Embed Size (px)
Citation preview

doi:10.1006/jmbi.2001.4499 available online at http://www.idealibrary.com on J. Mol. Biol. (2001) 307, 707±720
Structures of Bovine Glutamate DehydrogenaseComplexes Elucidate the Mechanism ofPurine Regulation
Thomas J. Smith1*, Peter E. Peterson1, Timothy Schmidt1, Jie Fang2
and Charles A. Stanley2
1Department of BiologicalSciences, Purdue UniversityWest Lafayette, IN 47907, USA2The Children`s Hospital ofPhiladelphia, EndocrinologyDivision, Rm. 410D, 3516Civic Center Blvd. PhiladelphiaPA 19104, USA
E-mail address of the [email protected]
0022-2836/01/0020707±14 $35.00/0
Glutamate dehydrogenase is found in all organisms and catalyses theoxidative deamination of L-glutamate to 2-oxoglutarate. However, onlyanimal GDH utilizes both NAD(H) or NADP(H) with comparable ef®-cacy and exhibits a complex pattern of allosteric inhibition by a wide var-iety of small molecules. The major allosteric inhibitors are GTP andNADH and the two main allosteric activators are ADP and NAD�. Thestructures presented here have re®ned and modi®ed the previous struc-tural model of allosteric regulation inferred from the originalboGDH �NADH �GLU �GTP complex. The boGDH �NAD��a-KG complexstructure clearly demonstrates that the second coenzyme-binding site liesdirectly under the ``pivot helix'' of the NAD� binding domain. In thiscomplex, phosphates are observed to occupy the inhibitory GTP site andmay be responsible for the previously observed structural stabilization bypolyanions. The boGDH �NADPH �GLU �GTP complex shows the locationof the additional phosphate on the active site coenzyme molecule and theGTP molecule bound to the GTP inhibitory site. As expected, sinceNADPH does not bind well to the second coenzyme site, no evidence ofa bound molecule is observed at the second coenzyme site under thepivot helix. Therefore, these results suggest that the inhibitory GTP site isas previously identi®ed. However, ADP, NAD�, and NADH all bindunder the pivot helix, but a second GTP molecule does not. Kineticanalysis of a hyperinsulinism/hyperammonemia mutant stronglysuggests that ATP can inhibit the reaction by binding to the GTP site.Finally, the fact that NADH, NAD�, and ADP all bind to the same siterequires a re-analysis of the previous models for NADH inhibition.
# 2001 Academic Press
Keywords: allostery; glutamate dehydrogenase; purine regulation;hyperinsulinism
*Corresponding authorIntroduction
Glutamate dehydrogenase plays a pivotal role innitrogen and carbon metabolism.1 In the oxidativedeamination reaction, GDH feeds the TCA cycle byconverting L-glutamate to 2-oxoglutarate, whereasthe reductive amination reaction may supply nitro-gen for several biosynthetic pathways. In yeast andmost bacteria, separate forms of enzyme are madeto utilize either NAD(H) or NADP(H) dependingupon the metabolic needs of the organisms.2 Somearchaebacteria are the exception to this rule as thecoenzyme Vmax/Km ratios differ by only about one
ing author:
order of magnitude.3,4 Mammalian GDH utilizeseither coenzyme with nearly identical ef®cacy.
Bovine GDH is the most extensively studied ver-tebrate form and has been shown to exhibit com-plex homotropic and heterotropic allostericregulation. Kinetic and binding analyses haveclearly shown that NAD� and NADP� exhibitnegatively cooperative binding in the presence ofglutamate or glutarate.5 ± 7 In the reductive amin-ation reaction, evidence for such negative coopera-tivity is only found in binding studies.6,8 NAD(P)Hbinding is clearly negatively cooperative at pH 8.0,but not at pH 7.0. In the presence of glutamate,NAD(P)H binding is greatly enhanced and exhibitsnegative cooperativity at both pHs. In the presence
# 2001 Academic Press

708 Purine Regulation of Glutamate Dehydrogenase
of glutarate, NAD(P)H binding is also enhanced,but does not promote observable negative coopera-tivity. This suggests that the substituent on the a-carbon of the substrate plays some role in theinduction of negative cooperativity.8
In both the oxidative deamination and the reduc-tive amination reactions, the data suggest that thekinetic mechanism of GDH is rapid equilibrium,random order.9 ± 12 The oxidative deamination reac-tion has been described as a four-phase process.13
The ®rst phase is marked by a burst of protonrelease after substrate binding. The second phase ishydride transfer. The third phase is very slow andis where the product, 2-oxoglutarate, is replaced byglutamate before the reduced coenzyme dis-sociates. The ®nal phase is the steady-state releaseof free, reduced coenzyme. At neutral to high pH,or when substrate concentrations are high, the res-olution of the non-catalytic complex in the thirdphase, GDH �glutamate �NAD(P)H, is much slowerthan hydride transfer. This is made evident by apre-steady-state burst of this red-shifted species. Atlow substrate concentrations or acidic pHs, theelimination of this pre-steady-state burst impliesthat the hydride transfer step becomes the rate-limiting step. During this catalytic process, theNAD domain undergoes a large conformationalchange that opens and closes the catalytic cleft.13,14
It has been suggested that this coenzyme/substratedriven conformational change facilitates catalysisby forcing the two ligands together and dehydrat-ing the catalytic cleft.13
Mammalian GDH is tightly controlled by a num-ber of allosteric regulators. GTP is a potent inhibi-tor of the enzyme and inhibits enzyme turnoverover a wide range of conditions by increasing theaf®nity of the enzyme for the reaction product,15
making product release rate-limiting under all con-ditions in the presence of GTP. In contrast, ADPactivates the enzyme by facilitating productrelease.15,16 When the enzyme is highly saturatedwith substrate, an inhibitory abortive complexforms in the active site: NAD(P)H �glutamate in theoxidative deamination reaction at high pH andNAD(P)��a-KG in the reductive amination reactionat low pH.17 Under these conditions, ADP isa potent activator by decreasing product af®nityand allowing the enzyme to reconcile these non-catalytic complexes.
In addition to the catalytic site, coenzymeappears bind to one or two additional sites persubunit. NADH has been shown to bind to asecond, inhibitory site.18,19 NADPH can also bindto this site, but with at least a tenfold higher dis-sociation constant.18,19 Binding studies havesuggested that when NADH is used as coenzyme,there are two GTP sites per subunit20 and GTPinhibition is agonized by synergistic bindingbetween the second coenzyme and one of the GTPbinding sites.21 Paradoxically, NAD� can also bindto a second, allosteric site, but instead causesactivation. Again, as with NADPH, NADP� bindswith much less af®nity to this activation site than
NAD�. These observations suggest that if coen-zyme binds to only one allosteric site, then acti-vation/inhibition is entirely controlled by theoxidative state of the nicotinamide ring. Alterna-tively, there may be two different allosteric sitesand binding selectivity is conferred by the coen-zyme oxidation state. It has been suggested thatthe second NAD� site is equivalent to the ADPactivation site.16,22
The structures of several bacterial forms of GDHand bovine GDH complexed with NADH, gluta-mate, and GTP have been determined.23 ± 28 Themajor difference between bovine GDH and thesebacterial forms was a 48 residue domain in bovineGDH that forms an ``antenna'' structure extendingfrom the top of the NAD domain and lies immedi-ately adjacent to the 3-fold axis of the hexamer. Itwas suggested that this antenna may be involvedin some or all of the allosteric regulation that isunique to mammalian GDH. From the proposedlocations of the GTP and ADP sites in the bovineGDH structure (Figure 1), it was suggested thatthese allosteric regulators exert their effects bychanging the energy required to open and closethe catalytic cleft during enzymatic turnover.28
Recent studies on a form of congenital hyperin-sulinism, that is characterized by hypoglycemiaand hyperammonemia, suggests that GDH plays arole in both ammonia metabolism and insulinhomeostasis.29 Infants with this disorder produceforms of GDH that are much less sensitive to inhi-bition by GTP. It was proposed that this alteredform of GDH would lead to higher oxidation ratesdue to increased levels of 2-oxoglutarate. Thisincrease in the ATP/ADP ratio would close the K�
channels and lead to depolarization of the pancrea-tic b-cells. This depolarization causes calciumin¯ux and the release of stored insulin granules.The mutated residues responsible for this pathol-ogy were found to lie mainly around the GTP no.1 site.28
Presented here is the re®ned structure of theboGDH �NADH �GLU �GTP complex and the struc-tures of the boGDH �NADPH �GLU �GTP andboGDH �NAD��a-KG complexes. While the overallconformations of these enzyme complexes arenearly identical, there are subtle differences in thebinding environments of the active site ligands.These complexes con®rm our previous assignmentof the inhibitory GTP site, however they refute ourprevious conclusion that a second GTP moleculebinds under the pivot helix.28 The data nowstrongly demonstrate that NADH, NAD�, andprobably ADP all bind to this site. The weak den-sity previously observed in the core of the hexameris present in all of these complexes and thereforedoes not represent a regulatory ligand. From theseresults, we present a model to explain the complexallosteric regulation of boGDH by purine nucleo-tides and a re-evaluation of the role of the secondcoenzyme-binding site.

Purine Regulation of Glutamate Dehydrogenase 709
Results
The boGDH �NADH �GLU �GTP complex
The previously reported boGDH �NADH �GLU �GTP complex was further re®ned using X-PLORand several changes were made in the model. Thecurrent re®nement statistics are shown in Table 1.For each subunit, NADH and glutamate have beenmodeled into the active site, one GTP and oneNADH molecule have been placed in their regulat-ory sites and 55 water molecules per subunit wereadded in the later stages of re®nement. All of theresidues with poor geometrical characteristics lie inrelatively disordered loops. All other geometricalparameters are within, or better than, thevalues found in other proteins determined to thisresolution.30
After further re®nement and real-space aver-aging, weak, but interpretable, density wasobserved for residues 1-5 and these residues arenow included in the structure. Due to weak den-sity, presumably because of a number of glycineresidues, the descending strand of the antenna(residues 421-444) was incorrectly built and hasnow been corrected. However, the residues at thevery top of the antenna are badly disordered andhave very high B values. During re®nement, theweak density around the loop comprising residues25-40 improved slightly and was rebuilt.
The substrate and coenzyme interactions withthe enzyme at the active site have only changedslightly during re®nement (Figure 2). The majordifference is that several water molecules are nowclearly visible in the 6-fold averaged, omit maps.One water molecule lies between the carbonyl oxy-gen of N374 and the carboxyl group of the boundglutamate. This is still different from what wasobserved in Clostridium symbiosum (csGDH) wherethe NZ atom of K126 and the carbonyl oxygen ofG92 (K125 and G91 in csGDH) directly interactwith the substrate and not via a bound watermolecule 14. Another water molecule is observedbetween the nicotinamide amide and the carbonyl
Table 1. Data and re®nement statistics
NADH �GLU �GTP
Resolution range (AÊ ) 30-2.5Completeness (%) 77 (33)R-factor (%) 10.8 (25.9)6-fold averaging
C.C. (%) 92R-factor (%) 18
Model Rwork (%) 17Model Rfree (%) 23RMS bond error (AÊ ) 0.008RMS angular error (�) 1.6Ramachandran
% favored 88.6% additional 9.7% generous 0.7% disallowed 0.9
The numbers in parenthesis denote the statistics in the highest reso
oxygen of S171. Finally, a water molecule is boundbetween the amide of M169 and the oxygen ofb-phosphate of the coenzyme.
In the prior model, the majority of the contactsbetween the enzyme and the bound GTP werewith the triphosphate moiety and relatively fewinteractions with the purine ring. However, in there®ned model E292 hydrogen bonds with the N1imino group and the C2 amino of the purine ring(Figure 3). In addition, K289 forms a hydrogenbond with the C6 carbonyl oxygen of the purinering. While it does not hydrogen bond to thebound GTP directly, the guanidinium group ofR261 stacks against the purine ring. Since themajority of the interactions between the enzymeand the bound GTP are with the g-phosphate oxy-gen atoms, it is clear that this site is an ``energysensor'' that preferentially binds triphosphatenucleotides. These newly observed interactionswith the guanosine ring would probably not pre-clude the binding of adenosine, but will clearlyfavor the binding of guanosine over adenosine.
Finally, the re®ned density combined with theresults with the GDH �NAD��aKG complex (seebelow), has shed some doubt on our previous con-clusion that the second NADH molecule binds tothe inner core of the enzyme. While there is stillisolated density within the core of the enzyme, itdid not improve with re®nement. In the previousmodel, a second GTP molecule was modeled intothe region below the pivot helix that lies behindthe NAD binding domain. Upon further re®ne-ment, weak density was observed to extend downinto the interface between each adjacent pair ofsubunits within the trimers and upwards from theb-phosphate. The strongest evidence that this wasnot a bound GTP molecule came from the fact thatnearly identical electron density was obtained fromthe GDH �NAD��aKG complex (Figure 4). This isin spite of the fact that the enzyme was dialyzedextensively against sodium phosphate buffer.Therefore, it appears that this is the second coen-zyme site rather than a second GTP site. It is
NAD��aKG NADPH �GLU �GTP
30-3.2 30-2.879 (80) 70.1 (77.6)
12.3 (47.5) 6.3(11.6)
82 8628 3023 2529 30
0.010 0.0101.6 1.7
82.8 83.125.5 12.81.2 3.50.5 0.7
lution shell.

Figure 1. Stereo image of thebovine GDH hexamer complexedwith NADH, glutamate, GTP. TheCa backbones of each of the sixsubunits are shown in different col-ors. The 3-fold axis of the hexamerlies along the vertical axis. Thebound GTP is represented by theyellow CPK model, the glutamateis orange, the active site NADH iscyan, and the second coenzyme siteis shown in mauve.
710 Purine Regulation of Glutamate Dehydrogenase
important to note that the atoms common to ADPhave the strongest electron density, causing theprevious error in interpretation.
The GDH �NAD��aaaKG complex
There were several reasons to examine the struc-ture of this complex. Firstly, it represents the abor-tive complex found in the reductive aminationreaction. Secondly, this complex should locate thesecond NAD� binding site and determine whether
it is the same as the location as the second, inhibi-tory NADH.
This complex was re®ned to a resolution of3.2 AÊ . The data and re®nement statistics are sum-marized in Table 1. Using the program MolView,31
the structure of one subunit of the NADH �GLU �GTP complex was compared to one of theNAD��aKG complex. Using all of the residues, theRMS deviation in the Ca backbone is 0.39 AÊ . Therewas no evidence that the domains of the varioussubunits adopted different orientations. In
Figure 2. Re®ned structure of theactive site ligands in the NADH �GLU �GTP complex. (a) Schematicdiagram of the bound ligands inthe active site and their contactswith the enzyme. In this orien-tation, the NAD binding domain istowards the top of the diagram, theglutamate towards the bottom, andthe 3-fold axis runs approximatelyin the vertical direction towards theleft side of the image. (b) 6-Foldaveraged, electron density wherethese ligands had been omittedfrom structure factor calculations(omit map). The stick model iscolor coded according to atomtypes; oxygen is read, nitrogen isblue, carbon is yellow, and phos-phates are green. Some of thewater molecules are also includedand represented by red balls.
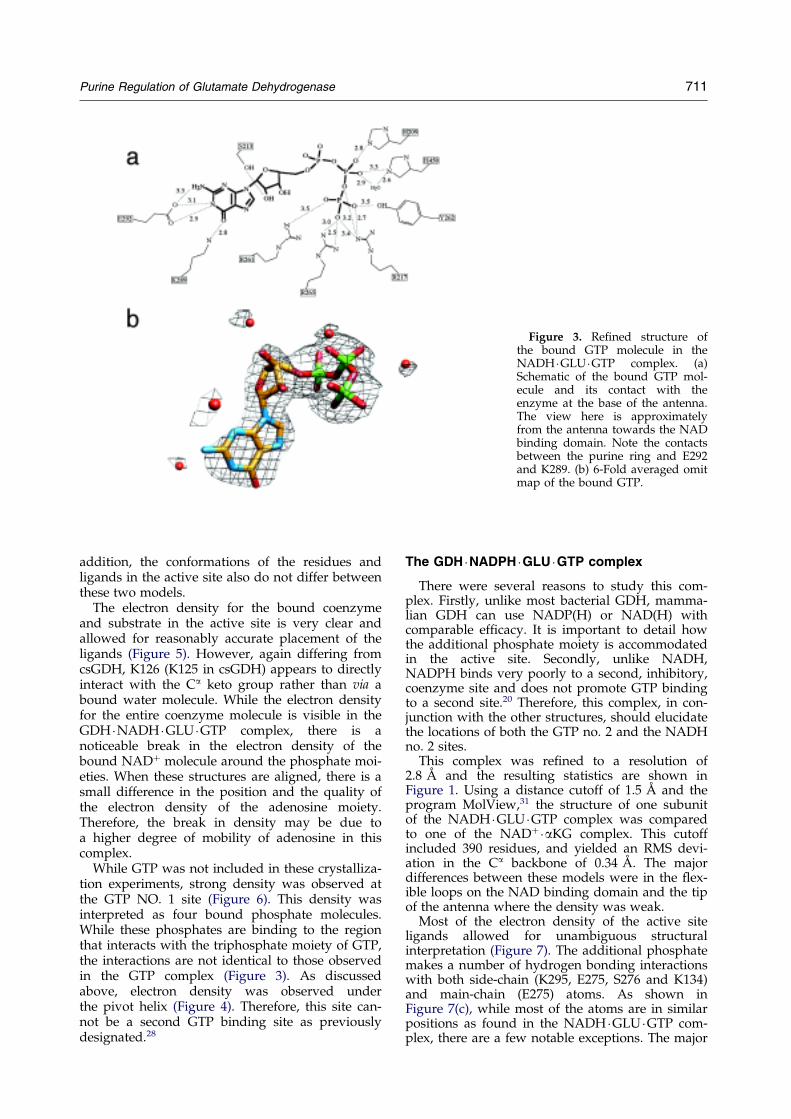
Figure 3. Re®ned structure ofthe bound GTP molecule in theNADH �GLU �GTP complex. (a)Schematic of the bound GTP mol-ecule and its contact with theenzyme at the base of the antenna.The view here is approximatelyfrom the antenna towards the NADbinding domain. Note the contactsbetween the purine ring and E292and K289. (b) 6-Fold averaged omitmap of the bound GTP.
Purine Regulation of Glutamate Dehydrogenase 711
addition, the conformations of the residues andligands in the active site also do not differ betweenthese two models.
The electron density for the bound coenzymeand substrate in the active site is very clear andallowed for reasonably accurate placement of theligands (Figure 5). However, again differing fromcsGDH, K126 (K125 in csGDH) appears to directlyinteract with the Ca keto group rather than via abound water molecule. While the electron densityfor the entire coenzyme molecule is visible in theGDH �NADH �GLU �GTP complex, there is anoticeable break in the electron density of thebound NAD� molecule around the phosphate moi-eties. When these structures are aligned, there is asmall difference in the position and the quality ofthe electron density of the adenosine moiety.Therefore, the break in density may be due toa higher degree of mobility of adenosine in thiscomplex.
While GTP was not included in these crystalliza-tion experiments, strong density was observed atthe GTP NO. 1 site (Figure 6). This density wasinterpreted as four bound phosphate molecules.While these phosphates are binding to the regionthat interacts with the triphosphate moiety of GTP,the interactions are not identical to those observedin the GTP complex (Figure 3). As discussedabove, electron density was observed underthe pivot helix (Figure 4). Therefore, this site can-not be a second GTP binding site as previouslydesignated.28
The GDH �NADPH �GLU �GTP complex
There were several reasons to study this com-plex. Firstly, unlike most bacterial GDH, mamma-lian GDH can use NADP(H) or NAD(H) withcomparable ef®cacy. It is important to detail howthe additional phosphate moiety is accommodatedin the active site. Secondly, unlike NADH,NADPH binds very poorly to a second, inhibitory,coenzyme site and does not promote GTP bindingto a second site.20 Therefore, this complex, in con-junction with the other structures, should elucidatethe locations of both the GTP no. 2 and the NADHno. 2 sites.
This complex was re®ned to a resolution of2.8 AÊ and the resulting statistics are shown inFigure 1. Using a distance cutoff of 1.5 AÊ and theprogram MolView,31 the structure of one subunitof the NADH �GLU �GTP complex was comparedto one of the NAD��aKG complex. This cutoffincluded 390 residues, and yielded an RMS devi-ation in the Ca backbone of 0.34 AÊ . The majordifferences between these models were in the ¯ex-ible loops on the NAD binding domain and the tipof the antenna where the density was weak.
Most of the electron density of the active siteligands allowed for unambiguous structuralinterpretation (Figure 7). The additional phosphatemakes a number of hydrogen bonding interactionswith both side-chain (K295, E275, S276 and K134)and main-chain (E275) atoms. As shown inFigure 7(c), while most of the atoms are in similarpositions as found in the NADH �GLU �GTP com-plex, there are a few notable exceptions. The major

Figure 4. Re®ned structure ofNADH bound at the second coen-zyme site beneath the pivot helix inthe NADH �GLU �GTP complex.(a) One of the two conformationsof the bound coenzyme. In thisorientation, the ribose-nicotinamidemoiety points down towards theintra-trimer subunit interface. Notethat due to the poor quality of thenicotinamide-ribose electron den-sity, the interactions in this regionshould only be used to denote theapproximate binding environment.(b) The 6-fold averaged omit mapelectron density (black lines) withthe two conformations of thebound NADH. The model with thenicotinamide-ribose moiety point-ing down is that which is rep-resented in (a). (c) The electrondensity and ®tted coenzyme modelfrom the NAD��a-KG complex.Note that the electron density ishighly similar for both complexes.
712 Purine Regulation of Glutamate Dehydrogenase
difference is in the position of the nicotinamidering. There appears to be two portions of electrondensity for this ring (only the stronger of the two isshown in Figure 7(b)) and neither is equivalent tothat observed with the other complexes. In thisposition, the nicotinamide ring appears to havepushed the glutamate into a slightly different con-formation, causing it to lose some interactions suchas with K114. This suggests that the conformationsof these abortive complexes are subtly different,but it is not clear whether these differences wouldbe also found in the true catalytic complexes.
The locations of the other ligands were consist-ent with results described above. The location andinteractions of the bound GTP were almost identi-cal to that observed in the NADH �GLU �GTP com-plex. However, the lack of observable differencesbetween these complexes implies that the NADH/GTP synergism may not be due to conformationalrearrangements. No density was observed at whatis now de®ned as the second coenzyme site. This isprobably due to the fact that there is not suf®cientspace in the second coenzyme-binding site toaccommodate the additional phosphate. As withthe other two complexes, diffuse density was
observed in the core of the hexamer. Since thiscomplex is not expected to bind either a secondcoenzyme or a second GTP molecule, it is unlikelythat this density represents a bound regulatoryligand in any of the complexes.
ATP/ADP effects on human GDH activity
Paradoxically, ADP and ATP have been shownto have opposite effects on GDH activity. At pH 8.0and high NADH concentrations, ADP increases thereductive amination reaction velocity by aboutthreefold, whereas ATP inhibits the reaction byabout fourfold.19 Since the g-phosphate of GTPdominates the GTP/GDH interactions (Figure 3), itis possible that ATP is binding to the GTP site. Totest this hypothesis, the steady-state velocity of thereductive amination reaction was measured in thepresence and absence of varying ATP or ADP con-centrations using wild-type and the H454Y (H450in boGDH) mutant forms of human GDH. TheH454Y mutation was observed in hyperinsulin-ism/hyperammonemia (HI/HA) patients anddesensitizes the enzyme to GTP inhibition.29 H454interacts with the b-phosphate of GTP (Figure 3)
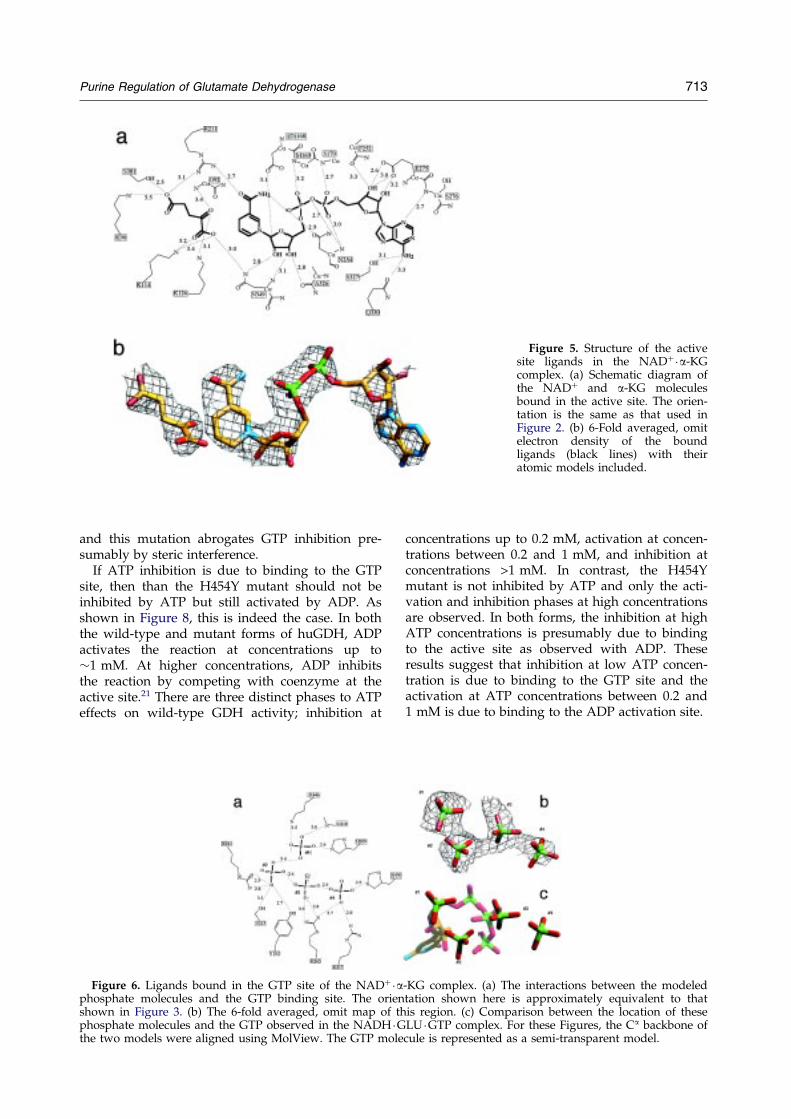
Figure 5. Structure of the activesite ligands in the NAD��a-KGcomplex. (a) Schematic diagram ofthe NAD� and a-KG moleculesbound in the active site. The orien-tation is the same as that used inFigure 2. (b) 6-Fold averaged, omitelectron density of the boundligands (black lines) with theiratomic models included.
Purine Regulation of Glutamate Dehydrogenase 713
and this mutation abrogates GTP inhibition pre-sumably by steric interference.
If ATP inhibition is due to binding to the GTPsite, then than the H454Y mutant should not beinhibited by ATP but still activated by ADP. Asshown in Figure 8, this is indeed the case. In boththe wild-type and mutant forms of huGDH, ADPactivates the reaction at concentrations up to�1 mM. At higher concentrations, ADP inhibitsthe reaction by competing with coenzyme at theactive site.21 There are three distinct phases to ATPeffects on wild-type GDH activity; inhibition at
Figure 6. Ligands bound in the GTP site of the NAD��aphosphate molecules and the GTP binding site. The orienshown in Figure 3. (b) The 6-fold averaged, omit map of thphosphate molecules and the GTP observed in the NADH �Gthe two models were aligned using MolView. The GTP mole
concentrations up to 0.2 mM, activation at concen-trations between 0.2 and 1 mM, and inhibition atconcentrations >1 mM. In contrast, the H454Ymutant is not inhibited by ATP and only the acti-vation and inhibition phases at high concentrationsare observed. In both forms, the inhibition at highATP concentrations is presumably due to bindingto the active site as observed with ADP. Theseresults suggest that inhibition at low ATP concen-tration is due to binding to the GTP site and theactivation at ATP concentrations between 0.2 and1 mM is due to binding to the ADP activation site.
-KG complex. (a) The interactions between the modeledtation shown here is approximately equivalent to thatis region. (c) Comparison between the location of theseLU �GTP complex. For these Figures, the Ca backbone ofcule is represented as a semi-transparent model.

Figure 7. Active site ligands inthe NADPH �GLU �GTP complex.(a) Schematic of the active site-ligand interactions. The orientationshown here is the same as thatused in Figures 2 and 5. (b) 6-Foldaveraged omit map of the activesite ligands. Note the differences inthe nicotinamide orientation andcontacts. There is a peak of isolateddensity above the nicotinamidering (not shown in this Figure) thatmay represent a second orientation.This orientation was chosen since itoffered a better ®t for the nicotina-mide and ribose moiety. (c) Com-parison between the conformationsof the NADH �GLU (transparentmodel) and the NADPH �GLUligands. For this comparison,MolView was used to align the Ca
backbones.
714 Purine Regulation of Glutamate Dehydrogenase
Discussion
Active site
When the Ca backbones of the three differentcomplexes were compared, few signi®cantdifferences were found. In all of these abortivecomplexes, K126 directly interacts with the sub-strate a-carbon substituent (Figures 2, 5 and 7).This is in contrast to the csGDH �GLU complex,where homologous residues interact via a boundwater molecule.14 In the boGDH �NADH �GLU �GTP complex, a water molecule is observed tointeract with the carbonyl group of N374. Previoussequence data had suggested that residue 198 wasAsn in boGDH.32 However, subsequent sequencedata on all other forms of GDH strongly suggestthat this residue is probably an Asp. Therefore, wewill designate this residue as (D)168. While D165in csGDH is observed to directly interact with thesubstrate, the side-chain of (D)168 in all of theseboGDH complexes is rotated somewhat away fromthe substrate and interacts more with the ribosemoiety. The current distance between (D)168 andthe substrate Ca moiety is >4 AÊ in all of these com-plexes. As previously noted,28 the active sites aremore ``closed'' in these boGDH abortive complexesthan the csGDH �GLU complex.14 This differencemay be imposed by the abortive complexes or due
to subtle differences in the timing of the hydridetransfer step.
An unexpected result was the difference in nico-tinamide position between the di- and triphosphateforms of coenzyme. In both the NAD� and theNADH complexes, the electron density of the nico-tinamide ring is clear. In contrast, there are two``islands'' of density for the nicotinamide ring inthe NADPH complex, suggesting that it binds intwo conformations. Neither of these islands corre-spond to the location of the ring in the NAD(H)complexes. Identical results were obtained usingan independent data set collected on this complex(data not shown). This may be a consequence ofthe subtle changes in coenzyme binding imposedby the additional phosphate or it may be that thesetwo forms of coenzyme form subtly different abor-tive complexes. This might also explain the slightpreference that the enzyme has for NAD(H) overNADP(H).33
GTP inhibition
The location of the HI/HA mutations that abro-gate GTP inhibition and the clarity of the densityof the bound GTP molecule unequivocally demon-strate that this is the GTP inhibition site. We pro-pose that this site is actually an ``energy sensor''.As shown in Figure 8, ATP inhibits the reductive
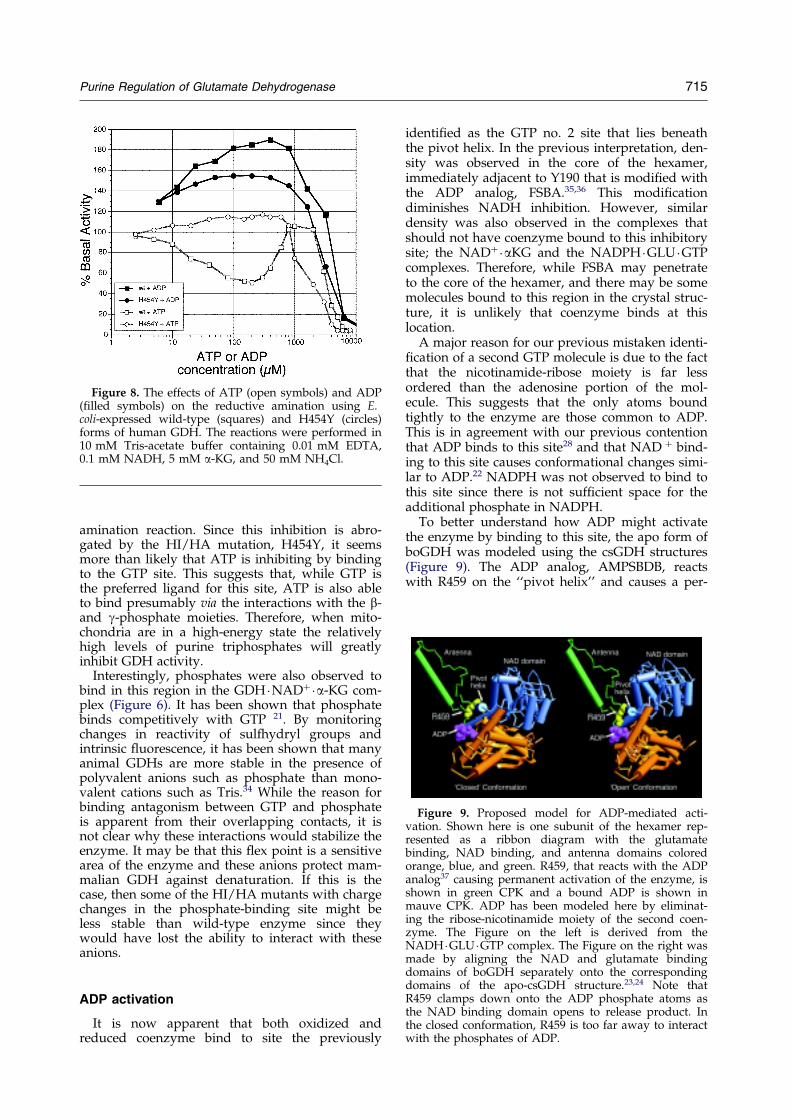
Figure 9. Proposed model for ADP-mediated acti-vation. Shown here is one subunit of the hexamer rep-resented as a ribbon diagram with the glutamatebinding, NAD binding, and antenna domains coloredorange, blue, and green. R459, that reacts with the ADPanalog37 causing permanent activation of the enzyme, isshown in green CPK and a bound ADP is shown inmauve CPK. ADP has been modeled here by eliminat-ing the ribose-nicotinamide moiety of the second coen-zyme. The Figure on the left is derived from theNADH �GLU �GTP complex. The Figure on the right wasmade by aligning the NAD and glutamate bindingdomains of boGDH separately onto the correspondingdomains of the apo-csGDH structure.23,24 Note thatR459 clamps down onto the ADP phosphate atoms asthe NAD binding domain opens to release product. Inthe closed conformation, R459 is too far away to interactwith the phosphates of ADP.
Figure 8. The effects of ATP (open symbols) and ADP(®lled symbols) on the reductive amination using E.coli-expressed wild-type (squares) and H454Y (circles)forms of human GDH. The reactions were performed in10 mM Tris-acetate buffer containing 0.01 mM EDTA,0.1 mM NADH, 5 mM a-KG, and 50 mM NH4Cl.
Purine Regulation of Glutamate Dehydrogenase 715
amination reaction. Since this inhibition is abro-gated by the HI/HA mutation, H454Y, it seemsmore than likely that ATP is inhibiting by bindingto the GTP site. This suggests that, while GTP isthe preferred ligand for this site, ATP is also ableto bind presumably via the interactions with the b-and g-phosphate moieties. Therefore, when mito-chondria are in a high-energy state the relativelyhigh levels of purine triphosphates will greatlyinhibit GDH activity.
Interestingly, phosphates were also observed tobind in this region in the GDH �NAD��a-KG com-plex (Figure 6). It has been shown that phosphatebinds competitively with GTP 21. By monitoringchanges in reactivity of sulfhydryl groups andintrinsic ¯uorescence, it has been shown that manyanimal GDHs are more stable in the presence ofpolyvalent anions such as phosphate than mono-valent cations such as Tris.34 While the reason forbinding antagonism between GTP and phosphateis apparent from their overlapping contacts, it isnot clear why these interactions would stabilize theenzyme. It may be that this ¯ex point is a sensitivearea of the enzyme and these anions protect mam-malian GDH against denaturation. If this is thecase, then some of the HI/HA mutants with chargechanges in the phosphate-binding site might beless stable than wild-type enzyme since theywould have lost the ability to interact with theseanions.
ADP activation
It is now apparent that both oxidized andreduced coenzyme bind to site the previously
identi®ed as the GTP no. 2 site that lies beneaththe pivot helix. In the previous interpretation, den-sity was observed in the core of the hexamer,immediately adjacent to Y190 that is modi®ed withthe ADP analog, FSBA.35,36 This modi®cationdiminishes NADH inhibition. However, similardensity was also observed in the complexes thatshould not have coenzyme bound to this inhibitorysite; the NAD��aKG and the NADPH �GLU �GTPcomplexes. Therefore, while FSBA may penetrateto the core of the hexamer, and there may be somemolecules bound to this region in the crystal struc-ture, it is unlikely that coenzyme binds at thislocation.
A major reason for our previous mistaken identi-®cation of a second GTP molecule is due to the factthat the nicotinamide-ribose moiety is far lessordered than the adenosine portion of the mol-ecule. This suggests that the only atoms boundtightly to the enzyme are those common to ADP.This is in agreement with our previous contentionthat ADP binds to this site28 and that NAD � bind-ing to this site causes conformational changes simi-lar to ADP.22 NADPH was not observed to bind tothis site since there is not suf®cient space for theadditional phosphate in NADPH.
To better understand how ADP might activatethe enzyme by binding to this site, the apo form ofboGDH was modeled using the csGDH structures(Figure 9). The ADP analog, AMPSBDB, reactswith R459 on the ``pivot helix'' and causes a per-

716 Purine Regulation of Glutamate Dehydrogenase
manently activated form of the enzyme.37 In the``closed mouth'' conformation found in all of thesecomplexes, R459 is distal to the ADP portion ofbound coenzyme. However, when the mouthopens to release product, our modeling resultssuggest that R459 rotates down and interacts withthe phosphate moieties of the coenzyme. We pro-pose that this interaction decreases the energyrequired to open the catalytic cleft and facilitatesproduct release. This would also explain the func-tional and binding antagonism between GTP andADP. ADP presumably binds better when themouth is open since R459 would clamp down onthe phosphate moieties. It can be further arguedthat the GTP inhibition site is made available whenthe catalytic cleft is closed and that GTP binding tothe ``hinge'' region of the NAD domain increasesthe energy required to open the catalytic cleft andrelease product. Therefore GTP and ADP bind inan antagonistic manner and cause opposite effectson substrate/coenzyme binding.
Second coenzyme regulation
Chemical modi®cation analysis, structural stu-dies, and the location of the hyperinsulinism/hyperammonemia mutations have clearly de®nedthe locations of the ADP and GTP sites. Thelocation and mechanism of action of the secondcoenzyme site(s) have been more dif®cult to under-stand. Shafer et al.38 demonstrated that NADHalone binds with a stoichiometry of seven to eightmolecules per hexamer. Glutamate enhancedNADH af®nity and increased the stoichiometry to12 per hexamer. Koberstein and Sund21 demon-strated that 0.5 mM GTP increased the stoichi-ometry of reduced coenzyme binding from 6 to 12per hexamer with NADH and NADPH binding tothis site with Kds of 57 and 700 mM, respectively.In the case of oxidized coenzyme, NAD�, twobinding sites were observed. The binding of one ofthese coenzyme molecules is blocked by theaddition of 1 mM ADP.7 Similar effects of ADP onNADH binding have also been observed.39 Thesebinding studies suggest that GTP and glutamateenhance binding of NADH to a second site andADP blocks binding of both NAD� and NADH toa second site. The competitive nature of theNAD(H) and ADP binding agree with the structur-al results presented here that suggest that ADPand NAD(H) all bind to the same site.
These results, however, result in a paradox;NADH, NAD�, ADP bind to the same site yetNADH inhibits while NAD� and ADP activate theenzyme. Furthermore, the nicotinamide ring ishighly mobile in both NADH and NAD� com-plexes, yet its oxidation state apparently dictatesallosteric activity. It should be noted that, unlikethat observed with ADP and GTP, a number ofchemical reagents affect NADH inhibition by bind-ing to disparate sites of the enzyme; the antenna(TNBS,40 FSBA35,36), the core of the hexamer(FSBAzA,41 FSBA35,36), and the outer portion of the
NAD binding domain (6-BDB-TADP42). Theseresults demonstrate that a number of regions areinvolved in NADH inhibition, yet none of theseresidues are near the bound coenzyme. Moreover,the ADP analog, AMPSBDB, reacts with R459 thatis adjacent to the coenzyme site. This modi®cationpermanently activates the enzyme, but does noteliminate NADH inhibition.37 Combined with ourstructural results, this suggests that coenzyme canbind to the ADP site, but this binding may not bewholly responsible for the observed NADHinhibition.
There are at least two possible models for howNADH inhibition might occur. The ®rst is whereoxidized and reduced coenzyme both bind to theADP site, but their effects are dependent upontheir binding conformations. One conformation ofthe nicotinamide-ribose moiety of the bound coen-zyme points down towards the subunit-subunitinterface whereas the other conformation points upaway from the surface of the enzyme and interactswith R459. It is possible that the oxidized nicotina-mide ring of NAD� preferentially binds down inthe interface allowing R459 on the pivot helix tointeract with the phosphates in a way analogous toADP. In contrast, the reduced nicotinamide moietyin NADH may preferentially bind in the upwardposition and interfere with the rotation of the pivothelix and therefore act synergistically with GTP.The major problem with this model is that there isnot a large difference between the electron den-sities of the bound NAD� no. 2 and NADH no. 2molecules (Figure 4). However, this could be dueto the fact that both complexes were crystallized asthe closed form because of the tightly bound abor-tive complexes.
Alternatively, it is possible that all of theseallosteric effects are mediated, at least partially,by coenzyme binding to the active site. In theoxidative deamination reaction, ADP activates athigh pH, but inhibits at low pH using eitherNAD� or NADP� as coenzyme. In the reductiveamination reaction, ADP is a potent activator atlow pH and low substrate concentration. AtpH 6.0, high concentrations of a-KG and NADH,but not NADPH, inhibit the reaction. Inaddition, the fact that NADH inhibition isaffected by modi®cation at so many differentsites also suggests that coenzyme regulation maynot be due to a unique allosteric site. Further-more, the Kd of NADH to this second site ismuch lower than the concentration required forsigni®cant NADH inhibition.21 Since abortivecomplexes form in the active site and greatlyinhibit catalytic turnover it is dif®cult to be cer-tain that high concentrations of coenzyme inhibitsolely by to binding to an allosteric site. A pro-blem with this model is that NADPH also bindsto the active site but does not cause measurableinhibition.17 However, is possible that this mightbe due to the subtle differences in the nicotina-mide binding orientations (Figure 7) or due todifferences in abortive complex stability. To test

Purine Regulation of Glutamate Dehydrogenase 717
these two hypotheses, we are currently usingour huGDH expression system to mutate theADP site and determine whether there is a con-comitant loss in NADH inhibition.
Perhaps a more important question is whetherNADH inhibition plays a signi®cant role in vivo. Inmammalian mitochondria, assuming a matrixvolume of 1 ml/mg of protein, the concentrationsof NAD(H) and NADP(H) are approximately 0.5-2.0 mM.43 However, activity of the transhydrogen-ase transfers much of the reductive power ofNADH to NADPH. Using metabolite indicators,the mitochondrial NADH/NAD � ratio was esti-mated to be �0.2 and the NADPH/NADP � ratiowas �200.44 In experiments on submitochondrialparticles, the energy-linked transhydrogenase wasfound to maintain NADP up to 500 times morereduced than NAD.45,46 These results suggest thatthe range of NADH concentration is �0.083-0.33 mM. NADH inhibition is observed at concen-trations above 0.2 mM (e.g. see42), but only reaches�50 % inhibition at 1 mM NADH. These results,combined with the enzyme's high Km forammonium, question the signi®cance of NADHinhibition in vivo.
Summary
From these results, we can add more details toour previously proposed model for GTP and ADPregulation. The ADP site (also the second NAD(H)site), with its deep purine binding pocket, is veryselective at the adenine-ribose end, but makes fewcontacts with the ligand beyond the phosphatemoieties. In contrast, it appears that the GTP sitefavors triphosphate binding with only marginalpurine selectivity.
We also propose that ADP and GTP have oppo-site conformational selectivity. It is possible thatwhen the catalytic mouth opens, the GTP site isdeformed and no longer available for ribose-tri-phosphate interactions. From our modeling exer-cise using the structures of csGDH, this motionrequires deformation in the loops that lie immedi-ately adjacent to the GTP site. In contrast, ADP canbind to either the open or closed conformation assuggested by the observed binding of a secondNAD� molecule in the closed mouth conformationof the boGDH �NAD��aKG complex. This bindingis enhanced as the mouth opens and R459 on thepivot helix is allowed to interact with ADP. Thiscontrol on NAD� domain motion might somehowbe related to the ADP abrogation of coenzymenegative cooperativity.5 In this way, ADP and GTPexhibit both functional and binding antagonism. Itis important to note that this ADP activation can-not be described as a simple T to R transition sincecatalytic turnover cannot occur if the mouth is keptopen. Therefore, we conclude that ADP decreasesthe energy required to open the catalytic mouthbut does not keep the enzyme in one particularconformation.
The more dif®cult allosteric process to under-stand is second-site coenzyme regulation. Eitherthe oxidation state of the nicotinamide ring isaffecting the binding conformation that in turndetermines its allosteric effects or the apparentcoenzyme allostery is due to direct effects in theactive site. From the current results, it seems poss-ible that binding of the second coenzyme to theADP site is due to structural overlap with ADPand may not play a role in vivo. These issues willbe addressed by site-directed mutagenesis on thehuGDH clone and these newly identi®ed contactresidues.
Materials and Methods
boGDH �NADPH �GLU �GTP complex data collection:
This complex was crystallized using the vapor diffu-sion method and the sitting drop apparatus at room tem-perature. The reservoir solution contained 0.1 M sodiumphosphate (pH 7.0), 1 % (w/v) octyl-b-glucopyranoside,5 % (w/v) methyl pentanediol, 0.5 M sodium chloride,9 % (w/v) polyethylene glycol 8000, and 1 mM sodiumazide. 4.5 ml of this reservoir solution was added to25.5 ml of the drop solution that contained ®nal concen-trations (after addition of the reservoir solution) of1.5 mM NADPH, 1.5 mM GTP, 20 mM L-sodium gluta-mate, and 2.5 mg/ml bovine glutamate dehydrogenase.Crystals were prepared for freezing by soaking 17minutes in a solution containing 33 % PEG 400, 0.1 Msodium phosphate (pH 7.0), 1 mM sodium azide, 12 %PEG 8000, 0.15 M sodium chloride, 4.5 mM GTP,and 60 mM L-glutamate. Data were collected froma single frozen crystal with dimensions of0.1 mm � 0.1 mm � 0.4 mm. The crystal belongs to theP21 space group with cell dimensions of a � 124.3 AÊ ,b � 102.5 AÊ , c � 169.2 AÊ , and b � 102.2 �.
Data were collected at the Cornell High Energy Syn-chrotron Source (CHESS) at the F1 beamline station. Thecrystal to ®lm distance was 250 mm, the oscillation anglewas 0.3 �, and the exposure time was 25 seconds perframe. Data were collected using the ADSC Quantum 4CCD detector and the re¯ections were integrated usingthe program DENZO.47 The 219 frames of data werethen merged using the SCALEPACK48 application.Re¯ections were observed to a resolution of 2.2 AÊ , butthe quality of the data degraded beyond 2.8 AÊ resolution(Table 1).
boGDH �NAD��aaa-KG complex data collection
Sitting drop apparatus and the vapor diffusion meth-od were used to crystallize the abortive complex of thereductive amination reaction at room temperature. Thereservoir solution contained 0.1 M sodium phosphate(pH 7.0), 1 % octyl-b-glucopyranoside, 5 % methyl penta-nediol, 0.5 M sodium chloride, 7 % polyethylene glycol8000, and 1 mM sodium azide. 9 ml of this solution wasadded to 21 ml of the drop solution that contained ®nalconcentrations (after the addition of the reservoir sol-ution) of 1.5 mM NAD�, 20 mM a-KG, and 3.25 mg/mlboGDH. These crystals also belong to the P21 spacegroup and have cell dimensions of a � 122.5 AÊ ,b � 101.0 AÊ , c � 164.6 AÊ , and b � 102.2 �. Data were col-lected from a single frozen crystal with dimensions of0.05 mm � 0.05 mm � 0.4 mm. To prepare the crystals

718 Purine Regulation of Glutamate Dehydrogenase
for freezing, two aliquots of 2 ml of PEG 400 were addeddirectly to the well and allowed to incubate for 15 min-utes after each addition. The crystal was then frozendirectly in the cryo-stream. The crystal to ®lm distancewas 150 mm, the exposure time was 20 minutes, and theoscillation angle was 0.5 � per frame. Diffraction maximawere collected using an R-Axis IV area detector attachedto a Rigaku generator, integrated using the programDENZO,47 and 117 frames of data were merged usingthe SCALEPACK48 application. The diffraction intensitydropped off sharply past 3.4 AÊ (Table 1).
Structure determination:
The structure of the boGDH �NADH �GLU �GTP com-plex structure, excluding the bound ligands, was used asa starting model for both structure determinations.Using the program X-PLOR,49 the starting model was®rst subjected to simulated-annealing protocols withoutapplying non-crystallographic symmetry (NCS). Thesemodels were then further re®ned using standard pos-itional and B-factor re®nement without non-crystallo-graphic symmetry followed by tightly restrained NCSre®nement. For real-space averaging, the programMAMA50 was used to calculate the mask of one of thesubunits, MolView31 to calculate the NCS matrices fromthe atomic model, the CCP4 package51 for structure fac-tor and Fourier calculations, and RAVE52 for real spaceaveraging.
Omit maps were used to rebuild dif®cult areas of theprotein and for re®ning the structures of the boundligands. To calculate omit maps, these portions of themodel were included in creating the masks, but were notincluded in the structure factor calculations. RAVE52 wasthen used to perform 6-fold averaging of these electrondensity maps. The electron densities shown in Figures 2-7 were all calculated in this manner.
Human GDH expression
Human GDH (huGDH) containing the hyperinsulin-ism/hyperammonemia mutation, H454Y (bovine GDHH450Y), was expressed as the mature enzyme with theexception of the N-terminal methionine (unpublishedresults). A cDNA clone for human GDH was obtainedfrom Dr Roberta Colman and modi®ed by site-directedmutagenesis to omit the mitochondrial targeting leadersequence. Mutant human GDH was co-expressed inEscherichia coli with pGroESL to provide chaperone pro-teins for stabilizing the expressed enzyme. The expressedenzyme migrated as a single band on PAGE after puri®-cation by anion exchange, hydrophobic interaction, andGTP af®nity chromatography. GDH activity wasmeasured spectrophotometrically in the reductive amin-ation direction.37
Atomic co-ordinates
The NADH �GLU �GTP, NAD��aKG, and theNADPH �GLU �GTP complexes have been deposited inthe Protein Data Bank; 1HWX, 1HWY, 1HWZ, respect-ively.
Acknowledgements
This work was supported by grants from the NationalInstitutes of Health (GM10704) to T.J.S. and from theAmerican Diabetes Association to C.A.S. and T.J.S.(grant no. 1320000179). The program MolView (http://bilbo.bio.purdue.edu/� tom) was used to createFigures 1, 2(b), 3(b), 4(b), 4(c), 5(b), 6(b), 6(c), 7(b), 7(c),and 9. We also thank Dr Roberta Colman and Dr J. EllisBell for extremely helpful discussions and support.
References
1. Hudson, R. C. & Daniel, R. M. (1993). L-Glutamatedehydrogenases: distribution, properties and mech-anism. Comp. Biochem. Physiol. B, 106, 767-792.
2. Smith, E. L., Austen, B. M., Blumenthal, K. M. &Nyc, J. F. (1975). Glutamate dehydrogenases. In TheEnzymes (Boyer, P. D., ed.), pp. 293-367, AcademicPress, New York.
3. Consalvi, V., Chiaraluce, R., Politi, L., Vaccaro, R.,DaRosa, M. & Scandurra, R. (1991). Extremelythermostable glutamate dehydrogenase from thehyperthermophilic archaebacterium Pyrococcus furio-sus. Eur. J. Biochem. 202, 1189-1196.
4. Consalvi, V., Chiaraluce, R., Politi, L., Gambacorta,A., DaRosa, M. & Scandurra, R. (1991). Glutamatedehydrogenase from the thermoacidophilic archae-bacterium Sulfolobus solfataricus. Eur. J. Biochem. 196,459-467.
5. Dalziel, K. & Egan, R. R. (1972). The binding ofoxidized coenzymes by glutamate dehydrogenaseand the effects of glutarate and purine nucleotides.Biochem. J. 126, 975-984.
6. Dalziel, K. (1975). Kinetics and mechanisms of nico-tinamide-nucleotide-linked dehydrogenases. In TheEnzymes (Boyer, P. D., ed.), pp. 1-60, AcademicPress, New York.
7. Limuti, C. M. (1983). Glutamate dehydrogenase:equilibrium and kinetic studies. PhD thesis, Depart-ment of Biochemistry University of Rochester,Rochester, New York.
8. Bell, E. T., LiMuti, C., Renz, C. L. & Bell, J. E. (1985).Negative co-operativity in glutamate dehydrogen-ase. Involvement of the 2-position in the inductionof conformational changes. Biochem. J. 225, 209-217.
9. Engel, P. & Dalziel, K. (1969). Kinetic studies ofglutamate dehydrogenase with glutamate andnorvaline as substrates. Biochem. J. 115, 621-631.
10. Engel, P. C. & Dalziel, K. (1970). Kinetic studies ofglutamate dehydrogenase. The reductive aminationof 2-oxoglutarate. Biochem. J. 118, 409-419.
11. Colen, A. H., Prough, R. A. & Fisher, H. F. (1972).The mechanism of glutamate dehydrogenase reac-tion. IV. Evidence for random and rapid binding ofsubstrate and coenzyme in the burst phase. J. Biol.Chem. 247, 7905-7909.
12. Engel, P. C. & Chen, S. (1975). A product-inhibitionstudy of bovine liver glutamate dehydrogenase. Bio-chem. J. 151, 305-318.
13. Singh, N., Maniscalco, S. J. & Fisher, H. F. (1993).The real-time resolution of proton-related transient-state steps in an enzymatic reaction. J. Biol. Chem.268, 21-28.
14. Stillman, T. J., Baker, P. J., Britton, K. L. & Rice,D. W. (1993). Conformational ¯exibility in glutamate

Purine Regulation of Glutamate Dehydrogenase 719
dehydrogenase. Role of water in substrate recog-nition and catalysis. J. Mol. Biol. 234, 1131-1139.
15. Iwatsubo, M. & Pantaloni, D. (1967). Regulation DeL' Activite' De La glutamate dehydrogenase par leseffecteurs GTP et ADP: ETUDE par ``stopped ¯ow''.Bull. Soc. Chem. Biol. 49, 1563-1572.
16. George, S. A. & Bell, J. E. (1980). Effects ofadenosine 50-diphosphate on bovine glutamatedehydrogenase: diethyl pyrocarbonate modi®cation.Biochemistry, 19, 6057-6061.
17. Bailey, J. S., Bell, E. T. & Bell, J. E. (1982). Regulationof bovine glutamate dehydrogenase. J. Biol. Chem.257, 5579-5583.
18. Frieden, C. (1959). Glutamic dehydrogenase I. Theeffect of coenzyme on the sedimentation velocityand kinetic mechanism. J. Biol. Chem. 234, 809-814.
19. Frieden, C. (1959). Glutamic dehydrogenase II Theeffect of various nucleotides on the association-disassociation and kinetic properties. J. Biol. Chem.234, 815-819.
20. Jacobson, M. A. & Colman, R. F. (1982). Labeling ofa guanosine 50-triphosphate site of glutamate by a¯uorescent nucleotide analog, 50-[p-(¯uorosulfonyl)-benzoyl]-1,n6-ethenoadenosine. Biochemistry, 21,2177-2186.
21. Koberstein, R. & Sund, H. (1973). The in¯uence ofADP, GTP and L-glutamate on the binding of thereduced coenzyme to beef-liver glutamate dehydro-genase. Eur. J. Biochem. 36, 545-552.
22. Bayley, P. M. & O'Neill, T. J. (1980). The binding ofoxidised coenzyme to bovine-liver glutamate dehy-drogenase studied by circular-difference spec-troscopy. Eur. J. Biochem. 112, 521-531.
23. Rice, D. W., Baker, P. J., Farrants, G. W. & Hornby,D. P. (1987). The crystal structure of glutamatedehydrogenase from Clostridium symbiosum at0.6 nm resolution. Biochem. J. 242, 789-795.
24. Baker, P. J., Britton, K. L., Engel, P. C., Farrants,G. W., Lilley, K. S., Rice, D. W. & Stillman, T. J.(1992). Subunit assembly and active site location inthe structure of glutamate dehydrogenase. Proteins:Struct. Funct. Genet. 12, 75-86.
25. Yip, K. S. P., Stillman, T. J., Britton, K. L., Artymiuk,P. J., Baker, P. J., Sedelnikova, S. E., Engel, P. C.,Pasquo, A., Chiaraluce, R., Consalvi, V., Scandurra,R. & Rice, D. W. (1995). The structure of Pyrococcusfuriosus glutamate dehydrogenase reveals a key rolefor ion-pair networks in maintaining enzyme stab-ility at extreme temperatures. Structure, 3, 1147-1158.
26. Smith, C. A., Norris, G. E. & Baker, E. N. (1996).The structure of the thermophilic glutamate dehy-drogenase from Thermococcus ANI. In IUCr XVIICongress and General Assembly, , Seattle, Washington.
27. Knapp, S., de Vos, W. M., Rice, D. & Ladenstein, R.(1997). Crystal structure of glutamate dehydrogen-ase from the hyperthermophilic eubacterium Thermo-toga maritima at 3.0 AÊ resolution. J. Mol. Biol. 267,916-932.
28. Peterson, P. E. & Smith, T. J. (1999). The structure ofbovine glutamate dehydrogenase provides insightsinto the mechanism of allostery. Structure, 7, 769-782.
29. Stanley, C. A., Lieu, Y. K., Hsu, B. Y. L., Burlina,A. B., Greenberg, C. R., Hopwood, N. J., Perlman,K., Rich, B. H., Zammarchi, E. & Poncz, M. (1998).Hyperinsulinism and hyperammonemia in infantswith regulatory mutations of the glutamate dehy-drogenase gene. New Engl. J. Med. 338, 1352-1357.
30. Laskowski, R. A., MacArthur, M. W., Moss, D. S. &Thornton, J. M. (1993). PROCHECK: a program tocheck the sterochemical quality of protein structures.J. Appl. Crystallog. 26, 283-291.
31. Smith, T. J. (1995). MolView: a program to analyzeand display atomic structures on the Macintosh per-sonal computer. J. Mol. Graphics, 13, 122-125.
32. Smith, E. J., Landon, M., Piszkiewicz, D., Brattin,W. J., Langley, T. J. & Melamed, M. D. (1970).Bovine liver glutamate dehydrogenase: tentativeamino acid sequence; identi®cation of a reactivelysine; nitration of a speci®c tyrosine and loss ofallosteric inhibition by guanosine triphosphate. Proc.Natl Acad. Sci. USA, 67, 724-730.
33. Frieden, C. (1963). L-Glutamate dehydrogenase. InThe Enzymes (Boyer, P. D., Lardy, H. & Myrback, K.,eds), 2nd edit., pp. 3-24, Academic Press, New York.
34. Frieden, C. (1963). Glutamate dehydrogenase IV.Studies on enzyme inactivation and coenzyme bind-ing. J. Biol. Chem. 238, 146-154.
35. Pal, P. K., Wechter, W. J. & Colman, R. F. (1975).Af®nity labeling of a regulatory site of bovine liverglutamate dehydrogenase. Biochemistry, 14, 707-715.
36. Schmidt, J. A. & Colman, R. F. (1984). Identi®cationof the lysine and tyrosine peptides labeled by 50-p-¯uorosulfonylbenzoyladenosine in the NADHinhibitory site of glutamate dehydrogenase. J. Biol.Chem. 259, 14515-14519.
37. Wrzeszczynski, K. O. & Colman, R. F. (1994). Acti-vation of bovine liver glutamate dehydrogenase bycovalent reaction of adenosine 50-O-[S-(4-bromo-2,3-dioxobutyl)thiophosphate] with arginine-459 at anADP regulatory site. Biochemistry, 33, 11544-11553.
38. Shafer, J. A., Chiancone, E., Vittorelli, L. M.,Spagnuolo, C., Machler, B. & Antonini, E. (1972).Binding of reduced cofactor to glutamate dehydro-genase. Eur. J. Biochem. 31, 166-171.
39. Dieter, H., Koberstein, R. & Sund, H. (1981). Studiesof glutamate dehydrogenase. The interaction ofADP, GTP, and NADPH in complexes with gluta-mate dehydrogenase. Eur. J. Biochem. 115, 217-226.
40. Goldin, B. R. & Frieden, C. (1971). Effect of trinitro-phenylation of speci®c lysyl residues on the cataly-tic, regulatory, and molecular properties of bovineliver glutamate dehydrogenase. Biochemistry, 10,3527-3534.
41. Dombrowski, K. E., Huang, Y.-C. & Colman, R. F.(1992). Identi®cation of amino acids modi®ed by thebifunctional af®nity label 50-(p-¯uorosulfonyl)ben-zoyl)-8-azidoadenosine in the reduced coenzymeregulatory site of bovine liver glutamate dehydro-genase. Biochemistry, 31, 3785-3793.
42. Batra, S. P. & Colman, R. F. (1986). Isolation andidenti®cation of cysteinyl peptide labeled by 6-[(4-bromo-2,3-dioxobutyl)thio]-6-deaminoadenosine 50-diphosphate in the reduced diphosphopyridinenucleotide inhibitory site of glutamate dehydrogen-ase. Biochemistry, 25, 3508-3515.
43. Lenartowicz, E. (1990). A complex effet of arseniteon the formatio of a-ketoglutarate in rate liver mito-chondria. Arch. Biochem. Biophys. 283, 388-396.
44. Hoek, J. B. & RydstroÈm, J. (1988). Physiologicalroles of nicotinamide nucleotide transhydrogenase.Biochem. J. 254, 1-10.
45. Lee, C. P. & Ernster, L. (1968). Studies of theenergy-transfer system of submitochondrial par-ticles. I. Competition between oxidative phosphoryl-ation and the energy-linked nicotinamide-adenine

720 Purine Regulation of Glutamate Dehydrogenase
dinucleotide transhydrogenase reaction. Eur. J. Bio-chem. 3, 385-390.
46. RydstroÈm, J., Teixeira da Cruz, A. & Ernster, L.(1970). Factors governing the kinetics and steadystate of the mitochondrial nicotinamide nucleotidetranshydrogenase system. Eur. J. Biochem. 17, 56-62.
47. Otwinowski, Z. (1993). DENZO. In Data Collectionand Processing (Sawyer, L., Isaacs, N. & Bailey, S.,eds), pp. 56-62, SERC Daresbury Laboratory,Warrington, UK.
48. Otwinowski, Z. & Minor, W. (1997). Processing ofX-ray diffraction data collected in oscillation mode.In Methods in Enzymology (Carter, C. W. J. & Sweet,R. M., eds), pp. 307-326, Academic Press.
49. BruÈ nger, A. T. (1992). X-plor (Version 3.1) User'sGuide, Yale University, New Haven, CT.
50. Kleywegt, G. J. & Jones, T. A. (1999). Software forhandling macromolecular envelopes. Acta Crystallog.sect. D, 55, 941-944.
51. Bailey, S. (1994). The CCP4 suite: programs forprotein crystallography. Acta Crystallog. sect. D, 50,760-763.
52. Kleywegt, G. J. & Jones, T. A. (1994). Halloween . . .masks and bones. In From First Map to FinalModel (Bailey, S., Hubbard, R. & Waller, D., eds),pp. 59-66, SERC Daresbury Laboratory, Daresbury,UK.
Edited by I. A. Wilson
(Received 9 November 2000; received in revised form 23 January 2001; accepted 23 January 2001)





























