Embed Size (px)
Citation preview

The Journal of Immunology
Purinergic Receptor Inhibition Prevents the Development ofSmoke-Induced Lung Injury and Emphysema
Sanja Cicko,*,1 Monica Lucattelli,†,1 Tobias Muller,*,1 Marek Lommatzsch,‡
Giovanna De Cunto,† Silvia Cardini,† William Sundas,† Melanine Grimm,*
Robert Zeiser,x Thorsten Durk,* Gernot Zissel,* Jean-Marie Boeynaems,{
Stephan Sorichter,* Davide Ferrari,‖ Francesco Di Virgilio,x J. Christian Virchow,‡
Giuseppe Lungarella,†,1 and Marco Idzko*,1
Extracellular ATP acts as a “danger signal” and can induce inflammation by binding to purinergic receptors. Chronic obstructive
pulmonary disease is one of the most common inflammatory diseases associated with cigarette smoke inhalation, but the underlying
mechanisms are incompletely understood. In this study, we show that endogenous pulmonary ATP levels are increased in a mouse
model of smoke-induced acute lung inflammation and emphysema. ATP neutralization or nonspecific P2R-blockade markedly
reduced smoke-induced lung inflammation and emphysema. We detected an upregulation the purinergic receptors subtypes on
neutrophils (e.g., P2Y2R), macrophages, and lung tissue from animals with smoke-induced lung inflammation. By using P2Y2R
deficient (2/2) animals, we show that ATP induces the recruitment of blood neutrophils to the lungs via P2Y2R. Moreover, P2Y2R
deficient animals had a reduced pulmonary inflammation following acute smoke-exposure. A series of experiments with P2Y2R2/2
andwild type chimera animals revealed that P2Y2R expression on hematopoietic cell plays the pivotal role in the observed effect. We
demonstrate, for the first time, that endogenous ATP contributes to smoke-induced lung inflammation and then development of
emphysema via activation of the purinergic receptor subtypes, such as P2Y2R. The Journal of Immunology, 2010, 185: 688–697.
Chronic obstructive pulmonary disease (COPD) is one ofthe leading causes of morbidity and mortality worldwide(1). COPD is associated with a pulmonary inflammation
and obstruction, particularly of the small airways, and the de-struction of lung parenchyma resulting in emphysema (2). Epi-demiologically, inhalation of cigarette smoke is the main riskfactor for the development of COPD. However, the precisemechanisms that initiate and perpetuate COPD and the underlyinginflammation are poorly understood (3). The major effector cellspresent in the lungs of patients with COPD are neutrophils,macrophages, CD4+ and CD8+ T lymphocytes, which are capableof releasing various mediators (including reactive oxygen
metabolites and proinflammatory cytokines), and tissue degradingenzymes (e.g., neutrophil elastase or matrix metalloproteinases)that contribute to tissue destruction, emphysema formation, andchronic inflammation (4, 5).Recently, extracellular ATP, which activates purinergic receptors
belonging to the P2Y family (P2Y1, P2Y2, P2Y4, P2Y6, P2Y11,P2Y12, P2Y13, and P2Y14) (6, 7) or the P2X family (P2X1–P2X7)(8–10), has gained attention as a mediator of inflammation (11, 12).Under physiologic conditions, extracellular concentrations of ATPare low and tightly regulated by ectonucleotidases, which de-phosphorylate ATP to ADP, AMP, and adenosine. However, underconditions such as hypoxia, trauma, infection, or inflammation,extracellular concentrations of ATP are elevated, because of activeor passive release from a number of cell types including airwayepithelial cells and inflammatory cells (11, 13) and/or because ofa concomitant downregulation of nucleotidases (14, 15). ATPmodulates mucin secretion and mucociliary clearance in humanlungs. The activation of the P2Y2 subtype improves mucociliaryclearance and clinical trials using P2Y2 agonists are in progress (16,17). ATP can also modify the recruitment and function of in-flammatory cells, such as neutrophils (18–22), macrophages (23–25), lymphocytes, and dendritic cells (13, 26–29). Finally, we re-cently demonstrated that endogenous ATP plays a role in thepathogenesis of acute asthmatic airway inflammation, where it hasbeen shown to activate dendritic cells (30).In this study, we provide in vivo evidence that ATP is linked to the
pathogenesis of smoke-induced lung emphysema. We demonstratethatATP levels are elevated in the lungs ofmicewith smoke-inducedpulmonary inflammation and emphysema and that these ATP levelscorrelate with pulmonary neutrophilia. During the preparation ofthis article, Mortaz et al. (31) reported increased ATP levels inthe bronchoalveolar lavage fluid (BALF) of animals with emphy-sema, suggesting that ATP might be involved in the pathogenesis ofCOPD. However, the effects of increased ATP levels in the
*Department of Pulmonary Medicine and xDepartment of Hematology and Oncology,University Hospital, Freiburg; ‡Department of Pulmonary Medicine, University Hos-pital, Rostock, Germany; †Department of Physiopathology and Experimental Medi-cine, University of Siena, Siena; ‖Section of General Pathology, Department ofExperimental and Diagnostic Medicine, Interdisciplinary Center for the Study ofInflammation, University of Ferrara, Ferrara, Italy; and {Institut de Recherche Inter-disciplinaire en Biologie Humaine et Moleculaire and Erasme Hospital, UniversiteLibre de Bruxelles, Brussels, Belgium
1S.C., M.L., T.M., G.L., and M.I. contributed equally to this work.
Received for publication December 22, 2009. Accepted for publication April 14,2010.
This work was supported by Emmy Noether Grant DFG ID7/4-1 from the DeutscheForschungsgemeinshaft (to M.I.).
Address correspondence and reprint requests to Dr. Marco Idzko, Department ofPulmonary Medicine, University Hospital Freiburg, Killianstrasse 5, 79106 Freiburg,Germany. E-mail address: [email protected]
The online version of this article contains supplemental material.
Abbreviations used in this paper: Air, exposure to room air; BAL, bronchoalveolarlavage; BALF, bronchoalveolar lavage fluid; COPD, chronic obstructive pulmonarydisease; ISA, internal surface area of the lungs; Lm, mean linear intercept; PPADS,pyridoxal-phosphate-6-azophenyl-29,49-disulfonic acid; ROS, reactive oxygen spe-cies; Vv, volume density; wt, wild type.
Copyright� 2010 by TheAmericanAssociation of Immunologists, Inc. 0022-1767/10/$16.00
www.jimmunol.org/cgi/doi/10.4049/jimmunol.0904042

development of lung emphysema are still unknown. In this study,we show for the first time that neutralizing intrapulmonary ATPlevels or blocking airway P2 receptors inhibits smoke-induced lunginflammation and confers protection from the development of em-physema. Moreover, in animals with smoke-induced lung injury weobserved a specific upregulation of the P2Y2R on blood and lungneutrophils and macrophages. The upregulation of the P2Y2R onneutrophils is of pathogenetic relevance, because ATP induces mi-gration of neutrophils into the lungs of naive mice by this receptorsubtype. Using P2Y2R deficient (2/2) and wild type (wt) chimeraanimals, we provide evidence that the expression of P2Y2R onhematopoietic cells contributes to the observed effects.
Materials and MethodsMice
C57/Bl/6 mice (6–8 wk old) were purchased from Charles River Labora-tories (Calco, Italy) or were bred at the animal facilities at the UniversityHospital of Freiburg or at the University of Siena. P2Y2 receptor deficientmice (P2Y2
2/2) on a C57/Bl6 background were generated as previouslydescribed (32). All experiments were performed according to institutionalguidelines of the animal ethics committee from the Italian or Germangovernments, respectively.
Generation of bone marrow chimera
Wt or P2Y2R2/2 recipients (both C57BL/6) were given 5 3 106 wt orP2Y2R2/2 bone marrow cells (C57BL/6) i.v. after lethal irradiation with 900cGy (23 450 cGy). The following donor–recipient pairs were combined: wt→wt, P2Y2R2/2→wt (hematopoietic system: P2Y2R2/2), wt→ P2Y2R2/2
(nonhematopoietic system: P2Y2R2/2), P2Y2R2/2 → P2Y2R2/2.
Lung inflammation induced by acute smoke exposure
Mice were exposed to whole smoke of five cigarettes (commercial Virginiafilter cigarettes [Marlboro Red, Philip Morris, Munich, Germany]: 12 mg tar,0.9mg nicotine) or room air for 20min on three consecutive days in speciallydesigned Makrolon cages (Tecniplast, Buguggiate, Italy), as previously de-scribed (33). The smoke was produced by burning a cigarette and was in-troduced into the chamber with the airflow generated by a mechanicalventilator (7025 Rodent Ventilator, Ugo Basile; Biological ResearchInstruments, Comerio, Italy), at a rate of 250 ml/min. A second mechanicalventilator was used to provide room air for dilution (1:8) of the smoke stream.
In some experiments, intratracheal treatment was performed prior to thesmoke exposure: animals were anesthetized by i.p. injection of ketamine-xylazine and received an intratracheal injection of the indicated compoundsin a total volume of 80 ml. One hour after the last smoke challenge, micewere sacrificed and bronchoalveolar lavage (BAL) was performed.
To study the effect of the P2R-antagonist in the ongoing smoke-inducedlung inflammation, mice were exposed to smoke on the days 1–3. Next, theywere randomized to receive either vehicle or P2R-antagonists (suramin orpyridoxal-phosphate-6-azophenyl-29,49-disulfonic acid [PPADS]) intratra-cheally 30 min before each of a series of three smoke or air challengesgiven at days 4–6. At days 3–6, 1 h after the last smoke or air challenge,mice were killed and the BALF was collected.
Chronic exposure to cigarette smoke
The methodology for chronic smoke exposure has been described previously(33).Micewere exposed to either the smoke of three cigarettes per day, 5 d perweek, or room air (controls) for 4, 6, or 7 mo, respectively. When indicated,animals received an oral treatment by gavagewith either suramin (1.6 mg/kg)or PPADS (0.6 mg/kg) 45 min prior to smoke exposure. At the end ofthe respective time period, animals were sacrificed; BAL was performed,followed by lung resection and fixation in formalin for histologic analysis.
Migration assay in vivo
To study neutrophil migration to the lungs, ATP, the stable analog ATPgS, orvehicle were administered intratracheally to wt and P2Y2(
2/2) mice.Twenty-four to 36 h after injection, the total number of neutrophils in theBALF was analyzed by FACS.
BAL in animals
BALF was collected by cannulating the trachea under deep pentobarbitalanesthesia and washing the lung three times with 1 ml PBS containing 0.1mM EDTA, as described previously (30). Total cell numbers were counted,and differential cell counts were performed on cytospin preparations after
staining with Diff-Quick (Medion Diagnostics, Dudingen, Switzerland).Differential cell counts were made on .200 cells using standardmorphologic criteria and FACS analysis.
Isolation of blood neutrophils
Blood neutrophils from smoke- and air-exposed animals were isolated aspreviously reported (21).
Flow cytometry and sorting
After counting and washing, BAL cells were stained for 30 min with anti-MHC class II (macrophages/DC), anti-7/4 (Caltag) FITC (neutrophils; Abd-Serotec, Dusseldorf, Germany), anti-CD3 and anti-CD19 cy-chrome (lym-phocytes), and anti-CD11cAPCmacrophages/DCs (eBioscience, SanDiego,CA) in PBS containing 0.5% BSA and 0.01% sodium azide. Differential cellcounts were analyzed by flow cytometry, as described previously (30, 34).
For the sorting of BAL neutrophils and BAL macrophages for real-timePCR, BAL cells were stained for 30 min with anti-CD68 PE (macrophages),anti-7/4-Caltag FITC (neutrophils), anti-CD3 and anti-CD19 cy-chrome(lymphocytes), and anti-I-Ad/I-Ed APC (macrophages/DCs) in PBS con-taining 0.5% BSA and 0.01% sodium azide. In all experiments, dead cellswere excluded from analysis using propidium iodide. Analysis was per-formed on a FACSCalibur flow cytometer (BD Biosciences, San Jose,CA) using Cellquest version 3.3 (BD Biosciences) and FlowJo version
FIGURE 1. Functional analysis of BALF-ATP concentrations following
acute smoke induced lung inflammation. A, Male C57BL/6 mice were left
either untreated or exposed to the smoke of five cigarettes on three con-
secutive days. One hour after the last smoke exposure, the animals were
killed and BALF was collected and analyzed for its ATP content. Data
from one representative experiment of four are shown. Data are shown as
mean 6 SEM. n = 5 mice in each group. pppp , 0.001. B, Correlation
between BALF-ATP levels and BALF-neutrophils in animals with acute
smoke-induced lung inflammation (r = 0.85; p = 0.00019). Data from three
experiments with n = 5 mice in each group are shown. C, Male C57BL/6
mice were left either untreated or exposed to the smoke of five cigarettes
on three consecutive days. Thirty minutes before smoke exposure, animals
received an intratracheal injection of vehicle, denatured apyrase, or apy-
rase (4 U/ml; 80 ml). One hour after the last smoke exposure, the animals
were killed and BALF was collected and analyzed for the number and
distribution of cells and (D) cytokine content. Data from one representative
experiment of four are shown. Data are shown as mean6 SEM. n = 5 mice
in each group. pp, 0.001, vehicle–air exposed versus vehicle or denatured
apyrase–smoke-exposed animals; #p , 0.01, vehicle or denatured-
apyrase–treated versus apyrase-treated smoke-exposed animals.
The Journal of Immunology 689

6.4.7 (TreeStar, Ashland, OR) software. The purity of the sorted BAL neu-trophils was .98% and was ,98% for the BAL macrophages.
ATP measurements in BAL fluid
To measure ATP concentrations in BALF in mice, fresh BALF supernatantswere used, according to manufacturer’s instructions (ATPlite Assay; Per-kinElmer, Wellesley, MA), but without the cell lysis step to avoid anycontaminating intracellular ATP, as described previously (30).
Cytokine measurements
Cytokine concentrations in BALF were measured using commerciallyavailable ELISA kits (R&D Systems, Minneapolis, MN) according to themanufacturer’s recommendations.
Real-time RT-PCR
Total RNAwas isolated from cell pellets or homogenized lung tissue, usingthe RNeasyMini-Kit (Qiagen, Hilden, Germany) according to the manufac-turer’s recommendations. Reverse transcription was performed using Stra-tascript reverse transcriptase (Stratagene, La Jolla, CA) and random primers(Invitrogen, Karlsruhe, Germany). Quantitative PCR was performed withTaqman Universal PCR Mastermix (Applied Biosystems, Foster City, CA)and preformulated primers and probe mixes (Assay on Demand; AppliedBiosystems). PCR conditions were 2 min at 50˚C and 10 min at 95˚C,followed by 45 cycles of 15 s at 95˚C and 60˚C for 1 min using a thermalcycler (iCycler; Bio-Rad, Hercules, CA). PCR amplification of the house-keeping gene encoding GADPH was performed during each run for eachsample to allow normalization between samples.
FIGURE 2. Effect of P2R-antagonists on the induction and maintenance of ongoing acute smoke-induced lung inflammation. A, Male C57BL/6 mice
were left untreated or exposed to the smoke of five cigarettes on three consecutive days. Thirty minutes before smoke exposure, animals received an
intratracheal injection of either vehicle or the P2-receptor antagonists suramin (10 mM, 80 ml) or PPADS (10 mM, 80 ml). One hour after the last smoke
exposure, animals were killed and BALF was analyzed for the number and distribution of cells and for cytokine content. Data are shown as mean 6 SEM,
n = 5 mice in each group. pp , 0.001, vehicle–air-exposed versus vehicle–smoke-exposed animals. #p , 0.001; xp , 0.05, vehicle-treated versus suramin-
or PPADS-treated smoke-exposed animals. B and C, Animals were left either untreated or exposed to the smoke of five cigarettes on three consecutive days.
From day 4, smoke exposure was either continued (upper panel) or changed to ambient air (lower panel). On day 4, day 4–5, or day 4–6, animals received
an intratracheal application of suramin (10 mM, 80 ml) 30 min prior to smoke or air exposure. On day 6, BALF (B) cell differentials and ATP concentrations
were analyzed. Data are shown as mean 6 SEM; n = 5 mice in each group. pp , 0.001, air-exposed versus smoke-exposed animals; #p , 0.05, smoke–
vehicle-treated versus smoke–suramin-treated animals at the same time points. C, Correlation of between BALF-ATP levels and BALF-neutrophils in
animals with ongoing smoke induced lung inflammation (r = 0.83; p , 0.0001). Data of the different animal groups are shown (total n = 70 mice).
690 PURINERGIC RECEPTOR IN SMOKE-INDUCED LUNG INJURY

Lung morphology and morphometry
At 4 and 7 mo after chronic exposure to room air or cigarette smoke, 8–12animals of each groupwere sacrificed and the lungswere fixed intratracheallywith formalin (5%) at a pressure of 20 cm H2O. Lung volume was measuredby water displacement. All lungs were then dehydrated, cleared in toluene,and embedded under vacuum in paraffin. Two 7-mm transversal sectionswere made and stained with H&E. Two pathologists blinded to theexposure protocol performed morphologic and morphometric evaluation.Morphometric assessment included determination of the mean linearintercept (Lm), which represents the average size of air space (alveolarducts, alveolar sacs, and alveoli), and of the internal surface area of thelungs (ISA). Lm is the length of a test line placed over histologic slides ofthe lung, divided by the number of times the line crosses alveolar walls (notsurfaces). Fields with bronchi, large bronchioli, or blood vessels wereexcluded from the measurements. Lm values were used to calculate theISA, necessary for evaluating the degree of emphysema, from the equation4V/Lm, where V is the postfixation lung volume. For the determination of theLm for each pair of lungs, 40 histologic fields were evaluated both verticallyand horizontally. Examination of this many fields meant that practically theentire lung area was evaluated.
The immunohistochemical analysis for the detection of CD4+ T cells,IFN-g, and IL-13–producing T cells, was performed as reported below.Sections were incubated with 3% BSA for 30 min at room temperature, toblock nonspecific Ab binding, and then exposed to rat monoclonal anti-mouseCD4-Ab (Abcam, Cambridge, U.K.), hamster monoclonal anti-mouse IFN-gAb (R&D Systems) or goat Ab to mouse IL-13 (R&D Systems) overnight.Next, sections were rinsed and incubated with goat polyclonal anti-rat bio-tinylated IgG (Abcam), or with mouse anti-hamster biotinylated IgG (BDPharmingen, Franklin Lakes, NJ) for the detection of CD4 and IFN-g orwith anti-goat IgG (Sigma-Aldrich, Milan, Italy) for IL-13. The stainingwas revealed by adding Streptavidin-HRP (BD Pharmingen, Buccinasco,Italy) for CD4 and IFN-g or peroxidase-antiperoxidase complex (Sigma-Aldrich) prepared from goat serum for IL-13 respectively. Detection wasaccomplished by incubation in diaminobenzidine freshly dissolved in0.03% H2O2 in 50 mM Tris/HCl (pH 7.6). As negative controls for theimmunostaining, the primary Ab was replaced with nonimmunized serum.
The volume fractions of the immunopositive cells were determined bypoint counting, using a grid with 48 points, a320 objective and a computerscreen for a final magnification of 3580. Twenty fields were examined foreach pair of lungs, for a total of 960 points. Next, the number of stainedcells per point were divided by the total number of points on the lungsection. This number was then multiplied by the volume of the lung cor-rected by the weight of the mouse, to give the total volume of the spe-cifically stained cells in the lung (35)
Statistical analysis
If not stated otherwise for most experiments, the statistical significanceof differences between samples was calculated using ANOVA, followedby Bonferroni comparison test. Differences were considered significant ifp , 0.05.
ResultsATP levels in BALF in animals with acute smoke-induced lunginflammation
C57BL/6animalswereexposedtothesmokeoffivecigarettesorairondays 1–3. Immediately after the last smoke or air exposure, animalswere killed and the BALF was collected (30). As shown in Fig. 1A,acute smoke-induced lung inflammation was associated with astrong increase of ATP levels in the BALF. As shown in Fig. 1B,ATP concentrations correlatedwith the number of BALFneutrophils(r = 0.85; p = 0.00019). In addition there was also a correlation withthe number of BALF macrophages (r = 0.62; p = 0.008).
Effect of ATP neutralization and purinergic receptorantagonists in an acute model of smoke-induced lunginflammation
Toevaluate the effects ofATPon smoke-induced lung inflammation,mice were treated intratracheally with the ATP/ADP hydrolyzingenzyme apyrase or denatured-apyrase 30min before each smoke ex-posure. Animals treated with apyrase, but not denatured-apyrase,had lowATP concentrations in the BALF (data not shown), a reduc-tion in the total cell count, and the number of macrophages and
neutrophils (Fig. 1C). In addition, there was a significant reductionin the concentrations of the cytokines IL-6, IL-1-b, IFN-g, KC, andMIP-2 in the BALF of apyrase-treated animals compared withanimals treated with vehicle alone or denatured apyrase (Fig.1D). Treatment of air-exposed animals with apyrase or dena-tured-apyrase did not significantly change the number of BALFcells or cytokine levels compared with vehicle treatment alone(data not shown).To test whether this observation was due to a reduced activation
of purinergic receptors, the nonspecific purinergic receptorantagonists suramin and PPADS were administered intratracheallyprior to each smoke exposure. Both antagonists reduced the numberof macrophages and neutrophils (Fig. 2A) and the concentrationsof IL-6, IL-1-b, KC, IFN-g, and MIP-2 (Fig. 2A) in BALF ina dose-dependent manner (Supplemental Table I). Of note, theeffects of suramin where also observed when suramin was admin-istered after the induction of smoke-induced lung inflammation(starting at the fourth cigarette smoke exposure), suggesting thatthis antagonist can prevent and treat early stages of establishedsmoke-induced lung inflammation (Fig. 2B). The latter treatmentscheme also led to a reduction in ATP levels, suggesting that thereis an ongoing release of ATP within the lungs (Fig. 2B). Again,ATP levels in BALF significantly correlated with the number ofneutrophils (Fig. 2C; r = 0.83; p , 0.0001) and to a lower degreewith macrophage numbers (r = 0.62; p , 0.001).
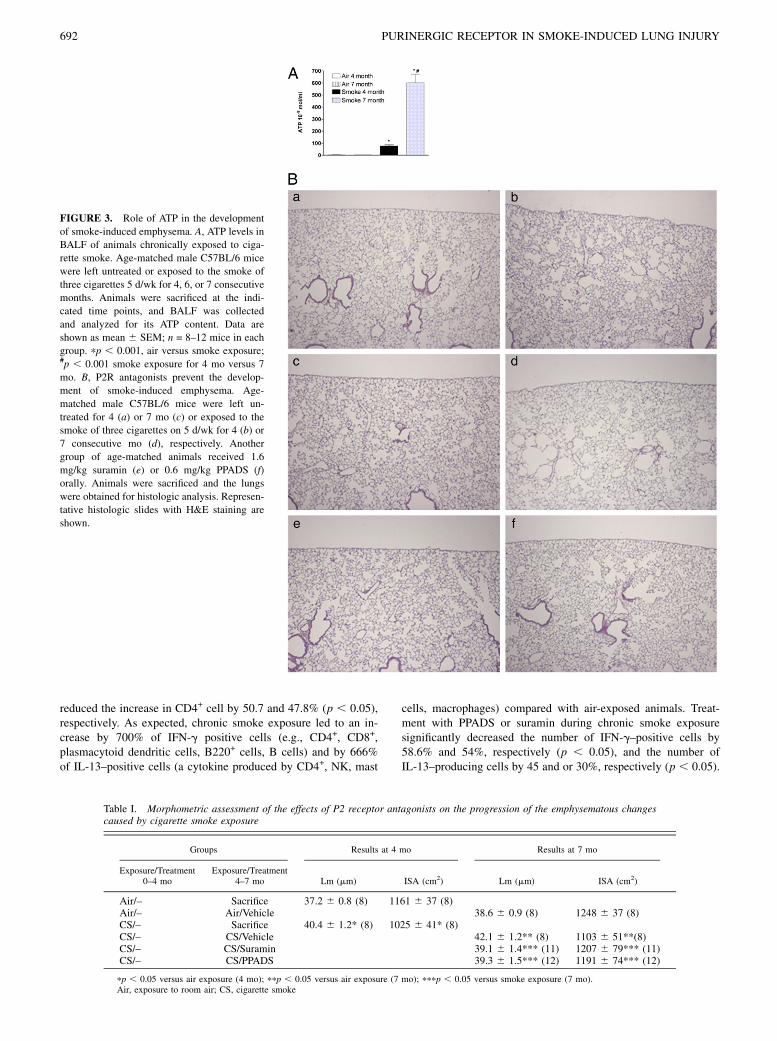
P2R-inhibition prevents the development of emphysema ina model of chronic smoke-induced lung inflammation
To better reflect the natural course of COPD, we used a model ofchronic smoke-induced lung inflammation characterized by the de-velopment of pulmonary emphysema. ATP concentrations were in-creased in BALF from animals chronically exposed to cigarettesmoke induced lung inflammation (Fig. 3A). Because repeatedanesthesia (3 d/wk for 4 mo) for intratracheal treatment wouldhave been fatal for the animals, oral treatment with the antagonistssuramin and PPADS was used. Prior to this experiment, the rela-tive potency of intratracheal versus oral treatment in the acutemodel of smoke-induced lung inflammation had to be compared.Oral treatment required higher concentrations of suramin andPPADS to reach a comparable reduction in lung inflammation(Supplemental Fig. 1). Lungs of mice exposed to room air for 4and 7 mo showed normal parenchyma and airways (Fig. 3Ba,3Bc), whereas mice exposed for 4 and 7 mo to cigarette smokeshowed foci of emphysema disseminated throughout the lung pa-renchyma (Fig. 3Bb, 3Bd). Oral treatment with suramin (1.6 mg/kg) or PPADS (0.6 mg/kg) prevented the development of emphy-sema in cigarette smoke-exposed mice. These mice showed nor-mal lung architecture (Fig. 3Be, 3Bf). Morphometric assessmentwas performed as previously described (36). The results of thevarious groups are given in Table I. After 4 and 7 mo of smokeexposure, the average interalveolar distance (i.e., Lm) was signif-icantly increased, and the ISA was significantly decreased com-pared with the respective air-exposed group. These effects werenot seen after suramin or PPADS treatment. The reduction of lunginflammation after treatment with suramin or PPADS was accom-panied by lower intrapulmonary ATP levels (data not shown). Inaddition, we also analyzed the number of CD4-positive cells andthe T cells producing IL-13 and IFN-g in the lungs of the animalsexposed to cigarette smoke and treated or not with PPADS orsuramin. As shown in Table II, the CD4+ T lymphocytes, whichwere mainly found perivascularly, in the airways and in thelymphoid follicles were increased in the lungs of the miceexposed to cigarette smoke by 670% (p , 0.05).Treatment ofsmoke-exposed animals with suramin and PPADS significantly
The Journal of Immunology 691
reduced the increase in CD4+ cell by 50.7 and 47.8% (p , 0.05),respectively. As expected, chronic smoke exposure led to an in-crease by 700% of IFN-g positive cells (e.g., CD4+, CD8+,plasmacytoid dendritic cells, B220+ cells, B cells) and by 666%of IL-13–positive cells (a cytokine produced by CD4+, NK, mast
cells, macrophages) compared with air-exposed animals. Treat-ment with PPADS or suramin during chronic smoke exposuresignificantly decreased the number of IFN-g–positive cells by58.6% and 54%, respectively (p , 0.05), and the number ofIL-13–producing cells by 45 and or 30%, respectively (p , 0.05).
Table I. Morphometric assessment of the effects of P2 receptor antagonists on the progression of the emphysematous changescaused by cigarette smoke exposure
Groups Results at 4 mo Results at 7 mo
Exposure/Treatment0–4 mo
Exposure/Treatment4–7 mo Lm (mm) ISA (cm2) Lm (mm) ISA (cm2)
Air/– Sacrifice 37.2 6 0.8 (8) 1161 6 37 (8)Air/– Air/Vehicle 38.6 6 0.9 (8) 1248 6 37 (8)CS/– Sacrifice 40.4 6 1.2* (8) 1025 6 41* (8)CS/– CS/Vehicle 42.1 6 1.2** (8) 1103 6 51**(8)CS/– CS/Suramin 39.1 6 1.4*** (11) 1207 6 79*** (11)CS/– CS/PPADS 39.3 6 1.5*** (12) 1191 6 74*** (12)
pp , 0.05 versus air exposure (4 mo); ppp , 0.05 versus air exposure (7 mo); pppp , 0.05 versus smoke exposure (7 mo).Air, exposure to room air; CS, cigarette smoke
FIGURE 3. Role of ATP in the development
of smoke-induced emphysema. A, ATP levels in
BALF of animals chronically exposed to ciga-
rette smoke. Age-matched male C57BL/6 mice
were left untreated or exposed to the smoke of
three cigarettes 5 d/wk for 4, 6, or 7 consecutive
months. Animals were sacrificed at the indi-
cated time points, and BALF was collected
and analyzed for its ATP content. Data are
shown as mean 6 SEM; n = 8–12 mice in each
group. pp , 0.001, air versus smoke exposure;#p , 0.001 smoke exposure for 4 mo versus 7
mo. B, P2R antagonists prevent the develop-
ment of smoke-induced emphysema. Age-
matched male C57BL/6 mice were left un-
treated for 4 (a) or 7 mo (c) or exposed to the
smoke of three cigarettes on 5 d/wk for 4 (b) or
7 consecutive mo (d), respectively. Another
group of age-matched animals received 1.6
mg/kg suramin (e) or 0.6 mg/kg PPADS (f)
orally. Animals were sacrificed and the lungs
were obtained for histologic analysis. Represen-
tative histologic slides with H&E staining are
shown.
692 PURINERGIC RECEPTOR IN SMOKE-INDUCED LUNG INJURY

Upregulation of purinergic receptors in animals with smoke-induced lung inflammation
To elucidate the regulation of purinergic receptors in acute smoke-induced lung inflammation, we analyzed the expression of puriner-
gic receptor subtypes in blood neutrophils, BALF neutrophils,
macrophages, and lung tissue after collection of the BALF. As
shown in Fig. 4A, smoke exposure led to an upregulation of the
P2X1, P2X4, P2X7 and P2Y2R subtypes in blood neutrophils, and
of the P2X7R and P2Y2R subtypes in BALF neutrophils (Fig. 4B).
In BALF macrophages, an upregulation could be observed for the
P2X7R, P2Y2R and P2Y13R (Fig. 4C). In the lung tissue of ani-
mals with smoke-induced lung inflammation, an upregulation of
the receptors P2Y4, P2Y6, P2Y13, P2X1, P2X4 and P2X7 was
observed (Fig. 4D).
FIGURE 4. Expression of P2R subtypes on blood
and BALF neutrophils, alveolar macrophages, and
lung tissue in mice with acute smoke-induced lung
inflammation. Male C57BL/6 mice were left either
untreated or exposed to the smoke of five cigarettes
on three consecutive days. One hour after the last
smoke exposure, animals were killed and blood and
BALF for the isolation of neutrophils and macro-
phages were collected and RNA was isolated from
whole lung tissue. Relative expression of the different
P2Y and P2X receptors compared with GAPDH in
triplicates were analyzed using quantitative RT-PCR.
A, Expression of P2Y and P2XR subtypes on pooled
blood neutrophils from 5 per group. B, Expression of
P2Yand P2XR subtypes on pooled BALF neutrophils
from 10 animals for smoke exposure and 20 animals
for air exposure. C, Expression of P2Y and P2XR
subtypes on pooled BALF macrophages from 10 ani-
mals for smoke exposure and 20 animals for air ex-
posure. D, Expression of P2Y and P2XR subtypes on
lung tissue of smoke or air exposed animals. Data
from one representative experiment of three are
shown. Data are shown as mean 6 SEM. n = 5 mice
in each group. pp , 0.05.
Table II. Volume density of CD4+, IFN-g, and IL-13–positive cells in the lungs of C57BL/6 mice exposed toroom air or cigarette smoke for 7 mo and treated or not with PPADS or suramin
Exposure/TreatmentCD4+ Positive Cells Vv
(3 1024 ml/g)IFN-g Positive CellsVv (3 1024 ml/g)
IL-13 Positive CellsVv (3 1024 ml/g)
Air/– 1.0 6 0.9 1.1 6 0.9 0.6 6 0.2CS/Vehicle 6.7 6 1.5* 7.0 6 1.9* 4.1 6 0.7*CS/Suramin 3.2 6 1.8** 3.8 6 0.8** 1.2 6 0.6**CS/PPADS 3.4 6 2.3** 4.1 6 0.6** 1.8 6 0.5**
pp , 0.05 versus air exposure; ppp , 0.05 versus smoke exposure.CS, cigarette smoke; Vv, volume density.
The Journal of Immunology 693

Effect of ATP on the migration of neutrophils
Based on the observation of a close correlation between neutrophilnumbers and ATP concentrations in BALF, we questioned whetherATP can enhance the recruitment of neutrophils into the lungsin vivo. Intratracheal treatment with ATP (100 mM) or non-hydrolysable stable ATP-analog ATPgS (100 mM) significantlyenhanced the number of neutrophils in BALF compared with ve-hicle treatment alone (Fig. 5A).To analyze whether the P2Y2 receptor is involved, P2Y2
2/2
mice were examined. Intratracheal application of the stable non-hydrolyzable ATP analog ATPgS failed to induce a recruitment ofneutrophils to the lung P2Y2
2/2 animals (Fig. 5B). The non-hydrolyzable ATPyS was used to exclude effects of the ATP-metabolite adenosine on cell migration, as reported previously(20, 21).
Role of P2Y2R on the hematopoietic system in smoke-inducedlung inflammation
To further elucidate the functional relevance of the upregulation ofthe P2Y2R in smoke-induced lung inflammation, P2Y2
2/2 micewere treated according to the acute smoke-induced lung inflam-mation model. P2Y2
2/2 deficiency was associated with a decreasein smoke-induced lung inflammation (reduced numbers of neutro-phils and macrophages; Fig. 6A) and lower levels of ATP, IL-6, IL-1b, KC, IFN-g, and MIP-2 (Fig. 6B, 6C) in the BALF of theseanimals.Finally, to discriminate between the relevance of P2Y2R ex-
pression on the hematopoietic system and nonhematopoietic sys-tem, experiments with bone marrow chimera were performed. Asshown in Fig. 6D and 6E, wt mice with a lack of P2Y2R expressionin the hematopoietic system displayed a significant decrease inlung inflammation, as evidenced by reduced numbers of neu-trophils and macrophages in BALF (Fig. 6D) and reduced BALFconcentrations of ATP, IL-6, IL-1b, KC, IFN-g, and MIP-2 (Fig.6E). In contrast, P2Y2R
2/2 animals reconstituted with a wthematopoietic system (wt → P2Y2R
2/2) showed no protectionagainst smoke induce lung inflammation.
DiscussionThe pathogenesis of smoke-induced lung inflammation and emphy-sema is still incompletely understood. In this study, we demonstrate
that extracellular ATP accumulates in the airways ofmicewith acute
smoke-induced lung inflammation and emphysema. ATP neutrali-
zation or unspecific purinergic blockade markedly reduced the in-
flammatory reaction to cigarette smoke and the development of
smoke-induced lung inflammation and emphysema. Mechanisti-
cally, we provide evidence that the observed effects might involve
the activation of the P2Y2 receptor on neutrophils and macro-
phages. Thus, our findings suggest that extracellular ATP, via bind-
ing to purinergic receptors, such as P2Y2R, is involved in the
pathogenesis of some of the pathologic features of smoke-
induced lung inflammation and emphysema.In healthy tissues, extracellular ATP levels are regulated by
a dynamic balance between ATP release and ATP degradation. ATP
release occurs via several tightly controlled mechanisms, such as
constitutive or stimulated exocytosis, membrane transporters, or
nonselective channels, whereas ATP degradation is due to ubiq-
uitous ectonucleotidases. Under physiologic conditions, the ex-
tracellular ATP concentration is kept in the low nanomolar range.
However, tissue perturbation, as it occurs in the presence of injury,
inflammation or cancer, may cause a massive accumulation of
extracellular ATP (up to micromolar levels) (37). We found
a strong increase of endobronchial ATP concentrations in mice
with smoke-induced lung inflammation. These findings were re-
cently confirmed by another group (31). The underlying reason for
this increase in ATP concentrations is currently unclear, because it
could be either smoke-induced ATP release or suppression of
ATP-degrading ectonucleotidases. Indeed, ectonucleotidases have
been shown to be downregulated by reactive oxygen species
(ROS), bacterial endotoxin or cytokines, such as TNF-a (15, 38,
39). Of note, cigarette smoke contains high concentrations of ROS
(40) and inflammatory cells such activated macrophages and
neutrophils can also generate substantial amounts of ROS (41).
Alternatively, direct toxic effects of the cigarette smoke on several
cell populations could lead to an enhanced release of intracellular
ATP into the extracellular space.It has been hypothesized that the accumulation of extracellular
ATP merely reflects damage and does not have pathogenetic
implications in inflammation or tissue destruction. However, our
data show that ATP neutralization or the inhibition of purinergic
receptors can prevent smoke-induced lung inflammation by reduc-
ing neutrophil and macrophage infiltration and the release of
proinflammatory cytokines, such as IL-1b, MIP-2, KC, IFN-g,
and IL-6 in BALF. A reduction of these parameters was also
observed when purinergic signaling was blocked during ongoing
inflammation. Thus, we provide evidence that ATP is actively
involved in the pathogenesis of several features associated with
smoke-induced lung inflammation. This finding is further sup-
ported by histomorphologic evidence indicating that inhibition
of purinergic receptors interfered with the induction of cigarette
smoke-induced emphysema. Interestingly, this effect was still ob-
served in mice with established inflammation and emphysema. It
has recently been reported that the lungs of patients with COPD
show more CD4+ cells and an upregulated expression of Th1
cytokines, such as IFN-g, and Th2 cytokines, such as IL-13 (3).
However, the specific role of the adaptive immune system in the
development of the disease is still a matter of discussion. Simi-
larly, the role of the innate immunity in the development of cig-
arette smoke-induced emphysema has not been clarified. Of
interest, P2R-blockade by suramin and PPADS also prevents the
development of emphysema in smoke-exposed mice by reducing
FIGURE 5. ATP induced migration of neutrophils to the lungs of naive
mice in vivo: involvement of P2Y2 receptors. Male C57BL/6 WT or P2Y2-
receptor–deficient mice were anesthetized and injected intratracheally with
ATP, ATPgS (100 mM, 80 ml), or solvent. Twenty-four hours later, animals
were sacrificed and the number of neutrophils in the BALF was determined
(A, B). Data from one representative experiment of three are shown. Data
are shown as mean 6 SEM. n = 6 mice in each group. pp , 0.05.
694 PURINERGIC RECEPTOR IN SMOKE-INDUCED LUNG INJURY

the influx into the lungs of the inflammatory cells of both theinnate and adaptive immune systems. With all the limitations ofsuch animal models, these data support the hypothesis that puri-nergic signaling plays a role in the pathogenesis of COPD.Inhibition of smoke-induced lung inflammation by blocking the
ATP/P2R pathway could involve an alteration of neutrophil func-tion, because ATP via binding to the P2Y2 receptor has strongchemotactic activity on neutrophils and other inflammatory cells(20, 21). Indirect effects of ATP on chemokines, such as IL-8 (42),might augment the influx of neutrophils into the lungs, as has beenshown in a mouse model of sepsis (22, 43). In addition, ATPincreases viability of human neutrophils by preventing apoptosis(44). In our study, exogenous administration of ATP to naive miceincreased the number of neutrophils in BALF via activation of P2Y2
receptors. In addition, there was a strong correlation betweenneutrophil counts and ATP levels in the BALF of animals withacute and ongoing smoke-induced lung inflammation. In mice trea-ted with purinergic receptor antagonists or apyrase, there were re-duced concentrations of KC and MIP-2, which play a role inneutrophil recruitment (45, 46). These data suggest that ATPmight also exert its effects indirectly. Neutrophil recruitment byATP might be harmful per se, because neutrophils accelerate
inflammation and alveolar destruction by secreting proteases,ROS, and proinflammatory cytokines (4, 5). However, ATP has alsobeen shown to directly induce the secretion of ROS, elastase, andLTB4 (19, 47, 48). Thus, the proinflammatory effect of ATP mightbe due to enhanced neutrophil recruitment and enhanced neutrophilactivation.The hypothesis that P2Y2R signaling contributes to the accu-
mulation of neutrophils in the lungs is supported by our findings ofstrong upregulation of the P2Y2R expression on blood and BALFneutrophils in mice with smoke-induced lung inflammation. Inaddition, we demonstrate that P2Y2 receptor-deficient animalshave reduced pulmonary inflammation after acute smoke expo-sure. Experiments with chimera P2Y2R
2/2 and wt animalsrevealed that expression of P2Y2R on hematopoietic cells mightaccount for this effect, as P2Y2R
2/2 reconstituted with wt bonemarrow did not show a reduction in smoke-induced lung inflam-mation compared with wt animals.Macrophages have been suggested to play a pivotal role in the
pathophysiology of COPD and can account for some of the featuresof the disease (5). ATP is a powerful macrophage stimulant thatinduces the secretion of cytokines (e.g., IL-6, IL-8, IL-1b, IL-18)of matrix metalloproteinase 9, the predominant elastolytic enzyme
FIGURE 6. Reduced smoke-induced lung inflamma-
tion in P2Y2R-deficient animals. A–C, Male P2Y2-
receptor2/2 mice or wt animals were left either
untreated or exposed to the smoke of five cigarettes
on three consecutive days. One hour after the last
smoke exposure, animals were killed and BALF was
analyzed for (A) the number and distribution of cells,
(B) ATP, and (C) cytokine content. Data are shown as
mean 6 SEM; n = 5 mice in each group; pp , 0.001,
smoke versus air exposure; #p , 0.05. P2Y2R2/2
smoke versus wt smoke. D and E, The different bone
marrow chimera animals were exposed to the smoke of
five cigarettes on three consecutive days. One hour after
the last smoke exposure, animals were killed and BALF
was analyzed for (D) the number and distribution of
cells and (E) cytokine content. Data are shown as
mean 6 SEM; n = 5 mice in each group; pp , 0.05
P2Y2R2/2 in P2Y2R2/2 versus wt in wt; #p , 0.05
P2Y2R2/2 in wt versus wt in wt.
The Journal of Immunology 695

in patients with COPD (5), and the production of ROS in macro-phages (13, 23, 29, 49, 50). Furthermore, extracellular ATP causesthe release of IL-8 and IL-6 via activation of P2YR-subtypes fromairway epithelial cells (42, 51, 52). Indeed, the involvement ofother P2R subtypes in ATP-mediated pulmonary damage is sup-ported by our finding that the administration of the unselectiveP2R-antagonist PPADS to P2Y2
2/2 mice exposed to cigarettesmoke led to a further reduction in smoke-induced pulmonaryinflammation (data not shown). Because suramin and PPADSblock several purinergic receptors (7, 9), the precise receptors thatmediate the observed features of smoke-induced lung inflamma-tion remain to be elucidated. The strong upregulation of the P2Y2
receptor in BALF neutrophils and BALF macrophages, the upre-gulation of the P2X1, P2X4, P2X7 and P2Y6 receptors in the lungsof mice with smoke-induced lung inflammation, and experimentsusing P2Y2
2/2 mice suggest that this receptor subtype might beinvolved. Therefore, further studies are needed to identify thespecific subtypes of particular relevance in the pathogenesis ofCOPD to develop selective purinergic receptor antagonists forthe treatment or prevention of COPD.These data suggest that enhanced endogenous ATP concen-
trations, via activation of purinergic receptors, such as P2Y2R,mightbe causally related to the pathogenesis of smoke-induced lung in-flammation and might be a new target for therapy of COPD.
DisclosuresThe authors have no financial conflicts of interest.
References1. Rabe, K. F., B. Beghe, F. Luppi, and L. M. Fabbri. 2007. Update in chronic
obstructive pulmonary disease 2006. Am. J. Respir. Crit. Care Med. 175: 1222–1232.
2. Hogg, J. C., F. Chu, S. Utokaparch, R. Woods, W. M. Elliott, L. Buzatu,R. M. Cherniack, R. M. Rogers, F. C. Sciurba, H. O. Coxson, and P. D. Pare.2004. The nature of small-airway obstruction in chronic obstructive pulmonarydisease. N. Engl. J. Med. 350: 2645–2653.
3. Hogg, J. C. 2004. Pathophysiology of airflow limitation in chronic obstructivepulmonary disease. Lancet 364: 709–721.
4. O’Donnell, R., D. Breen, S. Wilson, and R. Djukanovic. 2006. Inflammatorycells in the airways in COPD. Thorax 61: 448–454.
5. Barnes, P. J., S. D. Shapiro, and R. A. Pauwels. 2003. Chronic obstructivepulmonary disease: molecular and cellular mechanisms. Eur. Respir. J. 22:672–688.
6. von Kugelgen, I., and A. Wetter. 2000. Molecular pharmacology of P2Y-receptors. Naunyn Schmiedebergs Arch. Pharmacol. 362: 310–323.
7. Abbracchio, M. P., G. Burnstock, J. M. Boeynaems, E. A. Barnard, J. L. Boyer,C. Kennedy, G. E. Knight, M. Fumagalli, C. Gachet, K. A. Jacobson, andG. A. Weisman. 2006. International Union of Pharmacology LVIII: update on theP2Y G protein-coupled nucleotide receptors: from molecular mechanisms andpathophysiology to therapy. Pharmacol. Rev. 58: 281–341.
8. Humphrey, P. P., G. Buell, I. Kennedy, B. S. Khakh, A. D. Michel, A. Surprenant,and D. J. Trezise. 1995. New insights on P2X purinoceptors. Naunyn Schmie-debergs Arch. Pharmacol. 352: 585–596.
9. Gever, J. R., D. A. Cockayne, M. P. Dillon, G. Burnstock, and A. P. Ford. 2006.Pharmacology of P2X channels. Pflugers Arch. 452: 513–537.
10. Burnstock, G. 2006. Purinergic signalling--an overview. Novartis Found. Symp.276: 26–48; discussion 48–57, 275–281.
11. Burnstock, G. 2006. Pathophysiology and therapeutic potential of purinergicsignaling. Pharmacol. Rev. 58: 58–86.
12. Khakh, B. S., and R. A. North. 2006. P2X receptors as cell-surface ATP sensorsin health and disease. Nature 442: 527–532.
13. Bours, M. J., E. L. Swennen, F. Di Virgilio, B. N. Cronstein, and P. C. Dagnelie.2006. Adenosine 59-triphosphate and adenosine as endogenous signaling mole-cules in immunity and inflammation. Pharmacol. Ther. 112: 358–404.
14. Lazarowski, E. R., R. C. Boucher, and T. K. Harden. 2003. Mechanisms of re-lease of nucleotides and integration of their action as P2X- and P2Y-receptoractivating molecules. Mol. Pharmacol. 64: 785–795.
15. Robson, S. C., E. Kaczmarek, J. B. Siegel, D. Candinas, K. Koziak, M. Millan,W. W. Hancock, and F. H. Bach. 1997. Loss of ATP diphosphohydrolase activitywith endothelial cell activation. J. Exp. Med. 185: 153–163.
16. Kellerman, D., A. Rossi Mospan, J. Engels, A. Schaberg, J. Gorden, andL. Smiley. 2008. Denufosol: a review of studies with inhaled P2Y(2) agoniststhat led to Phase 3. Pulm. Pharmacol. Ther. 21: 600–607.
17. Donnelly, L. E., and D. F. Rogers. 2003. Therapy for chronic obstructive pul-monary disease in the 21st century. Drugs 63: 1973–1998.
18. Chen, Y., A. Shukla, S. Namiki, P. A. Insel, and W. G. Junger. 2004. A putativeosmoreceptor system that controls neutrophil function through the release ofATP, its conversion to adenosine, and activation of A2 adenosine and P2receptors. J. Leukoc. Biol. 76: 245–253.
19. Meshki, J., F. Tuluc, O. Bredetean, Z. Ding, and S. P. Kunapuli. 2004. Molecularmechanism of nucleotide-induced primary granule release in human neutrophils:role for the P2Y2 receptor. Am. J. Physiol. Cell Physiol. 286: C264–C271.
20. Verghese, M. W., T. B. Kneisler, and J. A. Boucheron. 1996. P2U agonists inducechemotaxis and actin polymerization in human neutrophils and differentiatedHL60 cells. J. Biol. Chem. 271: 15597–15601.
21. Chen, Y., R. Corriden, Y. Inoue, L. Yip, N. Hashiguchi, A. Zinkernagel, V. Nizet,P. A. Insel, and W. G. Junger. 2006. ATP release guides neutrophil chemotaxisvia P2Y2 and A3 receptors. Science 314: 1792–1795.
22. Inoue, Y., Y. Chen, M. I. Hirsh, L. Yip, and W. G. Junger. 2008. A3 and P2Y2receptors control the recruitment of neutrophils to the lungs in a mouse model ofsepsis. Shock 30:173–177.
23. Myrtek, D., T. Muller, V. Geyer, N. Derr, D. Ferrari, G. Zissel, T. Durk,S. Sorichter, W. Luttmann, M. Kuepper, et al. 2008. Activation of human al-veolar macrophages via P2 receptors: coupling to intracellular Ca2+ increasesand cytokine secretion. J. Immunol. 181: 2181–2188.
24. Sluyter, R., J. G. Dalitz, and J. S. Wiley. 2004. P2X7 receptor polymorphismimpairs extracellular adenosine 59-triphosphate-induced interleukin-18 releasefrom human monocytes. Genes Immun. 5: 588–591.
25. Hanley, P. J., B. Musset, V. Renigunta, S. H. Limberg, A. H. Dalpke, R. Sus,K. M. Heeg, R. Preisig-Muller, and J. Daut. 2004. Extracellular ATP inducesoscillations of intracellular Ca2+ and membrane potential and promotestranscription of IL-6 in macrophages. Proc. Natl. Acad. Sci. USA 101:9479–9484.
26. Idzko, M., S. Dichmann, D. Ferrari, F. Di Virgilio, A. la Sala, G. Girolomoni,E. Panther, and J. Norgauer. 2002. Nucleotides induce chemotaxis and actinpolymerization in immature but not mature human dendritic cells via activationof pertussis toxin-sensitive P2y receptors. Blood 100: 925–932.
27. Ferrari, D., C. Pizzirani, E. Adinolfi, R. M. Lemoli, A. Curti, M. Idzko,E. Panther, and F. Di Virgilio. 2006. The P2X7 receptor: a key player inIL-1 processing and release. J. Immunol. 176: 3877–3883.
28. Idzko, M., E. Panther, H. C. Bremer, S. Sorichter, W. Luttmann, C. J. Virchow,Jr., F. Di Virgilio, Y. Herouy, J. Norgauer, and D. Ferrari. 2003. Stimulation of P2purinergic receptors induces the release of eosinophil cationic protein andinterleukin-8 from human eosinophils. Br. J. Pharmacol. 138: 1244–1250.
29. Di Virgilio, F., P. Chiozzi, D. Ferrari, S. Falzoni, J. M. Sanz, A. Morelli,M. Torboli, G. Bolognesi, and O. R. Baricordi. 2001. Nucleotide receptors: anemerging family of regulatory molecules in blood cells. Blood 97: 587–600.
30. Idzko, M., H. Hammad, M. van Nimwegen, M. Kool, M. A. Willart, F. Muskens,H. C. Hoogsteden, W. Luttmann, D. Ferrari, F. Di Virgilio, et al. 2007. Extra-cellular ATP triggers and maintains asthmatic airway inflammation by activatingdendritic cells. Nat. Med. 13: 913–919.
31. Mortaz, E., S. Braber, M. Nazary, M. E. Givi, F. P. Nijkamp, and G. Folkerts. 2009.ATP in the pathogenesis of lung emphysema. Eur. J. Pharmacol. 619: 92–96.
32. Matos, J. E., B. Robaye, J. M. Boeynaems, R. Beauwens, and J. Leipziger. 2005.K+ secretion activated by luminal P2Y2 and P2Y4 receptors in mouse colon.J. Physiol. 564: 269–279.
33. Cavarra, E., M. Lucattelli, F. Gambelli, B. Bartalesi, S. Fineschi, A. Szarka,F. Giannerini, P. A. Martorana, and G. Lungarella. 2001. Human SLPI in-activation after cigarette smoke exposure in a new in vivo model of pulmonaryoxidative stress. Am. J. Physiol. 281: L412–L417.
34. Reutershan, J., A. Basit, E. V. Galkina, and K. Ley. 2005. Sequential recruitmentof neutrophils into lung and bronchoalveolar lavage fluid in LPS-induced acutelung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 289: L807–L815.
35. Meshi, B., T. Z. Vitalis, D. Ionescu, W. M. Elliott, C. Liu, X. D. Wang,S. Hayashi, and J. C. Hogg. 2002. Emphysematous lung destruction by cigarettesmoke. The effects of latent adenoviral infection on the lung inflammatory re-sponse. Am. J. Resp. Cell Mol. Biol. 26: 52–57.
36. Martorana, P. A., R. Beume, M. Lucattelli, L. Wollin, and G. Lungarella. 2005.Roflumilast fully prevents emphysema in mice chronically exposed to cigarettesmoke. Am. J. Respir. Crit. Care Med. 172: 848–853.
37. Pellegatti, P., L. Raffaghello, G. Bianchi, F. Piccardi, V. Pistoia, and F. Di Vir-gilio. 2008. Increased level of extracellular ATP at tumor sites: in vivo imagingwith plasma membrane luciferase. PLoS ONE 3: e2599.
38. Kishi, Y., S. Ohta, N. Kasuya, S. Y. Sakita, T. Ashikaga, and M. Isobe. 2003.Perindopril augments ecto-ATP diphosphohydrolase activity and enhancesendothelial anti-platelet function in human umbilical vein endothelial cells.J. Hypertens. 21: 1347–1353.
39. Vlaar, A. P., W. J. van Son, and W. W. Bakker. 2009. Histochemical detection ofischemia-like alterations induced in kidney tissue in vitro–different sensitivityto oxidant stress of glomerular ENTPD1 versus E5NT. Nephron. Physiol. 111:p1–p8.
40. Pryor, W. A., and K. Stone. 1993. Oxidants in cigarette smoke. Radicals, hydrogenperoxide, peroxynitrate, and peroxynitrite. Ann. N.Y. Acad. Sci. 686: 12–27; dis-cussion 27–18.
41. Klink, M., K. Jastrzembska, K. Bednarska, M. Banasik, and Z. Sulowska. 2009.Effect of nitric oxide donors on NADPH oxidase signaling pathway in humanneutrophils in vitro. Immunobiology 214: 692–702.
42. Theatre, E., V. Bours, and C. Oury. 2009. A P2X ion channel-triggered NF-kappaB Pathway Enhances TNF-alpha-induced IL-8 expression in airway epi-thelial cells. Am. J. Respir. Cell Mol. Biol. 41: 705–713.
696 PURINERGIC RECEPTOR IN SMOKE-INDUCED LUNG INJURY

43. Kukulski, F., F. Ben Yebdri, J. Lefebvre, M. Warny, P. A. Tessier, and J. Sevigny.2007. Extracellular nucleotides mediate LPS-induced neutrophil migrationin vitro and in vivo. J. Leukoc. Biol. 81: 1269–1275.
44. Vaughan, K. R., L. Stokes, L. R. Prince, H. M. Marriott, S. Meis, M. U. Kassack,C.D.Bingle, I. Sabroe,A.Surprenant, andM.K.Whyte. 2007. Inhibitionof neutrophilapoptosis by ATP is mediated by the P2Y11 receptor. J. Immunol. 179: 8544–8553.
45. Churg, A., R. D. Wang, H. Tai, X. Wang, C. Xie, and J. L. Wright. 2004. Tumornecrosis factor-alpha drives 70% of cigarette smoke-induced emphysema in themouse. Am. J. Respir. Crit. Care Med. 170: 492–498.
46. Stevenson, C. S., K. Coote, R. Webster, H. Johnston, H. C. Atherton, A. Nicholls,J. Giddings, R. Sugar, A. Jackson, N. J. Press, et al. 2005. Characterization ofcigarette smoke-induced inflammatory and mucus hypersecretory changes in ratlung and the role of CXCR2 ligands in mediating this effect. Am. J. Physiol.Lung Cell Mol. Physiol. 288: L514–L522.
47. Suh, B. C., J. S. Kim, U. Namgung, H. Ha, and K. T. Kim. 2001. P2X7 nu-cleotide receptor mediation of membrane pore formation and superoxide gen-eration in human promyelocytes and neutrophils. J. Immunol. 166: 6754–6763.
48. Tuluc, F., O. Bredetean, E. Brailoiu, J. Meshki, A. Garcia, N. J. Dun, andS. P. Kunapuli. 2005. The priming effect of extracellular UTP on human neu-trophils: Role of calcium released from thapsigargin-sensitive intracellularstores. Purinergic Signal. 1: 359–368.
49. Gu, B. J., and J. S. Wiley. 2006. Rapid ATP-induced release of matrix metal-loproteinase 9 is mediated by the P2X7 receptor. Blood 107: 4946–4953.
50. Murphy, J. K., F. R. Livingston, E. Gozal, M. Torres, and H. J. Forman. 1993.Stimulation of the rat alveolar macrophage respiratory burst by extracellularadenine nucleotides. Am. J. Respir. Cell Mol. Biol. 9: 505–510.
51. Muller, T., H. Bayer, D. Myrtek, D. Ferrari, S. Sorichter, M. W. Ziegenhagen,G. Zissel, J. C. Virchow, Jr., W. Luttmann, J. Norgauer, et al. 2005. The P2Y14receptor of airway epithelial cells: coupling to intracellular Ca2+ andIL-8 secretion. Am. J. Respir. Cell Mol. Biol. 33: 601–609.
52. Douillet, C. D., W. P. Robinson, 3rd, P. M. Milano, R. C. Boucher, andP. B. Rich. 2006. Nucleotides induce IL-6 release from human airway epitheliavia P2Y2 and p38 MAPK-dependent pathways. Am. J. Physiol. 291: L734–L746.
The Journal of Immunology 697





























