: +381 11 187 133.E-mail address: [email protected] (S. Mentus).
1 ISE member.
rimgsTa
013-4686/$ – see front matter © 2007 Elsevier Ltd. All rights reserved.oi:10.1016/j.electacta.2006.12.071
e; Titanium dioxide
lated many authors to investigate the electrochemical behaviourf metal/titania composite films synthesized in various ways5–17]. Composite, either oxide or metal-oxide, films werepplied on titanium surface mostly by thermal decompositionf unstable compounds of titanium and either noble, or tran-ition metals [1–10]. Furthermore, Kokkinidis et al. [11] usedeplacement reaction, based on very negative potential of tita-ium metal, while Tammeveski et al. [12] used platinum vacuumvaporation, to obtain Pt/TiO2 or Pt/Ti layers. Some authors con-idered the galvanostatic deposition of catalytically active metaln titanium. In this sense, Despic et al. [13], and Vukovic et al.14–17] used galvanostatic deposition of noble metals on tita-ium. Baez and Pletcher [18] compared the behavior of Au/TiO2omposite layers produced by several different ways previouslyescribed in the literature.
Thanks to a very negative potential of naked titanium,apid corrosion commences immediately upon its immersionnto any aqueous solution. In moderately alkaline, neutral and
oderately acidic aqueous solutions, according to Pourbaix dia-
rams [19], TiO2 is the most stable titanium oxide in waterolutions, and corrosion may be described by the equation:i + 2H2O = TiO2 + 2H2. This behaviour of titanium enableslso deposition of more noble metals from the solutions of their
4 ica A
siomoMd
ocmNcwbwpafep[
fatianctseeth[c4
ocwotia
2
gtnot(
lpg
iaw2
2id
tbe
iH
(aefp−0oc1
wt(
e0b
3
3
Hcmifm−To
582 D. Jasin et al. / Electrochim
alts, by replacement reaction [11]. The resistivity of TiO2 layers rather high and amounts to 108 � cm [20]. The formationf oxide layer is main reason why glavanostatically depositedetal show very poor adherence [13–17]. To compete to the
xide layer formation and improve adherence of metal deposit,entus developed potentiostatic technique which gave adherent
roplet like metals deposits incorporated into TiO2 film [21,22].The oxygen reduction reaction (ORR) is an important subject
f electrochemical investigations. This reaction is very sluggish,haracteristic of several different reaction paths, directed pri-arily by the catalytic effect of the electrode material [23,24].owadays this reaction is especially interesting as being a main
athodic reaction in fuel cells [25]. Its kinetics and mechanismas widely investigated primarily on platinum, known as itsest electrocatalyst [23,24,26]. To save platinum, much effortas invested to enhance its catalytic effectiveness, either by dis-ersing it on high surface area carbon support [27,28], or bylloying it with less noble metals [28], as well as by searchingor SMSI activation [1,10]. Furthermore, some other metals, forxample Ag and Au, under determined circumstances, may dis-lay effectiveness in oxygen reduction close to that of platinum29–31].
Oxygen reduction reaction was found to proceed on variousorms of titanium dioxide, although at much higher overvolt-ge in comparison to platinum. Clechet et al. evidenced thathis reaction takes place on Ti-oxide layer formed galvanostat-cally [32]. More recently, Clark et al. [33] published that onnodically formed TiO2, the overvoltage for ORR may be sig-ificantly reduced after an extensive activation of oxide layer byyclic polarization within a limited potential range. As regards tohe reaction mechanism of ORR, on TiO2 single crystal, Parkin-on et al. [34] and Clark et al. [33], by a ring-disk electrode,stablished nearly 4e− path in alkaline solutions, while Clechett al. [32], on the basis of qualitative chemical analysis, claimedhat oxygen reduction resulted generally in the formation ofydrogen peroxide (which, according to the relevant literature23,24], indicates an indirect, 2e− path). Mentus [35], on anodi-ally formed TiO2/Ti layer, observed the transition from 2e− toe− path with the transition from acidic to alkaline solutions.
In this work, both currentless and potentiodynamic depositionf gold on titanium were carried out to obtain thin Au/TiO2omposite layers over titanium metal. ORR in alkaline solutionas used as a test reaction to examine electrochemical behaviourf these layers. The theme is interesting in view of the fact thathe TiO2 surrounding may modify the catalytic behaviour ofncorporated Au particles, by influencing electronic propertiesnd adsorption of reactants and reaction intermediate species.
. Experimental
The electrochemical cell was a double-wall thermostatedlass cell, with the electrodes and gas inlet tube immersedhrough the top cover. The working electrode was a rod of tita-
ium (Aldrich Chem. Co., 99.99%). It was abraded to a diameterf 3 mm, and impressed into a PTFE cylinder of a rotating elec-rode. Reference electrode was a saturated calomel electrodeSCE), separated from the electrolyte bulk by a Luggin capil-ttea
cta 52 (2007) 4581–4588
ary filled with the same electrolyte. Counter-electrode was alatinum foil, separated from the electrolyte bulk by a sinteredlass disk.
The solutions were made from Merck or Aldrich p.a. chem-cals and redistilled water, and they were either purged byrgon stream, or saturated by oxygen. The purity of the gasesas 99.9995 vol.%. Electrolyte temperature was always kept at5 ◦C.
The dc measurements were carried out using a PAR Model73 Potentiostat/Galvanostat. The rotation speed of the work-ng electrode was controlled by a Beckman rotating electrodeevice.
To prepare the working electrode for electrochemical inves-igations, the exposed, disk-shaped Ti surface was dry-cleanedy emery paper no. 1200. Without any prolongation, sandedlectrode was subjected to further treatment.
For gold plating by replacement reaction, the freshly pol-shed titanium electrode was brought into the solution 2 g dm−3
AuCl4 in 0.1 M HClO4, and kept immersed 10 min.For potentiodynamic plating, cyanide plating solution
4 g dm−3 K[Au(CN)2] + 15 g dm−3 KCN) was used. Immedi-tely after dipping freshly polished Ti electrode, and closing thelectric circuit of the cell, potentiodynamic polarization was per-ormed at a polarization rate of 50 mV s−1, between a fixed initialotential of −1.2 V, and an variable anodic potential, which was0.9 V in the first cycle, and then steeply enlarged in steps of
.30 V, up to a maximum value of 0.6 V versus SCE. Massivexygen evolution limited the final anodic potential. Finally, theyclic sweeps between −1.2 and +0.6 V were repeated usually0 times.
The microphotographs of the electrode surface were recordedith JEOL JSM-840A scanning electron microscope (back scat-
ered electron mode) and with atomic force microscope (AFM)Topometrix Corp., contact mode).
For electrochemical characterization of Au/TiO2 layers, thelectrode was transferred from plating solutions into the solution.1 M NaOH either deaerated by pure argon stream, or saturatedy oxygen.
. Results and discussion
.1. The formation mode and appearance of Au/TiO2 layer
When freshly polished titanium is dipped in aqueous 0.1 MClO4 solution, its potential increased abruptly (Fig. 1, bottom
urve) from very negative (unknown) initial value of nakedetal, to approximately −0.1 V versus SCE during approx-
mately 1500 s, as a consequence of spontaneous TiO2 filmormation. This potential value is unexpectedly high, having inind that the standard potential of Ti/TiO2/H+ electrode is only0.86 V versus SHE. Probably the completing of insulatingiO2 layer along the whole titanium surface causes the lossf power of Ti/TiO2/H+ electrode, and some other processes
ake the responsibility for open circuit potential, for examplehe H+/H2 one, involving atomic or molecular hydrogenncapsulated by oxide. Upon immersion in tetrachloroauriccid + 0.1 M HClO4 solution, within only several seconds, open
D. Jasin et al. / Electrochimica A
F(
cpovtgh[
stvdicFsstoislcoettidw[36,37], captures gold and does not allow them to develop in any
FH
ig. 1. Time dependence of open circuit potential of polished Ti in HAuCl42 g dm−3) + 0.1 M HClO4 (top curve) and in 0.1 M HClO4 only (bottom curve).
ircuit potential rose up to over 0.8 V versus SCE, indicatingractically instantaneous establishment of a mixed potentialf Au/AuCl4− and Au/Au3+ electrodes (E0 = 0.994 and 1.42 Versus SCE, respectively). This is due to a displacement reac-ion: 4Au(III) + 3Ti(0) = 4Au(0) + 3Ti(IV) leading to a metallic
old on Ti surface, like to a displacement reaction observed inexachloroplatinic acid solution, leading to a platinum deposit11]. As a consequence of reactivity of titanium with waterfio
ig. 2. SEM (top) and AFM (bottom) microphotographs of mechanically polishedAuCl4 + 0.1 M HClO4.
cta 52 (2007) 4581–4588 4583
olutions, simultaneously TiO2 layer grows up. This means thatitanium loses its reducing power very quickly, and thereforeery limited amount of gold may be deposited by spontaneousisplacement reaction. After a 10 min period the electrode wasmmersed in tetrachlorauric acid solution, by naked eye, onean barely perceive slight gold-yellow nuance of the surface.ig. 2 presents the SEM and AFM microphotographs of Tiurface after spontaneous gold deposition. Obviously, on thecratched Ti surface, neither SEM nor AFM techniques enableo distinguish the features which could be described unequiv-cally to separate gold particles. X-ray diffraction was alsonsensitive to detect gold in this case, and gave a diffractogramimilar to that found by Pouilleau et al. for 10 nm thick TiO2ayer on Ti surface [36]. However, apart of the potential-timehange shown in Fig. 1, there are also other reliable evidencesf the presence of gold on the observed surface. Namely, SEMnergy dispersive X-ray analysis (SEM-EDX) evidenced thathe lighter fields are gold rich parts, while darker fields areitanium rich parts of the surface. Cyclovoltammetric evidences presented in Fig. 4, too. Therefore, one may conclude that,uring their simultaneous spontaneous growth, TiO2, which isell known to be amorphous in its early stages of development
orm which may be recognized clearly by SEM or AFM. This isn sound with the evidenced ability of gold to wet TiO2 surfacesf various crystallographic orientation [38], causing its epitaxial
(scratched) Ti surface immersed for 10 min in an aqueous solution 2 g dm−3
4584 D. Jasin et al. / Electrochimica Acta 52 (2007) 4581–4588
F poten5
grm
stducctprhoAtsb
3
tpvtas
Ac0wea0rp
pTclu[e
0ppswmately 400 �C cm . This charge density was accepted in theprocedures of real surface area determination of gold [42–44].In our case, the surface area of cathodic peak, for potentiody-namically formed Au/TiO2 layer, was found to correspond to
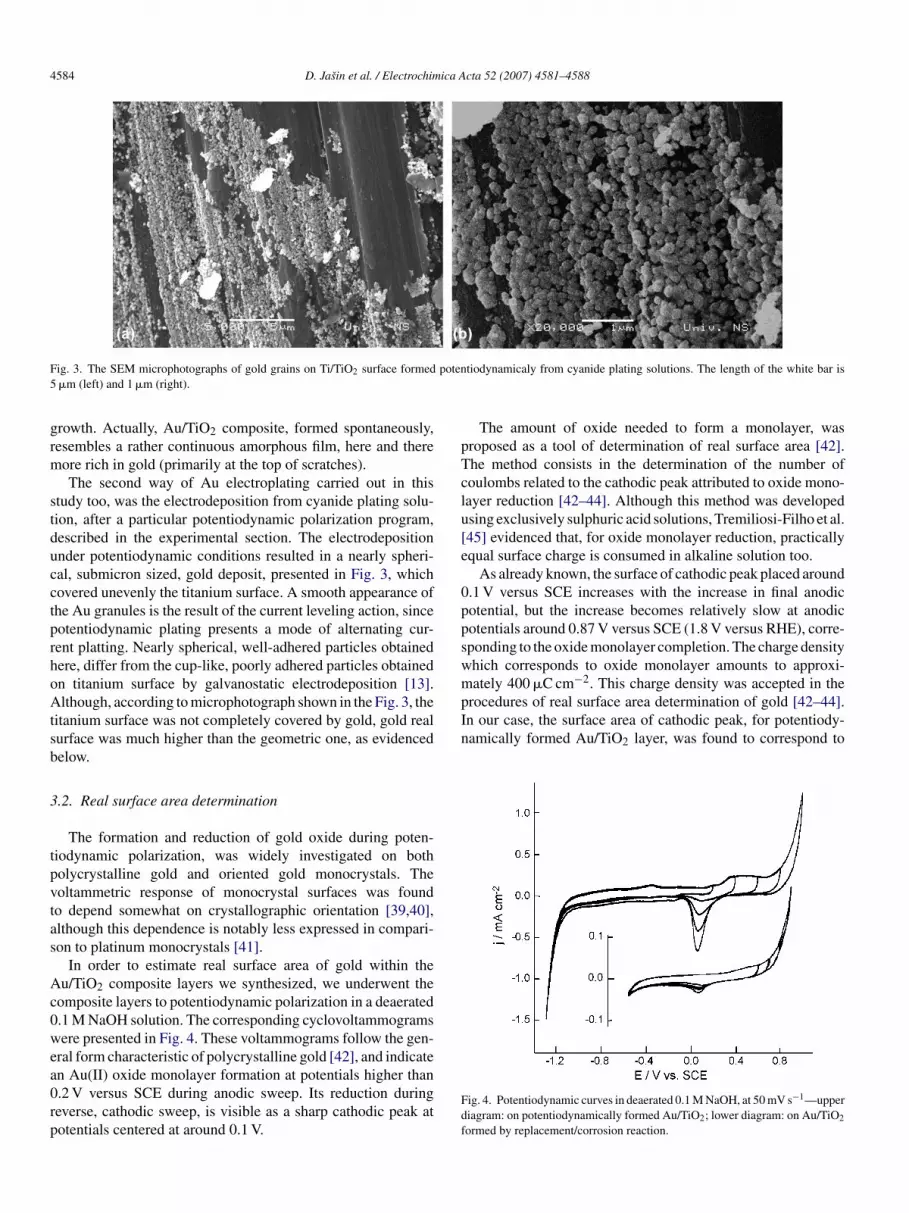
ig. 3. The SEM microphotographs of gold grains on Ti/TiO2 surface formed�m (left) and 1 �m (right).
rowth. Actually, Au/TiO2 composite, formed spontaneously,esembles a rather continuous amorphous film, here and thereore rich in gold (primarily at the top of scratches).The second way of Au electroplating carried out in this
tudy too, was the electrodeposition from cyanide plating solu-ion, after a particular potentiodynamic polarization program,escribed in the experimental section. The electrodepositionnder potentiodynamic conditions resulted in a nearly spheri-al, submicron sized, gold deposit, presented in Fig. 3, whichovered unevenly the titanium surface. A smooth appearance ofhe Au granules is the result of the current leveling action, sinceotentiodynamic plating presents a mode of alternating cur-ent platting. Nearly spherical, well-adhered particles obtainedere, differ from the cup-like, poorly adhered particles obtainedn titanium surface by galvanostatic electrodeposition [13].lthough, according to microphotograph shown in the Fig. 3, the
itanium surface was not completely covered by gold, gold realurface was much higher than the geometric one, as evidencedelow.
.2. Real surface area determination
The formation and reduction of gold oxide during poten-iodynamic polarization, was widely investigated on botholycrystalline gold and oriented gold monocrystals. Theoltammetric response of monocrystal surfaces was foundo depend somewhat on crystallographic orientation [39,40],lthough this dependence is notably less expressed in compari-on to platinum monocrystals [41].
In order to estimate real surface area of gold within theu/TiO2 composite layers we synthesized, we underwent the
omposite layers to potentiodynamic polarization in a deaerated.1 M NaOH solution. The corresponding cyclovoltammogramsere presented in Fig. 4. These voltammograms follow the gen-
ral form characteristic of polycrystalline gold [42], and indicaten Au(II) oxide monolayer formation at potentials higher than.2 V versus SCE during anodic sweep. Its reduction duringeverse, cathodic sweep, is visible as a sharp cathodic peak atotentials centered at around 0.1 V.
Fdf
tiodynamicaly from cyanide plating solutions. The length of the white bar is
The amount of oxide needed to form a monolayer, wasroposed as a tool of determination of real surface area [42].he method consists in the determination of the number ofoulombs related to the cathodic peak attributed to oxide mono-ayer reduction [42–44]. Although this method was developedsing exclusively sulphuric acid solutions, Tremiliosi-Filho et al.45] evidenced that, for oxide monolayer reduction, practicallyqual surface charge is consumed in alkaline solution too.
As already known, the surface of cathodic peak placed around.1 V versus SCE increases with the increase in final anodicotential, but the increase becomes relatively slow at anodicotentials around 0.87 V versus SCE (1.8 V versus RHE), corre-ponding to the oxide monolayer completion. The charge densityhich corresponds to oxide monolayer amounts to approxi-
−2
ig. 4. Potentiodynamic curves in deaerated 0.1 M NaOH, at 50 mV s−1—upperiagram: on potentiodynamically formed Au/TiO2; lower diagram: on Au/TiO2
ormed by replacement/corrosion reaction.

ica Acta 52 (2007) 4581–4588 4585
tc(pbocecIodlgmt
3
rrwacs
rwmorht
FTp7c(a
Fig. 6. The diffusion corrected Tafel plots of oxygen reduction reaction in oxy-gen saturated 0.1 M NaOH, on Au/TiO2 formed potentiodynamically (stars) andsrt
Otttlr(nft
D. Jasin et al. / Electrochim
he charge density of 1.9 mC cm−2, i.e. 4.75 times the amountorresponding to one monolayer. Therefore, the real surface areasurface roughness factor) may be considered to be 4.75 cm−2
ro unit of geometric surface area. For an Au/TiO2 layer formedy replacement reaction, the charge corresponding to the surfacef cathodic peak was much smaller, i.e. 0.05 mC cm−2, whichorresponds to the surface roughness factor of 0.13. Assumingven very flat Au surface, this calculation shows that in the lastase, about 90% of the electrode surface consisted of pure TiO2.ndeed, one should be somewhat reserved toward the accuracyf last figures, namely, it is not sure that the coulometry method,eveloped for massive gold, applies directly for the Au/TiO2ayer formed by replacement reaction, having in mind that theold layer may be somewhere only one-atom thick, and further-ore, some gold atoms or clusters may be screened by TiO2 and
hus non-exposed to oxidation.
.3. Kinetics of oxygen reduction
In Fig. 5, the voltammetric data for oxygen reductioneaction on pure TiO2, and Au/TiO2 obtained by both cor-osion/replacement reaction and potentiodynamic polarization,ere presented. The data for rotation frequency of 10 rps for
ll three systems was presented in Fig. 5. In Fig. 6, theurrent–potential dependencies for various surfaces were pre-ented in a form of Tafel (E versus log j) plot.
Generally, the oxygen reduction on different electrode mate-ials may proceed usually either as a 4e− reaction, with orithout the participation of peroxide ion as an absorbed inter-ediate, or as a 2e− reaction [23], recognizable by deliberation
f hydrogen peroxide from electrode surface. Rotating disc-ing electrode, which enables determination of the fraction ofydrogen peroxide deliberated, presents a powerful tool to dis-inguish easily between 4e− and 2e− reaction paths [40,41,45].
ig. 5. Current–potential dependence of oxygen reduction in 0.1 M NaOH oniO2 (2,3), and Au/TiO2 (5–8) rotating dics electrode. Au/TiO2 was formedotentiodynamically (7 and 8) and spontaneously (5,6). Full symbols (2,5 and) correspond to cathodic, empty simbols (3, 6 and 8) to anodic scan. Baselineurrents in deaerated solutions, for TiO2 (1) and spontaneously formed Au/TiO2
4). All voltammetry curves were recorded at a polarization rate of 20 mV s−1,nd rotation rate of 10 rps.
pcerAw
tf
j
wbw
e
j
Fvaoko1u
pontaneously (squares), and on pure TiO2 (circles) (anodic scans, as in Fig. 5,ecorded at a polarization rate 5 mV s−1). Empty triangles are estimated dataaken from ref. [46] for Au(1 0 0).
n gold surface, the rate of oxygen reduction is very sensitiveo crystallographic orientation [40,41]. In alkaline solutions, inhe potential region of mixed, charge transfer-diffusion control,he reduction proceeds as a 2e− proces on low-index crystal-ographic planes, (1 1 0) and (1 1 1), while on (1 0 0) plane theeduction proceeds primarily via 4e− process [24,40,41]. For1 0 0) plane, there is an intermediate voltage region in whichegatively charged peroxide ion may not withstand repulsionorces, and leaves the surface before it undergoes the reduc-ion, and thus, in this potential interval, the oxygen reductionroceeds via 2e− route without any exception. However, at highathodic overvoltages the rate of peroxide ion reduction becomesnough high to compete effectively with ion desorption, and theeduction becomes always 4e− one. This is the reason why, onu(1 0 0) plane, one may observe a drop of cathodic currentithin the region of diffusion limitation [40,41].In the case of a diffusion limited electrochemical reaction,
he disc current is a linear function of the square root of rotationrequency [8,12]:
l = 0.62nFD2/3ν−1/6ω1/2C (1)
here ν presents the kinematic viscosity, i.e. viscosity dividedy density, and ω is the angular rotation frequency (ω = 2πf,here f is rotation frequency).Assuming constant concentration of diffusing species, this
quation may be written in the following shorter form:
l = Bω1/2 (1′)
or diluted, oxygen saturated aqueous solutions, the values ofariables involved into the constant B do not depend consider-bly on the nature of supporting electrolyte. If the concentrationf supporting electrolyte does not exceed 0.1 M, commonly,
inematic viscosity is assumed to amount 0.01 cm2 s−1,xygen solubility 1.18 mmol dm−3 and diffusion coefficient.9 × 10−5 cm2 s−1 [8,12]. Therefore, for diluted, oxygen sat-rated solutions, the constant B depends exclusively on the
4 ica A
nawTt4
cω
laiabkfir
FlfvsTir
ireiiitIotetnpOstraoamotac
ibA
Atbcocdpgmbt
patbiifow
oli(spontdrfzchwme
rtitroofvcb
586 D. Jasin et al. / Electrochim
umber of electrons, n, consumed per one oxygen molecule, andmounts to 0.436 mA cm−2 (rad s−1)−1/2 for 4e− route [8,12],hile 2e− process is distinguishable by a half of this value.hus, in the case of O2 reduction, the constant B, if experimen-
ally determined, enables to distinguish easily between 2e− ande− processes.
With the known value of constant B, by means of Eq. (1), onean calculate the limiting diffusion current density for any given. For instance, for rotation frequency 10 rps (ω = 62.8 rad s−1
imiting current density amounts to 3.4 mA cm−2, for 4e− route,nd half of that value for 2e− route. On polycrystalline Au discn 0.1 M NaOH, at 26 ◦C and at rotation rate of 10 rps, Zurilla etl. [47] found the current density of 1.82 mA cm−2 (estimatedy us, for the sake of comparison to our data), in accordance withnown fact that on polycrystalline Au, in alkaline solutions, therst reduction wave correspond to a 2e− pathway of oxygeneduction.
As expectable on the basis of previous publication [35], inig. 5, one may see that among given surface layers, pure TiO2
ayer shows the highest overvoltage (the lowest catalytic action)or oxygen reduction. Deep cathodic polarization causes acti-ation of TiO2 layer [33,35], and thus, during reverse, anodic,weep the voltammogram shows always reduced overvoltage.he reaction attains diffusion control at deep cathodic polar-
zation (nearly 3.5 mA cm−2 at 10 rps) characteristic of oxygeneduction via 4e− route.
The reduction of O2 on polarized TiO2/aqueous solutionnterface was discussed elsewhere [35]. According to theecently published models [48,49], surface species on TiO2 areither H+, adsorbed on O2− ions, or OH−, adsorbed on Ti4+
ons. On cathodically polarized interface, reduced species, Ti3+
ons, are produced on the surface of TiO2 layer, which manifesttself by symmetric cyclovoltammograms with respect to poten-ial axis [50], characteristic otherwise of the surface processes.t is expected that O2 reduction proceeds via the interactionf oxygen with Ti3+ ions (the number of which increases withhe depth of cathodic polarization), which act as the electronxcess sites within a Ti4+ matrix of TiO2. In alkaline solutions,he blockage of active Ti3+ sites is ineffective since the hydro-ium ions are practically absent, and thus there is reasonablerobability that both O2 molecule, or intermediate peroxide ion2H− may be adsorbed on free Ti3+ sites. Doubtlessly, hystere-
is loop of cyclovoltammogarms of O2 reduction indicates thathe deeply pre-reduced TiO2 surface keeps (“remembers”) itseducing power in the course of anodic polarization. Most prob-bly, this is connected to the progressive thickening and thinningf Ti3+ rich part of titanium oxide layer during cathodic andnodic sweep, respectively, which is slow process due to a lowobility of both electrons and ions through insulating titanium
xide. This type of cathodic activation is different in nature fromhe activation of Pt [41] or Ag [29]; namely, these metals becomective toward O2 reduction only if purged (by reduction duringathodic polarization) from surface oxides.
If gold is present on Ti surface, it takes over the role of dom-nant active centers of O2 reduction. On Au/TiO2 layer formedy spontaneous, displacement/corrosion reaction, in spite of lowu coverage, an appreciable catalytic effect is visible (Fig. 5).
srte
cta 52 (2007) 4581–4588
long with the notably reduced overvoltage for oxygen reduc-ion, the corresponding polarization curve keeps somewhat theehaviour of pure TiO2, namely shows a pronounced activationaused by deep cathodic polarization. If, in Fig. 6, for a commonvervoltage of −0.7 V (where the reaction is preferably underharge-transfer control) one compares the current densities atifferent surface layers, one may see that the Au/TiO2 com-osite layer formed by replacement reaction, with coverage byold of approximately 10% only, enables even for two orders ofagnitude higher current density than pure TiO2. The catalytic
ehavior of the surface is in this case roughly middle betweenhat of pure TiO2 and that of pure gold.
As regards to the potentiodynamically formed Au/TiO2 com-osite layer, in both Figs. 5 and 6, one may see its excellentctivity, which is, according to the literature [30,31,46], closeo mostly active, Au(1 0 0) monocrystal plane. The distinctiveehavior of the Au/TiO2 layer in comparison to Au(1 0 0) plane,s very reduced drop in limiting current density in the potentialnterval (−0.8 to −0.4 V) related to the peroxide ion desorptionrom gold surface [41,46]. Since TiO2 alone does not supportxygen reduction in this voltage range, obviously one deals hereith a synergistic effect, which will be discussed later.From Fig. 6 it is visible that Tafel slope for O2 reduction
n Au/TiO2 composite surfaces amounts to 0.11 V per unit ofog j axis. This is consistent with the mechanism of reductionnvolving the reaction O2 + e = O2
− as the rate determining stepfollowing by O2
−ads + H2O + e− = HO2
− + OH−) [51]. For theake of comparison, the data for Au(1 0 0) monocrystal plane areresented in Fig. 6 too (taken from Ref. [46]), where the slopef Tafel line of 90 mV per unit of log j axis was reported. Onoble metals, somewhere the change from −60 mV per decadeo −120 mV per decade was observed on going from low currentensity (lcd) toward high current density (lcd) region. Bock-is et al. [51] explained this event in terms of the transitionrom Temkin to Langmuir conditions, caused by the drop toero in surface coverage by adsorbed oxygen, rather than by thehange in reaction mechanism. However, in some cases, muchigher Tafel slopes in hcd region were found experimentally,hich is not well explained thus far. For example, Tam-aveski et al. reported even −380 mV per decade for a Pt/TiO2
lectrode [12].Using the limiting diffusion current density as the criteria of
eaction path (about 3.5 mA cm−2 in Fig. 5), each of here inves-igated electrode/electrolyte boundaries enables 4e− reductionn the overvoltage region of pure diffusion control. In ordero examine the number of electrons involved in the overalleduction of oxygen at various sweep directions and variousvervoltages, involving also the overvoltages out of the regionf pure diffusion limitation, a series of K–L plots was calculatedor various overvoltages, using the voltammograms recorded atarious rotating rates. These plots were presented in Fig. 7, andompared to the ones calculated for n = 2 and 4. It is obvious thatoth TiO2 and Au/TiO2 composite layer in alkaline solution give
lopes which correspond to oxygen reduction preferably via 4e−oute. Only certain exception was observed in the case of spon-aneously formed Au/TiO2 layer at −0.7 V, for which somewhatnlarged K–L slope, corresponding to n = 3.7, was found.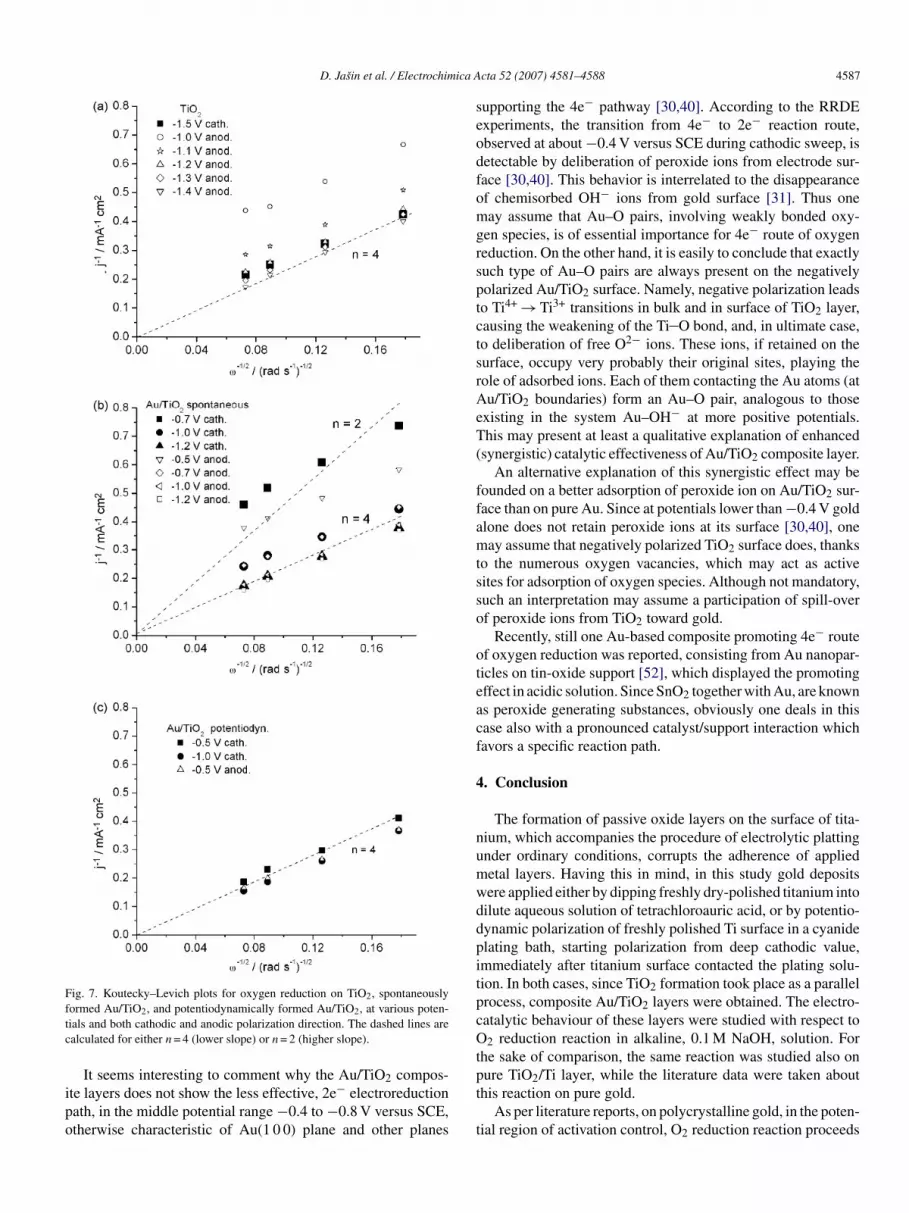
D. Jasin et al. / Electrochimica A
Fig. 7. Koutecky–Levich plots for oxygen reduction on TiO2, spontaneouslyftc
ipo
seodfomgrsptctsrAeT(
ffamtsso
oteacf
4
numwddpitpcOtpure TiO /Ti layer, while the literature data were taken about
ormed Au/TiO2, and potentiodynamically formed Au/TiO2, at various poten-ials and both cathodic and anodic polarization direction. The dashed lines arealculated for either n = 4 (lower slope) or n = 2 (higher slope).
It seems interesting to comment why the Au/TiO compos-
2te layers does not show the less effective, 2e− electroreductionath, in the middle potential range −0.4 to −0.8 V versus SCE,therwise characteristic of Au(1 0 0) plane and other planest
t
cta 52 (2007) 4581–4588 4587
upporting the 4e− pathway [30,40]. According to the RRDExperiments, the transition from 4e− to 2e− reaction route,bserved at about −0.4 V versus SCE during cathodic sweep, isetectable by deliberation of peroxide ions from electrode sur-ace [30,40]. This behavior is interrelated to the disappearancef chemisorbed OH− ions from gold surface [31]. Thus oneay assume that Au–O pairs, involving weakly bonded oxy-
en species, is of essential importance for 4e− route of oxygeneduction. On the other hand, it is easily to conclude that exactlyuch type of Au–O pairs are always present on the negativelyolarized Au/TiO2 surface. Namely, negative polarization leadso Ti4+ → Ti3+ transitions in bulk and in surface of TiO2 layer,ausing the weakening of the Ti O bond, and, in ultimate case,o deliberation of free O2− ions. These ions, if retained on theurface, occupy very probably their original sites, playing theole of adsorbed ions. Each of them contacting the Au atoms (atu/TiO2 boundaries) form an Au–O pair, analogous to those
xisting in the system Au–OH− at more positive potentials.his may present at least a qualitative explanation of enhanced
synergistic) catalytic effectiveness of Au/TiO2 composite layer.An alternative explanation of this synergistic effect may be
ounded on a better adsorption of peroxide ion on Au/TiO2 sur-ace than on pure Au. Since at potentials lower than −0.4 V goldlone does not retain peroxide ions at its surface [30,40], oneay assume that negatively polarized TiO2 surface does, thanks
o the numerous oxygen vacancies, which may act as activeites for adsorption of oxygen species. Although not mandatory,uch an interpretation may assume a participation of spill-overf peroxide ions from TiO2 toward gold.
Recently, still one Au-based composite promoting 4e− routef oxygen reduction was reported, consisting from Au nanopar-icles on tin-oxide support [52], which displayed the promotingffect in acidic solution. Since SnO2 together with Au, are knowns peroxide generating substances, obviously one deals in thisase also with a pronounced catalyst/support interaction whichavors a specific reaction path.
. Conclusion
The formation of passive oxide layers on the surface of tita-ium, which accompanies the procedure of electrolytic plattingnder ordinary conditions, corrupts the adherence of appliedetal layers. Having this in mind, in this study gold depositsere applied either by dipping freshly dry-polished titanium intoilute aqueous solution of tetrachloroauric acid, or by potentio-ynamic polarization of freshly polished Ti surface in a cyanidelating bath, starting polarization from deep cathodic value,mmediately after titanium surface contacted the plating solu-ion. In both cases, since TiO2 formation took place as a parallelrocess, composite Au/TiO2 layers were obtained. The electro-atalytic behaviour of these layers were studied with respect to2 reduction reaction in alkaline, 0.1 M NaOH, solution. For
he sake of comparison, the same reaction was studied also on
2his reaction on pure gold.As per literature reports, on polycrystalline gold, in the poten-
ial region of activation control, O2 reduction reaction proceeds

4 ica A
pAfOsebap
A
bReCE
R
[
[
[
[
[[[
[
[[
[
[[[
[
[[
[
[
[[
[[
[[
[[
[
[
[
[
[
[
[
[[
[
[[[
588 D. Jasin et al. / Electrochim
referably as a 2e− process. In this study, involving two types ofu/TiO2 composite layers with quite different surface roughnes
actor, namely 0.13 and 4.75, by Koutecky–Levich analysis, the2 reduction reaction was evidenced to proceed almost exclu-
ively via 4e− route. This synergistic effect was attempted toxplain in terms of Au–O pairs formed between the looselyonded oxygen anions within a TiO2 layer and the adjacent Automs, as well as by enhanced (compared to Au) adsorption oferoxide ions on negatively polarized TiO2 surface.
cknowledgements
AA-R is the participant in the contract TR6712B supportedy the Ministry of Science and Environmental Protection ofepublic Serbia. SM is a participant on the EU Project “Prom-teas”, Contract No. ICA2-CT-2001-10037, as well as in theontract No. 142047 supported by the Ministry of Science andnvironmental Protection of Republic Serbia.
eferences
[1] D. Pletcher, J. Appl. Electrochem. 14 (1984) 403.[2] M. Haruta, N. Yamada, T. Kobayashi, S. Iijima, J. Catal. 115 (1989) 301.[3] M. Haruta, T. Tsubota, S. Kobayashi, T. Kageyama, H. Genet, B. Delmon,
J. Catal. 144 (1993) 175.[4] M. Haruta, Catal. Today 36 (1997) 153.[5] B.C. Beard, P.N. Ross, J. Electrochem. Soc. 133 (1986) 1839.[6] T.A.F. Lassali, J.F.C. Boodts, S.C. de Castro, R. Landers, S. Trasatti, Elec-
trochim. Acta 39 (1994) 95.[7] L.A. Da Silva, V.A. Alves, M.A.P. Da Silva, S. Trasatti, J.F.C. Boodts,
Electrochim. Acta 41 (1996) 1279.[8] K. Tammeveski, T. Tenno, A. Rosental, P. Talonen, L.-S. Johansson, L.
Niinisto, J. Electrochem. Soc. 146 (1999) 669.[9] G. Foti, C. Mousty, K. Novy, Ch. Comninellis, V. Reid, J. Appl. Elec-
trochem. 30 (2000) 147.10] S.G. Neophytides, S.H. Zafeiratos, M.M. Jaksic, J. Electrochem. Soc. 150
(2003) E512.11] G. Kokkinidis, A. Paapoutsis, D. Stoychev, A. Milchev, J. Electroanal.
Chem. 486 (2000) 48.12] K. Tammeveski, M. Arulepp, T. Teno, C. Ferrater, J. Claret, Electrochim.
Acta 42 (1997) 2961.13] R. Zejnilovic, M. Pjescic, M. Sljukic, A.R. Despic, J. Appl. Electrochem.
14 (1984) 481.14] M. Vukovic, Electrochim. Acta 34 (1989) 287.15] D. Cukman, M. Vukovic, J. Electroanal. Chem. 279 (1990) 273.16] D. Cukman, M. Vukovic, M. Milun, J. Electroanal. Chem. 389 (1995)
209.
17] M. Vukovic, D. Marijan, D. Cukman, P. Pervan, M. Milun, J. Mater. Sci.34 (1999) 869.18] V.B. Baez, D. Pletcher, J. Electroanal. Chem. 377 (1994) 231.19] M. Pourbaix (Ed.), Atlas of Electrochemical Equilibria in Aqueous solu-
tions, Pergamon, New York, 1966.
[[
[
cta 52 (2007) 4581–4588
20] B. Rogers, R.D. Shannon, A.W. Sleight, J.L. Gillson, Inorg. Chem. 8 (1969)841.
21] S.V. Mentus, Electrochim. Acta 50 (2005) 3609.22] I. Boskovic, S. Mentus, M. Pjescic, Electrochem. Commun. 7 (2005) 797.23] H.S. Wroblowa, Y.C. Pan, G. Razumney, J. Electroanal. Chem. 69 (1976)
195.24] R. Adzic, in: J. Lipkowski, P.N. Ross (Eds.), Electrocatalysis, Wiley–WCH,
New York, 1998, p. 197.25] F.J. Luczak, D.A. Landsman, US Patent 4,677,092 (1987).26] M.R. Tarasevich, A. Sadkowski, E. Yeager, Comprehensive Treatise in
Electrochemistry, Plenum Press, New York, 1983, p. 301 (Chapter 6).27] H. Bonnemann, R. Brinkmann, P. Britz, U. Endruschat, R. Mortel, U.A.
Paulus, G.J. Feldmeyer, T.J. Schmidt, H.A. Gasteiger, R.J. Behm, J. NewMater. Electrochem. Syst. 3 (2000) 199.
28] U.A. Paulus, A. Wokaun, G.E. Scherer, T.J. Schmidt, V. Stamenkovic, N.N.Markovic, P.N. Ross, Electrochim. Acta 47 (2002) 3787.
29] I. Boskovic, S. Mentus, M. Pjescic, Electrochim. Acta 51 (2006) 2793.30] R.R. Adzic, N.N. Markovic, V.B. Vesovic, J. Electroanal. Chem. 165 (1984)
105.31] S. Strbac, R.R. Adzic, Electrochim. J. Electroanal. Chem. 403 (1996) 169.32] P. Clechet, C. Martelet, J.R. Martin, R. Olier, Electrochim. Acta 24 (1979)
457.33] T. Clark, D.C. Johnson, Electroanalysis 9 (1997) 273.34] B. Parkinson, F. Decker, J.F. Juliao, M. Abramovich, H.C. Chagas, Elec-
trochim. Acta 25 (1980) 521.35] S. Mentus, Electrochim. Acta 50 (2004) 27.36] J. Pouilleau, D. Devilliers, F. Garrido, S. Durand-Vidal, E. Mahe, Mater.
Sci. Eng. B 47 (1997) 235.37] J.L. Delplancke, A. Garnier, Y. Massiani, R. Winand, Electrochim. Acta
39 (1994) 1281.38] L. Meyer, C. Lemire, Sh.K. Shaikhutdinov, H.-J. Freunf, Gold Bull. 37
(2004) 72.39] A. Prieto, J. Hernandez, E. Herrero, J.M. Feliu, J. Solid State Electrochem.
7 (2003) 599.40] S. Strbac, N.A. Anastasijevic, R.R. Adzic, J. Electroanal. Chem. 323 (1992)
179.41] N.M. Markovic, P.N. Ross, in: A. Wieckowski (Ed.), Interfacial Elec-
trochmistry, Theory, Experiment and Applications, Marcel Dekker Inc.,New York, Basel, 1999, p. 821 (Chapter 46).
42] D.A.J. Rand, R. Woods, J. Electroanal. Chem. Interfacial Electrochem. 31(1971) 29.
43] H.A. Kozlovska, B.E. Convay, A. Hamelin, L. Stoicoviciu, J. Electroanal.Chem. Interfacial Electrochem. 228 (1987) 429.
44] S. Trasatti, O.A. Petrii, Pure Appl. Chem. 63 (1991) 711.45] G. Tremiliosi-Filho, L.H. Dall’Antonia, G. Jerkiewicz, J. Electroanal.
Chem. 422 (1997) 149.46] B.B. Blizanac, C.A. Lucas, M.E. Gallagher, M. Arenz, P.N. Ross, N.M.
Markovicı, J. Phys. Chem. B 108 (2004) 625.47] R.W. Zurilla, R.K. Sen, E. Yeager, J. Electrochem. Soc. 125 (1978) 1103.48] U. Diebold, Surf. Sci. Rep. 48 (2003) 53.49] U. Diebold, Appl. Phys. A76 (2002) 1.
50] V.B. Baez, J.E. Graves, D. Pletcher, J. Electroanal. Chem. 340 (1992) 273.51] J.O.’M. Bockris, S.U.M. Khan, Surface Electrochemistry, a MolecularLevel Approach, Plenum Press, New York and London, 1993, p. 331.52] W.S. Baker, J.J. Pietron, M.E. Teliska, P.J. Bouwman, D.E. Ramaker, K.E.
Swider-Lyons, J. Electrochem. Soc. 153 (2006) A1702.





























