Embed Size (px)
Citation preview

Report
Progesterone pre-treatment potentiates EGF pathway signaling in the breast cancer
cell line ZR-75w
A. Carvajal1, N. Espinoza1, S. Kato1, M. Pinto1, A. Sadarangani1, C. Monso1, E. Aranda1,M. Villalon1, J.K. Richer2, K.B. Horwitz2, J.J. Brosens3, and G.I. Owen11Unidad de Reproduccion y Desarrollo, Facultad de Ciencias Biologicas, Pontificia Universidad Catolica de Chile,Santiago, Chile; 2Department of Medicine, Division of Endocrinology, University of Colorado Health Sciences Center,Denver, Colorado, USA; 3Institute of Reproductive and Developmental Biology, Imperial College London,Hammersmith Hospital, London, United Kingdom
Key words: c-fos, EGFR, erk, HRT, progestin, STAT5
Summary
Progesterone in hormone replacement therapy (HRT) preparations increases, while hysterectomy greatly reduces,the incidence of breast cancer. Cross-talk between the progesterone and growth factor signaling pathways occurs atmultiple levels and this maybe a key factor in breast cancer survival and progression. To test this hypothesis, wecharacterized the effect of progesterone pre-treatment on the sensitization of the epidermal growth factor (EGF)signaling pathway to EGF in the breast cancer cell line ZR-75. For the first time in ZR-75 cells and in agreementwith previous work using synthetic progestins, we demonstrate that pre-treatment with the natural ligand pro-gesterone increases EGF receptor (EGFR) levels and subsequent ligand-dependent phosphorylation. Downstreamwe demonstrate that progesterone alone increases erk-1 + 2 phosphorylation, potentiates EGF-phosphorylatederk-1 + 2 and maintains these levels elevated for 24 h; over 20 h longer than in vehicle treated cells. Additionally,progesterone increased the levels of STAT5, another component of the EGF signaling cascade. Progesteroneincreased EGF mediated transcription of a c-fos promoter reporter and the nuclear localization of the native c-fosprotein. Furthermore, progesterone and EGF both alone and in combination, significantly increase cell prolifera-tion. Several results presented herein demonstrate the conformity between the action of the natural ligand pro-gesterone with that of synthetic progestins such as MPA and R5020 and allows the postulation that the progestin/progesterone-dependent increase of EGF signaling provides a survival advantage to burgeoning cancer cells and maycontribute to the breast cancer risk associated with endogenous progesterone and with progestin-containing HRT.
Abbreviations: C: vehicle control; EGF: epidermal growth factor; EGFR: EGF receptor; ER: estrogen receptor;hours: h; HRT: hormone replacement therapy; minutes: min; P4: Progesterone; PR: progesterone receptor; STAT:signal transducers and activators of transcription
Introduction
Progesterone is a critical steroid hormone that controlscell proliferation and differentiation. In normal mam-mary gland ductal epithelium, high levels of progester-one during the luteal phase mediate proliferation anddifferentiation in milk ducts in preparation for thepossibility of pregnancy [reviewed in 1]. These effects aremediated by progesterone through two progesteronereceptors (PR-A and PR-B) which arise from differentialpromoter usage [2,3]. Mice lacking progesterone recep-tors (PR) exhibit incomplete mammary gland ductal andfailure of lobulo-alveolar development [4].
The association between increased breast cancerincidence and the presence of endogenous ovarian hor-mones has been know since the nineteenth century [5].
Exogenous progestins are found in contraceptivesand in hormone replacement therapy (HRT). In womenreceiving estrogen replacement therapy (ERT), there is aslight increase in breast cancer; however, when proges-terone is added in conjunction with estrogen (combinedtherapy) the incidence of breast cancer increases dra-matically [6–8]. The risk of breast cancer also increaseswith combined hormone contraceptive preparations[reviewed in 9]. In agreement with these in vivo obser-vations, progesterone metabolites have been found toinduce proliferation in the breast cancer lines [10,11]. Inaddition, medroxyprogesterone acetate (MPA) inducesmammary adenocarcinomas in BALB/C mice [12].These clinical observations, coupled to laboratory
wThis work was supported by the National Chilean Fund for theDevelopment of Science and Technology (FONDECYT 1020715) anda Wellcome Trust International collaboration grant GR071469.
Breast Cancer Research and Treatment (2005) � Springer 2005DOI 10.1007/s10549-005-7726-6

findings, indicate that when progesterone is included inhormonal therapies there is an increase in the incidenceof breast cancer. However data is lacking on the effect ofthe natural ligand progesterone in breast cancer cells.One of the objectives of this paper is to ascertain if theeffects observed by the synthetic progestins are alsoexhibited by progesterone in the breast cancer cell lineZR-75.
Signaling through the EGFR is a requirement for thenormal development of the mammary ductal system [13].EGFR)/) mice only survive for up to 8 days after birthand show an incomplete proliferation of mammarystromal and epithelial cells [13,14]. The signaling path-ways activated by EGF and EGF receptors (EGFRs)regulate several cellular process including proliferation,differentiation, adhesion, migration, apoptosis and sur-vival [15]. The activation of EGFR has been associatedwith breast cancer development and progression. Hor-mone-resistant breast cancer is associated with an in-creased expression of both EGF receptor and ligand. Thisco-expression suggests the presence of an autocrinepathway of uncontrolled cell growth sustaining neoplastictransformation [16–18]. It was previously demonstratedthat in the breast cancer cell line T47Dco (ER)/PR+)progesterone up-regulates EGFR protein levels and sen-sitizes these cells to EGF-induced proliferative effects[19]. Progesterone is known to cross-talk with the EGFsignaling pathway in the control of breast cancer cellgrowth [20–23]. Incubation of the estrogen receptor (ER)and progesterone receptor (PR) positive (ER+/PR+)breast cancer cell line T-47D with the synthetic progestin,MPA, resulted in a time and dose-dependent increase inEGFR mRNA [24]. The synthetic progesterone agonist,R5020, has been reported to potentiate the effects of EGFby up-regulating EGFR, c-ErbB2 and c-ErbB3 receptors,and by enhancing EGF-stimulated tyrosine phosphory-lation of signaling molecules known to associate withactivated type I receptors [21]. EGF induces dimerizationbetween the EGFR and other ErbB family members[ErbB2-4; 25] with the subsequent activation of intrinsictyrosine kinase activity. In the clinic, ErbB family mem-ber overexpression is used as diagnostic marker in breastcancer and is associated with poor patient prognosis [26].
Auto-phosphorylation of tyrosine residues localizedin intracellular domain of EGFR leads to Ras-Raf-MEK-erk (MAPK) pathway activation among others.Erk1 and 2 (also known as p42/p44 mitogen-activatedprotein kinase), activation has been implicated in breastcarcinogenesis with cancers frequently containing anincreased proportion of cells with activated form of erk1and 2 [27]. Previous work has demonstrated that thesynthetic progestin R5020 potentiates EGF-stimulatederk-1 + 2, p38 MAP kinase, and JNK activities inT47D-YB (ER)/PRB+), a breast cancer cell lineexpressing PR-B but not PR-A [21,28].
EGFR signaling is also mediated by STATs (signaltransducers and activators of transcription), latentcytoplasmic transcription factors classically activated bycytokine signaling [15]. Following phosphotyrosine-
dependent dimerization, STAT family members canbypass classical kinase cascades by entering the nucleusdirectly to transciptionally regulate genes [29]. STAT5activity has been linked to alveolar proliferation andfunction [30]. During pregnancy, a period dominated byhigh levels of progesterone, mRNA and protein levels ofSTAT5 are raised [31]. Furthermore, as with erk-1 and2, STAT5 activation via phosphorylation has also beenimplicated in breast cancer progression [32]. Microarrayanalysis in T47Dco (ER)/PR+) cells demonstrated anincrease in STAT5 mRNA and protein expression afterR5020 addition [33].
The proto-oncogene c-fos is a known target gene ofthe EGF signaling pathway and contains promoterbinding sites for erk-1 and 2 substrates and STAT familymembers [34,35]. Expression of Fos family members iscorrelated with metalloprotease expression in primarytumors and the invasive potential of MCF7 breastcancer cells is enhanced by c-fos [36]. c-fos is rapidly buttransiently induced by synthetic progestin treatment inT-47D (ER+/PR+) [37] and progesterone synergizeswith EGF to increase transcriptional activity from thec-fos promoter fused to luciferase reporter in the T-47D-YB (ER)/PRB+) cell line [20].
As summarized above, previous work has concen-trated on the MCF7 and T47D variant breast cancer celllines in the presence of the synthetic progestins MPA orR5020. Herein, we demonstrate, in accordance withpublished results using synthetic progestins, that thenatural ligand progesterone increases EGFR expressionand this increase corresponds to a sensitization of the erkand STAT5 pathways downstream of EGF in the ZR-75breast cancer cell line. We furthermore demonstrate forthe first time, that progesterone pretreatment prolongs theerk phosphorylation state out to 24 h, after a single pulseof EGF. Sensitization or ‘priming’ of this pathway byprogesterone corresponds to an increase in transcriptionalactivity on the c-fos promoter and importantly, enhancednuclear localization of native c-fos protein.
As stated above, the EGFR pathway is involved intumor proliferation, adhesion and migration. We pos-tulate that the activated-state of EGFR pathway medi-ated by progesterone confers a survival advantage toburgeoning breast cancer cells. As progestins are widelyused in oral contraception, in HRT, and in cancertreatments, it is critically important that the subtleties oftheir mechanisms of action be clearly understood.
Materials and methods
Cell culture, hormonal and inhibitor treatments
ZR-75-1 breast cancer cells [38] were maintained inDMEM/F12 media (Gibco BRL, USA) supplementedwith 10% fetal bovine serum (Invitrogen, NY, USA). Forprotein experiments, cells were plated at 50% confluencein 10 cm2 Petri dishes (Falcon, Becton-Dickinson, NJ,USA), then the medium changed to charcoal-treated
A Carvajal et al.

medium containing 5% serum for 24 h before progester-one or EGF treatment. Progesterone (Sigma-Aldrich, St.Louis, USA) was dissolved in ethanol and added to thecells to a final concentration of 10 nM. EGF (UpstateBiotechnology, NY, USA) was added at a final concen-tration 60 ng/ml. An equal volume of ethanol or waterwas used as control and in EGF treatments. The MEK1/2, inhibitor U0126 (1 or 2.5 (M; Cell Signaling Tech, Inc.,USA) or vehicle (DMSO) were applied 40 min beforeEGF treatment. The antiprogestin RU486 (mifepristone100 nM; Sigma-Aldrich, St Louis, USA) or vehicle (eth-anol) was applied 30 min before progesterone treatment.
Western blotting
Cells were harvested in cold PBS and the pellet resus-pended in lysis buffer (0.4 M KCl, 20 mM Hepes pH7.4, 1 mM DTT, 20% glycerol). After sonification onice, the lysate was centrifuged at 14,000 g for 20 min at4 �C to obtain a total protein extract. The proteinconcentration was determined by Bradford assay.100 lg of crude total extract was loaded in each lane,separated by 8–10% polyacrylamide gel electrophoresisin the presence of sodium dodecylsulfate, transferred tonitrocellulose membranes, and incubated overnight withpurified anti-bodies to EGFR (1:1000; Santa Cruz Bio-tech., Inc., USA), phospho-EGFR (1:750; UpstateBiotech., NY, USA), ERK-2 (1:1500; Santa Cruz Bio-tech., Inc., USA), phospho-ERK (1:1000; Cell SignalingTech, Inc., USA), STAT5a (L-20) and STAT5b (G2)(both 1:1000; Santa Cruz Biotech., Inc., USA) andphospho-STAT5 (1:750; Upstate Biotech., NY, USA).Goat anti-rabbit or anti-mouse IgG secondary antibodycoupled to horse radish peroxidase (1:3000 or 1:5000,Bio-Rad Labs, CA, USA) was applied for 2 h at roomtemperature. The reaction was developed by chemilumi-nescence using ECL Western blot analysis system (NEN,Western lightning, Perkin-Elmer). Semi-quantitativedensitometry of the bands was performed using NIHImage 1.62c software package for Macintosh to deter-mine if significant differences between treatments existed.
Immunofluorescence and confocal microscopy
ZR-75-1 cells (60,000/well) were cultured on glass slidesin DMEM/F12 media supplemented with 10% fetalbovine serum. After 24 h in 5% charcoal treated fetalbovine serum cells were treated for a further 24 h withprogesterone (10 nM) or ethanol vehicle and then stim-ulated with EGF (60 ng/ml) or water vehicle for up to2 h. The slides were washed three times in Ca/Mg buffer(PBS supplemented with 0,2 mM CaCl2 and 1 mMMgCl2) and fixed 4% paraformaldehyde for 30 min atroom temperature. Slides were blocked with 5% BSAprotein block (IgG free, Sigma-Aldrich, St Louis, USA)for 1 h at room temperature. The primaries antibodies(1:500) to STAT5 (Santa Cruz Biotech., Inc., USA,phospho-STAT5 (Upstate Biotech., NY, USA), phos-pho-ERK (Cell Signaling Tech, Inc., USA) and c-fos
(Oncogene, USA) were added for 15 h at 4 �C. Negativecontrols were performed in the absence of primary anti-body. The slides were subsequently rinsed and incubatedin the dark for 1 h at 37 �C with goat anti-rabbit or anti-mouse IgG secondary antibody coupled to Cy2 or Cy3(1:500; Immunoresearch, France). The slides were rinsedand counterstained with propidium iodide (1 lg/ml) for5 min to distinguish the nucleus. Finally, the slides wererinsed and mounted in Fluoromount G and stored at)20 �C until analysis. The slides were observed on aPascal LSM 200 microscopy (Carl Zeiss Microscopy)using a 63· objective, zoom (2,3·) and laser (k 488 and546 nm; 460 and 543 nm). Images were analyzed usingConfocal Microscopy Software LSM Image Examiner.
Transient transfections
ZR-75-1 cells (80,000/well in triplicate) were cultured in24-wells plates in DMEM/F12 media supplemented with10% fetal bovine serum. After 6 h in 5% charcoal treatedfetal bovine serum cells were transiently co-transfectedin DMEM/F12 media with 0.35 lg of c-fos promoter(c-fos-TK-luc) and 0.05 lg of CMV-b-galactosidase inpA3 vector to check transfection efficiency using Lipo-fectamin plus (Invitrogen, USA). After 5 h, the mediumwas changed and the cells treated with progesterone(10 nM) or vehicle (ethanol) for 24 h and then stimu-lated with EGF (60 ng/ml) for a further 18 h. The cellswere harvested on ice in lysis buffer (Pharmingen BDBiosciences, San Diego, USA) and luciferase activity(Pharmingen BD Biosciences, San Diego, USA) wasmeasured by luminometer and normalized in respect tob-galactosidase activity.
Cell proliferation assay
ZR-75 cells were plated in DMEM/F12 media (GibcoBRL, USA) supplemented with 10% fetal bovineserum (Invitrogen, NY, USA) in 96 well plates (BectonDickinson, USA). After 24 h the medium was changed toDMEM/F12 containing 5% charcoal treated fetal bovineserum for a further 24 h. One pulse of progesterone(10 nM) or vehicle (equal volume of ethanol) was addedwith a change of medium. After 24 h, EGF (60 ng/ml) orvehicle (equal volume of H2O) was added without amedium change. Cells were harvested 72 h later for theCellTiter 96� AQueous Cell Proliferation Assay followingthe manufactures instructions (Promega, WI, USA). Thecell proliferation assay was repeated three times. In eachexperiment each treatment was performed in quintuplet.
Statistics
In replicate experiments we obtain mean values andstandard errors. Results are expressed as the percentageof control for graphical representation and statisticalanalysis. Differences between the means of these per-centages were analyzed using Mann–Whitney U-test,with statistical significance regarded as p<0.05.
Regulation of EGF pathway by progesterone
Results
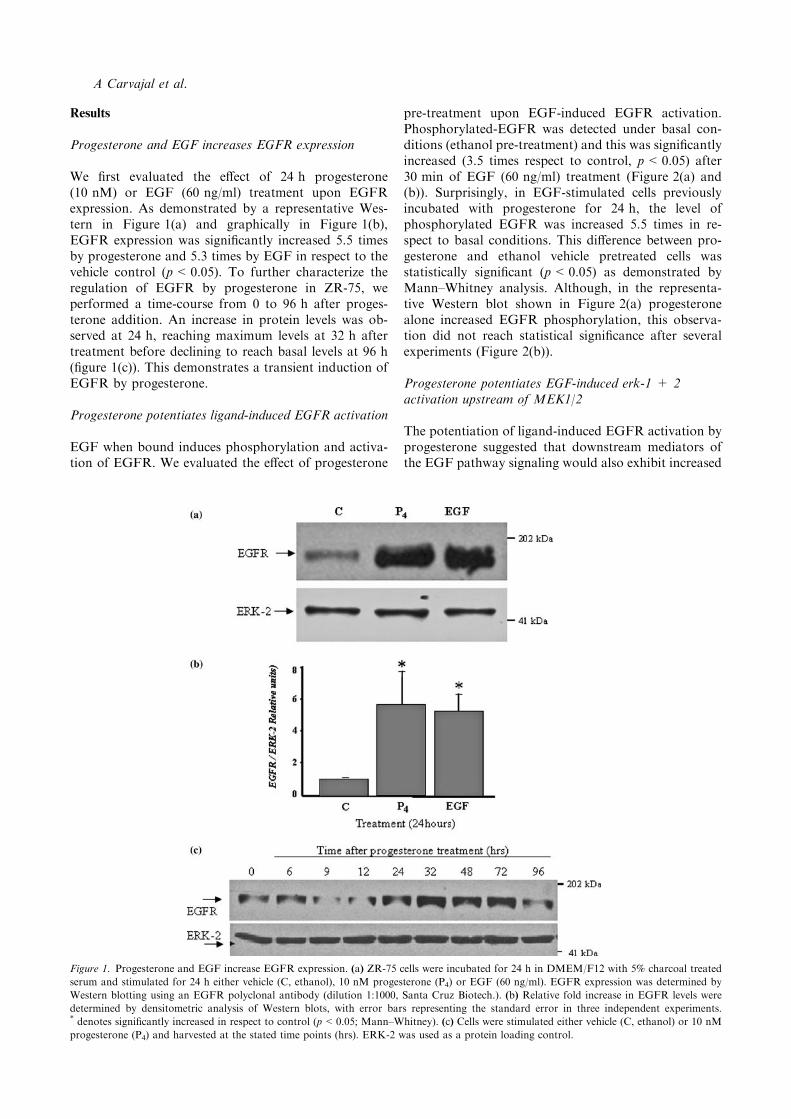
Progesterone and EGF increases EGFR expression
We first evaluated the effect of 24 h progesterone(10 nM) or EGF (60 ng/ml) treatment upon EGFRexpression. As demonstrated by a representative Wes-tern in Figure 1(a) and graphically in Figure 1(b),EGFR expression was significantly increased 5.5 timesby progesterone and 5.3 times by EGF in respect to thevehicle control (p<0.05). To further characterize theregulation of EGFR by progesterone in ZR-75, weperformed a time-course from 0 to 96 h after proges-terone addition. An increase in protein levels was ob-served at 24 h, reaching maximum levels at 32 h aftertreatment before declining to reach basal levels at 96 h(figure 1(c)). This demonstrates a transient induction ofEGFR by progesterone.
Progesterone potentiates ligand-induced EGFR activation
EGF when bound induces phosphorylation and activa-tion of EGFR. We evaluated the effect of progesterone
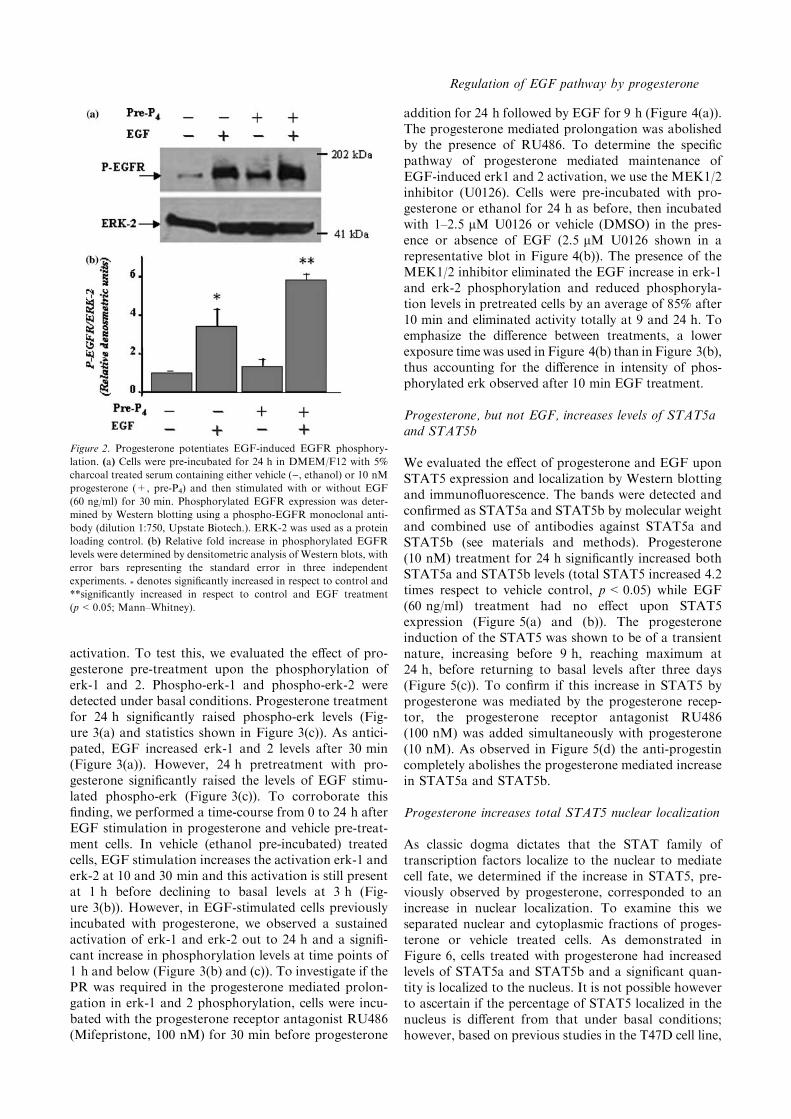
pre-treatment upon EGF-induced EGFR activation.Phosphorylated-EGFR was detected under basal con-ditions (ethanol pre-treatment) and this was significantlyincreased (3.5 times respect to control, p<0.05) after30 min of EGF (60 ng/ml) treatment (Figure 2(a) and(b)). Surprisingly, in EGF-stimulated cells previouslyincubated with progesterone for 24 h, the level ofphosphorylated EGFR was increased 5.5 times in re-spect to basal conditions. This difference between pro-gesterone and ethanol vehicle pretreated cells wasstatistically significant (p<0.05) as demonstrated byMann–Whitney analysis. Although, in the representa-tive Western blot shown in Figure 2(a) progesteronealone increased EGFR phosphorylation, this observa-tion did not reach statistical significance after severalexperiments (Figure 2(b)).
Progesterone potentiates EGF-induced erk-1 + 2activation upstream of MEK1/2
The potentiation of ligand-induced EGFR activation byprogesterone suggested that downstream mediators ofthe EGF pathway signaling would also exhibit increased
Figure 1. Progesterone and EGF increase EGFR expression. (a) ZR-75 cells were incubated for 24 h in DMEM/F12 with 5% charcoal treated
serum and stimulated for 24 h either vehicle (C, ethanol), 10 nM progesterone (P4) or EGF (60 ng/ml). EGFR expression was determined by
Western blotting using an EGFR polyclonal antibody (dilution 1:1000, Santa Cruz Biotech.). (b) Relative fold increase in EGFR levels were
determined by densitometric analysis of Western blots, with error bars representing the standard error in three independent experiments.* denotes significantly increased in respect to control (p<0.05; Mann–Whitney). (c) Cells were stimulated either vehicle (C, ethanol) or 10 nM
progesterone (P4) and harvested at the stated time points (hrs). ERK-2 was used as a protein loading control.
A Carvajal et al.
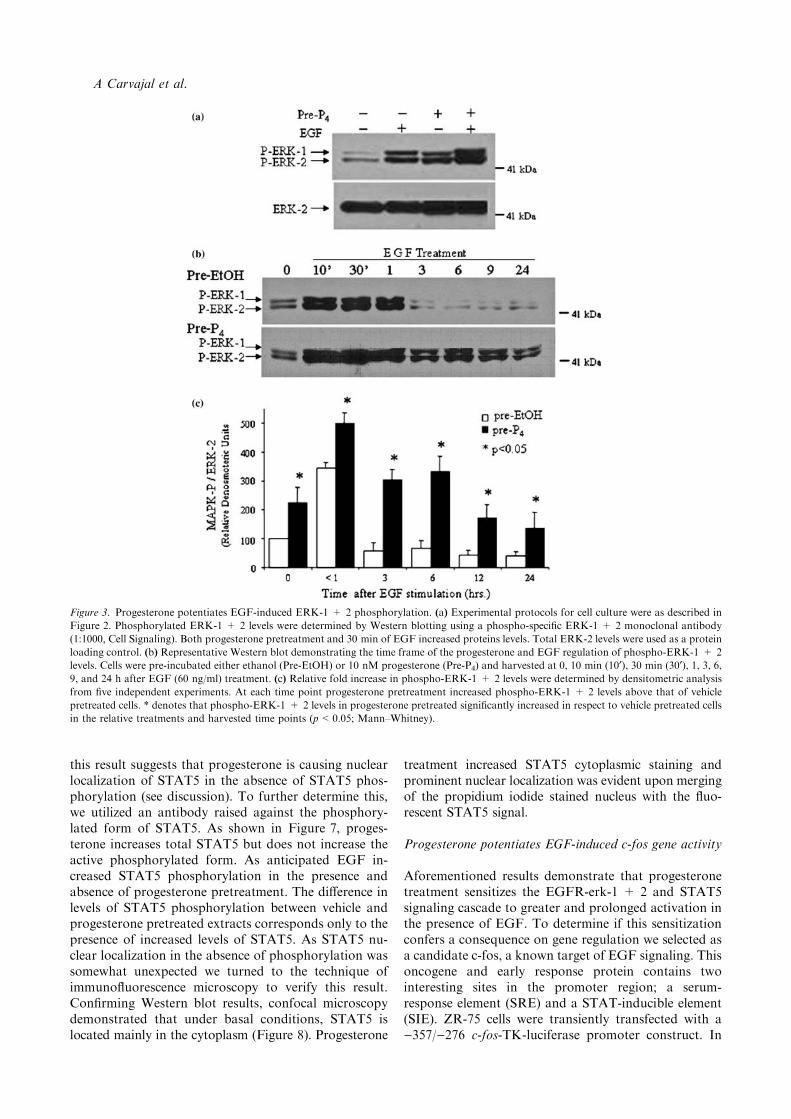
activation. To test this, we evaluated the effect of pro-gesterone pre-treatment upon the phosphorylation oferk-1 and 2. Phospho-erk-1 and phospho-erk-2 weredetected under basal conditions. Progesterone treatmentfor 24 h significantly raised phospho-erk levels (Fig-ure 3(a) and statistics shown in Figure 3(c)). As antici-pated, EGF increased erk-1 and 2 levels after 30 min(Figure 3(a)). However, 24 h pretreatment with pro-gesterone significantly raised the levels of EGF stimu-lated phospho-erk (Figure 3(c)). To corroborate thisfinding, we performed a time-course from 0 to 24 h afterEGF stimulation in progesterone and vehicle pre-treat-ment cells. In vehicle (ethanol pre-incubated) treatedcells, EGF stimulation increases the activation erk-1 anderk-2 at 10 and 30 min and this activation is still presentat 1 h before declining to basal levels at 3 h (Fig-ure 3(b)). However, in EGF-stimulated cells previouslyincubated with progesterone, we observed a sustainedactivation of erk-1 and erk-2 out to 24 h and a signifi-cant increase in phosphorylation levels at time points of1 h and below (Figure 3(b) and (c)). To investigate if thePR was required in the progesterone mediated prolon-gation in erk-1 and 2 phosphorylation, cells were incu-bated with the progesterone receptor antagonist RU486(Mifepristone, 100 nM) for 30 min before progesterone
addition for 24 h followed by EGF for 9 h (Figure 4(a)).The progesterone mediated prolongation was abolishedby the presence of RU486. To determine the specificpathway of progesterone mediated maintenance ofEGF-induced erk1 and 2 activation, we use the MEK1/2inhibitor (U0126). Cells were pre-incubated with pro-gesterone or ethanol for 24 h as before, then incubatedwith 1–2.5 lM U0126 or vehicle (DMSO) in the pres-ence or absence of EGF (2.5 lM U0126 shown in arepresentative blot in Figure 4(b)). The presence of theMEK1/2 inhibitor eliminated the EGF increase in erk-1and erk-2 phosphorylation and reduced phosphoryla-tion levels in pretreated cells by an average of 85% after10 min and eliminated activity totally at 9 and 24 h. Toemphasize the difference between treatments, a lowerexposure time was used in Figure 4(b) than in Figure 3(b),thus accounting for the difference in intensity of phos-phorylated erk observed after 10 min EGF treatment.
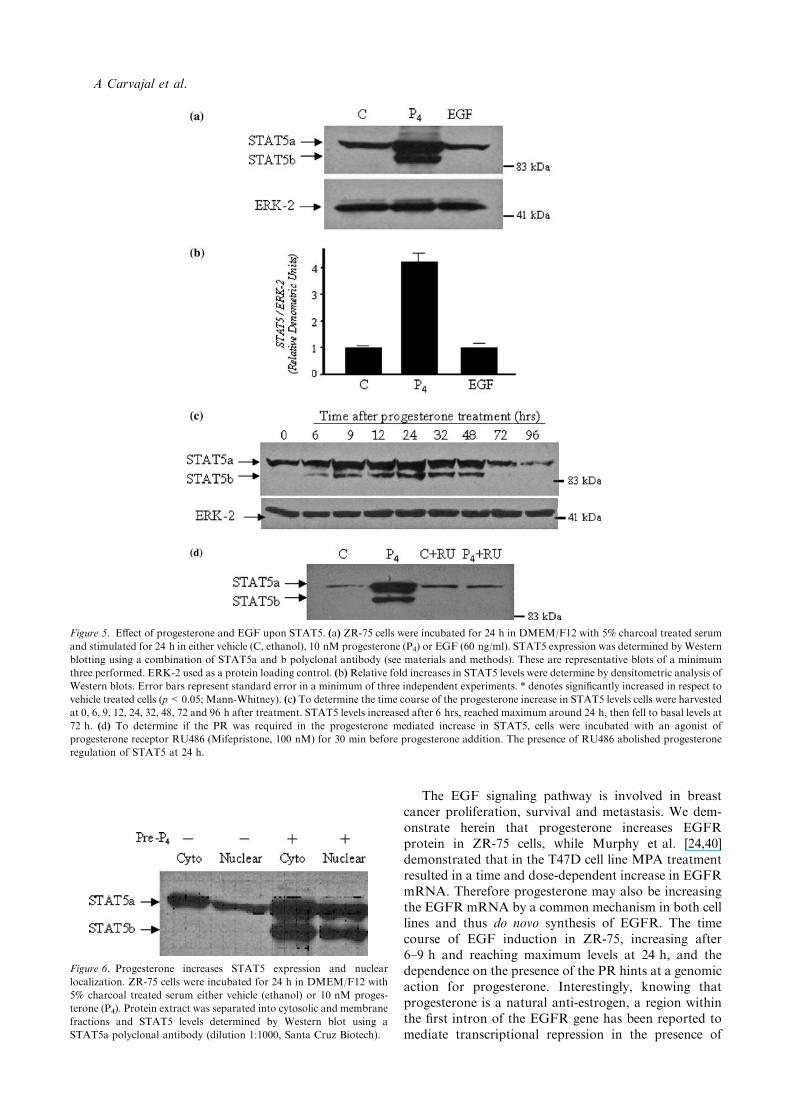
Progesterone, but not EGF, increases levels of STAT5aand STAT5b
We evaluated the effect of progesterone and EGF uponSTAT5 expression and localization by Western blottingand immunofluorescence. The bands were detected andconfirmed as STAT5a and STAT5b by molecular weightand combined use of antibodies against STAT5a andSTAT5b (see materials and methods). Progesterone(10 nM) treatment for 24 h significantly increased bothSTAT5a and STAT5b levels (total STAT5 increased 4.2times respect to vehicle control, p<0.05) while EGF(60 ng/ml) treatment had no effect upon STAT5expression (Figure 5(a) and (b)). The progesteroneinduction of the STAT5 was shown to be of a transientnature, increasing before 9 h, reaching maximum at24 h, before returning to basal levels after three days(Figure 5(c)). To confirm if this increase in STAT5 byprogesterone was mediated by the progesterone recep-tor, the progesterone receptor antagonist RU486(100 nM) was added simultaneously with progesterone(10 nM). As observed in Figure 5(d) the anti-progestincompletely abolishes the progesterone mediated increasein STAT5a and STAT5b.
Progesterone increases total STAT5 nuclear localization
As classic dogma dictates that the STAT family oftranscription factors localize to the nuclear to mediatecell fate, we determined if the increase in STAT5, pre-viously observed by progesterone, corresponded to anincrease in nuclear localization. To examine this weseparated nuclear and cytoplasmic fractions of proges-terone or vehicle treated cells. As demonstrated inFigure 6, cells treated with progesterone had increasedlevels of STAT5a and STAT5b and a significant quan-tity is localized to the nucleus. It is not possible howeverto ascertain if the percentage of STAT5 localized in thenucleus is different from that under basal conditions;however, based on previous studies in the T47D cell line,
Figure 2. Progesterone potentiates EGF-induced EGFR phosphory-
lation. (a) Cells were pre-incubated for 24 h in DMEM/F12 with 5%
charcoal treated serum containing either vehicle (), ethanol) or 10 nM
progesterone (+, pre-P4) and then stimulated with or without EGF
(60 ng/ml) for 30 min. Phosphorylated EGFR expression was deter-
mined by Western blotting using a phospho-EGFR monoclonal anti-
body (dilution 1:750, Upstate Biotech.). ERK-2 was used as a protein
loading control. (b) Relative fold increase in phosphorylated EGFR
levels were determined by densitometric analysis of Western blots, with
error bars representing the standard error in three independent
experiments. * denotes significantly increased in respect to control and
**significantly increased in respect to control and EGF treatment
(p<0.05; Mann–Whitney).
Regulation of EGF pathway by progesterone
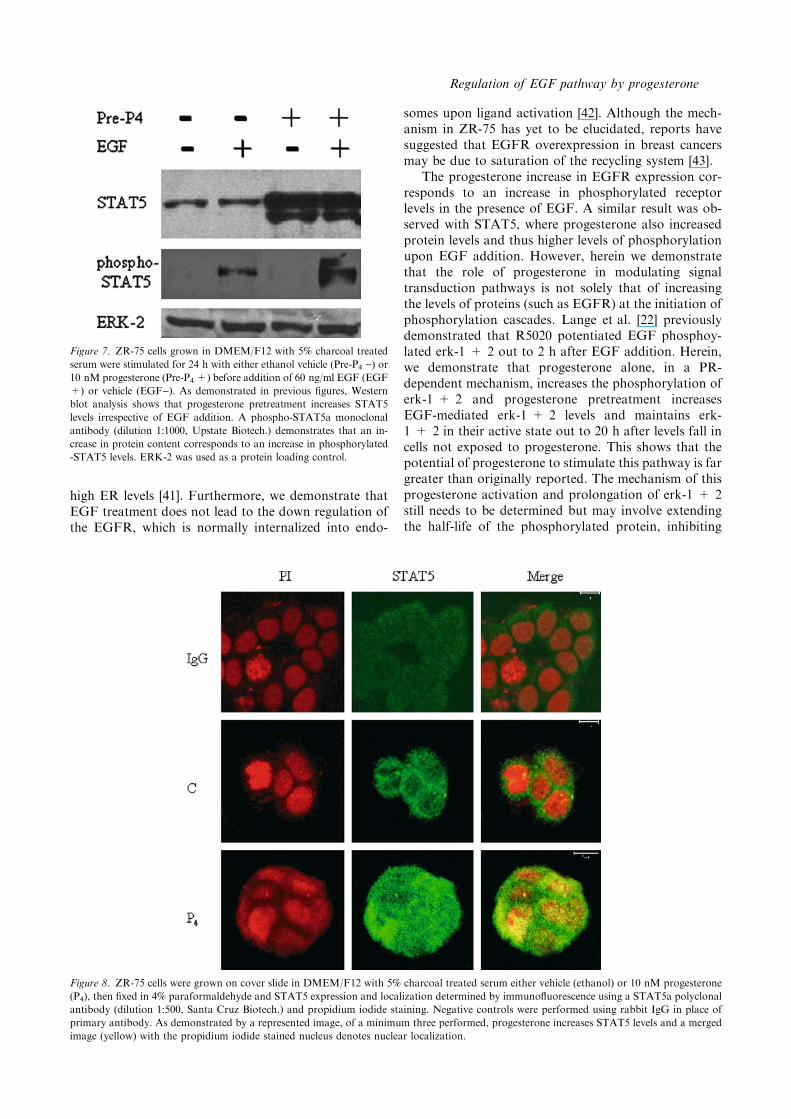
this result suggests that progesterone is causing nuclearlocalization of STAT5 in the absence of STAT5 phos-phorylation (see discussion). To further determine this,we utilized an antibody raised against the phosphory-lated form of STAT5. As shown in Figure 7, proges-terone increases total STAT5 but does not increase theactive phosphorylated form. As anticipated EGF in-creased STAT5 phosphorylation in the presence andabsence of progesterone pretreatment. The difference inlevels of STAT5 phosphorylation between vehicle andprogesterone pretreated extracts corresponds only to thepresence of increased levels of STAT5. As STAT5 nu-clear localization in the absence of phosphorylation wassomewhat unexpected we turned to the technique ofimmunofluorescence microscopy to verify this result.Confirming Western blot results, confocal microscopydemonstrated that under basal conditions, STAT5 islocated mainly in the cytoplasm (Figure 8). Progesterone
treatment increased STAT5 cytoplasmic staining andprominent nuclear localization was evident upon mergingof the propidium iodide stained nucleus with the fluo-rescent STAT5 signal.
Progesterone potentiates EGF-induced c-fos gene activity
Aforementioned results demonstrate that progesteronetreatment sensitizes the EGFR-erk-1 + 2 and STAT5signaling cascade to greater and prolonged activation inthe presence of EGF. To determine if this sensitizationconfers a consequence on gene regulation we selected asa candidate c-fos, a known target of EGF signaling. Thisoncogene and early response protein contains twointeresting sites in the promoter region; a serum-response element (SRE) and a STAT-inducible element(SIE). ZR-75 cells were transiently transfected with a)357/)276 c-fos-TK-luciferase promoter construct. In
Figure 3. Progesterone potentiates EGF-induced ERK-1 + 2 phosphorylation. (a) Experimental protocols for cell culture were as described in
Figure 2. Phosphorylated ERK-1 + 2 levels were determined by Western blotting using a phospho-specific ERK-1 + 2 monoclonal antibody
(1:1000, Cell Signaling). Both progesterone pretreatment and 30 min of EGF increased proteins levels. Total ERK-2 levels were used as a protein
loading control. (b) Representative Western blot demonstrating the time frame of the progesterone and EGF regulation of phospho-ERK-1 + 2
levels. Cells were pre-incubated either ethanol (Pre-EtOH) or 10 nM progesterone (Pre-P4) and harvested at 0, 10 min (10¢), 30 min (30¢), 1, 3, 6,9, and 24 h after EGF (60 ng/ml) treatment. (c) Relative fold increase in phospho-ERK-1 + 2 levels were determined by densitometric analysis
from five independent experiments. At each time point progesterone pretreatment increased phospho-ERK-1 + 2 levels above that of vehicle
pretreated cells. * denotes that phospho-ERK-1 + 2 levels in progesterone pretreated significantly increased in respect to vehicle pretreated cells
in the relative treatments and harvested time points (p<0.05; Mann–Whitney).
A Carvajal et al.

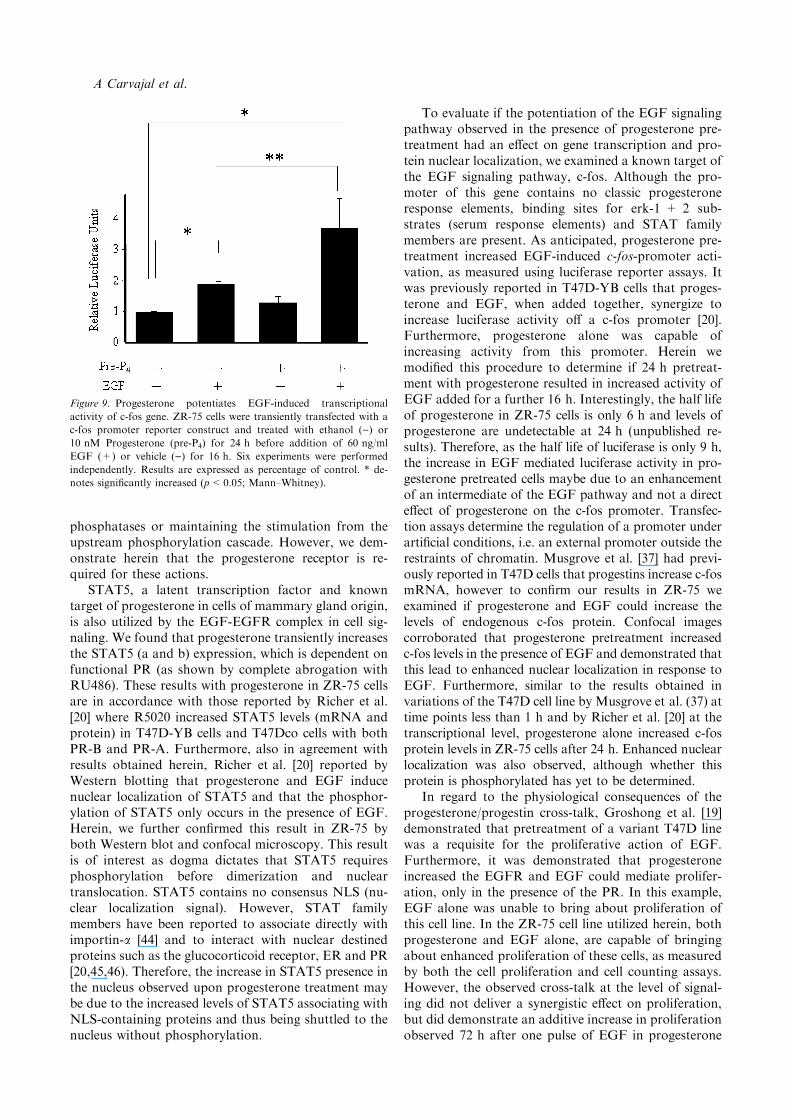
ethanol-pretreated conditions (24 h, as in previousexperiments) we observed basal transcriptional activitythat was significantly increased (1.8 times, p<0.005)after EGF (60 ng/ml) stimulation (Figure 9). Proges-terone pre-treatment for 24 h did not significantlyincrease luciferase activity, however a tendency to in-crease activity was present in most experiments. How-ever, the EGF mediated increase in luciferase activityincreased to 3.7 times over basal levels (p<0.005) incells pretreated with progesterone. The difference inEGF activity between vehicle and progesteronepretreated cells was statistically significant byMann–Whitney analysis (p<0.005). This experimentwas performed a minimum of six times and every pointwas performed in triplicate in each individual assay.
Progesterone potentiates basal and EGF-induced c-fosexpression
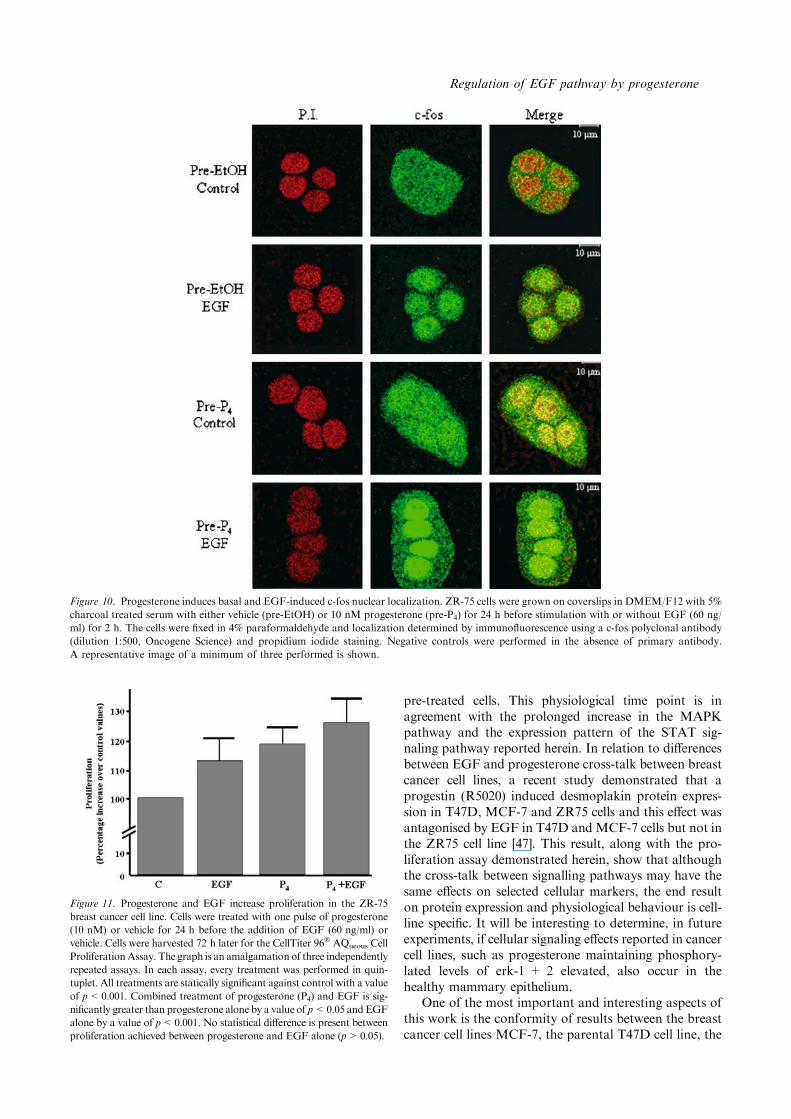
To examine if increased transcriptional activity observedoff the c-fos promoter construct was indicative of nativec-fos protein levels, we performed immunofluorescenceand confocal analysis. Under basal conditions (ethanolpre-treatment), we detected an immunoreactivity toc-fos in the cytoplasmic region that was increased andtranslocated to the nucleus after EGF (60 ng/ml) stim-ulation for 2 h (Figure 10). Surprising, progesteronetreatment for 24 h mediated an increase in cellular c-fos
levels and in co-staining with propidium iodide indi-cating a nuclear localization (Figure 10). Interestingly,by immunofluorescence (Figure 10) we observed an in-crease in c-fos protein by progesterone, despite only atendency to increase the c-fos promoter being previouslyobserved (compare Figures 9 and 10). EGF-stimulationafter progesterone pretreatment produced an intenseand virtually exclusive nuclear localization of c-fosprotein. The elevated levels of c-fos, and the moreexclusive nuclear localization after EGF stimulation inpretreated cells, demonstrates that pre-exposure toprogesterone is altering the EGF signaling cascade inthe breast cancer cell line ZR-75.
Progesterone and EGF mediated effects on cellproliferation
In search of a physiological role for the above reportedcross-talk between the progesterone and EGF signalingpathways we performed cell proliferation assays underthe same treatments conditions used previously. Asdemonstrated in Figure 11, both progesterone andEGF, alone and in combination, increase cell prolifer-ation by this assay (all treatments are statistically sig-nificant against control with a value of p<0.001). Anadditive effect on cell proliferation is observed at 72 hwhen cells are pretreated with progesterone before EGFtreatment. Here, combined progesterone and EGFtreatment is significantly greater than progesteronealone by a value of p<0.05 and EGF alone by a valueof p<0.001. The ability of progesterone and EGF aloneto increase the proliferation of ZR-75 cells was furtherdemonstrated in cell counting assays, where both agentssignificantly increased cell number in respect to vehiclecontrols after 6 and 8 days in culture (results notshown).
Discussion
We have previously hypothesized that progesteroneprimes breast cancer cells for cross-talk with prolifera-tive signals [19–23,39]. Previous publications showedthat in variant T47D cell lines the synthetic progestinR5020 increases STAT5 and EGFR levels. Regulationof the c-fos promoter by EGF was also demonstrated ina clone of T47D which stably expressed the B-form ofthe progesterone receptor. The purpose of this reportwas to (A) expand on these results and examine in closerdetail the regulation of the EGF signaling pathway, (B)to confirm that our previously published results werereproducible in other cells of breast cancer origin whichendogenously express both the ER along with the PRand (C) confirm that cells responded in same manner tothe natural ligand progesterone as to the previouslyutilized synthetic progestins R5020 and MPA. Herein,we characterize for the first time the potentiation ofEGF signal transduction pathway by progesterone inthe ER+/PR+ breast cancer cell line ZR75.
Figure 4. (a) To determine if the PR was required in the progesterone
mediated prolongation in ERK-1 + 2 phosphorylation, cells were
incubated with an agonist of progesterone receptor RU486 (Mifepri-
stone, 100 nM) for 30 min before progesterone addition. The presence
of RU486 abolished progesterone mediated prolongation at 9 h. (b)
Effect of MEK1/2 inhibitor upon ERK-1 + 2 phosphorylation. Cells
were pre-incubated for 24 h in either ethanol (pre-EtOH) or 10 nM
progesterone (pre-P4) and then stimulated with or without EGF
(60 ng/ml) for 0, 10 min (10¢), 9 and 24 h in the presence of vehicle
(DMSO) or MEK1/2 inhibitor (U0126, 2.5 lM). U0126 decreases
EGF increased phosphorylated ERK-1 + 2 levels in progesterone
pretreated cells by 86%, while in ethanol pretreated cells the levels were
not detectable on Western blots. A lower exposure time was used than
that in figure 3B to emphasize the difference between treatments and
thus accounts for the difference in intensity of phosphorylated ERK
observed after 10 min EGF treatment in the panels of 4B.
Regulation of EGF pathway by progesterone
The EGF signaling pathway is involved in breastcancer proliferation, survival and metastasis. We dem-onstrate herein that progesterone increases EGFRprotein in ZR-75 cells, while Murphy et al. [24,40]demonstrated that in the T47D cell line MPA treatmentresulted in a time and dose-dependent increase in EGFRmRNA. Therefore progesterone may also be increasingthe EGFR mRNA by a common mechanism in both celllines and thus do novo synthesis of EGFR. The timecourse of EGF induction in ZR-75, increasing after6–9 h and reaching maximum levels at 24 h, and thedependence on the presence of the PR hints at a genomicaction for progesterone. Interestingly, knowing thatprogesterone is a natural anti-estrogen, a region withinthe first intron of the EGFR gene has been reported tomediate transcriptional repression in the presence of
Figure 6. Progesterone increases STAT5 expression and nuclear
localization. ZR-75 cells were incubated for 24 h in DMEM/F12 with
5% charcoal treated serum either vehicle (ethanol) or 10 nM proges-
terone (P4). Protein extract was separated into cytosolic and membrane
fractions and STAT5 levels determined by Western blot using a
STAT5a polyclonal antibody (dilution 1:1000, Santa Cruz Biotech).
Figure 5. Effect of progesterone and EGF upon STAT5. (a) ZR-75 cells were incubated for 24 h in DMEM/F12 with 5% charcoal treated serum
and stimulated for 24 h in either vehicle (C, ethanol), 10 nM progesterone (P4) or EGF (60 ng/ml). STAT5 expression was determined by Western
blotting using a combination of STAT5a and b polyclonal antibody (see materials and methods). These are representative blots of a minimum
three performed. ERK-2 used as a protein loading control. (b)Relative fold increases in STAT5 levels were determine by densitometric analysis of
Western blots. Error bars represent standard error in a minimum of three independent experiments. * denotes significantly increased in respect to
vehicle treated cells (p<0.05; Mann-Whitney). (c) To determine the time course of the progesterone increase in STAT5 levels cells were harvested
at 0, 6, 9, 12, 24, 32, 48, 72 and 96 h after treatment. STAT5 levels increased after 6 hrs, reached maximum around 24 h, then fell to basal levels at
72 h. (d) To determine if the PR was required in the progesterone mediated increase in STAT5, cells were incubated with an agonist of
progesterone receptor RU486 (Mifepristone, 100 nM) for 30 min before progesterone addition. The presence of RU486 abolished progesterone
regulation of STAT5 at 24 h.
A Carvajal et al.
high ER levels [41]. Furthermore, we demonstrate thatEGF treatment does not lead to the down regulation ofthe EGFR, which is normally internalized into endo-
somes upon ligand activation [42]. Although the mech-anism in ZR-75 has yet to be elucidated, reports havesuggested that EGFR overexpression in breast cancersmay be due to saturation of the recycling system [43].
The progesterone increase in EGFR expression cor-responds to an increase in phosphorylated receptorlevels in the presence of EGF. A similar result was ob-served with STAT5, where progesterone also increasedprotein levels and thus higher levels of phosphorylationupon EGF addition. However, herein we demonstratethat the role of progesterone in modulating signaltransduction pathways is not solely that of increasingthe levels of proteins (such as EGFR) at the initiation ofphosphorylation cascades. Lange et al. [22] previouslydemonstrated that R5020 potentiated EGF phosphoy-lated erk-1 + 2 out to 2 h after EGF addition. Herein,we demonstrate that progesterone alone, in a PR-dependent mechanism, increases the phosphorylation oferk-1 + 2 and progesterone pretreatment increasesEGF-mediated erk-1 + 2 levels and maintains erk-1 + 2 in their active state out to 20 h after levels fall incells not exposed to progesterone. This shows that thepotential of progesterone to stimulate this pathway is fargreater than originally reported. The mechanism of thisprogesterone activation and prolongation of erk-1 + 2still needs to be determined but may involve extendingthe half-life of the phosphorylated protein, inhibiting
Figure 8. ZR-75 cells were grown on cover slide in DMEM/F12 with 5% charcoal treated serum either vehicle (ethanol) or 10 nM progesterone
(P4), then fixed in 4% paraformaldehyde and STAT5 expression and localization determined by immunofluorescence using a STAT5a polyclonal
antibody (dilution 1:500, Santa Cruz Biotech.) and propidium iodide staining. Negative controls were performed using rabbit IgG in place of
primary antibody. As demonstrated by a represented image, of a minimum three performed, progesterone increases STAT5 levels and a merged
image (yellow) with the propidium iodide stained nucleus denotes nuclear localization.
Figure 7. ZR-75 cells grown in DMEM/F12 with 5% charcoal treated
serum were stimulated for 24 h with either ethanol vehicle (Pre-P4 )) or10 nM progesterone (Pre-P4 +) before addition of 60 ng/ml EGF (EGF
+) or vehicle (EGF)). As demonstrated in previous figures, Western
blot analysis shows that progesterone pretreatment increases STAT5
levels irrespective of EGF addition. A phospho-STAT5a monoclonal
antibody (dilution 1:1000, Upstate Biotech.) demonstrates that an in-
crease in protein content corresponds to an increase in phosphorylated
-STAT5 levels. ERK-2 was used as a protein loading control.
Regulation of EGF pathway by progesterone
phosphatases or maintaining the stimulation from theupstream phosphorylation cascade. However, we dem-onstrate herein that the progesterone receptor is re-quired for these actions.
STAT5, a latent transcription factor and knowntarget of progesterone in cells of mammary gland origin,is also utilized by the EGF-EGFR complex in cell sig-naling. We found that progesterone transiently increasesthe STAT5 (a and b) expression, which is dependent onfunctional PR (as shown by complete abrogation withRU486). These results with progesterone in ZR-75 cellsare in accordance with those reported by Richer et al.[20] where R5020 increased STAT5 levels (mRNA andprotein) in T47D-YB cells and T47Dco cells with bothPR-B and PR-A. Furthermore, also in agreement withresults obtained herein, Richer et al. [20] reported byWestern blotting that progesterone and EGF inducenuclear localization of STAT5 and that the phosphor-ylation of STAT5 only occurs in the presence of EGF.Herein, we further confirmed this result in ZR-75 byboth Western blot and confocal microscopy. This resultis of interest as dogma dictates that STAT5 requiresphosphorylation before dimerization and nucleartranslocation. STAT5 contains no consensus NLS (nu-clear localization signal). However, STAT familymembers have been reported to associate directly withimportin-a [44] and to interact with nuclear destinedproteins such as the glucocorticoid receptor, ER and PR[20,45,46). Therefore, the increase in STAT5 presence inthe nucleus observed upon progesterone treatment maybe due to the increased levels of STAT5 associating withNLS-containing proteins and thus being shuttled to thenucleus without phosphorylation.
To evaluate if the potentiation of the EGF signalingpathway observed in the presence of progesterone pre-treatment had an effect on gene transcription and pro-tein nuclear localization, we examined a known target ofthe EGF signaling pathway, c-fos. Although the pro-moter of this gene contains no classic progesteroneresponse elements, binding sites for erk-1 + 2 sub-strates (serum response elements) and STAT familymembers are present. As anticipated, progesterone pre-treatment increased EGF-induced c-fos-promoter acti-vation, as measured using luciferase reporter assays. Itwas previously reported in T47D-YB cells that proges-terone and EGF, when added together, synergize toincrease luciferase activity off a c-fos promoter [20].Furthermore, progesterone alone was capable ofincreasing activity from this promoter. Herein wemodified this procedure to determine if 24 h pretreat-ment with progesterone resulted in increased activity ofEGF added for a further 16 h. Interestingly, the half lifeof progesterone in ZR-75 cells is only 6 h and levels ofprogesterone are undetectable at 24 h (unpublished re-sults). Therefore, as the half life of luciferase is only 9 h,the increase in EGF mediated luciferase activity in pro-gesterone pretreated cells maybe due to an enhancementof an intermediate of the EGF pathway and not a directeffect of progesterone on the c-fos promoter. Transfec-tion assays determine the regulation of a promoter underartificial conditions, i.e. an external promoter outside therestraints of chromatin. Musgrove et al. [37] had previ-ously reported in T47D cells that progestins increase c-fosmRNA, however to confirm our results in ZR-75 weexamined if progesterone and EGF could increase thelevels of endogenous c-fos protein. Confocal imagescorroborated that progesterone pretreatment increasedc-fos levels in the presence of EGF and demonstrated thatthis lead to enhanced nuclear localization in response toEGF. Furthermore, similar to the results obtained invariations of the T47D cell line by Musgrove et al. (37) attime points less than 1 h and by Richer et al. [20] at thetranscriptional level, progesterone alone increased c-fosprotein levels in ZR-75 cells after 24 h. Enhanced nuclearlocalization was also observed, although whether thisprotein is phosphorylated has yet to be determined.
In regard to the physiological consequences of theprogesterone/progestin cross-talk, Groshong et al. [19]demonstrated that pretreatment of a variant T47D linewas a requisite for the proliferative action of EGF.Furthermore, it was demonstrated that progesteroneincreased the EGFR and EGF could mediate prolifer-ation, only in the presence of the PR. In this example,EGF alone was unable to bring about proliferation ofthis cell line. In the ZR-75 cell line utilized herein, bothprogesterone and EGF alone, are capable of bringingabout enhanced proliferation of these cells, as measuredby both the cell proliferation and cell counting assays.However, the observed cross-talk at the level of signal-ing did not deliver a synergistic effect on proliferation,but did demonstrate an additive increase in proliferationobserved 72 h after one pulse of EGF in progesterone
Figure 9. Progesterone potentiates EGF-induced transcriptional
activity of c-fos gene. ZR-75 cells were transiently transfected with a
c-fos promoter reporter construct and treated with ethanol ()) or
10 nM Progesterone (pre-P4) for 24 h before addition of 60 ng/ml
EGF (+) or vehicle ()) for 16 h. Six experiments were performed
independently. Results are expressed as percentage of control. * de-
notes significantly increased (p<0.05; Mann–Whitney).
A Carvajal et al.
pre-treated cells. This physiological time point is inagreement with the prolonged increase in the MAPKpathway and the expression pattern of the STAT sig-naling pathway reported herein. In relation to differencesbetween EGF and progesterone cross-talk between breastcancer cell lines, a recent study demonstrated that aprogestin (R5020) induced desmoplakin protein expres-sion in T47D, MCF-7 and ZR75 cells and this effect wasantagonised by EGF in T47D and MCF-7 cells but not inthe ZR75 cell line [47]. This result, along with the pro-liferation assay demonstrated herein, show that althoughthe cross-talk between signalling pathways may have thesame effects on selected cellular markers, the end resulton protein expression and physiological behaviour is cell-line specific. It will be interesting to determine, in futureexperiments, if cellular signaling effects reported in cancercell lines, such as progesterone maintaining phosphory-lated levels of erk-1 + 2 elevated, also occur in thehealthy mammary epithelium.
One of the most important and interesting aspects ofthis work is the conformity of results between the breastcancer cell lines MCF-7, the parental T47D cell line, the
Figure 11. Progesterone and EGF increase proliferation in the ZR-75
breast cancer cell line. Cells were treated with one pulse of progesterone
(10 nM) or vehicle for 24 h before the addition of EGF (60 ng/ml) or
vehicle. Cells were harvested 72 h later for the CellTiter 96� AQueous Cell
ProliferationAssay. The graph is an amalgamation of three independently
repeated assays. In each assay, every treatment was performed in quin-
tuplet. All treatments are statically significant against control with a value
of p<0.001. Combined treatment of progesterone (P4) and EGF is sig-
nificantly greater than progesterone alone by a value of p<0.05 and EGF
alone by a value of p<0.001. No statistical difference is present between
proliferation achieved between progesterone and EGF alone (p>0.05).
Figure 10. Progesterone induces basal and EGF-induced c-fos nuclear localization. ZR-75 cells were grown on coverslips in DMEM/F12 with 5%
charcoal treated serum with either vehicle (pre-EtOH) or 10 nM progesterone (pre-P4) for 24 h before stimulation with or without EGF (60 ng/
ml) for 2 h. The cells were fixed in 4% paraformaldehyde and localization determined by immunofluorescence using a c-fos polyclonal antibody
(dilution 1:500, Oncogene Science) and propidium iodide staining. Negative controls were performed in the absence of primary antibody.
A representative image of a minimum of three performed is shown.
Regulation of EGF pathway by progesterone

T47Dco and T47DYB clonal lines and the cell line usedherein, ZR-75 in regard to intracellular cross-talkbetween signaling pathways. Furthermore, similar re-sults were observed in the presence of the natural ligandprogesterone as for the synthetic progestins MPA andR5020 in the regulation of the EGFR, the potentiationof erk-1 + 2 phosphorylation and the regulation ofc-fos. These results are encouraging as they demonstratethat breast cell lines are responding in similar mannersto hormonal treatment and that results obtained usingsynthetic progestins can be extrapolated to apply to theaction of the natural ligand, and vice versa.
In summary, we present for the first time, that pro-gesterone potentiates the EGF signaling pathway inZR-75-1 cells. We have determined that it does this byincreasing the total amount of Stat5 and by an increase inunphosphorylated Stat5 in the nucleus and also the nuclearlocalization of c-fos. We further show that progesteronepretreatment maintains EGF-increased erk-1 + 2 levelselevated, most strikingly, after three to nine hours of thegrowth factor treatment. Given the association betweenEGF and breast cancer progression, these results couldexplain, in part, the increased risk of breast cancer asso-ciated with progesterone containing HRT preparations.
Acknowledgments
We gratefully acknowledge Dr Fernando Torrealba(P. Universidad Catolica de Chile) for the kind donationof the anti-c-fos polynoclonal antibody, Dr VictoriaVelarde (P. Universidad Catolica de Chile) for theMEK1/2 inhibitor and Dr Horacio Croxatto (P. Uni-versidad Catolica de Chile) for the PR antagonistMifepristone. Thank you also to the laboratory of DrEnrique Brandan (P. Universidad Catolica de Chile) foruse of their luminometer. For their help and assistancewith the confocal analysis, thanks to Yasna Sanhueza(P. Universidad Catolica de Chile) and Daniel Valdes(Universidad de Santiago de Chile).
References
1. Graham D, Clarke C: Physiological action of progesterone in
target tissues. Endocrine Rev 18: 502–519, 1997
2. Conneely OM, Kettelberger DM, Tsai MJ, Schrader WT,
O’Malley BW: The chicken progesterone receptor A and B iso-
forms are products of an alternate translation initiation event. J
Biol Chem 264: 14062–14064, 1989
3. Bain DL, Franden MA, McManaman JL, Takimoto GS, Horwitz
KB: The N -terminal region of human progesterone B-receptors:
biophysical and biochemical comparison to A-receptors. J Biol
Chem. 276: 23825–23831, 2001
4. Lydon JP, DeMayo FJ, Funk CR, Mani SK, Hughes AR,
Montgomery CA Jr, Shyamala G, Conneely OM, O’Malley BW:
Mice lacking progesterone receptor exhibit pleiotropic reproduc-
tive abnormalities. Genes Dev 9: 2266–2278, 1995
5. Beatson G: On the treatment of inoperable cases of carcinoma of
the mamma: suggestions for a new method of treatment will
illustrative cases. Lancet ii: 104–107, 1896
6. Peerson I, Wliderpass E, Bergkrist L, Bergtrom R, Schairer C:
Risk of breast and endometrial cancer after estradiol and estradiol-
progestine replacement. Cancer Causes Control 10: 253–260, 1999
7. Schairer C, Lubin J, Troisi R, Sturgeon S, Brinton L, Hoover R:
Menopausal estrogen and estrogen-progestin replacement therapy
and breast cancer risk. JAMA 283: 485–491, 2000
8. Chlebowski RT, Hendrix SL, Langer RD, Stefanick ML, Gass M,
Lane D, Rodabough RJ, Gilligan MA, Cyr MG, Thomson CA,
Khandekar J, Petrovitch H, McTiernan AWHI Investigators
Influence of estrogen plus progestin on breast cancer and mam-
mography in healthy postmenopausal women: the Women’s
Health Initiative Randomized Trial. JAMA 289: 3243–3253, 2003
9. Key TJ, Verkasalo PK, Banks E: Epidemiology of breast cancer.
Lancet Oncol 2: 133–140, 2001
10. Catherino W, Jeng M, Jordan V: Norgestrel and gestodene stim-
ulate breast cancer cell growth through an oestrogen receptor
mediated mechanism. Br J Cancer 67: 945–952, 1993
11. Weiler PJ, Wiebe JP: Plasma membrane receptors for the cancer-
regulating progesterone metabolites, 5alpha-pregnane-3,20-dione
and 3alpha-hydroxy-4-pregnen-20-one in MCF-7 breast cancer
cells. Biochem Biophys Res Commun 272: 731–737, 2000
12. Montecchia M, Lamb C, Molinolo A, Luthy I, Pazos P, Charreau
E, Vanzulli S, Lanari C: Progesterone receptor involvement in
independent tumor growth in MPA-induced murine mammary
adenocarcinomas. J Steroid Biochem Mol Biol 68: 11–21, 1999
13. Wiesen J, Young P, Werb G, Cunha G: Signaling through the
stromal epidermal growth factor receptor is necessary for mam-
mary ductal development. Development 126: 335–344, 1999
14. Miettinen PJ, Berger JE, Meneses J, Phung Y, Pedersen RA, Werb
Z, Derynck R: Epithelial immaturity and multiorgan failure in
mice lacking epidermal growth factor receptor. Nature 376: 337–
341, 1995
15. Jorissen R, Walker F, Pouliot N, Garrett T, Ward C, Burgess A:
Epidermal growth factor receptor: mechanisms of activation and
signaling. Exp Cell Res 284: 31–53, 2003
16. Fox SB, Harris AL: The epidermal growth factor receptor in
breast cancer. J Mammary Gland Biol Neoplasia 2: 131–141, 1997
17. Normanno N, Ciardiello F: EGF-related peptides in the patho-
physiology of the mammary gland. J Mammary Gland Biol
Neoplasia 2: 143–151, 1997
18. Navolanic PM, Steelman LS, McCubrey JA: EGFR family sig-
naling and its association with breast cancer development and
resistance to chemotherapy (Review). Int J Oncol 22: 237–252,
2003
19. Groshong S, Owen G, Grimison B, Schauer I, Todd M, Langan T,
Sclafani R, Lange C, Horwitz K: Biphasic regulation of breast
cancer cell growth by progesterone: role of the cyclin-dependent
kinase inhibitors, p21 and p27(Kip1). Mol Endocrinol 11: 1593–
1607, 1997
20. Richer J, Lange N, Manning N, Owen G, Powell R, Horwitz K:
Convergence of progesterone with growth factor and cytokine
signaling in breast cancer. J Biol Chem 273: 31317–31326, 1998
21. Lange CA, Richer JK, Horwitz KB: Hypothesis: progesterone
primes breast cancer cells for cross-talk with proliferative or an-
tiproliferative signals. Mol Endocrinol 13: 829–836, 1999
22. Lange C, Richer J, Shen T, Horwitz K: Convergence of proges-
terone and epidermal growth factor signaling in breast cancer.
J Biol Chem 273: 31308–31316, 1998
23. Lange C: Making sense of cross-talk between steroid hormone
receptors and intracellular signaling pathways: who will have the
last word?. Mol Endocrinol 18: 269–278, 2004
24. Murphy LC, Murphy LJ, Dubik D, Bell GI, Shiu RP: Epidermal
growth factor gene expression in human breast cancer cells: regu-
lation of expression by progestins. Cancer Res 48: 4555–4560, 1998
25. Earp HS, Dawson TL, Li X, Yu H: Heterodimerization and
functional interaction between EGF receptor family members: a
new signalling paradigm with implications for breast cancer re-
search. Breast Cancer Res Treat 35: 115–132, 1995
26. Menard S, Fortis M, Castiglioni F, Agresti R, Balsari A: HER2 as
a prognostic factor in breast cancer. Oncology 61: 67–72, 2001
A Carvajal et al.

27. Santen R, Song R, McPherson R, Kumar R, Adam L, Jeng M,
Yue W: The role of mitogen-activated protein (MAP) kinase in
breast cancer. J Steoid Biochem Mol Biol 80: 239–256, 2002
28. Sartorius CA, Groshong SD, Miller LA, Powell RL, Tung L,
Takimoto GS, Horwitz KB: New T47D breast cancer cell lines for
the independent study of progesterone B- and A-receptors: only
antiprogestin-occupied B-receptors are switched to transcriptional
agonists by cAMP. Cancer Res 54: 3868–3877, 1994
29. Calo V, Migliavacca M, Bazan V, Macaluso M, Buscemi M,
Gebbia N, Russo A: STAT proteins: from normal control of cel-
lular events to tumorigenesis. J Cell Physiol 197: 157–168, 2003
30. Hennighausen L, Robinson GW, Wagner KU, Liu XJ: Develop-
ing a mammary gland is a STAT affair. Mammary Gland Biol
Neoplasia. 2: 365–372, 1997
31. Philp JA, Burdon TG, Watson CJ: Differential activation of
STATs 3 and 5 during mammary gland development. FEBS Lett
396: 77–80, 1996
32. Iavnilovitch E, Groner B, Barash I: Overexpression and forced
activation of STAT5 in mammary gland of transgenic mice pro-
motes cellular proliferation, enhances differentiation, and delays
postlactational apoptosis. Mol Cancer Res 1: 32–47, 2002
33. Richer J, Jacobsen B, Manning N, Abel G, Wolf D, Horwitz K:
Differential gene regulation by the two progesterone receptor
isoforms un human breast cancer cells. J Biol Chem 277: 5209–
5218, 2002
34. Hill CS, Treisman R: Differential activation of c-fos promoter
elements by serum, lysophosphatidic acid, G proteins and poly-
peptide growth factors. EMBO J 14: 5037–5047, 1995
35. Kim DW, Cheriyath V, Roy AL, Cochran BH: TFII-I enhances
activation of the c-fos promoter through interactions withup-
stream elements. Mol Cell Biol 18: 3310–3320, 1998
36. Milde-Langosch K, Roder H, Andritzky B, Aslan B, Hemminger
G, Brinkmann A, Bamberger CM, Loning T, Bamberger AM: The
role of the AP-1 transcription factors c-Fos, FosB, Fra-1 and Fra-
2 in the invasion process of mammary carcinomas. Breast Cancer
Res Treat 86: 139–152, 2004
37. Musgrove EA, Lee CS, Sutherland RL: Progestins both stimulate
and inhibit breast cancer cell cycle progression while increasing
expression of transforming growth factor alpha, epidermal growth
factor receptor, c-fos, and c-myc genes. Mol Cell Biol 11: 5032–
5043, 1991
38. Engel LW, Young NA, Tralka TS, Lippman ME, O’Brien SJ,
Joyce MJ: Establishment and characterization of three new con-
tinuous cell lines derived from human breast carcinomas. Cancer
Res 38: 3352–3364, 1978
39. Owen G, Richer J, Tung L, Takimoto G, Horwitz K: Progesterone
regulates transcription of the p21WAF1 cyclin-dependent kinase
inhibitor gene through Sp1 and CBP/p300. J Biol Chem 273:
10696–10701, 1998
40. Murphy L, Sutherland R, Stead B, Murphy LC, Lazarus L: Pro-
gestin regulation of epidermal growth factor receptor in human
mammary carcinoma cells. Cancer Res 46: 728–734, 1986
41. Wilson MA, Chrysogelos SA: Identification and characterization
of a negative regulatory element within the epidermal growth
factor receptor gene first intron in hormone-dependent breast
cancer cells. J Cell Biochem 85: 601–614, 2002
42. Holbro T, Civenni G, Hynes N: The ErbB receptors and their role
in cancer progression. Exp Cell Res 284: 99–110, 2003
43. Wiley HS: Trafficking of the ErbB receptors and its influence on
signaling. Exp Cell Res 284: 78–88, 2003
44. McBride K, Banninger G, McDonald C, Reich N: Regulated
nuclear import of the STAT1 transcription factor by direct binding
of importin-a. EMBO J 21: 1754–1763, 2002
45. Stocklin E, Wissler M, Gouilleux F, Groner B: Functional inter-
actions between Stat5 and the glucocorticoid receptor. Nature 383:
726–728, 1996
46. Bjornstrom L, Kilic E, Norman M, Parker M, Sjoberg M: Cross-
talk between STAT5b and estrogen receptor-a and -b in mammary
epithelial cells. J Mol Endocrinol 27: 93–106, 2001
47. Pang H, Rowan BG, Al-Dhaheri M, Faber LE: Epidermal growth
factor suppresses induction by progestin of the adhesion protein
desmoplakin in T47D breast cancer cells. Breast Cancer Res 6:
R239–R245, 2004
Address for offprints and correspondence: G.I. Owen, Unidad deReproduccion y Desarrollo, Facultad de Ciencias Biologicas, PontificiaUniversidad Catolica de Chile, Alameda 340, Santiago, Chile; Tel.: 56-2-686-2854; Fax: 56-2-222-5515; E-mail: [email protected]
Regulation of EGF pathway by progesterone












![Syntheses of [21-11C] and (21-13C) progesterone](https://img.pdfslide.net/doc/110x75/634d50fc7c06afa1b60d0a1c/syntheses-of-21-11c-and-21-13c-progesterone.jpg)
















