Embed Size (px)
Citation preview

The “Dark Side” of Endocannabinoids: A Neurotoxic Rolefor Anandamide
*Ibolja Cernak, *†Robert Vink, *JoAnne Natale, *Bogdan Stoica, *Paul M. Lea IV,*Vilen Movsesyan, *Farid Ahmed, *Susan M. Knoblach, *Stanley T. Fricke, and *Alan I. Faden
*Department of Neuroscience, Georgetown University Medical Center, Washington D.C., U.S.A.; and †Department of Pathology,University of Adelaide, Australia
Summary: Endocannabinoids, including 2-arachidonoylglyc-erol and anandamide (N-arachidonoylethanolamine; AEA),have neuroprotective effects in the brain through actions atCB1 receptors. However, AEA also binds to vanilloid (VR1)receptors and induces cell death in several cell lines. Here weshow that anandamide causes neuronal cell death in vitro andexacerbates cell loss caused by stretch-induced axonal injury ortrophic withdrawal in rat primary neuronal cultures. Adminis-tered intracerebroventricularly, AEA causes sustained cerebraledema, as reflected by diffusion-weighted magnetic resonance
imaging, regional cell loss, and impairment in long-term cog-nitive function. These effects are mediated, in part, throughVR1 as well as through calpain-dependent mechanisms, but notthrough CB1 receptors or caspases. Central administration ofAEA also significantly upregulates genes involved in pro-inflammatory/microglial-related responses. Thus, anandamideproduces neurotoxic effects both in vitro and in vivo throughmultiple mechanisms independent of the CB1 receptor. KeyWords: Endocannabinoids—Anandamide—Cell death—Microarray—Magnetic resonance imaging—Cognitive deficit.
It is now known that much of the tissue damagecaused by central nervous system (CNS) insults resultsfrom delayed biochemical mechanisms (Faden, 2001).One of the secondary processes that may contribute todelayed neuronal death is activation of phospholipase-mediated signaling pathways that leads to membranephospholipid degradation (Chan et al., 1989; Homayounet al., 2000).
Endocannabinoids are endogenous lipid ligands,which bind to the same cannabinoid receptors (CB1,CB2) that mediate the effects of �9-tetrahydrocannabi-nol, the active compound of cannabis (Mechoulam,2002). There are three members of the endocannabinoidfamily discovered to date: N-arachidonoylethanolamine(anandamide; AEA), 2-arachidonoylglycerol (2-AG),
and 2-arachidonoylglyceryl ether (2-AGE; noladin);these have different affinities for CB1 and CB2 receptorsand distinct biologic effects (Di Marzo et al., 2002). En-docannabinoid signaling system occurs in both neuronsand astrocytes; astrocytes may use this system to com-municate with surrounding neurons or other astrocytes(Walter et al., 2002). During the last decade, consider-able experimental work has demonstrated protective bio-logic effects of endocannabinoids after brain injury (vander Stelt et al., 2002). Indeed, neuroprotective effects ofcannabinoids have been shown in global and focal is-chemia, as well as in neuronal cultures subjected to is-chemic conditions (Nagayama et al., 1999; Sinor etal., 2000). Both 2-AG and AEA can protect cerebralrat cortical neurons from in vitro ischemia (Sinoret al., 2000), whereas 2-AG reduces cerebral brainedema and infarct volume, decreases hippocampal cellloss, and improves clinical outcome after traumaticbrain injury in mice (Panikashvili et al., 2001). How-ever, it should be noted that in contrast to the datasupporting cannabinoid-induced neuroprotection (Marsi-cano et al., 2003; Mechoulam and Lichtman, 2003), sev-eral studies have revealed that activation of CB1 recep-tors can induce cytotoxic effects in a number of culturedcell systems (Downer et al., 2003) including culturedhippocampal (Chan et al., 1998) and cortical neurons(Downer et al., 2001). Among suggested mechanisms of
Received August 14, 2003; final version received November 5,2003; accepted December 16, 2003.
Address and reprint requests to Dr. Ibolja Cernak, Department ofNeuroscience, Georgetown University Medical Center, 3970 ReservoirRoad, NW, Research Building, Room EP04, Washington, DC 20057,U.S.A.; e-mail: [email protected]
Abbreviations used: ADC, apparent diffusion coefficient; AEA, an-andamide; 2-AG, 2-arachidonoylglycerol; 2-AGE, 2-arachidonoylglyc-eryl ether; BDNF, brain-derived growth factor; CGC, cerebellar gran-ule cell; CPZ, capsazepine; ESTs, expression sequence tags; NAE,N-acylethanolamine; NAPE, N-acylated species of phosphatidyletha-nolamine; NM, neurobasal medium; TBI, traumatic brain injury;TIMP, tissue inhibitors of metalloproteinase.
Journal of Cerebral Blood Flow & Metabolism24:564–578 © 2004 The International Society for Cerebral Blood Flow and MetabolismPublished by Lippincott Williams & Wilkins, Baltimore
564 DOI: 10.1097/01.WCB.0000117813.35136.12

cannabinoid-induced neurotoxicity are activation ofcaspase-3-dependent apoptosis (Campbell, 2001; Downeret al., 2001), generation of reactive oxygen species (Chanet al., 1998), sustained ceramide accumulation (Galve-Roperh et al., 2002), activation of the JNK cascade (Sarkerand Maruyama, 2003), and sphingomyelin hydrolysis(Sanchez et al., 1998). It is highly possible that CB1activation may lead to both neurotoxicity and neuropro-tection, depending on a variety of influences such asnature and intensity of the toxic insult, as well as the celltype under study (Downer et al., 2003; Guzman, 2003).
Anandamide as an N-acylethanolamine (NAE) can besynthesized as a hydrolytic product of N-acylated speciesof phosphatidylethanolamine (NAPE) through a processcatalyzed by phospholipase D (Devane et al., 1992;Schmid, 2000). Under normal conditions, the levels ofNAPE and related NAE are very low, with their synthe-sis and metabolism strictly controlled (Schmid, 2000).However, accumulation of NAPE and NAE occurs incells undergoing degeneration and phospholipid degen-eration (Gray, 1976), as well as in conditions associatedwith membrane degradation (Epps et al., 1980). In-creased levels of NAPE and NAE may occur with neu-ronal death induced by glutamate or the mitochondrialrespiratory chain inhibitor, sodium azide (Hansen et al.,1997). Similarly, significant increases in both NAPE andNAE concentrations are found after glutamate-inducedneurotoxicity in vivo (Hansen et al., 2001) as well as inpost-decapitative ischemia (Natarajan et al., 1986).These lipid compounds, including AEA, may be formedin response to the high intracellular calcium concentra-tion that occurs in injured cells (Hampson et al., 1998).Although AEA originally was identified as an endog-enous ligand for cannabinoid receptors (Devane et al.,1992), more recent data suggest that it might also interactdirectly with other molecular targets, including non-CB1, non-CB2 G-protein coupled receptors (Di Marzo etal., 2000; Sagan et al., 1999), gap junctions (Venance etal., 1995), various ion channels (Hampson et al., 1998;Maingret et al., 2001; Szoke et al., 2000), and vanilloid(VR1) receptors (Zygmunt et al., 1999). Although a sub-stantial body of evidence demonstrates that activation ofCB1 receptors by endocannabinoids (Nagayama et al.,1999; Panikashvili et al., 2001), including AEA (van derStelt et al., 2002), has neuroprotective effects, stimula-tion of VR1 receptors has been found to increase intra-cellular Ca2+ and lead to subsequent cytotoxicity (Olah etal., 2001).
VR1 is a nonselective ligand-gated cation channelwith six-transmembrane domains and belongs to thefamily of transient release-potential ion-channels (Ben-ham et al., 2002). It may be activated by exogenouscompounds such as capsaicin, the pungent component ofchili peppers, and resiniferatoxin, a plant toxin (Caterinaet al., 1997; Szallasi and Blumberg, 1990; Szallasi,
2002). In the rat, VR1-positive neurons are locatedthroughout the neuroaxis (Szallasi and Di Marzo, 2000),and the distribution of AEA is mainly overlapping withthe localization of VR1 receptors (Szallasi and Di Marzo,2000). There is ample evidence now (Ross, 2003) thatthe interaction of AEA with VR1 receptors is specific,whereas the efficacy of AEA as a VR1 agonist dependson numerous factors including receptor reserve, phos-phorylation, CB1 receptor activation, voltage, tempera-ture, and pH, among others.
Although a decade has passed since the discovery ofAEA, conflicting data on its biologic effects are stillemerging, and the mechanisms of AEA actions remainunclear. The root of the controversy resides in the factthat AEA is an endogenous ligand for both cannabinoidand vanilloid receptors, which often manifest opposingeffects. For example, the neuroprotective effects of theendocannabinoids through CB1 have been demonstratedin numerous in vitro and in vivo models (Gomez DelPulgar et al., 2002; Maccarrone et al., 2000; Mechoulamet al., 2002b), whereas the activation of VR1 receptorsseems to be involved in various forms of neuronal celldeath (Hail and Lotan, 2002; Hail, 2003; Jambrina et al.,2003). Indeed, in recent studies, AEA was shown to in-duce apoptotic cell death in human neuroblastomaCHP100, lymphoma U937, and PC-12 cells (Maccarroneet al., 2000; Sarker et al., 2000); the formation of apo-ptotic bodies induced by AEA corresponds to increasesin intracellular calcium, mitochondrial uncoupling, andcytochrome c release (Maccarrone et al., 2000). Thesepro-apoptotic effects of AEA were mediated via VR1receptors (Maccarrone et al., 2000). However, it shouldbe noted that recent results (Veldhuis et al., 2003) dem-onstrated that arvanil, a ligand for both VR1 and CB1receptors, leads to neuroprotective effects acting at bothCB1 and VR1. Moreover, it has been shown that the invivo neuroprotective effects of AEA are mediated byCB1 but not by VR1 or by lipoxygenase metabolites(Veldhuis et al., 2003). Taken together, it is possible thatduring pathologic conditions such as inflammation orcell damage when pH is decreased, the PKC-dependentsignaling system is activated, and NAPE is increasinglysynthesized and released by cells; under these conditions,AEA may become more active at VR1 than CB1 recep-tors. We hypothesize that AEA may induce either neu-roprotection or neurotoxicity, depending on the balanceof its action on CB1 receptors on the one hand, and VR1receptors or calcium-mediated signal transduction path-ways on the other. We have addressed these questionsusing both in vitro and in vivo model systems.
METHODS
AnimalsAll protocols involving the use of animals were in com-
pliance with the Guide for the Care and Use of Laboratory
ANANDAMIDE NEUROTOXICITY 565
J Cereb Blood Flow Metab, Vol. 24, No. 5, 2004

Animals published by NIH (DHEW publication NIH 85-23-2985), and were approved by the Georgetown University Ani-mal Use Committee. Male Sprague-Dawley rats (340 to 380 g)for in vivo studies and female pregnant rats used to prepareneuronal and glial cultures were purchased from Harlan (India-napolis, IN, U.S.A.).
DrugsAnandamide (AEA) and capsazepine (CPZ) were purchased
from Tocris (Tocris Cookson, Ellisville, MO, U.S.A.), and dis-solved in a minimum of ethyl alcohol and diluted with saline(the final concentration of ethanol was 2% (v/v). Five micro-liters of AEA and CPZ contained 20 nmol/L and 35 nmol/L,respectively. AM251, a specific CB1 antagonist, (N-(piperidin-1-yl)-5-(4-iodophenyl)-1-(2, 4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide) was obtained from Tocris (TocrisCookson, Ellisville, MO, U.S.A.), first dissolved in dimethylsulfoxide (DMSO) and then diluted in saline (the final concen-tration of DMSO was 10%), whereas 5 �L contained 35nmol/L. z-DEVD-fmk (N-benzyl-oxycarbonyl-Asp(OMe)-Val-Asp(OMe)-fluoromethylketone) was purchased from EnzymeSystems (Livermore, CA, U.S.A.), whereas SJA6017 (N-(4-fluorophenylsulfonyl)-L-valyl-L-leucinal), the calpain inhibitorVI, was obtained from Calbiochem (San Diego, CA, U.S.A.).Both drugs were dissolved in DMSO and diluted in saline toaccommodate a dosing volume of 5 �L containing 160 ng and1 �g, respectively. The vehicle for the AEA- and CPZ-treatedanimals was saline containing 2% of ethyl alcohol, whereas thevehicle for AM251-, z-DEVD-fmk-, and SJA6017-treatedgroups consisted of saline with 10% DMSO. Injection of CPZ(35 nmol/L), AM251 (35 nmol/L), z-DEVD-fmk (160 ng), andSJA6017 (1 �g) did not induce significant changes in cognitiveoutcome or alteration in blood pressure, compared to vehiclecontrol (results not shown). Moreover, administration of thesedrugs did not alter the apparent diffusion coefficient (ADC),measured by diffusion-weighted magnetic resonance imaging(MRI) (results not shown); ADC significantly correlates withthe changes of extracellular water and as such reflects brainedema (Albensi et al., 2000). Because there were no significantdifferences between cognitive performances, blood pressure,ADC values, and gene profile in animals treated withsaline/ethyl alcohol or saline/DMSO vehicles, these animalswere pooled into one group.
Cell culturesGlia were prepared from 1- to 3-day-old rat cortices, and
neurons were prepared from 17- to 18-day-old rat embryoniccortices, as described previously in detail (Lea et al., 2002).Cortical neuronal cultures were used to examine AEA dose–response curves between 0.5 and 100 �M, and cytotoxicitymeasured by lactate dehydrogenase (LDH) release. Mixed neu-ronal–glial cultures were used to test the effects of AEA onmechanical (stretch) injury-induced LDH release. Cerebellargranule cell (CGC) cultures were prepared as previously de-scribed (Toman et al., 2002). In experiments using trophic sup-port withdrawal, CGCs cultured in neurobasal medium (NM)with 2% B27 supplement and 25 mmol/L KCl were washedonce in NM and placed in B27-free NM containing 5 mmol/LKCl.
Stretch injuryUsing the original method described by Ellis et al. (Ellis et
al., 1995), cells cultured on a deformable membrane werestretched with compressed gas. Stretch (7.5-mm deformationsof the membrane; 50-millisecond duration) was applied to the
cells using a cell injury controller (Biomedical EngineeringFacility, Medical College of Virginia, VA, U.S.A.).
Assay for in vitro cell deathLactate dehydrogenase activity cell culture growth media
was quantified as an index of cell death (Mukhin et al., 1997).Injury- and/or anandamide-induced release of LDH was mea-sured using a CytoTox-96 nonradioactive cytotoxicity assay kit(Promega, Madison, WI, U.S.A.) according to the manufactur-er’s protocol 24 hours after injury or application of AEA togrowth media. Relative absorbance was measured at 490 nmusing a Multiskan Ascent microplate reader (Labsystem Oy,Helsinki, Finland). LDH levels in injured or treated cultureswere expressed as a percentage of a mean value (100%) inuninjured cultures.
Intracerebroventricular injectionsSurgical anesthesia was induced and maintained with 4%
and 2% isoflurane, respectively, using a flow rate of 1.0 to 1.5L/min oxygen. A guide cannula for microinjection of drugs wasimplanted into right lateral cerebroventricle. The drugs wereadministered to rats in a volume of 5 �L for 2 minutes using amicroliter syringe. Administration of CPZ, AM251, SJA6017,or z-DEVD-fmk was performed 5 minutes after AEA injection.
Nuclear magnetic resonance imagingAt 24 hours, 48 hours, and 7 days after drug administration,
all animals (n � 6/group) were reanesthetized with isofluraneand subjected to magnetic resonance imaging examination us-ing a Bruker 7T/21 cm Biospec-Avance system (Bruker, Karls-ruhe, Germany) as previously described (Albensi et al., 2000).Briefly, animals were placed in the heated Plexiglas holder, anda respiratory motion detector was positioned over the thorax tofacilitate respiratory gating. The animal bed was positionedwith the head in the center of the magnet within a 72-mm 1Hbirdcage resonator (Bruker). Field homogeneity across thebrain was optimized and a sagittal scout image was acquired(RARE [rapid acquisition relaxation enhancement pulse se-quence] image, field of vision, 4 × 4 cm; 128 × 128 resolution;repetition time [TR] to echo time [TE], 1,500/10 millisecondswith a RARE factor of 8, making the effective TE 40 millisec-onds). Multislice T2-weighted images were then acquired toobtain eight contiguous slices commencing at the end of theolfactory bulb and working caudally (field of vision, 3 × 3 cm;slice thickness 2 mm; 256 × 256 resolution; TR/TE 2,000/20ms; four echo images and two averages). Diffusion weighted(DWI) images were then acquired with a spin-echo pulse se-quence that had diffusion gradients added before and after therefocusing pulse. Gradient strength was varied in six steps us-ing sensitization values ranging from 20 to 1,000 s/mm2. A 256× 256 matrix was used with a 3-cm field of view, TR 2.0seconds, TE 502 milliseconds, slice thickness of 2 mm, and 4echoes. Diffusion maps were generated by applying the Stej-skal-Tanner equation in association with a Marquart algorithmusing a commercially available Paravision software (Bruker,Billerica, MA, U.S.A.). ADCs were calculated for four regions:left cortex, right cortex, left subcortex, and right subcortex (Fig.1). ADCs were expressed as 10−5 mm2/s ± SD.
Nuclear magnetic resonance spectroscopyAnandamide-treated animals used in MRI experiments at 24
hours were also subject to phosphorus magnetic resonancespectroscopy using a Bruker 7T/21 cm Biospec-Avance system(Bruker, Karlsruhe, Germany) as previously described in detailelsewhere (Vink and McIntosh, 1990). Additionally, naïve orvehicle-treated animals were used as spectroscopy controls.
I. CERNAK ET AL.566
J Cereb Blood Flow Metab, Vol. 24, No. 5, 2004

Because there were no differences in vehicle-treated or naïveanimals (results not shown), they were pooled for statisticalanalysis (n � 6). Briefly, animals were placed in a speciallyconstructed, temperature-controlled Plexiglas holder and a5-mm × 9-mm surface coil was placed centrally over the ex-posed skull. Skin and muscle were retracted well clear of thecoil to prevent contributions from these tissues. The animalswere then inserted into the center of a 7.0-Tesla magnet inter-faced with a Bruker spectrometer and field homogeneity opti-mized on the water signal before acquisition of phosphorusspectra. Phosphorus spectra were obtained in 20-minute blocksusing a 90° pulse calibrated for a 2-mm cortical depth, a 700-millisecond delay time, and a 5,000-Hz spectral width contain-ing 2,048 data points. Rectal temperature and respiration weremonitored at all times. The anesthesia was maintained usingisoflurane. At the conclusion of the acquisition period, animalswere removed from the magnet, their wounds were closed, andthe animals were returned to their cages.
Phosphorus magnetic resonance spectra were analyzed usingthe resident Bruker computer software program. After convo-lution difference (400/20 Hz), chemical shifts and integrals ofthe individual peaks were determined following line fitting.Intracellular pH, brain free magnesium concentration, and cy-tosolic phosphorylation potential were then determined as de-scribed in detail elsewhere (Vink et al., 1994). Briefly, intra-cellular pH was determined from the chemical shift of theinorganic phosphate peak (�Pi) relative to phosphocreatine(PCr) in the magnetic resonance spectra using the equation
pH = 6.77 + log��Pi − 3.29
5.68 − �Pi� (1)
Similarly, free magnesium concentration was determined fromthe chemical shift difference between the � and � peaks of ATPusing the equation
�Mg2+� = Kd�10.82 − ��−�
��−� − 8.35 � (2)
where ��−� is the chemical shift difference between the � and� peaks of ATP. The Kd for MgATP was initially assumed tobe 50 �mol/L at pH 7.2 and 0.15 mol/L ionic strength and wascorrected for pH according to Bock et al. (Bock et al., 1987).Cytosolic phosphorylation potential (PP) was determined ac-cording to the equation
PP =��ATP�
��ADP���Pi�(3)
where � represents all the ionic forms of the free species. Theconcentration of ADP was calculated from the creatine kinaseequilibrium equation after correcting the equilibrium constantfor pH and free magnesium concentration as previously de-scribed in detail elsewhere (Vink et al., 1994). Concentrationsof the other metabolites were determined from the integratedpeak areas of the respective MRS peaks, assuming that prein-jury the normal values for PCr and ATP were 4.72 and 2.59�mol/g, respectively, and that the total creatine pool remainedconstant at 10.83 �mol/g (Siesjo, 1981; Veech et al., 1979).Brain water content was assumed to be 80%, with the intracel-lular compartment accounting for 78% of the total water(Siesjo, 1981).
Morris water maze testCognitive outcome (spatial learning) was determined using
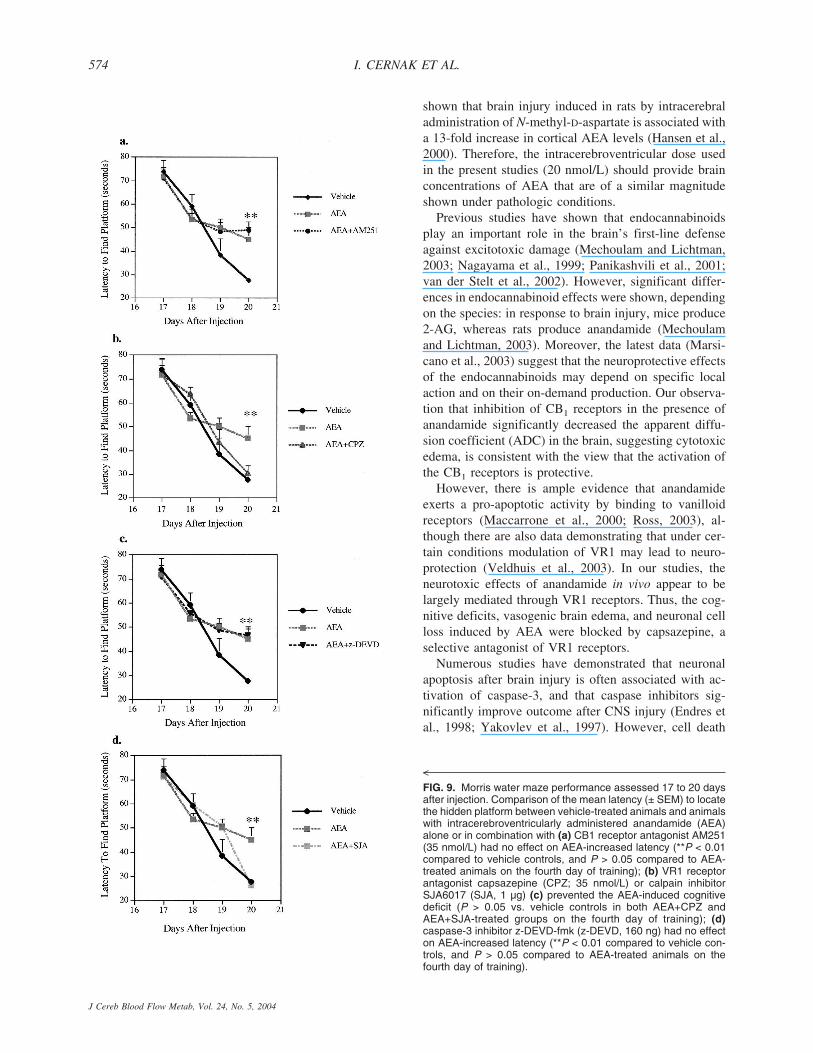
the hidden platform version of the Morris water maze as pre-viously described (Hamm et al., 1996). Briefly, rats (n �10/group) were trained to locate a hidden, submerged platformusing constant extra-maze visual information, while the moni-toring was performed using a PC-controlled video system (Ac-cuScan Instruments, Columbus, OH, U.S.A.). The apparatusconsists of a large, white circular pool (900-mm diameter, 500mm high, water temperature 24 ± 1°C) with a Plexiglas plat-form 76 mm in diameter painted white and submerged 15 mmbelow the surface of the water (225 mm high). The surface ofthe water is rendered opaque with the addition of dilute, white,nontoxic paint. During training, the platform remained in aconstant location hidden in one quadrant 14 cm from the side-wall. The rat was gently placed in the water facing the wall atone of four randomly chosen locations separated by 90°. Thelatency to find the hidden platform within a 90-second criteriontime was recorded by a masked observer. A series of 16 train-ing trials, administered in blocks of four, were conducted ondays 17, 18, 19, and 20 in rats after drug injection. To controlfor visual discriminative ability or motor impairment, the sameanimals were finally required to locate a clearly visible blackplatform (placed in a different location) raised 5 mm above thewater surface at least 2 hours after the last training trial. Theresults were expressed as a latency to find the platform (sec-onds) ± SD.
Western immunoblottingThe animals were killed and the brain was immediately re-
moved. After dissection, the brain samples were stored at−80°C. For analysis, the brain tissue was resuspended in lysisbuffer (60 mmol/L Tris-HCl, pH 7.8 containing 150 mmol/LNaCl, 5 mmol/L EDTA, 10% glycerol, 2 mmol/L Na3VO4, 25mmol/L NaF, 10 �g/ml leupeptin (Sigma), 10 �g/ml aprotinin(Sigma), 1 mmol/L AEBSF (Sigma), 1 mmol/L Pepstatin(Sigma), 1 mmol/L Microcystin LR (Sigma), 0.1% sodium do-decyl sulfate (SDS), 0.5% Na deoxycholate, and 1% TritonX-100 (Calbiochem, La Jolla, CA, U.S.A.). The samples wereincubated on ice for at least 30 minutes and centrifuged at20,000 g for 15 min. The soluble fraction representing total cellextracts was recovered and stored at −80°C until use. Proteinconcentration in the samples was determined with the BCAassay kit (Pierce, Rockford, IL, U.S.A.). Equal protein aliquots(25 to 50 �g) were resolved by SDS-polyacrylamide gel elec-trophoresis and transferred to nitrocellulose membranes (Hy-bond-C super; Amersham, Arlington Heights, IL, U.S.A.). Af-ter transfer, the gels were stained with GelCode blue stainreagent (Pierce) to verify equal protein loading. The mem-branes were probed with specific primary antibodies and the
FIG. 1. Diagrammatic representation of the different regions uti-lized for apparent diffusion coefficient determinations. LC, leftcortex; RC, right cortex; LSC, left subcortex; RSC, right subcor-tex. The right side is ipsilateral, whereas the left side is contra-lateral to the anandamide injection.
ANANDAMIDE NEUROTOXICITY 567
J Cereb Blood Flow Metab, Vol. 24, No. 5, 2004

immune complexes were detected using appropriate horserad-ish peroxidase–liked secondary antibodies (Amersham Phar-macia Biotech), chemiluminescence reagents (Super SignalWestDura, Pierce), and Kodak Biomax MR-1 films (Sigma).
Calpain activity assayTissue was homogenized in extraction buffer (K240-100,
Biovision), incubated on ice for 20 minutes, and then centri-fuged (10,000 rcf, 4°C). Supernatant was removed and centri-fuged again (10,000 rcf, 4°C). The final supernatant was ali-quoted and frozen. Later, protein concentration was determinedusing the method of Bradford (Bradford, 1976). Fifty micro-grams of protein was then diluted in reaction buffer (25% Ex-traction Buffer, 5% DMSO, 13.6 mmol/L Tris-HCL [pH 7.5],2.7 mmol/L dithiothreitol, 2.7 mmol/L CaCl2, and 500 �mol/LSuc-Leu-Tyr-AMC) and activity was measured at excitation of360 nm and an emission of 530 nm on a microplate fluores-cence reader (Cytofluor4000, PerSeptive Biosystems).
Caspase activity assayAssay for caspase-3- and caspase-9-like activity was per-
formed as previously described (Yakovlev et al., 1997). Ali-quots of cytosolic extracts (25 �g of protein in 100 �L ofextraction buffer) are preincubated at 37°C for 30 minutes, andthen mixed with an equal volume of 40 �mol/L fluorescenttetrapeptide substrate (Ac-DEVD-AMC or Ac-LEHD-AMC,respectively; Bachem, Torrance, CA, U.S.A.) in the samebuffer solution. Free aminomethylcoumarin (AMC) accumula-tion, which resulted from cleavage of the aspartate–AMC bond,is monitored continuously in each sample during 30 minutes in96-well microtiter plates, using a CytoFluor II fluorometer(PerSeptive Biosystems, Framingham, MA, U.S.A.) at 360-nmexcitation and 460 emission wavelengths. The emission fromeach well is plotted against time. Linear regression analysis ofthe initial velocity (slope) of each curve yielded an activity foreach sample. Data are expressed as a percentage of the caspaseactivity in samples from sham-treated control animals.
HistologyBrains, prefixed in 4% phosphate-saline buffered formalde-
hyde, were cut using a cryostat, and serial 6-�m, anterior-to-posterior sections were made. The sections were stained withhematoxylin and eosin. Cell count of neuronal profile was per-formed for the pyramidal cell layer of CA3 hippocampal area,in 10 randomly chosen sections from each brain, as previouslydescribed (Bramlett et al., 1997). All neurons were included,regardless of whether they were normal or with changed struc-tural characteristics. Hippocampal cell count in our vehicle-treated animals (254 ± 4 neurons in CA3 area) was comparableto values from the literature (Bramlett et al., 1997). The neuralprofile of the CA3 hippocampal sector of treated animals wasexpressed as a percentage of the total number of the neuronalcells in the CA3 region in vehicle-treated animals (100%). Cellcount data were analyzed statistically using the t-test for inde-pendent samples.
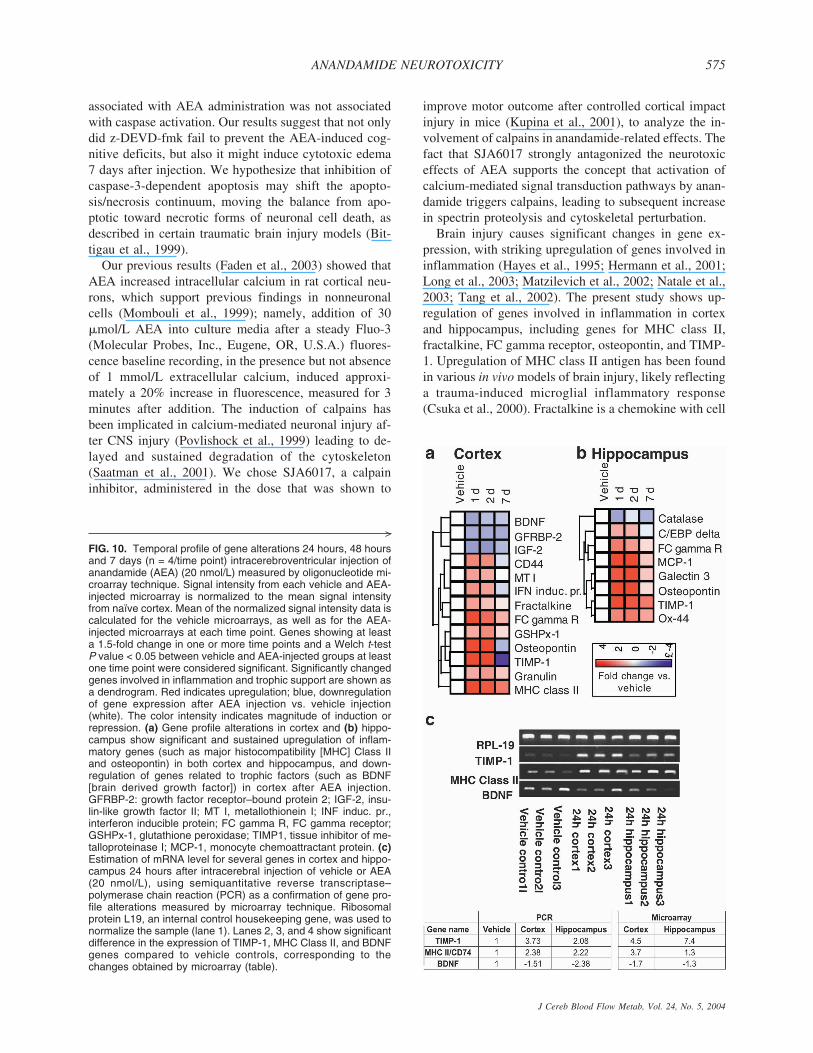
Gene profiling by oligonucleotide microarrayCortical and hippocampal samples were harvested at 24
hours, 48 hours, and 7 days after AEA (20 nmol/L, n � 3/timepoint) or vehicle administration (n � 3/time point). Total RNAfrom homogenized tissue was extracted using TRIzol reagent(Invitrogen Corporation, Carlsbad, CA, U.S.A.), and then con-verted to biotinylated cRNA and hybridized to 22 AffymetrixU34A oligonucleotide microarray containing 8,800 genes andexpression sequence tags (ESTs), as described by us (Di Gio-vanni et al., 2003). Signal intensity values for each oligonucleo-
tide probe set were calculated using Affymetrix GeneChipMAS 5.0 software. Any data not meeting stringent quality con-trol criteria were repeated to meet standards. Signal intensityfor each gene on the microarrays from AEA and vehicle-treatedanimals was normalized to levels in naïve cortex. Statisticalanalysis and hierarchic clustering was performed using Gene-Spring 5.0. Within each region, genes with significant changeswere grouped by the primary function of their gene product.
Semiquantitative reverse transcriptase-polymerasechain reaction
One �g of total RNA used for gene profiling was also usedfor cDNA synthesis using SuperScript reverse transcriptase(Gibco BRL, Bethesda, MD, U.S.A.) and oligo(dT)-primer.The amount of synthesized cDNA was evaluated by polymer-ase chain reaction (PCR) using primers specific for ribosomalprotein RPL19. PCR reactions were performed in a PTC-225Thermal Cycler (MJ Research, Waltham, MA, U.S.A.) usingAmpliTaq polymerase (Perkin Elmer Life Sciences, Torrance,CA, U.S.A.). Each PCR reaction was repeated at least twice.The thermal cycling parameters were as follows: 1 minute 30seconds at 94°C followed by 30 cycles of 30 seconds at 94°C,1 minute 30 seconds at 59°C, 1 minute at 72°C, and finalincubation for 5 minutes at 72°C. PCR reaction products wereanalyzed by agarose gel-electrophoresis. Intensity of injuredcortex and hippocampus (n � 3) was adjusted to respectivevehicle controls (n � 3) using a housekeeping gene RibosomalProtein (Invitrogen Corp., Carlsbad, CA, U.S.A.) L-19 (RPL-19). Normalized cDNA was then used to estimate the relativeabundances of tissue inhibitors of metalloproteinase (TIMP)-1,MHC class II and brain-derived growth factor (BDNF) to con-firm the array hybridization data. Primers for each gene werelocated in different exons. Different dilutions of cDNA sampleswere used for different genes to provide linear range of PCRreactions (Di Giovanni et al., 2003).
Data analysisContinuous variables subjected to repeated measurements
during a period of time (ADC, water maze studies) were ana-lyzed using a repeated-measures analysis of variance followedby Tukey pairwise comparison at each time point. MRS data,activity assays, and cell counts were analyzed with a t-test.Analysis of variance followed by t-test with Bonferroni correc-tions for multiple comparisons were used for comparisons ofanandamide- and vehicle-treated groups; P < 0.05 was consid-ered to reflect a statistically significant difference.
RESULTS
In vitro studyWe initially tested the effect of AEA on cell viability
measured by LDH release in primary cortical neuronalcultures, as previously described (Mukhin et al., 1997).Concentrations of AEA greater than 50 �mol/L signifi-cantly increased LDH release, indicating a neurotoxicaction (Fig. 2a). We subsequently examined the effect ofanandamide on rat cortical neurons exposed to a moder-ate stretch injury. This trauma model has been used tostudy diffuse axonal injury-induced molecular and bio-chemical responses in vitro (Ellis et al., 1995); it causesboth necrotic and apoptotic pathways of cell death (Pikeet al., 2000). Addition of 25 �mol/L AEA to cell culturemedia, a dose that did not cause cell death by itself,
I. CERNAK ET AL.568
J Cereb Blood Flow Metab, Vol. 24, No. 5, 2004
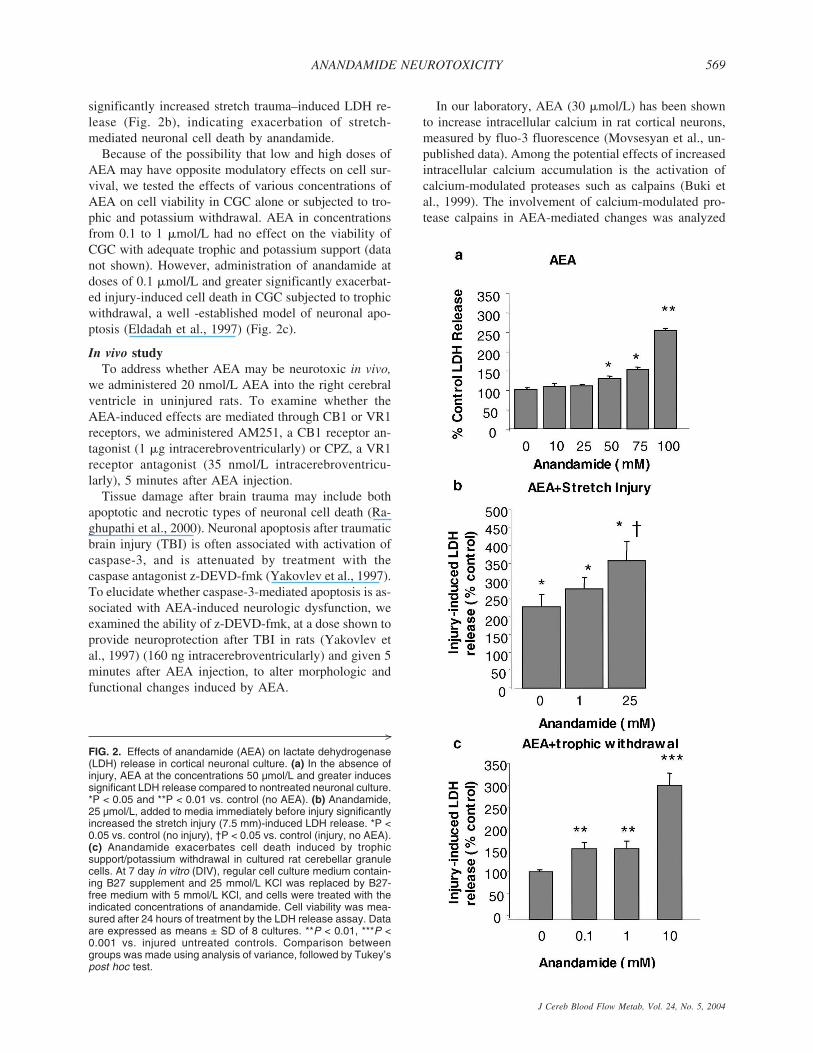
significantly increased stretch trauma–induced LDH re-lease (Fig. 2b), indicating exacerbation of stretch-mediated neuronal cell death by anandamide.
Because of the possibility that low and high doses ofAEA may have opposite modulatory effects on cell sur-vival, we tested the effects of various concentrations ofAEA on cell viability in CGC alone or subjected to tro-phic and potassium withdrawal. AEA in concentrationsfrom 0.1 to 1 �mol/L had no effect on the viability ofCGC with adequate trophic and potassium support (datanot shown). However, administration of anandamide atdoses of 0.1 �mol/L and greater significantly exacerbat-ed injury-induced cell death in CGC subjected to trophicwithdrawal, a well -established model of neuronal apo-ptosis (Eldadah et al., 1997) (Fig. 2c).
In vivo studyTo address whether AEA may be neurotoxic in vivo,
we administered 20 nmol/L AEA into the right cerebralventricle in uninjured rats. To examine whether theAEA-induced effects are mediated through CB1 or VR1receptors, we administered AM251, a CB1 receptor an-tagonist (1 �g intracerebroventricularly) or CPZ, a VR1receptor antagonist (35 nmol/L intracerebroventricu-larly), 5 minutes after AEA injection.
Tissue damage after brain trauma may include bothapoptotic and necrotic types of neuronal cell death (Ra-ghupathi et al., 2000). Neuronal apoptosis after traumaticbrain injury (TBI) is often associated with activation ofcaspase-3, and is attenuated by treatment with thecaspase antagonist z-DEVD-fmk (Yakovlev et al., 1997).To elucidate whether caspase-3-mediated apoptosis is as-sociated with AEA-induced neurologic dysfunction, weexamined the ability of z-DEVD-fmk, at a dose shown toprovide neuroprotection after TBI in rats (Yakovlev etal., 1997) (160 ng intracerebroventricularly) and given 5minutes after AEA injection, to alter morphologic andfunctional changes induced by AEA.
In our laboratory, AEA (30 �mol/L) has been shownto increase intracellular calcium in rat cortical neurons,measured by fluo-3 fluorescence (Movsesyan et al., un-published data). Among the potential effects of increasedintracellular calcium accumulation is the activation ofcalcium-modulated proteases such as calpains (Buki etal., 1999). The involvement of calcium-modulated pro-tease calpains in AEA-mediated changes was analyzed
>
FIG. 2. Effects of anandamide (AEA) on lactate dehydrogenase(LDH) release in cortical neuronal culture. (a) In the absence ofinjury, AEA at the concentrations 50 µmol/L and greater inducessignificant LDH release compared to nontreated neuronal culture.*P < 0.05 and **P < 0.01 vs. control (no AEA). (b) Anandamide,25 µmol/L, added to media immediately before injury significantlyincreased the stretch injury (7.5 mm)-induced LDH release. *P <0.05 vs. control (no injury), †P < 0.05 vs. control (injury, no AEA).(c) Anandamide exacerbates cell death induced by trophicsupport/potassium withdrawal in cultured rat cerebellar granulecells. At 7 day in vitro (DIV), regular cell culture medium contain-ing B27 supplement and 25 mmol/L KCl was replaced by B27-free medium with 5 mmol/L KCl, and cells were treated with theindicated concentrations of anandamide. Cell viability was mea-sured after 24 hours of treatment by the LDH release assay. Dataare expressed as means ± SD of 8 cultures. **P < 0.01, ***P <0.001 vs. injured untreated controls. Comparison betweengroups was made using analysis of variance, followed by Tukey’spost hoc test.
ANANDAMIDE NEUROTOXICITY 569
J Cereb Blood Flow Metab, Vol. 24, No. 5, 2004
by injecting a calpain inhibitor, SJA6017 (35 nmol/L),intracerebroventricularly 5 minutes after AEA adminis-tration.
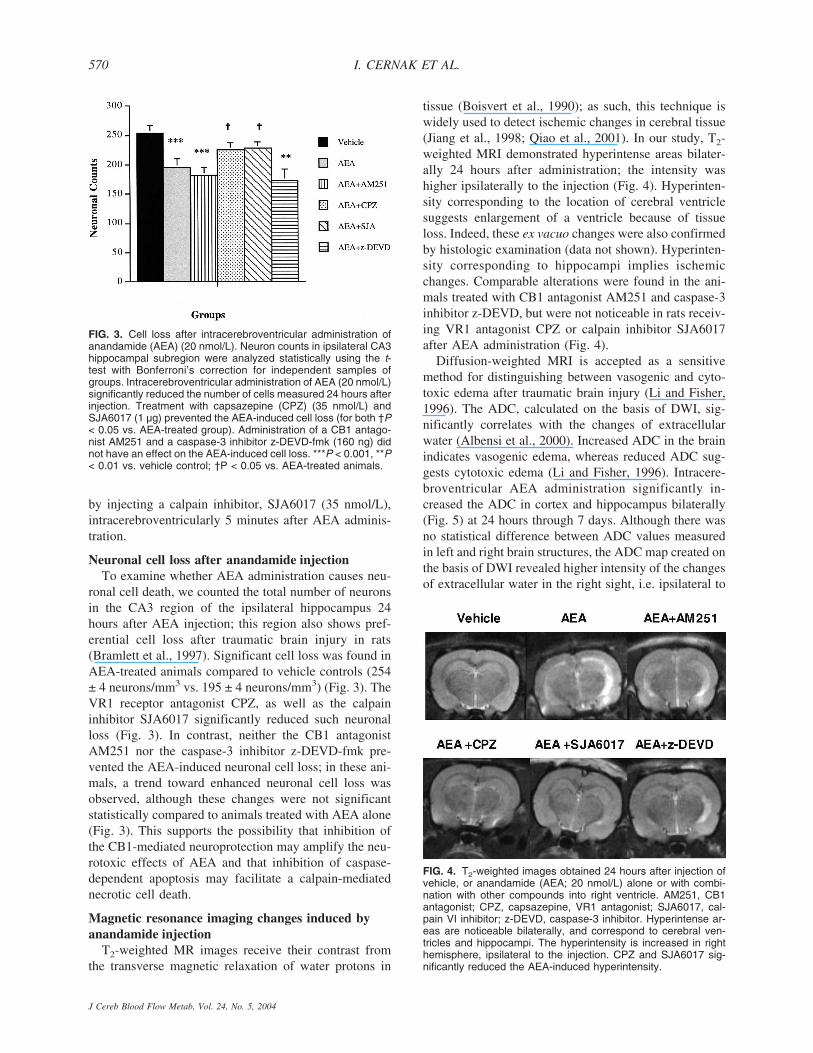
Neuronal cell loss after anandamide injectionTo examine whether AEA administration causes neu-
ronal cell death, we counted the total number of neuronsin the CA3 region of the ipsilateral hippocampus 24hours after AEA injection; this region also shows pref-erential cell loss after traumatic brain injury in rats(Bramlett et al., 1997). Significant cell loss was found inAEA-treated animals compared to vehicle controls (254± 4 neurons/mm3 vs. 195 ± 4 neurons/mm3) (Fig. 3). TheVR1 receptor antagonist CPZ, as well as the calpaininhibitor SJA6017 significantly reduced such neuronalloss (Fig. 3). In contrast, neither the CB1 antagonistAM251 nor the caspase-3 inhibitor z-DEVD-fmk pre-vented the AEA-induced neuronal cell loss; in these ani-mals, a trend toward enhanced neuronal cell loss wasobserved, although these changes were not significantstatistically compared to animals treated with AEA alone(Fig. 3). This supports the possibility that inhibition ofthe CB1-mediated neuroprotection may amplify the neu-rotoxic effects of AEA and that inhibition of caspase-dependent apoptosis may facilitate a calpain-mediatednecrotic cell death.
Magnetic resonance imaging changes induced byanandamide injection
T2-weighted MR images receive their contrast fromthe transverse magnetic relaxation of water protons in
tissue (Boisvert et al., 1990); as such, this technique iswidely used to detect ischemic changes in cerebral tissue(Jiang et al., 1998; Qiao et al., 2001). In our study, T2-weighted MRI demonstrated hyperintense areas bilater-ally 24 hours after administration; the intensity washigher ipsilaterally to the injection (Fig. 4). Hyperinten-sity corresponding to the location of cerebral ventriclesuggests enlargement of a ventricle because of tissueloss. Indeed, these ex vacuo changes were also confirmedby histologic examination (data not shown). Hyperinten-sity corresponding to hippocampi implies ischemicchanges. Comparable alterations were found in the ani-mals treated with CB1 antagonist AM251 and caspase-3inhibitor z-DEVD, but were not noticeable in rats receiv-ing VR1 antagonist CPZ or calpain inhibitor SJA6017after AEA administration (Fig. 4).
Diffusion-weighted MRI is accepted as a sensitivemethod for distinguishing between vasogenic and cyto-toxic edema after traumatic brain injury (Li and Fisher,1996). The ADC, calculated on the basis of DWI, sig-nificantly correlates with the changes of extracellularwater (Albensi et al., 2000). Increased ADC in the brainindicates vasogenic edema, whereas reduced ADC sug-gests cytotoxic edema (Li and Fisher, 1996). Intracere-broventricular AEA administration significantly in-creased the ADC in cortex and hippocampus bilaterally(Fig. 5) at 24 hours through 7 days. Although there wasno statistical difference between ADC values measuredin left and right brain structures, the ADC map created onthe basis of DWI revealed higher intensity of the changesof extracellular water in the right sight, i.e. ipsilateral to
FIG. 3. Cell loss after intracerebroventricular administration ofanandamide (AEA) (20 nmol/L). Neuron counts in ipsilateral CA3hippocampal subregion were analyzed statistically using the t-test with Bonferroni’s correction for independent samples ofgroups. Intracerebroventricular administration of AEA (20 nmol/L)significantly reduced the number of cells measured 24 hours afterinjection. Treatment with capsazepine (CPZ) (35 nmol/L) andSJA6017 (1 µg) prevented the AEA-induced cell loss (for both †P< 0.05 vs. AEA-treated group). Administration of a CB1 antago-nist AM251 and a caspase-3 inhibitor z-DEVD-fmk (160 ng) didnot have an effect on the AEA-induced cell loss. ***P < 0.001, **P< 0.01 vs. vehicle control; †P < 0.05 vs. AEA-treated animals.
FIG. 4. T2-weighted images obtained 24 hours after injection ofvehicle, or anandamide (AEA; 20 nmol/L) alone or with combi-nation with other compounds into right ventricle. AM251, CB1antagonist; CPZ, capsazepine, VR1 antagonist; SJA6017, cal-pain VI inhibitor; z-DEVD, caspase-3 inhibitor. Hyperintense ar-eas are noticeable bilaterally, and correspond to cerebral ven-tricles and hippocampi. The hyperintensity is increased in righthemisphere, ipsilateral to the injection. CPZ and SJA6017 sig-nificantly reduced the AEA-induced hyperintensity.
I. CERNAK ET AL.570
J Cereb Blood Flow Metab, Vol. 24, No. 5, 2004
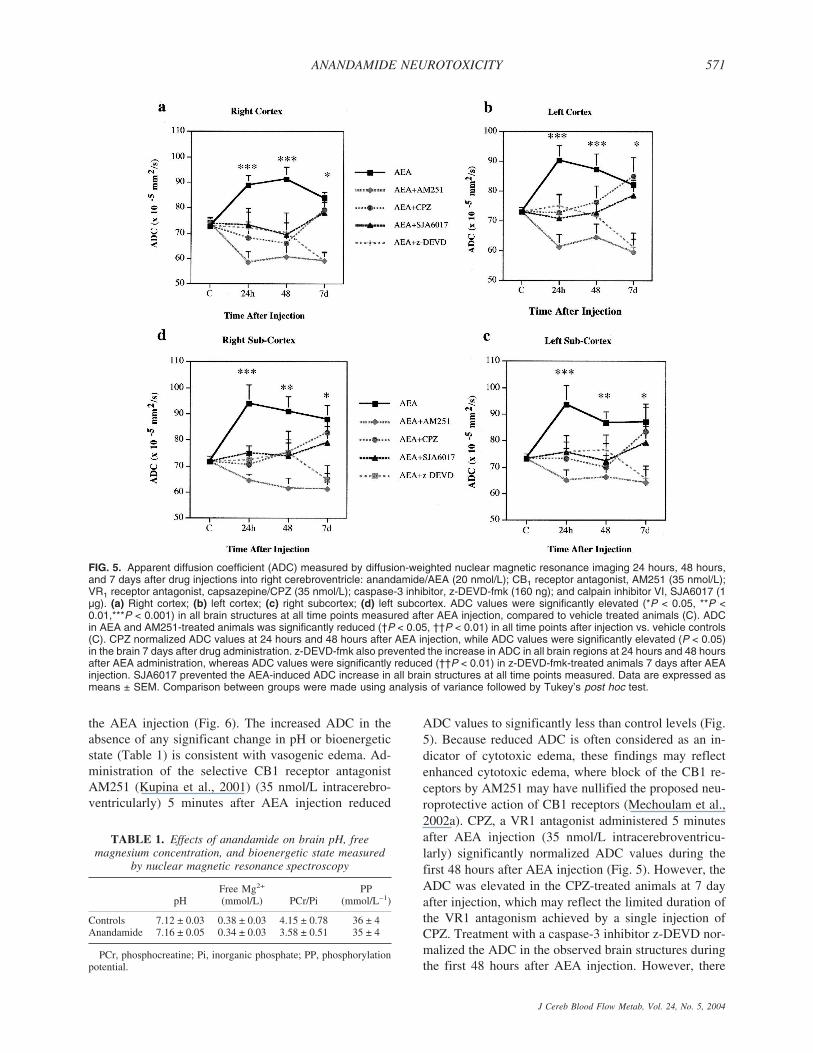
the AEA injection (Fig. 6). The increased ADC in theabsence of any significant change in pH or bioenergeticstate (Table 1) is consistent with vasogenic edema. Ad-ministration of the selective CB1 receptor antagonistAM251 (Kupina et al., 2001) (35 nmol/L intracerebro-ventricularly) 5 minutes after AEA injection reduced
ADC values to significantly less than control levels (Fig.5). Because reduced ADC is often considered as an in-dicator of cytotoxic edema, these findings may reflectenhanced cytotoxic edema, where block of the CB1 re-ceptors by AM251 may have nullified the proposed neu-roprotective action of CB1 receptors (Mechoulam et al.,2002a). CPZ, a VR1 antagonist administered 5 minutesafter AEA injection (35 nmol/L intracerebroventricu-larly) significantly normalized ADC values during thefirst 48 hours after AEA injection (Fig. 5). However, theADC was elevated in the CPZ-treated animals at 7 dayafter injection, which may reflect the limited duration ofthe VR1 antagonism achieved by a single injection ofCPZ. Treatment with a caspase-3 inhibitor z-DEVD nor-malized the ADC in the observed brain structures duringthe first 48 hours after AEA injection. However, there
FIG. 5. Apparent diffusion coefficient (ADC) measured by diffusion-weighted nuclear magnetic resonance imaging 24 hours, 48 hours,and 7 days after drug injections into right cerebroventricle: anandamide/AEA (20 nmol/L); CB1 receptor antagonist, AM251 (35 nmol/L);VR1 receptor antagonist, capsazepine/CPZ (35 nmol/L); caspase-3 inhibitor, z-DEVD-fmk (160 ng); and calpain inhibitor VI, SJA6017 (1µg). (a) Right cortex; (b) left cortex; (c) right subcortex; (d) left subcortex. ADC values were significantly elevated (*P < 0.05, **P <0.01,***P < 0.001) in all brain structures at all time points measured after AEA injection, compared to vehicle treated animals (C). ADCin AEA and AM251-treated animals was significantly reduced (†P < 0.05, ††P < 0.01) in all time points after injection vs. vehicle controls(C). CPZ normalized ADC values at 24 hours and 48 hours after AEA injection, while ADC values were significantly elevated (P < 0.05)in the brain 7 days after drug administration. z-DEVD-fmk also prevented the increase in ADC in all brain regions at 24 hours and 48 hoursafter AEA administration, whereas ADC values were significantly reduced (††P < 0.01) in z-DEVD-fmk-treated animals 7 days after AEAinjection. SJA6017 prevented the AEA-induced ADC increase in all brain structures at all time points measured. Data are expressed asmeans ± SEM. Comparison between groups were made using analysis of variance followed by Tukey’s post hoc test.
TABLE 1. Effects of anandamide on brain pH, freemagnesium concentration, and bioenergetic state measured
by nuclear magnetic resonance spectroscopy
pHFree Mg2+
(mmol/L) PCr/PiPP
(mmol/L−1)
Controls 7.12 ± 0.03 0.38 ± 0.03 4.15 ± 0.78 36 ± 4Anandamide 7.16 ± 0.05 0.34 ± 0.03 3.58 ± 0.51 35 ± 4
PCr, phosphocreatine; Pi, inorganic phosphate; PP, phosphorylationpotential.
ANANDAMIDE NEUROTOXICITY 571
J Cereb Blood Flow Metab, Vol. 24, No. 5, 2004
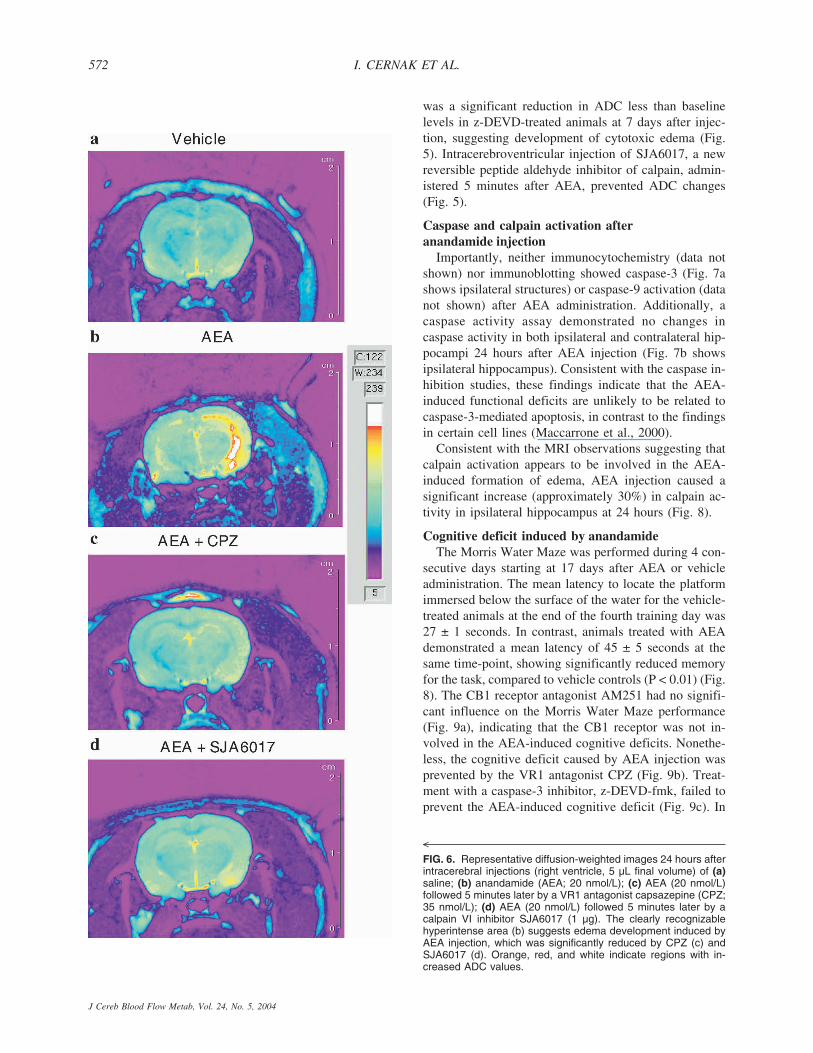
was a significant reduction in ADC less than baselinelevels in z-DEVD-treated animals at 7 days after injec-tion, suggesting development of cytotoxic edema (Fig.5). Intracerebroventricular injection of SJA6017, a newreversible peptide aldehyde inhibitor of calpain, admin-istered 5 minutes after AEA, prevented ADC changes(Fig. 5).
Caspase and calpain activation afteranandamide injection
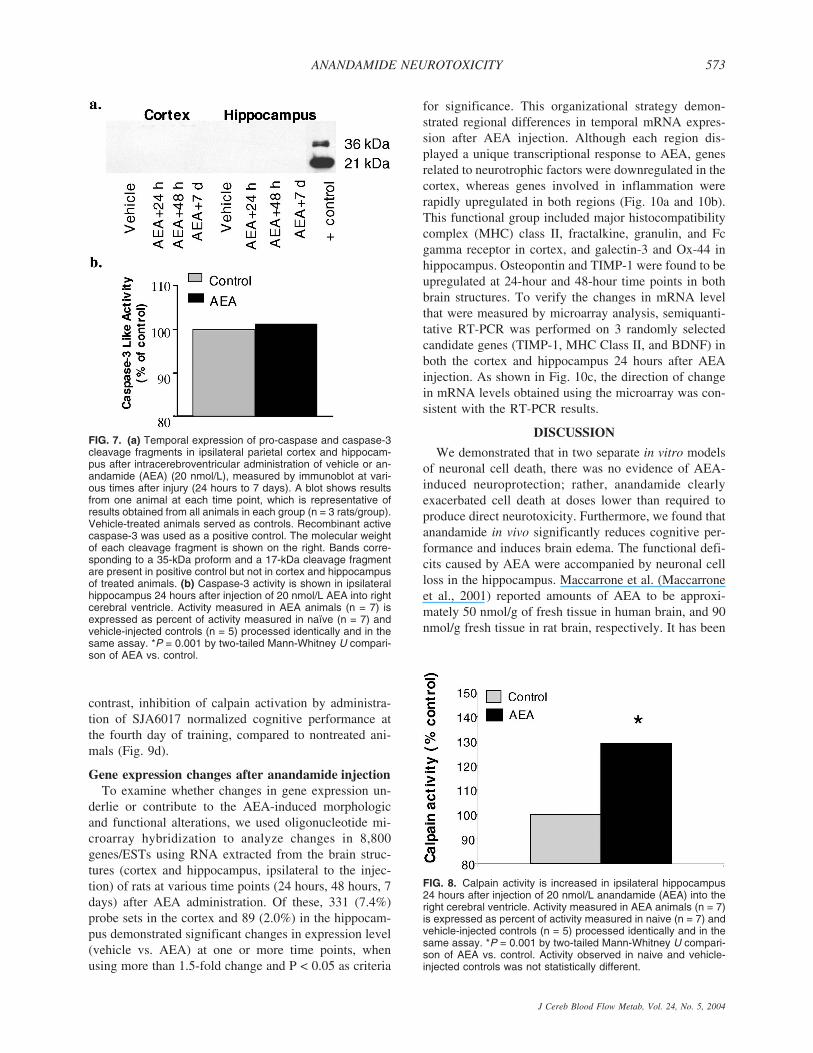
Importantly, neither immunocytochemistry (data notshown) nor immunoblotting showed caspase-3 (Fig. 7ashows ipsilateral structures) or caspase-9 activation (datanot shown) after AEA administration. Additionally, acaspase activity assay demonstrated no changes incaspase activity in both ipsilateral and contralateral hip-pocampi 24 hours after AEA injection (Fig. 7b showsipsilateral hippocampus). Consistent with the caspase in-hibition studies, these findings indicate that the AEA-induced functional deficits are unlikely to be related tocaspase-3-mediated apoptosis, in contrast to the findingsin certain cell lines (Maccarrone et al., 2000).
Consistent with the MRI observations suggesting thatcalpain activation appears to be involved in the AEA-induced formation of edema, AEA injection caused asignificant increase (approximately 30%) in calpain ac-tivity in ipsilateral hippocampus at 24 hours (Fig. 8).
Cognitive deficit induced by anandamideThe Morris Water Maze was performed during 4 con-
secutive days starting at 17 days after AEA or vehicleadministration. The mean latency to locate the platformimmersed below the surface of the water for the vehicle-treated animals at the end of the fourth training day was27 ± 1 seconds. In contrast, animals treated with AEAdemonstrated a mean latency of 45 ± 5 seconds at thesame time-point, showing significantly reduced memoryfor the task, compared to vehicle controls (P < 0.01) (Fig.8). The CB1 receptor antagonist AM251 had no signifi-cant influence on the Morris Water Maze performance(Fig. 9a), indicating that the CB1 receptor was not in-volved in the AEA-induced cognitive deficits. Nonethe-less, the cognitive deficit caused by AEA injection wasprevented by the VR1 antagonist CPZ (Fig. 9b). Treat-ment with a caspase-3 inhibitor, z-DEVD-fmk, failed toprevent the AEA-induced cognitive deficit (Fig. 9c). In
<
FIG. 6. Representative diffusion-weighted images 24 hours afterintracerebral injections (right ventricle, 5 µL final volume) of (a)saline; (b) anandamide (AEA; 20 nmol/L); (c) AEA (20 nmol/L)followed 5 minutes later by a VR1 antagonist capsazepine (CPZ;35 nmol/L); (d) AEA (20 nmol/L) followed 5 minutes later by acalpain VI inhibitor SJA6017 (1 µg). The clearly recognizablehyperintense area (b) suggests edema development induced byAEA injection, which was significantly reduced by CPZ (c) andSJA6017 (d). Orange, red, and white indicate regions with in-creased ADC values.
I. CERNAK ET AL.572
J Cereb Blood Flow Metab, Vol. 24, No. 5, 2004
contrast, inhibition of calpain activation by administra-tion of SJA6017 normalized cognitive performance atthe fourth day of training, compared to nontreated ani-mals (Fig. 9d).
Gene expression changes after anandamide injectionTo examine whether changes in gene expression un-
derlie or contribute to the AEA-induced morphologicand functional alterations, we used oligonucleotide mi-croarray hybridization to analyze changes in 8,800genes/ESTs using RNA extracted from the brain struc-tures (cortex and hippocampus, ipsilateral to the injec-tion) of rats at various time points (24 hours, 48 hours, 7days) after AEA administration. Of these, 331 (7.4%)probe sets in the cortex and 89 (2.0%) in the hippocam-pus demonstrated significant changes in expression level(vehicle vs. AEA) at one or more time points, whenusing more than 1.5-fold change and P < 0.05 as criteria
for significance. This organizational strategy demon-strated regional differences in temporal mRNA expres-sion after AEA injection. Although each region dis-played a unique transcriptional response to AEA, genesrelated to neurotrophic factors were downregulated in thecortex, whereas genes involved in inflammation wererapidly upregulated in both regions (Fig. 10a and 10b).This functional group included major histocompatibilitycomplex (MHC) class II, fractalkine, granulin, and Fcgamma receptor in cortex, and galectin-3 and Ox-44 inhippocampus. Osteopontin and TIMP-1 were found to beupregulated at 24-hour and 48-hour time points in bothbrain structures. To verify the changes in mRNA levelthat were measured by microarray analysis, semiquanti-tative RT-PCR was performed on 3 randomly selectedcandidate genes (TIMP-1, MHC Class II, and BDNF) inboth the cortex and hippocampus 24 hours after AEAinjection. As shown in Fig. 10c, the direction of changein mRNA levels obtained using the microarray was con-sistent with the RT-PCR results.
DISCUSSION
We demonstrated that in two separate in vitro modelsof neuronal cell death, there was no evidence of AEA-induced neuroprotection; rather, anandamide clearlyexacerbated cell death at doses lower than required toproduce direct neurotoxicity. Furthermore, we found thatanandamide in vivo significantly reduces cognitive per-formance and induces brain edema. The functional defi-cits caused by AEA were accompanied by neuronal cellloss in the hippocampus. Maccarrone et al. (Maccarroneet al., 2001) reported amounts of AEA to be approxi-mately 50 nmol/g of fresh tissue in human brain, and 90nmol/g fresh tissue in rat brain, respectively. It has been
FIG. 8. Calpain activity is increased in ipsilateral hippocampus24 hours after injection of 20 nmol/L anandamide (AEA) into theright cerebral ventricle. Activity measured in AEA animals (n = 7)is expressed as percent of activity measured in naive (n = 7) andvehicle-injected controls (n = 5) processed identically and in thesame assay. *P = 0.001 by two-tailed Mann-Whitney U compari-son of AEA vs. control. Activity observed in naive and vehicle-injected controls was not statistically different.
FIG. 7. (a) Temporal expression of pro-caspase and caspase-3cleavage fragments in ipsilateral parietal cortex and hippocam-pus after intracerebroventricular administration of vehicle or an-andamide (AEA) (20 nmol/L), measured by immunoblot at vari-ous times after injury (24 hours to 7 days). A blot shows resultsfrom one animal at each time point, which is representative ofresults obtained from all animals in each group (n = 3 rats/group).Vehicle-treated animals served as controls. Recombinant activecaspase-3 was used as a positive control. The molecular weightof each cleavage fragment is shown on the right. Bands corre-sponding to a 35-kDa proform and a 17-kDa cleavage fragmentare present in positive control but not in cortex and hippocampusof treated animals. (b) Caspase-3 activity is shown in ipsilateralhippocampus 24 hours after injection of 20 nmol/L AEA into rightcerebral ventricle. Activity measured in AEA animals (n = 7) isexpressed as percent of activity measured in naïve (n = 7) andvehicle-injected controls (n = 5) processed identically and in thesame assay. *P = 0.001 by two-tailed Mann-Whitney U compari-son of AEA vs. control.
ANANDAMIDE NEUROTOXICITY 573
J Cereb Blood Flow Metab, Vol. 24, No. 5, 2004
shown that brain injury induced in rats by intracerebraladministration of N-methyl-D-aspartate is associated witha 13-fold increase in cortical AEA levels (Hansen et al.,2000). Therefore, the intracerebroventricular dose usedin the present studies (20 nmol/L) should provide brainconcentrations of AEA that are of a similar magnitudeshown under pathologic conditions.
Previous studies have shown that endocannabinoidsplay an important role in the brain’s first-line defenseagainst excitotoxic damage (Mechoulam and Lichtman,2003; Nagayama et al., 1999; Panikashvili et al., 2001;van der Stelt et al., 2002). However, significant differ-ences in endocannabinoid effects were shown, dependingon the species: in response to brain injury, mice produce2-AG, whereas rats produce anandamide (Mechoulamand Lichtman, 2003). Moreover, the latest data (Marsi-cano et al., 2003) suggest that the neuroprotective effectsof the endocannabinoids may depend on specific localaction and on their on-demand production. Our observa-tion that inhibition of CB1 receptors in the presence ofanandamide significantly decreased the apparent diffu-sion coefficient (ADC) in the brain, suggesting cytotoxicedema, is consistent with the view that the activation ofthe CB1 receptors is protective.
However, there is ample evidence that anandamideexerts a pro-apoptotic activity by binding to vanilloidreceptors (Maccarrone et al., 2000; Ross, 2003), al-though there are also data demonstrating that under cer-tain conditions modulation of VR1 may lead to neuro-protection (Veldhuis et al., 2003). In our studies, theneurotoxic effects of anandamide in vivo appear to belargely mediated through VR1 receptors. Thus, the cog-nitive deficits, vasogenic brain edema, and neuronal cellloss induced by AEA were blocked by capsazepine, aselective antagonist of VR1 receptors.
Numerous studies have demonstrated that neuronalapoptosis after brain injury is often associated with ac-tivation of caspase-3, and that caspase inhibitors sig-nificantly improve outcome after CNS injury (Endres etal., 1998; Yakovlev et al., 1997). However, cell death
<
FIG. 9. Morris water maze performance assessed 17 to 20 daysafter injection. Comparison of the mean latency (± SEM) to locatethe hidden platform between vehicle-treated animals and animalswith intracerebroventricularly administered anandamide (AEA)alone or in combination with (a) CB1 receptor antagonist AM251(35 nmol/L) had no effect on AEA-increased latency (**P < 0.01compared to vehicle controls, and P > 0.05 compared to AEA-treated animals on the fourth day of training); (b) VR1 receptorantagonist capsazepine (CPZ; 35 nmol/L) or calpain inhibitorSJA6017 (SJA, 1 µg) (c) prevented the AEA-induced cognitivedeficit (P > 0.05 vs. vehicle controls in both AEA+CPZ andAEA+SJA-treated groups on the fourth day of training); (d)caspase-3 inhibitor z-DEVD-fmk (z-DEVD, 160 ng) had no effecton AEA-increased latency (**P < 0.01 compared to vehicle con-trols, and P > 0.05 compared to AEA-treated animals on thefourth day of training).
I. CERNAK ET AL.574
J Cereb Blood Flow Metab, Vol. 24, No. 5, 2004
associated with AEA administration was not associatedwith caspase activation. Our results suggest that not onlydid z-DEVD-fmk fail to prevent the AEA-induced cog-nitive deficits, but also it might induce cytotoxic edema7 days after injection. We hypothesize that inhibition ofcaspase-3-dependent apoptosis may shift the apopto-sis/necrosis continuum, moving the balance from apo-ptotic toward necrotic forms of neuronal cell death, asdescribed in certain traumatic brain injury models (Bit-tigau et al., 1999).
Our previous results (Faden et al., 2003) showed thatAEA increased intracellular calcium in rat cortical neu-rons, which support previous findings in nonneuronalcells (Mombouli et al., 1999); namely, addition of 30�mol/L AEA into culture media after a steady Fluo-3(Molecular Probes, Inc., Eugene, OR, U.S.A.) fluores-cence baseline recording, in the presence but not absenceof 1 mmol/L extracellular calcium, induced approxi-mately a 20% increase in fluorescence, measured for 3minutes after addition. The induction of calpains hasbeen implicated in calcium-mediated neuronal injury af-ter CNS injury (Povlishock et al., 1999) leading to de-layed and sustained degradation of the cytoskeleton(Saatman et al., 2001). We chose SJA6017, a calpaininhibitor, administered in the dose that was shown to
improve motor outcome after controlled cortical impactinjury in mice (Kupina et al., 2001), to analyze the in-volvement of calpains in anandamide-related effects. Thefact that SJA6017 strongly antagonized the neurotoxiceffects of AEA supports the concept that activation ofcalcium-mediated signal transduction pathways by anan-damide triggers calpains, leading to subsequent increasein spectrin proteolysis and cytoskeletal perturbation.
Brain injury causes significant changes in gene ex-pression, with striking upregulation of genes involved ininflammation (Hayes et al., 1995; Hermann et al., 2001;Long et al., 2003; Matzilevich et al., 2002; Natale et al.,2003; Tang et al., 2002). The present study shows up-regulation of genes involved in inflammation in cortexand hippocampus, including genes for MHC class II,fractalkine, FC gamma receptor, osteopontin, and TIMP-1. Upregulation of MHC class II antigen has been foundin various in vivo models of brain injury, likely reflectinga trauma-induced microglial inflammatory response(Csuka et al., 2000). Fractalkine is a chemokine with cell
>
FIG. 10. Temporal profile of gene alterations 24 hours, 48 hoursand 7 days (n = 4/time point) intracerebroventricular injection ofanandamide (AEA) (20 nmol/L) measured by oligonucleotide mi-croarray technique. Signal intensity from each vehicle and AEA-injected microarray is normalized to the mean signal intensityfrom naïve cortex. Mean of the normalized signal intensity data iscalculated for the vehicle microarrays, as well as for the AEA-injected microarrays at each time point. Genes showing at leasta 1.5-fold change in one or more time points and a Welch t-testP value < 0.05 between vehicle and AEA-injected groups at leastone time point were considered significant. Significantly changedgenes involved in inflammation and trophic support are shown asa dendrogram. Red indicates upregulation; blue, downregulationof gene expression after AEA injection vs. vehicle injection(white). The color intensity indicates magnitude of induction orrepression. (a) Gene profile alterations in cortex and (b) hippo-campus show significant and sustained upregulation of inflam-matory genes (such as major histocompatibility [MHC] Class IIand osteopontin) in both cortex and hippocampus, and down-regulation of genes related to trophic factors (such as BDNF[brain derived growth factor]) in cortex after AEA injection.GFRBP-2: growth factor receptor–bound protein 2; IGF-2, insu-lin-like growth factor II; MT I, metallothionein I; INF induc. pr.,interferon inducible protein; FC gamma R, FC gamma receptor;GSHPx-1, glutathione peroxidase; TIMP1, tissue inhibitor of me-talloproteinase I; MCP-1, monocyte chemoattractant protein. (c)Estimation of mRNA level for several genes in cortex and hippo-campus 24 hours after intracerebral injection of vehicle or AEA(20 nmol/L), using semiquantitative reverse transcriptase–polymerase chain reaction (PCR) as a confirmation of gene pro-file alterations measured by microarray technique. Ribosomalprotein L19, an internal control housekeeping gene, was used tonormalize the sample (lane 1). Lanes 2, 3, and 4 show significantdifference in the expression of TIMP-1, MHC Class II, and BDNFgenes compared to vehicle controls, corresponding to thechanges obtained by microarray (table).
ANANDAMIDE NEUROTOXICITY 575
J Cereb Blood Flow Metab, Vol. 24, No. 5, 2004

adhesion and chemoattractive properties, which has beenfound to facilitate inflammatory processes in the CNS(Chapman et al., 2000). It has been demonstrated thatfractalkine released from injured neurons can induce pro-liferation, activation, or migration of microglia, and assuch can participate in the inflammatory response to neu-ronal injury (Tarozzo et al., 2002). Aggregation of Fcgamma receptors has been shown to activate phospholi-pases, increase intracellular Ca2+, and participate in an-tibody-directed cytotoxicity (Melendez et al., 1998). Up-regulation of Fc gamma receptor has been reported in rathippocampus during the early posttraumatic period(Matzilevich et al., 2002) as well in mouse cortex 3 daysafter cortical compact injury (Kobori et al., 2002). Os-teopontin is a proinflammatory cytokine that enhancesinterferon-� and interleukin-12 production, decreases in-terleukin-10, and plays a role in autoimmune-mediateddemyelination (Chabas et al., 2001). Increased expres-sion of osteopontin mRNA has been demonstrated inactivated microglia as a response of the brain to kainic-acid induced excitotoxic injury (Kim et al., 2002).TIMPs that are specific inhibitors for metalloproteinaseshave been implicated in pathologic tissue remodelingsuch as scar formation (La Fleur et al., 1996). Significantupregulation of TIMP-1 has been found in reactiveastrocytes after stab injury to the mature rat brain (Jawor-ski, 2000) as well as in ischemic rat cortices after mid-dle cerebral occlusion (Rosenberg et al., 1998). Be-cause many of the genes mentioned here are involvedin inflammation and microglial activation, these re-sults suggest the possible role of AEA in neurogenicinflammation.
Our results show downregulation of genes related toneurotrophic factors in cortex after AEA administration.Changes in the gene expression profile of neurotrophicfactors have been found after brain injury (Hicks et al.,1999; Kobori et al., 2002; Rall et al., 2003), with BDNFbeing upregulated (Kobori et al., 2002). Downregulationof neuroprotective neurotrophins, including BDNF,may represent additional mechanism of AEA-inducedneurotoxicity.
In conclusion, the present study demonstrates that theendocannabinoid anandamide is not necessarily neuro-protective as previously believed (Mechoulam et al.,2002a; Panikashvili et al., 2001; Sinor et al., 2000), butrather may induce pathologic effects in the brain, medi-ated through VR1 receptors and calpain activation. Fur-thermore, the increased vasogenic edema and the geneprofiling changes induced by anandamide are consistentwith the hypothesis that AEA initiates a neurogenic in-flammatory response. Anandamide is also neurotoxic invitro, and exacerbates neuronal cell death induced byeither stretch injury or trophic withdrawal. Because AEAhas been shown to be neuroprotective in different in vitromodel systems, we suggest that AEA may, like Janus
facing in opposite directions, show opposite actions withregard to modulation of cell death, depending on whichreceptors or signal transduction cascades are activated.
REFERENCES
Albensi BC, Knoblach SM, Chew BG, O’Reilly MP, Faden AI, PekarJJ (2000) Diffusion and high resolution MRI of traumatic braininjury in rats: time course and correlation with histology. ExpNeurol 162:61–72
Benham CD, Davis JB, Randall AD (2002) Vanilloid and TRP chan-nels: a family of lipid-gated cation channels. Neuropharmacology2002;42:873–888
Bittigau P, Sifringer M, Pohl D, Stadthaus D, Ishimaru M, Shimizu H,Ikeda M, Lang D, Speer A, Olney JW, Ikonomidou C (1999)Apoptotic neurodegeneration following trauma is markedly en-hanced in the immature brain. Ann Neurol 45:724–735
Bock JL, Wenz B, Gupta RK (1987) Studies on the mechanism ofdecreased NMR-measured free magnesium in stored erythrocytes.Biochim Biophys Acta 928:8–12
Boisvert DP, Handa Y, Allen PS (1990) Proton relaxation in acute andsubacute ischemic brain edema. Adv Neurol 52:407–413
Bradford MM (1976) A rapid and sensitive method for the quantitationof microgram quantities of protein utilizing the principle of pro-tein-dye binding. Anal Biochem 72:248–254
Bramlett HM, Dietrich WD, Green EJ, Busto R (1997) Chronic histo-pathological consequences of fluid-percussion brain injury in rats:effects of post-traumatic hypothermia. Acta Neuropathol (Berl)93:190–199
Buki A, Siman R, Trojanowski JQ, Povlishock JT (1999) The role ofcalpain-mediated spectrin proteolysis in traumatically inducedaxonal injury. J Neuropathol Exp Neurol 58:365–375
Campbell VA (2001) Tetrahydrocannabinol-induced apoptosis of cul-tured cortical neurones is associated with cytochrome c release andcaspase-3 activation. Neuropharmacology 40:702–709
Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD,Julius D (1997) The capsaicin receptor: A heat-activated ion chan-nel in the pain pathway. Nature 389:816–824
Chabas D, Baranzini SE, Mitchell D, Bernard CC, Rittling SR, Den-hardt DT, Sobel RA, Lock C, Karpuj M, Pedotti R, Heller R,Oksenberg JR, Steinman L (2001) The influence of the proinflam-matory cytokine, osteopontin, on autoimmune demyelinating dis-ease. Science 294:1731–1735
Chan GC, Hinds TR, Impey S, Storm DR (1998) Hippocampal neuro-toxicity of Delta9-tetrahydrocannabinol. J Neurosci 18:5322–5332
Chan PH, Longar S, Chen S, Yu AC, Hillered L, Chu L, Imaizumi S,Pereira B, Moore K, Woolworth V, et al. (1989) The role of ara-chidonic acid and oxygen radical metabolites in the pathogenesisof vasogenic brain edema and astrocytic swelling. Ann N Y AcadSci 559:237–247
Chapman GA, Moores K, Harrison D, Campbell CA, Stewart BR,Strijbos PJ (2000) Fractalkine cleavage from neuronal membranesrepresents an acute event in the inflammatory response to excito-toxic brain damage. J Neurosci 20:RC87
Csuka E, Hans VH, Ammann E, Trentz O, Kossmann T, Morganti-Kossmann MC (2000) Cell activation and inflammatory responsefollowing traumatic axonal injury in the rat. Neuroreport 11:2587–2590
Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, GriffinG, Gibson D, Mandelbaum A, Etinger A, Mechoulam R (1992)Isolation and structure of a brain constituent that binds to thecannabinoid receptor. Science 1992;258:1946–1949
Di Giovanni S, Knoblach SM, Brandoli C, Aden SA, Hoffman EP,Faden AI (2003) Gene profiling in spinal cord injury shows role ofcell cycle in neuronal death. Ann Neurol 53:454–468
Di Marzo V, Breivogel CS, Tao Q, Bridgen DT, Razdan RK, ZimmerAM, Zimmer, A, Martin BR (2000) Levels, metabolism and phar-macological activity of anandamide in CB(1) cannabinoid receptorknockout mice: evidence for non-CB(1), non-CB(2) receptor-mediated actions of anandamide in mouse brain. J Neurochem75:2434–2444
I. CERNAK ET AL.576
J Cereb Blood Flow Metab, Vol. 24, No. 5, 2004

Di Marzo V, Blumberg PM, Szallasi A (2002) Endovanilloid signalingin pain. Curr Opin Neurobiol 12:372–379
Downer E, Boland B, Fogarty M, Campbell V (2001) Delta 9-tetrahy-drocannabinol induces the apoptotic pathway in cultured corticalneurones via activation of the CB1 receptor. Neuroreport 12:3973–3978
Downer EJ, Fogarty MP, Campbell VA (2003) Tetrahydrocannabinol-induced neurotoxicity depends on CB1 receptor-mediated c-JunN-terminal kinase activation in cultured cortical neurons. Br JPharmacol 140:547–557
Eldadah BA, Yakovlev AG, Faden AI (1997) The role of CED-3-related cysteine proteases in apoptosis of cerebellar granule cells.J Neurosci 17:6105–6113
Ellis EF, McKinney JS, Willoughby KA, Liang S, Povlishock JT(1995) A new model for rapid stretch-induced injury of cells inculture: characterization of the model using astrocytes. J Neuro-trauma 12:325–339
Endres M, Namura S, Shimizu-Sasamata M, Waeber C, Zhang L,Gomez-Isla T, Hyman BT, Moskowitz MA (1998) Attenuation ofdelayed neuronal death after mild focal ischemia in mice by inhi-bition of the caspase family. J Cereb Blood Flow Metab 18:238–247
Epps DE, Natarajan V, Schmid PC, Schmid HHO (1980) Accumulationof N-acylethanolamine glycerophospholipids in infarcted myocar-dium. Biochim Biophys Acta 618:420–430
Faden AI (2001) Neuroprotection and traumatic brain injury: the searchcontinues. Arch Neurol 58:1553–1555
Faden AI, Movsesyan V, Stoica BA, Knoblach SM, Lea PM, Ahmed F,Natale J-AE, Vink R, Cernak I (2003) The “dark side” of endo-cannabinoids: a neurotoxic role for anandamide. Ann Neurol54:S77
Galve-Roperh I, Rueda D, Gomez del Pulgar T, Velasco G, Guzman M(2002) Mechanism of extracellular signal-regulated kinase activa-tion by the CB(1) cannabinoid receptor. Mol Pharmacol 62:1385–1392
Gomez Del Pulgar T, De Ceballos ML, Guzman M, Velasco G (2002)Cannabinoids protect astrocytes from ceramide-induced apoptosisthrough the phosphatidylinositol 3-kinase/protein kinase B path-way. J Biol Chem 277:36527–36533
Gray GM (1976) Phosphatidyl-(N-acyl)-ethanolamine (1976) A lipidcomponent of mammalian epidermis. Biochim Biophys Acta431:1–8
Guzman M (2003) Neurons on cannabinoids: dead or alive? Br J Phar-macol 140:439–440
Hail N Jr, Lotan R (2002) Examining the role of mitochondrial respi-ration in vanilloid-induced apoptosis. J Natl Cancer Inst 94:1281–1292
Hail N Jr (2003) Mechanisms of vanilloid-induced apoptosis. Apoptosis8:251–262
Hamm RJ, Temple MD, Pike BR, O’Dell DM, Buck DL, Lyeth BG(1996) Working memory deficits following traumatic brain injuryin the rat. J Neurotrauma 13:317–323
Hampson AJ, Bornheim LM, Scanziani M, Yost CS, Gray AT, HansenBM, Leonoudakis DJ, Bickler PE (1998) Dual effects of anan-damide on NMDA receptor-mediated responses and neurotrans-mission. J Neurochem 70:671–676
Hansen HH, Ikonomidou C, Bittigau P, Hansen SH, Hansen HS (2001)Accumulation of the anandamide precursor and other N-acylethanolamine phospholipids in infant rat models of in vivonecrotic and apoptotic neuronal death. J Neurochem 76:39–46
Hansen HS, Lauritzen L, Strand AM, Vinggaard AM, Frandsen A,Schousboe A (1997) Characterization of glutamate-induced forma-tion of N-acylphosphatidylethanolamine and N-acylethanolaminein cultured neocortical neurons. J Neurochem 69:753–761
Hansen HS, Moesgaard B, Hansen HH, Petersen G (2000) N-Acylethanolamines and precursor phospholipids—relation to cellinjury. Chem Phys Lipids 108:135–150
Hayes RL, Yang K, Raghupathi R, McIntosh TK (1995) Changes ingene expression following traumatic brain injury in the rat. J Neu-rotrauma 12:779–790
Hermann DM, Kilic E, Hata R, Hossmann KA, Mies G (2001) Rela-tionship between metabolic dysfunctions, gene responses and de-
layed cell death after mild focal cerebral ischemia in mice. Neu-roscience 104:947–955
Hicks RR, Martin VB, Zhang L, Seroogy KB (1999) Mild experimentalbrain injury differentially alters the expression of neurotrophin andneurotrophin receptor mRNAs in the hippocampus. Exp Neurol160:469–478
Homayoun P, Parkins NE, Soblosky J, Carey ME, Rodriguez de TurcoEB, Bazan NG (2000) Cortical impact injury in rats promotes arapid and sustained increase in polyunsaturated free fatty acids anddiacylglycerols. Neurochem Res 25:269–276
Jambrina E, Alonso R, Alcalde M, del Carmen Rodriguez M, SerranoA, Martinez AC, Garcia-Sancho J, Izquierdo M (2003) Calciuminflux through receptor-operated channel induces mitochondria-triggered paraptotic cell death. J Biol Chem 278:14134–14145
Jaworski DM (2000) Differential regulation of tissue inhibitor of me-talloproteinase mRNA expression in response to intracranial in-jury. Glia 30:199–208
Jiang Q, Zhang RL, Zhang ZG, Ewing JR, Divine GW, Chopp M(1998) Diffusion-, T2-, and perfusion-weighted nuclear magneticresonance imaging of middle cerebral artery embolic stroke andrecombinant tissue plasminogen activator intervention in the rat. JCereb Blood Flow Metab 18:758–767
Kim SY, Choi YS, Choi JS, Cha JH, Kim ON, Lee SB, Chung JW,Chun MH, Lee MY (2002) Osteopontin in kainic acid-inducedmicroglial reactions in the rat brain. Mol Cells 13:429–435
Kobori N, Clifton GL, Dash P (2002) Altered expression of novelgenes in the cerebral cortex following experimental brain injury.Brain Res Mol Brain Res 104:148–158
Kupina NC, Nath R, Bernath EE, Inoue J, Mitsuyoshi A, Yuen PW,Wang KK, Hall ED (2001) The novel calpain inhibitor SJA6017improves functional outcome after delayed administration in amouse model of diffuse brain injury. J Neurotrauma 18:1229–1240
La Fleur M, Underwood JL, Rappolee DA, Werb Z (1996) Basementmembrane and repair of injury to peripheral nerve: defining apotential role for macrophages, matrix metalloproteinases, and tis-sue inhibitor of metalloproteinases-1. J Exp Med 184:2311–2326
Lea PM, Custer SJ, Vicini S, Faden AI (2002) Neuronal and glialmGluR5 modulation prevents stretch-induced enhancement ofNMDA receptor current. Pharmacol Biochem Behav 73:287–298
Li FH, Fisher M (1996) Diffusion-weighted and perfusion magneticresonance imaging and ischemic stroke. Drugs of Today 32:615–627
Long Y, Zou L, Liu H, Lu H, Yuan X, Robertson CS, Yang K (2003)Altered expression of randomly selected genes in mouse hippo-campus after traumatic brain injury. J Neurosci Res 71:710–720
Maccarrone M, Lorenzon T, Bari M, Melino G, Finazzi-Agro A (2000)Anandamide induces apoptosis in human cells via vanilloid recep-tors. Evidence for a protective role of cannabinoid receptors. J BiolChem 275:31938–31945
Maccarrone M, Attina M, Cartoni A, Bari M, Finazzi-Agro A (2001)Gas chromatography-mass spectrometry analysis of endogenouscannabinoids in healthy and tumoral human brain and human cellsin culture. J Neurochem 76:594–601
Maingret F, Patel AJ, Lazdunski M, Honore E (2001) The endocan-nabinoid anandamide is a direct and selective blocker of the back-ground K(+) channel TASK-1. EMBO J 20:47–54
Marsicano G, Goodenough S, Monory K, Hermann H, Eder M, Can-nich A, Azad SC, Cascio MG, Gutierrez SO, Van Der Stelt M,Lopez-Rodriguez ML, Casanova E, Schutz G, Zieglgansberger W,Di Marzo V, Behl C, Lutz B (2003) CB1 cannabinoid receptorsand on-demand defense against excitotoxicity. Science 302:84–88
Matzilevich DA, Rall JM, Moore AN, Grill RJ, Dash PK (2002) High-density microarray analysis of hippocampal gene expression fol-lowing experimental brain injury. J Neurosci Res 67:646–663
Mechoulam R (2002) Discovery of endocannabinoids and some ran-dom thoughts on their possible roles in neuroprotection and ag-gression. Prostaglandins Leukot Essent Fatty Acids 66:93–99
Mechoulam R, Panikashvili D, Shohami E (2002a) Cannabinoids andbrain injury: therapeutic implications. Trends Mol Med 8:58–61
Mechoulam R, Spatz M, Shohami E (2002b) Endocannabinoids andneuroprotection. SCI STKE 2002:RE5
ANANDAMIDE NEUROTOXICITY 577
J Cereb Blood Flow Metab, Vol. 24, No. 5, 2004

Mechoulam R, Lichtman AH (2003) Stout guards of the central ner-vous system. Science 302:65–67
Melendez A, Floto RA, Cameron AJ, Gilooly DJ, Harnett MM, AllenJM (1998) A molecular switch changes the signalling pathwayused by the Fc gamma RI antibody receptor to mobilize calcium.Curr Biol 8:210–221
Mombouli JV, Schaeffer G, Holzmann S, Kostner GM, Graier WF(1999) Anandamide-induced mobilization of cytosolic Ca2+ in en-dothelial cells. Br J Pharmacol 126:1593–1600
Mukhin AG, Ivanova SA, Knoblach SM, Faden AI (1997) New in vitromodel of traumatic neuronal injury: evaluation of secondary injuryand glutamate receptor-mediated neurotoxicity. J Neurotrauma14:651–663
Nagayama T, Sinor AD, Simon RP, Chen J, Graham SH, Jin K, Green-berg DA (1999) Cannabinoids and neuroprotection in global andfocal cerebral ischemia and in neuronal cultures. J Neurosci19:2987–2995
Natale JE, Ahmed F, Cernak I, Stoica B, Faden AI (2003) Gene ex-pression profile changes are commonly modulated across modelsand species after traumatic brain injury. J Neurotrauma 20:907–927
Natarajan V, Schmid PC, Schmid HHO (1986) N-acylethanolaminephospholipid metabolism in normal and ischemic rat brain. Bio-chim Biophys Acta 878:32–41
Olah Z, Szabo T, Karai L, Hough C, Fields RD, Caudle RM, BlumbergPM, Iadarola MJ (2001) Ligand-induced dynamic membranechanges and cell deletion conferred by vanilloid receptor 1. J BiolChem 276:11021–11030
Panikashvili D, Simeonidou C, Ben-Shabat S, Hanus L, Breuer A,Mechoulam R, Shohami E (2001) An endogenous cannabinoid(2-AG) is neuroprotective after brain injury. Nature 413:527–531
Pike BR, Zhao X, Newcomb JK, Glenn CC, Anderson DK, Hayes RL(2000) Stretch injury causes calpain and caspase-3 activation andnecrotic and apoptotic cell death in septo-hippocampal cell cul-tures. J Neurotrauma 17:283–298
Povlishock JT, Buki A, Koiziumi H, Stone J, Okonkwo DO (1999)Initiating mechanisms involved in the pathobiology of traumati-cally induced axonal injury and interventions targeted at bluntingtheir progression. Acta Neurochir Suppl (Wien) 73:15–20
Qiao M, Malisza KL, Del Bigio MR, Tuor UI (2001) Correlation ofcerebral hypoxic-ischemic T2 changes with tissue alterations inwater content and protein extravasation. Stroke 32:958–963
Raghupathi R, Graham DI, McIntosh TK (2000) Apoptosis after trau-matic brain injury. J Neurotrauma 17:927–938
Rall JM, Matzilevich DA, Dash PK (2003) Comparative analysis ofmRNA levels in the frontal cortex and the hippocampus in thebasal state and in response to experimental brain injury. Neuro-pathol Appl Neurobiol 29:118–131
Rosenberg GA, Estrada EY, Dencoff JE (1998) Matrix metalloprotein-ases and TIMPs are associated with the blood-brain barrier openingafter reperfusion in rat brain. Stroke 29:2189–2195
Ross RA (2003) Anandamide and vanilloid TRPV1 receptors. Br JPharmacol 140:790–801
Saatman KE, Bareyre FM, Grady MS, McIntosh TK (2001) Acutecytoskeletal alterations and cell death induced by experimentalbrain injury are attenuated by magnesium treatment and exacer-bated by magnesium deficiency. J Neuropathol Exp Neurol60:183–194
Sagan S, Venance L, Torrens Y, Cordier J, Glowinski J, Giaume C(1999) Anandamide and WIN 55212-2 inhibit cyclic AMP forma-tion through G-protein-coupled receptors distinct from CB1 can-nabinoid receptors in cultured astrocytes. Eur J Neurosci 11:691–699
Sanchez C, Galve-Roperh I, Canova C, Brachet P, Guzman M (1998)Delta9-tetrahydrocannabinol induces apoptosis in C6 glioma cells.FEBS Lett 436:6–10
Sarker KP, Obara S, Nakata M, Kitajima I, Maruyama I (2000) Anan-
damide induces apoptosis of PC-12 cells: involvement of super-oxide and caspase-3. FEBS Lett 472:39–44
Sarker KP, Maruyama I (2003) Anandamide induces cell death inde-pendently of cannabinoid receptors or vanilloid receptor 1: pos-sible involvement of lipid rafts. Cell Mol Life Sci 60:1200–1208
Schmid HHO (2000) Pathways and mechanisms of N-acylethanol-amine biosynthesis: can anandamide be generated selectively?Chem Phys Lipids 108:71–87
Siesjo BK (1981) Cell damage in the brain: a speculative synthesis. JCereb Blood Flow Metab 1:155–185
Sinor AD, Irvin SM, Greenberg DA (2000) Endocannabinoids protectcerebral cortical neurons from in vitro ischemia in rats. NeurosciLett 278:157–160
Szallasi A, Blumberg PM (1990) Resiniferatoxin and its analogs pro-vide novel insights into the pharmacology of the vanilloid (capsa-icin) receptor. Life Sci 47:1399–1408
Szallasi A, Di Marzo V (2000) New perspectives on enigmatic vanil-loid receptors. Trends Neurosci 23:491–497
Szallasi A (2002) Vanilloid (capsaicin) receptors in health and disease.Am J Clin Pathol 118:110–121
Szoke E, Balla Z, Csernoch L, Czeh G, Szolcsanyi J (2000) Interactingeffects of capsaicin and anandamide on intracellular calcium insensory neurones. Neuroreport 11:1949–1952
Tang Y, Lu A, Aronow BJ, Wagner KR, Sharp FR (2002) Genomicresponses of the brain to ischemic stroke, intracerebral hemor-rhage, kainate seizures, hypoglycemia, and hypoxia. Eur J Neuro-sci 15:1937–1952
Tarozzo G, Campanella M, Ghiani M, Bulfone A, Beltramo M (2002)Expression of fractalkine and its receptor, CX3CR1, in response toischemia-reperfusion brain injury in the rat. Eur J Neurosci15:1663–1668
Toman RE, Movsesyan V, Murthy SK, Milstien S, Spiegel S, Faden AI(2002) Ceramide-induced cell death in primary neuronal cultures:upregulation of ceramide levels during neuronal apoptosis. J Neu-rosci Res 68:323–330
van der Stelt M, Veldhuis WB, Maccarrone M, Bar PR, Nicolay K,Veldink GA, Di Marzo V, Vliegenthart JF (2002) Acute neuronalinjury, excitotoxicity, and the endocannabinoid system. Mol Neu-robiol 26:317–346
Veech RL, Lawson JW, Cornell NW, Krebs HA (1979) Cytosolicphosphorylation potential. J Biol Chem 254:6538–6547
Veldhuis WB, van der Stelt M, Wadman MW, van Zadelhoff G, Mac-carrone M, Fezza F, Veldink GA, Vliegenthart JF, Bar PR, NicolayK, Di Marzo V (2003) Neuroprotection by the endogenous canna-binoid anandamide and arvanil against in vivo excitotoxicity in therat: role of vanilloid receptors and lipoxygenases. J Neurosci23:4127–4133
Venance L, Piomelli D, Glowinski J, Giaume C (1995) Inhibition byanandamide of gap junctions and intercellular calcium signalling instriatal astrocytes. Nature 372:590–594
Vink R, McIntosh TK (1990) Pharmacological and physiological ef-fects of magnesium on experimental traumatic brain injury.Magnes Res 3:163–169
Vink R, Golding EM, Headrick JP (1994) Bioenergetic analysis ofoxidative metabolism following traumatic brain injury in rats. JNeurotrauma 11:265–274
Walter L, Franklin A, Witting A, Moller T, Stella N (2002) Astrocytesin culture produce anandamide and other acylethanolamides. J BiolChem 277:20869–20876
Yakovlev AG, Knoblach SM, Fan L, Fox GB, Goodnight R, Faden AI(1997) Activation of CPP32-like caspases contributes to neuronalapoptosis and neurological dysfunction after traumatic brain in-jury. J Neurosci 17:7415–7424
Zygmunt PM, Petersson J, Andersson DA, Chuang H, Sorgard M, DiMarzo V, Julius D, Hogestatt ED (1999) Vanilloid receptors onsensory nerves mediate the vasodilator action of anandamide. Na-ture 400:452–457
I. CERNAK ET AL.578
J Cereb Blood Flow Metab, Vol. 24, No. 5, 2004





























