Embed Size (px)
Citation preview

Protein Traffic Activates NF-kB Gene Signaling and PromotesMCP-1–Dependent Interstitial Inflammation
Roberta Donadelli, BiolSciD, Mauro Abbate, MD, Cristina Zanchi, Chemist,Daniela Corna, Chemist, Susanna Tomasoni, BiolSciD, Ariela Benigni, BiolSciD,
Giuseppe Remuzzi, MD, and Carla Zoja, BiolSciD
● Mononuclear cells accumulate in the renal interstitium and contribute to renal injury in proteinuric nephropa-thies. Angiotensin-converting enzyme (ACE) inhibitors reduce protein trafficking and also lessen renal structuraland functional damage. Many proinflammatory genes, including monocyte chemoattractant protein-1 (MCP-1), achemoattractant for monocytes and T lymphocytes, are transcriptionally regulated by nuclear factor-kappa B(NF-kB). We aimed to study NF-kB activation and MCP-1 expression over time in two models of progressiveproteinuric nephropathies (5/6 nephrectomy and passive Heymann nephritis [PHN]) and evaluate the effect ofantiproteinuric therapy with an ACE inhibitor on these factors. In both models, increased urinary protein excretionover time was associated with a remarkable increase in NF-kB activity, which was almost completely suppressed byreducing proteinuria with lisinopril. NF-kB activation was paralleled by upregulation of MCP-1 messenger RNA andinterstitial accumulation of ED-1–positive monocytes/macrophages and CD8-positive T cells. Lisinopril inhibitedMCP-1 upregulation and limited interstitial inflammation. In a group of PHN rats with advanced disease and severeproteinuria, a dose of lisinopril high enough to inhibit renal ACE activity failed to reduce proteinuria and also did notlimit NF-kB activation, which was sustained over time, along with MCP-1 gene overexpression and interstitialinflammation. These data suggest that NF-kB is activated in the presence of increased protein traffic, enhancing thenuclear transcription of the MCP-1 gene with potent chemotactic and inflammatory properties. This mechanismmay help explain the long-term renal toxicity of filtered proteins.© 2000 by the National Kidney Foundation, Inc.
INDEX WORDS: Proteinuria; nuclear factor kappa B (NF-kB) activation; monocyte chemoattractant protein-1(MCP-1); interstitial inflammation; angiotensin-converting enzyme (ACE) inhibitor.
CHRONIC NEPHROPATHIES with highlyenhanced glomerular permeability to pro-
teins are accompanied by tubulointerstitial inflam-mation and scarring responsible for the loss ofrenal function over time.1,2 The severity of pro-teinuria correlates with that of tubulointerstitialinjury and the rate of declining renal function inboth experimental and human proteinuric renaldiseases,2-7 and evidence in rat models showsthat angiotensin-converting enzyme (ACE) in-hibitors prevent proteinuria and ameliorate renalstructural and functional injury.8-14
Studies have convincingly documented thatexcessive filtration and tubular reabsorption ofproteins as a consequence of increased glomeru-lar permeability lead to the activation of tubular-dependent pathways of interstitial inflammation.Thus, in rats with Adriamycin (Pharmacia andUpjohn, Milan, Italy) nephrosis15 or age-relatedproteinuria,3 accumulation of filtered proteins inthe cytoplasm of proximal tubular cells wasconsistently followed by an interstitial inflamma-tory reaction. Similarly, in rats administered re-peated injections of albumin, excessive amountsof albumin in the tubular filtrate may act topromote macrophage and T-lymphocyte infiltra-tion into renal interstitium and subsequent accu-mulation of extracellular matrix components.16Anumber of proinflammatory molecules are be-lieved to be responsible for cell recruitment,including such chemokines as monocyte chemoat-tractant protein-1 (MCP-1), regulated upon acti-vation normal T-cell expressed and secreted(RANTES), and osteopontin, which are upregu-lated in vivo by protein overload of proximaltubular cells.17-19 The phenotypic character ofproximal tubular cells changes in vitro in re-sponse to protein overloading in such a way that
From the Mario Negri Institute for PharmacologicalResearch; and the Division of Nephrology and Dialysis,Azienda Ospedaliera, Ospedali Riuniti di Bergamo, Ber-gamo, Italy.
Received February 14, 2000; accepted in revised formJuly 14, 2000.
Presented in part at the 31st Annual Meeting of theAmerican Society of Nephrology, Philadelphia, PA, October25-28, 1998.
Address reprint requests to Carla Zoja, BiolSciD, MarioNegri Institute for Pharmacological Research, Via Gavaz-zeni 11, 24125 Bergamo, Italy. E-mail: [email protected]
© 2000 by the National Kidney Foundation, Inc.0272-6386/00/3606-0017$3.00/0doi:10.1053/ajkd.2000.19838
American Journal of Kidney Diseases, Vol 36, No 6 (December), 2000: pp 1226-12411226

genes encoding vasoactive and inflammatory sub-stances are upregulated. Thus, exposure to albu-min, immunoglobulin G (IgG), or transferrincaused concentration-dependent increases in therate of synthesis of endothelin-1.20 Similarly,high concentrations of proteins stimulated thetranscription of the genes for MCP-121 and RAN-TES,22 chemokines that share potent chemotacticproperties for monocytes/macrophages and Tlymphocytes. Enhanced secretion of endothe-lin-1 and chemokines was mainly polarized to-ward the basolateral compartment of the culturedcells.20-22The same events occurring to the sameextent in vivo would mean that excessive amountsof proinflammatory substances generated in re-sponse to protein overload can accumulate in therenal interstitium to initiate local inflammation.
Like many other proinflammatory genes, acti-vation of the MCP-1 and RANTES genes iscontrolled by the transcription factor, nuclearfactor kappa B (NF-kB).23,24NF-kB is composedof homodimeric or heterodimeric complexes ofthe Rel/NF-kB family of proteins, designatedp50, p52, p65, c-Rel, and RelB.25,26NF-kB dimersare present in the cytosol of unstimulated cells inan inactive form bound to the inhibitory protein,IkB.27 Cell activation by such triggers as cyto-kines, viruses, and oxidants leads to proteolyticdegradation of IkB, allowing NF-kB transloca-tion into the nucleus for binding to DNA motifsin gene promoters.25
We recently found that in cultured proximaltubular cells, albumin overloading activatedNF-kB in a dose-dependent manner; this corre-lated with increased production of RANTES.22
That this transcriptional pathway is responsiblefor protein-induced chemokine production is sup-ported by the finding that inhibitors of NF-kBactivation fully prevented the formation of RAN-TES22 and reduced MCP-1 upregulation28 in thissetting.
In the present study, we sought to (1) assessNF-kB activation and MCP-1 messenger RNA(mRNA) expression over time in two models ofprogressive proteinuric nephropathy (5/6 nephrec-tomy and passive Heymann nephritis [PHN]),and (2) evaluate the effects of reducing proteintraffic by means of an ACE inhibitor on NF-kBactivation, MCP-1 expression, and the extent ofinterstitial accumulation of mononuclear cells.
METHODS
Experimental Design
Male Sprague-Dawley rats (Charles River Italia spa, Calco,Italy) with initial body weights of 275 to 320 g were used inthese studies. Animal care and treatment were conducted inconformity with institutional guidelines, which comply withnational (Decreto Legislativo [DL] no. 116, Gazzetta Uffi-ciale [GU], suppl 40, 18 February 1992, Circolare no. 8, GU,14 Luglio 1994) and international laws and policies (Eco-nomic European Community [EEC] Council Directive 86/609, Official Journal L [OJL] 358, December 1987; NationalInstitutes of Health Guide for the Care and Use of Labora-tory Animals, US National Research Council, 1996). Allanimals were housed in a constant-temperature room with a12-hour–dark 12-hour–light cycle and fed a standard diet.Renal mass reduction (RMR) was obtained by right nephrec-tomy and ligation of two or three branches of the left renalartery.29 Four groups of rats (n5 8 in each group) werekilled 7, 30, 60, and 90 days after surgery. Sham-operatedrats killed day 60 were used as controls. To assess the effectsof an ACE inhibitor, two additional groups of rats with RMRwere administered lisinopril (Merck Sharp & Dohme, Rome,Italy), 2 mg/d, in the drinking water30 from day 7 aftersurgery until day 60 or 90 (n5 8 in each group); then theanimals were killed.
PHN was induced in nonanesthetized rats by a singleintravenous injection of 0.4 mL/100 g of body weight ofrabbit anti-Fx1A antibody. Seven days later, when protein-uria was present, animals underwent unilateral nephrectomyto accelerate the onset of renal damage.31 PHN rats werekilled day 7 or month 1, 2, 4, or 8 after disease induction(n 5 4 each time, except for month 8, n5 5); five rats weretreated with lisinopril, 2 mg/d, in the drinking water12 fromday 7 after PHN induction for 8 months. Age-matchedgroups of normal rats (n5 4 in each group) were used ascontrols for each time point. In an additional experiment, agroup of five rats with PHN at an advanced phase of thedisease, ie, 4 months, were administered a high dose oflisinopril (20 mg/d) in the drinking water until month 8; fiveuntreated PHN rats and four normal rats were studied for thesame period.
Twenty-four–hour urine samples were collected in meta-bolic cages before disease induction and at each subsequenttime point to determine urinary protein excretion. Protein-uria was determined by the modified Coomassie blue Gdye-binding assay for proteins, with bovine serum albumin(BSA) as the standard.32 Systolic blood pressure was re-corded by tail plethysmography in conscious rats with RMRat 0, 60, and 90 days and rats with PHN at 0, 4, and 8months.
The rats were anesthetized and killed, and the kidneyswere removed and further processed to determine NF-kBactivity and MCP-1 mRNA expression by Northern blotanalysis, in situ hybridization, and immunohistochemistry.ACE activity was measured in the renal tissue of 8-monthPHN rats untreated or treated with high-dose ACE inhibitorand in the corresponding controls.
ANTIPROTEINURIC THERAPY LIMITS NF-kB SIGNALING 1227

Preparation of Nuclear ExtractsNuclear extracts from whole-kidney tissue were prepared
using a modified method of Negoro et al.33 Frozen renaltissue was homogenized and resuspended in cold extractionbuffer containing 20 mmol/L ofN-2-hydroxyethylpiperazine-propanesulfonic acid (HEPES)-NaOH (pH 7.6), 20% glyc-erol, 0.35 mol/L of NaCl, 5 mmol/L of MgCl2, 0.1 mmol/Lof EDTA, 1 mmol/L of dithiothreitol (DTT), 0.5 mmol/L ofPefabloc (Boehringer Mannheim, Mannheim, Germany), 1mg/mL of pepstatin A, and 2mg/mL of aprotinin. Thehomogenate was shaken and rocked at 4°C for 30 minutes,and insoluble materials were precipitated by centrifugationat 40,000g for 30 minutes at 4°C. The supernatant wasdialyzed overnight against a buffer containing 20 mmol/L ofHEPES-NaOH (pH 7.6), 20% glycerol, 0.1 mol/L of NaCl, 5mmol/L of MgCl2, and 0.1 mmol/L of EDTA. The dialyzedsupernatant was cleared by centrifugation at 10,000g for 15minutes at 4°C. The supernatant collected (nuclear extract)was divided into aliquots and stored at –70°C for subsequentuse. Protein concentrations were determined by the Bradfordassay using the Biorad (Richmond, CA) protein assay re-agent.
Electrophoretic Mobility Shift AnalysisThe kB DNA sequence of the immunoglobulin gene was
used for the electrophoretic mobility shift analysis (EMSA;59-CCGGTCAGAGGGGACTTTCCGAGACT). The corekB sequence is underlined. Probe DNA (with 59 overhangs)was end-labeled by the kleenow enzyme witha-32P deoxycy-tidine triphosphate (dCTP) and separated from unincorpo-rated nucleotides over a G-50 Sephadex column (AmershamPharmacia Biotech, Uppsala, Sweden). Nuclear extracts (20mg) were equilibrated for 10 minutes at room temperature ina binding buffer containing 4% glycerol, 1 mmol/L ofMgCl2,, 0.5 mmol/L of EDTA, 0.5 mmol/L of DTT, 50mmol/L of NaCl, 10 mmol/L of Tris-HCl (pH 7.5), and 50mg/mL of poly (dI-dC). Labeled NF-kB oligonucleotide (50103 count per minute) was added to the binding reactionmixture and incubated for 20 minutes at room temperature.The DNA-protein complexes were electrophoresed on anondenaturing 5% polyacrylamide gel at 120 V for approxi-mately 3 hours to maximize the separation of proteins andachieve some degree of resolution between the migration ofdifferent homodimer and heterodimer complexes that couldbind to the labeled oligonucleotide. Consequently, the freeprobe migrated off the gel and was not present on theautoradiograph replicas. The gel then was dried and sub-jected to autoradiography.
To assay the specificity of the binding reaction, a 100-foldexcess of unlabeled NF-kB probe or an unlabeled nonspe-cific oligonucleotide (tissue plasminogen activator [TPA]regulatory element [TRE])34 was added to the binding reac-tion mixture 10 minutes before the addition of the labeledprobe. For densitometric analysis, an equal-sized box wasdrawn around each band, and the volume density wasdetermined in arbitrary units. The sum of the volume densityof bands for a single sample was used as an indirect measure
of NF-kB activation and expressed as a fold increase of themean NF-kB densitometry of control animals.
For antibody EMSA, the reaction mixture minus theprobe was incubated for 1 hour at room temperature withconcentrated rabbit antisera specific for p65 (sc-109), p50(sc-114), c-Rel (sc-71), RelB (sc-226), or p52 (sc-298; SantaCruz Biotechnology, Santa Cruz, CA). The labeled NF-kBoligonucleotide was then added, and incubation was contin-ued at room temperature for 20 minutes. The optimal anti-body concentrations (1mg for p50; 2 mg for p65, c-Rel,RelB, and p52) and incubation period necessary to super-shift or reduce the intensity of the complexes were deter-mined in preliminary experiments.
Northern Blot AnalysisTotal RNA was isolated from whole-kidney tissue by the
guanidium isothiocyanate/cesium chloride procedure, as pre-viously described.35 Twenty micrograms of total RNA wasthen fractionated on 1.6% agarose gel and blotted ontosynthetic membranes (Zeta-probe; Biorad). Plasmid contain-ing murine JE/MCP-1 probe was provided by Dr Charles D.Stiles (Harvard Medical School and Dana-Faber CancerInstitute, Boston, MA). MCP-1 mRNA was detected byusing 577 bp of an MCP-1 complementary DNA (cDNA).36
The cDNA fragment of MCP-1 was labeled witha-32P dCTPby random-primed method. Hybridization was performedovernight in 0.5 mol/L of Na2HPO4 (pH 7.2) and 7% sodiumdodecyl sulfate (SDS). Filters were washed twice for 30minutes with 40 mmol/L of Na2HPO4 (pH 7.2) and 5% SDSand twice for 10 minutes with 40 mmol/L of Na2HPO4 (pH7.2) and 1% SDS at 65°C. Membranes were subsequentlyprobed with a glyceraldehyde-3-phosphate dehydrogenase(GAPDH) cDNA, taken as the internal standard of equalloading of the samples on the membrane. MCP-1 mRNAoptical density was normalized to that of the constituentlyreleased GAPDH gene expression.
In Situ HybridizationThe murine MCP-1 antisense and sense RNA probes were
prepared and labeled by in vitro transcription using digoxige-nin-labeled uridine triphosphate (Boehringer Mannheim Bio-chemica, Mannheim, Germany).37 A 577-bp murine MCP-1cDNA was cloned into theEcoRI site of the pGEM-1 vectorbetween SP6 and T7 promoters. Fragments of renal cortexwere fixed in Dubosq-Brazil, dehydrated in alcohol, andembedded in paraffin. Sections were cut at 4mm andprocessed as previously described.37 Briefly, after permeabi-lization with proteinase K (40mg/mL; Sigma, St Louis,MO), the sections were hybridized with the RNA probes atthe final concentrations of 0.1 to 0.5 ng/mL in 23 sodiumchloride/sodium citrate (SSC), 10% dextran sulfate, 13Denhardt’s solution, 20 mmol/L of Vanadyl RibonucleosideComplex (Gibco BRL, Life Technologies, Gaithersburg,MD), and 0.1 mol/L of sodium phosphate and incubatedovernight in a moist chamber at 45°C. After being washed in0.23 SSC and blocked with a buffer-blocking solution (50mg/mL of skimmed dried milk, 150 mmol/L of NaCl in 100mmol/L of Tris HCL, pH 7.8) at room temperature for 30
DONADELLI ET AL1228
minutes, the sections were incubated with antidigoxigeninantibody conjugated with alkaline phosphatase (BoehringerMannheim Biochemica) at a dilution of 1:1,000 for 45minutes at 37°C. Colorimetric detection with nitro bluetetrazolium salt and 5-bromo-4-chloro-3-indolyl phosphate(Boehringer Mannheim Biochemica) was then performed,and the sections were mounted in 60% glycerol and exam-ined by light microscopy. The negative control includedhybridization step with the sense probe.
Immunohistochemical AnalysisThe p50 subunit of NF-kB was localized as described by
Morrissey and Klahr38 in kidneys from rats with RMR orsham operation at day 60 (n5 3). The kidneys wereperfused with ice-cold Hank’s balanced salt solution, fol-lowed by Histochoice (Amresco, Solon, OH). Four-micronsections of paraffin-embedded kidneys were deparaffinized,rehydrated, and treated with 3% hydrogen peroxide metha-nol (1:3) for 20 minutes. They were blocked in 5% goatserum and incubated with 20mg/mL of polyclonal rabbitanti-p50 antibody (Santa Cruz Biotechnology), diluted in1% BSA/phosphate-buffered saline (PBS) at 4°C overnight.The sections were washed in PBS and incubated withbiotin-conjugated goat antirabbit IgG (Vector Laboratories,Burlingame, CA) for 1 hour. After washing, they wereincubated with avidin biotin complex (ABC) reagent (VectorLaboratories) for 30 minutes and washed again. Diaminoben-zidine (DAB)-H2O2 solution (Merck, Darmstadt, Germany)was applied to reveal binding sites. The sections werewashed, dehydrated, and mounted using a nonaqueousmounted medium.
Mouse monoclonal antibodies were used for the detectionof ED-1 antigen expressed in rat monocytes and macro-phages (Chemicon, Temecula, CA) and rat CD8 cell-surfaceglycoprotein on T-suppressor cells (OX8; Chemicon). ED-1antigen was stained on paraffin sections using an alkalinephosphatase-Fast Red technique (Boehringer Mannheim), asdescribed.39 CD8 staining wasperformed by indirect immuno-fluorescence technique. Fragments of renal tissues were frozenin liquid nitrogen and cut at 5mm using a Mikrom 500 Ocryostat (Walldorf, Germany). The sections were blocked with1% PBS/BSA, incubated overnight at 4°C with primary anti-body (1:100), washed with PBS, and then incubated withCy3-conjugated donkey antimouse IgG antibodies (5mg/mL inPBS; Jackson ImmunoResearch Laboratories, West Grove, PA)for 1 hour at room temperature. For each marker, positive cellswere counted in at least 10 randomly selected high-power fields(original magnification3400) per each animal.
Measurement of ACE ActivityWhole-kidney tissue was homogenized in distilled water
and centrifuged at 12,000g for 10 minutes at 4°C. Theresulting supernatant was used for ACE activity determina-tion by a spectrophotometric method (Sigma), described byRuiz-Ortega et al.14 ACE activity was expressed as relativeunits per milligram of tissue protein.
Statistical AnalysisResults are expressed as mean6 SE. Data were analyzed
using the nonparametric Kruskal-Wallis test for multiple
comparisons. Statistical significance level is defined asPless than 0.05.
RESULTS
Activation of NF-kB in the Remnant Kidney andEffect of Antiproteinuric Drug Therapy
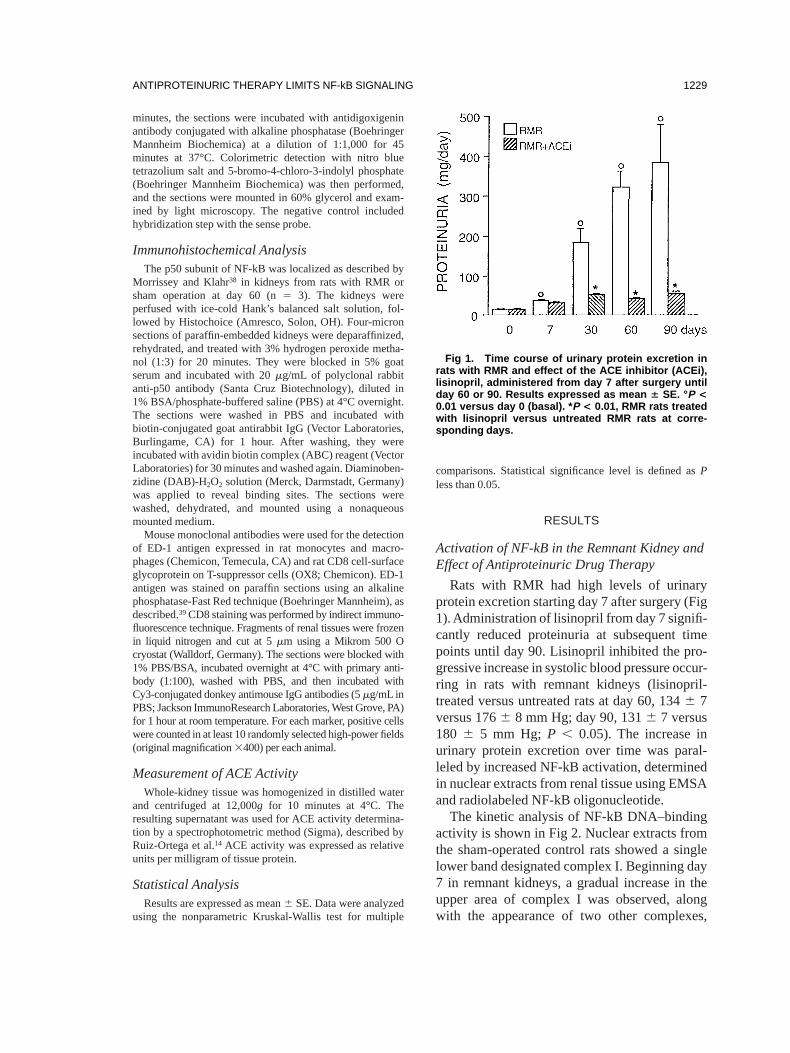
Rats with RMR had high levels of urinaryprotein excretion starting day 7 after surgery (Fig1).Administration of lisinopril from day 7 signifi-cantly reduced proteinuria at subsequent timepoints until day 90. Lisinopril inhibited the pro-gressive increase in systolic blood pressure occur-ring in rats with remnant kidneys (lisinopril-treated versus untreated rats at day 60, 1346 7versus 1766 8 mm Hg; day 90, 1316 7 versus180 6 5 mm Hg; P , 0.05). The increase inurinary protein excretion over time was paral-leled by increased NF-kB activation, determinedin nuclear extracts from renal tissue using EMSAand radiolabeled NF-kB oligonucleotide.
The kinetic analysis of NF-kB DNA–bindingactivity is shown in Fig 2. Nuclear extracts fromthe sham-operated control rats showed a singlelower band designated complex I. Beginning day7 in remnant kidneys, a gradual increase in theupper area of complex I was observed, alongwith the appearance of two other complexes,
Fig 1. Time course of urinary protein excretion inrats with RMR and effect of the ACE inhibitor (ACEi),lisinopril, administered from day 7 after surgery untilday 60 or 90. Results expressed as mean 6 SE. °P <0.01 versus day 0 (basal). * P < 0.01, RMR rats treatedwith lisinopril versus untreated RMR rats at corre-sponding days.
ANTIPROTEINURIC THERAPY LIMITS NF-kB SIGNALING 1229
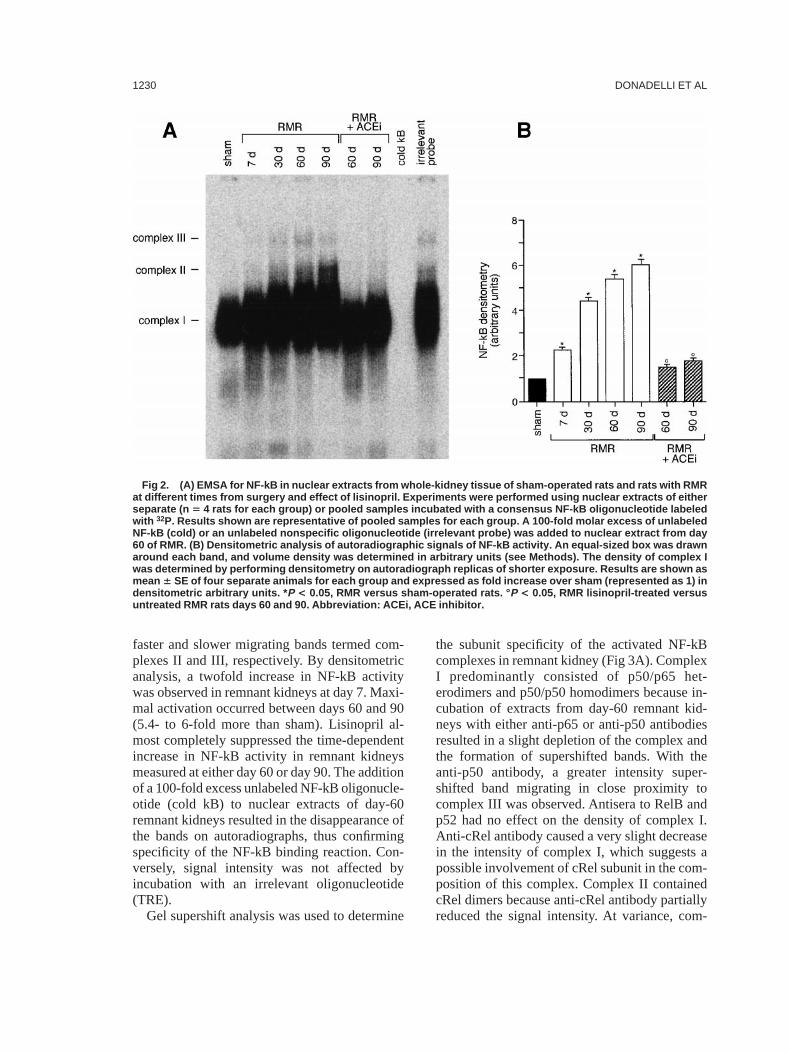
faster and slower migrating bands termed com-plexes II and III, respectively. By densitometricanalysis, a twofold increase in NF-kB activitywas observed in remnant kidneys at day 7. Maxi-mal activation occurred between days 60 and 90(5.4- to 6-fold more than sham). Lisinopril al-most completely suppressed the time-dependentincrease in NF-kB activity in remnant kidneysmeasured at either day 60 or day 90. The additionof a 100-fold excess unlabeled NF-kB oligonucle-otide (cold kB) to nuclear extracts of day-60remnant kidneys resulted in the disappearance ofthe bands on autoradiographs, thus confirmingspecificity of the NF-kB binding reaction. Con-versely, signal intensity was not affected byincubation with an irrelevant oligonucleotide(TRE).
Gel supershift analysis was used to determine
the subunit specificity of the activated NF-kBcomplexes in remnant kidney (Fig 3A). ComplexI predominantly consisted of p50/p65 het-erodimers and p50/p50 homodimers because in-cubation of extracts from day-60 remnant kid-neys with either anti-p65 or anti-p50 antibodiesresulted in a slight depletion of the complex andthe formation of supershifted bands. With theanti-p50 antibody, a greater intensity super-shifted band migrating in close proximity tocomplex III was observed. Antisera to RelB andp52 had no effect on the density of complex I.Anti-cRel antibody caused a very slight decreasein the intensity of complex I, which suggests apossible involvement of cRel subunit in the com-position of this complex. Complex II containedcRel dimers because anti-cRel antibody partiallyreduced the signal intensity. At variance, com-
Fig 2. (A) EMSA for NF-kB in nuclear extracts from whole-kidney tissue of sham-operated rats and rats with RMRat different times from surgery and effect of lisinopril. Experiments were performed using nuclear extracts of eitherseparate (n 5 4 rats for each group) or pooled samples incubated with a consensus NF-kB oligonucleotide labeledwith 32P. Results shown are representative of pooled samples for each group. A 100-fold molar excess of unlabeledNF-kB (cold) or an unlabeled nonspecific oligonucleotide (irrelevant probe) was added to nuclear extract from day60 of RMR. (B) Densitometric analysis of autoradiographic signals of NF-kB activity. An equal-sized box was drawnaround each band, and volume density was determined in arbitrary units (see Methods). The density of complex Iwas determined by performing densitometry on autoradiograph replicas of shorter exposure. Results are shown asmean 6 SE of four separate animals for each group and expressed as fold increase over sham (represented as 1) indensitometric arbitrary units. * P < 0.05, RMR versus sham-operated rats. ° P < 0.05, RMR lisinopril-treated versusuntreated RMR rats days 60 and 90. Abbreviation: ACEi, ACE inhibitor.
DONADELLI ET AL1230
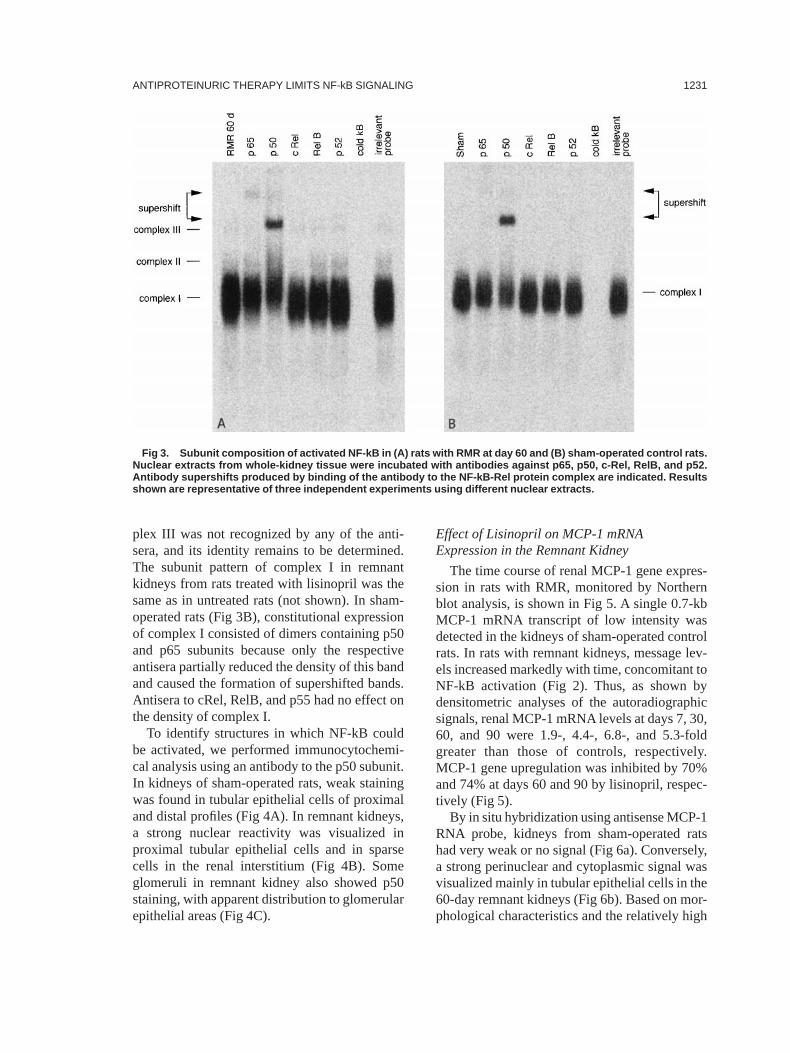
plex III was not recognized by any of the anti-sera, and its identity remains to be determined.The subunit pattern of complex I in remnantkidneys from rats treated with lisinopril was thesame as in untreated rats (not shown). In sham-operated rats (Fig 3B), constitutional expressionof complex I consisted of dimers containing p50and p65 subunits because only the respectiveantisera partially reduced the density of this bandand caused the formation of supershifted bands.Antisera to cRel, RelB, and p55 had no effect onthe density of complex I.
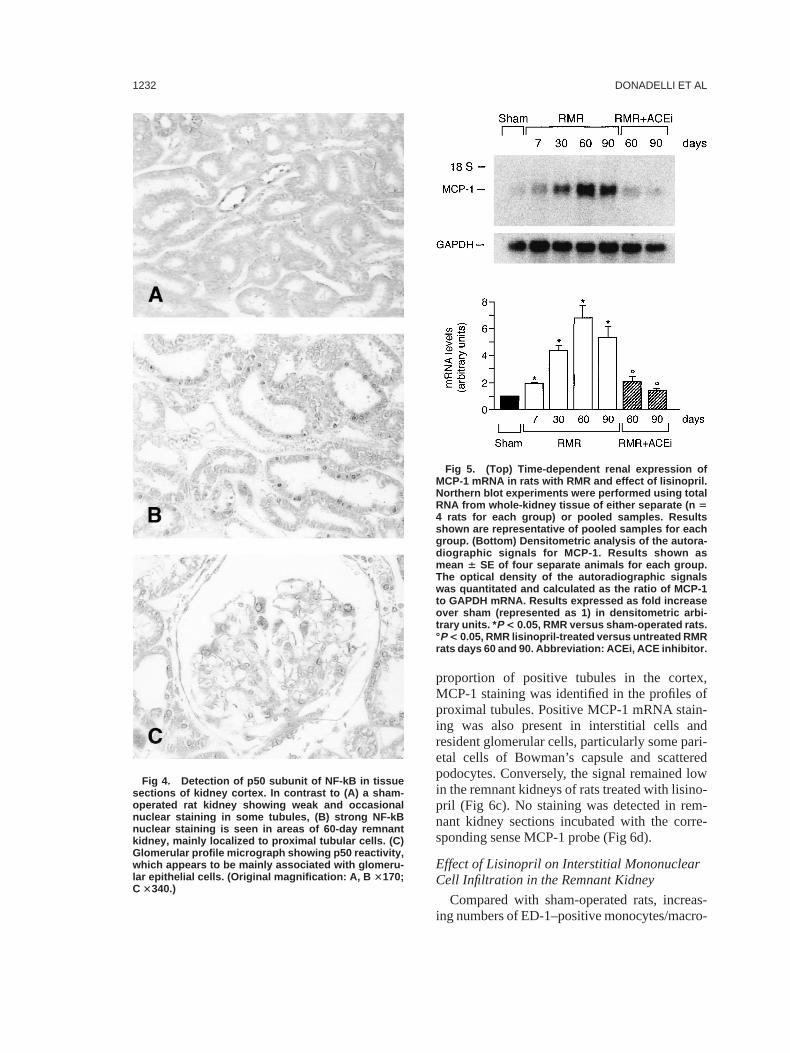
To identify structures in which NF-kB couldbe activated, we performed immunocytochemi-cal analysis using an antibody to the p50 subunit.In kidneys of sham-operated rats, weak stainingwas found in tubular epithelial cells of proximaland distal profiles (Fig 4A). In remnant kidneys,a strong nuclear reactivity was visualized inproximal tubular epithelial cells and in sparsecells in the renal interstitium (Fig 4B). Someglomeruli in remnant kidney also showed p50staining, with apparent distribution to glomerularepithelial areas (Fig 4C).
Effect of Lisinopril on MCP-1 mRNAExpression in the Remnant Kidney
The time course of renal MCP-1 gene expres-sion in rats with RMR, monitored by Northernblot analysis, is shown in Fig 5. A single 0.7-kbMCP-1 mRNA transcript of low intensity wasdetected in the kidneys of sham-operated controlrats. In rats with remnant kidneys, message lev-els increased markedly with time, concomitant toNF-kB activation (Fig 2). Thus, as shown bydensitometric analyses of the autoradiographicsignals, renal MCP-1 mRNA levels at days 7, 30,60, and 90 were 1.9-, 4.4-, 6.8-, and 5.3-foldgreater than those of controls, respectively.MCP-1 gene upregulation was inhibited by 70%and 74% at days 60 and 90 by lisinopril, respec-tively (Fig 5).
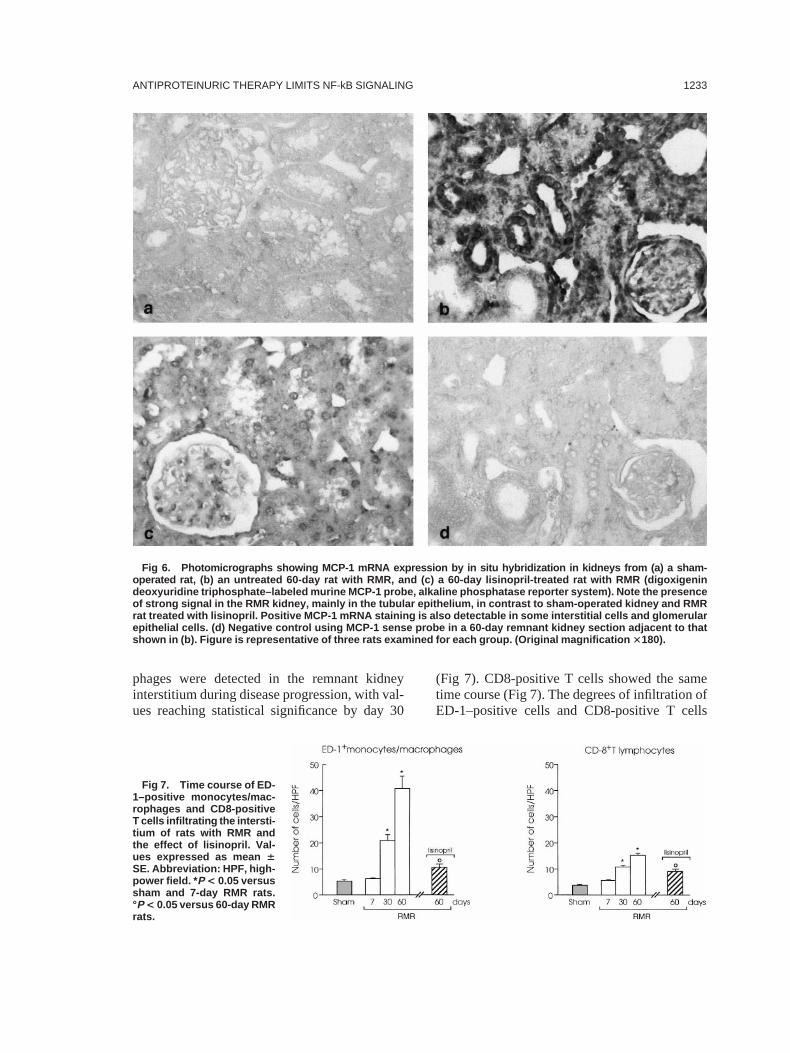
By in situ hybridization using antisense MCP-1RNA probe, kidneys from sham-operated ratshad very weak or no signal (Fig 6a). Conversely,a strong perinuclear and cytoplasmic signal wasvisualized mainly in tubular epithelial cells in the60-day remnant kidneys (Fig 6b). Based on mor-phological characteristics and the relatively high
Fig 3. Subunit composition of activated NF-kB in (A) rats with RMR at day 60 and (B) sham-operated control rats.Nuclear extracts from whole-kidney tissue were incubated with antibodies against p65, p50, c-Rel, RelB, and p52.Antibody supershifts produced by binding of the antibody to the NF-kB-Rel protein complex are indicated. Resultsshown are representative of three independent experiments using different nuclear extracts.
ANTIPROTEINURIC THERAPY LIMITS NF-kB SIGNALING 1231
proportion of positive tubules in the cortex,MCP-1 staining was identified in the profiles ofproximal tubules. Positive MCP-1 mRNA stain-ing was also present in interstitial cells andresident glomerular cells, particularly some pari-etal cells of Bowman’s capsule and scatteredpodocytes. Conversely, the signal remained lowin the remnant kidneys of rats treated with lisino-pril (Fig 6c). No staining was detected in rem-nant kidney sections incubated with the corre-sponding sense MCP-1 probe (Fig 6d).
Effect of Lisinopril on Interstitial MononuclearCell Infiltration in the Remnant Kidney
Compared with sham-operated rats, increas-ing numbers of ED-1–positive monocytes/macro-
Fig 4. Detection of p50 subunit of NF-kB in tissuesections of kidney cortex. In contrast to (A) a sham-operated rat kidney showing weak and occasionalnuclear staining in some tubules, (B) strong NF-kBnuclear staining is seen in areas of 60-day remnantkidney, mainly localized to proximal tubular cells. (C)Glomerular profile micrograph showing p50 reactivity,which appears to be mainly associated with glomeru-lar epithelial cells. (Original magnification: A, B 3170;C 3340.)
Fig 5. (Top) Time-dependent renal expression ofMCP-1 mRNA in rats with RMR and effect of lisinopril.Northern blot experiments were performed using totalRNA from whole-kidney tissue of either separate (n 54 rats for each group) or pooled samples. Resultsshown are representative of pooled samples for eachgroup. (Bottom) Densitometric analysis of the autora-diographic signals for MCP-1. Results shown asmean 6 SE of four separate animals for each group.The optical density of the autoradiographic signalswas quantitated and calculated as the ratio of MCP-1to GAPDH mRNA. Results expressed as fold increaseover sham (represented as 1) in densitometric arbi-trary units. * P < 0.05, RMR versus sham-operated rats.°P < 0.05, RMR lisinopril-treated versus untreated RMRrats days 60 and 90. Abbreviation: ACEi, ACE inhibitor.
DONADELLI ET AL1232
phages were detected in the remnant kidneyinterstitium during disease progression, with val-ues reaching statistical significance by day 30
(Fig 7). CD8-positive T cells showed the sametime course (Fig 7). The degrees of infiltration ofED-1–positive cells and CD8-positive T cells
Fig 7. Time course of ED-1–positive monocytes/mac-rophages and CD8-positiveT cells infiltrating the intersti-tium of rats with RMR andthe effect of lisinopril. Val-ues expressed as mean 6SE. Abbreviation: HPF, high-power field. * P < 0.05 versussham and 7-day RMR rats.°P < 0.05 versus 60-day RMRrats.
Fig 6. Photomicrographs showing MCP-1 mRNA expression by in situ hybridization in kidneys from (a) a sham-operated rat, (b) an untreated 60-day rat with RMR, and (c) a 60-day lisinopril-treated rat with RMR (digoxigenindeoxyuridine triphosphate–labeled murine MCP-1 probe, alkaline phosphatase reporter system). Note the presenceof strong signal in the RMR kidney, mainly in the tubular epithelium, in contrast to sham-operated kidney and RMRrat treated with lisinopril. Positive MCP-1 mRNA staining is also detectable in some interstitial cells and glomerularepithelial cells. (d) Negative control using MCP-1 sense probe in a 60-day remnant kidney section adjacent to thatshown in (b). Figure is representative of three rats examined for each group. (Original magnification 3180).
ANTIPROTEINURIC THERAPY LIMITS NF-kB SIGNALING 1233
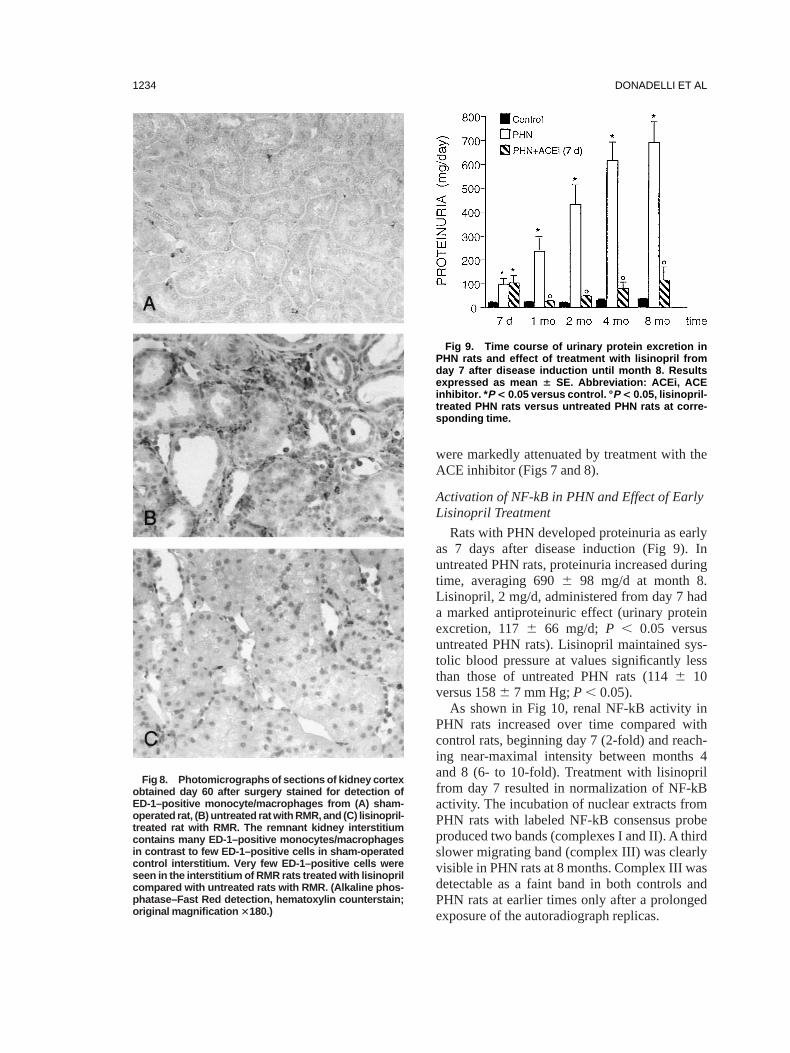
were markedly attenuated by treatment with theACE inhibitor (Figs 7 and 8).
Activation of NF-kB in PHN and Effect of EarlyLisinopril Treatment
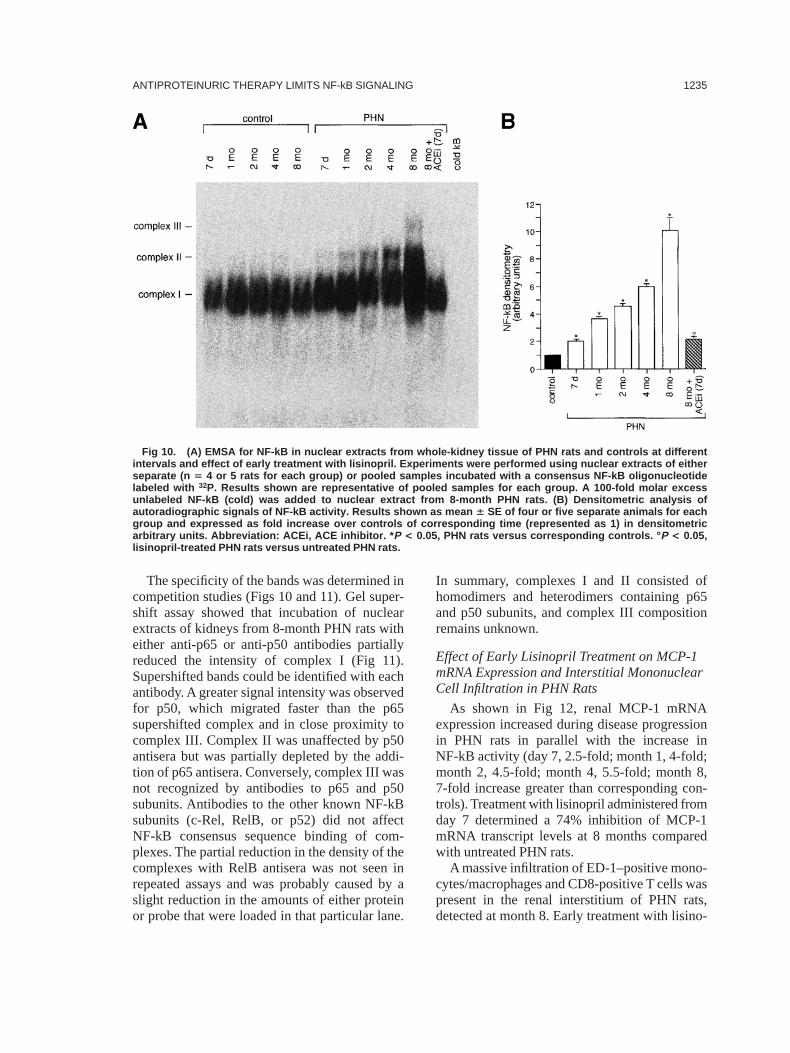
Rats with PHN developed proteinuria as earlyas 7 days after disease induction (Fig 9). Inuntreated PHN rats, proteinuria increased duringtime, averaging 6906 98 mg/d at month 8.Lisinopril, 2 mg/d, administered from day 7 hada marked antiproteinuric effect (urinary proteinexcretion, 1176 66 mg/d; P , 0.05 versusuntreated PHN rats). Lisinopril maintained sys-tolic blood pressure at values significantly lessthan those of untreated PHN rats (1146 10versus 1586 7 mm Hg;P , 0.05).
As shown in Fig 10, renal NF-kB activity inPHN rats increased over time compared withcontrol rats, beginning day 7 (2-fold) and reach-ing near-maximal intensity between months 4and 8 (6- to 10-fold). Treatment with lisinoprilfrom day 7 resulted in normalization of NF-kBactivity. The incubation of nuclear extracts fromPHN rats with labeled NF-kB consensus probeproduced two bands (complexes I and II). A thirdslower migrating band (complex III) was clearlyvisible in PHN rats at 8 months. Complex III wasdetectable as a faint band in both controls andPHN rats at earlier times only after a prolongedexposure of the autoradiograph replicas.
Fig 8. Photomicrographs of sections of kidney cortexobtained day 60 after surgery stained for detection ofED-1–positive monocyte/macrophages from (A) sham-operated rat, (B) untreated rat with RMR, and (C) lisinopril-treated rat with RMR. The remnant kidney interstitiumcontains many ED-1–positive monocytes/macrophagesin contrast to few ED-1–positive cells in sham-operatedcontrol interstitium. Very few ED-1–positive cells wereseen in the interstitium of RMR rats treated with lisinoprilcompared with untreated rats with RMR. (Alkaline phos-phatase–Fast Red detection, hematoxylin counterstain;original magnification 3180.)
Fig 9. Time course of urinary protein excretion inPHN rats and effect of treatment with lisinopril fromday 7 after disease induction until month 8. Resultsexpressed as mean 6 SE. Abbreviation: ACEi, ACEinhibitor. * P < 0.05 versus control. ° P < 0.05, lisinopril-treated PHN rats versus untreated PHN rats at corre-sponding time.
DONADELLI ET AL1234
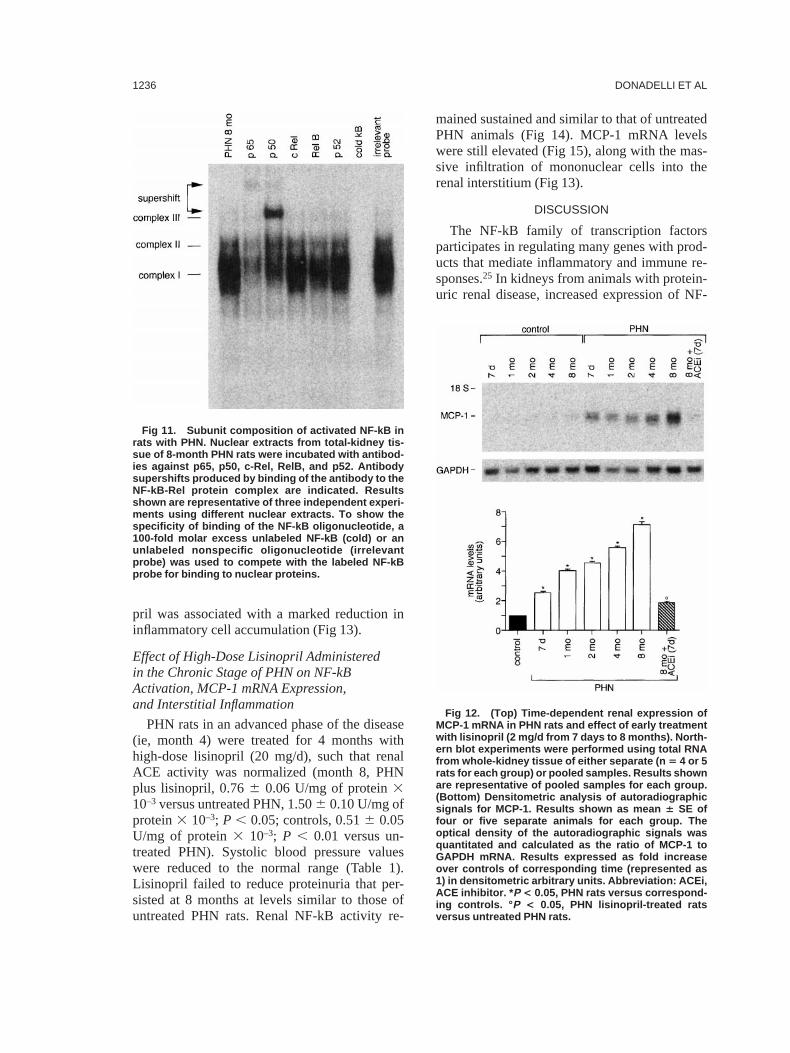
The specificity of the bands was determined incompetition studies (Figs 10 and 11). Gel super-shift assay showed that incubation of nuclearextracts of kidneys from 8-month PHN rats witheither anti-p65 or anti-p50 antibodies partiallyreduced the intensity of complex I (Fig 11).Supershifted bands could be identified with eachantibody. A greater signal intensity was observedfor p50, which migrated faster than the p65supershifted complex and in close proximity tocomplex III. Complex II was unaffected by p50antisera but was partially depleted by the addi-tion of p65 antisera. Conversely, complex III wasnot recognized by antibodies to p65 and p50subunits. Antibodies to the other known NF-kBsubunits (c-Rel, RelB, or p52) did not affectNF-kB consensus sequence binding of com-plexes. The partial reduction in the density of thecomplexes with RelB antisera was not seen inrepeated assays and was probably caused by aslight reduction in the amounts of either proteinor probe that were loaded in that particular lane.
In summary, complexes I and II consisted ofhomodimers and heterodimers containing p65and p50 subunits, and complex III compositionremains unknown.
Effect of Early Lisinopril Treatment on MCP-1mRNA Expression and Interstitial MononuclearCell Infiltration in PHN Rats
As shown in Fig 12, renal MCP-1 mRNAexpression increased during disease progressionin PHN rats in parallel with the increase inNF-kB activity (day 7, 2.5-fold; month 1, 4-fold;month 2, 4.5-fold; month 4, 5.5-fold; month 8,7-fold increase greater than corresponding con-trols). Treatment with lisinopril administered fromday 7 determined a 74% inhibition of MCP-1mRNA transcript levels at 8 months comparedwith untreated PHN rats.
A massive infiltration of ED-1–positive mono-cytes/macrophages and CD8-positive T cells waspresent in the renal interstitium of PHN rats,detected at month 8. Early treatment with lisino-
Fig 10. (A) EMSA for NF-kB in nuclear extracts from whole-kidney tissue of PHN rats and controls at differentintervals and effect of early treatment with lisinopril. Experiments were performed using nuclear extracts of eitherseparate (n 5 4 or 5 rats for each group) or pooled samples incubated with a consensus NF-kB oligonucleotidelabeled with 32P. Results shown are representative of pooled samples for each group. A 100-fold molar excessunlabeled NF-kB (cold) was added to nuclear extract from 8-month PHN rats. (B) Densitometric analysis ofautoradiographic signals of NF-kB activity. Results shown as mean 6 SE of four or five separate animals for eachgroup and expressed as fold increase over controls of corresponding time (represented as 1) in densitometricarbitrary units. Abbreviation: ACEi, ACE inhibitor. * P < 0.05, PHN rats versus corresponding controls. ° P < 0.05,lisinopril-treated PHN rats versus untreated PHN rats.
ANTIPROTEINURIC THERAPY LIMITS NF-kB SIGNALING 1235
pril was associated with a marked reduction ininflammatory cell accumulation (Fig 13).
Effect of High-Dose Lisinopril Administeredin the Chronic Stage of PHN on NF-kBActivation, MCP-1 mRNA Expression,and Interstitial Inflammation
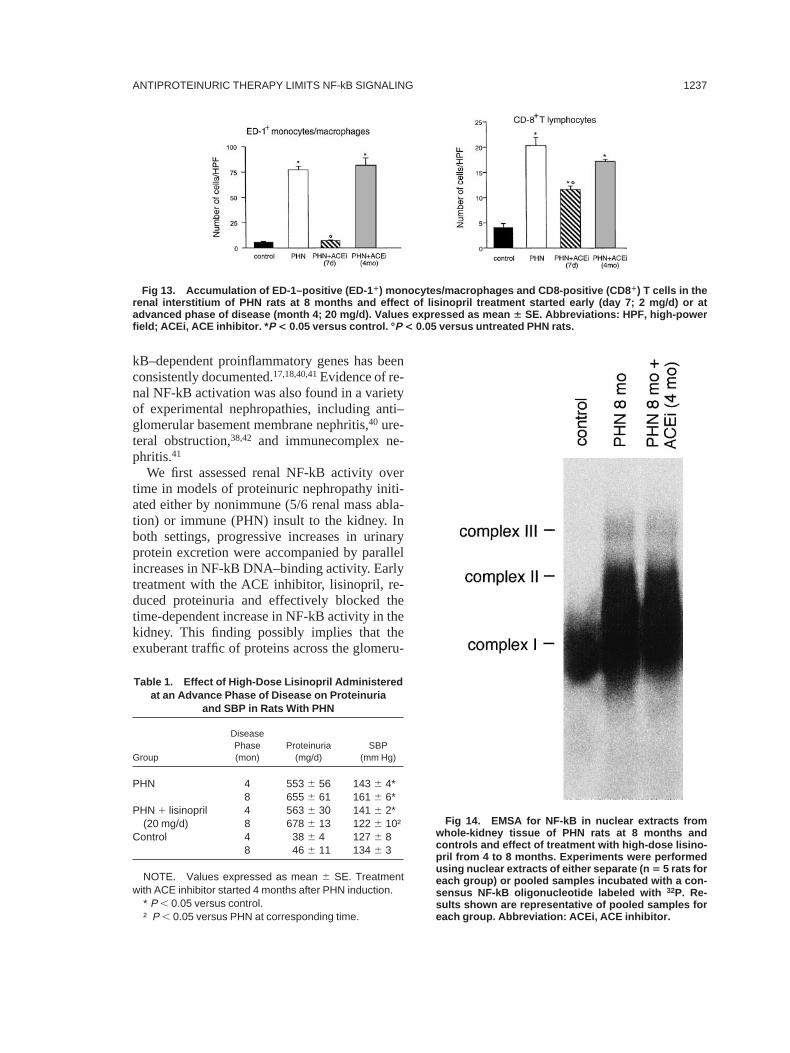
PHN rats in an advanced phase of the disease(ie, month 4) were treated for 4 months withhigh-dose lisinopril (20 mg/d), such that renalACE activity was normalized (month 8, PHNplus lisinopril, 0.766 0.06 U/mg of protein310–3 versus untreated PHN, 1.506 0.10 U/mg ofprotein3 10–3; P , 0.05; controls, 0.516 0.05U/mg of protein3 10–3; P , 0.01 versus un-treated PHN). Systolic blood pressure valueswere reduced to the normal range (Table 1).Lisinopril failed to reduce proteinuria that per-sisted at 8 months at levels similar to those ofuntreated PHN rats. Renal NF-kB activity re-
mained sustained and similar to that of untreatedPHN animals (Fig 14). MCP-1 mRNA levelswere still elevated (Fig 15), along with the mas-sive infiltration of mononuclear cells into therenal interstitium (Fig 13).
DISCUSSION
The NF-kB family of transcription factorsparticipates in regulating many genes with prod-ucts that mediate inflammatory and immune re-sponses.25 In kidneys from animals with protein-uric renal disease, increased expression of NF-
Fig 11. Subunit composition of activated NF-kB inrats with PHN. Nuclear extracts from total-kidney tis-sue of 8-month PHN rats were incubated with antibod-ies against p65, p50, c-Rel, RelB, and p52. Antibodysupershifts produced by binding of the antibody to theNF-kB-Rel protein complex are indicated. Resultsshown are representative of three independent experi-ments using different nuclear extracts. To show thespecificity of binding of the NF-kB oligonucleotide, a100-fold molar excess unlabeled NF-kB (cold) or anunlabeled nonspecific oligonucleotide (irrelevantprobe) was used to compete with the labeled NF-kBprobe for binding to nuclear proteins.
Fig 12. (Top) Time-dependent renal expression ofMCP-1 mRNA in PHN rats and effect of early treatmentwith lisinopril (2 mg/d from 7 days to 8 months). North-ern blot experiments were performed using total RNAfrom whole-kidney tissue of either separate (n 5 4 or 5rats for each group) or pooled samples. Results shownare representative of pooled samples for each group.(Bottom) Densitometric analysis of autoradiographicsignals for MCP-1. Results shown as mean 6 SE offour or five separate animals for each group. Theoptical density of the autoradiographic signals wasquantitated and calculated as the ratio of MCP-1 toGAPDH mRNA. Results expressed as fold increaseover controls of corresponding time (represented as1) in densitometric arbitrary units. Abbreviation: ACEi,ACE inhibitor. * P < 0.05, PHN rats versus correspond-ing controls. ° P < 0.05, PHN lisinopril-treated ratsversus untreated PHN rats.
DONADELLI ET AL1236
kB–dependent proinflammatory genes has beenconsistently documented.17,18,40,41Evidence of re-nal NF-kB activation was also found in a varietyof experimental nephropathies, including anti–glomerular basement membrane nephritis,40 ure-teral obstruction,38,42 and immunecomplex ne-phritis.41
We first assessed renal NF-kB activity overtime in models of proteinuric nephropathy initi-ated either by nonimmune (5/6 renal mass abla-tion) or immune (PHN) insult to the kidney. Inboth settings, progressive increases in urinaryprotein excretion were accompanied by parallelincreases in NF-kB DNA–binding activity. Earlytreatment with the ACE inhibitor, lisinopril, re-duced proteinuria and effectively blocked thetime-dependent increase in NF-kB activity in thekidney. This finding possibly implies that theexuberant traffic of proteins across the glomeru-
Fig 13. Accumulation of ED-1–positive (ED-1 1) monocytes/macrophages and CD8-positive (CD8 1) T cells in therenal interstitium of PHN rats at 8 months and effect of lisinopril treatment started early (day 7; 2 mg/d) or atadvanced phase of disease (month 4; 20 mg/d). Values expressed as mean 6 SE. Abbreviations: HPF, high-powerfield; ACEi, ACE inhibitor. * P < 0.05 versus control. ° P < 0.05 versus untreated PHN rats.
Table 1. Effect of High-Dose Lisinopril Administeredat an Advance Phase of Disease on Proteinuria
and SBP in Rats With PHN
Group
DiseasePhase(mon)
Proteinuria(mg/d)
SBP(mm Hg)
PHN 4 553 6 56 143 6 4*8 655 6 61 161 6 6*
PHN 1 lisinopril(20 mg/d)
4 563 6 30 141 6 2*8 678 6 13 122 6 10†
Control 4 38 6 4 127 6 88 46 6 11 134 6 3
NOTE. Values expressed as mean 6 SE. Treatmentwith ACE inhibitor started 4 months after PHN induction.
* P , 0.05 versus control.† P , 0.05 versus PHN at corresponding time.
Fig 14. EMSA for NF-kB in nuclear extracts fromwhole-kidney tissue of PHN rats at 8 months andcontrols and effect of treatment with high-dose lisino-pril from 4 to 8 months. Experiments were performedusing nuclear extracts of either separate (n 5 5 rats foreach group) or pooled samples incubated with a con-sensus NF-kB oligonucleotide labeled with 32P. Re-sults shown are representative of pooled samples foreach group. Abbreviation: ACEi, ACE inhibitor.
ANTIPROTEINURIC THERAPY LIMITS NF-kB SIGNALING 1237

lar barrier acts as a determinant of NF-kB activa-tion. That this transcriptional pathway can bedirectly activated by proteins abnormally filteredand reabsorbed in proximal tubular cells is sup-ported by in vitro evidence that cultured proxi-mal tubular cells exposed to high concentrationsof albumin showed a dose-dependent increase inNF-kB activity with augmented RANTES22 andMCP-128 synthesis.
Because distinct NF-kB/Rel complexes havedifferent sequence preferences and transcrip-tional activation properties,43 we characterizedthe pattern of proteins binding to the NF-kBmotif in kidney of rats with the previously men-tioned forms of renal injury. EMSA-supershiftexperiments showed that in both the renal massablation and PHN models, the activated NF-kBcomplexes contained p50 and p65 subunits. Inaddition, involvement of the cRel subunit wasobserved for remnant kidney. The finding thatthe slower migrating band (complex III) innuclear extracts from both models was unaf-fected by antibodies to the known NF-kB sub-units leads us to speculate that this complex maycontain novel NF-kB proteins or interact withother proteins44 that mask antibody binding.
Next, by immunocytochemical analysis per-formed with an antibody to the p50 subunit, wefound that numerous nuclei of proximal tubular
cells were strongly positive for p50 in remnantkidneys in contrast to only occasional tubularp50-positive nuclei in the control cortex. To amuch lesser extent, cells in the interstitium, in alllikelihood infiltrating inflammatory cells, andglomerular cells positively stained for p50 andthus may partially account for the increasedtranscription factor activity measured in the rem-nant kidney.
Another finding of the present study is thatconcomitant to the activation of NF-kB, a pro-gressive increase in MCP-1 gene expression oc-curred with time in the kidney of rats with RMRbeginning day 7. Intense MCP-1 mRNA signalswere detected in tubular epithelial cells and, to alesser extent, in interstitial infiltrating cells, docu-mented by in situ hybridization experiments.MCP-1 mRNA upregulation preceded the accu-mulation of ED-1–positive monocytes/macro-phages and CD8-positive T lymphocytes in theremnant kidney interstitium, suggesting that inthis model, initial mononuclear cell recruitmentmay occur at least in part by MCP-1–dependentmechanisms. As in remnant kidneys, the progres-sive increase in NF-kB activation in kidneys ofPHN rats was paralleled by increased MCP-1gene transcription and interstitial mononuclearcell infiltration. In other models of proteinuricnephropathies, renal MCP-1 upregulation hasbeen shown to precede or coincide with infiltra-tion of mononuclear cells into the renal intersti-tium.17,45 That MCP-1 is functionally importantin eliciting an interstitial inflammatory responsecan be derived from the evidence that administer-ing a neutralizing anti-MCP-1 antibody to ratswith tubulointerstitial nephritis significantly de-creased macrophage infiltration.45
If the interstitial inflammatory reaction ensuedas a consequence of excess ultrafiltration andproximal tubular reabsorption of proteins thatalso promoted NF-kB activation and chemokinesynthesis, limiting the enhanced protein trafficshould also limit NF-kB activation, MCP-1 geneupregulation, and inflammatory cell recruitment.The best strategy to date in experimental animalsand humans to reduce protein traffic rests onACE inhibitors, which additionally limit renaldisease progression.2,5,8-14,46To our knowledge,no other intervention is as effective as ACEinhibition in reducing proteinuria. Here, lisino-
Fig 15. Effect of delayed treatment with high-doselisinopril from 4 to 8 months on renal expression ofMCP-1 mRNA. Northern blot experiments were per-formed using total RNA of either separate (n 5 5 ratsfor each group) or pooled samples. Results shown arerepresentative of pooled samples for each group. Ab-breviation: ACEi, ACE inhibitor.
DONADELLI ET AL1238

pril administered to rats with remnant kidneysreduced urinary protein excretion, almost com-pletely suppressed NF-kB DNA–binding activ-ity, and reduced by approximately 70% MCP-1mRNA expression compared with untreated rem-nant kidney rats. The accumulation of ED-1–positive monocytes/macrophages and CD8-posi-tive T cells in the remnant kidney interstitiumwas also greatly limited by lisinopril. Similarly,in PHN rats in which the early administration oflisinopril effectively reduced proteinuria, we ob-served a dramatic decrease of NF-kB activationassociated with downregulation of MCP-1 expres-sion and reduction of interstitial mononuclearcell numbers. In line with our hypothesis thatNF-kB activation has a role in the downstreaminflammatory reaction rather than itself being apromoter of proteinuria are recent data showingthat in rats withAdriamycin nephropathy, chronictreatment with the NF-kB inhibitor, pyrrolidinedithiocarbamate (PDTC), started when animalshad overt proteinuria, suppressed cortical NF-kBactivation, and markedly reduced interstitialmonocyte infiltration, although it did not affecturinary protein excretion rate.47
That enhanced protein traffic is a determinantof NF-kB activation and downstream pathwaysof interstitial inflammation is supported furtherby the set of experiments using PHN rats withadvanced disease and severe proteinuria at thebeginning of treatment with high-dose ACE in-hibitor. In these animals, lisinopril failed to re-duce proteinuria and NF-kB activation. MCP-1mRNA transcript levels remained abnormallysustained, and inflammatory cell accumulationwas not limited by ACE inhibition despite theremarkable effect of high-dose lisinopril in nor-malizing renal ACE activity and blood pressure.
Previous studies have proposed enhanced an-giotensin II generation as a promoter of NF-kBactivation and monocyte infiltration in progres-sive nephropathies, based on findings that ACEinhibitors limited interstitial inflammation byblunting NF-kB activation and, in turn, MCP-1overexpression.41,48Although a concomitant roleof angiotensin II in promoting NF-kB–depen-dent pathways of interstitial inflammation cannotbe excluded, our present findings in PHN ratswith advanced disease that a dose of lisinoprilhigh enough to completely inhibit renal ACE
activity failed to reduce proteinuria and NF-kBactivation greatly favor the possibility of in-creased protein traffic as the direct trigger ofNF-kB activation.
In summary, we have shown that (1) NF-kBwas activated over time in kidneys of rats with5/6 nephrectomy or PHN, concomitant with theincrease in urinary protein excretion; (2) renalexpression of MCP-1 mRNA increased duringdisease progression in parallel with mononuclearcell accumulation in renal interstitium; (3) earlytreatment with an ACE inhibitor decreased pro-teinuria and reduced NF-kB activation andMCP-1 overexpression and greatly limited inter-stitial monocyte/macrophage and T-cell accumu-lation; and (4) high-dose ACE inhibitor treat-ment starting in the advanced phase of diseasefailed to reduce proteinuria, as well as NF-kBactivation and NF-kB–dependent pathways ofinterstitial inflammation.
ACKNOWLEDGMENTThe authors thank Dr Donatella Balducci, Dr Elena Gagli-
ardini, and Daniela Rottoli for technical assistance in immu-nohistochemical studies.
REFERENCES1. Remuzzi G, Bertani T: Is glomerulosclerosis a conse-
quence of altered glomerular permeability to macromol-ecules? Kidney Int 38:384-394, 1990
2. Remuzzi G, Bertani T: Pathophysiology of progressivenephropathies. N Engl J Med 339:1448-1456, 1998
3. Bertani T, Zoja C, Abbate M, Rossini M, Remuzzi G:Age-related nephropathy and proteinuria in rats with intactkidneys exposed to diets with different protein content. LabInvest 60:196-204, 1989
4. MagilAB: Tubulointerstitial lesions in human membra-nous glomerulonephritis: Relationship to proteinuria. Am JKidney Dis 25:375-379, 1995
5. The Gisen Group: Randomised placebo-controlled trialof effect of ramipril on decline in glomerular filtration rateand risk of terminal renal failure in proteinuric, non-diabeticnephropathy. Lancet 349:1857-1863, 1997
6. Wehrmann M, Bohle A, Held H, Schumm G, Kendzi-orra H, Pressler H: Long-term prognosis of focal sclerosingglomerulonephritis. An analysis of 250 cases with particularregard to tubulointerstitial changes. Clin Nephrol 33:115-122, 1990
7. Bohle A, Wehrmann M, Bogenschutz O, Batz C,Muller CA, Muller GA: The pathogenesis of chronic renalfailure in diabetic nephropathy. Investigation of 488 cases ofdiabetic glomerulosclerosis. Pathol Res Pract 187:251-259,1991
8. Anderson S, Rennke HG, Brenner BM: Therapeuticadvantage of converting enzyme inhibitors in arresting pro-gressive renal disease associated with systemic hypertensionin the rat. J Clin Invest 77:1993-2000, 1986
ANTIPROTEINURIC THERAPY LIMITS NF-kB SIGNALING 1239

9. Lafayette RA, Mayer G, Park SK, Meyer TW: Angio-tensin II receptor blockade limits glomerular injury in ratswith reduced renal mass. J Clin Invest 90:766-771, 1992
10. Ots M, Mackenzie HS, Troy JL, Rennke HG, BrennerBM: Effects of combination therapy with enalapril andlosartan on the rate of progression of renal injury in rats with5/6 renal mass ablation. J Am Soc Nephrol 9:224-230, 1998
11. Remuzzi A, Puntorieri S, Battaglia C, Bertani T,Remuzzi G: Angiotensin-converting enzyme inhibition ame-liorates glomerular filtration of macromolecules and waterand lessens glomerular injury in the rat. J Clin Invest85:541-549, 1990
12. Zoja C, Donadelli R, Corna D, Testa D, FacchinettiD, Maffi R, Luzzana E, Colosio V, Bertani T, Remuzzi G:The renoprotective properties of angiotensin-converting en-zyme inhibitors in a chronic model of membranous nephrop-athy are solely due to the inhibition of angiotensin II:Evidence based on comparative studies with a receptorantagonist. Am J Kidney Dis 29:254-264, 1997
13. Zoja C, Remuzzi A, Corna D, Perico N, Bertani T,Remuzzi G: Renal protective effect of angiotensin-convert-ing enzyme inhibition in aging rats. Am J Med 92:S60-S63,1992 (suppl 4B)
14. Ruiz-Ortega M, Gonzalez S, Seron D, Condom E,Bustos C, Largo R, Gonzales E, Ortiz A, Egido J: ACEinhibition reduces proteinuria, glomerular lesions and extra-cellular matrix production in a normotensive rat model ofimmune complex nephritis. Kidney Int 48:1778-1791, 1995
15. Bertani T, Cutillo F, Zoja C, Broggini M, Remuzzi G:Tubulo-interstitial lesions mediate renal damage in Adriamy-cin glomerulopathy. Kidney Int 30:488-496, 1986
16. Eddy AA: Interstitial nephritis induced by protein-overload proteinuria. Am J Pathol 135:719-733, 1989
17. Eddy AA, Giachelli CM: Renal expression of genesthat promote interstitial inflammation and fibrosis in ratswith protein-overload proteinuria. Kidney Int 47:1546-1557,1995
18. Lloyd CM, Minto AW, Dorf ME, Proudfoot A, WellsTN, Salant DJ, Gutierrez-Ramos JC: RANTES and mono-cyte chemoattractant protein-1 (MCP-1) play an importantrole in the inflammatory phase of crescentic nephritis, butonly MCP-1 is involved in crescent formation and interstitialfibrosis. J Exp Med 185:1371-1380, 1997
19. Abbate M, Zoja C, Corna D, Capitanio M, Bertani T,Remuzzi G: In progressive nephropathies, overload of tubu-lar cells with filtered proteins translates glomerular perme-ability dysfunction into cellular signals of interstitial inflam-mation. J Am Soc Nephrol 9:1213-1224, 1998
20. Zoja C, Morigi M, Figliuzzi M, Bruzzi I, Oldroyd S,Benigni A, Ronco P, Remuzzi G: Proximal tubular cellsynthesis and secretion of endothelin-1 on challenge withalbumin and other proteins. Am J Kidney Dis 26:934-941,1995
21. Wang Y, Chen J, Chen L, Tay YC, Rangan GK, HarrisDC: Induction of monocyte chemoattractant protein-1 inproximal tubule cells by urinary protein. J Am Soc Nephrol8:1537-1545, 1997
22. Zoja C, Donadelli R, Colleoni S, Figliuzzi M, Bonaz-zola S, Morigi M, Remuzzi G: Protein overload stimulatesRANTES production by proximal tubular cells dependingon NF-kB activation. Kidney Int 53:1608-1615, 1998
23. Ueda A, Okuda K, Ohno S, Shirai A, Igarashi T,Matsunaga K, Fukushima J, Kawamoto S, Ishigatsubo Y,Okubo T: NF-kB and Sp1 regulate transcription of thehuman monocyte chemoattractant protein-1 gene. J Immu-nol 153:2052-2063, 1994
24. Nelson PJ, Kim HT, Manning WC, Goralski TJ,KrenskyAM: Genomic organization and transcriptional regu-lation of the RANTES chemokine gene. J Immunol 151:2601-2612, 1993
25. Baeuerle PA, Henkel T: Function and activation ofNF-kB in the immune system. Annu Rev Immunol 12:141-179, 1994
26. Barnes PJ, Karin M: Nuclear factor-kB—A pivotaltranscription factor in chronic inflammatory diseases. N EnglJ Med 336:1066-1071, 1997
27. Henkel T, Machleidt T, Alkalay I, Kronke M, Ben-Neriah Y, Baeuerle PA: Rapid proteolysis of IkB-a is neces-sary for activation of the transcription factor NF-kB. Nature365:182-185, 1993
28. Wang Y, Rangan GK, Tay Y-C, Harris DCH: Induc-tion of monocyte chemoattractant protein-1 by albumin ismediated by nuclear factor kB in proximal tubule cells. J AmSoc Nephrol 10:1204-1213, 1999
29. Olson JL, Hostetter TH, Rennke HG, Brenner BM,Venkatachalam MA: Altered glomerular permselectivity andprogressive sclerosis following extreme ablation of renalmass. Kidney Int 22:112-126, 1982
30. Remuzzi G, Zoja C, Gagliardini E, Corna D, AbbateM, Benigni A: Combining an antiproteinuric approach withmycophenolate mofetil fully suppresses progressive nephrop-athy of experimental animals. J Am Soc Nephrol 10:1542-1549, 1999
31. Benigni A, Corna D, Maffi R, Benedetti G, Zoja C,Remuzzi G: Renoprotective effect of contemporary block-ing of angiotensin II and endothelin-1 in rats with membra-nous nephropathy. Kidney Int 54:353-359, 1998
32. Read SM, Northcote DH: Minimization of variationin the response to different proteins of the Coomassie blue Gdye-binding assay for protein. Anal Biochem 116:53-64, 1981
33. Negoro N, Kanayama Y, Haraguchi M, Umetani N,Nishimura M, Konishi Y, Iwai J, Okamura M, Inoue T,Takeda T: Blood pressure regulates platelet-derived growthfactor A-chain gene expression in vascular smooth musclecells in vivo. An autocrine mechanism promoting hyperten-sive vascular hypertrophy. J Clin Invest 95:1140-1150, 1995
34. Satriano J, Schlondorff D: Activation and attenuationof transcription factor NF-kB in mouse glomerular mesan-gial cells in response to tumor necrosis factor-a, immuno-globulin G, and adenosine 39:59-cyclic monophosphate. Evi-dence for involvement of reactive oxygen species. J ClinInvest 94:1629-1636, 1994
35. Rambaldi A, Young DC, Griffin JD: Expression of theM-CSF (CSF-1) gene by human monocytes. Blood 69:1409-1413, 1987
36. Rollins BJ, Morrison ED, Stiles CD: Cloning andexpression of JE, a gene inducible by platelet-derived growthfactor and whose product has cytokine-like properties. ProcNatl Acad Sci U S A 85:3738-3742, 1988
37. Zoja C, Liu XH, Donadelli R, Abbate M, Testa D,Corna D, Taraboletti G, Vecchi A, Dong QG, Rollins BJ,
DONADELLI ET AL1240

Bertani T, Remuzzi G: Renal expression of monocyte che-moattractant protein-1 in lupus autoimmune mice. J Am SocNephrol 8:720-729, 1997
38. Morrissey JJ, Klahr S: Enalapril decreases nuclearfactor kB activation in the kidney with ureteral obstruction.Kidney Int 52:926-933, 1997
39. Zoja C, Abbate M, Corna D, Capitanio M, DonadelliR, Bruzzi I, Oldroyd S, Benigni A, Remuzzi G: Pharmaco-logic control of angiotensin II ameliorates renal diseasewhile reducing renal TGF-a in experimental mesangioprolif-erative glomerulonephritis. Am J Kidney Dis 31:453-463,1998
40. Sakurai H, Hisada Y, Ueno M, Sugiura M, Ka-washima K, Sugita T: Activation of transcription factorNF-kB in experimental glomerulonephritis in rats. BiochimBiophys Acta 1316:132-138, 1996
41. Ruiz-Ortega M, Bustos C, Hernandez-Presa MA,Lorenzo O, Plaza JJ, Egido J: Angiotensin II participates inmononuclear cell recruitment in experimental immune com-plex nephritis through nuclear factor-kB activation and mono-cyte chemoattractant protein-1 synthesis. J Immunol 161:430-439, 1998
42. Wendt TH, Zhang YM, Bierhaus A, Kriegsmann J,Deng Y, Waldherr R, Teske T, Luther T, Fu¨nfstuck R,Nawroth PP, Stein G: Tissue factor expression in an animal
model of hydronephrosis. Nephrol Dial Transplant 10:1820-1828, 1995
43. Perkins ND, Schmid RM, Duckett CS, Leung K, RiceNR, Nabel GJ: Distinct combinations of NF-kB subunitsdetermine the specificity of transcriptional activation. ProcNatl Acad Sci U S A 89:1529-1533, 1992
44. Sheppard KA, Rose DW, Haque ZK, Kurokawa R,McInerney E, Westin S, Thanos D, Rosenfeld MG, GlassCK, Collins T: Transcriptional activation by NF-kB requiresmultiple coactivators. Mol Cell Biol 19:6367-6378, 1999
45. Tang WW, Qi M, Warren JS, Van GY: Chemokineexpression in experimental tubulointerstitial nephritis. J Im-munol 159:870-876, 1997
46. Ruggenenti P, Perna A, Gherardi G, Gaspari F, BeniniR, Remuzzi G, on behalf of Gruppo Italiano di StudiEpidemiologici in Nefrologia (GISEN): Renal function andrequirement for dialysis in chronic nephropathy patients onlong-term ramipril: REIN follow-up trial. Lancet 352:1252-1256, 1998
47. Rangan GK, Wang Y, Tay Y-C, Harris DCH: Inhibi-tion of nuclear factor-kB activation reduces cortical tubulo-interstitial injury in proteinuric rats. Kidney Int 56:118-134,1999
48. Klahr S, Morrissey J: Angiotensin II and gene expres-sion in the kidney. Am J Kidney Dis 31:171-176, 1998
ANTIPROTEINURIC THERAPY LIMITS NF-kB SIGNALING 1241














![anexo tecnología [7695 kb] - BOX](https://img.pdfslide.net/doc/110x75/631c33ea6c6907d368013292/anexo-tecnologia-7695-kb-box.jpg)














