Embed Size (px)
Citation preview

Torfron, Vol . 24, No . 3, pp . 259-272, 1986.
0041-0101/86 53 .00+ .00
Printed in Great Britain.
© 1986 Peraamon Press Ltd .
PHOSPHOLIPASE A2 FROM BOTHROPS ALTERNATUS (VfBORADE LA CRUZ) VENOM. PURIFICATION AND SOME
CHARACTERISTIC PROPERTIES
H. E . NISENBOM, t C. SEKI2 and J . C. VIDAL3*Instituto de Neurobiologia (IDNEU) Serrano 665, 1414 Buenos Aires, Argentina
(Acceptedforpublication 8 August 1985)
H. E . NISENBOM, C. SEKI and J. C. VIDAL . Phospholipase A, from Bothrops alternatus (vfbora dela cruz) venom. Purification and some characteristic properties . Toxicon 24, 259-272, 1986 . -One single protein species with phospholipase activity has been isolated from Bothrops alternatusvenom by a procedure involving gel-filtration on Sephadex G-50 (Step 1), chromatography on SP-Sephadex C-50 (Step 2) and gel-filtration on Sephadex G-75 (Step 3) . The purified sample behavedas a homogeneous, monodisperse protein with a molecular weight of 15,000 and isoelectric pointof 5.04 . The yield in enzyme activity was 48% of the starting material and the apparentpurification was 51-fold . When assayed on 1,2-diheptanoyl- or 1,2-dimyristoyl-sn-glycero-3-phosphorylcholine, fatty acids and lysolecithins were the only reaction products, in accordancewith the predicted stoichiometry. Studies on positional specificity suggested that the enzyme is aphospholipase A, . The enzyme requires Ca" ions for activity and exhibited stereochemicalspecificity, since the enantiomeric 2,3-diheptanoyl-sn-glycero-l-phosphorylcholine was nothydrolyzed . Under the experimental conditions employed, reaction products representative ofeither phospholipase B or C activities could not be detected . After Step 1, the phospholipaseactivity recovered was higher than the total activity in the crude venom sample, which is explainedby the separation of an inhibitor during enzyme purification . The inhibitor was responsible fortheinitial lag period that characterized the kinetics ofthe enzyme reaction with crude venom acting onaggregated substrates (lipoprotein, vesicles or micelles), while the rate of hydrolysis ofmonomericlecithins was not affected .
INTRODUCTION
PHOSPHOLIPASES A, (phosphatide acyl hydrolase, EC 3 .1 .1 .4) catalyze the hydrolysis ofthe fatty acyl ester bond in position 2 of the 1,2-diacyl(1-alkenyl-2-acyl or 1-alkyl-2-acyl)-sn-glycero-3-phosphoryl analogs releasing free fatty acids and 1-acyl(1-alkenyl or 1-alkyl)-sn-glycero-3-phosphoryl analog derivatives (lysophosphatides) (VAN DEENEN and DEHAAS, 1963) via an O-acyl cleavage (WELLS, 1971). Enzymes catalyzing this reaction arewidely distributed among animal tissues (GALLAI-HATCHARD and THOMPSON, 1965),however, the richest sources of this enzyme are snakevenoms . Thus, partial or completelypurified phospholipases A, have been obtained from a wide variety of venoms (forreviews see IWANAGA and SUZUKI, 1979 ; ROSENBERG, 1979).
'Research Fellow, Consejo Nacional de Investigaciones Cientffcas y T6cnicas (CONICET), Argentina .'Career Technician, Consejo Nacional de Investigaciones Cientfffcas y T6cnicas (CONICET), Argentina.'Career Investigator, Consejo Nacional de Investigaciones Cientificas y T6cnicas (CONICET), Argentina .`Present address : IQUIFIB (UBA-CONICET), Faculty of Pharmacy and Biochemistry, University of Buenos
Aires, Junin 956, 1113 Buenos Aires, Argentina .
259

260
H. E. NISENBOM et al.
Bothrops venoms have been usually considered to be poor sources of phospholipase Az(MARINETTI, 1965 ; MEBS, 1970), however, it has been shown (VIDAL and STOPPANI,1971 ; VIDAL et al., 1972) that the venoms of several Bothrops species contained highlyactive phospholipases Az. Within this group, the venom from Bothrops alternates hasnever been studied, although it exhibited some features which make it of particularinterest . It displays one of the lowest phospholipase activities among Bothrops venoms,and it has been reported to have phospholipase C activity (VIDAL BREARD and ELfAS,1950). In the present study, an adequate purification procedure as well as some relevantproperties of the purified phospholipase A from B. alternates venom are described.
MATERIALSAND METHODSMaterialsVenom was obtained at our laboratory from healthy specimens of Bothrops alternates from Entre Rios
(Argentina) . The venom was collected in ice-cooled Petri dishes, centrifuged at 16,000 S for 20 min and thesupernatant was carefully removed, lyophilized and kept at -60°C. Sephadex G-25, G-50 (fine grade), G-75(S .F . grade) were purchased from Pharmacia Ltd. (Uppsala, Sweden). The gels were swollen and prepared asrecommended by the manufacturers. The ion exchangers SP-Sephadex C-50 and DEAE-Sephadex A-50(Pharmacia) were washed as described by HIMMELHOCH and PETERSON (1966) . p-Tosyl-L-arginine methyl ester(TAME), calcium-bis4p-nitrophenyl}phosphate, ; adenyliçacid (AMP), bovine serum albumin (Type F, BSA)p-nitrophenyl phosphate and 4-bromophenacyl bromide (pBPB) were purchased from Sigma Chemical Co . (St.Louis, MO). Casein (vitamin-free) was purchased from Fluka A.G . (Buks, Switzerland) . Lysozyme, ß-lactoglobulin, ovalbumin and carbonic anhydrase were purchased from Polyscience Inc. (Warrington, PA).Ampholines were purchased from LKB Produkter AB (Bromma, Sweden) and Rhodamine 6G was purchasedfrom British Drug Houses (U.K .) . All the other reagents were analytical grade and used without furtherpurification .
Synthetic 1,2-dimyristoyl[PC(14 :0)1- and 1,2-diheptanoyl[PC(7 :0)]-sn-glycero-3-phosphorylcholines wereprepared according to the method of CuBERO-ROBLES and VAN DEN BERGH (1969) or purchased from AvantiPolar Lipids Inc. (Birmingham, AL). Aluminium oxide (Mallinckrodt) and silicic acid (E . Merck, Darmstadt,F.R.G .) were employed for chromatographic purification of the phospholipid samples according to the methoddescribed by ANSELL and HAWTHORNE (1964) . Synthetic 2,3-diheptanoyl sn-glycero-l-phosphorylcholine [D-PC(7:0)] was a gift from Dr G. H. De Haas, Utrecht State University, The Netherlands . The purity ofphospholipid samples was checked by thin layer chromatography on silica-gel plates . The chromatograms weredeveloped with chloroform- methanol -water (65:25:4 by volume for dimyristoyl lecithin or 65 :35:4 by volumefor diheptanoyl lecithins) and the spots were made visible by charring with sulfuric acid- formaldehyde (97:3 byvolume) (VIDAL et al., 1972) . Purified samples of dimyristoyl lecithin were stored in hexane, while diheptanoyllecithin and D-diheptanoyl lecithin were stored in absolute ethanol and kept at -20°C. Before use, aliquots ofdiheptanoyl lecithin or D-diheptanoyl lecithin solutions were dried under nitrogen and dissolved in anappropriate amount of water up to a final concentration of about 15 - 20-fold their critical micellarconcentrations . Clear micellar solutions were obtained with either diheptanoyl or D-diheptavoyl lecithins at20°C . Critical micellar concentration was determined by the spectral shift induced by the incorporation ofRhodamine 6G into phospholipid micelles according to the procedure described by BoNsEN et al. (1972). Thevalues obtained with either diheptanoyl or D-diheptanoyl lecithins were 1.6 x 10 -' M in 10 mM Tris-HCIbuffer, pH 8.0, and about 1 .2 x 10-' M in 0.15 M NaCl, 10 mM Tris-HCl buffer, pH 8.0 .
Aliquots of dimyristoyl lecithin solution were dried under nitrogen and dispersed in 0.15 M NaCl, 0.1 mMEDTA, 10 mM Tris-HCI buffer, pH 8.0, with a vortex mixer at 25°C to give a final concentration of 3.0 mM.The coarse dispersion was submitted to ultrasonic irradiation at 10°C in a MSE (Measuring and ScientificEquipment Ltd., U.K .) ultrasonic disintegrator at 20 kHz and cavitating intensity . After 20 min, the temperaturewas raised to 26°C for the remaining sonication (total time : 60 min). Thevesicles were allowed to anneal at 42°Cfor 1 hr (LAWAczEcx et al., 1976), and single bilayer vesicles were separated from the larger aggregates bycentrifugation, as described by BARENHOLz et al. (1977) . The critical micellar concentration of dimirystoyllecithin was 6 x 10-' M (VIDAL et al,_ 1978) .
Phospholipase A, from Bothrops neuwledl venom was prepared as described by VIDAL and SToPPAN1(1971) .The homogeneous sample had a specific activity of 2300 units per mg . Pancreatic lipase (37.03 UFIP/mg) was agift from Dr Alberto Iglesias (Laboratorios BYK-LIPRANDI, Argentina) .
Analytical methodsPhosphorus content was determined by the method of CHEN et al. (1956), as modified by ROUSER and
FLEISCHER (1964) . Protein concentration in the effluents from columns was followed spectrophotometrically at280 run. The collected active fractions were combined and concentrated by ultrafiltration under nitrogen

Bothrops alternatus Phospholipase A,
261
pressure in a 60 nil Amicon cell (AmiconCo ., Bloomington, MA) fitted with an IC-0 .5 Diaflo membrane . Finalprotein concentration in the samples was measured according to the method of LOWRY et al. (1951) using bovineserum albumin as standard .
Enzymatic activitiesPhospholipase activity was measured by the titrimetric method at 30°C using egg yolk lipoprotein as substrate
in a Radiometer TTT 2 sutotitrator equipped with syringe burette ABU-12 as dispenser of 0.1 N NaOH, asdescribed by CANziANI et al. (1982) . One unit of enzyme activity is defined as the consumption (release) of 1 peqalkali (fatty acid) per min at pH 8.0 . PhospholipaseA specific activity was determined as units per mg protein.When diheptanoyl lecithin monomers or micelles or dimyristoyl lecithin vesicles were employed as substrates, theinitial rates of hydrolysis were determined by the spectrophotometric method Of WELLS (1972), as described byCANZIANI et al. (1982) .When the degree of phospholipid hydrolysis had to be measured in buffered media, the substrate was
suspended in 1.0 nil (final volume) of a solution containing 0.15 M NaCl, 10 mM CaCl, and 50 mM buffer . Atdifferent intervals of time 0.1 nil aliquots were placed, in 10 pl of 0.3 MEDTA and 0.9 ml absolute ethanol wasadded. Control samples containing no enzyme or the enzyme plus EDTA were treated similarly. The sampleswere evaporated under nitrogen, taken up in 50 Ml of absolute ethanol and analyzed by thin layerchromatography . The spots of lecithin and lysolecithin were scraped off and the phosphorous content wasmeasured as described above.
Proteolytic activity was measured with casein as substrate (KuNITZ, 1947 ; WAGNER and PREscoTr, 1966);TAME-esterase activity was measured spectrophotometrically as described by HUMMEL (1959) ;phosphodiesterase was measured with calcium-bis-(p-nitrophenyl)-phosphate, according to the method ofSULKowsKI et al. (1963) .
SDSpolyacrylamide gel electrophoresisSamples (about 50 Ng protein) were precipitated with 10 volumes of acetone, kept overnight at -20°C and
centrifuged at 10,000g for 30 min. The pellets were dried under vacuum, resuspended in 50 pl of sample buffer(LAEMMLI, 1970) and boiled for 1 min. Aliquots (10 pl) were submitted to discontinuous electrophoresis in arunning gel consistingof a gradient offrom 10 to 3070 polyacrylamide containing 270 (weight in volume) sodiumdodecyl sulfate, as described by MINOR (1979) . Staining was performed with Coomassie Brilliant Blue (0.270 w/vin methanol-acetic acid 10 :1 :8 by volume). The gels were destained by washing with acetic acid-methanol(1 .5 :1 by volume).
IsoelectricfocusingThe gels were prepared as described by O'FARRELL (1977) using a mixture of ampholines (Pharmalite,
Pharmacia, Uppsala, Sweden), pH 3 -10, to give a final pH range from 3 .8 to 9.5 . Pre-runs were successivelyperformed at 200 V (15 min), 300 V (30 min) and 400 V (30 min) . After loading of the sample, isoelectricfocusing was performed for 17 hr at 400 V. Staining and subsequent destaining were performed as describedabove.
Two-dimensional gel electrophoresisThe gels were prepared as described above (O'FARRELL, 1975) and a non-equilibrium pH gradient was used to
separate the proteins in the first dimension. The gels were then sealed on the top of a 12.57o polyacrylamide gel(LAEMMLI, 1970) with 170 (w/v) agarose containing 1 pg/ml bromophenol blue as tracking dye. Electrophoresiswas performed at 20 mA . After electrophoretic separation, the running gel was soaked overnight inmethanol-water-acetic acid (5 :4 :1 by volume) followed by three successive washes (10, 20 and 30 min,respectively) with water at 60°C . Silver staining was performed according to the method Of MERRIL et al. (1981).The staining technique is sensitive enough to allow the detection of 0.16 ng when tested with human serumalbumin. When necessary, background was removed as described by MARSHALL (1983) .
RESULTS
Phospholipase A2 activity in crude venom samplesResults varied when theenzyme activity was measured with different methods. With the
titrimetric method using egg yolk lipoprotein as substrate an initial lag period was noted,which increased with the amount of venom added to the reaction mixture (i .e . 1 .0 minusing 30 jug protein to 2 .0 min with 106.5 ug protein) . The plot of enzyme activity as afunction of venom concentration departed from linearity and the enzyme specific activitydecreased with increase in venom concentration. With several batches of crude venom the

262
H. E. NISENBOM et aL
initial specific activity obtained with 50 IAg protein ranged from 35 to 45 units per mgprotein. A similar behavior was observed when either dimyristoyl lecithin vesicles ordiheptanoyl lecithin micelles were used as substrates . Conversely, when a monomericsolution of diheptanoyl lecithin was employed as substrate (i .e . well below the criticalmicellar concentration), no lag period was observed and the initial velocities were linearwith the venom concentration. These results with pure lecithins as substrates wereconfirmed by thin layer chromatographic analysis of the reaction mixtures .Although the use of monomeric lecithins as substrates seemed to be advisable, since it
avoided the anomalous behavior of the enzyme activity in crudeB. alternatus venom, it isnot suitable for fractionation purposes due to the extremely low activity displayed by theenzyme towards monomeric substrates . On the other hand, with purified fractions no lagwas noted and a strict linearity between enzyme activity and protein concentration wasobserved with all the different methods employed to determine phospholipase activity.
Enzyme stability in crude venomWhen samples of crude venom are incubated at 20°C in 0.15 MNaCI adjusted to pH
6.0 with sodium acetate (10 mM), there is an increase in phospholipase activity up to 33%of the initial activity after 20 min incubation, followed by a decrease up to about 45% ofthe maximum after 120 inin .Samples of crudevenom in 0.1 M buffers of different pH values were incubated at 4°C
and phospholipase A activity was measured in aliquots taken at different intervals oftime . The enzyme activity remained stable for 48 hr in the range of pH 3.0-4.0, while athigher pH values there was asignificant decrease, up to 5% of the original at pH 7.0- 8.0 .The addition of 2.0 mM EDTA fully stabilized the enzyme . The role of venom proteasesin enzyme inactivation was borne out, first by the stability of phospholipase activity atacidic pH values, at which venom proteases are inhibited (MANDELBAUM et al., 1984)and, secondly, by the protecting effect afforded by EDTA, which is a well-knowninactivator of venom proteases . These observations suggest an early inactivation ofproteolytic enzymes as the initial step in phospholipase A purification .
Fractionation methodAll steps were carried out at 2-4°C. Dried venom (300 mg) was suspended in 4 ml of
2 mM EDTA, 0.1 M ammonium formate buffer pH 4.45 and the suspension wascentrifuged at 10,000 g for 20 min. The yellowish, slightly turbid supernatant whichcontained all the phospholipase activity was the starting material . ,
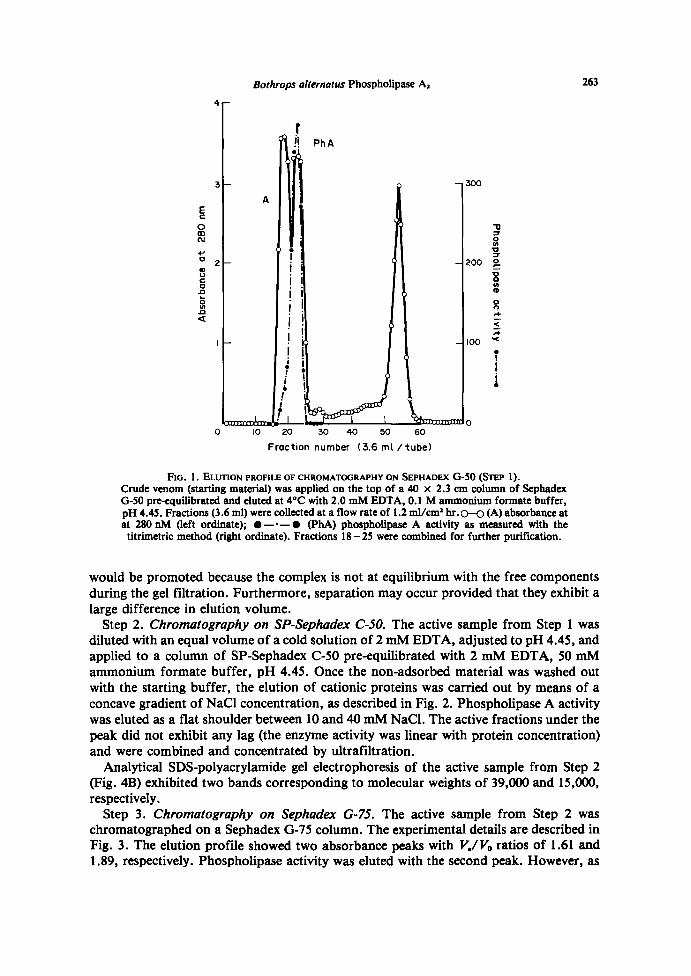
Step 1 . Chromatography on Sephadex G-S0 . The experimental details are described inFig. 1 . The enzyme activity is eluted as a single peak with a K� = 0.175, clearly retardedwith respect to the main absorbance peak which is eluted with the void volume . Noproteolytic activity was found in the fractions under the peak of phospholipaseA activity .The active fractions were combined and the sample was concentrated by ultrafiltration.Gel bed volume critically determined the elution profile of phospholipase A at this step,since the V,/V0 ratio, as well as the K� value, increased as the total gel bed (V,) wasincreased (not shown). This means that the chromatographic behavior of the enzyme waspartially determined by interaction with the gel matrix, which may be significant for theenzyme purification .The total activity units recovered after Step 1 were higher than those in the starting
material, which is consistent with the separation of a phospholipase A inhibitor. Theinhibitor would presumably form a reversible complex with the enzyme and dissociation
EcOa)N
O
ûô
aa
4
3
2
Bothrops alternatus Phospholipase A,
263
A
PhA
r
I
01010
2
30
40
50
60
Fraction number (3.6 ml /tube)
0
300
0
m0
100
FIG . 1 . ELUTION PROFILE OF CHROMATOGRAPHY ON SEPHADEx G-50 (STEP 1) .Crude venom (starting material) was applied on the top of a 40 x 2 .3 cm column of SephadexG-50 pre-equilibrated and eluted at 4°C with 2 .0 mM EDTA, 0.1 M ammonium formate buffer,pH 4.45 . Fractions (3 .6 ml) were collected at a flow rate of 1 .2 ml/cm' hr . o-G (A) absorbance atat 280 nM (left ordinate) ; "- "- " (PhA) phospholipase A activity as measured with thetitrimetric method (right ordinate) . Fractions 18-25 were combined for further purification .
would be promoted because the complex is not at equilibrium with the free componentsduring the gel filtration . Furthermore, separation may occur provided that they exhibit alarge difference in elution volume .
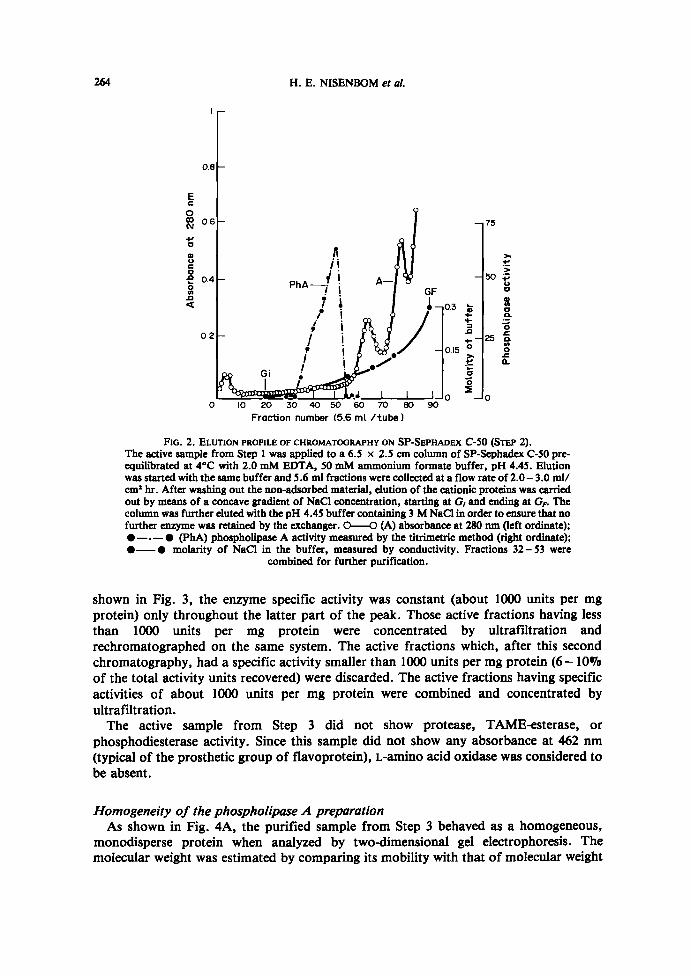
Step 2. Chromatography on SP-Sephadex C-S0 . The active sample from Step 1 wasdiluted with an equal volume of a cold solution of 2 mM EDTA, adjusted to pH 4.45, andapplied to a column of SP-Sephadex C-50 pre-equilibrated with 2 mM EDTA, 50 mMammonium formate buffer, pH 4.45 . Once the non-adsorbed material was washed outwith the starting buffer, the elution of cationic proteins was carried out by means of aconcave gradient of NaCl concentration, as described in Fig. 2. Phospholipase A activitywas eluted as a flat shoulder between 10 and 40 mM NaCl. The active fractions under thepeak did not exhibit any lag (the enzyme activity was linear with protein concentration)and were combined and concentrated by ultrafiltration.
Analytical SDS-polyacrylamide gel electrophoresis of the active sample from Step 2(Fig . 4B) exhibited two bands corresponding to molecular weights of 39,000 and 15,000,respectively .
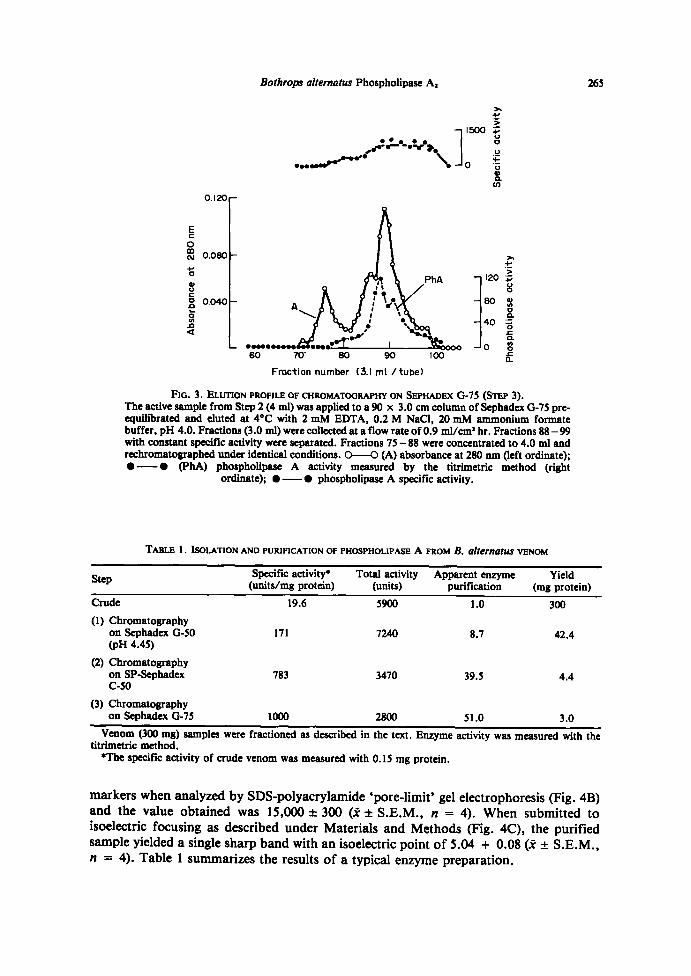
Step 3. Chromatography on Sephadex G-7S. The active sample from Step 2 waschromatographed on a Sephadex G-75 column . The experimental details are described inFig. 3. The elution profile showed two absorbante peaks with V,/Vo ratios of 1 .61 and1 .89, respectively . Phospholipase activity was eluted with the second peak . However, as
Fraction number (5 .6 mL /tube)
0.3
0.15
0
ôôî
75
50
0
T:t.>_
OmN0a.~ô
25 raa0c
FIG. 2. ELUTION PROFILE OF CHROMATOGRAPHY ON SP-SEPHADEx C-50 (STEP 2) .The active sample from Step 1 was applied to a 6.5 x 2.5 cm column of SP-Sephadex C-50 pre-equilibrated at 4°C with 2.0 mM EDTA, 50 mM ammonium formate buffer, pH 4.45. Elutionwas startedwith the same buffer and 5.6 ml fractions were collected at a flow rate of 2.0- 3 .0 ml/cm' hr . After washing out the non-adsorbed material, elution of the cationic proteins was carriedout by means of a concave gradient of NaCl concentration, starting at G, and ending at GF. Thecolumn was further eluted with the pH 4.45 buffer containing 3 MNaCl in order to ensure that nofurther enzyme was retained by the exchanger. 0-0 (A) absorbante at 280 run (left ordinate);0- "-" (PhA) phospholipaseA activity measured by the titrimetric method (right ordinate);0-0 molarity of NaCl in the buffer, measured by conductivity . Fractions 32-53 were
combined for further purification .
shown in Fig. 3, the enzyme specific activity was constant (about 1000 units per mgprotein) only throughout the latter part of the peak . Those active fractions having lessthan 1000 units per mg protein were concentrated by ultrafiltration andrechromatographed on the same system . The active fractions which, after this secondchromatography, had a specific activity smaller than 1000 units per mg protein (6-10%of the total activity units recovered) were discarded . The active fractions having specificactivities of about 1000 units per mg protein were combined and concentrated byultrafiltration.The active sample from Step 3 did not show protease, TAME-esterase, or
phosphodiesterase activity . Since this sample did not show any absorbance at 462 rim.(typical of the prosthetic group of flavoprotein), L-amino acid oxidase was considered tobe absent.
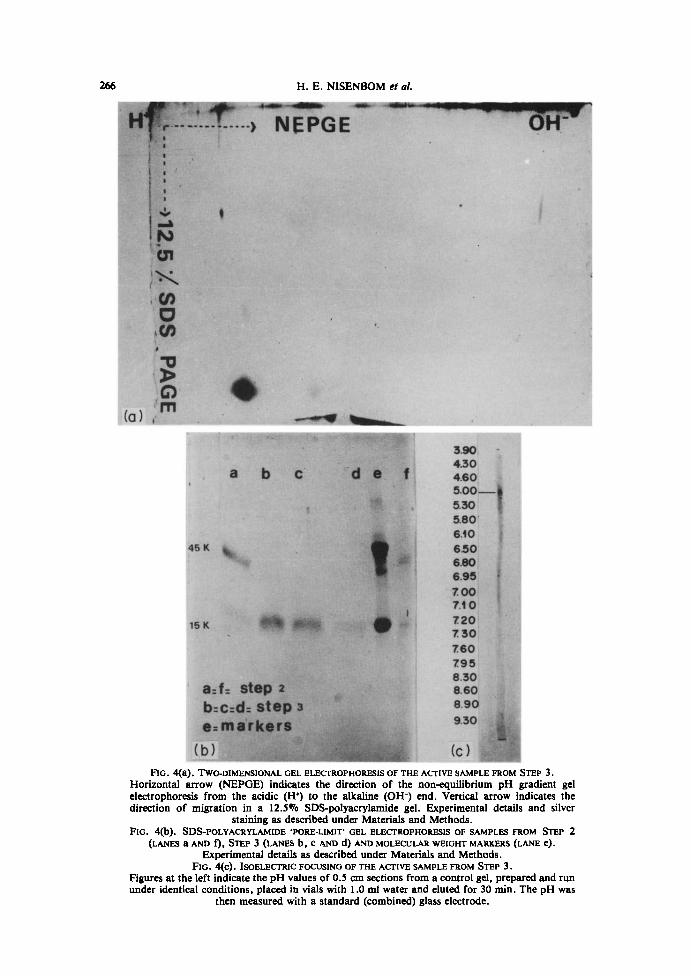
Homogeneity of the phospholipase A preparationAs shown in Fig. 4A, the purified sample from Step 3 behaved as a homogeneous,
monodisperse protein when analyzed by two-dimensional gel electrophoresis. Themolecular weight was estimated by comparing its mobility with that of molecular weight
EcON
mVCva
a
0.120
0.080
0.040
Bothrops alternates Phospholipase A,
L ""4"""""""60 70' 80 90 100
Fraction number (3.1 ml /tube)
so Svo.
40 'öL
FIG . 3 . ELUTION PROFILE OF CHROMATOGRAPHY ON SEPHADEX G-75 (STEP 3) .The active sample from Step 2 (4 ml) was applied to a 90 x 3 .0 cm column ofSephadex G-75 pre-equilibrated and eluted at 4°C with 2 mM EDTA, 0.2 M NaCl, 20 mM ammonium formatebuffer, pH 4.0 . Fractions (3 .0 ml) were collected at aflow rate of0.9 ml/cm' hr . Fractions 88 - 99with constant specific activity were separated . Fractions 75 - 88 were concentrated to 4.0 ml andrechromatographed under identical conditions . 0-0 (A) absorbante at 280 run (left ordinate);"- "
(PhA) phospholipase A activity measured by the titrimetric method (rightordinate) ; "-" phospholipase A specific activity .
TABLE 1 . ISOLATION AND PURIFICATION OF PHOSPHOLIPASE A FROM B . alternates VENOM
263
Venom (300 mg) samples were fractioned as described in the text . Enzyme activity was measured with thetitrimetric method .'The specific activity of crude venom was measured with 0.15 mg protein .
markers when analyzed by SDS-polyacrylamide 'pore-limit' gel electrophoresis (Fig . 413)and the value obtained was 15,000 t 300 Qc* S.E.M., n = 4) . When submitted toisoelectric focusing as described under Materials and Methods (Fig . 4C), the purifiedsample yielded a single sharp band with an isoelectric point of 5 .04 + 0.08 (z ± S.E.M.,n = 4) . Table 1 summarizes the results of a typical enzyme preparation.
Step Specific activity*(units/mg protein)
Total activity(units)
Apparent enzymepurification
Yield(mg protein)
Crude 19 .6 5900 1 .0 300(1) Chromatography
on Sephadex G-50 171 7240 8 .7 42.4(pH 4.45)
(2) Chromatographyon SP-Sephadex 783 3470 39 .5 4 .4C-50
(3) Chromatographyon Sephadex G-75 1000 2800 51 .0 3 .0
26 6
H. E. NISENBOM et al.
FIG . 4(a) . TWO-DIMENSIONAL GEL ELECTROPHORESIS OF THE ACTIVE SAMPLE FROM STEP 3 .Horizontal arrow (NEPGE) indicates the direction of the non-equilibrium pH gradient gelelectrophoresis from the acidic (H') to the alkaline (OH-) end. Vertical arrow indicates thedirection of migration in a 12.5% SDS-polyacrylamide gel . Experimental details and silver
staining as described under Materials and Methods.FIG . 4(b) . SDS-POLYACRYLAMIDE 'PORE-LIMIT' GEL ELECTROPHORESIS OF SAMPLES FROM STEP 2
(LANES a AND f), STEP 3 (LANES b, c AND d) AND MOLECULAR WEIGHT MARKERS (LANE e) .
Experimental details as described under Materials and Methods.FIG . 4(c) . ISOELECTRIC FOCUSING OF THE ACTIVE SAMPLE FROM STEP 3 .
Figures at the left indicate the pH values of 0.5 cm sections from a control gel, prepared and rununder identical conditions, placed in vials with 1 .0 ml water and eluted for 30 min. The pH was
then measured with a standard (combined) glass electrode .
Bothrops alternates Phospholipase A,
267
Characterization of phospholipase A from B. alternatus venomIn order to confirm the nature of the purified fraction, the enzyme reaction was
characterized as follows. Diheptanoyl lecithin micelles (4.67 mM) or dimyristoyl lecithinvesicles were incubated at 25°C, pH 8 .0, with 1 .25 Fig of purified enzyme from Step 3 .Aliquots taken at different intervals of time were analyzed by thin layer chromatographyas described under Materials and Methods . One mole of lecithin gave 1 mole oflysolecithin and 1 mole of fatty acid, thus confirming that the enzyme is a phospholipaseA. Lysolecithin and fatty acid were the only reaction products formed, even if the reactionwas allowed to proceed to completion and the samples incubated for a further 20 hr . Noother products attributable to phospholipase B or C were detected .The positional specificity was studied by incubating diheptanoyl lecithin micelles (4.67
mM) at pH 8.0 and 25°C with 0.5 jig of purified B. alternates enzyme . When 50076 of thesubstrate had been hydrolyzed, 9Mg of B. neuwiedi phospholipase A, were added and thehydrolysis proceeded to completion within a few minutes. The amount of lysolecithinformed was identical to that obtained when the hydrolysis was allowed to proceed tocompletion with either 15 lAg of pure B. alternatus enzyme or with 9 lAg of pure B. neuwiedienzyme separately . Only when samples completely hydrolyzed by these phospholipaseswere further treated with pancreatic lipase was there a time-dependent decrease in theamount of lysolecithin accompanied by the increase of a new phosphorus containing spotjust above the origin, at the position corresponding to glyceryl phosphorylcholine .
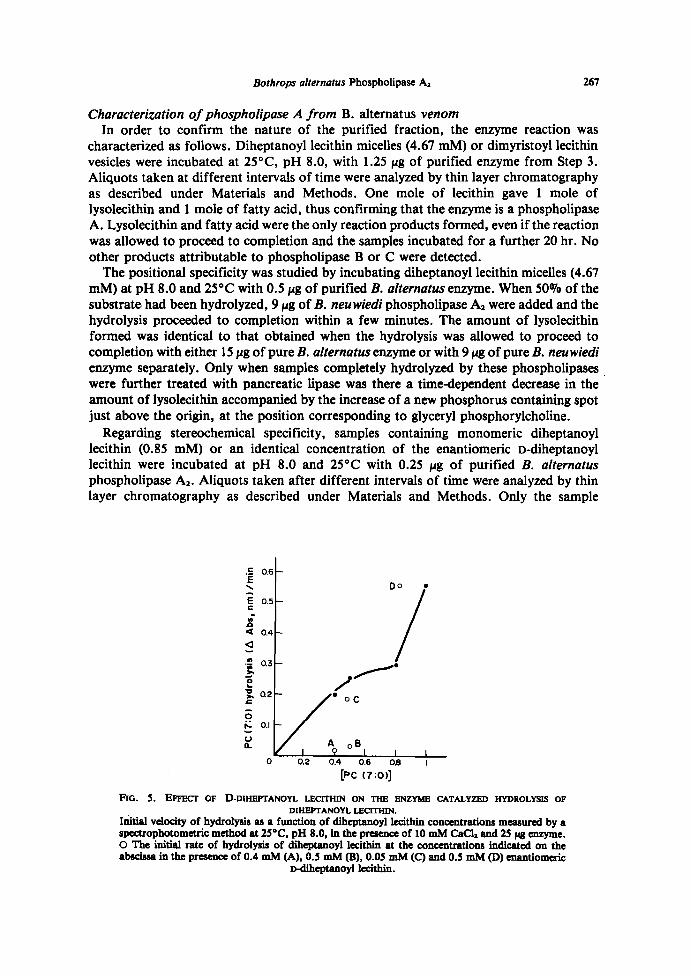
Regarding stereochemical specificity, samples containing monomeric diheptanoyllecithin (0.85 mM) or an identical concentration of the enantiomeric D-diheptanoyllecithin were incubated at pH 8.0 and 25°C with 0.25 lAg of purified B. alternatusphospholipase A, . Aliquots taken after different intervals of time were analyzed by thinlayer chromatography as described under Materials and Methods. Only the sample
0.6É
£ 0.5c
âQ 0.44
� 0.3
0«U, 02
Ua A o B
I
4
I
I
I0.2 0.4 0.6 O.6 1
[PC (7 :0)]
FIO. 5. EFFECT OF D-DIHEPTANOYL LECITHIN ON THE ENZYME CATALYZED HYDROLYSIS OFDIHEPTANOYL LECITHIN.
Initial velocity of hydrolysis as a function of diheptanoyl lecithin concentrations measured by aspectrophotometric method at 25°C, pH 8.0, in the presence of 10 mM CaCl, and 25 Erg enzyme .O The initial rate of hydrolysis of diheptanoyl lecithin at the concentrations indicated on theabscissa in the presence of 0.4 mM (A), 0.5 mM (B), 0.05 mM (C) and 0.5 mM (D) enantiomeric
D-diheptanoyl lecithin .
268
H. E. NISENBOM et al.
mâ0p
0Ei
La+. vmôa
0.3
0.2
Time (min)
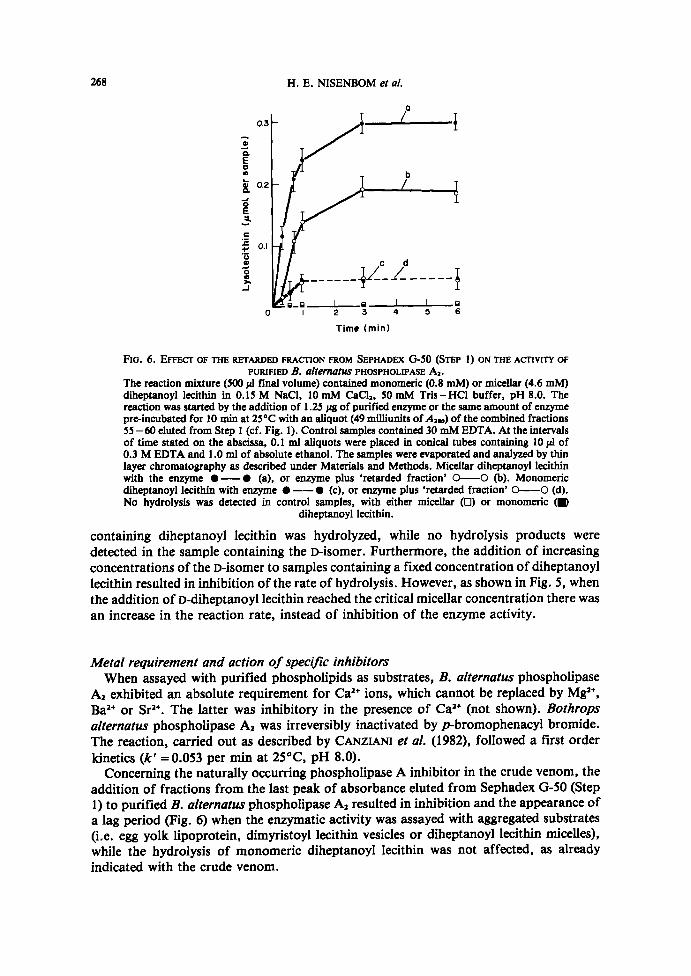
FIO . 6 . EFFECT OF THE RETARDED FRAcriON FROM SEPHADEX G-50 (STEP 1) ON THE ACTIVITY OFPURIFIED B . alternatus PHOSPHOLIPASE A, .
The reaction mixture (500 ;d final volume) contained monomeric (0 .8 mM) or micellar (4 .6 mM)diheptanoyl lecithin in 0.15 M NaCl, 10 mM CaCl,, 50 mM Tris-HC1 buffer, pH 8.0. Thereaction was started by the addition of 1 .25 Ng of purified enzyme or the same amount of enzymepre-incubated for 10 min at 25°C with an aliquot (49 milliunits ofA�,) ofthe combined fractions55-60 eluted from Step 1 (cf . Fig. 1) . Control samples contained 30 mM EDTA. At the intervalsof time stated on the abscissa, 0.1 ml aliquots were placed in conical tubes containing 10 11 of0.3 M EDTA and 1 .0 ml of absolute ethanol . The samples were evaporated and analyzed by thinlayer chromatography as described under Materials and Methods. Micellar diheptanoyl lecithinwith the enzyme "-" (a), or enzyme plus 'retarded fraction' O-O (b). Monomericdiheptanoyl lecithin with enzyme "-- " (c), or enzyme plus 'retarded fraction' O--O (d).No hydrolysis was detected in control samples, with either micellar (0) or monomeric (K)
diheptanoyl lecithin .
containing diheptanoyl lecithin was hydrolyzed, while no hydrolysis products weredetected in the sample containing the D-isomer . Furthermore, the addition of increasingconcentrations of the D-isomer to samples containing a fixed concentration of diheptanoyllecithin resulted in inhibition of the rate of hydrolysis . However, as shown in Fig. 5, whenthe addition of D-diheptanoyl lecithin reached the critical micellar concentration there wasan increase in the reaction rate, instead of inhibition of the enzyme activity .
Metal requirement and action ofspecific inhibitorsWhen assayed with purified phospholipids as substrates, B. alternatus phospholipase
A2 exhibited an absolute requirement for Ca2+ ions, which cannot be replaced by Mg=',Bat+ or Sr'+ . The latter was inhibitory in the presence of Ca" (not shown) . Bothropsalternatus phospholipase Az was irreversibly inactivated by p-bromophenacyl bromide.The reaction, carried out as described by CANZIANI et al. (1982), followed a first orderkinetics (k' =0.053 per min at 25°C, pH 8.0) .Concerning the naturally occurring phospholipase A inhibitor in the crude venom, the
addition of fractions from the last peak of absorbante eluted from Sephadex G-50 (Step1) to purified B. alternatus phospholipase A2 resulted in inhibition andthe appearance ofa lag period (Fig. 6) when the enzymatic activity was assayed with aggregated substrates(i .e . egg yolk lipoprotein, dimyristoyl lecithin vesicles or diheptanoyl lecithin micelles),while the hydrolysis of monomeric diheptanoyl lecithin was not affected, as alreadyindicated with the crude venom.

Bothrops alternates Phospholipase A,
269
DISCUSSION
Previous studies on Bothrops venoms (VIDAL and STOPPANI, 1971 ; VIDAL et al., 1971,1972) have indicated that the poor phospholipase A activity observed with these venomscould be explained by the presence of endogenous enzyme inhibitors . Theenzyme fromB.alternates venom represented an interesting problem, since it displayed the lowestphospholipase activity among Bothrops venoms and its phospholipase A2 has not beenstudied previously .The procedure described in the present paper yielded a single, highly purified
phospholipase A= which behaved as an homogeneous, monodisperse protein species whenanalyzed by two-dimensional polyacrylamide gel electrophoresis (Fig . 4A) with amolecular weight of 15,000 (Fig . 4B) and isoelectric point of about 5 .0 (Fig . 4C).
If the yield of this purification procedure is compared with the method employed withother Bothrops venoms (VIDAL and STOPPANI, 1971 ; VIDAL et al ., 1972), the finalrecovery of enzyme activity from B. alternatus venom was 39% of the total activity afterStep 1 and 48% of the initial activity (the difference is accounted for by the effect of theinhibitor) . The yields obtained with the venoms of B. jararaca, B. jararacussu and B.atroxwere 60% and 80%, respectively, while with the venomofB. neuwiedi the yield was265% of the initial activity . There are reasons to believe that the high yield obtained withthe latter venom is largely due to the relatively high content of phospholipase inhibitor.The purification value of 51-fold obtained with B. alternates is similar to those re-ported with other Bothrops venoms (VIDAL and STOPPANI, 1971). However, as shown inTable 1, purified phospholipase A3 from B. alternates venom had a specific activity ofabout 1000 units per mg protein, which is about one half of the specific activity obtainedwith purified phospholipase AZ from other Bothrops venoms (VIDAL and STOPPANI,1971). At present we do not know the reason for this difference . Nevertheless, it is clearthat the low phospholipase activity ofB. alternates venom cannot be ascribed to a highercontent of enzyme inhibitor.
Interestingly, the procedure currently employed for purification of phospholipase Azfrom other Bothrops venoms (VIDAL et al., 1972) failed when applied to B. alternatesvenom. For example, chromatography on DEAE-Sephadex A-50 gave rather poor results,in terms of both enzyme recovery and specific activity (i .e . 1 .6 mg protein with a specificactivity of 185 units per mg protein) .The behavior of B. alternates venom during the purification procedure is not observed
with other Bothrops venoms . For instance, the active sample from Step 2 exhibited twocomponents (Fig . 4B) of molecular weights 39,000 and 15,000, respectively, which, inspite of their large difference in size, were eluted together upon chromatography onSephadex G-50 (Step 1) with a K�, = 0.175 . This suggests the formation of a complexdisplaying a strong interaction with the gel matrix, since the minimal molecular weight(54,000, assuming 1 :1 stoichiometry) should be larger than the exclusion limit ofSephadex G-50 . This interpretation is further supported by the considerable overlappingof these two components upon chromatography on Sephadex G-75 (Step 3), as may beexpected for dissociation of a complex under non-equilibrium conditions . As shown inFig. 3, the V~/Vo ratios for the two components corresponded to molecular weights of25,000 and 15,000, respectively, indicating that the first peak displayed a strongerinteraction with the gel matrix than phospholipase A,.

270
H. E. NISENBOM et aL
Moreover, while phospholipases A, in other Bothrops venoms exist as two isoenzymes(VIDAL and STOPPANI, 1971 ; VIDAL et al ., 1972), B. alternatus phospholipase A, appearsto be a single protein species . This result seemed not to be a methodological artifact, sincewhen all the other venoms were submitted to chromatography on SE-Sephadex C-50under identical experimental conditions, each isoenzyme was eluted in a different peak(VIDAL et al., 1972).
Purified B. alternatus phospholipase, when assayed on diheptanoyl lecithin micelles ordimyristoyl lecithin vesicles as substrates yielded lysolecithin and free fatty acid as theonly reaction products, in accordance with the predicted stoichiometry for aphospholipase A. IfB. alternatus enzyme hydrolyzed the fatty acyl ester in position 1 ofdiheptanoyl lecithin, the product would be 2-heptanoyl-sn-glycero-3-phosphorylcholine,which, in turn, should be hydrolyzed by the B. neuwiedi enzyme, leading to heptanoicacid and glyceryl phosphorylcholine. This would result in a decrease in the amount oflysolecithin recovered, which was not observed experimentally . Therefore, it wasconcluded that B. alternatus enzyme is a phospholipase of the A, type . Furthermore, noreaction products representative of phospholipase C activity were detected .B. alternatus phospholipase A, required Ca" ions for activity and exhibited
stereospecificity, since the enantiomeric 2,3-diheptanoyl-sn-glycero-l-phosphorylcholine(D-diheptanoyl lecithin) was not hydrolyzed . As shown in Fig. 5, the D-isomer inhibits therate of hydrolysis of diheptanoyl lecithin monomers, which may reflect competitionbetween the two enantiomeric lecithins for the enzyme active site . This suggests thatstereospecificity is an expression of catalytic rather than binding specificity . The increasein rate observed when the total diheptanoyl lecithin concentration (i .e. L- plus D-isomer)reaches the critical micellar concentration can be explained by formation of mixedmicelles which provide a phospholipid - water interface to which the enzyme can beadsorbed . Thus, the rate of diheptanoyl lecithin hydrolysis will increase drastically,overcoming the inhibitory action of the D-isomer . The possibility that the enzyme alsodisplayed phospholipase B activity, with the latter being enhanced as a consequenceof the micelle formation (SHILOAH et al ., 1973a,b) is unlikely, since no glycerylphosphorylcholine was observed experimentally, even after 20 hr incubation .The anomalous behavior of the enzyme activity in crudeB. alternatus venom seems to
be attributable to the presence of an endogenous enzyme inhibitor . The inhibitorappeared to be inactivated, presumably by the action of venom proteases, thus explainingsatisfactorily the self-activation of phospholipaseAwhen the crude venom is incubated atpH 6.0, since the lag period disappears and, at the same time, the apparent enzymeactivity increases to an optimal value. A subsequent decrease in phospholipaseA activitycan be attributed to the further action of venom proteases on the free enzyme . Theimportance of venom proteases in autolytic activation is supported by the observationthat the presence of EDTA or an acidic pH (4.5 or lower) fully prevented the changes inphospholipase A activity upon incubation of crude venom.The addition of the last peak of absorbance eluted from Sephadex G-50 (Step 1; Fig. 1)
to purified phospholipase A, resulted in the appearance of the initial lag period andinhibition of the enzyme activity (Fig . 6) when assayed on aggregated substrates (i .e . eggyolk lipoprotein, dimyristoyl lecithin vesicles or diheptanoyl lecithin micelles) while, asoccurred with crude venom, the rate of hydrolysis of diheptanoyl lecithin monomers wasnot affected (Fig . 6) . No inhibitory fraction could be detected in samples of crude venomincubated at pH 6.0 up to maximal phospholipase A activation and this treatment did notaffect the chromatographic behavior of the enzyme . Moreover, the purified enzyme

Bothrops alternatus Phospholipase A,
27 1
obtained from pre-incubated venom samples retained its sensitivity to the inhibitoryfraction from crude venom (not shown) .The natural occurrence of peptidic inhibitors of phospholipases A2 seems to be a
common feature in Bothrops venoms . A peptidic inhibitor has been isolated from B.nettwiedi venom (VIDAL et al., 1972) and demonstrated in B. jararaca, B. jararacussuandB. atrox venoms (VIDAL et al., 1971 ; ROSENBERG, 1979) . The mechanism ofphospholipase A inhibition by these peptides seemed to be similar, i.e . formation of anenzyme- inhibitor complex which prevents the enzyme interaction with aggregatedsubstrates (VIDAL and STOPPANI, 1972), while the interaction with monomeric substratesis not affected . The structure, as well as the specificity, of these inhibitors for thehomologous enzyme, as well its for phospholipases A, from other Bothrops venoms, is atpresent under study.
Acknowledgements -The authors acknowledge many helpful discussions with Dr G. A. CANzIANI . We are alsoindebted to Dr J. M. PARISI for carrying out the PAGE, to Mr D. CAso for his expert technical assistance, toProf . M. MIRANDA and Mr G. COUTURIER for venom extractions, to Servicio de Fotograffa Cientffica fordrawingthe graphics, and to J. M. VERA for typing the paper. This study was supported byConsejo Nacional deInvestigaciones Cientificas y Técnicas (CONICET) and Fundaci6n Instituto de Neurobiologfa (FIDNEU).
REFERENCES
ANSELL, G. B. and HAWTHORNE, J. N. (1964) Preparation of phospholipids . In : Phospholipids, p. 88 .Amsterdam: Elsevier .
BARENHOLZ, Y. GIBBEs, D., LITMAN, B. J., GOLL, J., THOMPSON, T. E. and CARLSON, F. D. (1977) A simplemethod for the preparation of homogeneous phospholipid vesicles . Biochemistry 16, 2806 .
BONSEN, P. P. M., DE HAAS, G. H., PIETERsoN, W. A. and VAN DEENEN, L . L. M. (1972) Studies onphospholipase A and its zymogen from porcine pancreas . IV : The influence of chemical modification on thelecithin structure on substrate properties . Biochim. biophys . Acta 270, 364.
CANZIANI, G. A., SEKI, C. and VIDAL, J. C. (1982) Accessibility of the active site of crotoxin B in the crotoxincomplex. Toxicon 20, 890.
CHEN, P. S., TORIBARA, T. Y. and WAGNER, H. (1956) Microdetermination of phosphorus . Analyt. Chem. 28,1756 .
CUBERo-ROBLES, E. and VAN DEN BERGH, D. (1969) Synthesis of lecithins by acylation of O-(sn-glycero-3-phosphoryl)choline with fatty acid anhydrydes . Biochim. biophys. Acta 187, 520.
DE HAAS, G. H., PosTEMA, N. M., NIEWENH, W., VAN DEENEN, L. L. (1968) Purification and properties ofphospholipase A from porcine pancreas . Biochim. biophys. Acta 159, 103.
GALLAI-HATCHARD, J. J . and THOMPSON, R. H . S. (1965) Phospholipase A activity of mammalian tissues .Biochim. biophys. Acta 98, 128.
HALPERT, J., EATER,D. and KARISSON,E. (1976) The role of phospholipase activity in the action of presynapticneurotoxin from the venom of Notechis scutatus scutatus (Australian tiger snake) . FEBS Lett . 61, 72 .
HIMMELHOCH, S. R. and PETERSON, E. A. (1966) Experimental problems in the use of commercially preparedDEAF-celluloses for protein chromatography . Analyt. Biochem. 17, 383.
HUMMEL, B. C. W. (1959) A modified spectrophotometric determination of chymotrypsin, trypsin andthrombin. Can. J. Biochem. Physiol . 37, 1393 .
IWANAGA, S. and SuzuKi, T. (1979) Enzymes in snake venoms . In : Handbook ofExperimental Pharmacology,Vol. 52, p. 61 (CHEN-YUAN LEE, Ed .) . Berlin : Springer-Verlag .
KUNrrz, M. (1947) Crystalline soybean trypsin inhibitor. J. gen. Physiol . 40, 29 .LAEmMLI, U . K. (1970) Cleavage of structural proteins during the assembly of bacteriophage T, . Nature, Land.227,680.
I.AWACZECK, R., KAINOSHO, M. and CHAN, S. I . (1976) The formation and annealing of structural defects inlipid bilayer vesicles . Biochim. biophys. Acta 443, 313 .
LowRV, O.H., ROSEBROUGH, N. H., FARR, I. L. and RANDALL, R. J. (1951) Protein measurement with the Folinphenol reagent. J. biol. Chem. 193, 265 .
MANDELBAUM, F. R., AssAtnlRA, M. T. and REICHL, A. P. (1984) Characterization of two hemorrhagic factorsisolated from the venom of Bothrops neuwiedi (Jararaca pintada) . Toxicon 22, 193.
MARINETTI, G. V. (1965) Action of phospholipase A on lipoproteins . Biochim. biophys. Acta 98, 554.MARSHALL, T. (1983) Detection of DNA and lipopolysaccharide in thick polyacrylamide gels using an improved
silver stain . Electrophoresis 4, 269.

272
H. E. NISENBOM et al.
MARRILL, C. R., GOODMAN, D., SEDMAN, S. A. and EBERT, M. H. (1981) Ultrasensitive stain for proteins inpolyacrylamide gels shows regional variation in cerebrospinal fluid proteins . Science 211, 1437 .
MFRS, D. (1970) Comparative study of enzyme activities in snake venoms . Int. J. Biochem. 1, 335.MINOR, P. D. (1980) Comparative biochemical studies of type III poliovirus . J. Yirol. 34, 73 .O'FARRELL, P . H. (1975) High resolution two-dimensional electrophoresis ofproteins . J. biol. Chem. 250, 4007 .O'FARRELL, P. Z., GOODMAN, H. M. and O'FARRELL, P. H. (1977) High resolution two-dimensional
electrophoresis of basic as well as acidic proteins . Cell 12, 1133 .ROCK, C. O. and SNYDER, F. (1975) Rapid purification of phospholipase A, from Crotalus adamanteusvenomby affinity chromatography . J. biol. Chem. 250, 6564.
ROSENBERG, P. (1979) Pharmacology of phospholipase A, from snake venoms . In : Handbook ofExperimentalPharmacology, Vol. 52, p. 403 (CHEN-YUEN LEE, Ed .) . Berlin : Springer-Verlag .
ROUSER, G. and FLEISCHER, S. (1964) Isolation, characterization and determination of polar lipids ofmitochondria . Meth . Enzym. 10, 385.
SHILOAH, J ., KLIBANSKY, C. and DE VRIES, A. (1973a) Phospholipase isoenzymes from Nqja nqja venom. 1.Purification and partial characterization . Toxicon 11, 481 .
SHILOAH, J., KLIBANSKY, C. and DE VRIES, A. (19736) Phospholipase isoenzymes from Nqja nqja venom. 11 .PhospholipaseA and B activities . Toxicon 11, 491 .
SULKOWSKI, E., WORK, W. and LAsKowsKi, M. S. R. (1963) A specific and non-specific alkalinemonophosphatase in the venom of Bothrops atrox and their occurrence in the purified venomphosphodiesterase . J. biol. Chem. 238, 2477 .
VANDEENEN,L. L. andDE HAAS, G. H. (1963) The substrate specificity of phospholipase A. Biochim . biophys.Acta 70, 538.
VIDAL, J. C. and STOPPANI, A. O. M. (1971) Isolation and purification of two phospholipases A, from Bothropsvenoms . Archs Biochem . Biophys. 145, 543.
VIDAL, J. C. and STOPPANI, A. O. M. (1972) Isolation and properties of an inhibitor ofphospholipase AfromBothrops neuwiedi venom. Archs Biochem . Biophys. 147, 66 .
VIDAL, J. C., CATTANEo,P. and STOPPANI, A. O.M. (1971) Some characteristic properties of phospholipase A,from Bothrops neuwiedi venom. Archs Biochem. Biophys. 151, 168.
VIDAL, J. C., MOLINA, H. and STOPPANi, A. O. M. (1972) Ageneral procedure for the isolation and purificationof phospholipases A, isoenzymes from Bothrops venoms . Acta physiol. latinoam. 23, 91 .
VIDAL, J. C., GUGLIELMUCCI, E. A. and STOPPANi, A. O. M. (1978) Interaction of 3-D+)hydroxybutyrateapodehydrogenase with phospholipid . Archs Biochem. Biophys. 187, 138.
VIDAL BREARD, J. J . and ELfAS, V. E. (1950) Bioquimica de los venenos de serpientes 1. Actividad de lecitinasaC en veneno de Bothrops alternates. Archos Farm. BioquiM . Tucuman 5, 77.
VOLWERK, J. J., PIETERSON, W. A. and DE HAAS, G. H. (1974) Histidine at the active site of phospholipase A.Biochemistry 13, 1446.
WAGNER,F. W. and PRESCOTT, J. M. (1966) Comparative study of proteolytic activities in the venoms of someNorth American snakes . Comp. Biochem . Physiol. 17, 191.
WELLS, M. A. (1971) Evidence for O-acyl cleavage during hydrolysis of 1-2-diacyl-sn-glycero-3-phosphorylcholine by phospholipase A, of Crotalus adamanteus. Biochim . biophys. Acta 248, 80.
WELLS, M. A. (1972) A kinetic study of phospholipase A, (Crotalus adamanteus) catalyzed hydrolysis of 1,2-dibutyryl-sn-glycero-3-phosphorylcholine . Biochemistry 11, 1030 .
WELLS, M.A. (1975) A simple and high yield purification of Crotalus adamanteus phospholipase A, . Biochim.biophys. Acta 380, 357.





























