Embed Size (px)
Citation preview

Structural Abnormalities Develop in theBrain After Ablation of the GeneEncoding Nonmuscle Myosin II-B
Heavy Chain
ANTONELLA N. TULLIO,1 PAUL C. BRIDGMAN,2 NANCY J. TRESSER,3
CHI-CHAO CHAN,4 MARY ANNE CONTI,1 ROBERT S. ADELSTEIN,1* AND
YOSHINOBU HARA5*1Laboratory of Molecular Cardiology, National Heart, Lung, and Blood Institute,
National Institutes of Health, Bethesda, Maryland 208922Washington University School of Medicine, St. Louis, Missouri
3Office of the Clinical Director, National Institute of Neurological Disorders and Stroke4Laboratory of Immunology, National Eye Institute, National Institutes of Health,
Bethesda, Maryland 208925Laboratory of Molecular Neurobiology, Human Gene Sciences Center, Tokyo Medical &
Dental University, Tokyo, Japan
ABSTRACTAblation of nonmuscle myosin heavy chain II-B (NMHC-B) in mice results in severe
hydrocephalus with enlargement of the lateral and third ventricles. All B-/B- mice died eitherduring embryonic development or on the day of birth (PO). Neurons cultured from superiorcervical ganglia of B-/B- mice between embryonic day (E) 18 and P0 showed decreased ratesof neurite outgrowth, and their growth cones had a distinctive narrow morphology comparedwith those from normal mice. Serial sections of E12.5, E13.5, and E15 mouse brains identifieddevelopmental defects in the ventricular neuroepithelium. On E12.5, disruption of the co-herent ventricular surface and disordered cell migration of neuroepithelial and differentiatedcells were seen at various points in the ventricular walls. These abnormalities resulted in theformation of rosettes in various regions of the brain and spinal cord. On E13.5 and E15,disruption of the ventricular surface and aberrant protrusions of neural cells into theventricles became more prominent. By E18.5 and P0, the defects in cells lining the ventric-ular wall resulted in an obstructive hydrocephalus due to stenosis or occlusion of the thirdventricle and cerebral aqueduct. These defects may be caused by abnormalities in the celladhesive properties of neuroepithelial cells and suggest that NMHC-B is essential for bothearly and late developmental processes in the mammalian brain. J. Comp. Neurol. 433:62–74,2001. Published 2001 Wiley-Liss, Inc.†
Indexing terms: nonmuscle myosin II-B; hydrocephalus; cell adhesion; brain development;
cytoskeleton; rosette formation
Cell adhesion and migration are two important propertiesmanifested by neuroepithelial and differentiating neuralcells during development of the nervous system. These cellsmigrate over relatively large distances from the neuroepi-thelial zone. Proper cell-cell interactions between migratingcells play a major role in guiding neural cells to their ulti-mate destination (reviewed in Hatten, 1999). The ubiquitouscontractile proteins actin and nonmuscle myosin, the latterincluding both conventional (myosin II) and unconventionalmyosins, are thought to play a role in these two processes(Mermall et al., 1998; Sellers, 1999).
Drs. Tullio and Bridgman made equivalent contributions to this work.*Correspondence to: Dr. Robert S. Adelstein, National Institutes of
Health, Building 10, Room 8N202, 10 Center Drive, MSC 1762, Bethesda,MD 20892-1762. E-mail: [email protected] or Dr. Yoshinobu Hara,Human Gene Sciences Center, Tokyo Medical and Dental University,1-5-45 Yushima, Bunkyo-ku, Tokyo 113-8510, Japan.E-mail: [email protected]
Received 18 May 2000; Revised 17 January 2001; Accepted 17 January2001
THE JOURNAL OF COMPARATIVE NEUROLOGY 433:62–74 (2001)
PUBLISHED 2001 WILEY-LISS, INC. †This article is a USGovernment work and, as such, is in the public domain in theUnited States of America.

Nonmuscle myosin II-B and the closely related non-muscle myosin II-A are present in various amounts in allvertebrate cells. Both myosins are hexamers composed oftwo identical heavy chains (200 kDa) and two pairs of lightchains (20 and 17 kDa). The heavy chains are the productsof two different genes and, although most cells in the bodycontain both proteins, NMHC-B is predominant in neuralcells (Rochlin et al., 1995). Different cellular localizationsof NMHC-A and B in the growth cones of cultured neuro-nal cells suggest that these two molecules have distinctfunctions in the nervous system (Miller et al., 1992; Chenget al., 1992; Rochlin et al., 1995; Lin et al., 1996). How-ever, it has not been possible to identify the specific func-tion of NMHC-B, because multiple members of the myosinsuperfamily are coexpressed in the same cells. Further-more, there have been no direct studies on the function ofNMHC-B in the development of the complex architectureof the nervous system.
To overcome these difficulties, we used homologous re-combination to generate mice lacking NMHC-B (B-/B-)and reported that these mice died in utero or on the day ofbirth (P0) with cardiac defects (Tullio et al., 1997). All ofthe B-/B- mice also developed hydrocephalus during em-bryogenesis. To elucidate the underlying brain deformitiesthat lead to hydrocephalus, we analyzed the structure ofthe brains of B-/B- mice between E12.5 and P0 and theneurite outgrowth of explants of superior cervical ganglia(SCG) cultured between E18 and P0. At E12.5, before thedevelopment of overt hydrocephalus, focal disruption ofthe ventricular surface was identified with disordered cellmigration in the brain and spinal cord, suggesting a defectin adhesion of the neuroepithelial cells at the ventricularsurface. Between E15 and P0, most of these mice devel-oped a progressive brain deformity with an obstructivehydrocephalus due to stenosis and occlusion of the dorsalregion of the third ventricle and the cerebral aqueduct.
This study is the first to elucidate the essential roles ofNMHC-B in the establishment of the form and integrity ofthe ventricular walls in the developing nervous system. Itshows that a small disturbance of the ventricular surface atthe critical stage of E12.5 and E13.5, due to the absence ofNMHC-B, can cause a severe brain deformity at later stages.
MATERIALS AND METHODS
B-/B- null mutant mice were generated by homologousrecombination as previously described (Tullio et al., 1997).Heterozygous matings were used to generate embryos ofthe three genotypes. The day of the vaginal plug wasconsidered as E0.5. In all experiments, littermates werecompared and the genotypes were confirmed by Southernand immunoblot analysis.
For histology, the embryos were isolated in PBS (pH7.4), fixed in Bouin’s fixative for 1 day and embedded inparaffin. Serial sections of 5 mm thickness were stained byhematoxylin and eosin or thionine. For immunoenzymehistochemistry, the embryos were fixed in 4% paraformal-dehyde phosphate buffer (pH 7.4) at room temperature for16 hours (Fig. 1B) or 95% ethanol and 5% acetic acid at4°C for 16 hours. Paraffin sections of 5 mm thickness werestained by using a rabbit polyclonal antibody to thecarboxyl-terminal sequence of human NMHC-B (Phillipset al., 1995). NMHC-B protein was visualized as a reddishcolor by a biotinylated secondary antibody and alkalinephosphatase-labeled streptavidin by using New Fuchsin
as substrate and counterstained by hematoxylin. For Fig-ure 2, which was not counterstained, NMHC-B proteinwas visualized as a brown color by the Vectastain EliteABC peroxidase system by using diaminobenzidine assubstrate. For immunofluorescence histochemistry, weprepared paraffin sections of 5 mm thickness from wild-type E12.5 mouse embryos fixed in 95% ethanol and 5%acetic acid or fresh frozen sections of 10 mm thicknesspost-fixed in acetone. They were then stained with theappropriate antibodies: anti-NMHC-B or -NMHC-A,which is raised against human platelet myosin composedonly of myosin II-A and recognizes only NMHC-A, andanti-N-cadherin (Zymed, S. San Francisco, CA), -b-catenin(Zymed), and a fluorescein isothiocyanate–labeled second-ary antibody. A control experiment omitting the primaryantibody and substituting the appropriate IgG was car-ried out for each of the panels shown in Figure 3 and wasnegative. The affinity purified polyclonal antibodies de-tecting NMHC-B and the antibodies to platelet myosindetecting NMHC-A were characterized in previous publi-cations, including Maupin et al. (1994), Phillips et al.(1995), and most recently, Takeda et al. (2000). Fluores-cent images were acquired by using a Zeiss 510 confocalmicroscope at 103 magnification. The composite figures ofdigital images were made without enhancement by usingAdobe Photoshop.
Superior cervical ganglion (SCG) explants from E18–P0mice were cultured in vitro as previously described (Roch-lin et al., 1995). Potential myosin II-B knockout pups wereselected on the basis of their abnormal morphology andtheir genotype was confirmed by Southern blotting. Livingcultures were visualized by phase contrast or differentialinterference contrast (DIC) microscopy. Images weretaken by either standard film photography or recorded byvideo-enhanced methods and stored as digital image files.Determination of outgrowth rates by using radial out-growth measurements from SCG explants was performedas previously described (Rochlin et al., 1996). Briefly, thisrequired photographing the explants by using phase con-trast microscopy at two different time points and thenmeasuring the change in outgrowth length between thetime points. For each explant at each time point, thedistance from the edge of the explant to the main field ofgrowth cones was measured in three places around theperimeter. The average of the three measurements wasused as an index of outgrowth for that explant. The dif-ference between the two outgrowth indices was divided bythe elapsed time between the two time points to give therate of outgrowth.
Digitized video-enhanced images of growth cones wereused to determine growth cone area and perimeter. Im-ages of the growth cone were traced by using a Wacomtablet and Scanalytics Iplab software (Scanalytics, Fair-fax, VA). The area of the growth cone and perimeter of thetraced image were calculated in pixels and then convertedto micrometers by using a calibration slide.
RESULTS
Physical appearance andimmunohistochemistry of B
1/B
1and B
-/B
-
embryos
B-/B- mice showed abnormalities in the central nervoussystem and died throughout embryonic development or on
63NEURAL CELL DEFECTS IN B-/B- MICE

P0. B1/B- mice appeared normal and showed no differ-ences from B1/B1 mice. All of the B-/B- mice analyzedfrom E12.5 and later showed an enlarged head and dila-tion of the lateral and third ventricles to differing extents,suggesting that the volume of the cerebral spinal fluid wasincreased (Fig. 1A). The distribution of NMHC-B in thewhole embryo at E14 is shown in Figure 1B by usingimmunohistochemical staining. For these experiments,we used an affinity purified polyclonal antibody raised tothe carboxyl-terminal sequence of NMHC-B (Phillips etal., 1995). Wild-type embryos demonstrated ubiquitous,but not homogenous, distribution of NMHC-B proteinthroughout the entire embryo. As a control experiment,the immunohistochemical staining of NMHC-B in theB-/B- mouse is also shown (Fig. 1B). No signal was de-tected under the same conditions.
Embryonic distribution of NMHC-B in thenervous system
In the nervous system, NMHC-B is expressed in the cellbodies and processes of all neural and non-neural cells.However, the levels of expression vary with cell type andchange during development, as cells migrate and differen-tiate. At E10.5–12.5, when ventricular formation is veryactive in the nervous system, NMHC-B is most prominentat the ventricular surface and subpial surface. We definethe ventricular surface as the apical edge of the ventricu-lar zone and the subpial surface as the outer edge of themarginal zone. Figure 2 shows the prominent expressionof NMHC-B at the ventricular and subpial surfaces in thedorsal midbrain at E11.5 at both low and high magnifica-tion. Expression at these surfaces decreases progressivelyat later stages.
Whereas NMHC-B was widely expressed in all neuralcells and was most prominent on the ventricular andsubpial surfaces (Figs. 2, 3A), NMHC-A was predomi-nantly expressed in the capillary endothelial cells as dem-onstrated by immunofluorescent histochemistry (Fig. 3B).Both proteins were also detected in the meninges andskin. The intense staining of NMHC-B in the ventricularand subpial surfaces prompted us to stain these samesections with antibodies to N-cadherin and b-catenin,which also showed intense staining in these areas (Fig.3C,D). These results suggested that NMHC-B might playa role in cell adhesion in the cells lining the ventricularand subpial surfaces (see Discussion section).
Abnormalities in cultured neurons fromB
-/B
-mice
During or after neuronal cell migration to their properlocations, axons and dendrites extend to make properconnections between neurons. To determine whetherB-/B- neuronal cells show defects in axonal outgrowth, westudied the radial outgrowth of neuronal processes in anin vitro culture system and also observed the morphologyof the growth cones. Superior cervical ganglia (SCG) ex-plants from both B1/B1 and B-/B- mice, between E18 andP0, were used for these studies. Figure 4A compares theoutgrowth of a SCG explant from a B1/B1 and a B-/B-
mouse cultured for 16 hours on laminin-coated plates.Results from three separate experiments showed that therate of radial outgrowth of the B-/B- explants was, onaverage, 32% less than the rate of radial outgrowth of aB1/B1 control (see Table 1). Moreover, as shown in Figure4B and quantitated in the accompanying graph, growth
cones from B-/B- mice between E18 and P0 are narrower,have a more convoluted perimeter, and occupy a decreasedarea compared with the B1/B1 growth cones.
These data suggested that a careful study of the devel-oping brains of B-/B- mice should result in useful infor-mation on the role of NMHC-B in vivo as well as an insightinto the etiology of the hydrocephalus in these mice.Therefore, to analyze the in vivo defect in B-/B- mice,serial sections were studied from E12.5, 13.5, 15, 18.5, andP0 mice. Heterozygous and wild-type littermates wereused as controls.
Focal disruption of the ventricular surfacewith protrusion of neural cells into the
ventricles
In the B-/B- mouse at E12.5, major architecture of thebrain appeared normal in most regions. However, enlarge-ment of the brain with dilation of the ventricles is alreadyclearly seen at this stage in the absence of any obstructionof the ventricular system. Furthermore, closer analysisrevealed important cellular defects in the neuroepithelialcells which are lining the ventricular walls at this stage.The ventricular surface that is formed by the apical edgeof the cell membrane of neuroepithelial cells was focallydisrupted in some areas where groups of cells in the ven-tricular zone protruded into the ventricle (compare Fig.5B,D,F with 5A,C,E). The focal defects of the ventricularsurface were present throughout the brain and spinal cordas shown here at the level of the third (Fig. 5B, rightarrowhead) and fourth ventricles (left arrowhead).
In some cases, disruption of the ventricular surfaceresulted in the formation of rosettes in various regions ofthe brain and spinal cord as shown here in Figure 6 (seealso Figs. 7D, 8I). The process of neuroepithelial rosetteformation at E12.5 is shown at the level of the fourthventricle in Figure 6A,B,C. In the area where the limitingmembrane of the ventricular surface was retained, as seenin the center of the figures, the apical processes of neuro-epithelial cells were anchored to the ventricular surfaceand the cell bodies were radially oriented with narrowextracellular spaces. In contrast, in the areas where thelimiting membrane of the ventricular surface was de-stroyed, as seen in the right and left sides of the figures,the apical processes of neuroepithelial cells were retractedand the cell bodies were oriented in various directionswith wide extracellular spaces. These findings suggest adefect in adhesion between the neuroepithelial cells at theventricular surface. Cell migration of the neural cells fromthis site is not restricted by the ventricular surface, andthe cells are folded over in some cases to generate aventricular-like structure. In Figure 6C, the residual ven-tricular surface is seen at the bottom of the rosette withdisordered neuroepithelial cells at the top.
Rosette formation of a different type was found in theretina of all B-/B- mice at P0 (see Fig. 6D,E). In the retina,there was no destruction of the external surface of theneuroepithelial cell layer that corresponds to the ventric-ular surface of the brain and spinal cord. However, rosetteformation of immature neuroepithelial cells that have de-tached from the inner neuroepithelial layer was presentwithin the inner nuclear layer but not at the ventricularsurface. A ventricular-like structure can be seen in thecenter of the rosette on the right (Fig. 6D, arrow) and at ahigher magnification in Figure 6E.
64 A.N. TULLIO ET AL.

Fig. 1. Abnormal enlargement of the head in B-/B- mice and im-munohistochemical staining of nonmuscle myosin heavy chain II-B(NMHC-B) in B1/B1 and B-/B- mouse embryos at embryonic day (E)14. A: B1/B1 embryo and B-/B- embryo at E12.5. The latter showsenlargement of the head. B: NMHC-B was visualized as a reddish
color by using a specific antibody to the carboxyl terminus of NMHC-B(see Materials and Methods section). NMHC-B protein is distributedwidely but not homogeneously throughout the embryo of the B1/B1
mouse embryo. In contrast, the B-/B- embryo, used as a negativecontrol, does not stain for NMHC-B. Scale bars 5 1 mm in A and B.
65NEURAL CELL DEFECTS IN B-/B- MICE

In E13.5 B-/B- mice, the focal disruption of the ventric-ular surface and the disturbance of cell migration weremore prominent as shown at low and high magnificationin Figure 7. Figure 7A,B shows disruption of the hypotha-lamic sulcus of the third ventricle with the protrusion ofneural cells. The disruption of the ventricular surface isconfined to one side, with no abnormality on the otherside. Figure 7C,D shows severe destruction of the fourthventricle. A large mass composed mostly of pale stainedlarge size cells, possibly neurons, distinct from darkstained neuroepithelial cells is protruding from the dorsalregion of the brainstem (see arrowhead in Fig. 7D). Thedeformed ventricular surface of the neuroepithelial cells is
present under this cell mass. It is noteworthy that thisspecific site in the fourth ventricle consistently (10 of 10B-/B- mice) showed abnormalities of this type in B-/B-
mice. In other areas of the ventricular system, the locationand extent of the focal disruptions were variable.
Formation of an aberrant ventricle at E15in B
-/B
-brains
Figure 8 shows a different deformity in the B-/B- brainat E15, that is formation of a large, aberrant ventricle atthe level of the cerebral aqueduct. The abnormal forma-tion of the new ventricle dorsal to the cerebral aqueduct(labelled CA) was clearly seen from the anterior end (A) tothe posterior end (I) in a series of coronal sections. Thearrowhead in Figure 8A indicates the anterior end ofthe neuroepithelial cells lining this aberrant ventricle.The ventricle itself is seen in Figure 8B–I. Protrusion ofthe neuroepithelial cells into this abnormal ventricle isindicated by the asterisk (Fig. 8D–I). In addition, a large,misplaced blood vessel, which is penetrating into the ec-topically formed neuroepithelial cell zone, is seen just tothe left of the asterisk in Figure 8E,F. The cerebral aque-duct, which is slightly deformed in panel C, communicatesposteriorly with the aberrant ventricle in Figure 8D–I.Most of the normal ventricular surface is destroyed, and alarge number of dead cells are present in the subventricu-lar structure (Fig. 8B–I) and in the aqueduct andpseudoventricle (Fig. 8D–I). Numerous neuroepithelial ro-settes can be seen in the posterior midbrain (Fig. 8I).
Fig. 2. Immunohistochemical staining of nonmuscle myosin heavychain II-B (NMHC-B) protein in the ventricular and subpial surfacesin embryonic day (E) 11.5 brain. A: Low-magnification view of asagittal section of the midbrain at E11.5. The posterior is shown onthe left. NMHC-B protein is stained as brown without counterstain-ing. B: High-magnification view of the dorsal midbrain, indicated bythe arrowhead in A. In the nervous system, NMHC-B is expressed inthe cell bodies and processes of all neural and non-neural cells. It ismost prominent at the ventricular (V) and subpial (SP) surfaces atE11.5, when ventricular formation is very active. NMHC-B is alsoexpressed in non-neural cells as shown in the developing skin (SK)and meninges (M) in the head. Scale bar 5 100 mm in A; 20 mm in B.
Fig. 3. Immunofluorescent histochemistry of brain sections fromB1/B1 mice at embryonic day (E) 12.5. A: In B1/B1 mice, nonmusclemyosin heavy chain II-B (NMHC-B) protein is widely distributed atvarious levels in the brain, most prominently in the ventricular andsubpial surfaces. B: NMHC-A is predominantly expressed in capillaryendothelial cells in the brain. Both NMHC-B and NMHC-A can bedetected in the meninges. C: N-Cadherin. D: b-Catenin. Cell adhesionmolecules such as N-cadherin and b-catenin show a distribution sim-ilar to NMHC-B on the ventricular and subpial surfaces. Scale bar 550 mm in A (applies to A–D).
66 A.N. TULLIO ET AL.

These abnormalities indicate that loss of NMHC-B, whichresults in focal disruption in the ventricular wall, can alsoinduce a major deformity in the ventricular system. How-ever, the occurrence and shape of this abnormal ventricleis quite variable in B-/B- mice. Nonetheless, a similar typeof deformity, but with less severe structural defects, wasseen in the cerebral aqueduct of most B-/B- mice.
Hydrocephalus in E18.5 and P0 B-/B
-mice
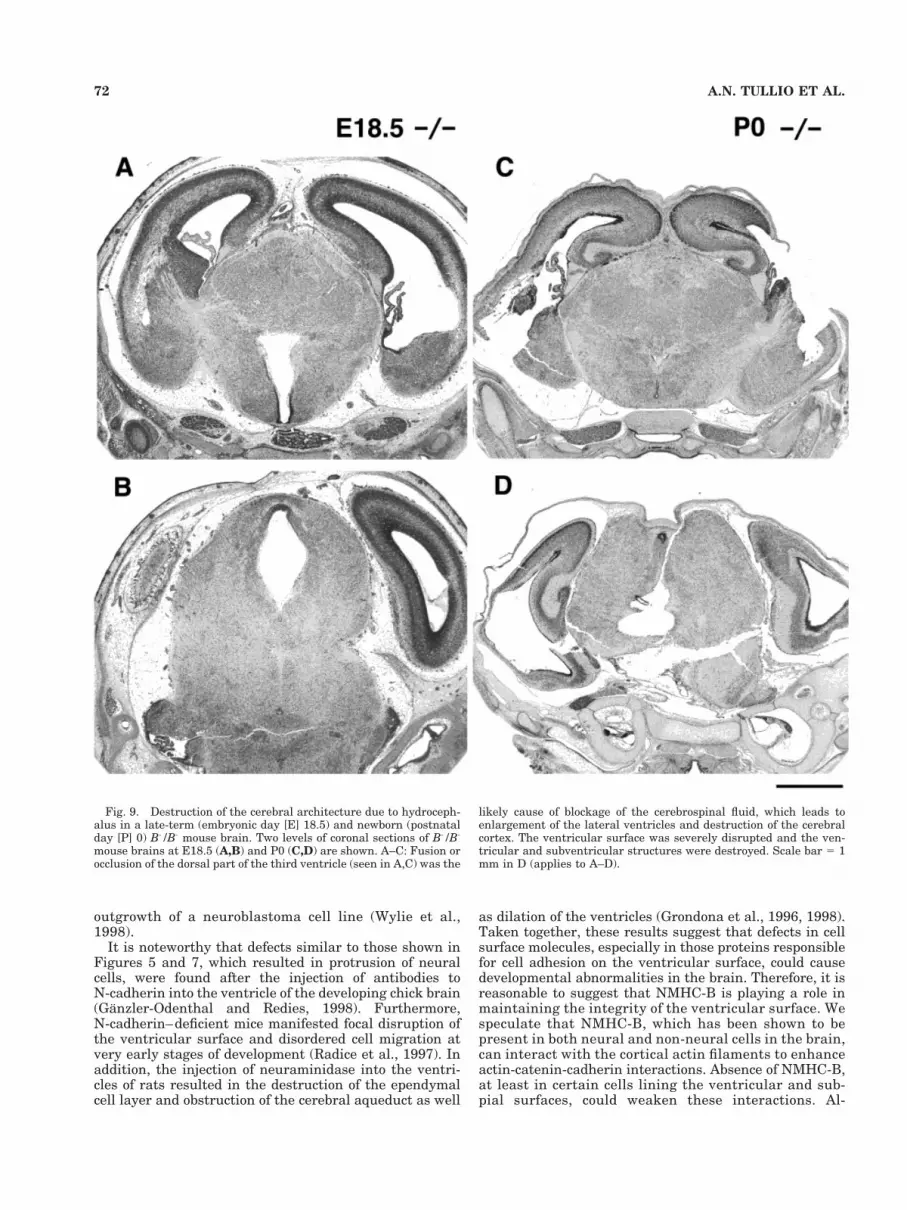
All of the B-/B- mice developed a severe hydrocephalusat the late embryonic stage from E18.5 to P0 and died (Fig.
9). Histologic analysis revealed deformity of the brain invarious regions. The most prominent abnormality is thestenosis or occlusion of the dorsal region of the thirdventricle and the cerebral aqueduct at this stage. In thetwo B-/B- mice shown in Figure 9, the midline of thedorsal regions of the posterior thalamus and the preopticarea appeared to be fused with complete loss of the ven-tricle from the posterior thalamus to the anterior cerebralaqueduct (Fig. 9A,C). This deformity could be the cause ofthe expansion of the lateral ventricle by blocking the ce-rebrospinal fluid (CSF) circulation. At E18.5, the ventric-ular surface of the cerebral cortex was destroyed and theventricular and subventricular structures were severelydamaged as seen in the right cerebral cortex (Fig. 9A). AtP0, the expansion of the lateral ventricle and the massivedestruction of the cerebral cortex reached to the pial sur-face and, ultimately, distorted the skull (Fig. 9C,D). Thetypical abnormalities found in severe hydrocephalus, thatis, the destruction of the deep structures caused by infarc-tion and edema, are seen in the left cerebral cortex (Fig.9A) and the ventral midbrain (Fig. 9D).
DISCUSSION
Defects in cell adhesion and migration inB
-/B
-mice
To understand the etiology of the abnormal brain devel-opment in the early stages, we studied embryos betweenE12.5 and E15, where we found evidence of defects thatwere consistent with alterations in cell adhesion and cellmigration of the neural cells. Of particular note were thedefects found in B-/B- embryos at E12.5 and E13.5 (Figs.5, 6A–C, 7). In these mice, the ventricular surface of theneuroepithelial cells was disrupted and the underlyingneural cells were seen to be protruding into the ventriclesat various points throughout the ventricular system, mostprominently in the third and fourth ventricles. At thesesites, the apical processes of the pseudostratified neuro-epithelial cells were retracted to the cell bodies, whichappeared loosely adherent to each other. These abnormal-ities were most likely due to defects in cell adhesion be-tween the cells lining the ventricular wall. As a result, amixture of mitotic and postmitotic neural cells grow outinto the ventricles, thereby preventing the postmitoticcells from migrating to their normal cell layer.
A second interesting finding was a decrease in neuriteoutgrowth and abnormal morphology of the growth conesof in vitro cultured SCG cells from P0 B-/B- mice. Growth
Fig. 4. Decreased radial outgrowth of neural processes and abnor-mal shape of growth cones of superior cervical ganglion (SCG) ex-plants from B-/B- mice. A: An explant from a B1/B1 mouse that wasgrown for 16 hours in culture on laminin (16 mg/ml). The radialoutgrowth (bracket) from the explant is 884 mm (left). An explant froma B-/B- mouse that was grown for 16 hours on laminin. The radialoutgrowth (bracket) is reduced to 661 mm (right). B: Video-enhanceddifferential interference contrast (DIC) images of living growth cones.In contrast to the growth cones from the B1/B1 mouse (left), thegrowth cone from the B-/B- mouse (right) is narrow and irregular inshape. C: The graph of perimeter versus area shows that the B-/B-
growth cones are smaller and have a more convoluted outline than theB1/B1 growth cones. Each point represents a single growth cone.R2 5 0.70 for the B1/B1 growth cone and 0.35 for the B-/B- growthcone. Scale bars 5 114 mm in A; 10 mm in B.
TABLE 1. Decreased Radial Outgrowth of Neural Processesfrom B2/B2 Explants
Experiment PhenotypeOutgrowth rate(mm/min. 6 SD) N
%difference
1 Cont1 0.43 6 0.05 2 30KO 0.30 6 0.06 22 Cont 0.63 6 0.02 2 35KO 0.41 6 0.04 23 Cont 0.65 6 0.05 5 32KO 0.44 6 0.05 3
1Control cultures were derived from wild-type and heterozygous pups. For experiment1, the time between measurements was 4 hours and the explants were from postnatalday 0 pups. The difference was not significant (t-test). In experiments 2 and 3, theexplants were from embryonic day 18–19 embryos, and the time between measure-ments was 9 and 10 hours, respectively (the differences were significant by t test; P ,0.02 and 0.01). In all experiments, the measurements were taken within 24 hours ofplating. Cont, control; KO, knockout.
67NEURAL CELL DEFECTS IN B-/B- MICE
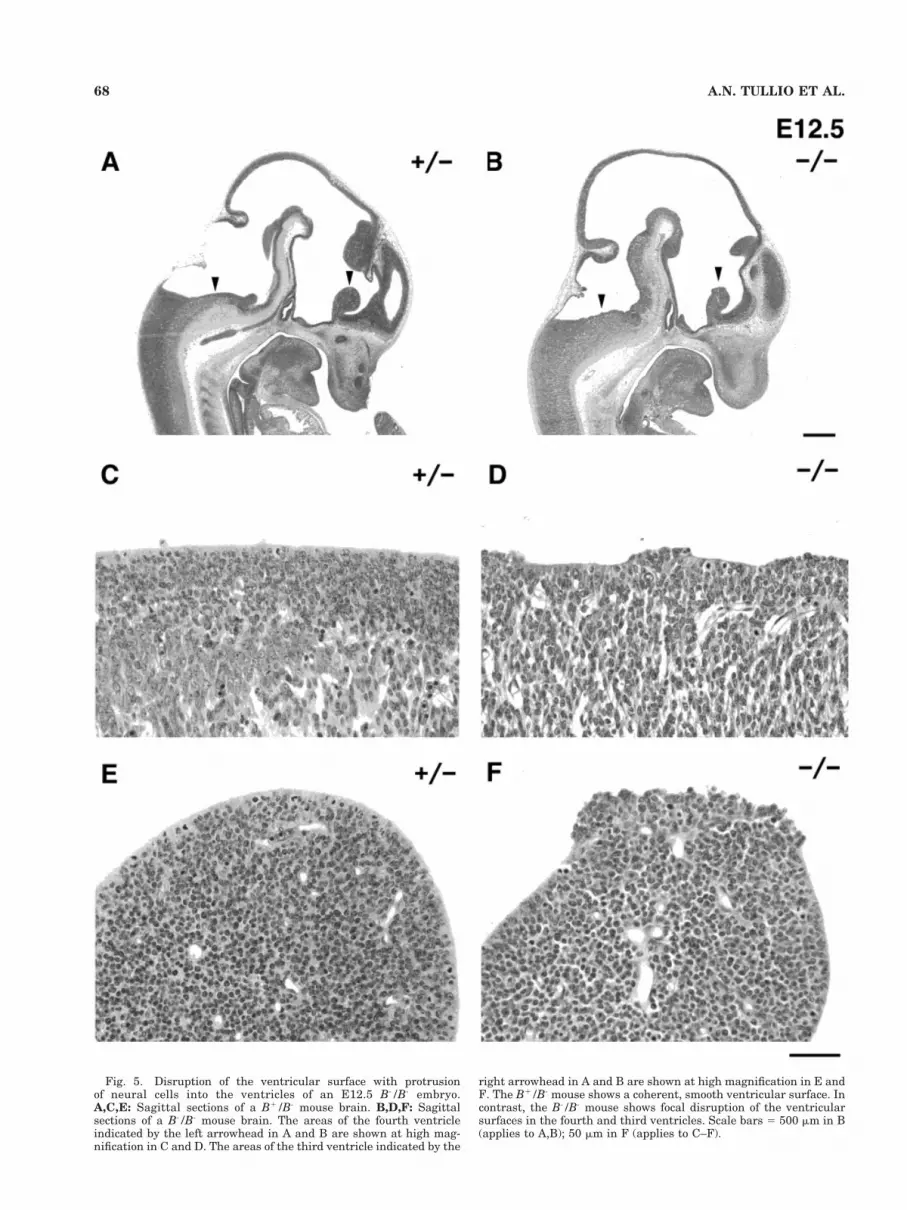
Fig. 5. Disruption of the ventricular surface with protrusionof neural cells into the ventricles of an E12.5 B-/B- embryo.A,C,E: Sagittal sections of a B1/B- mouse brain. B,D,F: Sagittalsections of a B-/B- mouse brain. The areas of the fourth ventricleindicated by the left arrowhead in A and B are shown at high mag-nification in C and D. The areas of the third ventricle indicated by the
right arrowhead in A and B are shown at high magnification in E andF. The B1/B- mouse shows a coherent, smooth ventricular surface. Incontrast, the B-/B- mouse shows focal disruption of the ventricularsurfaces in the fourth and third ventricles. Scale bars 5 500 mm in B(applies to A,B); 50 mm in F (applies to C–F).
68 A.N. TULLIO ET AL.

and retraction of neurites and the guidance of growthcones require cell adherence to the extracellular matrixand also require the dynamic reorganization of the actinfilaments (reviewed in Mueller, 1999). We have shownhere that the absence of NMHC-B results in disturbanceof these processes (Fig. 4), which is also consistent withthe work of others studying the function of nonmusclemyosin II and actin (reviewed in Burridge et al., 1997;Hall, 1998). Previous work has shown that both actin andmyosin II can be localized to the cortical region of neuronalcells (Rochlin et al., 1995) as well as other nonmuscle cells
(Kelley et al., 1996; Kolega, 1998). Moreover, neuronalcells are particularly enriched for NMHC-B comparedwith NMHC-A and the former has been shown to partici-pate in neurite retraction. This process can also be regu-lated through Rho GTPase signaling (reviewed in Hall,1998). For example, Hirose et al. (1998) demonstrated arequirement for Rho and the Rho-associated kinase inneurite remodeling. They showed that an increase in non-muscle myosin II light chain phosphorylation correlatedwith neurite retraction. Nonmuscle myosin II-B wasalso found to be responsible, at least in part, for neurite
Fig. 6. Rosette formations in the brain and retina of B-/B- mice.A–C: Rosette formation in the fourth ventricle in a brain of a B-/B-
mouse at embryonic day (E) 12.5. C: The residual ventricular surfacecan be seen at the bottom of the rosette. D,E: Rosettes in the retina of
a B-/B- mouse on postnatal day (P0). A ventricular-like structure canbe seen in the center of the rosette on the right in E. Hematoxylin andeosin stain. Scale bars 5 50 mm in C (applies to A–C), 335 mm in D, 85mm in E.
69NEURAL CELL DEFECTS IN B-/B- MICE

Fig. 7. Progression of protrusions of neural cells into the ventriclesof an embryonic day (E) 13.5 B-/B- brain. A: Coronal section of a B-/B-
brain at the level of the third ventricle. B: Large magnification view ofthe hypothalamic sulcus of the third ventricle indicated by an arrow-head in A. C: Coronal section of a B-/B- brain at the level of the fourthventricle. D: High-magnification view of the fourth ventricle indicated
by an arrowhead in C. In both cases, the ventricular surfaces areseverely disrupted and a mass of cells, composed of neuroepithelialand postmitotic cells, has protruded into the ventricle. It is notewor-thy that the abnormal protrusion of large neurons into the fourthventricle is highly reproducible in B-/B- mice. Hematoxylin and eosinstain. Scale bar 5 1 mm in A,C; 100 mm in B,D.
70 A.N. TULLIO ET AL.

Fig. 8. Formation of an aberrant ventricle at the level of thecerebral Aqueduct in an embryonic day (E) 15 B-/B- mouse. Coronalsections of the brain of a B-/B- mouse at E15 are shown at the level ofthe cerebral aqueduct from the anterior (A) to posterior (I) direction.Abnormal formation of the ventricle over the cerebral aqueduct (CA)is indicated by an arrowhead. Protrusion of neuroepithelial cells into
the aberrant ventricle is marked by an asterisk. An abnormal, largevessel has invaded this area (E–I). The ventricular surface was dis-rupted, and massive cell death is seen in the ventricular and subven-tricular zones. Note the presence of rosettes in the posterior midbrain(I). Hematoxylin and eosin stain. Scale bar 5 1 mm in I (applies toA–I).
outgrowth of a neuroblastoma cell line (Wylie et al.,1998).
It is noteworthy that defects similar to those shown inFigures 5 and 7, which resulted in protrusion of neuralcells, were found after the injection of antibodies toN-cadherin into the ventricle of the developing chick brain(Ganzler-Odenthal and Redies, 1998). Furthermore,N-cadherin–deficient mice manifested focal disruption ofthe ventricular surface and disordered cell migration atvery early stages of development (Radice et al., 1997). Inaddition, the injection of neuraminidase into the ventri-cles of rats resulted in the destruction of the ependymalcell layer and obstruction of the cerebral aqueduct as well
as dilation of the ventricles (Grondona et al., 1996, 1998).Taken together, these results suggest that defects in cellsurface molecules, especially in those proteins responsiblefor cell adhesion on the ventricular surface, could causedevelopmental abnormalities in the brain. Therefore, it isreasonable to suggest that NMHC-B is playing a role inmaintaining the integrity of the ventricular surface. Wespeculate that NMHC-B, which has been shown to bepresent in both neural and non-neural cells in the brain,can interact with the cortical actin filaments to enhanceactin-catenin-cadherin interactions. Absence of NMHC-B,at least in certain cells lining the ventricular and sub-pial surfaces, could weaken these interactions. Al-
Fig. 9. Destruction of the cerebral architecture due to hydroceph-alus in a late-term (embryonic day [E] 18.5) and newborn (postnatalday [P] 0) B-/B- mouse brain. Two levels of coronal sections of B-/B-
mouse brains at E18.5 (A,B) and P0 (C,D) are shown. A–C: Fusion orocclusion of the dorsal part of the third ventricle (seen in A,C) was the
likely cause of blockage of the cerebrospinal fluid, which leads toenlargement of the lateral ventricles and destruction of the cerebralcortex. The ventricular surface was severely disrupted and the ven-tricular and subventricular structures were destroyed. Scale bar 5 1mm in D (applies to A–D).
72 A.N. TULLIO ET AL.

though we have focused on N-cadherin and related pro-teins, we cannot exclude the possibility that absence ofNMHC-B might affect other adhesion proteins, such asN-CAM.
Pathogenesis of hydrocephalus in the earlyand late embryos
How do these findings help to explain the etiology ofhydrocephalus in the early embryos at E12.5 and E13.5 inB-/B- mice? At these stages, the ventricles are alreadyexpanded, although the ventricular system is still commu-nicating. One possibility is that NMHC-B, through inter-action with the actin cytoskeleton, plays an important rolein maintaining the cortical stiffness as well as in cell-celladhesion of the neuroepithelial cells. The reduction inmechanical tension in the B-/B- brain tissue and the de-crease of cell adherence could cause a disequilibrium be-tween the hydrostatic pressure of the CSF and the com-pliance of the tissue itself. As a result, the ventricles willbecome dilated (Van Essen, 1997; Kamiguchi et al., 1998).We also cannot rule out other possibilities such as de-creased absorption of the CSF. Because NMHC-B is highlyexpressed in the subarachnoid membrane, abnormal ab-sorption in this region is conceivable. However, no clearmorphologic abnormalities were identified at this stage inthis area. There is also the possibility of overproduction ofthe CSF. If this is the case, the sites of the defects may notbe in the choroidal plexus because the expansion of theventricle is seen before the development of the choroidalplexus, suggesting defects in other sites, such as the cap-illary endothelium. In any case, further work is needed tounderstand the etiology of hydrocephalus at the earlystages.
The development of hydrocephalus in the late embryo islikely due to the blockage of the CSF at the cerebralaqueduct or the dorsal part of the third ventricle. Typicalprogression of severe hydrocephalus was seen with a mas-sive dilation of the lateral and third ventricles and thedestruction of the ventricular and subventricular struc-tures in these late embryos. Most of the defects manifestedin the late embryos could be caused by a primary defect inthe ventricular surface of the neuroepithelial cells in theearly embryos. Early disruption of the ventricular surfaceof the cerebral aqueduct or the dorsal part of the thirdventricle could cause severe hydrocephalus in the latestages of development, because the ventricles in theseregions are very narrow.
Related models of hydrocephalus
Several animal models and human diseases that resultin dilated ventricles resemble, in some aspects, the abnor-malities described here for B-/B- mice. Targeted disrup-tion of two different proteins known to play a role incell-cell adhesion results in phenotypes similar to those wefound in B-/B- mice. These proteins are the neural celladhesion protein L1 and the actin-binding protein non-muscle filamin. L1 is a cell adhesion protein that interactswith the cytoskeletal linker protein ankyrin, which trans-duces signals from the extracellular environment to thecytoskeleton (Dahlin-Huppe et al., 1997; Kamiguchi et al.,1998; Fransen et al., 1998; Scotland et al., 1998). Muta-tions of the L1 gene in humans cause a variety of malfor-mations in the nervous system such as an X-linked hydro-cephalus (Fransen et al., 1998). A functional coupling,between L1 and the cytoskeletal protein ankyrin, was
suggested when ankyrin-ablated mice were shown to ex-hibit phenotypes similar to L1 (-/-) mice and human pa-tients with the L1 mutation (Scotland et al., 1998). Inaddition, mutations in nonmuscle filamin, a protein thatcrosslinks nonmuscle actin and, thereby, transducesligand-receptor binding into actin reorganization, hasbeen identified as a cause for the human disease, periven-tricular heterotopia (Fox et al., 1998). In this condition,neural cells fail to migrate to their target destinations andproliferate as nodules in the vicinity of the ventricles.Dilated ventricles are distinctive features of this disease.All these findings clearly indicate that disturbance of thecell adhesion complexes can cause developmental abnor-malities in the brain and can eventually lead to the com-mon phenotype of hydrocephalus. Of note is our findings ofdefects in neural cell adhesion and migration in a relatedmutant mouse model with only decreased amounts ofNMHC-B. Compared with B-/B- mice, the defects in theseNMHC-B hypomorphic mice were present with decreasedseverity and over a more protracted time course, whichincluded postnatal development (Uren et al., 2000).
In summary, our results suggest an important role fornonmuscle myosin II-B in cell adhesion and cell migrationin the developing brain. In the early embryo, we identifiedthe disruption of the ventricular surface of the neuroepi-thelial cells, possibly due to defects in cell adhesion. Thesedefects could lead to deformity of the cerebral aqueduct orthe dorsal part of the third ventricle, resulting in severehydrocephalus due to the blockage of CSF in late embryosor newborn mice. In later embryos or P0 mice, we alsoidentified a defect in axonal outgrowth. Taken together,these findings clearly indicate an essential role for non-muscle myosin II-B in brain development and suggest thatdefects in this gene could be responsible for similar devel-opmental diseases in humans.
ACKNOWLEDGMENTS
Y.H. is supported in part by SCF of STA for StrategicPromotion System for Brain Science and HSRG of MHWfor Specific Diseases (Intractable Hydrocephalus) in Ja-pan. This work was supported in part by a grant from NIH(NS26150) to P.C.B. The authors thank Alexander Grin-berg and Heiner Westphal for their help in generatingthese mice, Stefano Bertuzzi and Jerrold Ward for theirhelp and advice, and Catherine S. Magruder for experteditorial assistance. Antoine Smith provided expert tech-nical assistance.
LITERATURE CITED
Burridge K, Chrzanowska-Wodnicka M, Zhong CL. 1997. Focal adhesionassembly. Trends Cell Biol 7:342–347.
Cheng TPO, Murakami N, Elzinga M. 1992. Localization of myosin IIB atthe leading edge of growth cones from rat dorsal root ganglionic cells.FEBS Lett 311:91–94.
Dahlin-Huppe K, Berglund EO, Ranscht B, Stallcup WB. 1997. Mutationalanalysis of the L1 neuronal cell adhesion molecule identifiesmembrane-proximal amino acids of the cytoplasmic domain that arerequired for cytoskeletal anchorage. Mol Cell Neurosci 9:144–156.
Fox JW, Lamperti ED, Eksioglu YZ, Hong SE, Feng Y, Graham DA,Scheffer IE, Dobyns WB, Hirsch BA, Radtke RA, Berkovic SF, Hutten-locher PR, Walsh CA. 1998. Mutations in filamin 1 prevent migrationof cerebral cortical neurons in human periventricular heterotopia. Neu-ron 21:1315–1325.
Fransen E, D’Hooge R, Van Camp G, Verhoye M, Sijbers J, Reyniers E,Soriano P, Kamiguchi H, Willemsen R, Koekkoek SKE, De Zeeuw CI,
73NEURAL CELL DEFECTS IN B-/B- MICE

De Deyn PP, Van der Linden A, Lemmon V, Kooy RF, Willems PJ.1998. L1 knockout mice show dilated ventricles, vermis hypoplasia andimpaired exploration patterns. Hum Mol Genet 7:999–1009.
Ganzler-Odenthal SI, Redies C. 1998. Blocking N-cadherin function dis-rupts the epithelial structure of differentiating neural tissue in theembryonic chicken brain. J Neurosci 18:5415–5425.
Grondona JM, Perez-Martin M, Cifuentes M, Perez J, Jimenez AJ, Perez-Figares JM, Fernandez-Llebrez P. 1996. Ependymal denudation, aq-ueductal obliteration and hydrocephalus after a single injection ofneuraminidase into the lateral ventricle of adult rats. J Neuropath ExpNeurol 55:999–1008.
Grondona JM, Perez-Martin M, Cifuentes M, Perez J, Estivill-Torrus G,Perez-Figares JM, Fernandez-Llebrez P, Rodriguez EM. 1998. Neur-aminidase injected into the cerebrospinal fluid impairs the assembly ofthe glycoproteins secreted by the subcommissural organ preventing theformation of Reissner’s fiber. Histochem Cell Biol 109:391–398.
Hall A. 1998. RhoGTPases and the actin cytoskeleton. Science 279:509–514.
Hatten ME. 1999. Central nervous system neuronal migration. Annu RevNeurosci 22:511–539.
Hirose M, Ishizaki T, Watanabe N, Uehata M, Kranenburg O, MoolenaarWH, Matsumura F, Maekawa M, Bito H, Narumiya S. 1998. Moleculardissection of the rho-associated protein kinase (p160ROCK)-regulatedneurite remodeling in neuroblastoma N1E-115 cells. J Cell Biol 141:1625–1636.
Kamiguchi H, Hlavin ML, Lemmon V. 1998. Role of L1 in neural develop-ment: what the knockouts tell us. Mol Cell Neurosci 12:48–55.
Kelley CA, Sellers JR, Gard DL, Bui D, Adelstein RS, Baines IC. 1996.Xenopus nonmuscle myosin heavy chain isoforms have different sub-cellular localizations and enzymatic activities. J Cell Biol 134:675–687.
Kolega J. 1998. Cytoplasmic dynamics of myosin IIA and IIB: spatial“sorting” of isoforms in locomoting cells. J Cell Sci 111:2085–2095.
Lin CH, Espreafico EM, Mooseker MS, Forscher P. 1996. Myosin drivesretrograde F-actin flow in neuronal growth cones. Neuron 16:769–782.
Maupin P, Phillips CL, Adelstein RS, Pollard T. 1994. Differential local-ization of myosin-II isozymes in human cultured cells and blood cells.J Cell Sci 107:3077–3090.
Mermall V, Post PL, Mooseker MS. 1998. Unconventional myosins in cellmovement, membrane traffic and signal transduction. Science 279:527–533.
Miller M, Bower E, Levitt P, Li D, Chantler PD. 1992. Myosin II distribu-tion in neurons is consistent with a role in growth cone motility but notsynaptic vesicle mobilization. Neuron 8:25–44.
Mueller BK. 1999. Growth cone guidance: first steps towards a deeperunderstanding. Annu Rev Neurosci 22:351–388.
Phillips CL, Yamakawa K, Adelstein RS. 1995. Cloning of the cDNAencoding human nonmuscle myosin heavy chain-B and analysis ofhuman tissues with isoform-specific antibodies. J Muscle Res Cell Motil16:379–389.
Radice GL, Rayburn H, Matsunami H, Knudsen KA, Takeichi M, HynesRO. 1997. Developmental defects in mouse embryos lackingN-cadherin. Dev Biol 181:64–78.
Rochlin MW, Itoh K, Adelstein RS, Bridgman PC. 1995. Localization ofmyosin II A and B isoforms in cultured neurons. J Cell Sci 108:3661–3670.
Rochlin MW, Wickline KM, Bridgman PC. 1996. Microtubule stabilitydecreases axon elongation but not axoplasm production. J Neurosci16:3236–3246.
Scotland P, Zhow D, Benveniste H, Bennett V. 1998. Nervous systemdefects of ankyrin b (-/-) mice suggest functional overlap between thecell adhesion molecule L1 and 440-kD ankyrin b in premyelinatedaxons. J Cell Biol 143:1305–1315.
Sellers JR. 1999. Myosins, 2nd ed. Oxford, UK: Oxford University Press.Takeda K, Yu Z-X, Qian S, Chin TK, Adelstein RS, Ferrans VJ. 2000.
Nonmuscle myosin II localizes to the Z-lines and intercalated discs ofcardiac muscle and to the Z-line of skeletal muscle. Cell Motil Cytoskel46:59–68.
Tullio AN, Accili D, Ferrans VJ, Yu Z-X, Takeda K, Grinberg A, WestphalH, Preston YA, Adelstein RS. 1997. Nonmuscle myosin II-B is requiredfor normal development of the mouse heart. Proc Natl Acad Sci USA94:12407–12412.
Uren D, Hwang H-K, Hara Y, Takeda K, Kawamoto S, Tullio AN, Yu Z-X,Ferrans VJ, Tresser N, Grinberg A, Preston YA, Adelstein RS. 2000.Gene dosage affects the cardiac and brain phenotype in nonmusclemyosin II-B depleted mice. J Clin Invest 105:663–671.
Van Essen DC. 1997. A tension-based theory of morphogenesis and com-pact wiring in the central nervous system. Nature 385:313–318.
Wylie SR, Wu P-J, Patel H, Chantler PD. 1998. A conventional myosinmotor drives neurite outgrowth. Proc Natl Acad Sci USA 95:12967–12972.
74 A.N. TULLIO ET AL.





























