Embed Size (px)
Citation preview

Geochimica et Cosmochimica Acta, Vol. 69, No. 4, pp. 801–823, 2005Copyright © 2005 Elsevier Ltd
Printed in the USA. All rights reserved
doi:10.1016/j.gca.2004.09.006
Solubility and speciation of sulfur in silicate melts: The ConjugatedToop-Samis-Flood-Grjotheim (CTSFG) model
ROBERTO MORETTI1,* and GIULIO OTTONELLO
2
1Istituto Nazionale di Geofisica e Vulcanologia, sezione Osservatorio Vesuviano, Via Diocleziano 328, Napoli, 80124, Italy2Laboratorio di Geochimica, Dip. Te.Ris, Università di Genova, Corso Europa 26, Genova, 16132, Italy
(Received April 2, 2004; accepted in revised form September 9, 2004)
Abstract—A Conjugated Toop-Samis-Flood-Grjotheim (CTSFG) model is developed by combining theframework of the Toop-Samis polymeric approach with the Flood-Grjotheim theoretical treatment of silicatemelts and slags. Electrically equivalent ion fractions are computed over the appropriate matrixes (anionic andcationic) in a Temkin notation for fused salts, and are used to weigh the contribution of the variousdisproportionation reactions of type:
M2/�O�melt� � ½S2�gas� ⇔ M2/�S�melt� � ½O2�gas�
M2/�O�melt� � ½S2�gas� � 3⁄2O2,�gas� ⇔ M2/�SO4�melt�
� being the charge of the generic M�� cation. The extension of the anionic matrix is calculated in theframework of a previously developed polymeric model (Ottonello et al., 2001), based on a parameterizationof Lux-Flood acid-base properties of melt components. Model activities follow the Raoultian behavior implicitin the Temkin notation, without the needs of introducing adjustable parameters. The CTSFG model is basedon a large amount of data available in literature and exhibits a satisfactory heuristic capability, with virtuallyno compositional limits, as long as the structural role given to each oxide holds. The model may be employedto compute gas-melt equilibria involving sulfur and allows computing sulfide and sulfate contents of silicatemelts whenever the fugacity of a gaseous sulfur species and oxygen are known. Alternatively, the modelcalculates the oxidation state of the system (i.e., oxygen fugacity), whenever an analytical determination ofeither sulfide/sulfate or ferrous/ferric ratios in the melt is provided. Calculated sulfide and sulfate capacitiesallow the estimates of sulfur abundance in various melts of geological interest, both under anhydrous andhydrous conditions or, alternatively, of fS2, given fO2 and the bulk sulfur content. In this case, fSO2 and fH2Smay be eventually computed along the water-sulfur-melt boundary provided fH O is known. Copyright
0016-7037/05 $30.00 � .00
2
© 2005 Elsevier Ltd
1. INTRODUCTION
There is a widespread interest in understanding the occur-rence of the different types of sulfur compounds in magma-related environments. The transformations triggered by sulfurspecies, due to changes in the oxidation state, represent acrucial factor also in the cycling of other several elementsrelated to sulfur. Subduction settings, in particular, show thatthe heterogeneous distribution and partitioning of sulfur amongdifferent immiscible phases result in a variable sulfur concen-tration in magmas, strongly influenced by redox conditions(Métrich et al., 1999). The superimposition of different phys-icochemical processes, such as degassing, separation of a S-bearing phase and magma mixing may be eventually proven byisotopic investigations (Marini et al., 1998; De Hoog et al.,2001), but the origin and the way natural sulfur is released isbetter understood only through the assessment of its solubilityand speciation state in magmas. As for other volatile compo-nents in silicate melts, solubility properties of sulfur are ofextreme relevance in modern volcanology, to quantify mag-matic degassing and to reconstruct the P-T-X pathways ofvolatile exsolution. Because of the large number of oxidationstates (from 2� to 6�), sulfur may form several gaseous
* Author to whom correspondence should be addressed ([email protected]).
801
species. In particular, H2S and SO2, together with water andcarbon dioxide, are the most relevant species among naturalmagmatic gases in the C-H-O-S compositional tetrahedron(Gerlach and Nordlie, 1975a,b,c). Although present in volcanicgases in minor amounts with respect to water and carbondioxide, sulfur plays a pivotal role for the monitoring of activevolcanoes, being easily estimated through remote-sensing (Sy-monds et al., 1994 and references therein; Burton et al., 2003)thus furnishing important clues on magma supply and volcanodynamics. Moreover H2S and SO2 constitute an importantredox buffer of separated volcanic gases, especially in andesiticenvironments (Giggenbach, 1996). SO2, in particular, has beenrecognized to be a major gas species which greatly contributesto the pressurization of shallow magmatic chambers, such as inthe case of Pinatubo and St. Helens volcanoes (Westrich andGerlach, 1992; Scaillet and Evans, 1998). The huge exsolutionof sulfur from mixed dacitic and basaltic melts has been qual-itatively recognized to play an important role in triggering thevolcanic explosion of Mt. Pinatubo in 1991 (Kress, 1997a). Theso-called sulfur excess problem (Westrich and Gerlach, 1992;Keppler, 1999) is clearly related to the exsolution of a S-bearing gaseous phase at depth from gas-saturated magmaticreservoirs (Wallace, 2001). The large amount of SO2 emitted inthe atmosphere during large and essentially explosive volcanic
events has been also the object of extensive studies focused on
802 R. Moretti and G. Ottonello
environmental and metereological impacts (Rampino and Self,1984; Graf et al., 1993; Grainger and Highwood, 2003). Hugeamounts of sulfur are also released as SO2 by active volcanoes(Allard et al., 1991, 1994; Carn and Bluth, 2003; McGonigle etal., 2003) characterized by a persistent emission monitoredthrough remote-sensing techniques. The bulk of these volcanicemissions severely affects the atmospheric sulfur budget(Halmer et al., 2002), influencing the atmospheric circulation(Graf et al., 1997, 1998) and the backscatter of solar radiation(Rampino and Self, 1992) through formation of sulfate aerosols(Pinto et al., 1989, Pyle et al., 1996). Modeling of such phe-nomena depends upon a precise knowledge of sulfur solubilityin silicate melts and gas-phase speciation.
The solubility of sulfur is of primary interest also for mate-rial scientists: in metallurgy, for instance, the equilibrium be-tween silicate slag and a metallic phase is important for theremoval of sulfur, whereas in glass technology dissolved so-dium sulfate is often employed as a refining agent in glassbatches.
Similarly, immiscibility between silicate melts and FeS richliquids is a crucial step for the study of separation of S-bearingsolid phases such as pyrrothite (Mavrogenes and O’Neill, 1999)and a liquid Fe-O-S phase (Kress, 1997b, 2000), playing alsoan important role at low pressure in the formation of oredeposits.
Sulfur content in silicate melts is severely affected by meltcomposition (Carroll and Webster, 1994 and referencestherein), especially when moving from basic to acidic liquids.Besides an advancement in the qualitative appraisal of compo-sitional effects, so far, no models of general validity in asufficiently wide P-T-X domain have been proposed. Despitethe importance of the argument, metallurgists provided ther-mochemical models (Reddy and Blander, 1987; Pelton et al.,1993) dealing with at most three components, at 1 bar pressureand at low fO2 (i.e., solubility of sulfur as sulfide). Othercompositionally-weighted expressions of sulfide solubility aredeprived of any heuristic capability (Sosinsky and Sommer-ville, 1986; Young et al., 1992; Beckett, 2002), being based onsimple fitting procedures without any critical assessment of theadopted parametric scale (optical basicity; cf. Duffy, 1992 andreferences therein). O’Neill and Mavrogenes (2002) fitted sul-fide capacities of silicate melt compositions experimentallyinvestigated at 1400°C with an equation of the type:
lnCS � �i,Modifier
XiAi � BFe-TiXFeXTi � const,
where Ai terms are related to the difference in the free energiesof formation of sulfide and oxide components, and BFe-Ti maybe conceived as an interaction parameter for Fe-Ti bearingmelts.
No equations are available in literature for S dissolutionunder oxidizing conditions. More recently, Moretti and Ot-tonello (2003a) have shown that the 1 bar sulfide capacity ofsilicate melts is conveniently parameterized in terms of Flood-Grjotheim electrically equivalent ion fractions when polymericreactivity is also accounted for. In this work it is shown that,not only the sulfide capacity, but also the bulk solubility ofsulfur (sulfide plus sulfate) in silicate melts can be parameter-ized in a similar manner, as anticipated by Moretti et al. (2003).
The developed thermodynamic treatment can be indifferentlyapplied to both simple (i.e., metallurgical slags) and complexsilicate melts. This is an unconditional (but often overlooked)prerequisite of any thermodynamic parameterization of reactiveproperties of a given phase.
2. SULFUR SOLUBILITY AND POLYMERIC NATUREOF MELTS
In the case of components characterized by a reactive solu-bility in silicate melts, such as sulfur species, it is mandatory toassess melt polymerization and oxidation state. Mutual inter-actions of the constituting oxides may be described in theframework of Temkin’s fused salt approach (Temkin, 1945),with ionic species obeying a Raoultian behavior over theirrespective matrixes.
The polymeric nature of silicate melts implies a strongreactivity between charged species and functional groups (cat-ions, free anions and polymeric units or “structons,” accordingto Fraser, 1975a,b, 1977) which, in their turn, determine themixing properties of the phase. It is thus obvious that thedevelopment of a model based on functional reactivity in apolymeric structure is a prerequisite in approaching the solu-bility of sulfur in silicate melts.
The role played by compositional variables in dictating theacid-base properties of silicate melts has been extensivelydiscussed elsewhere (Ottonello, 1983; Ottonello et al., 2001;Moretti, 2002). Here, we simply recall that the speciation ofelements having multiple valence states such as iron and sulfuris governed by the bulk oxidation state of the melt and by itspolymerization, which, in turn, depend upon intensive variablesand acid-base properties of constituting oxides. In the case ofsulfur, for instance one writes:
O2� � 1⁄2S2 ⇔ S2� � 1⁄2O2 (1)
O2� � 1⁄2S2 � 3⁄2O2 ⇔ SO42� (2)
Equilibria 1 and 2 were introduced by Fincham and Richardson(1954) to account for dissolution of sulfur in melt as sulfide andsulfate.
Oxidation state and polymerization are intimately relatedthrough the normal oxygen electrode reaction ½O2 � 2e�NO2�
(Ottonello et al., 2001) and the polarization state of oxygen,conveniently expressed by the Fincham-Richardson notation
Oo � O2� ⇔ 2O� (3)
Equilibrium 3 has been extensively discussed by Toop andSamis (1962a,b), Hess (1971), Fraser (1975a,b, 1977), Ot-tonello (1983, 2001), and Ottonello et al. (2001). In particular,Ottonello et al. (2001) gave a parameterization of equilibrium3 based on optical basicity through the following equation:
K3,melt � exp[4.662 � (�i
XMi���Mi
����j
XTj���Tj
��) � 1.1445]
(4)
where XMi��, �Mi
�� and XTj��, �Tj
�� are respectively atom fractionand basicity moderating parameter of network modifiers andnetwork formers in one mole of the multicomponent melt orslag (Table 1 in Moretti and Ottonello, 2003b, with �H� � 2.50,in agreement with Young et al., 1992). Eqn. 4 establishes a
formal link between acid-base properties of the medium (ex-
803Sulfur solubility in melts
pressed as a balance between formers and modifiers’ basicities)and polymerization constant. In this conceptual framework,activities of oxide components are those of Temkin’s fused saltnotation (Temkin, 1945), which ascribes the activity of themolten component to the activity product of ionic fractionsover the cation and anion matrixes. Therefore, the standardstate adopted is for the completely dissociated form of eachcomponent.
According to equilibria 1 and 2, the concentration of sulfurdissolved in the melt initially decreases with increasing fO2,(equilibrium 1), and then increases again when equilibrium 2 isdominant. The solubility minimum depicted by this dual be-havior represents an important theoretical constraint for thecorrect interpretation of data on S-solubility at oxygen fugac-ities near the QFM buffer (Carroll and Webster, 1994). Nev-
Table 1. Arrhenius coefficients o
M��AO-S,M
A�O-S,M
BO-S,M
B�O-S,M
P5� 2.1987 �247861.7132 �247862.157 �24786
Si4� 2.0492 �3641812.752 �3641812.9577 �36418
Ti4� �4.7934 �16575.9�4.8018 �16575.9
Al3� 2.1838 �289267.7087 �289267.8105 �28926
Fe3� 15.0328 �200006.6613 �20000
Cr3� 0 0Fe2� 1.7787 �15265
�5.910 �8897.95.4414 �152658.4132 �15265
Mn2� �4.4868 �9420.8�1.1437 �8391.7�0.9170 �8391.7�1.0202 �8391.7
Ca2� �5.0645 �6804.2�4.0239 �6804.2�4.229 �6804.2
Mg2� �3.4254 �15181�4.1698 �15181�5.0762 �15181
Na� 14.7310 �1639614.530 �1639611.2533 �16396
K� 3.9100 �1643211.266 �1643210.6606 �16432
H� 0.427 �18764.14.7606 �18764.11.7851 �18764.
* Sources of data are as follows:a Barin and Knacke (1973);b Pelton et al. (1993);c recalculated from Ghiorso et al. (1983);d Barin et al. (1977);e Robie et al. (1978);f Moretti and Ottonello (2003a);g this study.
ertheless, we wish to stress here that constant theoretical slopes
on log(S wt%) vs. log fO2 plots are attained only if fSO2 (or thefugacity of any other S-bearing gas species) and free oxygenactivity, [O2�], are kept constant over the entire fO2 range(Moretti et al., 2003). In fact, by readjusting Eqns. 1 and 2 interms of SO2 one has:
log[S2�] � �3⁄2 logaO2� logaSO2
� logK � log[O2�] (5)
log[SO42�] � logaO2
� logaSO2� logK � log[O2�] (6)
Through the appropriate conversion factors, for a fixed fSO2
and a fixed melt composition (i.e., constant [O2�]), at anytemperature and pressure we have:
M and K�O–S,M (italics) relations.
Sources of data* Remarks
a,cfg
a,dfgfgafgfg
ebfgb
a,dfgbfgbfgafg
a,cfgd
Papale-model adoptedBurnham-model adopted
f KO–S,
logS�wt%� � �3⁄2 logfO2 � c (Reduced region) (5a)

fate solhon tra
804 R. Moretti and G. Ottonello
logS(wt%) � ½logfO2 � c �Oxidized region� (6a)
in which standard state fugacities for gaseous molecular oxy-gen and sulfur dioxide are embodied in the constant term c.Eqns. 5a and 6a also take into account the fact that S2�, O2�
and SO42� speciate over the same matrix, so that the eventual
activity coefficients cancel out.Matching theoretical slopes is a condition always achieved in
equilibrium experiments about sulfur solubility where ingoinggaseous mixtures with fixed stoichiometry are imposed. This ishowever not the natural case, since rarely the gas phase com-position is expected to buffer the system. Eqns. 5a and 6a maybe used—and they had been in literature—to test whether allsulfur is present either as sulfide or sulfate (e.g., Fincham andRichardson, 1954; Carroll and Webster, 1994, and referencestherein).
To give an appraisal of how complicated can be the sulfursolubility in melts, let us consider the data of Gorbachev andBezmen (2002) on a hydrous Ab35Di10An55 melt. On a logS(wt%) vs. log fO2 plot (Fig. 1; side a in upper part) one couldinterpret the increase of sulfur as related to an enhanced dis-solution of sulfur as sulfate. However, the log (S)wt% � log-fSO2 vs. log fO2 plot (side b, upper part of Fig. 1) demonstratesthat all points are disposed on the sulfide side where the
Fig. 1. Bulk sulfur solubility and relative sulfide-sulGorbachev and Bezmen, 2002) and in a remelted El-Chic
theoretical slope is �1.5 (Eqn. 5a). The fact that all points do
not strictly obey such an alignment is simply due to the fact thatdifferent pressures and water content at saturation locally hold.Part (c) and (d) of the same figure show that in the case of ElChichon trachyandesite (Luhr, 1990) theoretical slopes are alsoattained. In this case we considered fS2 instead fSO2. Slopevalues are obviously different according to the reaction stoi-chiometry (see reactions 1 and 2). At every P, T conditionsolubility points on the left side of the c-d plots can be attrib-uted to samples containing sulfide sulfur. The position of thesolubility minimum may also be roughly identified.
In the past, attention has focused on equilibrium 1 because ofthe metallurgical interests in refining steel and alloys fromsulfur. Because sulfur contents of slags is a complex function ofT, fO2 and fS2, through various reasonable assumptions, Fin-cham and Richardson (1954) defined a sulfur solubility func-tion (sulfide capacity) as follows:
CS2� � [S]wt%� fO2
fS2�1⁄2
(7)
The fact that Eqn. 7 descends from equilibrium 1 is evident,being both based on the same sort of considerations which yieldEqn. 5a. In Eqn. 7 [S]wt% is the weight percent of sulfurdissolved as sulfide in a melt or slag. Fincham and Richardson
ubilities in hydrous Ab35Di10An55 melt (T � 1200°C;chyandesite (T � 1000°C; P � 2–4 kbar; Luhr, 1990).
(1954) demonstrated that, within the limits of their measure-

805Sulfur solubility in melts
ments, CS2� is an empirically predictable property. It must bestressed here that the “sulfide capacity” is a pseudoequilibriumconstant and does not, in any way, locate the sulfur solubility atsaturation with another liquid or solid phase. Its nature is verysimilar to that of an equilibrium constant, inasmuch CS2� is aconstant for each composition (T and P being held constant) aslong as [S]wt% is relatively low (i.e., activities of oxide com-ponents in melts are nearly identical to those observed in thesulfur-free melt).
By analogy with Eqn. 7, we may also eventually define a“sulfate capacity” as
CS6� � [S]wt%fO2�3⁄2fS2
�1⁄2 (8)
The compositional control exerted on both sulfur capacities,may be easily understood if we accept that each molten sulfideand sulfate contributes to the solubility mechanisms 1 and 2through specific dissociation constants (Moretti and Ottonello,2003b). For example, in the case of components undergoing abasic dissociation the following reactions (on a one S atombasis) take place:
M2⁄�S ⇔2
�M�� � S2� (9)
M2⁄�SO4 ⇔2
�M�� � SO4
2� (10)
The same applies obviously to the corresponding basic moltenoxides, for which (on a one O atom basis)
M2⁄�O ⇔2
�M�� � O2� (11)
These equilibria involve “ionic quasi-chemical species” andtheir assessment requires standard state transpositions betweenthe standard states of pure melt component and completelydissociated component (Moretti and Ottonello, 2003a). Thethermochemical cycle of Flood and Grjotheim (1952) is theformal way adopted to take into account the actual thermody-namic complexes present in the silicate melt. A description ofthis treatment, following the pathline of Gaskell (2000) is givenin Appendix A.
3. THE CONJUGATED TOOP-SAMIS-FLOOD-GRJOTHEIMTREATMENT OF SILICATE MELTS
The polymeric model of Ottonello et al. (2001) was devel-oped in the framework of the quasi-chemical Toop-Samis ap-proach (Toop and Samis, 1962a,b). Moretti (2004) extended themodel to hydrous compositions, accounting for the evidencesof amphoteric behavior of water (Fraser, 1975a, 2003; Xue andKanzaki, 2003). Based on this approach water is completelydissociated and the following chemical equilibrium takes placebetween its dissociation products:
OH� ⇔ H� � O2� (12)
Only H�do contribute to the polymerization constant K3, en-tering the �i XMi
���Mi�� term in Eqn. 4, whereas OH� anions
(speciating over the anionic matrix as free anions) do not
explicitly operate on the acid-base balance.To define Temkin activities in a multicomponent melt wemust consider mixing of both cationic and anionic constituentsover two distinct sublattices: the cation matrix and the anionmatrix, the latter composed by free ions such as O2�, S2�
SO42� etc. and by the polymeric units or “structons” SiO4
4�,Si2O6
4� etc. The acid-base behavior of each dissolved oxidewill result in a disproportionation between “network formers”and “network modifiers.” Let us see the case of basic compo-nents: the Temkin activity of the generic completely dissoci-ated basic oxide M�O will be, after introduction of sulfurspecies and hydroxyls,
aM�O� � (M��)
� cations��
�� (O2�)
[(O2�) � (OH�) � (S2�) � (SO42�) � � structons]�
(13)
with (O2�) and �structons computed as shown in Ottonello etal. (2001) and Moretti and Ottonello (2003a). Similar equationsapply to sulfide and sulfate components. The Temkin standardstate is the reference standard state of both Toop-Samis andFlood-Grjotheim treatments.
3.1. Oxide-Sulfide and Oxide-Sulfate Disproportionation
Disproportionation equilibria of oxide and sulfide or sulfatecomponents (on a one-oxygen basis) within silicate melts areusually described in terms of reactivity with an externallybuffered gaseous phase, i.e.:
M2⁄�O�m� � 1⁄2S2�g� ⇔ M2⁄�S�m� � 1⁄2O2�g� (14)
M2⁄�O�m� � 1⁄2S2�g� � 3⁄2O2�g� ⇔ M2⁄�SO4�m� (15)
KO�S,M �aM2⁄�S
aM2⁄�O�aO2
aS2
�1⁄2
(14a)
KO�SO4,M �aM2⁄�SO4
aM2⁄�O�aO2
�3⁄2aS2
�1⁄2� (15a)
Although equilibria (14) and (15) are referred to some partic-ular oxide-forming cation M of charge ��, the oxide-sulfideand oxide-sulfate disproportionation in multicomponent slagsor melts may be readily generalized in terms of Flood-Grjotheim thermochemical cycle (Appendix A):
lnKO�S′ � �
i�1
n
XMi��
′ lnCO�S,Mi
anneal. KO�S,Mi(16)
lnKO�SO4
′ � �i�1
n
XMi��
′ lnCO�SO4,Mi
anneal. KO�SO4,Mi(17)
where X= are the electrically equivalent ion fractions, i.e.:
XMi��
′�
�i�nMi
��
�i�nMi
��
(18)
and CO-S,Mi
anneal and CO-SO4Mi
anneal are constants (entropies of annealing)accounting for energy gaps relating different standard states
(see later on and Appendix A). Electrically equivalent ion
806 R. Moretti and G. Ottonello
fractions are obviously referred to the appropriate matrix, eithercationic or anionic.
Let us now consider the single oxide-sulfide and oxide-sulfate disproportionation equilibria in the context of the poly-meric approach. As we mentioned, the reference state conditionfor M2/�O, M2/�S and M2/�SO4 consistent with the Toop-Samisdevelopment is that one of complete dissociation (either basicor acidic) of oxide, sulfide and sulfate components in solution,in which the (Temkin model) activity is one. Nevertheless, forsome particular cation M�� thermodynamic equilibria, referredto the melt component at P and T of interest as the standardstate, demand that:
aMv�aOmelt2�
aM2⁄�Omelt
� Kdiss.M2⁄�O;aMv�aSmelt
2�
aM2⁄�Smelt
� Kdiss.M2⁄�S;
aMv�aSO4melt
2�
aM2⁄�SO4melt
� Kdiss.M2⁄�SO4(19)
although the Temkin state of reference is such that Kdiss.M2/�O,
Kdiss.M2/�Sand Kdiss.M2/�SO4
reduce to one. We may address thisapparent inconsistency to an energy gap between the standardstate of completely dissociated (Temkin model) oxide, sulfideand sulfate components, for which
�M2⁄�O,melt � �*M2⁄�O,melt
� RTln�aMv� · aO2�� (20)
�M2⁄�S,melt � �*M2⁄�S,melt
� RTln�aMv� ·aS2�� (21)
�M2⁄�SO4,melt � �*M2⁄�SO4,melt
� RTln�aMv� · aSO42�� (22)
and the true molten oxide, sulfide and sulfate components at Tand P of interest, for which
�M2⁄�O,melt � �M2⁄�O,melt0 � RTlnaM2⁄�O,melt (23)
�M2⁄�S,melt � �M2⁄�S,melt0 � RTlnaM2⁄�S,melt (24)
�M2⁄�S,melt � �M2⁄�SO4,melt0 � RTlnaM2⁄�SO4,melt (25)
We have thus
exp��*M2⁄�O,melt
� �M2⁄�O,melt0
RT��
aM2⁄�O,melt
aMv� · aO2�
� Kdiss.M2⁄�O (26)
exp��*M2⁄�S,melt
� �M2⁄�S,melt0
RT��
aM2⁄�S,melt
aMv� ·aS2�
� Kdiss.M2⁄�S (27)
exp��*M2⁄�SO4,melt
� �M2⁄�SO4,melt0
RT��
aM2⁄�SO4,melt
aMv� ·aSO42�
� Kdiss.M2⁄�SO4
(28)
Oxide-sulfide and oxide-sulfate equilibria between melt and
components and a gaseous phase demand thus:(S2�)
(O2�)� KO�S,M exp
��M2⁄�S,melt0 � �M2⁄�O,melt
0 � �*M2⁄�O,melt
� �*M2⁄�S,melt
RT�� aS2
aO2
�1⁄2
� K′O�S,M� aS2
aO2
�1⁄2
(29)
(SO42�)
(O2�)� KO�SO4,M exp
��M2⁄�SO4,melt0 � �M2⁄�O,melt
0 � �*M2⁄�O,melt
� �*M2⁄�SO4,melt
RT�� aS2
1⁄2aO2
3⁄2
� KO�SO4,M′ aS2
1⁄2aO2
3⁄2 (30)
Since the amount of free oxygen is established through poly-meric equilibria by the Temkin activity of dissolved oxides,sulfides and sulfates (Eqn. 13), the molar amounts of sulfideand sulfate per unit mole of melt are also readily established(activity coefficients on the cation matrix cancel out and thoseof sulfide and sulfate ions and free oxygen in the anion matrixmay be assumed to be identical within a reasonable approxi-mation).
Evaluation of the bulk oxide-sulfide and oxide-sulfate dis-proportionation constants (Eqns. 16 and 17; K= terms in Eqns.29 and 30) requires the knowledge of the individual Gibbs freeenergy of reaction in single component equilibria at the variousT of interest. Thermodynamic data are insufficient to assesssuch amounts for all components of interest in this study.Moreover, the energy gaps between pure liquid oxides andsulfides and the Temkin-model potential at standard state ofreference must be accounted for. The oxide-sulfide and oxide-sulfate thermodynamic constants may be thus treated as inde-pendent variables in a nonlinear minimization routine thatcompares, through the appropriate stoichiometric conversions,the computed sulfide and sulfate solubility with measured “sul-fide and sulfate capacities.”
3.1.1. Single Component Oxide-Sulfide Equilibria
Deconvolution of the chemistry of the investigated systemsinto network formers and network modifiers was carried out asanticipated by Ottonello et al. (2001): P5�, Al3�, Fe3� arenetwork formers and speciate over the anionic matrix as long asthey are counterbalanced by network modifiers in originatingcomplexes of the type
�4 � v
�M��Tv��O4
4�
with M � network modifier (Na�, K�, Mg2�, Ca2�, Mn2�,Fe2�, H�, and eventually Fe3�).
Since in most circumstances only total iron is reported (e.g.,FeOtot), the appropriate FeII/FeIII ratio was first computedthrough the thermobarometric function described in Ottonelloet al. (2001) to ensure a complete internal consistency of themodel.
Oxide-sulfide disproportionation equilibrium constants (see
Eqn. 14a) were then allocated for all oxides of interest as
807Sulfur solubility in melts
described in Moretti and Ottonello (2003a) and the ensuingequilibrium constant were expanded in terms of an ArrhenianT-dependence:
ln KO�S,Mi
′ � AO�S,Mi
′ � BO�S,Mi
′ ⁄ T (31)
where KO�S′ accounts for the CO-S,Mi
anneal term in Eqn. 16, embodiedin the entropic AO�S
′ term. Although computational details maybe found in Moretti and Ottonello (2003a), it is worth to recallhere that guess values of slope and intercept coefficients A=, B=were first obtained from the examination of Gibbs free energiesof reaction between pure liquid M2/�O and M2/�S and thegaseous phase at various T conditions (Eqn. 17 rewritten forliquid components instead of molten ones). This implies that, atfirst sight, relaxation effects in the annealing of pure liquids inthe melt phase were neglected. This is practically equivalent toassume that differences in the standard states of pure liquid andpure melt components reduce to zero, hence the exponentialterm in Eqn. 29 reduces to one and KO�S,M
′ � KO-S,M.For a generic M2/�S salt, data for the liquid state of aggre-
gation are available in thermochemical compilations from T offusion to some high T (Barin and Knacke, 1973; Robie et al.,1978; Ghiorso et al., 1983). Gibbs free energies of metastable(supercooled and/or superheated) “liquid” M2/�S in a wider Trange may be obtained from the isobaric heat capacity of pureliquid M2/�S by applying
HMS,l � HMS,l,Tf
0 � �Tf
T
CpdT (32)
SMS,l � SMS,l,Tf
0 � �Tf
T
Cp
dT
T(33)
GMS,l � HMS,l,Tf� TSMS,l (34)
Individual exchange reactions of the type
½SiO2,liq � ½S2,gas ⇔ ½SiS2,liq � O2,gas (35)
resulted in lnK vs 1/T regressions of the type:
ln KO�S,Si � 2.0492 � 36418 ⁄ T (36)
which give us AO�S,Miand BO�S,Mi
parameters for each individ-ual oxide-sulfide exchange reaction referred to the pure liquidcomponent standard state. Stemming from these rough prelim-inary assessments, our estimates were further refined through anonlinear minimization technique involving a large data set(1081 anhydrous compositions in the T range 1100 to 1650°C,i.e., 354 exceeding those used in Moretti and Ottonello, 2003a)for a better appraisal of the actual annealing entropy,�SO�S,Mi
annealing � R · CO-S,Manneal � R�AO�S,Mi
′ � AO�S,Mi�, which is re-
quired in the standard state transposition between “pure liquidcomponent” to “pure melt component” (Appendix A).
The nonlinear minimization was conducted with a MonteCarlo exploration of the model space followed by SteepestDescent and Gradient Migration techniques (James and Roos,1977). The adopted misfit function was a chi-squared summa-
tion of logarithmic terms in the form:� �2 �
�logCS2�,c
CS2�,o�2
logCS2�,o
(37)
where CS2�,o is the observed sulphide capacity and CS2�,c is thecalculated sulfide capacity:
CS2�,c � KO�S,M′ �O2��� f°O2
f°S2�1⁄2
MS �moloxides (38)
with�moloxides � molecular summation of all oxides per 100
gr. of substance,MS � atomic weight of sulfur,
f°O2 and f°S2 � 1-bar standard state fugacities of gaseouscomponents.
Although the adopted database is quite extensive, the pre-liminary exploration of the model space showed that the vari-ous AO�S,M
′ ,BO�S,M′ parameters cannot be treated as truly inde-
pendent variables. The investigated temperatures span a narrowrange and the contouring of the misfit function indicates strongcorrelation between AO�S,M
′ ,BO�S,M′ terms, as shown for example
in Figure 2. The slope coefficients BO�S,M′ were then assumed to
be coincident with the BO�S,M terms (i.e., no enthalpic contri-butions involved in the standard state transposition) and theminimization was conducted on the entropic AO�S,M
′ terms(Table 1).
In the case of the H2O component, A and B parameters foroxide-sulfide exchange reaction were calculated consideringthe gas phase homogeneous equilibrium:
H2O(g) � ½S2 ⇔ H2S(g) � ½O2 (39)
The model furnishes A= � 14.7606 when estimating the H2Oamount in melt through the model of Papale (1997), or 11.7851if the Burnham’s model is applied (Burnham, 1975; Eggler andBurnham, 1984; Burnham and Nekvasil, 1986). B= is againassumed to be coincident with B, that is �18764.
3.1.2. Single Component Oxide-Sulfate Equilibria
The conceptual framework is similar to oxide-sulfide dispro-portionations, but with two main limits arising in the case of
Fig. 2. Contouring of the misfit function (Eqn. 37) for Mg2�
ion.
oxide-sulfate equilibria of type (15):

808 R. Moretti and G. Ottonello
i) very few thermodynamic data exist for pure liquid com-ponents M2/�SO4, so that Gibbs free energies of reaction be-tween pure liquid M2/�O and M2/�SO4 may be directly esti-mated only for Mg2�, Na� and K�;
ii) the database about sulfate solubility is not as extended asthat of sulfide. Only 221 data may be found in literature whenone disregards extrapolated data and experiments not reportingfugacities of gaseous species or related parameters (such ascomposition of the ingoing gas flux).
For the thermodynamic properties of liquid sulfates, currentcompilations report data about liquid sodium, potassium andmagnesium sulfate between, respectively, 1157 to 2000°C,1342 to 1700°C and 1400 to 2000°C (Barin and Knacke, 1973;Barin et al., 1977).
Maintaining the same Arrhenian T-dependency adoptedfor oxide-sulfide disproportionations, we used the sametechnique of nonlinear minimization, solving first for bina-ries and ternaries in the silica-alkalies system, stemmingfrom guess values of AO-SO4,Na,BO-SO4,Na,AO-SO4,K,BO-SO4,K
based on the thermochemical compilation of Barin andKnacke (1973). The choice to begin with these systems wasdictated by the consideration that they are quite detailed interms of i) temperature, ii) redox conditions in the oxidizeddomain, iii) silica concentration. In this way we settledvalues of AO�SO4,M
′ , BO�SO4,M
′ for silicon, sodium and potas-sium. Obviously A= and B= parameters for silicon wereunconstrained, as a result of the lack of thermodynamic datain literature for the component Si(SO4)2.
Also for oxide-sulfate disproportionation a preliminary ex-ploration of the model space showed that the various AO�SO4,M
′ ,BO�SO4,M
′ parameters for alkalies cannot be considered as trulyindependent variables. The slope coefficients BO�SO
′ for alka-
Table 2. Arrhenius coefficients of KO-
M��
AO-SO4,M
AO�S,M′
BO-SO4,M
BO�S,M′
P5� 0. 0.Si4� �26.8836 77291.1Ti4� �22.6614 98230Al3� �31.0955 85879.8Fe3� �31.9273 85879.8Cr3� 0 0Fe2� �42.0516 99540.8Mn2� �51.4008 106414.1Ca2� �43.5173 107754.9Mg2� �42.881 95831
�36.3719 95831Na� �21.812 100934
�21.6124 100934K� �31.642 100741
14.814 100741H� �37.695 67292
�60.3994 67292�116.805 67292�56.344 67292
�101.959 67292
* Sources of data are as follows:a Barin and Knacke (1973);b Barin et al. (1977);c this study.
4,M
lies and magnesium were then assumed to be coincident with
the BO-SO4,M and the minimization was conducted on the
AO�SO4,M
′ terms (Table 2).In the case of H2O, slope and intercept terms calculated for
the homogeneous gas-phase reaction at 1 bar P conditions,
H2O(g) � 3⁄2O2 � S2 ⇔ H2SO4 (40)
are respectively A � �37.695 and B � 67292. The B= term forH2O (slope of the Arrhenian dependence on T) has been setequal to B, consistently to what done for the other oxidecomponents in the melt phase.
We note a strong difference in the AO�SO4,M
′ terms relative toalkalies (�Sannealing is 1.66 J/mol K for sodium and 386.3 J/molK for potassium), with the A parameter of sodium practicallyunaffected in the transfer from the aggregation state of pureliquid to the melt phase.
To reduce the number of unconstrained parameters and tostabilize the numerical algorithms with respect to composi-tional variations, we adopted the following simplificationwhich guarantees a physically sound solution:
BMO�MSO4
′ � BMO�MS′
�BSi0.5O�Si0.5SO4
′ � BMgO�MgSO4
′ � BNa2O�Na2O4
′ � BK2O�K2SO4
′
4
�BSi0.5O�Si0.5S
′ � BMgO�MgS′ � BNa2O�Na2S
′ � BK2O�K2S′
4� const
(41)
Eqn. 41 simply states that the enthalpy of the various (iso-coulombic) sulfide-sulfate exchange reactions (M2/�S �2O N M SO may be approximated to a constant value
d KO�SO4,M′ KO�SO4,M
′ (italics) relations.
rces of data* Remarks
cccccccccacacacbc Papale-modelc Papale-model � volumetric annealingc Burnham-modelc Burnham-model � volumetric annealing
SO4,M an
Sou
2ó 2/� 4)
(114 kJ/mol), independently of the coordinating metal cation

rding t
809Sulfur solubility in melts
involved in reaction, in line to what already observed forsolid compounds (Jacob and Jyengar, 1982). We could fix inthis way the B= parameter for important oxide-sulfate cou-ples, such as those involving aluminum, calcium, titanium,manganese and divalent iron. Having fixed these parameters,we approached AO�SO4,M
′ for trivalent iron by imposing thesame B= slope of aluminum. Considered the structural simil-itude between aluminum and trivalent iron in melts, we
Fig. 3. Model performance. (a,b) bulk reproducibility offor hydrous compositions. (c,d) bulk reproducibility of sulEggler and Burnham (1984) and Burnham and Nekvasilsulfate solubility adopting either the Papale or Burnhamannealing entropies (i.e., annealing volumetric terms acco
preferred to follow this additional step since B= for the ferric
oxide-sulfide couple is a completely unconstrained parame-ter (Moretti and Ottonello, 2003a; see also Table 1). Phos-phorous and chromium were not treated due to lack of data.
3.1.3. Details on Model Extension to P � 1 bar
To define the solubility of sulfur at pressure, we must ac-count for volume terms in the Flood-Grjotheim thermochemi-
and sulfate solubility adopting the model of Papale (1997)sulfate solubility adopting the model of Burnham (1975),for hydrous compositions. (e,f) bulk reproducibility of
ls for hydrous compositions and assuming P-dependento Maxwell equation [Eqn. 44]).
sulfidefide and
(1986)mode
cal cycle. For molten oxides, we adopted the volumes given by

810 R. Moretti and G. Ottonello
Lange and Carmichael (1990) and Lange (1994). Volumes ofwater and sulfur trioxide were computed through a second-order regression of molar volumes against optical basicity(Moretti, 2002), which gives VSO3 � 57.71 cm3/mol and VH2O
� 16.44 cm3/mol at 1400°C. An optical basicity (�) value of0.33 was used for SO3 (Young et al., 1992). Volumes of puremolten sulfates were calculated isometrically as
VM2⁄��SO4� VM2⁄��O � VSO3
Molar volumes of sulfide components were obtained applying:
�VSO4�S2� VM2⁄��SO4
� VM2⁄��S � 41(cm3 ⁄ mol) (42)
This constant term was obtained by minimizing residualsbetween calculated and observed sulfide capacities of 61water-bearing compositions in the sulfide domain. Such avalue is akin to the average volume difference (40 cm3/mol)between fictive super-cooled molten components, assumingnegligible volume of melting at 298.15 K for sulfides (i.e.,VMS,crystal,298.15 VMS,melt,298.15; volumes of solid sulfidestaken from Naumov et al., 1971).
Figure 3 (b,c,e) depicts model results in terms of sulfatecapacity (log10 scale) for the hydrous database. The trend alongthe identity line is maintained, but some bias is visible at highCso4. Based on the observed biases, it could be plausible toinfer the existence of a P-dependent volumetric annealing termfor the various reactions of type (18). In fact, on the basis of the3rd Maxwell thermodynamic relationship we have:
� �Sannealing
P �T
� ��VP.Tannealing��P.T
annealing (43)
Sannealing, P,T � Sannealing,1, T � ��VP,Tannealing��P,T
annealing(P � 1)
� � (P � 1) (44)
Through the linear approximation of Eqn. 44, we assume theannealing reaction volume (�VP,T
annealing) times the thermal ex-pansion coefficient of annealing (��P,T
annealing) to be constant,with � 3.1 · 10�2 J/(mol · K · bar) by adopting the Papale(1997) model of water solubility and � 2.87 · 10�2 J/(mol ·K · bar) by adopting the Burnham model (Burnham, 1975;Eggler and Burnham, 1984; Burnham and Nekvasil, 1986). Inprinciple solubility data contain all the information needed torecast volumetric annealing terms for each oxide-sulfate cou-ple, provided the availability of a quite large database, wellcharacterized for all quantities involved in the definition ofsulfur capacities. In the case of sulfate-oxide equilibria, due topaucity of data and the high uncertainty on logfS2 estimates(Luhr, 1990) we prefer to adopt a unique volumetric annealingterm for all oxide-sulfate exchange equilibria. Assuming an-nealing terms to be P-dependent improves substantially thereproducibility.
Oxide-sulfide equilibria do not seem to require any signifi-cant volumetric contribution to the annealing entropy of reac-tion. Computational details about the adopted database forhydrous sulfide-bearing compositions (Clémente, 1998; Luhr,
1990) can be found in Moretti (2002).4. SULFUR SOLUBILITY
4.1. Sulfide and Sulfate Capacities
Figure 3 resumes computational results in terms of calcu-lated vs. observed sulfide and sulfate capacities for all investi-gated compositions.
In the case of anhydrous compositions, the mean error insulfide capacity is 0.2 log units and the correlation coefficientR2 is 0.946. More important, the error propagation is restrictedfor most samples within � 20%, independently of the localvalue attained by the computed magnitude, and does not evi-dence any systematic shift. A remarkable fit is displayed be-tween calculated and observed sulfate capacities (log scale, Fig.3b,d,e,f), with departures from the identity line within the 10%.
A more thorough statistical treatment of the data through aquantile-quantile representation (Fig. 4) shows that significantdepartures from identity in anhydrous sulfide-bearing systemstake place at the lowest Csulfide values. It is likely that depar-tures from identity depend upon analytical errors which are themore important i) the lower is the actual sulfide concentrationin solution, and ii) the higher is the so-called sulfurizing po-tential, fS2/fO2,of the equilibrium gas mixture. The data show-ing deviations at about �5.5 in the quantile-quantile represen-tation of Figure 4 belong to a single experimental study(Haughton et al., 1974) but involve only data collected atlogfO2 � �9.31, log fS2 � �0.98 and not all the data set givenby the Authors. The observed deviation is thus likely due toundetected variations of the sulfurizing potential or may even-tually be ascribed to analytical errors in sulfur determination.Inconsistencies in this subset were already recognized byO’Neill and Mavrogenes (2002).
4.2. Sulfur Solubility
It is now of interest to see the model reproducibility in termsof simple sulfur content, also compared to previous models.Since the amount of sulfur is calculated by means of Eqns. 29and 30, its assessment reflects—especially on a linear scale—all uncertainties encountered in the determination of molecularoxygen and sulfur fugacity in the equilibrated gas-phase. Ingeneral, the model reproduces pretty well the preferential as-
Fig. 4. Quantile-quantile representation of measured vs. observedsulfide capacity populations (anhydrous systems).
sociation of sulfide with network modifiers. In particular Figure
02) eig
811Sulfur solubility in melts
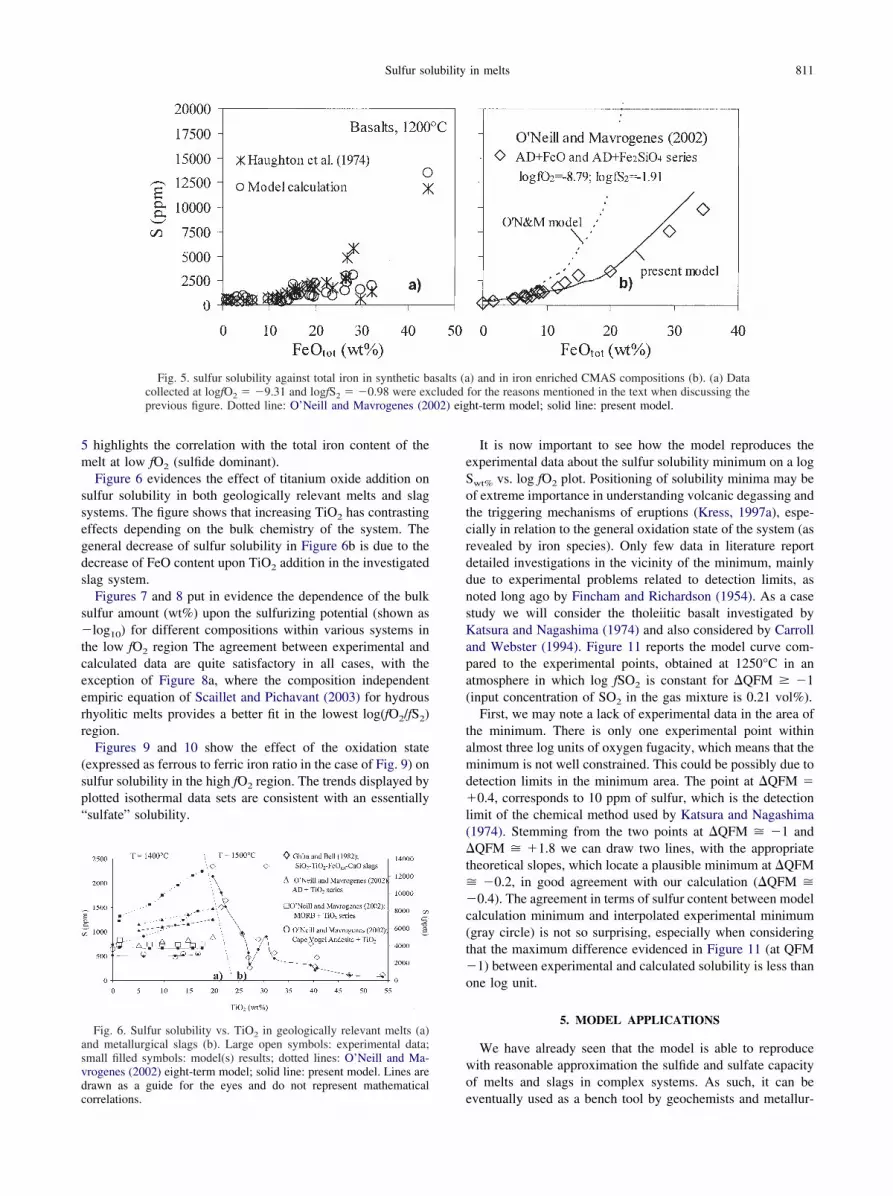
5 highlights the correlation with the total iron content of themelt at low fO2 (sulfide dominant).
Figure 6 evidences the effect of titanium oxide addition onsulfur solubility in both geologically relevant melts and slagsystems. The figure shows that increasing TiO2 has contrastingeffects depending on the bulk chemistry of the system. Thegeneral decrease of sulfur solubility in Figure 6b is due to thedecrease of FeO content upon TiO2 addition in the investigatedslag system.
Figures 7 and 8 put in evidence the dependence of the bulksulfur amount (wt%) upon the sulfurizing potential (shown as�log10) for different compositions within various systems inthe low fO2 region The agreement between experimental andcalculated data are quite satisfactory in all cases, with theexception of Figure 8a, where the composition independentempiric equation of Scaillet and Pichavant (2003) for hydrousrhyolitic melts provides a better fit in the lowest log(fO2/fS2)region.
Figures 9 and 10 show the effect of the oxidation state(expressed as ferrous to ferric iron ratio in the case of Fig. 9) onsulfur solubility in the high fO2 region. The trends displayed byplotted isothermal data sets are consistent with an essentially“sulfate” solubility.
Fig. 5. sulfur solubility against total iron in synthetic bcollected at logfO2 � �9.31 and logfS2 � �0.98 were exprevious figure. Dotted line: O’Neill and Mavrogenes (20
Fig. 6. Sulfur solubility vs. TiO2 in geologically relevant melts (a)and metallurgical slags (b). Large open symbols: experimental data;small filled symbols: model(s) results; dotted lines: O’Neill and Ma-vrogenes (2002) eight-term model; solid line: present model. Lines are
drawn as a guide for the eyes and do not represent mathematicalcorrelations.It is now important to see how the model reproduces theexperimental data about the sulfur solubility minimum on a logSwt% vs. log fO2 plot. Positioning of solubility minima may beof extreme importance in understanding volcanic degassing andthe triggering mechanisms of eruptions (Kress, 1997a), espe-cially in relation to the general oxidation state of the system (asrevealed by iron species). Only few data in literature reportdetailed investigations in the vicinity of the minimum, mainlydue to experimental problems related to detection limits, asnoted long ago by Fincham and Richardson (1954). As a casestudy we will consider the tholeiitic basalt investigated byKatsura and Nagashima (1974) and also considered by Carrolland Webster (1994). Figure 11 reports the model curve com-pared to the experimental points, obtained at 1250°C in anatmosphere in which log fSO2 is constant for �QFM � �1(input concentration of SO2 in the gas mixture is 0.21 vol%).
First, we may note a lack of experimental data in the area ofthe minimum. There is only one experimental point withinalmost three log units of oxygen fugacity, which means that theminimum is not well constrained. This could be possibly due todetection limits in the minimum area. The point at �QFM ��0.4, corresponds to 10 ppm of sulfur, which is the detectionlimit of the chemical method used by Katsura and Nagashima(1974). Stemming from the two points at �QFM �1 and�QFM �1.8 we can draw two lines, with the appropriatetheoretical slopes, which locate a plausible minimum at �QFM �0.2, in good agreement with our calculation (�QFM �0.4). The agreement in terms of sulfur content between modelcalculation minimum and interpolated experimental minimum(gray circle) is not so surprising, especially when consideringthat the maximum difference evidenced in Figure 11 (at QFM�1) between experimental and calculated solubility is less thanone log unit.
5. MODEL APPLICATIONS
We have already seen that the model is able to reproducewith reasonable approximation the sulfide and sulfate capacityof melts and slags in complex systems. As such, it can be
) and in iron enriched CMAS compositions (b). (a) Datafor the reasons mentioned in the text when discussing theht-term model; solid line: present model.
asalts (acluded
eventually used as a bench tool by geochemists and metallur-

awn as
812 R. Moretti and G. Ottonello
gists involved in the characterization of S-bearing systemsalso.1 To display the utility of the developed model in assistingalso volcanological investigations we focus here the attentionon two model by-products: the ability of depicting the specia-tion state of sulfur in complex silicate melts and to enlighten tosome extent the specific role of water in bulk sulfur solubility.
5.1. Assessment of the Speciation State of Sulfur inComplex Silicate Melts
The speciation state of sulfur is typically described throughthe following equilibrium, which is obtained by subtractingequilibrium 2 from equilibrium 1:
S2� � 2O2 ⇔ SO42� (45)
Sulfur speciation in quenched glasses, usually expressed as
nSO42� ⁄ � nStot
1 Copies of an executable F77 FORTRAN package may be obtainedupon request to the senior author. The authors decline any responsi-
Fig. 7. Relative sulfide solubility in a synthetic basalt aa; perhaps the oldest electron microprobe sulfur measuremLarge gray circles: experimental data; small circles: meight-term model; solid line: present model. Lines are drcorrelations.
bility for an improper use and/or application not finalized to purescientific goals.
may be obtained by wet chemical methods (Nagashima andKatsura, 1973; Katsura and Nagashima, 1974) or by means ofelectron microprobe, measuring the shift of the Sk� emission’swavelength (Carroll and Rutherford, 1988). In particular, thepeak shift method is successfully employed for analyzing sulfurspeciation of many quenched natural melts. In many volcanicglasses the amounts of sulfide and sulfate are comparable, asevident by reported S-speciation analyses. The effect of themelt composition is not explicit in Eqn. 45, although it isrequired by the various dissolution reactions of type (14–15).We must however be aware that an equilibrium between“quasi-chemical” species of sulfur requires transposition be-tween the standard state of pure melt component to the standardof completely dissociated melt component (Moretti and Ot-tonello, 2003b). Briefly, Eqn. 45 does not represent an equilib-rium between discrete sulfide and sulfate species as insteadassumed, for example, in Wallace and Carmichael (1994).
Since the CTSFG model furnishes Temkin activities of bothsulfide and sulfate in the melt, it is interesting to compare itsresults to literature data concerning the determination of thesulfide to sulfate ionic ratio and the consequent appraisal ofoxygen fugacity. Let us introduce the experiments of Na-
xperimental data from Buchanan and Nolan (1979) (partailable in literature) and O’Neill and Mavrogenes (2002).results; dotted lines: O’Neill and Mavrogenes (2002)a guide for the eyes and do not represent mathematical
nalog. Eents avodel(s)
gashima and Katsura (1973), Katsura and Nagashima (1974),

813Sulfur solubility in melts
Carroll and Rutherford (1988), Nilsson and Peach (1993), andWallace and Carmichael (1992, 1994) as a case study. Thecomparison is particularly stringent since these data do notbelong to the data set adopted to parameterize the model (withthe exception of the five data from Nagashima and Katsura,1973, and Katsura and Nagashima, 1974). We will compare
Fig. 8. Relative sulfide solubility in a synthetic hydrous rhyolite.Large gray circles: experimental data; small black circles: model(s)results; dotted lines: Scaillet and Pichavant (2003) equation; solid line:present model. Lines are drawn as a guide for the eyes and do notrepresent mathematical correlations.
Fig. 9. S-solubility dependence on the ferrous to ferric iron ratio (i.e.,oxidation state). Data are from Turkdogan and Darken (1961) and St.Pierre and Chipman (1956). Lines connecting model calculated points
(small gray symbols) are drawn as a guide for the eyes and do notrepresent mathematical correlations.experimental fO2 determinations to model calculations basedon reaction 45 and to analytical nSO4
2�⁄nStotratios. To have a
complete characterization of oxides in nominally anhydrousiron-bearing glasses we accepted the analytical value of FeIII/�Fe furnished by wet chemical analyses. In particular, datafrom Nilsson and Peach (1993) were considered after calculat-ing temperature with the empiric relationship given by Wallaceand Carmichael (1992) (their eqn. 1). The FeIII/�Fe ratio ofhydrous glasses of compositions given in Carroll and Ruther-ford (1988) was not available, probably due to highly crystal-lized material not suitable for wet-chemistry analyses. FeIII/�Fe values were thus estimated by the model accounting forthe presence of water as well (Papale, 1997).
Figure 12 puts in evidence the predictive property of themodel in depicting the experimental results of Carroll andRutherford (1988). When accounting for the presence of waterin the melt through the Papale (1997) model (part b of figure)the computed oxygen fugacity is more consistent than theanhydrous estimate (part a of the same figure) with the mea-sured one. The figure shows quite well that the computation issensitive to both pressure and water content, therefore requiring
Fig. 10. S as sulfate solubility dependence on fo21.5 · fs2
0.5 (see Eqn. 8).Data are from Turkdogan and Darken (1961). The line connectingmodel generated points (small gray circles) is drawn as a guide for theeyes and does not represent a mathematical correlation.
Fig. 11. Calculated bulk sulfur solubility in tholeiite, compared toexperimental results of Katsura and Nagashima (1974). The circle
shows the position of the expected minimum, based on theoreticalslopes.
814 R. Moretti and G. Ottonello
a complete characterization of the melt phase. This point wasalready addressed by Carroll and Rutherford (1988) who re-marked that their ��Sk� measurements “should not be taken toindicate that the speciation of dissolved sulfur is unaffected bypressure and melt composition” (Carroll and Rutherford, 1988).The underestimation of logfO2 for the more oxidized samplesfrom Nilsson and Peach (1993) could be partly ascribed to apoor characterization of the hydration state, but it could be alsodue to technical limits in the spectrometric �Sk� method. It isworth to remark, to this purpose, the good agreement with datafrom obtained through gravimetric titration by Nagashima andKatsura (1973) and Katsura and Nagashima (1974) (Fig. 12b).
When referred to as a deviation from a specific fO2 buffer,such as QFM, temperature effects become implicit and theresults of the model on the previously discussed data set displaythe behavior pictured in Figure 13, where data recalculatedthrough the equations of Wallace and Carmichael (1994) andMatthews et al. (1999), both calibrated adopting the presentdata set, are shown for comparative purpose.
The agreement between the results of the CTSFG model andthe equations of Wallace and Carmichael (1994) and Matthews
Fig. 12. Model calculated oxygen fugacities, based on analyticalsulfide/sulfate ratio, compared to experimental results. When accountedfor, water was calculated through the Papale (1997) model.
et al. (1999) is quite satisfactory. The mean error given by the
present model (0.721 log units) is lower than those of Wallaceand Carmichael (1994) (0.738) and Matthews et al. (1999)(0.874). As a major conclusion, we see that the method basedon the determination of the �Sk� X-ray emission shift worksfairly well between QFM-3 and QFM � 2, that is, in a range ofredox conditions which applies to most volcanological prob-lems. Deviations outside this fO2 range may be ascribed to thewell-known nonlinear dependence of the sulfate content on the�Sk� shift (De Hoog, 2000),2 which can be then assumed onlyto some extent. It is worth noting that the 5th order polynomialfunction of Matthews et al. (1999) works well even in thehigh-fO2 range. However, the sigmoid shape of the �Sk� vs.logfO2 diagram (Carroll and Rutherford, 1988) implies that anaccurate correlation of the analytical S speciation with logfO2 ispossible only between QFM and QFM � 2, i.e., well within thevalidity range defined in Figure 13.
If sulfate to sulfide ratios are recomputed through the model(Fig. 14), adopting the experimental fO2, we note that such anonlinear dependence takes place approximately at QFM. Onthe other hand, such a behavior is not displayed by data fromgravimetric analyses of Nagashima and Katsura (1973) andKatsura and Nagashima (1974), as already observed in Fig-ure 12.
The CTSFG model gives a higher sulfate content with re-spect to the Wallace and Carmichael (1994) equation, crossingthe region of variation of experimental points. Differences inthis case are larger than those observed in terms of fO2 due tothe lower algebraic impact of the sulfate to sulfide ratio on thecomputation of fO2 (Moretti and Ottonello, 2003b). Meanerrors are 1.444 log units for the CTSFG model and 1.971 forthe Wallace and Carmichael (1994) equation.
Neglecting the annealing volumetric terms (Eqn. 44) for thefew data at P � 1 bar does not affect appreciably the resultsshown in Figures 12 to 14.
2 Peak magnitudes and also band widths of “limiting” sulfide and
Fig. 13. �QFM estimates of the CTSFG model and of the Wallaceand Carmichael (1994) and Matthews et al. (1999) equations comparedto observations.
sulfate peaks shift progressively, so that their position may not be takenas a reliable estimate of the relative sulfate amount.

815Sulfur solubility in melts
5.2. Sulfide and Sulfate Capacities of Some Natural MeltCompositions
The CTSFG model has been applied to calculate the sulfideand sulfate capacity of some geologically-relevant melts with
Fig. 14. Sulfate/sulfide ratio (log scale) against relative oxygenfugacity. Solid lines locate qualitatively the range of maximum varia-tion of analytical data.
Fig. 15. Sulfide capacity along the melt-water saturation curcalculated with the model of Papale (1997).
compositions constrained at different P and T along the melt-water saturation boundary through the Papale (1997) model ofwater solubility. Application of the model reveals some com-plexities in the compositional dependencies of both sulfide andsulfate capacities. In Figures 15 to 17 we plotted isotherms andisobars of both sulfide and sulfate capacities vs. water contentfor the compositions listed in Table 3.
In Figure 15 we see that an increase in temperature increasesthe sulfide capacity for all compositions. Figure 15 evidencesalso that addition of water to the system enhances sulfidecapacity. Such an increase follows a logarithmic trend, beinglarger at lower water contents. The conformation of logCS
surfaces is similar for all compositions, but in the case of basicmelts (part a and b of figure) high-P isobars have an almostvertical trend, whereas in the case of acidic melts (part c and d)these are dependent on the water content. Things are quitedifferent when sulfate capacity is considered. Figure 16 showsthe sulfate capacity calculated without the introduction of P-dependent volumetric contributions to annealing entropies(Eqns. 43 and 44), while Figure 17 accounts for the P-depen-dence given by these terms.
In Figure 16, we see that sulfate capacity is inversely relatedto temperature, i.e., an increase in T leads to a decrease insulfate capacity. Also the addition of water to the system
ve for four different melt compositions. Water contents

ion cur
816 R. Moretti and G. Ottonello
decreases generally the sulfate capacity, although the modelreveals a more complex compositional control with respect tosulfide capacity. The relationship between water content andsulfate capacity is in fact inverse for all composition, althoughboth andesitic and rhyolitic melts display at every T a maxi-mum in log CSO4 for water contents between 1 and 1.6. wt%.Such a maximum is not due to volumetric effects on meltoxide-sulfate equilibria, which lead to the decrease of sulfatecapacity with increasing pressure, but is implicit in the chem-ical interaction of the H2Om-SO4,m couple. Similarly to sulfidecapacity, sulfate capacity isobars follow a roughly verticaltrend for the more basic compositions.
When sulfate capacity is calculated accounting for volu-metric annealing terms (Fig. 17), we see some importantchanges, especially in the high-P region, where sulfate ca-pacity tends to rise at every T, as an effect of this contribu-tion. Figure 17 shows how this increase is particularlymarked in the case of Kilauean tholeite, arc-basalt andandesite, which display an evident minimum between 4 and6 wt% H2O. The effect of the compositional control of waterdisappears for the andesitic melt (part c of figure), but is stillpresent in the case of rhyolite (part d of figure), which drawsa maximum in sulfate capacity for water contents of �0.4
Fig. 16. Sulfate capacity along the melt-water saturatcalculated with the model of Papale (1997).
wt%, corresponding to 50 bar of pressure. However, it is
worth remarking that the magnitude of this maximum islower than the analogous case of Figure 16.
The computed diagrams of Figures 15 to 17 may be adoptedto estimate values of CS and CSO4 relevant to petrologicalproblems, to retrieve either the amounts of sulfide and sulfate orfO2 and/or fS2. The following general equation may in fact beapplied:
S�wt%, tot � S�wt%, sulfide � S�wt%, sulfate
� CS6�fO23⁄2fS2
1⁄2 � CS2�(fS21⁄2 ⁄ fO2
1⁄2) (46)
6. DISCUSSION OF ACCURACY
Adopting the difference in the basicity moderating param-eters of network formers and network modifiers to quantifythe bulk extent of polymerization reactions in multicompo-nent melts has the same significance as adopting the differ-ence in electronegativity between constituent atoms in pre-dicting the heat of formation of simple components. To thispurpose, Eqn. 4 is rather brutal because it disregards theeffect of intensive variables on the polymerization extent inmulticomponent melts. Nevertheless, it seems to work prop-
ve for four different melt compositions. Water contents
erly also when applied to an extended database (1362 com-

curve fugh the
817Sulfur solubility in melts
positions) involving the bulk solubility of sulfur in silicatemelts at various P,T conditions. Previous attempts of corre-lating optical basicity with the sulfide capacity of silicatemelts (see for example O’Neill and Mavrogenes, 2002, foran application of the model of Young et al., 1992, to geo-logically relevant melts) proved to be unsuccessful becauseof the empirical nature of correlation algorithms, unable totranspose the basicity of the medium into sulfide solubility.
Fig. 17. Sulfate capacity along the melt-water saturationterms are here considered. Water contents calculated thro
Table 3. Compositions employed for calculation o
Oxide Kilauean Tholeite Rhyolite
SiO2 51.85 77.57TiO2 0. 0.07Al2O3 13.98 12.98Fe2O3 2.75 0.07FeO 9.91 0.31MnO 0. 0.MgO 6.81 0.05CaO 11.74 0.52Na2O 2.74 4.07K2O 0.15 4.18P2O5 0. 0.09Source Moretti et al. (2003) Moretti et al. (2
NotesFor such a reason, the use of optical basicity has beenlimited to very simple systems, such as 1-bar silicate slags(Sosinsky and Sommerville, 1986; Beckett, 2002; Young etal., 1992). A more rigorous expression for melt polymeriza-tion in the multicomponent space, based on a full character-ization of the Lux-Flood reactivity of oxides, carried outalong the guidelines established for simple binaries (Ot-tonello and Moretti, 2004), should provide a more precise
or four different melt compositions. Volumetric annealingPapale (1997) model.
e and sulfate capacity under hydrous conditions.
Andesite Arc basalt
59.00 47.760.74 0.98
18.1 16.531.36 1.965.44 5.880.1 0.162.9 4.426.7 12.493.5 2.421.84 1.730.22 0.
Whitford et al. (1979) Métrich et al. (2001)
f sulfid
003)
Average of 45 samples Glass inclusion
818 R. Moretti and G. Ottonello
quantification of the anionic matrix, especially in terms of(O2�).
Summarizing, the reliability of the CTSFG model restsmainly on two aspects:
i) the availability of high-quality solubility data of sulfur inmelts under controlled conditions of pressure, temperature, fO2
and fS2 and, in the case of hydrous systems, a good character-ization of the water content in the melt and of the ferric toferrous ratio,
ii) the availability of sound thermodynamic data on pureliquid and melt species.
The two aspects are obviously related, as the thermodynamicproperties of pure species may be retrieved by the thermo-chemical treatment of solubility data. On the other hand, thelack of basic thermodynamic data on liquid sulfide and inparticular of sulfate components requires the adoption of somegeneralizations. It is worth to remark, to this purpose, that acompletely unconstrained fitting procedure on A= parametersfor CaO, Al2O3, FeO, Fe2O3, MnO, TiO2, and H2O originatesimportant biases, and, although increasing the precision inreproducing the anhydrous part of the database, it gives unre-alistic energetics, especially for CaO-CaSO4 and Al2O3-Al2(SO4)3 exchange reactions.
Again, as most data (and particularly the metallurgical datasets) deal with “capacities,” which represent a combination offS2, [S]wt% and fO2, bulk sulfur solubility estimates are affectedin a complex fashion by the accuracy on the estimates of bothfO2 and fS2 and are resulting thus in larger errors.
It must be stressed that the role of volumetric terms ofannealing is not negligible, as shown by Figures 16 and 17. Amore rigorous treatment is not possible at present, requiring abetter appraisal of partial molar volumes of both oxide andsulfate species and of their topological evolution along thecompositional space of interest.
7. CONCLUSIONS
After having qualitatively assessed the basics of the disso-lution of sulfur as sulfide and sulfate in melts and slags, wehave developed a model (Conjugated Toop-Samis-Flood-Grjotheim model, CTSFG) for sulfur solubility on the basis ofa treatment previously developed for the oxidation state ofsilicate melts (Ottonello et al., 2001) in the framework of theToop-Samis polymeric approach (Toop and Samis, 1962a,b),combining it with the approach of Moretti and Ottonello(2003a) for sulfide capacity.
As remarked by Haughton et al. (1974), and more recentlyby O’Neill and Mavrogenes (2002), “the transposition of sul-fide capacities derived from binary compositions to multicom-ponent melts is complex” (Haughton et al., 1974), implyingcompositional criteria to define CS2� on the basis of oppor-tunely weighted oxide concentrations. These compositionalcriteria for CS2� were assessed by Moretti and Ottonello(2003a) and have been then introduced within a consistentthermodynamic treatment related to chemical equilibria be-tween oxides, sulfides and sulfates, avoiding empirical fittingprocedures and reexpressing reactive properties in terms of theFlood and Grjotheim (1952) thermochemical cycle. This ther-mochemical cycle has been shown to be a powerful tool in
deciphering equilibria between quasi-chemical species inchemically complex multicomponent spaces, since it allowsone to assess the pure components standard state potentialsconsistent both with a Temkin notation for fused salts and witha Toop-Samis polymeric treatment of melt reactivity. Indepen-dent volume terms have been introduced in the thermodynamictreatment for a model extension to high pressures, relevant tovolcanological investigation.
The performance of the CTSFG model, tested on an ex-tended database, indicates that the computed “electricallyequivalent fractions” and the computed molar amounts of freeoxygen (O2�) are sufficiently accurate and internally consistent(independently of the chemical complexity of the investigatedsystem) to allow a sound analysis of sulfur solubility in melts.
The contribution of each oxide-sulfide and oxide-sulfatecouple to the bulk sulfur solubility is weighted by the electricalequivalent ion fractions over both cation and anion matrixes.The central role of silica, as well as of aluminum, alreadyperceived by Vogt (1923; cf. Haughton et al., 1974) almost onecentury ago, is due to its large structural control in the anneal-ing of liquid components in the melt phase. To this purpose, itis interesting to remark that recent analyses of the local struc-ture of S-bearing alkali silicate glasses shows the existence ofsilicon atoms coordinating sulfur atoms instead of oxygen, andof alkali-sulfide coordination groups (Asahi et al., 2003).Therefore the thermochemical approach of the CTSFG modelis consistent with physical evidences at the atomic scale.
Concerning sulfur speciation, the model is consistent withthe gravimetric analyses of Nagashima and Katsura (1973) andKatsura and Nagashima (1974) and predicts fairly well the dataon S speciation in compositions investigated in literature. Nev-ertheless, the analytical trend of sulfur speciation obtained viaelectron microprobe with the method of Carroll and Rutherford(1988) diverges sensibly from theoretical predictions of theCTSFG model at logfO2 � QFM, showing a nonlinear depen-dence on oxygen fugacity. The CTSFG model indicates thatsuch discrepancies do not affect remarkably the calculation ofoxygen fugacity in the range �3 �QFM 2, in which theelectron microprobe-based technique may be safely employed.In general, the model shows heuristic capabilities for predictingoxygen fugacity somewhat better than semiempiric techniques,such as the Wallace and Carmichael (1994) and Matthews etal., (1999) equations. The CTSFG model provides the couplingof Fe and S oxidation states and may be utilized as a fO2
geobarometric tool whenever analytical determinations of bothsulfide and sulfate are available, a fact which may be ofparticular importance in the study of glass inclusions. Themodel may be also adopted as a geobarometer for fS2, providedthe achievement of S saturation (with respect to the gas phase)and an independent estimate of fO2. Some model-generatedplots of sulfide and sulfate capacities have been given for somesystems of petrological interest, with different water contents atsaturation. These data may be usefully employed for calculat-ing SO2 and H2S partial pressures along compositional paths,such as those depicted by a suite of glass inclusions, andquantifying the extent of sulfur degassing of magmatic systems.
The CTSFG model is obviously subjected to further amelio-ration. High-quality data and further experiments under con-trolled conditions of T, P, fO2, fS2, fH2O (and fCO2) areneeded, to better constrain the thermochemical properties of
liquid sulfur components. These will help, in their turn, to
819Sulfur solubility in melts
refine the S-H2O chemical interaction and to define that one ofS with CO2 in a next future.
Acknowledgments—R.M. is very grateful to P. Papale (INGV, Pisa) forhis continuous support, the stimulating discussions and for havingcalculated the water solubility data of compositions investigated in thiswork. The manuscript benefited of the helpful comments of B. Scailletand two other anonymous reviewers. J.K. Russell is acknowledged foreditorial handling. This study has been done partly in the framework ofthe Ph. D. thesis of R.M. at Dipartimento di Scienze della Terra,Università di Pisa, under a grant of Ministero dell’Università e RicercaScientifico-Tecnologica (MURST) of the Italian Government, andpartly with a contribution for basic research by Regione Campania,financed through Regional Law 5/2002.
Associate editor: J. K. Russell
REFERENCES
Abraham K. P. Davies M. W., and Richardson F. D. (1960) Sulphidecapacities of silicate melts. Part II. J. Iron Steel Inst. 196, 309–312.
Abraham K. P. and Richardson F. D. (1960) Sulphide capacities ofsilicate melts. Part I. J. Iron Steel Inst. 196, 313–317.
Allard P., Carbonelle J., Dajlevic D., Le Bronec J., Morel P., RobeM. C., Maurenas J. M., Faivre-Pierret R., Martin D., Sabroux J. C.,and Zettwoog P. (1991) Eruptive and diffusive emissions of CO2
from Mount Etna. Nature 351, 387–391.Allard P., Carbonnelle J., Metrich N., Loyer J., and Zetwoog P. (1994)
Sulphur output and magma degassing budget of Stromboli volcano.Nature 368, 326–330.
Asahi T., Miura Y., Yamashita H., and Aekawa T. (2003) Localstructure analysis of alkali silicate glasses containing sulfur (ab-stract). Geochim. Cosmochim. Acta 67 (18 Suppl.), A26.
Barin I. and Knacke O. (1973) Thermochemical Properties of Inor-ganic Substances. Springer-Verlag.
Barin I., Knacke O., and Kubaschewsky O. (1977) ThermochemicalProperties of Inorganic Substances. Supplement. Springer-Verlag.
Beckett J. R. (2002) Role of basicity and tetrahedral speciation incontrolling the thermodynamic properties of silicate liquids, part 1:The system CaO-MgO-Al2O3-SiO2. Geochim. Cosmochim. Acta66, 93–107.
Brown S. D., Roxburgh R. J., Ghita I., and Bell H. B. (1982) Sulphidecapacity of titania-containing slags. Ironmaking Steelmaking 9,163–67.
Buchanan D. L. and Nolan J. (1979) Solubility of sulfur and sulfideimmiscibility in synthetic tholeiitic melts and their relevance toBushveld-complex rocks. Can. Miner. 17, 483–494.
Burnham C. W. (1975) Water and magmas: A mixing model. Geochim.Cosmochim. Acta 39, 1077–1084.
Burnham C. W. and Nekvasil H. (1986) Equilibrium properties ofgranitic magmas. Am. Mineral. 71, 239–263.
Burton M., Allard P., Muré F. and Oppheneimer C. (2003) FTIRremote sensing of fractional magma degassing at Mount Etna,Sicily. In Volcanic Degassing (eds. C. Oppenheimer, D. M. Pyleand J. Barclay), pp. 281–293. Spec. Publ, 213. Geol. Soc. London.
Carn S. A. and Bluth G. J. S. (2003). Prodigious sulfur dioxideemission from Nyamuragira volcano, D.R. Congo. Geophys. Res.Lett. 30, doi:10.1029/2003GL018465.
Carroll M. R. and Rutherford M. J. (1988) Sulfur speciation in hydrousexperimental glasses of varying oxidation state: Results from mea-sured wavelength shifts of sulfur X-rays. Am. Mineral. 73, 845–849.
Carroll M. R. and Webster J. D. (1994) Solubilities of sulfur, noblegases, nitrogen, chlorine and fluorine in magmas. In Volatiles inMagmas (eds. M. R. Carroll and J. R. Holloway), pp. 231–279.Rev. Mineral. 30, Mineralogical Society of America.
Carter P. T. and MacFarlane T. G. (1957a) Thermodynamics of slagssystems: Part I—The thermodynamic properties of CaO-Al2O3
slags. J. Iron Steel Inst.185, 54–62.Carter P. T. and MacFarlane T. G. (1957b) Thermodynamics of slags
systems: Part II—The thermodynamic properties of CaO-SiO2
slags. J. Iron Steel Inst. 185, 62–66.
Clémente B. (1998) Etude expérimentale et modélisation de la solu-bilité du soufre dans les liquides magmatiques. Ph. D. thesis,Université de Orléans (in French).
Clémente B., Scaillet B., and Pichavant M. (2004) The solubility ofsulphur in hydrous rhyolitic melts. J. Petrol., 4S, 2171-2196.
Danckwerth P. A., Hess P. C., and Rutherford M. J. (1979) Thesolubility of sulfur in high-TiO2 mare basalts. In Proceedings of the10th Lunar Planetary Science Conference,pp. 517–530. LunaryPlanetary Science.
de Hoog J. C. M. (2000) Non-linearity of sulfur peak shift with fractionsulfate sulfur may result in underestimation of measured sulfurconcentrations in glasses and inaccurate oxidation state determina-tions (abstract). Eos 81–48, F1295.
de Hoog J. C. M., Taylor B. E., and van Bergen M. J. (2001) Sulfurisotope systematics of basaltic lavas from Indonesia: Implicationsfor the sulfur cycle in subduction zones. Earth Planet. Sci. Lett.189, 237–252.
Douglas R. W. and Zaman M. S. (1969) The chromophore in iron-sulphur amber glasses. Phys. Chem. Glass 10, 125–132.
Drakalivsky E., Sichen D., and Seetharaman S. (1997) An experimentalstudy of the sulphide capacities in the system Al2O3-CaO-SiO2.Can. Met. Q. 36, 115–120.
Duffy J. A. (1992) A review of optical basicity and its applications tooxidic systems. Geochim. Cosmochim. Acta 57, 3961–3970.
Eggler D. H. and Burnham C. W. (1984) Solution of H2O in diopsidemelts: A thermodynamic model. Contr. Mineral. Petrol. 85, 58–66.
Fincham C. J. B. and Richardson F. D. (1954) The behaviour of sulphurin silicate and aluminate melts. Proc. R. Soc. Lond. A223, 40–62.
Flood H. and Grjotheim T. (1952) Thermodynamic calculation of slagequilibria. J. Iron Steel Inst. 171, 64–80.
Fraser D. G. (1975a) Activities of trace elements in silicate melts.Geochim. Cosmochim. Acta 39, 1525–1530.
Fraser D. G. (1975b) An investigation of some long-chain oxi-acidsystems. Ph.D. thesis. University of Oxford.
Fraser D. G. (1977) Thermodynamic properties of silicate melts. InD.G. Fraser) Thermodynamics in Geology (ed. Reidel), 300–325.
Fraser D. G. (2003) Acid-base properties, structons and the thermody-namic properties of silicate melts (abstract). Geochim. Cosmochim.Acta 67 (18 Suppl.), A103.
Gaskell. (2000) Early model of the thermodynamic behavior of slagsand salts. In Proceedings of the Sixth International Conference onMolten Slags, Fluxes and Salts, Stockholm, Sweden—Helsinki,Finland 12-17 June 2000. Edited by S. Seetharaman and D.U.Sichen. Production coordinator R.E. Aune. © 2000 by Division ofMetallurgy, KTH, Sweden. ISBN 91-7170-606-2. Trita Met 85June 2000 August 2000.
Gerlach T. M. and Nordlie B. E. (1975a) The C-H-O-S gaseous system,Part I: Composition limits and trends in basaltic cases. Am. J. Sci.275, 353–376.
Gerlach T. M. and Nordlie B. E. (1975b) The C-H-O-S gaseous system,Part II: Temperature, atomic composition and molecular equilibriain volcanic gases. Am. J. Sci. 275, 377–394.
Gerlach T. M. and Nordlie B. E. (1975c) The C-H-O-S gaseous system,Part III: Magmatic gases compatible with oxides and sulfides inbasaltic magmas. Am. J. Sci. 275, 395–410.
Ghiorso M. S., Carmichael I. S. E., Rivers M. L., and Sack R. O. (1983)The Gibbs free energy of natural silicate liquids; an expandedregular solution approximation for the calculation of magmaticintensive variables. Contr. Mineral. Petrol. 84, 107–145.
Ghita I. and Bell H. B. (1982) Sulphide capacity of slags containingFeO, CaO, TiO2 and SiO2. Ironmaking Steelmaking 9 (6), 239–243.
Giggenbach W. F. (1996) Chem. composition of volcanic gases. InMonitoring and Mitigation of Volcano Hazards (eds. R. Scarpa andR. I. Tilling), pp. 202–226. Springer.
Gorbachev P. and Bezmen N. (2002) Solubility of sulfur in water-saturated An-Di melts under various conditions (abstract). EMPGIX, Zurich, J. Conf., 7, 40.
Graf H.-F., Kirchner I., Robock A., and Schult I. (1993) Pinatuboeruption winter climate effects: Model versus observation. Clim.Dyn. 9, 81–93.
Graf H.-F-, Feichter J., and Langmann B. (1997) Volcanic sulfur
emissions: Estimates of source strength and its contribution to theglobal sulfate distribution. J. Geophys. Res. 102, 10727–10738.
820 R. Moretti and G. Ottonello
Graf H.-F., Langmann B., and Feichter J. (1998) The contribution ofEarth degassing to the atmospheric sulfur budget. Chem. Geol. 147,131–145.
Grainger R. G. and Highwood E. J. (2003) Changes in stratosphericcomposition, chemistry, radiation and climate caused by volcaniceruptions. In Volcanic Degassing (eds. C. Oppenheimer, D. M. Pyleand J. Barclay), pp. 329–347. Spec. Publ. 213. Geol. Soc. London.
Halmer M. M., Schmincke H.-U., and Graf H.-F. (2002) The annualvolcanic gas input into the atmosphere, in particular into the strato-sphere: A global data set for the past 100 years. J. Volcanol.Geotherm. Res. 115, 511–528.
Haughton D. R., Roedder P. L., and Skinner B. J. (1974) Solubility ofsulfur in mafic magmas. Econ. Geol. 69, 451–467.
Hess P. C. (1971) Polymer model of silicate melts. Geochim. Cosmo-chim. Acta 35, 289–306.
Holmquist S. (1965) Oxygen ion activity and the solubility of sulfurtrioxide in sodium silicate melts. J. Am. Ceram. Soc. 49, 467–473.
Jacob K. T. and Jyengar G. N. K. (1982) Oxidation of alkaline earthsulfides to sulfates: Thermodynamic aspects. Metall. Trans. B 17B,387–390.
James F. and Roos M. (1977) MINUIT: A System for Function Min-imisation and Analysis of Parameter Errors and Correlations.CERN Computer Center, Geneva.
Kalyanram M. R., MacFarlane T. G., and Bell H. B. (1960) The activityof calcium oxide in slags in the systems CaO-MgO-Al2O3-SiO2 at1500°C. J. Iron Steel Inst. 195, 58–64.
Katsura T. and Nagashima S. (1974) Solubility of sulfur in somemagmas at 1 atmosphere. Geochim. Cosmochim. Acta 38, 517–531.
Keppler H. (1999) Experimental evidence for the source of excesssulfur in explosive volcanic eruptions. Science 284, 1652–1654.
Kor G. J. W. and Richardson F. D. (1968) Sulphur in lime-aluminamixtures. J. Iron Steel Inst. 206, 700–704.
Kress V. (1997a) Magma mixing as a source for Pinatubo sulphur.Nature 389, 591–593.
Kress V. (1997b) Thermochemistry of sulfide liquids. I. The systemO-S-Fe at 1 bar. Contr. Mineral. Petrol. 127, 176–186.
Kress V. (2000) Thermochemistry of sulfide liquids. II. Associatedsolution model for sulfide liquids in the system O-S-Fe. Contr.Mineral. Petrol. 139, 316–325.
Lange R. A. (1994) The effect of H2O, CO2 and F on the density andviscosity of silicate melts. In Volatiles in Magmas(eds. M. R.Carroll and J. R. Holloway), pp. 331–369. Rev. Mineral. 30, Min-eralogical Society of America.
Lange R. A. and Carmichael I. S. E. (1990) Thermodynamic propertiesof silicate liquids with emphasis on density, thermal expansion andcompressibility. In Modern Methods of Igneous Petrology (eds. J.Nicholls and J. K. Russell), pp. 25–64. Rev. Mineral. 24, Miner-alogical Society of America.
Luhr J. (1990) Experimental phase relations of water and sulfur-saturated arc magmas and the 1982 eruption of El-Chichon volcano.J. Petrol. 31, 1071–1114.
Marini L., Chiappini V., Cioni R., Cortecci G., Dinelli E., Principe C.,and Ferrara G. (1998) Effect of degassing on sulfur contents and�34S values in Somma-Vesuvius magmas. Bull. Volcanol. 60, 187–194.
Matthews S. J., Moncrieff D. H. S., and Carroll M. R. (1999) Empiricalcalibration of the sulphur valence oxygen barometer from naturaland experimental glasses: Methos and applications. Min. Mag. 63,421–431.
Mavrogenes J. A. and O’Neill H. St. C. (1999) The relative effects ofpressure, temperature and oxygen fugacity on the solubility ofsulfide in mafic magmas. Geochim. Cosmochim. Acta 63, 1173–1180.
McGonigle A. J. S., Oppenheimer C., Hayes A. R., Galle B., EdmondsM., Caltabiano T., Salerno G., Burton M., and Mather T. A. (2003)Sulphur dioxide fluxes from Mount Etna, Vulcano and Strombolimeasured with an automated scanning ultraviolet spectrometer. J.Geophys. Res. 108(B9), doi:10.1029/2002JB002261.
Métrich N., Schiano P., Clocchiatti R., and Maury R. C. (1999) Trans-fer of sulfur in subduction settings: An example from Batan Island(Luzon volcanic arc, Philippines). Earth Planet. Sci. Lett. 167,
1–14.Métrich N., Bertagnini A., Landi P., and Rosi M. (2001) Crystallizationdriven by decompression and water loss at Stromboli volcano(Aeolian Islands, Italy). J. Petrol. 42, 1471–1490.
Moretti R. (2002) Solubilità di volatili nei fusi silicatici con particolareriferimento alle specie dello zolfo: Aspetti teorici e applicazioni allevulcanite etnee. Ph.D. thesis. Università di Pisa (in English).
Moretti R. (2004) Polymerisation, basicity, oxidation state and theirrole in ionic modelling of silicate melts. Ann. Geophys.,in press.
Moretti R. and Ottonello G. (2003a) A polymeric approach to thesulfide capacity of silicate slags and melts. Metall. Mater. Trans. B34B, 399–410.
Moretti R. and Ottonello G. (2003b) Polymerization and disproportion-ation of iron and sulfur in silicate melts: Insights from an opticalbasicity-based approach. J. Non-Cryst. Sol. 323, 111–119.
Moretti R., Papale P., and Ottonello G. (2003) A model for thesaturation of C-O-H-S fluids in silicate melts. In Volcanic Degas-sing ((C. Oppenheimer, D.M. Pyle, and J. Barclay), pp. 81–101.Spec. Publ. 213. Geol. Soc. London.
Nagashima S. and Katsura T. (1973) The Solubility of sulfur in Na2O-SiO2 melts under various oxygen partial pressures at 1100°C,1250°C and 1300°C. Bull. Chem. Soc. Jpn. 46, 3099–3103.
Naumov G. B., Rhyzenko B., and Khodakovsky I. L. (1971) Handbookof Thermodynamic Data. Atomizdat, Moscow.
Nilsson K. and Peach C. L. (1993) Sulfur speciation, oxidation stateand sulfur concentration in backarc magmas. Geochim. et Cosmo-chim. Acta 57, 3807–3813.
O’Neill H. St. C. and Mavrogenes J. A. (2002) The sulfide capacity andthe sulfur content at sulfide saturation of silicate melts at 1400°Cand 1 bar. J. Petrol. 43, 1049–1087.
Ottonello G. (1983) Trace elements as monitors of magmatic processes(1) limits imposed by Henry’s law problem and (2) compositionaleffect of silicate liquid. In The Significance of Trace Elements inSolving Petrogenetic Problems and Controversies (ed. S. Augus-thitis). Theophrastus Publications, Athens, 39–82.
Ottonello G. (2001) Thermodynamic constraints arising from the poly-meric approach to silicate slags: The system CaO-FeO-SiO2 as anexample. J. Non-Cryst. Sol. 282, 72–85.
Ottonello G., Moretti R., Marini L., and Vetuschi Zuccolini M. (2001)On the oxidation state of iron in silicate melts and glasses: Athermochemical model. Chem. Geol. 174, 157–179.
Ottonello G. and Moretti R. (2004) Lux-Flood basicity of binarysilicate melts. J. Phys. Chem. Sol. 65, 1609–1614.
Papadopoulos K. (1973) The solubility of SO3 in soda-lime-silica-melts. Phys. Chem. Glass. 14, 61–65.
Papale P. (1997) Modeling of the solubility of one component H2O orCO2 fluid in silicate liquid, Contr. Mineral. Petrol. 126, 237–251.
Pelton A. D., Eriksson G., and Romero-Serrano A. (1993) Calculationof sulfide capacities of multicomponent slags. Metall. Trans. B24B, 817–825.
Pinto J. P., Turco R. P., and Toon O. B. (1989) Self-limiting physicaland chemical effects in volcanic eruption clouds. J. Geophys. Res.94 (D8), 11165–11174.
Pyle D. M., Beattie P. D., and Bluth G. J. S. (1996) Sulphur emissionsto the stratosphere from explosive volcanic eruptions. Bull. Volca-nol. 57, 663–671.
Rampino M. R. and Self S. (1984) Sulphur-rich volcanic eruptions andstratospheric aerosols. Nature 310, 677–679.
Rampino M. R. and Self S. (1992) Volcanic winter and acceleratedglaciation following the Toba super-eruption. Nature 359, 50–52.
Reddy R. G. and Blander M. (1987) Modeling of sulfide capacities ofsilicate melts. Metall. Trans. B 18B, 591–596.
Richardson F. D. and Fincham C. J. B. (1956) Sulphur in silicate andaluminate slags. J. Iron Steel Inst. 178, 4–15.
Robie R. A., Hemingway B. S., and Fisher J. R. (1978) Thermody-namic properties of minerals and related substances at 298.15 K and1 bar (105 pascal) pressure and at higher temperatures. USGS Bull.1452.
Scaillet B. and Evans B. W. (1998) The June 15, 1991 eruption ofMount Pinatubo. I: Phase equilibria and pre-eruption P-T-fO2-fH2Oconditions of the dacite magma. J. Petrol. 40, 381–411.
Scaillet B. and Pichavant M. (2003) Experimental constraints on vol-
atile abundances in arc magmas and their implications for degas-sing processes. In Volcanic Degassing (C. Oppenheimer, D. M.
821Sulfur solubility in melts
Pyle, and J. Barclay), pp. 23–52. Spec. Publ. 213. Geol. Soc.London.
Sharma R. A. and Richardson F. D. (1962) The solubility of calciumsulphide and activities in lime-silica melts. J. Iron Steel Inst. 200,373–379.
Sharma R. A. and Richardson F. D. (1965) Activities of manganeseoxide, sulfide capacities and activity coefficients in aluminate andsilicate melt. Trans. AIME 233, 1586–1592.
Sosinsky D. J. and Sommerville I. D. (1986) The composition andtemperature dependence of the sulfide capacity of metallurgicalslags. Metall. Trans. B 17B, 331–337.
St. Pierre G. R. and Chipman J. (1956) Sulfur equilibria between gasesand slags containing FeO. J. Metals / Trans. AIME 206, 1474–1483.
Symonds R. B., Rose W. I., Bluth G. J. S. and Gerlach T. M. (1994)Volcanic gas studies: Methods results and applications. In Volatilesin Magmas (eds. M. R. Carroll and J. R. Holloway), pp. 1–60. Rev.Mineral. 30, Mineralogical Society of America.
Temkin M. (1945) Mixtures of fused salts as ionic solutions. ActaPhys.Chim. URSS 20, 411–420.
Toop G. W. and Samis C. S. (1962a) Some new ionic concepts ofsilicate slags. Can. Met. Q. 1, 129–152.
Toop G. W. and Samis C. S. (1962b) Activities of ions in silicate melts.Trans. Met. Soc. AIME 224, 878–887.
Turkdogan E. T. and Darken L. S. (1961) Sulfur equilibria betweengases and calcium ferrite melts. Trans. AIME 221, 464–474.
Vogt J. H. L. (1923) Nickel in igneous rocks. Econ. Geol. 18, 307–353.Wallace P. J. (2001) Volcanic SO2 emissions and the abundance and
distribution of exsolved gas in magma bodies. J. Volcanol. Geo-therm. Res. 108, 85–106.
Wallace P. J. and Carmichael I. S. E. (1994) S speciation in submarinebasaltic glasses as determined by measurements of Ska X-raywavelength shifts. Am. Mineral. 79, 161–167.
Wallace P. J. and Carmichael I. S. E. (1992) Sulfur in basaltic magmas.Geochim. Cosmochim. Acta. 56, 1863–1874.
Westrich H. R. and Gerlach T. M. (1992) Magmatic gas source for thestratospheric SO2 cloud from the June 15, 1991, eruption of MountPinatubo. Geology 20, 867–870.
Whitford D. J., Nicholls D. A., and Taylor S. R. (1979) Spatialvariations in the geochemistry of quaternary lavas across the SundaArc in Java and Bali. Contr. Mineral. Petrol. 70, 341–356.
Xue X. and Kanzaki M. (2003) The dissolution mechanism of water inalkaline earth silicate and aluminosilicate melts: One view from 1HMAS NMR (abstract). Geochim. Cosmochim. Acta 67, (18 Suppl.)A543.
Young R. W., Duffy J. A., Hassall G. J., and Xu Z. (1992) Use ofoptical basicity concept for determining phosphorous and sulphurslag-metal partitions. Ironmaking Steelmaking 19, 201–219.
APPENDIX A
The Flood-Grjotheim Thermochemical Treatment of SilicateSlags and Melts
The Flood-Grjotheim treatment of silicate slags was first devisedfor slag-metal equilibria (Flood and Grjotheim, 1952). Here it isrevisited following a general scheme proposed by Gaskell (2000).Slag-gas equilibrium involving the simple case of sulfide and oxidecomponents of two generic divalent cations (M2� and N2�) is hereexplored.
Four binary joins and one pseudobinary mixture existing in themelt phase are relevant to our purposes: i) M(O,S), ii) N(O,S), iii)(M,N)O, iv), (M,N)S, v) (M,N)(O,S). Join (v) is a mixture of sulfideand oxide, each component having mixed cations in the same rationM
2�/nN2�.
Consider now the following equilibrium, at P and T of interest
(M,N)Omelt � ½S2, gas ⇔ (M,N)Smelt � ½O2, gas (A1)
whose shorthand representation is given by Eqn. 1. The equilibriumconstant for this reaction is
Kv �a(M,N)SaO2
1⁄2
a(M,N)OaS2
1⁄2�
X(M,N)SaO2
1⁄2
X(M,N)OaS2
1⁄2
�(M,N)S
�(M,N)O
� Kv′ �(M,N)S
�(M,N)O
(A2)
SinceX�M,N�S
X�M,N�O
�XS2�
XO2�
, Kv′ in Eqn. A2 is the equilibrium constant, on a
one site basis, of the “shorthand” form represented by Eqn. 1. Similarlythe corresponding equilibrium constants of the two joins i) and ii) willbe:
Ki �aMSaO2
1⁄2
aMOaS2
1⁄2�
XMSaO2
1⁄2
XMOaS2
1⁄2
�MS∗
�MO∗ � Ki
′ �MS
�MO
(A3)
Kii �aNSaO2
1⁄2
aNOaS2
1⁄2�
XNSaO2
1⁄2
XNOaS2
1⁄2
�NS∗
�NO∗ � Ki
′ �NS
�NO
(A4)
Equilibria A3 and A4 apply, respectively, only along the pseudobina-ries M(O,S) and N(O,S) with activity coefficients relative to the stan-dard state of pure liquid MS and MO components (Eqn. A3) and pureliquid NS and NO components (Eqn. A4).
Reaction scheme is as follows1) with slag and gas at equilibrium, half a mole of molecular
sulfur is reversibly transferred from the gas to the slag and half amole of molecular oxygen is reversibly transferred in the reversedirection, in accordance with equilibrium A1. The amounts of slagand gas are large enough to make the above exchanges composi-tionally irrelevant;
2) one mole of pseudobinary component (M,N)S is revers-ibly extracted from the large quantity of the (M,N)O-(M,N)Spseudobinary mixture, therefore without modification of slag com-position;
3) the extracted mole of (M,N)S is unmixed to produce XM2� moles
of pure component MS and XN2� moles of pure component NS;
4) half a mole of gaseous molecular oxygen is reversibly nucleatedfrom the gas phase, without modification of the gas composition;
5) XM2� moles of unmixed MS and XN
2� moles of unmixed NS aretransformed into pure MO and NO components through reaction withpure gaseous molecular oxygen, with production of half a mole of puregaseous molecular sulfur;
6) XM2� moles of pure MO and XN
2� moles of pure NO arereversibly mixed to form one mole of (M,N)O;
7) (M,N)O is transferred to the large quantity of the (M,N)O-(M,N)Spseudobinary mixture slag, therefore without modification of slag com-position;
8) half a mole of gaseous molecular sulfur is transferred to the largequantity of the gaseous phase, without modification of gas composition.
Based on Hess additivity rule, the overall change in free energy ofthe cycle is zero.
Let us now detail the Gibbs free energy changes involved in eachstep of the cycle.
Step 1: �G1 � 0 (A5)
as the reaction is conducted reversibly at equilibrium.
Step 2: �G2 � ��G(M,N)Smixing (A6)
�G2 � �RT ln a(M,N)S (A7)
�G2 � �RT ln X(M,N)Sslag �(M,N)S (A8)
�G2 � �RT ln XS2��(M,N)S (A9)
�Gmixing,(M,N)S is the partial molar free energy of mixing of (M,N)Sin the (M,N)O-(M,N)S pseudobinary mixture. �(M,N)S is the activitycoefficient of (M,N)S in the slag relative to pure (M,N)S as standardstate.
Step 3: �G3 � ��Gf(M,N)So (A10)
As X(M)S � XM2� and X(N)S XN2�, it follows that
�G3 � �RTXM2�lnaMS � XN2�lnaNS� (A11)

822 R. Moretti and G. Ottonello
�G3 � �RTXM2�lnXMS�MS∗ � XM2�lnXNS�NS
∗ � (A12)
�G3 � �RTXM2�ln XM2� � XN2�ln XN2�
� XM2�ln �MS∗ � XN2�ln �NS
∗ � (A13)
�G°f,(M,N)S is the molar free energy of formation of (M,N)S from itscomponents MS and NS. Asterisks indicate that activity coefficients inthe pseudobinary (M,N)S are relative to pure MS and pure NS asstandard state.
Step 4: �G4 � �½�GO2gasmixing (A14)
�G4 � �½RT lnaO2
gas (A15)
�GO2gasmixing is the partial molar free energy of mixing of half a mole of O2
in the gas mixture.
Step 5: �G5 � �XM2��G(A3)0 � XN2��G(A4)
0 (A16)
�G5 � XM2�RT ln Ki � XN2�RT ln Kii (A17)
�G5, the free energy change, is the negative of the sum of the Gibbsfree energy changes involved in equilibria (A3) and (A4).
Step 6: �G6 � �Gf(M,N)Oo (A18)
�G6 � RTXM2�ln aMO � XN2�ln aNO� (A19)
�G6 � RTXM2�ln XMO�MO∗ � XN2�ln XNO�NO
∗ � (A20)
�G6 � RTXM2�ln XM2� � XN2�ln XN2�
� XM2�ln�MO∗ � XN2�ln �NO
∗ � (A21)
�G6 is the molar Gibbs free energy of formation of (M,N)O from itscomponents MO and NO. Asterisks indicate that activity coefficients inthe pseudobinary (M,N)O are relative to pure MO and NO componentsas standard state.
Step 7: �G7 � �G(M,N)Omixing (A22)
�G7 � RT lna(M,N)Oslag (A23)
�G7 � RT lnX(M,N)Oslag �(M,N)O (A24)
�G(M,N)Omixing is the partial molar Gibbs free energy of mixing of (M,N)O
in the pseudobinary (M,N)O-(M,N)S mixture. �(M,N)O is the activitycoefficient of (M,N)O in the slag relative to pure (M,N)O as standardstate.
Step 8: �G8 � ½�GS2, gas
mixing (A25)
�G8 � �½RT lnaS2
gas (A26)
�GS2gasmixing is the partial molar Gibbs free energy of mixing of S2 in the gas
mixture.
As already noted, the overall change of free energy is zero; hence, byrearranging the various terms of the cycle we obtain:
lnXS2�O2
1⁄2
XO2�S21⁄2
� ln Kv′ � XM2�ln Ki � XN2�ln Kii � ln
��M,N�O
��M,N�S
� XM2�ln�MO
∗
�NS∗ � XN2�ln
�MO∗
�NS∗ (A27)
The first activity coefficient ratio of this expression can be very rea-sonably set equal 1, since mixing between (M,N)O and (M,N)S underthe same nM
2�/nN2� ratio is nearly ideal, at least in the low sulfur
concentration range (Flood and Grjotheim, 1952). Since S2� and O2�
speciate over the same matrix, this implies that the pseudobinarymixture (M,N)O-(M,N)S behaves as an ideal Temkin mixture. It fol-lows that:
ln Kv′ � XM2��ln Ki � ln
�MO∗
�MS∗ �� XN2��ln Kii � ln
�NO∗
�NS∗ � (A28)
which may be rearranged as
ln Kv′ � XM2��ln CO�S,M
anneal. Ki��XN2��ln CO�S,Nanneal. Kii� (A29)
CO�S,Manneal �
�MO∗
�MS∗ , CO�S,N
anneal .��NO
∗
�NS∗ in Eqn. A28 may be, at first approxima-
tion, set constant for every gas-melt exchange reaction of the type MO� ½S2 N MS � ½O2.
The presence of oxides of cations of differing charge is accounted forby means of electrically equivalent ion-fractions, therefore by substi-tuting X with X= (Eqn. 18 in the text).
In the presence of network forming oxides, polymerization must beconsidered to calculate the concentration of O2� and the Flood andGrjotheim cycle is developed also accounting for interactions of sulfuron the anionic matrix.
The terms CO-S,Manneal. and CO-S,N
anneal. introduced in Eqn. A29 representthe standard state transposition from “pure liquid component” to“pure slag/melt component.” These additional terms may be seen asGibbs free energies of annealing of oxides and sulfide componentsin the slag structure or, assuming athermal behavior for the inves-tigated exchange reactions, as annealing entropies, that is configu-rational entropy contributions, usually measured through Differen-tial-Scanning-Calorimetry (DSC) in well-relaxed network-formingmaterials and originated by the cooperative rearrangements of theirmolecules or polymeric units.
Because of the ideal behavior (Temkin-like) of melts/slags, so far asthe exchange of S2� and O2� is concerned, it follows that these termsalso reconcile the developed computation with the standard state of theTemkin model. This is in fact the standard state implicit in ionic modelsof silicate melts when treating components as completely dissociatedand admitting Raoultian behavior over ionic sites, as in the case of theFlood and Grjotheim thermochemical cycle.
APPENDIX B
In the following tables we resume some information concerning theemployed database.

823Sulfur solubility in melts
Table B1. Sulfide solubility data set.
Reference Type of experiment T (°C) P (kbar) Melt composition
Abraham and gas-flow furnace 1500 & 1650 0.001 SiO2-CaO-Al2O3-MgO, CaO-P2O5, FeO-SiO2, CaO-P2O5-SiO2,Richardson (1960)a CaO-P2O5-Al2O3
Abraham et al. (1960) gas-flow furnace 1500 & 1650 0.001 MnO-SiO2, MnO-SiO2-CaO, MgO-SiO2-CaOBrown et al. (1982) gas-flow furnace 1500 0.001 CaO-TiO2-SiO2, CaO-MgO-TiO2-SiO2, CaO-TiO2-SiO2-Al2O3
Buchanan and Nolan(1979)
gas-flow furnace 1200 0.001 synthetic tholeiitic melt
Carter and MacFarlane(1957a,b)
gas-flow furnace 1430 & 1520 0.001 CaO-Al2O3-CaO-SiO2
Clemente (1998) andClemente et al. (2004)
hydrothermal capsule 798 to 992 2-2.1 dacitic-rhyolitic melt
Drakalivsky et al. (1997) gas-flow furnace 150 to 1650 0.001 CaO-Al2O3-SiO2
Danckwerth et al. (1979) gas-flow furnace 1200 & 1300 0.001 synthetic lunar mare basaltFincham and Richardson
(1954)agas-flow furnace 1350 & 1650 0.001 FeO-SiO2, CaO-Al2O3, CaO-SiO2, CaO-SiO2-Al2O3
Douglas and Zaman(1969)
gas-flow furnace 1400 0.001 SiO2-K2O
Ghita and Bell (1982) gas-flow furnace 1500 0.001 FeO-TiO2-SiO2, FeO-TiO2-CaO, FeO-CaO-TiO2-SiO2
Gorbachev and Bezmen(2002)
High gas pressureequipment
1200 2.25, 2.53,3.37 & 4.76
An35Ab10Di55mol% � H2O
Haughton et al. (1974) gas-flow furnace 1200 0.001 synthetic basaltic meltKalyanram et al. (1960) gas-flow furnace 1500 0.001 CaO-Al2O3-SiO2, CaO-MgO-SiO2, CaO-MgO-Al2O3-SiO2
Katsura and Nagashima(1974)
gas-flow furnace 1100 & 1300 0.001 tholeiitic, hawaiitic, Rhyodacitic melts
Kor and Richardson(1968)
gas-flow furnace 1500 0.001 CaO-Al2O3
Luhr (1990) hydrothermal capsule 800 to 1000 1 to 4 trachyandesitic meltNagashima and Katsura
(1973)gas-flow furnace 1100 to 1300 0.001 SiO2-Na2O
O’Neill and Mavrogenes(2002)
gas-flow furnace 1400 0.001 CMAS (� Fe, Ti, Na, K) system
Richardson and Fincham(1956)b
gas-flow furnace 1450 & 1650 0.001 CaO-Al2O3, CaO-SiO2, CaO-SiO2,-Al2O3
Sharma and Richardson(1962)
gas-flow furnace 1500 & 1550 0.001 SiO2-CaO
Sharma and Richardson(1965)
gas-flow furnace 1650 0.001 MnO-Al2O3, SiO2-MnO-Al2O3, MnO-SiO2, MgO-SiO2-Al2O3,MgO-SiO2, SiO2-Al2O3, MgO-MnO-SiO2
a The authors furnish both new data and data recalculated from Fincham and Richardson (1954) and Richardson and Fincham (1956), whose sulfidecapacities are recalculated with up-to-date thermodynamic data for gaseous species.
b These data have not been used for model calibration because of the systematic overestimation due to old thermodynamic data of sulfur gaseous
species (see above).Table B2. Sulfate solubility data set.
Reference Type of experiment T (°C) P (kbar) Melt composition
Fincham and gas-flow furnace 1350 & 1650 0.001 FeO-SiO2, CaO-Al2O3, CaO-SiO2, CaO-SiO2-Al2O3
Richardson (1954)Douglas and Zaman
(1969)gas-flow furnace 1400 0.001 SiO2-K2O
Holmquist (1965) gas-flow furnace 1150 to 1250 0.001 SiO2-Na2OKatsura and
Nagashima (1974)gas-flow furnace 1100 & 1300 0.001 tholeiitic, hawaiitic, Rhyodacitic melts
Luhr (1990) hydrothermal capsule 800 to 1000 1 to 4 trachyandesitic meltNagashima and
Katsura (1973)gas-flow furnace 1100 to 1300 0.001 SiO2-Na2O
Papadopoulos (1973) gas-flow furnace 1370 to 1483 0.001 SiO2-Na2OSt. Pierre and
Chipman (1956)gas-flow furnace 1500 & 1550 0.001 CaO-FeO-Fe2O3, CaO-FeO-Fe2O3-SiO2
Richardson andFincham (1956)
gas-flow furnace 1450 & 1650 0.001 CaO-Al2O3, CaO-SiO2, CaO-SiO2,-Al2O3
Turkdogan and gas-flow furnace 1408 to 1630 0.001 CaO-FeO-Fe2O3, Open Hearth Slag
Darken (1961)




























