Embed Size (px)
Citation preview

p73 functionally replaces p53 in Adriamycin-treated, p53-deficient breast
cancer cells
Muriel Vayssade1,3, Hedi Haddada2, Laetitia Faridoni-Laurens1,3, Sophie Tourpin1,3, Alexandre Valent4,Jean B�enard1 and Jean-Charles Ahomadegbe1,3,5*
1Unit�e de G�en�etique Tumorale, D�epartement de Biologie Clinique, Institut Gustave Roussy, Villejuif C�edex, France2Inserm U362, Institut Gustave Roussy, Villejuif C�edex, France3Upress 3535 ‘‘Pharmacologie et nouveaux traitements des cancers’’, Institut Gustave Roussy, Villejuif C�edex, France4Laboratoire de G�enomique Cellulaire des Cancers CNRS-UMR 1599, Institut Gustave Roussy, Villejuif C�edex, France5Facult�e de Pharmacie, UPJV, Amiens C�edex, France
p53-related genes, p73 and p63, encode 2 classes of proteins, TA-p73/p63 and DN-p73/p63. TA-p73/p63 demonstrate p53-like prop-erties including gene transactivation and cell death promotion,whereas DN-p73/p63 lack these p53-like functions. Although p53-deficient cancer cells are often less responsive to chemotherapy,they are not completely drug resistant, suggesting that other apop-totic pathways are at work. Here, we compared for the first timeto our knowledge p73 and p63 activation in various breast cancer(BC) cell lines after Adriamycin (ADR) treatment, an agent con-sidered as mandatory in breast cancer chemotherapy. Our studywas carried out using 1 p53-proficient BC cell line (MCF7 cells)and 3 BC cell lines deficient in p53 response (MCF7/ADR
IGR,
MDA-MB157 and T47D) after ADR-induced genotoxic stress. Wereport that in cells with no p53 response after ADR treatment,TAp73, but not TAp63 or DN-p73/p63, may replace p53 in trigger-ing not only apoptosis but also cell cycle arrest or DNA repaireffectors such as p21, GADD45, 14-3-3r and p53R2. We also dem-onstrate that TAp73 siRNA inhibits the accumulation of TAp73 inresponse to ADR treatment in MDA-MB157 cells and confers pro-tection against ADR. ADR-induced downregulation of the DNp73isoform in the T47D cell line with nonfunctional mutant p53 fur-ther supports anti-apoptotic function of the isoform antagonisticto both p53 and TA-p73/p63. Exogenous TAp73 and DNp73 over-expression in p53-response-deficient cell lines further confirmsthese results. cDNA microarray techniques demonstrated that thecellular response induced by p73 during ADR treatment couldinvolve specific genes.' 2005 Wiley-Liss, Inc.
Key words: p53; p63; p73; Adriamycin; breast cancer
The p53 protein is a transcription factor that regulates the cellcycle and apoptosis via transactivation of canonical genes, such asp21, 14-3-3r, Gadd45 and Bax.1–4 In response to cellular stressescaused by a number of anticancer agents, the p53 protein accumu-lates and transactivates these target genes, causing cell cyclearrest, DNA repair or apoptosis.5 However, cell cycle arrest orapoptosis can occur in p53-deficient cells. Indeed, Adriamycin(ADR) has been found to induce p21 overexpression and apoptosisin p53-/- human lymphoid cells.6 Moreover, it has been suggestedthat the inactivation of the p53 pathway could improve theresponse to chemotherapy in high-grade breast cancers.7 Thesedata suggest the existence of p53-independent pathways involvedin the cellular response to anticancer drugs like Adriamycin.
Two new genes, which structurally resemble p53, have beendescribed and named: p73 and p40/p51/p63.8–12 Both p73 and p63genes encode proteins that possess transactivation, DNA bindingand oligomerization domains that present marked similarities tocorresponding p53 domains. However, unlike p53, p73 and p63genes, each contain 2 independent promoters and make significantuse of differential splicing at the gene’s 30 end, hence yielding anarray of at least 6 unique proteins that form 2 distinct classes:those containing an amino acid terminus (called TAp73 andTAp63) and those that are truncated (called DNp73 and DNp63)with no amino-terminus region.12–16 In experimental systems,when exogenously overexpressed in cells, TAp73 and TAp63were shown to transactivate many p53 target genes.17–19
It has been demonstrated that genotoxic stresses (actinomycinD, UV, Adriamycin, cisplatin, taxol and mitomycin C) do not trig-ger p73 protein induction in p53-proficient cells.8,20–21 The origi-nal, common notion that p73 is not upregulated in response toDNA damage was then challenged by several reports that clearlydemonstrated that p73 is a target for c-abl tyrosine kinase inresponse to DNA damages caused by cisplatin and g-radiation.22–24
Flores et al. then observed that the p632/2; p732/2 double nullmouse embryo fibroblasts (MEF) were as resistant to apoptosis asp532/2 MEF cells, pointing out that p63 and p73 are required forp53-induced apoptosis and delineating functional interplay betweenp53 family members.25 More recent studies have shown that p73could be implicated in the cellular response to drugs in cancer cellssuch as squamous cell carcinomas or osteosarcoma cell lines.26–27
Today, there are no data regarding a hypothetical role of p73and p63 in cellular response to ADR in p53-deficient breast can-cers. Here, we have analyzed mRNA and protein levels of p53-family members and p53-target genes, and cell apoptosis in 4ADR-treated breast cancer cell lines, with varying p53 statuses.Our results show that p73, but not p63, plays a major role in thecellular response to ADR treatment and that in ADR-treated p53-deficient breast cancer cells, p73 could take p53’s role in apopto-sis, cell growth and DNA repair.
Material and methods
Cell culture conditions
The MCF7, MCF7/ADRIGR, MDA-MB157 and T47D breastcancer cell lines were cultured as monolayers as previouslydescribed.28 The MCF7/ADRIGR cell line had been selected bysubmitting parental MCF7 strain, subcultured from a batch of theAmerican Type Tissue Collection certified line, to continuousstepwise ADR exposures (from 0.02 lM to 10 lM of drug). Thestudies used the resistant cells cultured for a single passage with-out the drug and showed a similar resistance index to that of thecontinuously treated subline.
Twenty-four hours after plating, cells were treated for 24 hrwith Adriamycin (ADR, Adriblastine1, Pharmacia, Milan, Italy)or cisplatin (CDDP, Cisplatine, Merck, Lyon, France) beforebeing harvested and counted using the Trypan blue exclusionmethod. The remaining cells were maintained in ADR-freemedium and harvested 48 hr after the ADR treatment.
Grant sponsor: Association pour la Recherche contre le Cancer (ARC-Villejuif); Grant number: 4381; Grant sponsor: La Ligue Contre le Cancer,Comit�e de la Somme.*Correspondence to: Unit�e de G�en�etique Tumorale, UPRESS 3535
‘‘Pharmacologie et nouveaux traitements des cancers’’ (IFR 54), InstitutGustave Roussy, 39 rue Camille Desmoulins, 94 805 Villejuif C�edex,France. Fax:133-1-42-11-52-80. E-mail: [email protected] 30 January 2004; Accepted after revision 13 January 2005DOI 10.1002/ijc.21033Published online 22 April 2005 in Wiley InterScience (www.interscience.
wiley.com).
Int. J. Cancer: 116, 860–869 (2005)' 2005 Wiley-Liss, Inc.
Publication of the International Union Against Cancer

DNA and RNA preparations
The cell monolayers fixed on the bottom of the flasks were tryp-sinized; the cells were counted and then lysed using sarkosyl andguanidinium isothiocyanate (GTC). DNA and total RNAs wereprepared as previously described.29
p53 gene sequencing
The p53 gene was sequenced as previously described.29
Semiquantification of p53, p73, p21, GADD45 and14-3-3r transcripts levels
cDNA was obtained by reverse transcription (RT) of RNA(1 lg) using Moloney Murine Leukemia Virus Reverse Transcriptase(SuperScriptTM RNase H, MMLV-RT Kit, Invitrogen, Rockville,MD) and random hexamers to prime the synthesis in conditionsspecified by the manufacturer.
p53 cDNA was amplified using primers as indicated in Table I.To semiquantitatively determine the transcript levels of thesegenes, preliminary 1-cycle stepwise amplifications were per-formed, and amplification kinetic curves were plotted; these dem-onstrated that a plateau phase was reached after a different numberof cycles for each gene. The transcript level was therefore detectedat a number of cycles preceding the plateau phase. GAPDH cDNAwas amplified and used as the internal control (Table I).
cDNA (1 ll) was amplified in a 20 ll reaction mixture contain-ing 100 ng of each primer as previously described.28
Two microliters of amplified cDNA fragments were electro-phoresed on polyacrylamide gels. Densitometric quantificationwas performed using the Joyce-Loebl chromoscan.
Immunoblotting
Cells (106) excluding Trypan blue were lysed in 50 ll ofLaemmli buffer (Biorad, Hercules, CA), and heated to 100�C for5 min. Lysates (10 ll) were electrophoresed in sodium dodecylsulfate (SDS)-polyacrylamide gel and then transferred to nitrocel-lulose membranes (Amersham, Buckinghamshire, UK) in a semi-dry transfer cell (Biorad). The filters were incubated with 5 lg/mlof rabbit anti-mdr (Ab-1, Oncogene Research, San Diego, CA), 1lg/ml of mouse anti-p53 (DO7, DAKO, Copenhagen, Denmark),2 lg/ml of mouse anti-p21 (Ab1, Oncogene Research), rabbit anti-p73a diluted to 1/1,000 (generously donated by Dr. Caput, SanofiLabege, France), 2 lg/ml of mouse anti-p63 (4A4, Santa Cruz,Santa Cruz, CA), 2 lg/ml of rabbit anti-GADD45 (H165, SantaCruz), 2 lg/ml of goat anti-14-3-3r (C18, Sc 7683, Santa Cruz),0.4 lg/ml of goat anti-p53R2 (C18, Sc 10843, Santa Cruz), 0.2lg/ml of mouse anti-PCNA (PC10, Santa Cruz), and 1 lg/ml ofrabbit anti-SOD1 (FL54, Santa Cruz) antibodies.
Proteins were visualized using an enhanced chemiluminescencedetection system (Amersham). Membranes were then washed in0.5% TBS-Tween, and incubated with 0.2 lg/ml of mouse anti-actin (C4, Chemicon, Temecula, CA) or 5 lg/ml of mouse anti-tubulin antibody (CP06, Oncogene Research) to quantify and nor-
malize the results.28
Recombinant p73-adenovirus constructs and cell infection
The plasmid constructs pcDNA-TAp73a and pcDNA-DNp73awere generously donated by Dr. Daniel Caput (Sanofi Recherche,Labege). The recombinant adenoviral vector expressing full-length TAp73a (Ad-TAp73a) and DN-p73a (Ad-DNp73a) wereproduced by Dr. H Haddada (IGR-Villejuif-France).
Cells (5 3 105) were plated-out in 6-well plates and infected24 hr later with an empty adenovirus vector or with adenovirusharboring TAp73a or DNp73a cDNA (12.5 pfu). Cells were har-vested 48 hr later and lysed in Laemmli buffer to be analyzed byWestern blotting.
To assess the influence of TAp73a or DNp73 on ADR toxicity,the cells infected with TAp73a or DNp73 were treated with ADRand their ADR IC50 were compared to those obtained in unin-fected cells.
siRNA studies
The oligonucleotide sequences for TAp73 inhibition were:50r(CGGAUUCCAGCAUGGACGU)d(TT)30 and 50r(ACGUCC-ACGUCCAUGCUGGAAUCCG d(TT)30.27
siRNA were obtained from Qiagen (Venlo, The Netherlands)and transfected using the Oligofectamine transfection reagent(Invitrogen). In preliminary studies, cells were treated with50–200 nM siRNA according to the manufacturer’s instructionsto determine optimum concentrations for the downregulation ofTAp73. In later studies, cells were transfected with 200 nM siRNAat approximately 30–40% confluence. Forty-eight hours later, cellswere treated with 0.05 or 0.2 lM of ADR. The effects of siRNAtreatment on cell viability or induction of the TAp73 target genewere assessed 24 hr (indicated as cells ‘‘Day 4’’) or 48 hr (indi-cated as cells ‘‘Day 5’’) after drug treatment. Viability was com-pared to cells receiving siRNA alone.
Microarray membrane technique
Hybridization was performed, as recommended by the manufac-turer, onto membranes containing 1176 cDNA (Atlas HumanCancer 1.2, Clontech, BD Biosciences, Palo Alto, CA). In brief,labeled cDNA were obtained by reverse transcription of RNA (2.5lg) in a reaction mixture containing 13 buffer, 0.5 mM dNTP,13 specific primers, 5 mM DTT, 50 U reverse transcriptaseMMLV and 30 lCi [a-32P]ATP for 30 min at 48�C. cDNA werethen purified on Nucleospin columns (Clontech), and probes weredenatured in denaturing solution for 20 min at 68�C. Clontechmembranes were prehybridized at 68�C in Expresshyb buffer sup-plemented with salmon sperm DNA for 30 min. Probes were thenadded, and the membranes were incubated overnight at 68�C. Themembranes were washed 3 times for 30 min in 23 SSC, 1% SDSat 68�C and 1 time for 30 min in 0.13 SSC, 0.5% SDS, beforebeing placed in a PhosphorImager cassette. Films were scannedand spot hybridization analyzed using AtlasImage 1.01 software(Clontech).
TABLE I – PRIMER PAIRS USED FOR AMPLIFICATION REACTIONS
Gene Primers Annealing Cycles
p53 p1: 50 CCC CTC CTC AGC ATC TTA TCC 30 59�C 25p2: 50 CAC CTC AAA GCT GTT CCG TCC 30
p73 p3: 50 TGG ATG ACC CTG TCA CCG GC 30 65�C 32p4: 50 TGC TCC CGG TAG TGG TCC TCA 30p5: 50 CCG ACC CCA GCC TCG TCA 30 65�C 32p6: 50 CTG AGC CGC CGA TGG AGA T 30
p21 p7: 50 GCG ACT GTG ATG GCG TAA TG 30 60�C 22p8: 50 AGA AGA TCA GCC GGC GTT TG 30
GADD45 p9: 50 GAA GAC CGA AAG GAT GG 30 55�C 28p10: 50 GGG AGA TTA ATC ACT GG 30
14-3-3r p11: 50 AAG GGC TCC GTG GAG AGG G 30 58�C 25p12: 50 AGA GGG GAA CTT TAT TGA GAG G 30
GAPDH g1: 50 CTG CAC CAC CAA CTG CTT AG 30 65�C 22g2: 50 AGG TCC ACC ACT GAC ACG TT 30
861p73 INVOLVEMENT IN BREAST CANCER CELLS CHEMOTHERAPY

DAPI staining and cell apoptosis study
Cell apoptosis was analyzed using DAPI staining. Cells weregrown on culture slides (Labtek, Nunc, Rochester, NY) and treatedwith 0.5 lM ADR for 24 hr and 48 hr. Cells were washed twice withPBS, fixed over 10 min in 3.8% formaldehyde and stained with 40 60diamino 2 phenyl indol (Sigma, St. Louis, MO) over 30 min.
Results
p53 status and ADR sensitivity in breast cancer cell lines
Four breast cancer cell lines were tested in our study: MCF7,MCF7/ADRIGR, MDA-MB157 and T47D. MCF7/ADRIGR is anew, ADR-resistant MCF7 subline that was isolated in our labora-tory. This MCF7/ADRIGR cell line exhibits an ADR resistanceindex of around 1900.
MCF7 and MCF7/ADRIGR demonstrated a wild p53 genotype.In the MDA-MB157 cell line, we have recently described a 26 bpdeletion in p53 exon 4, associated with a lack of p53 protein(Fig. 1).28 Finally, a mutation at codon 194 (exon 6) of the p53gene has previously been reported in the T47D cell line, resultingin stabilization and overexpression of mutant p53 protein incapa-ble of transactivating p53 target genes such as p21.30
The MCF7, MCF7/ADRIGR, MDA-MB157 and T47D cell lineswere analyzed at ADR concentrations yielding similar toxicity.The ADR IC50 was 0.08 lM in MCF7, 150 lM in MCF7/ADRIGR, 0.16 lM in MDA-MB157 and 0.25 lM in T47D.
An overexpression of MDR protein, a protein involved in multi-drug resistance and also termed Pgp170, was found in MCF7/ADRIGR cells, suggesting that MDR is a major drug-resistancemechanism used by MCF7/ADRIGR against ADR (Fig. 2).31 TheMDR protein was not affected by ADR treatment (data notshown).
p53 expression in ADR-treated breast cancer cell lines
p53 mRNA and protein expressions were measured in treatedcell lines using semiquantitative RT-PCR and Western blot,respectively.
At an equivalent cytotoxic index, no variation in p53 mRNAlevels could be detected in ADR-treated MCF7 and MCF7/ADRIGR cells, whereas in ADR-treated MDA-MB157 cells, therewas a complete lack of p53 mRNA transcript. Significant p53 pro-tein accumulation was found in ADR-treated MCF7 cells, in con-trast to a very weak p53 protein increase detected in treatedMCF7/ADRIGR cells (Fig. 1a; Table II). These results show a sig-nificant variation in the extent of p53 activation in response toDNA-damaging agents between the MCF7 cell line and its resist-ant counterpart MCF7/ADRIGR. No p53 protein was detected intreated-MDA-MB157 cells, even after prolonged exposure ofautoradiography film (Fig. 1a and data not shown). As statedabove, mutation at codon 194 provokes stabilization and accumu-lation of p53 protein in untreated T47D cells. We found that ADR
FIGURE 2 – MDR protein expression in breast cancer cell lines. Anal-ysis of MDRPgp170 protein expression by Western blot, using a mono-clonal antibody (clone Ab-1, Oncogene Research). No MDR (Pgp170)protein was expressed in MCF7, MDA-MB157 and T47D cells,whereas MDR (Pgp170) was overexpressed in MCF7/ADRIGR cells.
FIGURE 1 – p53, p73 and p63 protein expression in p53-response-deficient human breast cancer cells. (a) Analysis of p53 protein expressionby Western blot, using a monoclonal antibody (clone DO7, Dako) in ADR-treated MCF7, MCF7/ADRIGR, MDA-MB157 and T47D cells. p53protein accumulated in ADR-treated MCF7 cells and very weakly increased in ADR-treated MCF7/ADRIGR cells. As expected, no p53 proteinwas detected in the p53-deficient cell line MDA-MB157, even after prolonged exposure of the film (data not shown). The p53 mutant T47D cellline exhibited a strong p53 protein accumulation but no variation in p53 protein level with exposure to ADR. (b) Analysis of p73a proteinexpression by Western blotting, using a polyclonal antibody (Sanofi, Labge). Upon ADR treatment, TAp73a protein level increased in MCF7,MCF7/ADRIGR and MDA-MB157 cells, but TAp73a and DNp73a protein levels decreased in T47D cells. SK-N-AS cells transfected (SK-N-AS-Tr) with TAp73a or DNp73a were used as positive controls to discriminate between the 2 isoforms. (c) Analysis of p63a protein expressionby Western blotting, using a monoclonal antibody (clone 4A4, Santa Cruz). No significant TAp63a protein level variation was observed inADR-treated MCF7, MCF7/ADRIGR and MDA-MB157 cells. In contrast, p63a protein decreased in ADR-treated T47D cells; note the decreaseof another p63 isoform migrating faster than TAp63a protein, possibly DNp63a. The blot was rehybridized with anti-actin monoclonal antibody(clone C4, Chemicon) used as loading control.
862 VAYSSADE ET AL.

did not cause any further increase in p53 protein levels in theT47D cell line (Fig. 1a).
Either due to very weak basal p53 induction (MCF/ADRIGR), atotal lack of p53 (MDA-MB157) or a constitutive mutation givingrise to a nonfunctional p53 (T47D), in contrast to MCF7, these 3cell lines did not show any significant p53 activation in responseto ADR treatment and are referred to as ‘‘p53-nonresponsive orp53-response deficient’’ hereafter.
ADR treatment induces p73 overexpression in breast cancercell lines
Testing the assumption that p73 could replace p53 functions inADR-treated p53-deficient cells, we analyzed p73 total transcriptlevels using semiquantitative RT-PCR in ADR-treated breast can-cer cell lines. Primers were designed so as to be capable of detect-ing both a and b isoforms or distinguishing individual isoforms.
The level of p73 transcript was unchanged in ADR-treatedMCF7 and MCF7/ADRIGR cells (Fig. 3a, Table II). However, intreated p53-deficient MDA-MB157 cells, p73 gene expressionincreased up to 2.5-fold after ADR treatment (Fig. 3a, Table II).These results were confirmed by p73a and p73b mRNA expres-sion analysis. Remarkably, in ADR-treated MDA-MB157 cells,the level of p73b mRNA was increased to a greater extent thanp73a (6 vs. 2.5-fold) (Fig. 3b, Table II). Intriguingly, p73 tran-script levels were downregulated in ADR-treated T47D cells(Fig. 3, Table II and data not shown).
p73 protein expression was analyzed using Western blot, with apolyclonal antibody directed against the p73a isoform-COOH ter-minal domain. Significant TAp73a protein accumulation wasfound in ADR-treated MCF7 and MCF7/ADRIGR cells (Fig. 1b,Table II), whereas no change in p73 transcript levels was found inthese cells, as described above. These data suggest an increase in
the half-life of the TAp73a protein. In accordance with theincrease in p73 transcript levels, MDA-MB157 cells demonstratedTAp73a protein overexpression after ADR treatment, as statedabove. Among the 4 breast cancer cell lines tested in our study,only T47D expressed a high level of DNp73a, the p73 isoformwith no transactivation domain. A sharp decrease in TAp73a andDNp73a protein expression was observed in T47D at high ADRconcentrations (Fig. 1b).
We have also treated cell lines with CDDP and found TAp73aprotein accumulation in treated-MCF7 and MDA-MB157 cells(data not shown).
p63a expression in ADR-treated breast cancer cell lines
p63, the third member of the p53 family, was tested as a poten-tial candidate for activation after ADR treatment. No significantvariation in TAp63a expression could be detected in ADR-treatedMCF7, MCF7/ADRIGR or MDA-MB157 cells (Fig. 1c, Table II).In contrast, ADR-treated T47D cells exhibited a decrease inTAp63a and in another p63 isoform, which migrated faster thanTAp63a and was possibly DNp63a (Fig. 1c, Table II).
Together, these data clearly indicate that, in contrast to p53 andp73, p63a protein does not play an active role in the cellularresponse to ADR.
p53-family target gene expression and cell apoptosis inADR-treated breast cancer cell lines
To assess whether p73 accumulation in weakly p53-responsiveMCF7/ADRIGR cells and in constitutively null p53 MDA-MB157cells could lead to the induction of p53-target genes, we analyzedp21, GADD45, 14-3-3r and p53R2 mRNA and protein expres-sions in ADR-treated breast cancer cell lines.
TABLE II – p53-FAMILY MEMBERS AND TARGET GENES mRNA AND PROTEIN EXPRESSION IN ADRIAMYCIN (ADR) TREATEDBREAST CANCER CELL LINES1
p53 p73
mRNA Proteinp73a p73b p63 p21 GADD45 14-3-3r p53R2
mRNA Protein mRNA Protein mRNA Protein mRNA Protein mRNA Protein Protein
MCF7 5 ƒƒ 5 ƒƒ 5 5 ƒƒ ƒƒ ƒ ƒ 5 ƒ 5MCF7/ADRIGR 5 ƒ 5 ƒƒ 5 5 ƒ ƒƒ ƒ ƒ 5 ƒ ƒMDA-MB157 0 0 ƒ ƒƒ ƒƒ 5 ƒ ƒƒ ƒ ƒ 5 0 ƒƒT47D ND 5 # # # # # # # n.d. 5 n.d. 5 n.d. ƒ 5
1Transcript levels were assessed by semiquantitative RT-PCR. PCR fragments were separated on a polyacrylamide gel, quantitated using aChromoscan 3 Joyce Loebl and normalised to GAPDH. Proteins levels were measured by Western blotting. 0, no detectable expression; 5, nochange in expression; ƒ, moderately increased expression; ƒƒ, markedly increased expression; # , slightly decreased expression; # # , mark-edly decreased expression; n.d., not determined.
FIGURE 3 – p73 mRNA analysis in ADR-treated breast cancer cell lines. (a) Semiquantitative RT-PCR analysis of p73 gene transcriptlevels using primers located in DNA binding domain (exons 6–8), common to all p73 transcript isoforms. p73 transcript levels weredetermined after 32 cycles. GAPDH cDNA was amplified after 22 cycles and used as internal control. For each sample, 2 ll of amplifiedp73 cDNA and 2 ll of amplified GAPDH cDNA were mixed and electrophoresed on polyacrylamide gels. Overall, p73 transcript levelwas unchanged in ADR-treated MCF7 and MCF7/ADRIGR but increased in MDA-MB157 cells and decreased in T47D cells. (b) Semi-quantitative RT-PCR analysis of p73a and p73b transcripts using primers encompassing exons 12 and 14. p73a and p73b expressionremained unchanged in ADR-treated MCF7 and MCF7/ADRIGR cells but increased in ADR-treated MDA-MB157 cells (2.5-fold and 6-fold, respectively) and decreased in T47D cells.
863p73 INVOLVEMENT IN BREAST CANCER CELLS CHEMOTHERAPY

p21 and GADD45 mRNA and protein levels were increasedin ADR-treated MCF7, MCF7/ADRIGR and MDA-MB157 cells(Figs. 4 and 5; Table II) but remained unchanged in T47D cells.
14-3-3r transcript level was only slightly increased in bothADR-treated MCF7 and MCF7/ADRIGR cells (Fig. 4c, Table II).The 14-3-3r transcript could only be detected in MDA-MB157cells after numerous PCR cycles (34 cycles instead of 25 for
MCF7 and MCF7ADRIGR). No variation in the level of 14-3-3rmRNA was seen in these ADR-treated cells, in contrast to thestrong (9-fold) increase detected after 72 hr of treatment (Fig. 4c,Table II and data not shown). 14-3-3r protein level was increasedin ADR-treated MCF7 and MCF7/ADRIGR cells (Fig. 5c,Table II) but remained undetectable in MDA-MB157 cells, evenafter ADR treatment. In T47D cells, significant induction of
FIGURE 4 – p21 (a), GADD45 (b) and 14-3-3r (c) mRNA expression in ADR-treated MCF7, MCF7/ADRIGR and MDA-MB157 cells. Semi-quantitative RT-PCR analysis of gene transcripts using GAPDH gene expression as internal control. (a) Upon ADR treatment, p21 gene expres-sion was increased in MCF7 cells, whereas a very weak increase had been found in MCF7/ADRIGR and MDA-MB157 cells. (b) GADD45 tran-script level was weakly increased in ADR-treated MCF7, MCF7/ADRIGR and MDA-MB157 cells. (c) 14-3-3r transcript level was unchanged inADR-treated MCF7 and MCF7/ADRIGR cells and undetectable in MDA-MB157 cells after 25 cycles of PCR.
FIGURE 5 – p21, GADD45, 14-3-3r and p53R2 protein levels in ADR-treated MCF7, MCF7/ADRIGR, MDA-MB157 and T47D cells. Proteinexpression was detected using antibodies as described in Material and Methods. (a) p21 protein level was strongly increased in ADR-treatedMCF7, MCF7/ADRIGR and MDA-MB157 cells. (b) GADD45 protein levels increase in ADR-treated MCF7, MCF7/ADRIGR and MDA-MB157cells. (c) Immunoblotting revealed an increase of 14-3-3r protein in ADR-treated MCF7 and MCF7/ADRIGR cells, whereas no 14-3-3r expres-sion was detected in MDA-MB157, probably due to the low constitutive level in this cell line. Analysis of ADR-treated T47D cells exhibitedincreased 14-3-3r protein level. (d) Upregulation of p53R2 protein was present in ADR-treated MCF7/ADRIGR and MDA-MB157 cells. Theblot was rehybridized with anti-actin monoclonal antibody (clone C4, Chemicon) used as loading control.
864 VAYSSADE ET AL.

14-3-3r protein expression was observed after ADR treatment,contrasting with the steady protein levels from other p53-induciblegenes.
p53R2, a ribonucleotide reductase gene, is a direct target forp53 that is also implicated in DNA repair activity after genotoxicstresses.32 Intriguingly, our data demonstrate that p53R2 expres-sion is strongly increased in MCF7/ADRIGR and MDA-MB157cells after ADR treatment, in contrast to the lack of induction seenin treated MCF7 and T47D cells (Fig. 5d, Table II).
In conclusion, we have shown that p53 target genes could beinduced in MCF7/ADRIGR and MDA-MB157 cells that lack p53activation but in which TAp73a is overexpressed after ADR treat-ment.
Finally, the DAPI staining technique was used to demonstratethat ADR treatment triggers cell apoptosis in p53-deficient MDA-MB157 cells. Our data revealed that MDA-MB157 cells displaynuclear fragmentation after ADR treatment (Fig. 6).
TAp73 cDNA infection induces p53-target gene expression inp53-deficient cell lines and increases ADR toxicity
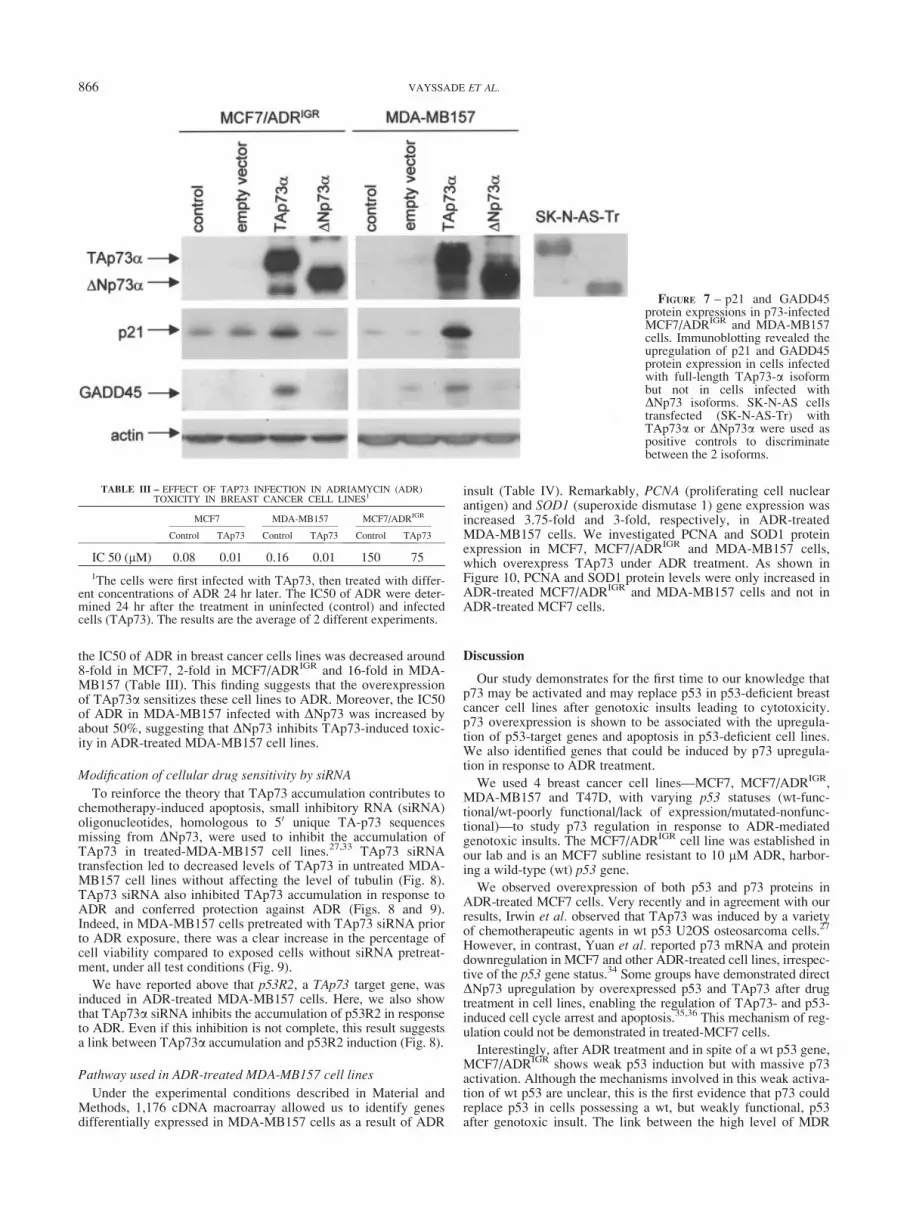
To emphasize our observation of p53-target gene induction inADR-treated MCF7/ADRIGR and MDA-MB157 cells associatedwith endogenous p73 overexpression, we induced exogenous p73overexpression. This was achieved by infecting these p53-response-deficient cells with an adenovirus containing full-lengthTA or truncated DN p73a isoform cDNA (Fig. 7). p21 andGADD45 protein levels were upregulated in cells infected withfull-length TAp73, suggesting that, in contrast to the DN isoform,TAp73a protein is able to induce p21 and GADD45 expressionsin our system (Fig. 7). Similar results were obtained when MCF7/ADRIGR and MDA-MB157 cells were infected with the TA andDN p73b isoforms (data not shown).
To test the effect of TAp73 in ADR toxicity, MCF7, MCF7/ADRIGR and MDA-MB157 cell lines were infected with TAp73acDNA and then treated with ADR. As expected, after infection,
FIGURE 6 – Apoptosis at cellularlevel in ADR-treated MDA-MB157 cells using DAPI stainingtechnique. MDA-MB157 cellswere cultured for 48 hr without (a)or with 0.5 lM ADR (b,c) andthereafter fixed and stained withDAPI. (a) Control cells withoutany treatment coloration withDAPI (magnification 4003). (b1,b2) Cells treated with 0.5 lMADR, with visible signs of thebeginning of nucleus fragmenta-tion (start of apoptosis); magnifica-tion 4003 (b1) and 1,000x (b2).(c1–c3) The end of the apoptosiswith clearly visible apoptoticbodies (magnification 1,0003).
865p73 INVOLVEMENT IN BREAST CANCER CELLS CHEMOTHERAPY
the IC50 of ADR in breast cancer cells lines was decreased around8-fold in MCF7, 2-fold in MCF7/ADRIGR and 16-fold in MDA-MB157 (Table III). This finding suggests that the overexpressionof TAp73a sensitizes these cell lines to ADR. Moreover, the IC50of ADR in MDA-MB157 infected with DNp73 was increased byabout 50%, suggesting that DNp73 inhibits TAp73-induced toxic-ity in ADR-treated MDA-MB157 cell lines.
Modification of cellular drug sensitivity by siRNA
To reinforce the theory that TAp73 accumulation contributes tochemotherapy-induced apoptosis, small inhibitory RNA (siRNA)oligonucleotides, homologous to 50 unique TA-p73 sequencesmissing from DNp73, were used to inhibit the accumulation ofTAp73 in treated-MDA-MB157 cell lines.27,33 TAp73 siRNAtransfection led to decreased levels of TAp73 in untreated MDA-MB157 cell lines without affecting the level of tubulin (Fig. 8).TAp73 siRNA also inhibited TAp73 accumulation in response toADR and conferred protection against ADR (Figs. 8 and 9).Indeed, in MDA-MB157 cells pretreated with TAp73 siRNA priorto ADR exposure, there was a clear increase in the percentage ofcell viability compared to exposed cells without siRNA pretreat-ment, under all test conditions (Fig. 9).
We have reported above that p53R2, a TAp73 target gene, wasinduced in ADR-treated MDA-MB157 cells. Here, we also showthat TAp73a siRNA inhibits the accumulation of p53R2 in responseto ADR. Even if this inhibition is not complete, this result suggestsa link between TAp73a accumulation and p53R2 induction (Fig. 8).
Pathway used in ADR-treated MDA-MB157 cell lines
Under the experimental conditions described in Material andMethods, 1,176 cDNA macroarray allowed us to identify genesdifferentially expressed in MDA-MB157 cells as a result of ADR
insult (Table IV). Remarkably, PCNA (proliferating cell nuclearantigen) and SOD1 (superoxide dismutase 1) gene expression wasincreased 3.75-fold and 3-fold, respectively, in ADR-treatedMDA-MB157 cells. We investigated PCNA and SOD1 proteinexpression in MCF7, MCF7/ADRIGR and MDA-MB157 cells,which overexpress TAp73 under ADR treatment. As shown inFigure 10, PCNA and SOD1 protein levels were only increased inADR-treated MCF7/ADRIGR and MDA-MB157 cells and not inADR-treated MCF7 cells.
Discussion
Our study demonstrates for the first time to our knowledge thatp73 may be activated and may replace p53 in p53-deficient breastcancer cell lines after genotoxic insults leading to cytotoxicity.p73 overexpression is shown to be associated with the upregula-tion of p53-target genes and apoptosis in p53-deficient cell lines.We also identified genes that could be induced by p73 upregula-tion in response to ADR treatment.
We used 4 breast cancer cell lines—MCF7, MCF7/ADRIGR,MDA-MB157 and T47D, with varying p53 statuses (wt-func-tional/wt-poorly functional/lack of expression/mutated-nonfunc-tional)—to study p73 regulation in response to ADR-mediatedgenotoxic insults. The MCF7/ADRIGR cell line was established inour lab and is an MCF7 subline resistant to 10 lM ADR, harbor-ing a wild-type (wt) p53 gene.
We observed overexpression of both p53 and p73 proteins inADR-treated MCF7 cells. Very recently and in agreement with ourresults, Irwin et al. observed that TAp73 was induced by a varietyof chemotherapeutic agents in wt p53 U2OS osteosarcoma cells.27
However, in contrast, Yuan et al. reported p73 mRNA and proteindownregulation in MCF7 and other ADR-treated cell lines, irrespec-tive of the p53 gene status.34 Some groups have demonstrated directDNp73 upregulation by overexpressed p53 and TAp73 after drugtreatment in cell lines, enabling the regulation of TAp73- and p53-induced cell cycle arrest and apoptosis.35,36 This mechanism of reg-ulation could not be demonstrated in treated-MCF7 cells.
Interestingly, after ADR treatment and in spite of a wt p53 gene,MCF7/ADRIGR shows weak p53 induction but with massive p73activation. Although the mechanisms involved in this weak activa-tion of wt p53 are unclear, this is the first evidence that p73 couldreplace p53 in cells possessing a wt, but weakly functional, p53after genotoxic insult. The link between the high level of MDR
FIGURE 7 – p21 and GADD45protein expressions in p73-infectedMCF7/ADRIGR and MDA-MB157cells. Immunoblotting revealed theupregulation of p21 and GADD45protein expression in cells infectedwith full-length TAp73-a isoformbut not in cells infected withDNp73 isoforms. SK-N-AS cellstransfected (SK-N-AS-Tr) withTAp73a or DNp73a were used aspositive controls to discriminatebetween the 2 isoforms.
TABLE III – EFFECT OF TAP73 INFECTION IN ADRIAMYCIN (ADR)TOXICITY IN BREAST CANCER CELL LINES1
MCF7 MDA-MB157 MCF7/ADRIGR
Control TAp73 Control TAp73 Control TAp73
IC 50 (lM) 0.08 0.01 0.16 0.01 150 75
1The cells were first infected with TAp73, then treated with differ-ent concentrations of ADR 24 hr later. The IC50 of ADR were deter-mined 24 hr after the treatment in uninfected (control) and infectedcells (TAp73). The results are the average of 2 different experiments.
866 VAYSSADE ET AL.

protein in MCF7/ADRIGR and the noninduction of wild-type p53after ADR treatment needs clarification. One could hypothesizethat the overexpression of MDR protein blocks p53 activation,which is rescued by p73 induction.
In MDA-MB157 cells, which do not express p53 at all, it is alsolikely that p73 functions could take over p53 functions. The mod-est (2–6-fold) upregulation of p73 mRNA expression and the sig-nificant (around 25–50-fold) induction of p73 protein in ADR-treated MDA-MB157 cells strongly suggests the stabilization ofp73 protein as described above for MCF7 and MCF7/ADRIGR.
Our results show that TAp73 protein overexpression inADR-treated MDA-MB157 and MCF7/ADRIGR cells is associ-ated with the induction of apoptosis and the upregulation ofp53 target gene, suggesting that p73 may have some p53 func-tions in certain cell lines that are unable to activate p53. Thisassumption is further supported by the fact that TAp73 siRNAconfers protection against ADR in MDA-MB157, indicatingthat TAp73 accumulation is involved in chemotherapy-induced
apoptosis. In agreement with these results, Irwin et al. havereported that, in U2OS osteosarcoma cells, chemoresistance isenhanced by blocking TAp73 function using TAp73 siRNAtransfection.27 Moreover, we demonstrate that exogenousTAp73 overexpression in MDA-MB157 and MCF7/ADRIGR
induces the upregulation of p53 target genes and sensitizes thecell lines to ADR (Fig. 7, Table III), whereas exogenous over-expression of DNp73, an anti-apoptotic TAp73-antagonistic iso-form, induces ADR resistance, supporting the theory thatTAp73 could be implicated in ADR toxicity.
T47D cells with a mutated and nonfunctional p53 were found tooverexpress DNp73. Recently, it has been reported that the induc-tion or overexpression of DNp73 promoted cell survival but com-petition with p53 and TAp73.13,14 The authors also found thatcamptothecin-induced apoptosis was enhanced after the abolitionof DNp73 expression using antisense oligonucleotides. In agree-ment with these data, we demonstrated that ADR induces thedownregulation of DNp73 mRNA transcript and protein levels in
FIGURE 8 – Inhibition of TAp73a and p53R2 expressions in ADR-treated MDA-MB 157 cells by TAp73a siRNA. MDA-MB157 cell linewas transfected with TAp73 siRNA and subsequently treated with ADR. Immunoblotting revealed that TAp73 siRNA blocks the induction ofTAp73 and weakly inhibits p53R2 expression in ADR-treated MDA-MB157. The blot was rehybridized with anti-tubulin monoclonal antibody(CP06, Oncogene Research) used as loading control.
FIGURE 9 – Analysis of ADR toxicity in MDA-MB157 transfected with TAp73 siRNA. Twenty-four hours after plating, cells were treatedwith (black bars) or without (white bars) TAp73 siRNA. Forty-eight hours later, cells were exposed to 0.05 or 0.2 lM ADR. After 24 hr (Day 4)or 48 hr (Day 5), cells were harvested and the viability was calculated relative to control cells without any ADR treatment. After pretreatmentwith TAp73 siRNA (black bars), there was a clear increase of viability of MDA-MB157 cells exposed to ADR compared to cells exposed todrug without siRNA pretreatment (white bars) under all test conditions.
867p73 INVOLVEMENT IN BREAST CANCER CELLS CHEMOTHERAPY

T47D and that this process is associated with cell apoptosis andtoxicity. Currently, we are trying to clarify the mechanismsunderlying DN degradation, since the concurrent decrease inTAp73 in ADR-treated T47D reveals a complex regulation ofp73 isoform expression. Our findings appear to differ from thoseof Bergamaschi et al., who reported an increase in the levels ofendogenous TAp73 in all p53-mutant squamous cell carcinomacell lines after exposure to doxorubicin.26 However, none of thecell lines used in Bergamaschi’s study constitutively overex-pressed DNp73. Therefore, our data suggests that the inductionand activity of TAp73 in mutant p53 cells could also depend onwhether the anti-apoptotic isoform, DNp73, is expressed in thesecells lines. Finally, our findings prompt an investigation intowhether other chemotherapeutic drugs are able to suppressendogenous DNp73 expression, thereby increasing the ability ofthese drugs to induce apoptosis.
The p53R2 gene is a new p53-inducible gene encoding a ribo-nucleotide reductase, a protein shown to play a role in the repairof DNA damage.32 Interestingly, we found an overexpression ofp53R2 in ADR-treated MDA-MB157 and MCF7/ADRIGR cellsbut not in ADR-treated MCF7 and T47D cells, suggesting that thisprotein has a role in mediating certain p53-independent DNAdamage-response pathways. Remarkably and directly linked tothis assumption, TAp73 siRNA inhibits p53R2 induction in ADR-treated MDA-MB157 cell lines, suggesting a link between TAp73overexpression and p53R2 accumulation. However, this inhibitionwas not complete, suggesting that additional pathways could be
involved in p53R2 transactivation in ADR-treated MDA-MB157cell lines. PCNA and SOD1 transcript levels were also increasedin ADR-treated MDA-MB157 cells. The overexpression of PCNAand SOD1 proteins in ADR-treated MCF7/ADRIGR and MDA-MB157 cells, but not in ADR-treated MCF7 cells, supports thetheory that p73 could participate in the induction of certain genesin ‘‘p53-deficient’’ cell lines in response to some drugs. Strength-ening this assumption, we demonstrated SOD1 and PCNA upregu-lation in MCF7/ADRIGR and MDA-MB157 ectopically overex-pressing TAp73 after adenoviral TAp73 infection (data notshown). We are currently trying to clarify the induction mecha8-nisms for these genes.
Taken together, our data indicate that p73 may assume the func-tions of p53 in breast cancer cell lines that lack drug-induced p53activation. However, p73 may not be able to assume all of p53’sfunctions, particularly in cells with both p53 mutation and DNp73isoform overexpression. Further studies are needed to determinethe link between the presence of DNp73, TAp73 and the cellularresponse to chemotherapy in breast cancer cells with p53mutations.
Acknowledgements
We thank Dr. D. Caput and Dr. M. Kaghad (Sanofi-Labege) forproviding p73 antibody, and Dr. D. Cappellen, Gwenaelle Lerouxand Etienne Blanc for their expert technical assistance.
TABLE IV – GENES DIFFERENTIALLY EXPRESSED IN MDA-MB157 CELLS TREATED WITH ADRIAMYCIN (ADR)
Gene/protein name Ratio1
Cyclin-dependent kinase inhibitor 1A (p21) 2Growth arrest and DNA damage-inducible protein (GADD45) 2Proliferating cyclic nuclear antigen (PCNA); cyclin 3.75Tyrosine kinase receptor tie-1 precursor 2.49Cytosolic superoxide dismutase 1 (SOD1) 3.02Histone H4 2.09Neural cell adhesion molecule L1 precursor (N-CAM L1); MIC5 0.4660S ribosomal protein L32 2.04Ribonucleoside-diphosphate reductase M2 subunit; ribonucleotide reductase 2.54
1After hybridization, the genes’ expressions were normalized to 9 housekeeping genes, and the profilesof the genes’ expressions in untreated and ADR-treated MDA-MB157 cells were then compared usingAtlasImage 1.01 software (Clontech). For each gene, the ratio represents ADR-treated MDA-MB157 cellgene expression over untreated MDA-MB157 cell gene expression. Ratios > 2 or < 0.5 are shown.
FIGURE 10 – PCNA and SOD protein expressions in ADR-treated MCF7, MCF7/ADRIGR and MDA-MB157 cells. Western blotting revealedPCNA and SOD1 expression induction in ADR-treated MCF7/ADRIGR and MDA-MB157 cells.
868 VAYSSADE ET AL.

References
1. El-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, TrentJM, Lin D, Mercer EW, Kinzler KW, Vogelstein B. WAF1, a poten-tial mediator of p53 tumor suppression. Cell 1993;75:817–25.
2. Hermeking H, Lengauer C, Polyak K, He TC, Zhang L, ThiagalingamS, Kinzler KW, Vogelstein B. 14-3-3r is a p53-regulated inhibitor ofG2/M progression. Mol Cell 1997;1:3–11.
3. Kastan MB, Zhan Q, El-Deiry WS, Carrier F, Jacks T, Walsh WV,Plunkett BS, Vogelstein B, Fornace AJ Jr. A mammalian cell cyclecheckpoint pathway utilizing p53 and GADD45 is defective in ataxia-telengiectasia. Cell 1992;71:587–97.
4. Miyashita T, Krajewski S, Krajewska M, Wang HG, Lin HK, Lieber-mann DA, Hoffman B, Reed JC. Tumor suppressor p53 is a regulatorof bcl-2 and bax gene expression in vitro and in vivo. Oncogene1994;9:1799–805.
5. Levine AJ. p53, the cellular gatekeeper for growth and division. Cell1997;88:323–31.
6. Gartenhaus RB, Wang P, Hoffmann P. Induction of the WAF1/CIP1protein and apoptosis in human T-cell leukemia virus type I-trans-formed lymphocytes after treatment with Adriamycin by using a p53-independent pathway. Proc Natl Acad Sci USA 1996;93:265–8.
7. Bertheau P, Plassa F, Espie M, Turpin E, de Roquancourt A, MartyM, Lerebours F, Beuzard Y, Janin A, de The H. Effect of mutatedTP53 on response of advanced breast cancers to high-dose chemother-apy. Lancet 2002;360:852–4.
8. Kaghad M, Bonnet H, Yang A, Creancier L, Biscan JC, Valent A, MintyA, Chalon P, Lellas JM, Dumont X, Ferrara P, McKeon F, et al. Mono-allelically expressed gene related to p53 at 1p36, a region frequent-ly deleted in neuroblastoma and other human cancers. Cell 1997;90:809–19.
9. Osada M, Ohba M, Kawahara C, Ishioka C, Kanamaru R, Katoh I,Ikawa Y, Nimura Y, Nakagawara A, Obinata M, Ikawa S. Cloningand functional analysis of human p51, which structurally and func-tionally resembles p53. Nat Med 1998;4:839–43.
10. Trink B, Okami K, Wu L, Sriuranpong V, Jen J, Sidransky D. A newhuman p53 homologue. Nat Med 1998;4:747–8.
11. Yang A, Kaghad M, Wang Y, Gillett E, Fleming MD, D€otsch V,Andrews NC, Caput D, McKeon F. p63, a p53 homolog at 3q27-29,encodes multiples products with transactivating, death-inducing, anddominant-negative activities. Mol Cell 1998;2:305–16.
12. Yang A, Walker N, Bronson R, Kaghad M, Oosterwegel M, Bonnin J,Vagner C, Bonnet H, Dikkes P, Sharpe A, McKeon F, Caput D.p73-deficient mice have neurological, pheromonal and inflammatorydefects but lack spontaneous tumours. Nature 2000;404:99–103.
13. Ishimoto O, Kawahara C, Enjo K, Obinata M, Nukiwa T, Ikawa S.Possible oncogenic potential of DeltaNp73: a newly identified isoformof human p73. Cancer Res 2002;62:636–41.
14. Zaika AI, Slade N, Erster SH, Sansone C, Joseph TW, Pearl M, Cha-las E, Moll U. DeltaNp73, a dominant-negative inhibitor of wild-typep53 and TAp73 is up-regulated in human tumors. J Exp Med 2002;196:765–80.
15. B�enard J, Douc-Rasy S, Ahomadegbe JC. TP53 family members andhuman cancers. Human Mutat 2003;21:182–91.
16. Jost CA, Marin MC, Kaelin WG Jr. p73 is a human p53-related pro-tein that can induce apoptosis. Nature 1997;389:191–4.
17. Zhu J, Jiang J, Zhou W, Chen X. The potential tumor suppressor p73differentially regulates cellular p53 target genes. Cancer Res 1998;58:5061–5.
18. Shimada A, Kato S, Enjo K, Osada M, Ikawa Y, Kohno K, ObinataM, Kanamaru R, Ikawa S, Ishioka C. The transcriptional activities ofp53 and its homologue p51/p63: similarities and differences. CancerRes 1999;59:2781–6.
19. Miro-Mur F, Meiller A, Haddada H, May E. p73a expression inducesboth accumulation and activation of wt-p53 independent of the p73atranscriptional activity. Oncogene 2003;22:5451–6.
20. Fang L, Lee SW, Aaronson SA. Comparative analysis of p73 and p53regulation and effector functions. J Cell Biol 1999;147:823–30.
21. Vikhanskaya F, D’Incalci M, Broggini M. p73 competes with p53 andattenuates its response in a human ovarian cancer cell line. NucleicAcids Res 2000;28:513–9.
22. Agami R, Blandino G, Oren M, Shaul Y. Interaction of c-Abl andp73a and their collaboration to induce apoptosis. Nature 1999;399:809–13.
23. Gong JG, Costanzo A, Yang HQ, Melino G, Kaelin WG Jr, LevreroM, Wang JYG. The tyrosine kinase c-Abl regulates p73 in apop-totic response to cisplatin-induced DNA damage. Nature 1999;399:806–9.
24. Yuan ZM, Shioya H, Ishiko T, Sun X, Gu J, Huang YY, Lu H,Kharbanda S, Weichselbaum R, Kufe D. p73 is regulated by tyrosinekinase c-Abl in the apoptotic response to DNA damage. Nature 1999;399:814–7.
25. Flores ER, Tsai KY, Crowley D, Sengupta S, Yang A, McKeon F,Jacks T. p63 and p73 are required for p53-dependent apoptosis inresponse to DNA damage. Nature 2002;416:560–4.
26. Bergamaschi D, Gasco M, Hiller L, Sullivan A, Syed N, Trigiante G,Yulug I, Merlano M, Numico G, Comino A, Attard M, Reelfs O,et al. p53 polymorphism influences response in cancer chemotherapyvia modulation of p73-dependent apoptosis. Cancer Cell 2003;3:387–402.
27. Irwin MS, Kondo K, Marin MC, Cheng LS, Hahn WC, Kaelin WG Jr.Chemosensitivity linked to p73 function. Cancer Cell 2003;3:403–10.
28. Vayssade M, Faridoni-Laurens L, B�enard J, Ahomadegbe JC. Expres-sion of p53-family members and associated target molecules in breastcancer cell lines in response to vincristine treatment. Biochem Phar-macol 2002;63:1611–9.
29. Ahomadegbe JC, Barrois M, Fogel S, Le Bihan ML, Douc-Rasy S,Duvillard P, Armand JP, Riou G. High incidence of p53 alterations(mutation, deletion, overexpression) in head and neck primary tumorsand metastases; absence of correlation with clinical outcome. Fre-quent protein overexpression in normal epithelium and in early non-invasive lesions. Oncogene 1995;10:1217–27.
30. Guillot C, Falette N, Paperin MP, Courtois S, Gentil-Perret A,Treilleux I, Ozturk M, Puisieux A. p21WAF1/CIP1 response to geno-toxic agents in wild-type TP53 expressing breast primary tumours.Oncogene 1997;14:45–52.
31. Fairchild CR, Ivy SP, Kao-Shan CS, Whang-Peng J, Rosen N, IsraelMA, Melera PW, Cowan KH, Goldsmith ME. Isolation of amplifiedand overexpressed DNA sequences from Adriamycin-resistant humanbreast cancer cells. Cancer Res 1987;47:5141–8.
32. Nakano K, Balint E, Ashcroft M, Vousden KH. A ribonucleotidereductase gene is a transcriptional target of p53 and p73. Oncogene2000;19:4283–9.
33. Elbashir SM, Harboth J, Lendeckel W, Yalcin A, Weber K, Tuschl T.Duplexes of 21-nucleotide RNAs mediate RNA interference in cul-tured mammalian cells. Nature 2001;411:494–8.
34. Yuan R, Meng Q, Hu H, Goldberd ID, Rosen EM, Fan S. p53-inde-pendent downregulation of p73 in human cancer cells treated withAdriamycin. Cancer Chemother Pharmacol 2001;47:161–9.
35. Nakagawa T, Takahashi M, Ozaki T, Watanabe Ki K, Todo S,Mizuguchi H, Hayakawa T, Nakagawara A. Autoinhibitory regulationof p73 by delta Np73 to modulate cell survival and death through ap73-specific target element within the delta Np73 promoter. Mol CellBiol 2002;22:2575–85.
36. Vossio S, Palescandolo E, Pediconi N, Moretti F, Balsano C, LevreroM, Costanzo A. DNp73 is activated after DNA damage in a p53-dependent manner to regulate p53-induced cell cycle arrest. Oncogene2002;23:3796–803.
869p73 INVOLVEMENT IN BREAST CANCER CELLS CHEMOTHERAPY





























