Embed Size (px)
Citation preview

Journal of Critical Care (2011) xx, xxx–xxx
Sepsis: Something old, something new, and a systems viewRami Namas MDa, Ruben Zamora PhDa, Rajaie Namas MDa,1, Gary An MDb,j,John Doyle PhD c, Thomas E. Dick PhDd, Frank J. Jacono MDd,e,Ioannis P. Androulakis PhD f, Gary F. Nieman PhDg,Steve Chang MSh, Timothy R. Billiar MDa, John A. Kellum MD i,Derek C. Angus MD, MPH, FRCP i, Yoram Vodovotz PhDa,j,⁎
aDepartment of Surgery, University of Pittsburgh, Pittsburgh, PA 15213, USAbDepartment of Surgery, University of Chicago, Chicago, IL 60637, USAcControl and Dynamical Systems, California Institute of Technology, Pasadena, CA 91125, USAdDepartment of Medicine, Case Western University, Cleveland, OH 44106, USAeLouis Stokes Cleveland VA Medical Center, Cleveland, OH 44106, USAfDepartment of Chemical and Biochemical Engineering and Department of Biomedical Engineering, Rutgers University,Piscataway, NJ 08854, USAgDepartment of Surgery, Upstate Medical University, Syracuse, 13210 NY, USAhImmunetrics, Inc, Pittsburgh, PA 15203, USAiDepartment of Critical Care Medicine, University of Pittsburgh, Pittsburgh, PA 15213, USAjCenter for Inflammation and Regenerative Modeling, McGowan Institute for Regenerative Medicine, University of Pittsburgh,Pittsburgh, PA 15219, USA
fa
0d
Keywords:Sepsis;Inflammatory response;Physiologic variability;Mathematical model
Abstract Sepsis is a clinical syndrome characterized by a multisystem response to a microbialpathogenic insult consisting of a mosaic of interconnected biochemical, cellular, and organ-organinteraction networks. A central thread that connects these responses is inflammation that, whileattempting to defend the body and prevent further harm, causes further damage through the feed-forward, proinflammatory effects of damage-associated molecular pattern molecules. In this review, weaddress the epidemiology and current definitions of sepsis and focus specifically on the biologiccascades that comprise the inflammatory response to sepsis. We suggest that attempts to improveclinical outcomes by targeting specific components of this network have been unsuccessful due to thelack of an integrative, predictive, and individualized systems-based approach to define the time-varying,multidimensional state of the patient. We highlight the translational impact of computational modelingand other complex systems approaches as applied to sepsis, including in silico clinical trials, patient-specific models, and complexity-based assessments of physiology.© 2011 Elsevier Inc. All rights reserved.
⁎ Corresponding author. Department of Surgery, University of Pittsburgh, W944 Biomedical Sciences Tower, Pittsburgh, PA 15213. Tel.: +1 412 647 5609;x: +1 412 383 5946.E-mail address: [email protected] (Y. Vodovotz).1 Currently at Department of Internal Medicine, Hurley Medical Center, Michigan State University, Flint, MI 48503.
883-9441/$ – see front matter © 2011 Elsevier Inc. All rights reserved.oi:10.1016/j.jcrc.2011.05.025

2 R. Namas et al.
1. Introduction
Sepsis is a significant public health concern [1-3].Although “septicemia” accounts for approximately 1% ofoverall deaths in the United States [4], the number is muchlarger (nearly 10%) when factoring in deaths frompneumonia and other causes of severe sepsis [1,2]. Sepsisaffects persons of all ages [5], is the leading cause ofmorbidity and mortality for patients admitted to an intensivecare unit [1,6-8], and may be considered the 10th leadingcause of death overall in the United States [2,9]. Theincidence of sepsis is projected to increase by 1.5% per year[1,2,5,10], rising to more than 1 110 000 cases or moreannually by 2020 [2]. Sepsis also reduces the quality of lifeof many of those who survive [2,11,12].
Despite a large body of scientific literature concerningindividual mechanisms of disease in sepsis—implicatingorgan dysfunction caused by failure of key processes inepithelial cells [13,14] and involving various biologicmechanisms from endothelial defects [15,16] to dysregulatedinflammation and the associated complement and coagula-tion networks [17,18]—there are few therapies and relativelyimprecise diagnostics for sepsis. In the present review, wesuggest that our view of sepsis has evolved from a generalconcept, to that of rigidly defined (but seldom absolute)diagnoses, to a more fluid perception of sepsis as a dynamicprogression of host-pathogen interactions that can beassessed by examining the dynamic, multidimensional stateof the patient (Fig. 1). By “dynamic,” we mean time varying,and by “state,” we mean literally the compendium of allrelevant variables that can inform a clinician about thepathophysiology and biologic state of a patient at a giveninstant. Importantly, a characterization of this “dynamicstate” could be considered sufficiently complete when thedata contained therein are sufficient for prediction of thefuture course of the patient. Thus, the concept of dynamicstate encompasses the idea that the data that make up thisstate are sufficient to predict the progression of a patient for aclinically appropriate future duration and that these data areof sufficient granularity to reflect all relevant biologic andphysiologic processes and therefore amenable for analysis
/ Sepsis DiagnosisMulti-
DimensionalState
20th CenturyB.C. 21st Century
Germ Theory
19th Century
Fig. 1 Sepsis: a brief history. Our concept of sepsis hasprogressed from phenomenological description (antiquity) to rigiddiagnostic criteria (20th century). The future holds the potential forindividualized, predictive, and multidimensional description of thepatient's state.
using predictive computational models and algorithms.Although potentially daunting in scope, we suggest that theconcept of the dynamic state underlies a modern mechanisticview of a very old disease, one that better encompasses theprogression of this disease in individual patients rather thanfocusing on rigid, predefined diagnoses.
2. Sepsis: a brief history
The difficulty in developing mechanistically baseddiagnoses has hampered our understanding of the patho-physiology of sepsis and, to a certain degree, has impairedthe development of successful therapies. In beginning tounravel the mechanistic underpinnings of sepsis with thegoals of improved diagnosis and therapy, it is useful to tracethe dynamic evolution of our concepts concerning thisdisease. Starting from the origins of the word “sepsis”approximately 2700 years ago in the Greek word “σηψιs”(the “decomposition of animal or vegetable organic matter inthe presence of bacteria” [19]), our view of sepsis hasprogressed through various stages. The initial view of theprocess was encapsulated in the germ theory in whichpathogens were the sole causes of sepsis [20-22]. Subsequentadvances led to the development of fairly rigid diagnosticguidelines based on the host's response to infection,guidelines developed, in part, in response to the inability tocurb sepsis solely through therapy aimed at the pathogen (seebelow). The most recent development has been theemergence of a multiscale systems perspective of host-pathogen interactions at the organ, tissue, cellular, andmolecular levels (Fig. 1) [23-31]. Below, we focus on thismost recent development.
By 1990, after approximately 20 years of intensive careand 40 years of antisepsis, therapies based on the germtheory suggested that many patients could die despiteantibiotics and life support; the notion emerged that thehost's intertwined inflammatory and physiologic responsesto the pathogen were at least as much to blame as thepathogen itself [32]. In 1991, the American College of ChestPhysicians and the Society of Critical Care Medicine agreedon a new definition of sepsis as the development of asystemic inflammatory response syndrome (SIRS) due toinfection. The severity of sepsis was graded based on thedevelopment of hemodynamic compromise and associatedorgan dysfunction as follows: severe sepsis, septic shock,and multiple organ dysfunction syndrome [33]. Thesedefinitions served as a more uniform reference point forperforming clinical trials, facilitating hypothesis generationand establishing guidelines for the care of the septic patient(Fig. 1). The 1991 North American Consensus Conferenceconcept of SIRS is now considered outdated, however[21,34,35], and the 4 SIRS criteria have been expanded to alonger list of possible signs of sepsis in the latest definitions[34]. These were developed in 2001 under the auspices of the

3A systems view of sepsis
Society of Critical Care Medicine, the European Society ofIntensive Care Medicine, the American College of ChestPhysicians, and the Surgical Infection Societies. Theconferees concluded that the diagnostic criteria for SIRSwere overly sensitive and nonspecific and that a morecomprehensive list of signs and symptoms that mayaccompany sepsis would better reflect the clinical responseto infection [34]. In addition, a staging system was proposedfor the purpose of incorporating both host factors andresponse to a particular infectious insult. This concept,termed predisposition, infection, response, organ dysfunc-tion [34], represents an attempt to include the patient'sresponse to treatment in the diagnosis.
As important as these advances have been, we suggestthat these metrics and criteria remain too imprecise to movebeyond identifying population tendencies and are removedfrom the increasing mechanistic knowledge being generated.We have progressed in our understanding of sepsis to includehigh-dimensional genomic and proteomic data sets [36,37],signal processing techniques that assist in creating diagnosticsense from chaotic physiologic data [24,30,38-40], andmechanistic mathematical modeling based on preclinicaland clinical data [41,42]. This increased resolution ofknowledge regarding the pathophysiology of sepsis hasoffered the promise of more precise characterization of thedisease [43,44]. These advances have also raised thepossibility of defining the multidimensional state of anindividual patient with sepsis based on direct measure-ments of the molecules that orchestrate the interplayamong infection, inflammation, and organ dysfunction(Fig. 1) [45]. As we discuss below, these emergingapproaches may help define sepsis in a more precisefashion (Fig. 2) that includes detailed, dynamic, physio-logic, and molecular characteristics of patient subgroupsand, eventually, of individuals.
3. Sepsis: a process flow
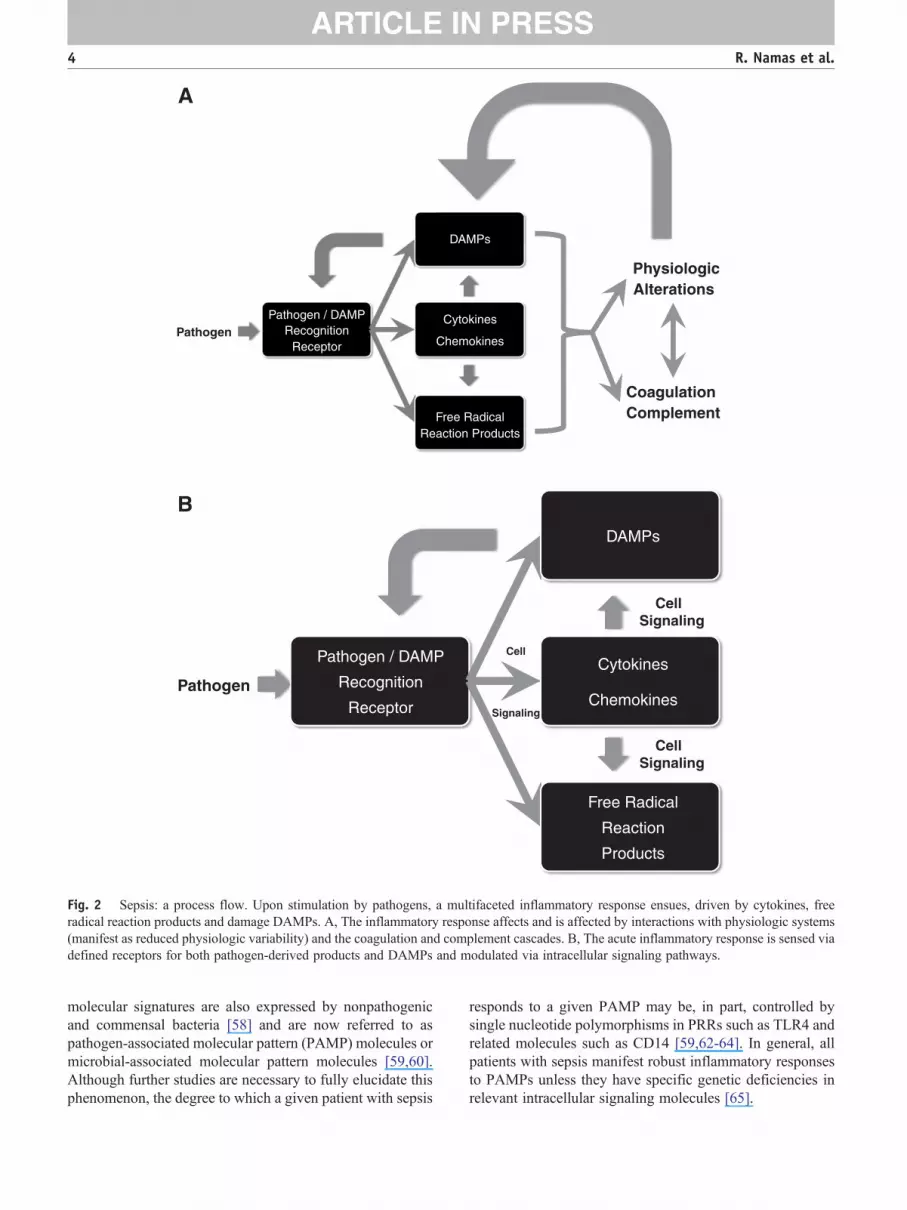
Just as the conceptual evolution of sepsis has beendynamic, we now appreciate that the pathogenesis of sepsisinvolves a dynamic, complex process of cellular activationresulting in the activation of neutrophils, monocytes, andmicrovascular endothelial cells; the triggering of neuroen-docrine mechanisms; and activation of the complement,coagulation, and fibrinolytic systems [46,47]. Acute inflam-mation is a central mechanism that helps connect theseprocesses across time and space (Fig. 2A). The innateimmune response recognizes the presence of invadingpathogens; acts toward initial containment; recruits addi-tional cells to eliminate the pathogens; and, concurrently,involves feedback mechanisms that serve to limit and restrictthe proinflammatory component such that homeostaticdynamic equilibrium can be re-established [48]. Thesefactors function in a series of interlinked and overlapping
networks, suggesting that “inflammation is communication”[49]. Like any situation that involves communication, thecontent, tone, and context matter a great deal. On the onehand, an appropriately robust inflammatory response isnecessary to survive diverse insults both in the very short andlong term [50]. Although organs obtained from patients withsepsis postmortem may not exhibit histologic damage [51],these organs are nonetheless dysfunctional because ofvarious defects that manifest, at the cellular level, in bothepithelial [52] and endothelial cells [15,16]. We suggest thatthis dysfunction occurs due to a positive feedback loop inwhich inflammation induced by pathogen-derived signalsleads to the release from epithelial and endothelial cells ofdamage-associated molecular pattern (DAMP) molecules,the molecular messengers of tissue damage. In turn, these“danger signals” stimulate nearby inflammatory cells toproduce more of the classical inflammatory mediators,leading to further release of DAMPs and ultimately to self-maintaining inflammation even after the pathogen has beencleared (Fig. 2B). The body is equipped to suppressinflammation and drive cell/tissue/organ healing boththrough the production of anti-inflammatory mediators aswell as through an inherent suppression of proinflammatorysignaling (referred to as tolerance or desensitization).However, in progressive sepsis, these anti-inflammatoryinfluences are either insufficient to suppress self-maintaininginflammation or are overproduced and lead to an immuno-suppressed state [48,53-55].
In the following sections, we will describe some of thesecomponents and place them into an appropriate context. Itshould be noted that presenting the information requires alinear structure; this should in no way obscure the complexdynamic actuality of the system in reality (Fig. 2). Wesuggest that the key to developing effective diagnostics andtreatments for sepsis requires effective characterization of thearchitecture and dynamics of the inflammatory system from amechanistic standpoint.
3.1. Pathogen recognition
The innate immune system is a highly evolutionarilyconserved host defense mechanism against pathogens [56],although an alternative viewpoint suggests that this systemevolved to respond to trauma and injury (see below) [57].Innate immune responses to pathogens are initiated bypattern recognition receptors (PRRs) that recognize specificstructures of microorganisms (Fig. 2). At least 4 families ofPRRs are recognized: Toll-like receptors (TLRs), nucleotideoligomerization domain leucine-rich repeat proteins, cyto-plasmic caspase activation and recruiting domain helicasessuch as retinoic acid-inducible gene I–like helicases, and C-type lectin receptors expressed on dendritic and myeloidcells [56,58-61]. Bacteria and viruses have molecularstructures that are generally not shared with their host,common among related pathogens, and invariant. These
A
PhysiologicAlterations
CoagulationComplement
PathogenPathogen / DAMP
RecognitionReceptor
DAMPs
Cytokines
Chemokines
Free RadicalReaction Products
Pathogen
Pathogen / DAMP
Recognition
Receptor
DAMPs
Cytokines
Chemokines
Free Radical
Reaction
Products
CellSignaling
CellSignaling
Cell
Signaling
B
Fig. 2 Sepsis: a process flow. Upon stimulation by pathogens, a multifaceted inflammatory response ensues, driven by cytokines, freeradical reaction products and damage DAMPs. A, The inflammatory response affects and is affected by interactions with physiologic systems(manifest as reduced physiologic variability) and the coagulation and complement cascades. B, The acute inflammatory response is sensed viadefined receptors for both pathogen-derived products and DAMPs and modulated via intracellular signaling pathways.
4 R. Namas et al.
molecular signatures are also expressed by nonpathogenicand commensal bacteria [58] and are now referred to aspathogen-associated molecular pattern (PAMP) molecules ormicrobial-associated molecular pattern molecules [59,60].Although further studies are necessary to fully elucidate thisphenomenon, the degree to which a given patient with sepsis
responds to a given PAMP may be, in part, controlled bysingle nucleotide polymorphisms in PRRs such as TLR4 andrelated molecules such as CD14 [59,62-64]. In general, allpatients with sepsis manifest robust inflammatory responsesto PAMPs unless they have specific genetic deficiencies inrelevant intracellular signaling molecules [65].

5A systems view of sepsis
3.2. Inflammatory signal transduction
Inflammatory signals are transduced by a series of adaptormolecules that bind to the PRRs and protein kinases andphosphatases that control signal propagation in the cyto-plasm, culminating either in the rapid, posttranscriptional, orposttranslational modulation of a variety of inflammatorymediators or in the activation of various transcription factors(Fig. 2B). These factors include nuclear factor-κB, activatorprotein 1, members of the CCAAT enhancer-binding proteinfamily, early growth response protein 1, p53, and signaltransducer and activator of transcription 1. These mecha-nisms have been the subject of considerable study and havebeen reviewed extensively elsewhere [59,66].
3.3. Production of “classical” inflammatorymediators
A wide variety of cytokines and effector molecules suchas reactive oxygen and nitrogen species are produced as aconsequence of the receptor binding and signaling eventsdescribed above (Fig. 2B). Many of these mediators and theiractions in sepsis have been studied for 2 decades and havebeen discussed extensively elsewhere [18,47,67-73]. Amongthe earliest mediators to be tested both experimentally andclinically in sepsis, tumor necrosis factor α (TNF-α)continues to be of interest from a systems perspectivebecause it is central to a well-studied positive feedback loopthat augments both further production of TNF-α as well asnumerous other inflammatory mediators [74,75]. Tumornecrosis factor α also helps drive the production of anti-inflammatory cytokines. In patients with established SIRS,both proinflammatory cytokines and anti-inflammatoryspecies coexist in the circulation in markedly increasedamounts; the simultaneous presence of both proinflamma-tory cytokines and their counter-regulators has beenassociated with adverse outcomes [1,76-78]. Although thephenotype of systemic inflammation can be recapitulated byexogenous administration of TNF-α, a series of failedanticytokine clinical trials [79,80]—as well as subsequentstudies demonstrating the beneficial and necessary roles ofTNF-α in a well-balanced inflammatory response [50,81]—has led to a retrospective recognition of the individual- andcontext-specific interplay of pro- and anti-inflammatorymediators.
3.4. Damage-associated molecular patterns and“late mediators” of sepsis
As has been recently appreciated, intracellular molecules(eg, intracellular proteins or fragments thereof, DNA, andeven inorganic crystals) that are expressed or released afterhost tissue injury are endogenous equivalents of PAMPs.These molecules are known as alarmins or DAMPs [59,82].Damage-associated molecular patterns, like PAMPs, also
bind to PRRs either expressed on the surface of immune cellsor present intracellularly (Fig. 2) [59,83]. In the setting ofsepsis, high-mobility group protein B1 (HMGB1), a nuclearprotein that stabilizes nucleosome formation in almost alleukaryotic cells, is a key DAMP [84-86] and a therapeutictarget [84,87]. High-mobility group protein B1 is but 1member of a growing list of DAMPs [88]. For example,mitochondrial DNA and other products were recentlyreported to be DAMPs in the setting of trauma [89].
Although extensive, reductionist studies have yielded atremendous amount of data and the potential for sepsistherapies are directed against inflammatory cytokines [73],TLRs [90-92], DAMPs such as HMGB1 [93-95], andmediators of inflammatory signal transduction [96-98], wesuggest that the development and implementation of suchtherapies will require an understanding of the complexity ofthe myriad actions and interactions of these ligands andreceptors (Fig. 3) [99-101].
4. Changes in physiology during sepsis:insights from complex systems
Inflammation-induced organ dysfunction is a hallmark ofsepsis. From a systems perspective, it has been hypothesizedfor more than 15 years that these oscillatory systems arecoupled and that the disruption of this coupling is a hallmarkof sepsis [38]. Both experimental and clinical studies havesuggested that 1 measure of this disrupted oscillatorycoupling is reduced variability (or increased regularity) invarious physiologic signals, chief among them being heartrate (Fig. 3) [102-104]. Time-domain analysis of heart ratevariability (HRV) has subsequently evolved as a potentialnoninvasive diagnostic modality for sepsis [105]. These datacan also be used indirectly to detect variability attributed tosympathetic and parasympathetic branches of the autonomicnervous system as well as other physiologic processes thataffect heart rate, including respiration, blood pressure, andtemperature [105]. Using these sophisticated signal proces-sing techniques, various studies have reported that a decreasein HRV indices may be potentially diagnostic of highermorbidity and mortality in critically ill patients [103,106-113]. In addition to HRV, examination of other physiologicparameters from a complex systems approach has alsoyielded valuable insights into the physiology of sepsis. Forexample, changes in ventilation and breath-to-breath vari-ability occur with sepsis, particularly in the setting ofrespiratory failure. Multiple mechanisms have been impli-cated including increased central drive and increasedmetabolic requirements [114] as well the cyclooxygenasepathway [115]. Furthermore, these changes in breathing andHRV have implications for heart-lung interactions in sepsis.However, we are still far from a complete understanding ofthe iterative, recursive interactions between inflammationand HRV. Based on prior work in animal models of sepsis

PhysiologicAlterations
Systems-BasedDiagnosis
PersonalizedTherapy
Systems-Based Drug Design and In Silico Clinical Trials
PathogenPathogen / DAMP
RecognitionReceptor
DAMPs
Cytokines
Chemokines
Free RadicalReaction Products
Fig. 3 Toward multidimensional, individualized description of patient state. The future of sepsis diagnosis and therapy will depend on agrowing understanding of the cellular and molecular mechanisms of inflammation by which pathogens are sensed and eliminated alongwith the effects of inflammation on physiology and vice versa. These interactions will form the basis of computational models used forrational design of drugs and the clinical trials by which those drugs are tested. Multidimensional analysis of inflammation biomarkers andphysiologic waveforms along with mechanistic mathematical modeling may aid in discerning individual patient states for the purposes ofdiagnosis and therapy.
6 R. Namas et al.
(F.J. Jacono and T.E. Dick, unpublished observations), wehypothesize that proinflammatory cytokines such as inter-leukin 1β and TNF-α act to decrease HRV and breathingpattern variability by affecting the central nervous system inseptic patients and that, in turn, reduced physiologicvariability further stimulates inflammation. If this hypothesisis correct, a systems understanding may allow us to unify thepattern-based, diagnostically relevant use of physiologicwaveforms with the increasingly detailed, mechanisticunderstanding of acute inflammation to improve therapyfor sepsis (Fig. 3).
Renewed interest in the diagnostic utility of metrics suchas HRV in the setting of trauma and sepsis [111,116]suggests that these methods may reach clinical utility in thenear future (see the recent 9th International Conference onComplexity in Acute Illness; http://www.iccai.org/sci_info_2010.php). We suggest that, in order for theanalyses of physiologic variability to progress beyondpattern analysis, a mechanistic understanding of how sepsisresults in reduced physiologic variability is required. Wefurther suggest that the inflammatory mechanisms describedabove will affect physiologic function (and will thereforemanifest as changes in indices of physiologic variability); inturn, these changes in physiology will impact the inflamma-tory response (Fig. 3).
5. Deciphering the nonlinear process flow ofsepsis via computational simulations
Despite promising results at the basic science andpreclinical level, large-scale trials of therapies targeted atinhibiting specific inflammatory mediators have generallyfailed to improve survival [117]. Above, we have discussedthe growing recognition that inflammation and the physiol-ogy to which it is coupled demonstrates complex, nonlinearbehavior. This property significantly limits the intuitiveextrapolation to system/patient level effects of mechanisticknowledge derived from basic science [24,26-28,38,118].Reductionism has been successful when applied to systemswhose behavior can be reduced to a “linear” (ie, single directrelationship) representation such that the results of variousindependent experiments can be aggregated additively toobtain and predict the behavior of the system as a whole [28].However, systems such as the acute inflammatory responsethat have multiple feedback loops and saturating doseresponse kinetics are inherently nonlinear. The nonlinearinteractions among pathogen recognition elements, signaltransduction pathways, inflammatory mediators, DAMPs,and the physiologic processes that they collectively impact(Fig. 2A) require more sophisticated mathematical represen-tation for their characterization [23-31].

7A systems view of sepsis
Systems biology approaches [25,119] may offer asolution to understanding these interactions. For addressingcomplex biologic processes such as the acute inflammatoryresponse in sepsis [28], both the National Institutes of Healthin its Roadmap Initiative (http://nihroadmap.nih.gov/) andthe Food and Drug Administration in its “Critical Path”document have called for the use of in silico (computer)models to augment preclinical animal studies to developnovel therapies [119]. In silico studies use the growing powerof digital computers to mine large databases in search ofpatterns that either elucidate mechanisms or are diagnosti-cally useful [120]. Mathematical models of physiologycharacterize the evolution of observables over time and aretherefore dynamic. Their purpose is predictive description—to provide entailment and insight into what the future state ofthe system might be, given the knowledge of the current stateof the system. This dynamic property suggests thatmathematical models can be considered as testable hypoth-eses. When a mathematical model predicts measurablebehavior that emulates the physiology under study, one canreasonably infer that the mathematical model has, in fact,captured potentially useful interrelations [28]. Conversely,when model and experiment disagree, the assumptionsencapsulated in the model must be reassessed (it should benoted that this process is not limited to mathematicalmodels). Therefore, transparency in model construction iscritical such that the assumptions underlying the model canbe examined in detail. Because behavioral nonlinearities andhigh-dimensional parameter spaces represent challenges inthe calibration of such models to experimental data andbecause such data fitting is further impaired by uncertaintyand variability of sparse observations (especially in settingsof preclinical and clinical studies) [121], the formalismsassociated with mathematical models may provide aframework in which underlying hypotheses can be moreeffectively examined and modified.
There have been notable successes in the translationalapplications of mechanistic mathematical models of acuteinflammation as applied to sepsis, trauma, and woundhealing. On a purely theoretical level, simple models ofacute inflammation have suggested that morbidity andmortality in sepsis may arise from diverse insult- andpatient-specific circumstances such as pathogen numberand virulence (ie, degree to which pathogens stimulate aproinflammatory response) as well as the degree to whichDAMPs are produced in response to both the pathogen andproinflammatory mediators [122]. Mathematical models ofsome of the inflammatory signal-transduction cascadesdescribed above may help drive mechanism-based drugdiscovery and device development, namely, for thedemonstration of likely efficacy throughout the develop-ment process; augmentation of and integration with existingexperimental data sets directed toward drug/device devel-opment; and the execution of simulated clinical trials, bothto facilitate the planning of future clinical trials [123-125](Fig. 3). Other mathematical models were used to yield
insights into the acute inflammatory response in diverseshock states (most importantly, the suggestion that acommon “wiring framework” but different initial conditionscould account for diverse manifestations of endotoxemic vshemorrhagic shock) [126-131] as well as the responses toanthrax [132] and necrotizing enterocolitis [133]. At thepreclinical level, mathematical modeling has helped defineand predict the acute inflammatory responses of experi-mental animals [126,129,134-136] and humans [137], allcrucial advances if we are to bridge the gap from imperfectpreclinical animal models to the setting of human sepsis.
Initial translational successes of mathematical modelsinvolved the ability to reproduce (and suggest improvementsto) clinical trials in sepsis [31,138] (Fig. 3); these successeshave been extended to the design of prospective clinical trials[41,49,100,101,139]. One in silico clinical trial platform(Immunetrics, Inc, Pittsburgh, PA) was recently augmentedto include a multiscale, equation-based mechanistic simula-tion that encompasses dynamic interactions among multipletissues, immune cells, and inflammatory mediators alongwith a “virtual clinician” (an automated system to examinesimulated patients' status at clinically relevant intervals andadminister standard-of-care interventions as necessary). Thismathematical model was fit to time course data consisting ofvarious biomarkers and clinical markers, both inflammatoryand physiologic variables from published human endotox-emia studies [140] as well and patients with community-acquired severe sepsis in the Genetic and InflammatoryMarkers of Sepsis [76] study. This model was capable ofreproducing the entire spectrum of patients in the Geneticand Inflammatory Markers of Sepsis study by altering only ahandful of parameters related to pathogen, antibioticefficacy, and baseline patient status. The model simulationsprovided evidence of changes in disease progression andlikelihood of survival as a function of various treatments,patient stratification based on outcome and time of death, andpredictive ability for patient outcome beyond hospitaldischarge. Model training and optimization led to theidentification of a minimum set of temporal analyte datarequired to predict future trajectories and outcomes ofpatients (S. Chang, Y. Vodovotz, J.A. Kellum, and D.C.Angus, unpublished observations). We suggest that compu-tational platforms such as this one could usher in a new era ofrationally designed drugs as well as informing the design offuture clinical trials (Fig. 3).
From a potentially diagnostic standpoint (and in accordwith clinical findings described above), studies involvingmechanistic mathematical models of sepsis suggest thatdetrimental outcomes are accompanied by the simultaneouselevation of both pro- and anti-inflammatory mediators[127,132,138]. Moreover, recent mathematical modelingstudies have begun to lay the foundation for a similarmechanistic underpinning for the interactions betweeninflammation and HRV [137,141]. Furthermore, insightsprovided by data-driven modeling approaches such asprincipal component analysis in individual patients with

8 R. Namas et al.
trauma/sepsis suggest that characteristic profiles of pro- andanti-inflammatory cytokines are both central drivers of thepathology of severely ill patients and that principal cytokine“barcodes” may be of diagnostic potential, although rawcytokine data may not [49,101]. These advances in the use ofmathematical modeling and related systems approaches holdthe potential to change the way drugs and diagnostics aredeveloped and clinical trials are designed and carried out, allbased on rational, mechanistic, and, in a sense, “predictable”underpinnings [41,42,49,99-101,142].
6. Conclusions and future prospects
We have come quite a long way since the early attempts atmathematical modeling in sepsis were met with both hope[143] and skepticism [120]. The septic response, a complexchain of events involving pro- and anti-inflammatoryprocesses, humoral and cellular reactions, and microcircula-tory alterations [51,144], requires that we move away from abiomarker search [43,44] (whether biologic [76,145-155] orphysiologic [156-158]). Although many of these individualmarkers have shown merit in defined cohorts, we suggestthat merely sorting through an ever-growing array ofbiomarkers and metrics of physiologic signals will notsolve the problem of accurate diagnosis or prediction oftreatment efficacy. Rather, mechanistically oriented compu-tational simulation and modeling may be a means forreconciling the diverse attempts at diagnosis of sepsis as wellas providing a rational framework for the design of new,personalized therapies (Fig. 3). Although substantial addi-tional work is needed, we suggest that computationalmodeling can facilitate the transition from static diagnosistoward a dynamic definition of the state of the individualseptic patient.
7. Disclosures
Drs Vodovotz and Billiar are cofounders of andstakeholders in Immunetrics, Inc, which has licensed fromthe University of Pittsburgh the rights to commercializeaspects of the mathematical modeling of inflammation. DrAn is a consultant to Immunetrics, Inc.
Acknowledgments
This work was supported, in part, by the NationalInstitutes of Health grants R01GM67240, P50GM53789,R33HL089082 , R01HL080926 , R01AI080799 ,R01HL76157, 3R01GM034695-25S1, and R01GM082974;National Institute on Disability and Rehabilitation Researchgrant H133E070024; the Veterans Administration Research
Service; as well as grants from the Commonwealth ofPennsylvania, the Pittsburgh Lifesciences Greenhouse, andthe Pittsburgh Tissue Engineering Initiative.
References
[1] Angus DC, Linde-Zwirble WT, Lidicker J, et al. Epidemiology ofsevere sepsis in the United States: analysis of incidence, outcome, andassociated costs of care. Crit Care Med 2001;29:1303-10.
[2] Martin GS, Mannino DM, Eaton S, et al. The epidemiology of sepsisin the United States from 1979 through 2000. N Engl J Med2003;348:1546-54.
[3] Vincent JL, Sakr Y, Sprung CL, et al. Sepsis in European intensivecare units: results of the SOAP study. Crit Care Med 2006;34:344-53.
[4] Heron M, Hoyert DL, Murphy SL, et al. Deaths: final data for 2006.Natl Vital Stat Rep 2009;57:1-134.
[5] Weycker D, Akhras KS, Edelsberg J, et al. Long-term mortality andmedical care charges in patients with severe sepsis. Crit Care Med2003;31:2316-23.
[6] Wheeler AP, Bernard GR. Treating patients with severe sepsis.N Engl J Med 1999;340:207-14.
[7] Angus DC, Wax RS. Epidemiology of sepsis: an update. Crit CareMed 2001;29:S109-16.
[8] Sands KE, Bates DW, Lanken PN, et al. Epidemiology of sepsissyndrome in 8 academic medical centers. JAMA 1997;278:234-40.
[9] Hoyert DL, Arias E, Smith BL, et al. Deaths: final data for 1999. NatlVital Stat Rep 2001;49:1-113.
[10] Increase in National Hospital Discharge Survey rates for septicemia—United States, 1979-1987. MMWR Morb Mortal Wkly Rep 1990;39:31-4.
[11] Heyland DK, Hopman W, Coo H, et al. Long-term health-relatedquality of life in survivors of sepsis. Short Form 36: a valid andreliable measure of health-related quality of life. Crit Care Med2000;28:3599-605.
[12] Perl TM, Dvorak L, Hwang T, et al. Long-term survival and functionafter suspected gram-negative sepsis. JAMA 1995;274:338-45.
[13] Protti A, Singer M. Bench-to-bedside review: potential strategies toprotect or reverse mitochondrial dysfunction in sepsis-induced organfailure. Crit Care 2006;10:228.
[14] Balestra GM, Legrand M, Ince C. Microcirculation and mitochondriain sepsis: getting out of breath. Curr Opin Anaesthesiol 2009;22:184-90.
[15] Aird WC. The role of the endothelium in severe sepsis and multipleorgan dysfunction syndrome. Blood 2003;101:3765-77.
[16] Ait-Oufella H, Maury E, Lehoux S, et al. The endothelium:physiological functions and role in microcirculatory failure duringsevere sepsis. Intensive Care Med 2010;36:1286-98.
[17] Rittirsch D, Flierl MA, Ward PA. Harmful molecular mechanisms insepsis. Nat Rev Immunol 2008;8:776-87.
[18] Levi M, van der Poll T. Inflammation and coagulation. Crit Care Med2010;38:S26-34.
[19] Geroulanos S, Douka ET. Historical perspective of the word “sepsis”.Intensive Care Med 2006;32:2077.
[20] Ewald PW. Evolution of virulence. Infect Dis Clin North Am2004;18:1-15.
[21] Vincent JL. Clinical sepsis and septic shock—definition, diagnosisand management principles. Langenbecks Arch Surg 2008;393:817-24.
[22] Schottmueller H. Wesen und Behandlung der Sepsis. Inn Med2009;31:257-80.
[23] An G. Agent-based computer simulation and sirs: building a bridgebetween basic science and clinical trials. Shock 2001;16:266-73.
[24] Buchman TG, Cobb JP, Lapedes AS, et al. Complex systemsanalysis: a tool for shock research. Shock 2001;16:248-51.

9A systems view of sepsis
[25] Kitano H. Systems biology: a brief overview. Science 2002;295:1662-4.
[26] Neugebauer EA, Willy C, Sauerland S. Complexity and non-linearityin shock research: reductionism or synthesis? Shock 2001;16:252-8.
[27] Tjardes T, Neugebauer E. Sepsis research in the next millennium:concentrate on the software rather than the hardware. Shock 2002;17:1-8.
[28] Vodovotz Y, Clermont G, Chow C, et al. Mathematical models of theacute inflammatory response. Curr Opin Crit Care 2004;10:383-90.
[29] Vodovotz Y, Csete M, Bartels J, et al. Translational systems biologyof inflammation. PLoS Comput Biol 2008;4:e1000014.
[30] Cohen MJ, Grossman AD, Morabito D, et al. Identification ofcomplex metabolic states in critically injured patients usingbioinformatic cluster analysis. Crit Care 2010;14:R10.
[31] An G. In-silico experiments of existing and hypothetical cytokine-directed clinical trials using agent based modeling. Crit Care Med2004;32:2050-60.
[32] Strieter RM, Lynch III JP, Basha MA, et al. Host responses inmediating sepsis and adult respiratory distress syndrome. SeminRespir Infect 1990;5:233-47.
[33] American College of Chest Physicians/Society of Critical CareMedicine Consensus Conference: definitions for sepsis and organfailure and guidelines for the use of innovative therapies in sepsis.Crit Care Med 1992;20:864-74.
[34] Levy MM, Fink MP, Marshall JC, et al. 2001 SCCM/ESICM/ACCP/ATS/SIS International Sepsis Definitions Conference. Crit Care Med2003;31:1250-6.
[35] Bone RC, Balk RA, Cerra FB, et al. Definitions for sepsis and organfailure and guidelines for the use of innovative therapies in sepsis.The ACCP/SCCM Consensus Conference Committee. AmericanCollege of Chest Physicians/Society of Critical Care Medicine. Chest1992;101:1644-55.
[36] Chung TP, Laramie JM, Province M, et al. Functional genomics ofcritical illness and injury. Crit Care Med 2002;30:S51-7.
[37] Cobb JP, O'Keefe GE. Injury research in the genomic era. Lancet2004;363:2076-83.
[38] Godin PJ, Buchman TG. Uncoupling of biological oscillators: acomplementary hypothesis concerning the pathogenesis of multipleorgan dysfunction syndrome. Crit Care Med 1996;24:1107-16.
[39] Buchman TG. Nonlinear dynamics, complex systems, and thepathobiology of critical illness. Curr Opin Crit Care 2004;10:378-82.
[40] Seely AJ, Macklem PT. Complex systems and the technology ofvariability analysis. Crit Care 2004;8:R367-84.
[41] Vodovotz Y, Csete M, Bartels J, et al. Translational systems biologyof inflammation. PLoS Comput Biol 2008;4:1-6.
[42] Vodovotz Y, Constantine G, Rubin J, et al. Mechanistic simulationsof inflammation: current state and future prospects. Math Biosci2009;217:1-10.
[43] Ventetuolo CE, Levy MM. Biomarkers: diagnosis and riskassessment in sepsis. Clin Chest Med 2008;29:591-603, vii.
[44] Claus RA, Otto GP, Deigner HP, et al. Approaching clinical reality:markers for monitoring systemic inflammation and sepsis. Curr MolMed 2010;10:227-35.
[45] Bone RC, Grodzin CJ, Balk RA. Sepsis: a new hypothesis forpathogenesis of the disease process. Chest 1997;112:235-43.
[46] Vincent JL, Abraham E. The last 100 years of sepsis. Am J RespirCrit Care Med 2006;173:256-63.
[47] Cinel I, Opal SM. Molecular biology of inflammation and sepsis: aprimer. Crit Care Med 2009;37:291-304.
[48] Vodovotz Y, Chow CC, Bartels J, et al. In silico models of acuteinflammation in animals. Shock 2006;26:235-44.
[49] Mi Q, Li NYK, Ziraldo C, et al. Translational systems biology ofinflammation: potential applications to personalized medicine.Personalized Med 2010;7:549-59.
[50] Namas R,GhumaA, Torres A, et al. An adequately robust early TNF—a response is a hallmark of survival following trauma/hemorrhage.PLoS ONE 2009;4:e8406.
[51] Hotchkiss RS, Karl IE. The pathophysiology and treatment of sepsis.N Engl J Med 2003;348:138-50.
[52] FinkMP, Delude RL. Epithelial barrier dysfunction: a unifying themeto explain the pathogenesis of multiple organ dysfunction at thecellular level. Crit Care Clin 2005;21:177-96.
[53] Marshall JC. Inflammation, coagulopathy, and the pathogenesis ofmultiple organ dysfunction syndrome. Crit Care Med 2001;29:S99-106.
[54] Jarrar D, Chaudry IH, Wang P. Organ dysfunction followinghemorrhage and sepsis: mechanisms and therapeutic approaches(review). Int J Mol Med 1999;4:575-83.
[55] Crouser E, Exline M, Knoell D, et al. Sepsis: links between pathogensensing and organ damage. Curr Pharm Des 2008;14:1840-52.
[56] Creagh EM, O'Neill LA. TLRs, NLRs and RLRs: a trinity ofpathogen sensors that co-operate in innate immunity. TrendsImmunol 2006;27:352-7.
[57] Matzinger P. The danger model: a renewed sense of self. Science2002;296:301-5.
[58] Granucci F, Foti M, Ricciardi-Castagnoli P. Dendritic cell biology.Adv Immunol 2005;88:193-233.
[59] Nduka OO, Parrillo JE. The pathophysiology of septic shock. CritCare Clin 2009;25:677-702, vii.
[60] Cinel I, Dellinger RP. Advances in pathogenesis and management ofsepsis. Curr Opin Infect Dis 2007;20:345-52.
[61] Opitz B, Eitel J, Meixenberger K, et al. Role of Toll-like receptors,NOD-like receptors and RIG-I-like receptors in endothelial cells andsystemic infections. Thromb Haemost 2009;102:1103-9.
[62] Arcaroli J, Fessler MB, Abraham E. Genetic polymorphisms andsepsis. Shock 2005;24:300-12.
[63] Gibot S, Cariou A, Drouet L, et al. Association between a genomicpolymorphism within the CD14 locus and septic shock susceptibilityand mortality rate. Crit Care Med 2002;30:969-73.
[64] Paterson HM, Murphy TJ, Purcell EJ, et al. Injury primes the innateimmune system for enhanced Toll-like receptor reactivity. J Immunol2003;171:1473-83.
[65] Vogel SN, Awomoyi AA, Rallabhandi P, et al. Mutations in TLR4signaling that lead to increased susceptibility to infection in humans:an overview. J Endotoxin Res 2005;11:333-9.
[66] Pritts T, Hungness E, Wang Q, et al. Mucosal and enterocyte IL-6production during sepsis and endotoxemia—role of transcriptionfactors and regulation by the stress response. Am J Surg 2002;183:372-83.
[67] van der Poll T, van Deventer SJ. Cytokines and anticytokines in thepathogenesis of sepsis. Infect Dis Clin North Am 1999;13:413-26, ix.
[68] Adrie C, Pinsky MR. The inflammatory balance in human sepsis.Intensive Care Med 2000;26:364-75.
[69] Maier RV. Pathogenesis of multiple organ dysfunction syndrome—endotoxin, inflammatory cells, and their mediators: cytokines andreactive oxygen species. Surg Infect(Larchmt) 2000;1:197-205.
[70] Ulloa L, Tracey KJ. The “cytokine profile”: a code for sepsis. TrendsMol Med 2005;11:56-63.
[71] Jawa RS, Kulaylat MN, Baumann H, et al. What is new in cytokineresearch related to trauma/critical care. J Intensive Care Med2006;21:63-85.
[72] Pinsky MR. Sepsis and multiple organ failure. Contrib Nephrol2007;156:47-63.
[73] Cai B, Deitch EA, Ulloa L. Novel insights for systemic inflammationin sepsis and hemorrhage. Mediators Inflamm 2010;2010:642462.
[74] Chen G, Goeddel DV. TNF-R1 signaling: a beautiful pathway.Science 2002;296:1634-5.
[75] Hoffmann A, Levchenko A, Scott ML, et al. The IkB-NFkB signalingmodule: temporal control and selective gene activation. Science2002;298:1241-5.
[76] Kellum JA, Kong L, Fink MP, et al. Understanding the inflammatorycytokine response in pneumonia and sepsis: results of the Genetic andInflammatory Markers of Sepsis (GenIMS) Study. Arch Intern Med2007;167:1655-63.

10 R. Namas et al.
[77] Goldie AS, Fearon KC, Ross JA, et al. Natural cytokine antagonistsand endogenous antiendotoxin core antibodies in sepsis syndrome.The Sepsis Intervention Group. JAMA 1995;274:172-7.
[78] van der Poll T, de Waal MR, Coyle SM, et al. Antiinflammatorycytokine responses during clinical sepsis and experimental endotox-emia: sequential measurements of plasma soluble interleukin (IL)-1receptor type II, IL-10, and IL-13. J Infect Dis 1997;175:118-22.
[79] Abraham E, Anzueto A, Gutierrez G, et al. Double-blind randomisedcontrolled trial of monoclonal antibody to human tumour necrosisfactor in treatment of septic shock. NORASEPT II Study Group.Lancet 1998;351:929-33.
[80] Reinhart K, Menges T, Gardlund B, et al. Randomized, placebo-controlled trial of the anti-tumor necrosis factor antibody fragmentafelimomab in hyperinflammatory response during severe sepsis: theRAMSES Study. Crit Care Med 2001;29:765-9.
[81] Pinsky MR. Dysregulation of the immune response in severe sepsis.Am J Med Sci 2004;328:220-9.
[82] van der Poll T, Opal SM. Host-pathogen interactions in sepsis. LancetInfect Dis 2008;8:32-43.
[83] dib-Conquy M, Cavaillon JM. Stress molecules in sepsis andsystemic inflammatory response syndrome. FEBS Lett 2007;581:3723-33.
[84] Wang H, Bloom O, Zhang M, et al. HMG-1 as a late mediator ofendotoxin lethality in mice. Science 1999;285:248-51.
[85] Sunden-Cullberg J, Norrby-Teglund A, Rouhiainen A, et al.Persistent elevation of high mobility group box-1 protein(HMGB1) in patients with severe sepsis and septic shock. CritCare Med 2005;33:564-73.
[86] Angus DC, Yang L, Kong L, et al. Circulating high-mobility groupbox 1 (HMGB1) concentrations are elevated in both uncomplicatedpneumonia and pneumonia with severe sepsis. Crit Care Med2007;35:1061-7.
[87] Yang H, Ochani M, Li J, et al. Reversing established sepsis withantagonists of endogenous high-mobility group box 1. Proc NatlAcad Sci U S A 2004;101:296-301.
[88] Skoberne M, Beignon AS, Bhardwaj N. Danger signals: a time andspace continuum. Trends Mol Med 2004;10:251-7.
[89] Zhang Q, Raoof M, Chen Y, et al. Circulating mitochondrial DAMPscause inflammatory responses to injury. Nature 2010;464:104-7.
[90] Leon CG, Tory R, Jia J, et al. Discovery and development of toll-likereceptor 4 (TLR4) antagonists: a new paradigm for treating sepsis andother diseases. PharmRes 2008;25:1751-61.
[91] Hennessy EJ, Parker AE, O'Neill LA. Targeting Toll-like receptors:emerging therapeutics? Nat Rev Drug Discov 2010;9:293-307.
[92] Wittebole X, Castanares-Zapatero D, Laterre PF. Toll-like receptor 4modulation as a strategy to treat sepsis. Mediators Inflamm2010;2010:568396.
[93] Klune JR, Dhupar R, Cardinal J, et al. HMGB1: endogenous dangersignaling. Mol Med 2008;14:476-84.
[94] Wang H, Ward MF, Sama AE. Novel HMGB1-inhibiting therapeuticagents for experimental sepsis. Shock 2009;32:348-57.
[95] Yang H, Tracey KJ. Targeting HMGB1 in inflammation. BiochimBiophys Acta 2010;1799:149-56.
[96] Adams JL, Badger AM, Kumar S, et al. p38 MAP kinase: moleculartarget for the inhibition of pro-inflammatory cytokines. Prog MedChem 2001;38:1-60.
[97] Calzado MA, Bacher S, Schmitz ML. NF-kappaB inhibitors for thetreatment of inflammatory diseases and cancer. Curr Med Chem2007;14:367-76.
[98] Uwe S. Anti-inflammatory interventions of NF-kappaB signaling:potential applications and risks. Biochem Pharmacol 2008;75:1567-79.
[99] Namas R, Ghuma A, Hermus L, et al. The acute inflammatoryresponse in trauma/hemorrhage and traumatic brain injury: currentstate and emerging prospects. Libyan J Med 2009;4:136-48.
[100] Vodovotz Y, An G. Systems biology and inflammation. In: Yan Q,editor. Systems Biology in Drug Discovery and Development:
Methods and Protocols. Totowa (NJ): Springer Science & BusinessMedia; 2009. p. 181-201.
[101] Vodovotz Y, Constantine G, Faeder J, et al. Translational systemsapproaches to the biology of inflammation and healing. Immuno-pharmacol Immunotoxicol 2010;32:181-95.
[102] Annane D, Trabold F, Sharshar T, et al. Inappropriate sympatheticactivation at onset of septic shock: a spectral analysis approach. Am JRespir Crit Care Med 1999;160:458-65.
[103] Korach M, Sharshar T, Jarrin I, et al. Cardiac variability in criticallyill adults: influence of sepsis. Crit Care Med 2001;29:1380-5.
[104] Piepoli M, Garrard CS, Kontoyannis DA, et al. Autonomic control ofthe heart and peripheral vessels in human septic shock. Intensive CareMed 1995;21:112-9.
[105] Fairchild KD, Saucerman JJ, Raynor LL, et al. Endotoxin depressesheart rate variability in mice: cytokine and steroid effects. Am JPhysiol Regul Integr Comp Physiol 2009;297:R1019-27.
[106] Heart rate variability. Standards of measurement, physiologicalinterpretation, and clinical use. Task Force of the European Societyof Cardiology and the North American Society of Pacing andElectrophysiology. Eur Heart J 1996;17:354-81.
[107] Kleiger RE, Stein PK, Bigger Jr JT. Heart rate variability:measurement and clinical utility. Ann Noninvasive Electrocardiol2005;10:88-101.
[108] Pomeranz B, Macaulay RJ, Caudill MA, et al. Assessment ofautonomic function in humans by heart rate spectral analysis. Am JPhysiol 1985;248:H151-3.
[109] Godin PJ, Fleisher LA, Eidsath A, et al. Experimental humanendotoxemia increases cardiac regularity: results from a prospective,randomized, crossover trial. Crit Care Med 1996;24:1117-24.
[110] Barnaby D, Ferrick K, Kaplan DT, et al. Heart rate variability inemergency department patients with sepsis. Acad Emerg Med2002;9:661-70.
[111] Chen WL, Kuo CD. Characteristics of heart rate variability canpredict impending septic shock in emergency department patientswith sepsis. Acad Emerg Med 2007;14:392-7.
[112] Pontet J, Contreras P, Curbelo A, et al. Heart rate variability as earlymarker of multiple organ dysfunction syndrome in septic patients.J Crit Care 2003;18:156-63.
[113] Ahmad S, Ramsay T, Huebsch L, et al. Continuous multi-parameterheart rate variability analysis heralds onset of sepsis in adults. PLoSONE 2009;4:e6642.
[114] Magder S. Bench-to-bedside review: ventilatory abnormalities insepsis. Crit Care 2009;13:202.
[115] Preas HL, Jubran A, Vandivier RW, et al. Effect of endotoxin onventilation and breath variability: role of cyclooxygenase pathway.Am J Respir Crit Care Med 2001;164:620-6.
[116] Cancio LC, Batchinsky AI, Salinas J, et al. Heart-rate complexity forprediction of prehospital lifesaving interventions in trauma patients.J Trauma 2008;65:813-9.
[117] Bone RC. Why sepsis trials fail. JAMA 1996;276:565-6.[118] Seely AJ, Christou NV. Multiple organ dysfunction syndrome:
exploring the paradigm of complex nonlinear systems. Crit Care Med2000;28:2193-200.
[119] Vodovotz Y. Deciphering the complexity of acute inflammationusing mathematical models. Immunol Res 2006;36:237-45.
[120] Marshall JC. Through a glass darkly: the brave new world of in silicomodeling. Crit Care Med 2004;32:2157-9.
[121] Vodovotz Y, Clermont G, Hunt CA, et al. Evidence-based modelingof critical illness: an initial consensus from the Society forComplexity in Acute Illness. J Crit Care 2007;22:77-84.
[122] Kumar R, Clermont G, Vodovotz Y, et al. The dynamics of acuteinflammation. J Theor Biol 2004;230:145-55.
[123] Gibbs JB. Mechanism-based target identification and drug discoveryin cancer research. Science 2000;287:1969-73.
[124] Faeder JR, Hlavacek WS, Reischl I, et al. Investigation of earlyevents in Fc epsilon RI-mediated signaling using a detailedmathematical model. J Immunol 2003;170:3769-81.

11A systems view of sepsis
[125] RiviŠre B, Epshteyn Y, Swigon D, et al. A simple mathematicalmodel of signaling resulting from the binding of lipopolysaccharidewith Toll-like receptor 4 demonstrates inherent preconditioningbehavior. Math Biosci 2009;217:19-26.
[126] Chow CC, Clermont G, Kumar R, et al. The acute inflammatoryresponse in diverse shock states. Shock 2005;24:74-84.
[127] Reynolds A, Rubin J, Clermont G, et al. A reduced mathematicalmodel of the acute inflammatory response: I. Derivation of model andanalysis of anti-inflammation. J Theor Biol 2006;242:220-36.
[128] Day J, Rubin J, Vodovotz Y, et al. A reduced mathematical model ofthe acute inflammatory response: II. Capturing scenarios of repeatedendotoxin administration. J Theor Biol 2006;242:237-56.
[129] Torres A, Bentley T, Bartels J, et al. Mathematical modeling of post-hemorrhage inflammation in mice: studies using a novel, computer-controlled, closed-loop hemorrhage apparatus. Shock 2009;32:172-8.
[130] Foteinou PT, Calvano SE, Lowry SF, et al. Modeling endotoxin-induced systemic inflammation using an indirect response approach.Math Biosci 2009;217:27-42.
[131] Foteinou PT, Calvano SE, Lowry SF, et al. In silico simulation ofcorticosteroids effect on an NFkB- dependent physicochemicalmodel of systemic inflammation. PLoS ONE 2009;4:e4706.
[132] Kumar R, Chow CC, Bartels J, et al. A mathematical simulation of theinflammatory response to anthrax infection. Shock 2008;29:104-11.
[133] Arciero J, Rubin J, Upperman J, et al. Using a mathematical model toanalyze the role of probiotics and inflammation in necrotizingenterocolitis. PLoS ONE 2010;5:e10066.
[134] Prince JM, Levy RM, Bartels J, et al. In silico and in vivo approach toelucidate the inflammatory complexity of CD14-deficient mice. MolMed 2006;12:88-96.
[135] Lagoa CE, Bartels J, Baratt A, et al. The role of initial trauma in thehost's response to injury and hemorrhage: Insights from a comparisonof mathematical simulations and hepatic transcriptomic analysis.Shock 2006;26:592-600.
[136] Daun S, Rubin J, Vodovotz Y, et al. An ensemble of models of the acuteinflammatory response to bacterial lipopolysaccharide in rats: resultsfrom parameter space reduction. J Theor Biol 2008;253:843-53.
[137] Foteinou PT, Calvano SE, Lowry SF, et al. Multiscale model for theassessment of autonomic dysfunction in human endotoxemia. PhysiolGenomics 2010;42:5-19.
[138] Clermont G, Bartels J, Kumar R, et al. In silico design of clinicaltrials: a method coming of age. Crit Care Med 2004;32:2061-70.
[139] An G, Bartels J, Vodovotz Y. In silico augmentation of the drugdevelopment pipeline: examples from the study of acute inflamma-tion. Drug Dev Res 2010;72:1-14.
[140] Andreasen AS, Krabbe KS, Krogh-Madsen R, et al. Humanendotoxemia as a model of systemic inflammation. Curr MedChem 2008;15:1697-705.
[141] Foteinou PT, Calvano SE, Lowry SF, et al. A physiological model forautonomic heart rate regulation in human endotoxemia. Shock 2011(in press).
[142] Vodovotz Y. Translational systems biology of inflammation andhealing. Wound Repair Regen 2010;18:3-7.
[143] Buchman TG. In vivo, in vitro, in silico. Crit Care Med 2004;32:2159-60.
[144] Gullo A, Bianco N, Berlot G. Management of severe sepsis and septicshock: challenges and recommendations. Crit Care Clin 2006;22:489-501, ix.
[145] Lobo SM, Lobo FR, Bota DP, et al. C-reactive protein levels correlatewith mortality and organ failure in critically ill patients. Chest2003;123:2043-9.
[146] Ugarte H, Silva E, Mercan D, et al. Procalcitonin used as a marker ofinfection in the intensive care unit. Crit Care Med 1999;27:498-504.
[147] Casey LC, Balk RA, Bone RC. Plasma cytokine and endotoxin levelscorrelate with survival in patients with the sepsis syndrome. AnnIntern Med 1993;119:771-8.
[148] Damas P, Ledoux D, Nys M, et al. Cytokine serum level duringsevere sepsis in human IL-6 as a marker of severity. Ann Surg1992;215:356-62.
[149] Friedman G, Jankowski S, Marchant A, et al. Blood interleukin 10levels parallel the severity of septic shock. J Crit Care 1997;12:183-7.
[150] Pinsky MR, Vincent JL, Deviere J, et al. Serum cytokine levels inhuman septic shock. Relation to multiple-system organ failure andmortality. Chest 1993;103:565-75.
[151] Marshall JC, Foster D, Vincent JL, et al. Diagnostic and prognosticimplications of endotoxemia in critical illness: results of the MEDICstudy. J Infect Dis 2004;190:527-34.
[152] Gibot S, Kolopp-Sarda MN, Bene MC, et al. Plasma level of atriggering receptor expressed on myeloid cells-1: its diagnosticaccuracy in patients with suspected sepsis. Ann Intern Med 2004;141:9-15.
[153] Dempfle CE, Lorenz S, Smolinski M, et al. Utility of activated partialthromboplastin time waveform analysis for identification of sepsisand overt disseminated intravascular coagulation in patients admittedto a surgical intensive care unit. Crit Care Med 2004;32:520-4.
[154] Toh CH, Ticknor LO, Downey C, et al. Early identification of sepsisand mortality risks through simple, rapid clot-waveform analysis.Implications of lipoprotein-complexed C reactive protein formation.Intensive Care Med 2003;29:55-61.
[155] Seely AJE, Christou NV. Multiple organ dysfunction syndrome:exploring the paradigm of complex nonlinear systems. [Review]CritCare Med 2000;28:2193-200.
[156] Schmidt HB, Werdan K, Muller-Werdan U. Autonomic dysfunctionin the ICU patient. Curr Opin Crit Care 2001;7:314-22.
[157] Moorman JR, Lake DE, Griffin MP. Heart rate characteristicsmonitoring for neonatal sepsis. IEEE Trans Biomed Eng 2006;53:126-32.
[158] Werdan K, Schmidt H, Ebelt H, et al. Impaired regulation of cardiacfunction in sepsis, SIRS, and MODS. Can J Physiol Pharmacol2009;87:266-74.





























