Embed Size (px)
Citation preview

Rs
EG1
a
ARRAA
KMDINA
1
fpohbcsa1ltiphtcn
h1
Journal of Molecular Catalysis A: Chemical 387 (2014) 76–85
Contents lists available at ScienceDirect
Journal of Molecular Catalysis A: Chemical
jou rna l h om epa ge: www.elsev ier .com/ locate /molcata
ole of support in deoxygenation and isomerization of methyltearate over nickel–molybdenum catalysts
ika W. Qian ∗, Ning Chen, Shaofeng Gongraduate School of Bio-Applications and Systems Engineering, Tokyo University of Agriculture and Technology, Nakacho, 2-24-16, Koganei, Tokyo84-8588, Japan
r t i c l e i n f o
rticle history:eceived 26 December 2013eceived in revised form 26 February 2014ccepted 26 February 2014vailable online 7 March 2014
eywords:ethyl stearate
a b s t r a c t
Microporous SAPO-11 and highly ordered mesoporous AlSBA-15 with different aluminum contents (withSi/Al ratio of 5 and 10) were synthesized. Thus prepared samples were characterized by BET, pyridine-FTIRand NH3-TPD to investigate their structural and acidic properties. The samples were then transformedinto bifunctional catalysts by loading with molybdenum and nickel. Their activities were tested in thehydroconversion of methyl stearate using a fixed bed flow reactor system. The sulfided NiMo catalystsexhibited high conversion and deoxygenation activities. High isomerization activities observed for bothNiMo/SAPO-11 and NiMo/AlSBA-15 catalysts, similar to the isomerization of light naphtha, was attributed
eoxygenationsomerizationiMo catalystcidic support
to the acidity of supports. However, the acidity of supports was not the only factor influencing the isomer-ization of long chain molecules. AlSBA-15 had a large specific surface area that contained more acidic sitesinside of its channels, promoting the formation of cracking products; SAPO-11 had a suitable pore sizeand contained fewer acidic sites inside the pore channels, promoting the formation of mono-branchedisomers while suppressing cracking reactions.
© 2014 Elsevier B.V. All rights reserved.
. Introduction
Recently, much attention has been focused on producing greenuels or chemicals from triglycerides which are the main com-onents of vegetable oils [1,2]. As biodiesel fuel oil, reductionf oxygen in triglycerides is required. Therefore, recent researchas focused on hydroconversion of triglycerides into hydrocar-ons [3,4]. The catalysts commonly employed to lower the oxygenontent are cobalt- or nickel-promoted Mo(W)S2 on �-aluminaupport [5–7]. The thus obtained deoxygenated products, however,re mainly the mixture of n-paraffins with the carbon numbers of5–18, whose cold flow properties were so unfavorable that strictly
imited their application as diesel fuels. Hence, utilizing an addi-ional catalytic process, such as the isomerization of n-paraffins,s recommended to solve this problem [8]. Currently, a two-steprocess is used by Neste Oil Company [9] on an industrial scale;owever, the complexity and high cost of the process necessitatehe development of a simpler and more economical solution that
an remove the oxygen atoms from the feedstock, while simulta-eously isomerizing the deoxygenated products.∗ Corresponding author. Tel.: +81 42 388 7410; fax: +81 42 388 7410.E-mail address: [email protected] (E.W. Qian).
ttp://dx.doi.org/10.1016/j.molcata.2014.02.031381-1169/© 2014 Elsevier B.V. All rights reserved.
In catalysis, acid catalyzed reactions are valuable industrialchemical process used to produce various important chemicals[10]. The key step during n-alkane isomerization is the branchedalkylcarbenium ion formation, and this step depends on theBrønsted acid site. Among the family of porous materials, zeo-lites containing bridging hydroxyl groups, i.e. the Brønsted acidsites, could be suitable for promoting isomerization reactions [11].Recently, Pt/SAPO-11 has demonstrated exhibited high activityduring the hydrotreatment of Jatropha oil to produce isomers.We discussed the effects of the Si/Al ratio and Pt loading on thecatalytic activity [12]. Wang and co-workers designed a one-stephydrotreatment of soybean oil into isomerous alkanes by usingPt/Zeolite (SAPO-11 and ZSM-22) [13]. The effects of acid andalkali treatment on the nature of supports and property of cata-lysts have been investigated. Kikhtyanin and co-workers observedgood isomerization activity with Pd/SAPO-31 while hydrotreatingof sunflower oil [14]. They discussed the effect of reaction condi-tion on the product distribution. Although high quality fuel oil canbe produced with complete deoxygenation and high isomerizationselectivity using catalysts containing noble metals loaded on acidicsupports, the effect of support on the deoxygenation and isomeri-
zation pathways remain ambiguous. Furthermore, few studies havedescribed the effect of support on isomerization, cracking reactionsor the isomer distribution in hydroconversion of vegetable oil orFAMEs.
r Cata
Pmap[macasttsbwYpirPnp
tbtretfdNSnod
2
2
maabhdpaitTwac
(atsHtq
E.W. Qian et al. / Journal of Molecula
Numerous reports have investigated using zeolites loaded witht or Pd to isomerize light naphtha (C5–C8) into branched iso-ers [15]. Among these materials, SAPO-11 molecular sieves have
n AlPO4-11 (AEL) topology, and exhibit superior isomerizationerformance due to their shape selectivity and moderate acidity16]. SBA-15 was synthesized by Zhao et al. [17]; in contrast to
icroporous zeolites, this mesoporous silica has a pore size up topproximately 30 nm, allowing bulky molecules to enter. The dis-overy of SBA-15 helped design new materials that were applicables catalysts and adsorbents for bulkier molecules. However, purelyiliceous SBA-15 materials have few silanol groups on their surface;hese groups are weak acids and catalytically inactive. Therefore,o make these materials useful for acidic catalysis, stronger acidicites must be introduced into their framework. Two methods haveeen developed to incorporate aluminum atoms into the frame-ork of mesoporous materials: direct synthesis and post-synthesis.ue et al. have proposed directly synthesizing AlSBA-15 at a lowH (close to 1) [18], but recent studies have demonstrated that
ncorporating Al into the tetrahedral position of these supportsequired very careful control over the synthetic conditions [19,20].ost-synthesis alumination may take place either in aqueous oron-aqueous solution and is a promising alternative method forreparing AlSBA-15 supports.
The activity of bifunctional catalysts during isomeriza-ion depends on the balance of metallic versus acidic sitesecause the interaction between these sites might governhe hydrogenation–dehydrogenation, isomerization and crackingeactions over the corresponding catalysts [21–24]. The mostxtensively used metals for isomerizing n-alkanes are Pt and Pd dueo their good hydrogenation–dehydrogenation activities. However,ew studies have described supported NiMoS during simultaneouseoxygenation and isomerization process. In this work, a series ofi–Mo sulfide bimetallic catalysts loaded on acidic supports, i.e.,APO-11 and AlSBA-15, were prepared using sequential impreg-ation method. The aim of the work is to investigate the effectsf acidity and pore structure of SAPO-11 and AlBSA-15 during theeoxygenation and isomerization behavior of methyl stearate.
. Experimental
.1. Synthesis of supports
SAPO-11 was hydrothermally synthesized from an aqueousedium using a conventional method [25]. The synthesis processed
s follows: aluminum isopropoxide was mixed with phosphoriccid and distilled water, and the mixture was stirred in aeaker heated in a water-bath at 35 ◦C for 4 h. Subsequently, aomogeneous mixture containing di-n-propylamine (DPA) andi-iso-propylamine (DIPA) in a molar ratio of 2 were added as tem-lating agents. Subsequently, silica gel was added to the beakernd the mixture was stirred thoroughly for 2 h. Finally, the result-ng gel had a mole composition of 1.0 P2O5:1.0 Al2O3:0.4 SiO2:1.5emplate mixture:60 H2O and was transferred to a stainless-steeleflon-lined autoclave; the mixture was heated to 200 ◦C for 24 hithout stirring. The as-synthesized SAPO-11 sample was washed
nd filtered before being dried at 120 ◦C for 2 h and subsequentlyalcined at 550 ◦C in flowing air for 4 h.
Pure siliceous SBA-15 was synthesized using Pluronic P123EO20PO70EO20, BASF, Mw = 5800) as the structure directing agentnd tetraethyl orthosilicate (TEOS) as the silicon source accordingo a previously reported procedure [17,26]. In a typical synthe-
is, 20 g Pluronic P123 was dissolved in 150 g water and 600 g 2 MCl solution at 35 ◦C. Subsequently, 42.5 g TEOS was added intohe solution. The mixture was stirred at 35 ◦C for 20 h and subse-uently aged at 95 ◦C for 48 h without stirring. The as-synthesized
lysis A: Chemical 387 (2014) 76–85 77
SBA-15 sample was washed, filtered and dried at 80 ◦C for 2 h;finally, the sample was calcined at 550 ◦C in flowing air for 6 h.The aluminum was incorporated into the SBA-15 lattice post-synthetically by using tetramethylammonium hydroxide (TMAOH,10% in methanol; Tokyo Chemical Industry Co., Ltd.) as templateagent. To incorporate the calculated amount of Al into SBA-15needed for Si/Al mole ratios of 5 and 10, Eq. (1) was used to calculatethe necessary amount of TMAOH [27].
13Al(H2O)63+ + 32OH− � [AlO0Al12(OH)24(H2O)20]7+ + 62H2O
(1)
where the mole ratio of TMAOH to Al should be 32/13 ≈ 2.5Modified SBA-15 samples were labeled as AlSBA-15(x), while x
represents the Si/Al ratio (5 and 10). For a typical synthesis of AlSBA-15, 12 ml aqueous AlCl3·H2O solution and an appropriate amountof TMAOH (TAMOH/Al = 2.5) were heated at 80 ◦C with stirring togenerate a clear solution. Afterward, 6 g pure SBA-15 was addedwith stirring. The mixture was stirred for 2 h at the same tempera-ture. After filtration, washing and drying at 80 ◦C, the product wascalcined at 500 ◦C for 3 h in flowing air.
The mechanical mixtures, Al-SAPO-11 and Al-AlSBA-15, wereprepared by blending powdered SAPO-11 or AlSBA-15(5) withAl2O3 (Nippon Ketjen) and alumina sol (Nissan Chemical Indus-tries) in a 3:6:1 mass ratio. First, the supports were ground into apowder and a small amount of water was added. Subsequently, ahard dough was extruded using a hand extruder. The extrudateswere first dried at room temperature overnight and later at 105 ◦Cfor 2 h, before being calcined at 500 ◦C for 6 h.
2.2. Preparation of the Ni–Mo catalysts
All catalysts were prepared using conventional impregnationmethod procedures as reported [28]. The respective supportswere loaded with Mo and Ni using aqueous solutions of(NH4)6Mo7O24·4H2O and Ni(NO3)2·6H2O (Wako Pure Chem. Co.)to achieve a loading amount of 20 wt.% MoO3 and 3.5 wt.% NiO, inwhich Mo was introduced first. After impregnation, the sampleswere dried at 105 ◦C for 2 h and afterward calcined at 450 ◦C for10 h in flowing air.
2.3. Characterization of supports and catalysts
Powder X-ray diffraction (XRD) patterns were recorded usingan X-ray diffractometer (RAD-IIC; Rigaku Corp.) with Cu-Ka radi-ation operated at 40 kV and 20 mA to identify the phase structureof supports. Elemental analysis was performed using an X-ray flu-orescence instrument (EDX-800, Shimadzu Corp.).
The textural properties of supports and catalysts were mea-sured using nitrogen adsorption–desorption isotherms recordedat −196 ◦C (Belsorp-mini II; Bel Japan Co.). Before measurements,the samples were degassed under vacuum at 400 ◦C for 1 h. Thespecific surface areas (SBET) and pore volumes (Vtotal) were calcu-lated using the Brunauer–Emmett–Teller (BET) method; the poresize (dP) distributions were obtained from the desorption isothermusing the Barrett–Joyner–Halenda (BJH) method for mesoporoussamples and the MP method for SAPO-11 samples.
Ammonia temperature-programmed desorption (NH3-TPD)which was used to examine the acidity of the catalysts was con-ducted using a chemisorption–physisorption analyzer (ChemBETPULSAR TPR/TPD, Quantachrome Instruments Co.) according to thefollowing procedure. Typically, an approximately 200 mg sample
was pretreated at 500 ◦C in a highly pure helium gas atmosphere(15 mL/min) for 3 h and subsequently cooled to room temperatureunder the helium atmosphere. The ammonia adsorption was con-ducted for 40 min under 15 mL/min ammonia flow. The physically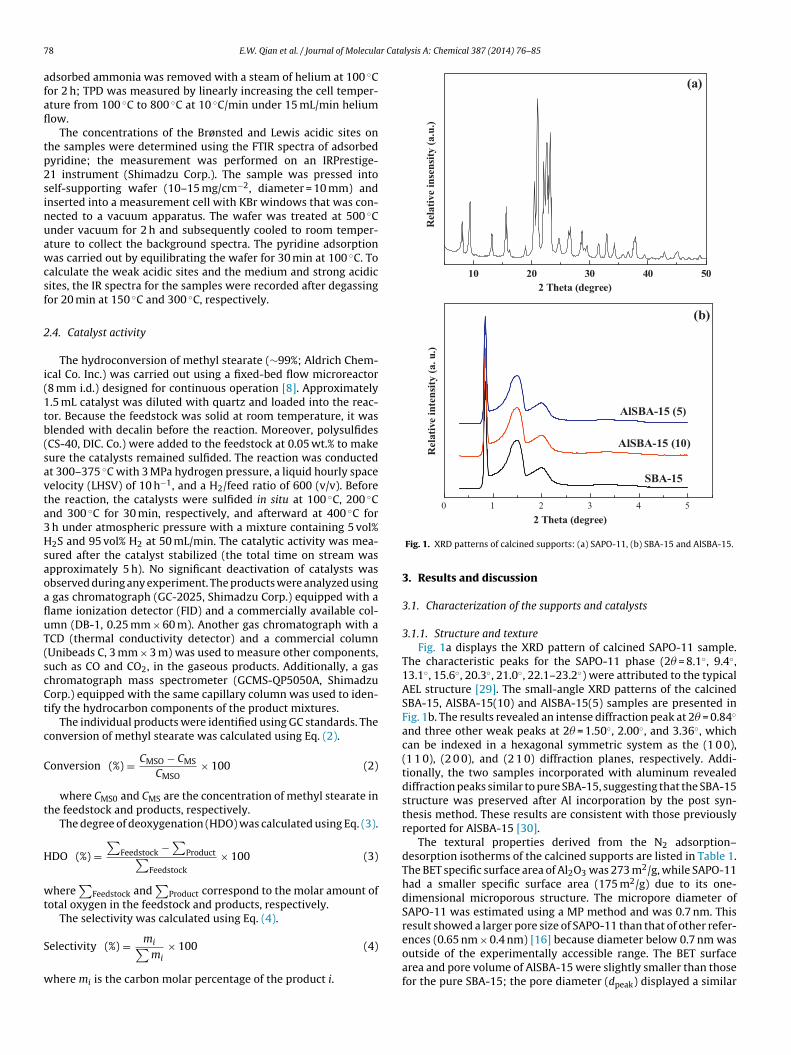
7 r Catalysis A: Chemical 387 (2014) 76–85
afafl
tp2sinuawcsf
2
i(1tb(savta3HsaoafluT(scCt
c
C
t
H
wt
S
w
10 20 30 40 50
Rel
ati
ve
inse
nsi
ty (
a.u
.)
2 Theta (degr ee)
(a)
0 1 2 3 4 5
SBA-15
AlSBA-15 (10)
Rela
tive i
nte
nsi
ty (
a. u
.)
(b)
AlSBA- 15 (5)
8 E.W. Qian et al. / Journal of Molecula
dsorbed ammonia was removed with a steam of helium at 100 ◦Cor 2 h; TPD was measured by linearly increasing the cell temper-ture from 100 ◦C to 800 ◦C at 10 ◦C/min under 15 mL/min heliumow.
The concentrations of the Brønsted and Lewis acidic sites onhe samples were determined using the FTIR spectra of adsorbedyridine; the measurement was performed on an IRPrestige-1 instrument (Shimadzu Corp.). The sample was pressed intoelf-supporting wafer (10–15 mg/cm−2, diameter = 10 mm) andnserted into a measurement cell with KBr windows that was con-ected to a vacuum apparatus. The wafer was treated at 500 ◦Cnder vacuum for 2 h and subsequently cooled to room temper-ture to collect the background spectra. The pyridine adsorptionas carried out by equilibrating the wafer for 30 min at 100 ◦C. To
alculate the weak acidic sites and the medium and strong acidicites, the IR spectra for the samples were recorded after degassingor 20 min at 150 ◦C and 300 ◦C, respectively.
.4. Catalyst activity
The hydroconversion of methyl stearate (∼99%; Aldrich Chem-cal Co. Inc.) was carried out using a fixed-bed flow microreactor8 mm i.d.) designed for continuous operation [8]. Approximately.5 mL catalyst was diluted with quartz and loaded into the reac-or. Because the feedstock was solid at room temperature, it waslended with decalin before the reaction. Moreover, polysulfidesCS-40, DIC. Co.) were added to the feedstock at 0.05 wt.% to makeure the catalysts remained sulfided. The reaction was conductedt 300–375 ◦C with 3 MPa hydrogen pressure, a liquid hourly spaceelocity (LHSV) of 10 h−1, and a H2/feed ratio of 600 (v/v). Beforehe reaction, the catalysts were sulfided in situ at 100 ◦C, 200 ◦Cnd 300 ◦C for 30 min, respectively, and afterward at 400 ◦C for
h under atmospheric pressure with a mixture containing 5 vol%2S and 95 vol% H2 at 50 mL/min. The catalytic activity was mea-
ured after the catalyst stabilized (the total time on stream waspproximately 5 h). No significant deactivation of catalysts wasbserved during any experiment. The products were analyzed using
gas chromatograph (GC-2025, Shimadzu Corp.) equipped with aame ionization detector (FID) and a commercially available col-mn (DB-1, 0.25 mm × 60 m). Another gas chromatograph with aCD (thermal conductivity detector) and a commercial columnUnibeads C, 3 mm × 3 m) was used to measure other components,uch as CO and CO2, in the gaseous products. Additionally, a gashromatograph mass spectrometer (GCMS-QP5050A, Shimadzuorp.) equipped with the same capillary column was used to iden-ify the hydrocarbon components of the product mixtures.
The individual products were identified using GC standards. Theonversion of methyl stearate was calculated using Eq. (2).
onversion (%) = CMSO − CMS
CMSO× 100 (2)
where CMS0 and CMS are the concentration of methyl stearate inhe feedstock and products, respectively.
The degree of deoxygenation (HDO) was calculated using Eq. (3).
DO (%) =∑
Feedstock −∑
Product∑Feedstock
× 100 (3)
here∑
Feedstock and∑
Product correspond to the molar amount ofotal oxygen in the feedstock and products, respectively.
The selectivity was calculated using Eq. (4).
electivity (%) = mi∑mi
× 100 (4)
here mi is the carbon molar percentage of the product i.
2 Theta (degr ee)
Fig. 1. XRD patterns of calcined supports: (a) SAPO-11, (b) SBA-15 and AlSBA-15.
3. Results and discussion
3.1. Characterization of the supports and catalysts
3.1.1. Structure and textureFig. 1a displays the XRD pattern of calcined SAPO-11 sample.
The characteristic peaks for the SAPO-11 phase (2� = 8.1◦, 9.4◦,13.1◦, 15.6◦, 20.3◦, 21.0◦, 22.1–23.2◦) were attributed to the typicalAEL structure [29]. The small-angle XRD patterns of the calcinedSBA-15, AlSBA-15(10) and AlSBA-15(5) samples are presented inFig. 1b. The results revealed an intense diffraction peak at 2� = 0.84◦
and three other weak peaks at 2� = 1.50◦, 2.00◦, and 3.36◦, whichcan be indexed in a hexagonal symmetric system as the (1 0 0),(1 1 0), (2 0 0), and (2 1 0) diffraction planes, respectively. Addi-tionally, the two samples incorporated with aluminum revealeddiffraction peaks similar to pure SBA-15, suggesting that the SBA-15structure was preserved after Al incorporation by the post syn-thesis method. These results are consistent with those previouslyreported for AlSBA-15 [30].
The textural properties derived from the N2 adsorption–desorption isotherms of the calcined supports are listed in Table 1.The BET specific surface area of Al2O3 was 273 m2/g, while SAPO-11had a smaller specific surface area (175 m2/g) due to its one-dimensional microporous structure. The micropore diameter ofSAPO-11 was estimated using a MP method and was 0.7 nm. Thisresult showed a larger pore size of SAPO-11 than that of other refer-
ences (0.65 nm × 0.4 nm) [16] because diameter below 0.7 nm wasoutside of the experimentally accessible range. The BET surfacearea and pore volume of AlSBA-15 were slightly smaller than thosefor the pure SBA-15; the pore diameter (dpeak) displayed a similar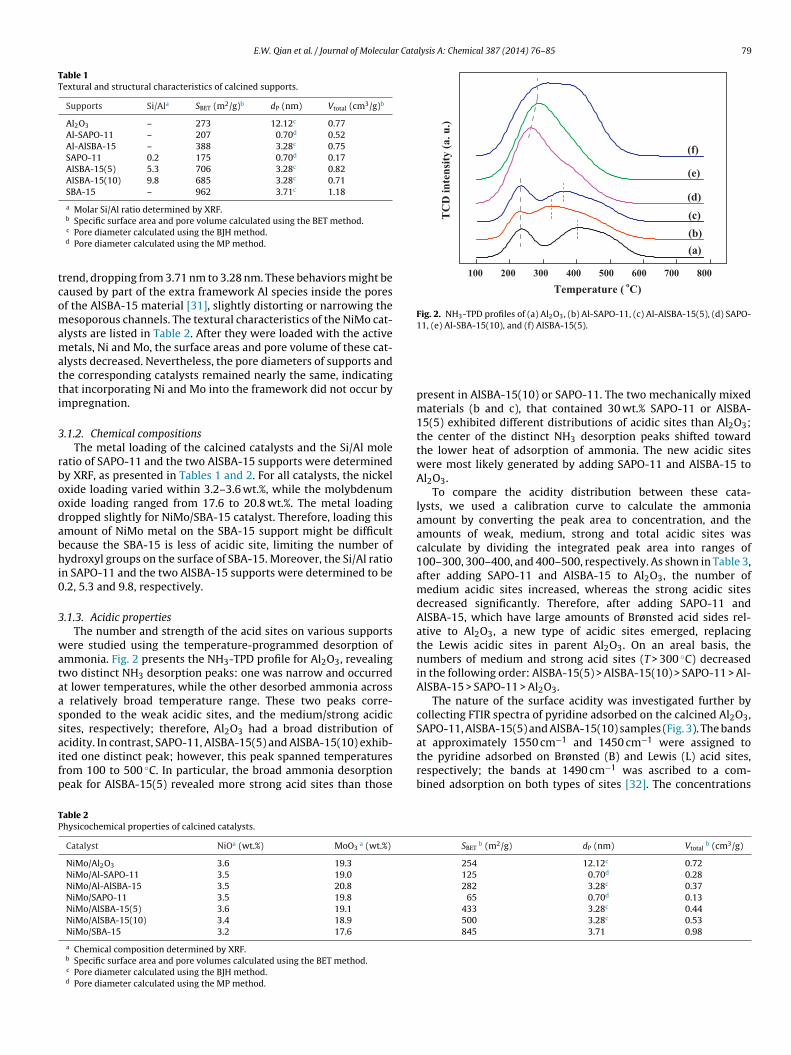
E.W. Qian et al. / Journal of Molecular Catalysis A: Chemical 387 (2014) 76–85 79
Table 1Textural and structural characteristics of calcined supports.
Supports Si/Ala SBET (m2/g)b dP (nm) Vtotal (cm3/g)b
Al2O3 – 273 12.12c 0.77Al-SAPO-11 – 207 0.70d 0.52Al-AlSBA-15 – 388 3.28c 0.75SAPO-11 0.2 175 0.70d 0.17AlSBA-15(5) 5.3 706 3.28c 0.82AlSBA-15(10) 9.8 685 3.28c 0.71SBA-15 – 962 3.71c 1.18
a Molar Si/Al ratio determined by XRF.b
tcomamatti
3
rboodabhi0
3
wataassaifp
800700600500400300200100
(f)
(e)
(d)
(c)
(b)
TC
D i
nte
nsi
ty (
a. u
.)
Temperature (oC)
(a)
Fig. 2. NH3-TPD profiles of (a) Al2O3, (b) Al-SAPO-11, (c) Al-AlSBA-15(5), (d) SAPO-11, (e) Al-SBA-15(10), and (f) AlSBA-15(5).
TP
Specific surface area and pore volume calculated using the BET method.c Pore diameter calculated using the BJH method.d Pore diameter calculated using the MP method.
rend, dropping from 3.71 nm to 3.28 nm. These behaviors might beaused by part of the extra framework Al species inside the poresf the AlSBA-15 material [31], slightly distorting or narrowing theesoporous channels. The textural characteristics of the NiMo cat-
lysts are listed in Table 2. After they were loaded with the activeetals, Ni and Mo, the surface areas and pore volume of these cat-
lysts decreased. Nevertheless, the pore diameters of supports andhe corresponding catalysts remained nearly the same, indicatinghat incorporating Ni and Mo into the framework did not occur bympregnation.
.1.2. Chemical compositionsThe metal loading of the calcined catalysts and the Si/Al mole
atio of SAPO-11 and the two AlSBA-15 supports were determinedy XRF, as presented in Tables 1 and 2. For all catalysts, the nickelxide loading varied within 3.2–3.6 wt.%, while the molybdenumxide loading ranged from 17.6 to 20.8 wt.%. The metal loadingropped slightly for NiMo/SBA-15 catalyst. Therefore, loading thismount of NiMo metal on the SBA-15 support might be difficultecause the SBA-15 is less of acidic site, limiting the number ofydroxyl groups on the surface of SBA-15. Moreover, the Si/Al ratio
n SAPO-11 and the two AlSBA-15 supports were determined to be.2, 5.3 and 9.8, respectively.
.1.3. Acidic propertiesThe number and strength of the acid sites on various supports
ere studied using the temperature-programmed desorption ofmmonia. Fig. 2 presents the NH3-TPD profile for Al2O3, revealingwo distinct NH3 desorption peaks: one was narrow and occurredt lower temperatures, while the other desorbed ammonia across
relatively broad temperature range. These two peaks corre-ponded to the weak acidic sites, and the medium/strong acidicites, respectively; therefore, Al2O3 had a broad distribution of
cidity. In contrast, SAPO-11, AlSBA-15(5) and AlSBA-15(10) exhib-ted one distinct peak; however, this peak spanned temperaturesrom 100 to 500 ◦C. In particular, the broad ammonia desorptioneak for AlSBA-15(5) revealed more strong acid sites than thoseable 2hysicochemical properties of calcined catalysts.
Catalyst NiOa (wt.%) MoO3a (wt.%)
NiMo/Al2O3 3.6 19.3
NiMo/Al-SAPO-11 3.5 19.0
NiMo/Al-AlSBA-15 3.5 20.8
NiMo/SAPO-11 3.5 19.8
NiMo/AlSBA-15(5) 3.6 19.1
NiMo/AlSBA-15(10) 3.4 18.9
NiMo/SBA-15 3.2 17.6
a Chemical composition determined by XRF.b Specific surface area and pore volumes calculated using the BET method.c Pore diameter calculated using the BJH method.d Pore diameter calculated using the MP method.
present in AlSBA-15(10) or SAPO-11. The two mechanically mixedmaterials (b and c), that contained 30 wt.% SAPO-11 or AlSBA-15(5) exhibited different distributions of acidic sites than Al2O3;the center of the distinct NH3 desorption peaks shifted towardthe lower heat of adsorption of ammonia. The new acidic siteswere most likely generated by adding SAPO-11 and AlSBA-15 toAl2O3.
To compare the acidity distribution between these cata-lysts, we used a calibration curve to calculate the ammoniaamount by converting the peak area to concentration, and theamounts of weak, medium, strong and total acidic sites wascalculate by dividing the integrated peak area into ranges of100–300, 300–400, and 400–500, respectively. As shown in Table 3,after adding SAPO-11 and AlSBA-15 to Al2O3, the number ofmedium acidic sites increased, whereas the strong acidic sitesdecreased significantly. Therefore, after adding SAPO-11 andAlSBA-15, which have large amounts of Brønsted acid sides rel-ative to Al2O3, a new type of acidic sites emerged, replacingthe Lewis acidic sites in parent Al2O3. On an areal basis, thenumbers of medium and strong acid sites (T > 300 ◦C) decreasedin the following order: AlSBA-15(5) > AlSBA-15(10) > SAPO-11 > Al-AlSBA-15 > SAPO-11 > Al2O3.
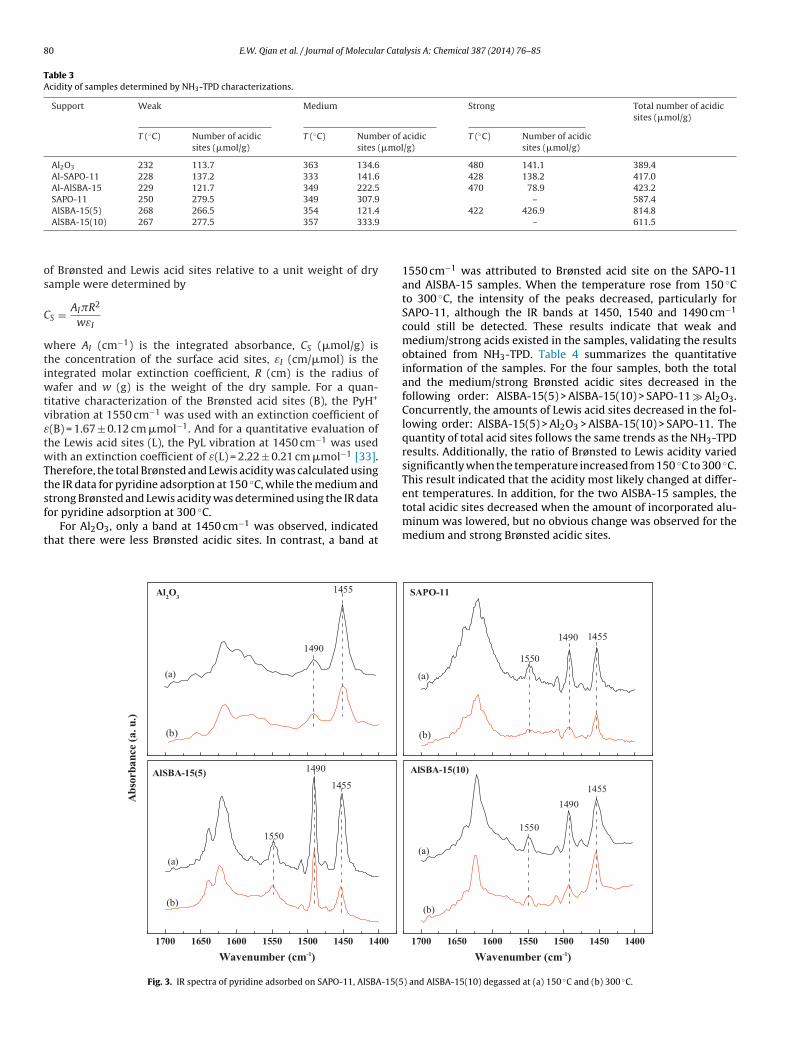
The nature of the surface acidity was investigated further bycollecting FTIR spectra of pyridine adsorbed on the calcined Al2O3,SAPO-11, AlSBA-15(5) and AlSBA-15(10) samples (Fig. 3). The bandsat approximately 1550 cm−1 and 1450 cm−1 were assigned tothe pyridine adsorbed on Brønsted (B) and Lewis (L) acid sites,respectively; the bands at 1490 cm−1 was ascribed to a com-
bined adsorption on both types of sites [32]. The concentrationsSBETb (m2/g) dP (nm) Vtotal
b (cm3/g)
254 12.12c 0.72125 0.70d 0.28282 3.28c 0.37
65 0.70d 0.13433 3.28c 0.44500 3.28c 0.53845 3.71 0.98
80 E.W. Qian et al. / Journal of Molecular Catalysis A: Chemical 387 (2014) 76–85
Table 3Acidity of samples determined by NH3-TPD characterizations.
Support Weak Medium Strong Total number of acidicsites (�mol/g)
T (◦C) Number of acidicsites (�mol/g)
T (◦C) Number of acidicsites (�mol/g)
T (◦C) Number of acidicsites (�mol/g)
Al2O3 232 113.7 363 134.6 480 141.1 389.4Al-SAPO-11 228 137.2 333 141.6 428 138.2 417.0Al-AlSBA-15 229 121.7 349 222.5 470 78.9 423.2SAPO-11 250 279.5 349 307.9 – 587.4AlSBA-15(5) 268 266.5 354 121.4 422 426.9 814.8
os
C
wtiwtvεtwTtsf
t
AlSBA-15(10) 267 277.5 357 333.9
f Brønsted and Lewis acid sites relative to a unit weight of dryample were determined by
S = AI�R2
wεI
here AI (cm−1) is the integrated absorbance, CS (�mol/g) ishe concentration of the surface acid sites, εI (cm/�mol) is thentegrated molar extinction coefficient, R (cm) is the radius of
afer and w (g) is the weight of the dry sample. For a quan-itative characterization of the Brønsted acid sites (B), the PyH+
ibration at 1550 cm−1 was used with an extinction coefficient of(B) = 1.67 ± 0.12 cm �mol−1. And for a quantitative evaluation ofhe Lewis acid sites (L), the PyL vibration at 1450 cm−1 was usedith an extinction coefficient of ε(L) = 2.22 ± 0.21 cm �mol−1 [33].
herefore, the total Brønsted and Lewis acidity was calculated usinghe IR data for pyridine adsorption at 150 ◦C, while the medium and
trong Brønsted and Lewis acidity was determined using the IR dataor pyridine adsorption at 300 ◦C.For Al2O3, only a band at 1450 cm−1 was observed, indicatedhat there were less Brønsted acidic sites. In contrast, a band at
1700 1650 1600 1550 15 00 1450 14 00
Al2O
3
1490
1455
(b)
(a)
1490
1550
(b)
Ab
sorb
an
ce
(a. u
.)
Wavenu mber (c m-1)
(a)
1455
AlSBA-15(5)
Fig. 3. IR spectra of pyridine adsorbed on SAPO-11, AlSBA-15(5
– 611.5
1550 cm−1 was attributed to Brønsted acid site on the SAPO-11and AlSBA-15 samples. When the temperature rose from 150 ◦Cto 300 ◦C, the intensity of the peaks decreased, particularly forSAPO-11, although the IR bands at 1450, 1540 and 1490 cm−1
could still be detected. These results indicate that weak andmedium/strong acids existed in the samples, validating the resultsobtained from NH3-TPD. Table 4 summarizes the quantitativeinformation of the samples. For the four samples, both the totaland the medium/strong Brønsted acidic sites decreased in thefollowing order: AlSBA-15(5) > AlSBA-15(10) > SAPO-11 � Al2O3.Concurrently, the amounts of Lewis acid sites decreased in the fol-lowing order: AlSBA-15(5) > Al2O3 > AlSBA-15(10) > SAPO-11. Thequantity of total acid sites follows the same trends as the NH3-TPDresults. Additionally, the ratio of Brønsted to Lewis acidity variedsignificantly when the temperature increased from 150 ◦C to 300 ◦C.This result indicated that the acidity most likely changed at differ-
ent temperatures. In addition, for the two AlSBA-15 samples, thetotal acidic sites decreased when the amount of incorporated alu-minum was lowered, but no obvious change was observed for themedium and strong Brønsted acidic sites.1700 1650 1600 1550 1500 1450 1400
SAPO-11
(b)
(a)
1490
1550
145 5
(b)
Wavenumber (cm-1)
(a)
1455
1490
1550
AlSBA -15 (10)
) and AlSBA-15(10) degassed at (a) 150 ◦C and (b) 300 ◦C.
E.W. Qian et al. / Journal of Molecular Catalysis A: Chemical 387 (2014) 76–85 81
Table 4Acidity of samples determined by pyridine FTIR characterizations.
Support Number of acidic sites (�mol/g)
Total acidity (150 ◦C) Medium and strong acidity (300 ◦C)
Lewis Brønsted Total B/L Lewis Brønsted Total B/L
Al2O3 103.5 0 103.5 – 73.7 0 73.7 –SAPO-11 65.6 43.9 109.5 0.7 48.3 8.6 56.9 0.2
3
1rhv
3
a3cisnuowmoz
baamiittt
TH
R
AlSBA-15(5) 203.8 82.1 285.9AlSBA-15(10) 86.3 51.3 137.6
.2. Catalytic test
A series of Ni–Mo catalysts were prepared with Al2O3, Al-SAPO-1, Al-AlSBA-15, SAPO-11, AlSBA-15(5), AlSBA-15(10) and SBA-15,espectively; these catalysts are denoted A–G, and assessed byydrotreating methyl stearate (10 wt.% dissolved in decalin sol-ent) as a model reaction.
.2.1. Deoxygenation of methyl stearateTable 5 shows the conversion, deoxygenation degree (HDO)
nd detailed product distributions for all the studied catalysts at00 ◦C. The conversion, as well as the deoxygenation degree, waslose to 100%, indicated that all the catalysts exhibited high activ-ty under the operating conditions. The hydroconversion of methyltearate mainly led to CH4, CO and CO2 as gaseous products, whileormal and isomerized C17H36 and C18H38 hydrocarbons as liq-id products. In addition, small amounts of C2–C16 hydrocarbons,xygenates, and hydrocarbons with carbon number more than 18ere observed. As a matter of fact, the catalytic hydroconversion ofethyl stearate is a complex process comprising the hydrogenation
f ester, deoxygenation of intermediate oxygenates, and isomeri-ation of deoxygenated hydrocarbons.
Light esters, e.g. methyl hexanoate, can be converted to car-oxylic acids by means of hydrolysis, which is the reverse of thecid-catalyzed esterification reaction by which a carboxylic acidnd an alcohol form an ester and water [34]. However, ther-odynamic calculations using the HSC chemistry® software [35]
ndicate that transformation of methyl stearate into stearic acid
s thermodynamically unfavorable under our experimental condi-ions (Eq. (5)). Therefore, another explanation could be proposed:ransformation of methyl stearate into stearic acid by hydrogena-ion and generate methane. As shown in Eq. (6), this reaction isable 5ydroconversion results for methyl stearate over various NiMo catalysts.
Catalyst
A B C
The yields of gaseous products (wt.%)CH4 5.16 5.09 5.16
C2H6–C4H10 0.05 0.06 0.29
CO 0.76 2.46 4.53
CO2 1.92 1.20 1.02
Lumped composition of the liquid product (C%)Oxygenates 0.52 0.76 1.03
C5H12–C16H34 0.25 0.21 3.87
i-C17H36 – 0.89 5.83
n-C17H36 28.32 24.72 34.09
i-C18H38 0.08 1.51 7.99
n-C18H38 70.37 71.75 46.81
C18+ 0.46 0.17 0.39
Conversion (mol%) 100 ∼100 ∼100
HDO (mol%) 99.48 99.24 98.97
eaction conditions: 300 ◦C, 3 MPa, LHSV = 10 h−1, H2/feed = 600 (v/v).
0.4 91 49.9 140.9 0.50.6 58.6 46.7 95.3 0.8
thermodynamic favorable under the experimental conditions:
C18H35COOCH3 (l) + H2O (g) = C18H35COOH (l) + CH3OH (g)
�G573k = 2.6 kJ (5)
C18H35COOCH3 (l) + H2 (g) = C18H35COOH (l) + CH4 (g)
�G573k = −115.0 kJ (6)
Deoxygenation was used as a general term for oxygenelimination; this process proceeds via two pathways: hydrodeoxy-genation (HDO) and decarbonylation/decarboxylation (DCO). Forthe hydrodeoxygenation pathway, oxygen is removed as waterand the resulting hydrocarbons retain the same number of car-bon atoms (C18H38) as their corresponding fatty acid (stearicacid) bound in the original methyl stearate (Eq. (7)). On theother hand, decarbonylation refers to a process where the oxy-gen in the carboxylic acid group is removed as CO and H2O,generating hydrocarbons (C17H36) with one carbon atom lessthan the corresponding fatty acid bound in the original methylstearate (Eq. (8)). In the decarboxylation pathway, the oxygenin the carboxylic acid group is removed as CO2 without con-suming hydrogen, forming of hydrocarbons that have the samenumber of carbon atoms as the products of decarbonylation (Eq.(9)). Trace amount of H2O could be found in the liquid products,but it could not be separated because too little was gener-ated.
C17H35COOH + 3H2 = C18H38 + 2H2O (7)
C17H35COOH + H2 = C17H36 + CO + H2O (8)
C17H35COOH = C17H36 + CO2 (9)
D E F G
5.11 5.19 5.11 5.120.13 0.34 0.19 –5.42 5.97 5.77 3.450.40 0.16 0.12 –
0.39 0.97 0.19 0.531.80 12.73 11.23 0.351.36 17.88 12.02 0.22
36.38 46.08 49.86 25.864.17 5.97 4.78 2.55
55.25 15.78 21.07 70.080.66 0.59 0.86 0.41
100 ∼100 ∼100 ∼10099.61 99.03 99.81 99.47
8 r Catalysis A: Chemical 387 (2014) 76–85
oiboccbttitailiostoRrwcaotiwscr
eNiof1Nrsrbptbmptaawwtttcb
tHzior
300 325 350 3750
10
20
30
40
50
60
Sele
cti
vit
y t
oiso -
C5
-18 (
C%
)
Temperat ure (oC)
Al2O
3
AlSBA-1 5(5 )
AlSBA -15( 10)
SAPO- 11
Al-AlSB A-1 5
Al- SAPO-1 1
2 E.W. Qian et al. / Journal of Molecula
It is now well accepted that the nature of active sites presentn the edges of the NiMoS and MoS2 phase in sulfided catalystss a sulfur vacancy. A possible reaction mechanism was proposedy Brillouet et al. [36] to interpret the HDO and DCO pathwaysver Mo/Al2O3 catalyst. In the case of sulfided NiMo catalyst, thearbonyl group (C O) bound in carboxylic acid was first disso-iative adsorbed on the sulfur vacancy sites. The next step coulde a protonation of hydroxide group bound in stearic acid dueo the acidity of the SH group and elimination of water. Afterhat, the adsorbed carbocation could be a key intermediate. Thisntermediate can undergo either a hydride addition step leadingo aldehydes (C17H35CHO) or an elimination step and generatelkenes (C17H34) with one carbon atom less than the correspond-ng carboxylic acid. No alkene, however, was identified in theiquid products. This is because the alkenes could be isomer-zed/cracked or hydrogenated immediately due to the acid sitesn the supports and the hydrogenation activity of sulfided NiMoites, respectively. The aldehyde generated from the hydride addi-ion step was then adsorbed on the vacancy site similar to the casef carboxylic acid and eventually transformed into hydrocarbons.uinart de Brimont et al. [37] demonstrated that the deoxygenationeaction follows preferentially the HDO pathway when aldehydeas used as the sole reactant due to a stronger adsorption of
arboxylic acid than that of aldehyde. Consequently, it could bessumed that the main hydrocarbon obtained from hydrogenationf aldehyde was C18H38. Additionally, Kubicka et al. [38] inves-igated the effect of support on deoxygenation of triglycerides,n particular on the selectivity toward the HDO and DCO path-
ays. Their results provided clear evidence that even with theame metallic sites, i.e. sulfided NiMo, the selectivity of HDO/DCOould be fine-turned by support, which were consistent with ouresults.
When comparing the formation of CO and CO2 over differ-nt catalysts, only that obtained from hydroconversion overiMo/Al2O3 exhibited higher CO2 formation than that of CO,
ndicating that the decarboxylation pathway was the dominantne in DCO mechanism. The amount of CO increased in theollowing order: NiMo/Al2O3 < NiMo/Al-SAPO-11 < NiMo/SAPO-1, NiMo/Al2O3 < NiMo/Al-AlSBA-15 < NiMo/AlSBA-15(5) andiMo/SBA-15 < NiMo/AlSBA-15(10) < NiMo/AlSBA-15(5). The
esults of acidity characterization demonstrated that the acidity ofupports increased by adding acid supports into Al2O3. Numerouseports have noted that the acidity of support could be changedy introducing additives to the �-alumina, such as boron, fluorine,hosphorus, silicon and zeolites [39–41]. Chen et al. [42] studiedhe catalytic behavior of CoMo and Mo on Al2O3 with differentoron loadings in 4,6-DMDBT HDS. The boron addition couldodulate the electronic properties of the sulfided Mo and CoMo
hases, enhancing the hydrogenation activity of catalysts. Duringhe deoxygenation of methyl stearate, increasing the acidity bydding acid support might accelerate the formation of alcohols orldehydes from stearic acid. Thus, the decarboxylation pathwayould be suppressed. However, a different trend of CO2 formationas obtained from the hydrotreating over the SBA-15 catalysts;
he amount of CO2 increased with the acidity of the supports. Fromhe acidity results, the AlSBA-15 supports have much strong acidityhan other supports. The formation of CO2 over NiMo/AlSBA-15atalysts might have been due to thermal decomposition causedy the strong acidic sites.
The amounts of these oxygenates were very low under allhe reaction conditions, indicating that these catalysts had highDO activities (with a HDO ratio > 98%). In contrast, the isomeri-
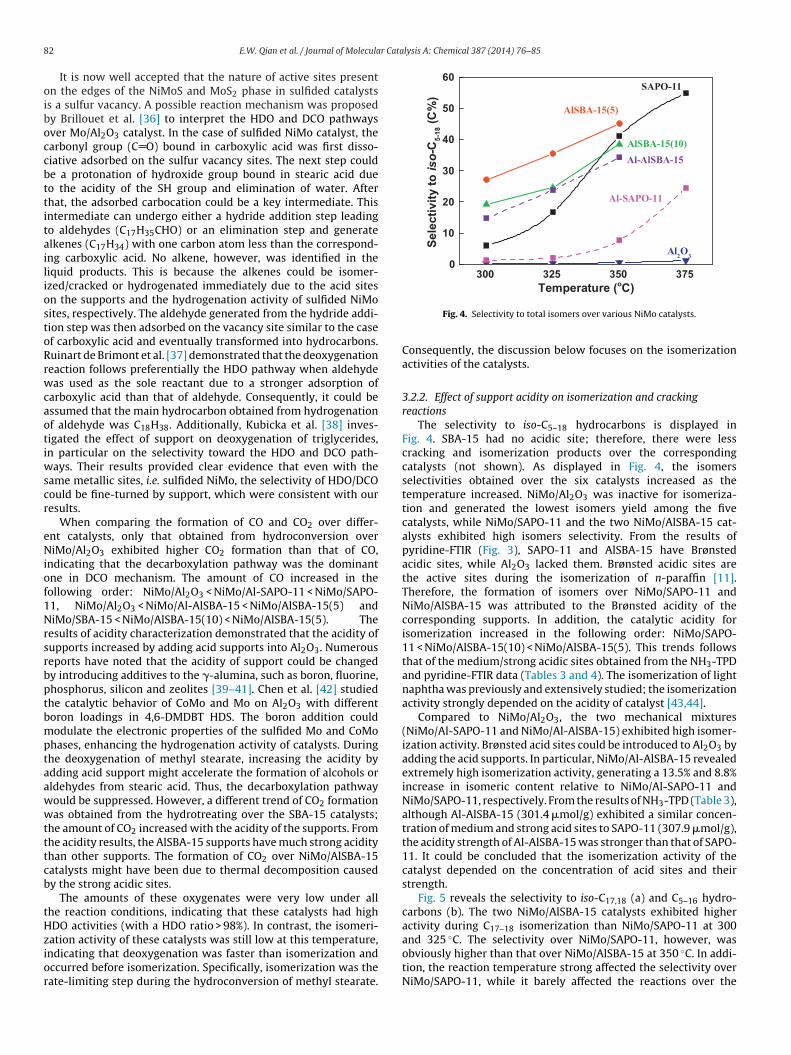
ation activity of these catalysts was still low at this temperature,ndicating that deoxygenation was faster than isomerization andccurred before isomerization. Specifically, isomerization was theate-limiting step during the hydroconversion of methyl stearate.Fig. 4. Selectivity to total isomers over various NiMo catalysts.
Consequently, the discussion below focuses on the isomerizationactivities of the catalysts.
3.2.2. Effect of support acidity on isomerization and crackingreactions
The selectivity to iso-C5–18 hydrocarbons is displayed inFig. 4. SBA-15 had no acidic site; therefore, there were lesscracking and isomerization products over the correspondingcatalysts (not shown). As displayed in Fig. 4, the isomersselectivities obtained over the six catalysts increased as thetemperature increased. NiMo/Al2O3 was inactive for isomeriza-tion and generated the lowest isomers yield among the fivecatalysts, while NiMo/SAPO-11 and the two NiMo/AlSBA-15 cat-alysts exhibited high isomers selectivity. From the results ofpyridine-FTIR (Fig. 3), SAPO-11 and AlSBA-15 have Brønstedacidic sites, while Al2O3 lacked them. Brønsted acidic sites arethe active sites during the isomerization of n-paraffin [11].Therefore, the formation of isomers over NiMo/SAPO-11 andNiMo/AlSBA-15 was attributed to the Brønsted acidity of thecorresponding supports. In addition, the catalytic acidity forisomerization increased in the following order: NiMo/SAPO-11 < NiMo/AlSBA-15(10) < NiMo/AlSBA-15(5). This trends followsthat of the medium/strong acidic sites obtained from the NH3-TPDand pyridine-FTIR data (Tables 3 and 4). The isomerization of lightnaphtha was previously and extensively studied; the isomerizationactivity strongly depended on the acidity of catalyst [43,44].
Compared to NiMo/Al2O3, the two mechanical mixtures(NiMo/Al-SAPO-11 and NiMo/Al-AlSBA-15) exhibited high isomer-ization activity. Brønsted acid sites could be introduced to Al2O3 byadding the acid supports. In particular, NiMo/Al-AlSBA-15 revealedextremely high isomerization activity, generating a 13.5% and 8.8%increase in isomeric content relative to NiMo/Al-SAPO-11 andNiMo/SAPO-11, respectively. From the results of NH3-TPD (Table 3),although Al-AlSBA-15 (301.4 �mol/g) exhibited a similar concen-tration of medium and strong acid sites to SAPO-11 (307.9 �mol/g),the acidity strength of Al-AlSBA-15 was stronger than that of SAPO-11. It could be concluded that the isomerization activity of thecatalyst depended on the concentration of acid sites and theirstrength.
Fig. 5 reveals the selectivity to iso-C17,18 (a) and C5–16 hydro-carbons (b). The two NiMo/AlSBA-15 catalysts exhibited higheractivity during C17–18 isomerization than NiMo/SAPO-11 at 300
◦
and 325 C. The selectivity over NiMo/SAPO-11, however, wasobviously higher than that over NiMo/AlSBA-15 at 350 ◦C. In addi-tion, the reaction temperature strong affected the selectivity overNiMo/SAPO-11, while it barely affected the reactions over the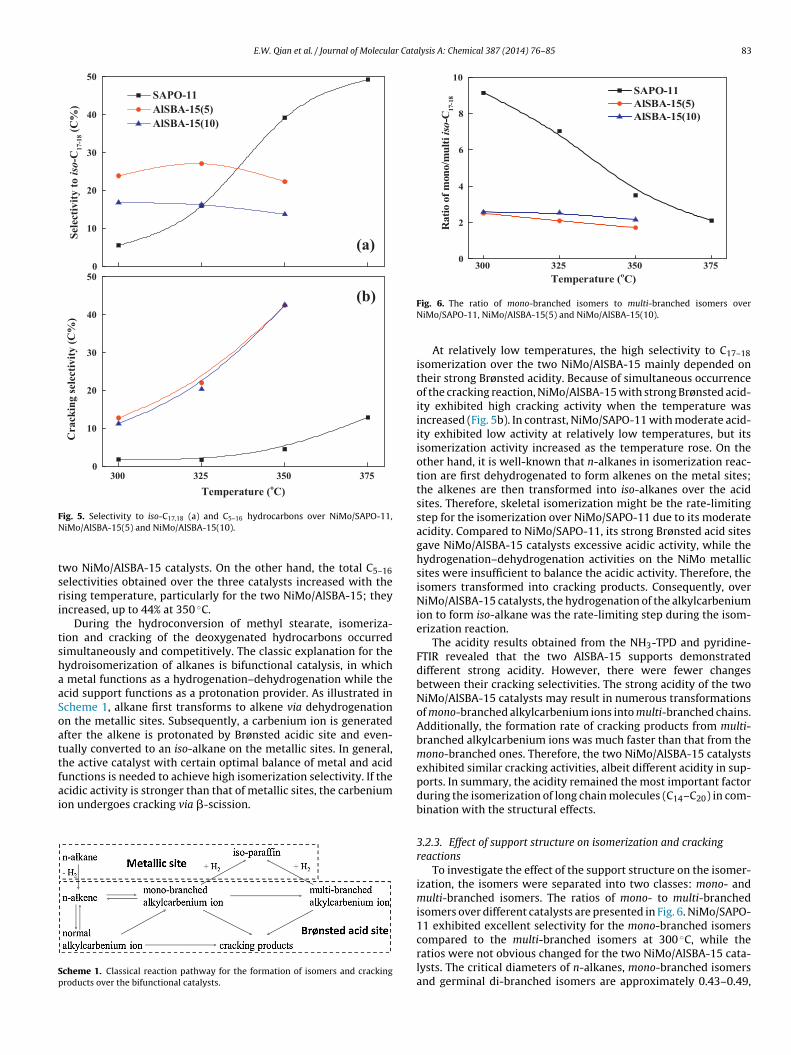
E.W. Qian et al. / Journal of Molecular Catalysis A: Chemical 387 (2014) 76–85 83
0
10
20
30
40
50
300 325 350 3750
10
20
30
40
50
Sel
ecti
vit
y t
oiso
-C17-1
8 (
C%
)
SAPO-11
AlSBA-15(5)
AlSBA-15(10)
Cra
ckin
g s
elec
tivit
y (
C%
)
Temperature (oC)
(a)
(b)
FN
tsri
tshaaSoattfai
Sp
300 325 350 3750
2
4
6
8
10
Rati
o o
f m
on
o/m
ult
iiso-
C17-1
8
Temperat ure (oC)
SAP O-1 1
AlSBA-15(5)
AlSBA-15(10)
ig. 5. Selectivity to iso-C17,18 (a) and C5–16 hydrocarbons over NiMo/SAPO-11,iMo/AlSBA-15(5) and NiMo/AlSBA-15(10).
wo NiMo/AlSBA-15 catalysts. On the other hand, the total C5–16electivities obtained over the three catalysts increased with theising temperature, particularly for the two NiMo/AlSBA-15; theyncreased, up to 44% at 350 ◦C.
During the hydroconversion of methyl stearate, isomeriza-ion and cracking of the deoxygenated hydrocarbons occurredimultaneously and competitively. The classic explanation for theydroisomerization of alkanes is bifunctional catalysis, in which
metal functions as a hydrogenation–dehydrogenation while thecid support functions as a protonation provider. As illustrated incheme 1, alkane first transforms to alkene via dehydrogenationn the metallic sites. Subsequently, a carbenium ion is generatedfter the alkene is protonated by Brønsted acidic site and even-ually converted to an iso-alkane on the metallic sites. In general,he active catalyst with certain optimal balance of metal and acidunctions is needed to achieve high isomerization selectivity. If the
cidic activity is stronger than that of metallic sites, the carbeniumon undergoes cracking via �-scission.cheme 1. Classical reaction pathway for the formation of isomers and crackingroducts over the bifunctional catalysts.
Fig. 6. The ratio of mono-branched isomers to multi-branched isomers overNiMo/SAPO-11, NiMo/AlSBA-15(5) and NiMo/AlSBA-15(10).
At relatively low temperatures, the high selectivity to C17–18isomerization over the two NiMo/AlSBA-15 mainly depended ontheir strong Brønsted acidity. Because of simultaneous occurrenceof the cracking reaction, NiMo/AlSBA-15 with strong Brønsted acid-ity exhibited high cracking activity when the temperature wasincreased (Fig. 5b). In contrast, NiMo/SAPO-11 with moderate acid-ity exhibited low activity at relatively low temperatures, but itsisomerization activity increased as the temperature rose. On theother hand, it is well-known that n-alkanes in isomerization reac-tion are first dehydrogenated to form alkenes on the metal sites;the alkenes are then transformed into iso-alkanes over the acidsites. Therefore, skeletal isomerization might be the rate-limitingstep for the isomerization over NiMo/SAPO-11 due to its moderateacidity. Compared to NiMo/SAPO-11, its strong Brønsted acid sitesgave NiMo/AlSBA-15 catalysts excessive acidic activity, while thehydrogenation–dehydrogenation activities on the NiMo metallicsites were insufficient to balance the acidic activity. Therefore, theisomers transformed into cracking products. Consequently, overNiMo/AlSBA-15 catalysts, the hydrogenation of the alkylcarbeniumion to form iso-alkane was the rate-limiting step during the isom-erization reaction.
The acidity results obtained from the NH3-TPD and pyridine-FTIR revealed that the two AlSBA-15 supports demonstrateddifferent strong acidity. However, there were fewer changesbetween their cracking selectivities. The strong acidity of the twoNiMo/AlSBA-15 catalysts may result in numerous transformationsof mono-branched alkylcarbenium ions into multi-branched chains.Additionally, the formation rate of cracking products from multi-branched alkylcarbenium ions was much faster than that from themono-branched ones. Therefore, the two NiMo/AlSBA-15 catalystsexhibited similar cracking activities, albeit different acidity in sup-ports. In summary, the acidity remained the most important factorduring the isomerization of long chain molecules (C14–C20) in com-bination with the structural effects.
3.2.3. Effect of support structure on isomerization and crackingreactions
To investigate the effect of the support structure on the isomer-ization, the isomers were separated into two classes: mono- andmulti-branched isomers. The ratios of mono- to multi-branchedisomers over different catalysts are presented in Fig. 6. NiMo/SAPO-11 exhibited excellent selectivity for the mono-branched isomers
◦
compared to the multi-branched isomers at 300 C, while theratios were not obvious changed for the two NiMo/AlSBA-15 cata-lysts. The critical diameters of n-alkanes, mono-branched isomersand germinal di-branched isomers are approximately 0.43–0.49,
8 r Catalysis A: Chemical 387 (2014) 76–85
0Smmcosi
oitiomtkodoN
rmfomsa
pNsiBihobtst
3s
aiiwto
mNtgbgifimcmt
[[[
[
[
[[
[
[
4 E.W. Qian et al. / Journal of Molecula
.50–0.62 and 0.7 nm, respectively [45]. The pore diameter ofAPO-11 was below 0.70 nm. Consequently, the transformation ofono-branched isomers into bulky molecules (multi-branched iso-ers) in the pore channels was limited over NiMo/SAPO-11. In
ontrast, the two NiMo/AlSBA-15 catalysts exhibited low ratiosf mono- to multi-branched isomers; AlSBA-15 had a large poreize (3.28 nm), providing enough space to generate multi-branchedsomers inside the pore channels.
On the other hand, the ratio of mono- to multi-branched isomersbtained over NiMo/SAPO-11 decreased rapidly as the temperaturencreased. Considering that increasing the temperature accelerateshe diffusion without varying the contact time, the mono-branchedsomers could have a better chance of reaching the acidic sitesn the external surface of catalyst and further transferred intoulti-branched isomers. Claude and Martens [46] proposed that
he multi-branched isomers were generated by pore mouth orey lock modes during the isomerization of long chain alkanesver Pt/H-ZSM-22. SAPO-11 has a similar pore structure (one-imensional 10-ring pores) to ZSM-22. Therefore, the formationf multi-branched isomers might occur at the pore mouths withiMo/SAPO-11.
For NiMo/AlSBA-15, there was no obvious variation relative toeaction temperature for the ratio of mono- to multi-branched iso-ers. The pore mouths of AlSBA-15 were too large to suppress the
ormation of multi-branched isomers. In addition, the pore mouthr key lock modes of isomerization could not be generated in theesoporous materials due to their large pore mouths. Therefore,
harp selectivity was not observed on the two NiMo/AlSBA-15 cat-lysts.
When comparing NiMo/SAPO-11 to NiMo/AlSBA-15, the meso-orous AlSBA-15 had a larger specific surface area. Therefore,iMo/SAPO-11 with the lower surface area had less Brønsted acid
ites inside its pore channels, hindering the formation of crack-ng products. In contrast, AlSBA-15 had abundant and accessiblerønsted acid sites inside the channel; mono-branched carbenium
on could undergo cracking through �-scission to generate lightydrocarbons. Consequently, it could be reasonably inferred that tobtain better isomerization activity, the acidity of AlSBA-15 shoulde reduced further to balance the Ni–Mo metallic sites. Based onhe results above, it can be concluded that structure of the supporttrongly influenced the isomerization of long chain molecules overhe NiMo catalysts.
.2.4. Reaction pathway for the hydroconversion of methyltearate to isomerized alkanes
At relatively low temperature, i.e. 300 ◦C, complete deoxygen-tion of methyl stearate was achieved, although the yields of thesomers were still low (Table 5). Additionally, all of the oxygenatedntermediates obtained from the deoxygenation of methyl stearate
ere identified as straight chain molecules. These results indicatedhat the isomerization reaction occurred after the deoxygenationf methyl stearate.
Scheme 2 displays the reaction pathway for the long chainolecules over NiMo/SAPO-11 and NiMo/AlSBA-15 catalysts.iMo/SAPO-11 had appropriate acidity and pore size, allowing
he deoxygenated n-alkanes to diffuse into the pore channels andenerate mono-branched carbenium ions. Subsequently, the mono-ranched carbenium ions could follow two different routes toenerate isomers: (i) directly hydrogenate to form mono-branchedsomers on the NiMo metallic sites or (ii) diffuse into the sur-ace of SAPO-11 and isomerize to form multi-branched carbeniumons on the Brønsted acidic sites at the pore mouths. Finally, the
ulti-branched carbenium ions were hydrogenated, forming theorresponding alkanes. Note that the formation of multi-branchedolecules was suppressed inside the pore channels of SAPO-11 due
o its small pore size. For NiMo/AlSBA-15, due to the large pore size,
[
[
Scheme 2. Reaction pathways for the isomerization of long chain paraffin overNiMo/SAPO-11 and NiMo/AlSBA-15 catalysts.
multi-branched carbenium ions were generated inside the porechannels. In addition, AlSBA-15 had a large specific surface areathat allowed numerous Brønsted acidic sites to remain accessibleinside the pore channel; therefore, the mono-branched carbeniumion could undergo cracking, generating light hydrocarbons.
4. Conclusions
The hydroconversion of methyl stearate over sulfided Ni–Mocatalysts was conducted in flow reactor, mainly generating normaland isomerous C18 and C17 hydrocarbons. The deoxygenation ofmethyl stearate occurred before isomerization; isomerization is therate-limiting step during the hydroconversion of methyl stearate.The two mechanical mixtures demonstrated a higher isomeriza-tion activity than NiMo/Al2O3 because they had more Brønstedacidic sites. While isomerizing long chain molecules, structure ofthe support had a strong influence over the NiMo catalysts’ isom-erization behavior, even though the catalyst acidity remains animportant factor. SAPO-11 has a moderate acidity and a suitablepore size, imparting superior mono-branched isomers selectivityand minimal cracking activity to the corresponding catalyst. ForNiMo/AlSBA-15, numerous cracking products were generated dueto its strong acidity and large pore sizes.
References
[1] S.M. Geyer, M.J. Jacobus, S.S. Lestz, Trans. ASAE 27 (1984) 0375–0381.[2] D. Kubicka, L. Kaluza, J. Cejka, Catal. Rev. Sci. Eng. 55 (2013) 1–78.[3] S. Jain, M.P. Sharma, Renew. Sust. Energy Rev. 14 (2010) 667–678.[4] H. Aatola, M. Larmi, T. Sarjovaara, S. Mikkonen, SAE Int. 1 (2008) 2500.[5] I. Kubickova, D. Kubicka, Waste Biomass Valorizat. 1 (2010) 293–308.[6] D. Kubicka, L. Kaluza, Appl. Catal. A 372 (2010) 199–208.[7] S. Gong, A. Shinozaki, M. Shi, E.W. Qian, Energy Fuels 26 (2012) 2394–2399.[8] S. Gong, N. Chen, S. Nakayama, E.W. Qian, J. Mol. Catal. A 370 (2013) 14–21.[9] D.Y. Murzin, European Patent 1 681 337 A1 (2006), to Neste Oil Oyj.10] T. Okuhara, Chem. Rev. 102 (2002) 3641–3665.11] V.M. Akhmedov, S.H. Al-Khowaiter, Catal. Rev. 49 (2007) 33–139.12] N. Chen, S. Gong, H. Shirai, T. Watanabe, E.W. Qian, Appl. Catal. A 466 (2013)
105–115.13] C. Wang, Z. Tian, L. Wang, R. Xu, Q. Liu, W. Qu, H. Ma, B. Wang, ChemSusChem
5 (2012) 1974–1984.14] O.V. Kikhtyanin, A.E. Rubanov, A.B. Ayupov, G.V. Echevsky, Fuel 89 (2010)
3085–3092.15] H. Deldari, Appl. Catal. A 293 (2005) 1–10.16] M. Alfonzo, J. Goldwasser, C.M. Lopez, F.J. Machado, M. Matjushin, B. Mendez,
J. Mol. Catal. A 98 (1995) 35–48.17] D. Zhao, J. Feng, Q. Huo, N. Melosh, G.H. Fredrickson, B.F. Chmelka, G.D. Stucky,
Science 279 (1998) 548–552.18] Y. Yue, A. Gedeon, J.-L. Bonardet, J.-B.D. Espinose, J. Fraissard, N. Melosh, Chem.
Commun. (1999) 1967–1968.19] A. Vinu, V. Murugesan, W. Boehlmann, M. Hartmann, J. Phys. Chem. B 108 (2004)
11496–11505.20] Y. Li, W. Zhang, L. Zhang, Q. Yang, Z. Wei, Z. Feng, C. Li, J. Phys. Chem. B 108
(2004) 9739–9744.

r Cata
[
[[[
[
[
[
[[[
[[
[[
[
[
[
[
[
[
[
[[43] A-G.A. Ali, L.I. Ali, S.M. Aboul-Fotouh, A.K. Aboul-Gheit, Appl. Catal. A: Gen. 215
E.W. Qian et al. / Journal of Molecula
21] B.T. Carvill, B.A. Lerner, B.J. Adelman, D.C. Tomczak, W.M. Sachtler, J. Catal. 144(1993) 1–8.
22] E. Blomsma, J.A. Martens, P.A. Jacobs, J. Catal. 155 (1995) 141–147.23] E. Blomsma, J.A. Martens, P.A. Jacobs, J. Catal. 159 (1996) 323–331.24] M.J. Remy, D. Stanica, G. Poncelet, E.J.P. Feijen, P.J. Grobet, J.A. Martens, P.A.
Jacobs, J. Phys. Chem. 100 (1996) 12440–12447.25] B.M. Lok, C.A. Messina, R.L. Patton, R.T. Gajek, T.R. Cannan, E.M. Flanigen, U.S.
Patent 4,440,871 (1984).26] D. Zhao, Q. Huo, J. Feng, B.F. Chmelka, G.D. Stucky, J. Am. Chem. Soc. 120 (1998)
6024–6036.27] S. Zeng, J. Blanchard, M. Breysse, Y. Shi, X. Shu, H. Nie, D. Li, Microporous Meso-
porous Mater. 85 (2005) 297–304.28] T. Kabe, W. Qian, Y. Hirai, L. Li, A. Ishihara, J. Catal. 190 (2000) 191–198.29] S. Zhang, S. Chen, P. Dong, G. Yuan, K.Q. Xu, Appl. Catal. A 332 (2007) 46–55.30] B. Dragoi, E. Dumitriu, C. Guimon, A. Auroux, Microporous Mesoporous Mater.
121 (2009) 7–17.31] L.Y. Lizama, T.E. Klimova, J. Mater. Sci. 44 (2009) 6617–6628.
32] T. Barzetti, E. Selli, D. Moscotti, L. Forni, J. Chem. Soc. Faraday Trans. 92 (1996)1401–1407.33] C.A. Emeis, J. Catal. 141 (1993) 347–354.34] O.I. Senol, E.-M. Ryymin, T.-R. Viljava, A.O.I. Krause, J. Mol. Catal. A: Chem. 268
(2007) 1–8.
[[[
lysis A: Chemical 387 (2014) 76–85 85
35] A. Roine, HSC Chemistry® for Windows, Chemical Reaction and EquilibriumSoftware with Extensive Thermochemical Database and Flowsheet Simulation,Version 6.0, Outokumpu Research Oy, Pori, Finland, 2006.
36] S. Brillouet, E. Baltag, S. Brunet, F. Richard, Appl. Catal. B 148 (2014)201–211.
37] M. Ruinart de Brimont, C. Dupont, A. Daudin, C. Geantet, P. Raybaud, J. Catal.286 (2012) 153–164.
38] D. Kubicka, J. Horacek, M. Setnicka, R. Bulanek, A. Zukal, I. Kubickova, Appl.Catal. B: Environ. 145 (2014) 101–107.
39] G. Murali Dhar, B.N. Srinivas, M.S. Rana, M. Kumar, S.K. Maity, Catal. Today 86(2003) 45–60.
40] R. Palcheva, L. KaluZA., A. Spojakina, K. JirÁ.TovÁ., G. Tyuliev, Chin. J. Catal. 33(2012) 952–961.
41] R. Iwamoto, J. Grimblot, B.C.G. Werner, O. Haag, K. Helmut, Adv. Catal. 44 (1999)417.
42] W. Chen, F. Mauge, J. Gestel, H. Nie, D. Li, X. Long, J. Catal. 304 (2013) 47–62.
(2001) 161–173.44] J.M. Campelo, F. Lafont, J.M. Marinas, J. Catal. 156 (1995) 11–18.45] G.C. Bond, R. Yahya, J. Mol. Catal. 68 (1991) 243–254.46] M.C. Claude, J.A. Martens, J. Catal. 190 (2000) 39–48.





























