Embed Size (px)
Citation preview

1
Specific Cationic Photoinitiators for Near UV and visible LEDs:
Iodonium vs. Ferrocenium Structures.
Haifaa Mokbel1, 2, Joumana Toufaily2, Tayssir Hamieh2, Frederic Dumur,3 Damien Campolo,3
Didier Gigmes,3 Jean Pierre Fouassier, Joanna Ortyl*4,*Jacques Lalevée1
1) Institut de Science des Matériaux de Mulhouse IS2M – UMR CNRS 7361 – UHA; 15, rue Jean Starcky, 68057 Mulhouse Cedex – France.
2) Laboratoire de Matériaux, Catalyse, Environnement et Méthodes analytiques (MCEMA-CHAMSI), EDST, Université Libanaise, Campus Hariri, Hadath, Beyrouth, Liban.
3) Aix-Marseille Université, CNRS, Institut de Chimie Radicalaire ICR, UMR 7273, F-13397 Marseille, France.
4) Faculty of Chemical Engineering and Technology, Cracow University of Technology, Warszawska 24, Cracow 31-155, Poland.
e-mails: [email protected] ; [email protected]
Abstract:
Two iodonium salts based on a coumarin chromophore are investigated for
polymerization upon light emitting diode irradiations (LEDs). They work as one-component
photoinitiators. They initiate the cationic polymerization of epoxides (under air) and
vinylethers (laminate) upon exposure to violet LEDs (385 and 405 nm). Excellent
polymerization profiles are recorded. Their efficiency is quite similar to that of a ferrocenium
salt. Interpenetrating polymer networks can also be obtained through a concomitant
cationic/radical photopolymerization of an epoxy/acrylate blend monomer. The light
absorption properties of these new salts as well as the involved photochemical mechanisms
are investigated for the first time through electron spin resonance, laser flash photolysis,
steady state photolysis experiments. Molecular orbital calculations are also used to shed some
light on the initiation mechanisms.
Keywords: iodonium salt, ferrocenium salt, cationic polymerization, photoinitiators, light
emitting diode (LED), interpenetrated polymer network (IPN).

2
Introduction
Over the last decades, there has been a growing interest for the industrial application of
photoinitiated cationic polymerization (CP) reactions (see e.g. in [1,2,3,4]) which have several
advantages over the free radical polymerization (FRP). Epoxides and vinyl ethers that exhibit
a low volatility and a negligible toxicity and lead to polymers with good adhesion and
mechanical properties are the most classical and widely used cationic monomers. In contrast
to FRP, molecular oxygen does not inhibit the polymerization and aerated photosensitive
films can be easily photocured. In a dry environment, however, the termination rate is rather
low, permitting the continuation of the polymerization even if the irradiation is stopped
thereby leading to an interesting dark polymerization reaction.
Today, the Light Emitting Diode (LED) technology appears as an alternative to the
traditional UV lamps used in the UV-Curing technology. LEDs are characterized by strong
advantages: low heat generation, low energy consumption, low operating costs, less
maintenance, long life, portability, compact design, easy and safe handling, and possible
incorporation in robots... They have already found a high potential for e.g. digital inkjet
printing, adhesive curing, curing of thin heat-sensitive plastic foils, spot curing and dentistry.
The performance of LEDs vs. conventional light sources in UV-curing experiments has been
evaluated (mostly for FRP [5]).
Onium salts [1,2,3,4,6,7,8] have been extensively studied as photoinitiators (PI) for CP.
Among them, diaryliodonium and triarylsulfonium are commercially important. They consist
in a cationic moiety and a counter anion [6]. The nucleophilicity of the anion is an important
parameter affecting the performance: high nucleophilic anions prematurely terminate the
cationic chain reaction. Consequently, for the commonly used anions, the polymerization
rates usually follow the order: BF4- < PF6
- < SbF6- [1,2,9,10], ~ (C6H5F)4B
- [11]. A large
variety of many other onium salts has been developed (see examples in ref [1a,1b,2,3,4,12]).
Most structures are characterized by high photolysis quantum yields and are efficient when
the irradiation is carried out using lights in the UV region (230-300 nm) [12]. Extending their
spectral response to visible wavelengths requires the presence of photosensitizers and/or free-
radical photoinitiators [1,2,3,4,12,13]. Rather complex multicomponent photoinitiating
systems were thus successfully proposed e.g. photosensitizer/cationic PI; radical PI/cationic
PI; radical PI/cationic PI/additive (see e.g. in a recent review [2]). On the opposite, the search
of more or less colored structures have led to the design of one-component systems sensitive
to higher wavelengths (see e.g. in [1,2,3,4]) and more recently to onium polyoxometalate salts
[14]. The simple introduction of chromophoric moieties on the iodonium scaffold allows

3
interesting red-shifted absorptions (e.g. using a fluorenone [6,15] or a coumarin [16]). Apart
onium salts, organometallic compounds have been known for a long time [17]. In particular,
ferrocenium salt derivatives possessing various ligands (allowing suitable visible light
absorptions) and different nucleophilic anions are efficient (see examples in [3,17,18]).
The design and development of novel high-performance cationic PIs directly adapted to
light-emitting diodes (LEDs) as irradiation devices (especially near UV and visible LEDs) are
still a challenge and clearly attract a great attention in the photopolymerization field [2,19-21].
In the present paper, we focus on a study of two coumarin-based iodonium salts. Indeed, two-
component systems containing Coumarin-153 or Coumarin-7/diphenyl iodonium
hexafluorophosphate Iod for the CP of oxiranes [22] or 7-diethylamino-3-(2’-benz-
imidazolyl)coumarin/Iod for two-photon polymerization [23] are already known. A recent
report was concerned with the design of novel coumarin moiety containing iodonium salts
[16]: replacing one phenyl moiety by the coumarin scaffold red-shifts the absorption.
Promising results for the CP of epoxides and divinyl ethers have been gained upon exposure
to LED @315 nm and LED @365 nm [16] (the polymerization profile was determined using
a fluorescence probe technique); but no excited state processes and no mechanistic approach
are available. Therefore, herein, we propose to use the two derivatives (P3C-P and P3C-Sb)
shown in Scheme 1, monitor the polymerization profiles of a diepoxide, a divinylether and a
diepoxide/acrylate blend in aerated films under violet LEDs (centered at 385 and 405 nm) by
real time FTIR, exemplify the role of the anion and the dark reaction, investigate the excited
state processes by laser flash photolysis and electron spin resonance spin trapping, understand
the absorption properties and the bond cleavage by molecular orbital calculations, carry out
steady state photolysis and, finally, elaborate a mechanism for the initiation step. The
achieved performance will be compared to that of a ferrocenium salt.
Scheme 1. Investigated compounds.
Experimental Section:
i) Synthesis of the different photoinitiators:

4
P3C-P (GalOrti-7M-P) and P3C-Sb (GalOrti-7M-S) were obtained from (Photo HiTech
Ltd., Poland) and used with the best purity available. The synthesis is described in detail in
[16].
The ferrocenium salt - [(1,2,3,4,4a,8a-η)-naphthalene][(1,2,3,4,5-η)-2,4-
cyclopentadien-1-yl]-iron(l+) hexafluorophosphate(1-) - was synthesized according to the
procedure described in the literature [18b].
ii) Chemical compounds:
Diphenyliodonium hexafluorophosphate (Ph2I+ or Iod) and triethyleneglycol divinyl
ether (DVE-3) were obtained from Sigma-Aldrich and used with the best purity available
(Scheme 2). (3,4-epoxycyclohexane)methyl 3,4-epoxycyclohexylcarboxylate (EPOX;
Uvacure 1500) and trimethylolpropane triacrylate (TMPTA) were obtained from Allnex
(Scheme 2). EPOX, DVE-3 and TMPTA were selected as representative monomers: these
monomers are well known in the photopolymerization field and represent excellent structures
to evaluate the initiating ability of the new photoinitiating systems.
3
PF6
DVE-3
_
EPOX Iod (salt) TMPTA
Scheme 2. Chemical structures of the used compounds.
iii) Irradiation sources:
Several lights were used: i) polychromatic light from a halogen lamp (Fiber-Lite, DC-
950 - incident light intensity: I0 ≈ 12 mW cm-2; in the 370-800 nm range); ii) monochromatic
light delivered by LED@385 nm (M385L2- ThorLabs; I0 ≈ 25 mW/cm2 or Hamamatsu : 500
mW/cm2), LED@405 nm (M405L2 - ThorLabs; I0 ≈ 100 mW/cm2), blue LED@455 nm
(M455L3 - ThorLabs; I0 ≈ 80 mW/cm2) and LED@530 nm (ThorLabs; I0 ≈ 60 mW/cm2). The
emission spectra for these irradiation devices are given in [20].
iv) Cationic Photopolymerization:
The experimental conditions are given in the Figure captions. The weight content for the
PI is related to the monomer. The photosensitive formulations (25 µm thick) were deposited

5
on a BaF2 pellet under air for irradiation with different light sources under air. The evolution
of the epoxy group (for EPOX) or vinyl ether (for DVE-3) contents were continuously
followed by real time FTIR spectroscopy (JASCO FTIR 4100) at about 790 cm-1 and 1620
cm-1, respectively [24].
v) Synthesis of Interpenetrated Polymer Networks (IPNs):
The synthesis of interpenetrated polymer networks (IPN) based on EPOX/TMPTA
blend (50%/50% w/w) was carried out in laminate. The evolution of the epoxy group and
double bond content of the film (25 µm thick) was continuously followed by real time FTIR
spectroscopy (JASCO FTIR 4100) at about 790 and 1630 cm-1, respectively as in [24].
vi) Computational Procedure:
Molecular orbital calculations were carried out with the Gaussian 03 suite of programs
[25]. The HOMO and LUMO of the different compounds were calculated with the time-
dependent density functional theory at B3LYP/LANL2DZ level on the relaxed geometries
calculated at UB3LYP/LANL2DZ level. The C-I bond dissociation energy was calculated
from the fully optimized reactants and the free radicals generated.
vii) ESR spin trapping (ESR-ST) experiments:
The ESR-ST experiments were carried out using an X-Band spectrometer (MS 400
Magnettech). The radicals were produced at RT upon the LED@385nm exposure under N2
and trapped by phenyl-N-tbutylnitrone (PBN) according to a procedure described in detail in
[26]. The ESR spectra simulations were carried out with the PEST WINSIM program.
viii) Laser Flash Photolysis
Nanosecond laser flash photolysis (LFP) experiments were carried out using a Q-
switched nanosecond Nd/YAG laser (λexc = 355 nm, 9 ns pulses; energy reduced down to 10
mJ) from Continuum (Minilite) and an analyzing system consisted of a ceramic xenon lamp, a
monochromator, a fast photomultiplier and a transient digitizer (Luzchem LFP 212) [27].
Results and Discussion:
1/ Absorption properties of the photoinitiators:
The absorption spectra of the three salts in acetonitrile are depicted in Figure 1. Their
absorption maxima as well as their molar extinction coefficient ε are compared to those of the
commercial iodonium salt (Iod) in Table 1. Remarkably, the maxima for P3C-Sb, P3C-P, and
DC 1200 are located in the near-UV visible region. As known, Iod does not significantly

6
absorb light above 300 nm [6]. When one of the phenyl substituent of Iod is changed for a
coumarin chromophore, the corresponding absorption spectra are shifted to longer
wavelengths i.e. the maximum absorption being close to 350 nm for P3C-Sb and P3C-P vs.
230 nm for Iod. Compared to the iodonium salts, DC1200 exhibits lower molar extinction
coefficients in the UV but the absorption spreads in the 400-550 nm range. Interestingly, the
absorption spectra exhibit an interesting overlapping with the emission spectra of the
LED@385 and LED@405 nm (for P3C-Sb, P3C-P and DC 1200) and the LED@455 nm and
LED@530 nm (for DC 1200 only).
Figure 1. UV-visible absorption spectra of the different salts in acetonitrile. Insert: absorption
of DC1200 (expanded scale).
200 300 400 500
0
10000
20000
30000
εε εε (M
-1 c
m-1)
λλλλ (nm)
7MP - 009 7MS - 008 DC 1200
P3C-Sb
P3C-P
DC1200
350 400 450 500 550 6000
400
800
1200
1600
2000
λ (nm)
ε(M
-1cm
-1)
200 300 400 500
0
10000
20000
30000
εε εε (M
-1 c
m-1)
λλλλ (nm)
7MP - 009 7MS - 008 DC 1200
P3C-Sb
P3C-P
DC1200200 300 400 500
0
10000
20000
30000
εε εε (M
-1 c
m-1)
λλλλ (nm)
7MP - 009 7MS - 008 DC 1200
P3C-Sb
P3C-P
DC1200
350 400 450 500 550 6000
400
800
1200
1600
2000
λ (nm)
ε(M
-1cm
-1)
Table 1. Comparison of the light absorption properties of the three salts (PC3-P, PC3-Sb,
DC1200) vs. Iod.
Photoinitiators λλλλmax (nm) / ε ε ε ε (M-1cm-1)
PC3-P 347/~17 000
PC3-Sb 347/~17 000
Iod 237/~15 000
DC1200 350-490/1100-200

7
The MOs involved in the lowest HOMO → LUMO energy transition are depicted in
Figure 2. In Iod, the HOMO and LUMO are localized on the phenyl rings. The absorption
red-shift of P3C-P (or PC3-Sb) vs. Iod is due to an enhanced π electron delocalization on the
coumarin moiety as well as to the presence of a charge transfer transition (i.e the highest
HOMO strongly involves the coumarin moiety whereas the LUMO is located in the C-I bond
and presents an antibonding character.
Figure 2. Highest Occupied Molecular Orbital (HOMO) and Lowest Unoccupied Molecular Orbital (LUMO) of the iodonium salts. At UB3LYP/LANL2DZ level; isovalue: 0.04.
Iod Coumarin-based iodonium salt
LUMO
HOMO
2/ Cationic photopolymerization of a diepoxide:
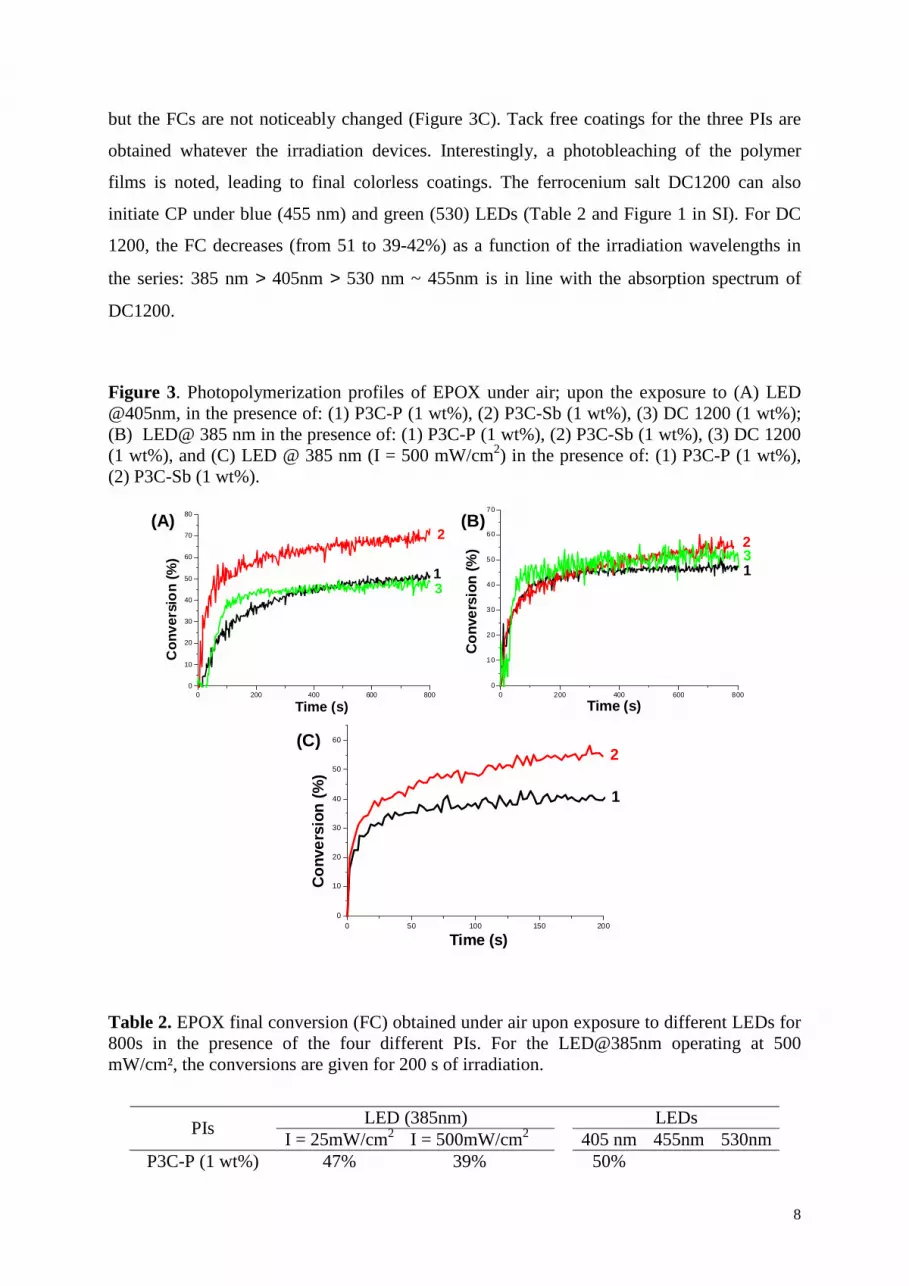
Typical conversion-time profiles for the CP of EPOX under air are depicted in Figure 3.
The final conversions (FC) after 800s of irradiation are given in Table 2. The well known Iod
used as a reference is completely ineffective, which is obviously due to its extremely poor
absorption under the LEDs used here. By contrast, upon exposure to the LED@405nm
(Figure 3A), P3C-Sb appeared as an excellent PI: a high conversion (69%) together with a
high rate of polymerization is reached. P3C-P is less efficient (FC = 50%; as expected, this is
due to the nucleophilicity difference of the anion) and behaves as DC1200 (FC = 47%). Upon
the LED@385nm, at low intensity (25 mW/cm2), almost similar FCs are obtained (Figure 3B,
Table 2). Under a higher intensity (500 mW/cm2), the polymerization rates are much higher
I+
O
I+
OO
8
but the FCs are not noticeably changed (Figure 3C). Tack free coatings for the three PIs are
obtained whatever the irradiation devices. Interestingly, a photobleaching of the polymer
films is noted, leading to final colorless coatings. The ferrocenium salt DC1200 can also
initiate CP under blue (455 nm) and green (530) LEDs (Table 2 and Figure 1 in SI). For DC
1200, the FC decreases (from 51 to 39-42%) as a function of the irradiation wavelengths in
the series: 385 nm > 405nm > 530 nm ~ 455nm is in line with the absorption spectrum of
DC1200.
Figure 3. Photopolymerization profiles of EPOX under air; upon the exposure to (A) LED @405nm, in the presence of: (1) P3C-P (1 wt%), (2) P3C-Sb (1 wt%), (3) DC 1200 (1 wt%); (B) LED@ 385 nm in the presence of: (1) P3C-P (1 wt%), (2) P3C-Sb (1 wt%), (3) DC 1200 (1 wt%), and (C) LED @ 385 nm (I = 500 mW/cm2) in the presence of: (1) P3C-P (1 wt%), (2) P3C-Sb (1 wt%).
0 200 400 600 8000
10
20
30
40
50
60
70
80
3
2
1
Co
nver
sio
n (%
)
Time (s)
0 200 400 600 8000
10
20
30
40
50
60
70
32
1
Co
nver
sion
(%)
Time (s)
0 50 100 150 2000
10
20
30
40
50
60
2
1
Co
nver
sion
(%
)
Time (s)
(A) (B)
(C)
Table 2. EPOX final conversion (FC) obtained under air upon exposure to different LEDs for 800s in the presence of the four different PIs. For the LED@385nm operating at 500 mW/cm², the conversions are given for 200 s of irradiation.
LED (385nm) LEDs PIs
I = 25mW/cm2 I = 500mW/cm2
405 nm 455nm 530nm P3C-P (1 wt%) 47% 39% 50%

9
P3C-Sb (1 wt%) 55% 55% 69% npa npa DC 1200 (1 wt%) 51% 47% 39% 42%
Iod (1 wt%) npa npa npa npa npa ano polymerization (<5%).
Concentration effects of the PIs have also been carried out under the LED@405nm
exposure. P3C-Sb appears to be excellent PIs whatever the concentration (the change of FC is
insignificant: ∼2%; Figure 4A). On the opposite, a strong effect is observed when using P3C-
P. Using [P3C-P] = 1%wt and 2%wt, the FC dramatically increases: FC = 50% (Figure 4B
curve 2 vs. curve 1) and FC = 65% (Figure 4B curve 3 vs. curve 2), respectively, instead of
FC = 37%) for [P3C-P] = 0.5%wt. In both cases, as a result of a higher amount of absorbed
light, the improvement on the polymerization rate is noticeable. Tack free coatings are still
obtained.
Figure 4. Photoinitiator concentration effects. Photopolymerization profiles of EPOX under air; upon the exposure to LED @405 nm in the presence of: (A) P3C-Sb (1) 0.5 wt%; (2) 1 wt%; (3) 2 wt%; (B) P3C-P (1) 0.5 wt%, (2) 1 wt%, (3) 2 wt%.
0 200 400 600 8000
20
40
60
80
100
1
23
Con
vers
ion
(%)
Time (s)
0 200 400 600 8000
10
20
30
40
50
60
70
80
3
2
1
Co
nve
rsio
n (%
)
Time (s)
(A) (B)
3/ Dark polymerization:
The presence of a possible dark polymerization after a short irradiation period can be an
advantage of CP compared to FRP [1,28]. Indeed, the reaction can continue through a living
process. The dark polymerization efficiency which is an important parameter for the global
performance of an initiating system decreases here in the series P3C-Sb >> DC1200 ~ P3C-P.
Under irradiation at 385nm or 405nm, using DC1200 or PC3-P, the dark polymerization has a
low efficiency (< 5 %; Figure 5B, Figure 5A). On the opposite, with P3C-Sb, the
polymerization proceeds after the light it switched off (200s of irradiation): FC increase =

10
18% in the dark (after 1800s). This better behavior is also ascribed to the lowest
nucleophilicity of its counter anion.
Figure 5. Photopolymerization profiles of EPOX under air: (A) under LED @405nm exposure (1) P3C-Sb (1%wt), (2) P3C-P (1%wt), and (B) in the presence of DC 1200 (1%wt) (1) LED@385nm, (2) LED @405nm.
0 500 1000 1500 20000
10
20
30
40
50
60
70
Light switched off
2
1
Co
nve
rsio
n (
%)
Time (s)0 500 1000 1500 2000
0
10
20
30
40
50
Light switched off
21
Co
nve
rsio
n (
%)
Time (s)
(A) (B)
0 500 1000 1500 20000
10
20
30
40
50
60
70
Light switched off
2
1
Co
nve
rsio
n (
%)
Time (s)0 500 1000 1500 2000
0
10
20
30
40
50
Light switched off
21
Co
nve
rsio
n (
%)
Time (s)
(A) (B)
0 500 1000 1500 20000
10
20
30
40
50
60
70
Light switched off
2
1
Co
nve
rsio
n (
%)
Time (s)0 500 1000 1500 2000
0
10
20
30
40
50
Light switched off
21
Co
nve
rsio
n (
%)
Time (s)
(A) (B)
4/ Synthesis of Interpenetrating Polymer Networks (IPNs):
The synthesis of interpenetrating polymer networks (IPN) from EPOX/TMPTA blends
(50%/50% w/w) in laminate upon the LED@405nm exposure is feasible (Figure 6). The FCs
of EPOX and TMPTA are not significantly affected by the kind of iodonium salt (47% vs.
44% in Figure 6A curve 1 vs. curve 2 and 37% vs. 32% in Figure 6B curve 2 vs. curve 1).
Tack free coatings are obtained.
Figure 6. Photopolymerization profiles of the TMPTA/EPOX blend (50%/50% w/w) in laminate upon a LED@405nm exposure, using (1) P3C-P (1% wt); (2) P3C-Sb (1% wt); (A) epoxy conversions and (B) acrylate conversions.

11
0 200 400 600 8000
10
20
30
40
50
60
21
Con
vers
ion
(%)
Time (s)
0 200 400 600 8000
10
20
30
40
50
21
Co
nver
sion
(%)
Time (s)
(A) (B)
5/ The cationic photopolymerization of a divinyl ether:
The cationic photopolymerization of DVE-3 was carried out in laminate upon
LED@385 and @405 nm using P3C-P as photoinitiator. Very high polymerization rates as
well as very high final conversions are found ((> 90%; Figure 7); tack free coatings are also
obtained. The polymerization is slightly faster upon LED@385 nm than LED@405 nm due to
the better absorption properties for P3C-P at 385 nm.
Figure 7. Photopolymerization profiles of DVE-3 in laminate upon the exposure to (1) LED @385 nm or (2) LED@405 nm in the presence of P3C-P (1 wt%).
0 200 400 600 8000
20
40
60
80
100
2
1
Co
nve
rsio
n (
%)
Time (s)0 200 400 600 800
0
20
40
60
80
100
2
1
Co
nve
rsio
n (
%)
Time (s)
6/ Chemical mechanisms
In ESR spin trapping experiments (Figure 8(A) and (B)), phenyl radicals Ph● are clearly
observed during the photolysis of P3C-P and P3C-Sb (hyperfine coupling constants: aN = 14.1
G; aH = 2.2 G, for the Ph●/PBN adducts; reference values in [24a,26]). These Ph● radicals
finally result from a cleavage of the C-I bond (reaction (1a) where Ph-I+-coum stands for

12
P3C-P and P3C-Sb). Another C-I bond breaking might be considered (1b). Molecular orbital
calculations (Scheme 3) show that the corresponding bond dissociation energy BDE (C-I)A
and BDE (Ph-I)B are 46.66 and 61.37 kcal.mol-1, respectively. This suggests a rather selective
I-Ph cleavage reaction in full agreement with the detection of only one kind of carbon
centered radical (Ph●) in ESR-ST. In line with the photolysis of iodonium salts studied in
[12], the primary radicals and cation radicals might be formed here through a singlet or a
triplet radical pair where homolytic and heterolytic cleavages occur. Then (as in [12]), out-of-
cage or/and in-cage processes leading to the release of protons should be possible. These
protons initiate the CP.
Ph-I+-coum (hν) → Ph● + +●I-coum (A) (1a)
Ph-I+-coum (hν) → PhI●+ + ●coum (B) (1b)
BDE(C-I)A
BDE(C-I)B
= 45.66 kcal mol-1
= 61.37 kcal mol-1A
B
PhI●+ Coum●
Ph● Coum-I●+
BDE(C-I)A
BDE(C-I)B
= 45.66 kcal mol-1
= 61.37 kcal mol-1A
B
BDE(C-I)A
BDE(C-I)B
= 45.66 kcal mol-1
= 61.37 kcal mol-1A
B
PhI●+ Coum●
Ph● Coum-I●+
Scheme 3. Possible dissociation mechanisms (A vs. B) of the coumarin-based Iod salts calculated at uB3LYP/LANL2DZ level.
Figure 8. ESR-Spin Trapping spectrum of the radical generated in (A) P3C-Sb upon LED@385 nm exposure; (B) P3C-P upon LED@385 nm exposure; and (C) DC 1200 upon UV exposure; in tert-butylbenzene; under N2; experimental (a) and simulated (b) spectra. Phenyl-N-tert-butylnitrone (PBN) is used as spin trap.

13
3440 3460 3480 3500 3520
(b)
(a)
B (G)
3440 3460 3480 3500 3520
(b)
(a)
B (G)
3300 3310 3320 3330 3340 3350 3360 3370 3380
(b)
(a)
B (G)
(A) (B)
(C)
In steady state photolysis (Figure 9; halogen lamp exposure; under air), the ground state
absorption bands of P3C-P and P3C-Sb decreased at ~ 350 nm as resulting from (1a) and
concomitantly, the absorption at 270 nm markedly increases (formation of photoproducts).
Interestingly, the presence of isobestic points show that no side reaction occurs.
Figure 9. Photolysis of P3C-P (A) P3C-Sb (B) and DC1200 (C) in acetonitrile; under air; halogen lamp exposure.
250 300 350 400 4500,0
0,2
0,4
0,6
30 min
0 min
O.D
.
λλλλ (nm)
250 300 350 400 4500,0
0,2
0,4
30 min
0 min
O.D
.
λλλλ (nm)
300 400 5000
2
4
200 s
0 s
O.D
.
λλλλ (nm)
(A) (B)
(C)

14
In laser flash photolysis (LFP) experiments on e.g. P3C-Sb, a strong bleaching of the
ground state absorption is found at 350 nm demonstrating the high photosensitivity of this
compound (Figure 10). The very short depletion time (<< 10 ns) is limited here by the
resolution time of the device. A weak transient is observed with a maximum absorption in the
500-600 nm range: it can be ascribed to the iodinium radical cation generated after C-I
cleavage process as already noted in the LFP of Iod [29].
Figure 10. (A) Transient absorption recorded at 350 nm for a laser excitation at 355nm for P3C-Sb in nitrogen saturated acetonitrile. (B) UV-vis spectra recorded before and after laser excitation.
0 20 40 60 80-0,005
-0,004
-0,003
-0,002
-0,001
0,000
0,001
0,002
∆∆ ∆∆O
.D.
Time (µµµµs)
200 250 300 350 400 450 5000,0
0,2
0,4
0,6
0,8
1,0
O.D
.
λλλλ (nm)
specre avant LFP spectre apres LFP-- Before LFP-- After LFP
B
A
0 20 40 60 80-0,005
-0,004
-0,003
-0,002
-0,001
0,000
0,001
0,002
∆∆ ∆∆O
.D.
Time (µµµµs)0 20 40 60 80
-0,005
-0,004
-0,003
-0,002
-0,001
0,000
0,001
0,002
∆∆ ∆∆O
.D.
Time (µµµµs)
200 250 300 350 400 450 5000,0
0,2
0,4
0,6
0,8
1,0
O.D
.
λλλλ (nm)
specre avant LFP spectre apres LFP-- Before LFP-- After LFP
200 250 300 350 400 450 5000,0
0,2
0,4
0,6
0,8
1,0
O.D
.
λλλλ (nm)
specre avant LFP spectre apres LFP-- Before LFP-- After LFP
B
A
In the case of DC 1200, cyclopentadienyl (Cp●) radicals are observed upon irradiation
in ESR experiments (Figure 8C; hyperfine coupling constants: aN = 14.3 G; aH = 2.6 G;
reference value in [30]). They are attributed to a Fe-Cp bond dissociation as already proposed
[31]. Under identical light irradiation conditions, DC 1200 exhibits a faster bleaching than the
two iodonium salts. The decrease of the absorption at 280 nm is accompanied by the

15
appearance of two new peaks at 280 nm and 350nm ascribed to the formation of naphthalene
(Figure 9C; isobestic point; no side reaction). As known [18e], the ring opening
polymerization of an epoxide in the presence of such ferrocenium salts is assumed to take
place through a ligand transfer reaction where the arene moiety is replaced by three molecules
of epoxide.
Conclusion:
In the present paper, we have proposed two modified iodonium salts for exposure to
violet LEDs. They work as PIs for the ring opening polymerization of an epoxide under air
and exhibit an efficiency similar to that of a ferrocenium salt; interpenetrating polymer
networks can also be synthesized; the photopolymerization of divinylethers is also easily
achieved. Their behavior as one-component photoinitiators is expected to be of interest, the
use of a photosensitization process (that is always tricky) becoming unnecessary. Other
iodonium salts applicable as high-performance PIs operating under blue or green LEDs
deserve to be developed.
Supporting Information:
This information is available free of charge via the Internet.

16
References:
[1] a) J.V. Crivello, Photoinitiators for Free Radical, Cationic; Anionic Photopolymerization, 2nd ed.; John Wiley & Sons: Chichester,1998; b) Photoinitiated Polymerization, K.D. Belfied, J.V. Crivello, Eds., ACS Symp. Ser. 847, Washington DC, 2003; c) J. P. Fouassier, Photoinitiator, Photopolymerization and Photocuring: Fundamentals and Applications; Hanser Publishers, Munich Vienna: New York, 1995; d) S. Davidson, Exploring the Science, Technology and Application of UV and EB Curing; Sita Technology Ltd: London, 1999; e) Photochemistry and Photophysics of Polymer Materials, N. S. Allen Ed., Wiley: New York, 2010. [2] J.P. Fouassier, J. Lalevée, Photoinitiators for Polymer Synthesis-Scope, Reactivity, and Efficiency; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, 2012. [3] a) Y. Yagci, Macromol. Symp.2006, 240, 93–101; b) M.U. Kahveci, A.G. Gilmaz, Y. Yagci, in Photochemistry and Photophysics of Polymer Materials, N.S. Allen, Eds, Wiley, USA, 421-478, 2010; c) Y. Yagci, S. Jockusch, N. J. Turro, Macromolecules 2010, 43, 624–639 [4] a) J.V. Crivello, in Dyes and Chromophores in Polymer Science, J. Lalevée, J.P. Fouassier, Eds., ISTE Wiley, London, 2015; b) M. Sangermano, N. Razza, J.V. Crivello, Macromolecular Materials and Engineering, 2014, 299, 775-793. [5] a) K. D. Jandt and R. W. Mills, Dent. Mater. , 2013, 29, 605-617; b) F. Robert and J. Karlicek, in UV-LED, eds. C. Cordon and C. Miller, RadTech International, Bethesda, MD, 2013; c) C. Rahiotis, K. Patsouri, N. Silikas and A. Kakaboura, J. Oral Sci., 2010, 52, 187-195; d) D. L. Leonard, D. G. Charlton, H. W. Roberts and M. E. Cohen, J. Esthet. Restor. Dent. , 2002, 14, 286-295; e) R. Karsten and M. Beck, Radtech Report Winter, 2011, 26-31. [6] a) J.V. Crivello, J.H.W. Lam Macromolecules, 1977, 10, 1307-1315; b) J.V. Crivello, J.H.W. J. Polym. Sci., Part A: Polym. Chem, 1978, 16, 2441-2451; c) J.V. Crivello, J.H.W. Lam J. Polym. Sci., Part A: Polym. Chem, 1979, 17, 977-999; d) A. Hartwing, A. Harder, A. Luhring, H. Schroder, European Polymer Journal, 37, 2001, 1449-1455. [7] S. P. Pappas, In Photopolymerization and Photoimaging Science and Technology; N. S. Allen, Ed.; Elsevier: London, 1989; 55–73 [8] a) J.D. Cho, J.W. Hong, J. Appl. Polym. Sci. 2005,97, 1345–1351; b) M. Sangermano, G. Malucelli, R. Bongiovanni, A. Priola, U. Annby, N. Rehnberg, Eur. Polym. J., 2002, 38, 655–659. [9] J.V. Crivello, Adv. Polym. Sci. 1984, 62, 1-48. [10] J.V. Crivello J. Polym. Sci.: Part A: Polym. Chem.1999, 37, 4241-4254. [11] F. Castellanos, J.P. Fouassier, C. Priou, J. Cavezzan, J. Appl. Polym. Sci., 1996, 60, 705-713. [12] a) Y. Yagci, A. Kornowski, W. Schnabel, J. Polym. Sci. Part A: Polym. Chem. 1992, 30, 1987–1991; b) S. Denizligil, Y. Yagci, C. McArdle, Polymer 1995, 36, 3093–3098; c) S. Denizligil, R. Resul, Y. Yagci, C. McArdle, J. P. Fouassier, Macromol. Chem. Phys. 1996, 197, 1233–1240; d) F. Kasapoglu, A. Onem, N. Bicak, Y. Yagci, Polymer 2002, 43, 2575–2579; e) J. V. Crivello, S. Q. Kong, Macromolecules 2000, 33, 825–832; f) Z. Gomurashvili, J. V. Crivello, J. Polym. Sci. Part A: Polym.Chem. 2001, 39, 1187–1197; g) Kasapoglu, F.; Yagci, Y. Macromol. Rapid Commun.2002, 23, 567–570; h) B. Aydogan, G. E. Gunbas, A. Durmus, L. Toppare and Y. Yagci, Macromolecules, 2009, 43, 101-106; i) B. Aydogan, B. Gacal, A. Yildirim, N. Yonet, Y.Yuksel, Y. Yagci, in Photochemistry and UV Curing, J.P. Fouassier, Ed., Research Signpost, Trivandrum India, 187-201, 2006; j) Y.Y. Durmaz, G. Yilmaz, M.A. Tasleden, B. Aydogan, B. Koz, Y. Yagci, in Basics and Applications of Photopolymerization Reactions, J.P. Fouassier, X. Allonas, Eds., Research Signpost, Trivandrum India, 7-22, 2010; k) G. Liu, X. Zhu, B. Xu, X. Qian, G. Song, J. Nie, J. Appl.

17
Polym. Sci., 2013, 130, 3698–3703; l) O. S. Taskin, I. Erel-Goktepe, M. Alyaan A. Khan, S. Pispas, Y. Yagci, J. Photochem. Photobiol. A: Chemistry, 2014, 285, 30-36; m) Durmaz YY, Moszner N, Yagci Y., Macromolecules. 2008;41:6714-8; n) B. Aydogan, Y. Yagci, L. Toppare, S. Jockusch, N.J. Turro Macromolecules 2012, 45, 7829-34; o) U. Bulut, G.E. Gunbas, L. Toppare J. Polym. Sci., Part A: Polym. Chem. 2010, 48, 209-13; p) J.V. Crivello J. Macromol. Sci. Part A Pure Appl Chem. 2009, 46, 474-483; q) J.V. Crivello J. Polym. Sci., Part A: Polym Chem. 2009, 47, 866-75; r) R. Podsiadły J. Photochem. Photobiol. A. 2009, 208, 147-53; s) J.V. Crivello, in Radiation Curing in Polymer Science and Technology, J.P. Fouassier, J.F. Rabek Eds., Elsevier, Barking, UK, vol.2, 435-472, 1993; t) N.P. Hacker, in Radiation Curing in Polymer Science and Technology, J.P. Fouassier, J.F. Rabek Eds., Elsevier, Barking, UK, vol.2, 473-504, 1993. [13] a) S. P. Pappas, J. H. Jilek, Photogr. Sci. Eng. 1979, 23,140–143; b) J. L. Dektar, N. P. Hacker, J. Am. Chem. Soc. 1990,112, 6004–6015. [14] H. Mokbel, P. Xiao, C. Simonnet-Jégat, F. Dumur, D. Gigmes, J. Toufaily, T. Hamieh, J-P. Fouassier, J. Lalevée, J. Polym. Sci. A Polym. Chem. 2015, DOI: 10.1002/pola.27526 [15] J.V. Crivello, J.P. Fouassier, D. Burr, Macromol. Sci., Pure and Appl. Chem., 1994, A31, 677-685. [16] a) J. Ortyl, R. Popielarz, Polymery, 2012, 57, 7-8; b) Polish Pat. Appl. P-393 501, 2010; c) Polish Pat. Appl. P-395 515, 2011. [17] A. F. Cunningham and V. Desobry, in Radiation Curing in Polymer Science and Technology, eds. J. P. Fouassier and J. F. Rabek, Elsevier, Barking UK, 1993, vol. 2, pp. 323-374. [18] a) T. Wang, Z. Li, Y. Zhang, K. Hassan and X. Wang, Polym. Eng. Sci. , 2009, 49, 613-618; b) T. Wang, B. S. Li and L. X. Zhang, Polym. Int., 2005, 54, 1251-1255; c) M. Li, Y. Chen, H. Zhang and T. Wang, Prog. Org. Coat., 2010, 68, 234-239; d) T. Wang, J. W. Chen, Z. Q. Li and P. Y. Wan, J. Photochem. Photobiol., A 2007, 187, 389-394. e) F. Lohse, H. Zweifel, Adv. Polym. Sci., 1986, 78, 62-69. [19] a) M. A. Tasdelen, M. Ciftci, Y. Yagci, Macromol. Chem. Phys. 2012, 213, 1391–1399; b) M. A. Tasdelen, M. Uygun, Y. Yagci, Macromol. Chem. Phys. 2011, 212, 2036–2042; c) S. Banerjee, E. B. Veale, C. M. Phelan, S. A. Murphy, G. M. Tocci, L. J. Gillespie, D. O. Frimannsson, J. M. Kelly, T. Gunnlaugsson, Chem. Soc. Rev. 2013, 42, 1601–1618; d) V. B. Bojinov, D. B.; Simeonov, Polym. Degrad. Stab. 2010, 95, 43–52; e) S. Noppakundilograt, S. Suzuki, T. Urano, M. Miyagawa, S. Takahara, T. Yamaoka, Polym. Adv. Technol. 2002, 13, 527–533; f) C. Coenjarts, O. Garcıa, L. Llauger, J. Palfreyman, A. L. Vinette, J. C. Scaiano, J. Am. Chem. Soc. 2003, 125, 620–621; g) V. Bojinov, I. Grabchev, Dyes Pigm. 2003, 59, 277–283; h) I. Grabchev, T. Philipova, Des Monomers Polym. 2000, 3, 479–488; i) S. Sharifi, H. Mirzadeh, M. Imani, F. Ziaee, M. Tajabadi, A. Jamshidi, M. Atai, Polym. Adv. Technol. 2008, 19, 1828–1838. [20] P. Xiao, J. Zhang, F. Dumur, M. A. Tehfe, F. Morlet-Savary, B. Graff, D. Gigmes, J. Fouassier, P. Lalevée, J. Prog. Polym. Sci., 2015, 41, 32-66 [21] C. Dietlin, S. Schweizer, P. Xiao, J. Zhang, F. Morlet-Savary, B. Graff, J.P. Fouassier, J. Lalevée, Polym. Chem., 2015, DOI: 10.1039/C5PY00258C. [22] Q.Q. Zhu, W. Schnabel, Polymer, 1996, 37, 4129-4133. [23] C. Li, L. Luo, S.Wang, W. Huang, Q. Gong, Y. Yang, S. Feng, Chemical Physics Letters 340 ,2001, 444-448. [24] Tehfe, M.A.; Lalevée, J.; Telitel, S.; Contal, E.; Dumur, F.; Gigmes, D.; Bertin, D.; Nechab, M.; Graff, B.; Morlet-Savary, F.; Fouassier, J.P. Macromolecules, 2012, 45, 4454−4460. [25] a) Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Zakrzewski, V. G.; Montgomery, J. A.; Stratmann, J. R. E.; Burant, J. C.;

18
Dapprich, S.; Millam, J. M.; Daniels, A. D.; Kudin, K. N.; Strain, M. C.; Farkas, O.; Tomasi, J.; Barone, V.; Cossi, M.; Cammi, R.; Mennucci, B.; Pomelli, C.; Adamo, C.; Clifford, S.; Ochterski, J.; Petersson, G. A.; Ayala, P. Y.; Cui, Q.; Morokuma, K.; Salvador, P.; Dannenberg, J. J.; Malick, D. K.; Rabuck, A. D.; Raghavachari, K.; Foresman, J. B.; Cioslowski, J.; Ortiz, J. V.; Baboul, A. G.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Gomperts, R.; Martin, R. L.; Fox, D. J.; Keith, T.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Challacombe, M.; Gill, P. M. W.; Johnson, B.; Chen, W.; Wong, M. W.; Andres, J. L.; Gonzalez, C.; Head-Gordon, M.; Replogle, E. S.; Pople, J. A., Gaussian 03, Revision B-2. Gaussian, Inc., Pittsburgh PA: 2003; b) Foresman, J. B.; Frisch, A., Exploring Chemistry with Electronic Structure Methods. Second ed.; Gaussian. Inc.: 1996. [26] a) J. Lalevée, N. Blanchard, M.A. Tehfe, F. Morlet-Savary, J.P. Fouassier, Macromolecules, 2011, 43, 10191−10195; b) P. Tordo, Spin-trapping: recent developments and applications; N.M. Atherton, M.J. Davies, B.C. Gilbert, Eds.; Electron Spin Resonance 16; The Royal Society of Chemistry: Cambridge, U.K., 1998; c) H. Chandra, I.M.T. Davidson, M.C.R. Symons, J. Chem. Soc. Faraday Trans. 1, 1983, 79, 2705-2711; d) A. Alberti, R. Leardini, G.F. Pedulli, A. Tundo, G. Zanardi, Gazz. Chim. Ital. 1983, 113, 869-871. [27] J. Lalevée, N. Blanchard, M. A. Tehfe, M. Peter, F. Morlet-Savary, D. Gigmes and J. P. Fouassier, Polym. Chem., 2011, 2, 1986-1991. [28] B. Golaz, V. Michaud, Y. Leterrier, J.-A.E. Månson, Polymer, 2012, 53, 2038-2048. [28] B. Golaz, V. Michaud, Y. Leterrier, J.-A.E. Månson, Polymer, 2012, 53, 2038-2048. [29] a) S.P. Pappas, B.C. Pappas, L.R. Gatechair, W. Schnabel, Journal of Polymer Science: Polymer Chemistry Edition., 1984, 22, 69-76; b) S. Peter Pappas, Betty C. Pappas, Leslie R. Gatechair, Josef H. Jilek, Polymer Photochemistry, 1984, 5, 1-22. [30] P. J. Barker, S. R. Stobart and P. R. West, J. Chem. Soc., Perkin Trans. 2, 1986, 127-130. [31] a) R. Bowser and R. Stephen Davidson, J. Photochem. Photobiol., A, 1994, 77, 269-276; b) M. Tanabe and I. Manners, J. Am. Chem. Soc. , 2004, 126, 11434-11435; c) G. Tabbì, C. Cassino, G. Cavigiolio, D. Colangelo, A. Ghiglia, I. Viano and D. Osella, J. Med. Chem. , 2002, 45, 5786-5796; d) N. S. Ieong and I. Manners, J. Organomet. Chem. , 2008, 693, 802-807.

19
Graphical Abstract:
BDE(C-I)A
BDE(C-I)B
= 45.66 kcal mol-1
= 61.37 kcal mol-1A
B
PhI●+ Coum●
Ph● Coum-I●+
BDE(C-I)A
BDE(C-I)B
= 45.66 kcal mol-1
= 61.37 kcal mol-1A
B
BDE(C-I)A
BDE(C-I)B
= 45.66 kcal mol-1
= 61.37 kcal mol-1A
B
PhI●+ Coum●
Ph● Coum-I●+
Cationic Polymerization: epoxy, vinylether
Interpenetrated Polymer Network Synthesis (IPN)
Specific Cationic Photoinitiators for Near UV
and visible LEDs: Iodonium vs. Ferrocenium
Structures.
BDE(C-I)A
BDE(C-I)B
= 45.66 kcal mol-1
= 61.37 kcal mol-1A
B
PhI●+ Coum●
Ph● Coum-I●+
BDE(C-I)A
BDE(C-I)B
= 45.66 kcal mol-1
= 61.37 kcal mol-1A
B
BDE(C-I)A
BDE(C-I)B
= 45.66 kcal mol-1
= 61.37 kcal mol-1A
B
PhI●+ Coum●
Ph● Coum-I●+
Cationic Polymerization: epoxy, vinylether
Interpenetrated Polymer Network Synthesis (IPN)
Specific Cationic Photoinitiators for Near UV
and visible LEDs: Iodonium vs. Ferrocenium
Structures.





























