Embed Size (px)
Citation preview

The EMBO Journal vol. 1 1 no.3 pp. 1 131 - 1139, 1992
Domain structure of the human immunodeficiency virusreverse transcriptase
Hermann Lederer1"7, Octavian Schatz2'8,Roland May3, Henry Crespi4, Jean-Luc Darlix5,Stuart F.J.Le Grice6 and Hermann Heumann1'91Max Planck Institute of Biochemistry, D-8033 Martinsried, FRG,2Central Research Units, F.Hoffmann-La Roche Ltd, CH-4002 Basel,Switzerland, 3Institut Laue-Langevin, BP 156, F-38042 GrenobleCedex 9, France, 4Argonne National Laboratory, IL 60439, USA,5Ecole Normale Superieure, Laboratoire de Biologie, 46, All6ed'Italie, F-69367 Lyon Cedex 07, France and 6Division of InfectiousDiseases, Case Western Reserve University School of Medicine,Cleveland, OH 44106, USA
7Present address: Max Planck Institute of Plasma Physics, Garching,FRG
8Present address: Dept. of Oncology and Virology, Ciba-Geigy Ltd.,CH-4002 Basel, Switzerland
Communicated by W.Saenger
The spatial arrangement of subunits p51 and p66 of theHIV-1 reverse transcriptase and the position of the RNaseH containing domain, p15, have been determined bymeans of neutron small-angle scattering. The reversetranscriptase (p66/p5i) is a flat molecule, which can beapproximated by an ellipsoid with the half axes of5.2 mm, 4.8 mm and 1.4 mm. The two subunits pSi andp66 having a centre-to-centre distance of 3.3 0.3 nmare attached at their flat sides, slightly shifted sideways.The p15 domain is located at the long axis of the ellip-soidal reverse transcriptase having a distance of5.0 0.5 am to the centre of the p5ld domain, whichis part of the p66 subunit, and a distance of5.3 + 1.2 mm to the centre of the neighbouring P51ssubunit.Key words: HIV-1/neutron scattering/reconstitution/reversetranscriptase/structure
IntroductionReverse transcriptase (RT) catalyses replication of viralgenomic RNA. Location of the RNA primer, elongation ofthe primer by synthesis of a DNA strand complementaryto the sequence of the RNA template strand and coordinatedigestion of the template are the functional steps mediatedby RT, leading to formation of minus strand DNA. Plusstrand formation is catalysed by the same enzyme (Varmusand Swanstrom, 1984; Varmus, 1987; Goff, 1990). Thefunctional form of type 1 human immune deficiency virus(HIV-1) is a heterodimer comprising the subunits pSi(molecular weight: 51 kDa) and p66 (molecular weight:66 kDa; Di Marzo Veronese et al., 1986; Lightfoote et al.,1986; Lowe et al., 1988). The p66 subunit consists of twodomains, the p5 Id domain (the subscripts s and d,respectively, denote the subunit and the domain, p51) andthe p15 domain. The latter contains the RNase H activityHansen et al., 1988), whose structure was recently solved
Oxford University Press
at atomic resolution (Davies et al., 1991). In this paper wepresent a model of the solution structure of the isolated HIV-1reverse transcriptase at low resolution using neutron smallangle scattering. This method provides information aboutthe quaternary structure of biopolymeric complexes aspreviously shown, e.g. for the Escherichia coli RNApolymerase-DNA complex (Heumann et al., 1988), thetetracycline repressor-DNA complex (Lederer et al., 1989)and the 30S and the 50S ribosomal subunits of E. coli (Capelet al., 1987; Nierhaus et al., 1988). The main advantageof this method is that a single component in a complex canbe highlighted by deuteration and the scattering contribu-tion of the remaining part can be matched by contrastvariation of the solution buffer with D20 (Crichton et al.,1977).We applied this technique to the isolated RT and visualized
either the p5 1s subunit or the p66 subunit in the heterodimercomplex for obtaining the scattering functions of thecorresponding subunits in situ. This information, togetherwith the scattering functions of the heterodimer (p66/pS 1)and the homodimer (p66/p66) allowed us to determine thespatial arrangement of the subunits p66 and p51s, and thep15 domain, respectively, within the heterodimeric RT.
ResultsScattering parameters of the heterodimeric RTp66/p51Solutions of free RT with the subunits p66 and p5 1, in a1:1 stoichiometry were subjected to small angle neutronscattering in H20 (see Materials and methods). Thesemeasurements provided the scattering curve of RT (Figurela), from which the corresponding Fourier transform, thedistance distribution function, p(r) (Figure lb), wascalculated. This function represents the frequency of thedistances between any two volume elements of the scatteringmolecule. An assessment of the quality of the data is ofteneasier by analysing the distance distribution function insteadof the original scattering curve. From the distance distribu-tion function the following parameters were derived: (i) themolecular weight (M) of RT, (ii) the maximum dimension(dmax), which is read from the p(r) function at r = rmax,(iii) the radius of gyration (Rg) and the volume (v) (seeMaterials and methods). These data are compiled in Table I.The parameters d,a, RG and V were used to calculate the
shape parameters of RT in terms of a scattering equivalentellipsoid. According to the procedure described in Materialsand methods, an oblate ellipsoid was obtained having thehalf axes 5.2 nm, 4.8 nm and 1.4 nm. Model calculationshave verified that an ellipsoid with these half axes is a goodapproximation for the shape of the RT (see Figure la).The information about the shape of RT serves as a
constraint for the more detailed analysis of the spatialarrangement of the components of RT, the subunits p66 andp5 l, and within the p66 subunit the spatial arrangement ofthe p5ld domain and the p15 domain.
1131

H.Lederer et al.
a
0.0 1.0
Q (1/nm)
b
'.4
2.0
r(nm)Fig. 1. (a) The scattering curve of the protonated heterodimeric HIV-1 RT. Filled squares, scattering curve determined experimentally. The errorbars lie within the symbols used for data representation. Dashed line, scattering curve calculated for an ellipsoid with the half axes 5.2 nm, 4.8 nmand 1.4 nm. (b) The distance distribution functions, p(r), of the HIV RT components. Filled squares, p(r) of the protonated p5l/p66 heterodimer(RT) in H20. Filled triangles, p(r) of the protonated p66/p66 homodimer in H20. Open triangles, p(r) of the p5lS subunit in situ, obtained from RT(H-D-hybrid) containing a deuterated pSl subunit and a protonated p66 subunit, whose scattering contribution was matched using a 41 % D20 buffer.Open squares, p(r) of the p66 subunit in situ, extrapolated from a set of scattering curves of H-D-hybrid RT at different D20 concentrations (seeFigure 3 and Materials and methods).
Scattering parameters of the RT componentsDetermination of the spatial arrangement of the subunits/domains requires information about the shape of thecomponents, their centre-to-centre distances and theirorientations. These parameters can be obtained by neutronscattering, provided a single component or a sub-assemblycan be visualized by deuteration. For that purpose wereconstituted an active RT from a deuterated and a protonated(natural abundance hydrogens) subunit.
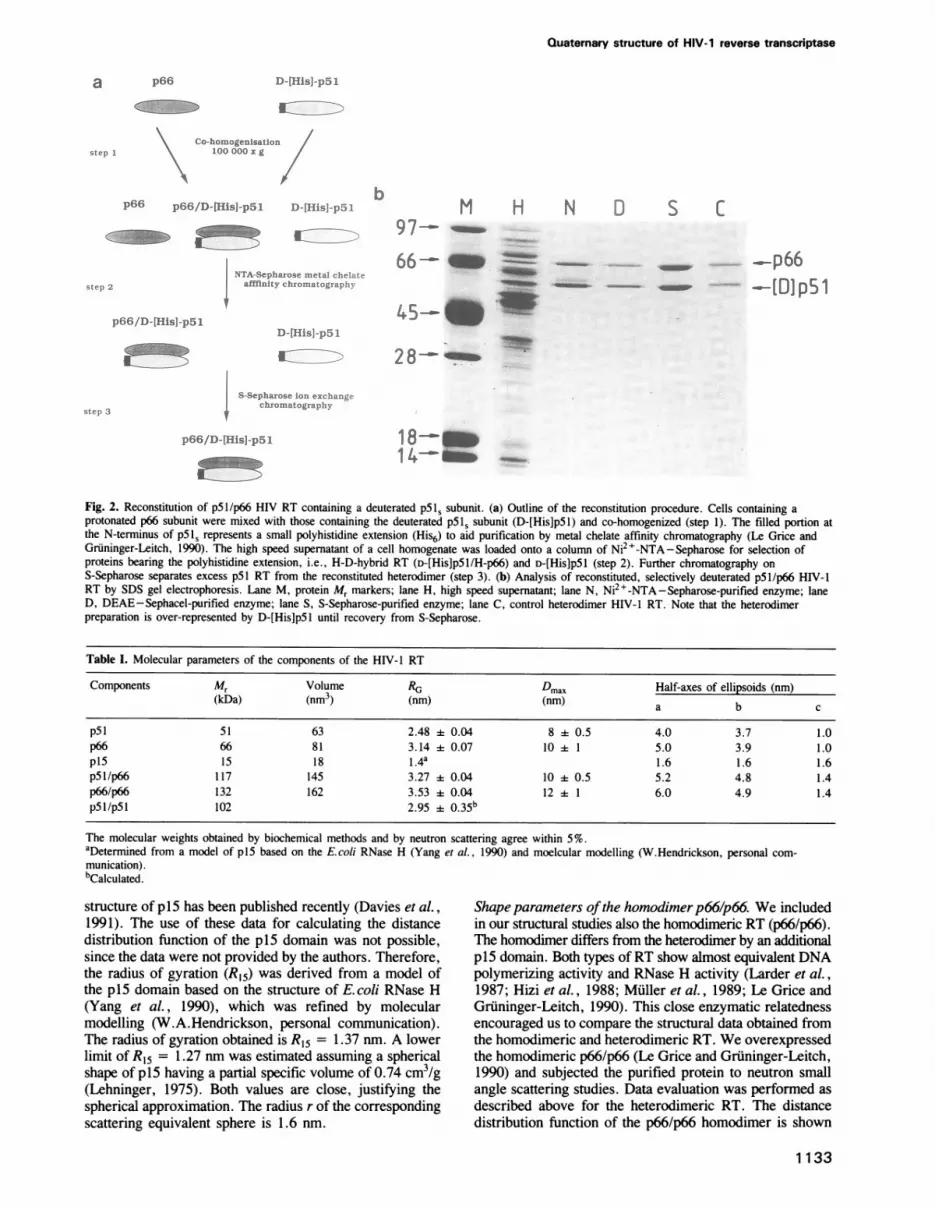
Reconstitution of selectively deuterated heterodimer RT.Reconstitution partners were the protonated p66 anddeuterated subunits p5l,. The latter had an extension of sixhistidines at the N-terminus. Due to a dynamic equilibriumbetween subunits in the dimeric and the monomeric form,a mixture of p66 subunits and p51, subunits containspopulations of monomers, homodimers and heterodimers inratios determined by their equilibrium constants. The yieldof heterodimers is high due to the higher affinity constantfor the formation of heterodimers (Restle et al., 1991).Figure 2 shows the outline of the reconstitution procedure.Since attempts to reconstitute heterodimer from purifiedsubunits were unsuccessful (S.Le Grice, unpublished) theapproach we followed involved co-homogenization of theappropriately induced cultures and metal chelate affinitychromatography (Le Grice et al., 1991). RT reconstitutedfrom protonated p66 and deuterated p51s, the H-D-hybridcontained both subunits in a 1:1 stoichiometry, followingchromatography on Ni2+-NTA- and S-Sepharose (Figure2b). In the present study (data not shown) and related work(Le Grice et al., 1991; Howard et al., 1991), we havedemonstrated that reconstitution does not affect the enzymaticproperties or tRNALYs 3 binding of the resulting heterodimer(Barat et al., 1989; Darlix, unpublished results).
Shape parameters of the subunit pSJs in situ. HeterodimericRT with a deuterated pSi kDa subunit was subjected toneutron scattering studies with the aim of determining the
scattering curve of the p51, subunit in situ, i.e. as abuilding block of RT. The contrast of the protonated p66subunit was matched by using a buffer containing 41% D20(data not shown). Under these conditions only the deuteratedp51 subunit is 'visible' for neutrons. The distance distribu-tion function of the pS5 subunit in situ, which is shown inFigure lb, was obtained by Fourier transformation of thescattering curve. The parameters derived from the distancedistribution function are presented in Table I.
Shape parameters ofthe p66 subunit in situ. The scatteringcurve of the p66 subunit in situ was obtained using the sameRT preparation as in the previous experiment. Theprotonated p66 subunit was visualized after matching thedeuterated p5 1 subunit. The contrast of deuterated proteincan be eliminated in 1 18% D20. This value of 1 8 1%was obtained by an approach described by Lederer et al.(1986) taking into account the 'zero scattering intensity' (IO)of the H-D-hybrid RT determined at different D20concentrations (see Figure 3a), as described in Materials andmethods. 118% D20 obviously cannot be achievedexperimentally. However, an extrapolation proceduredescribed by Stuhrmann permits the determination of thescattering curve of the deuterated p66 subunit from thescattering curves of the H-D-hybrid RT measured at differentcontrasts (Ibel and Stuhrmann, 1975). We have determinedthe scattering curves at different D20 concentrations (0, 41,74 and 92%) and used these curves for obtaining thescattering curve of the complex in 118% D20. Figure 3billustrates the procedure for obtaining the radius of gyrationof the subunit p66 in situ and Figure lb shows the distancedistribution function of the p66 subunit obtained accordingto the Stuhrmann protocol.
7he p15, RNase H-containing domain. The scattering curveof the p15 domain in situ could not be obtained by neutronscattering, since this domain, as part of the p66 subunit,could not be selectively deuterated. The X-ray crystal
1132
Quaternary structure of HIV-1 reverse transcriptase
a p66 D
\ Co-homogenisationstep I 100 000 a g
p66 p66/D-[His]-p51 D-lHis]-p5
NTA-Se2 afffib
p66/D-[His]-p5 1
'3- _---
b
epha-rose metal chc:nity chromatograph:
D-[His]-p5 1
M H N
45-0- al~~~~~~~~~~~~~~~~~~*e;-F.:97-2..8 mmm66-~~~ _
S c
.... ._.-p66poo -[D] 1
S-Sepharose ion exchanLchromatographi
ste) 3
p66/D-[Hisl-p5 1 18 -_s14- am
Fig. 2. Reconstitution of p5l/p66 HIV RT containing a deuterated p5IS subunit. (a) Outline of the reconstitution procedure. Cells containing aprotonated p66 subunit were mixed with those containing the deuterated pSls subunit (D-[His]pSl) and co-homogenized (step 1). The filled portion atthe N-terminus of p51S represents a small polyhistidine extension (His6) to aid purification by metal chelate affinity chromatography (Le Grice andGruninger-Leitch, 1990). The high speed supematant of a cell homogenate was loaded onto a column of Ni2+-NTA-Sepharose for selection ofproteins bearing the polyhistidine extension, i.e., H-D-hybrid RT (D-[His]pS5/H-p66) and D-[His]pSl (step 2). Further chromatography on
S-Sepharose separates excess p51 RT from the reconstituted heterodimer (step 3). (b) Analysis of reconstituted, selectively deuterated p5l/p66 HIV-1RT by SDS gel electrophoresis. Lane M, protein Mr markers; lane H, high speed supernatant; lane N, Ni2+-NTA-Sepharose-purified enzyme; laneD, DEAE-Sephacel-purified enzyme; lane S, S-Sepharose-purified enzyme; lane C, control heterodimer HIV-1 RT. Note that the heterodimerpreparation is over-represented by D-[His]pSl until recovery from S-Sepharose.
Table I. Molecular parameters of the components of the HIV- 1 RT
Components Mr Volume RG Dmax Half-axes of ellipsoids (nm)(kDa) (nm3) (nm) (nm) a b c
p5l 51 63 2.48 + 0.04 8 + 0.5 4.0 3.7 1.0p66 66 81 3.14 0.07 10 1 5.0 3.9 1.0p15 15 18 1.4a 1.6 1.6 1.6p5l/p66 117 145 3.27 + 0.04 10 + 0.5 5.2 4.8 1.4p66/p66 132 162 3.53 + 0.04 12 ± 1 6.0 4.9 1.4p5l/pSI 102 2.95 0.35b
The molecular weights obtained by biochemical methods and by neutron scattering agree within 5%.aDetermined from a model of p15 based on the E.coli RNase H (Yang et al., 1990) and moelcular modelling (W.Hendrickson, personal com-munication).bCalculated.
structure ofp5 has been published recently (Davies et al.,1991). The use of these data for calculating the distancedistribution function of the p15 domain was not possible,since the data were not provided by the authors. Therefore,the radius of gyration (R15) was derived from a model ofthe p15 domain based on the structure of E.coli RNase H(Yang et al., 1990), which was refined by molecularmodelling (W.A.Hendrickson, personal communication).The radius of gyration obtained is R15 = 1.37 nm. A lowerlimit of R15 = 1.27 nm was estimated assuming a sphericalshape of p15 having a partial specific volume of 0.74 cm3/g(Lehninger, 1975). Both values are close, justifying thespherical approximation. The radius r of the correspondingscattering equivalent sphere is 1.6 nm.
Shape parameters ofthe homodimerp66/p66. We includedin our structural studies also the homodimeric RT (p66/p66).The homodimer differs from the heterodimer by an additionalpl5 domain. Both types of RT show almost equivalent DNApolymerizing activity and RNase H activity (Larder et al.,1987; Hizi et al., 1988; Muller et al., 1989; Le Grice andGruninger-Leitch, 1990). This close enzymatic relatednessencouraged us to compare the structural data obtained fromthe homodimeric and heterodimeric RT. We overexpressedthe homodimeric p66/p66 (Le Grice and Gruninger-Leitch,1990) and subjected the purified protein to neutron smallangle scattering studies. Data evaluation was performed asdescribed above for the heterodimeric RT. The distancedistribution function of the p66/p66 homodimer is shown
1133
)-[Hisl-p5 1
step

H.Lederer et at.
b 12
1 0
u-i 8
EC 6
CMa
4
2
020 40 60
CD20CH20 C D20
Ia-T --T
-1.5 - 1 -0.5 0 0.5
1 /Ap [10 nmm]
Fig. 3. (a) Contrast variation of the H-D-RT. The normalized scattering amplitudeAS= (ION)/2*signAQ was plotted against the normalized D20concentration, with Io being the normalized 'zero scattering intensity' of the scattering curves determined at the corresponding D20 concentrations, asdescribed in Materials and methods. (b) 'Sturhmann plot' of H-D-hybrid RT for obtaining the distance between the subunits p66 and p5is, and theradius of gyration of the p66 subunit. From a set of scattering curves at different D20 concentrations the radii of gyration (RG) of the H-D-RT weredetermined by Guinier plots (not shown). The squares of these RC, were plotted against the inverse scattering contrasts 1/AQ and fitted to theparabolic equation (4). From the parameters of this parabola the centre-to-centre distance between the protonated p66 subunit and the deuterated pSlssubunit was determined, as shown in Materials and methods. The error bars lie within the size of the symbols used for data representation.
Table 11. Centre-to-centre distances of the components of the HIV-1 RT
(a) Calculated from the parallel axes theorem of mechanics
Components Within zi Z2 Distance(nm)
P51d p15 p66 as part of theheterodimer p5l/p66 0.773 0.227 5.0 ± 0.5
p5ls p66 heterodimer p5l/p66 0.436 0.564 3.25 ± 0.37
p5Id/P51s= p15 heterodimer p5l/p66 0.872 0.128 5.1 : 1.2p66 p66 homodimer p66/p66 0.5 0.5 3.38 ± 0.42
p51s/p51dd pl5/pl5 homodimer p66/p66 0.773 0.227 5.1 ± 0.9
Zi are the scattering mass ratios (see Materials and methods).
(b) Calculated by other methods
Components Within Method DistanceI
pSi = p66 p66 as part of theheterodimer p5l/p66 Stuhrmann plot 3.28 i 0.3
p5ld p66 heterodimer p5l/p66 mass low 1.14 i 0.11p51d pSl heterodimer p5I/p66 see Results 3.0 + 0.9p5Sl p15 heterodimer p51/p66 geometrical construction 5.3 f 1.2
in Figure lb, the radius of gyration and the derivedparameters in Table I.
Centre-to-centre distances of the RT componentsApproximation of the shape of the components by triaxialbodies was the first step in evluating the domain structureof RT. The positioning of these components requiresinformation about their centre-to-centre distances. Theneutron scattering approach can give this informationprovided that the centre of scattering mass determined byneutron scattering coincides with the centre of gravitationalmass. This is the case in the approximation applied here,which assumes that subunits have a homogeneous mass
distribution.The distance between two components was calculated
using three parameters, the radii of gyration of each singlecomponent and that of the united components. Assuming that
1134
the scattering parameters obtained for the p5 ls subunitapply also for the pSld domain (which is part of the p66subunit), the following centre-to-centre distances were
calculated by means of the parallel axes theorem ofmechanics (see Materials and methods and Table IIa). Thedistance between the p5ld domain and the p15 domain,d5ld.15, is 5.0 0.5 nm, and the distance between thep51s subunit and the p66 subunit, d5ls-66, is 3.25 il0.37 nm. This value was verified by an independent distancedetermination using the 'Stuhrmann plot' (see Materials andmethods, Table IIb and Figure 3b) obtaining d5j,_66 valueof 3.28 ±E 0.3 nm. The centre-to-centre distance betweenthe p5 Id domain and the p51s subunit in the heterodimer,d5ld-51s, could not be determined directly, since thesecomponents could not be deuterated selectively. However,the information about the radius of gyration of the sub-assembly p5ld/p5ls is contained in the complete p66/pSl,
a
._
ce
a-
2
0
-1I
1

Quaternary structure of HIV-1 reverse transcriptase
a)
_4 - --_-.-
7.4r Fr 2nFig. 4. Reconstruction of the spatial arrangement of the p5ld and the p15, RNase H domain within the p66 subunit. Projection of the ellipsoidal p66subunit (a) along the two longer axes and (b) along the shortest and the longest axes. 'S' indicates the centre of gravity of the correspondingcomponent. dmax is the maximum dimension of the p66 subunit. The concentric shaded rings indicate the error margins of dmax and of the distancedI5-51d, respectively. The thickest line shows the overall dimension of the p66 subunit represented by a scattering equivalent ellipsoid.
molecule, whose spatial arrangement is known. Using thisinformation an estimation of the distance d5ld-51s ispossible. As shown in Materials and methods, we obtained:
2.1 nm < d5ls-51d < 3.9 nm,
and for the radius of gyration R5Sd,51s: i
2.6 nm < R5ld,51s < 3.3 nm. S5Id 3
I Usin2 this radius of evration. the distance between the centre Pa\of the p5ld/p5li sub-assembly and the p15 domain,d5d5 Is -15, was calculated using the parallel axes theoremof mechanics as 5.1 1.2 nm.
I
Proposed model for p5 1/p66 HIV- 1 RTThe centre-to-centre distances of the components in TableII and their shape parameters in Table I were the basis formodelling the domain structure of heterodimeric RT. Westarted with the spatial arrangement of the components ofthe p66 subunit, i.e. the p15 domain and the p5Id domain,and then arranged the p51s subunit so that the resultingstructure fulfils the distance and shape parameters listed inTables I and II.
The p15 domain and the pS1d domain within the p66subunit. Figure 4 shows how the spatial arrangement of thedomains of RT was reconstructed. The mass centre of thep15 domain is located on a sphere of radius dI5-5Id = 5.0
0.5 nm around the centre of the p5ld domain. Theposition of the p5 domain on the sphere is constrained bythe maximum dimension of the p66 subunit dmax (p66) =
10 i 1 nm. Figure 4a and b show the geometricalreconstruction in two planes with the longest axis incommon. The requirement of contact between the p15- andthe p5ld domain restricts the position of p15 to the polesof the ellipsoidal pSld domain. This is obvious in theprojection along the longest and shortest axes of the p5lddomain displayed in Figure 4b. It is less obvious in the otherprojection displayed in Figure 4a. Due to the error marginsof the distance parameters, an exact positioning of the p15
-:unm
I
v S51 p5
Fig. 5. Reconstruction of the p5 I/p66 heterodimer. Projection of theheterodimer (a) along the two longest axes and (b) along the twoshortest axes but without the p15 domain. Arrows represent thedistances between the centres of gravity of the components. Thethickest line shows the overall dimension of the p66/pSl RTrepresented by a scattering equivalent ellipsoid.
domain was not possible. Using in addition the distanced5sld..S (3.0 ± 0.9 nm) and d51s_15 (5.1 4 0.9 nm), theposition of the p15 domain was determined by triangulation.
Fixation of the distance triangle within the RT moleculerequired information about the orientation of the two subunitspSi and p66 within the RT.
Spatial arrangement of the p5ls subunit with respect to thep66 subunit. The mass centre of the p51s subunit is locatedon the distance sphere of radius d66-51s = 3.25 ± 0.37 nmaround the centre of the p66 subunit. The position of the
1135

H.Lederer et al.
RNA cutting site
Polymensation siteFig. 6. Quaternary structure of the heterodimeric HIV- 1 RT with the template docked at a putative binding site. The model of RT as obtained fromneutron solution scattering was docked to the template, taking into account the following information: (i) the distance between the RNA cutting siteand the polymerization site is 15-18 bases (Furfine and Reardon, 1991; Wohrl and Molling, 1990), (ii) the RNA cutting site is located within thep15 domain (this study), (iii) the template binds to the interface between the pSi domain and the p5l subunit (Beccera et al., 1990; Painter et al.,1990).
p51, subunit is further constrained by the overall shapeparameters of the complex p66/pS5s. The shortest half axisof the p66/pS Is complex is only 0.4 nm larger than that ofa single p51 component, which is 1.0 nm. This is a strongconstraint for the position and the orientation of the p51ssubunit on the distance sphere. Both subunits must bearranged side by side on their flat side in order to fit intothe overall dimension of the complete RT as shown in FigureSb. Figure 5a shows a 900 turned projection of the spatialarrangement of the domains of RT. This projection showsthat the derived domain structure fits well into the dimensionof RT.
Spatial arrangement of the two p66 subunits in thehomodimer p66/p66. The distance between the two p66subunits, d66-66, is 3.38 ±i 0.42 nm (Table Ila). Thisparameter was calculated using the parallel axes theorem,assuming that the radius of gyration of the single p66 subunitobtained from the heterodimeric RT also holds for thesubunits in the homodimer. Concerning the orientation, twoarrangements were considered, a parallel alignment of thetwo p66 subunit in a mirror-symmetrical fashion, whichmeans that the two p15 domains would be positioned in closeproximity, and an antiparallel arrangement in a point-symmetrical fashion, which means that the two p15 domainswould be positioned on opposite sides of the homodimer.A distance between the centre of gravity of the p5Is/pS5 dsub-assembly and the piS/pIS sub-assembly was calculatedusing the parallel axes theorem. Here the assumption wasmade that there is no drastic difference between the spatialarrangement of the p5 Is/p5 1d sub-assembly in the hetero-and homodimers. A similar value of d51,5Id-15/15 = 5.1 i
0.9 nm was obtained for both the parallel and the antiparallelarrangements (Table Ila). This finding is an independentverification of the corresponding parameter in theheterodimer, the distance d5s515d -15, which was 5.1 ±fi1.2 nm. Even if data analysis showed that the antiparallelarrangement is more likely, it was not possible to decidewhich of the two arrangements resides in the homodimer.
DiscussionRT is one of the major targets in drug design against AIDS(Jacobo-Molina and Arnold, 1991; Mitsuya et al., 1990;Merluzzi et al., 1990; Pauwels et al., 1990). A betterknowledge of the structure and function of this enzyme isessential in the search for suitable drugs. Many models havebeen proposed in the literature on the basis of speculationand suggestions. Our neutron small angle study is the firstapproach which provides structural information about RTon the basis of experimental data. A crystallographicapproach would have been preferable due to higherresolution, but this approach will have to await theavailability of suitable crystals. In contrast to the crystallo-graphic approach, the data interpretation of neutron smallangle scattering is less straightforward. Detailed conclusionsabout the shape would require sophisticated fitting procedureswhich, in general, do not yield unique results. We avoidedthis by confining the conclusions about the shapes of thecomponents to ellipsoids, since their half axes can becalculated directly from the experimentally obtainedscattering parameters. The strength of neutron small anglescattering is the possibility of gettting information about thedistances of the components within a molecule. This
1136

Quaternary structure of HIV-1 reverse transcriptase
information is obtained by solution studies without destroyingthe molecules. According to the capacity of neutron solutionscattering it is the aim of this study to determine the spatialarrangement of the domains of RT rather than their shape.The distance determination between two components is
possible if the radii of gyration of these components and theirassembly are known. The measurement of the radii ofgyration of the components in situ requires selectivevisualization of the components. This is achieved by isotopiclabelling of the single components with deuterium. Wesucceeded in preparing p5 l/p66 HIV-l RT from a protonatedp66 subunit and a deuterated p51s subunit using a novelreconstitution procedure (Figure 2, Le Grice et al., 1991;Howard et al., 1991). Subjection of the hybrid H-D-RT tocontrast variation with D20 allowed the determination ofthe radii of gyration of the p66 and p5l, subunits. Theradius of gyration of the p15, RNase H-containing domainwas derived from molecular modelling procedures using theE. coli RNase H as a basis (Hendrickson, personalcommunication, see Results).Using the six radii of gyration of the components p15,
p51s and p5ld and the assemblies p51s/p66 (the completeheterodimer), p5ld/P15 (the p66 subunit) and the sub-assembly, p51d/p51,, the centre-to-centre distances betweenthe components were calculated by the parallel axes theorem.Here the assumption was made that there is no greatstructural difference between the p51s subunit and the p51ddomain in situ, justifying the use of the same radius ofgyration for both components. We are aware that minorstructural differences between the p5ls subunit and the p51ddomain must exist, since both components reside within theRT in a different structural context. However, a drasticconformational change which could affect our conclusionson the RT model is rather unlikely, since the expectedstructural changes are a consequence of a proteolytic exci-sion of one of the p15 components of the homodimeric p66.The relative error of the distances is - 10% . The distance
between the p66 and p51s subunits is best determined andverified by an independent approach according to Stuhrmannand Miller (1978). The distance between the p15 domainand the sub-assembly, p51d/p51s, shows the least accuracy.This is due to the small influence of the low molecular weightcomponent, p15, on the radius of gyration of the complex.However, this distance is confirmed by an independentdistance determination of these components in the p55/p66homodimer. Despite this, it could not be decided whetherthe two p66 subunits are aligned parallel or antiparallel.Therefore, the p5ld domain and the p5ls subunit aredisplayed in the model in Figure 6 as symmetrical bodies.The centres of gravity of the components are located at
the corners of the distance triangle formed by the centre-to-centre distances of the components. The orientation of thecomponents was constrained by the overall shape parametersof the heterodimer. Figure 6 shows that RT is a flat moleculewith axes of 10 nm, 9 nm and 3 nm. The two p5icomponents are positioned side by side, attached at their flatsides. The RNase H domain is positioned at the far end ofthe ellipsoidal RT, most probably having contacts to bothp5l components.
Figure 6 shows the model of RT in the complex with itstemplate fitting the nucleic acid template in A-form. Thismodel has to be considered as a tentative model with respectto the position of the nucleic acid. The template is docked
to RT between the two subunits p66 and p5 1, as proposedpreviously (Jacobo-Molina and Arnold, 1991; Arnold andArnold, 1991). This position was proposed, taking intoaccount the following information: (i) the functionally activeform of HIV RT is the dimeric form (Restle et al., 1990)although the p66 subunit alone contains all functionalactivities (Cheng et al., 1991; Wu et al., 1991), (ii) thereis only one template binding site on the heterodimeric RT(Painter et al., 1990) and (iii) the two subunits p5Is and p66interact via a hydrophobic region (Becerra et al., 1990;Painter et al., 1990), which might serve as the templatebinding site.The orientation of RT shown in Figure 6 is based on our
knowledge that the RNA template in a DNA-RNA hybridis cut upstream of the polymerization site (Schatz et al.,1990; Furfine and Reardon, 1991). The number of basesbetween the RNA cutting site and the polymerization sitevaries between 7 and 18 bases (Oyama et al., 1989; Schatzet al., 1990; Furfine and Reardon, 1991; Wohrl and Molling,1990). We used the greatest value of 16-18 bases in orderto map the polymerization site in our model.
ConclusionOur neutron scattering studies on selectively deuteratedp5I/p66 HIV-1 RT provide information about the spatialarrangement of the components of RT, i.e. the subunit p51sand the domains p51d and p15, in solution. The modelderived is considered as a working model, which can serveas a basis for designing new experiments concerning RTstructure and function e.g. (i) the role of the p51 subunitin comparison to the p66 subunit, which carries all functionalactivities of RT, (ii) the conformational change induced inthe template following RT binding, (iii) the portion of thetemplate which interacts with RT. Answers to these questionsmight also be significant for the design of a nucleic acidfragment suitable for co-crystallization of RT with itstemplate.
Materials and methodsPreparation of RTHeterodimeric HIV-1 RT was prepared by metal chelate affinitychromatography from E coli strain M 15:: pDMI. 1:: pRT6H-PROT, whichdirects expression of p66 RT and HIV-1 protease (Le Grice and Gruninger-Leitch, 1990). Enzyme thus prepared bears a polyhistidine extension onthe C-terminus of the 66 kDa subunit. Reconstituted, selectively deuteratedHIV-1 RT was prepared according to published procedures (Le Grice et al.,1991), with the exception that (i) bacteria expressing His-pSI (Schatz et al.,1990) were grown and induced in deuterated algal hydrolysate and (ii)DEAE-Sephacel and S-Sepharose chromatography followedNi2+-NTA-Sepharose purification. The protocol for preparation of thisenzyme, D-[HisIpSl/Hp66, is outlined schematically in Figure 2a.Homodimeric p66/p66 R was prepared from the strain M 15:: pDM-
1. 1:: p6HRT as previously described (Le Grice and Gruninger-Leitch, 1990).All enzyme preparations were judged by SDS electrophoresis to be at least95% homogeneous.Growth and induction of the recombinant E. coli strains was as previously
described (Le Grice et al., 1991). In the experiments reported here andrelated studies (Le Grice et al., 1991; Howard et al., 1991), p5l/p66prepared by reconstitution retained full polymerase and RNase H activities.Furthermore, tRNALYS.3 binding to the reconstituted, selectively deuteratedenzyme was uanffected (J.-L.Darlix, unpublished). The concentration ofeach protein was determined using an extinction coefficient e1% of 18 forboth the homodimer and heterodimer.
Data collectionThe small-angle neutron scattering measurements were performed on theDII small-angle diffractometer of the Institut Laue-Langevin (Grenoble,
1137

H.Lederer et al.
France). The samples were dialysed against buffers containing 0.05 M or0.5 M NaCl, pH 7.9, 10 mM 13-mercaptoehanol in H20, D20 or mixturesof both. During beam exposure the samples were equilibrated to 8°C.The scattering data were processed and corrected as described by Heumann
et al. (1988).Concentration effects were eliminated by linearly extrapolating the
scattering curves, which were measured at two different concentrations,usually 6 mg/mi and 3 mg/ml, to concentration zero. From the scatteringcurves the distance distribution functions p(r) were calculated [with a Fortranprogram of Glatter (1977)], from which the maximum dimensions of theparticles were obtained. Forward scattered intensities and radii of gyrationwere determined from the area and second moment of the p(r) functions,as well as from Guinier plots of ln[I(Q)] versus Q2, where Q is themomentum transfer,
Q = (4) X sin ex
where 20 is the full scattering angle.The half axes (a, b, and c) of a scattering equivalent ellipsoid were
calculated from the radius of gyration RG and the maximum dimension dmax= 2c using the following equations:
R 2 = a2 + h2 +((1)
andV= 4 r X a x b x c (2)3
The volume (V) of a protein component was calculated from its molecularweight assuming a partial specific volume of v = 0.74 cm3/g (Lehninger,1975).
Distance determinationParallel axes theorem. The centre-to-centre distance d, 2 of twocomponents with the radii of gyration RI and R2 was determined from theradius of gyration R112 of the complexed particle according to the parallelaxes theorem of mechanics applied to scattering:
Rj,2 = zi x RI +Z2 -z2 x Z2 x dl2 (3)
z1 and Z2 denote the scattering fraction of each subunit
Ml + M2
where Mi = VjiQ, and Qo is the scattering length density.
Determination ofthe 'match point' ofthe D-pSI within the H-D-hybrid RTby contrast variation. The match point of the D-p5 1 in situ was determinedfollowing the formalism of Lederer et al. (1986) taking into account theinformation of the match point of the H-D-hybrid RT. This point was derivedfrom a series of measurements of the H-D-hybrid at different D20concentrations, by extrapolation of the scattering intensities (10) to thescattering angle zero and by determination of the normalized scatteringamplitude AO = (40),)"12*sign AQ, where 10,N = 10/(cDl) and signAQ isthe sign of the scattering length density of the hybrid RT. c is the massconcentration of RT, D is the path length of the cuvette and T is thetransmission. Interpolation of the normalized scattering amplitude Ao versusthe normalized D20 concentration to Ao = 0 yielded the match point at75% D20 (see Figure 3a). This value was used to determine the degreeof deuteration of the subunit p51, taking into account the fraction ofexchangeable protons determined from the match point of non-deuteratedRT of 41 % D20 (data not shown). Using the 'Lederer' approach thefraction of the exchangeable non-C bonded hydrogen atoms was calculatedas 83 4% and the concentration of D20 in the solution buffer, wherethe scattering contribution of the D-pS is suppressed, was determined as118 ± 1% D20.
7he 'Stuhrmann 'plot. Alternatively, the centre-to-centre distance betweenthe subunits p66 and p51s was obtained from a 'Stuhrmann' plot (Ibel andStuhrmann, 1975). This approach allows the determination of the distancebetween the centres of gravity of the scattering masses of a deuterated anda protonated component, such as the D-pS 1, subunit and the H-p66 subunit.Radii of gyration (RG) were determined from a series of measurements atdifferent D20 concentrations on the hybrid RT. RG2 was plotted versusl/AL and fitted to the parabolic equation:
RG2 = RC2 + aI/Ap - 3/ASL (4)
1138
with RC = 10.93 nm, the radius of gyration at infinite contrast, and thecoefficients a = -2.58 x 10-4 and 3 = 1.31 x 10-7 nm-2. The qualityof the fit is indicated by the correlation coefficient of 0.999. Using thecoefficient ( the distance between the components H-p66 and D-pSI isobtained according to the equation
dsl, - 66 = ( ) + ( 2 )AQi Q2
(5)
AQ and AQ2 are the contrasts at which the subunits H-p66 and D-p51 arematched, respectively.The centre-to-centre distance obtained is d5l,566 = 3.28 + 0.3 nm.
Extrapolation of the plot to the contrast matching of deuterated p51 in abuffer containing the hypothetical D20 concentraton of 118% D20, yieldedthe radius of gyration, R66 of 3.14 + 0.07 nm.
The mass law. Within the p66 subunit, the distance d66-51d between thecentres of the component p66 and the p5ld is correlated with the knowndistance, d66-15, between p66 and p15 according to their molecular weightsM51 and M15 as follows:
d66-51d x M51 = d66-15 x M15 (6)
Estimation of the distance between the centres of the pSJd domain and thepSI, subunit, and of the radius ofgyration Rs5d/s,5. The centre of the p51ddomain lies on the sphere of radius d51d-66 = 1.1 0.1 nm centredaround the p66 subunit. This distance is calculated according to the masslaw (6). An upper and lower limit of this distance is therefore, d515_51d= d5is_66 d5ld-66 = 3.3 i 1.2 nm. The upper limit, (d5l 51d) ..)maxis 4.5 nm. This can be further restricted to a value of 3.9 nm using theparallel axes theorem (3) by taking into account that an upper limit of theradius of gyration of the p5ld/p5l, sub-assembly is the radius of gyrationof the complete heterodimer, R66/51s = 3.27 + 0.04 nm. It then followsthat:
2.1 nm < d5iS-51d < 3.9 nm.
Using equation (3) we obtain for the radius of gyration:
2.6 nm < Rs5d5sIs < 3.3 nm.
AcknowledgementsH. L. and H.H. thank the Bundesministerium fur Forschung und Technologiefor support (Research Grant 11-108-90). S.Le G. is supported by a NATOCollaborative Research Grant (CRG 900471), and a grant from the NationalInstitutes of Health (ROI Al/GM 31147-01). J.-L.D. is supported by theFrench effort against AIDS. The support of Hoffmann-La Roche Basel,Switzerland in early experiments involving HIV RT reconstitution isgratefully acknowledged.
ReferencesArnold,E. and Arnold,G.F. (1991) Adv. Virus Res., 39, 1 -87.Barat,C., Lullien,V., Schatz,O., Keith,G., Nugeyre,M.T., Gruninger-
Leitch,F., Barre-Sinoussi,F., Le Grice,S.F.J., Darlix,J.-L. (1989) EMBOJ., 8, 3279-3285.
Becerra,S.P., Kumar,A., Widen,S.G., Karawya,E.M., Abbotts,J.,Hughes,S.H., Shiloach,J. and Wilson,S.H. (1990) Fourth Meeting ofGroups Studying the Structure of AIDS-Related Systems and 7heirApplication to Targeted Drug Design. NIGMS, NIH, Bethesda, MD.
Capel,M.S. et al. (1987) Science, 238, 1403-1406.Cheng,N., Painter,G.R. and Furman,P.A. (1991) Biochem. Biophys. Res.
Commun., 174, 785-789.Crichton,R.R., Engelman,D.M., Haas,J., Koch,M.H.J., Moore,P.B.,
Parfait,R. and Stuhrmann,H.B. (1977) Proc. Nall. Acad. Sci. USA, 74,5547-5550.
Davies,J.F.,ll, Hostomska,Z., Hostomsky,Z., Jordan,S.R. andMatthews,D.A. (1991) Science, 252, 88-95.
Di Marzo Veronese,F.D., Copeland,T.D., DeVico,A.L., Rahman,R.,Oroszlan,S., Gallo,R.C. and Sarngadharan,M.G. (1986) Science, 231,1289- 1291.
Furfine,E.S. and Reardon,J.E. (1991) J. Biol. Chem., 266, 406-412.Glatter,O. (1977) J. Appl. Crystallogr., 10, 415-421.Goff,S.P. 1990) J. Acquired Immun. Dc/ic. Syndr., 3, 817- 831.Hansen,J., Schulze,T., Mellert,W. and Molling,K. (1988) EMBO J., 7,239-243.

Quaternary structure of HIV-1 reverse transcriptase
Heumann,H., Lederer,H., Baer,G., May,R.P, Kjems,J.K. and Crespi,H.L.(1988) J. Mol. Biol., 201, 115-125.
Hizi,A., McGill,C. and Hughes,S.H. (1988) Proc. Natl. Acad. Sci. USA,85, 1218-1222.
Howard,K.J., Frank,K.B., Sim,I.S. and Le Grice,S.F.J. (1991) J. Biol.Chem., in press.
Ibel,K. and Stuhrmann,H.B. (1975) J. Mol. Biol., 93, 255-265.Jacobo-Molina,A. and Arnold,E. (1991) Biochemistry, 30, 6351-6361.Larder,B.A., Purifoy,D.J.M., Powell,K.L. and Darby,G. (1987) EMBO
J., 6, 3133-3137.Le Grice,S.F.J. and Gruninger-Leitch,F. (1990) Eur. J. Biochem., 187,
307-314.Le Grice,S.F.J., Naas,T., Wohlgensinger,B. and Schatz,O. (1991) EMBO
J., 10, 3905-3911.Le Grice,S.F.J., Zehnle,R. and Mous,J. (1988) J. Virol., 62, 2525 -2529.Lederer,H., May,R.P., Kjems,J.K., Schafer,W., Crespi,H.L. andHeumann,H. (1986) Eur. J. Biochem., 156, 655-659.
Lederer,H., Tovar,K., Baer,G., May,R.P., Hillen,W. and Heumann,H.(1989) EMBO J., 8, 1257-1263.
Lehninger,A.L. (1975) Biochemistry, 2nd edition. Worth, New York,p. 176.
Lightfoote,M.M., Coligan,J.E., Folks,T.M., FauciA.S., Martin,M.A. andVenkatesan,S. (1986) J. Virol., 60, 771-775.
Lowe,D.M., Aitken,A., Bradley,C., Darby,G.K., Larder,B.A.,Powell,K.L., Purifoy,D.J.M., Tisdale,M. and Stammers,D.K. (1988)Biochemistry, 27, 8884-8889.
Merluzzi,V.J. et al. (1990) Science, 250, 1411-1413.Mitsuya,H., Yarchoan,R. and Broder,S. (1990) Science, 249, 1533- 1544.Muller,B., Restle,T., Weiss,S., Gautel,M., Sczakiel,G. and Goody,R.
(1989) J. Biol. Chem., 264, 18808-18817.Nierhaus,K.H., Lietzke,R., May,R.P., Nowotny,V., Schulze,H.,
Simpson,K., Wurmbach,P. and Stuhrmann,H.B. (1983) Proc. Natl. Acad.Sci. USA, 80, 2889-2893.
Oyama,F., Kikuchi,R., Crouch,R.J. and Uchida,T. (1989) J. Biol. Chem.,264, 18808-18817.
Painter,G.R., Wright,L.L., Hopkins,S. and Furman,P.A. (1990) J. Cell.Biochem. UCLA Symposium HIV and AIDS: Pathogenesis, Therapy andVaccine, Abstract L140 S14D.
Pauwels,R. et al. (1990) Nature, 343, 470-474.Restle,T., Muller,B. and Goody,R.S. (1990) J. Biol. Chem., 265,
8986-8988.Schatz,O., MousJ. and Le Grice,F.S.J. (1990) EMBO J., 9, 1171-1176.Stuhrmann,H.B. and Miller.A. (1978) J. Appl. Crnstallogr., 11. 325-345.Varmus,H. (1987) Sci. Am., 257, 56-64.Varmus,H. and Swanstrom,R. (1984) In Weiss,R., Teich,N., Varmus,H.
and Coffinj. (eds), RNA Tumor Viruses. Cold Spring Harbor LaboratoryPress, Cold Spring Harbor, NY.
Wohrl,B.M. and Molling,K. (1990) Biochemistrn. 29, 10141-10147.Wu,J.C., Warren,T.C., Adams,J., Proudfoot,J., Skiles,J.. Raghavan.P.,
Perry,C., Potocki.I., Farina,P.R. and Grob,P.M. (1991) Biochelistry.30, 2022-2026.
Yang,W., Hendrickson,W.A., Crouch,R.J. and Satow,Y. (1990) Science.249, 1398-1405.
Received on November 8, 1991; revised on December 16, 1991
1139





























