Embed Size (px)
Citation preview

Simvastatin does not inhibit intimal hyperplasia andrestenosis but promotes plaque regression innormocholesterolemic patients undergoingcoronary stenting: A randomized study withintravascular ultrasoundAnna Sonia Petronio, MD, Giovanni Amoroso, MD, PhD, Ugo Limbruno, MD, PhD, Barbara Papini, RT,Marco De Carlo, MD, Andrea Micheli, MD, Nicola Ciabatti, MD, and Mario Mariani, MD Pisa, Italy
Background Restenosis after coronary stenting is mainly caused by intimal hyperplasia. Both experimental andclinical studies suggest that statins may be able to inhibit intimal hyperplasia and, therefore, in-stent restenosis (ISR), bymechanisms beyond lipid lowering.
Methods In a 12-month study, we randomized 71 normocholesterolemic patients to 20 mg simvastatin or no treatment,2 weeks before elective coronary stenting. Patients were evaluated by quantitative coronary angiography and intravascularultrasound, immediately after the index procedure and at the 12-month catheterization.
Results Binary ISR was present in 15% and in 18% of simvastatin-treated patients and controls, respectively ( P = NS).Intimal hyperplasia did not differ significantly between the 2 groups (3.6 F 1.8 vs 3.8 F 2.3 mm3/mm, 34% F 15% vs35% F 23% for simvastatin vs controls, P = NS). However, peristent plaque decreased with simvastatin but increased incontrols (�4.0 F 4.0 vs +1.6 F 3.8 mm3/mm, �14% F 10% vs +6% F 12%, P b .05). The same behavior was shownby intermediate plaques at nonstented sites (�2.5 F 3.0 vs +1.0 F 3.0 mm3/mm, �10% F 8% vs +9% F 9%, P b .05).Major adverse events at 12 months were present in 11% and 24% of simvastatin-treated patients and controls, respectively( P = .20).
Conclusions In normocholesterolemic patients undergoing coronary stenting, simvastatin does not prevent intimalhyperplasia or ISR, but it promotes atherosclerotic regression both at stented and at nonstented sites. (Am Heart J2005;149:520-6.)
See related Editorial on page 384.
Bare metal stents have been a breakthrough in
interventional cardiology because they have been prov-
en to reduce restenosis in comparison with balloon
angioplasty.1,2 However, they still carry a risk of
restenosis, accounting for 20% to 40% of all procedures.
Restenosis after coronary stenting is mainly caused by an
exaggerated hyperplasia of the intimal layer because of
smooth muscle cell proliferation and extracellular
From the Cardiothoracic Department, University of Pisa, Italy.
Submitted August 5, 2004; accepted October 9, 2004.
Reprint requests: Giovanni Amoroso, MD, PhD, Dipartimento Cardiotoracico, Ospedale
di Cisanello, via Paradisa 2, 56124 Pisa, Italy.
E-mail: [email protected]
0002-8703/$ - see front matter
n 2005, Published by Elsevier Inc.
doi:10.1016/j.ahj.2004.10.032
matrix deposition.3 Very recently, stents, which are
capable of reducing the incidence of in-stent restenosis
(ISR) by eluting antiproliferative drugs (sirolimus and
taxol), have been developed.4,5 Yet, the effect of drug-
eluting stents (DES) onto the underlying atherosclerotic
disease seems minimal.6
The 3-hydroxy-3-methylglutaryl coenzyme A inhibitors
(statins) have been clearly shown to reduce mortality
after coronary stenting.7 The beneficial effects of statins,
beyond their lipid-lowering action, mostly rely on their
anti-inflammatory properties.8 Simvastatin, but not other
molecules, has also proven to inhibit smooth muscle
cells proliferation.9
When previously tested to prevent restenosis after
balloon angioplasty, statins have given conflicting
results.10 Recently, Walter et al11 have suggested that
statins can reduce the incidence of angiographic ISR.
Their studies were, however, not randomized and
included only hypercholesterolemic patients. Moreover,
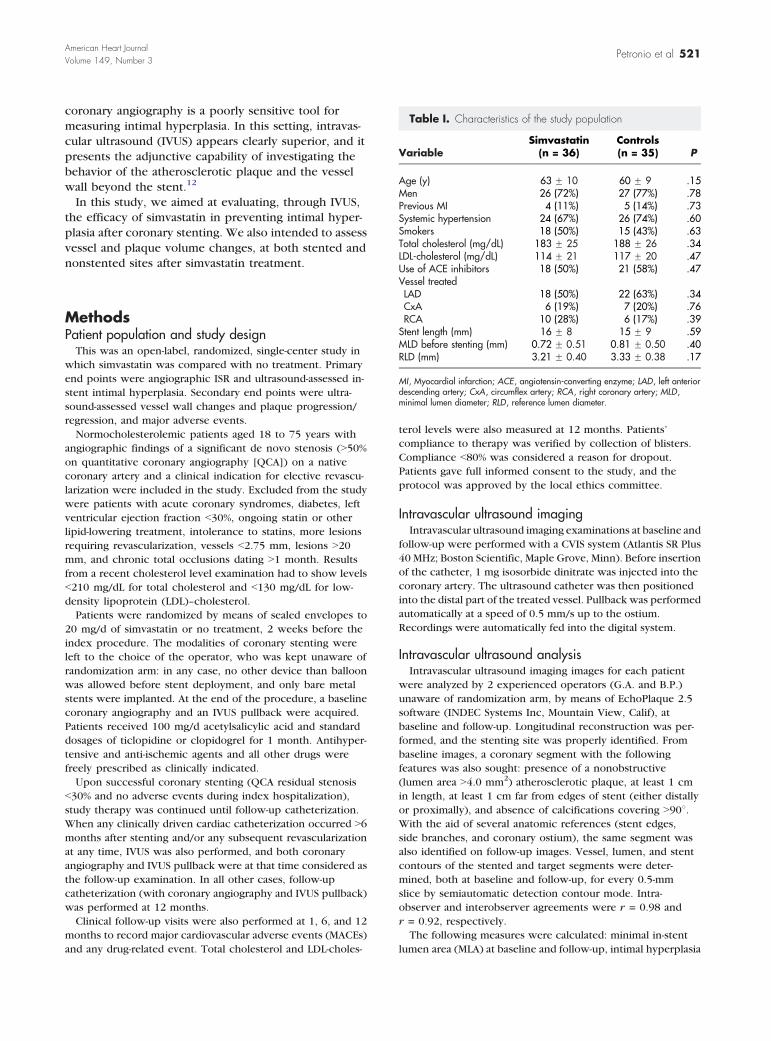
Table I. Characteristics of the study population
VariableSimvastatin
(n = 36)Controls(n = 35) P
Age (y) 63 F 10 60 F 9 .15Men 26 (72%) 27 (77%) .78Previous MI 4 (11%) 5 (14%) .73Systemic hypertension 24 (67%) 26 (74%) .60Smokers 18 (50%) 15 (43%) .63Total cholesterol (mg/dL) 183 F 25 188 F 26 .34LDL-cholesterol (mg/dL) 114 F 21 117 F 20 .47Use of ACE inhibitors 18 (50%) 21 (58%) .47
American Heart Journal
Volume 149, Number 3Petronio et al 521
coronary angiography is a poorly sensitive tool for
measuring intimal hyperplasia. In this setting, intravas-
cular ultrasound (IVUS) appears clearly superior, and it
presents the adjunctive capability of investigating the
behavior of the atherosclerotic plaque and the vessel
wall beyond the stent.12
In this study, we aimed at evaluating, through IVUS,
the efficacy of simvastatin in preventing intimal hyper-
plasia after coronary stenting. We also intended to assess
vessel and plaque volume changes, at both stented and
nonstented sites after simvastatin treatment.
Vessel treatedLAD 18 (50%) 22 (63%) .34CxA 6 (19%) 7 (20%) .76RCA 10 (28%) 6 (17%) .39
Stent length (mm) 16 F 8 15 F 9 .59MLD before stenting (mm) 0.72 F 0.51 0.81 F 0.50 .40RLD (mm) 3.21 F 0.40 3.33 F 0.38 .17
MI, Myocardial infarction; ACE, angiotensin-converting enzyme; LAD, left anteriordescending artery; CxA, circumflex artery; RCA, right coronary artery; MLD,minimal lumen diameter; RLD, reference lumen diameter.
MethodsPatient population and study design
This was an open-label, randomized, single-center study in
which simvastatin was compared with no treatment. Primary
end points were angiographic ISR and ultrasound-assessed in-
stent intimal hyperplasia. Secondary end points were ultra-
sound-assessed vessel wall changes and plaque progression/
regression, and major adverse events.
Normocholesterolemic patients aged 18 to 75 years with
angiographic findings of a significant de novo stenosis (N50%
on quantitative coronary angiography [QCA]) on a native
coronary artery and a clinical indication for elective revascu-
larization were included in the study. Excluded from the study
were patients with acute coronary syndromes, diabetes, left
ventricular ejection fraction b30%, ongoing statin or other
lipid-lowering treatment, intolerance to statins, more lesions
requiring revascularization, vessels b2.75 mm, lesions N20
mm, and chronic total occlusions dating N1 month. Results
from a recent cholesterol level examination had to show levels
b210 mg/dL for total cholesterol and b130 mg/dL for low-
density lipoprotein (LDL)–cholesterol.
Patients were randomized by means of sealed envelopes to
20 mg/d of simvastatin or no treatment, 2 weeks before the
index procedure. The modalities of coronary stenting were
left to the choice of the operator, who was kept unaware of
randomization arm: in any case, no other device than balloon
was allowed before stent deployment, and only bare metal
stents were implanted. At the end of the procedure, a baseline
coronary angiography and an IVUS pullback were acquired.
Patients received 100 mg/d acetylsalicylic acid and standard
dosages of ticlopidine or clopidogrel for 1 month. Antihyper-
tensive and anti-ischemic agents and all other drugs were
freely prescribed as clinically indicated.
Upon successful coronary stenting (QCA residual stenosis
b30% and no adverse events during index hospitalization),
study therapy was continued until follow-up catheterization.
When any clinically driven cardiac catheterization occurred N6
months after stenting and/or any subsequent revascularization
at any time, IVUS was also performed, and both coronary
angiography and IVUS pullback were at that time considered as
the follow-up examination. In all other cases, follow-up
catheterization (with coronary angiography and IVUS pullback)
was performed at 12 months.
Clinical follow-up visits were also performed at 1, 6, and 12
months to record major cardiovascular adverse events (MACEs)
and any drug-related event. Total cholesterol and LDL-choles-
terol levels were also measured at 12 months. Patients’
compliance to therapy was verified by collection of blisters.
Compliance b80% was considered a reason for dropout.
Patients gave full informed consent to the study, and the
protocol was approved by the local ethics committee.
Intravascular ultrasound imagingIntravascular ultrasound imaging examinations at baseline and
follow-up were performed with a CVIS system (Atlantis SR Plus
40 MHz; Boston Scientific, Maple Grove, Minn). Before insertion
of the catheter, 1 mg isosorbide dinitrate was injected into the
coronary artery. The ultrasound catheter was then positioned
into the distal part of the treated vessel. Pullback was performed
automatically at a speed of 0.5 mm/s up to the ostium.
Recordings were automatically fed into the digital system.
Intravascular ultrasound analysisIntravascular ultrasound imaging images for each patient
were analyzed by 2 experienced operators (G.A. and B.P.)
unaware of randomization arm, by means of EchoPlaque 2.5
software (INDEC Systems Inc, Mountain View, Calif), at
baseline and follow-up. Longitudinal reconstruction was per-
formed, and the stenting site was properly identified. From
baseline images, a coronary segment with the following
features was also sought: presence of a nonobstructive
(lumen area N4.0 mm2) atherosclerotic plaque, at least 1 cm
in length, at least 1 cm far from edges of stent (either distally
or proximally), and absence of calcifications covering N908.With the aid of several anatomic references (stent edges,
side branches, and coronary ostium), the same segment was
also identified on follow-up images. Vessel, lumen, and stent
contours of the stented and target segments were deter-
mined, both at baseline and follow-up, for every 0.5-mm
slice by semiautomatic detection contour mode. Intra-
observer and interobserver agreements were r = 0.98 and
r = 0.92, respectively.
The following measures were calculated: minimal in-stent
lumen area (MLA) at baseline and follow-up, intimal hyperplasia
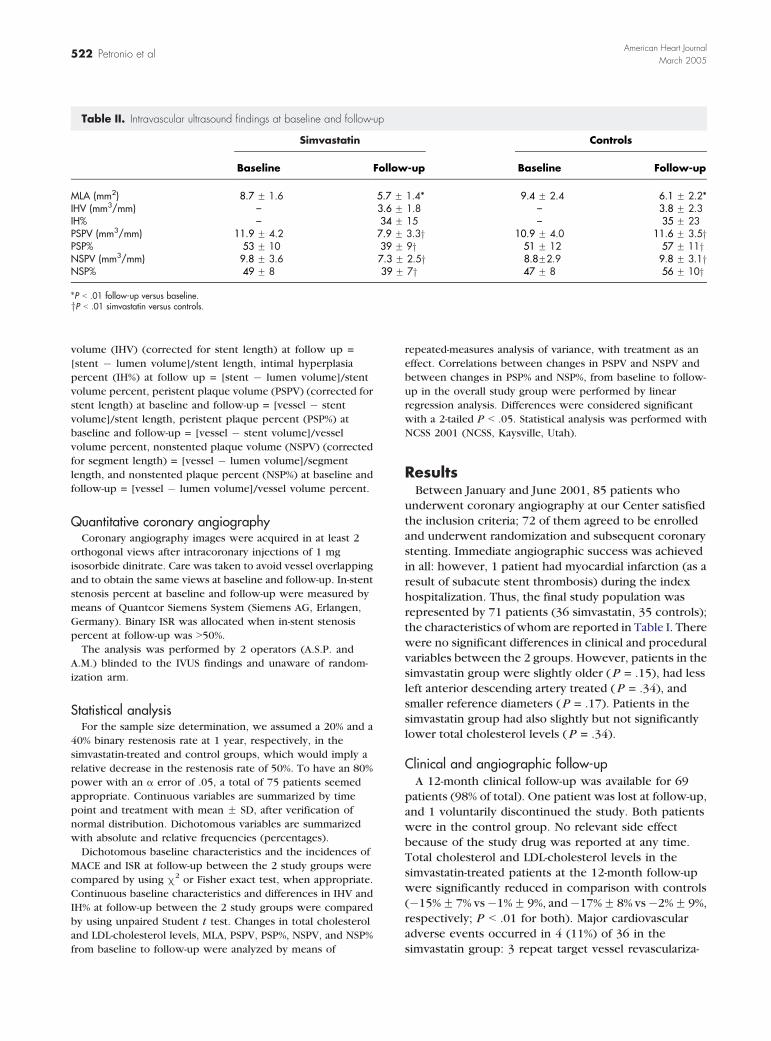
Table II. Intravascular ultrasound findings at baseline and follow-up
Simvastatin Controls
Baseline Follow-up Baseline Follow-up
MLA (mm2) 8.7 F 1.6 5.7 F 1.4* 9.4 F 2.4 6.1 F 2.2*IHV (mm3/mm) – 3.6 F 1.8 – 3.8 F 2.3IH% – 34 F 15 – 35 F 23PSPV (mm3/mm) 11.9 F 4.2 7.9 F 3.3y 10.9 F 4.0 11.6 F 3.5yPSP% 53 F 10 39 F 9y 51 F 12 57 F 11yNSPV (mm3/mm) 9.8 F 3.6 7.3 F 2.5y 8.8F2.9 9.8 F 3.1yNSP% 49 F 8 39 F 7y 47 F 8 56 F 10y
*P b .01 follow-up versus baseline.yP b .01 simvastatin versus controls.
American Heart Journal
March 2005522 Petronio et al
volume (IHV) (corrected for stent length) at follow up =
[stent � lumen volume]/stent length, intimal hyperplasia
percent (IH%) at follow up = [stent � lumen volume]/stent
volume percent, peristent plaque volume (PSPV) (corrected for
stent length) at baseline and follow-up = [vessel � stent
volume]/stent length, peristent plaque percent (PSP%) at
baseline and follow-up = [vessel � stent volume]/vessel
volume percent, nonstented plaque volume (NSPV) (corrected
for segment length) = [vessel � lumen volume]/segment
length, and nonstented plaque percent (NSP%) at baseline and
follow-up = [vessel � lumen volume]/vessel volume percent.
Quantitative coronary angiographyCoronary angiography images were acquired in at least 2
orthogonal views after intracoronary injections of 1 mg
isosorbide dinitrate. Care was taken to avoid vessel overlapping
and to obtain the same views at baseline and follow-up. In-stent
stenosis percent at baseline and follow-up were measured by
means of Quantcor Siemens System (Siemens AG, Erlangen,
Germany). Binary ISR was allocated when in-stent stenosis
percent at follow-up was N50%.
The analysis was performed by 2 operators (A.S.P. and
A.M.) blinded to the IVUS findings and unaware of random-
ization arm.
Statistical analysisFor the sample size determination, we assumed a 20% and a
40% binary restenosis rate at 1 year, respectively, in the
simvastatin-treated and control groups, which would imply a
relative decrease in the restenosis rate of 50%. To have an 80%
power with an a error of .05, a total of 75 patients seemed
appropriate. Continuous variables are summarized by time
point and treatment with mean F SD, after verification of
normal distribution. Dichotomous variables are summarized
with absolute and relative frequencies (percentages).
Dichotomous baseline characteristics and the incidences of
MACE and ISR at follow-up between the 2 study groups were
compared by using m2 or Fisher exact test, when appropriate.
Continuous baseline characteristics and differences in IHV and
IH% at follow-up between the 2 study groups were compared
by using unpaired Student t test. Changes in total cholesterol
and LDL-cholesterol levels, MLA, PSPV, PSP%, NSPV, and NSP%
from baseline to follow-up were analyzed by means of
repeated-measures analysis of variance, with treatment as an
effect. Correlations between changes in PSPV and NSPV and
between changes in PSP% and NSP%, from baseline to follow-
up in the overall study group were performed by linear
regression analysis. Differences were considered significant
with a 2-tailed P b .05. Statistical analysis was performed with
NCSS 2001 (NCSS, Kaysville, Utah).
ResultsBetween January and June 2001, 85 patients who
underwent coronary angiography at our Center satisfied
the inclusion criteria; 72 of them agreed to be enrolled
and underwent randomization and subsequent coronary
stenting. Immediate angiographic success was achieved
in all: however, 1 patient had myocardial infarction (as a
result of subacute stent thrombosis) during the index
hospitalization. Thus, the final study population was
represented by 71 patients (36 simvastatin, 35 controls);
the characteristics of whom are reported in Table I. There
were no significant differences in clinical and procedural
variables between the 2 groups. However, patients in the
simvastatin group were slightly older ( P = .15), had less
left anterior descending artery treated ( P = .34), and
smaller reference diameters ( P = .17). Patients in the
simvastatin group had also slightly but not significantly
lower total cholesterol levels ( P = .34).
Clinical and angiographic follow-upA 12-month clinical follow-up was available for 69
patients (98% of total). One patient was lost at follow-up,
and 1 voluntarily discontinued the study. Both patients
were in the control group. No relevant side effect
because of the study drug was reported at any time.
Total cholesterol and LDL-cholesterol levels in the
simvastatin-treated patients at the 12-month follow-up
were significantly reduced in comparison with controls
(�15% F 7% vs�1%F 9%, and�17%F 8% vs�2%F 9%,
respectively; P b .01 for both). Major cardiovascular
adverse events occurred in 4 (11%) of 36 in the
simvastatin group: 3 repeat target vessel revasculariza-
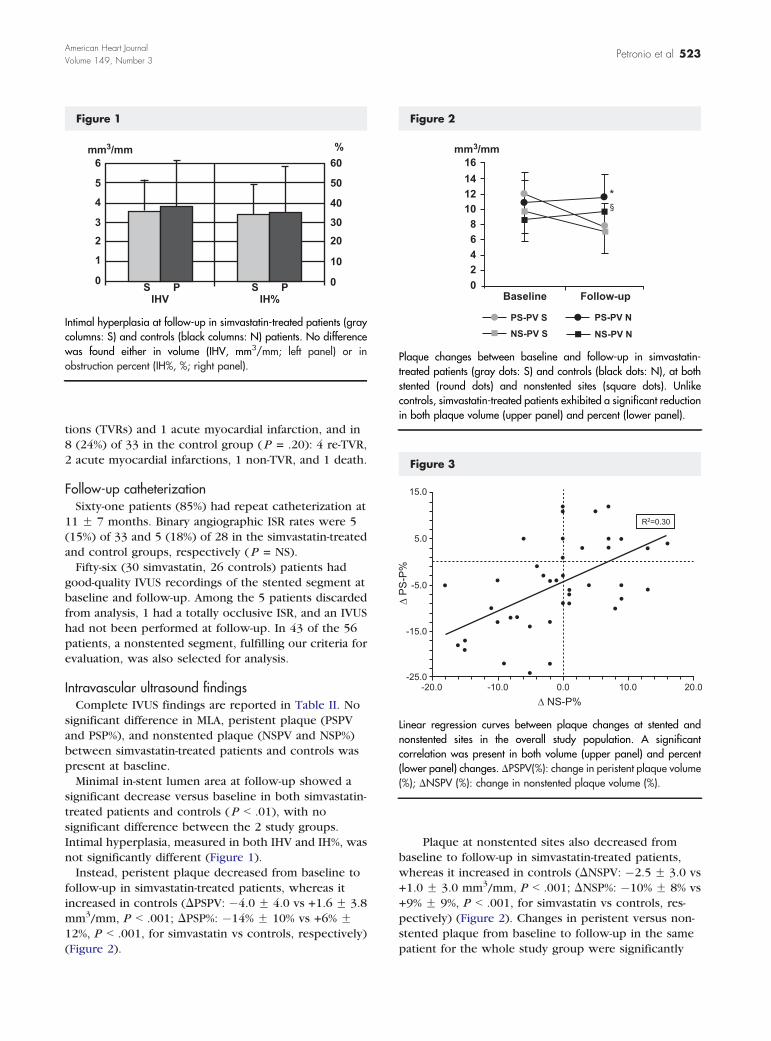
Figure 1
Intimal hyperplasia at follow-up in simvastatin-treated patients (graycolumns: S) and controls (black columns: N) patients. No differencewas found either in volume (IHV, mm3/mm; left panel) or inobstruction percent (IH%, %; right panel).
Figure 2
Plaque changes between baseline and follow-up in simvastatin-treated patients (gray dots: S) and controls (black dots: N), at bothstented (round dots) and nonstented sites (square dots). Unlikecontrols, simvastatin-treated patients exhibited a significant reductionin both plaque volume (upper panel) and percent (lower panel).
Figure 3
Linear regression curves between plaque changes at stented andnonstented sites in the overall study population. A significantcorrelation was present in both volume (upper panel) and percent(lower panel) changes. DPSPV(%): change in peristent plaque volume(%); DNSPV (%): change in nonstented plaque volume (%).
American Heart Journal
Volume 149, Number 3Petronio et al 523
tions (TVRs) and 1 acute myocardial infarction, and in
8 (24%) of 33 in the control group ( P = .20): 4 re-TVR,
2 acute myocardial infarctions, 1 non-TVR, and 1 death.
Follow-up catheterizationSixty-one patients (85%) had repeat catheterization at
11 F 7 months. Binary angiographic ISR rates were 5
(15%) of 33 and 5 (18%) of 28 in the simvastatin-treated
and control groups, respectively ( P = NS).
Fifty-six (30 simvastatin, 26 controls) patients had
good-quality IVUS recordings of the stented segment at
baseline and follow-up. Among the 5 patients discarded
from analysis, 1 had a totally occlusive ISR, and an IVUS
had not been performed at follow-up. In 43 of the 56
patients, a nonstented segment, fulfilling our criteria for
evaluation, was also selected for analysis.
Intravascular ultrasound findingsComplete IVUS findings are reported in Table II. No
significant difference in MLA, peristent plaque (PSPV
and PSP%), and nonstented plaque (NSPV and NSP%)
between simvastatin-treated patients and controls was
present at baseline.
Minimal in-stent lumen area at follow-up showed a
significant decrease versus baseline in both simvastatin-
treated patients and controls ( P b .01), with no
significant difference between the 2 study groups.
Intimal hyperplasia, measured in both IHV and IH%, was
not significantly different (Figure 1).
Instead, peristent plaque decreased from baseline to
follow-up in simvastatin-treated patients, whereas it
increased in controls (DPSPV: �4.0 F 4.0 vs +1.6 F 3.8
mm3/mm, P b .001; DPSP%: �14% F 10% vs +6% F12%, P b .001, for simvastatin vs controls, respectively)
(Figure 2).
Plaque at nonstented sites also decreased from
baseline to follow-up in simvastatin-treated patients,
whereas it increased in controls (DNSPV: �2.5 F 3.0 vs
+1.0 F 3.0 mm3/mm, P b .001; DNSP%: �10% F 8% vs
+9% F 9%, P b .001, for simvastatin vs controls, res-
pectively) (Figure 2). Changes in peristent versus non-
stented plaque from baseline to follow-up in the same
patient for the whole study group were significantly

American Heart Journal
March 2005524 Petronio et al
correlated (DPSPV vs DNSPV: R2 = 0.35, P b .001;
DPSP% vs DNSP%: R2 = 0.30, P b .001) (Figure 3).
DiscussionTo our knowledge, this is the first randomized study in
which the effect of simvastatin on ISR and intimal
hyperplasia after coronary stenting has been investigated
in normocholesterolemic patients.
The main findings of our study are (1) a similar
incidence of angiographic ISR and intimal hyperplasia in
simvastatin-treated patients and controls and (2) a
significant atherosclerotic regression both at stented and
nonstented sites with simvastatin.
Statins and in-stent restenosisPrevious studies have suggested that statins may have
a beneficial effect in preventing restenosis after coronary
stenting. Walter et al11 have found that patients
receiving prolonged statin therapy developed lower ISR
rates in comparison with nonreceivers (25% vs 38%).
The same authors also demonstrated that this beneficial
effect of statins was particularly evident in patients
carrying the PlA2 allele of the platelet GpIIIa receptor
(28.6% vs 50.9%).13
Unlike the findings of Walter et al, our study failed to
demonstrate a significant difference in angiographic
ISR (15% vs 18%) and intimal hyperplasia (3.6 F 1.8 vs
3.8 F 2.3 mm3/mm, 34% F 15% vs 35% F 23%)
between simvastatin-treated patients and controls. The
reasons for this discrepancy may be found in that the
study of Walter et al was indeed retrospective, enrolled
only hypercholesterolemic patients, and included a
high rate (22%) of patients with acute symptoms,
already known to respond at best to statin treatment.
Yet, the remarkably low rate of ISR in our overall
population, likely caused by the restrictive inclusion
criteria, may have hindered the potential effect of
statins, which may surface only among populations at a
higher risk of restenosis.
Indeed, the issue of pharmacological prevention of ISR
could soon become outmoded, when the favorable
evidences on DES provided by clinical trials will be
confirmed in daily practice. Yet, the RESEARCH study,
the first registry on the extensive use of DES in real-life
patients, reports a 65% reduction in 12-month clinically
driven TVRs (4.1% vs 10.9%).14
Statins and plaque progression/regressionSeminal studies on coronary disease regression sug-
gested the sole capability of statins of inhibiting the
progression of atherosclerotic disease and not of
promoting plaque regression.15 Because the usage of
IVUS proved otherwise,16 this apparent failure has
probably to be ascribed to the inability of coronary
angiography to assess atherosclerotic plaque changes
beyond vessel lumen. Yet, the REVERSAL trial,17 dem-
onstrated either a bzero progressionQ or a progression of
intermediate atherosclerotic plaques after 18-month
treatment with atorvastatin or with pravastatin, respec-
tively. In our study instead, we reported a significant
volumetric reduction of intermediate plaques not sub-
jected to invasive treatment with simvastatin in com-
parison with controls (�10% vs +9%). These results are
in agreement with those of Jensen et al,18 who found a
plaque regression of the same entity (�6.3%, as assessed
by IVUS) after 12-month treatment with simvastatin.
Cholesterol lowering reduces the accumulation of
macrophages into the atherosclerotic plaque and the
subsequent degradation of collagen, thus conferring
stability to the coronary lesion.8 -19
However, the plaque regression induced by simvas-
tatin cannot be explained by lipid lowering alone. In
fact, our study group was constituted only by
normocholesterolemic patients, who showed no ex-
treme reduction in total and LDL cholesterol by
treatment. Apparently, pleiotropic effects are mole-
cule-specific rather than class-specific. Simvastatin,
because of its lipophylic features, is entitled to many
pleiotropic effects, such as to preserve endothelial
function, to exert an anti-inflammatory and antiproli-
ferative action, to maintain the fibrinolytic imbal-
ance.8,20 Qualitatively, it appears that statin treatment
turns coronary plaques toward a hyperechogenic
appearance21 which, in term, is caused by a conver-
sion from a lipidic to a fibrotic pattern.22
The fate of atherosclerotic plaques beyond the stentIs there still a role for adjunctive antiproliferative after
invasive treatment in the DES era?
Our study demonstrated that, mirroring the behavior
of intermediate plaques not subjected to invasive
treatment, in simvastatin-treated patients, also peristent
plaques showed a volumetric reduction in both
absolute (�4.0 F 4.0 vs +1.6 F 3.8 mm3/mm) and
percent values (�14% F 10% vs +6% F 12%). Both
plaque fibrosis and shrinkage, and inhibition of out-
growing intimal hyperplasia can be possibly advocated
as the reasons for our findings.
As recently demonstrated by Degertekin et al6 by
means of IVUS, DES almost abolishes 8-month intimal
hyperplasia in comparison with bare metal stents (0.7 vs
33 mm3). On the other hand, they have no significant
impact on the regression/progression of the preexisting
atherosclerotic plaque surrounding the stent (+3.4% vs
+2.5%). Some authors have speculated that, because of a
slower healing response in human beings than in animal
models, DES may only postpone the occurrence of
restenosis,23,24 a fact which has not been fully excluded
by follow-up data available until now.25
Theoretically, a huge plaque burden surrounding the
stent could contribute to a late catch-up of ISR, thus

American Heart Journal
Volume 149, Number 3Petronio et al 525
jeopardizing the long-term efficacy of DES implantation.
At least for bare metal stents, a significant correlation
has been proven by Alfonso et al26 between external
plaque burden and late restenosis: when plaque exceeds
50% of the lumen area, a 4.4-fold increase in ISR
can be predicted.
Clinical implications for simvastatin therapy aftercoronary stenting
When considered globally, our data suggest that the
effect of simvastatin on coronary disease is substantially
untied from the implantation of stents but relies on a
direct action on the atherosclerotic plaque, which takes
place at both stented and nonstented sites, with
mechanisms which go beyond lipid lowering.
Indeed, the clinical potentials for the use of statins in
normocholesterolemic patients undergoing coronary
stenting are several. First, the bpharmacological
debulkingQ which, as we proved, can be obtained with
simvastatin at stented sites could help improving the
long-term efficacy of coronary stenting, even if DES
would be implanted. Yet, simvastatin-eluting stents are
currently under investigation.27 Second, it must not be
underestimated the adjunctive benefit of plaque regres-
sion at nonstented sites that can be achieved with
simvastatin in patients undergoing invasive procedures,
regardless of their baseline cholesterol levels.
Finally, simvastatin can prevent plaque instabilization,
being it related or not to the invasive procedure, and its
sequelae.28,29 Our study was not sized for clinical end
points: nevertheless, we reported a complete safety in
treatment and, although not statistically significant, a
reduction in adverse events by treatment (11% vs 24%).
These findings go along with those of Chan et al30 and
Schomig et al,7 who proved that statins are responsible
for a significant reduction in mortality after either
coronary intervention (2.4% vs 3.6%) or coronary
stenting (2.6% vs 5.6%).
Study limitationsAfter the Heart Protection Study stated that 40 mg
simvastatin is safe and efficacious in a broad popula-
tion,31 20 mg simvastatin can be considered at present a
low-dose regimen, and its pleiotropic effects on intimal
hyperplasia may have been hindered by so in our study.
However, when it was designed, concerns were raised
by our ethics committee about the safety of a more
aggressive statin regimen.32
Because an automated software for gray-scale analysis
is not implemented in our IVUS equipment, we were
unable to perform a qualitative analysis of the athero-
sclerotic plaque, so we can only speculate about the
reason for its volumetric reduction after treatment with
simvastatin (fibrosis, lipid pool reduction, and cell
growth inhibition), both at stented and nonstented sites.
ConclusionsPreventive treatment with 20 mg simvastatin does not
inhibit intimal hyperplasia and ISR, but it does induce a
volumetric reduction of both the atherosclerotic plaque
surrounding the stent and the intermediate atheroscle-
rotic plaques not otherwise treated, in normocholester-
olemic patients undergoing coronary stenting.
References1. Serruys PW, de Jaegere P, Kiemeneij F, et al. A comparison of
balloon-expandable–stent implantation with balloon angioplasty inpatients with coronary artery disease. Benestent Study Group.N Engl J Med 1994;331:489 -95.
2. Fischman DL, Leon MB, Baim DS, et al. A randomized comparisonof coronary-stent placement and balloon angioplasty in thetreatment of coronary artery disease. The Stent Restenosis StudyInvestigators. N Engl J Med 1994;331:496 -501.
3. Hoffmann R, Mintz GS, Dussaillant GR, et al. Patterns andmechanisms of in-stent restenosis. A serial intravascular ultrasoundstudy. Circulation 1996;94:1247 -54.
4. Moses JW, Leon MB, Popma JJ, et al. Sirolimus-eluting stents versusstandard stents in patients with stenosis in a native coronary artery.N Engl J Med 2003;349:1315-23.
5. Stone GW, Ellis SG, Cox DA, et al. One-year clinical results with theslow-release, polymer-based, paclitaxel-eluting TAXUS stent: theTAXUS-IV trial. Circulation 2004;109:1942 -7.
6. Degertekin M, Regar E, Tanabe K, et al. Evaluation of coronaryremodelling after sirolimus-eluting stent implantation by serialthree-dimensional intravascular ultrasound. Am J Cardiol2003;91:1046 -50.
7. Schomig A, Mehilli J, Holle H, et al. Statin treatment followingcoronary artery stenting and one-year survival. J Am Coll Cardiol2002;40:854 -61.
8. Libby P, Aikawa M. Mechanisms of plaque stabilization with statins.Am J Cardiol 2003;91:4B -8B.
9. Corsini A, Raiteri M, Soma MR, et al. Simvastatin but not pravastatinhas a direct inhibitory effect on rat and human myocyte prolifera-tion. Clin Biochem 1992;25:399 -400.
10. Weintraub WS, Boccuzzi SJ, Klein JL, et al. Lack of effect oflovastatin on restenosis after coronary angioplasty. LovastatinRestenosis Trial Study Group. N Engl J Med 1994;331:1331 -7.
11. Walter DH, Schachinger V, Elsner M, et al. Effect of statin therapyon restenosis after coronary stent implantation. Am J Cardiol2000;85:962 -8.
12. Topol EJ, Nissen SE. Our preoccupation with coronary luminology.The dissociation between clinical and angiographic findings inischemic heart disease. Circulation 1995;92:2333 -42.
13. Walter DH, Schachinger V, Elsner M, et al. Statin therapy isassociated with reduced restenosis rates after coronary stentimplantation in carriers of the Pl(A2) allele of the plateletglycoprotein IIIa gene. Eur Heart J 2001;22:587 -95.
14. Lemos PA, Hoye A, Goedhart D, et al. Clinical, angiographic, andprocedural predictors of angiographic restenosis after sirolimus-eluting stent implantation in complex patients: an evaluation fromthe Rapamycin-Eluting Stent Evaluated at Rotterdam CardiologyHospital (RESEARCH) study. Circulation 2004;109:1366 -70.
15. Jukema JW, Bruschke AV, van Boven AJ, et al. Effects of lipidlowering by pravastatin on progression and regression of coronaryartery disease in symptomatic men with normal to moderately

American Heart Journal
March 2005526 Petronio et al
elevated serum cholesterol levels. The Regression Growth EvaluationStatin Study (REGRESS). Circulation 1995;91:2528 -40.
16. Ishikawa K, Tani S, Watanabe I, et al. Effect of pravastatin oncoronary plaque volume. Am J Cardiol 2003;92:975 -7.
17. Nissen SE, Tuzcu EM, Schoenhagen P, et al. Effect of intensivecompared with moderate lipid-lowering therapy on progression ofcoronary atherosclerosis: a randomized controlled trial. JAMA2004;291:1071 -80.
18. Jensen LO, Thayssen P, Pedersen KE, et al. Regression of coronaryatherosclerosis by simvastatin. A serial intravascular ultrasoundstudy. Circulation 2004;110:265 -70.
19. Okazaki S, Yokoyama T, Miyauchi K, et al. Early statin treatment inpatients with acute coronary syndrome: demonstration of thebeneficial effect on atherosclerotic lesions by serial volumetricintravascular ultrasound analysis during half a year after coronaryevent: the ESTABLISH Study. Circulation 2004;110:1061-8.
20. Corsini A, Bellosta S, Baetta R, et al. New insights into thepharmacodynamic and pharmacokinetic properties of statins.Pharmacol Ther 1999;84:413 -28.
21. Schartl M, Bocksch W, Koschyk DH, et al. For the GermanAtorvastatin Intravascular Ultrasound Study Investigators (GAIN)use of intravascular ultrasound to compare effects of differentstrategies of lipid-lowering therapy on plaque volume and compo-sition in patients with coronary artery disease. Circulation2001;104:387 -92.
22. Crisby M, Nordin-Fredriksson G, Shah PK, et al. Pravastatintreatment increases collagen content and decreases lipid content,inflammation, metalloproteinases, and cell death in human carotidplaques: implications for plaque stabilization. Circulation2001;103:926 -33.
23. Virmani R, Kolodgie FD, Farb A, et al. Drug eluting stents: arehuman and animal studies comparable? Heart 2003;89:133 -8.
24. Farb A, Sangiorgi G, Carter AJ, et al. Pathology of acute andchronic coronary stenting in humans. Circulation 1999;99:44 -52.
25. Costa MA, Sousa AGMR, Abizaid AC, et al. Two-year angio-graphic and intravascular ultrasound follow-up after implantation ofSirolimus-eluting stents in human coronary arteries. Circulation2003;107:381-3.
26. Alfonso F, Garcia P, Pimentel G, et al. Predictors and implications ofresidual plaque burden after coronary stenting: an intravascularultrasound study. Am Heart J 2003;145:254 -61.
27. Terumo statin-releasing stent—rationale and animal results.Available at: http://www.tctmd.com/expert-presentations/table-2.html?product_id=3666. TCT 2002.
28. Cannon CP, Braunwald E, McCabe C, et al. Intensive versusmoderate lipid lowering with statins after acute coronary syn-dromes. N Engl J Med 2004;350:1495-504.
29. Pasceri V, Patti G, Nusca A, et al. Randomized trial of atorvastatinfor reduction of myocardial damage during coronary intervention:results from the ARMYDA (Atorvastatin for Reduction of MYocardialDamage during Angioplasty) study. Circulation 2004;110:674 -8.
30. Chan AW, Bhatt DL, Chew DP, et al. Early and sustained survivalbenefit associated with statin therapy at the time of percutaneouscoronary intervention. Circulation 2002;105:691 -6.
31. Heart Protection Study Collaborative Group. MRC/BHF heartprotection study of cholesterol lowering with simvastatin in 20,536high-risk individuals: a randomised placebo-controlled trial. Lancet2002;360:7 -22.
32. Staffa JA, Chang J, Green L. Cerivastatin and reports of fatalrhabdomyolysis. N Engl J Med 2002;346:539 -40.





























