Embed Size (px)
Citation preview

ORIGINAL ARTICLE
RNAi-mediated ERK2 knockdown inhibits growth of tumor cells in vitro
and in vivo
A Bessard1, C Fremin1, F Ezan1, A Fautrel2, L Gailhouste1 and G Baffet1
1INSERM U522, IFR 140, Hopital Pontchaillou, Universite de Rennes1, Rennes, France and 2Plate-forme d’Histo-Pathologie,IFR140, Universite de Rennes 1 CHU de Rennes, France
The MAPK MEK/ERK pathway is often upregulated incancer cells and represents an attractive target fordevelopment of anticancer drugs. Only few data concern-ing the specific functions of ERK1 and 2 are reported inthe literature. In this report, we investigated the specificrole of ERK1 and 2 in liver tumor growth both in vitro andin vivo. DNA synthesis and cells in S phase analysed byflow cytometry, correlated with strong inhibition of Cdk1and cyclin E levels, are strongly reduced after exposure tothe MEK inhibitor, U0126. We obtained a significantreduction of colony formation in soft agar assays and areduction in the size of tumor xenografts in nude micetreated with U0126. Then, we could specifically abolishedERK1 or 2 expression by small-interfering RNA (siRNA)and demonstrated that ERK2 knockdown but not ERK1interferes with the process of replication. Moreover, wefound that colony formation and tumor growth in vivo
were significantly inhibited by targeting ERK2 usingstable chemically modified siRNA. Taken together, ourresults emphasize the importance of the MEK/ERKpathway in liver cancer cell growth in vitro and in vivo
and argue for a crucial role of ERK2 in this regulation.Oncogene (2008) 27, 5315–5325; doi:10.1038/onc.2008.163;published online 2 June 2008
Keywords: MEK/ERK pathway; MEK inhibitorU0126; RNAi; tumor growth; in vivo
Introduction
Neoplasia is characterized by deregulated cell growth,survival and motility. Among the signaling pathwaysinvolved in these regulations, the MAPK MEK/ERKcascade seems to be a major component implicated inthese biological processes (including proliferation,apoptosis and differentiation). Therefore, both enzymesof the cascade are considered attractive targets forcancer therapy. The ERK1/2 signaling pathwayhas been implicated in the regulation of both G1/S and
G2/M transitions and mitosis in somatic cells. Disrup-tion of ERK2 locus leads to embryonic lethality earlyin mouse development after the implantation stage(Saba-El-Leil et al., 2003), whereas ERK1 mice areviable and fertile (Pages et al., 1999), arguing fordifferent roles of each kinase or a threshold of totalERK activity for viability. The lack of compensationof ERK2 deficiency by ERK1 raises the possibility thatERK2 could be specific in the developing embryo.
In addition, studies using pharmacological inhibitorsof MEK (Alessi et al., 1995; Dudley et al., 1995) oractivated MAPK phosphatase 1, a negative regulator ofMEK (Brondello et al., 1995), significantly altered theability of these cells to proliferate in response to growthfactor stimulation. A number of pharmacological agentshave been described to inhibit MEK/ERK signalingin mammalian cells including PD098059, U0126 andCI-1040 (Wang et al., 2007). A drug-targeting ERK activitydecreases the incidence of colon carcinoma developmentin vivo (Sebolt-Leopold et al., 1999). In a recent report, anovel orally active MEK inhibitor, PD184161, has beentested on hepatocellular carcinoma (HCC) xenograftsand induced partial antitumoral effects in vivo (Kleinet al., 2006). PD184161 significantly suppressed tumorengraftment and initial growth but established tumorswere not significantly affected. Validation of inhibitorsof the MAPK pathway as anticancer therapeutics hasbeen long awaited and CI-1040 became the first MEKinhibitor moved into a clinical trial (Wabnitz et al.,2004; Lorusso et al., 2005). In addition, recent studiessuggest that the antitumor activity of sorafenib in HCCmodels may be attributed to direct effects on tumor cellproliferation/survival in a MEK-dependent and -inde-pendent manners (Liu et al., 2006). Other data stronglysupport the role of the MEK/MAPK pathway in theresistance of breast cancer cells to gefitinib and providethe rationale for novel therapeutic approaches basedon combinations of signal transduction inhibitors(Normanno et al., 2006).
Some recent studies have been conducted onMEK/ERK pathway by RNA interference (RNAi) intumor cell lines suggesting a specific role of each isoform(ERK1/2) to contribute to a regulated cell cycle, motilityand apoptosis progression (Vantaggiato et al., 2006;Bessard et al., 2007). Dominant-negative forms of ERK,and also ERK antisense nucleotides, inhibited prolifera-tion of NIH 3T3 fibroblasts (Pages et al., 1993).
Received 30 October 2007; revised 3 March 2008; accepted 14 April2008; published online 2 June 2008
Correspondence: Dr G Baffet, INSERM U522, IFR 140, HopitalPontchaillou, Universite de Rennes1, rue h le Guillou, Rennes Cedex35033, France.E-mail: [email protected]
Oncogene (2008) 27, 5315–5325& 2008 Macmillan Publishers Limited All rights reserved 0950-9232/08 $32.00
www.nature.com/onc

In FT210 cells, ERK1 depletion caused cell-cycle arrestat G2, whereas the loss of ERK2 induced arrest at G1
(Liu et al., 2004). In NIH 3T3, knockdown of ERK2almost completely abolished normal and Ras-dependentcell proliferation. ERK1 probably affects the overallsignaling output of the cell by antagonizing ERK2activity (Vantaggiato et al., 2006). Recently, we estab-lished that RNAi-mediated inhibition of ERK2 inhuman hepatocarcinoma cells led to strongly reducedcell motility (Bessard et al., 2007). Moreover, inhepatoma cells, activation of nucleotide excision repairpathway through its leading gene ERCC1 was specifi-cally inhibited by knockdown of ERK2 in G1 phase(Andrieux et al., 2007). In normal hepatocytes, wehighlighted that DNA replication is regulated by anERK2-dependent mechanism (Fremin et al., 2007) andERK1 cannot rescue ERK2 deficiency. All these resultssuggest that targeting ERK2 by RNAi could be anattractive target for cancer therapy.
In the present study, we investigated the role of theMAPK cascade in proliferation and regulation of thecell cycle in two liver cancer cell lines: HCC cells (FAO)in vitro and a biliary epithelial cell line (FI) in vitro andin vivo. The results provide evidence that treatment ofthese cells by the chemical inhibitor U0126, ledto inhibition of ERK activity with a G1 cell-cyclearrest. Moreover, using stable RNAi, we assessed theconsequence of silencing ERK2 on tumor cell prolifera-tion. Our results emphasize the importance of theMEK/ERK pathway in tumor growth and argue fora crucial role of ERK2 in this regulation in vitro andin vivo.
Results
Proliferation of hepatocarcinoma cells is mediated by theMEK/ERK cascadeRat hepatocarcinoma cells (FAO) were stimulated ornot by 10% fetal calf serum (FCS) in presence orabsence of the MEK inhibitor, U0126. We firstobserved, after serum stimulation, that ERK1/2 weretransiently phosphorylated (Figures 1a and b) andsecond that ERK1/2 activation was completely abol-ished in presence of the MEK inhibitor (Figure 1b).DNA replication was strongly inhibited in presence ofMEK inhibitor (Figure 1c). This inhibition is not toxicand reversible as DNA synthesis was restored afterU0126 removal. We ascertained that MEK-inhibitedcells did not progress in apoptosis because U0126 didnot induce caspase-3/7 activity measured through theproteolytic DEVD-AMC cleavage (Figure 1d).
Flow cytometry was used to assess the cell cycle phasedistributions of the cells treated or not with U0126(Figure 1e). Cells have been collected 30 h afterstimulation by FCS in presence or absence of U0126or 15 h after removal of the inhibitor (U0126 reversion45 h). Cells stimulated by FCS exhibited a significantproportion in S phase (32.62%) whereas U0126 stronglydecreased the proportion of cells in S phase (9.92%) andincreased the proportion of cells in G0–G1 phase and to
a lesser extent in G2/M. We confirmed that theblockade is reversible because 15 h after U0126 removal,cells progressed in S phase (27.15%). All these resultsprovide evidence that the MAPK cascade MEK/ERKis mostly involved in S phase progression of hepato-carcinoma cells.
In primary culture of normal hepatocytes, expressionof Cdk1, one marker of G1/S phase transition, isregulated by the MEK/ERK pathway (Albrecht et al.,1993; Rescan et al., 2001; Huynh et al., 2003). Therefore,we looked at the regulation of this protein in trans-formed FAO cells and showed that Cdk1 expression wassignificantly blocked when cells were treated with U0126(Figure 1b). Another marker of progression through Sphase, cyclin E, showed the same MAPK-dependentregulation.
ERK2 is required for DNA replicationTo estimate the specific role of ERK1 and 2 in hepatomacell cycle replication, MAPK inhibition was performedby siRNA. At 72 and 96 h after transfection, the siERK2inhibited protein expression by 85±5% compared withcontrol transfection experiments. Moreover, the levelof phospho-ERK2 was markedly reduced in these cells.ERK1 expression/phosphorylation was not affectedshowing the specificity of this inhibition for ERK2.The decrease in ERK2 expression/phosphorylationcorrelated with a decrease of Cdk1 expression. Interest-ingly, a decrease in DNA synthesis (50 and 20%,48–72 h and 72–96 h, respectively, after transfection) hasbeen obtained (Figure 2a), indicating that ERK2contributes at least partly, to the replicating propertyof the pathway.
We targeted ERK1 by the same way. Western blotanalysis showed a fall of 80% in ERK1 expression,without affecting ERK2, and a strong decrease ofERK1 phosphorylation 72 and 96 h after transfection(Figure 2b). ERK1 knockdown had no effect on Cdk1expression and DNA replication was not affectedshowing that ERK1 is dispensable for full DNA synthesisas previously demonstrated in normal wild-type (wt) andERK1�/� hepatocytes (Fremin et al., 2007).
Inhibition of MEK/ERK cascade activation acts on tumorcell growth in vivoTo study the role of MEK/ERK in the regulation ofthe proliferation of transformed liver cells in vivo, weanalysed this regulation in a highly tumorigenic cell line.Indeed, FI cells are able to grow in soft agar and givetumors in vivo after subcutaneous injection in mice.First, we confirmed the transformed phenotype of theFI cells by looking at colony formation in soft agar(Figure 3a). Then, the MEK inhibitor was added or notto cell culture 1 h prior FCS stimulation (Figure 3b). Weperformed dose–response experiments in presence ofU0126 (Figure 3c). A high to a complete inhibition ofERK phosphorylation was obtained in presence of 10and 50 mM, respectively. Moreover, phosphorylation ofp38 was never affected by U0126 treatment, showing theabsence of cross talks between MEK/ERK and p38
ERK2 targeting inhibits tumor growth
A Bessard et al
5316
Oncogene

pathways (Figure 3c). Then, results evidenced thatreplication of FI cells was regulated by the MAPKcascade (Figure 3d), and a dose–response inhibition wasobtained with a complete inhibition at 50 mM. Weconfirmed this role of MEK/ERK with the analysis ofcolony formation in presence of U0126 (Figure 3e).Indeed, the MEK inhibitor induced a strong inhibitionon the ability of FI cells to form colonies in soft agar.
The promising finding that U0126 strongly inhibitedERK1/2 activation, replication and colony formation
in vitro prompted us to investigate its effect on tumorformation in nude mice. We analysed, first, thetumorigenicity of FI cells in vivo. Rapidly, 2–3 daysafter inoculation, mice developed solid spheroid tumors.Thus, 20 mm sections of tumors confirmed the presenceof DiA-labeled cells (Figure 4a).
Second, we tried to determine whether intraperitonealinjections of U0126 could potentially inhibit tumorengraftment and growth (Figure 4b). At 3 h after cellinjection subcutaneously, mice were treated daily with
Figure 1 Inhibition of ERK2 activation and DNA replication by the specific MEK/ERK inhibitor U0126. FAO cells werepreincubated for 1 h with solvent control (0.5% dimethyl sulfoxide, DMSO) or MEK inhibitor (50 mM U0126) prior to stimulation by10% fetal calf serum (FCS). (a, b) Total cell lysates were collected at the indicated times after stimulation, T0. The blot was probedwith antiphospho-ERK, -Cdk1, -cyclin E and with a mixture of equal ratios of anti-ERK1 and -ERK2 antibodies. (c) [3H] Methyl-thymidine incorporation was analysed at the indicated times after stimulation. Inhibitor was removed 30 h after treatment (reversion,Rev) and then, cells were cultured in the presence of FCS alone (U0126 Rev 30 h). (d) DEVD-AMC caspase activity was investigated atthe indicated times after stimulation. Data represent the mean±s.d. Control are normal rat hepatocytes cultures in absence of growthfactor (basal), stimulated with epidermal growth factor (EGF) or treated by U0126 (EGFþU0126). (e) Cells were harvested 30 and45 h after treatment, fixed, stained with propidium iodide and analysed for DNA staining profiles by flow cytometry. For the reversionexperiment, inhibitor was removed 30 h after treatment and cells were analysed 15 h later in presence of FCS (U0126 Rev 45 h).
ERK2 targeting inhibits tumor growth
A Bessard et al
5317
Oncogene

U0126 (10.5mg/kg) or solvent control (DMSO) byintraperitoneal injection. In control experiment, tumorsizes were constant or slightly increase all over thekinetic. At the opposite, in all U0126 experiments,engraftment and early tumor growth were significantlydecreased. Furthermore, a 60–70% reduction in thevolume of tumors treated with U0126 was obtained 9days after injection and thereafter. No treatment-relateddeaths of mice or toxicity were associated with U0126or dimethyl sulfoxide (DMSO) injections. Western blotanalysis of tumors indicated that U0126 induceda decrease in ERK1/2 phosphorylation (Figure 4c).Moreover, Cdk1 expression was also strongly reduced inU0126-treated mice confirming lower S phase entry inMAPK-inhibited tumor xenografts.
Our results showed that inhibition of tumor growthwas reversible and 3 days after U0126 removal, anincrease of tumor growth started to be observed(Figure 4d). Moreover, 10 days after the beginning ofreversion experiments (Figure 4e, right), ERK1/2 werephosphorylated and Cdk1 expressed in tumors,confirming the reversion of the U0126 treatment(Figure 4e, left).
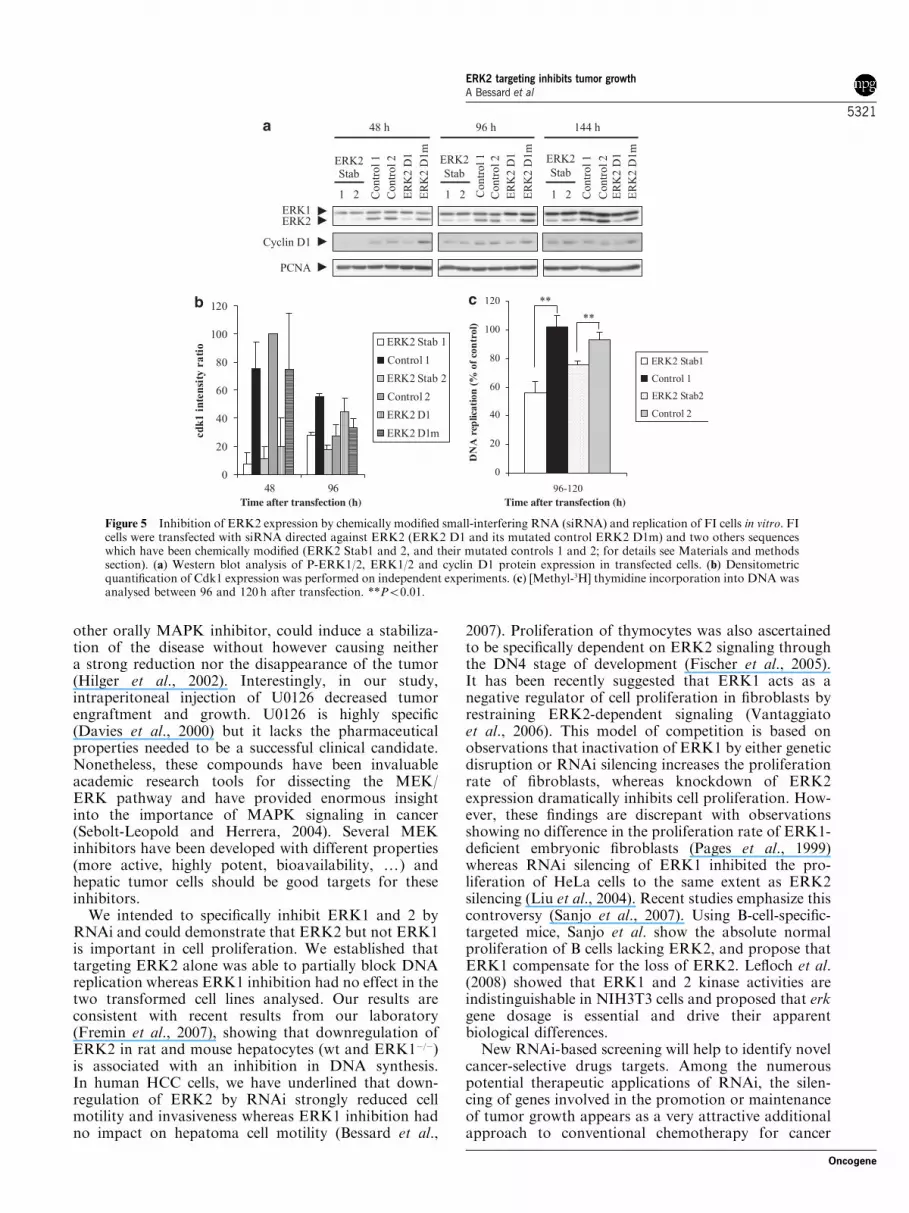
Specific ERK2 targeting by RNA interference inhibitstumor growth in vivoTo assess the role of ERK2 in tumor cell growth in vivo,FI cells were transfected with siRNA directed against
ERK2 (ERK2 D1 and its mutated control ERK2 D1m)and two others sequences against ERK2 that have beenchemically modified (ERK2 Stab1 and 2, and theirmutated control, controls 1 and 2). These locked nucleicacid (LNA) contain a methylene bridge connectingthe 20 oxygen of the ribose with the 40 carbon. Thebicyclic structure locks the furanose ring of the LNAmolecule in a 30-endo conformation, thereby structurallymimicking the standard RNA monomers. Westernblot analysis (Figure 5a) showed the high efficiencyof ERK2 silencing 2, 4 and 6 days after transfection.Quantification studies of ERK2/PCNA indicatedthat ERK2 inhibitions were improved (92±5% ofinhibition) when cells have been transfected withLNA sequences (ERK2 Stab1 and 2) as comparedto unmodified sequence (ERK2 D1; 40±3% of ERK2inhibition), 144 h after transfection. No significantdifferences could be noticed between the two chemicallymodified sequences and no changes with all controlmutated sequences. In parallel, we looked at cellcycle markers expressions of cyclin D1 (Figure 5a) andCdk1 (Figure 5b), two markers which are expressed,respectively, at late G1 and S phase in normalhepatocytes and in HCC (Albrecht et al., 1993, 1995;Ito et al., 1998; Rescan et al., 2001). Our resultsshowed that cyclin D1 inhibition was partially inhibitedin transfected cells. Moreover, Cdk1 expressionsignificantly dropped 48 and 96 h after ERK2 silencingwith ERK2 Stab1.
Figure 2 Influence of siERK2 or siERK1 on ERK1/2 protein expression and DNA synthesis in hepatocarcinoma cells. (a, b) FAOcells were transfected with siERK2 (a) or siERK1 (b) duplex or small-interfering RNA (siRNA) control, and harvested 72 and 96 hafter transfection. Immunoblots were probed with antiphospho-ERK1/2, -ERK1/2 and -Cdk1 antibodies (left panel). Proliferating cellnuclear antigen (PCNA) was used as a loading control. DNA synthesis were analysed at the indicated times after transfection andexpressed as % of control (right panel). Statistical significance from the control, *Po0.05.
ERK2 targeting inhibits tumor growth
A Bessard et al
5318
Oncogene

Then, we determined the effects of ERK2 inhibition inthe replication process (Figure 5c). A decrease of 45 and30% has been obtained with Stab1 and 2, respectively,showing that the reduction of ERK2 expression in FIcells could be related with a decrease of cell proliferationas determined in FAO cells. The effects of ERK2silencing on cellular growth were evidenced by colonyformation assays. Transfection with ERK2 RNAiinduced a high decrease on the ability to form colonies:a 85 and 70% decrease for Stab1 and 2, respectively,but only 6% reduction for the non-LNA duplex(ERK2 D1), 13 days after seeding (Figures 6A and B).Control mutated sequences had no effect on theclonogenic capacity of the transfected cells.
The promising findings that inhibition of ERK2expression by RNAi strongly inhibited colony forma-tion led us to investigate its effect on tumor formation innude mice, in vivo. ERK2 Stab1 siRNA and its mutatedcontrol were transfected, and 48 h after transfection,2� 106 cells were inoculated subcutaneous into nudemice. Interestingly, cells transfected with Stab1 grew atlower rates than those transfected with the mutatedcontrol (Figure 6C) and a 90% reduction in the volume
was measured 13 days postinjection. Finally, tumorcells were laser capture microdissected from sectionsof tumor using a Veritas LCM system (Arcturus)(Figure 6D). Proteins were extracted from the capturedcells (Figure 6D, c) and phospho-ERK1/2 analysed byimmunoblotting (Figure 6E). Our results showed thatthe level of phospho-ERK2 was markedly reduced intumors, 13 days after injection in nude mice of cellstransfected with the ERK2 Stab1 siRNA and arguedfor a sustained inhibition of ERK2 after transfectionwith LNA.
Discussion
In the present report, we focused on the specific andcritical role of ERK1 and 2 in the regulation ofproliferation in vitro and tumorigenicity in vivo, usingtwo experimental approaches: MEK inhibition bychemical inhibitor, ERK1 or 2 knockdown by siRNAs.The p44 ERK1 and p42 ERK2 have attracted intenseresearch interest because of their involvement in theregulation of cell proliferation and survival (Pages et al.,
Figure 3 U0126 inhibits growth of highly tumorigenic liver cells in vitro, and colony formation in soft agar. (a) Colony formation ofliver epithelial cells (FI) in soft agar, 7 and 12 days after seeding as described in Materials and methods section. (b) Kinetic of ERK1/2phosphorylation after MEK inhibition. FI cells were treated or not with U0126 (50mM) and analysed at the indicated times aftertreatment. (c) ERK1/2 (P-ERK) and p38 (P-p38) activations were analysed 0.5 h after fetal calf serum (FCS) stimulation in presence ofincreasing concentrations of U0126 (from 10 to 50 mM). (d) For DNA synthesis experiment, a dose-dependent inhibition was performedat the indicated times after U0126 treatment. (e) Cells were seeded in soft agar in presence or absence of U0126 (50mM). MEK inhibitoror solvent control (dimethyl sulfoxide, DMSO) was renewed each day and colony formation was observed 2 and 7 days after seeding.
ERK2 targeting inhibits tumor growth
A Bessard et al
5319
Oncogene

1993; Vantaggiato et al., 2006). There is increasingevidence that this pathway is abnormally regulated intransformed cells and could be important in tumorigen-esis and maintenance of liver tumor growth (Schmidtet al., 1997; Huynh et al., 2003). In addition, werecently demonstrated that motility and invasiveness ofhepatocarcinoma cells required MEK/ERK activation(Bessard et al., 2007). In this work, we demonstrated theimportance of the MEK/ERK pathway in cancer livercells proliferation in vitro and in vivo and argued for acrucial role of ERK2 in this regulation.
First, proliferation have been analysed in vitro bytargeting MEK with the chemical inhibitor, U0126. Ourresults confirm the importance of the MAPK ERK1/2pathway in the regulation of proliferation of the twotransformed hepatic cell lines analysed, similar to thatpreviously described in normal rat and mouse hepato-cytes (Talarmin et al., 1999; Fremin et al., 2007).Moreover, this inhibition of proliferation and theblockade in G1/S phase are reversible and not toxicin agreement with a nonapoptotic engagement of
MEK-inhibited cells as measured by terminal caspasesactivity. Similar observations were obtained inpancreatic cancer cells lines where MEK inhibition ledto a cessation of cell proliferation accompanied by aG0–G1 cell-cycle arrest without a significant short-termincrease in apoptosis (Gysin et al., 2005).
Much of the focus in the development of noveltherapeutics has involved inhibitors of signal transduc-tion molecules, in particular protein kinases (Arslanet al., 2006; Sebolt-Leopold and English, 2006). MEK-targeted treatment has been studied in colon, pancreatic,breast or melanoma tumors (Sebolt-Leopold et al., 1999;Duesbery et al., 2001; Yeh et al., 2007; Haass et al.,2008) and recently in human HCC tumors (Klein et al.,2006). Targeting MEK with PD184161, which has theobvious advantages of solubility and oral bioavailabil-ity, in vivo resulted in a significant delay in HCC tumorengraftment and inhibition of early tumor growth. Incontrast, PD184161 was ineffective at suppressingestablished HCC tumor growth. This result is inagreement with the report showing that PD184352, an
Figure 4 Effect of MEK inhibition by U0126 treatment on tumor cell growth in vivo. FI cells were labeled with a stable fluorescent dye(DiA) before subcutaneous injection into female nude mice (see Materials and methods section). (a) Under a fluorescent microscope20 mm sections of tumors were observed (� 20). (b) At 3 h after cells injection, mice were treated with U0126 (10.5mg/kg per day) orsolvent control (dimethyl sulfoxide, DMSO) in i.p. for 11 consecutive days. Tumor volumes were daily measured with calliper andcalculated from width (W) and length (L) by the equation (V¼W2
�L/2) as described in Materials and methods section. The meansand s.d. values of tumor volumes were calculated from nine mice for each condition. **Po0.01, statistical significance from thecontrol. (c) Western blot analysis of P-ERK1/2, ERK1/2 and Cdk1 expressions performed on protein samples isolated from fourindividual tumors coming from control (left) or U0126 (right)-treated mice, 11 days after injection. (d, e) Reversion experiments:treatment with the inhibitor was stopped 10 days postinjection and tumor sizes were measured up to 10 days after inhibitor removal(that is, 19 days after injection) (d). (e) Homogenates were prepared from one tumor coming from U0126 treated mice 9 days afterinjection (left) and two individual tumors 10 days after inhibitor removal (19 days after injection) (right) and assessed by western blotanalysis for phosphorylated ERK1/2, ERK1/2 and Cdk1 levels. All samples are from the same blot.
ERK2 targeting inhibits tumor growth
A Bessard et al
5320
Oncogene
other orally MAPK inhibitor, could induce a stabiliza-tion of the disease without however causing neithera strong reduction nor the disappearance of the tumor(Hilger et al., 2002). Interestingly, in our study,intraperitoneal injection of U0126 decreased tumorengraftment and growth. U0126 is highly specific(Davies et al., 2000) but it lacks the pharmaceuticalproperties needed to be a successful clinical candidate.Nonetheless, these compounds have been invaluableacademic research tools for dissecting the MEK/ERK pathway and have provided enormous insightinto the importance of MAPK signaling in cancer(Sebolt-Leopold and Herrera, 2004). Several MEKinhibitors have been developed with different properties(more active, highly potent, bioavailability, y) andhepatic tumor cells should be good targets for theseinhibitors.
We intended to specifically inhibit ERK1 and 2 byRNAi and could demonstrate that ERK2 but not ERK1is important in cell proliferation. We established thattargeting ERK2 alone was able to partially block DNAreplication whereas ERK1 inhibition had no effect in thetwo transformed cell lines analysed. Our results areconsistent with recent results from our laboratory(Fremin et al., 2007), showing that downregulation ofERK2 in rat and mouse hepatocytes (wt and ERK1�/�)is associated with an inhibition in DNA synthesis.In human HCC cells, we have underlined that down-regulation of ERK2 by RNAi strongly reduced cellmotility and invasiveness whereas ERK1 inhibition hadno impact on hepatoma cell motility (Bessard et al.,
2007). Proliferation of thymocytes was also ascertainedto be specifically dependent on ERK2 signaling throughthe DN4 stage of development (Fischer et al., 2005).It has been recently suggested that ERK1 acts as anegative regulator of cell proliferation in fibroblasts byrestraining ERK2-dependent signaling (Vantaggiatoet al., 2006). This model of competition is based onobservations that inactivation of ERK1 by either geneticdisruption or RNAi silencing increases the proliferationrate of fibroblasts, whereas knockdown of ERK2expression dramatically inhibits cell proliferation. How-ever, these findings are discrepant with observationsshowing no difference in the proliferation rate of ERK1-deficient embryonic fibroblasts (Pages et al., 1999)whereas RNAi silencing of ERK1 inhibited the pro-liferation of HeLa cells to the same extent as ERK2silencing (Liu et al., 2004). Recent studies emphasize thiscontroversy (Sanjo et al., 2007). Using B-cell-specific-targeted mice, Sanjo et al. show the absolute normalproliferation of B cells lacking ERK2, and propose thatERK1 compensate for the loss of ERK2. Lefloch et al.(2008) showed that ERK1 and 2 kinase activities areindistinguishable in NIH3T3 cells and proposed that erkgene dosage is essential and drive their apparentbiological differences.
New RNAi-based screening will help to identify novelcancer-selective drugs targets. Among the numerouspotential therapeutic applications of RNAi, the silen-cing of genes involved in the promotion or maintenanceof tumor growth appears as a very attractive additionalapproach to conventional chemotherapy for cancer
Figure 5 Inhibition of ERK2 expression by chemically modified small-interfering RNA (siRNA) and replication of FI cells in vitro. FIcells were transfected with siRNA directed against ERK2 (ERK2 D1 and its mutated control ERK2 D1m) and two others sequenceswhich have been chemically modified (ERK2 Stab1 and 2, and their mutated controls 1 and 2; for details see Materials and methodssection). (a) Western blot analysis of P-ERK1/2, ERK1/2 and cyclin D1 protein expression in transfected cells. (b) Densitometricquantification of Cdk1 expression was performed on independent experiments. (c) [Methyl-3H] thymidine incorporation into DNA wasanalysed between 96 and 120h after transfection. **Po0.01.
ERK2 targeting inhibits tumor growth
A Bessard et al
5321
Oncogene

treatment (Leung and Whittaker, 2005). However, fewdata have been published reporting the successful use ofsiRNA for gene silencing in developing tumors in vivo in
rodent models systems (Aharinejad et al., 2004; Takeiet al., 2004). Indeed, the biggest obstacle to developingsiRNA-based therapies is the delivery of the siRNA
Figure 6 ERK2 targeting by RNA interference inhibits colony formation in soft agar and tumor growth in vivo. (A) Representativephotographs of colonies in soft agar after transfection with small-interfering RNA (siRNA) against ERK2 or mutated controls.(B) Quantitative analysis of colony area in all conditions of transfection at the indicated times after seeding. Student’s t-test was appliedfor statistical analysis between ERK2 Stab1 and control 1 or ERK2 Stab2 and control 2. *Po0.05, **Po0.01 and ***Po0.001.(C) FI cells transfected with siRNA chemically modified sequence (ERK2 Stab1) and its mutated control (control 1) were injectedsubcutaneously into nude mice. Tumor volumes were daily measured with calliper and calculated as previously described. The meansand s.d. values of tumor volumes were calculated from nine mice for each condition. *Po0.05, **Po0.01 and ***Po0.001.(D) Tumor cells were laser capture microdissected from fluorescent tumor sections. Images of precapture (a), post-capture (b) andthe captured cells (c) are shown. (E) Western blot analysis of ERK1/2 activations in tumors, 13 days after injection in nude mice, thatis, 15 days after transfection of cells with chemically modified siRNA (ERK2 Stab1) and its mutated control (control 1). HSC 70 wasused as a loading control.
ERK2 targeting inhibits tumor growth
A Bessard et al
5322
Oncogene

molecules to the target tissue. Experiments have beendone in vivo including hydrodynamic injection ofsynthetic siRNA, delivery using lipid-based agents andvarious carriers such as atelocollagen and polyethyle-neimine, and also local administration (Behlke, 2006). Inour study, we improved strength and effectiveness ofinhibition in vivo with LNA chemical modificationsof siRNA. LNAs showed extraordinary thermalstabilities when hybridized with DNA, RNA or LNA itself.Moreover, introduction of LNA into siRNA couldresult in significantly less off target-regulated genes(Elmen et al., 2005; Dahlgren et al., 2006; Mook et al.,2007). In agreement with other reports (Braasch et al.,2003; Elmen et al., 2005; Mook et al., 2007), we showedthat our ERK2 LNA are substantially compatible withthe siRNA machinery, exhibit improved biostability andenhance inhibition in vivo. Indeed, at least 15 days aftertransfection, phospho-ERK2 inhibition was observedwith the ERK2 Stab1, in vivo. Our data strengthen theusefulness of LNA-modified siRNA as an effective toolin functional genomics and possible future therapeuticsapplications in vivo.
In summary, this study shows that silencing ERK2 iseffective against a model of tumoral growth in nudemice. Importantly, the reduction and the blockade oftumor growth observed after effective silencing ofendogenous ERK2, reported here, strongly supportsits potential therapeutic application. Obviously, identi-fication of new molecules like siRNA that couldinterfere with specific targeted protein kinase shouldprovide better and more rational treatments of cancer,in association with conventional treatment such aschemotherapy and/or radiotherapy.
Materials and methods
Cell lines and treatmentsRat hepatoma cell line FAO (Reuber, 1961) was maintainedin a mixture of 50% Ham’s F12 and 50% NCTC 135,supplemented with penicillin (100 IU/ml), streptomycin(100mg/ml), glutamin (100mg/ml) and 10% of FCS. FI ratbiliary epithelial cells were obtained from liver of 10-day-oldFischer (Morel-Chany et al., 1978). FI cells were grown inWilliams’ E medium containing sodium carbonate (22mg/ml),penicillin (100 IU/ml), streptomycin (100mg/ml) and 7.5%of FCS. All cells were maintained at 37 1C under a humidified5% CO2 atmosphere.
Immunoblotting analysisTotal protein (30 mg) was resolved on a 10% polyacrylamidegel and transferred to nitrocellulose membranes for 1 h aspreviously described (Rescan et al., 2001). Membranes wereincubated overnight at 4 1C with the following primaryantibody in Tris-buffered saline (pH 7,4): a-phospho-ERK1/2 and a-phospho-p38 (Thr180/Tyr182) (Cell Signaling, SaintQuentin en Yvelines, France); a-pan ERK1 (sc-94) and ERK2(sc-154), a-cyclin E (sc-481) and a-HSC 70 (sc-7298) (SantaCruz Biotechnology, Santa Cruz, CA, USA); a-PCNA (Dako,Trappes, France); a-cyclin D1 (Neomarkers, Westinghouse,CA, USA). Anti-Cdk1 is a polyclonal antiserum specificallydirected against the C-terminal part of human p34 (Loyeret al., 1996). Antigen–antibody complexes were visualized
using the SuperSignal Ultra Chemiluminescent Substrateprocedure (Pierce, Rockford, IL, USA). Densitometricanalysis of the bands was performed using Bioprofil Bio-1D(Vilbert-Lourmat, Marne-La-Vallee, France) software.
[3H] Thymidine incorporationThe rate of DNA synthesis was measured in cells, by adding2 mCi of [methyl-3H] thymidine (5Ci/mmol) (Amersham,Buckinghamshire, UK) for given periods of time prior totreatments and precipitation with ice-cold trichloroaceticacid (5%).
Caspase activity assayCells were lysed in the caspase activity buffer as previouslydescribed (Stennicke and Salvesen, 1997). Caspase-mediatedcleavage of substrate AMC was measured by spectrofluoro-metry (Molecular Devices, Wokingham, UK) at the excitation/emission wavelength pair of 380/440 nm. The caspase activitywas expressed in Vmax.
Flow cytometry cell-cycle analysisCell-cycle phase distribution was determined by flowcytometry. Cells were plated at a density of 3� 105 cells per35mm diameter dishes and treated or not with MEK inhibitor,U0126 (Promega, Charbonnieres, France) for indicatedtimes. DNA stained with propidium iodide (CycleTest PlusDNA reagent kit, BD Biosciences, San Jose, CA, USA) wasthen quantitated through the FACScalibur flow cytometer.Each measurement was conducted on 10 000 events andanalysed on Cell Quest and Modfit Mac V2 Softwares(BD Biosciences).
Transfection and small interference experimentsCells were plated at the density of 3.5� 105 FAO cellsor 2� 105 FI cells in 35mm diameter dishes and after24 h, transfected for 15 h, for FAO or 5 h for FI cellline according to manufacturer’s recommendations with200 ml OPTIMEM, 5 ml transfectine (Bio-Rad, Hercules, CA,USA), 5% FCS and 5 ml of 20 mM small-interfering RNA(siRNA).Two siRNA against murin ERK2 (ERK2: 50-GUGCUGUG
UCUUCAAGAGC-30; control: 50-GGUGCCAUGGAACAGGUUG-30) and two against murin ERK1 (ERK1: 50-UCCAAGGGCUACACCAAAU-30; control. 50-UGUUAUAGGCAUCCGAGAC-30) were established according to Elbashiret al. (2002) and purchased from Eurogentec (Seraing,Belgium).Two LNA oligonucleotides against ERK2 (ERK2
Stab1 and 2) and their mutated control (controls 1 and 2)were kindly provided by Heike Lehrmann, Sigma-Aldrich(St Louis, MO, USA). These sequences contain two [20-O,40-C methylene bridge] on the 50 end of the sense strand whichrestrict the flexibility of the ribofuranose ring and lock thestructure into a rigid bicyclic formation.
Colony formation assayColony formation assay was determined by a two-layer agarsystem. Biliary epithelial cells were plated in a 35mm diameterdishes at the density of 1.5� 105 cells per dish. Colonies werevisualized daily. In U0126 experiments, cells were suspended inpresence of 50 mM U0126 or DMSO (solvent control) (Sigma,St Louis, MO, USA) at a final concentration of 0.5%. Everyday, U0126 or DMSO was added.
ERK2 targeting inhibits tumor growth
A Bessard et al
5323
Oncogene

AnimalsAthymic female nude mice (SWISS, nu/nu, Iffa-Credo,L’Arbresle, France) were purchased from germ free at 2–3weeks old. Mice were housed in microisolator units undercontrolled humidity and temperature, and 12 h light–darkcycle. Animals received a standard sterilizable laboratory diet(UAR, France). Experiments were carried out in accordancewith French laws and regulations.
Primary tumor growthPrior to injection, FI cells were labeled with a stablefluorescent dye molecule, DiA (Molecular Probes, CergyPontoise, France) at 10mg/ml for 5 h at 37 1C. After washingto remove free DiA, cells were trypsinized for inoculation(U0126 experiments) or transfection (RNAi experiments).Biliary epithelial cells were injected subcutaneously, at theindicated times, into the tibia of nude mice. In the chemicalexperiments, 3 h after inoculation, mice were treated withU0126 (10.5mg/kg) daily by intraperitoneal injection. Thelength and width of each tumor were measured every day byusing a caliper. The following formula was used to calculatetumor volumes¼width2
� length/2. Mice were killed at the endof experiment. Tumors were immediately frozen in liquidnitrogen.
Laser capture microdissectionTumor cells were laser capture microdissected by using aVeritas LCM system (Arcturus) from fluorescent tumorsections. Microdissected samples were dropped by gravityinto cap-tubes under microscope inspection. Total protein
was extracted from the captured cells and analysed byimmunoblotting.
Statistical analysisData are presented as means±s.d. from at least threeexperiments. The statistical analyses were carried outusing Student’s t-test. A P-value o0.05 was considered asstatistically significant.
Abbreviations
cdk, cyclin-dependent kinase; ERK, extracellular signal-regulated kinase; HCC, hepatocellular carcinoma; LNA,locked nucleic acid; MEK, mitogen-activated protein kinase;siRNA, small-interfering RNA.
Acknowledgements
We thank Dr P Loyer for flow cytometry advices, Sigma-Aldrich and Dr Heike Lehrmann for giving us the LNA ERK2oligonucleotides, and C Ribault for technical assistance. Thisresearch was supported by the Institut National de la Santeet de la Recherche Medicale and the Association pour laRecherche sur le Cancer (ARC). A Bessard and C Fremin are arecipient of fellowship from the Region Bretagne/INSERMand the Ministere de l0Education Nationale et de laTechnologie, respectively, and the ARC.
References
Aharinejad S, Paulus P, Sioud M, Hofmann M, Zins K, Schafer R
et al. (2004). Colony-stimulating factor-1 blockade by antisense
oligonucleotides and small interfering RNAs suppresses growth
of human mammary tumor xenografts in mice. Cancer Res 64:
5378–5384.
Albrecht JH, Hoffman JS, Kren BT, Steer CJ. (1993). Cyclin and
cyclin-dependent kinase 1 mRNA expression in models of regener-
ating liver and human liver diseases. Am J Physiol 265: G857–G864.
Albrecht JH, Hu MY, Cerra FB. (1995). Distinct patterns of cyclin D1
regulation in models of liver regeneration and human liver. Biochem
Biophys Res Commun 209: 648–655.
Alessi DR, Cuenda A, Cohen P, Dudley DT, Saltiel AR. (1995). PD
098059 is a specific inhibitor of the activation of mitogen-activated
protein kinase kinase in vitro and in vivo. J Biol Chem 270:
27489–27494.
Andrieux LO, Fautrel A, Bessard A, Guillouzo A, Baffet G,
Langouet S. (2007). GATA-1 is essential in EGF-mediated
induction of nucleotide excision repair activity and ERCC1
expression through ERK2 in human hepatoma cells. Cancer Res
67: 2114–2123.
Arslan MA, Kutuk O, Basaga H. (2006). Protein kinases as drug
targets in cancer. Curr Cancer Drug Targets 6: 623–634.
Behlke MA. (2006). Progress towards in vivo use of siRNAs.
Mol Ther 13: 644–670.
Bessard A, Fremin C, Ezan F, Coutant A, Baffet G. (2007).
MEK/ERK-dependent uPAR expression is required for motility
via phosphorylation of P70S6K in human hepatocarcinoma cells.
J Cell Physiol 212: 526–536.
Braasch DA, Jensen S, Liu Y, Kaur K, Arar K, White MA et al.
(2003). RNA interference in mammalian cells by chemically
modified RNA. Biochemistry 42: 7967–7975.
Brondello JM, McKenzie FR, Sun H, Tonks NK, Pouyssegur J.
(1995). Constitutive MAP kinase phosphatase (MKP-1) expression
blocks G1 specific gene transcription and S-phase entry in
fibroblasts. Oncogene 10: 1895–1904.
Dahlgren C, Wahlestedt C, Thonberg H. (2006). No induction of
anti-viral responses in human cell lines HeLa and MCF-7 when
transfecting with siRNA or siLNA. Biochem Biophys Res Commun
341: 1211–1217.
Davies SP, Reddy H, Caivano M, Cohen P. (2000). Specificity and
mechanism of action of some commonly used protein kinase
inhibitors. Biochem J 351: 95–105.
Dudley DT, Pang L, Decker SJ, Bridges AJ, Saltiel AR. (1995). A
synthetic inhibitor of the mitogen-activated protein kinase cascade.
Proc Natl Acad Sci USA 92: 7686–7689.
Duesbery NS, Resau J, Webb CP, Koochekpour S, Koo HM, Leppla
SH et al. (2001). Suppression of ras-mediated transformation and
inhibition of tumor growth and angiogenesis by anthrax lethal
factor, a proteolytic inhibitor of multiple MEK pathways. Proc Natl
Acad Sci USA 98: 4089–4094.
Elbashir SM, Harborth J, Weber K, Tuschl T. (2002). Analysis of gene
function in somatic mammalian cells using small interfering RNAs.
Methods 26: 199–213.
Elmen J, Thonberg H, Ljungberg K, Frieden M, Westergaard M, Xu
Y et al. (2005). Locked nucleic acid (LNA) mediated improvements
in siRNA stability and functionality. Nucleic Acids Res 33: 439–447.
Fischer AM, Katayama CD, Pages G, Pouyssegur J, Hedrick SM.
(2005). The role of erk1 and erk2 in multiple stages of T cell
development. Immunity 23: 431–443.
Fremin C, Ezan F, Boisselier P, Bessard A, Pages G, Pouyssegur J
et al. (2007). ERK2 but not ERK1 plays a key role in hepatocyte
replication: an RNAi-mediated ERK2 knockdown approach in
wild-type and ERK1 null hepatocytes. Hepatology 45: 1035–1045.
Gysin S, Lee SH, Dean NM, McMahon M. (2005). Pharmacologic
inhibition of RAF–MEK–ERK signaling elicits pancreatic cancer
cell cycle arrest through induced expression of p27Kip1. Cancer Res
65: 4870–4880.
Haass NK, Sproesser K, Nguyen TK, Contractor R, Medina CA,
Nathanson KL et al. (2008). The mitogen-activated protein/
extracellular signal-regulated kinase kinase inhibitor AZD6244
ERK2 targeting inhibits tumor growth
A Bessard et al
5324
Oncogene

(ARRY-142886) induces growth arrest in melanoma cells and
tumor regression when combined with docetaxel. Clin Cancer Res
14: 230–239.
Hilger RA, Scheulen ME, Strumberg D. (2002). The Ras-Raf-MEK-
ERK pathway in the treatment of cancer. Onkologie 25: 511–518.
Huynh H, Nguyen TT, Chow KH, Tan PH, Soo KC, Tran E. (2003).
Over-expression of the mitogen-activated protein kinase (MAPK)
kinase (MEK)-MAPK in hepatocellular carcinoma: its role in tumor
progression and apoptosis. BMC Gastroenterol 3: 19.
Ito Y, Sasaki Y, Horimoto M, Wada S, Tanaka Y, Kasahara A et al.
(1998). Activation of mitogen-activated protein kinases/extracellular
signal-regulated kinases in human hepatocellular carcinoma.
Hepatology 27: 951–958.
Klein PJ, Schmidt CM, Wiesenauer CA, Choi JN, Gage EA,
Yip-Schneider MT et al. (2006). The effects of a novel MEK
inhibitor PD184161 on MEK-ERK signaling and growth in human
liver cancer. Neoplasia 8: 1–8.
Lefloch R, Pouyssegur J, Lenormand P. (2008). Single and combined
silencing of ERK1 and ERK2 reveals their positive contribution to
growth signaling depending on their expression levels. Mol Cell Biol
28: 511–527.
Leung RK, Whittaker PA. (2005). RNA interference: from gene
silencing to gene-specific therapeutics. Pharmacol Ther 107: 222–239.
Liu L, Cao Y, Chen C, Zhang X, McNabola A, Wilkie D et al. (2006).
Sorafenib blocks the RAF/MEK/ERK pathway, inhibits tumor
angiogenesis, and induces tumor cell apoptosis in hepatocellular
carcinoma model PLC/PRF/5. Cancer Res 66: 11851–11858.
Liu X, Yan S, Zhou T, Terada Y, Erikson RL. (2004). The MAP
kinase pathway is required for entry into mitosis and cell survival.
Oncogene 23: 763–776.
Lorusso PM, Adjei AA, Varterasian M, Gadgeel S, Reid J, Mitchell
DY et al. (2005). Phase I and pharmacodynamic study of the oral
MEK inhibitor CI-1040 in patients with advanced malignancies.
J Clin Oncol 23: 5281–5293.
Loyer P, Cariou S, Glaise D, Bilodeau M, Baffet G,
Guguen-Guillouzo C. (1996). Growth factor dependence of
progression through G1 and S phases of adult rat hepatocytes
in vitro. Evidence of a mitogen restriction point in mid-late G1.
J Biol Chem 271: 11484–11492.
Mook OR, Baas F, de Wissel MB, Fluiter K. (2007). Evaluation of
locked nucleic acid-modified small interfering RNA in vitro and in
vivo. Mol Cancer Ther 6: 833–843.
Morel-Chany E, Guillouzo C, Trincal G, Szajnert MF. (1978).
Spontaneous neoplastic transformation in vitro of epithelial cell
strains of rat liver: cytology, growth and enzymatic activities. Eur J
Cancer 14: 1341–1352.
Normanno N, De Luca A, Maiello MR, Campiglio M, Napolitano M,
Mancino M et al. (2006). The MEK/MAPK pathway is involved in
the resistance of breast cancer cells to the EGFR tyrosine kinase
inhibitor gefitinib. J Cell Physiol 207: 420–427.
Pages G, Guerin S, Grall D, Bonino F, Smith A, Anjuere F et al.
(1999). Defective thymocyte maturation in p44 MAP kinase (Erk 1)
knockout mice. Science 286: 1374–1377.
Pages G, Lenormand P, L’Allemain G, Chambard JC, Meloche S,
Pouyssegur J. (1993). Mitogen-activated protein kinases p42mapk
and p44mapk are required for fibroblast proliferation. Proc Natl
Acad Sci USA 90: 8319–8323.
Rescan C, Coutant A, Talarmin H, Theret N, Glaise D, Guguen-
Guillouzo C et al. (2001). Mechanism in the sequential control of
cell morphology and S phase entry by epidermal growth factor
involves distinct MEK/ERK activations. Mol Biol Cell 12: 725–738.
Reuber MD. (1961). A transplantable bile-secreting hepatocellular
carcinoma in the rat. J Natl Cancer Inst 26: 891–899.
Saba-El-Leil MK, Vella FD, Vernay B, Voisin L, Chen L, Labrecque
N et al. (2003). An essential function of the mitogen-activated
protein kinase Erk2 in mouse trophoblast development. EMBO Rep
4: 964–968.
Sanjo H, Hikida M, Aiba Y, Mori Y, Hatano N, Ogata M et al.
(2007). Extracellular signal-regulated protein kinase 2 is required for
efficient generation of B cells bearing antigen-specific immunoglo-
bulin G. Mol Cell Biol 27: 1236–1246.
Schmidt CM, McKillop IH, Cahill PA, Sitzmann JV. (1997). Increased
MAPK expression and activity in primary human hepatocellular
carcinoma. Biochem Biophys Res Commun 236: 54–58.
Sebolt-Leopold JS, Dudley DT, Herrera R, Van Becelaere K, Wiland
A, Gowan RC et al. (1999). Blockade of the MAP kinase pathway
suppresses growth of colon tumors in vivo [see comments]. Nat Med
5: 810–816.
Sebolt-Leopold JS, English JM. (2006). Mechanisms of drug inhibition
of signalling molecules. Nature 441: 457–462.
Sebolt-Leopold JS, Herrera R. (2004). Targeting the mitogen-activated
protein kinase cascade to treat cancer. Nat Rev Cancer 4: 937–947.
Stennicke HR, Salvesen GS. (1997). Biochemical characteristics of
caspases-3, -6, -7, and -8. J Biol Chem 272: 25719–25723.
Takei Y, Kadomatsu K, Yuzawa Y, Matsuo S, Muramatsu T. (2004).
A small interfering RNA targeting vascular endothelial growth
factor as cancer therapeutics. Cancer Res 64: 3365–3370.
Talarmin H, Rescan C, Cariou S, Glaise D, Zanninelli G, Bilodeau M
et al. (1999). The mitogen-activated protein kinase kinase/extra-
cellular signal-regulated kinase cascade activation is a key signalling
pathway involved in the regulation of G(1) phase progression in
proliferating hepatocytes. Mol Cell Biol 19: 6003–6011.
Vantaggiato C, Formentini I, Bondanza A, Bonini C, Naldini L,
Brambilla R. (2006). ERK1 and ERK2 mitogen-activated protein
kinases affect Ras-dependent cell signaling differentially. J Biol
5: 14.
Wabnitz PA, Mitchell D, Wabnitz DA. (2004). In vitro and in vivo
metabolism of the anti-cancer agent CI-1040, a MEK inhibitor, in
rat, monkey, and human. Pharm Res 21: 1670–1679.
Wang D, Boerner SA, Winkler JD, Lorusso PM. (2007). Clinical
experience of MEK inhibitors in cancer therapy. Biochim Biophys
Acta 1773: 1248–1255.
Yeh TC, Marsh V, Bernat BA, Ballard J, Colwell H, Evans RJ et al.
(2007). Biological characterization of ARRY-142886 (AZD6244), a
potent, highly selective mitogen-activated protein kinase kinase 1/2
inhibitor. Clin Cancer Res 13: 1576–1583.
ERK2 targeting inhibits tumor growth
A Bessard et al
5325
Oncogene





























