Embed Size (px)
Citation preview

BASIC AND TRANSLATIONAL SCIENCE
Role of Pyrimidine Depletion in the MitochondrialCardiotoxicity of Nucleoside Analogue Reverse
Transcriptase Inhibitors
Kerstin Balcarek, MD,* Nils Venhoff, MD,*† Catherine Deveaud, PhD,‡
Bertrand Beauvoit, PhD,‡ Jacques Bonnet, MD,‡ Janbernd Kirschner, MD,§
Ana C. Venhoff, PhD,* Dirk Lebrecht, PhD,* and Ulrich A. Walker, MD*k
Objective: Long-term antiretroviral treatment with nucleoside
analogue reverse transcriptase inhibitors (NRTI) may result in
a cardiomyopathy due to mitochondrial DNA (mtDNA) depletion. An
intact mitochondrial function is required for the synthesis of
intramyocardial pyrimidine nucleotides, which in turn are building
blocks of mtDNA. We investigated if NRTI-related cardiomyopathy
can be prevented with pyrimidine precursors.
Methods: Mice were fed with zidovudine or zalcitabine with or
without simultaneous Mitocnol, a dietary supplement with high
uridine bioavailability. Myocardia were examined after 9 weeks.
Results: Both NRTI induced a cardiomyopathy with mitochondrial
enlargement, a disrupted cristal architecture on electron microscopy
and diminished myocardial mtDNA copy numbers. The myocardial
mtDNA-encoded cytochrome c-oxidase I subunit was impaired more
profoundly than the nucleus-encoded cytochrome c-oxidase IV
subunit. The myocardial formation of reactive oxygen species and
mtDNA mutations was enhanced in zidovudine and zalcitabine
treated animals. Mitocnol attenuated or normalized all myocardial
pathology when given with both NRTI, but by itself had no intrinsic
effects and no apparent adverse effects.
Conclusions: Zidovudine and zalcitabine induce a mitochondrial car-
diomyopathy, which is antagonized with uridine supplementation, impli-
cating pyrimidine pool depletion in its pathogenesis. Pyrimidine pool
replenishment may be exploited clinically because uridine is well tolerated.
Key Words: cardiomyopathy, mitochondria, oxidative phosphoryla-
tion, uridine, zidovudine
(J Acquir Immune Defic Syndr 2010;55:550–557)
INTRODUCTIONNucleoside analogue reverse transcriptase inhibitors
(NRTI) are widely used in the antiretroviral treatment of HIV-infected individuals. A major limitation of their long-term useis their mitochondrial toxicity, which may affect liver,1 adiposetissue,2 skeletal muscle,3 and other organs. Animal modelssuggest that thymidine analogue NRTI also cause a mitochon-drial cardiomyopathy4,5 although in clinical practice overtmyocardial failure is only occasionally observed,6–8 NRTI may,however, cause more subtle cardiac abnormalities. Zidovudinefor example has been associated with left ventricularhypertrophy in adults,9 and fetal and perinatal exposure isassociated with reduced left ventricular mass, septal wallthickness and increased left ventricular afterload.10
The currently prevailing hypothesis for this form ofmyocardial damage is that triphosphorylated NRTI interferewith the metabolism of cardiac mitochondria by inhibitingpolymerase gamma, the enzyme which replicates mitochon-drial DNA (mtDNA).11 Zalcitabine for example, is a relativelypotent NRTI inhibitor of polymerase gamma in clinicallyrelevant concentrations, whereas zidovudine in contrast is onlya relatively weak inhibitor.11 Recent investigations suggest thatzidovudine may predominantly cause mtDNA depletion bya second mechanism, namely by interfering with thymidinekinase 2 (TK2), a mitochondrial enzyme which is the majorthymidine kinase in nonreplicating tissues such as theheart.12,13 Through inhibition of TK2, zidovudine may limitthe supply of thymidine phosphates as mtDNA buildingblocks, also resulting in mtDNA depletion.12,13 It has also beenshown that zidovudine induces oxidative stress,11,14 an effectwhich may be secondary to respiratory chain failure and byitself also impairs mtDNA replication.15
Last but not least, severe mtDNA depletion andconsequent respiratory chain failure can also induce myocar-dial pyrimidine depletion because a normal electron fluxthrough the respiratory chain is essential for the activityof dihydroorotate dehydrogenase, an enzyme which is
Received for publication February 24, 2010; accepted July 14, 2010.From the *Department of Rheumatology and Clinical Immunology,
Medizinische Universitatsklinik, Freiburg, Germany; †Centre of ChronicImmunodeficiency (CCI), Freiburg, Germany; ‡INSERM U828, Pessac,France; §Department of Neuropediatrics and Muscle Disorders, Medi-zinische Universitatsklinik, Freiburg, Germany; and kDepartment ofRheumatology, Basel University, Basel, Switzerland.
Supported exclusively by the Deutsche Forschungsgemeinschaft (D.F.G.),grant Number WA 1387 2-1.
Parts of the data have been presented at the following meetings: 10thInternational Workshop on Adverse Drug Reactions and Lipodystrophy inHIV, November 6–8, 2008, London, United Kingdom; 9th InternationalCongress on Drug Therapy in HIV Infection, November 9–13, 2008,Glasgow, United Kingdom.
The authors K.B. and N.V. contributed equally.Correspondence to: Ulrich A. Walker, MD, Department of Rheumatology,
Basel University, Felix Platter Spital, Burgfelderstr. 101, 4012 Basel,Switzerland (e-mail: [email protected]).
Copyright � 2010 by Lippincott Williams & Wilkins
550 | www.jaids.com J Acquir Immune Defic Syndr � Volume 55, Number 5, December 15, 2010

indispensable for pyrimidine de novo synthesis16 and criticalfor cell survival.
As it has been recently shown in vitro that pyrimidineloss sensitizes and contributes to the mitochondrial toxicity ofthymidine analogues,17 we aimed to explore the role ofpyrimidine depletion in the pathogenesis of the NRTI-inducedmitochondrial cardiomyopathy and to study if uridine,a nucleoside which replenishes pyrimidines via the salvagepathway, may be therapeutically employed to antagonize theonset of zidovudine cardiomyopathy. As a source ofexogenous uridine, we used Mitocnol, a dietary supplementwith a high uridine bioavailability.18
METHODS
AnimalsSeven-week-old BALB/c mice (Janvier, France) were
divided into 6 groups. Group A animals (n = 10) were keptwithout any treatment and served as controls. Group B mice (n= 10) received 340 mg/kg/d of Mitocnol (Pharma Nord,Vojens, Denmark) in the drinking water. Groups C and D (n =10 each) received zidovudine (100 mg/kg/d; MP Biomedicals,Strasbourg, France) in the drinking water. Groups E and F (n =9 each) received 13 mg/kg/d of zalcitabine (Sigma, Taufkirch-en, Germany). The dosages of zidovudine and zalcitabinecorresponded to human dosage adjusted for body area and thehigher drug disposal rate in rodents and was calculated on thebasis of a daily liquid consumption of 5 mL.19 Groups D and Fwere cotreated with 340 mg/kg/d of Mitocnol in the drinkingwater.
Mice were killed by cervical dislocation at 16 weeks ofage. Left ventricle, apex, and septum were snap frozen andcryopreserved. Aliquots were fixed in glutaraldehyde (3%) forelectron microscopy. All work was approved by our animalwelfare board and conformed to its guidelines.
Histopathology andMitochondrial Ultrastructure
The severity and extent of the myocardial lesions wasscored on a qualitative/quantitative morphological gradingscale on apical heart sections (5 mm) stained with hematoxylinand eosin.20 Two randomly selected samples from each groupwere examined with electron microscopy by a person blindedto the treatment status of the animals.
Myocellular LipidsCardiomyocyte steatosis was assessed in oil red O stains.
Lipids were also extracted from the myocardia usingmethanol/chloroform/water and quantified spectrophotomet-rically using a sulfo-phospho-vanillin reaction on lipidstandards (Sigma, Taufkirchen, Germany) as described.21
MtDNA Copy NumberTotal DNA was extracted with the QIAamp DNA
isolation kit (Qiagen, Hilden, Germany). MtDNA and nDNAcopy numbers were determined on a Roche LightCycler 480real-time polymerase chain reaction (PCR) system, asdescribed.22 Amplifications of mitochondrial and nuclearproducts were performed in triplicates. Absolute mtDNA and
nDNA copy numbers were calculated using serial dilutions ofplasmids with known copy numbers.
MtDNA-Encoded Respiratory Chain ProteinThe mtDNA-encoded subunit I of cytochrome c-oxidase
(COX I) was quantified by immunoblot. The COX I signal wasnormalized to the expression of the subunit IV of cytochromec-oxidase (COX IV), which is encoded by nuclear DNA.23
Lipid PeroxidationMalondialdehyde (MDA) is one of the end products of
lipid peroxidation and an indicator of free radical productionand oxidative stress. MDA was spectrophotometricallyquantified in cardiac tissue using an assay for material reactivewith thiobarbituric acid.24
Detection of the ‘‘Common’’ mtDNA DeletionMtDNA contains direct repeats between which base
pairs may be deleted by slipped mispairing during replica-tion.25 In humans, an age-related 4977 base pair (bp) deletionis the most frequent, so-called ‘‘common’’ deletion.25 In mice,a deletion almost identical in size to the human ‘‘common’’deletion has been described.26
We probed for the murine ‘‘common’’ mtDNA deletionby amplifying 10 ng of genomic DNA with extradeletionalprimers F8265 (5#-AATTACAGGCTTCCGACACA-3#) andB13703 (5#-GAGATTGGTTGATGTATGAG-3#) in a PCR.By choosing a short extension time (30 seconds), the deletedmolecule was preferentially amplified as a 445 bp product. Theidentity of the amplicon was confirmed by sequencing (MWGBiotech, Ebersberg, Germany).
The ‘‘common’’ mtDNA deletion was quantified onagarose gels with Scion Image, using defined DNA ladderamounts as standard.
StatisticsGroup means were compared by unpaired t test or
Mann–Whitney analysis, as appropriate. Regressions werecomputed by exponential regression analysis (Sigma Plot 9.0,SPSS Inc, Chicago, IL).
RESULTS
Macroscopic and Microscopic PathologyDaily fluid consumption, body weight, and heart weight
were unaffected by zidovudine or zalcitabine treatment (datanot shown). On post mortem examination, no clinical signs ofcardiomyopathy were detected.
The histopathological cardiomyopathy score was in-creased after treatment with zidovudine (540% of untreatedcontrol values; P , 0.001) and with zalcitabine (313% ofcontrol values; P , 0.001; Table 1). Cotreatment withMitocnol significantly attenuated the zidovudine-induced andzalcitabine-induced histopathological lesions but did not havean intrinsic effect (Fig. 1).
On ultrastructural investigations, the mitochondria in theMitocnol-only hearts did not show abnormalities (Fig. 2). Themyofibrillar lattice of the cardiomyocytes of animals treatedwith zidovudine or zalcitabine in the absence of Mitocnol was
q 2010 Lippincott Williams & Wilkins www.jaids.com | 551
J Acquir Immune Defic Syndr � Volume 55, Number 5, December 15, 2010 Pyrimidine Depletion in NRTI Cardiotoxicity

in disarray, the cardiomyoctes contained enlarged mitochon-dria in which the cristal architecture was disrupted. Cotreat-ment with Mitocnol normalized the myocardial ultrastructure.
Myocellular LipidsNeither zidovudine nor zalcitabine augmented myocel-
lular lipid deposition compared with untreated controls asevidenced by oil red O staining (data not shown) andbiochemical quantification (Table 1). Zalcitabine, however,induced a slight, but quantitatively significant decrease ofcardiac lipids. Mitocnol treatment per se did not altermyocardial lipids.
MtDNA Copy NumberIn mice treated with zidovudine alone, the mean mtDNA
copy numbers were diminished by 13% compared with controlvalues (P = 0.02). Zalcitabine caused a similar mtDNAdepletion (14%; P = 0.01). Whereas Mitocnol did not have anintrinsic effect on mtDNA copy numbers, this pyrimidineprecursor fully normalized mtDNA copy numbers inzidovudine-treated and zalcitabine-treated animals. Amongall animals, mtDNA copy numbers were inversely correlatedwith the cardiomyopathy score (r = 20.67; P , 0.001;Fig. 3A).
MtDNA-Encoded Respiratory Chain SubunitsThe mean COX I/COX IV ratio was substantially
reduced in the zidovudine group (57% of control values; P ,0.001) and to a lesser extent in the zalcitabine group (82%; P =0.005) (Table 1). The COX I protein expression relative toCOX IV was not altered in mice treated with Mitocnol alone.Mitocnol however attenuated the zidovudine-induced down-regulation of COX I expression relative to COX IV (P = 0.03).Furthermore, Mitocnol normalized the COX I expression inmice cotreated with zalcitabine. Among all mice, the COXI/COX IV ratio was inversely correlated with the cardiomy-opathy score (r = 20.51; P , 0.001; Fig. 3B).
MDA Levels and Lipid PeroxidationMDA levels are an indirect indicator of lipid perox-
idation due to reactive oxygen species (ROS). Comparedwith controls, MDA levels were increased by 70% (P , 0.001)in hearts exposed to zidovudine, and by 106% (P , 0.001)in hearts exposed to zalcitabine (Table 1). Uridine supple-mentation normalized cellular MDA formation in bothzidovudine-treated and zalcitabine-treated animals (bothP , 0.05).
The cardiac MDA content was positively correlated withthe cardiomyopathy score (r = 0.68; P , 0.001; Fig. 3C);mtDNA copy numbers were inversely correlated with MDAlevels (r = 20.70; P , 0.001; Fig. 3D).
MtDNA DeletionsA 445 bp PCR product, corresponding to the
‘‘common’’ mtDNA 4974 bp deletion in mice,26 was identifiedin all zidovudine and zalcitabine-treated animals. In contrast,70% of the myocardia of the control animals and 80% ofMitocnol-only myocardia did not harbor detectable amounts ofthe deletion. In all cases in which the ‘‘common’’ mtDNAdeletion was detectable in the control or Mitocnol-onlymyocardia, the deletion quantity was lower than the lowestdeletion quantity in the zidovudine or zalcitabine hearts.
On average, the quantity of the mtDNA mutation wasincreased by 433% in the zidovudine group compared withcontrols (P , 0.001), and by 454% in the zalcitabine group (P, 0.001; Table 1; Fig. 4). With Mitocnol cotreatment, thefrequency of the mtDNA deletion relative to controls was305% in the zidovudine group (P = 0.001) and 313% in thezalcitabine group (P = 0.002). Thus cotreatment with Mitocnoldiminished the presence of the mtDNA common deletionsignificantly compared with NRTI without Mitocnol (bothP , 0.001).
Among mice in which the ‘‘common’’ mtDNA deletionwas detectable, its intensity was positively correlated withthe myocardial MDA content (r = 0.60; P , 0.001; Fig. 3E).
TABLE 1. Effect of Uridine Supplementation With Mitocnol on Cardiac Muscle Histology, mtDNA-Encoded Respiratory ChainSubunits, mtDNA Content, Lipid Peroxidation (MDA Levels) and the ‘‘common’’ mtDNA Deletion in Mice Treated withZidovudine or Zalcitabine
Group A B C D E F
Treatment Control Mitocnol340 mg/kg/d
Zidovudine100 mg/kg/d
Zidovudine100 mg/kg/d +Mitocnol 340 mg/kg/d
Zalcitabine13 mg/kg/d
Zalcitabine13 mg/kg/d +Mitocnol 340 mg/kg/d
n 10 10 10 10 9 9
Cardiomyopathy score 1.0 6 0.6 1.0 6 0.2 6.4 6 2.1* 4.1 6 2.0*§ 4.1 6 2.2* 2.3 6 1.1†§
Myocardial lipids (mg/g tissue) 2.2 6 0.6 2.0 6 0.6 1.7 6 0.5 1.8 6 0.4 1.5 6 0.4† 1.9 6 0.7
mtDNA copy number/cardiomyocyte 628 6 67 619 6 62 548 6 76† 601 6 67 543 6 54† 583 6 79
COX I/COX IV-ratio (% of control mean) 100 6 14 93 6 10 57 6 17* 73 6 12*§ 82 6 9† 92 6 20
MDA (mmol/g tissue) 1.19 6 0.37 1.02 6 0.43 2.03 6 0.60* 1.29 6 0.47§ 2.45 6 0.40* 1.62 6 0.51§
‘‘Common’’ mtDNA deletion (arbitrary units) 0.21 6 0.35 0.16 6 0.33 1.13 6 0.05* 0.86 6 0.13*‡ 1.18 6 0.16* 0.88 6 0.11*‡
Values represent group means (6SD).*P , 0.001 vs. controls.†P , 0.05 vs. controls.‡P , 0.001 vs. mice treated without Mitocnol.§P , 0.05 vs. mice treated without Mitocnol.
552 | www.jaids.com q 2010 Lippincott Williams & Wilkins
Balcarek et al J Acquir Immune Defic Syndr � Volume 55, Number 5, December 15, 2010

The intensity of the ‘‘common’’ mtDNA deletion was inverselycorrelated with the COX I/COX IV ratio (r = 20.52; P ,0.001) and positively correlated with the cardiomyopathyscore (r = 0.70; P , 0.001).
DISCUSSIONThe present study analyzed the effects of uridine
supplementation on NRTI-mediated mitochondrial toxicityin cardiac muscle. Our findings demonstrate that zidovudineand zalcitabine induce a cardiomyopathy in which mtDNA-encoded respiratory chain deficiency plays a significant role.The reduction of COX subunit I expression suggests thatquantitative and qualitative mtDNA lesions are important ifnot pivotal in this form of respiratory dysfunction and does notsupport mitochondrial toxicity secondary to other mechanismssuch as increased ROS formation.
Our results also exhibit that the marked mitochondrialcardiotoxicity can be antagonized by oral uridine supplemen-tation and support a central role of intracellular pyrimidinepools in its pathogenesis. As discussed above, mtDNAdepletion by zidovudine may result from the inhibition ofmitochondrial TK2, with consecutive depletion of normaldeoxypyrimidine pools as mtDNA building blocks.12,13 Thismechanism operates predominantly in postmitotic tissues such
as the cardiac muscle in which thymidine kinase 1 activity asan alternate pathway of thymidine phosphorylation is down-regulated.12,27
Thymidine triphosphate concentrations are critical andrate limiting for mitochondrial DNA replication28 and reducedpyrimidine pools promote mutagenesis by reducing replicationfidelity.28 Thus, the diminished thymidine triphosphate poolsresulting from the inhibition of TK2 can explain the reducedwild-type mtDNA content and the increased prevalence ofmtDNA deletions in zidovudine-treated myocardia.
The murine ‘‘common’’ mtDNA deletion26 was alsopresent in the hearts of mice treated with zalcitabine,a polymerase gamma inhibitor not known to antagonizeTK2 activity. MtDNA mutations with zalcitabine may arisefrom ROS which are produced by the defective respiratorychain.29,30 Mutagenic pyrimidine nucleotide depletion mayalso arise from defective de novo synthesis by dihydroorotatedehydrogenase secondary to the dysfunctional oxidativephosphorylation.28 Both mechanisms can explain our obser-vation that ROS formation as assessed by myocardial MDAcontent was fully normalized with uridine supplementation.
Most importantly our study demonstrates that uridinesupplementation attenuates all aspects of NRTI-relatedcardiomyopathy. The exact mechanism of the beneficial effectof uridine is not fully delineated, but it is likely that uridine
FIGURE 1. Hematoxylin and eosin stains of representative apical heart muscle sections. Size bars 20 mm.
q 2010 Lippincott Williams & Wilkins www.jaids.com | 553
J Acquir Immune Defic Syndr � Volume 55, Number 5, December 15, 2010 Pyrimidine Depletion in NRTI Cardiotoxicity

itself or its metabolites disinhibit mtDNA replication bycompeting with zidovudine and zalcitabine as pyrimidinenucleotides either at TK2 or at polymerase gamma.Alternatively, uridine may compete with pyrimidine NRTI atother steps of intracellular NRTI transport or phosphoryla-tion.31,32 Uridine may in addition correct an intracellularpyrimidine deficit which in itself is sufficient to induceapoptosis.17,33
The dose of the uridine supplement used in this studydeserves some consideration. The dosage of Mitocnoladministered to mice in our study corresponded on a perbody weight basis to a human dose of 24 g/70 kg, and on a perbody surface area basis to 13 g/m2 of Mitocnol.34 Mitocnolconsists of 0.6 g of uridine plus 5.4 g of triacetyl uridine,a uridine precursor with several-fold higher uridine bio-availability over conventional uridine.35 Mitocnol constitutesthe active ingredient of a commercially available dietarysupplement named NucleomaxX. NucleomaxX is delivered insachets (36 g), each of which contain 6 g of Mitocnol plusother substances to camouflage the bitter taste of uridine.Continuous administration of 3 sachets of NucleomaxX perday to men yields mean uridine plasma levels of 163 mM about1 hour after administration. These uridine plasma levels are ina similar order of magnitude as those found to be protectiveagainst the mitochondrial toxicity of pyrimidine nucleoside
analogues in vitro.32 This was the rationale for the doseadministered to the mice in this study. In vitro studies have alsoindicated that it took considerably longer for mitochondrialtoxicity to develop in the absence of uridine (weeks), than ittook for uridine to abrogate established mitochondrial toxicity(days).32 This relatively quick therapeutic effect of uridinerelative to the more prolonged development of mitochondrialtoxicities in these in vitro studies may allow for intermittentdosing to ‘‘reset the mitochondrial clock’’, an approach whichhas been tested in clinical trials of NucleomaxX for NRTI-related hepatotoxicity and lipoatrophy.36,37 A number of studiesin indications not related to myocardial disease or HIVinfection have demonstrated the safety and good tolerability ofuridine and triacetyl uridine.18,32 High doses of uridine werealso shown to not compete with NRTI at HIV reversetranscriptase.31,36,38–40 Thus our work may not only open a newavenue for the therapy of NRTI-related cardiotoxicity in HIVpatients and potentially also for the cardiomyopathiesassociated with inherited mtDNA mutations.41–43
ACKNOWLEDGMENTThe authors thank Pharma Nord (Vojens, Denmark) for
providing Mitocnol and Karin Sutter and Carmen Kopp forexpert technical assistance.
FIGURE 2. Representative electron micrographs of murine cardiomyocytes and their mitochondria (*). Size bars 1.7 mm.
554 | www.jaids.com q 2010 Lippincott Williams & Wilkins
Balcarek et al J Acquir Immune Defic Syndr � Volume 55, Number 5, December 15, 2010
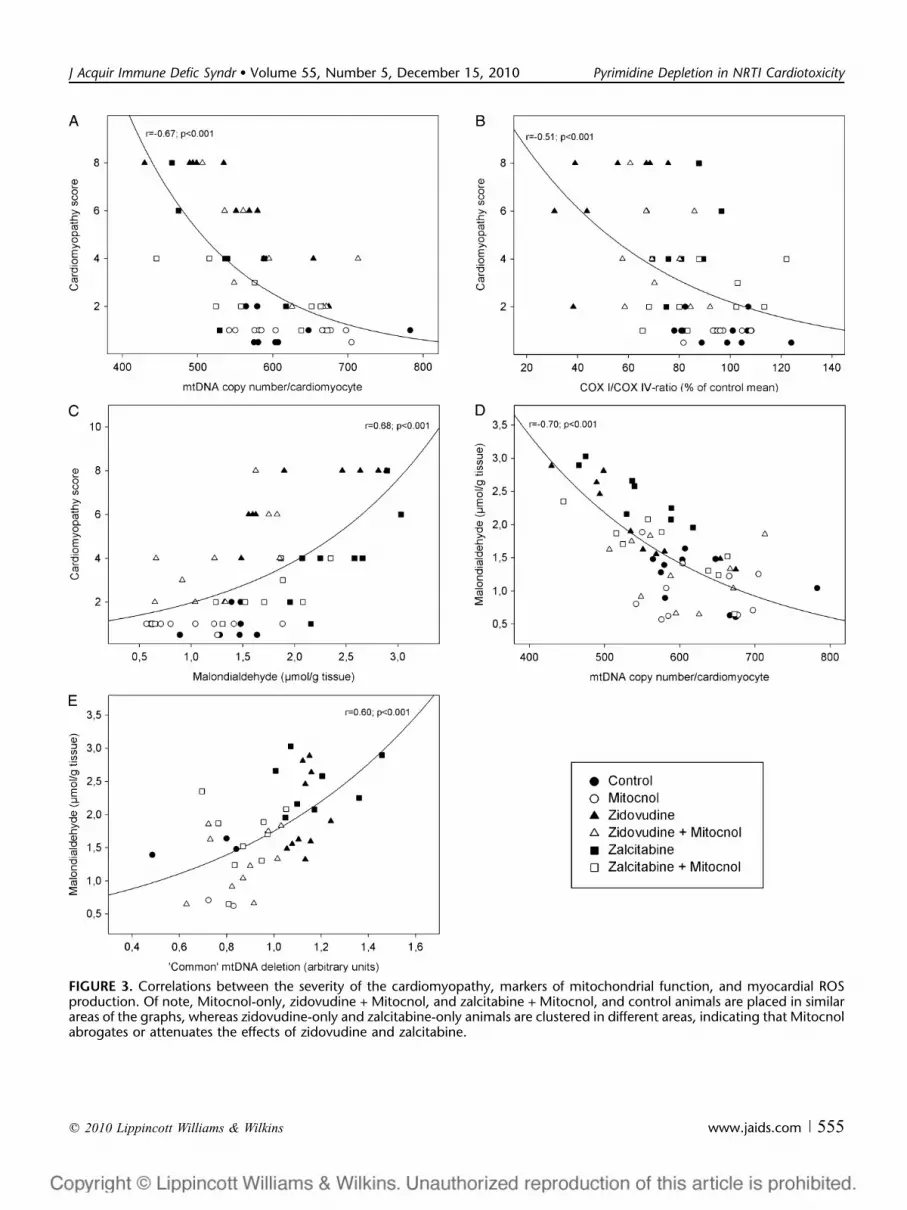
FIGURE 3. Correlations between the severity of the cardiomyopathy, markers of mitochondrial function, and myocardial ROSproduction. Of note, Mitocnol-only, zidovudine + Mitocnol, and zalcitabine + Mitocnol, and control animals are placed in similarareas of the graphs, whereas zidovudine-only and zalcitabine-only animals are clustered in different areas, indicating that Mitocnolabrogates or attenuates the effects of zidovudine and zalcitabine.
q 2010 Lippincott Williams & Wilkins www.jaids.com | 555
J Acquir Immune Defic Syndr � Volume 55, Number 5, December 15, 2010 Pyrimidine Depletion in NRTI Cardiotoxicity

REFERENCES1. Walker UA, Bauerle J, Laguno M, et al. Depletion of mitochondrial DNA
in liver under antiretroviral therapy with didanosine, stavudine, orzalcitabine. Hepatology. 2004;39:311–317.
2. Walker UA, Bickel M, Lutke Volksbeck SI, et al. Evidence of nucleosideanalogue reverse transcriptase inhibitor-associated genetic and structuraldefects of mitochondria in adipose tissue of HIV-infected patients.J Acquir Immune Defic Syndr. 2002;29:117–121.
3. Arnaudo E, Dalakas M, Shanske S, et al. Depletion of musclemitochondrial DNA in AIDS patients with zidovudine-induced myopathy.Lancet. 1991;337:508–510.
4. Lewis W, Day BJ, Kohler JJ, et al. Decreased mtDNA, oxidative stress,cardiomyopathy, and death from transgenic cardiac targeted humanmutant polymerase gamma. Lab Invest. 2007;87:326–335.
5. Lewis W, Kohler JJ, Hosseini SH, et al. Antiretroviral nucleosides,deoxynucleotide carrier and mitochondrial DNA: evidence supporting theDNA poly hypothesis. AIDS. 2006;20:675–684.
6. Brogly SB, Ylitalo N, Mofenson LM, et al. In utero nucleoside reversetranscriptase inhibitor exposure and signs of possible mitochondrialdysfunction in HIV-uninfected children. AIDS. 2007;21:929–938.
7. Frerichs FC, Dingemans KP, Brinkman K. Cardiomyopathy withmitochondrial damage associated with nucleoside reverse-transcriptaseinhibitors. N Engl J Med. 2002;347:1895–1896.
8. Thoden J, Lebrecht D, Venhoff N, et al. Highly active antiretroviral HIVtherapy-associated fatal lactic acidosis: quantitative and qualitativemitochondrial DNA lesions with mitochondrial dysfunction in multipleorgans. AIDS. 2008;22:1093–1094.
9. Mondy K, Gottdiener J, Overton ET, et al, and SUN study investigators.Prevalence and risk factors in HIV-infected persons for echocardiographicabnormalities in the era of modern HAART. Presented at: 15thConference on Retroviruses and Opportunistic Infections; 2008; Boston,MA. Abstract 978.
10. Dube MP, Lipshultz SE, Fichtenbaum CJ, et al. Effects of HIV infectionand antiretroviral therapy on the heart and vasculature. Circulation. 2008;118:e36–e40.
11. Kakuda TN. Pharmacology of nucleoside and nucleotide reversetranscriptase inhibitor-induced mitochondrial toxicity. Clin Ther. 2000;22:685–708.
12. Lynx MD, McKee EE. 3#-Azido-3#-deoxythymidine (AZT) is a compet-itive inhibitor of thymidine phosphorylation in isolated rat heart and livermitochondria. Biochem Pharmacol. 2006;72:239–243.
13. Susan-Resiga D, Bentley AT, Lynx MD, et al. Zidovudine inhibitsthymidine phosphorylation in the isolated perfused rat heart. AntimicrobAgents Chemother. 2007;51:1142–1149.
14. de la Asuncion JG, del Olmo ML, Sastre J, et al. AZT treatment inducesmolecular and ultrastructural oxidative damage to muscle mitochondria.Prevention by antioxidant vitamins. J Clin Invest. 1998;102:4–9.
15. Pursell ZF, McDonald JT, Mathews CK, et al. Trace amounts of 8-oxo-dGTP in mitochondrial dNTP pools reduce DNA polymerase gammareplication fidelity. Nucleic Acids Res. 2008;36:2174–2181.
16. Gattermann N, Dadak M, Hofhaus G, et al. Severe impairment ofnucleotide synthesis through inhibition of mitochondrial respiration.Nucleosides Nucleotides Nucleic Acids. 2004;23:1275–1279.
17. Setzer B, Lebrecht D, Walker UA. Pyrimidine nucleoside depletionsensitizes to the mitochondrial hepatotoxicity of the reverse transcriptaseinhibitor stavudine. Am J Pathol. 2008;172:681–690.
18. Venhoff N, Zilly M, Lebrecht D, et al. Uridine pharmacokinetics ofMitocnol, a sugar cane extract. AIDS. 2005;19:739–740.
19. Note R, Maisonneuve C, Letteron P, et al. Mitochondrial and metaboliceffects of nucleoside reverse transcriptase inhibitors (NRTIs) in micereceiving one of five single- and three dual-NRTI treatments. AntimicrobAgents Chemother. 2003;47:3384–3392.
20. Lebrecht D, Setzer B, Ketelsen UP, et al. Time-dependent and tissue-specific accumulation of mtDNA and respiratory chain defects in chronicdoxorubicin cardiomyopathy. Circulation. 2003;108:2423–2429.
21. Frings CS, Fendley TW, Dunn RT, et al. Improved determination of totalserum lipids by the sulfo-phospho-vanillin reaction. Clin Chem. 1972;18:673–674.
22. Lebrecht D, Vargas Infante YA, Setzer B, et al. Uridine supplementationantagonizes zalcitabine-induced microvesicular steatohepatitis in mice.Hepatology. 2007;45:72–79.
23. Walker UA, Setzer B, Venhoff N. Increased long-term mitochondrialtoxicity in combinations of nucleoside analogue reverse-transcriptaseinhibitors. AIDS. 2002;16:2165–2173.
24. Angel MF, Ramasastry SS, Swartz WM, et al. The critical relationshipbetween free radicals and degrees of ischemia: evidence for tissueintolerance of marginal perfusion. Plast Reconstr Surg. 1988;81:233–239.
25. Samuels DC, Schon EA, Chinnery PF. Two direct repeats cause mosthuman mtDNA deletions. Trends Genet. 2004;20:393–398.
26. Tanhauser SM, Laipis PJ. Multiple deletions are detectable inmitochondrial DNA of aging mice. J Biol Chem. 1995;270:24769–24775.
27. Rylova SN, Albertioni F, Flygh G, et al. Activity profiles of deoxynucleo-side kinases and 5#-nucleotidases in cultured adipocytes and myoblasticcells: insights into mitochondrial toxicity of nucleoside analogs. BiochemPharmacol. 2005;69:951–960.
28. Song S, Pursell ZF, Copeland WC, et al. DNA precursor asymmetries inmammalian tissue mitochondria and possible contribution to mutagenesisthrough reduced replication fidelity. Proc Natl Acad Sci U S A. 2005;102:4990–4995.
29. Corral-Debrinski M, Stepien G, Shoffner JM, et al. Hypoxemia isassociated with mitochondrial DNA damage and gene induction.Implications for cardiac disease. JAMA. 1991;266:1812–1816.
30. Larsson NG, Holme E, Kristiansson B, et al. Progressive increase of themutated mitochondrial DNA fraction in Kearns-Sayre syndrome. PediatrRes. 1990;28:131–136.
31. Walker UA, Venhoff N, Koch E, et al. Uridine abrogates mitochondrialtoxicity related to nucleoside analogue reverse transcriptase inhibitors inHepG2 cells. Antivir Ther. 2003;8:463–470.
32. Walker UA, Venhoff N. Uridine in the prevention and treatment of NRTI-related mitochondrial toxicity. Antivir Ther. 2005;10:M117–M123.
33. Fox RI. Mechanism of action of leflunomide in rheumatoid arthritis.J Rheumatol Suppl. 1998;53:20–26.
34. Lebrecht D, Deveaud C, Beauvoit B, et al. Uridine supplementationantagonizes zidovudine-induced mitochondrial myopathy and hyper-lactatemia in mice. Arthritis Rheum. 2008;58:318–326.
35. Weinberg ME, Roman MC, Jacob P, et al. Single-dose and cumulativepharmacokinetis of the food supplement NucleomaxX� and mechanismfor enhanced bioavailability of uridine. Antivir. Ther. 2007;12:(Suppl 2):L25.
36. Sutinen J, Walker UA, Sevastianova K, et al. Uridine supplementation forthe treatment of antiretroviral therapy-associated lipoatrophy: a random-ized, double-blind, placebo-controlled trial. Antivir Ther. 2007;12:97–105.
FIGURE 4. Representative agarose gel electrophoresis of PCRproducts amplified from 10 ng of genomic DNA for thedetection of the ‘‘common’’ mtDNA deletion. Lane 1: 100 bpDNA ladder standard. Compared with untreated control (lane2) or Mitocnol-only–exposed hearts (lane 3), PCR productsfrom zidovudine (lane 4), zidovudine + Mitocnol (lane 5),zalcitabine (lane 6) and zalcitabine + Mitocnol (lane 7) heartswere more abundant. Mitocnol cotreatment, however, dimin-ishes the prevalence of the ‘‘common’’ mtDNA deletion. Lane8: PCR without DNA template.
556 | www.jaids.com q 2010 Lippincott Williams & Wilkins
Balcarek et al J Acquir Immune Defic Syndr � Volume 55, Number 5, December 15, 2010

37. Banasch M, Goetze O, Knyhala K, et al. Uridine supplementationenhances hepatic mitochondrial function in thymidine-analogue treatedHIV-infected patients. AIDS. 2006;20:1554–1556.
38. Koch EC, Schneider J, Weiss R, et al. Uridine excess does not interferewith the antiretroviral efficacy of nucleoside analogue reverse transcrip-tase inhibitors. Antivir Ther. 2003;8:485–487.
39. McComsey GA, O#Riordan M, Setzer B, et al. Uridine supplementation inHIV lipoatrophy: pilot trial on safety and effect on mitochondrial indices.Eur J Clin Nutr. 2008;62:1031–1037.
40. Sommadossi JP, Carlisle R, Schinazi RF, et al. Uridine reverses thetoxicity of 3#-azido-3#-deoxythymidine in normal human granulocyte-
macrophage progenitor cells in vitro without impairment of antiretroviralactivity. Antimicrob Agents Chemother. 1988;32:997–1001.
41. Schon EA, Rizzuto R, Moraes CT, et al. A direct repeat is a hotspot forlarge-scale deletion of human mitochondrial DNA. Science. 1989;244:346–349.
42. Tsutsui H, Kinugawa S, Matsushima S. Mitochondrial oxidative stress anddysfunction in myocardial remodelling. Cardiovasc Res. 2009;81:449–456.
43. Holt IJ, Harding AE, Morgan-Hughes JA. Deletions of musclemitochondrial DNA in patients with mitochondrial myopathies. Nature.1988;331:717–719.
q 2010 Lippincott Williams & Wilkins www.jaids.com | 557
J Acquir Immune Defic Syndr � Volume 55, Number 5, December 15, 2010 Pyrimidine Depletion in NRTI Cardiotoxicity





![New one-pot synthesis of spiro[furo[2,3-d]pyrimidine-6,5-pyrimidine]pentaones and their sulfur analogues](https://img.pdfslide.net/doc/110x75/63188db8b41f9c8c6e095871/new-one-pot-synthesis-of-spirofuro23-dpyrimidine-65-pyrimidinepentaones-and.jpg)















![Pyrazolo[1,5-a]pyrimidine-based inhibitors of HCV polymerase](https://img.pdfslide.net/doc/110x75/6351adc189352761b10649d4/pyrazolo15-apyrimidine-based-inhibitors-of-hcv-polymerase.jpg)







