Embed Size (px)
Citation preview

Page 25
1186th Conferenceconferenceseries.com
Physical Chemistry 2017
4th International Conference on
Physical and Theoretical ChemistrySeptember 18-19, 2017 Dublin, Ireland
Day 1Scientific Tracks & Abstracts

Page 26
Physical Chemistry 2017
Day 1 September 18, 2017
Sessions:
Physical Chemistry: A Molecular Approach | Physical Chemistry of MacromoleculesSession ChairElena G KovalevaUral Federal University, Russia
Session Co-ChairAlex BOEGLINUniversité de Strasbourg, France
Session IntroductionTitle: Sum-frequency generation from chiral bisoxazoline metal complexes: Experiments and dft
calculationsAlex BOEGLIN, Université de Strasbourg, France
Title: The Role of anharmonicity in the confinement effect in zeolites: Structure, spectroscopy and adsorption free energy of ethanol in H-ZSM-5Roger Rousseau, Pacific Northwest National Laboratory, USA
Title: Electron coupling and electron transfer between two bridged dimolybdenum unitsChun Yuan Liu, Jinan University, China
Title: Experimental and theoretical studies of coordination fullerene polymers conductivityKrzysztof Winkler, University of Bialystok, Poland
Title: Soft based hypersonic phononicsGeorge Fytas (Talk will be delivered by Yu Cang), Max Planck Institute for Polymer Research, Germany
Title: Theoretical study of the chemical reactions by the combination of quantum mechanical and molecular dynamics methodsToshiaki Matsubara, Kanagawa University, Japan
Title: Recent advances in quantum monte carlo: Applications to lithium ion – stockmayer clusters, hydrogen isotopic separation, and the investigation of excited state manifoldsEmanuele Curotto, Arcadia University, USA
Title: What is hidden behind a phase diagram?Fabienne Berthier, Paris-Sud University-CNRS, France
Session Introduction

Page 27
conferenceseries.com
Volume 7, Issue 4 (Suppl)J Phys Chem Biophys, an open access journal
ISSN: 2161-0398Physical Chemistry 2017
September 18-19, 2017
4th International Conference on
Physical and Theoretical ChemistrySeptember 18-19, 2017 Dublin, Ireland
Sum-frequency generation from chiral bisoxazoline metal complexes: Experiments and DFT calculationsAlex Boeglin, Grégory Taupier, Aline Maisse-François, Thierry Achard, Stéphane Bellemin-Laponnaz and Kokou Dodzi DorkenooInstitut de Physique et Chimie des Matériaux de Strasbourg, France
Today, metallopolymeric entities consisting of chains of metal ions interlinked with ambidentate ligands provide an attractive route towards so called smart materials capable of adjusting their physical properties in response to changes in environmental
factors and/or to external stimuli. The dynamic nature of the metal ligand bond in solution may readily be assessed through titration experiments based on linear optical properties and through vibrational spectroscopy. However, it is much more difficult to study the molecular motions persisting in the condensed phase although these are essential to the properties and performance of this class of emergent materials. C2-symmetric bisoxazoline units have attracted much attention because they have been successfully used in enantioselective catalysis when suitably substituted at the 4-position of the oxazoline heterocyclic rings to make it a chiral centre. When two isopropyl substituted oxazoline units are attached to a vinyl group, a bidentate monotopic ligand is achieved which shows a strong absorption band near 300 nm but no circular dichroism in methanol. Optical activity is detected however as soon as transition metal ions are added to the solutions. It attests to the formation of complexes expressing their axial chirality in their electronic transitions. Moreover, after the slow evaporation of the solvent, liquid casted homonuclear homoleptic complexes are capable of generating detectable levels of sum-frequency (SFG) signals which are specific to isotropic chiral media, i.e. which lack inversion symmetry only at the molecular level. In order to interpret these results, density functional theory (DFT) based electronic structure calculations is performed on individual metal complexes to determine the possible arrangements of the ligands around the metal ions and evaluate their relative energies. Time dependent (TD-DFT) calculations are used to establish the relationships between conformational structure and optical properties. Our results carry over to the related ditopic monomers containing bisoxazoline ligand units. But now, the formation of homoleptic homochiral species generates a chiral metallopolymer and lead to the formation of films of improved quality. The possibilities of optically active SFG based microscopy to study the formation of the metallopolymeric material obtained from chiral enantiopure components holds the promise of a sensitive technique where the optical expression of chirality can be used to probe self-assembling processes which may be relevant to other types of metallopolymer.
BiographyAlex Boeglin obtained his PhD in condensed matter Physics from the University of Strasbourg (France) in 1987 on the theoretical modeling of laser induced processes in molecular systems. After Post-Doctoral work with Prof. S. H. Lin at the Department of Chemistry of the Arizona State University in Tempe (AZ), he joined the Department of Nonlinear Optics of the then newly formed Institute of Physics and Chemistry of Materials of Strasbourg as a research specialist of the CNRS. In addition to modeling the effects of relaxation and dissipation phenomena on various nonlinear optical processes in molecules and aggregates, he became gradually more involved in the evaluation of molecular hyperpolarizabilities combining semi-empirical and DFT methods. Recently, he has also been carrying out DFT calculations aimed at understanding the vibronic features observed in low temperature single molecule STM induced luminescence experiments conducted at IPCMS.
Alex Boeglin et al., J Phys Chem Biophys 2017, 7:4(Suppl)DOI: 10.4172/2161-0398-C1-025

Page 28
Notes:
conferenceseries.com
Volume 7, Issue 4 (Suppl)J Phys Chem Biophys, an open access journal
ISSN: 2161-0398Physical Chemistry 2017
September 18-19, 2017
4th International Conference on
Physical and Theoretical ChemistrySeptember 18-19, 2017 Dublin, Ireland
The role of anharmonicity in the confinement effect in zeolites: Structure, spectroscopy and adsorption free energy of ethanol in H-ZSM-5Roger Rousseau1, Mal-Soon Lee1, Konstantinos Alexopoulos2, Marie-Françoise Reyniers2, Guy B Marin2, Vassiliki-Alexandra Glezakou1 and Johannes A Lercher1
1Pacific Northwest National Laboratory, USA2Ghent University, Belgium
Zeolite is one of promising solid acid catalysts for the conversion of renewable biomass-derived alcohols into fuels and chemicals. Dehydration of alcohols to alkenes is a well-known prototypical acid catalyzed reaction, where confinement and entropic effects
impact the rates of these reactions. For such conversions, HZSM-5 zeolite is commonly used as a platform for acid catalyzed reactions due to its strong acidity and enhancement of reaction rates due to confinement in pores. In this talk, we present the structure and thermochemistry of ethanol adsorption on the Brønsted acid site of the HZMS-5 by means of ab inito molecular dynamics (AIMD) simulations directly compared with in situ IR spectroscopy and thermochemical measurements on the same material. Simulations were performed using two different ethanol loadings (with/without deuterium substitution) at different temperatures (100 ≤ T ≤ 700). This enables us to take into account enthalpic and entropic effects caused by the dynamics of the motion of the reaction intermediates. AIMD simulations show that hydrogen transfer from the zeolite scaffold to ethanol occurs as temperature increases. In the simulations with higher ethanol loading, proton transfer occurs via relay between H-bonded ethanol molecules. Calculated projected vibrational density of states (VDOS) obtained from velocity autocorrelation function show a broad peak around 1600 cm-1 related to H-O-H bending mode which is also observed experimentally. We estimated entropy and enthalpy of adsorption using the computed VDSO along with a quasi-harmonic approximation, which shows good agreement with experimental measurement conversely, the more commonly employed harmonic vibrations lead to free energy estimates that deviate from experiment substantially. Overall, this study exemplifies how enharmonic effects, as capture by AIMD, are critical for the quantitative modeling of the free energetics of zeolite-catalyzed processes.
BiographyRoger Rousseau is awarded PhD in Inorganic Chemistry from The University of Michigan, USA in the 1995. He holds a Master Degree in Inorganic Chemistry from The University of Michigan, USA in the 1994, followed by a Bachelor’s Degree in Chemistry from University of Windsor, Canada in the 1991. Currently, he is working as a Senior Staff Scientist for Pacific Northwest National Laboratory, Richland, Washington. His research interests are focused on the application of quantum mechanical methods in simulations of the properties and reactivity of molecules, solids, and surfaces of relevance to catalysis for energy applications. Currently, He is working on the application and development of ab initio molecular dynamics methods to the study of heterogeneous and homogeneous catalysis reaction mechanisms. This includes participation in the Center on Molecular Electrocatalysis, an Energy Frontier Research Center, and PNNL's Institute for Integrated Catalysis.
Roger Rousseau et al., J Phys Chem Biophys 2017, 7:4(Suppl)DOI: 10.4172/2161-0398-C1-025

Page 29
conferenceseries.com
Volume 7, Issue 4 (Suppl)J Phys Chem Biophys, an open access journal
ISSN: 2161-0398Physical Chemistry 2017
September 18-19, 2017
4th International Conference on
Physical and Theoretical ChemistrySeptember 18-19, 2017 Dublin, Ireland
Notes:
Electron coupling and electron transfer between two bridged dimolybdenum unitsChun Y Liu, Miao Meng and Tao Cheng Jinan University, China
Bridged dimolybdenum dimers, denoted as [Mo2]-bridge-[Mo2], are favorable model compounds for study of electronic coupling and electron transfer because of the unique electronic structure of the [Mo2] unit. The formation of the metal-metal multiple
bond removes the d orbital degeneracy, rendering the Mo2 unit a σ2π4δ2 electronic configuration. Therefore, as an electronic donor-bridge-acceptor system, the transferring electron is specified to be the δ electrons and donor-acceptor electron transfer can be probed by the metal (δ) to bridging ligand (π*) and bridging ligand (π) to metal (δ) and vibronic (δ) metal to metal (δ) charge transfer absorptions in accordance with sup exchange theory. By varying the ancillary ligands and the bridging ligands, a diverse of complexes of this type have been synthesized and studied. The electronic coupling matrix elements (H) are evaluated according to the Mulliken-Hush and CNS theories, which give consistent results. The mixed-valence properties are discussed in terms of Robin-Day's scheme. System transition from Class II to Class III via Class II-III is examined in a series of four complexes with subtle structural differences. The δ→δ electron transfer kinetics in symmetrical as well as asymmetrical systems has been investigated, conforming well to the semi-classical two-state model and the Marcus-Hush theory. In study of the photo-induced electron transfer, we found the δ*→δ back electron transfer is faster than the process from the bridge to the Mo2 center by one order of magnitude, while in the latter case, the electron transfer distance is shorter but the electronic coupling is much stronger. This controversial electron transfer phenomenon is tentatively attributed to a quantum incoherent pathway.
BiographyChun Y Liu completed his PhD in 2005 at Texas A&M University. Currently, he is a Full Professor at Jinan University. His research has been focused on study of electronic coupling (EC) and electron transfer (ET) between two charge bearing sites by taking a molecular approach. He uses quadruply bonded Mo-Mo complex units as the electron donor (D) and acceptor (A) and a diverse of organic bridging ligand (B) to construct D-B-A experimental models, in which the transferring electron is identified to be the δ electrons. Quantitative evaluations of the EC and ET properties are achieved with the physical chemical parameters (Hab, ΔG°, ΔG*, λ, Ket…) derived from electrochemical and spectroscopic data under the semi-classical theories. In study of photo-induced electron transfer, it is found that the δ*→δ back electron transfer is quite different from the δ→δ ET reaction in the mixed-valence systems, thus, a quantum incoherent pathway is proposed.
Chun Y Liu et al., J Phys Chem Biophys 2017, 7:4(Suppl)DOI: 10.4172/2161-0398-C1-025

Page 30
Notes:
conferenceseries.com
Volume 7, Issue 4 (Suppl)J Phys Chem Biophys, an open access journal
ISSN: 2161-0398Physical Chemistry 2017
September 18-19, 2017
4th International Conference on
Physical and Theoretical ChemistrySeptember 18-19, 2017 Dublin, Ireland
Experimental and theoretical studies of coordination fullerene polymers conductivityKrzysztof Winkler, Emilia Grodzka, Monika Wysocka-Zolopa and Jakub GoclonUniversity of Bialystok, Poland
In coordination fullerene polymers, fullerene moieties are covalently bonded to transition metal atoms or their complexes to form a polymeric network. The polymer of fullerene C60 and palladium, poly (C60Pd3), was most intensively investigated. This polymer
can be synthesized electrochemically or chemically. The electrochemical synthesis results in the formation of thin and uniform film on the electrode surface. The film is electrochemically active at negative potentials due to the fullerene cages reduction. During switching between neutral and reduced state, the polymeric film is doped with supporting electrolyte cations. Such transition also results in sharp increase of the film conductivity. The conductivity of poly (C60Pd3) thin films was experimentally investigated with interdigitated array electrodes. The poly (C60Pd3) doping level and, therefore, charge carrier density depends on the size of counter-ions incorporated into polymeric structure during its reduction. The negative polaron-type carriers generated during the film reduction are responsible for film conductivity. The charge propagation through the polymeric film can be quantitatively described by electron-hopping model. The specific conductivity of poly (C60Pd3) and electron diffusion coefficient are in the same order of magnitude as these values reported for typical p-doped conducting polymers. The conductivity properties of the composite of poly (C60Pd3) polymer and palladium nanoparticles were also investigated. Metallic nanoparticles participate in the charge transport within the film also in the potential range of the polymer neutral state. Therefore, poly (C60Pd3)/Pd composite exhibits large potential window of good conductivity. The structure and conducting properties of poly (C60Pd3) polymers were also predicted applying DFT calculations. The isolated negative polarons are the preferred electronic states for reduced polymers.
BiographyKrzysztof Winkler completed his MSc (1982) and PhD (1989) degrees in Chemistry at Warsaw University, Poland. He was a Post-doctoral fellow at University of Saskatchewan, Saskatoon (1989-1991) and University of California, Davis (1995-1997). He is currently a Professor at Institute of Chemistry, University of Bialystok, Poland. He served as Head of this Institute (2004-2017). His research interests include “Kinetics of electrochemical processes, electro-deposition and properties of low-dimensional crystals, the synthesis, properties and application of fullerene-based polymers.
Krzysztof Winkler et al., J Phys Chem Biophys 2017, 7:4(Suppl)DOI: 10.4172/2161-0398-C1-025

Page 31
conferenceseries.com
Volume 7, Issue 4 (Suppl)J Phys Chem Biophys, an open access journal
ISSN: 2161-0398Physical Chemistry 2017
September 18-19, 2017
4th International Conference on
Physical and Theoretical ChemistrySeptember 18-19, 2017 Dublin, Ireland
Soft based hypersonic phononicsGeorge Fytas, Yu Cang and Bartlomiej Graczykowski Max Planck Institute for Polymer Research, Germany
Phononic structures (composite materials) in which a periodic distribution of elastic parameters facilitates control of the propagation of phonons, hold the promise to enable transformative material technologies in areas ranging from acoustic and thermal cloaking
to thermoelectric devices. This requires strategies to deliberately engineer the phononic band structure of materials in the frequency range of interest. Phononics, the acoustic equivalents of the photonics are controlled by a larger number of material parameters, as phonon cannot propagate in vacuum. The study of hypersonic phononics (hPnC) imposes substantial demand on fabrication and characterization techniques. Colloid and polymer science offer methods to create novel materials that possess periodic variations of density and elastic properties at length scales commensurate with the wave length of hypersonic phonons and hence visible photons. The key quantity is the dispersion ω(q) of high frequency (GHz) acoustic excitations with wave vector q which is measured by the noninvasive high resolution Brillouin light scattering. The approach involves the exploitation of Bragg-type band gaps (BGs) that result from the destructive interference of waves in periodic media. However, the sensitivity of BG formation to structural disorder limits the application of self-assembly methods that are susceptible to defect formation. Hybridization gaps (HG), originating from the anti-crossing between local resonant and propagating modes, are robust to structural disorder and occur at wavelengths much larger than the size of the resonant unit. Here, examples based on hierarchical structures will be highlighted: 1D-hPnC to acquire comprehensive understanding, while the incorporation of defects holds a wealth of opportunities to engineer ω(q); in colloid based phononics, ω(q) has revealed both types of band gabs; particle brush materials with controlled architecture of the grafted chains enable a new strategy to realize HG’s and; hierarchically nanostructured matter can involve unprecedented phonon phono propagation mechanisms.
Figure 1: Schematic phononic band diagram for structure directed interference (BG in a) and particle-resonance induced hybridization (HG in b) band gap where the wave vector q* is along the Brillouin zone. a, L are the lattice constant and the particle dimension and c, cp the sound velocity in the structure and particle, respectively.
BiographyGeorge Fytas is a Professor of Physical Chemistry in Department of Materials Science and Technology at University of Crete and External Member of the Max Planck Society since 1998. He holds PhD degree in Physical Chemistry at Technical University of Hannover, Germany; completed his Post-doctorate research at Stony Brook in USA and; Habilitation at University of Bielefeld in Germany. He has received Humboldt Senior Research Award (2002), became a Fellow of the American Physical Society (2004) and he was Adjunct Professor at University of Akron in 2013.
George Fytas et al., J Phys Chem Biophys 2017, 7:4(Suppl)DOI: 10.4172/2161-0398-C1-025

Page 32
conferenceseries.com
Volume 7, Issue 4 (Suppl)J Phys Chem Biophys, an open access journal
ISSN: 2161-0398Physical Chemistry 2017
September 18-19, 2017
4th International Conference on
Physical and Theoretical ChemistrySeptember 18-19, 2017 Dublin, Ireland
Theoretical study of the chemical reactions by the combination of quantum mechanical and molecular dynamics methodsToshiaki Matsubara Kanagawa University, Japan
I will introduce two subjects1,2 we have examined by the combination of quantum mechanical and molecular dynamics methods I. It has been known that the nucleophilic substitution at the Si atom, SiH3Cl + Cl*- → SiH3Cl* + Cl-, proceeds by two steps with the
inversion or retention of the configuration passing through an intermediate with the trigonal bipyramid (TBP) structure (Figure 1), although the conventional SN2 reaction at the C atom proceeds by one step with the inversion of the configuration passing through a transition state with the TBP structure. We followed by the QM method all the possible paths and found that TBPcis produced with a high probability is readily transformed to the energetically more stable TBPtrans. In order to obtain more information concerning the trajectory of Cl- on the dissociation from TBPtrans, which we cannot clarify on the basis of the energy profile determined by the QM method, we conducted the MD simulations with and without the water solvent. The QM-MD3 simulations without the water solvent revealed that the dissociation of Cl- from TBPtrans occurs without passing through TBPcis. The ONIOM-MD3 simulations with the water solvent further suggested that the thermal fluctuation of the water solvent significantly affects the oscillation of the kinetic and potential energies of the substrate to facilitate the isomerization of the TBP intermediate from the cis form to the trans form and the subsequent dissociation of Cl- from TBPtrans.II. Germanone R2Ge=O have not been isolated until recently, because it easily olymerizes due to its unstability. However, Tamao et al. recently succeeded to isolate the germanone (Eind)2Ge=O by the incorporation of bulky substituents called Eind. The isolated (Eind)2Ge=O is very reactive and the reactions that do not proceed in the case of ketone easily proceed in the case of germanone. For example, the σ bond of water adds to the Ge=O of germanone to form the germanediol at room temperature. In this study, we examine the mechanism of the σ bond cleavage of the substrate on the Ge=O bond. In the case of H2O, the QM calculations showed that the H2O coordinates to the Ge before the σ bond cleavage (Figure 2) and this coordination induces a heterolytic σ bond cleavage. We further performed the QM-MD3 simulations and found that the kinetic energy concentrates on the coordinated H2O oxygen to strongly oscillate the coordinate bond. This oscillation further enlarges just before the O-H σ bond cleavage. The kinetic energy of this oscillation would be transmitted to the normal mode of the O-H bond breaking. Thus, the coordination and the vibration of the H2O oxygen was thought to be an important driving force of the O-H σ bond cleavage.
BiographyToshiaki Matsubara is working as a Professor in Department of Chemistry, Faculty of Science, at Kanagawa University, Japan.
Toshiaki Matsubara, J Phys Chem Biophys 2017, 7:4(Suppl)DOI: 10.4172/2161-0398-C1-025

Page 33
Notes:
conferenceseries.com
Volume 7, Issue 4 (Suppl)J Phys Chem Biophys, an open access journal
ISSN: 2161-0398Physical Chemistry 2017
September 18-19, 2017
4th International Conference on
Physical and Theoretical ChemistrySeptember 18-19, 2017 Dublin, Ireland
Recent advances in quantum Monte Carlo: Applications to lithium ion – Stockmayer clusters, hydrogen isotopic separation, and the investigation of excited state manifoldsEmanuele Curotto1 and Massimo Mella2 1Arcadia University, USA 2Università degli Studi dell'Insubria, Italy
Over the past two decades, our group at Arcadia University and the Mella group at Universita` degli Studi dell'insubria, have been closely collaborating in the development and implementation of quantum Monte Carlo methods, primarily to estimate
nuclear quantum effects in condensed matter and clusters. The most recent advances include the formal development of path integral and diffusion Monte Carlo methods that permit enhanced convergence when rigid constraints are included in simulations, the introduction of Smart–Darting–like techniques to enhance the sampling of the ground state density in extremely frustrated systems, and the exploration of the mathematical property of the Langevin equation in manifolds with boundaries and gradient torsion associated with nodal surfaces of excited states. Application examples include the determination of coupling effects on the ground state of gas phase molecules between rotation and torsional degrees of freedom, the determination of the ground state energy and wave functions of lithium-ion Stockmayer clusters, the enhancement of hydrogen isotopic separation offered by the surface of ammonia clusters, the ring polymer molecular dynamics from the Brownian bridge representation of the path integral, and the simulation of multi-electronic states in the Kustaanheimo-Stiefel Space by Diffusion Monte Carlo.
Fig. 1 The capped pentagonal bipyramid global minimum of the 5-D PES of (H2)10
BiographyEmanuele Curotto completed his Bachelor of Science at University of Massachusetts Lowell; Doctorate degree at Yale University in 1996 under the direction of Dr. J Cross, and; was a Postdoctoral fellow at University of Rhode Island under the direction of Dr. D L Freeman. He currently serves as Professor and Department Chair at Arcadia University (formerly Beaver College).
Emanuele Curotto et al., J Phys Chem Biophys 2017, 7:4(Suppl)DOI: 10.4172/2161-0398-C1-025

Page 34
Notes:
conferenceseries.com
Volume 7, Issue 4 (Suppl)J Phys Chem Biophys, an open access journal
ISSN: 2161-0398Physical Chemistry 2017
September 18-19, 2017
4th International Conference on
Physical and Theoretical ChemistrySeptember 18-19, 2017 Dublin, Ireland
What is hidden behind a phase diagram?Fabienne Berthier1, Jérôme Creuze1 and Bernard Legrand2 1Paris-Sud University, France 2CEA Nuclear Energy Division, Section of Physical Metallurgy, France
The thermodynamics of binary alloys is still far from being well understood despite numerous studies, in particular when the two constituents have very different atomic volumes. That is the case for the Au-Ni and Ag-Cu alloys that tend to phase
separate and possess a large size mismatch. The phase diagrams of the two systems are characterized by a large miscibility gap. This apparent simplicity is, nevertheless, undermined by studies on the local order (short-range-order SRO). The ordering SRO observed experimentally is agreement with the phase diagram for Ag-Cu whereas it remains controversial for Au-Ni. We present a novel energetic model that takes into account atomistic relaxations to describe the thermodynamic properties of binary alloys . It involves of the calculation of site energies in a relaxed random solid solution as a function of the local composition and of the nominal concentration. The numerical results are obtained using N-body interatomic potentials derived on the second moment approximation (SMA) of the tight-binding scheme. This new model allows us to determine the effective pair interactions (EPI) that drive the SRO and to evaluate their contribution to the mixing enthalpy, as well as that of related to the lattice mismatch between the components. We apply this formalism to the Au-Ni and Ag-Cu alloys. Monte Carlo (MC) simulations on rigid lattice using this energetic model lead to phase diagrams that are in remarkable agreement with that obtained with SMA-MC simulations and the experimental ones. We show that the phase separation is mainly driven by the elastic contribution for Au-Ni and by the EPI’s contribution for Ag-Cu. Furthermore for Au-Ni, SRO which are related to the EPIs, display a sign change as a function of the concentration.
BiographyFabienne Berthier completed her education from Engineering School at Grenoble in Materials and Electrochemistry, 1982-1985. Her research interests include Atomistic simulations, multiscale modelling, thermodynamic properties of alloy at interfaces (surfaces, grain boundaries) and nanoalloys, aging kinetics. Her expertise is in modeling the thermodynamical properties of alloys to predict phase diagrams. She has developed a methodology that mixes a rigid lattice using model with off lattice Monte Carlo simulations. This mixed approach is very efficient to predict and analyze the interfacial segregation as for example at grain boundaries, surfaces and nanoparticles. She has also an expertise in Growth Kinetics and Ageing Kinetics.
Fabienne Berthier et al., J Phys Chem Biophys 2017, 7:4(Suppl)DOI: 10.4172/2161-0398-C1-025

Page 35
Day 1 September 18, 2017
Sessions:
Theoretical and Computational Chemistry | Chemical Physics | FemtochemistrySession ChairVassiliki-Alexandra GlezakouPacific Northwest National Laboratory, USA
Session Co-ChairIvan ŠtichSlovak Academy of Sciences, Slovakia
Session IntroductionTitle: Molecular design strategies for task-specific solvent technologies
Vassiliki-Alexandra Glezakou, Pacific Northwest National Laboratory, USATitle: Magnetism and spin transport in transition metal organometallic clusters
Ivan Štich, Slovak Academy of Sciences, Slovakia Title: Threshold photoelectron and electron-ion coincidence spectroscopies: Past, present and
futureRichard Tuckett, University of Birmingham, UK
Title: Homogeneous nucleation of solid, liquid and glass phases close to revolutionRobert F Tournier, University of Grenoble, France
Title: Recent advances in theoretical spectroscopy from ab initio molecular dynamicsSandra Luber, University of Zurich, Switzerland
Title: Ab initio theory for computing sum frequency generation spectra at aqueous interfacesTatsuhiko Ohto, Osaka University, Japan
Title: Furious and tranquil radicals: A computational study of sulfur-centred radical chemistryIsa Degirmenci, Ondokuz Mayis University, Turkey
Title: Design of novel imidazole-based corrosion inhibitors - molecular dynamics simulations and electrochemical studiesRichard M W Wong, National University of Singapore, Singapore
Title: Fragility of metallic liquids manifest in the high temperature structure and cohesive energyAnup K Gangopadhyay, Washington University in St. Louis, USA
Title: Low energy electrons induced damage to selected dna fragmentsManabendra Sarma, Indian Institute of Technology Guwahati, India
Title: Simulation of DBS, DBS-COOH and DBS-CONHNH2 as hydrogelatorsDafna Knani, ORT Braude College, Israel
Title: An appropriate quantum mechanical approach to understand the anomalous behaviors of liquid alkali metals and group- IV alloysAlok Satpathy, Bir Bikram Memorial College, India
Title: Atoms and dimers in rare gas crystals: Modelling of the stable trapping sitesAlexei A Buchachenko, Skolkovo Institute of Science and Technology, Russia
Title: Matrix isolation infrared spectroscopy and structures of weak (O-H···π) and strongly bound (OH···O) binary hydrogen bonded complexesPujarini Banerjee, Indian Association for the Cultivation of Science, India
Title: Determination of the isotopic composition of aqueous solutions radiospectroscopic methodRostislav Y Gerasimov, The Bauman Moscow State Technical University, Russia
Title: High-frequency radiations in water under the action of high-voltage short-wave pulses Elchin J Gurbanov, “Azersu” OJSC, Azerbaijan
Session Introduction

Page 36
conferenceseries.com
Volume 7, Issue 4 (Suppl)J Phys Chem Biophys, an open access journal
ISSN: 2161-0398Physical Chemistry 2017
September 18-19, 2017
4th International Conference on
Physical and Theoretical ChemistrySeptember 18-19, 2017 Dublin, Ireland
Molecular design strategies for task-specific solvent technologiesVassiliki-Alexandra GlezakouPacific Northwest National Laboratory, USA
CO2 capture from power plant exhaust is a complex problem that requires the capture and removal of massive quantities of gases. Solvent technologies for CO2 capture and conversion have become one of the most promising solutions with aqueous amines
being one of the industrial standards. However, their high regeneration costs render them prohibitive for many of the large-scale applications in power generation. My presentation will outline the computational approach used toward the deliberate design of single-molecule CO2-binding transformational solvents. These types of solvents constitute an attractive alternative to the water-based solvents, but are hampered by exponentially increasing viscosities at high CO2 saturation. Using state-of-the-art computational methods, like enhanced sampling methods for reaction free energetics in explicit solvent models using ab initio molecular dynamics, we describe the key structural parameters that allowed us to create reduced models for fast screening of solvent libraries. This approach led to tangible hypotheses as to the synthetic protocols that have already identified candidate molecules with appreciable viscosity reductions at target loading levels.
BiographyVassiliki-Alexandra Glezakou is a Computational Chemist with over 20 years of experience in atomistic simulations, with particular emphasis in transition metals chemistry and condensed phase systems relevant to carbon capture an conversion, catalysis and materials properties. Her focus of recent research is on the structure, vibrational spectroscopy and structure/activity correlations in a diverse ensemble of problems such as catalytic activity of metal clusters and oxide supports, transformative solvents for post-combustion carbon dioxide separations, mechanistic studies of MOF nucleation.
Vassiliki-Alexandra Glezakou, J Phys Chem Biophys 2017, 7:4(Suppl)DOI: 10.4172/2161-0398-C1-025
Notes:
Page 37
conferenceseries.com
Volume 7, Issue 4 (Suppl)J Phys Chem Biophys, an open access journal
ISSN: 2161-0398Physical Chemistry 2017
September 18-19, 2017
4th International Conference on
Physical and Theoretical ChemistrySeptember 18-19, 2017 Dublin, Ireland
Magnetism and spin transport in transition metal organometallic clustersIvan StichInstitute of Physics, Slovak Academy of Sciences, Slovakia
Transition metal organometallics has recently attracted much attention due to its potential for applications in catalysis, molecular recognition, high-density storage, quantum computing, and spintronics. Despite these applications, reliable theoretical
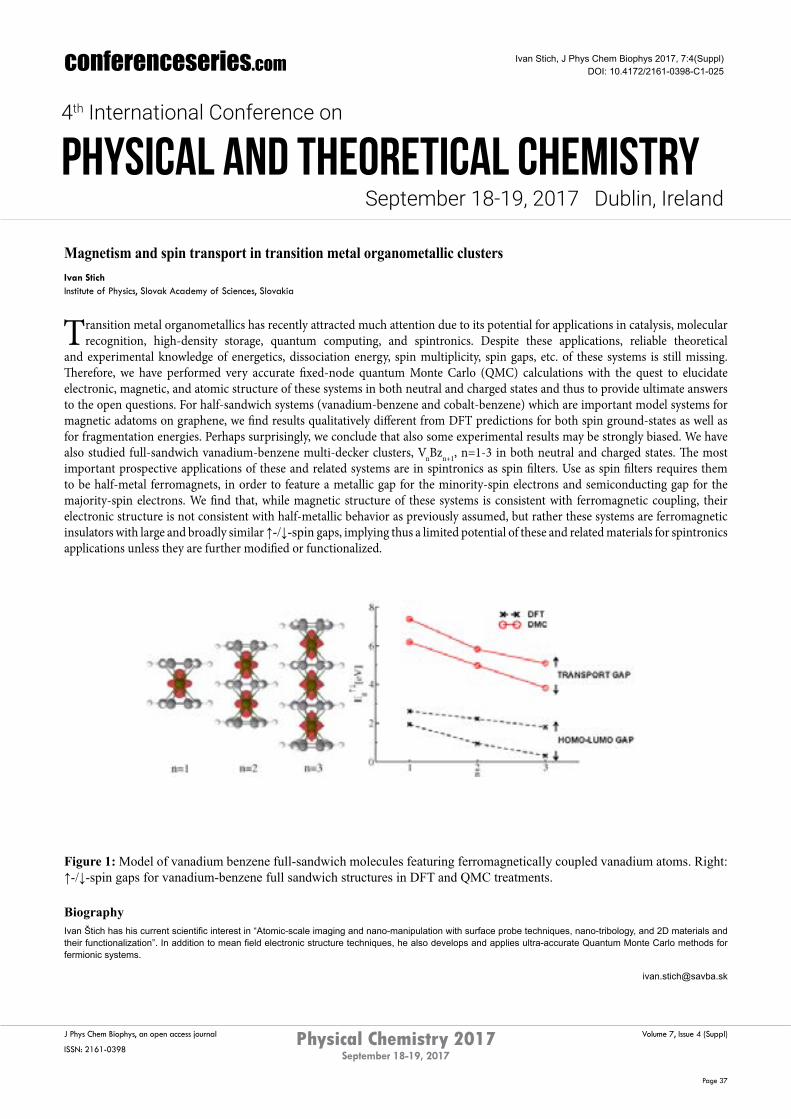
and experimental knowledge of energetics, dissociation energy, spin multiplicity, spin gaps, etc. of these systems is still missing. Therefore, we have performed very accurate fixed-node quantum Monte Carlo (QMC) calculations with the quest to elucidate electronic, magnetic, and atomic structure of these systems in both neutral and charged states and thus to provide ultimate answers to the open questions. For half-sandwich systems (vanadium-benzene and cobalt-benzene) which are important model systems for magnetic adatoms on graphene, we find results qualitatively different from DFT predictions for both spin ground-states as well as for fragmentation energies. Perhaps surprisingly, we conclude that also some experimental results may be strongly biased. We have also studied full-sandwich vanadium-benzene multi-decker clusters, VnBzn+1, n=1-3 in both neutral and charged states. The most important prospective applications of these and related systems are in spintronics as spin filters. Use as spin filters requires them to be half-metal ferromagnets, in order to feature a metallic gap for the minority-spin electrons and semiconducting gap for the majority-spin electrons. We find that, while magnetic structure of these systems is consistent with ferromagnetic coupling, their electronic structure is not consistent with half-metallic behavior as previously assumed, but rather these systems are ferromagnetic insulators with large and broadly similar ↑-/↓-spingaps, implying thus a limited potential of these and related materials for spintronics applications unless they are further modified or functionalized.
Figure 1: Model of vanadium benzene full-sandwich molecules featuring ferromagnetically coupled vanadium atoms. Right: ↑-/↓-spingapsforvanadium-benzenefullsandwichstructuresinDFTandQMCtreatments.
BiographyIvan Štich has his current scientific interest in “Atomic-scale imaging and nano-manipulation with surface probe techniques, nano-tribology, and 2D materials and their functionalization”. In addition to mean field electronic structure techniques, he also develops and applies ultra-accurate Quantum Monte Carlo methods for fermionic systems.
Ivan Stich, J Phys Chem Biophys 2017, 7:4(Suppl)DOI: 10.4172/2161-0398-C1-025

Page 38
conferenceseries.com
Volume 7, Issue 4 (Suppl)J Phys Chem Biophys, an open access journal
ISSN: 2161-0398Physical Chemistry 2017
September 18-19, 2017
4th International Conference on
Physical and Theoretical ChemistrySeptember 18-19, 2017 Dublin, Ireland
Threshold photoelectron and electron-ion coincidence spectroscopies: Past, present and futureRichard Tuckett1 and Tom Baer2
1University of Birmingham, UK2University of North Carolina, USA
The history and evolution of molecular threshold photoelectron spectroscopy and threshold photoelectron photo-ion coincidence spectroscopy (TPEPICO) in the gas phase over the last fifty years is reviewed. Emphasis is given on instrumentation and the
extraction of dynamical information about energy selected ion dissociation, not on the detailed spectroscopy of certain molecules. Three important advances have greatly expanded the power of the technique, and permitted its implementation in modern synchrotron radiation beam lines. (a) The use of velocity focusing of threshold electrons onto an imaging detector in the 1990s simultaneously improved the sensitivity and electron energy resolution, and also facilitated the subtraction of hot electron background in both threshold electron spectroscopy and TPEPICO studies. (b) The development of multi-start multi-stop collection detectors for both electrons and ions in the 2000s permitted the use of the full intensity of modern synchrotron radiation thereby greatly improving the signal-to-noise ratio. (c) Finally, recent developments involving imaging electrons in a range of energies as well as ions onto separate position-sensitive detectors has further improved the collection sensitivity, so that low density samples found in a variety of studies can be investigated. As a result, photoelectron photo ion coincidence spectroscopy is now well positioned to address a range of challenging problems that include the quantitative determination of compositions of isomer mixtures, the detection and spectroscopy of free radicals produced in pyrolysis or discharge sources as well as in combustion studies.
Deity of Science: Award winning original art work by Dr Jonelle Harvey (PhD student of RT). It depicts the imaging PEPICOend-stationattheSwissLightSource(wheresomeofthedatadescribedinPaper1(below)weretaken),andtheconstant stream of information produced from reactions generated from within.
BiographyRichard Tuckett completed his PhD in near-infrared spectroscopy from University of Cambridge in 1979. He first worked in electronic fluorescence spectroscopy of free radicals and molecular cations, often using supersonic beams and non-resonant electron excitation. From the late 1980s, he started using tunable vacuum-ultraviolet photon excitation from a synchrotron as a resonant ionisation source. He also developed an interest in threshold photoelectron spectroscopy and related coincidence techniques, particularly threshold photoelectron photo-ion coincidence studies.
Richard Tuckett et al., J Phys Chem Biophys 2017, 7:4(Suppl)DOI: 10.4172/2161-0398-C1-025

Page 39
Notes:
conferenceseries.com
Volume 7, Issue 4 (Suppl)J Phys Chem Biophys, an open access journal
ISSN: 2161-0398Physical Chemistry 2017
September 18-19, 2017
4th International Conference on
Physical and Theoretical ChemistrySeptember 18-19, 2017 Dublin, Ireland
Homogeneous nucleation of solid, liquid and glass phases close to revolution Robert F Tournier1, 2
1Grenoble Alps University, France2Institut Néel, France
The classical nucleation equation fails to predict the glass phases, the liquid-to-liquid phase transitions (LLPT), Lindemann’s constant, the presence of intrinsic growth nuclei above Tm inducing magnetic texturing by cooling magnetic melts in high
magnetic fields, and the first-order glass transitions of liquid helium under pressure. These problems are solved adding in (1) an enthalpy saving ε×ΔHm associated with the formation of spherical growth nuclei (super clusters) having the same melting temperature Tm and melting enthalpy ΔHm per mole whatever their radius R is: 3
21
4 ( ) 4 (1 )3 m mRG H R Hπ θ ε π ε σ∆ = ∆ × − + + ∆ (1 where ε(θ)= ε0(1-θ2x θo-2) is
a numerical coefficient equal to εls or εgs or Δε1g=(ε1s-εgs) with the indexes l for liquid, s for solid and g for glass phases, θ =(T-Tm)/Tm and θ0 is θ0l or θ0g. The glass transition is viewed as a LLPT from Phase 1 above Tg to Phase 2 below Tg. εls×ΔHm is reduced at the glass transition with εls replaced by εgs and θ0m by θog, εls and εgs being the enthalpy saving maximum coefficients associated with the homogeneous nucleation of nuclei inducing crystallization. The enthalpy change at Tg giving rise to the glass phase obeys (1) with ε replaced by εlg. All thermodynamic properties are calculated when Tg, εls=εls0 at Tm and θ0l are known. Phase 3 is relaxing in Phase 2. An enthalpy excess, due to quenching the melt or to vapor deposition, can induce sharp transitions to Phase 3. Two homogeneous nucleation temperatures θ =ε above Tm and θ =(ε-2)/3 below Tm are expected minimizing the surface energy. Values of ε have been obtained in pure liquid elements, strong and fragile glass-forming melts. In pure liquid elements, the smallest value εls0=0.217 leads to Lindemann’s constant equal to 0.103 at Tm. Δεlg, εls and εgs are used to predict LLPT between phase 1 and phase 2 above and below Tm even in water.
BiographyRobert F Tournier was Research Director at CNRS Grenoble in 2000. His group showed the appearance of magnetic moments in clusters of transition atoms and the existence of scaling laws for diluted spin glasses. The magnetic susceptibility of isolated impurities submitted to Kondo effect was separated from that of magnetic clusters. The disappearance of Kondo effect by antiparallel coupling of nuclear and electronic spins ½, the local spin fluctuations and electronic moments induced by nuclear moments in Praseodymium Van Vleck compounds were also discovered. The coexistence between superconductivity and ferromagnetism was studied or discovered in few compounds.
Robert F Tournier, J Phys Chem Biophys 2017, 7:4(Suppl)DOI: 10.4172/2161-0398-C1-025

Page 40
Notes:
conferenceseries.com
Volume 7, Issue 4 (Suppl)J Phys Chem Biophys, an open access journal
ISSN: 2161-0398Physical Chemistry 2017
September 18-19, 2017
4th International Conference on
Physical and Theoretical ChemistrySeptember 18-19, 2017 Dublin, Ireland
Recent advances in theoretical spectroscopy from ab initio molecular dynamicsSandra LuberUniversity of Zurich, Switzerland
Knowledge about properties of liquid is extremely helpful for the analysis of molecular structures and interactions. Moreover, it is a valuable source of information for the characterization of dynamic processes and facilitates the interpretation of experimental
data. Calculations provide additional insight allowing the targeted study of specific structures. In this way, it is possible to quantify the contributions of, e.g., solute and solvent molecules or adsorbates on solids. We present innovative methods for the calculation of spectroscopic and local properties for periodic systems such as liquids, which can efficiently be employed in density functional theory-based molecular dynamics. Moreover, computationally efficient approaches for the calculation of Raman and sum frequency generation spectroscopy have been developed as well as the first method for Raman optical activity spectroscopy from ab initio molecular dynamics. Recently studied systems include a gas-semiconductor interface as well as ionic liquids.
BiographySandra Luber completed her MSc and PhD degree from ETH Zurich in 2007 and 2009, respectively. After Post-doctoral studies at University of Basel and Yale University, she joined BASF SE in 2012. Afterwards, she became Project Group Leader at University of Zurich. Her Habilitation thesis was completed in 2016 and she is currently an SNSF Professor at University of Zurich.
Sandra Luber, J Phys Chem Biophys 2017, 7:4(Suppl)DOI: 10.4172/2161-0398-C1-025

Page 41
conferenceseries.com
Volume 7, Issue 4 (Suppl)J Phys Chem Biophys, an open access journal
ISSN: 2161-0398Physical Chemistry 2017
September 18-19, 2017
4th International Conference on
Physical and Theoretical ChemistrySeptember 18-19, 2017 Dublin, Ireland
Ab initio theory for computing sum frequency generation spectra at aqueous interfacesTatsuhiko OhtoOsaka University, Japan
Understanding aqueous interfaces at the molecular level is not only fundamentally important, but also highly relevant for a variety of disciplines. For instance, electrode–water interfaces are relevant for electrochemistry, as mineral–water interfaces
for geochemistry and air–water interfaces for environmental chemistry; lipid–water interfaces constitute the boundaries of the cell membrane, and are thus relevant for biochemistry. One of the major challenges in these fields is to link macroscopic properties such as interfacial reactivity, solubility, and permeability as well as macroscopic thermodynamic and spectroscopic observables to the structure, structural changes, and dynamics of molecules at these interfaces. Simulations, by themselves, or in conjunction with appropriate experiments, can provide such molecular-level insights into aqueous interfaces. We study aqueous interfaces, by assessing computations of the sum-frequency generation (SFG) spectra, which selectively detect the interfacial molecules, at aqueous interfaces. To avoid bias in the computational results and interpretation originating from the choices of the details of FF models, applying a parameter-free ab initio molecular dynamics (AIMD) simulation technique to the SFG calculation seems to be a promising route. However, the huge computational cost required for AIMD simulation has prohibited the widespread use of AIMD simulations for computing the SFG spectra. We have recently presented an efficient calculation algorithm for computing the SFG spectra of the water O–H stretch mode based on the surface-specific velocity–velocity correlation function, by separating degrees of freedom of the nuclei from solvation effects such as the induced dipole and polarizability. This methodology has been applied to the fundamental water-air, water-lipid, and aqueous solution-air interfaces. We are going to extend our method to solid-liquid interfaces.
Figure: Trimethylamine-N-oxide solution-air interface
BiographyTatsuhiko Ohto is an Assistant Professor at Osaka University, Osaka, Japan. He received his PhD degree from University of Tokyo in 2013. During his PhD course, he spent five months as a visiting student at Max Planck Institute for Polymer Research, Mainz, Germany. After Post-doctoral research at Advanced Institute of Science and Technology, Tsukuba, Japan, he joined Osaka University. His research interest is in theoretical modeling, primarily based on first-principles calculations of the structure and dynamics of molecules at interfaces and the electron transport of metal–molecule–metal systems.
Tatsuhiko Ohto, J Phys Chem Biophys 2017, 7:4(Suppl)DOI: 10.4172/2161-0398-C1-025

Page 42
Notes:
conferenceseries.com
Volume 7, Issue 4 (Suppl)J Phys Chem Biophys, an open access journal
ISSN: 2161-0398Physical Chemistry 2017
September 18-19, 2017
4th International Conference on
Physical and Theoretical ChemistrySeptember 18-19, 2017 Dublin, Ireland
Furious and tranquil radicals: A computational study of sulfur-centered radical chemistryIsa Degirmenci1 and Michelle L Coote2
1Ondokuz Mayis University, Turkey2Australian National University, Australia
Recently, there has been growing interest in both the thiol-ene (Scheme 1) and thiol-yne sister reactions in various application areas from polymer field to bioconjugative materials due to the extraordinary advantages of these reaction techniques in, for example,
forming uniform polymer networks with narrow Tg values and low shrinkage stress. The most outstanding feature of these reactions is combination of the advantages of both step growth and chain growth polymerization reactions. Another attractive subject is self-healing polymeric materials that have been brand-new of interest in polymer science. Some of these materials utilize trithiocarbonate or thiuram disulfide units in the polymer structure which allows healable backbone of polymer chain. It is well known that the sulfur-centered radicals play a vital role in these thiol reactions or self-healing processes. To better understand the structure-reactivity trends of these extraordinary radicals, we have used computational chemistry (at the G3(MP2)-RAD//MP2/6-31G(d) level of theory) to study the highly reactive alkyl thiyl radical addition reaction to the C=C and S=C double bonds of various compounds. In addition to this, the high stability of the sulfur-centered radicals has been extensively studied to elaborate controversial behavior of these radical species. We find that the high SOMO energy of the radicals has the ability to undergo resonance interactions with π* of the substrate and this allows formation of stabilized transition state structure in radical addition reactions. The same effect is account for enormous stability of sulfur-centered radicals which are more effectively conjugated with heavier lone-pair donor and π-acceptor substituents than carbon-centered radical analogues.
BiographyIsa Degirmenci completed his under-graduated from Marmara University in 1998-2002 and graduated from Boğaziçi University in 2002-2005 respectively. He was awarded PhD from Gent University, Belgium and completed Post-doctoral studies from Australian National University. He has his expertise in “Structure-reactivity relationships in the free radical polymerization reactions”. His investigations combine the structure properties of radical species and monomers with application of quantum chemical tools. His recent studies have simply explained the extraordinary reactivity and stability behavior of sulfur-centered radicals. These findings can be considered as paving the way for further utilizing recently emerged thiol-ene and thiol-yne polymerization reactions.
Isa Degirmenci et al., J Phys Chem Biophys 2017, 7:4(Suppl)DOI: 10.4172/2161-0398-C1-025

Page 43
conferenceseries.com
Volume 7, Issue 4 (Suppl)J Phys Chem Biophys, an open access journal
ISSN: 2161-0398Physical Chemistry 2017
September 18-19, 2017
4th International Conference on
Physical and Theoretical ChemistrySeptember 18-19, 2017 Dublin, Ireland
Design of novel imidazole-based corrosion inhibitors - molecular dynamics simulations and electrochemical studiesRichard M W Wong1, Choon Wee Kee1, Ismail Abdulazeea2, Abdulaziz A Al-Saadi2 and Mazen Khaled2
1National University of Singapore, Singapore2King Fahd University of Petroleum & Mineral, Saudi Arabia
Obtaining a detailed insight into the mechanism of the protective action of various organic corrosion inhibitors on mild steel corrosion has remained an active area of research over the years. The use of computational chemistry as a tool in this aspect have
greatly enhanced the prediction of the inhibition efficiencies of these inhibitors based on their electronic and molecular properties and reactivity indices, which are subsequently validated by experimental measurements. In the present study, we investigated the corrosion inhibition efficiency of mild steel in 1.0 M HCl of the following compounds; imidazole (Imz), 2-bromo-1H-imidazole (2-Br-Imz), 2-chloro-1H-imidazole (2-Cl-Imz), 2-iodo-1H-imidazole (2-I-Imz) and 4-phenyl imidazole (4-Ph-Imz). Density functional theory (DFT) calculations showed that the inhibitor molecules dissociate to form a network of protective film on the iron surface. The equilibrium adsorption energies obtained from DFT calculations decreases in the order 2-I-Imz>4-Ph-Imz>2-Br-Imz>Imz>2-Cl-Imz. Electrochemical studies of the two extreme cases showed that 2-I-Imz exhibited the best inhibition efficiency of 82.95% at 10 mM concentration acting as anodic-type inhibitor while 2-Cl-Imz exhibited an efficiency of 50.70% at the same concentration acting as cathodic-type inhibitor. Both inhibitors were found to fit the Langmuir adsorption isotherm with Gibbs free energy of adsorptions -27.34 KJ/mol and -25.24 KJ/mol at 25oC for 2-I-Imz and 2-Cl-Imz respectively. SEM of the steel samples after immersion in the inhibitors for 24 h revealed a significant formation of pits on the 2-Cl-Imz sample possibly due to chloride attack, and the absence of such in the 2-I-Imz sample indicating its ability to form a protective film. XPS analysis confirmed the adsorption of the inhibitor molecules on the metal surface from the functional group analysis of the peaks obtained. AFM analysis showed a decrease in surface roughness of the 2-I-Imz sample as compared to the 2-Cl-Imz, indicative of a better adsorption and consequent inhibition efficiency observed of both inhibitor molecules.
Figure 1: AFMimagesof(a)baresteelandaftertreatmentwith(b)2-Cl-Imzand(c)2-I-Imz.
BiographyRichard M W Wong received his PhD. degree from Australian National University (1989). Subsequently, he held Post-doctoral position at IBM Kingston and Yale University. In 1992, he took up an Australian Research Fellowship, hosted in University of Queensland. He joined the National University of Singapore in 1997 and is currently a Full Professor and Head of Department. He was the recipient of Fukui Award recently for his outstanding work in theoretical and computational chemistry. He has published about 200 scientific publications, which received over 9000 citations and H-index of 42. His research interests include application of computational quantum chemistry to a range of chemical problems, reactive intermediates, catalysis, materials design, chemical sensors, weak intermolecular interactions and drug design.
Richard M W Wong et al., J Phys Chem Biophys 2017, 7:4(Suppl)DOI: 10.4172/2161-0398-C1-025

Page 44
Notes:
conferenceseries.com
Volume 7, Issue 4 (Suppl)J Phys Chem Biophys, an open access journal
ISSN: 2161-0398Physical Chemistry 2017
September 18-19, 2017
4th International Conference on
Physical and Theoretical ChemistrySeptember 18-19, 2017 Dublin, Ireland
Fragility of metallic liquids manifest in the high temperature structure and cohesive energyAnup K GangopadhyayWashington University in St. Louis, USA
Glasses are traditionally classified as fragile or strong depending on the rates of change of dynamical properties (viscosity, diffusion coefficient) with temperature near the glass-transition temperature. It will be shown that the temperature dependence of viscosity
far above the liquidus is an equally good measure of fragility. From the measurements of liquid structures of a large number of metallic glass-forming liquids using the electrostatic levitation (ESL) technique, combined with synchrotron X-rays, it will be demonstrated that the rates of structural changes of equilibrium and super cooled (metastable liquid below the liquidus) liquids with temperature are intimately connected with the liquid/glass fragilities. A strong connection between fragilities and average cohesive energies for metallic liquids will also be demonstrated.
BiographyAnup K Gangopadhyay is working as a Research Scientist at Washington University in St. Louis, USA.
Anup K Gangopadhyay, J Phys Chem Biophys 2017, 7:4(Suppl)DOI: 10.4172/2161-0398-C1-025

Page 45
conferenceseries.com
Volume 7, Issue 4 (Suppl)J Phys Chem Biophys, an open access journal
ISSN: 2161-0398Physical Chemistry 2017
September 18-19, 2017
4th International Conference on
Physical and Theoretical ChemistrySeptember 18-19, 2017 Dublin, Ireland
Low energy electrons induced damage to selected DNA fragmentsManabendra Sarma and Renjith BhaskaranIndian Institute of Technology Guwahati, India
We have theoretically and computationally investigated the mechanism of low energy electron (LEE) induced DNA damages such as single strand breaks (SSBs) and glycosidic bond cleavage in some selected DNA fragments (Fig. 1) viz., 2'-deoxycytidine-
3'-monophosphate (3'-dCMPH) [Fig. 1(a)], 2'-deoxycytidine-5'-monophosphate (5'-dCMPH) [Fig. 1(b)] and sugar-phosphate-sugar (SPS) [Fig. 1(c)]. In this regard, we have used electronic structure theory and our newly implemented local complex potential based time dependent wave packet (LCP-TDWP) approach. Results from our calculations show that in 3'-dCMPH and 5'-dCMPH DNA fragments SSB predicted near 1 eV whereas in SPS moiety it appears around 0.6 eV. Further, in case of SPS moiety there are two dissociation channels namely 3' C-O and 5' C-O bond lesions. Our calculations show that the activation energy barrier for 5' C-O bond dissociation is less than of 3'C-O bond dissociation pathway. It has also been found that the metastable anion formed after electron attachment to SPS moiety is more long lived (~40-55 fs) than that to 3'-dCMPH and 5'-dCMPH fragments (~18-20 fs). On the other hand, the glycosidic bond cleavage in 3'-dCMPH moiety [Fig. 1(d)] requires higher activation energy than of the SSB in the same fragment and thus least preferred channel compared to SSB.
Figure 1: SomeoftheselectedDNAfragments.Foreachfragmentbondsusceptibleforcleavageismarkedwithanarrow.
BiographyManabendra Sarma received his BSc and MSc degrees in the year 2000 and 2002 respectively from Goalpara College and Indian Institute of Technology Guwahati, Assam, India. After completing his MSc degree, he moved to Indian Institute of Technology Bombay, Mumbai, India in 2002 to pursue his PhD under Professor Manoj K. Mishra. He completed his PhD in 2008 and subsequently joined the Department of Chemistry at Indian Institute of Technology Guwahati, Assam, India as a Senior Lecturer in the same year. In the year 2011, he received the prestigious BOYSCAST Fellowship of India to work with Professor Lorenz S Cederbaum of University of Heidelberg, Germany for a year. His current research interests include development of new theoretical approaches to laser assisted control of chemical reactions and resonances in electron-molecule scattering. Currently he is an Associate Professor in Chemistry at Indian Institute of Technology Guwahati, Assam, India.
Manabendra Sarma et al., J Phys Chem Biophys 2017, 7:4(Suppl)DOI: 10.4172/2161-0398-C1-025

Page 46
conferenceseries.com
Volume 7, Issue 4 (Suppl)J Phys Chem Biophys, an open access journal
ISSN: 2161-0398Physical Chemistry 2017
September 18-19, 2017
4th International Conference on
Physical and Theoretical ChemistrySeptember 18-19, 2017 Dublin, Ireland
Simulation of DBS, DBS-COOH and DBS-CONHNH2 as hydrogelators Dafna Knani and David AlpersteinORT Braude College, Israel
The organic gelator 1,3(R):2,4(S)-dibenzylidene-D-sorbitol (DBS) self-organizes to form a 3-D network at relatively low concentrations in a variety of nonpolar organic solvents and polymer melt. DBS could be transformed into a hydrogelators
by introduction of hydrophilic groups, which facilitate its self-assembly in aqueous medium. In this work, we have investigated the hydrogelators DBS-COOH and DBS-CONHNH2 and the organogelator DBS by molecular modeling. We have used quantum mechanics (QM) to elucidate the preferred geometry of one molecule and a dimer of each of the gelators, and molecular dynamics (MD) to simulate the pure gelators and their mixtures with water. The results of the simulation indicate that the interaction between DBS-COOH molecules is the strongest of the three and its water compatibility is the highest. Therefore, DBS-COOH seems to be a better hydrogelator than DBS-CONHNH2 and DBS. Intermolecular H-bonding interactions are formed between DBS, DBS-COOH and DBS-CONHNH2 molecules as pure substances, and they dramatically decrease in the presence of water. In contrast, the intramolecular interactions increase in water. This result indicates that in aqueous environment the molecular structure tend to be more rigid and fixed in the preferred conformation. The most significant intramolecular interaction is formed between O3 acetal and H-O6 groups. Due to the H-bonds, DBS, DBS-COOH and DBS-CONHNH2 molecules form a rigid structure similar to liquid crystal forming molecules, which might explain their tendency to create nano fibrils. It was found that the aromatic rings did not contribute significantly to the inter-and intra-molecular interactions. Their main role is probably to stiffen the molecular structure.
Figure 1: DBS,DBS-COOHandDBS-CONHNH2 molecular structure.
BiographyDafna Knani is a Senior Lecturer in Department of Biotechnology Engineering, ORT Braude College. She is an Organic Polymer Chemist, graduated from Chemistry Faculty, Technion- Israel Institute of Technology. In the past, she worked for surgical bio-polymeric materials start-up company as a Research Polymer Chemist (developing adhesives for hard tissues) and as a Research Chemist and Project Leader at Israel Chemicals Ltd. (ICL), IMI-Institute for R&D. Her current research focuses on “Molecular modeling of materials and biomaterials”. Some of her research topics are simulation of systems used for controlled drug release and tissue engineering and computational design of polymer additives.
Dafna Knani et al., J Phys Chem Biophys 2017, 7:4(Suppl)DOI: 10.4172/2161-0398-C1-025

Page 47
Notes:
conferenceseries.com
Volume 7, Issue 4 (Suppl)J Phys Chem Biophys, an open access journal
ISSN: 2161-0398Physical Chemistry 2017
September 18-19, 2017
4th International Conference on
Physical and Theoretical ChemistrySeptember 18-19, 2017 Dublin, Ireland
An appropriate quantum mechanical approach to understand the anomalous behaviors of liquid alkali metals and group-IV alloys Alok SatpathyBir Bikram Memorial College, India
The anomalous properties of liquid alloys of alkali metals with group III, IV,V and VI elements (post transition metals or semi metals), particularly alkali metals and group-IV (usually Pb and Sn) alloys at and around the stoichiometric compositions have
tempted internationally to thrust research works in these materials for the last three and half decades. The observed peculiar properties i.e. departure from ideality in the structure, thermodynamic and other related properties in these systems have been attempted to explain with the hypothesis of the existence of polyanions i.e., zintl ions such as (Pb4)
4-, (Sn4)4- and (Te2)
2- etc. But, the existence of these ions or complex compounds is not experimentally confirmed and some of the observed phenomena are also self-contradictory to the basis of zintl principle. Hence, concluded as paradox by many authors. So, a new bonding scheme of loose overlapping of the atomic orbitals is introduced to tackle this paradox. Hence, a significant amount of effective charge transfer takes place between the atoms in the stoichiometric compositions. The chemical short range order (CSRO) in these alloys has been explained by coulomb interaction in lieu of zintl hypothesis. The generalized models for structure and thermodynamics of charged –hard-spheres mixture of arbitrary charge and size has been developed through sustain efforts of decades’ research. These models are employed successfully to evaluate the structure and thermodynamic properties i.e., entropies and entropies of mixing etc., treating the samples as partially charge transferred systems. Thus, internationally proclaimed paradox has been resolved. Some of the excellent research outcomes, achievements of my decades’ struggle along with the future plan would be delivered in this invited speech.
BiographyAlok Satpathy has a passion for research in basic sciences. He has been pursuing theoretical research for more than two decades in the field of soft condensed matter physics, in spite of limited resources and facilities. Being an expert in the Statistical-Mechanical Model studies of structure, thermodynamics and transport properties of liquids, metallic glasses and amorphous substances such as molten metals, alloys and ionic (full, partial and complex) liquids, he has developed generalized theoretical models for investigating structural and thermodynamic properties of ionic liquids having mixtures of arbitrary charge and size. He has been popularizing science and its philosophy in society through active involvements in innumerable seminars, talks and other activities spanning throughout his career.
Alok Satpathy, J Phys Chem Biophys 2017, 7:4(Suppl)DOI: 10.4172/2161-0398-C1-025
Page 48
conferenceseries.com
Volume 7, Issue 4 (Suppl)J Phys Chem Biophys, an open access journal
ISSN: 2161-0398Physical Chemistry 2017
September 18-19, 2017
4th International Conference on
Physical and Theoretical ChemistrySeptember 18-19, 2017 Dublin, Ireland
Atoms and dimers in rare gas crystals: Modeling of the stable trapping sites Alexei BuchachenkoSkolkovo Institute of Science and Technology, Russia
Matrix isolation spectroscopy of atomic and small molecular species regularly and reproducibly reveals the existence of distinct trapping sites, sometimes undergoing reversible and irreversible interconversion processes upon heating or irradiation. While
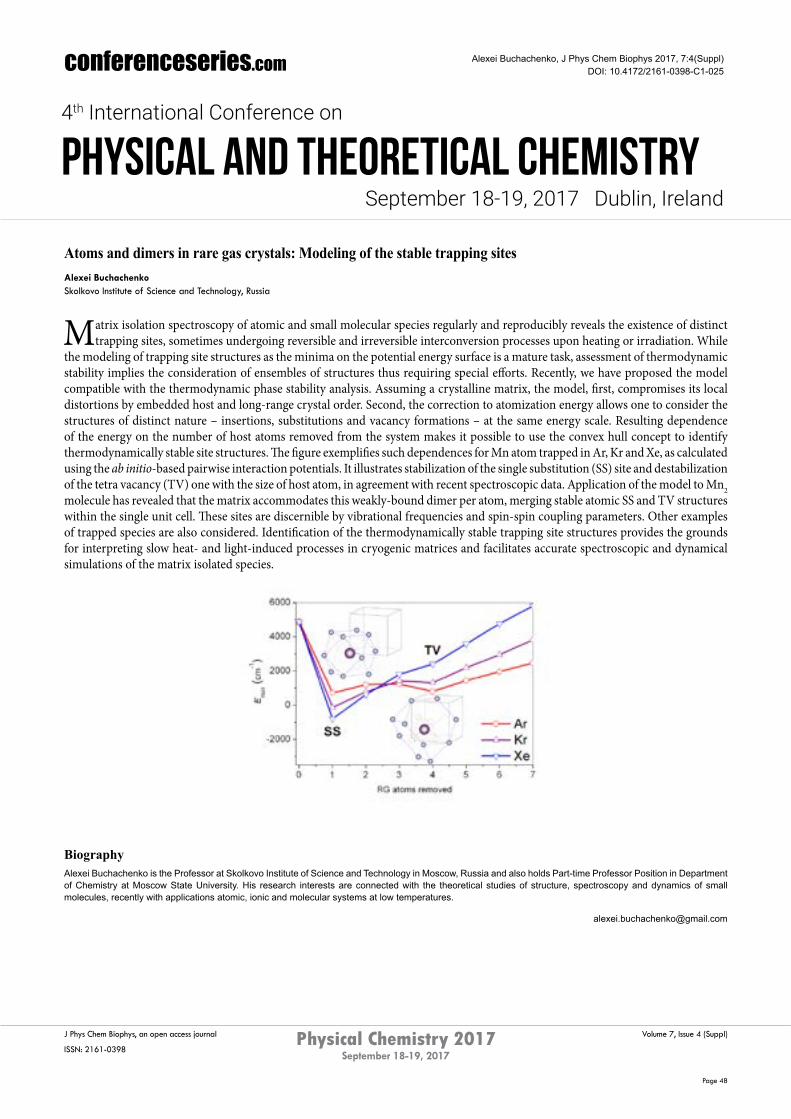
the modeling of trapping site structures as the minima on the potential energy surface is a mature task, assessment of thermodynamic stability implies the consideration of ensembles of structures thus requiring special efforts. Recently, we have proposed the model compatible with the thermodynamic phase stability analysis. Assuming a crystalline matrix, the model, first, compromises its local distortions by embedded host and long-range crystal order. Second, the correction to atomization energy allows one to consider the structures of distinct nature – insertions, substitutions and vacancy formations – at the same energy scale. Resulting dependence of the energy on the number of host atoms removed from the system makes it possible to use the convex hull concept to identify thermodynamically stable site structures. The figure exemplifies such dependences for Mn atom trapped in Ar, Kr and Xe, as calculated using the ab initio-based pairwise interaction potentials. It illustrates stabilization of the single substitution (SS) site and destabilization of the tetra vacancy (TV) one with the size of host atom, in agreement with recent spectroscopic data. Application of the model to Mn2 molecule has revealed that the matrix accommodates this weakly-bound dimer per atom, merging stable atomic SS and TV structures within the single unit cell. These sites are discernible by vibrational frequencies and spin-spin coupling parameters. Other examples of trapped species are also considered. Identification of the thermodynamically stable trapping site structures provides the grounds for interpreting slow heat- and light-induced processes in cryogenic matrices and facilitates accurate spectroscopic and dynamical simulations of the matrix isolated species.
BiographyAlexei Buchachenko is the Professor at Skolkovo Institute of Science and Technology in Moscow, Russia and also holds Part-time Professor Position in Department of Chemistry at Moscow State University. His research interests are connected with the theoretical studies of structure, spectroscopy and dynamics of small molecules, recently with applications atomic, ionic and molecular systems at low temperatures.
Alexei Buchachenko, J Phys Chem Biophys 2017, 7:4(Suppl)DOI: 10.4172/2161-0398-C1-025

Page 49
conferenceseries.com
Volume 7, Issue 4 (Suppl)J Phys Chem Biophys, an open access journal
ISSN: 2161-0398Physical Chemistry 2017
September 18-19, 2017
4th International Conference on
Physical and Theoretical ChemistrySeptember 18-19, 2017 Dublin, Ireland
Matrix isolation infrared spectroscopy and structures of weak (O-H•••π) and strongly bound (O-H•••O) binary hydrogen bonded complexes Pujarini Banerjee and Tapas ChakrabortyIndian Association for the Cultivation of Science, India
Matrix isolation infrared spectroscopic studies of two binary O-H•••π hydrogen bonded complexes of formic acid (FA) and phenol (Ph) with benzene (Bz), and a series of binary O-H•••O hydrogen bonded fluoro-phenol-water complexes will be reported.
In the first category, complexation results in red shifts of O-H stretching fundamentals (νOH) of Ph and FA by ~78 and ~120 cm-1, respectively, and the latter is the largest shift known so far for a binary complex of an O-H donor with π-orbitals of Bz as acceptor. We propose to use these observed red-shifts as benchmarks to test the accuracy of electronic structure methods in predicting geometries of the two complexes. We have noted that popularly used electronic structure theory methods with larger sized basis sets do not always predict structures that are consistent with the observed νOH shifts. This holds especially for the complex of Bz with FA, which will be discussed in detail. In the case of binary fluoro-phenol-water complexes, we have observed systematic νOH red-shifts of the fluoro-phenol-donors, which increase by ~90% from phenol to pentafluorophenol. Surprisingly, the magnitudes of the spectral shifts of the binary complexes display excellent linear correlation with the aqueous phase pKa values of the fluorophenols. Furthermore, the shifts display poor correlation with the total binding energies of the complexes, as signatures of deviation from the well-known Badger-Bauer rules. On the other hand, it has been shown that the spectral shifts relate nicely with the local quantum-chemical charge transfer (CT) interactions at the site of hydrogen bonding. We infer that this local interaction is the primary determining factor for spectral red-shifts of the donor in such binary complexes, and the same also holds for O-H•••π hydrogen bonded dimers.
BiographyPujarini Banerjee has expertise in the technique of matrix isolation spectroscopy, and her research interests include the infrared spectroscopic probing of binary hydrogen bonded complexes. She completed her Doctoral degree from University of Calcutta, and is currently working as a Post-doctoral Researcher under the supervision of Prof. Tapas Chakraborty in the Department of Physical Chemistry, Indian Association for the Cultivation of Science, Kolkata, India.
Pujarini Banerjee et al., J Phys Chem Biophys 2017, 7:4(Suppl)DOI: 10.4172/2161-0398-C1-025

Page 50
conferenceseries.com
Volume 7, Issue 4 (Suppl)J Phys Chem Biophys, an open access journal
ISSN: 2161-0398Physical Chemistry 2017
September 18-19, 2017
4th International Conference on
Physical and Theoretical ChemistrySeptember 18-19, 2017 Dublin, Ireland
Determination of the isotopic composition of aqueous solutions radio-spectroscopic method Rostislav Gerasimov, Artemiy G Maslov, German N Fadeev and Yuri V GerasimovBauman Moscow State Technical University, Russia
The proposed new method is based on the spectra of radio wave radiation of the microwave range wideband scanning receiver. It is experimentally proved that this method has a high accuracy in the determination of the frequency spectrum. This allows
reaching a sensitivity of 5-6 Hz/nm. The obtained spectra are analyzed for the particular program hardware-software complex. Practice has shown that the proposed method allows not only to detect the difference of the substances qualitative composition and concentration, but also to determine the presence of heavy and super heavy hydrogen isotopes in water. The method for determining the presence of substances in nanoscopic amount is based on the registration of radio-wave emission spectra in the microwave region using a scanning wideband receiver. This radiation arises owing to the excitation of alternate high-frequency displacement and conduction currents in an object subjected to testing between the flexible plates of a capacitive working sensor. The results from measurements (the obtained spectra) are analyzed using a hardware–software complex. For the research of aqueous solutions it is important to establish how the structure of water is influenced by temperature. In Fig. 1 shows the changes in the spectra of water temperature. The spectral pattern changes when the water is frozen to ice (spectra 2). Additional peaks emerge in the right side of the spectrum, beginning at a frequency of 2456.54 MHz. After the water returns to the liquid state (spectra 3) at the reference temperature (25°C), the spectra differ from the initial one by shifting toward an increase in the frequency of the spectra and a drop in the amplitudes of all peaks. Finally, it is concluded that: The method of radioscopy in the microwave range allows us to explore the fluid system, by comparing the spectra of the pure solvent (reference samples) and solutions; in this work, the parameters of the spectra of the standard with bi-distilled water and aqueous solutions: individual substances in the analyzed water; the difference of the concentrations of dissolved substances; evaluation of isotopic composition in the system of hydrogen isotopes and; it was established experimentally that this method can detect the presence of small quantities of heavy (D) and super heavy (T) hydrogen isotopes in ordinary (H2O) and heavy (D2O) water.
Fig. 1 Spectra of water in different states: (1) initial state at 299 K, (2) frozen state at 273 K, (3) thawed state at 299 K.
BiographyRostislav Gerasimov has his expertise in radioscopy of liquids, solids and nondestructive control of construction materials. His universal non-destructive testing method in microwave range is based on comparing the research object to the reference pattern.
Rostislav Gerasimov et al., J Phys Chem Biophys 2017, 7:4(Suppl)DOI: 10.4172/2161-0398-C1-025

Page 56
1186th Conferenceconferenceseries.com
Physical Chemistry 2017
4th International Conference on
Physical and Theoretical ChemistrySeptember 18-19, 2017 Dublin, Ireland
Day 2Scientific Tracks & Abstracts

Page 57
Physical Chemistry 2017
Day 2 September 19, 2017
Sessions:
Photochemistry | Solid-state Chemistry | Spectroscopy | Surface Science | Quantum Chemistry | Biophysical ChemistrySession ChairWerner PAULUSUniversity of Montpellier, France
Session Co-ChairHideaki ShirotaChiba University, Japan
Session IntroductionTitle: Temperature dependent spectral features of room temperature ionic liquids: Aromatic
vs.nonaromaticHideaki Shirota, Chiba University, Japan
Title: Complex magnetic phases and photo-enhanced ferromagnetism in nano-sized core-shell Prussian blue analogue cubesWen-Hsien Li, National Central University, Taiwan
Title: Photoinduced electron transfer through silicon bridge: The source for blue-green emissionMalgorzata Bayda, Adam Mickiewicz University, Poland
Title: Why is gas phase photolysis of 2-nitrophenol a significant source of OH in the polluted atmosphere?Lei Zhu, University at Albany, USA
Title: Intra- and Intermolecular strategies to improving photoluminescence quantum yields of n-π* fluorophores capable of harvesting triplet excitonsYoungmin You, Ewha Womans University, South Korea
Title: A computational investigation of the photochemical oxaziridine conversion process of some experimentally analyzed small-chain conjugated nitronesAnjan Chattopadhyay, BITS Pilani, India
Title: Topotactic synthesis of mixed-anion oxide epitaxial thin filmsAkira Chikamatsu, The University of Tokyo, Japan
Title: On the structure and tribological effect of interfacial water between a graphite surface and metallic or semiconducting counter bodiesArnaud Caron, KoreaTech - Korea University of Technology and Education, South Korea
Title: Anion and cation diffusion in complex oxidesManfred Martin, RWTH Aachen University, Germany
Title: New Aspects of an old class of compounds: Tetrelphosphides and their thermoelectric performanceUlrich Wedig, Max Planck Institute for Solid State Research, Germany
Title: Gold nanoparticles characterization by scattering correlation spectroscopyNadia Djaker, Université Paris 13, France
Title: The golden doxorubicin: A tunable design of gold (III)-Doxorubicin complex – PEGylated nanocarrier for oncologicalJolanda Spadavecchia, Université Paris 13, France
Title: Particle growth, assembly and extended particle-solvent interactions in deep eutectic solventsJoshua Hammons, Lawrence Livermore National Laboratory, USA
Session Introduction
Page 58
conferenceseries.com
Volume 7, Issue 4 (Suppl)J Phys Chem Biophys, an open access journal
ISSN: 2161-0398Physical Chemistry 2017
September 18-19, 2017
4th International Conference on
Physical and Theoretical ChemistrySeptember 18-19, 2017 Dublin, Ireland
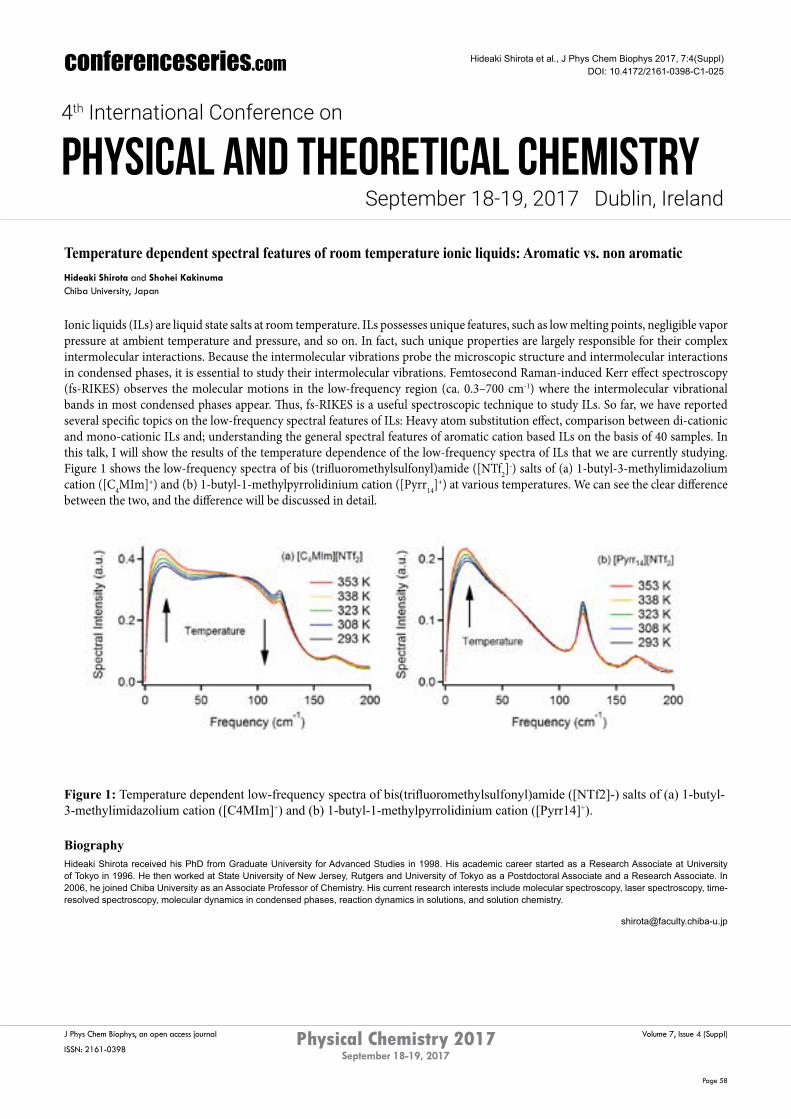
Temperature dependent spectral features of room temperature ionic liquids: Aromatic vs. non aromatic Hideaki Shirota and Shohei KakinumaChiba University, Japan
Ionic liquids (ILs) are liquid state salts at room temperature. ILs possesses unique features, such as low melting points, negligible vapor pressure at ambient temperature and pressure, and so on. In fact, such unique properties are largely responsible for their complex intermolecular interactions. Because the intermolecular vibrations probe the microscopic structure and intermolecular interactions in condensed phases, it is essential to study their intermolecular vibrations. Femtosecond Raman-induced Kerr effect spectroscopy (fs-RIKES) observes the molecular motions in the low-frequency region (ca. 0.3–700 cm-1) where the intermolecular vibrational bands in most condensed phases appear. Thus, fs-RIKES is a useful spectroscopic technique to study ILs. So far, we have reported several specific topics on the low-frequency spectral features of ILs: Heavy atom substitution effect, comparison between di-cationic and mono-cationic ILs and; understanding the general spectral features of aromatic cation based ILs on the basis of 40 samples. In this talk, I will show the results of the temperature dependence of the low-frequency spectra of ILs that we are currently studying. Figure 1 shows the low-frequency spectra of bis (trifluoromethylsulfonyl)amide ([NTf2]
-) salts of (a) 1-butyl-3-methylimidazolium cation ([C4MIm]+) and (b) 1-butyl-1-methylpyrrolidinium cation ([Pyrr14]
+) at various temperatures. We can see the clear difference between the two, and the difference will be discussed in detail.
Figure 1: Temperaturedependentlow-frequencyspectraofbis(trifluoromethylsulfonyl)amide([NTf2]-)saltsof(a)1-butyl-3-methylimidazoliumcation([C4MIm]+) and (b) 1-butyl-1-methylpyrrolidinium cation ([Pyrr14]+).
BiographyHideaki Shirota received his PhD from Graduate University for Advanced Studies in 1998. His academic career started as a Research Associate at University of Tokyo in 1996. He then worked at State University of New Jersey, Rutgers and University of Tokyo as a Postdoctoral Associate and a Research Associate. In 2006, he joined Chiba University as an Associate Professor of Chemistry. His current research interests include molecular spectroscopy, laser spectroscopy, time-resolved spectroscopy, molecular dynamics in condensed phases, reaction dynamics in solutions, and solution chemistry.
Hideaki Shirota et al., J Phys Chem Biophys 2017, 7:4(Suppl)DOI: 10.4172/2161-0398-C1-025

Page 59
Notes:
conferenceseries.com
Volume 7, Issue 4 (Suppl)J Phys Chem Biophys, an open access journal
ISSN: 2161-0398Physical Chemistry 2017
September 18-19, 2017
4th International Conference on
Physical and Theoretical ChemistrySeptember 18-19, 2017 Dublin, Ireland
Complex magnetic phases and photo-enhanced ferromagnetism in nano-sized core-shell Prussian blue analogue cubes Wen-Hsien Li1, Chi-Hung Lee1 and Daniel R Talham1National Central University, Taiwan2University of Florida, USA
Fruitful magnetic behaviors, such as light-induced magnetism, metastable ferrimagnetism, negative magnetization, spin crossover and spin delocalization have been identified in the hexacyanoferrate poly-nuclear complexes known as Prussian blue (PB) and
its analogues (PBA). The PBA’s are composed of alternately stacked MN6 and M'C6 octahedra along the three crystallographic axes, where M and M' can be divalent or trivalent transition metal ions. The flexibility to accommodate either divalent or trivalent ions at the M and M' sites has led to a large family of analogues and applications. For example, the structure builds up three-dimensional open channels to accommodate weakly bonded ions able to migrate through the channels and the framework has been exploited as electrode materials for secondary batteries, providing housing for ions to leave the framework during charging and reenter during discharging. In the present studies, different magnetic phases have been identified in nano-sized core/shell PBA cubes, with a 250 nm Rb-Co-Fe phase (Rb0.48Co[Fe(CN)6]0.75[(H2O)6]0.25•0.34H2O) in the core coated by a 45 nm K-Ni-Cr phase (K0.36Ni[Cr(CN)6]0.74[(H2O)6]0.26•0.11H2O) on the shell. Three separate characteristic temperatures at 86, 69, and 67 K are associated with magnetic phases in the K-Ni-Cr shell. Two magnetic exchange paths are identified. One propagates along the three crystallographic axis directions. The other propagates along the [110] crystallographic direction for the associated Ni-Ni interactions, but Cr-Cr interactions. The severe Cr-deficiency and the appearance of direct Ni-Ni exchange are used to understand the appearance of two separate transitions associated with magnetic ordering. A weak moment develops in the core at low temperature, corresponding to separate ordering of the Co-Fe PBA network.
BiographyWen-Hsien Li is a Full Professor in Physics department at National Central University, Taiwan. He has been the Director since the Center for Neutron Beam Applications of National Central University was found in 2006. He contributed to the birth of Taiwan Neutron Science Society (TWNSS) and Center for Neutron Beam Applications of National Central University. His current research is focused on quantum nanoparticle and multiferroic, using neutron scattering, Raman scattering, and other techniques to elucidate the interplay between the superconducting and magnetic degrees-of-freedom of these systems.
Wen-Hsien Li et al., J Phys Chem Biophys 2017, 7:4(Suppl)DOI: 10.4172/2161-0398-C1-025

Page 60
conferenceseries.com
Volume 7, Issue 4 (Suppl)J Phys Chem Biophys, an open access journal
ISSN: 2161-0398Physical Chemistry 2017
September 18-19, 2017
4th International Conference on
Physical and Theoretical ChemistrySeptember 18-19, 2017 Dublin, Ireland
Photo-induced electron transfer through silicon-bridge: The source for blue-green emission Malgorzata Bayda1, Gonzalo Angulo2, Gordon L Hug3, Monika Ludwiczak1, Jerzy Karolczak1, Jacek Koput1, Jacek Dobkowski2 and Bronislaw Marciniak1
1AMU/CAT, Poland 2PAS, Poland3NDRL, USA
There are still few examples of materials emitting blue and green light. Coming forward to meet these needs, we proposed silicon-bridged chromophores as sources for blue-green emission. Although the individual chromophores chosen [N-isopropylcarbazole
(CBL) and 1,4-divinylbenzene (DVB)] emit in the UV range, linking them through the silylene bridge switches on the colored emission which originates from an intramolecular charge transfer reaction (ICT). This phenomenon was observed not only for the polymer but also for its bichromophoric model compound representing the repeating unit of the polymer. This finding indicates that the ICT occurs between adjacent chromophores through the silylene bridge. If so, it was justified to use a model to describe in detail excited-state processes in this kind of substances to give rise to rational molecular design of new light-emitting materials. The questions raised in this work are: what are the nuclear motions essential to intramolecular charge transfer? Is the ICT process solvent-controlled or is the driving force some geometric change of a solute in the excited state? To answer these questions, we studied ICT on a model compound. We found that in nonpolar solvents, emission arises from the local excited state (LE) of carbazole whereas in more polar solvents dual emission was detected (LE+ICT). The CT character of the additional emission band was concluded from the linear dependence of the fluorescence maxima on solvent polarity. Electron transfer from CBL to DVB resulted in a large excited-state dipole moment (37.3 D) as determined from a solvatochromic plot and DFT calculations. Steady-state and picosecond time-resolved fluorescence experiments performed in butyronitrile (293-173 K) showed that the ICT excited state arises from the LE state of carbazole. These results were analyzed and found to consistent with an adiabatic version of Marcus theory including solvent relaxation.
BiographyMalgorzata Bayda received her PhD in Chemistry from Adam Mickiewicz University, Poznan, Poland. She has four external positions: short-term research positions at Radiation Laboratory, University of Notre Dame, USA working with Dr. Gordon L Hug (2008, 2015) and Postdoctoral Associate positions at Professor Jack Saltiel’s research laboratory at Florida State University, USA (2010-2011, 2012). Since 2009, she works as an Assistant Professor at AMU. She has her expertise in Photo-physics and Photochemistry of organosilicon compounds using steady-state and time-resolved absorption and emission spectroscopy. Her earlier work was focused on a cis/trans photo-isomerization of p-phenylene-silylene-vinylene polymers. Recently, her scientific interest turned toward searching for an attractive organosilicon light-emitting materials. To improve performance of such materials through rational molecular design, she investigates excited-state processes between silylene-bridged chromophores focusing on a role of silicon atom in these processes.
Malgorzata Bayda et al., J Phys Chem Biophys 2017, 7:4(Suppl)DOI: 10.4172/2161-0398-C1-025

Page 61
conferenceseries.com
Volume 7, Issue 4 (Suppl)J Phys Chem Biophys, an open access journal
ISSN: 2161-0398Physical Chemistry 2017
September 18-19, 2017
4th International Conference on
Physical and Theoretical ChemistrySeptember 18-19, 2017 Dublin, Ireland
Why is gas phase photolysis of 2-nitrophenol a significant source of OH in the polluted atmosphere? Lei Zhu1, 2
1Wadsworth Center, USA 2University at Albany, USA
The significantly elevated day time nitrous acid (HONO) concentrations compared to those predicted based on the photochemical stationary state between HONO sources and sinks leads to postulate that HONO is produced by photochemical sources. One
proposed HONO source is gas phase photolysis of 2-nitrophenol (o-C6H4(NO2)OH) over the 300-500 nm region. 2-Nitrophenol is also an important component of brown carbon in the atmosphere. The concentration of 2-nitrophenol is expected to be high in polluted areas where there are increased emissions of aromatic hydrocarbons. To assess the air quality impacts of pollutant emissions, it is important to determine oxidant formation potential of the emitted species. Although photo dissociation dynamics studies of 2-nitrophenol have reported OH formation at photolysis wavelengths of 266 nm, 355 nm, and over the 361-390 nm range, and HONO was observed as a product from 2-nitrophenol photolysis in a smog chamber, the lack of quantitative absorption cross section and product quantum yield information has prevented quantitative assessment of the extent of oxidant formation from 2-nitrophenol photolysis in the atmosphere. The purpose of this study is to determine quantitatively the gas phase absorption cross sections of 2-nitrophenol over the 295-400 nm range, to investigate the HONO and OH formation channels following the 308 and 351 nm photolysis of 2-nitrophenol, and to obtain the OH and the HONO quantum yields. We have estimated the atmospheric oxidant formation rate constants following the gas phase photolysis of 2-nitrophenol using 2-nitrophenol near UV absorption cross sections, and OH and HONO formation quantum yields obtained from this study. Gas phase photolysis rate constant of 2-nitrophenol is about twice that of NO2 and the sum of OH and HONO formation quantum yields are about unity at 308 nm and 351 nm. OH formation rate constant is fast from the gas phase photolysis of 2-nitrophenol. Recommendations are made to include gas phase 2-nitrophenol photolysis as a significant missing source of OH in the modeling of the chemistry of the polluted atmosphere.
Figure 1: Modeled atmospheric photo dissociation rate constants of 2-nitrophenol as a function of solar zenith angle
BiographyLei Zhu is a Research Scientist at Wadsworth Center, New York State Department of Health, and a Professor in the Department of Environmental Health Sciences at SUNY-Albany. His research program has been designed to investigate and understand what controls the atmosphere’s energy balance and how chemical reactions impact composition, pollutant and oxidant formation in the earth’s environment. Her research interests include “Kinetics and photochemistry of homogeneous and heterogeneous atmospheric reactions, atmospheric application of cavity ring-down spectroscopy and its variants, and atmospheric application of time-resolved FT-IR”.
Lei Zhu, J Phys Chem Biophys 2017, 7:4(Suppl)DOI: 10.4172/2161-0398-C1-025
Lei Zhu, J Phys Chem Biophys 2017, 7:4(Suppl)DOI: 10.4172/2161-0398-C1-025

Page 62
Notes:
conferenceseries.com
Volume 7, Issue 4 (Suppl)J Phys Chem Biophys, an open access journal
ISSN: 2161-0398Physical Chemistry 2017
September 18-19, 2017
4th International Conference on
Physical and Theoretical ChemistrySeptember 18-19, 2017 Dublin, Ireland
Intra- and intermolecular strategies to improve photoluminescence quantum yields of n-π* fluorophores capable of harvesting triplet excitons Youngmin You Ewha Womans University, South Korea
Spin statics of excitons is the key factor that determines the efficiency for interconversion between photon and electron. For example, internal quantum yields of electro-fluorescence devices are limited by 25% in the absence of processes that aid intersystem crossing.
Charge collection efficiencies in photovoltaic devices are also intimately associated with spin distributions. Efforts have, thus, been paid to develop the materials that overcome the spin selection rule. Notable examples include organometallic complexes of Ir(III) or Pt(II) which exhibit strong spin-orbit coupling. Of recent interest are dipolar organic molecules and coordination compounds of Cu(I). These compounds possess charge-separated excited states with small exchange energies. This electronic structure allows for thermally activated reverse intersystem crossing, leading to exciton-harvested fluorescence emission. Our group was intrigued by the key role of the excitonic spin states in electroluminescence devices. We investigated the fluorescence properties of chromophores bearing n-π* transitions. Although n-π* molecules can serve as electroluminescent materials because of the harvesting of singlet and triplet excitons through El-Sayed-rule-allowed reverse intersystem crossing, the weak fluorescence emissions of such molecules have prevented applications into devices. To enable systematic studies, we prepared a series of electron-deficient coumarin compounds having aryl substituents with different band gap energies. We observed two orders of magnitude improvement in fluorescence quantum yields upon facilitating intra- and intermolecular electron transfer to the coumarins. Special focus has been paid to understand the electron-transfer processes and the molecular factors that controlled the kinetic steps. The mechanistic studies revealed that judicious control over excited-state potentials was crucial to achieve efficient fluorescence.
BiographyYoungmin You is an Associate Professor of the Division of Chemical Engineering and Materials Science at Ewha Womans University. He completed his Bachelor and Master degrees in Chemical Engineering at Seoul National University in 1997 and 2003, respectively. He obtained his PhD degree in 2007. He started his independent carrier as an Assistant Professor at Kyung Hee University in 2013, and moved to Ewha Womans University in 2015. He focuses on the development of novel molecules and photo-electro-functions. His current research interests include luminescent molecules for exciton harvesting and circularly polarized emission, photo-redox catalysis, and photo-luminescent bio-probes. He published 54 papers, including six review articles.
Youngmin You, J Phys Chem Biophys 2017, 7:4(Suppl)DOI: 10.4172/2161-0398-C1-025

Page 63
conferenceseries.com
Volume 7, Issue 4 (Suppl)J Phys Chem Biophys, an open access journal
ISSN: 2161-0398Physical Chemistry 2017
September 18-19, 2017
4th International Conference on
Physical and Theoretical ChemistrySeptember 18-19, 2017 Dublin, Ireland
Topotactic synthesis of mixed-anion oxide epitaxial thin filmsAkira Chikamatsu and Tetsuya Hasegawa University of Tokyo, Japan
Transition metal oxides exhibit fascinating physical and chemical properties, including superconductivity, colossal magnetoresistance, ferroelectricity, and photocatalytic abilities. Since these properties are strongly affected by bonding interactions between the d
orbital of transition metal cations and the p orbital of oxide anions, moderate replacement of O2- by H- or F- can drastically change the characters. One of the most excellent methods to obtain oxyhydrides and oxyfluorides is topotactic synthesis using reagents, where guest species can be introduced into a host crystalline structure without destroying the initial crystalline matrix, for example, insulating BaTiO3 changes into oxide hydride BaTiO2.4H0.6 with metallic nature by CaH2 treatment and bulk crystal of SrFeO3-δ changes into SrFeO2F by annealing with poly-CH2CF2 (PVDF) at 400°C. Though this method has mainly been applied to powder bulk samples, the reaction on thin-film samples is expected to have several advantages over bulk: considerably higher reactivity owing to the larger surface area/volume ratio, stabilization of the crystal framework by epitaxial effect, and modification of physical properties by epitaxial strain. In this study, we examined four types of topotactic reactions for various transition-metal oxide epitaxial thin films, i.e., hydridation and strong reduction using CaH2, fluorination using PVDF, and strong oxidation using NaClO solution, as schematically illustrated in Figure 1. Furthermore, we found interesting electronic properties in the obtained mixed-anion oxide thin films, such as ferromagnetic metal to antiferromagnetic insulator transition. These reactions will be useful for designing and synthesizing novel mixed-anion compounds in epitaxial thin film form.
Figure 1:SchematicimageofthetopotacticreactionsofhydridationandstrongreductionusingCaH2,fluorinationusingPVDF,andstrongoxidationusingNaClOsolution.
BiographyAkira Chikamatsu holds a PhD degree from University of Tokyo in 2008, and currently works at University of Tokyo as Assistant Professor. He is an expert in “Solid-state chemistry and physics, thin-film growth, and electron spectroscopy”. He developed new thin-film growth techniques of mixed-anion transition metal oxides, and succeeded in creating some new materials by these methods. His current research interests include “Searching for new functionalities and new phenomena in mixed-anion transition metal oxides by using layer-by-layer thin film growth and topotactic reaction technologies”.
Akira Chikamatsu et al., J Phys Chem Biophys 2017, 7:4(Suppl)DOI: 10.4172/2161-0398-C1-025

Page 64
conferenceseries.com
Volume 7, Issue 4 (Suppl)J Phys Chem Biophys, an open access journal
ISSN: 2161-0398Physical Chemistry 2017
September 18-19, 2017
4th International Conference on
Physical and Theoretical ChemistrySeptember 18-19, 2017 Dublin, Ireland
On the structure and tribological effect of interfacial water between a graphite surface and metallic or semiconducting counter bodiesArnaud Caron Korea University of Technology and Education, South Korea
In this work, we apply atomic force microscopy/spectroscopy (AFM/S) and friction force microscopy (FFM) in immersed conditions to probe the structure of water at the interface of highly oriented pyrolytic graphite (HOPG) and AFM tips with different metallic
coatings. While AFS measurements allow the observation of the layering of water molecules as a function of the distance from the HOPG surface, FFM measurements and the occurrence of molecular scale stick-slip provide new insights in the two-dimensional distribution of interfacial water molecules. The layering of water is found to be significantly affected by the chemistry of the AFM tip approaching the HOPG surface. Beside the periodicity of the graphitic honeycomb structure, statistical analysis of the stick slip friction behavior reveals characteristic structural lengths that also depend on the chemistry of the AFM-tip sliding on HOPG. We discuss these observations based on the conformation of different ice structures at the interfaces between an HOPG surface and different counter bodies.
BiographyArnaud Caron is a Materials Scientist with expertise in “The multi-scale mechanical behavior of materials, surfaces and micro-components”. Since 2015, he is an Assistant Professor at Korea University of Technology and Education, Republic of Korea. He obtained his engineering degree in Materials Science in 2004 at University of Saarland, Germany and was awarded with the Schiebold Medal. In 2009, he completed his Doctoral degree in Materials Science at University of Saarland, Germany. From 2007 to 2015, he worked as a Research Associate at Institute of Micro- and Nanomaterials of the University of Ulm, Germany.
Arnaud Caron, J Phys Chem Biophys 2017, 7:4(Suppl)DOI: 10.4172/2161-0398-C1-025

Page 65
Notes:
conferenceseries.com
Volume 7, Issue 4 (Suppl)J Phys Chem Biophys, an open access journal
ISSN: 2161-0398Physical Chemistry 2017
September 18-19, 2017
4th International Conference on
Physical and Theoretical ChemistrySeptember 18-19, 2017 Dublin, Ireland
Anion and cation diffusion in complex oxidesManfred Martin RWTH Aachen University, Germany
Oxygen diffusion in complex oxide materials is of great importance for applications, e.g. in fuel cells (oxygen ion conductivity) or oxygen permeation membranes (ambipolar diffusion of oxygen). For heavily doped oxides, such as doped zirconia, ceria or
lanthanum gallate, we give a qualitative and quantitative explanation of the observed maximum of the conductivity as a function of the dopant fraction by combining DFT calculations of energies and entropies with kinetic Monte simulations of the oxygen ion conductivity. Concerning cation diffusion in complex oxides, we report our recent findings in perovskites, with a special focus on doped lanthanum gallate, barium titanate, and BSCF. Our experimental results indicate that the cation diffusion mechanisms are more complicated than simple vacancy mechanisms. We show that the experimental observations can be explained well by A- and B-site cation vacancies that are strongly bound in defect clusters and perform a highly correlated motion.
BiographyManfred Martin is a Professor and Head of the Institute of Physical Chemistry of RWTH Aachen University, Germany. He has more than 30 years of experience in education and research of physical chemistry of solids. His current research focusses on materials for energy conversion, resistive switching, solid-state reactions, secondary ion mass spectrometry, and computer simulations as well. He has published >200 scientific papers in international, refereed journals. He received Carl-Wagner Award and has been elected as member of the Royal Society of Chemistry. He has supervised more than 50 PhD students and more than 20 postdoctoral fellows.
Manfred Martin, J Phys Chem Biophys 2017, 7:4(Suppl)DOI: 10.4172/2161-0398-C1-025

Page 66
conferenceseries.com
Volume 7, Issue 4 (Suppl)J Phys Chem Biophys, an open access journal
ISSN: 2161-0398Physical Chemistry 2017
September 18-19, 2017
4th International Conference on
Physical and Theoretical ChemistrySeptember 18-19, 2017 Dublin, Ireland
New aspects of an old class of compounds: Tetrelphosphides and their thermoelectric performanceUlrich Wedig and Jürgen Nuss Max Planck Institute for Solid State Research, Germany
The tetrelphosphides Ag6Ge10P12 is the prototype of a class of compounds which is known since the mid of the 1970s. In the subsequent years, various experimental results were published. However, due to a lack of accurate quantum chemical investigations,
the interpretation of the data was not unambiguous at this time. After three decades of silence, these compounds attracted attention again, due to the promising thermoelectric performance. The figure of merit (zT) of pristine Ag6Ge10P12 is already 0.6 at 700 K, leaving room for improvements in this class of compounds. Reason for this relatively large zT value is the small thermal conductivity, k<1 W·m-1·K-1, which is related to the exceptional bonding characteristics. According to recent density functional calculation and a thorough bonding analysis, the crystal structure consists of a zinc blende like arrangement of germanium and phosphorus atoms with large voids. This covalent framework encloses subvalent silver octahedra. Four of the faces of the Ag6
4+ clusters are capped by another germanium atom, respectively. The atoms within the voids are weakly bound. Concerning the bonding types, there exists a hierarchy with a wide range of bond strength, giving rise to local, low-frequency phonon modes which lead to the reduced lattice contribution to the thermal conductivity. The electronic as well as the dynamic properties of the compound can be modified by substituting elements at the various tetrel sites in the crystal. The covalent framework becomes more rigid when replacing germanium by silicon. Tin as capping atoms of the silver octahedral resulted in a blue shift of the low-lying frequencies and a smaller band gap. By these controlled modifications, new insight can be gained into the complex interplay of electrical and thermal transport properties in thermoelectric materials.
BiographyUlrich Wedig research interests include Solid State Chemistry and Physics. Having a sound background in quantum chemistry, he collaborates with experimenters in order to elaborate a deeper understanding of the behaviour and properties of molecular and solid state systems. Special emphasis is put on the relation between quantum chemical data and chemical concepts, bridging the gap between more or less rigorous ab initio calculations and a local description of bonds in chemistry.
Ulrich Wedig et al., J Phys Chem Biophys 2017, 7:4(Suppl)DOI: 10.4172/2161-0398-C1-025

Page 67
conferenceseries.com
Volume 7, Issue 4 (Suppl)J Phys Chem Biophys, an open access journal
ISSN: 2161-0398Physical Chemistry 2017
September 18-19, 2017
4th International Conference on
Physical and Theoretical ChemistrySeptember 18-19, 2017 Dublin, Ireland
Gold nanoparticles characterization by scattering correlation spectroscopyNadia Djaker, Jolanda Spadavecchia, Hanane Moustaoui and Marc Lamy de la Chapelle Université Paris 13, France
Gold nanoparticles (GNP) are widely used in many fields, such as analytical chemistry, catalysis and biomedical applications. The geometrical and optical characterization of these GNP is an inevitable step before any practical application. For example,
plasmonic properties such as absorption and scattering and electromagnetic field enhancement have been explored for different type of GNP, with several techniques, like UV-Vis spectroscopy, surface enhanced Raman scattering (SERS) or correlation spectroscopy. Geometrical properties such as size and shape were mostly explored by electronic microscopy and have a strong influence on their optical properties. In addition, other properties like the surface area and volume are very important before GNP functionalization, especially for branched nanoparticles such as nano-stars, nano-flowers or nano-urchins. Recently, scattering correlation spectroscopy (SCS) is one of the most used techniques for GNP characterization. As fluorescence correlation spectroscopy, the SCS technique is based on the analysis of intensity fluctuations within a well-defined confocal volume (~ 1 fL). The correlation curve is directly related to the hydrodynamic radius of molecules or nanoparticles, to their diffusion coefficient, concentration and shape. The SCS is very sensitive to GNP morphology and brightness since the scattering intensity depends on the GNP volume. SCS technique will be presented to characterize the hydrodynamic sizes of different shapes of GNP (spheres, urchins and flowers), with different surface chemistries (PEG, thiophenol) and different sizes (20-80 nm) at very low concentrations (~pM) and with very high precision (~0.2 nm). We explored the scattering properties of these GNP at different wavelengths, close and far from their plasmon resonances. As predicted by Mie theory, we demonstrated that the increase in GNP size leads to the increase of the scattered intensity with the excitation power. In the case of nano-flowers, we observed a large increase of the scattered signal due to their specific surface morphology. Such results make this type of nanoparticles a better candidate for both cell imaging and photothermal therapy.
Figure 1: (Left)TEMimageof50nmsizedGNPs:(a)nano-spheres,(d)nano-flowers.(Right)Normalizedcross-correlationcurvesofthescatteredlightbyGNSs(b,c)andbyGNFs(e,f).
BiographyNadia Djaker is an Assistant-Professor in a Medical Faculty at Paris 13 University. She teaches optical techniques for biological media characterization to Master and PhD students. Her research expertise involves optical linear (fluorescence, Raman) and nonlinear (SHG, CARS) spectroscopy techniques. Recently, she has developed a project on nanoparticles toxicity, especially in nanoparticles and biological media interactions study by correlation spectroscopy with a collaboration of several national and international research teams.
Nadia Djaker et al., J Phys Chem Biophys 2017, 7:4(Suppl)DOI: 10.4172/2161-0398-C1-025

Page 68
conferenceseries.com
Volume 7, Issue 4 (Suppl)J Phys Chem Biophys, an open access journal
ISSN: 2161-0398Physical Chemistry 2017
September 18-19, 2017
4th International Conference on
Physical and Theoretical ChemistrySeptember 18-19, 2017 Dublin, Ireland
The golden doxorubicin: A tunable design of gold (III)-doxorubicin complex – PEGylated nanocarrier for oncologicalJolanda Spadavecchia, Hanane Moustaoui, Nadia Djaker and Marc Lamy de la Chapelle Université Paris 13, France
In the last five decades, metal complexes and organometallic compounds have been gaining importance in cancer therapy. Based on their structural and electronic similarity to cisplatin and cisplatin-related anti-tumor drugs, Au (III) species represent a promising
class of potential anticancer agents. In this study, we report the synthesis, physico-chemical characterization and results of the biological behavior of doxorubicin-complex –gold COOH-terminated PEG-coated NPs (DOXO IN-PEG-AuNPs) before and after conjugation with antibody (anti-Kv11.1-pAb) to evaluate the influence of the nanocarrier and of the active targeting functionality on the anti-tumor efficacy of doxorubicin, with respect to its half-maximal effective concentration (EC50) and to drug-triggered changes in the cell cycle. The anti-Kv11.1-pAb recognized specifically the Kv11.1 subunit of the hERG1 channel aberrantly expressed on the membrane of pancreatic cancer cells. The synthetic approach consist in four steps (Figure 1): Complexation between doxorubicin (DOXO) and tetrachoric acid (HAuCl4) to form gold clusters; adsorption of COOH-terminated PEG molecules (PEG) onto DOXO-Au complex; reduction of metal ions in that vicinity, growth of gold particles and colloidal stabilization and bio-conjugation of anti-Kv11.1-pAb. Raman spectroscopies were performed for the vibrational characterization of each step of the synthesis of doxorubicin-nanocarrier, distinguishing them from the free drug, protonated or not on the phenolic part of its chromophores. The calculated characterization DOXO IN-PEG-AuNPs vibrational bands show qualitative agreement with the experimental observations. Although preliminary, data gathered from this study have a considerable potential in the application of gold complexes with high stability, for the treatment of PDAC, a disease with a dismal prognosis and one of the main current burdens of today healthcare bill of industrialized countries. Further studies are still envisaged, focused on assessing the in vivo assessment toxicity, pharmacokinetics and dynamics on relevant.
Figure 1: SchematicrepresentationofthesynthesisofDOXIN-PEG-AuNPs
BiographyJolanda Spadavecchia is a Senior Researcher. Her research activities are focused on the realization of nanoparticles and biosensors. In particular, she is interested in the processes responsible for the bio-conjugation of protein, macromolecules or DNA oligonucleotides onto gold nanoparticles and substrates for the creation of optical biosensors. She is currently involved in the synthesis of polymeric nanoparticles and the development of nano-hybrid materials for nanomedicine. Actually, she has an active collaboration with Berlin and Louvre Museum in order to establish the mechanism responsible of the AuNP formation at the surface of ancient ivory objects from different archaeological and historical contexts.
jolanda.spadavecchia.univ-paris13.fr
Jolanda Spadavecchia et al., J Phys Chem Biophys 2017, 7:4(Suppl)DOI: 10.4172/2161-0398-C1-025

Page 69
Physical Chemistry 2017
Day 2 September 19, 2017
Sessions:
Photochemistry | Solid-state Chemistry | Spectroscopy | Surface Science | Quantum Chemistry | Biophysical ChemistrySession ChairTitus A BeuBabeş-Bolyai University, Romania
Session Co-ChairShigeharu KittakaOkayama University of Science, Japan
Session IntroductionTitle: CHARMM force field and molecular dynamics simulations of polyethylenimine chains
Titus A Beu, Babeş-Bolyai University, RomaniaTitle: Neutron scattering study of super cooled water confined in mesoporous silicas, MCM-
41 and SBA-16: Role of component pores and their sizeShigeharu Kittaka, Okayama University of Science, Japan
Title: 57-Fe mössbauer spectroscopy on Fe-Mg-O nanocomposite particles grown by a novel chemical vapor synthesis methodWerner Lottermoser, Salzburg University, Austria
Title: In situ hard X-ray photoelectron study of O2 and H2O adsorption on Pt nanoparticlesMasaharu Oshima, University of Tokyo, Japan
Title: Pyridinium salts as photoinduced electron trapsR Marshall Wilson, Bowling Green State University, USA
Title: Unpicking vibrational and vibrational torsional couplings in substituted benzenesTimothy G Wright, University of Nottingham, UK
Title: Reactive surface sites at metal oxide nanoparticles: From fundamental studies to potential medical applicationSlavica Stankic, Paris Institute of Nanosciences-CNRS, France
Title: An embedding technique based on a strategic use of atomic pseudo potentialsYannick Carissan, Aix-Marseille University, France
Title: Theoretical study of magnetic properties in redox-active ruthenium complexesCorentin BOILLEAU, Polish Academy of Sciences, Poland
Title: Water interaction and dissociation on the (0001) hematite surface: A DFT+U approachFabio R Negreiros, Federal University of ABC, Brazil
Title: Polymer-brush lubricationTorsten Kreer, Leibniz Institute for Polymer Research Dresden e. V. (IPF), Germany
Title: Excited-state symmetry breaking of linear quadrupolar chromophores: A transient absorption studyNadia Dozova, Pierre and Marie Curie University, France
Title: Phase equilibrium of the melt-vapor in the tellurium-sulfur systemValery N Volodin, Institute of Metallurgy and Ore Benefication, Kazakstan
Session Introduction

Page 70
conferenceseries.com
Volume 7, Issue 4 (Suppl)J Phys Chem Biophys, an open access journal
ISSN: 2161-0398Physical Chemistry 2017
September 18-19, 2017
4th International Conference on
Physical and Theoretical ChemistrySeptember 18-19, 2017 Dublin, Ireland
CHARMM force field and molecular dynamics simulations of polyethylenimine chainsTitus Adrian Beu and Alexandra FarcaşBabeş-Bolyai University, Romania
Over more than a decade, cationic polymers have been validated as excellent gene delivery vectors, not in the least, due to their accessible chemistry, cost effectiveness, and controllable toxicity. Polyethylenimine (PEI), in particular, is one of the most
commonly employed synthetic poly-cations. The predominant electrostatic interactions between the positive amino groups of these polymers and the negative phosphate groups of DNA lead to condensed polyplexes, which protect DNA from degradation and are able to enter cells via endocytosis. The specific charge pattern of protonated PEI is widely considered to be responsible for the release of the polyplexes from the endosome (via proton sponge effect), and, finally, for the release of DNA from polyplexes (prior to being processed by the nucleus). Our investigations aim to provide a new, realistic molecular mechanics force field for PEI, to be used in detailed atomistic simulations of DNA-PEI condensation. Accordingly, we tackle two major issues: (1) we develop a new atomistic CHARMM force field for PEI of arbitrary length and protonation patterns rigorously derived from high-quality ab initio calculations on model polymers, and (2) we perform molecular dynamics simulations, investigating the dynamic structuring of solvated PEI chains in dependence of their size and protonation state. We characterize the dynamic structure in terms of gyration radius, end-to-end distance, persistence length, radial distribution functions, coordination numbers, and diffusion coefficients. Altogether, the developed force field leads to more rigid PEI chains than other computational studies. Notably, the calculated diffusion coefficients are in excellent agreement with experimental data and validate the force field for the realistic modeling of the size and protonation behavior of linear PEI chains, whether individually or as part of polyplexes.
Figure 1:ProtonatedPEItetramerusedasmodelfortheforcefieldparametrization.
BiographyTitus Adrian Beu is Professor of Theoretical and Computational Physics at Babeş-Bolyai University, Romania. He has been actively involved in computational physics, material science, and chemical physics for more than 30 years. His research topics have evolved from tokamak plasma and nuclear reactor calculations in the 1980s, collision theory and molecular cluster spectroscopy in the 1990s, to simulations of fullerenes, nano-fluidic systems and biopolymers in recent years. He taught courses in general programming techniques and advanced numerical methods, general simulation methods and advanced molecular dynamics.
Titus Adrian Beu et al., J Phys Chem Biophys 2017, 7:4(Suppl)DOI: 10.4172/2161-0398-C1-025

Page 71
conferenceseries.com
Volume 7, Issue 4 (Suppl)J Phys Chem Biophys, an open access journal
ISSN: 2161-0398Physical Chemistry 2017
September 18-19, 2017
4th International Conference on
Physical and Theoretical ChemistrySeptember 18-19, 2017 Dublin, Ireland
Neutron scattering study of super cooled water confined in mesoporous silicas, MCM-41 and SBA-16: Role of component pores and their sizeShigeharu Kittaka1, K Yoshida2, T Yamaguchi2, M Mizuno3, M C Bellissent Funel4 and P Fouquet5 1Okayama University of Science, Japan 2Fukuoka University, Japan3Kanazawa University, Japan4University of Paris-Saclay, France5Institute Laue Langevin, France
The nature of super-cooled water in confinement is an important subject that involves many aspects of natural sciences. Strong hydrogen bonds lead to the formation of structured collective entities in liquid water, finally resulting in crystallization of ice.
Since the development of synthetic methods of well-defined porous silicas of various sizes, cylindrical, spherical, etc., experimental and theoretical analysis of pore water has significantly developed. The present interest is to find how the collective entities of water grow in fine pores of different shapes and what dynamic motions are there. Neutron spin echo (NSE) measurements were conducted on heavy water confined in cylindrically porous MCM-41 and spherically porous SBA-16 in the temperature range 210–290 K. Deuterium has a nuclear spin of 1 and thus has a highly coherent scattering cross-section that is convenient for the study of the dynamics of collective entities of heavy water. In the spherical pores of SBA-16, the translational motion of heavy water was strongly inhibited, even at 290 K. Rotational motion, however, was observed clearly in the temperature range 230–290 K and was analysed by the Vogel–Fulcher–Tammann relation. The relaxation time of the rotational motion of heavy water increased with a decrease in temperature. For heavy water in the cylindrically porous MCM-41, the relaxation time increased with reducing the temperature, as in SBA-16, but much more sharply. The larger value for the former is ascribed to the linear growth of hydrogen bonds in the cylindrical pores. In contrast, in the spherical space of SBA-16, spherical growth of heavy water clusters could permit a faster dynamic rotational motion. NSE measurements of light water in SBA-16 showed the translational diffusion of discrete water molecules there, indicative of the occurrence of breaking and recombination of hydrogen bonds in the collective entities.
BiographyShigeharu Kittaka has his expertise in surface chemistry of metal oxide-water systems: electrification of metal oxides in water, fine spherical particle formation and surface structure, layer structure of V2O5·nH2O by intercalation of water and organic molecules and electrical properties, phase changes of molecular liquids: water, ammonia, alcohol etc.
Shigeharu Kittaka et al., J Phys Chem Biophys 2017, 7:4(Suppl)DOI: 10.4172/2161-0398-C1-025

Page 72
Notes:
conferenceseries.com
Volume 7, Issue 4 (Suppl)J Phys Chem Biophys, an open access journal
ISSN: 2161-0398Physical Chemistry 2017
September 18-19, 2017
4th International Conference on
Physical and Theoretical ChemistrySeptember 18-19, 2017 Dublin, Ireland
57-Fe Mössbauer spectroscopy on Fe-Mg-O nanocomposite particles grown by a novel chemical vapor synthesis methodWerner Lottermoser1, Matthias Niedermaier1, Amir R Gheisi2, Gerold Tippelt1, Johannes Bernardi3 and Oliver Diwald1 1Salzburg University, Austria 2University of Erlangen-Nürnberg, Germany3Vienna University of Technology, Austria
Statement of the Problem: The admixture of 3d transition metals to particles and ceramic structures of non-reducible metal oxides has given rise to a variety of functionalities used in industrial applications. However, it is not easy to control the impurity localization and the nanomaterials functional properties.
Methodology & Theoretical Orientation: Powders of Fe-Mg-O nanocomposite particles have been grown using a novel chemical vapor synthesis approach which involves metalorganic precursor decomposition inside the combustion flame. After annealing in controlled gas atmosphere composition distribution functions, structure and phase stability of the obtained magnesiowüstite nanoparticles were measured with a combination of methods.
Findings: 57-Mössbauer spectroscopy measurements revealed that - depending on Fe loading and annealing temperature - either metastable and superparamagnetic solid solutions of Fe III ions in periclase MgO or phase separated mixtures of MgO and antiferromagnetic magnesioferrite MgFe2O4 nanoparticles can be obtained.
Conclusion & Significance: The combination of the present hybrid combustion technique with annealing protocols emphasizes the great potential of vapor phase grown non-equilibrium solids. Applying this method, phase separation, disproportionation and the appearance of magnetic properties can be tuned intentionally. Different from their bulk counterpart, MgFe2O4 nanoparticles with identical composition and structure are superparamagnetic and are promising material components for magnetic resonance imaging (MRI) as high density information storage materials or for magneto-caloric refrigeration.
BiographyWerner Lottermoser is a Solid State Physicist. He has completed his thesis work on neutron diffraction and magnetism of special silicates at CNRS, CENG and ILL Grenoble, France, and University of Frankfurt, Germany. He obtained qualification to become Professor after studying Single Crystal Mössbauer Spectroscopy at Salzburg University, Austria, and was working in different scientific projects granted by the Austrian Fund of Scientific Research (FWF).
Werner Lottermoser et al., J Phys Chem Biophys 2017, 7:4(Suppl)DOI: 10.4172/2161-0398-C1-025

Page 73
conferenceseries.com
Volume 7, Issue 4 (Suppl)J Phys Chem Biophys, an open access journal
ISSN: 2161-0398Physical Chemistry 2017
September 18-19, 2017
4th International Conference on
Physical and Theoretical ChemistrySeptember 18-19, 2017 Dublin, Ireland
In situ hard X-ray photoelectron study of O2 and H2O adsorption on Pt nanoparticlesMasaharu Oshima1, Yitao Cui1, Yoshihisa Harada1, Tatsuya Hatanaka2, Naoki Nakamura3 and Masaki Ando3 1Institute for Solid State Physics-University of Tokyo, Japan 2Toyota Central R&D Labs Inc., Japan3Toyota Motor Corp., Japan
In order to clarify the effect of water adsorption on fuel cell cathode catalysis surface, we have investigated electronic structure of Pt and Pt-Co nano-particles with O2 adsorption and O2/H2O co-adsorption by in situ hard X-ray photoelectron spectroscopy
(HAXPES) together with in situ high resolution fluorescence detected X-ray absorption (HERFD-XAS). The valence band (mainly Pt 5d) and Pt 4f spectra were successfully obtained under up to 1 mbar with an ambient cell for the first time by in situ HAXPES. Both valence band and Pt 4f spectra show that O2/H2O co-adsorption hindered oxygen adsorption. Based on our first principles calculation of valence band density-of-states (DOS) we have found that H2O molecules may occupy the oxygen adsorption sites on Pt surface more easily than oxygen, resulting in hindering the successive oxygen adsorption. However, under the more realistic condition at atmospheric pressure the formation of higher oxidation states of Pt in Pt L3-edge absorption spectra was enhanced by water adsorption, which was obtained by high resolution (Pt M5 FWHM about 2.5 eV) in situ HERFD-XAS. These changes in white line cannot be observed by conventional XAFS spectra due to large life-time broadening of Pt L3 (FWHM about 5.2 eV). At 1 bar more frequent attack by oxygen molecules onto water-adsorbed Pt surface may occur, resulting in the formation of hydrated hydroxyl intermediates and higher oxidation states. This enhanced oxygen adsorption is more clearly observed for Pt than Pt3Co nano-particles, probably because Pt nano-particles with stronger Pt-O bonding than Pt-Co nano-particles may further stabilize Pt-O bonding by additionally adsorbed water leading to less water effect on oxygen adsorption on Pt-Co. These results would be helpful to understand the reason why Pt-Co nano particles show higher ORR activity than Pt nano particles.
Figure 1: (a) Valence band spectra under in situ/ex situ reductions, (b)CalculatedprojectedPtd-pDOSsofbarePt(111)surface with various adsorbates, (c) experimental difference spectra of ex situ reduced, H2O, andO2 adsorbed condition (solid lines) obtained by subtracting the in situ reduced spectrum, together with calculated difference spectra of H2OandO2 adsorption (dashed lines).
BiographyMasaharu Oshima is a Project Researcher of Institute for Solid State Physics (ISSP), University of Tokyo. He completed his Bachelor degree in Dpt. of Industrial Chemistry, University of Tokyo in 1972, and Doctor of Engineering degree at University of Tokyo in 1984. After he started his experience at Stanford University in 1981-82, he is continuing synchrotron radiation science for semiconductors, magnetic materials and catalysts for more than 35 years. He became a Professor in Dpt. of Applied Chemistry, University of Tokyo in 1995. He was the President of the Japanese Society for Synchrotron Radiation Research (JSSRR) in 2009-2011, and the President of the Surface Science Society of Japan (SSSJ) in 2013-2015. He had received many research awards including ECS Best Paper Award in 2010 and Ministry of Education, Science and Technology (MEXT) Award in 2014.
Masaharu Oshima et al., J Phys Chem Biophys 2017, 7:4(Suppl)DOI: 10.4172/2161-0398-C1-025

Page 74
Notes:
conferenceseries.com
Volume 7, Issue 4 (Suppl)J Phys Chem Biophys, an open access journal
ISSN: 2161-0398Physical Chemistry 2017
September 18-19, 2017
4th International Conference on
Physical and Theoretical ChemistrySeptember 18-19, 2017 Dublin, Ireland
Pyridinium salts as photo-induced electron trapsR Marshall Wilson Bowling Green State University, USA
When two or more pyridinium salts are held face-to-face to each other they will share electrons equally between the rings. Calculations show this distribution of the trapped electron for the dimethyl 1,2-(di-4-pyridinium) ethane and the tetramethyl
1,1,2,2-(tetra-4- pyridinium) ethane as shown below. The electron trapping properties as characterized in ultrafast transient absorption spectroscopy and theoretical calculations for a variety of polypyridinium salts will be discussed.
BiographyR Marshall Wilson is a Research Professor in Chemistry department at Bowling Green State University, USA. He was awarded PhD in the year 1965 from Massachusetts Institute of Technology. His research interests are directed towards photochemical application of lasers, primarily argon ion lasers, and fall into two broad categories: the laser synthesis of new materials and the development of reagents for the photochemical manipulation of biological systems.
R Marshall Wilson, J Phys Chem Biophys 2017, 7:4(Suppl)DOI: 10.4172/2161-0398-C1-025

Page 75
Notes:
conferenceseries.com
Volume 7, Issue 4 (Suppl)J Phys Chem Biophys, an open access journal
ISSN: 2161-0398Physical Chemistry 2017
September 18-19, 2017
4th International Conference on
Physical and Theoretical ChemistrySeptember 18-19, 2017 Dublin, Ireland
Unpicking vibrational and vibrational torsional couplings in substituted benzenesTimothy G Wright, William D Tuttle, Adrian M Gardner and Laura E Whalley University of Nottingham, UK
We initially present vibrationally-resolved electronic spectra obtained using resonance-enhanced multi-photon ionization (REMPI) spectroscopy. The spectra are obtained from jet-cooled seeded expansion using lasers. The spectra exhibit many bands,
identifying the energetic positions of vibrational levels in the S1 electronic state; a number of these are found to arise from overlapped and/or interacting vibrational levels. By fixing one laser at the energy of one of those levels, we then ionize the electronically-excited molecule and record zero-kinetic-energy (ZEKE) spectra, whose assignment allows the deduction of the make-up of the intermediate S1 vibrational levels. In many cases, we can identify the so-called zero-order states (ZOSs) which have coupled to give the resultant eigenstate; this coupling occurs as a result of Fermi resonance. As well as pure vibrations, we find that these ZOSs may be torsional levels or vibration-torsion (vibtor) levels. The coupling of the ZOSs leads to levels whose motions are more delocalized across the molecule. This has implications for photo-stability and chemical control. Assignment of the spectra is aided by recording ZEKE spectra at different energies through a REMPI feature that corresponds to couple ZOSs. In this way, we can see activity move in and out of resonance through the feature. By plotting these spectra together, we obtain a two-dimensional ZEKE spectrum. Quantum chemical calculations are used to aid in the assignments. The treatment of the torsional levels requires the use of molecular symmetry groups: G12 for toluene and para-fluorotoluene; G72 for para-xylene.
BiographyTim G Wright has been working in the field of Spectroscopy covering electronic and photoelectron spectroscopy since 1988. He has used both conventional and laser-based methods. His work has always been underpinned by appropriate quantum chemical calculations and these often provide the foothold that allows the assignment of the spectra.
Timothy G Wright et al., J Phys Chem Biophys 2017, 7:4(Suppl)DOI: 10.4172/2161-0398-C1-025

Page 76
conferenceseries.com
Volume 7, Issue 4 (Suppl)J Phys Chem Biophys, an open access journal
ISSN: 2161-0398Physical Chemistry 2017
September 18-19, 2017
4th International Conference on
Physical and Theoretical ChemistrySeptember 18-19, 2017 Dublin, Ireland
Reactive surface sites at metal oxide nanoparticles: From fundamental studies to potential medical applicationSlavica StankicINSP-CNRS, France
The ubiquity of oxides in dispersed form has prompted research strategies in two directions: understanding the existing materials by means of appropriate reference systems and tailoring the desired properties through innovative syntheses. In this talk, author
will show examples of two prototype oxides, ZnO and MgO, to which extent they can be used as model systems for probing surface reactivity. When studied in parallel with DFT calculations, surface hydroxylation, provided either by adsorbing H2O or H2, turned to be a win-win combination for precise surface site identification. In that manner, we have demonstrated that ZnO nano-powders behave as multi-facet single crystals involving (10-10), (11-20), (0001) and (000-1) surfaces with the polar orientations corresponding to 25% of the total surface area. Moreover, we were able to report on water structures on ZnO(11-20) for the first time. Similarly, combining DFT and H2-infrared spectroscopy on MgO nanocubes, we proposed a model in which multisite dissociation of hydrogen is suggested to occur on mono- and di-atomic steps at (001) MgO surface. Nanoparticles of a well-defined size, shape, and surface termination are required for studying the reactions occurring over their surface. A strong emphasis in our work is, therefore, given to govern the synthesis pathways when producing desired nanoparticles, either in pure or doped form. Accordingly, an example of ruling the particles surface termination by controlling synthesis parameters will be presented in this talk. Finally, author will also show how the interactions between water and nanoparticles surface can be used for studying particles dissolution as a function of their size. This is especially important in case of mixed form of ZnO and MgO (ZnMgO) which exhibits a promising potential for medical applications as an alternative to existing antibiotics.
BiographySlavica Stankic has her expertise in synthesis and surface characterization of pure and multi metal oxide nanoparticles. After few years of experience of nanomaterials research from well-known international institutions (TU Vienna, Austria; INSP-CNRS, Paris, France), she has established new pathways for determining reactivity of surface sites or improving surface doping. Beside fundamental studies that involved photo-induced processes on oxide surfaces with a strong focus on the effects of particles size, shape and/or surface termination, she furthermore developed an interdisciplinary-based research project. Herein, metal oxide nanoparticles are used as model systems for studying their interaction with living organisms with a goal to assess their potential for medical applications as an alternative to existing antibiotics.
Slavica Stankic, J Phys Chem Biophys 2017, 7:4(Suppl)DOI: 10.4172/2161-0398-C1-025

Page 77
conferenceseries.com
Volume 7, Issue 4 (Suppl)J Phys Chem Biophys, an open access journal
ISSN: 2161-0398Physical Chemistry 2017
September 18-19, 2017
4th International Conference on
Physical and Theoretical ChemistrySeptember 18-19, 2017 Dublin, Ireland
An embedding technique based on a strategic use of atomic pseudo potentialsYannick Carissan, Alexander Punter and Paola Nava Aix-Marseille University, France
Atomic pseudo potentials were primarily used to replace core electrons in quantum chemistry calculations. Since 2013, we decided to use pseudo potentials to model core and valence electrons for hybridized atoms. In this work, we focus on the sp2 carbon
atom. We decided to begin with a pseudo-carbon, and used the CH3 radical as a reference to which we tried to optimize the model. Starting with a pseudo-carbon with a charge of one, and one electron, we were able to use potentials to force the occupation of specific orbitals, and to manipulate the energy levels of these orbitals. In practice, the only way to make the s potentials affect the orbitals was not to place them on the molecular plane itself, thus we ended up with a scheme that had potentials above and below the plane. After confirming the model worked on the ethene molecule, we were able to reproduce good accuracy characteristics such as ionisation and excitation energies across a range of molecules including chain alkenes and aromatic, cyclic compounds. Unlike in previous attempts, we are now able to extract atom based pseudo potentials: no bond centered potential is now required making the scope of use of these potentials extremely large. In order to be useful, these potentials must be able to replicate their results across other systems. Testing them with some of the systems used in Carissan & Drujon, we have met with success. A single set of optimized potentials give results within ~0.5 eV of reference calculations over HF, DFT and TD-DFT calculations.
BiographyYannick Carissan is an Assistant Professor at Aix-Marseille University. He is an expert in Theoretical Chemistry and focuses on the interaction between research and teaching. His model based on chemically relevant concepts is an attempt to fill the gap between empirical methods and ab initio full electron quantum chemistry calculations.
Yannick Carissan et al., J Phys Chem Biophys 2017, 7:4(Suppl)DOI: 10.4172/2161-0398-C1-025

Page 78
Notes:
conferenceseries.com
Volume 7, Issue 4 (Suppl)J Phys Chem Biophys, an open access journal
ISSN: 2161-0398Physical Chemistry 2017
September 18-19, 2017
4th International Conference on
Physical and Theoretical ChemistrySeptember 18-19, 2017 Dublin, Ireland
Theoretical study of magnetic properties in redox-active ruthenium complexesCorentin Boilleau1 and Karine Costuas2 1Institute of Physics, Polish Academy of Sciences, Poland 2Institut des Sciences Chimiques de Rennes, France
Due to the slowdown of the information technology development, a great challenge of present-day applied science is to develop new electronic devices at the molecular scale. Indeed, molecular spintronic offers great potential multifunctional molecules
performing new properties or operations unreachable by conventional semi-conductor technology. This project takes place in this quest of tomorrow's technologies conquest in the new field of molecular spintronic. The goal is to provide multifunctional compounds made from bricks with remarkable properties for storage or manipulation of information across a single molecule. This work use an uncommon strategy based on redox properties of ruthenium compounds associated with magnetic centers in order to obtain a device allowing a modulation of the magnetic properties. The aim is to study the inter-molecular interactions, to understand the interplay of the components in view of obtaining their synergistic working mode. Considering the crucial role of the electronic correlation in magnetic systems and the strong geometrical and electronic coupling existing between the different functional elements, modeling of such systems is a tough task. A correct description of these systems requires taking into account the couplings between the subunits. The nature of the interactions studied the presence of transition metals and the need for investigation of both ground and excited states suggested the use of post Hartree Fock methods. But, considering their computational cost and the size of our systems, they are here prohibited. Therefore, the use of DFT with hybrid functional is suitable. Standard and broken symmetry calculations have been performed to determine the magnetic coupling. The supramolecular assembly proposed present efficient switching properties allowing the realization of logical functions. Depending on their composition, shape, physical and chemical properties, they can be used as data processing devices (molecular wires, transistors, circuits) as information storage devices (molecular switchers) or as molecular machines.
BiographyCorentin Boilleau has his expertise in electronic structures and magnetic properties. He completed his PhD and then joined Vincenzo Barone's laboratory in Pisa for two years. He also joined Karine Costuas in Rennes for one year in order to study ruthenium based compounds using DFT methods and model Hamiltonians. Currently, he works at Institute of Physics in Warsaw where he obtained a grant for three years to carry out a study on multifunctional compounds allowing a modulation of their magnetic properties.
Corentin Boilleau et al., J Phys Chem Biophys 2017, 7:4(Suppl)DOI: 10.4172/2161-0398-C1-025
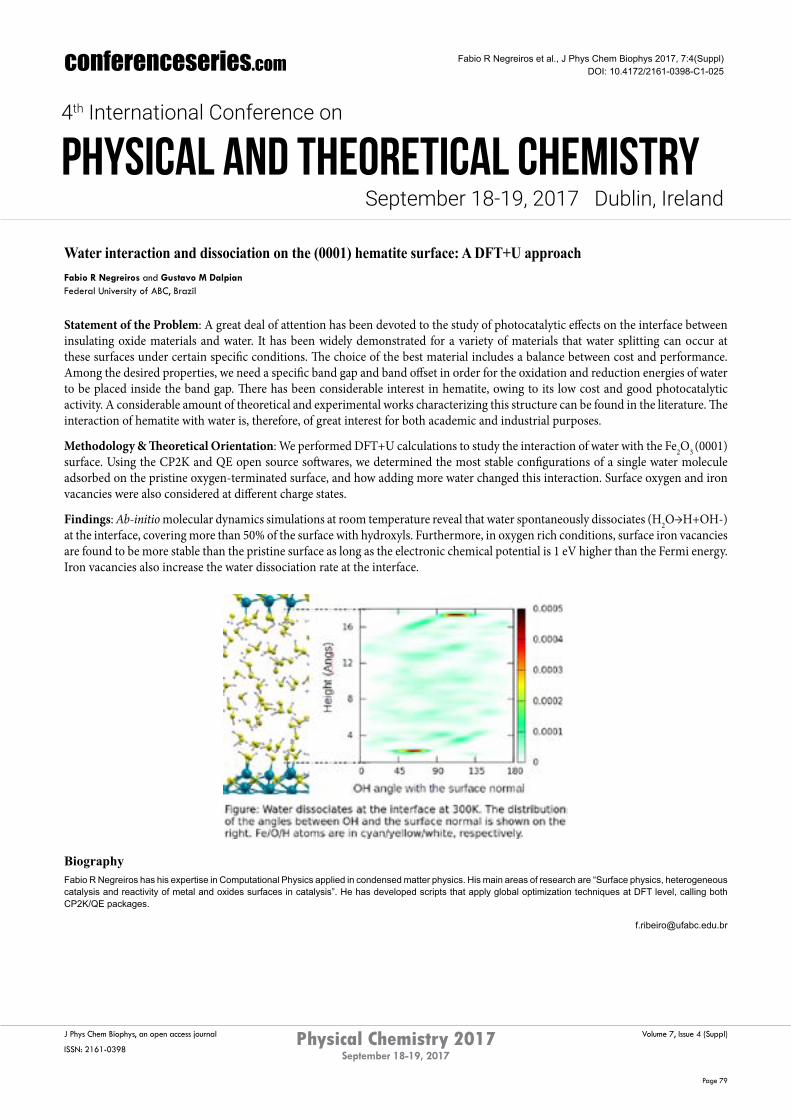
Page 79
conferenceseries.com
Volume 7, Issue 4 (Suppl)J Phys Chem Biophys, an open access journal
ISSN: 2161-0398Physical Chemistry 2017
September 18-19, 2017
4th International Conference on
Physical and Theoretical ChemistrySeptember 18-19, 2017 Dublin, Ireland
Water interaction and dissociation on the (0001) hematite surface: A DFT+U approachFabio R Negreiros and Gustavo M Dalpian Federal University of ABC, Brazil
Statement of the Problem: A great deal of attention has been devoted to the study of photocatalytic effects on the interface between insulating oxide materials and water. It has been widely demonstrated for a variety of materials that water splitting can occur at these surfaces under certain specific conditions. The choice of the best material includes a balance between cost and performance. Among the desired properties, we need a specific band gap and band offset in order for the oxidation and reduction energies of water to be placed inside the band gap. There has been considerable interest in hematite, owing to its low cost and good photocatalytic activity. A considerable amount of theoretical and experimental works characterizing this structure can be found in the literature. The interaction of hematite with water is, therefore, of great interest for both academic and industrial purposes.
Methodology & Theoretical Orientation: We performed DFT+U calculations to study the interaction of water with the Fe2O3 (0001) surface. Using the CP2K and QE open source softwares, we determined the most stable configurations of a single water molecule adsorbed on the pristine oxygen-terminated surface, and how adding more water changed this interaction. Surface oxygen and iron vacancies were also considered at different charge states.
Findings: Ab-initio molecular dynamics simulations at room temperature reveal that water spontaneously dissociates (H2O→H+OH-) at the interface, covering more than 50% of the surface with hydroxyls. Furthermore, in oxygen rich conditions, surface iron vacancies are found to be more stable than the pristine surface as long as the electronic chemical potential is 1 eV higher than the Fermi energy. Iron vacancies also increase the water dissociation rate at the interface.
BiographyFabio R Negreiros has his expertise in Computational Physics applied in condensed matter physics. His main areas of research are “Surface physics, heterogeneous catalysis and reactivity of metal and oxides surfaces in catalysis”. He has developed scripts that apply global optimization techniques at DFT level, calling both CP2K/QE packages.
Fabio R Negreiros et al., J Phys Chem Biophys 2017, 7:4(Suppl)DOI: 10.4172/2161-0398-C1-025
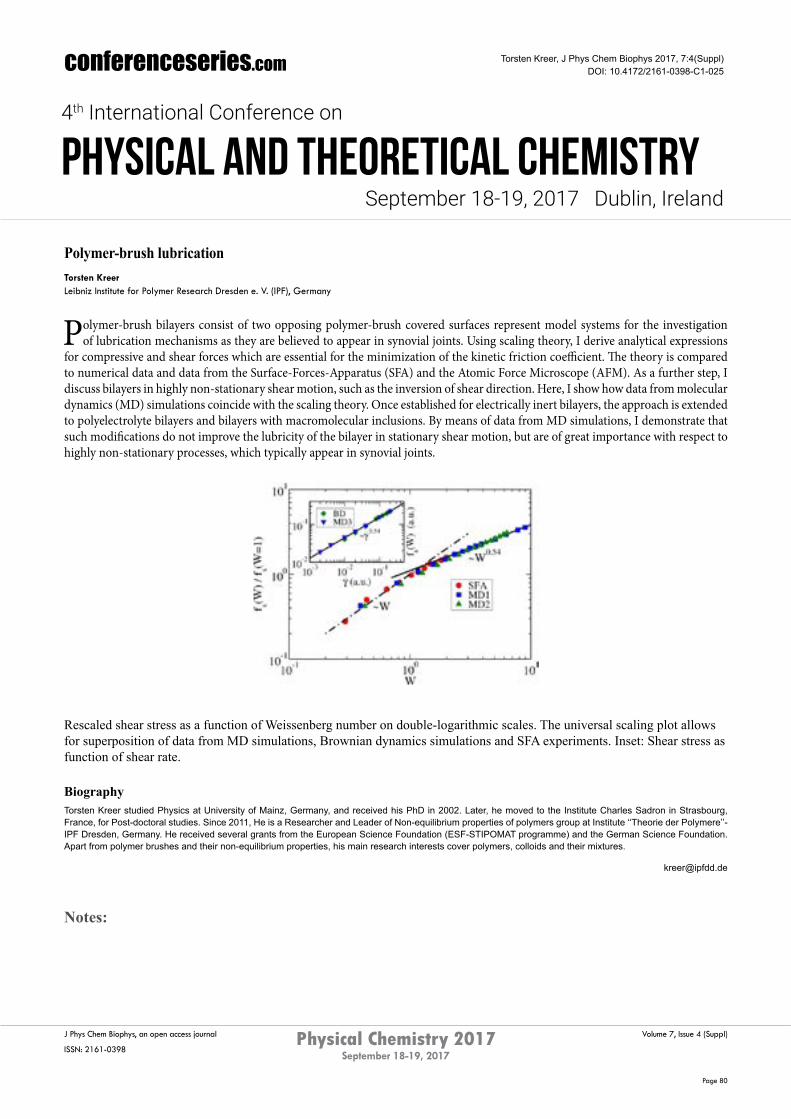
Page 80
Notes:
conferenceseries.com
Volume 7, Issue 4 (Suppl)J Phys Chem Biophys, an open access journal
ISSN: 2161-0398Physical Chemistry 2017
September 18-19, 2017
4th International Conference on
Physical and Theoretical ChemistrySeptember 18-19, 2017 Dublin, Ireland
Polymer-brush lubricationTorsten Kreer Leibniz Institute for Polymer Research Dresden e. V. (IPF), Germany
Polymer-brush bilayers consist of two opposing polymer-brush covered surfaces represent model systems for the investigation of lubrication mechanisms as they are believed to appear in synovial joints. Using scaling theory, I derive analytical expressions
for compressive and shear forces which are essential for the minimization of the kinetic friction coefficient. The theory is compared to numerical data and data from the Surface-Forces-Apparatus (SFA) and the Atomic Force Microscope (AFM). As a further step, I discuss bilayers in highly non-stationary shear motion, such as the inversion of shear direction. Here, I show how data from molecular dynamics (MD) simulations coincide with the scaling theory. Once established for electrically inert bilayers, the approach is extended to polyelectrolyte bilayers and bilayers with macromolecular inclusions. By means of data from MD simulations, I demonstrate that such modifications do not improve the lubricity of the bilayer in stationary shear motion, but are of great importance with respect to highly non-stationary processes, which typically appear in synovial joints.
Rescaled shear stress as a function of Weissenberg number on double-logarithmic scales. The universal scaling plot allows forsuperpositionofdatafromMDsimulations,BrowniandynamicssimulationsandSFAexperiments.Inset:Shearstressasfunction of shear rate.
BiographyTorsten Kreer studied Physics at University of Mainz, Germany, and received his PhD in 2002. Later, he moved to the Institute Charles Sadron in Strasbourg, France, for Post-doctoral studies. Since 2011, He is a Researcher and Leader of Non-equilibrium properties of polymers group at Institute ‘‘Theorie der Polymere’’- IPF Dresden, Germany. He received several grants from the European Science Foundation (ESF-STIPOMAT programme) and the German Science Foundation. Apart from polymer brushes and their non-equilibrium properties, his main research interests cover polymers, colloids and their mixtures.
Torsten Kreer, J Phys Chem Biophys 2017, 7:4(Suppl)DOI: 10.4172/2161-0398-C1-025

Page 81
Notes:
conferenceseries.com
Volume 7, Issue 4 (Suppl)J Phys Chem Biophys, an open access journal
ISSN: 2161-0398Physical Chemistry 2017
September 18-19, 2017
4th International Conference on
Physical and Theoretical ChemistrySeptember 18-19, 2017 Dublin, Ireland
Excited-state symmetry breaking of linear quadrupolar chromophores: A transient absorption studyNadia Dozova1, 2 1PSL Research University, France 2Sorbonne Universités, France
The photophysical properties of two highly symmetrical quadrupolar chromophores were studied by both steady-state and transient absorption spectroscopy. Their excited-state behavior is dominated by the solvent-induced Stokes shift of the stimulated-emission
band. The origin of this shift is attributed to symmetry breaking that confers a non-vanishing dipole moment to the excited state of both compounds. This dipole moment is large and constant in DMSO, whereas symmetry breaking appears significantly slower and leading to smaller excited-state dipole in toluene. Time-dependent increase of the excited-state dipole moment induced by weak solvation is proposed to explain the results in toluene.
BiographyNadia Dozova obtained her PhD in Physical Chemistry at Université Pierre et Marie Curie in 2006. She is an Assistant Professor in the Pasteur lab (Université Pierre et Marie Curie, École Normale Supérieure, CNRS) since 2009. Her current research interests focus on ultrafast spectroscopy (broadband transient absorption, fluorescence up-conversion). She is interested in photo-induced processes in supramolecular constructs and photoactive proteins.
Nadia Dozova, J Phys Chem Biophys 2017, 7:4(Suppl)DOI: 10.4172/2161-0398-C1-025
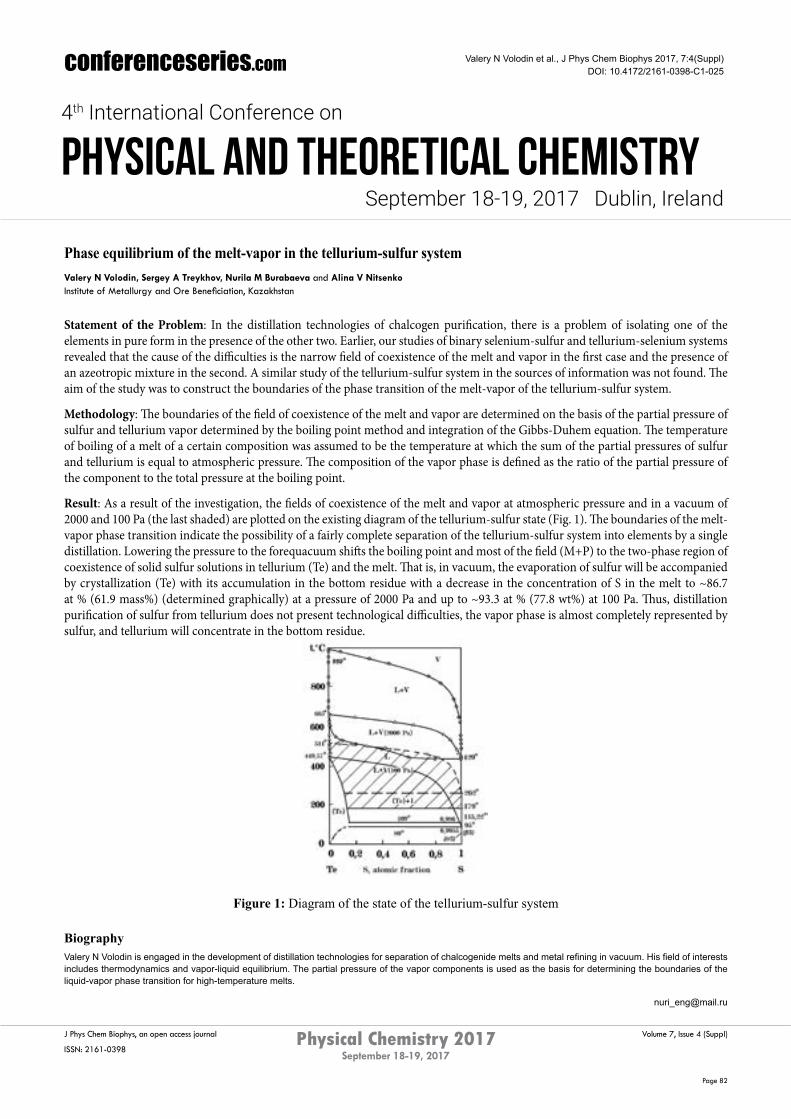
Page 82
conferenceseries.com
Volume 7, Issue 4 (Suppl)J Phys Chem Biophys, an open access journal
ISSN: 2161-0398Physical Chemistry 2017
September 18-19, 2017
4th International Conference on
Physical and Theoretical ChemistrySeptember 18-19, 2017 Dublin, Ireland
Phase equilibrium of the melt-vapor in the tellurium-sulfur systemValery N Volodin, Sergey A Treykhov, Nurila M Burabaeva and Alina V Nitsenko Institute of Metallurgy and Ore Beneficiation, Kazakhstan
Statement of the Problem: In the distillation technologies of chalcogen purification, there is a problem of isolating one of the elements in pure form in the presence of the other two. Earlier, our studies of binary selenium-sulfur and tellurium-selenium systems revealed that the cause of the difficulties is the narrow field of coexistence of the melt and vapor in the first case and the presence of an azeotropic mixture in the second. A similar study of the tellurium-sulfur system in the sources of information was not found. The aim of the study was to construct the boundaries of the phase transition of the melt-vapor of the tellurium-sulfur system.
Methodology: The boundaries of the field of coexistence of the melt and vapor are determined on the basis of the partial pressure of sulfur and tellurium vapor determined by the boiling point method and integration of the Gibbs-Duhem equation. The temperature of boiling of a melt of a certain composition was assumed to be the temperature at which the sum of the partial pressures of sulfur and tellurium is equal to atmospheric pressure. The composition of the vapor phase is defined as the ratio of the partial pressure of the component to the total pressure at the boiling point.
Result: As a result of the investigation, the fields of coexistence of the melt and vapor at atmospheric pressure and in a vacuum of 2000 and 100 Pa (the last shaded) are plotted on the existing diagram of the tellurium-sulfur state (Fig. 1). The boundaries of the melt-vapor phase transition indicate the possibility of a fairly complete separation of the tellurium-sulfur system into elements by a single distillation. Lowering the pressure to the forequacuum shifts the boiling point and most of the field (M+P) to the two-phase region of coexistence of solid sulfur solutions in tellurium (Te) and the melt. That is, in vacuum, the evaporation of sulfur will be accompanied by crystallization (Te) with its accumulation in the bottom residue with a decrease in the concentration of S in the melt to ~86.7 at % (61.9 mass%) (determined graphically) at a pressure of 2000 Pa and up to ~93.3 at % (77.8 wt%) at 100 Pa. Thus, distillation purification of sulfur from tellurium does not present technological difficulties, the vapor phase is almost completely represented by sulfur, and tellurium will concentrate in the bottom residue.
Figure 1: Diagram of the state of the tellurium-sulfur system
BiographyValery N Volodin is engaged in the development of distillation technologies for separation of chalcogenide melts and metal refining in vacuum. His field of interests includes thermodynamics and vapor-liquid equilibrium. The partial pressure of the vapor components is used as the basis for determining the boundaries of the liquid-vapor phase transition for high-temperature melts.
Valery N Volodin et al., J Phys Chem Biophys 2017, 7:4(Suppl)DOI: 10.4172/2161-0398-C1-025

Page 84
1186th Conferenceconferenceseries.com
Physical Chemistry 2017
4th International Conference on
Physical and Theoretical ChemistrySeptember 18-19, 2017 Dublin, Ireland
Young Researchers Forum

Page 85
Notes:
conferenceseries.com
Volume 7, Issue 4 (Suppl)J Phys Chem Biophys, an open access journal
ISSN: 2161-0398Physical Chemistry 2017
September 18-19, 2017
4th International Conference on
Physical and Theoretical ChemistrySeptember 18-19, 2017 Dublin, Ireland
Interactions between sodium fire aerosols and fission products - a theoretical chemistry and experimental approachAnkita JadonUniversity of Lille 1, France
Within the framework of generation IV nuclear reactor safety assessment, the objective of this research work is to investigate the radiological and chemical source term in case of a core disruptive accident in case of a sodium-cooled fast nuclear reactor.
This work investigates the interactions between sodium aerosols, formed after primary system sodium ejection in the containment, and gaseous iodine. Understanding the complex behaviour of surface reaction requires detailed knowledge of both macroscopic and microscopic processes that take place. To link these processes, we followed a combined theoretical and experimental approach. Firstly, methods to theoretically understand the thermodynamics of the heterogeneous reaction between sodium carbonate aerosols and fission products: I2, NaI and HI are proposed. Ab initio, density functional theory (DFT) calculations using Vienna ab initio simulation package are carried out. Secondly, interactions between (I2) g and Na2CO3 were investigated experimentally. (I2) g was generated by heating permeation tubes containing (I2)s, and, passing it through a reaction chamber containing Na2CO3 sorbent. The concentration of unreacted iodine was then measured at the exit of reaction chamber. DFT calculations show that for defect-free surfaces of Υ- Na2CO3 phase, the (001) facet is the most stable. This ideal surface reacts very strongly with HI and NaI, at T<300°C, a low partial pressure of these species (10-7 bar) is sufficient for achieving a surface coverage greater than 50%. However, I2 (g) would react weakly with this surface: to have a surface coverage of 10%, a high partial pressure of iodine is required (10-2 bar). Experimental investigations suggest a stronger reactivity of iodine with exposed Na2CO3 sorbent, at T<100°C; a partial pressure limited to 10-6 bar is sufficient to obtain 10% surface coverage. Both theoretical and experimental approaches indicate very low gas phase capture of I2 (g) by Na2CO3. In summary, we aim to combine computational and experimental studies to increase our understanding of complex surface adsorption phenomena.
BiographyAnkita Jadon is a young Nuclear Safety Researcher focused on challenges that sodium cooled reactor technology still offers. Her research lies primarily in the field of sodium fire aerosols produced in a severe accident. After completing her Master studies specializing in Nuclear Engineering at Ecole des Mines de Nantes, France, she started her professional career as Researcher at the French National Institute for Nuclear Safety (IRSN). In the laboratory, where she is a Doctoral Researcher, she is simulating and modeling interactions between sodium fire aerosols with iodine species in case of a severe accident in an SFR for a computer code which would be used to simulate severe accidents.
Ankita Jadon, J Phys Chem Biophys 2017, 7:4(Suppl)DOI: 10.4172/2161-0398-C1-025

Page 86
Notes:
conferenceseries.com
Volume 7, Issue 4 (Suppl)J Phys Chem Biophys, an open access journal
ISSN: 2161-0398Physical Chemistry 2017
September 18-19, 2017
4th International Conference on
Physical and Theoretical ChemistrySeptember 18-19, 2017 Dublin, Ireland
Strongly red-shifted photoluminescence band induced by molecular twisting in cyanine (Cy3) dye filmsSurendra Anantharaman1, 2, Sergii Yakunin1, 2, Chuyao Peng1, Marcus Vinícius Gonçalves Vismara4, Carlos F O Graeff3, Frank A Nüesch1, 2, Sandra Jenatsch1, 2, Roland Hany1, Maksym V Kovalenko1, 3 and Jakob Heier1
1Swiss Federal Laboratories for Materials Science and Technology, Switzerland2École Polytechnique Fédérale de Lausanne Switzerland3ETH Zurich, CH-8093 Zürich, Switzerland4Universidade Estadual Paulista Júlio de Mesquita Filho (UNESP), Brazil
Cyanine dye molecules, used as monomers or in aggregate form, find interesting applications in opto-electronic devices. Among the various aggregate species incorporating organic dyes, centrosymmetric dimers are known as non-luminescent. They can act
as exciton quenchers due to a low energy optically forbidden excited state. In this study, however, we show that a dimer species in thin films exhibits efficient and strongly red-shifted photoluminescence. When the films were excited, a monomer emission at 590 nm along with a second emission peak at 680 nm was observed. Temperature dependent fluorescence was studied for cyanine films. The dimer emission increases with decreasing temperature due to reduced non-radiative process becoming less effective. A close relation between the dye concentration and the emission showed that a new emission at 680 nm corresponds to the dimer emission. Circular dichroism (CD) spectroscopy reveals that a fraction of the dimers exists in a twisted dimer configuration. Stable, long-lived and quenchable fluorescence with high quantum yield are attributed to this dimer emission. Organic light emitting electrochemical cells (OLECs) fabricated with this dye showed a higher luminance owing to the dimer emission.
BiographySurendra Anantharaman has his expertise in growth and optical characterization of organic dye films. He completed his Bachelor degree in Physics at Anna University, India; Master’s degree in Materials Science and; M S at Indian Institute of Technology Madras, India. His Master’s thesis was on ‘Electrolyte materials for intermediate temperature solid oxide fuel cells’. His area of research interest lies in oxides, nitrides and organic molecules focusing on energy harvesting applications. After working as an Engineer at Taiwan Semiconductor Manufacturing Company (TSMC), Taiwan; he joined as PhD student at École Polytechnique Fédérale de Lausanne (EPFL) in 2015 under the guidance of Prof. Dr. Frank Nüesch and Dr. Jakob Heier. As a PhD student, he is working on understanding the growth of organic crystals and its methodology from the morphology and surface-molecule interactions in the Laboratory for Functional Polymers at Swiss Federal Laboratories for Materials Science and Technology, Switzerland.
Surendra Anantharaman et al., J Phys Chem Biophys 2017, 7:4(Suppl)DOI: 10.4172/2161-0398-C1-025

Page 87
Notes:
conferenceseries.com
Volume 7, Issue 4 (Suppl)J Phys Chem Biophys, an open access journal
ISSN: 2161-0398Physical Chemistry 2017
September 18-19, 2017
4th International Conference on
Physical and Theoretical ChemistrySeptember 18-19, 2017 Dublin, Ireland
Mechanistic insight towards the activation of aerobic oxidative coupling reactions of alcohols on nanoporous goldWilke Dononelli1, Lyudmila Moskaleva2 and Thorsten Klüner1
1University of Oldenburg, Germany2Universität Bremen, Germany
Bulk gold has been known as an inert material without any specific catalytic activity for almost a century. But then in the 1970s Bond et al. presented small gold particles placed on a SiO2 support that could be used for the hydrogenation of alkenes and
alkynes. Since this decade a lot of research has been done on nanostructured gold. These Au-based catalysts can be used for fuel cells, the synthesis of esters or the selective oxidation of alcohols. The selectivity of gold to partial oxidation products is higher than the selectivity of other metal catalysts, so there is a high interest in this gold based catalyst. A problem of gold nanoparticles as catalyst is that the efficiency increases if the average particle size is reduced so in most cases the major part of the surface area of the supporting material is not used for the catalytic processes. In addition to this supported forms of gold catalysts an unsupported form of gold, the nanoporous gold (np-Au), characterized by Zielasek et al. has recently attracted considerable interest due to its potential use in catalysis. Compared to support gold nanoparticles the complete entire surface of the material can be possibly usable as a catalytic material. The most prominent example for the use of np-Au as a catalyst is the selective oxidation of methanol. Although this reaction has been investigated by several groups, the origin of the catalytic activity of np-Au has not been understood completely. The main remaining question that we try to answer is the nature of the active sites of the np-Au. Within DFT (density functional theory) calculations, we describe the influence of residual silver atoms in the material and try to explain some possible pathways for the activation of oxygen, the most essential step of most of oxidative coupling reactions.
Figure 1: Possible reactant, intermediate species and reaction products for methanol oxidation on nanoporous gold.
BiographyWilke Dononelli has his expertise in quantum chemical modeling of catalytic reactions at surfaces. In 2010, he received his Bachelor degree in Mathematics and Chemistry and he started his PhD in Theoretical Chemistry at University of Oldenburg in Germany in 2014. He has experienced in density functional theory and high level ab initio calculations. His main focus lies on bridging from theoretical calculations to model experiments.
Wilke Dononelli et al., J Phys Chem Biophys 2017, 7:4(Suppl)DOI: 10.4172/2161-0398-C1-025
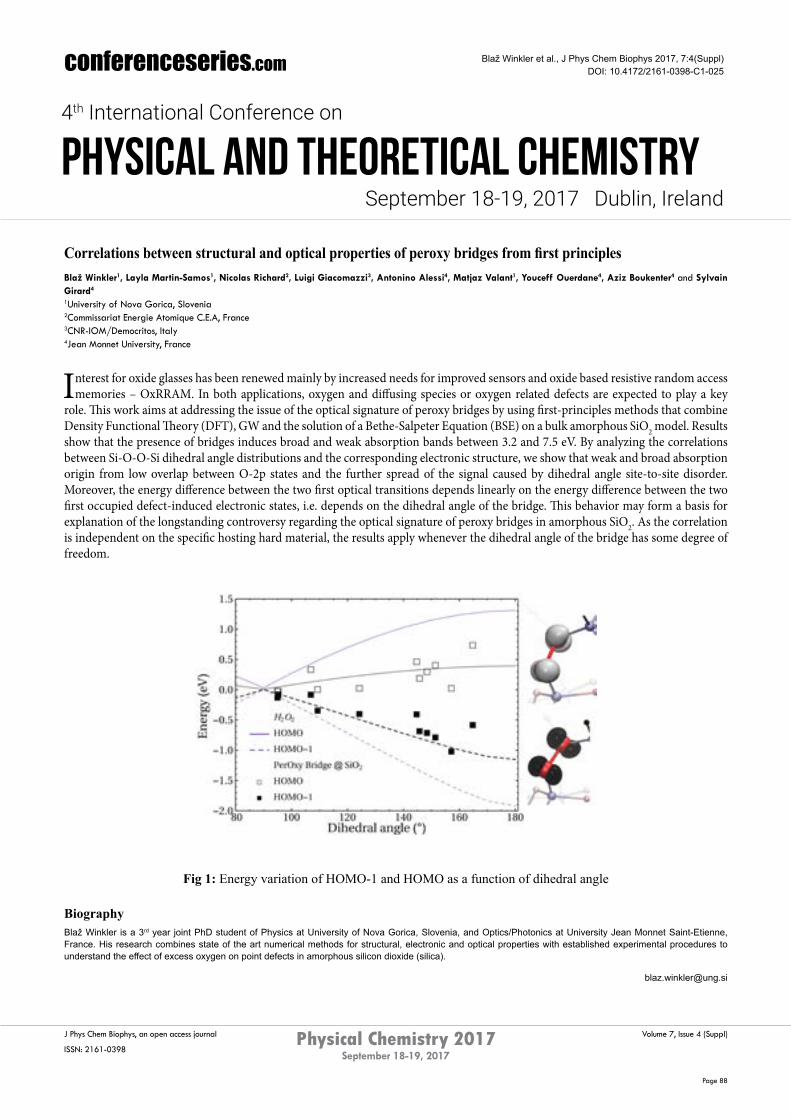
Page 88
conferenceseries.com
Volume 7, Issue 4 (Suppl)J Phys Chem Biophys, an open access journal
ISSN: 2161-0398Physical Chemistry 2017
September 18-19, 2017
4th International Conference on
Physical and Theoretical ChemistrySeptember 18-19, 2017 Dublin, Ireland
Correlations between structural and optical properties of peroxy bridges from first principlesBlaž Winkler1, Layla Martin-Samos1, Nicolas Richard2, Luigi Giacomazzi3, Antonino Alessi4, Matjaz Valant1, Youceff Ouerdane4, Aziz Boukenter4 and Sylvain Girard4
1University of Nova Gorica, Slovenia2Commissariat Energie Atomique C.E.A, France 3CNR-IOM/Democritos, Italy4Jean Monnet University, France
Interest for oxide glasses has been renewed mainly by increased needs for improved sensors and oxide based resistive random access memories – OxRRAM. In both applications, oxygen and diffusing species or oxygen related defects are expected to play a key
role. This work aims at addressing the issue of the optical signature of peroxy bridges by using first-principles methods that combine Density Functional Theory (DFT), GW and the solution of a Bethe-Salpeter Equation (BSE) on a bulk amorphous SiO2 model. Results show that the presence of bridges induces broad and weak absorption bands between 3.2 and 7.5 eV. By analyzing the correlations between Si-O-O-Si dihedral angle distributions and the corresponding electronic structure, we show that weak and broad absorption origin from low overlap between O-2p states and the further spread of the signal caused by dihedral angle site-to-site disorder. Moreover, the energy difference between the two first optical transitions depends linearly on the energy difference between the two first occupied defect-induced electronic states, i.e. depends on the dihedral angle of the bridge. This behavior may form a basis for explanation of the longstanding controversy regarding the optical signature of peroxy bridges in amorphous SiO2. As the correlation is independent on the specific hosting hard material, the results apply whenever the dihedral angle of the bridge has some degree of freedom.
Fig 1: EnergyvariationofHOMO-1andHOMOasafunctionofdihedralangle
BiographyBlaž Winkler is a 3rd year joint PhD student of Physics at University of Nova Gorica, Slovenia, and Optics/Photonics at University Jean Monnet Saint-Etienne, France. His research combines state of the art numerical methods for structural, electronic and optical properties with established experimental procedures to understand the effect of excess oxygen on point defects in amorphous silicon dioxide (silica).
Blaž Winkler et al., J Phys Chem Biophys 2017, 7:4(Suppl)DOI: 10.4172/2161-0398-C1-025

Page 89
conferenceseries.com
Volume 7, Issue 4 (Suppl)J Phys Chem Biophys, an open access journal
ISSN: 2161-0398Physical Chemistry 2017
September 18-19, 2017
4th International Conference on
Physical and Theoretical ChemistrySeptember 18-19, 2017 Dublin, Ireland
Tolerance factors for organic-inorganic perovskites: Applicability only for high temperature phasesMarkus Becker, Thorsten Kluener and Michael WarkUniversity of Oldenburg, Germany
A valuable geometrical concept for the prediction of stable perovskite compounds was found in the Goldschmidt Tolerance Factor (TF), which relates the ionic radii of the constituents in a semi-empirical manner ( TF=γA+γX/√2(γB+γX)). However, due to the
chemically soft nature of the halide anions in the organic-inorganic perovskites and the accompanied higher co-valency, it is necessary to consider the ability of coordinating the different individual metal ions. This is accomplished by introducing the octahedral factor (μ=γB/γX) allowing together with the TF, a 2D mapping of ABX3 stability criteria. The main difficulty lies in the determination of an effective ionic radius for the organic cation, since van-der-Waals interactions and hydrogen bonding give rise to alternated steric demands and varying bond lengths with respect to the inorganic framework. Affirmative, theoretical and experimental values for radii of commonly used cations methyl ammonium (MA+) and formamidinium (FA+), range with a quite large variation from 1.8 to 2.7 Å for MA+ and from 2.2 to 2.79 Å for FA+. In the present work, steric sizes of molecular mono-ammonium cations were calculated by concerning free rotation of the electron density around the center of mass of the molecule. Thereby, structural optimizations were intensively investigated regarding the level of theory and basis sets. A thorough literature study about existing hybrid perovskite compounds revealed a high success rate of predicted stability criteria and 3D phase formation. Furthermore, a case study including the smaller hydroxyl ammonium (HA+) replacing MA+, confirmed the key role of the cationic size on the structural stability and revealed negligible energy barriers associated with preferred molecular orientations in the cuboctahedron. The newly developed computational approach is well suited for high temperature phases, since it considers thermally enabled movements of the central cation and the associated averaging of inorganic deformations.
Figure 1: Scheme of determining the effective ionic radii of molecular cations for the calculation of tolerance factors in ABX3 perovskites.
BiographyMarkus Becker has his expertise in the development and improvement of sequentially deposited planar perovskite solar cells. One of his main focuses lies on the computational investigation of alternative absorber materials. Therefore, a contextual model has been built which allows the prescreening of possible three-dimensional perovskite phases. Combined with more elaborate DFT protocols, new combinations can be investigated considering the thermally enabled movement of the central cation. He has built this model after years of experience in research at University of Oldenburg, Germany.
Markus Becker et al., J Phys Chem Biophys 2017, 7:4(Suppl)DOI: 10.4172/2161-0398-C1-025

Page 90
Notes:
conferenceseries.com
Volume 7, Issue 4 (Suppl)J Phys Chem Biophys, an open access journal
ISSN: 2161-0398Physical Chemistry 2017
September 18-19, 2017
4th International Conference on
Physical and Theoretical ChemistrySeptember 18-19, 2017 Dublin, Ireland
Multi-photo-fragmentation of molecules: REMPI and VMI of HBrArnar HafliðasonUniversity of Iceland, Reykjavik, Iceland
Analysis of mass resolved spectra as well as velocity map images derived from resonance enhanced multi-photon ionization (REMPI) of HBr via resonance excitations to mixed Rydberg and valence (ion-pair) states allows characterization of the effect of
a triplet-to-singlet1,5 and singlet-singlet2,3,4,5 state interaction on further photo-excitation and photo-ionization processes. The analysis makes use of rotational spectra line shifts, line intensity alterations, kinetic energy release spectra as well as angular distributions. Energy-level-dependent state mixing of the resonance excited states is quantified and photo-excitation processes, leading to H+ formation are characterized in terms of the states and fragmentation processes involved, depending on the state mixing.
Figure 1: Schematic representation of themain channels involving excitation, fragmentation and ionization of the HBrmolecule.KERsarrows indicatekineticenergy releaseof fragment species.Otherarrowsshowexcitationand ionizationprocesses involved.
BiographyArnar Hafliðason is a PhD student at University of Iceland. He completed his BSc in Chemistry at University of Iceland, with emphasis on Physical Chemistry and Inorganic Chemistry. His PhD project is in the field of Physical Chemistry with focus on photochemistry. He has published three articles, with main emphasis on photo-dissociation, photoionization and state interaction.
Arnar Hafliðason, J Phys Chem Biophys 2017, 7:4(Suppl)DOI: 10.4172/2161-0398-C1-025

Page 91
Notes:
conferenceseries.com
Volume 7, Issue 4 (Suppl)J Phys Chem Biophys, an open access journal
ISSN: 2161-0398Physical Chemistry 2017
September 18-19, 2017
4th International Conference on
Physical and Theoretical ChemistrySeptember 18-19, 2017 Dublin, Ireland
Characterization of protein absorption on gold nanoparticles by scattering correlation spectroscopyHanane Moustaoui, Jolanda Spadavecchia, Marc Lamy de la Chapelle and Nadia DjakerUniversité Paris 13, France
Gold nanoparticles (GNP) are of great interest for several applications in nanomedicine, especially in imaging and sensing, drug delivery and photothermal therapy. In the case of therapy by nano vector or hyperthermia therapy, GNP interacts with blood
proteins after injection. This interaction induced coating of GNP by proteins namely protein corona. The GNP physicochemical properties like: size, shape and surface charge affect directly the structure and composition of the protein corona. To understand this interaction, protein corona has been explored for different size, shape of GNP, with several techniques, like UV-Vis spectroscopy, zeta potential and especially, correlation spectroscopy. Recently, scattering correlation spectroscopy (SCS) is one of the most used techniques for GNP characterization. The SCS technique is based on the temporal analysis of the scattered intensity fluctuations and the correlation curve is directly related to the hydrodynamic radius of GNP, to their diffusion coefficient, concentration and shape. The SCS is very sensitive to GNP morphology and brightness since the scattering intensity depends on the GNP volume. The characterization of protein corona by SCS technique will be presented for different sized GNP with different shapes (spheres, urchins and flowers), in presence of different concentrations of proteins (albumin, lysozyme and hemoglobin), at very low concentrations (~pM) and with very high precision. Such results show how protein cover gold nanoparticle (amount, conformation) and the specific adsorption of this protein according to shape and size of gold nanoparticles.
BiographyHanane Moustaoui completed her degree in Chemistry from Institut Galilée, University Paris 13, Villetaneuse, France. Since June 2015, she is a PhD student under the supervision of Doctor Nadia Djaker and Doctor Jolanda Spadavecchia of Laboratory CSPBAT, University Paris 13, France. Her research interests include “The synthesis of gold nanoparticles using biocompatible surfactant and study their toxicity in biological media by spectroscopy method”.
Hanane Moustaoui et al., J Phys Chem Biophys 2017, 7:4(Suppl)DOI: 10.4172/2161-0398-C1-025





























