Embed Size (px)
Citation preview

THE HORMONAL CONTROL OF NEUROPEPTIDE Y AND GONADOTROPIN-RELEASING HORMONE
HYPOTHALAMIC NEURONS
by
Sandeep S. Dhillon
A thesis submitted in conformity with the requirements for the degree of Doctor of Philosophy
Department of Physiology University of Toronto
© Copyright by Sandeep S. Dhillon 2010

ii
The Hormonal Control of Neuropeptide Y and Gonadotropin-
Releasing Hormone Hypothalamic Neurons
Sandeep S. Dhillon
Doctor of Philosophy
Department of Physiology University of Toronto
2010
Abstract
The physiological mechanisms that control energy homeostasis are reciprocally
linked to reproduction. However, the neuroendocrine circuitry that registers endocrine
cues to direct homeostatic responses in energy balance and reproduction remain unknown.
Neuropeptide Y (NPY) neurons have emerged as a key central target of estrogen and
leptin that are capable of modulating both reproduction and energy balance. The
hypothesis was generated that NPY neuronal subpopulations act as an integration centre to
regulate the effects of estrogen and leptin on these important physiological processes
through specific signaling pathways. Using hypothalamic cell lines that express the leptin
receptor (Ob-R), estrogen receptor (ER) and NPY, this hypothesis was tested in three
aims.
17β-estradiol (E2) was previously demonstrated to biphasically regulate NPY
mRNA in the mHypoE-38 neuronal cell line; where 24 h E2 exposure induced NPY gene
expression that our group proposed may be involved in the gonadotropin-releasing
hormone (GnRH) preovulatory surge. E2 also acts as an anorexigenic hormone through
unknown hypothalamic targets. E2 directly decreased NPY secretion in the mHypoE-42
and mHypoA-2/12 neuronal cell lines through ER-α. The anorexigenic action of E2 was

iii
mediated through the energy sensing 5’ AMP-activated protein kinase (AMPK) and the
phosphoinositide-3-kinase (PI3K) pathway. NPY secretion was also decreased by leptin in
mHypoA-59 and NPY-GFP cell models through AMPK- and PI3K-dependent
mechanisms. Prolonged exposure to leptin in NPY-GFP cell lines prevented AMPK
signaling and the leptin-mediated reduction in NPY secretion, indicating NPY neuronal
resistance with prolonged leptin exposure. Leptin also stimulated NPY secretion in
mHypoE-38 neurons, which was blocked by pharmacological inhibitors of the mitogen-
activated protein kinase (MAPK) and PI3K pathways. Importantly, conditioned medium
from the mHypoE-38 NPY neuronal cells induced GnRH transcripts in GT1-7 neurons,
which was inhibited by Y1-receptor antagonists. Pharmacological inhibitors of the MAPK
and PKA signal transduction pathways attenuated the NPY-mediated increase in GnRH
transcription.
Based upon these findings, I propose NPY neurons in the hypothalamus consist of
a heterogeneous population of neurons, and provide the first evidence of intrinsically
different responses to function as physiological integrators for two different systems: NPY
secretion can be suppressed to decrease food intake and induced to stimulate GnRH
neurons.

iv
Acknowledgments
I owe my deepest gratitude to Dr. Denise Belsham, who has been a significant
presence in my life. I am indebted to Dr. Belsham whose patience, kindness and academic
experience have allowed me to become the person I am today. Thank you Denise.
I would also like to thank my committee members, Dr. Isabella Canniggia and Dr.
Theodore Brown. I have benefited greatly from their guidance, mentorship and
encouragement, which have been instrumental to the completion of this degree.
I would like to thank the members of the Belsham lab for their valuable discussion,
encouragement and making the lab such an enjoyable experience. I also want to specially
thank Ginah Kim. You have become my best friend both inside and outside the laboratory.
Thank you for always being there.
Finally, I would like to thank my family for their constant support, patience and
love. I would not have made it here with out them. This accomplishment is as much yours
as it is mine.

v
Table of Contents
Acknowledgments ........................................................................................................................ iv
Table of Contents ...........................................................................................................................v
List of Tables ..................................................................................................................................x
List of Figures............................................................................................................................... xi
List of Abbreviations ................................................................................................................. xiv
1 Chapter 1 Relevant literature reviews ....................................................................................1
1.1 Introduction........................................................................................................................2
1.2 Reproductive function .......................................................................................................4
1.2.1 The hypothalamic-pituitary-gonadal axis ................................................................4
1.2.2 Gonadotropin-releasing hormone (GnRH) in the hypothalamus.............................6
1.2.3 Regulation of GnRH neurons...................................................................................7
1.3 Energy homeostasis and reproductive function ..............................................................9
1.3.1 Hypothalamic nuclei associated with regulation of food intake..............................9
1.3.2 Energy homeostasis and reproductive function .....................................................10
1.4 Neuropeptide Y ................................................................................................................13
1.4.1 Synthesis ................................................................................................................13
1.4.2 NPY receptors........................................................................................................14
1.4.3 Signaling pathways activated by NPY...................................................................14
1.4.4 NPY effects on energy homeostasis and reproduction ..........................................15
1.5 Estrogen ............................................................................................................................18
1.5.1 Synthesis and metabolism......................................................................................18
1.5.2 Estrogen receptors..................................................................................................19
1.5.3 Signaling pathways activated by estrogen .............................................................22
1.5.4 Effects of estrogen on reproduction and feeding behaviour ..................................27

vi
1.5.5 Estrogen-mediated regulation of NPY neurons .....................................................29
1.6 Leptin ................................................................................................................................30
1.6.1 Synthesis and metabolism......................................................................................30
1.6.2 Leptin receptors and signaling events....................................................................31
1.6.3 Effects of leptin on feeding behaviour and reproduction.......................................34
1.6.4 Leptin-mediated regulation of NPY neurons.........................................................35
1.6.5 Leptin Resistance ...................................................................................................37
1.7 Cell models........................................................................................................................38
1.7.1 GnRH-expressing GT1-7 neurons .........................................................................39
1.7.2 Embryonic hypothalamic cell lines – mHypoE-xx................................................41
1.7.3 Adult hypothalamic cell lines – mHypoA-xx ........................................................42
1.7.4 NPY-GFP cell line .................................................................................................43
1.8 Hypothesis and aims ........................................................................................................44
2 Chapter 2 .................................................................................................................................47
Materials and methods ................................................................................................................47
2.1 Cell culture and reagents.................................................................................................48
2.2 Semi-quantitative RT-PCR .............................................................................................49
2.2.1 One step RT-PCR ..................................................................................................49
2.2.2 Two step RT-PCR..................................................................................................49
2.3 Real-Time RT-PCR .........................................................................................................51
2.4 Enzyme Immunoassay .....................................................................................................51
2.5 Fluorescence-activated cell sorting (FACS)...................................................................52
2.6 Radioactive Immunoassay ..............................................................................................53
2.7 Western Blot Analysis .....................................................................................................53
2.8 Immunocytochemistry .....................................................................................................55
2.9 Co-culture .........................................................................................................................56

vii
2.10 Statistics ...........................................................................................................................56
Chapter 3 ......................................................................................................................................57
3 17β-estradiol inhibits NPY secretion through membrane-associated estrogen receptor (ER)-α in clonal, immortalized hypothalamic neurons .......................................57
3.1 Abstract.............................................................................................................................58
3.2 Introduction......................................................................................................................59
3.3 Results ...............................................................................................................................61
3.3.1 Expression of ER-α in FAC-sorted NPY-GFP neurons ........................................61
3.3.2 Expression of the ER subtypes and other hypothalamic markers in mHypoE-42 and mHypoA-2/12 neurons ...............................................................61
3.3.3 Regulation of NPY secretion by E2 in mHypoE-42 and mHypoA-2/12 neurons...................................................................................................................63
3.3.4 E2-mediated regulation of NPY secretion is dependent on ER-α in the mHypoE-42 and mHypoA-2/12 neurons ...............................................................63
3.3.5 E2 decreases NPY secretion via membrane-bound ER-α ......................................65
3.3.6 Inhibition of the PI3K and AMPK pathways affect the E2-mediated regulation of NPY secretion...................................................................................67
3.4 Discussion .........................................................................................................................71
4 Chapter 4 .................................................................................................................................79
Leptin differentially regulates NPY secretion in NPY-expressing hypothalamic cell lines through distinct intracellular signal transduction pathways ..............................79
4.1 Abstract.............................................................................................................................80
4.2 Introduction......................................................................................................................81
4.3 Results ...............................................................................................................................83
4.3.1 Expression of the Ob-R and other markers in mHypoE-38, mHypoE-42, mHypoA-59 and NPY-GFP neurons ...............................................................83
4.3.2 Regulation of NPY secretion by leptin in mHypoE-38, mHypoE-42, mHypoA-59 and NPY-GFP neurons .....................................................................85
4.3.3 Leptin increases NPY secretion in the mHypoE-38 neurons via PI3K and MAPK pathways .............................................................................................85

viii
4.3.4 Leptin decreases NPY secretion in the mHypoA-59 and NPY-GFP neurons via AMPK and PI3K pathways ................................................................87
4.3.5 AICAR directly stimulates NPY secretion in mHypoA-59 and NPY-GFP neurons ..........................................................................................................89
4.3.6 Leptin pre-treatment attenuates leptin-mediated phosphorylation of AMPK in NPY-GFP neurons ................................................................................89
4.3.7 Leptin pre-treatment attenuates the leptin-mediated decrease in NPY secretion in NPY-GFP neurons..............................................................................93
4.4 Discussion .........................................................................................................................93
5 Chapter 5 ...............................................................................................................................103
Neuropeptide Y induces gonadotropin-releasing hormone gene expression directly and through conditioned medium from mHypoE-38 NPY neurons ..................103
5.1 Abstract...........................................................................................................................104
5.2 Introduction....................................................................................................................104
5.3 Results .............................................................................................................................106
5.3.1 Expression of NPY receptor subtypes in GT1-7 neurons and hypothalamic markers in mHypoE-38 neurons ...................................................106
5.3.2 Regulation of GnRH mRNA expression by NPY in GT1-7 neurons ..................107
5.3.3 Effect of NPY receptor agonists on cAMP activity.............................................107
5.3.4 NPY rapidly phosphorylates PKA, ATF-1 and CREB in GT1-7 neurons ..........110
5.3.5 Inhibition of MAPK and PKA-C signaling pathways affects NPY-mediated regulation of GnRH mRNA expression in GT1-7 neurons..................113
5.3.6 Regulation of GnRH transcription by conditioned media from NPY-secreting mHypoE-38 neurons is mediated through the NPY Y1 receptor subtype ...................................................................................................113
5.4 Discussion .......................................................................................................................117
6 Chapter 6 ...............................................................................................................................125
Overall Discussion and Future Directions ...............................................................................125
6.1 Overall Discussion..........................................................................................................126
6.2 Limitations......................................................................................................................134

ix
6.3 Future directions of study .............................................................................................136
7 Chapter 7 – References .........................................................................................................140

x
List of Tables
Table 1.1 Peptides implicated in regulation of food intake ................................................11
Table 1.2 NPY receptors characteristics.............................................................................15
Table 1.3 Co-activators in estrogen receptor physiology ...................................................24
Table 2.1 Primer sequences ................................................................................................49
Table 3.1 Expression of ER, NPY, Ob-R and AgRP in hypothalamic nuclei ....................77

xi
List of Figures
Figure 1.1 Schematic illustration of the hypothalamic-pituitary-gonadal axis.....................5
Figure 1.2 Schematic illustration of the signal transduction mechanisms activated by NPY
.............................................................................................................................................16
Figure 1.3 Schematic illustration of estrogen signaling mechanisms.................................22
Figure 1.4 Schematic illustration of leptin receptor signaling............................................32
Figure 1.5 Immortalization of hypothalamic cell models...................................................39
Figure 1.6 Schematic illustration of the objectives of the current thesis ............................45
Figure 3.1 Expression of ER-α mRNA in NPY-GFP neurons using RT-PCR ..................61
Figure 3.2 Expression of ERs and other hypothalamic markers in mHypoE-42 and
mHypoA-2/12 neurons........................................................................................................62
Figure 3.3 Estrogen directly decreases NPY secretion in the mHypoE-42 and mHypoA-
2/12 neurons........................................................................................................................64
Figure 3.4 Estrogen attenuates NPY secretion via ER-α in mHypoE-42 and mHypoA-2/12
neurons................................................................................................................................66
Figure 3.5 ER-α localized at the cell membrane with caveolin-1 protein ..........................68
Figure 3.6 ER-α localized at the cell membrane is required for the estrogen-mediated
decrease in the mHypoE-42 and mHypoA-2/12 neurons ...................................................69
Figure 3.7 PI3K inhibitor LY294002 and AMPK inhibitor Compound C attenuates NPY-
mediated regulation of NPY secretion in mHypoE-42 neurons .........................................70
Figure 3.8 Model of the potential cellular signaling pathways involved in estrogen
regulation of NPY secretion………………………………………………………………78
Figure 4.1 Characterization of NPY-expressing hypothalamic cell models.......................84

xii
Figure 4.2 Leptin directly regulates NPY secretion in NPY-expressing hypothalamic cell
lines .....................................................................................................................................86
Figure 4.3 Leptin increases NPY secretion in the mHypoE-38 cell lines via PI3K and
MAPK pathways.................................................................................................................88
Figure 4.4 Leptin decreases NPY secretion in the NPY-GFP cell line via AMPK and PI3K
pathways. ............................................................................................................................90
Figure 4.5 Leptin decreases NPY secretion in the mHypoA-59 cell line via AMPK and
PI3K pathways ....................................................................................................................91
Figure 4.6 AICAR increases NPY secretion in the NPY-GFP and mHypoA-59 cell lines
.............................................................................................................................................92
Figure 4.7 Prolonged leptin exposure prevents the leptin-mediated decrease in phospho-
AMPK in the NPY-GFP cell line........................................................................................94
Figure 4.8 Prolonged leptin exposure prevents the leptin-mediated decrease in NPY
secretion in the NPY-GFP cell line.....................................................................................95
Figure 4.9 Model of the potential cellular signaling pathways involved in leptin regulation
of NPY secretion...............................................................................................................102
Figure 5.1 Expression of NPY Y1, Y2 and Y4 receptor mRNA transcripts in GT1-7
neurons..............................................................................................................................108
Figure 5.2 NPY-mediated regulation of GnRH gene expression in GT1-7 neurons ........109
Figure 5.3 NPY Y1 or Y4 receptor-mediated cAMP activity in the GT1-7 neurons .......111
Figure 5.4 NPY activates signal transduction second messengers in GT1-7 neurons......112
Figure 5.5 NPY Y1 antagonist BIBP-3226, MEK and PKA inhibitors attenuate NPY-
mediated regulation of GnRH mRNA levels in GT1-7 neurons.......................................114

xiii
Figure 5.6 Conditioned media from NPY-expressing mHypoE-38 neurons increases
GnRH mRNA expression in GT1-7 cells and can be blocked by the NPY Y1 antagonist
BIBP-3226 ........................................................................................................................116
Figure 5.7 Neuron-specific gene promoter for GnRH…………………………………..122
Figure 5.8 Model of the potential cellular signaling pathways involved in NPY regulation
of GnRH mRNA expression .............................................................................................124
Figure 6.1 Overall findings and future directions.............................................................133

xiv
List of Abbreviations
AC adenylyl cyclase
AF-1 transactivating function-1
AF-2 transactivating function-2
AgRP agouti-related peptide
AICAR 5-aminoimidazole-4-carboxamide ribonucleotide
AMP adenosine monophosphate
AMPK 5' AMP-activated protein kinase
AP-1 activator protein-1
ARC arcuate nucleus
ATP adenosine-5’-triphosphate
AVPV anteroventricular paraventricular nucleus
Bp base pairs
BSA bovine serum albumin
cAMP cyclic adenosine monophosphate
cDNA complementary deoxyribonucleic acid
CNS central nervous system
CPON C-terminal of neuropeptide Y
CRE cAMP response element
CREB cAMP response element binding protein
CRH corticotropin-releasing hormone
CT cycle threshold
DBD DNA-binding domain
DMEM Dulbecco’s modified eagle medium

xv
DMN dorsomedial nucleus
DMSO dimethyl sulfoxide
DNA deoxyribonucleic acid
DPN 2,3-bis(4-Hydroxyphenyl)-propionitrile
E1 estrone
E2 17β-estradiol
E3 estriol
ER estrogen receptor
ERE estrogen response element
ERK extracellular-related kinase
FBS fetal bovine serum
FITC fluorescein isothiocyanate
GABA γ-aminobutyric acid
GnRH gonadotropin-releasing hormone
GPCR G-protein couple receptor
h hours
HEK human embryonic kidney
HFD high fat diet
HPA hypothalamic-pituitary adrenal axis
HPG hypothalamic-pituitary gonadal axis
ICC immunocytochemistry
ICV intracerebroventricular
JNK c-Jun NH2 terminal kinase
kb kilobases

xvi
kDa kilodaltons
LH luteinizing hormone
LHa lateral hypothalamus
LBD ligand binding domain
MAPK mitogen activated protein kinase
ME median eminence
MCH melanin-concentrating hormone receptor 1
MNAR modulator of non-genomic activity of estrogen receptor
mRNA messenger ribonucleic acid
α-MSH melanocyte-stimulating hormone
MPP methyl-piperidinopyrazole
NE norepinephrine
NPY neuropeptide Y
NSE neuron specific enolase
NT neurotensin
NTC no template control
ODN oligonucleotide
OVX ovariectomized
PBS phosphate buffered saline
PCR polymerase chain reaction
PI3K phosphatidylinositol 3-kinase
PKC protein kinase C
POA preoptic area
PP pancreatic peptide

xvii
PPT 4,4’,4’’-(4-Propyl-[1H]-pyrazole-1,3,5-triyl)trisphenol
PVN paraventricular nucleus
PYY peptide YY
RNA ribonucleic acid
(R,R)-THC (R,R)-5,11-Diethyl-5,6,11,12-tetrahydro-2,8-chrysenediol
RT-PCR reverse transcriptase polymerase chain reaction
siRNA small interfering ribonucleic acid
STAT signal transducer activator of transcription
SV40 simian virus 40
T-Ag T-antigen
VMN ventromedial nucleus

1
1
1 Chapter 1
Introduction

2
2
1.1 Introduction
Appetite is regulated by an interplay of neuropeptides, neurotransmitters and
hormones produced from both central and peripheral sites. Centrally, appetite and energy
homeostasis is chiefly regulated in the hypothalamus by a complex neural circuitry
comprised of over 100 putative orexigenic (appetite stimulating) and anorexigenic
(appetite inhibiting) neuropeptides – including neuropeptide Y (NPY), melanin-
concentrating hormone (MCH), galanin, orexin, α-melanocyte-stimulating hormone (α-
MSH), neurotensin (NT) and corticotropin-releasing hormone (CRH) (1-7). These
appetite-stimulatory and appetite-inhibitory circuits of the hypothalamus are, in turn,
controlled by peripheral endocrine signals. Two key hormones, estrogens and leptin, are
thought to be involved in regulating these peptidergic feeding circuits by altering
secretion and gene expression of the feeding-related neuropeptides (8, 9). Additionally,
reduced nutritional status or body mass results in infertility and delayed reproductive
maturation while disturbing the course of the ovarian cycle (10). Perturbed gonadotropin-
releasing hormone (GnRH) secretion, the central regulator of the hypothalamic-pituitary-
gonadal (HPG) axis, is postulated as the most important etiological factor for
nutritionally-induced reproductive disorders (9). Although the relationship between
nutrition and reproductive success has been extensively studied, the exact mechanism
linking the two physiological processes is yet to be determined. NPY neuromodulators of
the hypothalamus have emerged as key factors involved in regulating both feeding
behaviour and reproductive homeostasis (11-13). Although several research groups have
documented the importance of NPY in the regulation of the reproductive axis and energy
homeostasis, the peripheral hormonal regulators of NPY neurons have not been fully

3
3
investigated. Recently, estrogen and leptin have emerged as key modulators of the NPY
neuron.
Estrogen and leptin receptors are expressed in overlapping NPY neuronal
populations in the arcuate nucleus (ARC) and ventromedial nucleus (VMN) (14-18).
Previous studies have demonstrated deficiencies in either leptin or estrogen levels can
result in an upregulation of hypothalamic NPY mRNA in the ARC (19-21). The
hypothesis that 17β-estradiol (E2) and leptin can positively or negatively regulate specific
NPY subpopulations to control feeding and reproductive physiology is yet to be verified.
The use of hypothalamic cell models provides a novel tool to study the direct regulation
of these neuropeptides by E2 and leptin. In this thesis, the first and second studies
characterize the E2- and leptin-mediated regulation of NPY secretion in embryonic- and
adult-derived mouse hypothalamic cell lines. The molecular mechanisms involved in the
differential regulation of NPY secretion and receptors responsible were investigated. In
addition, as GnRH is one of the most important peptides required for normal reproductive
function, the third study in this thesis characterizes the direct effect of NPY on GnRH
gene expression. GnRH-synthesizing GT1-7 neurons were used to study the receptors,
signaling pathways and transcriptional events of NPY and conditioned medium
treatments from NPY-synthesizing cell lines in a GnRH neuronal cell model.
The purpose of this thesis was to evaluate the ability of NPY to regulate GnRH
neuronal populations and describe the regulation of NPY-expressing neuronal cell lines
by peripheral endocrine hormones - E2 and leptin - that are required to maintain normal
feeding and reproductive homeostasis. Using clonal immortalized hypothalamic neuronal
cell lines, I provide detailed mechanics of this circuit and describe the differential
regulation of the NPY neuron. In addition, these studies provide further and more

4
4
comprehensive evidence of the significance of NPY in the regulation of the GnRH
neuron and overall reproductive function. These studies contribute to our understanding
of both leptin and estrogen physiology, and provide evidence that NPY neurons are
heterogeneous in nature with intrinsically different responses to these hormones.
1.2 Reproductive function
1.2.1 The hypothalamic-pituitary-gonadal axis
The key to species survival depends on the ability to reproduce. In mammals,
reproduction is maintained by a delicate feedback loop involving the hypothalamus,
pituitary gland and gonads, which form the HPG axis (Figure 1.1) (22, 23). At the
pinnacle of the HPG axis are GnRH neurons. GnRH is secreted into the median eminence
(ME) of the hypothalamus in pulses, and through the hypophyseal portal vasculature,
signals the anterior pituitary to secrete gonadotropin hormones, luteinizing hormone (LH)
and follicle stimulating hormone (FSH) (24). These hormones are released into the
systemic blood system and act at the gonads to regulate steroid hormone production and
gametogenesis. Specifically, FSH allows for the maturation of ovarian follicles in
females and spermatogenesis in males, and LH induces sex steroid hormone synthesis
and release, gametogenesis and regulates ovulation (25). The sex steroids produced by
the gonads include estrogens, progestins and androgens, which are produced in sexually
dimorphic quantities (26). Gonadal steroids indirectly control their own secretion through
positive and negative feedback loops, which maintain hormonal levels within a narrow
range and enable normal reproductive function (27, 28). In particular, gonadal steroids
modulate both the hypothalamus and pituitary to control the release of GnRH and
gonadotropin hormones. The regulation of normal secretory mechanisms, transcription

5
5
Hypothalamus:
Pituitary:
Gonads:
GnRH
Gonadotrope
LH, FSH
Estrogen
GnRH
The HPG
AxisAfferent neuronal
input
Figure 1.1. Schematic illustration of the hypothalamic-pituitary-gonadal axis.Adapted from Kim 2010.
The hypothalamus produces gonadotropin-releasing hormone (GnRH), which is released in a pulsa-tile fashion to control the synthesis and secretion of pituitary hormones, luteinizing hormone (LH) and follicle stimulating hormone (FSH). LH and FSH in turn are secreted into systemic circulation to act on the ovaries and testes to regulate steroidogenesis. The gonadal steroids estrogen and testos-terone are then secreted into the systemic circulation to regulate production and secretion of GnRH through positive and negative feedback loops, which occurs both directly at the GnRH neuron and indirectly through afferent neuronal circuitry upstream of GnRH neurons.

6
6
levels, receptor activation and cellular signaling cascades in the HPG axis are essential to
maintain normal reproductive function.
1.2.2 Gonadotropin-releasing hormone (GnRH) in the hypothalamus
There are two GnRH genes in mammals. GnRH-1 (GnRH) was originally
discovered by McCann and colleagues in the 1960’s, when extracts from rat
hypothalamic cultures were able to stimulate the release of LH (24). Additional studies
identified the active factor in the hypothalamic extracts to be a decapeptide that was
capable of stimulating both LH and FSH release from the anterior pituitary (29). This
decapeptide, now known as GnRH, is highly conserved in mammals (30). The gene
encoding GnRH spans 4.5 kb of DNA on chromosome 8 in humans and consists of three
introns and 4 exons encoding a 92 amino acid propeptide (31, 32). Post-translational
processing of the 92 amino acid propeptide is completed by endopeptidase,
carboxypeptidase E and peptidyl-glycine α-amidating monooxygenase to generate a 23
amino acid signal peptide, a 59 amino acid peptide product called GnRH-associated
peptide and the decapeptide GnRH (33-37). A second form of GnRH, termed chicken
GnRH-2, is expressed in mammals and found in the brainstem and medial hypothalamus
(38). This highly conserved peptide is present on chromosome 20 in humans, but its
function appears to be silenced in mice, humans, cattle and rat (39, 40). As a result
reproductive cycles studied by endocrinologists have focused almost exclusively on
GnRH.
In the rodent, GnRH is synthesized by a small population of GnRH-expressing
neurons (400-1000) mainly localized in the anterior hypothalamus, specifically in the
medial preoptic nucleus (POA) (36). GnRH neurons originate from the olfactory placode

7
7
during fetal life and migrate rostrally along the cribiform plate, through the nasal septum
towards the anterior hypothalamus in the developing brain (30). GnRH is secreted in a
pulsatile manner into the hypophyseal portal vasculature to reach the anterior pituitary
gonadotrophs where it stimulates the secretion of LH and FSH (41).
1.2.3 Regulation of GnRH neurons
GnRH neurons are under the control of a number of regulatory neuromodulators
including γ-aminobutyric acid (GABA) (42), kisspeptins (43), NPY (44, 45), neurotensin
(NT) (46), dopamine (47), norepinephrine (NE) (48), nitric oxide (49), activin (50),
histamine (51), androgen (52), estrogen (53) and melatonin (54), among many others
(55). Some of the latest and likely most relevant regulators of the GnRH neuron are
discussed in greater detail below.
GABA-expressing neurons have been shown to be a prominent inhibitory
regulator of GnRH neuronal function (56). GnRH neurons express the GABA receptor
and are directly innervated by GABA-expressing neurons (57). Both the down-regulation
of GABA neurotransmitter levels in pre-synaptic terminals innervating GnRH cell bodies
and decreases in GABA receptor expression in GnRH-expressing neurons are thought to
be key events in enabling the pre-ovulatory surge (58). Together, GABA-expressing
neurons are thought to have a prominent role in regulating GnRH neuronal function and
the reproductive axis.
Kisspeptin-expressing neurons of the hypothalamus represent another set of
neurons that are critical to reproductive function (59). Kisspeptin is the natural ligand of
the previously orphan receptor, GPR54 (60, 61). In 2003, two groups independently
identified an absence of puberty onset and hypogonadotropic hypogonadism in patients
with a loss of function mutation in the GPR54 gene (62, 63). Additional studies

8
8
demonstrated that kisspeptin is a key regulator of the GnRH neuron and thereby the HPG
axis (59, 61, 64). Kisspeptin has been demonstrated to directly depolarize and increase
the firing rate of GnRH neurons, which have been shown to express GPR54 (43).
Furthermore, Kisspeptin treatment results in an increase in GnRH release in a number of
species (59, 60, 65, 66). Although the cellular mechanisms of kisspeptin hormonal
regulation are incomplete, kisspeptin has emerged as an important regulator of
reproductive function.
NT-expressing neurons can also stimulate GnRH mRNA and release in vivo (46).
NT neurons from the anteroventral periventricular nucleus (AVPV) innervate and act on
GnRH neurons in a similar manner to NPY (67). NT has also been shown to regulate the
amplitude of GnRH secretion, which is seen in rats where centrally infused NT resulted
in increased LH release (46, 68). The precise role of NT-expressing neurons in the
regulation of the HPG axis remains to be determined; although it is likely that NT
neurons play a synergistic role to other neuronal populations such as kisspeptin, GABA
and NPY to regulate GnRH neuronal function.
NPY has been acknowledged for many years as a major afferent regulator of
reproductive function (69). NPY neurons in the ARC project to GnRH cell bodies in the
POA and to GnRH pre-synaptic terminals in the ME (44, 70). Additionally,
immunohistochemical evidence indicates co-localization of NPY receptors in GnRH
neurons (71). This neuroanatomical evidence for connections between NPY and GnRH
neurons establishes a possible mechanism in which NPY can directly influence the
reproductive axis. Several studies, both in vivo and in vitro, have shown NPY stimulates
GnRH secretion (45, 72, 73). In ewes, NPY infusion into the third ventricle substantially
increases GnRH secretion in the ME (74). However, depending upon the steroidal

9
9
environment and species, NPY can also down-regulate the reproductive axis through
GnRH (75). NPY injections into the third ventricle in ovariectomized rats led to a
reduction in plasma LH (76). Chronic NPY administration inhibited gonadotropin
secretion in intact female rats (76). In ovariectomized rabbits, NPY perfusion
significantly decreased mean levels of GnRH, while the same NPY perfusion stimulated
mean levels of GnRH in intact rabbits (77, 78). Overall, the role of NPY on the
reproductive axis has produced conflicting evidence, with both stimulatory and inhibitory
effects published. Although a number of studies have investigated the role of NPY on
GnRH secretion, the effect of NPY on GnRH neurons at the transcriptional level has not
been elucidated.
GnRH neurons act as the final channel for a number of neuroendocrine signals
that regulate reproductive function. However, classical in vivo approaches cannot firmly
establish the direct action of key neuromodulators on the GnRH neuron, primarily
because the GnRH system receives input from numerous sources (79). As a result, the
mechanism underlying the regulation of GnRH neurons by these neuromodulators are
still being investigated.
1.3 Energy homeostasis and reproductive function
1.3.1 Hypothalamic nuclei associated with regulation of food intake
The energy balance equation states that body mass remains constant when caloric
intake equals caloric expenditure. Chronic deviations from this balance results in a
change in body mass (80). Although the energy balance equation may appear simplistic
in nature, in reality, energy homeostasis involves the interaction of an array of
neuropeptides, neurotransmitters and hormones produced at both central and peripheral
sites (9, 81). The hypothalamus is the central regulator of feeding behaviour and energy

10
10
homeostasis, integrating neuronal, metabolic and endocrine signals. Several distinct
hypothalamic nuclei are involved in this process including, the lateral hypothalamic area
(LHA), paraventricular nucleus (PVN), dorsomedial nucleus (DMN), VMN and ARC
(82, 83). It is now well known that these feeding nuclei are controlled by a neural
circuitry comprised of orexigenic and anorexigenic neuropeptides including NPY, MCH,
galanin, orexin, galanin-like peptide (GALP), α-MSH, NT and CRH (8, 84). Table 1.1
provides an overview of the feeding neuropeptides and overall effect on feeding
behaviour. Energy homeostasis is carefully maintained through the regulation of these
neuropeptides by both peripheral and central signals.
1.3.2 Energy homeostasis and reproductive function
Reproduction is an energy intensive process that is metabolically gated. In
humans, under-nutrition or excessive exercise results in reproductive dysregulation (81).
Conversely, obesity and diabetes can also impair reproductive function. This relationship
has been demonstrated in mammals where a deficiency or excess of nutrients results in
impaired gonadal function, delayed reproductive maturation, and a disturbance in the
course of the ovarian cycle (85, 86). The hypothalamus plays a crucial role in integrating
these two physiological processes, where metabolic sensory stimuli including hormonal
neuromodulators and hypothalamic neuropeptides that control energy homeostasis can
also influence the HPG axis. A decrease in GnRH and/or LH secretion is postulated to be
the most important etiological factor for nutritionally induced reproductive disorders (9,
86). Although the relationship between nutrition and reproductive success has been
described, the exact mechanisms and neuropeptides linking these two physiological
processes has yet to be determined. NPY neurons of the hypothalamus have emerged as a

11
11
key neuromodulator involved in regulating both feeding behaviour and reproductive
homeostasis (13, 78, 87, 88).

12
12
Anorexigenic peptides Orexigenic peptides
Adrenomedullin Neuropeptide Y (NPY)
Bombesin Agouti-related peptide (AgRP)
Cholecystokinin (CCK) Galanin
Calcitonin Galanin-like peptide (GALP)
Cocaine-and amphetamine regulated transcript (CART)
Growth hormone-releasing hormone (GHRH)
Corticotropin releasing hormone (CRH)
Growth hormone (GH)
Ciliary neurotropic factor (CNTF)
Ghrelin
Gastrin-releasing peptide (GRP)
Melanin-concentrating hormone (MCH)
Galanin-like peptide (GALP) Opioids
Glucagon-like peptide-1 (GLP-1)
Orexin-A/B
Glucagon Prolactin
Melanocortin Peptide YY (PYY)
Melanocyte-stimulating hormone (MSH)
Neuromedin U
Neurotensin
Oxytocin
Pituitary adenylate cyclase-activating polypeptide (PACAP)
Pro-opiomelanocortin (POMC)
Somatostatin
Thyrotropin-releasing hormone (TRH)
Urocortin
Vasoactive intestinal polypeptide (VIP)
Table 1.1. Feeding-related peptides. Adapted from Ueta et al. Exp Biol Med, 228:1168-1174 (2003)

13
13
1.4 Neuropeptide Y
1.4.1 Synthesis
NPY is a 36 amino acid neuropeptide first discovered in the porcine brain by
Tatemoto et al. in 1982 (89). NPY is highly conserved in many species and forms the
pancreatic peptide family with Peptide YY (PYY) and pancreatic polypeptide (PP) (69,
89, 90). The NPY gene is located on human chromosome 7 at the locus 7p15.1 (69). The
NPY gene contains 3 introns that are separated by 4 exons (69). The coding region of the
prepro-NPY gene is 551 bp, which encodes a 97 amino acid prepropeptide. This prepro-
NPY peptide includes a signal peptide, the 36 amino acid NPY peptide product, and the
C-terminal peptide of NPY (CPON) (69). After translation, prepro-NPY is directed to the
endoplasmic reticulum where the signal peptide is cleaved to form pro-NPY. Prohormone
convertase then cleaves Pro-NPY at a dibasic site to generate NPY1-39 and CPON. Two
further truncations at the C-terminal end of Pro-NPY by a carboxypeptidase and the
peptidylglycine α-amidating monooxygenase lead to the biologically active 36 amino
acid peptide product. The mature NPY product can be further processed by two enzymes,
dipeptidyl peptidase 4 and aminopeptidase P, resulting in NPY3-36 and NPY2-36,
respectively (90). NPY is widely expressed in various regions of the brain, including the
hypothalamus, amygdala, hippocampus, nucleus of the solitary tract (NTS), locus
coeruleus, nucleus accumbens and cerebral cortex (69). Additionally, NPY fibres have
been shown to innervate a number of brain regions including the PVN, supraoptic
nucleus (SON), POA, DMH and ME (91-93). Peripheral NPY expression is seen in
endothelial cells, spleen, heart, adrenal medulla and liver (69). In accordance with its
wide distribution, NPY participates in the control of several physiological functions,

14
14
including feeding behaviour, water consumption, learning and memory, locomotion,
cardiovascular homeostasis, hormone secretion, emotional behaviour, reproduction and
circadian rhythms (94).
1.4.2 NPY receptors
NPY exerts its biological effects through G-protein coupled receptors, which have
been characterized based on their downstream physiological effects. The NPY receptor
family includes: 1) the Y1 receptor, a post-synaptic receptor involved in feeding,
vasoconstriction and reproduction (95), 2) the Y2 receptor, a pre-synaptic PYY
preferring-receptor expressed abundantly in the periphery and implicated with metabolic
syndrome (96), 3) the Y3 receptor, a controversial putative NPY-preferring receptor (97),
4) the Y4 receptor, a PP preferring-receptor involved in food intake (98), 5) the Y5
receptor, which is also involved in feeding, neuroprotection and reproduction, 6) the
putative Y6 receptor, which has been cloned but the physiological function remains to be
discovered (90, 94). Much of NPY receptor functionality has been studied using
pharmacological truncated and substituted NPY analogs that are receptor specific (94,
99). The pharmacological characteristics, binding preferences and functional significance
of each NPY receptor subtype are described in Table 1.2.
1.4.3 Signaling pathways activated by NPY
NPY receptors are, in most cases, coupled to pertussis toxin-sensitive G-proteins
(i.e. Gi and Go proteins). Therefore, the most common response to NPY is the
inactivation of adenylyl cyclase, which in turn, results in the inhibition of 3’,5’-cyclic
monophosphate (cAMP) synthesis (100). However, NPY has also been demonstrated to
induce cAMP production by inhibiting cAMP degradation by inactivating
phosphodiesterases in neuroblastoma cell lines (101).

15
15
In many cell types, NPY has also been linked to the induction of calcium
mobilization and calcium signaling pathways. NPY raises intracellular calcium
concentrations, which occurs through inositol 1,4,5-phosphate-dependent (IP3) or -
independent pathways and activating or blocking membrane bound calcium channels
(102). In addition, NPY has been demonstrated to activate the mitogen-activated protein
kinases (MAPK), as shown in erythroleukemia cells (92, 102). In particular, NPY has
been confirmed to induce the phosphorylation of p42/p44 (ERK1/2). This activation of
the MAPK pathway by NPY is suspected to occur through a phosphoinositide 3-kinase
(PI3K)-dependent or pertussis toxin sensitive mechanism (Figure 1.2). Finally, NPY has
also been demonstrated to induce vasodilation of human subcutaneous arteries via
intracellular events involving nitric oxide (103, 104). Notably, NPY signaling systems
activated are not helpful in providing the NPY-receptor subtype activated, since each
receptor subtype has been demonstrated to be capable of activating the same intracellular
pathways in transfected cells and tissue-specific studies.
The study of NPY second messenger systems has been essential to the
understanding of anxiety, memory, hypertension and drug addiction (94). However, the
NPY signaling events activated in the GnRH neuron, ultimately controlling the
reproductive axis, have not yet been fully described.
1.4.4 NPY effects on energy homeostasis and reproduction
NPY neurons are a prime candidate linking energy balance and reproduction in
the hypothalamus (10, 13, 81). The ability of NPY to regulate feeding behaviour was
reported when intracerebroventricular (ICV) injection of NPY elicited a strong feeding

16
16
Receptor Agonist affinity Distribution Physiological role
Y1 NPY > [Leu31,Pro34] >> NPY (13-36) >> PP
Vascular smooth muscle, adipocytes, cerebral cortex, colon, hypothalamus
Food inake, blood pressure, seizure regulation, anxiety, pain, depression, ethanol consumption, reproduction?
Y2 PYY > NPY > NPY (13-36) Hippocampus, adipocytes, renal proximal tubular cells, hypothalamus, amygdala
Food intake, blood pressure, seizure regulation, anxiety, bone formation, pain, GI motility, angiogenesis
Y4 PP > [Leu31,Pro34] > PYY > NPY
Brain, heart, coronary artery, ileum, testis, lungs Food intake, GI motility, reproduction?
Y5 NPY > NPY (3-36) = [D-Trp32] Dentate gyrus, hypothalamus, thalamus, cortex Food intake, seizure regulation, anxiety, reproduction?
Table 1.2. NPY receptor characteristics. Adapted from Gehlert. Neuropeptides, 38; 135-140 (2004)

17
17
Figure 1.2. Schematic illustration of the signal transduction mechanisms activated by NPY.
NPY can modulate a variety of pathways through the activation of G-protein coupled receptors, result-ing in: adenylyl cyclase inhibition and thus inhibition of PKA. Conversly, NPY can also increase cAMP levels and stimulate PKA activity by inhibiting PDE3B. NPY can also activate PLC and PI3K activity in a tissue specific manner. Mitogenic signals are stimulated through redundant mechanisms including the nitric oxide pathway (not depicted here).
ER
NPY
Ca2+
Endoplasmicreticulum
PKC
MEK1/2
CREB
PI3k
IRS1/2
PPDE3B
PLC
Gene transcription
MAPK
P
AC
cAMP
PKA P

18
18
response, even in satiated animals eventually leading to obesity (105-107). Subsequent
studies supported NPY as a natural orexigenic neuropeptide. NPY expression in the
hypothalamus dramatically increased in rats under poor metabolic conditions (108).
Additional studies demonstrated that peripheral administration of NPY stimulates feeding
and chronically administered doses results in obesity (109). Similarly, leptin-deficient
mice have strongly elevated hypothalamic NPY levels and are morbidly obese (110, 111).
Finally, NPY mRNA expression is increased during times of food restriction, further
indicating that the activation of the NPY system is involved in the stimulation of feeding
(106, 109). Moreover, increased NPY levels return to baseline levels after re-feeding,
indicating hypothalamic NPY is a key physiological signal for energy homeostasis.
Shortly after NPY was identified as an orexigenic neuropeptide, additional studies
implicated NPY in reproductive physiology. NPY neurons in the ARC project to GnRH
cell bodies in the POA and to GnRH pre-synaptic terminals in the ME (70, 93). Further
studies demonstrated NPY knock-out (NPY-KO) mice are not capable of generating a
normal LH surge, necessary stage for ovulation (13). Importantly, NPY mRNA has been
shown to accumulate in the ARC just prior to the LH surge, signifying that NPY may
regulate the LH surge (88, 112). Although the importance of NPY in the reproductive
axis has been demonstrated, the direct effect of NPY on GnRH neurons at the
transcriptional level has not been elucidated.
1.5 Estrogen
1.5.1 Synthesis and metabolism
Estrogens are steroid hormones that are important endocrine effectors of
reproduction, cardiovascular physiology, neuronal growth and differentiation,
neuroprotection, cognition, sexual differentiation and regulation of mood (113-115). The

19
19
most common forms of estrogen found in the body are: estrone, E2, 17α-estradiol and
estriol (113, 116, 117). Estrone, estriol and 17α-estradiol are considered short acting
estrogens with a much lower binding affinity to ERs than E2 (118). However, both 17α-
estradiol and E2 have high ER affinity in the brain, and have been implicated in the
control of hippocampal synaptic plasticity and neuroprotective effects in the brain (119).
Although 17α-estradiol is emerging as a key regulator of neural development, it is not
found in the circulation and is unaffected by ovariectomy, castration and/or
adrenalectomy, suggesting 17α-estradiol is produced in the brain (120, 121). As a result,
E2 is considered to be the most potent and dominant form of estrogen in the body (117,
122). Estrogens are synthesized from a microsomal member of the p450 superfamily,
aromatase cytochrome p450 (aromatase) (123). Through a series of reactions, aromatase
in granulosa cells catalyzes the conversion of C19 androgenic steroid substrates produced
from thecal cells to form a phenolic A ring structure, which is characteristic of estrogenic
compounds (113). Estrogens can be produced in a wide range of tissues (124). In
premenopausal women, the ovaries are the principal source of estrogens that circulates to
act on distal tissues. However, in men and women, estrogenic compounds are also
produced from extragonadal sites that operate primarily via paracrine mechanisms (115).
These sites include the mesenchymal cells of adipose tissue, osteoblasts and chondrocytes
of bone, vasculature endothelium, aortic smooth muscle and in the brain (115).
1.5.2 Estrogen receptors
The biological actions of E2 are primarily mediated through two specific nuclear
estrogen receptors (ER), ER-α and ER-β, which are part of the nuclear receptor
superfamily (19, 120, 125, 126). There are two distinct genes that encode ERs, which
may encode isoforms generated by alternative splicing (127, 128). In particular, data

20
20
supports that ER-β has multiple splice variants at the protein level (128, 129). Thus far,
studies have largely focused on ER-β1 (ER-β), the originally cloned sequence (130).
Additional isoforms ER-β2 and ER-β5 are derived from alternative splicing of the last
coding exon (exon 8) (131). Studies examining the putative functions of ER-β2 and ER-
β5 isoforms have found that only ER-β can bind to ligand and induce conformational
changes as determined by protease digestion assays (132). As a result, current studies are
examining the biological role of ER-β2 and ER-β5 isoforms. Presently, ER-β2 and ER-β5
isoforms are hypothesized to exist in an activated state, as these isoforms cannot bind to
E2 (133).
ERs are multidomain proteins that are comprised of: 1) A-B domain containing
activating function 1 (AF-1), 2) C region that contains a highly conserved DNA-binding
domain (DBD) and two zinc fingers critical for DNA-binding, 3) D region that acts as a
hinge, and 4) E domain, which contains the AF-2 region and is responsible for ligand
recognition and binding (134). ER-α and ER-β contain evolutionary conserved DBDs,
which are critical for receptor-DNA recognition and specificity, and C-terminal ligand
binding domains (LBD) that recognize specific estrogenic compounds to exert the
appropriate biological response (135). Although ER-α and ER-β show considerable
homology in their DBD (90%), the receptor subtypes share only 53% amino acid identity
at the carboxyl-terminal LBD (136). Additionally, the ER subtypes are products of
different genes and studies have indicated that each ER can have unique and overlapping
biological functions in a tissue- and cell context-dependent manner (136, 137). This is
exemplified in studies that compared the phenotypes observed in the individual lines of
ER knockout mice (ERKO), the αERKO and βERKO, which exhibit phenotypes that
generally mirror the respective ER expression patterns (19, 114). The most notable

21
21
phenotypes in the female αERKO mice include an underdeveloped reproductive tract,
hypergonadotropic hypergonadism, lack of pubertal onset, and excess adipose tissue,
whereas in the male, testicular degeneration and epididymal dysfunction are major factors
(114). These phenotypes combined with deficits in sexual behavior result in infertility in
both sexes of the αERKO mice. In contrast, βERKO males are fertile but demonstrate
neuronal deficits and an abundance of astroglial cells; however, βERKO females exhibit
inefficient ovarian function and subfertility (114, 138).
ERs are widely distributed throughout the body and display overlapping
expression in a number of tissues (120, 136). ER-α is expressed in the uterus, liver,
kidney and heart, whereas ER-β is expressed in the ovary, prostate, lung, gastrointestinal
tract, bladder and hematopoetic cells. ER-α and β are co-expressed in the mammary
glands, epididymis, thyroid, adrenal glands, bones, and in regions of the brain (120, 136).
The binding of E2 to ER induces conformational changes in the receptor that leads to
dimerization, protein-DNA interaction, recruitment of co-regulators/transcription factors
and ultimately the formation of a pre-initiation complex, as described in greater detail in
the next section (116, 136).
Evidence is accumulating that non-genomic E2 signaling may also be mediated by
the seven-transmembrane domain G-protein coupled receptor, GPR30 (139). Filardo and
colleagues identified E2 as a natural ligand for GPR30 as E2 activated the MAPK
pathway in MCF-7 cells in the absence of the classical ERs, ER-α or -β (139, 140).
Subsequent studies further implicated the novel GPR30 receptor to be directly involved
in mediating cellular responses of E2 (141-144). Transfection of GPR30 into MDA-MB-
231 cells induced E2 responsiveness in the ER-deficient cell line (140). In 2005, Thomas
et al. described the specific binding of E2 to GPR30 in transfected HEK293 with a Kd of

22
22
3 nM (145). This binding was eliminated by GPR30 silencing in HEK293 cells that were
treated with small interfering RNA (siRNA) specific to GPR30 (145). GPR30 is now
suspected to act as a scaffold, recruiting kinases for other signaling molecules that could
regulate the expression of conventional ERs (140, 142, 144).
Using green fluorescent protein (GFP) chimeric construct fusing GFP to the
carboxy-terminus of GPR30, GPR30 has been localized to the endoplasmic reticulum and
the plasma membrane (142). GPR30 transcripts are reported to be widely expressed in
humans in various tissues including heart, lung, ovary, liver and brain (140, 142, 143). In
the brain, immunohistochemical (IHC) studies have revealed GPR30 is expressed
throughout neurons in the hypothalamus, pituitary, hippocampus and brainstem. These
studies indicate that in addition to ERs, GPR30 is an endogenous receptor of E2 and is
involved in the activation and regulation of cellular physiology.
1.5.3 Signaling pathways activated by estrogen
1.5.3.1 Genomic mode of estrogen action
The genomic mechanism of E2 signaling involves E2 binding to cytosolic or
nuclear ERs producing a conformational change in their AF-2 domains, which allows ER
homodimerization and subsequent nuclear translocation (116, 146). In the nucleus, ER
acts as a ligand-dependent transcription factor, binding with high affinity to E2 responsive
elements, which are cis-acting enhancers/repressors located within the regulatory regions
of target genes (136, 147) (Figure 1.3). However, subsequent studies demonstrated ERs
inhibit progesterone receptor (PR) and glucocorticoid receptor (GR) activation on
promoters lacking EREs (148, 149). These observations suggested ERs could form ER

23
23
E2
E2
GPR30
Endoplasmicreticulum cAMP
Grb2
Sos
PKAP
Ras
MEK1/2
Raf
MAPK
Src
SHC
ER
ER ER Fos Jun
PI3k
IRS1/2
AKTP
PCREB
AMPKK
AMPKP
ACC P
PLC
Ca2+
Gene transcription
ER
Figure 1.3. Schematic illustration of estrogen signaling mechanisms.
The classical pathway includes estrogen directly binding to cytosolic ER, resulting in ER dimerization and protein-protein interactions with other transcription factors to regulate transcription. Nonclassical estrogen signaling results in the rapid activation of intracellular signaling kinases of the MAPK, PI3K, AMPK, PKA and PLC pathways. Rapid signaling cascades can occur through estrogen binding to cyto-solic and membrane-bound ERs or through the GPR30 receptor localized at the endoplasimic reticulum or cellular membrane. Estrogen is freely permeable, gaining access to intracellular ERs and GPR30 receptors.

24
24
multiprotein complexes with coregulatory proteins that can bind to non-ERE promoter
sites to directly regulate gene expression (116). Additional studies led to the
identification of a host of coregulatory proteins that interacted with the LBD, AF-1 and
AF-2 domains of ERs. One of the first ER-interacting proteins was identified after the
cloning and characterization of steroid receptor coactivator-1 (SRC-1) gene (122, 150).
SRC-1 was initially demonstrated to directly interact with ERs using yeast two-hybrid
screening and has been shown to enhance transcriptional activities in the presence of E2
using luciferase assays. Sheppard et al. identified a conserved motif within SRC-1 called
the nuclear receptor box (LXXLL; L = leucine, X = any amino acid), which is necessary
and sufficient for coactivator binding to activated ERs (151). The LXXLL structural
motif is also found in other ER coactivators, including TRAP220, CREB-binding protein
(CBP), p300, and the activating signal cointegrators (ASC-1 and ASC-2), which can also
modulate ER activity (150). Coregulatory proteins of ERs contain intrinsic histone
acetylase activity (HAT), which is known to facilitate chromatin remodeling at target
promoters and ER activity (122, 150). A number of ER coregulatory proteins have been
identified since the original identification of SRC-1, of which several have been
identified, including GRIP1, AIB1, CBP/p300, TRAP220, PGC-1, p68 RNA helicase,
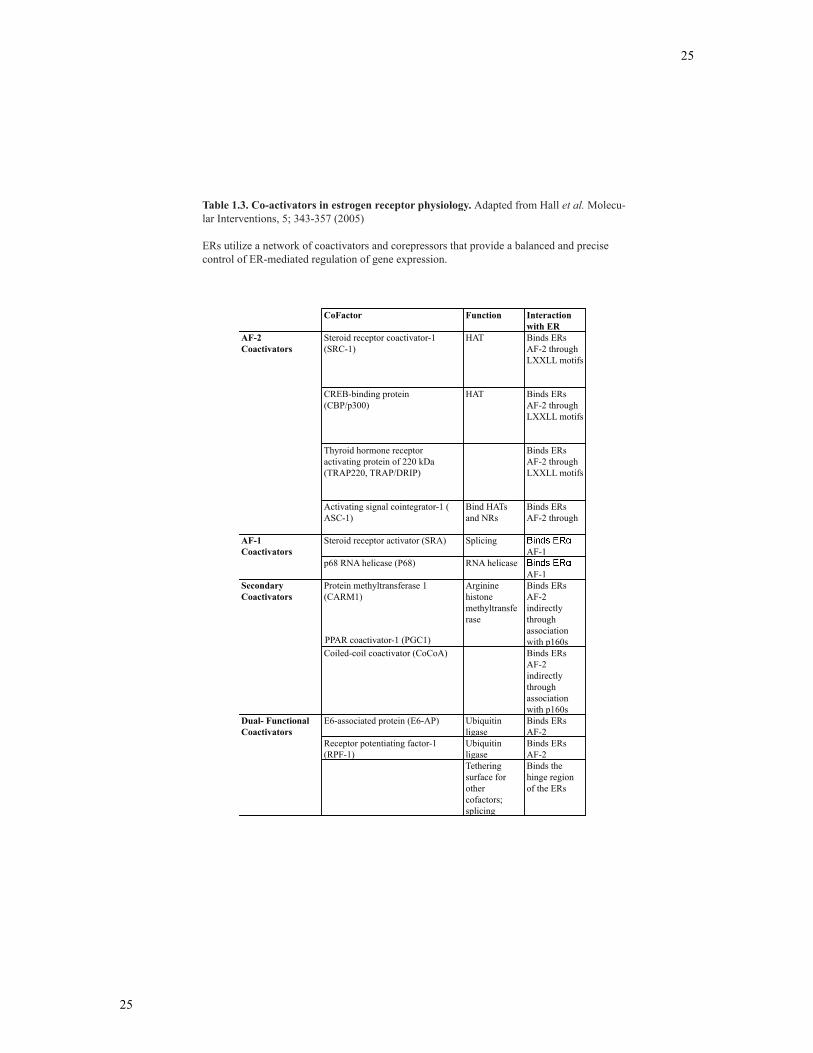
and SRA (152). Table 1.3 lists some of these coregulatory proteins, including their
function and interaction with ERs.
The classical actions of E2 ultimately result in the regulation of a number of
target genes, including matrix and structural proteins, regulatory enzymes, surface
receptors, ion channels, transcription factors and peptides. Depending on the cell,
promoter context, co-regulators expressed and ratio of ERα:ERβ, the DNA-bound
25
25
CoFactor Function Interaction with ER
Steroid receptor coactivator-1 (SRC-1)
HAT Binds ERs AF-2 through LXXLL motifs
CREB-binding protein (CBP/p300)
HAT Binds ERs AF-2 through LXXLL motifs
Thyroid hormone receptor activating protein of 220 kDa (TRAP220, TRAP/DRIP)
Binds ERs AF-2 through LXXLL motifs
Activating signal cointegrator-1 ( ASC-1)
Bind HATs and NRs
Binds ERs AF-2 through
Steroid receptor activator (SRA) SplicingAF-1
p68 RNA helicase (P68) RNA helicaseAF-1
Protein methyltransferase 1 (CARM1)
Arginine histone methyltransferase
Binds ERs AF-2 indirectly through association with p160s
Coiled-coil coactivator (CoCoA) Binds ERs AF-2 indirectly through association with p160s
E6-associated protein (E6-AP) Ubiquitin ligase
Binds ERs AF-2
Receptor potentiating factor-1 (RPF-1)
Ubiquitin ligase
Binds ERs AF-2
Tethering surface for other cofactors; splicing
Binds the hinge region of the ERs
AF-2 Coactivators
AF-1 Coactivators
Secondary Coactivators
Dual- Functional Coactivators
PPAR coactivator-1 (PGC1)
Table 1.3. Co-activators in estrogen receptor physiology. Adapted from Hall et al. Molecu-lar Interventions, 5; 343-357 (2005)
ERs utilize a network of coactivators and corepressors that provide a balanced and precise control of ER-mediated regulation of gene expression.

26
26
receptor exerts either a positive or negative effect on expression of the downstream target
gene.
1.5.3.2 Non-genomic mode of estrogen action
E2 also acts through non-genomic, rapid signaling mechanisms by binding to
plasma membrane-bound or cytoplasmic ERs or GPR30, which activate signal cascades
that can directly lead to cell-specific biological effects (120, 143, 146). The concept of
non-classical E2 signaling was originally suggested because E2 could induce cellular
changes that were far too rapid to be accounted for by classical E2 signaling (153). This
was first observed in 1967 by Szego and Davis, where E2 exposure increased cAMP in
the uterus of OVX mice in 15 seconds, which is considered too rapid for genomic
responses of E2, which often take hours for final changes in protein expression to take
place (154). Additional studies found E2 could bind to receptors located at the cell
membrane and initiate rapid cAMP accumulation in endometrial cells (155). Since then,
investigators have demonstrated that non-genomic E2 signaling involves the mobilization
of intracellular calcium, stimulation of adenylate cyclase activity and activation of the
MAPK and PI3K signaling pathways (156, 157). The non-genomic actions of E2 are
mediated through functional domains of the receptor that likely interact with scaffold
proteins such as the modulator of non-genomic action estrogen receptor (MNAR),
caveolin-1 and proximal signaling molecules including: G proteins, striatin, p130Cas, ras,
p85α and Shc (158-161). These ER-interacting proteins have been shown to couple ER to
kinases such as Src and PI3K to mediate rapid E2 activation of AKT and ERK. The
activation of signal transduction pathways may then enhance the activation of
downstream signaling components to ultimately elicit genomic responses. For instance,

27
27
transcription factors Elk-1, C/EBPβ and CREB are all targets for phosphorylation by the
MAPK signaling pathway (151, 158-161).
Although there is an abundance of research conducted on E2 signaling, the
relative contribution of genomic and non-genomic actions to certain gene responses
remains undetermined. These signaling mechanisms may occur concurrently or in series,
but subsequently converge at the level of transcription. The molecular responses to E2 are
likely to vary depending upon a number of conditions, such as the differential expression
of ERs, interaction of coregulatory/scaffold proteins, cell membrane or nuclear ER
content, steroidal/hormonal milieu and duration of E2 exposure.
1.5.4 Effects of estrogen on reproduction and feeding behaviour
Estrogens have been demonstrated to regulate both energy homeostasis and
reproductive function in the hypothalamus (162, 163). E2 is critical for the synthesis and
secretion of GnRH, paradoxically exerting both a stimulatory and inhibitory effect on
GnRH release (53, 79, 164). Estrogens act in the central nervous system to directly
inhibit GnRH transcription and secretion, in both male and female mice (27). However,
positive estrogen action is also required for the generation of the preovulatory LH surge
in females (165). Despite the importance of estrogen action on reproductive function, the
mechanisms regulating the hormonal response to estrogen are poorly understood. This is
primarily due to the complex circuitry of the hypothalamus and scattered distribution of
GnRH neurons in the brain (32, 166). However, carefully designed in vivo studies have
shed light on the importance of estrogen in the regulation of GnRH release. In OVX
rhesus monkeys and mice, exogenous E2 administration that reflect physiological levels
were sufficient to induce a GnRH surge (167). However, E2 directly reduces GnRH
expression and release in the homogenous GT1-7 cell lines, and thus, the repressive

28
28
effects of E2 on GnRH neurons are thought to be overcome by afferent neuronal fibres
stimulated by estrogen (53). Estrogens could potentially stimulate a number of afferent
fibres that are intimately associated with dendrites, cell bodies or nerve terminals of
GnRH neurons. Excitatory neuropeptides and neurotransmitters that have been implicated
in GnRH regulation are NPY, NT, norepinephrine, glutamate and aspartate (79).
Continuous and increasing estrogen exposure could enhance the synthesis and release of
these stimulatory factors and may overcome the direct inhibitory effect of estrogen on
GnRH neurons. As a result, estrogen is hypothesized to regulate the HPG axis through a
complex cellular network that is not yet fully characterized.
Estrogens are also well-recognized negative regulators of energy homeostasis and
feeding behaviour. Donohoe et al. found that subcutaneous injections of E2 and 17α-
estradiol reduced food intake in OVX rats (168). However, E2 treatments reduced food
intake significantly more compared to 17α-estradiol treatments (168). In addition, 17α-
estradiol is synthesized in the brain and not the ovaries, which suggests that the OVX
mice that have increased weight gain and food intake occurs through 17α-estradiol
independent mechanisms (121). In fact, in OVX mice brain 17α-estradiol levels are
significantly higher compared to wild type controls, suggesting E2 may negatively
regulate 17α-estradiol syntheses in the brain (121). Together, these studies have led to a
larger focus on E2 in the regulation of body weight and food intake.
Postmenopausal women display increased weight gain, visceral obesity and have
an increased risk of diabetes (169, 170). E2 replacement therapy normalizes these
abnormalities. This negative effect on energy homeostasis by E2 has also been
demonstrated in rodent studies. During the estrous cycle, peak levels of E2 observed
during the afternoon of proestrous results in significantly reduced food intake (171).

29
29
Ovariectomized (OVX) rats display increases in food intake and increases in adipose
tissue deposition, which can be readily reversed with the re-introduction of estrogen (20,
172). E2 is thought to regulate energy homeostasis through two major pathways: an
anorectic action through the central nervous system and a direct action on tissue
metabolism (162, 173). The central action of E2 that regulates feeding behaviour appears
to be mediated through the regulation of multiple hypothalamic orexigenic and
anorexigenic neuropeptides. One study found that castration of female mice resulted in a
decrease in anorexigenic pro-opiomelanocortin (POMC) and CRH mRNA expression,
which was normalized after E2 injections (162). Another study found that E2 was required
for normal action of the gut derived peptides cholecystokinin (CCK) and ghrelin, the
adipocyte derived hormone leptin, and the hypothalamic neuropeptide melanin-
concentrating hormone (MCH) on satiety signals in the hypothalamus (174-176). Overall,
E2 has a well-characterized anorectic effect in mammals, but the mechanisms and
hypothalamic targets have yet to be fully elucidated. Recent studies, however, have
implicated NPY neurons as a key central target of E2 in the hypothalamus.
1.5.5 Estrogen-mediated regulation of NPY neurons
E2 acts as a homeostatic feedback molecule between the periphery and the brain,
regulating energy balance and reproduction. The feedback mechanisms employed by E2
could occur through the modulation of several neuropeptidergic circuits. NPY neurons of
the hypothalamus have emerged as a key target of estrogen, as NPY has a potent role in
regulating both reproductive function and energy homeostasis. NPY neurons express both
ER-α and ER-β in vivo and in vitro (164, 177-179). Studies suggest E2 can modulate both
reproduction and feeding by regulating NPY mRNA expression in clonal, immortalized
hypothalamic cell models (164, 178). Here, E2 differentially regulated NPY mRNA

30
30
expression in two distinct NPY-expressing hypothalamic cell lines. In the mHypoE-42
NPY cell line, E2 tonically downregulated NPY mRNA throughout a 72 h time course
(164). In the mHypoE-38 NPY cell line, 8 h E2 treatment resulted in a decrease in NPY
mRNA expression, whereas a 24 h E2 treatment in the same cell line resulted in a 4-fold
increase in NPY mRNA that corresponded with increased ER-β mRNA levels (164).
Together, this study suggests E2 may differentially modulate NPY-expressing neurons,
which may result in an anorectic and reproductive effect. Although the transcriptional
regulation of E2 on NPY-expressing cell lines has shed light on the dual role of E2, a
number of questions remain unanswered including the E2-mediated control of NPY
secretion.
1.6 Leptin
1.6.1 Synthesis and metabolism
In 1994, Friedman and colleagues discovered and characterized an obese gene
(ob) mutated in the mouse strain ob/ob (180). The ob gene was later found to encode a
4.5 kb mRNA sequence that was predominantly expressed in adipose tissue (111, 181).
This 4.5 kb sequence is comprised of three exons that span 15 kb of genomic DNA.
Analysis of cloned sequences upstream of the transcriptional start site revealed that the
217 bp sequence upstream of the 5’ region is required for basal leptin gene expression in
adipocytes (181). The evolutionary conserved ob gene encodes for a 167 amino acid 16
kDa protein called leptin (181). Leptin plasma concentrations are directly proportional to
one’s body fat or body mass index (BMI), although this can vary with gender, nutritional
status and time of day (15, 182). Physiological leptin concentrations in males and females
range from 4 ng/ml to 100 ng/ml, observed in lean and obese subjects, respectively (182).

31
31
1.6.2 Leptin receptors and signaling events
The gene for the leptin receptor (Ob-R) is encoded by the db gene and is
alternatively spliced into several different receptor isoforms; one full length (isoform Ob-
Rb), and several shorter isoforms spliced at the C-terminal coding exon (Ob-Ra, Ob-Rc,
Ob-Rd, Ob-Re, Ob-Rf) (173, 174). These receptor proteins have identical sequences in
their extracellular and transmembrane domains, and also share the first 29 amino acids of
the cytoplasmic domain. However, the Ob-Rb isoform has a 302 amino acid
intracytoplasmic domain that includes several motifs for protein-protein interactions
(175, 176). The remaining shorter isoforms have truncated intracytoplasmic regions (173,
176). Ob-Rs are a member of the class 1 cytokine receptor family. This receptor family
uses an assortment of cystolic-signaling transducers to mediate changes in gene
transcription and cellular events (183). Ob-Rb is predominately expressed in the
hypothalamus (21, 176); however, the truncated isoforms are expressed in a wide range
of tissues, including hypothalamus, thymus, heart, lung, liver, spleen, kidney, stomach
and adipose tissue (184). The activation of cellular signaling cascades is thought to occur
primarily through the Ob-Rb isoform, as the short leptin receptor isoform does not
contain the post-receptor signaling machinery (185). The full-length Ob-Rb is known to
activate the JAK-STAT cascade as a major pathway in leptin signaling. Recent studies
have demonstrated continuous leptin injections result in the activation of STAT3 in
hypothalamic nuclei (179). Studies in peripheral tissues have also suggested alternate
leptin signaling mechanisms. Leptin has been shown to induce activation of the PI3K-
phosphodiesterase type 3B-(PDE3B)-cAMP pathway, resulting in a decrease in cAMP
levels in the hypothalamus (186). The MAPK family has also been proposed in leptin
receptor signaling, as leptin treatments induce MAPK/ERK activity in the hypothalamus

32
32
(181, 182). Most recently, AMPK, a fuel-sensing enzyme responsible for maintaining
metabolic homeostasis, has been shown to play a critical role in leptin action in the
hypothalamus (183, 184) (Figure 1.4). To add to the complexity of leptin signaling,
several groups have identified negative regulators of leptin signaling. Specifically,
suppressor of cytokine signaling (SOCS)-3, a leptin induced signaling protein, negatively
regulates leptin signaling by binding to SH2 binding domains on the Ob-R (185). In
addition, protein tyrosine phosphatase (PTP1B), originally identified as a negative
regulator of insulin signaling, was found to also reduce leptin signaling by impairing
leptin-induced JAK/STAT signaling as demonstrated in cells overexpressing PTP1B
(186). The discovery of the negative regulators of leptin signaling has led scientists to
hypothesize that overreactivity of the negative regulators of leptin signaling is a potential
causal mechanism of leptin-resistant obesity.
The role of the short form of leptin receptors remains to be defined; however,
studies have suggested the short isoforms may act as a free cytosolic binding protein for
leptin, thus regulating free leptin concentrations. Despite the functional uncertainty of the
short leptin receptor, the distinct tissue distribution and abundance of short leptin receptor
isoforms suggests that these receptors play an important role in the biological action of
leptin.

33
33
Ras
Leptin
JAK2P
Grb2
STAT3P
STAT3P
STAT3P
MEK1/2
PI3k
PP
socs3
Gene transcription and ion channels
JAK2PP
P
Sos
Shc Raf
MAPK
PTP1B IRS1/2
AMPKK
AKTAMPK
PP
ACCP
Figure 1.4. Schematic illustration of leptin receptor signaling. Adapted from Harvard and Ash-ford. Neuropharmacology, 44, 947-849 (2003)
Leptin binds to the Ob-R, which leads to increased activity of intracellular JAK2 kinases associated with membrane-proximal regions of the Ob-R. JAK2 phosphorylates Ob-R tyrosine residues that lead to the activation of the STAT3 and MAPK signaling pathways. PI3K is also involved in the regulation of rapid signaling kinases and membrane polarity. Through unknown mechanisms, leptin can directly regulate the activity of AMPK signaling. SOCS3 and PTP1B negatively regulate leptin signaling by binding to Ob-R tyrosine phosphorylated sites to prevent STAT3 activity and by dephosphorylating tyrosine residues, respectively.

34
34
1.6.3 Effects of leptin on feeding behaviour and reproduction
The idea of a blood-borne factor produced from fat mass that relays information
to the central nervous system was originally conceptualized by Kennedy and colleagues
as early as 1953 (187). This factor, leptin, was finally identified by Friedman’s group in
1994 and has since been extensively studied and characterized (180). Leptin is a blood
borne signal released in proportion to fat stores that informs the hypothalamus about
peripheral energy stores. Specifically, leptin has an appetite suppressing effect (181).
Leptin administration centrally or peripherally decreases food intake and increases energy
expenditure (182, 188). Lack of functional leptin receptors or signaling in mice results in
severe obesity (111, 184). Alternatively, the administration of leptin to obese mice with
defects in the ob gene (resulting in leptin insufficiency) can correct this abnormal
phenotype (189, 190). Leptin also regulates energy homeostasis in the periphery by
regulating muscle metabolism (191). Specifically, leptin stimulates beta-oxidation in
skeletal muscle (191). In addition, ob/ob mice depict a large increase in their triglyceride
content resulting in impaired beta cell function. Together, leptin regulates energy
homeostasis centrally by regulating food intake and peripherally by regulating metabolic
expenditure.
Adequate leptin levels are also required for normal reproductive function. The
link between leptin and reproduction became apparent when ob/ob mice were reported to
have a number of reproductive abnormalities (192). These mice are infertile, and similar
to the obese phenotype, abnormal reproductive function can be normalized by peripheral
injections of recombinant leptin (193-195). The reproductive malfunction in ob/ob mice
is thought to be due to reduced gonadal steroids and reduced HPG activity (196).
Prolonged and continuous leptin treatment increases uterine and ovarian weight in

35
35
females, and increases seminal vesicle and testicular weight in males (196). Leptin has
also been implicated in triggering the onset of puberty. Exogenous leptin treatments
result in precocious vaginal opening, an indicator for pubertal status (194). Together, the
evidence available thus far suggests leptin is a key peripheral signal that is required for
normal reproductive function and energetic status, as inert or absent leptin results in
reproductively deficient and obese animals. Although leptin is thought to control these
physiological processes through specific nuclei located in the hypothalamus, the
intermediary signals between leptin, feeding and reproductive homeostasis remains
unclear.
1.6.4 Leptin-mediated regulation of NPY neurons
Evidence clearly indicates that the central nervous system, particularly the
hypothalamus, is a major site of leptin action (197, 198). Ob-Rb is critical for
intracellular leptin signaling and has been localized to a number of hypothalamic nuclei
responsible for reproductive and metabolic function, including the ARC, DMH, LHA and
VMH (21, 197, 198). Two important neuropeptides, POMC and NPY, which are
involved in both feeding and reproductive homeostasis, have emerged as key
hypothalamic targets of leptin.
Neurons containing anorexigenic peptide-products of the POMC gene express the
Ob-R and make direct synaptic connections on GnRH-expressing neurons (199).
Interestingly, POMC gene expression is modified with varying leptin levels, where
untreated ob/ob mice show a 50-70% decrease in POMC gene expression, which can be
restored to wild-type levels with the treatment of leptin (200, 201). The most promising
peptide product of POMC that may be implicated in the leptin-mediated control of
feeding and reproduction is α-MSH (202). The leptin-mediated inhibitory action on food

36
36
intake was shown to be at least partially mediated through the α-MSH receptor,
melanocortin type 4 (MC4) (203). However, studies using MC4 specific
agonist/antagonist studies have failed to demonstrate that MC4 receptor plays a crucial
role on reproductive parameters despite the apparent synaptic connection between
POMC- and GnRH-expressing neurons (204). As a result, MC4 activity is suspected to
regulate GnRH synthesis and release indirectly by modulating receptors and
responsiveness to other key GnRH stimulators. Thus, although POMC/α-MSH plays an
important role in the control of energy homeostasis, studies have yet to conclusively link
MSH to reproductive function.
The orexigen NPY is a neuropeptide that also plays a dual role in regulating both
feeding and reproduction in the hypothalamus. NPY can directly stimulate GnRH
secretion, as demonstrated in push-pull cannulae studies in vivo and GT1-7 neuronal cell
lines in vitro (45, 78, 88). NPY is also a potent orexigenic compound, as peripheral and
central injections cause increased food intake (205). NPY-expressing neurons in the ARC
also contain Ob-R, suggesting NPY is directly responsive to circulating leptin (198).
Additionally, NPY mRNA is elevated in both fasted and ob/ob mice (206). Previous
work in our lab has demonstrated that leptin can directly down-regulate NPY mRNA in
hypothalamic cell lines; however, ensuing studies on whether leptin can directly regulate
NPY secretion to control energy balance and reproduction have yet to be completed
(164). The hypothesis that leptin can positively or negatively regulate neuropeptide
synthesis of afferent neuronal populations to feeding and reproductive nuclei have yet to
be verified. The use of hypothalamic cell lines will provide new models that allow the
study of the regulation of these neuropeptides by critical steroids and peripheral
hormones.

37
37
1.6.5 Leptin Resistance
A deficit in leptin does not underlie most cases of obesity in humans. In fact,
obese individuals exhibit elevated circulating leptin levels due to an increase in adipose
tissue mass (207, 208). Paradoxically, the elevated leptin level observed does not result in
a decrease in feeding. As a result, this failure of high leptin levels to suppress feeding and
decrease body weight suggests the presence of resistance to the anorexigenic effect of
leptin (209). Although leptin has a profound effect on regulating appetite and body
weight, the mechanisms of leptin resistance are not understood. To date, scientists have
proposed two mechanisms of leptin resistance and are actively investigating these
hypotheses (208). In the first hypothesis, impaired transport of leptin across the blood
brain barrier (BBB) is suspected to result in central leptin resistance (207, 210). In
support of this hypothesis is the finding that the concentration of leptin in the
cerebrospinal fluid (CSF) from obese humans is not increased in proportion to their
elevated serum leptin levels (207). The second and more likely hypothesis points to
downstream signaling defects in hypothalamic neurons as a primary cause of leptin
resistance. Munzberg et al. showed that in diet induced obese (DIO) mice (a classical
mouse model prone to leptin resistance and obesity when provided an experimental high-
fat diet) recombinant leptin completely failed to activate STAT3 in hypothalamic
extracts, indicating leptin resistance in the hypothalamus of DIO mice (211, 212).
Because Munzberg et al. used whole hypothalamic extracts, the precise neuronal
populations that develop leptin resistance remain unknown. Further studies localized
leptin resistance specifically to the ARC. After 16 weeks of high-fat-diet feeding, leptin-
activated phospho-STAT3 staining within the ARC was dramatically decreased. This
decrease in phospho-STAT3 was correlated with significantly higher SOCS3 levels in the

38
38
ARC (211). This study suggests that the ARC is selectively leptin resistant in DIO mice
(compared to wild-type control) and this may be caused by elevated SOCS3 in this
hypothalamic nucleus (211, 212).
The characteristics of specific neuronal populations that become leptin-resistant in
the ARC are unknown. However, the specific neurons that exist in the ARC that express
leptin receptors include, NPY, POMC and GALP neurons (213, 214). Based on this
knowledge, additional studies should examine the individual neuronal subpopulations to
determine the precise cell type and mechanisms involved in leptin resistance.
1.7 Cell models
Classical in vivo approaches have been instrumental in establishing synaptic
connectivity between distinct hypothalamic nuclei and the functional purpose of
numerous neuropeptides and neurotransmitters. However, the inherent complexity of the
neuronal circuitry comprising the hypothalamus presents a highly complex environment
to study the direct regulation of neuropeptides and cellular events by neuromodulators
(215). Additionally, the limited number of specific neuronal cell populations that are
responsible for key physiological effects such as feeding and reproductive function calls
for new complementary tools and methods to study the exact hormonal regulation of
distinct hypothalamic cell populations. Non-transformed hypothalamic primary cultures
are difficult to maintain, have scarce functional peptidergic neurons and are comprised of
a heterogeneous cell population. For these reasons, researchers have turned to
immortalized, clonal, hypothalamic cell lines to generate a comprehensive picture of the
molecular biology involved with the regulation of specific neuroendocrine hypothalamic
neurons. Cell lines provide a homogeneous population of cells that allow for the study of
the direct regulation of signaling pathways and molecular events (Figure 1.5) (216).

39
39
However, it is important to note there are limitations to the use of cell lines. The lack of
complexity, afferent innervations and endogenous stimuli prevents researchers from
concluding whether cell lines will respond identically to cells in vivo. Despite these
limitations, cell lines can be used to further develop theories of biological function prior
to completing studies in vivo. Studies reported using cell lines to date have been
instrumental in several novel findings that have been replicated in vivo (53, 217). In
particular, cell models created by both Belsham et al. and Mellon et al. have been
instrumental in the study of neuroendocrine regulation of key hypothalamic
neuropeptides (218-220).
1.7.1 GnRH-expressing GT1-7 neurons
GnRH neurons are a scarce population of neurons that are widely distributed
throughout the hypothalamus (79). Because of this, there are limited studies that examine
the cellular and molecular mechanisms that regulate GnRH neurons. There are now four
GnRH-expressing cell models used to study these cellular events: the GT1 (218), GN
(221), GnV (222) and GRT (223) cells. Each cell model was created using well-
characterized oncogenes targeting tumorigenesis in GnRH-expressing neurons. Of the
cell lines listed, the GT1 cells have proven to be the most similar to endogenous GnRH-
expressing neurons. GT1 neurons were immortalized by directing tumorigenesis in
GnRH-expressing neurons in transgenic mice through the expression of the simian virus
40 (SV40) T-antigen oncogene at the 5’ regulatory region of the GnRH gene (218).
Tumors from two offspring were cultured and cloned into GT1 cells, which were further
subcloned into three homogeneous cell populations labeled as GT1-1, GT1-3 and GT1-7.
Most studies to date have been performed using the GT1-7 cell lines because of the high
level of GnRH mRNA, expression of mature neuronal markers and classic neuronal
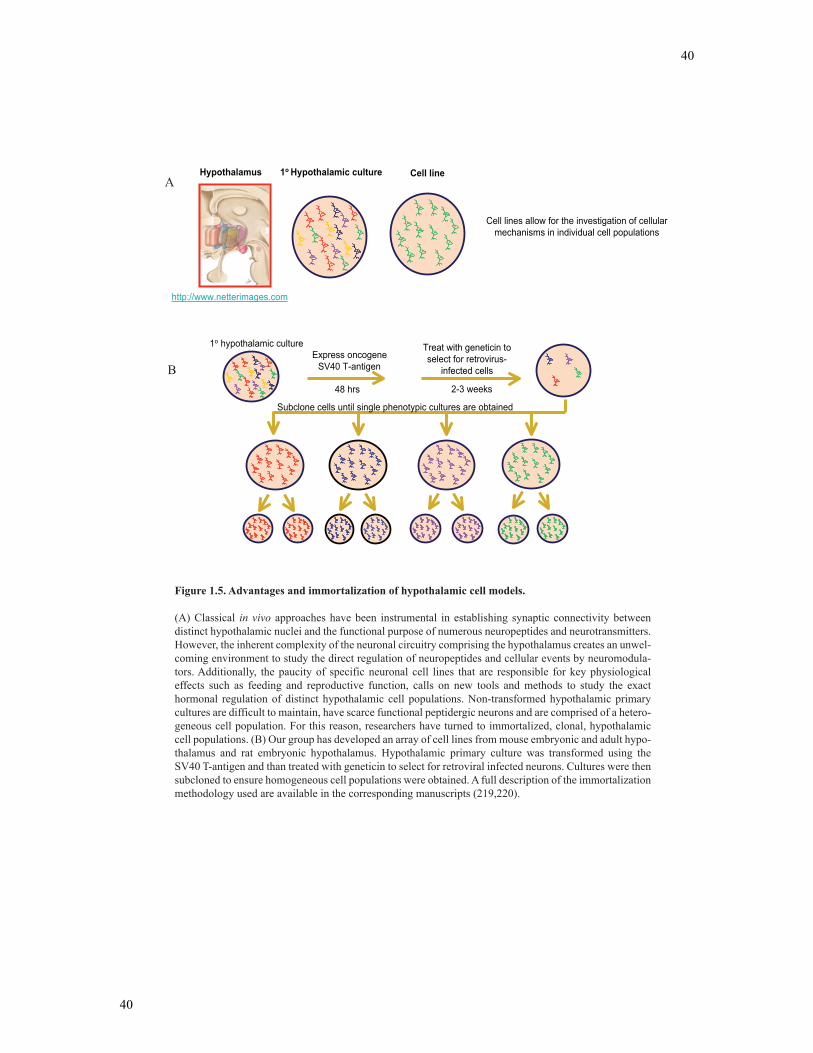
40
40
Figure 1.5. Advantages and immortalization of hypothalamic cell models. (A) Classical in vivo approaches have been instrumental in establishing synaptic connectivity between distinct hypothalamic nuclei and the functional purpose of numerous neuropeptides and neurotransmitters. However, the inherent complexity of the neuronal circuitry comprising the hypothalamus creates an unwel-coming environment to study the direct regulation of neuropeptides and cellular events by neuromodula-tors. Additionally, the paucity of specific neuronal cell lines that are responsible for key physiological effects such as feeding and reproductive function, calls on new tools and methods to study the exact hormonal regulation of distinct hypothalamic cell populations. Non-transformed hypothalamic primary cultures are difficult to maintain, have scarce functional peptidergic neurons and are comprised of a hetero-geneous cell population. For this reason, researchers have turned to immortalized, clonal, hypothalamic cell populations. (B) Our group has developed an array of cell lines from mouse embryonic and adult hypo-thalamus and rat embryonic hypothalamus. Hypothalamic primary culture was transformed using the SV40 T-antigen and than treated with geneticin to select for retroviral infected neurons. Cultures were then subcloned to ensure homogeneous cell populations were obtained. A full description of the immortalization methodology used are available in the corresponding manuscripts (219,220).
Hypothalamus 1o Hypothalamic culture Cell line
Cell lines allow for the investigation of cellularmechanisms in individual cell populations
http://www.netterimages.com
1o hypothalamic cultureExpress oncogene
SV40 T-antigen
48 hrs
Treat with geneticin toselect for retrovirus-
infected cells
2-3 weeks
Subclone cells until single phenotypic cultures are obtained
A
B

41
41
morphology (218, 224, 225). Additionally, GnRH is secreted in a pulsatile manner and
responds appropriately to neurotransmitters, and intrahypothalamic injections of GT1-7
cells can rescue fertility in hypogonadal mice (hpg mouse) (226). Together, studies
completed to date suggest the GT1-7 neurons are representative of GnRH neurons in
vivo, and thus provide the most valid and well-characterized model to study the cellular
and molecular regulation of GnRH neurons. In the third study of this thesis, the GT1-7
cell model was analyzed for the transcriptional regulation of GnRH mRNA levels in
response to NPY stimulation.
1.7.2 Embryonic hypothalamic cell lines – mHypoE-xx
The GnRH-expressing cell lines described above represent only one cell type from
the enormous range of cell types in the hypothalamus. Thus a number of laboratories
have set out to develop hypothalamic cell lines to compensate for this lack of
representation. Cell lines representative of the central nervous system have been created
by subcloning tumorigenic cell populations. CNS cell lines, Neuro2A and PC12 were
isolated from naturally occurring neuroblastoma and pheochromocytomas, respectively
(227). However, these cell models are not truly representative of fully differentiated
neurons. Other groups have utilized retroviral gene transfer of the SV40 T-antigen
oncogene to mass immortalize hypothalamic cells. In 1990, Rasmussen and colleagues
developed the RCF-8, RCF-12 and RCA-6 cell lines from rat embryonic hypothalamic
cultures (228). However, few studies have employed these cell lines, as they have not
been fully characterized. Kasckow et al. in 2003 also retrovirally transferred embryonic
hypothalamic cultures with the SV40 T-antigen to produce one cell line, IVB, which was
found to be a parvocellular CRH-expressing cell line (229). The lack of a representative
collection of hypothalamic cells that have been thoroughly characterized for the study of

42
42
hypothalamic physiology prompted the Belsham group to generate an array of
immortalized cell models from the hypothalamus (216). Using the retroviral transfer of
the SV40 T-antigen to primary hypothalamic cell cultures from fetal mice on days 15, 17
and 18, Belsham et al. were able to subclone over 60 embryonic cell lines labeled as
mHypoE-‘clone number’ (219). These cell lines express neuronal cell markers,
neurosecretory machinery, have clearly defined perikarya and neurites and have been
thoroughly characterized with over 100 neuroendocrine markers. Importantly, a number
of these cell lines generated express neuropeptides linked to energy and reproductive
homeostasis, providing new models to study the molecular biology of hypothalamic
neurons (216, 230). In the studies presented in this thesis, two embryonic, NPY-
expressing cell lines were used: mHypoE-38 and mHypoE-42. The cell lines express a
complement of neuropeptides listed in Figure 3.2 and Figure 4.1.
1.7.3 Adult hypothalamic cell lines – mHypoA-xx
Because the embryonic hypothalamic cell lines generated contributed to an
immense wealth of knowledge in the neuroregulation of hypothalamic neurons, the
Belsham group immortalized adult hypothalamic cell models to understand key
mechanisms involved in adult neuroendocrine cell types. In order to immortalize adult
neurons, cells were treated with ciliary neurotrophic factor (CNTF) to induce
proliferation, thus predisposing cell cultures to the retroviral transfer of the SV40 T-
antigen oncogene (220). Over 50 adult mouse cell lines were established labeled as
mHypoA-‘clone number’. Similar to the embryonic neuronal cells, the adult cell lines
express mature neuronal markers, exhibit neuronal morphology and have been
characterized for the expression of various neuropeptides and receptors. These cell lines
will be key to understanding hypothalamic physiology and can be used for the direct

43
43
comparison to embryonic neuronal cell lines. Overall, the hypothalamic cell lines now
made available by our lab and others, allows the study of mechanisms by which
peripheral hormones and neuromodulators can regulate hypothalamic neuroactivity. In
particular, I have taken advantage of these novel hypothalamic cell lines to piece together
the complex circuitry and cell-mediated responses that regulate energy and reproductive
homeostasis. In the studies outlined in this thesis, two adult hypothalamic cell models
were used: mHypoA-2/12 and mHypoA-59. Studies were completed in the mHypoA-
2/12 and mHypoA-59 as they express robust levels of NPY and hormone receptors of
interest. The phenotypic characterization of these cell lines were completed and listed in
Figure 3.2 and Figure 4.1.
1.7.4 NPY-GFP cell line
The Belsham group has recently devised a novel method of immortalizing NPY-
expressing neurons from the adult hypothalamus. NPY-GFP transgenic mouse
hypothalamii were dissected individually and immortalized as described above in section
1.7.3 (Belsham, unpublished data). Hypothalamii are from the adult transgenic mice
NPY-GFP mouse (strain B6.FVB-Tg(Npy-hrGFP)1Lowl/J generated by Dr. Bradford
Lowell, Beth Israel Deaconess Medical Center, Boston, MA, and available through The
Jackson Laboratory, Bar Harbor, ME). NPY neurons were than selected using flow
cytometry. NPY-GFP cell lines have been thoroughly characterized using RT-PCR, ICC
and NPY secretion assays. These cell lines will be important to understand the control
mechanisms utilized by mature NPY neurons in terms of basic physiological functions
and stimulus control. In this thesis, the NPY-GFP cell model is used in conjunction with
other hypothalamic cell models to describe the leptin-mediated regulation of NPY
secretion. The phenotypic characterization of these cell lines is described in Figure 4.2.

44
44
1.8 Hypothesis and aims
Neural networks of the hypothalamus and brainstem structures have been identified as
the homeostatic control systems of food intake. This homeostatic control system is comprised
of neuropeptides, neurotransmitters and hormones and is a crucial determinant of feeding
behaviour. Another extensive cortico-limbic neural system, the hypothalamus-ventral
tegmental-accumbens pathway, processes food intake in response to cues from the
environment. This non-homeostatic mechanism of food intake is undoubtedly a powerful
regulator of appetite that contributes to the obesity epidemic observed today. Importantly, in
this thesis I have focused on the homeostatic mechanisms of food intake in relation to the
control of reproductive function with a specific interest in neuropeptides involved in this
regulation.
Severe metabolic challenges or hormonal deficits disrupt reproductive function at all
three levels of the HPG axis, with reduced GnRH neuronal activity as a primary cause of
reproductive impairment. To effectively synchronize reproductive effects with energy
homeostasis, neuronal populations must be receptive to peripheral hormonal cues regulating
these processes. Two key endocrine hormones, E2 and leptin, can reduce food intake by
regulating the expression and secretion of hypothalamic neuropeptides involved with feeding,
and also positively regulate the reproductive system through afferent neuronal systems that
synapse on GnRH neurons. NPY neuronal populations have emerged as key central targets of
these hormones and may integrate both their feeding and reproductive effect. Studies
completed to date demonstrate that leptin and E2 treatments decrease NPY mRNA in the
whole hypothalamus in vivo. Although, these studies do not indicate whether these NPY
neuronal populations directly respond to leptin and E2 treatments, the NPY secretory
responses to hormonal stimulation, nor do they fully elucidate the mechanisms by which this
hormonal regulation occurs. Importantly, NPY released from neurons upstream of GnRH

45
45
neurons have been demonstrated to stimulate GnRH secretion in vitro, a suspected link
between the feeding and reproductive axis. However, the transcriptional regulation of GnRH
mRNA and NPY receptor(s) and signaling kinases involved remain to be determined.
It was therefore hypothesized that NPY neurons serve as integration centres to
modulate the effects of estrogen and leptin on reproduction and food intake. This
integration at the level of the NPY neuron is likely reflected by differential responses to
these hormones with regard to NPY secretion and signaling mechanisms. These
differential signaling and secretory responses should reflect the ability of these NPY
neuronal populations to decrease food intake and stimulate GnRH neuronal
populations.
Using well-characterized hypothalamic cell lines that endogenously express key
receptors of interest and secrete basal levels of NPY and GnRH, the hypothesis was tested
through 3 specific aims (Figure 1.6):
Aim 1: Determine the receptors, signaling mechanisms and regulation of NPY
secretion by E2 in individual hypothalamic NPY-synthesizing cell lines. These results are
presented in Chapter 3.
Aim 2: Determine the NPY secretory responses to leptin in individual NPY-secreting
cell lines and elucidate potential signaling mechanisms involved. In addition, this aim will
analyze the effect of prolonged leptin exposure on NPY secretory responses and intracellular
signaling mechanisms activated. These results are discussed in Chapter 4.
Aim 3: Determine the effects of NPY on GnRH mRNA levels in the GT1-7 GnRH-
expressing neurons and identify potential signaling mechanisms and receptors involved. In
addition, this aim will examine the effect of conditioned media treatments from NPY-
synthesizing cell lines on GnRH mRNA transcript levels. These results are described in
Chapter 5.
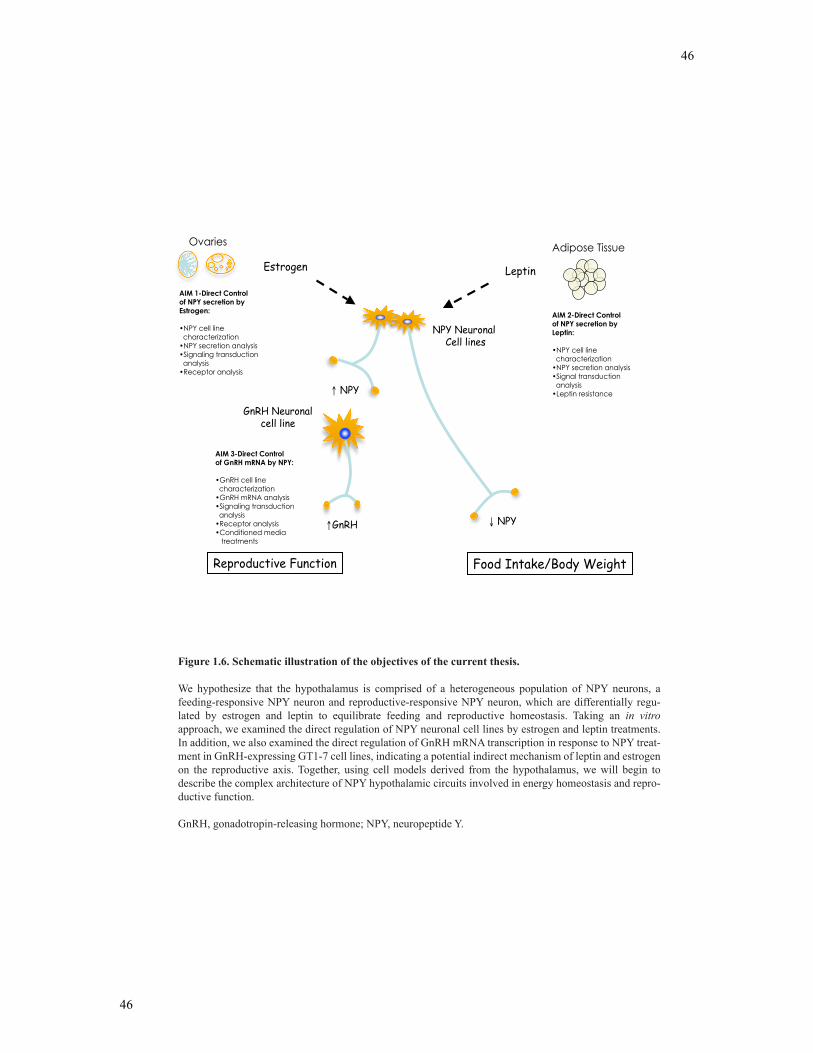
46
46
Figure 1.6. Schematic illustration of the objectives of the current thesis.
We hypothesize that the hypothalamus is comprised of a heterogeneous population of NPY neurons, a feeding-responsive NPY neuron and reproductive-responsive NPY neuron, which are differentially regu-lated by estrogen and leptin to equilibrate feeding and reproductive homeostasis. Taking an in vitro approach, we examined the direct regulation of NPY neuronal cell lines by estrogen and leptin treatments. In addition, we also examined the direct regulation of GnRH mRNA transcription in response to NPY treat-ment in GnRH-expressing GT1-7 cell lines, indicating a potential indirect mechanism of leptin and estrogen on the reproductive axis. Together, using cell models derived from the hypothalamus, we will begin to describe the complex architecture of NPY hypothalamic circuits involved in energy homeostasis and repro-ductive function.
GnRH, gonadotropin-releasing hormone; NPY, neuropeptide Y.
NPY
NPY
Food Intake/Body WeightReproductive Function
LeptinEstrogen
Ovaries Adipose Tissue
AIM 2-Direct Controlof NPY secretion byLeptin:
NPY cell linecharacterizationNPY secretion analysisSignal transductionanalysisLeptin resistance
NPY NeuronalCell lines
AIM 1-Direct Controlof NPY secretion byEstrogen:
NPY cell line characterization
NPY secretion analysisSignaling transductionanalysisReceptor analysis
GnRH Neuronalcell line
GnRH
AIM 3-Direct Controlof GnRH mRNA by NPY:
GnRH cell linecharacterizationGnRH mRNA analysisSignaling transductionanalysisReceptor analysisConditioned mediatreatments

47
47
2 Chapter 2
Materials and methods

48
48
2.1 Cell culture and reagents
Immortalized cells were grown in monolayer in Dulbecco’s modified Eagle
medium (DMEM) (Sigma-Aldrich Inc, Oakville, ON) supplemented with 5% fetal bovine
serum (FBS) (Hyclone Laboratories Inc., Logan, UT), 4.5mg/ml glucose and 1%
penicillin/streptomycin and maintained at 37oC in an atmosphere of 5% CO2 21% O2 and
74% N2. GT1-7 neurons were grown in the same conditions above, except in 10% FBS.
NPY was obtained from American Peptide (Sunnyvale, CA), while NPY receptor
specific agonists Peptide YY, D-[Trp32]-Neuropeptide Y (human, rat), [Leu31, Pro34]-
Neuropeptide Y, Neuropeptide Y (13-36), and Pancreatic Polypeptide (rat) were
obtained from Bachem (Torrance, CA). 17β-Estradiol was obtained from Sigma-Aldrich.
Additional pharmacological agents ER-α selective agonist propylpyrazole triol (PPT),
ER-ß-selective agonist diarylpropionitrile (DPN), ER-α selective antagonist methyl-
piperidino-pyrazole (MPP) and ER-ß antagonist R,R-tetrahydrochrysene (R,R)-THC
were obtained from Tocris Bioscience. β-Estradiol 6-(O-carboxy-methyl)oxime: BSA
(estrogen-BSA) was obtained from Sigma-Aldrich. Leptin was obtained from the
National Hormone and Peptide Program (Torrence, California). The MAPK family
MEK1/2 inhibitor U0126 [1,4-Diamino-2,3-dicyano-1,4-bis(o-aminophenylmercapto)
butadiene ethanolate] and PI3-K inhibitor LY294002 [2-(4-morpholinyl)-8-phenyl-
1(4H)-1-benzopyran-4-one-hydrochloride] were obtained from Cell Signaling
Technologies Inc. (Danvers, MA) and dissolved in dimethylsulfoxide (DMSO) (Sigma-
Aldrich). The selective PKA inhibitor H89 [N-[2-(p-Bromocinnamylamino)ethyl]-5-
isoquinolinesulfonamide dihydrochloride] was obtained from Sigma Aldrich and
dissolved in H20. LY294002 and UO126 were applied to neurons one hour before NPY

49
49
treatment and had final concentrations of 25 µM (optimal concentrations were determined
in previous studies) (164, 178, 231). H89 was applied in the same manner as above at a
final concentration of 30 µM (232). The NPY Y1 subtype receptor was blocked with 1 h,
1 µM pretreatments of the NPY Y1 antagonist, Diphenylacetyl-D-Arg-4-
hydroxybenzylamide (BIBP-3226) (Bachem) (233). The AMPK inhibitor Compound C
6-[4-[2-(1-Piperidinyl)ethoxy]phenyl]-3-(4-pyridinyl)-pyrazolo[1,5-a]pyrimidine
dihydrochloride was obtained from Tocris Bioscience (Ellisville, MS) and treated at a
concentration of 20 µM (234).
2.2 Semi-quantitative RT-PCR
2.2.1 One step RT-PCR
Total cellular RNA was isolated using the guanidinium isothyiocyanate phenol
choloroform extraction method. RNA was treated with Turbo DNase (Ambion, Austin,
TX) and one step RT-PCR was performed using the OneStep RT-PCR kit (Qiagen,
Mississauga, ON) according to the manufacturers protocol. The primer sequences are
listed in Table 2.1 along with the utilized annealing temperature.
2.2.2 Two step RT-PCR
Total RNA was isolated using the guanidinium isothyiocyanate phenol
choloroform extraction method. cDNA was made using the Applied Biosystems High
Capacity cDNA Reverse Transcriptase Kit (Foster City, CA). For RT-PCR, Mango Taq
polymerase was used according to the manufacturer’s protocol (Bioline, Taunton, MA).
PCR products were electrophoresed in 2% agarose gels and stained in an ethidium
bromide solution [10mg/ml ethidium bromide in 300 ml 1X TAE (Tris-acetic acid-
EDTA)]. Gels were visualized under UV light and quantified by densitometry. Primer
sequences used are listed in Table 2.1 along with the utilized annealing temperature.
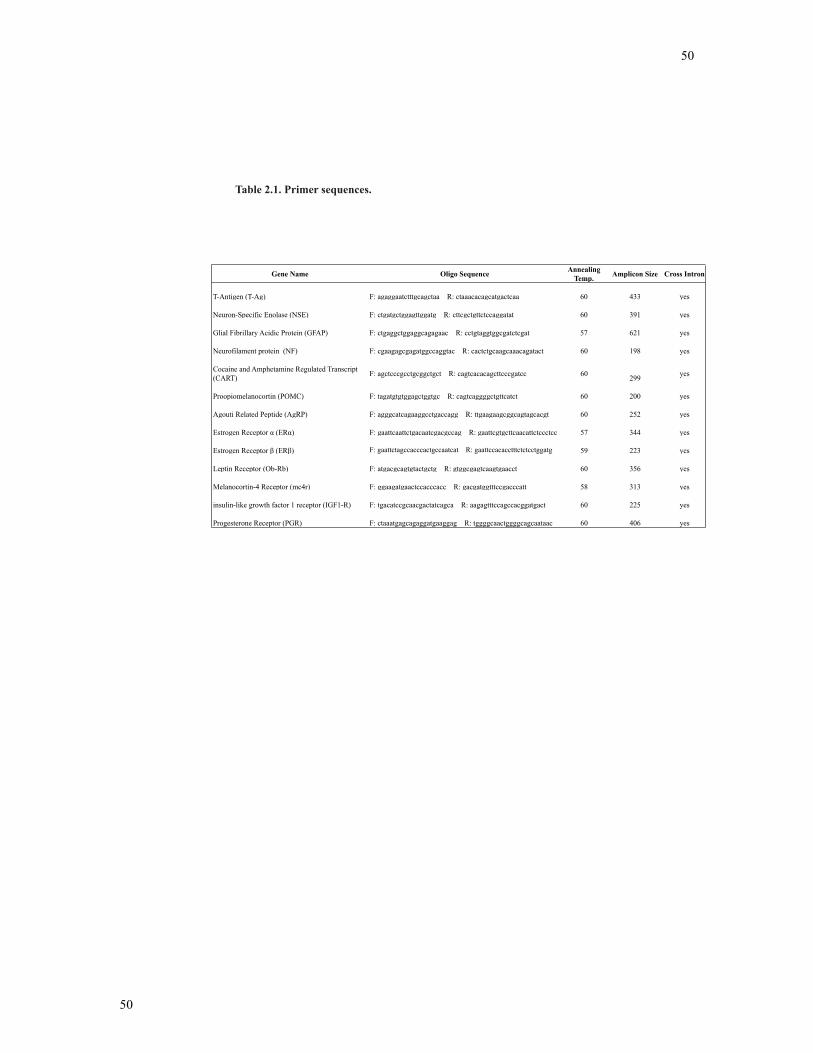
50
50
Table 2.1. Primer sequences.
Gene Name Oligo Sequence Annealing Temp. Amplicon Size Cross Intron
T-Antigen (T-Ag) F: agaggaatctttgcagctaa R: ctaaacacagcatgactcaa 60 433 yes
Neuron-Specific Enolase (NSE) F: ctgatgctggagttggatg R: cttcgctgttctccaggatat 60 391 yes
Glial Fibrillary Acidic Protein (GFAP) F: ctgaggctggaggcagagaac R: cctgtaggtggcgatctcgat 57 621 yes
Neurofilament protein (NF) F: cgaagagcgagatggccaggtac R: cactctgcaagcaaacagatact 60 198 yes
Cocaine and Amphetamine Regulated Transcript (CART) F: agctcccgcctgcggctgct R: cagtcacacagcttcccgatcc 60 299 yes
Proopiomelanocortin (POMC) F: tagatgtgtggagctggtgc R: cagtcaggggctgttcatct 60 200 yes
Agouti Related Peptide (AgRP) F: agggcatcagaaggcctgaccagg R: ttgaagaagcggcagtagcacgt 60 252 yes
Estrogen Receptor (ER ) F: gaattcaattctgacaatcgacgccag R: gaattcgtgcttcaacattctccctcc 57 344 yes
Estrogen Receptor (ER ) F: gaattctagccacccactgccaatcat R: gaattccacacctttctctcctggatg 59 223 yes
Leptin Receptor (Ob-Rb) F: atgacgcagtgtactgctg R: gtggcgagtcaagtgaacct 60 356 yes
Melanocortin-4 Receptor (mc4r) F: ggaagatgaactccacccacc R: gacgatggtttccgacccatt 58 313 yes
insulin-like growth factor 1 receptor (IGF1-R) F: tgacatccgcaacgactatcagca R: aagagtttccagccacggatgact 60 225 yes
Progesterone Receptor (PGR) F: ctaaatgagcagaggatgaaggag R: tggggcaactggggcagcaataac 60 406 yes

51
51
2.3 Real-Time RT-PCR
RNA from time-course and co-culture experiments was isolated by the guanidium
isothyiocyanate phenol choloroform extraction method. Analysis of GnRH mRNA levels
was completed using real-time RT-PCR. cDNA was made using the Applied Biosystems
High Capacity cDNA Reverse Transcriptase Kit (Foster City, CA). Real-time RT-PCR
reactions were performed with 100 ng of cDNA template using SYBR green PCR master
mix and run on the Applied Biosystems Prism 7000 real-time PCR machine. GnRH
primer sequences were as follows: GnRH SYBR sense - 5’ CGT TCA CCC CTC AGG
GAT CT -3’ : and SYBR anti-sense- 5’ CTC TTC AAT CAG ACT TTC CAG AGC -3’;
Amplicon size – 51 bp. Gamma-actin sequences are as follows: actin SYBR sense, 5’-
CTT CCC CAC GCC ATC TTG -3’ and SYBR antisense, 5’- CCC GTT CAG TCA
GAT CTT CAT -3’; Amplicon size – 79 bp. Real-time RT-PCR values were calculated
using the relative standard curve method and normalized to gamma-actin at the
corresponding time points.
2.4 Enzyme Immunoassay
In Chapter 3, mHypoE-42 and mHypoA-2/12 neurons were grown to 90%
confluency and serum starved in phenol-red free DMEM for 4 hours prior to incubation
with 10 nM E2, PPT, DPN, E2-BSA or vehicle alone for 1 hour at 37oC. 60 mM KCl were
completed for 15 minutes. KCl treatments were completed in all secretion studies as a
positive control. KCl treatment results in membrane depolarization and a large influx of
Ca(2+) for tens of minutes to induce exocytosis (235). In the pharmacological inhibitor
and ER antagonist studies, mHypoE-42 and mHypoA-2/12 neurons were treated with
vehicle or with E2 (10 nM) in the presence or absence of pharmacological inhibitors
directed against the PI3K, MAPK and AMPK pathways or ER specific antagonists MPP

52
52
and (R,R)-THC. Inhibitors and antagonists were applied for 1 hour prior to E2 treatments.
Cell suspensions were collected (in triplicate) and NPY-like immunoreactivity was
measured by an enzyme immunoassay (Phoenix Pharmaceuticals, CA) according to the
manufacturer’s protocol (assay sensitivity 0.09 ng/ml).
In Chapter 4, mHypoE-38, mHypoA-59 and NPY-GFP neurons were grown to
90% confluency and then serum starved in DMEM for 4 hours prior to incubation with
10 nM leptin or vehicle for 1 hour at 37oC. 60 mM KCl were completed for 15 minutes.
Pharmacological inhibitors used in these experiments were applied as described above.
For the leptin-resistance studies, the cells were pretreated with 10 nM leptin or vehicle
for 8 or 24 hours. At time zero, the cells were washed with 1xPBS and placed in fresh
serum-free medium for 2 hours. Cells were then re-challenged with leptin and media was
collected 8 or 24 hours later. Cell suspensions were collected (in triplicate) and NPY-like
immunoreactivity was measured by an EIA (Phoenix Pharmaceuticals, CA) according to
the manufacturer’s protocol (assay sensitivity 0.09 ng/ml).
2.5 Fluorescence-activated cell sorting (FACS)
The NPY-GFP mice (strain B6.FVB-Tg(Npy-hrGFP)1Lowl/J generated by Dr.
Bradford Lowell, Beth Israel Deaconess Medical Center, Boston, MA, and available
through The Jackson Laboratory, Bar Harbor, ME) were housed under standard viviarium
conditions in LD 12:12 light cycle with food and water available ad libitum. All
procedures were conducted in accordance with the regulations of the Canadian Council
on Animal Care and approved by the University of Toronto Animal Care Committee.
NPY-GFP transgenic mouse hypothalamii from 10-20 wk old mice were dissected
individually and stored in Hank’s balanced salt solution supplemented with 0.5 mM
EDTA and 1% bovine serum albumin (BSA). Cells were dispersed by trituration through

53
53
a 21 gauge needle and passed through 40 µM filter tubes. Cells were sorted on a BD
FACSAria cell sorter (Becton Dickinson, Franklin Lakes, NJ) with a 100-micron nozzle
tip and sheath pressure at 20 psi with a purity greater than 95%. NPY-GFP cells were
sorted by GFP fluorescence after gating to remove cell aggregates. All FACS was
completed by the Faculty of Medicine Flow Cytometry Facility, University of Toronto.
2.6 Radioactive Immunoassay
GT1-7 cells were grown to 90% confluence and cell medium was replaced with
serum-free opti-MEM containing 100 uM IBMX (Sigma-Aldrich) for 4 hours prior to
incubation with 100 nM NPY, 100 nM NPY receptor specific agonist, 30 uM forskolin or
vehicle alone for 15 min at 37oC. To extract cAMP from the cells, 1 ml of ice-cold
ethanol was added to each plate and left for 10 min. Cell suspensions were collected and
centrifuged at 20,800g and 4oC for 5 min. The supernatant was divided into 100 uL
aliquots, and were dried down using Labconco Centrivap DNA concentrator for 3-5
hours. cAMP levels were determined by radioimmunoassay (RIA) (Biomedical
Technologies Inc, MA) according to the manufacturers protocol.
2.7 Western Blot Analysis
In Chapter 5, GT1-7 were treated with 100 nM NPY, 30 uM forskolin or vehicle
alone for a 1 h time course. Cells were washed with ice-cold PBS and lysed in a high-salt
buffer (20 mM Tris hCl, 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% Triton, 2.5
mM sodium pyrophosphate, 1 mM β-glycerophosphate ,1 mM Na3VO4, 1 mM leupeptin)
(Cell Signaling Technology, MA) supplemented with 1% protease cocktail inhibitor (Cell
Signaling Technology) and 1 mM PMSF. Cell lysates were centrifuged at 20,800g for 10
min at 4oC and the collected supernatant was stored at -80oC. Protein concentration was
determined using the bicinchonicnic acid (BCA) protein assay kit (Pierce Biotechnology,

54
54
IL). Total protein (50 µg) was resolved on a 8% SDS-PAGE and blotted onto immuno-
Blot PVDF membrane (Bio-Rad Laboratories, Hercules, CA). Membranes were blocked
with 5% BSA (Sigma) in Tris-buffered saline containing 0.2% Tween 20 (TBS-T),
supplemented with a phosphatase inhibitor cocktail (Sigma) for 30 to 60 min and then
washed 3 times with TBS-T. Blots were then incubated with primary antibodies against
phospho-AKT (Ser473, 1:1000 Cell Signaling technology), phospho-ERK
(Thr202/Tyr204, 1:1000: Cell Signaling Technology) phospho-CREB (Ser133, 1:1000:
Cell Signaling Technology), phospho-PKA (Thr197, 1:1000: Cell Signaling Technology)
or Gβ (1:5000: Santa Cruz Biotechnology Inc, Santa Cruz, CA) overnight at 4 oC
followed by 3 washes in TBS-T and then incubated with secondary horseradish
peroxidase-labeled secondary goat anti-rabbit at 1:5000 dilution for 1-2 hours at room
temperature. Membranes were visualized with enhanced chemiluminescence (ECL kit,
GE Healthcare, UK) on the Kodak Imager 2000R.
In Chapter 4, mHypoE-38, mHypoA-59 and NPY-GFP neurons were treated with
leptin (10 nM) or vehicle alone over a 30-minute time course and protein was harvested
as described above and blotted onto PVDF membrane as described above. For leptin
resistance experiments, cells were pretreated with 10 nM leptin for 8 and 24 hours before
leptin treatment media was changed to serum-free DMEM for 2 hours. Cells were re-
challenged with 10 nM leptin and protein was isolated after 15 and 30 minutes. Blots
were then incubated with primary antibodies phospho-JAK2 (Tyr1007/1008, 1:1000, Cell
Signaling technology), phospho-AMPK (Thr172, 1:1000, Cell Signaling Technology),
phospho-ACC (Ser79, 1:1000, Cell Signaling Technology), phospho-STAT3 (Tyr705,
1:1000, Cell Signaling Technology), phospho-ERK1/2 (Thr202/Tyr204) 1:1000, Cell
Signaling Technology), phospho-JNK (Thr183/Tyr185, 1:1000, Cell Signaling

55
55
Technology), phospho-CREB (Ser133, 1:1000, Cell Signaling Technology), phospho-
AKT (Ser473, 1:1000, Cell Signaling Technology), Gβ (1:5000, Santa Cruz
Biotechnology Inc, Santa Cruz, CA) or leptin receptor (SC-1834, 1:1000, Santa Cruz
Biotechnology Inc) overnight at 4 oC followed by 3 washes in TBS-T. Blots were then
treated as illustrated above.
For Western blot studies, phospho-proteins were normalized to the loading
control G-beta. Although total protein comparisons are ideal, our group has found in
previous studies that Gbeta is a reliable indicator of loading status (164, 178, 236-241).
2.8 Immunocytochemistry
In Chapter 3, mHypoE-42 and mHypoA-2/12 cells were plated on eight-well
chamber slides (BD Biosciences) in DMEM overnight. Cells were fixed in 2%
paraformaldehyde, blocked with 1% BSA–PBS and incubated with primary antibody
overnight at 4°C. No antibody wells served as controls. The primary ER-α antibody was
used at a 1:50 1% BSA–PBS dilution of mouse monoclonal anti-human for ER-α
(DakoCytomation). For colocalization, primary antibody for rabbit polyclonal caveolin-1
(Santa Cruz Biotechnology) was used at a 1:50 dilution. Cells were washed with PBS and
incubated with a FITC-conjugated AffiniPure goat anti-mouse secondary antibody
(Jackson ImmunoResearch) in a 1:100 dilution at room temperature for 90 min. Cells
were then washed with PBS and incubated with rhodamine-labeled (Texas Red)
biotinylated goat anti-rabbit IgG secondary antibody (Vector Laboratories) in a 1:100
dilution at room temperature for 30 min. After washing cells with PBS, gaskets were
removed from the chamber slides and mounted with DakoCytomation fluorescent
mounting media. Fixed cells were then visualized with a confocal laser scanning
microscope at a magnification of 400x (LSM 510; Carl Zeiss). FITC fluorescence was

56
56
excited by the 488 nm argon laser line, whereas rhodamine was excited by the 543 nm
helium–neon laser line.
2.9 Co-culture
In Chapter 5, NPY-containing media from mHypoE-38 neurons grown to 80-90%
confluence was harvested from 60 mM KCl or vehicle treated cells. Media were
collected after treatment and run through a Zeba de-salting spin column (Thermo
Scientific, IL). As a control, GT1-7 neurons were treated with KCl- or vehicle-treated
medium from mHypoE-38 neurons and RNA from GT1-7 neurons was harvested after 4
hours and subjected to real-time RT-PCR, as described above.
2.10 Statistics
Data are presented as the mean ± the standard error of the mean (SEM). Data
were analyzed by one- or two-way analysis of variance (ANOVA) followed by a
Bonferroni multiple comparisons test or by a Student’s t-test using Graphpad Prism
(Graphpad Software Inc., CA), as indicated in the figure legends, with the exception of
the three-way ANOVA. Three-way ANOVAs were performed on SigmaStat version 2.01
for Windows (Jandel Scientific, San Rafael, CA). Experiments were performed on three
to twelve separate occasions. Comparisons were considered statistically significant at p
<0.05.

57
57
Chapter 3
3 17β-estradiol inhibits NPY secretion through membrane-associated estrogen receptor
(ER)-α in clonal, immortalized hypothalamic neurons
Manuscript in press within the International Journal of Obesity.
Citation:
Estrogen inhibits NPY secretion through membrane-associated estrogen receptor (ER)-α in clonal, immortalized hypothalamic neurons Dhillon SS, Belsham DD. Int J Obes. Accepted May 12 2010 Manuscript # 2010IJO00022RR
Contributions:
• SSD completed experiments and wrote the manuscript • DDB edited the manuscript and provided scientific input, direction and funding

58
58
3.1 Abstract
17β-estradiol (E2) has an inhibitory effect on food intake by acting centrally in the
hypothalamus; although it is not clear which neuronal cell types are functionally required
for this effect. Previous studies from our lab and others have implicated NPY neurons as
an important central anorectic target of E2. The present study was designed to investigate
whether E2 can directly regulate NPY secretion and examine the cellular mechanisms and
receptors responsible for this anorexigenic action of E2. Clonal, murine, hypothalamic
neuronal cell models, mHypoE-42 and mHypoA-2/12, were investigated for NPY
secretory responses to E2 in the presence or absence of pharmacological inhibitors
directed against the PI3K, MAPK and AMPK pathways or to ER specific
agonists/antagonists. E2 significantly decreased NPY secretion in both the mHypoE-42
and mHypoA-2/12 neurons. The E2-mediated repression of NPY secretion in the
mHypoE-42 and mHypoA-2/12 neurons required ER-α, but not ER-β, as demonstrated
by studies using ER-specific agonist/antagonists. Additionally, using
immunocytochemistry (ICC) I detected colocalization of ER-α and the cell membrane-
associated scaffold protein caveolin-1. Importantly, using E2-conjugated BSA (E2-BSA)
and ER antagonists, I was able to demonstrate that the E2-mediated decrease in NPY
secretion occurred through cell membrane-bound ER-α. Finally, using a combination of
pharmacological inhibitors, I found that inhibition of the PI3K or AMPK pathway
blocked the E2-mediated decrease in NPY secretion. These findings indicate that the
central anorectic action of E2 occurs at least partially through hypothalamic NPY-
synthesizing neurons. This regulation of NPY secretion occurs through non-genomic
signaling mechanisms and possibly through cell membrane-bound ER-α.

59
59
3.2 Introduction
E2 is thought to negatively regulate energy homeostasis through two pathways: an
anorectic action through the central nervous system and a direct action on tissue
metabolism (242). The central action of E2 is hypothesized to occur through the
regulation of multiple orexigenic and anorexigenic neuropeptides implicated in feeding
behaviour (243-245). A large body of evidence suggests NPY is involved with the E2-
mediated decrease in feeding behaviour (245). NPY is a potent orexigenic peptide, as the
central administration of NPY stimulates feeding and repeated doses results in an
increase in body weight (107, 246). Interestingly, the withdrawal of E2 by OVX in mice
results in significantly greater levels of NPY mRNA expression in the ARC (20, 245).
This effect was reversed by E2 administration. Additional studies found E2 significantly
decreased the sensitivity to the orexigenic effect of NPY (247). Finally, E2 treatment
resulted in a decrease in NPY release from microdissected hypothalamic sites and PVN
cultures (244). Although E2 appears to regulate NPY mRNA and release, whether E2 acts
directly on NPY neurons or solely through afferent neuronal connectivity to influence
NPY levels remains to be determined.
The actions of E2 are thought to be primarily mediated through two specific
nuclear receptor isoforms, ER-α and ER-β (117, 125). Studies undertaken to delineate the
ER subtype responsible for the central anorexigenic action of E2 have produced
conflicting data, implicating both ER-α and ER-β as the main receptor subtype. A study
of food intake and body weight demonstrated systemic injections of E2 or the ER-α
agonist 4,4',4''-(4-propyl-[1H]-pyrazole-1,3,5-triyl)trisphenol (PPT) significantly reduced
food intake, but not the ER-β agonist 2,3-bis(4-hydroxyphenyl)-propionitrile (DPN)
(248). In contrast, ICV injection of E2 and anti-sense oligonucleotides (ODN) directed

60
60
against ER-β attenuated the E2-mediated decrease in food intake, whereas ER-α ODN
had no effect (249). Thus, it remains unclear whether E2 acts through ER-α or ER-β in
the hypothalamus to influence food intake. To complicate matters, recent studies have
suggested NPY neurons do not express ER-α in the hypothalamus (171). In this study, I
confirmed the presence of ER-α in NPY-GFP FAC-sorted cells.
The aim of the current study is to determine the secretory events, rapid signal
transduction cascades and the E2 receptor subtype responsible for the anorectic action of
E2 on NPY-expressing neurons. To examine the mechanisms by which E2 regulates NPY
within an individual NPY-expressing neuron, I used hypothalamic neuronal cell models,
mHypoE-42 (219) and mHypoA-2/12 (220), recently generated by our lab. These cell
lines have been thoroughly characterized, demonstrate neurosecretory properties, express
neuron specific markers and have classical neuronal morphology (216, 230). In the
present study, I demonstrate that 17β-estradiol (E2) directly decreases NPY secretion in
both the mHypoE-42 and mHypoA-2/12 neurons. Using pharmacological ER-α and ER-
β receptor specific agonists/antagonists, I was able to determine the E2-mediated decrease
occurred via ER-α in both hypothalamic cell lines. Additionally, I detected the
colocalization of ER-α and the membrane-associated signaling protein caveolin-1. I
found this cell membrane-bound ER-α to be directly involved in the E2-mediated
decrease in NPY secretion using a combination of E2-BSA and ER antagonists. Finally, I
provide evidence that the PI3K and AMPK signaling pathways play an important role in
the E2 regulation of NPY secretion.

61
61
3.3 Results
3.3.1 Expression of ER-α in FAC-sorted NPY-GFP neurons
A recent study by Olofsson et al. demonstrated ER-α is not expressed by NPY
neurons of the hypothalamus in vivo (171). This result is in contrast to previous studies
that colocalized ER-α in NPY neurons. As a result, it is currently unclear whether NPY
neurons express ER-α and are a direct target of E2 in vivo. Thus I sought to determine
whether NPY neurons express ER-α in vivo using transgenic mice that express
humanized Renilla-GFP (hrGFP) driven by the NPY promoter. NPY-GFP neurons were
FAC-sorted (96% accuracy in sorting for GFP fluorescence) from the hypothalamus and
ER-α expression was measured/detected using RT-PCR (Figure 3.1). I confirmed the
expression of ER-α in NPY neurons, which suggests E2 may directly act on NPY neurons
in vivo.
3.3.2 Expression of the ER subtypes and other hypothalamic markers in mHypoE-
42 and mHypoA-2/12 neurons
The mHypoE-42 and mHypoA-2/12 neurons display neuronal morphology in
culture. A partial list of markers expressed in the cell lines is presented (Figure 3.2A),
including neuropeptides, receptors, and signaling molecules. RNA isolated from the
mouse hypothalamus was used as a positive control. The presence of these receptors and
the expression and secretion of NPY (Figure 3.2B) at appreciable levels indicates that the
mHypoE-42 (NPY concentration: 0.55±0.034 ng/mL) and mHypoA-2/12 (NPY
concentration: 0.77±0.027 ng/mL) neurons have the appropriate machinery to respond to
E2 and are suitable models to study the E2 mediated regulation of NPY secretion. Media
from GT1-7 neurons was used as a negative control as they do not synthesize NPY.

62
62
Figure 3.1. Expression of ER- mRNA in NPY-GFP neurons using RT-PCR
NPY-GFP neurons were isolated from the hypothalamus of the NPY-GFP transgenic mouse using fluorescence-activated cell sorting (FACS). Hypothalamus, Hypo; no-template control, NTC. ER- amplicon size: 344 bp.
DN
A la
dder
Hyp
o
NPY
-GFP
cells
NTC
DN
A la
dder
500 bp
250 bp

63
63
3.3.3 Regulation of NPY secretion by E2 in mHypoE-42 and mHypoA-2/12 neurons
Although evidence indicates that E2 can regulate NPY mRNA and release in vivo,
it is not yet known whether this regulation can occur directly at the level of the NPY
neuron (164, 178). mHypoE-42 and mHypoA-2/12 neurons were exposed to E2 (10 nM)
for 1 h. This dose was found to be the optimal treatment concentration in our cell models
in previous studies (164). As a control to ensure accurate NPY measurements, cells were
treated with the well-characterized depolarizing agent, KCl. 60 mM KCl treatments
resulted in a ~1.5-fold increase in NPY secretion in both mHypoE-42 and mHypoA-2/12
neurons. Analysis of the results in the embryonic mHypoE-42 neurons indicates that
NPY secretion was significantly reduced by E2 (vehicle, 1.02±0.085; E2, 0.83±0.10)
(Figure 3.3A). Similarly, in the adult hypothalamic cells, mHypoA-2/12, NPY secretion
was significantly inhibited by E2 exposure (vehicle, 1.09±0.0381; E2, 0.78±0.052)
(Figure 3.3B).
3.3.4 E2-mediated regulation of NPY secretion is dependent on ER-α in the
mHypoE-42 and mHypoA-2/12 neurons
I next examined the role of ER-α and ER-β in the E2-mediated down-regulation
of NPY secretion. The effects of vehicle, E2, ER-α selective agonist PPT, ER-ß-selective
agonist DPN, ER-α selective antagonist MPP and ER-ß antagonist (R,R)-THC on NPY
secretion are shown in Figure 3.4. In the mHypoE-42 neurons, 1 h 10 nM PPT treatment
(0.72±0.033) reduced NPY secretion compared to vehicle control (1.0±0.034) (Figure
3.4A). In agreement with the embryonic cell lines, the mHypoA-2/12 PPT treatment
group (0.61±0.11) had a significant reduction in NPY secretion when compared to the
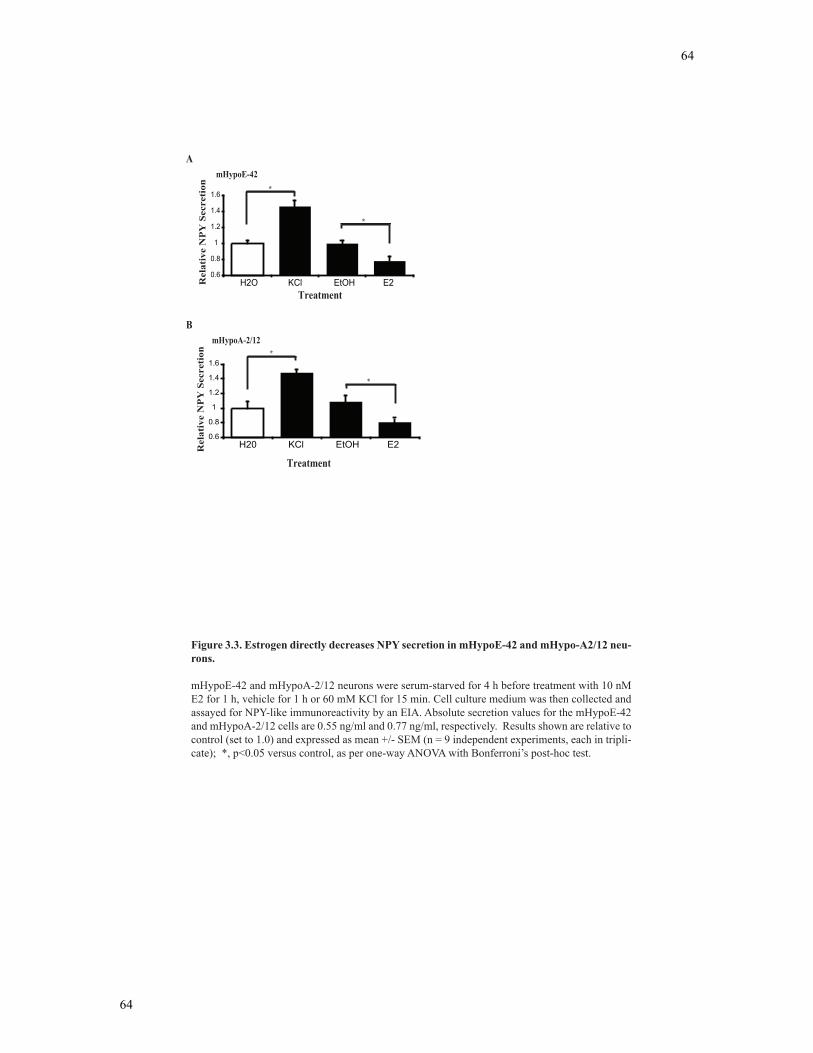
64
64
0.6
0.8
1
1.2
1.4
1.6
H20 KCl EtOH E2
0.6
0.8
1
1.2
1.4
1.6
H2O KCl EtOH E2
noi terce SYPNe vi tal e
R
Treatment
mHypoE-42
mHypoA-2/12
*
*
*
*noiterceS
YPNe vi tal e
R
Treatment
A
B
Figure 3.3. Estrogen directly decreases NPY secretion in mHypoE-42 and mHypo-A2/12 neu-rons.
mHypoE-42 and mHypoA-2/12 neurons were serum-starved for 4 h before treatment with 10 nM E2 for 1 h, vehicle for 1 h or 60 mM KCl for 15 min. Cell culture medium was then collected and assayed for NPY-like immunoreactivity by an EIA. Absolute secretion values for the mHypoE-42 and mHypoA-2/12 cells are 0.55 ng/ml and 0.77 ng/ml, respectively. Results shown are relative to control (set to 1.0) and expressed as mean +/- SEM (n = 9 independent experiments, each in tripli-cate); *, p<0.05 versus control, as per one-way ANOVA with Bonferroni’s post-hoc test.

65
65
vehicle control group (1.0±0.059) (Figure 3.4B). In contrast to PPT treatments, the ER-β
agonist DPN failed to elicit any changes in NPY secretion in both cell lines. Additionally,
ER-α antagonist MPP (1 µM) blocked the E2-mediated decrease in NPY secretion in both
the mHypoE-42 (E2, 0.83±0.10; MPP+E2, 1.02±0.074) (Figure 3.4C) and mHypoA-2/12
(E2, 0.78±0.052; MPP+ E2, 0.98±0.042) (Figure 3.4D) neurons. Conversely, in the
presence of the ER-β antagonist (R,R)-THC (1 µM), E2 still reduced NPY secretion in
both the mHypoE-42 ((R,R)-THC, 0.97±0.0179; (R,R)-THC+E2, 0.81±0.11) and the
mHypoA-2/12 ((R,R)-THC, 1.03±0.19; (R,R)-THC+E2, 0.72±0.084) (Figure 3.4E and
3.4F). Importantly, ER antagonist treatments alone did not significantly change NPY
release levels compared to controls. ER agonist/antagonist concentrations used in this
study are optimal treatment concentrations established in previous studies (136, 248, 250,
251). These results indicate that activation of ER-α mediates the effects of E2 on the
repression of NPY secretion.
3.3.5 E2 decreases NPY secretion via membrane-bound ER-α
There is some evidence that classical nuclear ER-α can localize to the membrane
and lead to the activation of intracellular signaling cascades (non-genomic E2 signaling)
(116, 117, 158). I attempted to detect ER-α at the level of the cell membrane by
colocalizing ER-α with the cell membrane protein caveolin-1. Using ICC with ER-α-and
caveolin-1-specific antibodies, I found ER-α could be colocalized at the cell membrane
(Figure 3.5). In order to implicate the membrane bound ER-α in the E2-mediated
decrease in NPY secretion, I used cell membrane impermeable E2-BSA in the presence of
ER antagonists MPP and (R,R)-THC. Interestingly, I observed that E2-BSA failed to
decrease NPY secretion in the presence of the ER-α antagonist, MPP (1 µM), in both
embryonic (MPP+E2-BSA, 1.01±0.032) (Figure 3.6A) and adult (MPP+E2-BSA,
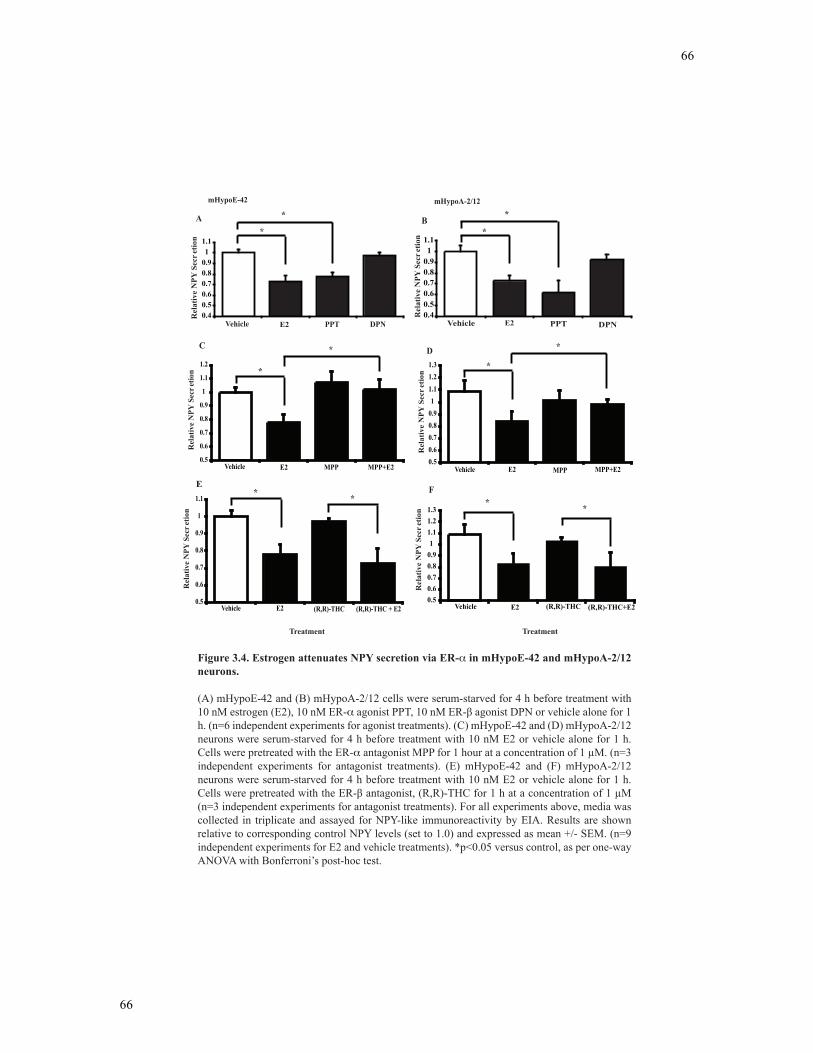
66
66
Figure 3.4. Estrogen attenuates NPY secretion via ER- in mHypoE-42 and mHypoA-2/12 neurons.
(A) mHypoE-42 and (B) mHypoA-2/12 cells were serum-starved for 4 h before treatment with 10 nM estrogen (E2), 10 nM ER-
neurons were serum-starved for 4 h before treatment with 10 nM E2 or vehicle alone for 1 h. Cells were pretreated with the ER- antagonist MPP for 1 hour at a concentration of 1 !M. (n=3 independent experiments for antagonist treatments). (E) mHypoE-42 and (F) mHypoA-2/12 neurons were serum-starved for 4 h before treatment with 10 nM E2 or vehicle alone for 1 h.
(n=3 independent experiments for antagonist treatments). For all experiments above, media was
independent experiments for E2 and vehicle treatments). *p<0.05 versus control, as per one-way
0.40.50.60.70.80.91
1.1
Vehicle PPT DPN
mHypoE-42
*
*
0.40.50.60.70.80.91
1.1
Vehicle PPT DPN
mHypoA-2/12
A B
Rel
ativ
e N
PY S
ecre
tion
Rel
ativ
e N
PY S
ecre
tion
Rel
ativ
e N
PY S
ecre
tion
C D
F
Treatment
E2 E2
0.5
0.6
0.7
0.8
0.9
1
1.1
1.2
Vehicle E2 MPP MPP+E2
*
*
*
0.5
0.6
0.7
0.8
0.9
1
1.1
Vehicle E2 (R,R)-THC (R,R)-THC + E2
**
E
0.50.60.70.80.91
1.11.21.3
Vehicle E2 (R,R)-THC (R,R)-THC+E2
**
Rel
ativ
e N
PY S
ecre
tion
0.50.60.70.80.91
1.11.21.3
Vehicle E2 MPP MPP+E2
Rel
ativ
e N
PY
Sec
reti
onR
elat
ive
NPY
Sec
retio
n
Treatment
*
*
*

67
67
0.98±0.042) (Figure 3.6B) hypothalamic cell lines. However, E2-BSA decreased NPY
secretion in the presence of the ER-β antagonist (R,R)-THC (1 µM) in the mHypoE-42
((R,R)-THC, 0.97±0.0179; (R,R)-THC+E2-BSA, 0.82±0.0808) (Figure 3.6C) and the
mHypoA-2/12 ((R,R)-THC, 1.03±0.019; (R,R)-THC+E2-BSA, 0.88±0.082) (Figure
3.6D) cell lines. Therefore, it is evident that membrane-bound ER-α mediates the effect
of E2 on NPY secretion.
3.3.6 Inhibition of the PI3K and AMPK pathways affect the E2-mediated
regulation of NPY secretion
I next determined whether the regulation of NPY by E2 is dependent on non-
genomic signal transduction pathways, MAPK, PI3K and AMPK, using pharmacological
inhibitors specific to these pathways. Neurons were treated with or without E2 (10 nM) in
the presence or absence of pharmacological inhibitors LY294002 (25 µM), UO126 (25
µM) or Compound C (20 µM). As shown in Figure 3.7A and 3.7B, the E2-mediated
decrease in NPY was blocked in the presence of the AMPK inhibitor, Compound C
dihydrochloride, in both the mHypoE-42 (E2, 0.83±0.10; Compound C+E2, 1.05±0.075)
and the mHypoA-2/12 (E2, 0.78±0.052; Compound C+E2, 1.03±0.030) neurons. Next, I
found E2 co-treatment with the PI3K inhibitor, LY294002, also prevented the E2-
mediated decrease in NPY secretion in both the embryonic (E2, 0.83±0.10;
LY294002+E2, 0.98±0.059) and adult (E2, 0.78±0.052; LY294002+E2, 1.09±0.018) cell
lines. In addition, I found that U0126 treatment, a MEK1/2 inhibitor, attenuated the E2-
mediated decrease in NPY secretion, but this effect was not found to be statistically
significant. Importantly, the pharmacological inhibitor treatments alone had no affect on
basal NPY secretory levels. Together, our data indicates that activation of the AMPK and
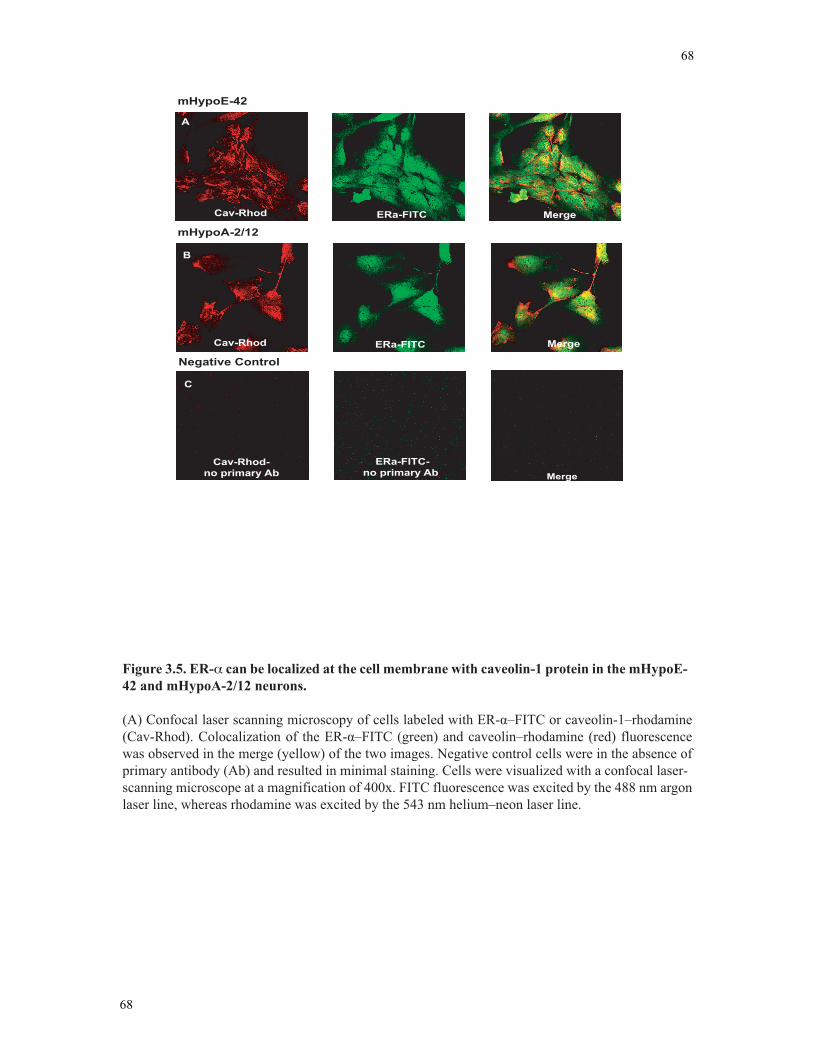
68
68
A
Cav-Rhod ERa-FITC Merge
mHypoE-42
Cav-Rhod ERa-FITC Merge
mHypoA-2/12
B
Merge Cav-Rhod- no primary Ab
ERa-FITC- no primary Ab
Negative Control
C
Figure 3.5. ER-! can be localized at the cell membrane with caveolin-1 protein in the mHypoE-42 and mHypoA-2/12 neurons.
(A) Confocal laser scanning microscopy of cells labeled with ER-!–FITC or caveolin-1–rhodamine (Cav-Rhod). Colocalization of the ER-!–FITC (green) and caveolin–rhodamine (red) fluorescence was observed in the merge (yellow) of the two images. Negative control cells were in the absence of primary antibody (Ab) and resulted in minimal staining. Cells were visualized with a confocal laser-scanning microscope at a magnification of 400x. FITC fluorescence was excited by the 488 nm argon laser line, whereas rhodamine was excited by the 543 nm helium–neon laser line.
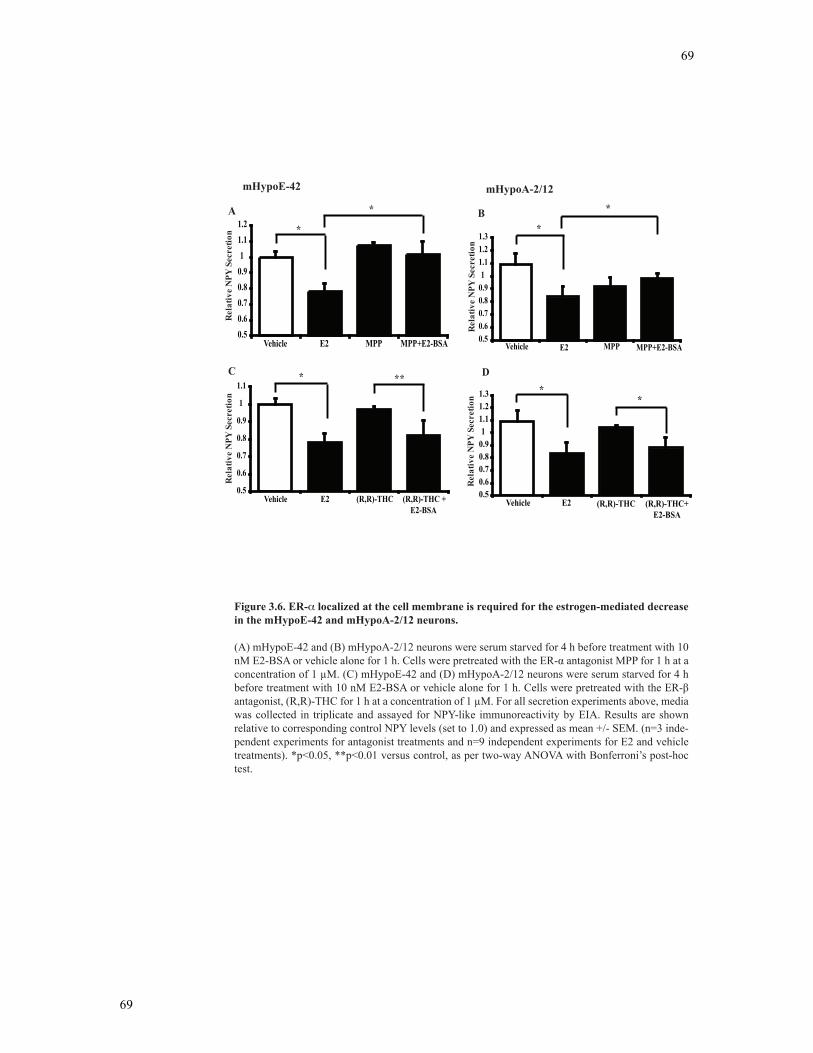
69
69
RelativeNPYSecretion
A B
C D
0.5
0.6
0.7
0.8
0.9
1
1.1
Vehicle E2 (R,R)-THC (R,R)-THC + E2-BSA
0.50.60.70.80.91
1.11.2
Vehicle E2 MPP MPP+E2-BSA
RelativeNPYSecretion
RelativeNPYSecretion
0.50.60.70.80.91
1.11.21.3
Vehicle E2 (R,R)-THC (R,R)-THC+ E2-BSA
RelativeNPYSecretion
0.50.60.70.80.91
1.11.21.3
Vehicle E2 MPP MPP+E2-BSA
*
* *
*
* **
*
*
Figure 3.6. ER- localized at the cell membrane is required for the estrogen-mediated decrease in the mHypoE-42 and mHypoA-2/12 neurons.
(A) mHypoE-42 and (B) mHypoA-2/12 neurons were serum starved for 4 h before treatment with 10
concentration of 1 !M. (C) mHypoE-42 and (D) mHypoA-2/12 neurons were serum starved for 4 h
-
test.
mHypoE-42 mHypoA-2/12
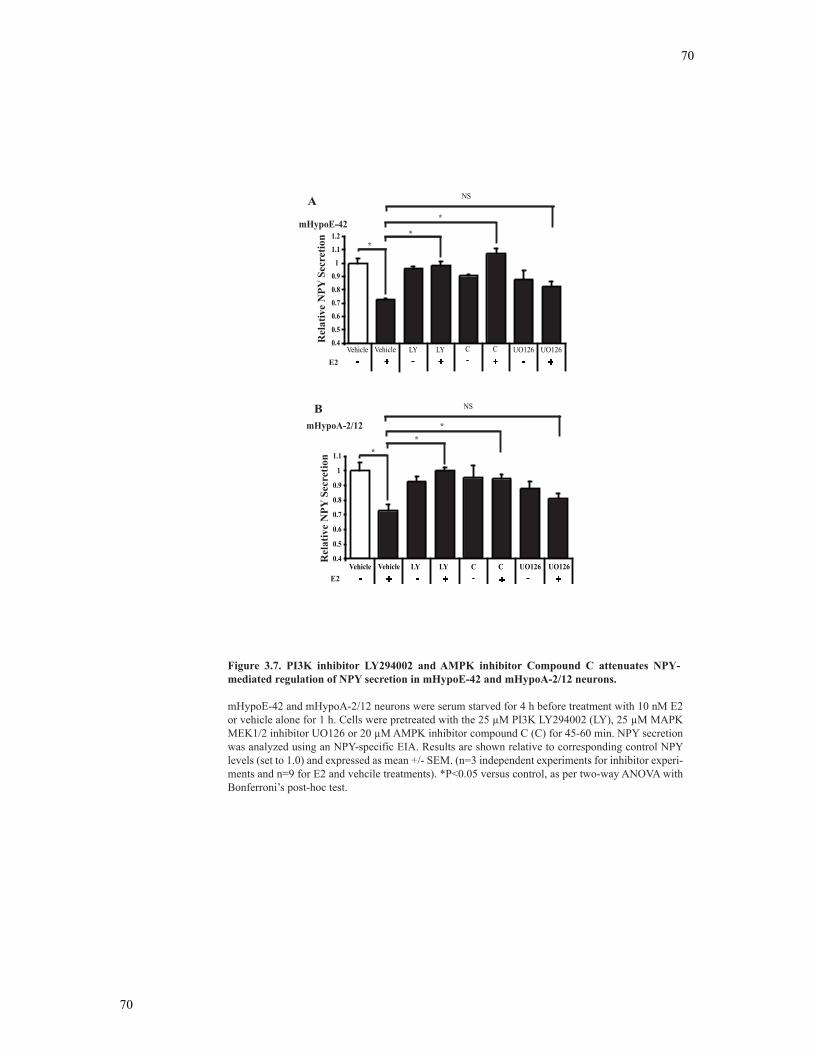
70
70
0.4
0.5
0.6
0.7
0.8
0.9
1
1.1
1.2
Vehicle Vehicle LY LY C C UO126 UO126E2 - + + + +- - -
Rel
ativ
e N
PY S
ecre
tion
**
*mHypoE-42
0.4
0.5
0.6
0.7
0.8
0.9
1
1.1
Relative NPY Secretion
Vehicle Vehicle LY LY C C UO126 UO126E2 - - - -+ + + +
mHypoA-2/12
**
Rel
ativ
e N
PY S
ecre
tion
Figure 3.7. PI3K inhibitor LY294002 and AMPK inhibitor Compound C attenuates NPY-mediated regulation of NPY secretion in mHypoE-42 and mHypoA-2/12 neurons.
mHypoE-42 and mHypoA-2/12 neurons were serum starved for 4 h before treatment with 10 nM E2 or vehicle alone for 1 h. Cells were pretreated with the 25 !M PI3K LY294002 (LY), 25 !M MAPK MEK1/2 inhibitor UO126 or 20 !M AMPK inhibitor compound C (C) for 45-60 min. NPY secretion was analyzed using an NPY-specific EIA. Results are shown relative to corresponding control NPY levels (set to 1.0) and expressed as mean +/- SEM. (n=3 independent experiments for inhibitor experi-ments and n=9 for E2 and vehcile treatments). *P<0.05 versus control, as per two-way ANOVA with Bonferroni’s post-hoc test.
*
A
B
NS
NS

71
71
PI3K pathways are critical for E2 to decrease NPY secretion in mHypoE-42 and
mHypoA-2/12 neurons.
3.4 Discussion
E2 is a well-known negative regulator of energy balance, acting peripherally to
increase metabolic activity and centrally to reduce food intake. The experiments
completed in this study were inspired by previous work from our lab that demonstrated
E2 decreased NPY mRNA expression in the mHypoE-38 and mHypoE-42 neurons (164,
178). Interestingly, the NPY mRNA response to E2 treatment observed in the neuronal
cell lines depended on the ratio of ER-α and ER-β. Where ER-β was linked to the
induction of NPY mRNA expression by E2 and ER-α was responsible for the reduction in
NPY mRNA (164). Additional studies using pharmacological inhibitors found the E2-
mediated decrease in NPY mRNA observed at 8 hours required both the PI3K and
MAPK pathway (178). Our findings extend these studies by providing the first evidence
that E2 exposure can rapidly regulate NPY secretion. I confirmed the expression of ER-α
in NPY neurons using FAC-sorted NPY-GFP neurons. Next, using clonal, immortalized,
hypothalamic, NPY-expressing cell lines I investigated the direct regulation of the
orexigenic neuropeptide NPY as a central target for E2. One hour E2 exposure in both the
embryonic mHypoE-42 and adult mHypoA-2/12 hypothalamic cell lines resulted in a
~20% decrease in NPY secretion. I propose the E2-mediated decrease in NPY occurs
through ER-α, and not ER-β. Further, I found that ER-α can be localized to the cell
membrane in our hypothalamic cell lines, and that this membrane-bound receptor was
responsible for the reduction in NPY secretion. Finally, through the use of
pharmacological inhibitors, I show that the PI3K and AMPK pathways are necessary for

72
72
the E2-mediated decrease in NPY secretion. This study adds to the growing list of
evidence that E2 can regulate NPY levels to modulate feeding behaviour.
E2 acts a homeostatic feedback molecule between the periphery and the brain
regulating reproduction and energy homeostasis (162, 217). E2 has a verified central
anorectic role to suppress hyperphagia, as ICV E2 treatment reduces food intake in OVX
mice (172, 252, 253). This anorexigenic effect could occur through the modulation of
several orexigenic and anorexigenic signals. One study found that the castration of
female mice resulted in a decrease in gene expression of the anorexigenic neuropeptides
POMC and CRH, which normalized after 12 and 24-hour E2 injections (245, 254).
Another study found E2 was required for normal action of the gut-derived peptides CCK
and ghrelin, the adipocyte-derived hormone leptin, and hypothalamic neuropeptide MCH
on satiety signaling in the hypothalamus (175, 176, 247). NPY can be added to the
growing list of neuropeptides and hormones that are regulated by E2. OVX mice have
increased NPY mRNA levels in the ARC and E2 treatment in the PVN results in a
decrease in NPY release (20, 244). Finally, using hypothalamic neuronal cell lines, our
lab has previously shown E2 can directly decrease NPY mRNA as a potential
anorexigenic mechanism of E2 in the hypothalamus (164). Our study corroborates these
gene expression studies and provides evidence that E2 exposure to hypothalamic
embryonic and adult cell lines can rapidly decrease NPY secretion. Our findings present
the first demonstration that the anorexigenic action of E2 can act directly through
hypothalamic NPY-synthesizing neurons by reducing NPY release.
ER-α and ER-β are widely distributed throughout the hypothalamus, with both
receptor subtypes present in the ARC and POA (14, 16). Although ER-α and ER-β show
considerable homology in their DBD and carboxyl-terminal LBD domains, they are

73
73
products of different genes and studies have indicated that each ER may have a unique
physiological role in a tissue- and cell context-dependent manner (122). Studies to date
have produced conflicting evidence in identifying the ER that is largely responsible for
the central anorectic role of E2. One report demonstrated the inhibitory effect of E2 on
feeding was blocked with the ICV administration of ODN directed against ER-β, but not
ODN directed against ER-α (249). However, the ER-α knockout mice display an obese
phenotype with increased food intake and decreased energy expenditure, implicating the
ER-α subtype in the regulation of feeding (173, 176). Conversely, ER-β knockout mice
display normal energy intake and expenditure (114). Additional studies found the ER-α
agonist, PPT, but not ER-β agonist, DPN, reduced total food intake in OVX rats (255).
Previous studies in our lab found ER-α to be critical in the decrease in NPY mRNA;
however, changes in mRNA expression may not translate into changes in protein content
(164). To examine which ER subtype is responsible for the E2-mediated reduction in
NPY secretion, I treated our neuronal cell lines with ER agonists PPT and DPN, and ER
antagonists MPP and (R,R)-THC. I found the ER-α agonist PPT significantly decreased
NPY secretion in both the mHypoE-42 and mHypoA-2/12 neurons, whereas DPN failed
to invoke a change in NPY secretion. These results were further corroborated by the
results of the ER antagonist studies. The ER-α antagonist MPP blocked the E2-mediated
decrease in NPY secretion, whereas the ER-β antagonist (R,R)-THC, did not. Although I
do not implicate ER-β in the regulation of NPY secretion, it does not rule out the
possibility that ER-β can regulate other key feeding-related hypothalamic neuropeptides.
Accordingly, these studies propose that E2 invokes an anorectic response by decreasing
NPY directly via ER-α in the NPY cell models.

74
74
Recent studies have shown E2 can mediate non-genomic signaling cascades
through a subpopulation of classical ERs located at the plasma membrane (256, 257).
Caveolae are thought to facilitate E2 signal transduction by providing a location for
numerous signaling proteins (258). In this study, I was able to co-localize ER-α to the cell
membrane invagination scaffold protein caveolin-1, suggesting E2 can activate non-
genomic membrane bound signaling kinases. In order to investigate whether membrane
bound ER-α plays a role in the regulation of NPY secretion, I examined the effect of the
cell membrane-impermeable E2-BSA in the presence and absence of ER antagonists on
NPY release. Here, I demonstrated a similar ER-α-dependent decrease in NPY secretion,
indicating E2 attenuates NPY release through membrane-bound ER-α. These studies
parallel previous work that supports ER-α as the anorexigenic target of E2 in the
hypothalamus and present the first line of evidence of the necessary role of membrane
bound ER-α in directly regulating NPY levels.
To date, E2 has been reported to activate a number of intracellular signaling
cascades through the interaction of various scaffold proteins in a tissue- and cell-specific
manner, including: 1) the mobilization of intracellular calcium, 2) the activation of
adenylate cyclase and cAMP synthesis, 3) the MAPK pathway and finally 4) the PI3K
pathway (116, 146, 256, 259). Interestingly, previous gene expression studies completed
in our lab directly linked the PI3K and MAPK pathways to the E2-mediated decrease in
NPY gene expression (164). I therefore used pharmacological inhibitors against these
pathways to determine their role in the E2-mediated regulation of NPY secretion. I found
that the 1 hour E2-mediated decrease in NPY secretion was significantly attenuated with
the PI3K inhibitor, but not the MAPK inhibitor. However, co-treatment of E2 and the
MAPK inhibitor failed to reduce NPY secretion to levels comparable to E2 treatment

75
75
alone, suggesting the MAPK pathway may play some role in this effect. I next assessed
the role of a novel fuel-sensing enzyme responsible for maintaining metabolic
homeostasis, AMPK. AMPK is involved in regulating a number of feeding-related
neuropeptides and is a well-characterized anorectic signaling target of leptin in the
hypothalamus (260, 261). I therefore hypothesized that E2 may also exert its anorectic
decrease in NPY secretion via the AMPK signaling pathway. To assess the potential role
of AMPK in mediating the decrease in NPY secretion by E2, I co-treated the mHypoE-42
and mHypoA-2/12 neurons with E2 and the pharmacological inhibitor Compound C. I
found inhibition of the AMPK pathway was sufficient to block the E2-mediated
repression of NPY release. These results support the hypothesis that AMPK is required
for the anorectic action of E2 on the NPY neuron. Taken together, these data outline the
potential signaling mechanisms that contribute to the reduction in NPY secretion by E2.
Considering that the mHypoE-42 and mHypoA-2/12 cell lines are neurons taken
from the entire hypothalamus, I can only speculate on the precise nuclei of the
hypothalamus from which each clonal cell line originated. To date, a number of feeding-
related hypothalamic nuclei have been implicated in the E2-mediated decrease in food
intake. Kalra and colleagues (7) have shown the anorectic action of E2 may be mediated
through NPY neurons in the PVN of the hypothalamus, although other areas of the
hypothalamus, especially the ARC where the vast majority of NPY-synthesizing neurons
are located, can not be ruled out from these studies. Other rodent studies have found E2
decreases food intake via the VMN (252). Interestingly, the POA has been shown to be
important in the anorectic action of E2 in the rat, although the POA is mainly comprised
of GnRH, NT and CRH-expressing neurons (254). In order to speculate where the
mHypoE-42 and mHypoA-2/12 neurons originated from in vivo, I reviewed published

76
76
data that reported the expression of specific neuropeptides and receptors in these feeding-
related hypothalamic nuclei. I found that: 1) in situ hybridization studies implicate ER-β
to be the predominant receptor of the PVN and ER-α to be the predominant receptor in
the VMH, suggesting the mHypoE-42 and mHypoA-2/12 neurons did not originate from
the either of these nuclei (14, 16); 2) 95% of neurons that co-express AgRP/NPY are
localized to the ARC (246); 3) The POA does not express NPY (254); and 4) ER-α, ER-
β and the leptin receptor (Ob-R) are expressed abundantly in the ARC in vivo. Taken
together, the ARC is the only hypothalamic nuclei known to express ER-α, ER-β, NPY,
AgRP and Ob-R. Overall, these observations suggest that the hypothalamic anorectic
target of E2 may be NPY-synthesizing neurons that reside in the ARC nucleus and
additional studies in vivo could confirm this (Table 3.1).
Intact E2 signaling is essential for the maintenance of energy homeostasis.
Delineating the hypothalamic targets and signal transduction cascades of E2 is therefore
critical for our understanding of E2-related feeding disorders. Our studies demonstrate
novel non-genomic mechanisms by which E2 can directly regulate the release of the most
potent orexigenic feeding-related peptide, NPY, in embryonic and adult hypothalamic
cell lines. I have found that E2 can rapidly reduce NPY secretion within 1 hour of
exposure through neurons likely located in the ARC nucleus. Furthermore, I demonstrate
that this decrease in NPY secretion is mediated through non-genomic E2 signaling via
ER-α localized at the cell membrane. Specifically, I found that E2 directly reduces NPY
secretion through a PI3K and AMPK dependent mechanism. These findings provide new
information towards our understanding of the central role of E2 that may be involved in
the anorexigenic activity of this steroid hormone (see Figure 3.8).
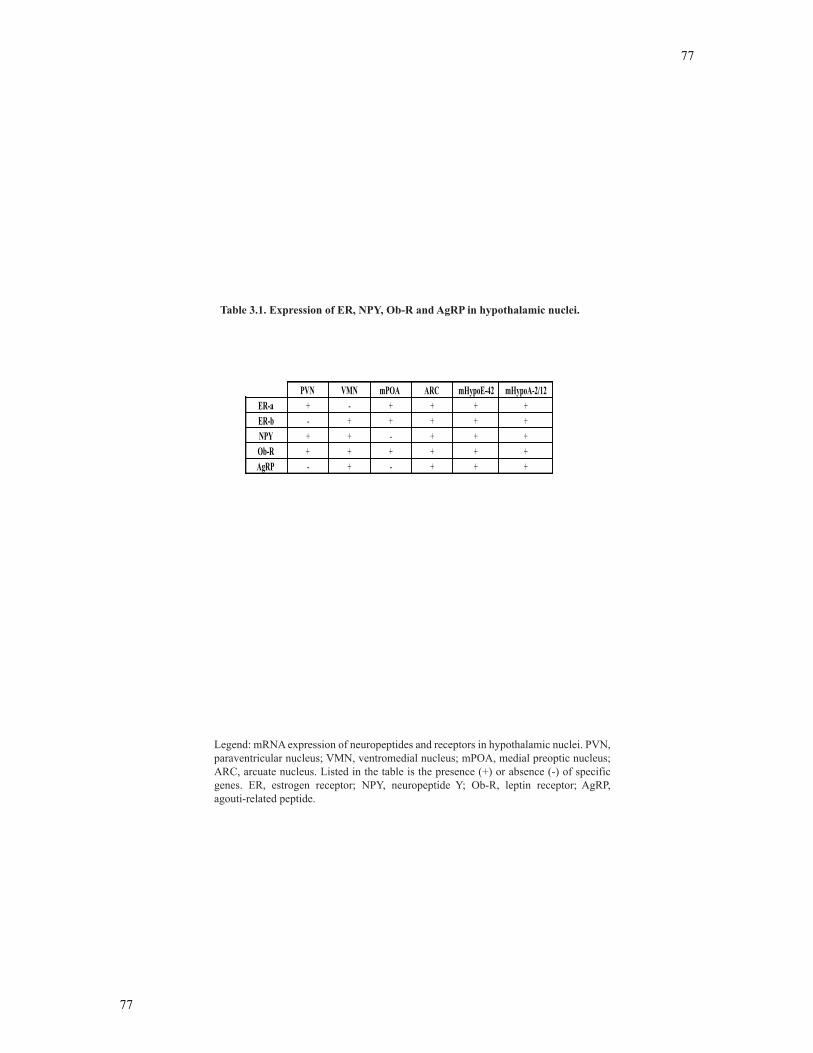
77
77
PVN VMN mPOA ARC mHypoE-42 mHypoA-2/12ER-a + - + + + +ER-b - + + + + +NPY + + - + + +Ob-R + + + + + +AgRP - + - + + +
Legend: mRNA expression of neuropeptides and receptors in hypothalamic nuclei. PVN, paraventricular nucleus; VMN, ventromedial nucleus; mPOA, medial preoptic nucleus; ARC, arcuate nucleus. Listed in the table is the presence (+) or absence (-) of specific genes. ER estrogen receptor NPY, neuropeptide Y; Ob-R, leptin receptor; AgRP, agouti-related peptide.
Table 3.1. Expression of ER, NPY, Ob-R and AgRP in hypothalamic nuclei.

78
78
NPY neuron
Grb2
Sos Src
SHC
PI3k
IRS1/2
AMPKK
AMPKP
E2
ER-ER-
Gene transcriptionER- ER-
ER- NPY mRNAER-
NPY secretion
Compound C
Ly294002
Titolo et al. 2006. MolEndocrinol.
Dhillon et al. 2010. Int JObes.
E2
Figure 3.8. Model of the potential cellular signaling pathways involved in estrogen regula-tion of NPY secretion.
Previous experiments completed by the Belsham group using siRNA directed against ER- and ER-ß found that estrogen-mediated repression of NPY mRNA levels required bot ER- and ER-ß or ER- alone. We have extended these studies and directly linked NPY secretory responses to estrogen treatments. Using receptor specific agonists/antagonists and E2-BSA, we found membrane-bound ER- was resonsible for the repressive effect of estrogen on NPY secretion. Additionally, pharmacological inhibitor studies found this effect to occur through the PI3K and AMPK signaling pathways.

79
79
4 Chapter 4
Leptin differentially regulates NPY secretion in NPY-expressing hypothalamic cell lines
through distinct intracellular signal transduction pathways
Manuscript is in preparation.
Citation:
Leptin directly decreases NPY secretion via AMPK- and PI3K-dependent mechanisms that is blocked after induction of leptin resistance in hypothalamic NPY neuronal cell models. Dhillon SS, Centeno ML, Kim GL, Belsham DD. To be submitted June 2010.
Contributions:
• SSD completed experiments and wrote the manuscript • MLC immortalized and characterized NPY-GFP cell line • GLK characterized the NPY-GFP cell line (RT-PCR) • DDB edited the manuscript and provided scientific input, direction and funding

80
80
4.1 Abstract
Leptin acts as a key peripheral hormone in the hypothalamus regulating food
intake and metabolism. Leptin also conveys metabolic information to the reproductive
axis through unknown mechanisms. Accumulating evidence shows that NPY neurons are
involved in mediating the anorexigenic and reproductive effects of leptin via
hypothalamic neuronal circuits. To determine the molecular and cellular basis for this
dual role of leptin, I determined the ability of leptin to differentially regulate NPY
neurons in vitro. mHypoE-38, mHypoE-42, mHypoA-59 and NPY-GFP cell lines were
characterized for leptin receptors, neuronal markers and NPY secretion using RT-PCR
and EIA. In the mHypoE-38 neurons, 1 h leptin treatment directly increased NPY
secretion, and this effect was directly linked to the MAPK and PI3K signaling pathways
using pharmacological inhibitors. Treatment with leptin increased the phosphorylation
status of Jak2, JNK and CREB as shown by Western blot analysis. In the mHypoE-42
neurons, leptin failed to change NPY secretion. However, in the mHypoA-59 and NPY-
GFP neurons, leptin decreased NPY secretion, which was linked to the AMPK and PI3K
pathways. Western blot analysis further demonstrated that leptin reduced the
phosphorylation status of AMPK in these two cell models. Additionally, AMPK
activation via (aminoimidazole carboxamide ribonucleotide) AICAR treatment directly
increased NPY secretion, highlighting the importance of AMPK activity in NPY neuronal
function. Prolonged leptin exposure in the NPY-GFP cells prevented leptin-induced
changes in AMPK phosphorylation and reductions in NPY secretion, indicating NPY
neurons are susceptible to leptin resistance. This is the first demonstration that leptin can
regulate individual NPY neuronal cell models through distinct intracellular signaling

81
81
pathways and secretory responses, which may serve as a metabolic signal for food intake
and a stimulatory signal for the reproductive axis.
4.2 Introduction
Leptin, an adipocyte-derived hormone and product of the ob gene, acts on its
receptor in the hypothalamus to reduce appetite and body weight (188, 189). Several
studies have demonstrated this anorexigenic role of leptin, as mutations in the ob gene
(ob/ob mice) or leptin receptor (db/db mice) are associated with morbid obesity, and both
central and peripheral administration of leptin decreases food intake in a number of
species (184). Although this leptin-induced reduction in appetite has been found to occur
through hypothalamic circuitry comprised of both orexigenic (i.e. NPY) and anorexigenic
(i.e. POMC) peptides, the mechanisms involved have yet to be defined (6, 7, 262). In
addition to the role of leptin in energy homeostasis, leptin has recently been recognized
as a key regulator of the reproductive axis (196, 204). This reproductive effect of leptin
was first observed in ob/ob mice, which have lower levels of LH and are infertile, a
phenotype that can be reversed with leptin administration (195). Conversely, both
transgenic mice overexpressing leptin and the peripheral administration of leptin results
in accelerated puberty (193). This action of leptin on the reproductive axis is believed to
occur through the GnRH neuron, as leptin administration elevates GnRH levels in vivo
(263). However, immunohistochemistry (IHC) studies have demonstrated that Ob-R are
not present in GnRH-expressing neurons in the rat hypothalamus, indicating that leptin
may indirectly stimulate GnRH activity through interneurons synapsing on GnRH-
expressing neurons (264). Although the anorexigenic and reproductive effects of leptin
have been well described, the identity of leptin-responsive target cells that reduce
appetite and stimulate the HPG axis remains unclear.

82
82
NPY neurons have emerged as a key target of leptin in the hypothalamus, as NPY
neurons within the ARC express Ob-R mRNA and protein (6, 265). NPY is a potent
orexigenic peptide that stimulates feeding upon central administration and increases body
weight after repeated doses (107). NPY has also been acknowledged for many years as a
major regulator of the reproductive axis. Morphological evidence indicates co-
localization of NPY receptors on GnRH neurons and studies in vivo and in vitro have
shown that NPY stimulates GnRH secretion (45, 70, 266). Given the dual role of NPY as
an orexigenic and reproductive signal, NPY neurons may act as an intermediary target of
leptin to regulate both the metabolic and reproductive axis. Although ICV leptin
administration results in a decrease in NPY mRNA levels in the whole hypothalamus, the
direct effect of leptin on individual NPY neurons and the signaling mechanisms through
which leptin acts have yet to be documented.
To directly study the effects of leptin on NPY neuronal function, I used
hypothalamic NPY-expressing neuronal cell models; mHypoE-38, mHypoE-42,
mHypoA-59 and NPY-GFP, which were immortalized using the retroviral transfer of
primary hypothalamic cell culture with SV40 T-antigen (219, 220). These cell lines have
been thoroughly characterized, demonstrate neurosecretory properties, express neuron
specific markers and have classical neuronal morphology (216). In the present study, I
hypothesized that NPY neuronal cell lines can be differentially regulated by leptin to
regulate the pleiotropic neuroendocrine responses of leptin. Based on this evidence, I
propose that leptin stimulates reproductive NPY neuronal cell lines that would impinge
on GnRH-expressing neurons to stimulate the reproductive axis, and inhibits feeding-
related NPY neuronal cell lines to reduce food intake. I have found that leptin exposure to
the mHypoE-38 cell lines stimulated NPY secretion through a MAPK- and PI3K-

83
83
dependent mechanism. However, in the mHypoA-59 and NPY-GFP cell lines, leptin
directly decreased NPY secretion via AMPK- and PI3K-dependent mechanisms. AMPK
phosphorylation was inhibited with leptin exposure in these two cell lines. Additionally,
prolonged leptin exposure prevented the leptin-mediated decrease in AMPK
phosphorylation and NPY secretion in the NPY-GFP cell line, suggesting NPY neuronal
populations are susceptible to leptin resistance. These data indicate that leptin can
differentially regulate NPY neuronal cell lines through the activation of intrinsically
different pathways. This differential leptin regulation of NPY may act to stimulate the
reproductive axis and reduce food intake, thereby providing a mechanism by which leptin
controls both of these important physiological processes.
4.3 Results
4.3.1 Expression of the Ob-R and other markers in mHypoE-38, mHypoE-42,
mHypoA-59 and NPY-GFP neurons
mHypoE-38, mHypoE-42 and mHypoA-59 cells were immortalized as previously
described. NPY-GFP cells were immortalized and then FAC-sorted by GFP fluorescence.
Dividing cells were retrovirally infected with the SV40 T-antigen cDNA sequence. NPY-
GFP cell lines were probed for NPY and GFP protein using specific antibodies and
visualized using ICC to confirm cell phenotype (Figure 4.1A). Cell lines were further
characterized for neuronal markers, receptors and neuropeptides (Figure 4.1B). The cells
secrete NPY as detected by EIA (Figure 4.2A-D).
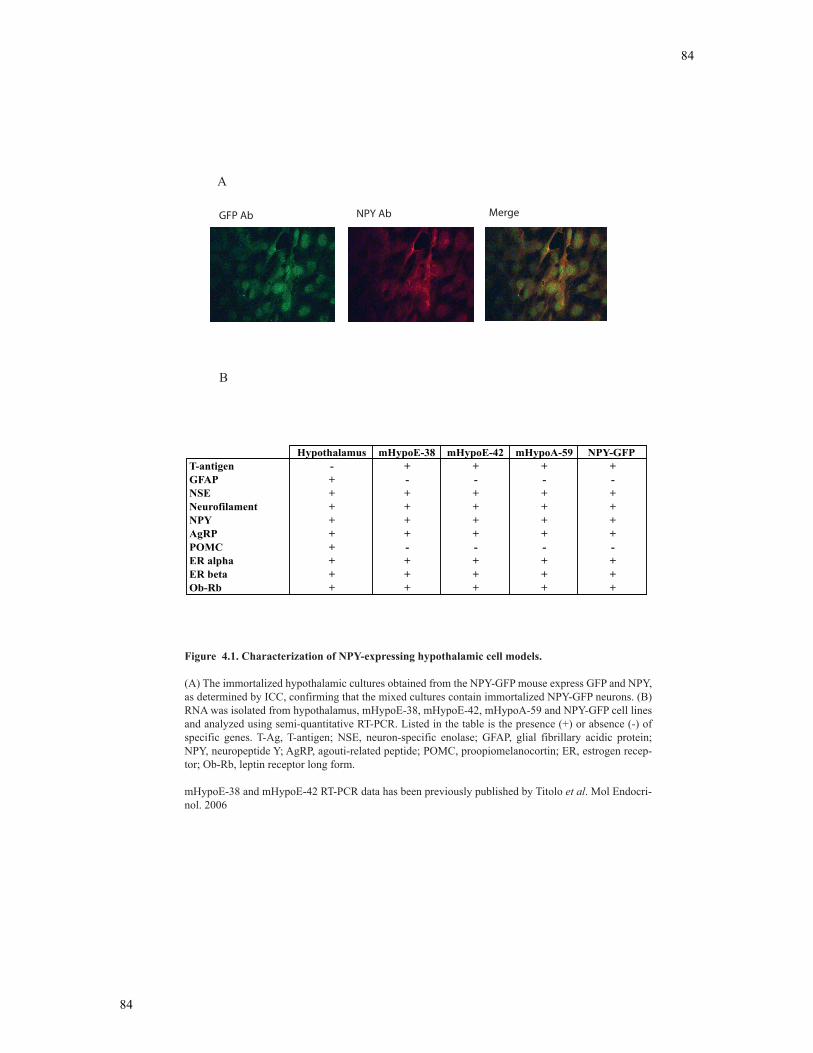
84
84
A
B
GFP Ab NPY Ab Merge
Figure 4.1. Characterization of NPY-expressing hypothalamic cell models.
(A) The immortalized hypothalamic cultures obtained from the NPY-GFP mouse express GFP and NPY, as determined by ICC, confirming that the mixed cultures contain immortalized NPY-GFP neurons. (B) RNA was isolated from hypothalamus, mHypoE-38, mHypoE-42, mHypoA-59 and NPY-GFP cell lines and analyzed using semi-quantitative RT-PCR. Listed in the table is the presence (+) or absence (-) of specific genes. T-Ag, T-antigen; NSE, neuron-specific enolase; GFAP, glial fibrillary acidic protein; NPY, neuropeptide Y; AgRP, agouti-related peptide; POMC, proopiomelanocortin; ER, estrogen recep-tor; Ob-Rb, leptin receptor long form.
mHypoE-38 and mHypoE-42 RT-PCR data has been previously published by Titolo et al. Mol Endocri-nol. 2006
Hypothalamus mHypoE-38 mHypoE-42 mHypoA-59 NPY-GFPT-antigen - + + + +GFAP + - - - -NSE + + + + +Neurofilament + + + + +NPY + + + + +AgRP + + + + +POMC + - - - -ER alpha + + + + +ER beta + + + + +Ob-Rb + + + + +

85
85
4.3.2 Regulation of NPY secretion by leptin in mHypoE-38, mHypoE-42,
mHypoA-59 and NPY-GFP neurons
Although evidence indicates that leptin can regulate NPY mRNA in vivo, it is not
known whether leptin can directly regulate NPY secretion (265). mHypoE-38, mHypoE-
42, mHypoA-59 and NPY-GFP neurons were exposed to leptin (10 nM) for 1 h. KCl
treatments resulted in a ~1.4-fold increase in NPY secretion in all cell lines. Analysis of
the results indicates that in the mHypoA-59 and NPY-GFP neurons, NPY secretion is
reduced by leptin (mHypoA-59: vehicle, 1.0±0.06: leptin, 0.78±0.03) (NPY-GFP: vehicle
1±0.14: leptin, 0.8±0.04) (Figure 4.2A,B). In the mHypoE-42 neurons, leptin treatments
failed to produce a change in NPY secretion compared to vehicle control (Figure 4.2C).
On the other hand, NPY secretion in mHypoE-38 neurons increased in response to leptin
treatment (vehicle, 1.0±0.07: leptin, 1.21±0.06) (Figure 4.2D). When comparing the cell
lines, it is evident that leptin can differentially regulate NPY secretion, indicating an
inherent difference in phenotype between the cell lines.
4.3.3 Leptin increases NPY secretion in the mHypoE-38 neurons via PI3K and
MAPK pathways
Leptin has been reported to act through the PI3K, AMPK, MAPK and JAK/STAT
pathways in the whole hypothalamus (213). In an attempt to delineate the molecular
mechanisms responsible for the differential regulation of NPY secretion by leptin, I
utilized Western blot analysis to examine the activity of key signaling kinases in the
mHypoE-38, mHypoA-59 and NPY-GFP neurons. The present and subsequent studies
were not completed using the mHypoE-42 neurons due the failure of leptin to elicit a
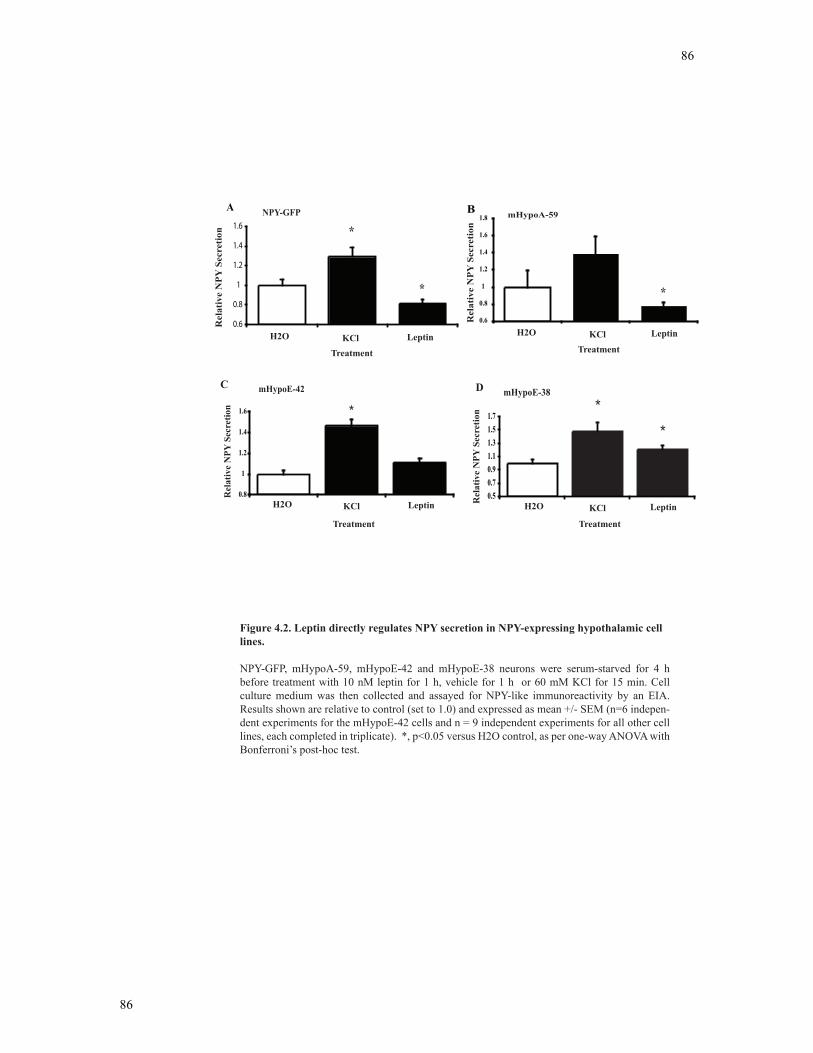
86
86
0.50.70.91.11.31.51.7
*
*
0.8
1
1.2
1.4
1.6 *
C
Rel
ativ
e N
PY S
ecre
tion
mHypoE-42 D
Rel
ativ
e N
PY S
ecre
tion
mHypoE-38
0.6
0.8
1
1.2
1.4
1.6
1.8 mHypoA-59BR
elat
ive
NPY
Sec
retio
nA
Figure 4.2. Leptin directly regulates NPY secretion in NPY-expressing hypothalamic cell lines.
NPY-GFP, mHypoA-59, mHypoE-42 and mHypoE-38 neurons were serum-starved for 4 h before treatment with 10 nM leptin for 1 h, vehicle for 1 h or 60 mM KCl for 15 min. Cell culture medium was then collected and assayed for NPY-like immunoreactivity by an EIA. Results shown are relative to control (set to 1.0) and expressed as mean +/- SEM (n=6 indepen-dent experiments for the mHypoE-42 cells and n = 9 independent experiments for all other cell lines, each completed in triplicate). *, p<0.05 versus H2O control, as per one-way ANOVA with Bonferroni’s post-hoc test.
*
*
0.6
0.8
1
1.2
1.4
1.6
Relative NPY secretion
H2O KCl Leptin
Treatment
H2O KCl Leptin
Treatment
H2O KCl Leptin
Treatment
H2O KCl Leptin
Treatment
*
NPY-GFP
Rel
ativ
e N
PY S
ecre
tion

87
87
change in NPY secretion. In the mHypoE-38 neurons, leptin significantly induced the
phosphorylation of Jak2 (Figure 4.3A), CREB (Figure 4.3B) and JNK (Figure 4.3C).
Leptin failed to change the phosphorylation status of AMPK in the mHypoE-38 cells. To
investigate the signaling kinases directly implicated in the leptin-mediated increase in
NPY secretion, pharmacological inhibitors directed against PI3K (LY294002), MAPK
(U0126) and AMPK (Compound C) pathways were applied for 1 h prior to 1 h leptin
treatments. I observed that when mHypoE-38 neurons were co-treated with leptin and the
PI3K or MAPK inhibitors, the leptin-mediated increase in NPY secretion was
significantly attenuated (Figure 4.3D). The inhibitors alone had no effect on basal levels
of NPY secretion. These results indicate that in mHypoE-38 neurons, leptin acts through
the MAPK and PI3K pathways to directly increase NPY secretion.
4.3.4 Leptin decreases NPY secretion in the mHypoA-59 and NPY-GFP neurons
via AMPK and PI3K pathways
Accumulating evidence shows that NPY neurons are involved in the anorexigenic
action of leptin (6, 200, 267, 268). To determine the signaling mechanisms activated by
leptin in the NPY-GFP (Figure 4.4) and mHypoA-59 (Figure 4.5) cell lines, I used
Western blot analysis to analyze phosphorylation activity of key signaling kinases. Leptin
transiently decreased phospho-AMPK, indicating AMPK inhibition. To determine
whether the PI3K, MAPK or AMPK pathways are required for the leptin-mediated
decrease in NPY secretion, mHypoA-59 and NPY-GFP neurons were pre-treated with
inhibitors for 1 h, followed by a 1 h leptin co-treatment.
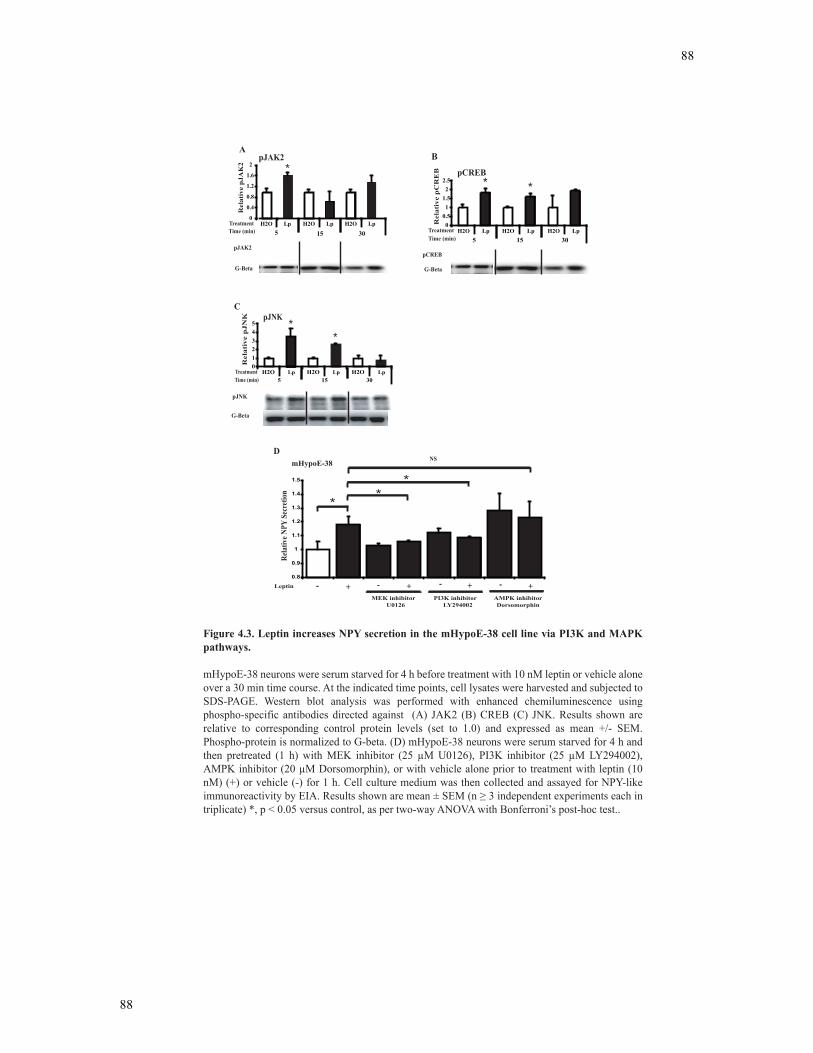
88
88
0
0.4
0.8
1.2
1.6
2
H2O Lp H2O Lp H2O Lp 5 15 30
TreatmentTime (min)
Rel
ativ
e pJ
AK
2
pJAK2*
A
pJAK2
G-Beta
012345
H2O Lp H2O Lp H2O Lp5 15 30
TreatmentTime (min)
Rel
ativ
e p
JNK pJNK
**
C
pJNK
G-Beta
* *
00.51
1.52
2.5
H2O Lp H2O Lp H2O Lp 5 15 30
TreatmentTime (min)
pCREB
B
Rel
ativ
e pC
RE
B
pCREB
G-Beta
0.8
0.9
1
1.1
1.2
1.3
1.4
1.5
Leptin - + + +- - -+MEK inhibitor
U0126PI3K inhibitor
LY294002AMPK inhibitorDorsomorphin
**
Rela
tive N
PY S
ecre
tion
mHypoE-38
*
D
Figure 4.3. Leptin increases NPY secretion in the mHypoE-38 cell line via PI3K and MAPK pathways.
mHypoE-38 neurons were serum starved for 4 h before treatment with 10 nM leptin or vehicle alone over a 30 min time course. At the indicated time points, cell lysates were harvested and subjected to SDS-PAGE. Western blot analysis was performed with enhanced chemiluminescence using phospho-specific antibodies directed against (A) JAK2 (B) CREB (C) JNK. Results shown are relative to corresponding control protein levels (set to 1.0) and expressed as mean +/- SEM. Phospho-protein is normalized to G-beta. (D) mHypoE-38 neurons were serum starved for 4 h and then pretreated (1 h) with MEK inhibitor (25 !M U0126), PI3K inhibitor (25 !M LY294002), AMPK inhibitor (20 !M Dorsomorphin), or with vehicle alone prior to treatment with leptin (10 nM) (+) or vehicle (-) for 1 h. Cell culture medium was then collected and assayed for NPY-like
triplicate) *, p < 0.05 versus control, as per two-way ANOVA with Bonferroni’s post-hoc test..
NS

89
89
I found that inhibiting either AMPK with Compound C or the PI3K pathway with
LY294002 prevented the leptin-mediated decrease in NPY secretion. These data provide
evidence that shows that AMPK and PI3K pathways are important mediators of the
anorexigenic action of leptin in the putative feeding-related NPY cell lines mHypoA-59
and NPY-GFP.
4.3.5 AICAR directly stimulates NPY secretion in mHypoA-59 and NPY-GFP
neurons
Treatment with AICAR activates AMPK activity in numerous cell types,
including neuronal cells (269, 270). I analyzed the effect of AICAR on NPY secretion in
the presence and absence of leptin in the mHypoA-59 and NPY-GFP neurons (Figure
4.6). AICAR alone directly increased NPY secretion in the mHypoA-59 and NPY-GFP
neurons. Together, these results indicate that AMPK activity is critical for NPY secretory
release and provides a potential mechanism by which orexigenic compounds (i.e. ghrelin)
can stimulate NPY secretion.
4.3.6 Leptin pre-treatment attenuates leptin-mediated phosphorylation of AMPK
in NPY-GFP neurons
High fat diets (HFD) or hyperleptinemia often result in impaired leptin signaling,
a state defined as leptin resistance. To determine the effects of prolonged leptin exposure
on neuronal signaling, I pre-treated NPY-GFP neurons with leptin for 8 and 24 h.
Following leptin pre-treatment, cells were washed with PBS, placed in fresh medium for
2 h and then re-challenged with leptin to determine neuronal responsiveness. Using
Western blot analysis, I determined that a 24 h leptin pre-treatment prevented the leptin-
induced
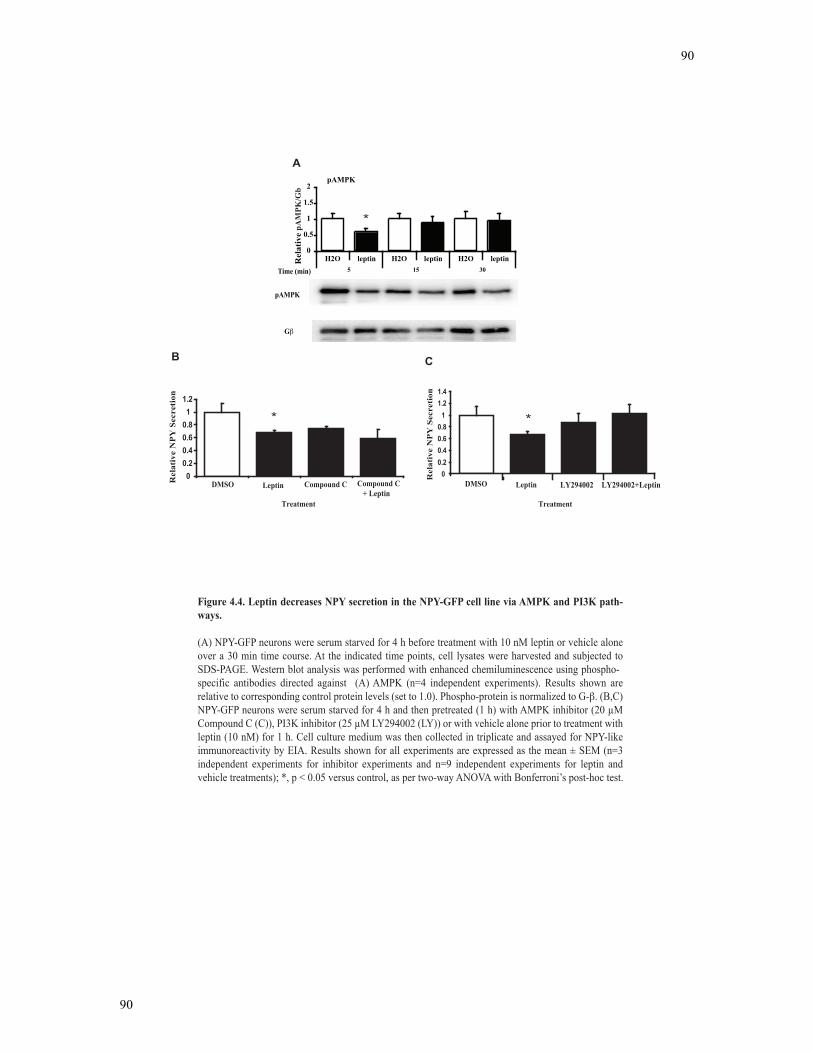
90
90
0
0.5
1
1.5
2
H2O leptin H2O leptin H2O leptin5 15 30
Relative pAMPK/gBeta
pAMPK
Rel
ativ
e pA
MPK
/Gb
00.20.40.60.811.2
DMSO Leptin Compound C Compound C + Leptin
Treatment
Rel
ativ
e N
PY
Sec
reti
onA
00.20.40.60.811.21.4
Rel
ativ
e N
PY
Sec
reti
on
DMSO Leptin
Treatment
B C
*
* *
Figure 4.4. Leptin decreases NPY secretion in the NPY-GFP cell line via AMPK and PI3K path-ways.
(A) NPY-GFP neurons were serum starved for 4 h before treatment with 10 nM leptin or vehicle alone over a 30 min time course. At the indicated time points, cell lysates were harvested and subjected to SDS-PAGE. Western blot analysis was performed with enhanced chemiluminescence using phospho-specific antibodies directed against (A) AMPK (n=4 independent experiments). Results shown are relative to corresponding control protein levels (set to 1.0). Phospho-protein is normalized to G- . (B,C) NPY-GFP neurons were serum starved for 4 h and then pretreated (1 h) with AMPK inhibitor (20 !M Compound C (C)), PI3K inhibitor (25 !M LY294002 (LY)) or with vehicle alone prior to treatment with leptin (10 nM) for 1 h. Cell culture medium was then collected in triplicate and assayed for NPY-like immunoreactivity by EIA. Results shown for all experiments are expressed as the mean ± SEM (n=3 independent experiments for inhibitor experiments and n=9 independent experiments for leptin and vehicle treatments); *, p < 0.05 versus control, as per two-way ANOVA with Bonferroni’s post-hoc test.
pAMPK
G
Time (min)
LY294002 LY294002+Leptin
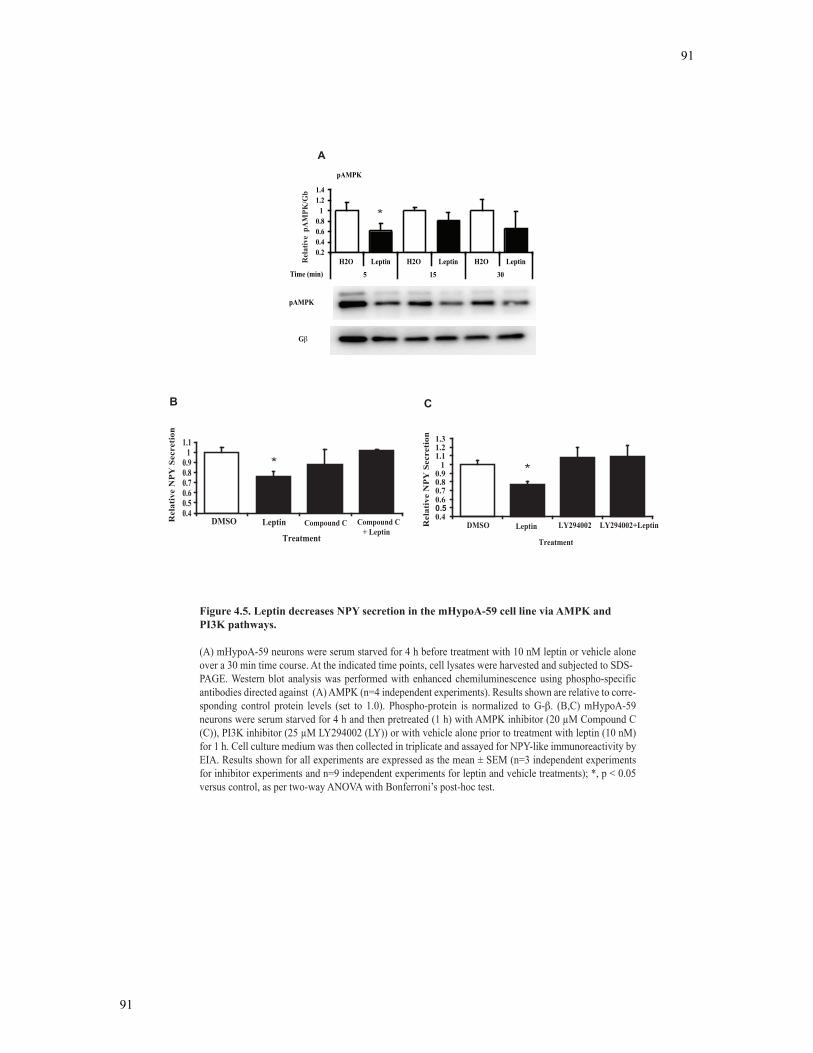
91
91
0.20.40.60.8
11.21.4
H2O Leptin H2O Leptin H2O Leptin
5 15 30
Relative pAMPK/gBeta
Rel
ativ
e p
AM
PK/G
b
Time (min)
pAMPK
0.40.50.60.70.80.9
11.1
Rel
ativ
e N
PY
Sec
reti
on
DMSO Leptin
Treatment
0.40.50.60.70.80.9
11.11.21.3
Rel
ativ
e N
PY
Sec
reti
on
DMSO Leptin LY294002 LY294002+Leptin
Treatment
B C
A
Figure 4.5. Leptin decreases NPY secretion in the mHypoA-59 cell line via AMPK and PI3K pathways.
(A) mHypoA-59 neurons were serum starved for 4 h before treatment with 10 nM leptin or vehicle alone over a 30 min time course. At the indicated time points, cell lysates were harvested and subjected to SDS-PAGE. Western blot analysis was performed with enhanced chemiluminescence using phospho-specific antibodies directed against (A) AMPK (n=4 independent experiments). Results shown are relative to corre-sponding control protein levels (set to 1.0). Phospho-protein is normalized to G- . (B,C) mHypoA-59 neurons were serum starved for 4 h and then pretreated (1 h) with AMPK inhibitor (20 !M Compound C (C)), PI3K inhibitor (25 !M LY294002 (LY)) or with vehicle alone prior to treatment with leptin (10 nM) for 1 h. Cell culture medium was then collected in triplicate and assayed for NPY-like immunoreactivity by EIA. Results shown for all experiments are expressed as the mean ± SEM (n=3 independent experiments for inhibitor experiments and n=9 independent experiments for leptin and vehicle treatments); *, p < 0.05 versus control, as per two-way ANOVA with Bonferroni’s post-hoc test.
*
**
pAMPK
G
Compound C Compound C + Leptin
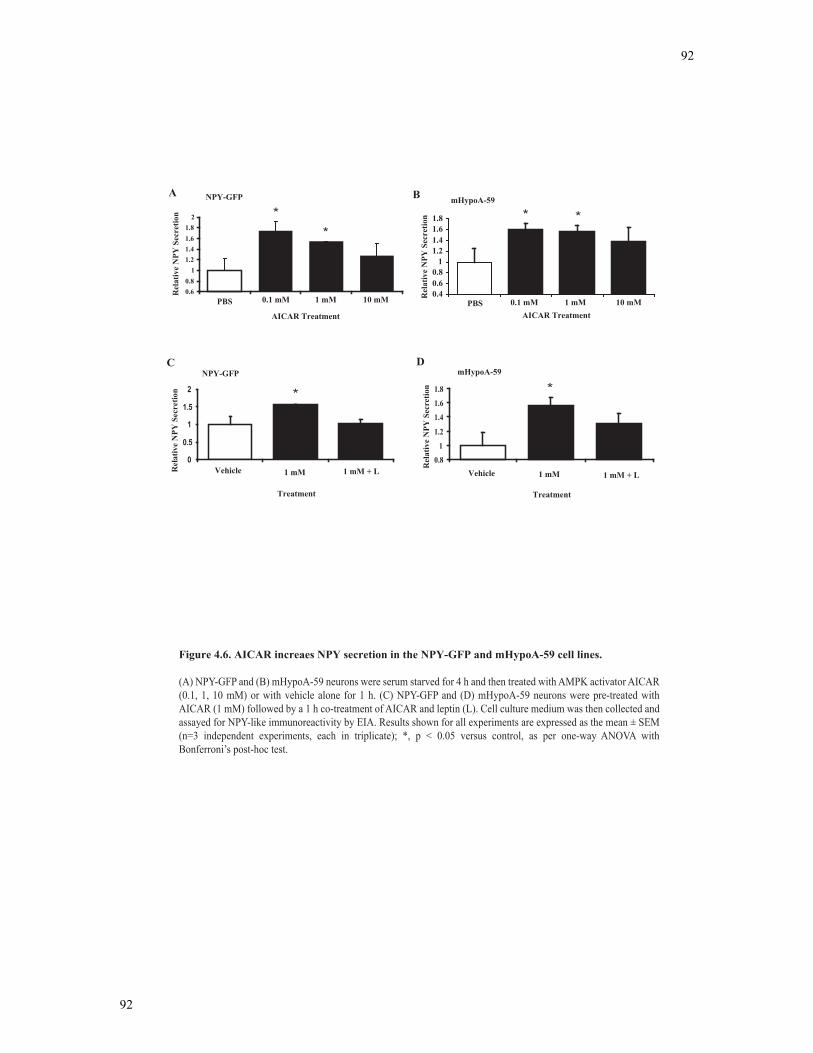
92
92
mHypoA-59
0.40.60.8
11.21.41.61.8
PBS 0.1 mM 1 mM 10 mMAICAR Treatment
Rel
ativ
e N
PY S
ecre
tion
NPY-GFP
0.60.8
11.21.41.61.8
2
PBS 0.1 mM 1 mM 10 mM
AICAR Treatment
A B
0.8
1
1.2
1.4
1.6
1.8
Rel
ativ
e N
PY S
ecre
tion
Rel
ativ
e N
PY S
ecre
tion
mHypoA-59
Treatment
Vehicle 1 mM 1 mM + L
D
0
0.5
1
1.5
2
NPY-GFP
Treatment
Vehicle 1 mM 1 mM + L
C
Rel
ativ
e N
PY S
ecre
tion
Figure 4.6. AICAR increaes NPY secretion in the NPY-GFP and mHypoA-59 cell lines.
(A) NPY-GFP and (B) mHypoA-59 neurons were serum starved for 4 h and then treated with AMPK activator AICAR (0.1, 1, 10 mM) or with vehicle alone for 1 h. (C) NPY-GFP and (D) mHypoA-59 neurons were pre-treated with AICAR (1 mM) followed by a 1 h co-treatment of AICAR and leptin (L). Cell culture medium was then collected and assayed for NPY-like immunoreactivity by EIA. Results shown for all experiments are expressed as the mean ± SEM (n=3 independent experiments, each in triplicate); *, p < 0.05 versus control, as per one-way ANOVA with Bonferroni’s post-hoc test.
*
*
* *
* *

93
93
reduction in phospho-AMPK (Figure 4.7). However, 8 or 24 h leptin pre-treatment
prevented leptin signaling. These data indicate that the endogenous leptin signaling in a
NPY cell model is impaired with prolonged leptin exposure.
4.3.7 Leptin pre-treatment attenuates the leptin-mediated decrease in NPY
secretion in NPY-GFP neurons
To address whether impaired leptin signaling induced from prolonged leptin
exposure can prevent the leptin-mediated reduction in NPY secretion, NPY-GFP cells
were pre-treated with leptin for 8 or 24 h. Following leptin pre-treatment, cells were
washed with PBS, placed in fresh medium for 2 h and then re-challenged with leptin for 1
h. Media were collected and NPY immunoreactivity was measured using an enzyme
immunoassay. I found that 8 and 24 h leptin pre-treatment prevented the leptin-mediated
decrease in NPY (Figure 4.8). These results suggest that leptin responsiveness decreases
in NPY neurons after experiencing hyperleptinemic conditions, and is consistent with the
possibility that cellular leptin resistance occurs at least partially through hypothalamic
NPY neuronal subtypes.
4.4 Discussion
The hypothalamus is a key site for the integration of both central and peripheral
endocrine signals involved in circadian rhythms, thermogenesis, satiety and reproduction
(215, 216). The diverse functions of the hypothalamus are controlled by a number of
heterogeneous, specialized cell populations. Recent evidence strongly supports the notion
that key peripheral hormones may differentially regulate specific hypothalamic cell
populations. For instance, NPY and POMC neurons respond differently to leptin
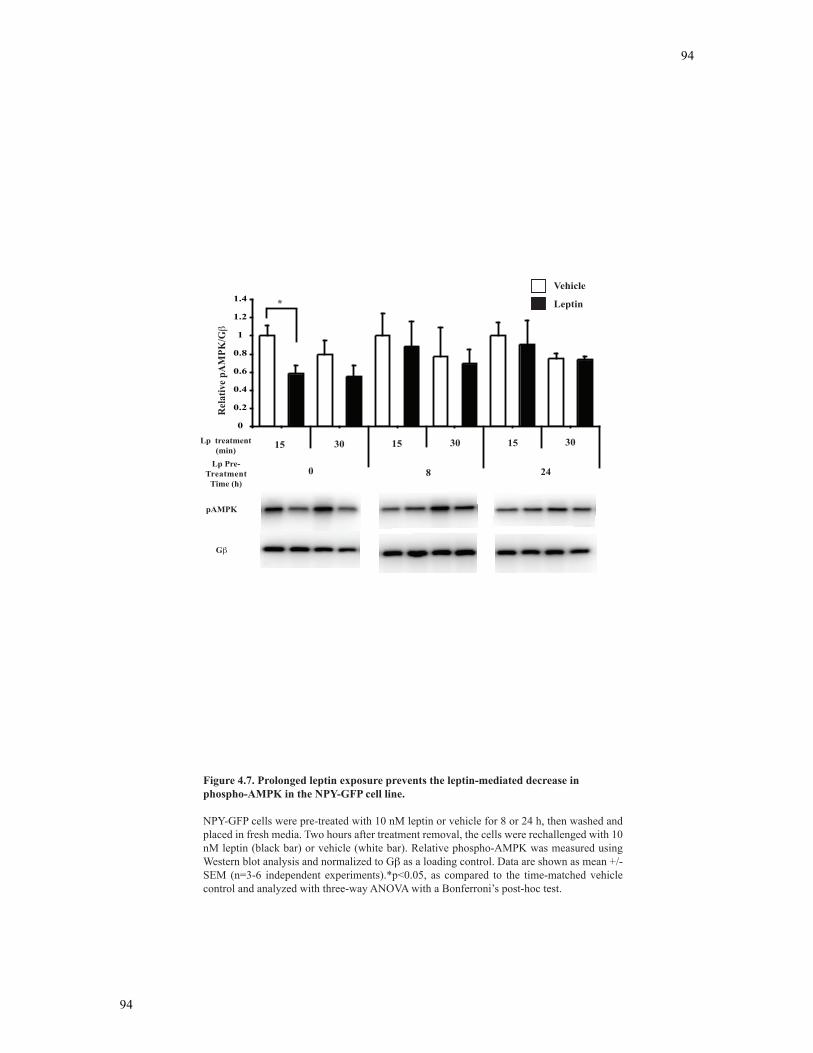
94
94
Figure 4.7. Prolonged leptin exposure prevents the leptin-mediated decrease in phospho-AMPK in the NPY-GFP cell line.
NPY-GFP cells were pre-treated with 10 nM leptin or vehicle for 8 or 24 h, then washed and placed in fresh media. Two hours after treatment removal, the cells were rechallenged with 10 nM leptin (black bar) or vehicle (white bar). Relative phospho-AMPK was measured using Western blot analysis and normalized to G as a loading control. Data are shown as mean +/- SEM (n=3-6 independent experiments).*p<0.05, as compared to the time-matched vehicle control and analyzed with three-way ANOVA with a Bonferroni’s post-hoc test.
0
0.2
0.4
0.6
0.8
1
1.2
1.4
Rel
ativ
e pA
MPK
/G
Vehicle
Leptin*
Lp Pre-Treatment
Time (h)0 8 24
15 30 30 3015 15Lp treatment (min)
pAMPK
G
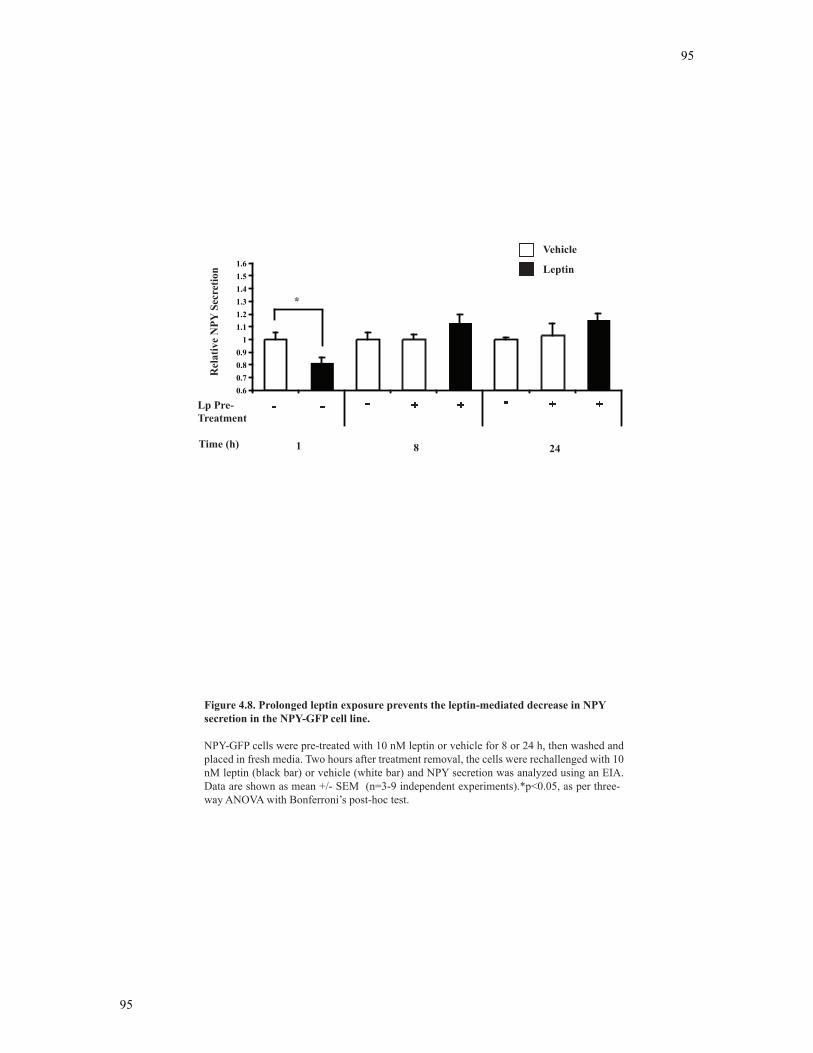
95
95
0.60.70.80.9
11.11.21.31.41.51.6
NPY Secretion
Lp Pre-Treatment
+ + + +
Time (h) 1 8 24
Rel
ativ
e N
PY S
ecre
tion
- -
*
Figure 4.8. Prolonged leptin exposure prevents the leptin-mediated decrease in NPY secretion in the NPY-GFP cell line.
NPY-GFP cells were pre-treated with 10 nM leptin or vehicle for 8 or 24 h, then washed and placed in fresh media. Two hours after treatment removal, the cells were rechallenged with 10 nM leptin (black bar) or vehicle (white bar) and NPY secretion was analyzed using an EIA. Data are shown as mean +/- SEM (n=3-9 independent experiments).*p<0.05, as per three-way ANOVA with Bonferroni’s post-hoc test.
Vehicle
Leptin
- -

96
96
treatments in the LHA: leptin induces SOCS-3 mRNA in NPY neurons, but induces both
SOCS-3 and Fos expression in POMC neurons (6). Interestingly, individual NPY neurons
may also respond differently to endocrine signals, depending on the function, location
and efferent projections of the neuron. Titolo et al. demonstrated that under the same E2
treatment, two separate NPY neuronal cell lines show distinct responses in NPY mRNA
expression (164, 178). This differential response to E2 was found to be dependent on the
ratio of ER-α to ER-β and demonstrates that different subpopulations of NPY neurons
respond uniquely to E2, and in doing so, may convey opposing messages to their
respective feeding and reproductive projections (164, 178). I hypothesized that NPY
neuronal cell lines may be differentially regulated by leptin, which in turn would regulate
multiple neuroendocrine processes. Specifically, I propose that 1) the reproductive role of
leptin may act through a specific subpopulation of Ob-Rb-containing NPY neurons to
achieve the GnRH pulse generator/surge, and 2) the anorexigenic role of leptin may act
by inhibiting NPY neuropetidergic circuitry that project to feeding-related hypothalamic
nuclei. Studying this hypothesis in vivo would be exceptionally difficult; thus, I have
taken an in vitro approach to demonstrate that individual NPY cell lines can be
differentially regulated by leptin treatment. Leptin treatment resulted in an increase in
NPY secretion in the mHypoE-38 neurons. Interestingly, NPY-containing conditioned
media treatments from the mHypoE-38 neurons induced an increase in GnRH mRNA
levels from the GT1-7 neurons as discussed in Chapter 5. Thus, the increase in NPY
peptide induced by leptin in the mHypoE-38 neurons may act on GnRH neurons to
stimulate the reproductive axis. Conversely, in the mHypoA-59 and NPY-GFP neurons,
leptin decreased NPY secretion, which would contribute to an anorexigenic effect of
leptin. These results suggest a novel mechanism in which circulating leptin can act on

97
97
different hypothalamic NPY subpopulations to regulate both metabolic and reproductive
functions.
Cell-based studies and in vivo experiments have led to a relatively detailed
understanding of the intracellular signaling pathways activated by leptin within the
hypothalamus in relation to specific biological function (7, 15, 82, 241). Bates et al.
determined that the hypothalamic control of reproduction by leptin occurs through
STAT3 independent mechanisms (271). In addition, indirect evidence implicated a
reproductive role of the MAPK pathway in leptin action, as tyrosine 985 mutations, a key
amino acid phosphorylation site that leads to the activation of the MAPK pathway, leads
to infertility (271, 272). Studies have yet to examine whether the PI3K pathway is
involved in the reproductive effect of leptin. Moreover, the individual signaling pathways
implicated in the role of leptin in reproductive function have yet to be attributed to a
specific neuronal population(s). I have found that leptin directly increases NPY secretion
in the mHypoE-38 neurons. This increase in NPY secretion may stimulate the
reproductive axis through downstream efferent projections to GnRH neurons.
Interestingly, the leptin-mediated increase in NPY secretion was dependent on the MAPK
pathway, which was previously demonstrated to be directly linked to the reproductive
role of leptin in vivo (272). For the first time, our studies also implicate the PI3K pathway
in the leptin-mediated increase in NPY secretion, as demonstrated by inhibitor studies
using LY294002. Together, these data implicate the MAPK and PI3K pathways in the
leptin-mediated increase in NPY neuronal activity in NPY-expressing mHypoE-38 cell
lines, which may in turn act to stimulate the reproductive axis through GnRH neurons.
Leptin also activates multiple non-genomic signaling pathways that are likely
critical in the release of hypothalamic neuropeptides to regulate appetite. Leptin activates

98
98
STAT3 proteins that bind to the phosphorylated 1138 tyrosine residue sites on Ob-Rb,
which are key signaling molecules in POMC neurons (201, 273). However, deletion of
STAT3 from NPY neurons does not alter NPY mRNA expression responses to leptin in
vivo (271, 273). PI3K stimulation by leptin has been observed in cultured hypothalamic
cells, and inhibition of hypothalamic PI3K activity in mice prevents leptin-mediated
decreases in appetite (274-276). Interestingly, in hypothalamic slices, leptin appears to
elicit cell depolarization via PI3K activity in POMC neurons; in contrast, leptin
withdrawal activates the PI3K pathway in NPY neurons, suggesting leptin hyperpolarizes
NPY neurons via PI3K-dependent mechanisms (277, 278). In fact, leptin has previously
been demonstrated to inhibit hypothalamic cell lines by hyperpolarization via KATP- and
PI3K-dependent mechanisms (276, 279). Most recently, considerable leptin research has
focused on AMPK, an energy sensor activated by an increasing ratio of AMP:ATP in
multiple cell types (270, 280). AMPK phosphorylates and inactivates ACC, a key
enzyme in fatty acid synthesis (281, 282). Leptin has been demonstrated to inhibit AMPK
activity in multiple regions of the hypothalamus (283). In addition, inhibition of
hypothalamic AMPK is sufficient to reduce appetite and weight gain (191, 281). Taken
together, leptin is hypothesized to stimulate ACC activity by inhibiting AMPK,
ultimately decreasing food intake (283). Although the anorectic signaling mechanisms of
leptin in the whole hypothalamus or specific hypothalamic nuclei have been studied, it is
unclear which cell types leptin is acting through and whether leptin directly or indirectly
regulates these neuronal cell types. In our studies, leptin directly decreased the
phosphorylation status of AMPK, suggesting an increase in ACC activity in our NPY
neuronal cell models; mHypoA-59 and NPY-GFP. Through inhibitor analysis, I
demonstrate that both the AMPK and PI3K pathways are required for the leptin-mediated

99
99
reduction in NPY secretion. Conversely, stimulating AMPK activity through AICAR
treatment directly stimulated NPY secretion. Together, this is the first study indicating
that leptin directly decreases NPY secretion in NPY neuronal cell lines via AMPK- and
PI3K-dependent mechanisms, which would ultimately reduce food intake and appetite in
vivo.
Studies document that obese individuals with high levels of leptin undergo a
failure to suppress feeding and decrease body weight (208, 209, 284). This suggests that
hyperleptinemia may cause leptin resistance. To date, four mechanisms of leptin
resistance have been hypothesized: 1) impaired leptin transport across the blood brain
barrier (BBB) (207), 2) alterations in leptin signaling (284), 3) perturbations in
developmental programming (212) and 4) increased Ob-R degradation (183, 285).
Although each of these mechanisms may contribute to the entirety of leptin resistance, I
examined the effect of prolonged leptin exposure on leptin signaling and leptin regulation
of NPY secretion in the NPY-GFP neuronal cell line. I found that impaired leptin
signaling might contribute to the leptin resistant state, as prolonged leptin exposure (8
and 24 h) prevented the leptin-mediated decrease in AMPK phosphorylation. In addition
to attenuated leptin signaling in the NPY-GFP cell line, sustained leptin treatments also
prevented the leptin-mediated decrease in NPY secretion. These results support the
notion of neuronal resistance to prolonged hyperleptinemia and indicate for the first time
that defective AMPK signaling and impaired NPY secretion may contribute to the leptin
resistant state.
I used three cell lines obtained from mice during different stages of development
for the studies in Chapter 4. The cell lines contain both clonal and mixed neuronal cells.
The use of cell lines from both embryonic and adult sources could conceivably produce a

100
100
differential response to hormonal stimulants in vitro, since hormones such as leptin
impose different physiological roles during different times of development (286, 287).
However, our characterization of these cell lines demonstrates that these cell models are
functionally similar to intact adult hypothalamic neurons. Importantly, the
characterization of these cell lines for specific receptors and neuropeptides has allowed us
to speculate on the nuclei from which these NPY neuronal cell lines are derived from in
vivo.
The mHypoE-38 cell line that I propose is upstream of GnRH neurons express
markers indicative of ARC origins. The expression of ER-α, ER-β, NPY and AgRP all
suggest that this cell line is from the ARC, as ICC studies have identified that these
markers are co-expressed almost exclusively in the ARC (17, 69, 163, 288, 289).
Importantly, these cell lines do not express tyrosine hydroxylase (TH), a marker of NPY
neurons that reside outside the ARC (290). In addition, IHC and retrograde analysis have
found that 49% of NPY fibres that innervate GnRH neurons originate from the ARC (88,
291). Together, these studies are consistent with the notion that the mHypoE-38 cell line
is derived from an ARC NPY neuronal population.
The adult mHypoA-59 neuronal cell line also displays markers that suggest an
ARC origin, including ER-α, ER-β, NPY, AgRP and Ob-R (17, 69, 163, 288, 289). Even
though both the hypothalamic cell lines seem to originate from the ARC, NPY neurons of
the ARC may be functionally heterogeneous. Anatomical evidence now exists for a
population of NPY neurons that also express the inhibitory neurotransmitter GABA in the
dorsomedial part of the ARC, whereas a subset of non-GABAergic NPY cells exists in
the ventral ARC (292). After additional research characterizes these cell lines and
identifies bona fide markers specific to each NPY neuronal cell line, future studies could

101
101
be completed in vivo. Finally, NPY-GFP cell lines represent a mixed population of NPY
neurons from the entire hypothalamus, and thus these cells represent multiple nuclei. The
leptin-mediated decrease in NPY secretion corroborates in vivo studies that demonstrate
that leptin treatments result in a decrease in NPY mRNA in the intact hypothalamus (265,
293). Together, our studies using a combination of embryonic, adult, clonal and mixed
NPY neuronal populations has allowed for a detailed mechanistic understanding of the
regulation of NPY secretory responses to leptin.
Based on our findings that leptin differentially regulates intracellular signaling
cascades and NPY secretory responses in NPY-synthesizing cell lines, I postulate that
leptin-mediated increases in NPY secretion is a possible indirect mechanism by which
leptin can exert a positive effect on the reproductive axis. However, leptin can also down-
regulate NPY secretion from NPY neuronal cell lines, which may achieve the
anorexigenic effect of leptin in the hypothalamus. During our experiments, prolonged
leptin exposure impaired AMPK signaling and was accompanied by an impaired NPY
secretory response to leptin. This finding implicates NPY neurons in the development of
central leptin resistance. Additionally, leptin failed to alter NPY secretion in the
mHypoE-42 neurons, suggesting that leptin does not affect all NPY-producing neurons.
These data support the hypothesis that hypothalamic NPY neurons add another level of
heterogeneity to the hypothalamus. Because the four cell lines responded to leptin
differentially, and have a distinctive markers expressed, it seems likely that they come
from distinct NPY neuronal subpopulations in the hypothalamus. The studies therefore
provide a novel mechanism of leptin action in specific cell populations responsible for
reproductive function and energy homeostasis (see Figure 4.9).
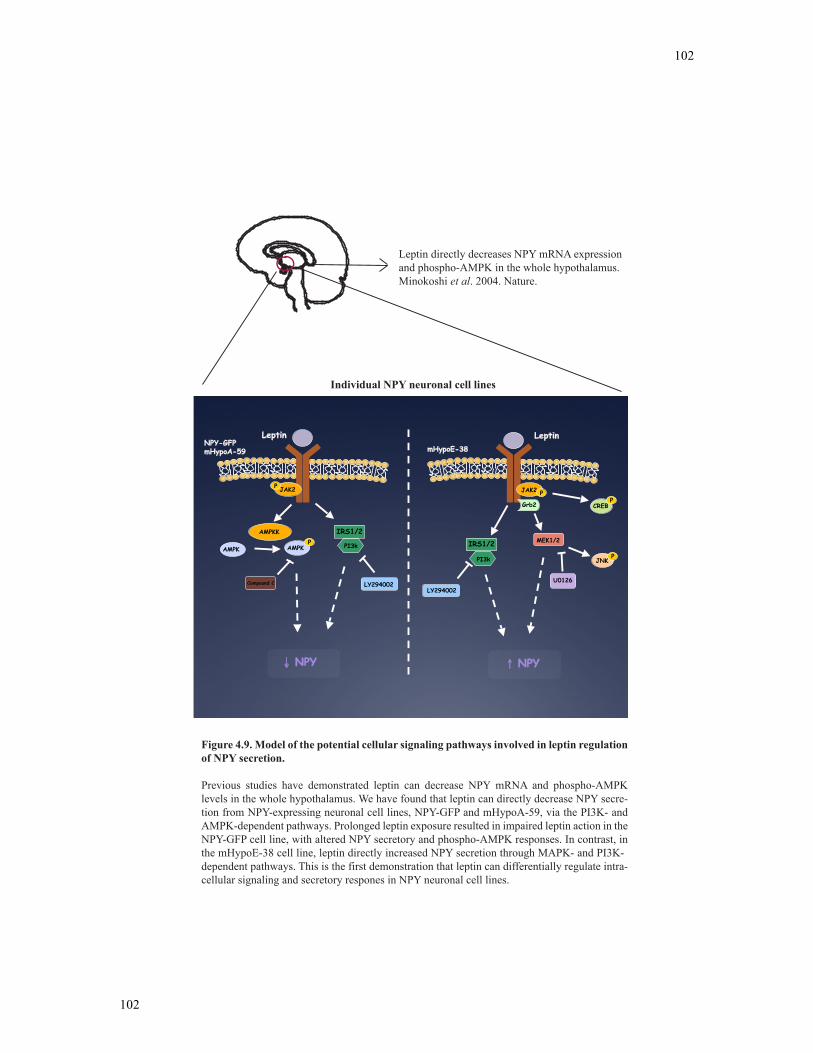
102
102
Leptin directly decreases NPY mRNA expression and phospho-AMPK in the whole hypothalamus.Minokoshi et al. 2004. Nature.
JNK
JAK2
Leptin
P
AMPK AMPKP
NPY-GFPmHypoA-59 mHypoE-38
Leptin
JAK2 P
NPY
Grb2 CREBP
P
MEK1/2
U0126
NPY
LY294002
PI3k
IRS1/2AMPKK
Compound C LY294002
IRS1/2
PI3k
Individual NPY neuronal cell lines
Figure 4.9. Model of the potential cellular signaling pathways involved in leptin regulation of NPY secretion.
Previous studies have demonstrated leptin can decrease NPY mRNA and phospho-AMPK levels in the whole hypothalamus. We have found that leptin can directly decrease NPY secre-tion from NPY-expressing neuronal cell lines, NPY-GFP and mHypoA-59, via the PI3K- and AMPK-dependent pathways. Prolonged leptin exposure resulted in impaired leptin action in the NPY-GFP cell line, with altered NPY secretory and phospho-AMPK responses. In contrast, in the mHypoE-38 cell line, leptin directly increased NPY secretion through MAPK- and PI3K-dependent pathways. This is the first demonstration that leptin can differentially regulate intra-cellular signaling and secretory respones in NPY neuronal cell lines.

103
103
5 Chapter 5
Neuropeptide Y induces gonadotropin-releasing hormone gene expression directly and
through conditioned medium from mHypoE-38 NPY neurons
Manuscript is published in Regulatory Peptides.
Citation:
Neuropeptide Y induces gonadotropin-releasing hormone gene expression directly and through conditioned medium from mHypoE-38 NPY neurons. Dhillon SS, Gingerich S, Belsham DD. Regul Pept. 2009 Aug 7;156(1-3):96-103. Epub 2009 Apr 14. PMID: 19371763
Contributions:
• SSD completed experiments and wrote the manuscript • SG completed experiments for figure 6 and edited the manuscript • DDB edited the manuscript and provided scientific input, direction and funding

104
104
5.1 Abstract
NPY regulates reproductive function at the level of the hypothalamus through
control of GnRH secretion. However, the direct control of GnRH gene expression by
NPY has not yet been studied. GT1-7 neurons were treated with 100 nM of NPY over a
36 h time-course. GnRH mRNA levels were significantly increased by NPY up to 12 h. I
determined that GT1-7 neurons expressed Y1, Y2, and Y4 NPY receptors, but not Y5.
Functional analysis of NPY receptor activation indicated that the Y1/Y4/Y5 receptor
agonist [Leu31, Pro34] significantly induced cAMP accumulation in the GT1-7 neurons.
Western blot studies demonstrated changes in the phosphorylation status of AKT,
ERK1/2, CREB and ATF-1 after NPY exposure. Pharmacological inhibitors of the
MAPK and PKA signal transduction pathways attenuated the NPY-mediated increase in
GnRH transcription. This NPY-mediated increase in GnRH mRNA was also inhibited
with the Y1-receptor specific antagonist BIBP-3326. The mHypoE-38 neurons secrete
detectable levels of NPY and can be used to study the effect of NPY in the presence of
other hypothalamic neuropeptides secreted. Conditioned medium from mHypoE-38
neurons induced an increase in GnRH mRNA, which was inhibited by the Y1 receptor
antagonist BIBP-3326. Our findings highlight the importance of NPY in the
transcriptional regulation of the GnRH gene and identify the receptors and signaling
pathways required for the stimulation of the reproductive axis.
5.2 Introduction
NPY has been acknowledged for many years as a major afferent regulator of
reproductive function (12, 13, 45, 112, 294). GnRH is a key decapeptide that sits at the
pinnacle of the HPG, and is released from a small population of neurons. NPY neurons in
the ARC project to GnRH cell bodies in the POA and to GnRH pre-synaptic terminals in

105
105
the ME (70, 295, 296). Additionally, morphological evidence indicates co-localization of
NPY receptors and GnRH neurons (70). This neuroanatomical evidence for connections
between NPY and GnRH neurons establishes a possible mechanism by which NPY
influences the reproductive axis. Several studies both in vivo and in vitro have shown
NPY stimulates GnRH secretion (44, 45, 72, 88, 106). In ewes, NPY infusion into the
third ventricle increases GnRH secretion substantially in the ME (74). Additionally, GT1-
7 neurons exposed to NPY significantly increased GnRH secretion (45). Further studies
demonstrated NPY-KO mice are not capable of generating a normal LH surge, a
necessary stage of the estrous cycle (13). However, depending upon the steroidal
environment and species, NPY can also downregulate the reproductive axis through
GnRH (75, 76, 297-299). NPY injections into the third ventricle in OVX rats led to a
reduction in plasma leutinizing hormone (LH) (300). Chronic NPY administration
inhibited gonadotropin secretion and sexual function in intact female rats (297, 301). In
OVX rabbits, NPY perfusion significantly decreases mean levels of GnRH. However,
similar NPY perfusion stimulated mean levels of GnRH in intact rabbits (77). Overall,
the physiological role of NPY on the reproductive axis remains unclear, with both
stimulatory and inhibitory effects published depending on the steroidal milieu. Although
a number of studies have investigated the role of NPY on GnRH secretion, the effect of
NPY on GnRH neurons at the transcriptional level in the absence of steroid hormones has
not been elucidated.
In the present study, I performed a series of experiments aimed to determine the
direct effects of NPY on GnRH expression and define the underlying signaling
mechanisms in the absence of steroidal modulators. These studies are difficult to perform
in the intact animal as GnRH neurons receive input from many afferent neuronal cell

106
106
types (302). To directly study the effects of NPY on GnRH expression, I used two cell
models: the clonal, GT1-7 GnRH-expressing neurons immortalized through targeted
tumorigenesis (218); and the clonal, hypothalamic NPY-expressing neurons, mHypoE-
38, immortalized using the retroviral transfer of a primary hypothalamic cell culture with
simian virus 40 T-antigen (219). Both cell lines have been thoroughly characterized, and
demonstrate neurosecretory properties, express neuron specific markers and have
classical neuronal morphology (164, 219). I demonstrate that both commercial NPY
peptide and NPY from mHypoE-38 (formerly called N-38) neurons increase GnRH
expression. I further examined the cellular mechanisms that are involved and second
messenger pathways. Finally, I demonstrate NPY induces GnRH gene expression, which
may be involved in the realization of the preovulatory surge.
5.3 Results
5.3.1 Expression of NPY receptor subtypes in GT1-7 neurons and hypothalamic
markers in mHypoE-38 neurons
The exact NPY receptor subtypes expressed in GT1-7 neurons has not been
confirmed. However there is evidence that GnRH neurons in vivo and in vitro express
some of the known NPY receptor subtypes (264). The presence of NPY receptor subtypes
Y1, Y2 and Y4 mRNA was detected in GT1-7 neurons using RT-PCR; however, the Y5
subtype was not found to be expressed (Figure 5.1). Mouse hypothalamic RNA was used
as a positive control as it confirms that the primers are specific for all receptor subtypes.
The presence of these receptors indicates that GT1-7 neurons are sensitive to NPY and
are therefore an appropriate model to study NPY-mediated regulation of GnRH mRNA
expression. mHypoE-38 neurons were found to express an extensive list of
neuropeptides, receptors and enzymes characteristic of neuroendocrine NPY

107
107
hypothalamic neurons (164, 219). Notably, the mHypoE-38 neurons express and secrete
NPY at appreciable levels and are therefore an appropriate model to study the effects of
NPY in the presence of other hypothalamic neuropeptides secreted by these hypothalamic
neurons on GnRH mRNA expression in GT1-7 neurons.
5.3.2 Regulation of GnRH mRNA expression by NPY in GT1-7 neurons
There is some evidence that NPY upregulates GnRH gene expression in vivo
(303); however, the direct transcriptional regulation of GnRH mRNA by NPY has not yet
been studied in detail. Time course studies revealed that 100 nM NPY exposure prompted
an increase in GnRH mRNA levels throughout the 36 h time course. GnRH mRNA was
significantly up-regulated at the 2, 4 and 12 h time points compared to time matched
controls (Figure 5.2A). Although all time points reveal an increase in GnRH mRNA
expression, the 4 h time point was subsequently used thereafter as the standard NPY
treatment time. A dose-response experiment was subsequently carried out where GT1-7
neurons were treated with 0.1 nM, 1 nM, 10 nM or 100 nM NPY for 4 h (Figure 5.2B).
All treatment concentrations resulted in an increase in GnRH mRNA levels compared to
vehicle control. However, the 100 nM NPY treatment concentration resulted in the most
robust response observed. These findings demonstrate that NPY substantially increases
GnRH mRNA expression in the GT1-7 GnRH neuron.
5.3.3 Effect of NPY receptor agonists on cAMP activity
NPY potently binds with differing affinities to a family of G-protein coupled
receptors; Y1-Y5, that belong to the rhodopsin-like superfamily of receptors. To
distinguish the precise receptor subtype(s) activated by NPY in the GnRH GT1-7 neuron,

108
108
Figure 5.1. Expression of NPY Y1, Y2 and Y4 receptor mRNA transcripts in GT1-7 neurons.
RNA harvested from GT1-7 neurons was used as a template for RT-PCR with primers specifically designed to amplify NPY receptor subtypes. Fragment sizes were NPY Y1- 481 bp, NPY Y2- 236 bp, NPY Y4- 530 bp, and NPY Y5- 291 bp. NTC, No template control. Hypothalamus was used as a positive control for all receptor subtypes.
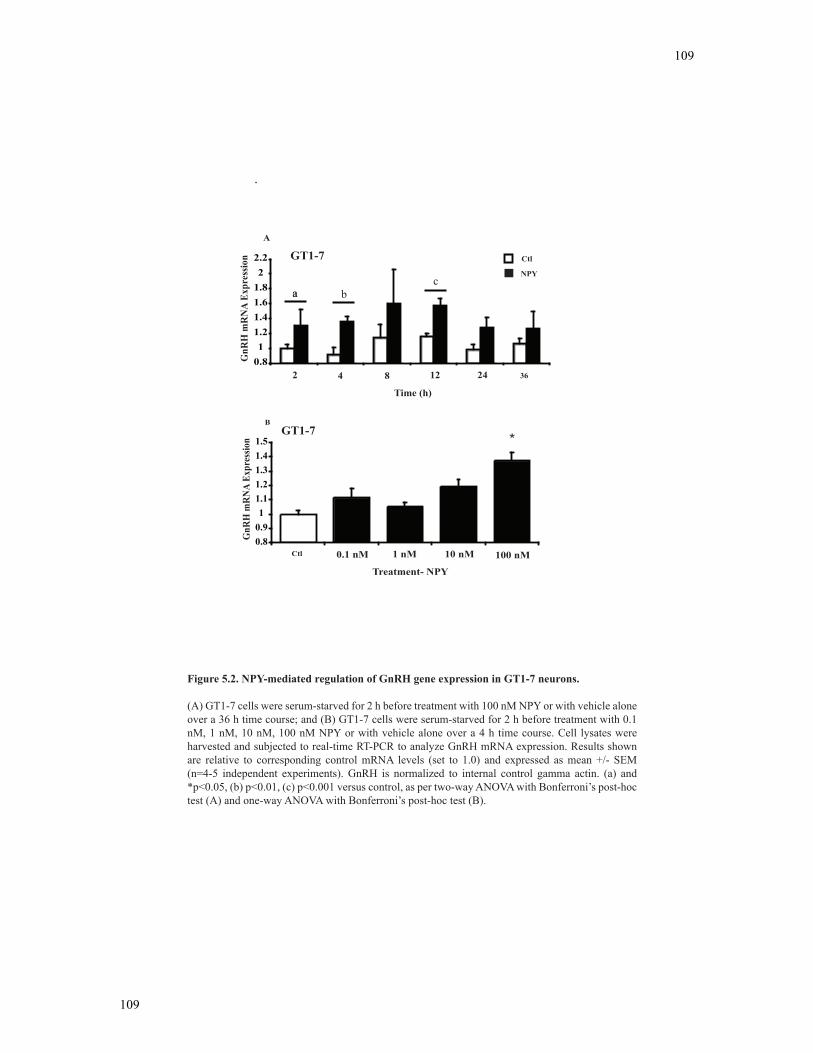
109
109
Time (h)
2 4 8 12 24 36
GnR
H m
RN
A Ex
pres
sion GT1-7 Ctl
NPY
0.81
1.21.41.61.82
2.2
Relative GnRH mRNA Levels
A
0.80.91
1.11.21.31.41.5
Relative GnRH Expression
GnR
H m
RN
A Ex
pres
sion
0.1 nM 1 nM 10 nM 100 nMCtl
Treatment- NPY
B
*GT1-7
Figure 5.2. NPY-mediated regulation of GnRH gene expression in GT1-7 neurons.
(A) GT1-7 cells were serum-starved for 2 h before treatment with 100 nM NPY or with vehicle alone over a 36 h time course; and (B) GT1-7 cells were serum-starved for 2 h before treatment with 0.1 nM, 1 nM, 10 nM, 100 nM NPY or with vehicle alone over a 4 h time course. Cell lysates were harvested and subjected to real-time RT-PCR to analyze GnRH mRNA expression. Results shown are relative to corresponding control mRNA levels (set to 1.0) and expressed as mean +/- SEM (n=4-5 independent experiments). GnRH is normalized to internal control gamma actin. (a) and *p<0.05, (b) p<0.01, (c) p<0.001 versus control, as per two-way ANOVA with Bonferroni’s post-hoc test (A) and one-way ANOVA with Bonferroni’s post-hoc test (B).
aa bc

110
110
specific NPY receptor subtype agonists were administered for 15 min and cAMP
synthesis, an indicator of GPCR activation was measured. As a control for cAMP
synthesis and dependability of RIA measurements, GT1-7 neurons were treated with the
well-characterized adenylyl cyclase activator, forskolin. 30 uM forskolin treatment
resulted in a 2.5-fold increase in cAMP immunoreactivity compared to vehicle control
(Figure 5.3). NPY (100 nM) and the Y1/ Y4/Y5 receptor agonist (100 nM) [Leu31, Pro34]
potently increased cAMP activity in GT1-7 neurons. Conversely, cAMP activity was not
significantly changed when compared to vehicle control upon treatment with 100 nM of
the Y2 selective agonist NPY13-36, Y4 selective agonist rat Pancreatic Polypeptide (rPP)
or the Y5 agonist D-[Trp32]. Thus, the cAMP sensitivity to the selective Y1/Y4 agonist
[Leu31, Pro34] highlights the importance of the Y1 and potentially Y4 receptors in NPY-
induced cAMP synthesis.
5.3.4 NPY rapidly phosphorylates PKA, ATF-1 and CREB in GT1-7 neurons
Because I found the MAPK and PKA pathways to be involved in NPY-mediated
stimulation in GnRH mRNA expression, I then assessed phosphorylation of signaling
proteins PKA and ERK1/2, and transcription factors CREB and activating transcription
factor 1 (ATF-1) downstream of the PKA and MAPK pathways. To elucidate whether
NPY activates specific signaling cascades in the GT1-7 neuron, 100 nM NPY was used
over a 1 h time-course. Western blot analysis using phospho-specific antibodies
demonstrated that NPY exposure induces the phosphorylation of PKA (Figure 5.4A),
ERK (Figure 5.4B), ATF-1 (Figure 5.4C) and CREB (Figure 5.4D) at 5 min and
returned to basal levels by 15 min post-treatment. These data suggest that the PKA and
MAPK signal transduction pathway, as well as transcription factors CREB and ATF-1
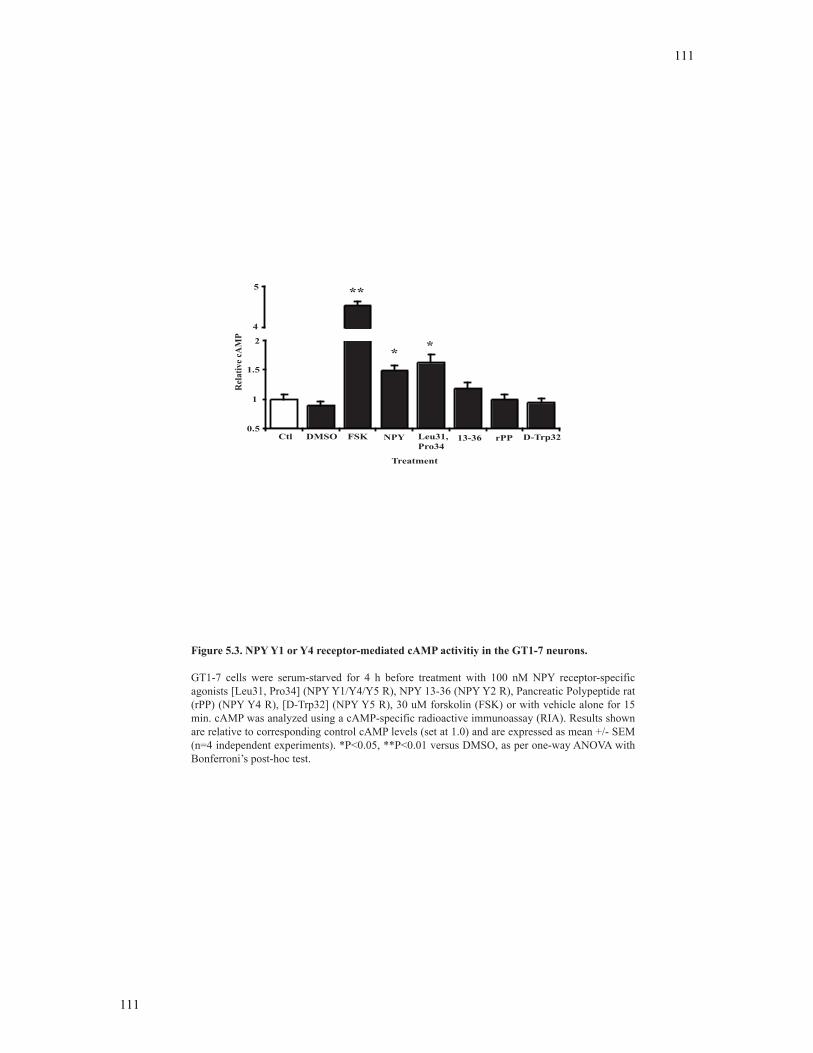
111
111
0.5
1
1.5
2
4
5
Ctl DMSO Leu31, Pro34
FSK 13-36 rPPNPY D-Trp32
**
* *
Treatment
Rel
ativ
e cA
MP
Figure 5.3. NPY Y1 or Y4 receptor-mediated cAMP activitiy in the GT1-7 neurons.
GT1-7 cells were serum-starved for 4 h before treatment with 100 nM NPY receptor-specific agonists [Leu31, Pro34] (NPY Y1/Y4/Y5 R), NPY 13-36 (NPY Y2 R), Pancreatic Polypeptide rat (rPP) (NPY Y4 R), [D-Trp32] (NPY Y5 R), 30 uM forskolin (FSK) or with vehicle alone for 15 min. cAMP was analyzed using a cAMP-specific radioactive immunoassay (RIA). Results shown are relative to corresponding control cAMP levels (set at 1.0) and are expressed as mean +/- SEM (n=4 independent experiments). *P<0.05, **P<0.01 versus DMSO, as per one-way ANOVA with Bonferroni’s post-hoc test.
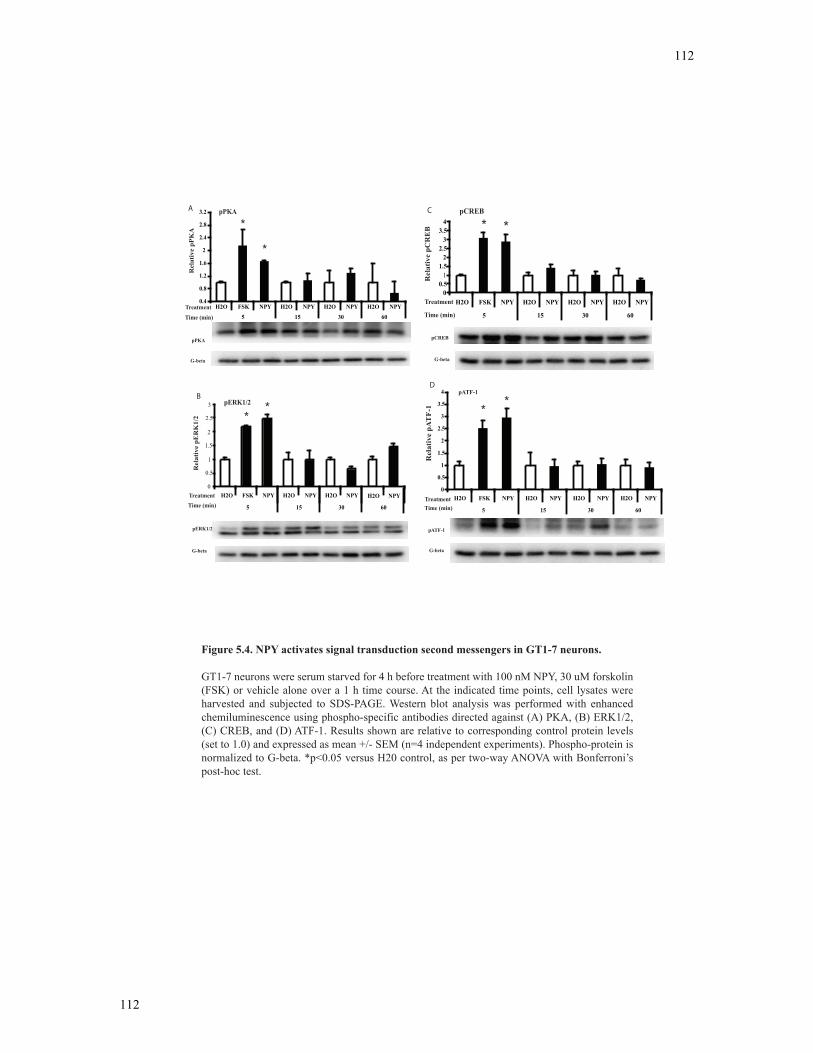
112
112
0.4
0.8
1.2
1.6
2
2.4
2.8
3.2
H2O FSK NPY H2O NPY H2O NPY H2O NPY5 15 30 60
pPKA
Rel
ativ
e pP
KA
TreatmentTime (min)
*
*
A
B
Relative pERK
H2O FSK NPY H2O NPY H2O NPY H2O NPY0
0.5
1
1.5
2
2.5
3 pERK1/2
TreatmentTime (min) 5 15 30 60
**
Rel
ativ
e pE
RK
1/2
pERK1/2
G-beta
pPKA
G-beta
0
0.5
1
1.5
2
2.5
3
3.5
4
H2O FSK NPY H2O NPY H2O NPY H2O NPY
5 15 30 60
pATF-1
**
TreatmentTime (min)
Rel
ativ
e pA
TF-
1
D
00.5
11.5
22.5
33.5
4
H2O FSK NPY H2O NPY H2O NPY H2O NPY
5 15 30 60
Relative pCREB
Treatment
Time (min)
pCREB
Rel
ativ
e pC
RE
B * *
pATF-1
G-beta
pCREB
G-beta
C
Figure 5.4. NPY activates signal transduction second messengers in GT1-7 neurons.
GT1-7 neurons were serum starved for 4 h before treatment with 100 nM NPY, 30 uM forskolin (FSK) or vehicle alone over a 1 h time course. At the indicated time points, cell lysates were harvested and subjected to SDS-PAGE. Western blot analysis was performed with enhanced chemiluminescence using phospho-specific antibodies directed against (A) PKA, (B) ERK1/2, (C) CREB, and (D) ATF-1. Results shown are relative to corresponding control protein levels (set to 1.0) and expressed as mean +/- SEM (n=4 independent experiments). Phospho-protein is normalized to G-beta. *p<0.05 versus H20 control, as per two-way ANOVA with Bonferroni’s post-hoc test.

113
113
plays a key role in the regulation of GT1-7 GnRH neurons.
5.3.5 Inhibition of MAPK and PKA-C signaling pathways affects NPY-mediated
regulation of GnRH mRNA expression in GT1-7 neurons
As reported in the time course study above, treatment with 100 nM NPY resulted
in increased GnRH mRNA expression in GT1-7 neurons over a 36 h time course (Figure
5.2A). To determine whether or not the increase in GnRH mRNA by NPY is mediated
through the NPY Y1 receptor subtype, I used a NPY Y1 receptor specific antagonist. I
demonstrate that the NPY Y1 antagonist BIBP-3326 inhibited the NPY-mediated
increase in GnRH mRNA, suggesting NPY acts through the NPY Y1 receptor to increase
GnRH mRNA (Figure 5.5A). Because I have shown NPY activates the MAPK and PKA
pathways, I assessed their role in the NPY-mediated increase in GnRH mRNA seen at 4 h
using pharmacological inhibitors. The co-treatment of NPY with the MAPK inhibitor
U0126 or the PKA inhibitor H89, resulted in attenuation of the NPY-mediated increase in
GnRH mRNA expression in the GT1-7 neurons compared to NPY treatment alone
(Figure 5.5B). H89 alone did not significantly change GnRH mRNA expression,
although U0126 moderately repressed basal levels of GnRH mRNA, but this was not
statistically significant. These results indicate that both the MAPK and PKA pathways
are critical for the regulation of GnRH mRNA by NPY in GT1-7 neurons.
5.3.6 Regulation of GnRH transcription by conditioned media from NPY-secreting
mHypoE-38 neurons is mediated through the NPY Y1 receptor subtype
Although NPY induces a significant increase in GnRH mRNA expression in GT1-7
neurons over a 36 h time course, it is unclear whether other secreted neuropeptides from
hypothalamic NPY neurons contribute to the regulation of GnRH mRNA levels.
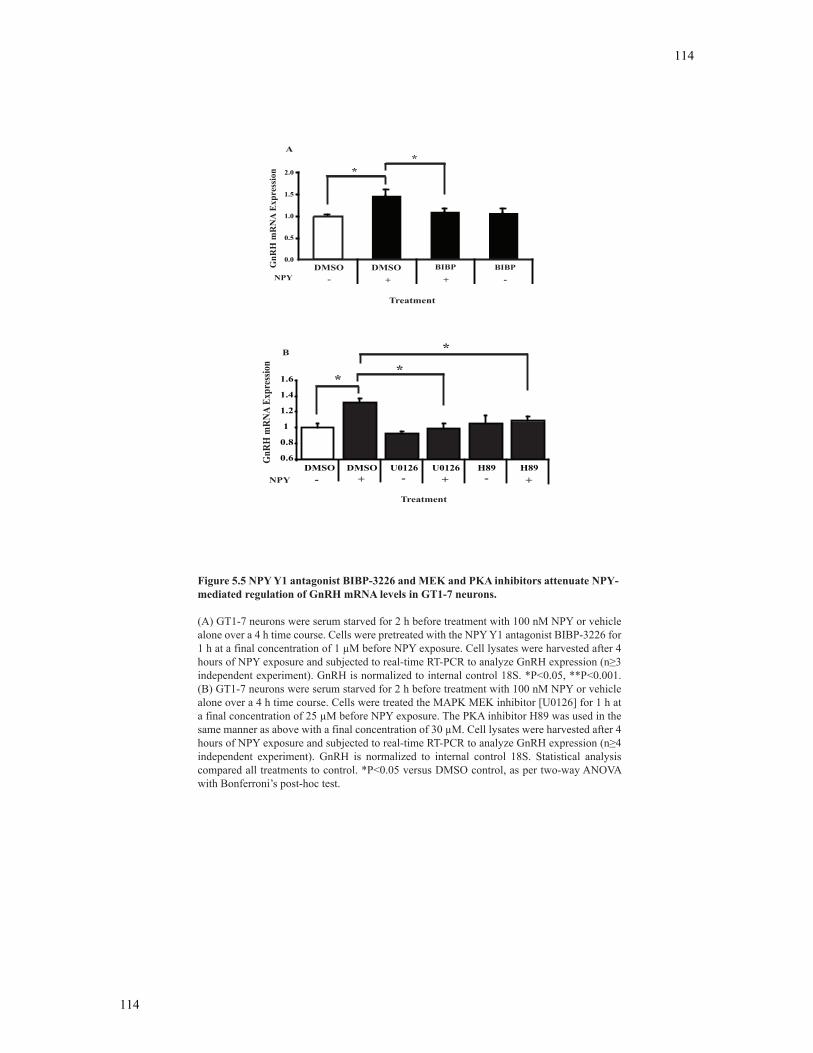
114
114
0.6
0.8
1
1.2
1.4
1.6 *
GnR
H m
RNA
Expr
essio
n
DMSO U0126 U0126 H89 H89DMSONPY +- - + +-
Treatment
0.0
0.5
1.0
1.5
2.0
GnR
H m
RN
A E
xpre
ssio
nNPY
DMSO BIBP BIBPDMSO- + -+
**
*
*
A
B
Treatment
Figure 5.5 NPY Y1 antagonist BIBP-3226 and MEK and PKA inhibitors attenuate NPY-mediated regulation of GnRH mRNA levels in GT1-7 neurons.
(A) GT1-7 neurons were serum starved for 2 h before treatment with 100 nM NPY or vehicle alone over a 4 h time course. Cells were pretreated with the NPY Y1 antagonist BIBP-3226 for 1 h at a final concentration of 1 !M before NPY exposure. Cell lysates were harvested after 4
independent experiment). GnRH is normalized to internal control 18S. *P<0.05, **P<0.001. (B) GT1-7 neurons were serum starved for 2 h before treatment with 100 nM NPY or vehicle alone over a 4 h time course. Cells were treated the MAPK MEK inhibitor [U0126] for 1 h at a final concentration of 25 !M before NPY exposure. The PKA inhibitor H89 was used in the same manner as above with a final concentration of 30 !M. Cell lysates were harvested after 4
independent experiment). GnRH is normalized to internal control 18S. Statistical analysis compared all treatments to control. *P<0.05 versus DMSO control, as per two-way ANOVA with Bonferroni’s post-hoc test.

115
115
To determine the effect of additional neuropeptides on GT1-7 neurons, conditioned
medium was collected from the NPY-secreting mHypoE-38 neurons. mHypoE-38
neurons were treated with 60 mM potassium chloride (KCl) or vehicle alone for 15 min.
KCl treatments resulted in a 1.5 fold increase in NPY secretion from the mHypoE-38
neurons compared to vehicle controls (Figure 5.6A). In order to assess whether
conditioning medium may regulate GT1-7 neurons, conditioned media from the
mHypoE-38 neurons treated with vehicle or NPY-contained media was directly
administered to GT1-7 neurons for 4 h. The conditioned medium was desalted (to remove
KCl) using a Zeba desalting spin column to avoid changes in GT1-7 neurons. Real-time
RT-PCR demonstrated that NPY-containing conditioned media significantly increased
GnRH mRNA expression in GT1-7 neurons (1.25 fold increase) compared to GT1-7
neurons treated with vehicle conditioned media (set to 1) (Figure 5.6B). According to the
secretion data in the mHypoE-38 neurons (Figure 5.6A), I calculated the concentration of
NPY in the conditioned medium to be approximately 0.27 nM. Attempts to concentrate
the medium, even 2-fold, resulted in complete cell death due to contamination or
excessive salt concentrations (concentration of media was completed by drying down
media contents in a centrifugal concentrator). The level of induction of GnRH by the
conditioned medium therefore should be relatively lower than that of 100 nM NPY. This
is confirmed by our dose curve (Figure 5.2B). As a control for desalting efficiency, GT1-
7 cells were treated directly with KCl and gene expression was assessed at 4 h. No
significant change in GnRH gene expression was detected with KCl alone (Figure 5.6B).
This indicates that NPY secreted by mHypoE-38 NPY neurons is sufficient to induce
GnRH mRNA levels in GT1-7 neurons, in a similar manner to which
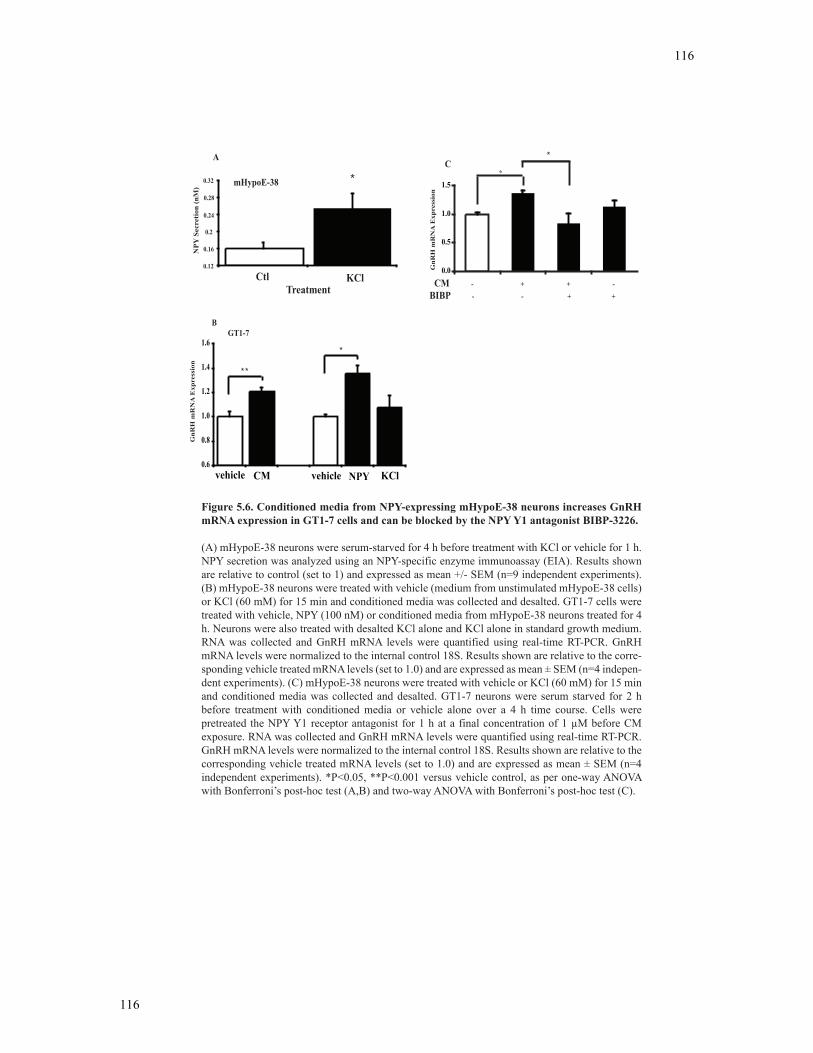
116
116
NP
Y S
ecre
tion
(nM
)
Relative NPY Secretion
0.12
0.16
0.2
0.24
0.28
0.32
TreatmentCtl KCl
mHypoE-38 *
A
GT1-7B
0.6
0.8
1.0
1.2
1.4
1.6
GnR
H m
RN
A E
xpre
ssio
n
vehicle CM vehicle NPY KCl
* *
*
0.0
0.5
1.0
1.5
GnR
H m
RN
A E
xpre
ssio
n
CM - + + -
BIBP - - + +
*C
*
Figure 5.6. Conditioned media from NPY-expressing mHypoE-38 neurons increases GnRH mRNA expression in GT1-7 cells and can be blocked by the NPY Y1 antagonist BIBP-3226.
(A) mHypoE-38 neurons were serum-starved for 4 h before treatment with KCl or vehicle for 1 h. NPY secretion was analyzed using an NPY-specific enzyme immunoassay (EIA). Results shown are relative to control (set to 1) and expressed as mean +/- SEM (n=9 independent experiments). (B) mHypoE-38 neurons were treated with vehicle (medium from unstimulated mHypoE-38 cells) or KCl (60 mM) for 15 min and conditioned media was collected and desalted. GT1-7 cells were treated with vehicle, NPY (100 nM) or conditioned media from mHypoE-38 neurons treated for 4 h. Neurons were also treated with desalted KCl alone and KCl alone in standard growth medium. RNA was collected and GnRH mRNA levels were quantified using real-time RT-PCR. GnRH mRNA levels were normalized to the internal control 18S. Results shown are relative to the corre-sponding vehicle treated mRNA levels (set to 1.0) and are expressed as mean ± SEM (n=4 indepen-dent experiments). (C) mHypoE-38 neurons were treated with vehicle or KCl (60 mM) for 15 min and conditioned media was collected and desalted. GT1-7 neurons were serum starved for 2 h before treatment with conditioned media or vehicle alone over a 4 h time course. Cells were pretreated the NPY Y1 receptor antagonist for 1 h at a final concentration of 1 !M before CM exposure. RNA was collected and GnRH mRNA levels were quantified using real-time RT-PCR. GnRH mRNA levels were normalized to the internal control 18S. Results shown are relative to the corresponding vehicle treated mRNA levels (set to 1.0) and are expressed as mean ± SEM (n=4 independent experiments). *P<0.05, **P<0.001 versus vehicle control, as per one-way ANOVA with Bonferroni’s post-hoc test (A,B) and two-way ANOVA with Bonferroni’s post-hoc test (C).

117
117
NPY peptide alone stimulates GnRH mRNA expression. To ensure NPY secreted from
the mHypoE-38 neurons mediates the increase in GnRH transcription and not other
potential neurosecretory factors from these neurons, I pretreated cells with the highly
selective NPY Y1 antagonist BIBP-3326. The co-treatment of NPY with BIBP-3326,
resulted in attenuation of the NPY-mediated increase in GnRH mRNA expression in the
GT1-7 neurons compared to NPY treatment alone, again suggesting NPY mediates its
increase in GnRH mRNA through the Y1 receptor subtype. Together, these studies
demonstrate that NPY is the key peptide released from the mHypoE-38 cell line required
to stimulate GnRH mRNA levels from GT1-7 cells.
5.4 Discussion
The most recognized functions of NPY include the regulation of endocrine
function, circadian rhythms, and satiety (90). Central administration of NPY stimulates
feeding and repeated doses results in an increase in body weight (107). NPY gene
expression and accumulation increase immediately before the preovulatory GnRH surge
(112, 294). Much insight into the exact role of NPY in reproductive physiology has been
achieved through the use of antisense oligonucleotides directed against NPY mRNA,
injected into the ARC of mice or primates (294, 304, 305). If NPY de novo synthesis is
blocked before the steroid-induced preovulatory rise in GnRH, the surge release of LH
causing ovulation does not occur (304) . Although several groups have documented the
importance of NPY regulating the reproductive axis, the transcriptional regulation and
cellular mechanisms governing the effect of NPY on GnRH transcription are not yet
described. Interestingly, a brief study of the in vivo effects of NPY on GnRH gene
expression found that GnRH mRNA levels were increased at 4 h (303), similar to what I
have found in the GT1-7 neurons. This study speculated that the Y1 receptor was

118
118
responsible for the increase in GnRH mRNA levels using the Y1/Y4/Y5 receptor agonist
[Leu31, Pro34], however the involvement of Y4 was not considered. In our study, I reveal
that NPY exposure increases GnRH over a 36 h time course possibly through the Y1 or
Y4 receptor in GT1-7 neurons.
NPY binds to at least four different receptor subtypes (Y1, Y2, Y4, Y5) belonging
to the seven-transmembrane domain G protein-coupled receptor (GPCR) superfamily
(101). Studies have attempted to elucidate which receptor is responsible for the
stimulatory effects of NPY on GnRH neurons (45, 70, 306). Previous studies using either
the GT1 cell line or GnRH neurons in situ have indicated that NPY can directly regulate
GnRH secretion through receptor subtypes NPY-Y1 (45, 73, 307), NPY-Y2 (45, 308), or
NPY-Y5 (266, 306). RT-PCR of GT1-7 total RNA failed to detect NPY Y5 receptor
mRNA transcript. Co-localization studies completed in vivo have demonstrated that 55%
of GnRH neurons express the Y5 receptor, indicating the Y5 receptor is differentially
expressed in GnRH neurons (309). A number of studies have supported the Y1 receptor
as the main receptor responsible for mediating the reproductive effects of NPY (45, 70,
75, 306, 310), although none have definitively ruled out the Y4 receptor. Most recently,
Klenke et al. found that NPY inhibits GnRH activity through the Y1 receptor subtype in
an explant model (311). The evidence that NPY acts through the Y1 receptor to mediate
its effects on GnRH neurons may not be decisive, as pharmacological profiles have
indicated that the selective Y1 agonist [Leu31, Pro34] also displays a significant affinity
for the Y4 and Y5 receptor subtypes (101). I do not detect the expression of Y5 in GT1-7
cells. Recent evidence has provided compelling reasons to include NPY Y4 receptors as
potential mediators of NPY action on the GnRH neuron. The NPY Y4 receptor, also
known as the primary receptor for pancreatic polypeptide (PP), is the least studied NPY

119
119
receptor subtype. Nonetheless, ICV injection of the Y1 antagonist and Y4 agonist
1229U91 rapidly and robustly increase GnRH secretion (312, 313). Since PP is not
found in the brain, NPY may therefore act as a central, albeit lower affinity ligand for the
Y4 receptor. Interestingly, the NPY Y4 receptor knockout restores fertility in the leptin-
deficient ob/ob mouse. However, ICV injections of human PP decreased plasma levels of
LH in ovariectomized rats (314). It appears that high basal levels of NPY in the ob/ob
mouse downregulates the HPG axis, specifically through Y4 receptor signaling (315).
The study implicating Y1 action in GnRH release in GT1 cells did not analyze expression
of the Y1 receptor nor the role of NPY Y4 (it was not yet cloned at the time) (45). To
approach which receptor subtype was active in the GT1-7 neurons, I have used specific
agonists of NPY receptor subtypes and assessed cAMP levels using an RIA. Previous
studies have demonstrated NPY primarily inhibits cAMP synthesis through actions on Gi
(102). The Y1/Y4/Y5 selective agonist [Leu31, Pro34] resulted in a surprisingly significant
accumulation of cAMP compared to vehicle control, an indication of receptor activation.
Nevertheless, cAMP activity was unchanged with the Y2 agonist NPY13-36, Y4 agonist
rPP, or the Y5 agonist D-[Trp32], suggesting that Leu31, Pro34 likely acted through the Y1
receptor. However, I cannot rule out the functional activity of the Y4 receptor, as rPP
may have decreased activity in the mouse; thus I could not detect a significant elevation
of cAMP in the GT1-7 neurons at the concentration used. Further, it has been suggested
that rPP does not have high affinity for the Y4 receptor (312, 313). Our results
demonstrating NPY can increase cAMP levels, suggesting NPY regulates cAMP in a
neuron-specific mechanism. Interestingly, pretreatments with the NPY Y1 antagonist
attenuated the NPY-mediated increase in GnRH mRNA, suggesting the Y1 subtype is the
main receptor for the NPY-mediated transcriptional regulation of GT1-7 neurons. Despite

120
120
this, additional studies will be performed once more specific agonists/antagonists are
available.
GPCR activation results in the dissociation of G-protein subunits that can either
stimulate or inhibit adenylyl cyclase activity (100). The stimulation of adenylyl cyclase
results in the conversion of ATP to cAMP. PKA becomes catalytically active in the
presence of cAMP and mediates most of cAMP’s actions. Interestingly, PKA activity has
been implicated in the stimulation of GnRH biosynthesis and secretion (316-318) and
also has been shown to either have no effect or to decrease GnRH mRNA in GT1-7
neurons (318, 319). Fittingly, in our study, NPY significantly up-regulates PKA
phosphorylation, suggesting the immediate increase in GnRH release could be a result of
rapid PKA phosphorylation. Our results demonstrate treatment with the PKA inhibitor
H89 alone did not alter basal GnRH mRNA levels, suggesting the basal levels of GnRH
mRNA operate independently of the PKA pathway in GT1-7 neurons. However, in the
presence of H89, NPY-mediated increases in GnRH mRNA were abolished, signifying
that the PKA pathway was required for the NPY-mediated increase in GnRH mRNA.
GPCRs also have the ability to activate the MAPK signaling cascade through the
interaction of specialized sub-domains (Gi and Gq/11) that dissociate with GPCR
activation (and also through PKC or activation sites such as Ras or Raf) (320-322).
Previous studies have demonstrated the activation of the MAPK pathway can regulate
GnRH mRNA expression (222, 319). Insulin treatment increases GnRH mRNA levels in
the Gnv-3 GnRH expressing neuron through the MAPK signaling cascade (222).
Pharmacological inhibitor studies further demonstrated the MAPK pathway is critical for
the melatonin-mediated down-regulation of GnRH mRNA expression (319). I show that
basal levels of GnRH mRNA expression are slightly repressed with the treatment of the

121
121
MEK1/2 inhibitor U0126. In contrast, in the presence of U0126, NPY-mediated increases
in GnRH mRNA levels are attenuated; implicating the MAPK pathway in NPY-induced
increases in GnRH mRNA.
Two regions within the GnRH gene, known as the promoter and enhancer region,
control GnRH transcription (79, 166, 323) (Figure 5.7). The promoter region is found
proximal to the transcriptional start site of the GnRH gene, and is particularly important
for basal GnRH gene expression (324). This region in the mouse and human is AT rich,
and therefore could be a critical region for a number of transcription factor binding motifs
(324). The enhancer region, found distal to the promoter region, was demonstrated to
mediate neuron-specific expression through deletion analysis studies (225). This 300-bp
enhancer site is located at the 5’ flanking site and binds to numerous nuclear proteins (79,
166, 323). The nuclear protein and immediate early gene cFos has been implicated in
controlling GnRH mRNA levels, as its protein product dimerizes with cJun to form the
transcription factor complex referred to as activator protein-1 (AP1). In order to elucidate
potential factors involved in the regulation of GnRH transcription, I analyzed
downstream effectors of the PKA and MAPK pathway, such as the transcription factors
ATF-1 and CREB. Both are phosphorylated upon NPY stimulation in GT1-7 neurons.
Interestingly, CREB may directly activate cFos, a component of the AP-1 protein critical
for GnRH transcriptional regulation. Further studies are required to determine whether or
not NPY regulates GnRH mRNA expression indirectly through the AP-1 complex or
through other elements within the promoter or GnRH enhancer region.
I describe the regulation of GnRH gene expression by NPY in GT1-7 neurons,
and importantly that conditioned medium from NPY-expressing mHypoE-38 neurons
mimic this response. I demonstrated that the transcriptional regulation of GnRH mRNA
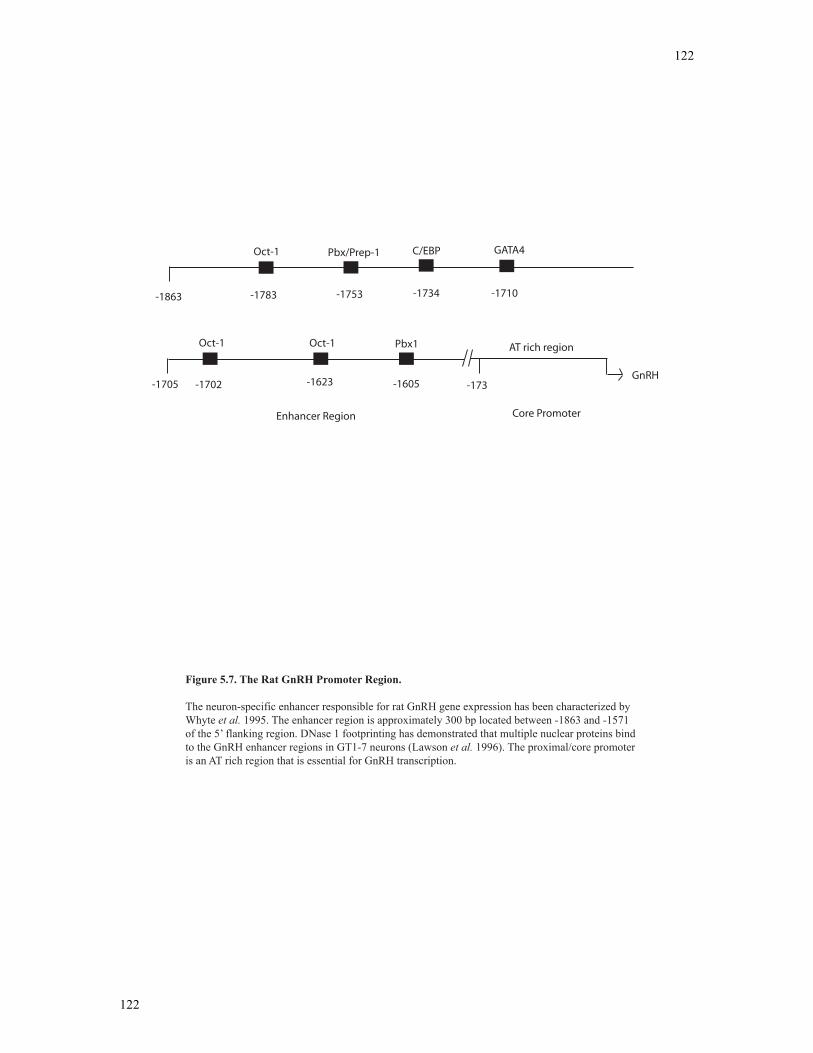
122
122
-1863
Oct-1
-1783
Pbx/Prep-1
-1753 -1734
C/EBP
-1710
GATA4
Oct-1 Oct-1 Pbx1
-1702 -1623 -1605-1705 -173
AT rich region
GnRH
Core PromoterEnhancer Region
Figure 5.7. The Rat GnRH Promoter Region.
The neuron-specific enhancer responsible for rat GnRH gene expression has been characterized by Whyte et al. 1995. The enhancer region is approximately 300 bp located between -1863 and -1571 of the 5’ flanking region. DNase 1 footprinting has demonstrated that multiple nuclear proteins bind to the GnRH enhancer regions in GT1-7 neurons (Lawson et al. 1996). The proximal/core promoter is an AT rich region that is essential for GnRH transcription.

123
123
in GT1-7 neurons responds to NPY predominantly through either the Y1 or Y4 receptor,
and that NPY exposure increased GnRH mRNA over a 36 h time course in the absence of
steroidal modulators. Western blot analysis revealed that NPY activates the PKA and
MAPK signal transduction pathways, as well as transcription factors CREB and ATF-1
that are potential transcriptional mediators of the NPY response. Through the use of
pharmacological inhibitors, I determined that the NPY-mediated induction of GnRH
transcription is dependent on the signal transduction cascades PKA and MAPK pathways.
These studies provide the paradigm in which to study the potential downstream
transcriptional mechanisms involved in this response. Importantly, I also demonstrate
NPY secreted from the mHypoE-38 neurons stimulates GnRH mRNA expression in
GT1-7 neurons. Finally, with receptor specific antagonist studies, I demonstrated that the
effects of NPY are mediated through the NPY Y1 or Y4 receptor subtype. Finally,
conditioned media treatment experiments indicate that NPY and not other hypothalamic
factors secreted by the mHypoE-38 cell lines was required to stimulate GnRH
transcription in the GT1-7 cell lines. These studies provide expanded evidence of the
significance of NPY in the regulation of the GnRH neuron itself in the absence of
steroidal modulators (see Figure 5.8).

124
124
GnRHneuron
PKA
CREB/ATF-1
P
P
Regulatory Region Core Promoter GnRH Gene
cAMP
ERKP
Y4
Y1
U0126
H89
GnRHmRNACREB/
ATF-1
P
NPYneuron NPY
BIBP-3326
Figure 5.8. Model of the potential cellular signaling pathways involved in NPY regulation of GnRH mRNA levels.
NPY directly increases GnRH mRNA levels in the GnRH-expressing GT1-7 GnRH neurons over an extended time-course. Conditioned media treatments from KCl-stimulated mHypoE-38 neurons also directly stimulates GnRH mRNA levels. This effect was found to occur through the NPY Y1/Y4 receptor subtype. NPY was found to stimulate the cAMP/PKA and MAPK pathways through Western blot analysis. The NPY-mediated increase in GnRH mRNA levels was linked to the MAPK and PKA signaling pathways using pharmacological inhibitors against key signaling molecules.

125
125
6 Chapter 6
Overall Discussion and Future Directions

126
126
6.1 Overall Discussion
Nutritional status can have marked effects on all aspects of physiology, particularly
on growth, metabolism and reproduction (85-87, 325). Although the consequential effects
of nutritional deprivation are well documented, the mechanisms by which the body
perceives changes in nutritional status and uses this information to regulate reproductive
processes and neuroendocrine function are less understood. Leptin and E2 are endocrine
cues that link energy status to hypothalamic pathways regulating appetite and
reproduction (7, 204, 259). To date, the preponderance of literature describing the E2- and
leptin-mediated regulation of hypothalamic neuropeptides is based on whole
hypothalamic extracts/tissue. Characterization of the direct regulation by E2 and leptin on
individual neuropeptidergic neurons, however, lags considerably behind. NPY, a
hypothalamic orexigenic peptide involved in the regulation of appetite and reproduction,
work in concert with leptin and E2 (81, 291). Although scientists have studied NPY for
over 20 years, much remains to be determined about the peripheral hormonal stimulants
and the cellular mechanisms through which NPY neurons are regulated to maintain
energy homeostasis and reproductive function. In this thesis, I examined the direct
regulation of NPY neurons by endocrine hormones, E2 and leptin to evaluate the cellular
signaling mechanisms and the secretory responses of individual NPY neuronal cell
models. Interestingly, food deprivation that is accompanied by abnormal
neuropeptidergic function inhibits the HPG axis at all levels, including the secretion of
GnRH from the hypothalamus, the secretion of LH and FSH from the anterior pituitary
and the secretion of gonadal steroids (85, 86, 326, 327). In my final studies, I determined
the influence of NPY neuropeptide content from NPY-synthesizing neurons on the direct
regulation of the HPG axis at the level of the GnRH neuron. This dissertation provides

127
127
additional mechanistic evidence of the relationship between feeding and reproductive
homeostasis in individual hypothalamic neuropeptidergic cell lines.
Menopause, the age-related loss of estrogen and progestins, is associated with an
increased risk of obesity and adiposity (169, 170, 328). E2 replacement therapy decreases
body weight and reduces appetite in menopausal women, suggesting that E2 plays an
important role in appetite regulation (329). Studies examining the suppressive effect of E2
on food intake have focused on the two main ER subtypes, ER-α and ER-β. Results from
studies using ER specific agonists, ER-α knockdown and gene knockout mice studies
provide evidence that ER-α mediates the anorectic action of E2 (19, 120, 248, 330).
However, studies using an ER-β ODN knockdown approach also inhibited the
anorexigenic effect of E2 (249). In addition to the uncertainty regarding which ERs are
responsible for the anorectic action of E2, there have been no reports on the direct action
of E2 on hypothalamic neuropeptide secretion. A recent study published by Olofsson et
al. found that E2 inhibits NPY mRNA expression in hypothalamic explants through an
ER-α-dependent mechanism (171). The study failed to co-localize ER-α in NPY
neurons, suggesting that E2 works through an indirect mechanism.
Taking an alternative approach to confirm ER-α expression in NPY neurons, I used
FACS to isolate NPY-GFP cells from the hypothalamus of the NPY-GFP transgenic
mouse, and RT-PCR to detect ER-α. The result concurs with previous reports that
demonstrate that ER-α and NPY are likely co-expressed in the same neurons in vivo
(163, 164). Using a combination of ER specific agonists/antagonists and E2-conjugated
BSA, I discerned that membrane-bound ER-α is directly responsible for the E2-mediated
decrease in NPY secretion in two NPY neuronal cell lines, mHypoE-42 and mHypoA-
2/12. Although feeding is regulated by a number of orexigenic and anorexigenic feeding

128
128
circuits, NPY is the most potent orexigenic compound (331). Therefore this E2-mediated
decrease in NPY secretion observed in our neuronal cell model may indicate that NPY
plays a large role in the anorectic action of E2.
Recent studies have demonstrated that AMPK and PI3K signaling are essential in
the maintenance of food intake at the level of the hypothalamus (283). AMPK is a
cellular energy sensor activated by peripheral endocrine signals during low energy status
periods (280). Minokoshi et al. have demonstrated that exogenous leptin administration
inhibits AMPK activity in the ARC (191). Inhibitor studies have further demonstrated
that hypothalamic AMPK activity is essential for the anorectic action of leptin (281). In
addition, studies on peripheral tissue have linked estrogen to increases in AMPK activity
that translates into enhanced beta-oxidation and metabolism (332). However, such studies
have not examined whether or not AMPK signaling is involved in the anorectic action of
estrogen at the level of the hypothalamus. Studies have also strongly implicated the PI3K
pathway in the hypothalamic neurons that influence food intake and body weight (209,
268, 275). The PI3K pathway has been considered a major signaling cascade in
mediating the action of metabolic hormones insulin and leptin as demonstrated both in
vitro and in vivo (276). E2 is also a major regulator of the PI3K pathway as demonstrated
in vitro (178, 333). However, studies have yet to conclusively link estrogen and the PI3K
pathway to energy homeostasis. In this thesis, our neuronal cell models have
demonstrated for the first time that leptin directly decreases NPY secretion through an
AMPK- and PI3K-dependent mechanism. In addition, I have linked the anorexigenic
action of E2 to AMPK and PI3K activity in NPY cell models. A noteworthy observation
here is that both leptin and E2 act through identical signaling mechanisms in order to alter
NPY neuronal activity. The altered NPY neuronal activity, in turn, could reduce food

129
129
intake. These identical signaling mechanisms could prove to be a pharmacological target
for menopausal women and/or leptin deficient individuals.
The failure of leptin to reduce appetite and weight loss in obese individuals gives
rise to the notion of leptin resistance, which is impaired leptin action in obese states (208-
211, 213, 284, 334). Leptin resistance is a complex subject that appears to occur at
multiple levels of physiology. At the hypothalamic level, the mechanisms underlying
leptin resistance have included impaired STAT3, PI3K, SOCS3 and PTP1B function in
the ARC (214). High concentrations of leptin have effectively limited the activity of these
signaling molecules in vivo (213, 335-337). Studies have yet to confirm the specific
neuronal populations in the ARC that are susceptible to leptin resistance and whether or
not the energy sensing kinase, AMPK, is altered in leptin resistant states. In this study, I
have demonstrated that the leptin-mediated decrease in AMPK phosphorylation is greatly
attenuated in NPY neuronal cell lines with prolonged exposure to leptin. This impaired
AMPK signaling was accompanied by the inability of the NPY cell lines to respond
appropriately to the leptin-mediated decrease in NPY secretion. This is the first
demonstration of impaired AMPK signaling and NPY secretory response to prolonged
leptin exposure. It provides essential information for potential neuroanatomical
determinants of leptin resistance and of the underlying impaired signaling mechanisms
that contribute to obese states.
In an unexpected finding, leptin was found to increase NPY secretion from the
mHypoE-38 neuronal cell line after a 1 h treatment. This leptin-mediated increase in
NPY secretion occurred through a MAPK-dependent mechanism. Previous studies have
implicated MAPK signaling with the stimulatory role of leptin in reproduction and
growth in mice (206, 272). Additional studies have also implicated the mHypoE-38 cell

130
130
line as a putative reproductive NPY neuronal cell line. Using this model, Titolo et al.
demonstrated that E2 induced a surge of NPY mRNA transcript levels after 24 h estrogen
exposure that was proposed to be involved in the GnRH preovulatory surge (163, 164).
Based on this evidence, I suspected that the leptin-mediated MAPK-dependent increase
in NPY secretion could stimulate GnRH neuronal activity. I used GT1-7 GnRH-
expressing cell lines and treated these cells with conditioned media from stimulated
mHypoE-38 neurons. Interestingly, media taken from the stimulated mHypoE-38 NPY
cell lines induced a significant induction in GnRH mRNA levels that occurred
exclusively through the Y1 receptor subtype. As a result, I propose that leptin may
stimulate a subset of NPY neuronal cells that project to GnRH cell bodies in order to
stimulate the reproductive axis. Future in vivo studies will be instrumental in verifying
and substantiating this hypothesis.
NPY has a paradoxical effect on the reproductive axis. Depending on the mode of
its administration, the hormonal status of the experimental animal and the time course of
the treatment, NPY can exert both suppressive and excitatory effects on the HPG axis
(45, 75, 78, 88, 294). Besecke et al. have previously investigated the secretory responses
of GnRH to NPY treatment in the GT1-7 immortalized cell line using a flow-through
superfusion system (45). In this study, NPY directly stimulated the release of GnRH in a
dose-dependent manner primarily through Ca(2+) dependent mechanisms. I have
extended this research by showing that NPY can directly stimulate GnRH mRNA levels
in the GT1-7 cell models. I am the first to show through inhibitor analysis that this occurs
through a MAPK- and PKA-dependent mechanism. NPY is an essential neurotransmitter
that serves as a communication bridge between the neural processes that regulate
reproduction and those that maintain energy homeostasis. The studies presented herein

131
131
provide additional mechanistic descriptions of the control of GnRH-expressing cell lines
by NPY in the absence of steroidal modulators.
The functional heterogeneity revealed in the NPY secretory responses to hormonal
treatments in our NPY-expressing cell lines provides a cellular basis for the role of NPY
neuronal subpopulations in regulating diverse neuroendocrine processes. This
observation coincides with studies in vivo that support the notion of functionally diverse
NPY neuronal populations. One of the first studies to identify biochemically distinct
NPY neuronal populations used fluorescent double-immunolabeling for NPY and
glutamic acid decarboxylase (292). This study found that only one third of ARC NPY
neuronal perikarya co-expressed the glutamic acid decarboxylase. Ensuing studies found
two distinct NPY subpopulations: 1) a subset of NPY and glutamic acid decarboxylase
co-producing cells located in the dorsomedial ARC, and 2) a subset of NPY cells in the
ventral ARC (292). Additional studies found heterogeneous electrophysiological
properties of NPY neurons in vivo. Using patch-clamp recordings, Fioramonti et al.
demonstrated that only 40% of NPY neurons are glucose-inhibited, indicating that a
distinct population of NPY neurons that are glucose-sensing (338). Most recently, Padilla
et al. demonstrated using a combination of fluorescence in situ hybridization (FISH) and
lineage tracing studies that approximately 17% of NPY neurons are derived from POMC
progenitor cells (202). This study indicated that a subset of NPY neurons in the
hypothalamus is derived from a specific population of progenitor cells that remain
distinct from other hypothalamic NPY neurons. This finding suggests NPY neuronal
heterogeneity. Together, studies in vivo strongly suggest a heterogeneity amongst
hypothalamic NPY cell populations, which aligns with our observations in vitro that
NPY-expressing cell lines respond uniquely to hormonal treatments, and together these

132
132
results begin to describe the cellular basis for the multifaceted control of NPY in energy
homeostasis and reproduction (see Figure 6.1).

133
133
E2
Leptin
ER
ObR
NPY
NPY
GnRHneuron
Intracellularsignalingpathways
Intracellularsignalingpathways
GnRH geneexpression
Preopticnucleus
VMH/LH/PVN
Feeding
Reproduction
1
2
3
Figure 6.1. Summary of findings.
(1) Chapter 3 describes the anorexigenic action of estrogen in NPY neuronal cell lines, mHypoE-42 and mHypoA-2/12. Here, estrogen decreases NPY secretion through membrane-bound ER- and through the PI3K and AMPK signaling pathways. (2) In Chapter 4, leptin differentially regulates NPY neuronal cell lines. Leptin decreases NPY secretion in the NPY-GFP and mHypoA-59 cell lines, which was also mediated through the PI3K and AMPK path-ways. Pronlonged leptin exposure prevents the decrease in NPY secretion and phospho-AMPK, indicating NPY neurons are susceptible to leptin-induced leptin resistance. Conversly, leptin increases NPY secretion in the mHypoE-38 cell line, occuring through the MAPK and PI3K pathways. (3) In Chapter 5, KCl-stimulated mHypoE-38 media directly placed onto GT1-7 cells increases GnRH mRNA levels through the Y1 receptor subtype. These studies provide addi-tional evidence that NPY neurons play a role in both reproductive function and appetite regulation,which may be mediated by spatially segregated circuitries projecting to medial preoptic GnRH neurons or feeding related nuclei including the PVN, LH and VMH.

134
134
6.2 Limitations
The strength of using immortalized hypothalamic clonal cell lines is their capacity
to demonstrate the cause-and-effect relationships of specific neuromodulators. However,
when taking this reductionist approach, scientists must avoid overstating their
conclusions since immortalized cell lines lack the heterogeneous and complex neuronal
architecture observed in vivo. The cells used in this study were isolated from both
embryonic and adult hypothalamic sources. The embryonic cell lines were derived from
mice at days embryonic 15 (E15), E17 and E18 during significant hypothalamic
neurogenesis (219). At this time of development, hypothalamic circuits are not fully
formed, which could result in functionally or phenotypically underdeveloped cells
compared to mature adult neurons. However, numerous studies using these embryonic
neuronal cell models have found that these neurons function similar to native adult
neurons (54, 216, 236-241, 339). In addition, I corroborated these studies by using a
combination of clonal and mixed adult hypothalamic cell lines. Collectively, the use of
multiple neuronal phenotypes from both embryonic and adult sources has allowed
scientists to analyze the direct effect of neuromodulators on individual neuronal cell
populations.
Although cell lines allow for the elucidation of molecular processes, a major
disadvantage is that the immortalizing gene SV40 T-antigen can interfere with cellular
processes. This might affect the pathways studied. Preliminary data from our laboratory
using the shRNA knockdown of T-antigen demonstrates that T-antigen expression
increases the basal activity of cellular signaling kinases AKT, Jak2 and STAT3
(Belsham, unpublished data). As a result, I have attempted to reduce the basal activity of

135
135
these signaling kinases using serum starvations, low glucose and/or pH modifications
during experimentation. Importantly, I found that T-antigen expression did alter key
genes of interest including ER-α and NPY in our hypothalamic cell lines. Additional
studies completed by May et al. demonstrate that T-antigen expression in murine
embryonic fibroblasts have only 379 genes altered (out of 22,600 probes in total) altered
compared to nontransformed cells, with the majority of these genes involved with
proliferation and nucleotide synthesis (340). Together, these studies show that
immortalized cell lines comprise a useful tool for unraveling complex signaling pathways
and for studying the direct action of neuromodulators on specific neuronal phenotypes.
Of course, researchers should design studies carefully in order to optimize experimental
conditions and to avoid overstating conclusions.
In Chapter 3, the membrane impermeable conjugate, E2-BSA, was used to analyze
the role of cell membrane-bound ERs in the regulation of NPY secretion. However,
recent studies have demonstrated that E2-BSA data should be interpreted cautiously. This
study analyzed the forms of commercially available E2-BSA conjugates and found that 3-
5 % of E2-BSA is available in the free unconjugated E2 form (341). Taguchi et al. have
also provided evidence that E2-BSA can induce ERE-driven luciferase gene activity in
neuroblastoma cells (341). This finding suggests the involvement of nuclear ER.
However, other studies have demonstrated that E2-BSA does not increase the
transcription of ERE-based reporter genes in human neuroblastoma cells (342). These
studies suggest that E2-BSA does not enter the cell and is unable to activate nuclear ER.
For this reason, it is advisable to filter E2-BSA to remove free E2 or to use the newly
generated and more stable E2 conjugate, E2 dendrimers (EDCs), produced by the
Katzenellenbogen lab (343).

136
136
Pharmacological inhibitors have been widely used in the study of signal
transduction and have provided invaluable insight into the function of signaling kinases
in various cell types. However, a substantial body of evidence indicates that inhibitors
can have non target effects, which are independent of inhibiting intended proteins.
Although these actions are varied, the inhibitors used in our studies have been
demonstrated to act on several unintended proteins. For example, LY294002 can
potentially inhibit glycogen synthase kinase and casein kinase 2; H89 can inhibit non-
PKA kinases including MAPK and calcium signaling pathways; U0126 can also inhibit
calcium signaling and calmodulin-dependent kinase (344). As a result, although
pharmacological inhibitors are valuable tools scientists use to dissect intracellular
signaling mechanisms, the potential of the inhibitors to act on a variety of other cellular
processes limits the conclusions that can be drawn. Additional studies utilizing RNA
interference or the introduction of dominant negative versions of protein kinases could be
used to study the role of specific signaling pathways. Although these methods can be
accompanied by transfection difficulties and inadequate construct expression, the results
of these techniques have an extremely high specificity compared to the results of
techniques using pharmacological agents. This high specificity allows for much more
decisive conclusions.
6.3 Future directions of study
This thesis describes the cellular mechanisms that leptin, E2 and NPY act through
to regulate neuropeptide mRNA and secretion levels from hypothalamic neuronal cell
lines. Although these studies identify important relationships between signal kinase
activity and neuronal function, several new questions arose during the completion of
these studies that warrant further investigation.

137
137
Studies have demonstrated that E2 deficient mice have decreased leptin
responsiveness, suggesting an interplay between estrogen and leptin signaling, although
how this occurs is unclear (345). Our group and others have found co-expression of ER
(both ER-α and ER-β) and Ob-R in NPY hypothalamic cell lines and our group is
currently in the process of confirming this finding in vivo (346). Additionally, Thorn et
al. has demonstrated that E2 can modulate Ob-R mRNA in specific E2-responsive tissue
(347). Most recently, preliminary studies from our laboratory indicate that the biphasic
regulation of NPY mRNA by E2 in the mHypoE-38 cell line can be attenuated with leptin
co-treatment (Belsham, unpublished data). Utilizing the NPY-expressing cell lines that
are responsive to hormonal treatments identified in this thesis, I could determine whether
or not E2 and leptin co-treatments can modify the NPY secretory responses to the
individual hormonal treatments. These experiments will allow us to determine whether
leptin or E2 pre/co-treatments can enhance the negative or positive effect on NPY release.
In order to identify signaling pathways involved in potential E2 and leptin cross-talk,
assays which contain high throughout signaling kinase analysis of protein
phosphorylation such as the R&D Systems Proteome Array profiler could be utilized.
These studies are of immense interest given the identification of the identical signaling
mechanisms involved in the leptin and E2 regulation of NPY secretion in hypothalamic
cell lines and given the paucity of information known about the interaction of these
hormones in the hypothalamus.
Although leptin is linked to the neuroendocrine regulation of the reproductive axis,
the direct regulation of the GnRH neuron by leptin has not been described. To date, leptin
receptors have not conclusively been found on GnRH neurons in vivo (264). This may be
due to technological limitations and inadequate staining sensitivity. Using RT-PCR, I

138
138
have found Ob-R mRNA in the newly generated GnRH-GFP immortalized cell lines and
our group is currently in the process of confirming these findings in vivo using a newly
available Ob-R antibody and GnRH-GFP hypothalamic slices (Belsham lab, unpublished
data). Additionally, I have preliminary data that demonstrates that leptin treatments can
activate the newly generated GnRH-GFP cell model by measuring cFos mRNA levels in
response to leptin (Belsham, unpublished data). Future studies could further analyze the
involvement of leptin action on the GnRH-expressing cell lines in these newly generated
GnRH-GFP cell populations by examining the GnRH secretory responses, transcriptional
changes and signaling mechanisms involved. In addition, E2 has been demonstrated to
enhance the GnRH neuronal responses to NPY (20). Further experimentation could
determine whether leptin requires the steroid hormone, E2, to exert this stimulatory effect
on GnRH neuronal cell lines. These studies would indicate that like E2, leptin might
modulate the reproductive axis through multiple mechanisms.
I found that prolonged treatment with leptin prevented the leptin-mediated
regulation of the AMPK phosphorylation and NPY secretion. To fully understand the
cellular mechanisms of leptin resistance and obesity, additional mechanistic studies could
be completed to decipher how hyperleptinemia results in desensitization in NPY neuronal
cell lines. Initially, experiments could determine the minimum concentration that is
required to induce leptin resistance in NPY neuronal cell lines. Dose response curves
using 0.1 pM to 100 nM leptin treatments could be used over a 4, 8 and 24 h time course.
Identifying the minimum length of exposure and concentration of leptin that is required
to induce leptin resistance would be valuable information for future experimental
paradigms. Leptin signaling is under the negative feedback control of SOCS3 and PTP1B
(335, 336). Thus, in order to begin to understand the potential role of these negative

139
139
regulators in hyperleptinemic conditions, studies could examine the effect of the
overexpression of SOCS3 and PTP1B and its effect on the signaling kinases, AMPK, in
NPY-neuronal hypothalamic cell lines. The commencement of leptin resistance with
SOCS3 and PTP1B overexpression would suggest such a mechanism. Through Western
blot analysis, studies could also examine whether or not prolonged leptin exposure
increases Ob-R degradation (285). Recent studies demonstrate that Ob-R can be degraded
via an ubiquitin-dependent endocytosis mechanism (183, 285). Studying leptin resistance
in NPY neuronal cell populations would therefore allow for a detailed description of
neuronal resistance in hyperleptinemic cases.
The differential regulation of NPY secretory responses from the immortalized
NPY-synthesizing cell lines should be confirmed in vivo. Initial studies should attempt to
identify bona fide gene markers specific to each NPY subpopulation. The identification
of cellular markers specific to each NPY subpopulation could be completed using
microarray analysis or RT-PCR. These newly identified cellular markers would allow for
the confirmation of these NPY subpopulations in vivo using IHC double-labeling
techniques. Ideally, NPY subpopulations from the ARC that project to GnRH neurons in
the anterior hypothalamus would display mHypoE-38 specific markers, and NPY
subpopulations that project to feeding related nuclei including the PVN and LH from the
ARC would selectively express markers identified in the NPY-GFP and mHypoA-59 cell
lines. The completion of these studies in vivo would substantially strengthen our
hypothesis of NPY neurons acting as the integration centre for peripheral hormones to
regulate food intake and reproduction.

140
140
7 Chapter 7 – References

141
141
References
1. Ahima RS, Osei SY. Molecular regulation of eating behavior: new insights and prospects for therapeutic strategies. Trends Mol Med. [2001 May;7(5):205-13.
2. Richy S, Burlet A, Max J, Burlet C, Beck B. Effect of chronic intraperitoneal injections of leptin on hypothalamic neurotensin content and food intake. Brain Res. [2000;862:276-9.
3. Sahu A, Carraway RE, Wang YP. Evidence that neurotensin mediates the central effect of leptin on food intake in rat. Brain Res. [2001;888:343-7.
4. Sahu A. Evidence suggesting that galanin (GAL), melanin-concentrating hormone (MCH), neurotensin (NT), proopiomelanocortin (POMC) and neuropeptide Y (NPY) are targets of leptin signaling in the hypothalamus. Endocrinology. [1998 Feb;139(2):795-8.
5. Watanobe H, Habu S. Leptin regulates growth hormone-releasing factor, somatostatin, and alpha-melanocyte-stimulating hormone but not neuropeptide Y release in rat hypothalamus in vivo: relation with growth hormone secretion. J Neurosci. [2002;22:6265-71.
6. Elias CF, Aschkenasi C, Lee C, Kelly J, Ahima RS, Bjorbaek C, et al. Leptin differentially regulates NPY and POMC neurons projecting to the lateral hypothalamic area. Neuron. [1999 Aug;23(4):775-86.
7. Kalra SP, Dube MG, Pu S, Xu B, Horvath TL, Kalra PS. Interacting appetite-regulating pathways in the hypothalamic regulation of body weight. Endocr Rev. [1999 Feb;20(1):68-100.
8. Thorburn AW, Proietto J. Neuropeptides, the hypothalamus and obesity: insights into the central control of body weight. Pathology. [1998 Aug;30(3):229-36.
9. Fernandez-Fernandez R, Martini AC, Navarro VM, Castellano JM, Dieguez C, Aguilar E, et al. Novel signals for the integration of energy balance and reproduction. Molecular and cellular endocrinology. [2006 Jul 25;254-255:127-32.
10. Kennedy GC. The relation between the central control of appetite, growth and sexual maturation. Guy's Hospital reports. [1969;118(3):315-27.
11. Sabatino FD, Murnane JM, Hoffman RA, McDonald JK. Distribution of neuropeptide Y-like immunoreactivity in the hypothalamus of the adult golden hamster. J Comp Neurol. [1987 Mar 1;257(1):93-104.
12. Woller MJ, McDonald JK, Reboussin DM, Terasawa E. Neuropeptide Y is a neuromodulator of pulsatile luteinizing hormone-releasing hormone release in the gonadectomized rhesus monkey. Endocrinology. [1992;130:2333-42.
13. Xu M, Hill JW, Levine JE. Attenuation of luteinizing hormone surges in neuropeptide Y knockout mice. Neuroendocrinology. [2000;72:263-71.

142
142
14. Osterlund M, Kuiper GG, Gustafsson JA, Hurd YL. Differential distribution and regulation of estrogen receptor-alpha and -beta mRNA within the female rat brain. Brain Res Mol Brain Res. [1998 Feb;54(1):175-80.
15. Friedman JM, Halaas JL. Leptin and the regulation of body weight in mammals. Nature. [1998 Oct 22;395(6704):763-70.
16. Shughrue PJ, Lane MV, Merchenthaler I. Comparative distribution of estrogen receptor-alpha and -beta mRNA in the rat central nervous system. The Journal of comparative neurology. [1997 Dec 1;388(4):507-25.
17. Mercer JG, Hoggard N, Williams LM, Lawrence CB, Hannah LT, Morgan PJ, et al. Coexpression of leptin receptor and preproneuropeptide Y mRNA in arcuate nucleus of mouse hypothalamus. Journal of neuroendocrinology. [1996 Oct;8(10):733-5.
18. Mercer JG, Hoggard N, Williams LM, Lawrence CB, Hannah LT, Trayhurn P. Localization of leptin receptor mRNA and the long form splice variant (Ob-Rb) in mouse hypothalamus and adjacent brain regions by in situ hybridization. FEBS Lett. [1996 Jun 3;387(2-3):113-6.
19. Couse JF, Korach KS. Estrogen receptor null mice: what have we learned and where will they lead us? Endocr Rev. [1999 Jun;20(3):358-417.
20. Shimizu H, Ohtani K, Kato Y, Tanaka Y, Mori M. Withdrawal of [corrected] estrogen increases hypothalamic neuropeptide Y (NPY) mRNA expression in ovariectomized obese rat. Neuroscience letters. [1996 Feb 2;204(1-2):81-4.
21. Elmquist JK, Bjorbaek C, Ahima RS, Flier JS, Saper CB. Distributions of leptin receptor mRNA isoforms in the rat brain. J Comp Neurol. [1998;395:535-47.
22. Filicori M, Crowley WF, Jr. The study of GnRH control of reproductive function. Upsala journal of medical sciences. [1984;89(1):13-8.
23. Harris GW. Sex Hormones, Brain Development and Brain Function. Endocrinology. [1964 Oct;75:627-48.
24. McCann SM, Taleisnik S, Friedman HM. LH-releasing activity in hypothalamic extracts. Proc Soc Exp Biol Med. [1960;104:432-43.
25. Hillier SG. Gonadotropic control of ovarian follicular growth and development. Molecular and cellular endocrinology. [2001 Jun 20;179(1-2):39-46.
26. Santoro N, Filicori M, Spratt D, Crowley WF, Jr. Gonadotropin-releasing hormone (GnRH) physiology in men and women. Acta Med Hung. [1986;43(2):201-21.
27. Kalra PS, Kalra SP. Steroidal modulation of the regulatory neuropeptides: luteinizing hormone releasing hormone, neuropeptide Y and endogenous opiod peptides. J steroid Biochem. [1986;25:733-40.

143
143
28. Kalra SP, Kalra PS. Do testosterone and estradiol-17β enforce inhibition or stimulation of luteinizing hormone-releasing hormone secretion? Biol Reprod. [1989;41:559-70.
29. Matsuo H, Baba Y, Nair RM, Arimura A, Schally AV. Structure of the porcine LH- and FSH-releasing hormone. I. The proposed amino acid sequence. Biochem Biophys Res Commun. [1971;43(6):1334-9.
30. Hoffman GE, Berghorn KA. Gonadotropin-releasing hormone neurons: their structure and function. Semin Reprod Endocrinol. [1997;15(1):5-17.
31. Adelman JP, Mason AJ, Hayflick JS, Seeburg PH. Isolation of the gene and hypothalamic cDNA for the common precursor of gonadotropin-releasing hormone and prolactin release-inhibiting factor in human and rat. Proceedings of the National Academy of Sciences of the United States of America. [1986 Jan;83(1):179-83.
32. Bond CT, Hayflick JS, Seeburg PH, Adelman JP. The rat gonadotropin-releasing hormone: SH locus: structure and hypothalamic expression. Molecular endocrinology (Baltimore, Md. [1989 Aug;3(8):1257-62.
33. Conn PM, Crowley WF, Jr. Gonadotropin-releasing hormone and its analogues. The New England journal of medicine. [1991 Jan 10;324(2):93-103.
34. Sagrillo CA, Grattan DR, McCarthy MM, Selmanoff M. Hormonal and neurotransmitter regulation of GnRH gene expression and related reproductive behaviors. Behav Genet. [1996 May;26(3):241-77.
35. Wray S, Grant P, Gainer H. Evidence that cells expressing luteinizing hormone-releasing hormone mRNA in the mouse are derived from progenitor cells in the olfactory placode. Proceedings of the National Academy of Sciences of the United States of America. [1989 Oct;86(20):8132-6.
36. Wray S, Nieburgs A, Elkabes S. Spatiotemporal cell expression of luteinizing hormone-releasing hormone in the prenatal mouse: evidence for an embryonic origin in the olfactory placode. Brain Res Dev Brain Res. [1989 Apr 1;46(2):309-18.
37. Hayflick JS, Adelman JP, Seeburg PH. The complete nucleotide sequence of the human gonadotropin-releasing hormone gene. Nucleic acids research. [1989 Aug 11;17(15):6403-4.
38. King JA, Millar RP. Isolation and structural characterization of chicken hypothalamic luteinizing hormone releasing hormone. J Exp Zool. [1984 Dec;232(3):419-23.
39. Lovejoy DA, Fischer WH, Ngamvongchon S, Craig AG, Nahorniak CS, Peter RE, et al. Distinct sequence of gonadotropin-releasing hormone (GnRH) in dogfish brain provides insight into GnRH evolution. Proceedings of the National Academy of Sciences of the United States of America. [1992 Jul 15;89(14):6373-7.

144
144
40. Ngamvongchon S, Lovejoy DA, Fischer WH, Craig AG, Nahorniak CS, Peter RE, et al. Primary structures of two forms of gonadotropin-releasing hormone, one distinct and one conserved, from catfish brain. Mol Cell Neurosci. [1992 Feb;3(1):17-22.
41. Goldsmith PC, Ganong WF. Ultrastructural localization of luteinizing hormone-releasing hormone in the median eminence of the rat. Brain Res. [1975;97:181-93.
42. Martinez de la Escalera G, Choi AL, Weiner RI. Biphasic GABAergic regulation of GnRH secretion in GT1 cell lines. Neuroendocrinology. [1994;59:420-5.
43. Colledge WH. Kisspeptins and GnRH neuronal signalling. Trends in endocrinology and metabolism: TEM. [2009 Apr;20(3):115-21.
44. Crowley WR, Hassid A, Kalra SP. Neuropeptide Y enhances the release of luteinizing hormone (LH) induced by LH-releasing hormone. Endocrinology. [1987 Mar;120(3):941-5.
45. Besecke LM, Wolfe AM, Pierce ME, Takahashi JS, Levine JE. Neuropeptide Y stimulates luteinizing hormone-releasing hormone release from supefused hypothalamic GT1-7 cells. Endocrinology. [1994;135:1621-7.
46. Ferris CF, Pan JX, Singer EA, Boyd ND, Carraway RE, Leeman SE. Stimulation of luteinizing hormone release after stereotaxic microinjection of neurotensin into the medial preoptic area of rats. Neuroendocrinology. [1984 Feb;38(2):145-51.
47. Martinez de la Escalera G, Gallo F, Choi ALH, Weiner RI. Dopaminergic regulation of the GT1 gonadotropin-releasing hormone (GnRH) neuronal cell lines: stimulation of GnRH release via D1-receptors positively coupled to adenylate cyclase. Endocrinology. [1992;131:2965-71.
48. Martinez de la Escalera G, Choi AL, Weiner RI. Beta 1-adrenergic regulation of the GT1 gonadotropin-releasing hormone (GnRH) neuronal cell lines: stimulation of GnRH release via receptors positively coupled to adenylate cyclase. Endocrinology. [1992 Sep;131(3):1397-402.
49. Moretto M, Lopez FJ, Negro-Vilar A. Nitric oxide regulates luteinizing hormone-releasing hormone secretion. Endocrinology. [1993;133(5):2399-402.
50. Gonzalez-Manchon C, Bilezikjian LM, Corrigan AZ, Mellon PL, Vale W. Activin-A modulates GnRH secretion from a GnRH-secreting neuronal cell line. Neuroendocr. [1991;54:373-7.
51. Noris G, Hol D, Clapp C, Martinez de la Escalera G. Histamine directly stimulates gonadotropin-releasing hormone secretion from GT1-1 cells via H1 receptors coupled to phosphoinositide hydrolysis. Endocrinology. [1995;136:2967-74.
52. Shakil T, Hoque AN, Husain M, Belsham DD. Differential regulation of gonadotropin-releasing hormone secretion and gene expression by androgen: membrane

145
145
versus nuclear receptor activation. Molecular endocrinology (Baltimore, Md. [2002 Nov;16(11):2592-602.
53. Roy D, Angelini N, Belsham DD. Estrogen directly represses gonadotropin-releasing hormone (GnRH) mRNA synthesis in GT1-7 GnRH-secreting hypothalamic neurons expressing estrogen receptor (ER) α and ERβ. Endocrinology. [1999;140:5045-53.
54. Gillespie JM, Roy D, Cui H, Belsham DD. Repression of gonadotropin-releasing hormone (GnRH) gene expression by melatonin may involve transcription factors COUP-TFI and C/EBP beta binding at the GnRH enhancer. Neuroendocrinology. [2004 Feb;79(2):63-72.
55. Smith MJ, Jennes L. Neural signals that regulate GnRH neurones directly during the oestrous cycle. Reproduction. [2001 Jul;122(1):1-10.
56. Herbison AE, Chapman C, Dyer RG. Role of medial preoptic GABA neurones in regulating luteinising hormone secretion in the ovariectomised rat. Exp Brain Res. [1991;87(2):345-52.
57. Leranth C, MacLusky NJ, Sakamoto H, Shanabrough M, Naftolin F. Glutamic acid decarboxylase-containing axons synapse on LHRH neurons in the rat medial preoptic area. Neuroendocrinology. [1985 Jun;40(6):536-9.
58. Robinson JE, Kendrick KM, Lambart CE. Changes in the release of gamma-aminobutyric Acid and catecholamines in the preoptic/septal area prior to and during the preovulatory surge of luteinizing hormone in the ewe. J Neuroendocrinol. [1991 Aug 1;3(4):393-9.
59. Smith JT, Clarke IJ. Kisspeptin expression in the brain: catalyst for the initiation of puberty. Reviews in endocrine & metabolic disorders. [2007 Mar;8(1):1-9.
60. Tena-Sempere M. The roles of kisspeptins and G protein-coupled receptor-54 in pubertal development. Curr Opin Pediatr. [2006 Aug;18(4):442-7.
61. Tena-Sempere M. GPR54 and kisspeptin in reproduction. Hum Reprod Update. [2006 Sep-Oct;12(5):631-9.
62. de Roux N, Genin E, Carel JC, Matsuda F, Chaussain JL, Milgrom E. Hypogonadotropic hypogonadism due to loss of function of the KiSS1-derived peptide receptor GPR54. Proceedings of the National Academy of Sciences of the United States of America. [2003 Sep 16;100(19):10972-6.
63. Seminara SB, Messager S, Chatzidaki EE, Thresher RR, Acierno JS, Jr., Shagoury JK, et al. The GPR54 gene as a regulator of puberty. The New England journal of medicine. [2003 Oct 23;349(17):1614-27.
64. Seminara SB. Kisspeptin in reproduction. Semin Reprod Med. [2007 Sep;25(5):337-43.

146
146
65. Gianetti E, Seminara S. Kisspeptin and KISS1R: a critical pathway in the reproductive system. Reproduction. [2008 Sep;136(3):295-301.
66. Plant TM, Ramaswamy S. Kisspeptin and the regulation of the hypothalamic-pituitary-gonadal axis in the rhesus monkey (Macaca mulatta). Peptides. [2009 Jan;30(1):67-75.
67. Simerly RB, Swanson LW. The distribution of neurotransmitter-specific cells and fibers in the anteroventral periventricular nucleus: implications for the control of gonadotropin secretion in the rat. Brain research. [1987 Jan 1;400(1):11-34.
68. Alexander MJ, Mahoney PD, Ferris CF, Carraway RE, Leeman SE. Evidence that neurotensin participates in the central regulation of the preovulatory surge of luteinizing hormone in the rat. Endocrinology. [1989 Feb;124(2):783-8.
69. Cerda-Reverter JM, Larhammar D. Neuropeptide Y family of peptides: structure, anatomical expression, function, and molecular evolution. Biochemistry and cell biology = Biochimie et biologie cellulaire. [2000;78(3):371-92.
70. Li C, Chen P, Smith MS. Morphological evidence for direct interaction between arcuate nucleus neuropeptide Y (NPY) neurons and gonadotropin-releasing hormone neurons and the possible involvement of NPY Y1 receptors. Endocrinology. [1999;140:5382-90.
71. Fetissov SO, Kopp J, Hokfelt T. Distribution of NPY receptors in the hypothalamus. Neuropeptides. [2004 Aug;38(4):175-88.
72. Bauer-Dantoin AC, McDonald JK, Levine JE. Neuropeptide Y potentiates luteinizing hormone (LH)-releasing hormone-induced LH secretion only under conditions leading to preovulatory LH surges. Endocrinology. [1992 Dec;131(6):2946-52.
73. Leupen SM, Besecke LM, Levine JE. Neuropeptide Y Y1-receptor stimulation is required for physiological amplification of preovulatory luteinizing hormone surges. Endocrinology. [1997;138:2735-9.
74. Advis JP, Klein J, Kuljis RO, Sarkar DK, McDonald JM, Conover CA. Regulation of gonadotropin releasing hormone release by neuropeptide Y at the median eminence during the preovulatory period in ewes. Neuroendocrinology. [2003 Apr;77(4):246-57.
75. Gonzales C, Voirol MJ, Giacomini M, Gaillard RC, Pedrazzini T, Pralong FP. The neuropeptide Y Y1 receptor mediates NPY-induced inhibition of the gonadotrope axis under poor metabolic conditions. Faseb J. [2004 Jan;18(1):137-9.
76. Kaynard AH, Pau KY, Hess DL, Spies HG. Third-ventricular infusion of neuropeptide Y suppresses luteinizing hormone secretion in ovariectomized rhesus macaques. Endocrinology. [1990 Nov;127(5):2437-44.

147
147
77. Khorram O, Pau KY, Spies HG. Bimodal effects of neuropeptide Y on hypothalamic release of gonadotropin-releasing hormone in conscious rabbits. Neuroendocrinology. [1987 Apr;45(4):290-7.
78. Khorram O, Pau KY, Spies HG. Release of hypothalamic neuropeptide Y and effects of exogenous NPY on the release of hypothalamic GnRH and pituitary gonadotropins in intact and ovariectomized does in vitro. Peptides. [1988 Mar-Apr;9(2):411-7.
79. Belsham DD, Lovejoy DA. Gonadotropin-releasing hormone: gene evolution, expression, and regulation. Vitam Horm. [2005;71:59-94.
80. Swinburn B, Ravussin E. Energy balance or fat balance? The American journal of clinical nutrition. [1993 May;57(5 Suppl):766S-70S; discussion 70S-71S.
81. Hill JW, Elmquist JK, Elias CF. Hypothalamic pathways linking energy balance and reproduction. American journal of physiology. [2008 May;294(5):E827-32.
82. Elmquist JK, Elias CF, Saper CB. From lesions to leptin: hypothalamic control of food intake and body weight. Neuron. [1999;22:221-32.
83. Sainsbury A, Cooney GJ, Herzog H. Hypothalamic regulation of energy homeostasis. Best Pract Res Clin Endocrinol Metab. [2002 Dec;16(4):623-37.
84. Arora S, Anubhuti. Role of neuropeptides in appetite regulation and obesity--a review. Neuropeptides. [2006 Dec;40(6):375-401.
85. I'Anson H, Foster DL, Foxcroft GR, Booth PJ. Nutrition and reproduction. Oxford reviews of reproductive biology. [1991;13:239-311.
86. Temple JL, Rissman EF. Nutrition, reproduction, and behavior. Progress in brain research. [2002;141:303-14.
87. Kalra SP, Kalra PS. Nutritional infertility: the role of the interconnected hypothalamic neuropeptide Y-galanin-opioid network. Frontiers in neuroendocrinology. [1996 Oct;17(4):371-401.
88. Sutton SW, Toyama TT, Otto S, Plotsky PM. Evidence that neuropeptide Y (NPY) released into the hypophysial-portal circulation participates in priming gonadotropes to the effects of gonadotropin releasing hormone (GnRH). Endocrinology. [1988 Aug;123(2):1208-10.
89. Tatemoto K, Carlquist M, Mutt V. Neuropeptide Y--a novel brain peptide with structural similarities to peptide YY and pancreatic polypeptide. Nature. [1982;296:659-60.
90. Wahlestedt C, Heilig M. Neuropeptide Y and related peptides. In: Bloom F, editor. Psychopharmacology-the fourth generation of progress, on-line edition.

148
148
Philadelphia, PA: Lippincott Williams & Wilkins; 1994. p. www.acnp.org/g4/GN401000052/CH052.html.
91. Sato N, Ogino Y, Mashiko S, Ando M. Modulation of neuropeptide Y receptors for the treatment of obesity. Expert Opin Ther Pat. [2009 Oct;19(10):1401-15.
92. Lin S, Boey D, Herzog H. NPY and Y receptors: lessons from transgenic and knockout models. Neuropeptides. [2004 Aug;38(4):189-200.
93. Sahu A, Kalra SP, Crowley WR, Kalra PS. Evidence that NPY-containing neurons in the brainstem project into selected hypothalamic nuclei: implication in feeding behavior. Brain research. [1988 Aug 9;457(2):376-8.
94. Pedrazzini T, Pralong F, Grouzmann E. Neuropeptide Y: the universal soldier. Cell Mol Life Sci. [2003 Feb;60(2):350-77.
95. Larhammar D, Blomqvist AG, Yee F, Jazin E, Yoo H, Wahlested C. Cloning and functional expression of a human neuropeptide Y/peptide YY receptor of the Y1 type. J Biol Chem. [1992 Jun 5;267(16):10935-8.
96. Rose PM, Fernandes P, Lynch JS, Frazier ST, Fisher SM, Kodukula K, et al. Cloning and functional expression of a cDNA encoding a human type 2 neuropeptide Y receptor. J Biol Chem. [1995 Dec 1;270(48):29038.
97. Lundell I, Blomqvist AG, Berglund MM, Schober DA, Johnson D, Statnick MA, et al. Cloning of a human receptor of the NPY receptor family with high affinity for pancreatic polypeptide and peptide YY. J Biol Chem. [1995 Dec 8;270(49):29123-8.
98. Michel MC, Beck-Sickinger A, Cox H, Doods HN, Herzog H, Larhammar D, et al. XVI. International Union of Pharmacology recommendations for the nomenclature of neuropeptide Y, peptide YY, and pancreatic polypeptide receptors. Pharmacological reviews. [1998 Mar;50(1):143-50.
99. Dumont Y, Quirion R. An overview of neuropeptide Y: pharmacology to molecular biology and receptor localization. EXS. [2006(95):7-33.
100. Baldwin JM. Structure and function of receptors coupled to G proteins. Current Opinions in Cellular Biology. [1994;6:180-90.
101. Pheng LH, Regoli D. Bioassays for NPY receptors: old and new. Regul Pept. [1998 Sep 25;75-76:79-87.
102. Motulsky HJ, Michel MC. Neuropeptide Y mobilizes Ca2+ and inhibits adenylate cyclase in human erythroleukemia cells. The American journal of physiology. [1988 Dec;255(6 Pt 1):E880-5.
103. Jung O, Marklund SL, Geiger H, Pedrazzini T, Busse R, Brandes RP. Extracellular superoxide dismutase is a major determinant of nitric oxide bioavailability:

149
149
in vivo and ex vivo evidence from ecSOD-deficient mice. Circ Res. [2003 Oct 3;93(7):622-9.
104. Nilsson T, Lind H, Brunkvall J, Edvinsson L. Vasodilation in human subcutaneous arteries induced by neuropeptide Y is mediated by neuropeptide Y Y1 receptors and is nitric oxide dependent. Canadian journal of physiology and pharmacology. [2000 Mar;78(3):251-5.
105. Sainsbury A, Rohner-Jeanrenaud F, Grouzmann E, Jeanrenaud B. Acute intracerebroventricular administration of neuropeptide Y stimulates corticosterone output and feeding but not insulin output in normal rats. Neuroendocrinology. [1996 Apr;63(4):318-26.
106. Kalra SP, Dube MG, Sahu A, Phelps CP, Kalra PS. Neuropeptide Y secretion increases in the paraventricular nucleus in association with increased appetite for food. Proceedings of the National Academy of Sciences of the United States of America. [1991 Dec 1;88(23):10931-5.
107. Stanley BG, Kyrkouli S, Lampert S, Leibowitz S. Neuropeptide Y chronically injected into the hypothalamus: a powerful neurochemical inducer of hyperphagia and obesity. Peptides. [1986;7:1189-92.
108. Kask A, Rago L, Harro J. Evidence for involvement of neuropeptide Y receptors in the regulation of food intake: studies with Y1-selective antagonist BIBP3226. Br J Pharmacol. [1998 Aug;124(7):1507-15.
109. Sahu A, Kalra SP. Neuropeptidergic regulation of feeding behavior Neuropeptide Y. Trends in endocrinology and metabolism: TEM. [1993 Sep;4(7):217-24.
110. Wilding JP, Gilbey SG, Bailey CJ, Batt RA, Williams G, Ghatei MA, et al. Increased neuropeptide-Y messenger ribonucleic acid (mRNA) and decreased neurotensin mRNA in the hypothalamus of the obese (ob/ob) mouse. Endocrinology. [1993 May;132(5):1939-44.
111. Lee GH, Proenca R, Montez JM, Carroll KM, Darvishzadeh JG, Lee JI, et al. Abnormal splicing of the leptin receptor in diabetic mice. Nature. [1996 Feb 15;379(6566):632-5.
112. Sahu A, Crowley WR, Kalra SP. Evidence that hypothalamic neuropeptide Y gene expression increases before the onset of the preovulatory LH surge. J Neuroendocrinol. [1995;7:291-6.
113. Gruber CJ, Tschugguel W, Schneeberger C, Huber JC. Production and actions of estrogens. The New England journal of medicine. [2002 Jan 31;346(5):340-52.
114. Korach KS. Estrogen receptor knock-out mice: molecular and endocrine phenotypes. Journal of the Society for Gynecologic Investigation. [2000 Jan-Feb;7(1 Suppl):S16-7.

150
150
115. Labrie F. Extragonadal synthesis of sex steroids: intracrinology. Annales d'endocrinologie. [2003 Apr;64(2):95-107.
116. Acconcia F, Kumar R. Signaling regulation of genomic and nongenomic functions of estrogen receptors. Cancer letters. [2006 Jul 8;238(1):1-14.
117. Cosman F, Lindsay R. Selective estrogen receptor modulators: clinical spectrum. Endocr Rev. [1999 Jun;20(3):418-34.
118. McNatty KP, Makris A, DeGrazia C, Osathanondh R, Ryan KJ. The production of progesterone, androgens, and estrogens by granulosa cells, thecal tissue, and stromal
tissue from human ovaries in vitro. J Clin Endocrinol Metab. [1979;49:687-99.
119. Zhu BT, Conney AH. Functional role of estrogen metabolism in target cells: review and perspectives. Carcinogenesis. [1998 Jan;19(1):1-27.
120. Matthews J, Gustafsson JA. Estrogen signaling: a subtle balance between ER alpha and ER beta. Molecular interventions. [2003 Aug;3(5):281-92.
121. Toran-Allerand CD, Tinnikov AA, Singh RJ, Nethrapalli IS. 17alpha-estradiol: a brain-active estrogen? Endocrinology. [2005 Sep;146(9):3843-50.
122. Nilsson S, Makela S, Treuter E, Tujague M, Thomsen J, Andersson G, et al. Mechanisms of estrogen action. Physiological reviews. [2001 Oct;81(4):1535-65.
123. Simpson ER, Davis SR. Minireview: aromatase and the regulation of estrogen biosynthesis--some new perspectives. Endocrinology. [2001 Nov;142(11):4589-94.
124. Jefcoate CR, Liehr JG, Santen RJ, Sutter TR, Yager JD, Yue W, et al. Tissue-specific synthesis and oxidative metabolism of estrogens. Journal of the National Cancer Institute. [2000(27):95-112.
125. Levin E. Cellular functions of the plasma membrane estrogen receptor. Trends Endocrinol Metab. [1999;10:374-7.
126. Tremblay GB, Tremblay A, Copeland NG, Gilbert DJ, Jenkins NA, Labrie F, et al. Cloning, chromosomal localization, and functional analysis of the murine estrogen receptor beta. Molecular endocrinology (Baltimore, Md. [1997 Mar;11(3):353-65.
127. Lu B, Leygue E, Dotzlaw H, Murphy LJ, Murphy LC, Watson PH. Estrogen receptor-beta mRNA variants in human and murine tissues. Molecular and cellular endocrinology. [1998 Mar 16;138(1-2):199-203.
128. Moore JT, McKee DD, Slentz-Kesler K, Moore LB, Jones SA, Horne EL, et al. Cloning and characterization of human estrogen receptor beta isoforms. Biochemical and biophysical research communications. [1998 Jun 9;247(1):75-8.

151
151
129. Fuqua SA, Schiff R, Parra I, Friedrichs WE, Su JL, McKee DD, et al. Expression of wild-type estrogen receptor beta and variant isoforms in human breast cancer. Cancer research. [1999 Nov 1;59(21):5425-8.
130. Mosselman S, Polman J, Dijkema R. ER beta: identification and characterization of a novel human estrogen receptor. FEBS Lett. [1996 Aug 19;392(1):49-53.
131. Ogawa S, Inoue S, Watanabe T, Orimo A, Hosoi T, Ouchi Y, et al. Molecular cloning and characterization of human estrogen receptor betacx: a potential inhibitor ofestrogen action in human. Nucleic acids research. [1998 Aug 1;26(15):3505-12.
132. Peng B, Lu B, Leygue E, Murphy LC. Putative functional characteristics of human estrogen receptor-beta isoforms. J Mol Endocrinol. [2003 Feb;30(1):13-29.
133. Leung YK, Mak P, Hassan S, Ho SM. Estrogen receptor (ER)-beta isoforms: a key to understanding ER-beta signaling. Proceedings of the National Academy of Sciences of the United States of America. [2006 Aug 29;103(35):13162-7.
134. Hall JM, Couse JF, Korach KS. The multifaceted mechanisms of estradiol and estrogen receptor signaling. J Biol Chem. [2001 Oct 5;276(40):36869-72.
135. Webb M, Thomas JO. Structure-specific binding of the two tandem HMG boxes of HMG1 to four-way junction DNA is mediated by the A domain. J Mol Biol. [1999 Nov 26;294(2):373-87.
136. Levin ER. Cell localization, physiology, and nongenomic actions of estrogen receptors. J Appl Physiol. [2001 Oct;91(4):1860-7.
137. Kuiper GG, Carlsson B, Grandien K, Enmark E, Haggblad J, Nilsson S, et al. Comparison of the ligand binding specificity and transcript tissue distribution of estrogen receptors alpha and beta. Endocrinology. [1997 Mar;138(3):863-70.
138. Rosenfeld CS, Ganjam VK, Taylor JA, Yuan X, Stiehr JR, Hardy MP, et al. Transcription and translation of estrogen receptor-beta in the male reproductive tract of estrogen receptor-alpha knock-out and wild-type mice. Endocrinology. [1998 Jun;139(6):2982-7.
139. Filardo EJ, Quinn JA, Frackelton AR, Jr., Bland KI. Estrogen action via the G protein-coupled receptor, GPR30: stimulation of adenylyl cyclase and cAMP-mediated attenuation of the epidermal growth factor receptor-to-MAPK signaling axis. Molecular endocrinology (Baltimore, Md. [2002 Jan;16(1):70-84.
140. Filardo EJ, Thomas P. GPR30: a seven-transmembrane-spanning estrogen receptor that triggers EGF release. Trends in endocrinology and metabolism: TEM. [2005 Oct;16(8):362-7.
141. Albanito L, Madeo A, Lappano R, Vivacqua A, Rago V, Carpino A, et al. G protein-coupled receptor 30 (GPR30) mediates gene expression changes and growth

152
152
response to 17beta-estradiol and selective GPR30 ligand G-1 in ovarian cancer cells. Cancer research. [2007 Feb 15;67(4):1859-66.
142. Prossnitz ER, Arterburn JB, Sklar LA. GPR30: A G protein-coupled receptor for estrogen. Molecular and cellular endocrinology. [2007 Feb;265-266:138-42.
143. Prossnitz ER, Arterburn JB, Smith HO, Oprea TI, Sklar LA, Hathaway HJ. Estrogen signaling through the transmembrane G protein-coupled receptor GPR30. Annual review of physiology. [2008;70:165-90.
144. Prossnitz ER, Oprea TI, Sklar LA, Arterburn JB. The ins and outs of GPR30: a transmembrane estrogen receptor. The Journal of steroid biochemistry and molecular biology. [2008 Apr;109(3-5):350-3.
145. Thomas P, Pang Y, Filardo EJ, Dong J. Identity of an estrogen membrane receptor coupled to a G protein in human breast cancer cells. Endocrinology. [2005 Feb;146(2):624-32.
146. Marino M, Galluzzo P, Ascenzi P. Estrogen signaling multiple pathways to impact gene transcription. Current genomics. [2006;7(8):497-508.
147. Bjornstrom L, Sjoberg M. Mechanisms of estrogen receptor signaling: convergence of genomic and nongenomic actions on target genes. Molecular endocrinology (Baltimore, Md. [2005 Apr;19(4):833-42.
148. Stormshak F, Bishop CV. Board-invited review: Estrogen and progesterone signaling: genomic and nongenomic actions in domestic ruminants. Journal of animal science. [2008 Feb;86(2):299-315.
149. Bishop CV, Stormshak F. Non-genomic actions of progesterone and estrogens in regulating reproductive events in domestic animals. Vet J. [2008 Jun;176(3):270-80.
150. Hall JM, McDonnell DP. Coregulators in nuclear estrogen receptor action: from concept to therapeutic targeting. Molecular interventions. [2005 Dec;5(6):343-57.
151. Sheppard HM, Matsuda S, Harries JC, Kindle KB, Heery DM. Transcriptional activation by estrogen receptor (ERalpha) and steroid receptor coactivator (SRC1) involves distinct mechanisms in yeast and mammalian cells. J Mol Endocrinol. [2003 Jun;30(3):411-22.
152. Cheung CC, Clifton DK, Steiner RA. Proopiomelanocortin neurons are direct targets for leptin in the hypothalamus. Endocrinology. [1997 Oct;138(10):4489-92.
153. Seeley RJ, Yagaloff KA, Fisher SL, Burn P, Thiele TE, van Dijk G, et al. Melanocortin receptors in leptin effects. Nature. [1997 Nov 27;390(6658):349.
154. Szego CM, Davis JS. Adenosine 3',5'-monophosphate in rat uterus: acute elevation by estrogen. Proc Natl Acad Sci U S A. [1967 Oct;58(4):1711-8.

153
153
155. Pietras RJ, Szego CM. Specific binding sites for oestrogen at the outer surfaces of isolated endometrial cells. Nature. [1977 Jan 6;265(5589):69-72.
156. Valverde MA, Rojas P, Amigo J, Cosmelli D, Orio P, Bahamonde MI, et al. Acute activation of Maxi-K channels (hSlo) by estradiol binding to the beta subunit. Science (New York, NY. [1999 Sep 17;285(5435):1929-31.
157. Hennessy BA, Harvey BJ, Healy V. 17beta-Estradiol rapidly stimulates c-fos expression via the MAPK pathway in T84 cells. Molecular and cellular endocrinology. [2005 Jan 14;229(1-2):39-47.
158. Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science (New York, NY. [2005 Mar 11;307(5715):1625-30.
159. Nadal A, Ropero AB, Fuentes E, Soria B. The plasma membrane estrogen receptor: nuclear or unclear? Trends Pharmacol Sci. [2001 Dec;22(12):597-9.
160. Nadal A, Diaz M, Valverde MA. The estrogen trinity: membrane, cytosolic, and nuclear effects. News Physiol Sci. [2001 Dec;16:251-5.
161. Nadal A, Ropero AB, Laribi O, Maillet M, Fuentes E, Soria B. Nongenomic actions of estrogens and xenoestrogens by binding at a plasma membrane receptor unrelated to estrogen receptor alpha and estrogen receptor beta. Proceedings of the National Academy of Sciences of the United States of America. [2000 Oct 10;97(21):11603-8.
162. Simpson E, Jones M, Misso M, Hewitt K, Hill R, Maffei L, et al. Estrogen, a fundamental player in energy homeostasis. The Journal of steroid biochemistry and molecular biology. [2005 May;95(1-5):3-8.
163. Acosta-Martinez M, Horton T, Levine JE. Estrogen receptors in neuropeptide Y neurons: at the crossroads of feeding and reproduction. Trends in endocrinology and metabolism: TEM. [2007 Mar;18(2):48-50.
164. Titolo D, Cai F, Belsham DD. Coordinate regulation of neuropeptide Y and agouti-related peptide gene expression by estrogen depends on the ratio of estrogen receptor (ER) alpha to ERbeta in clonal hypothalamic neurons. Molecular endocrinology (Baltimore, Md. [2006 Sep;20(9):2080-92.
165. Caraty A, Locatelli A, Martin GB. Biphasic response in the secretion of gonadotrophin-releasing hormone in ovariectomized ewes injected with oestradiol. The Journal of endocrinology. [1989 Dec;123(3):375-82.
166. Clark ME, Lawson MA, Belsham DD, Eraly SA, Mellon PL. Molecular aspects of GnRH gene expression. LeRoith D, editor. Greenwich, CN: JAI Press, Inc.; 1997.

154
154
167. Golub MS, Hogrefe CE, Germann SL, Lasley BL, Natarajan K, Tarantal AF. Effects of exogenous estrogenic agents on pubertal growth and reproductive system maturation in female rhesus monkeys. Toxicol Sci. [2003 Jul;74(1):103-13.
168. Donohoe TP, Stevens R, Johnson NJ, Barker S. Effects of stereoisomers of estradiol on food intake, body weight and hoarding behavior in female rats. Physiology & behavior. [1984 Apr;32(4):589-92.
169. Carr MC. The emergence of the metabolic syndrome with menopause. The Journal of clinical endocrinology and metabolism. [2003 Jun;88(6):2404-11.
170. Morimoto LM, White E, Chen Z, Chlebowski RT, Hays J, Kuller L, et al. Obesity, body size, and risk of postmenopausal breast cancer: the Women's Health Initiative (United States). Cancer Causes Control. [2002 Oct;13(8):741-51.
171. Olofsson LE, Pierce AA, Xu AW. Functional requirement of AgRP and NPY neurons in ovarian cycle-dependent regulation of food intake. Proc Natl Acad Sci U S A. [2009 Sep 15;106(37):15932-7.
172. Beatty WW, O'Briant DA, Vilberg TR. Effects of ovariectomy and estradiol injections on food intake and body weight in rats with ventromedial hypothalamic lesions. Pharmacology, biochemistry, and behavior. [1975 Jul-Aug;3(4):539-44.
173. Heine PA, Taylor JA, Iwamoto GA, Lubahn DB, Cooke PS. Increased adipose tissue in male and female estrogen receptor-alpha knockout mice. Proceedings of the National Academy of Sciences of the United States of America. [2000 Nov 7;97(23):12729-34.
174. Butera PC, Bradway DM, Cataldo NJ. Modulation of the satiety effect of cholecystokinin by estradiol. Physiology & behavior. [1993 Jun;53(6):1235-8.
175. Clegg DJ, Brown LM, Zigman JM, Kemp CJ, Strader AD, Benoit SC, et al. Estradiol-dependent decrease in the orexigenic potency of ghrelin in female rats. Diabetes. [2007 Apr;56(4):1051-8.
176. Geary N, Asarian L, Korach KS, Pfaff DW, Ogawa S. Deficits in E2-dependent control of feeding, weight gain, and cholecystokinin satiation in ER-alpha null mice. Endocrinology. [2001 Nov;142(11):4751-7.
177. Sar M, Sahu A, Crowley WR, Kalra SP. Localization of neuropeptide-Y immunoreactivity in estradiol-concentrating cells in the hypothalamus. Endocrinology. [1990 Dec;127(6):2752-6.
178. Titolo D, Mayer CM, Dhillon SS, Cai F, Belsham DD. Estrogen facilitates both phosphatidylinositol 3-kinase/Akt and ERK1/2 mitogen-activated protein kinase membrane signaling required for long-term neuropeptide Y transcriptional regulation in clonal, immortalized neurons. J Neurosci. [2008 Jun 18;28(25):6473-82.

155
155
179. Skinner DC, Herbison AE. Effects of photoperiod on estrogen receptor, tyrosine hydroxylase, neuropeptide Y, and beta-endorphin immunoreactivity in the ewe hypothalamus. Endocrinology. [1997 Jun;138(6):2585-95.
180. Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. [1994 Dec 1;372(6505):425-32.
181. Halaas JL, Gajiwala KS, Maffei M, Cohen SL, Chait BT, Rabinowitz D, et al. Weight-reducing effects of the plasma protein encoded by the obese gene. Science (New York, NY. [1995 Jul 28;269(5223):543-6.
182. Considine RV, Sinha MK, Heiman ML, Kriauciunas A, Stephens TW, Nyce MR, et al. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. The New England journal of medicine. [1996 Feb 1;334(5):292-5.
183. Wilcke M, Walum E. Characterization of leptin intracellular trafficking. Eur J Histochem. [2000;44(4):325-34.
184. Margetic S, Gazzola C, Pegg GG, Hill RA. Leptin: a review of its peripheral actions and interactions. Int J Obes Relat Metab Disord. [2002 Nov;26(11):1407-33.
185. Tartaglia LA. The leptin receptor. J Biol Chem. [1997 Mar 7;272(10):6093-6.
186. Zhao AZ, Huan JN, Gupta S, Pal R, Sahu A. A phosphatidylinositol 3-kinase phosphodiesterase 3B-cyclic AMP pathway in hypothalamic action of leptin on feeding. Nat Neurosci. [2002;5:727-8.
187. Kennedy GC. The role of depot fat in the hypothalamic control of food intake in the rat. Proceedings of the Royal Society of London Series B, Containing papers of a Biological character. [1953 Jan 15;140(901):578-96.
188. Ahima RS, Prabakaran D, Mantzoros C, Qu D, Lowell B, Maratos-Flier E, et al. Role of leptin in the neuroendocrine response to fasting. Nature. [1996 Jul 18;382(6588):250-2.
189. Campfield LA, Smith FJ, Guisez Y, Devos R, Burn P. Recombinant mouse OB protein: evidence for a peripheral signal linking adiposity and central neural networks. Science (New York, NY. [1995 Jul 28;269(5223):546-9.
190. Thornton JE, Cheung CC, Clifton DK, Steiner RA. Regulation of hypothalamic proopiomelanocortin mRNA by leptin in ob/ob mice. Endocrinology. [1997 Nov;138(11):5063-6.
191. Minokoshi Y, Kim YB, Peroni OD, Fryer LG, Muller C, Carling D, et al. Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature. [2002 Jan 17;415(6869):339-43.

156
156
192. Barash IA, Cheung CC, Weigle DS, Ren H, Kabigting EB, Kuijper JL, et al. Leptin is a metabolic signal to the reproductive system. Endocrinology. [1996 Jul;137(7):3144-7.
193. Ahima RS, Dushay J, Flier SN, Prabakaran D, Flier JS. Leptin accelerates the onset of puberty in normal female mice. The Journal of clinical investigation. [1997 Feb 1;99(3):391-5.
194. Chehab FF, Mounzih K, Lu R, Lim ME. Early onset of reproductive function in normal female mice treated with leptin. Science (New York, NY. [1997 Jan 3;275(5296):88-90.
195. Mounzih K, Lu R, Chehab FF. Leptin treatment rescues the sterility of genetically obese ob/ob males. Endocrinology. [1997 Mar;138(3):1190-3.
196. Cunningham MJ, Clifton DK, Steiner RA. Leptin's actions on the reproductive axis: perspectives and mechanisms. Biology of reproduction. [1999 Feb;60(2):216-22.
197. Schwartz MW, Seeley RJ, Campfield LA, Burn P, Baskin DG. Identification of targets of leptin action in rat hypothalamus. The Journal of clinical investigation. [1996 Sep 1;98(5):1101-6.
198. Baskin DG, Schwartz MW, Seeley RJ, Woods SC, Porte D, Jr., Breininger JF, et al. Leptin receptor long-form splice-variant protein expression in neuron cell bodies of the brain and co-localization with neuropeptide Y mRNA in the arcuate nucleus. J Histochem Cytochem. [1999 Mar;47(3):353-62.
199. Leshan RL, Louis GW, Jo YH, Rhodes CJ, Munzberg H, Myers MG, Jr. Direct innervation of GnRH neurons by metabolic- and sexual odorant-sensing leptin receptor neurons in the hypothalamic ventral premammillary nucleus. J Neurosci. [2009 Mar 11;29(10):3138-47.
200. Swart I, Jahng JW, Overton JM, Houpt TA. Hypothalamic NPY, AGRP, and POMC mRNA responses to leptin and refeeding in mice. Am J Physiol Regul Integr Comp Physiol. [2002 Nov;283(5):R1020-6.
201. Balthasar N, Coppari R, McMinn J, Liu SM, Lee CE, Tang V, et al. Leptin receptor signaling in POMC neurons is required for normal body weight homeostasis. Neuron. [2004 Jun 24;42(6):983-91.
202. Padilla SL, Carmody JS, Zeltser LM. Pomc-expressing progenitors give rise to antagonistic neuronal populations in hypothalamic feeding circuits. Nature medicine. [Apr;16(4):403-5.
203. Kask A, Rago L, Wikberg JE, Schioth HB. Evidence for involvement of the melanocortin MC4 receptor in the effects of leptin on food intake and body weight. Eur J Pharmacol. [1998 Oct 30;360(1):15-9.

157
157
204. Clarke IJ, Henry BA. Leptin and reproduction. Reviews of reproduction. [1999 Jan;4(1):48-55.
205. Edwards CM, Abusnana S, Sunter D, Murphy KG, Ghatei MA, Bloom SR. The effect of the orexins on food intake: comparison with neuropeptide Y, melanin-concentrating hormone and galanin. The Journal of endocrinology. [1999 Mar;160(3):R7-12.
206. Myers MG, Jr. Leptin receptor signaling and the regulation of mammalian physiology. Recent progress in hormone research. [2004;59:287-304.
207. Caro JF, Kolaczynski JW, Nyce MR, Ohannesian JP, Opentanova I, Goldman WH, et al. Decreased cerebrospinal-fluid/serum leptin ratio in obesity: a possible mechanism for leptin resistance. Lancet. [1996 Jul 20;348(9021):159-61.
208. El-Haschimi K, Lehnert H. Leptin resistance - or why leptin fails to work in obesity. Exp Clin Endocrinol Diabetes. [2003 Feb;111(1):2-7.
209. Sahu A. Leptin signaling in the hypothalamus: emphasis on energy homeostasis and leptin resistance. Frontiers in neuroendocrinology. [2003 Dec;24(4):225-53.
210. Banks WA, Coon AB, Robinson SM, Moinuddin A, Shultz JM, Nakaoke R, et al. Triglycerides induce leptin resistance at the blood-brain barrier. Diabetes. [2004 May;53(5):1253-60.
211. Munzberg H, Flier JS, Bjorbaek C. Region-specific leptin resistance within the hypothalamus of diet-induced obese mice. Endocrinology. [2004 Nov;145(11):4880-9.
212. Munzberg H, Myers MG, Jr. Molecular and anatomical determinants of central leptin resistance. Nat Neurosci. [2005 May;8(5):566-70.
213. Bjorbaek C, Kahn BB. Leptin signaling in the central nervous system and the periphery. Recent progress in hormone research. [2004;59:305-31.
214. Leshan RL, Bjornholm M, Munzberg H, Myers MG, Jr. Leptin receptor signaling and action in the central nervous system. Obesity (Silver Spring, Md. [2006 Aug;14 Suppl 5:208S-12S.
215. Everitt BJ, Hokfelt T. Neuroendocrine anatomy of the hypothalamus. Acta Neurochir Suppl (Wien). [1990;47:1-15.
216. Mayer CM, Fick LJ, Gingerich S, Belsham DD. Hypothalamic cell lines to investigate neuroendocrine control mechanisms. Frontiers in neuroendocrinology. [2009 Aug;30(3):405-23.
217. Skynner MJ, Sim JA, Herbison AE. Detection of estrogen receptor alpha and beta messenger ribonucleic acids in adult gonadotropin-releasing hormone neurons. Endocrinology. [1999 Nov;140(11):5195-201.

158
158
218. Mellon PL, Windle JJ, Goldsmith PC, Padula CA, Roberts JL, Weiner RI. Immortalization of hypothalamic GnRH neurons by genetically targeted tumorigenesis. Neuron. [1990 Jul;5(1):1-10.
219. Belsham DD, Cai F, Cui H, Smukler SR, Salapatek AM, Shkreta L. Generation of a phenotypic array of hypothalamic neuronal cell models to study complex neuroendocrine disorders. Endocrinology. [2004 Jan;145(1):393-400.
220. Belsham DD, Fick LJ, Dalvi PS, Centeno ML, Chalmers JA, Lee PK, et al. Ciliary neurotrophic factor recruitment of glucagon-like peptide-1 mediates neurogenesis, allowing immortalization of adult murine hypothalamic neurons. FASEB J. [2009 Dec;23(12):4256-65.
221. Radovick S, Wray S, Lee E, Nicols DK, Nakayama Y, Weintraub BD, et al. Migratory arrest of gonadotropin-releasing hormone neurons in transgenic mice. Proceedings of the National Academy of Sciences of the United States of America. [1991 Apr 15;88(8):3402-6.
222. Salvi R, Castillo E, Voirol MJ, Glauser M, Rey JP, Gaillard RC, et al. Gonadotropin-releasing hormone-expressing neurons immortalized conditionally are activated by insulin: implication of the mitogen-activated protein kinase pathway. Endocrinology. [2006 Feb;147(2):816-26.
223. Wolfe A, Ng Y, Divall SA, Singh SP, Radovick S. Development of an immortalised, post-pubertal gonadotrophin-releasing hormone neuronal cell line. J Neuroendocrinol. [2008 Sep;20(9):1029-37.
224. Wetsel WC, Valença MM, Merchenthaler I, Liposits Z, López FJ, Weiner RI, et al. Intrinsic pulsatile secretory activity of immortalized LHRH secreting neurons. Proc Natl Acad Sci USA. [1992;89:4149-53.
225. Whyte DB, Lawson MA, Belsham DD, Eraly SA, Bond CT, Adelman JP, et al. A neuron-specific enhancer targets expression of the gonadotropin-releasing hormone gene to hypothalamic neurosecretory neurons. Mol Endocrinol. [1995;9:467-77.
226. Silverman AJ, Roberts JL, Dong KW, Miller GM, Gibson MJ. Intrahypothalamic injection of a cell line secreting gonadotropin-releasing hormone results in cellular differentiation and reversal of hypogonadism in mutant mice. Proceedings of the National Academy of Sciences of the United States of America. [1992 Nov 15;89(22):10668-72.
227. Augusti-Tocco G, Sato G. Establishment of functional clonal lines of neurons from mouse neuroblastoma. Proceedings of the National Academy of Sciences of the United States of America. [1969 Sep;64(1):311-5.
228. Rasmussen DD, Gambacciani M, Swartz W, Tueros VS, Yen SS. Pulsatile gonadotropin-releasing hormone release from the human mediobasal hypothalamus in vitro:opiate receptor-mediated suppression. Neuroendocrinology. [1989;49:150-6.

159
159
229. Kasckow J, Mulchahey JJ, Aguilera G, Pisarska M, Nikodemova M, Chen HC, et al. Corticotropin-releasing hormone (CRH) expression and protein kinase A mediated CRH receptor signalling in an immortalized hypothalamic cell line. J Neuroendocrinol. [2003 May;15(5):521-9.
230. Belsham DD. Hormonal regulation of clonal, immortalized hypothalamic neurons expressing neuropeptides involved in reproduction and feeding. Molecular neurobiology. [2007 Apr;35(2):182-94.
231. Lee CM, Fuhrman CB, Planelles V, Peltier MR, Gaffney DK, Soisson AP, et al. Phosphatidylinositol 3-kinase inhibition by LY294002 radiosensitizes human cervical cancer cell lines. Clin Cancer Res. [2006 Jan 1;12(1):250-6.
232. Xie Y, Wolff DW, Lin MF, Tu Y. Vasoactive intestinal peptide transactivates the androgen receptor through a protein kinase A-dependent extracellular signal-regulated kinase pathway in prostate cancer LNCaP cells. Mol Pharmacol. [2007 Jul;72(1):73-85.
233. Shaw JL, Gackenheimer SL, Gehlert DR. Functional autoradiography of neuropeptide Y Y1 and Y2 receptor subtypes in rat brain using agonist stimulated [35S]GTPgammaS binding. J Chem Neuroanat. [2003 Nov;26(3):179-93.
234. Peairs A, Radjavi A, Davis S, Li L, Ahmed A, Giri S, et al. Activation of AMPK inhibits inflammation in MRL/lpr mouse mesangial cells. Clin Exp Immunol. [2009 Jun;156(3):542-51.
235. Di Virgilio F, Milani D, Leon A, Meldolesi J, Pozzan T. Voltage-dependent activation and inactivation of calcium channels in PC12 cells. Correlation with neurotransmitter release. J Biol Chem. [1987 Jul 5;262(19):9189-95.
236. Mayer CM, Belsham DD. Palmitate attenuates insulin signaling and induces endoplasmic reticulum stress and apoptosis in hypothalamic neurons: rescue of resistance and apoptosis through adenosine 5' monophosphate-activated protein kinase activation. Endocrinology. [Feb;151(2):576-85.
237. Mayer CM, Belsham DD. Central insulin signaling is attenuated by long-term insulin exposure via insulin receptor substrate-1 serine phosphorylation, proteasomal degradation, and lysosomal insulin receptor degradation. Endocrinology. [Jan;151(1):75-84.
238. Mayer CM, Belsham DD. Insulin directly regulates NPY and AgRP gene expression via the MAPK MEK/ERK signal transduction pathway in mHypoE-46 hypothalamic neurons. Molecular and cellular endocrinology. [2009 Aug 13;307(1-2):99-108.
239. Fick LJ, Cai F, Belsham DD. Hypothalamic preproghrelin gene expression is repressed by insulin via both PI3-K/Akt and ERK1/2 MAPK pathways in immortalized, hypothalamic neurons. Neuroendocrinology. [2009;89(3):267-75.

160
160
240. Cui H, Cai F, Belsham DD. Anorexigenic hormones leptin, insulin, and alpha-melanocyte-stimulating hormone directly induce neurotensin (NT) gene expression in novel NT-expressing cell models. J Neurosci. [2005 Oct 12;25(41):9497-506.
241. Cui H, Cai F, Belsham DD. Leptin signaling in neurotensin neurons involves STAT, MAP kinases ERK1/2, and p38 through c-Fos and ATF1. FASEB J. [2006 Dec;20(14):2654-6.
242. Toth MJ, Poehlman ET, Matthews DE, Tchernof A, MacCoss MJ. Effects of estradiol and progesterone on body composition, protein synthesis, and lipoprotein lipase in rats. American journal of physiology. [2001 Mar;280(3):E496-501.
243. Santollo J, Eckel LA. Estradiol decreases the orexigenic effect of neuropeptide Y, but not agouti-related protein, in ovariectomized rats. Behavioural brain research. [2008 Aug 22;191(2):173-7.
244. Bonavera JJ, Dube MG, Kalra PS, Kalra SP. Anorectic effects of estrogen may be mediated by decreased neuropeptide-Y release in the hypothalamic paraventricular nucleus. Endocrinology. [1994 Jun;134(6):2367-70.
245. Pelletier G, Li S, Luu-The V, Labrie F. Oestrogenic regulation of pro-opiomelanocortin, neuropeptide Y and corticotrophin-releasing hormone mRNAs in mouse hypothalamus. J Neuroendocrinol. [2007 Jun;19(6):426-31.
246. Broberger C, Johansen J, Johansson C, Schalling M, Hokfelt T. The neuropeptide Y/agouti gene-related protein (AGRP) brain circuitry in normal, anorectic, and monosodium glutamate-treated mice. Proceedings of the National Academy of Sciences of the United States of America. [1998 Dec 8;95(25):15043-8.
247. Ainslie DA, Morris MJ, Wittert G, Turnbull H, Proietto J, Thorburn AW. Estrogen deficiency causes central leptin insensitivity and increased hypothalamic neuropeptide Y. Int J Obes Relat Metab Disord. [2001 Nov;25(11):1680-8.
248. Santollo J, Wiley MD, Eckel LA. Acute activation of ER alpha decreases food intake, meal size, and body weight in ovariectomized rats. Am J Physiol Regul Integr Comp Physiol. [2007 Dec;293(6):R2194-201.
249. Liang YQ, Akishita M, Kim S, Ako J, Hashimoto M, Iijima K, et al. Estrogen receptor beta is involved in the anorectic action of estrogen. Int J Obes Relat Metab Disord. [2002 Aug;26(8):1103-9.
250. Gingerich S, Krukoff TL. Estrogen modulates endothelial and neuronal nitric oxide synthase expression via an estrogen receptor beta-dependent mechanism in hypothalamic slice cultures. Endocrinology. [2005 Jul;146(7):2933-41.
251. Gingerich S, Krukoff TL. Estrogen in the paraventricular nucleus attenuates L-glutamate-induced increases in mean arterial pressure through estrogen receptor beta and NO. Hypertension. [2006 Dec;48(6):1130-6.

161
161
252. Butera PC, Czaja JA. Intracranial estradiol in ovariectomized guinea pigs: effects on ingestive behaviors and body weight. Brain research. [1984 Nov 19;322(1):41-8.
253. Butera PC, Willard DM, Raymond SA. Effects of PVN lesions on the responsiveness of female rats to estradiol. Brain research. [1992 Apr 3;576(2):304-10.
254. Dagnault A, Richard D. Involvement of the medial preoptic area in the anorectic action of estrogens. The American journal of physiology. [1997 Jan;272(1 Pt 2):R311-7.
255. Roesch DM. Effects of selective estrogen receptor agonists on food intake and body weight gain in rats. Physiology & behavior. [2006 Jan 30;87(1):39-44.
256. Razandi M, Pedram A, Greene G, Levin E. Cell membrane and nuclear estrogen receptors (ERs) originate from a single transcript: Studies of ERα and ERβ expressed in CHO cells. Mol Endocrinol. [1999;13:307-19.
257. Pappas TC, Gametchu B, Watson CS. Membrane estrogen receptors identified by multiple antibody labeling and impeded-ligand binding. Faseb J. [1995 Mar;9(5):404-10.
258. Shaul PW, Anderson RG. Role of plasmalemmal caveolae in signal transduction. The American journal of physiology. [1998 Nov;275(5 Pt 1):L843-51.
259. Kelly MJ, Qiu J, Ronnekleiv OK. Estrogen signaling in the hypothalamus. Vitam Horm. [2005;71:123-45.
260. Andersson U, Filipsson K, Abbott CR, Woods A, Smith K, Bloom SR, et al. AMP-activated protein kinase plays a role in the control of food intake. J Biol Chem. [2004 Mar 26;279(13):12005-8.
261. Han SM, Namkoong C, Jang PG, Park IS, Hong SW, Katakami H, et al. Hypothalamic AMP-activated protein kinase mediates counter-regulatory responses to hypoglycaemia in rats. Diabetologia. [2005 Oct;48(10):2170-8.
262. Schwartz MW, Woods SC, Porte D, Jr., Seeley RJ, Baskin DG. Central nervous system control of food intake. Nature. [2000 Apr 6;404(6778):661-71.
263. Nagatani S, Guthikonda P, Thompson RC, Tsukamura H, Maeda KI, Foster DL. Evidence for GnRH regulation by leptin: leptin administration prevents reduced pulsatile LH secretion during fasting. Neuroendocrinology. [1998 Jun;67(6):370-6.
264. Yang Y, Zhou LB, Liu SQ, Tang JF, Li FY, Li RY, et al. Expression of feeding-related peptide receptors mRNA in GT1-7 cell line and roles of leptin and orexins in control of GnRH secretion. Acta Pharmacol Sin. [2005 Aug;26(8):976-81.
265. Korner J, Savontaus E, Chua SC, Jr., Leibel RL, Wardlaw SL. Leptin regulation of Agrp and Npy mRNA in the rat hypothalamus. J Neuroendocrinol. [2001 Nov;13(11):959-66.

162
162
266. Smith MS, Grove KL. Integration of the regulation of reproductive function and energy balance: lactation as a model. Frontiers in neuroendocrinology. [2002 Jul;23(3):225-56.
267. Sahu A. Leptin decreases food intake induced by melanin-concentrating hormone (MCH), galanin (GAL) and neuropeptide Y (NPY) in the rat. Endocrinology. [1998 Nov;139(11):4739-42.
268. Morrison CD, Morton GJ, Niswender KD, Gelling RW, Schwartz MW. Leptin inhibits hypothalamic Npy and Agrp gene expression via a mechanism that requires phosphatidylinositol 3-OH-kinase signaling. American journal of physiology. [2005 Dec;289(6):E1051-7.
269. Sakamoto K, Goransson O, Hardie DG, Alessi DR. Activity of LKB1 and AMPK-related kinases in skeletal muscle: effects of contraction, phenformin, and AICAR. American journal of physiology. [2004 Aug;287(2):E310-7.
270. Hardie DG. AMPK: a key regulator of energy balance in the single cell and the whole organism. International journal of obesity (2005). [2008 Sep;32 Suppl 4:S7-12.
271. Bates SH, Stearns WH, Dundon TA, Schubert M, Tso AW, Wang Y, et al. STAT3 signalling is required for leptin regulation of energy balance but not reproduction. Nature. [2003;421:856-9.
272. Bjorbaek C, Buchholz RM, Davis SM, Bates SH, Pierroz DD, Gu H, et al. Divergent roles of SHP-2 in ERK activation by leptin receptors. J Biol Chem. [2001 Feb 16;276(7):4747-55.
273. Bates SH, Myers MG. The role of leptin-->STAT3 signaling in neuroendocrine function: an integrative perspective. Journal of molecular medicine (Berlin, Germany). [2004 Jan;82(1):12-20.
274. Metlakunta AS, Sahu M, Sahu A. Hypothalamic phosphatidylinositol 3-kinase pathway of leptin signaling is impaired during the development of diet-induced obesity in FVB/N mice. Endocrinology. [2008 Mar;149(3):1121-8.
275. Sahu A, Metlakunta AS. Hypothalamic phosphatidylinositol 3-kinase-phosphodiesterase 3B-cyclic AMP pathway of leptin signalling is impaired following chronic central leptin infusion. Journal of neuroendocrinology. [2005 Nov;17(11):720-6.
276. Mirshamsi S, Laidlaw HA, Ning K, Anderson E, Burgess LA, Gray A, et al. Leptin and insulin stimulation of signalling pathways in arcuate nucleus neurones: PI3K dependent actin reorganization and KATP channel activation. BMC neuroscience. [2004 Dec 6;5:54.
277. Hill JW, Williams KW, Ye C, Luo J, Balthasar N, Coppari R, et al. Acute effects of leptin require PI3K signaling in hypothalamic proopiomelanocortin neurons in mice. The Journal of clinical investigation. [2008 May;118(5):1796-805.

163
163
278. Xu AW, Kaelin CB, Takeda K, Akira S, Schwartz MW, Barsh GS. PI3K integrates the action of insulin and leptin on hypothalamic neurons. The Journal of clinical investigation. [2005 Apr;115(4):951-8.
279. Ning K, Miller LC, Laidlaw HA, Burgess LA, Perera NM, Downes CP, et al. A novel leptin signalling pathway via PTEN inhibition in hypothalamic cell lines and pancreatic beta-cells. The EMBO journal. [2006 Jun 7;25(11):2377-87.
280. Carling D. The AMP-activated protein kinase cascade--a unifying system for energy control. Trends in biochemical sciences. [2004 Jan;29(1):18-24.
281. Kola B, Boscaro M, Rutter GA, Grossman AB, Korbonits M. Expanding role of AMPK in endocrinology. Trends in endocrinology and metabolism: TEM. [2006 Jul;17(5):205-15.
282. Hutber CA, Hardie DG, Winder WW. Electrical stimulation inactivates muscle acetyl-CoA carboxylase and increases AMP-activated protein kinase. The American journal of physiology. [1997 Feb;272(2 Pt 1):E262-6.
283. Xue B, Kahn BB. AMPK integrates nutrient and hormonal signals to regulate food intake and energy balance through effects in the hypothalamus and peripheral tissues. The Journal of physiology. [2006 Jul 1;574(Pt 1):73-83.
284. El-Haschimi K, Pierroz DD, Hileman SM, Bjorbaek C, Flier JS. Two defects contribute to hypothalamic leptin resistance in mice with diet-induced obesity. The Journal of clinical investigation. [2000 Jun;105(12):1827-32.
285. Uotani S, Bjorbaek C, Tornoe J, Flier JS. Functional properties of leptin receptor isoforms: internalization and degradation of leptin and ligand-induced receptor downregulation. Diabetes. [1999 Feb;48(2):279-86.
286. Simerly RB. Wired on hormones: endocrine regulation of hypothalamic development. Current opinion in neurobiology. [2005 Feb;15(1):81-5.
287. Hassink SG, de Lancey E, Sheslow DV, Smith-Kirwin SM, O'Connor DM, Considine RV, et al. Placental leptin: an important new growth factor in intrauterine and neonatal development? Pediatrics. [1997 Jul;100(1):E1.
288. Luquet S, Perez FA, Hnasko TS, Palmiter RD. NPY/AgRP neurons are essential for feeding in adult mice but can be ablated in neonates. Science (New York, NY. [2005 Oct 28;310(5748):683-5.
289. Simonian SX, Spratt DP, Herbison AE. Identification and characterization of estrogen receptor alpha-containing neurons projecting to the vicinity of the gonadotropin-releasing hormone perikarya in the rostral preoptic area of the rat. J Comp Neurol. [1999 Aug 23;411(2):346-58.

164
164
290. Vanhatalo S. Comparison of the Distributions of Neuropeptide Y-, Tyrosine Hydroxylase-, and Tryptophan Hydroxylase-Expressing Neurons in the Hypothalamic Arcuate Nucleus Nutritional Neuroscience. [2000 February 2000;3(1):11-7.
291. Turi GF, Liposits Z, Moenter SM, Fekete C, Hrabovszky E. Origin of neuropeptide Y-containing afferents to gonadotropin-releasing hormone neurons in male mice. Endocrinology. [2003 Nov;144(11):4967-74.
292. Horvath TL, Bechmann I, Naftolin F, Kalra SP, Leranth C. Heterogeneity in the neuropeptide Y-containing neurons of the rat arcuate nucleus: GABAergic and non-GABAergic subpopulations. Brain research. [1997 May 9;756(1-2):283-6.
293. Baskin DG, Hahn TM, Schwartz MW. Leptin sensitive neurons in the hypothalamus. Horm Metab Res. [1999 May;31(5):345-50.
294. Kalra PS, Bonavera JJ, Kalra SP. Central administration of antisense oligodeoxynucleotides to neuropeptide Y (NPY) mRNA reveals the critical role of newly synthesized NPY in regulation of LHRH release. Regul Pept. [1995;59:215-20.
295. Dudas B, Mihaly A, Merchenthaler I. Topography and associations of luteinizing hormone-releasing hormone and neuropeptide Y-immunoreactive neuronal systems in the human diencephalon. J Comp Neurol. [2000;427:593-603.
296. Dufourny L, Caraty A, Clarke IJ, Robinson JE, Skinner DC. Progesterone-receptive dopaminergic and neuropeptide Y neurons project from the arcuate nucleus to gonadotropin-releasing hormone-rich regions of the ovine preoptic area. Neuroendocrinology. [2005;82(1):21-31.
297. Pierroz DD, Gruaz NM, d'Alieves V, Aubert ML. Chronic administration of neuropeptide Y into the lateral ventricle starting at 30 days of life delays sexual maturation in the female rat. Neuroendocrinology. [1995 Mar;61(3):293-300.
298. McDonald JK, Lumpkin MD, DePaolo LV. Neuropeptide-Y suppresses pulsatile secretion of luteinizing hormone in ovariectomized rats: possible site of action. Endocrinology. [1989 Jul;125(1):186-91.
299. Kalra SP, Crowley WR. Differential effects of pancreatic polypeptide on luteinizing hormone release in female rats. Neuroendocrinology. [1984 Jun;38(6):511-3.
300. McDonald JK, Lumpkin MD, Samson WK, McCann SM. Neuropeptide Y affects secretion of luteinizing hormone and growth hormone in ovariectomized rats. Proceedings of the National Academy of Sciences of the United States of America. [1985 Jan;82(2):561-4.
301. Catzeflis C, Pierroz DD, Rohner-Jeanrenaud F, Rivier JE, Sizonenko PC, Aubert ML. Neuropeptide Y administered chronically into the lateral ventricle profoundly inhibits both the gonadotropic and the somatotropic axis in intact adult female rats. Endocrinology. [1993 Jan;132(1):224-34.

165
165
302. Herbison AE. Multimodal influence of estrogen upon gonadotropin-releasing hormone neurons. Endocrine Rev. [1998;19:302-30.
303. Li S, Hong M, Fournier A, St-Pierre S, Pelletier G. Role of neuropeptide Y in the regulation of gonadotropin-releasing hormone gene expression in the rat preoptic area. Brain Res Mol Brain Res. [1994;26:69-73.
304. Kalra PS, Kalra SP. Use of antisense oligodeoxynucleotides to study the physiological functions of neuropeptide Y. Methods. [2000;22:249-54.
305. Kasuya E, Mizuno M, Watanabe G, Terasawa E. Effects of an antisense oligodeoxynucleotide for neuropeptide Y mRNA on in vivo luteinizing hormone-releasing hormone release in ovariectomized female rhesus monkeys. Regul Pept. [1998;75-76:319-25.
306. Campbell RE, ffrench-Mullen JM, Cowley MA, Smith MS, Grove KL. Hypothalamic circuitry of neuropeptide Y regulation of neuroendocrine function and food intake via the Y5 receptor subtype. Neuroendocrinology. [2001;74:106-19.
307. Kalra SP, Fuentes M, Fournier A, Parker SL, Crowley WR. Involvement of the Y-1 receptor subtype in the regulation of luteinizing hormone secretion by neuropeptide Y in rats. Endocrinology. [1992;130:3323-30.
308. Knauf C, Chuoi MM, Jirou-Najou JL, Mortreux G, Beauvillain JC, Croix D. Involvement of NPY Y2 receptor subtype in the control of the spontaneous NO/GnRH release at the rat median eminence. Neuroreport. [2001;12:3365-9.
309. Campbell RE, ffrench-Mullen JM, Cowley MA, Smith MS, Grove KL. Hypothalamic circuitry of neuropeptide Y regulation of neuroendocrine function and food intake via the Y5 receptor subtype. Neuroendocrinology. [2001 Aug;74(2):106-19.
310. Pralong FP, Gonzales C, Voirol MJ, Palmiter RD, Brunner HR, Gaillard RC, et al. The neuropeptide Y Y1 receptor regulates leptin-mediated control of energy homeostasis and reproductive functions. Faseb J. [2002 May;16(7):712-4.
311. Klenke U, Constantin S, Wray S. Neuropeptide Y directly inhibits neuronal activity in a subpopulation of gonadotropin-releasing hormone-1 neurons via Y1 receptors. Endocrinology. [2010 Jun;151(6):2736-46.
312. Jain MR, Pu S, Kalra PS, Kalra SP. Evidence that stimulation of two modalities of pituitary luteinizing hormone release in ovarian steroid-primed ovariectomized rats may involve neuropeptide Y Y1 and Y4 receptors. Endocrinology. [1999;140:5171-7.
313. Raposinho PD, Broqua P, Hayward A, Akinsanya K, Galyean R, Schteingart C, et al. Stimulation of the gonadotropic axis by the neuropeptide Y receptor Y1 antagonist/Y4 agonist 1229U91 in the male rat. Neuroendocrinology. [2000;71:2-7.

166
166
314. McDonald JK, Lumpkin MD, Samson WK, McCann SM. Pancreatic polypeptides affect luteinizing and growth hormone secretion in rats. Peptides. [1985 Jan-Feb;6(1):79-84.
315. Sainsbury A, Schwarzer C, Couzens M, Jenkins A, Oakes SR, Ormandy CJ, et al. Y4 receptor knockout rescues fertility in ob/ob mice. Genes Dev. [2002;16:1077-88.
316. Kim K, Ramirez VD. Dibutyryl cyclic adenosine monophosphate stimulates in vitro luteinizing hormone-releasing hormone release only from median eminence derived from ovariectomized, estradiol-primed rats. Brain Res. [1985;342(1):154-7.
317. Lee BJ, Kim K, Cho WK. Activation of intracellular pathways with forskolin and phorbol ester increases LHRH mRNA level in the rat hypothalamus superfused in vitro. Brain Res Mol Brain Res. [1990;8(3):185-91.
318. Wetsel WC, Eraly SA, Whyte DB, Mellon PL. Regulation of gonadotropin-releasing hormone by protein kinase-A and -C in immortalized hypothalamic neurons. Endocrinology. [1993 Jun;132(6):2360-70.
319. Roy D, Belsham DD. Melatonin receptor activation regulates gonadotropin-releasing hormone (GnRH) gene expression and secretion in GT1-7 GnRH neurons: Signal transduction mechanisms. J Biol Chem. [2002;277:251-8.
320. van Biesen T, Luttrell LM, Hawes BE, Lefkowitz RJ. Mitogenic Signaling via G Protein-Coupled Receptors. Endocrine Reviews. [1996;17:698-714.
321. Bokoch GM. Interplay between Ras-related and heterotrmeric GTP binding proteins: lifestyles of the big and little. FASEB. [1996;10:1290-5.
322. Rozengurt E. Signal Transduction Pathways in the Mitogenic Response to G Protein-Coupled Neuropeptide Receptor Agonists. Journal of Cellular Physiology. [1998;177:507-17.
323. Wetsel WC. Immortalized hypothalamic luteinizing hormone-releasing hormone (LHRH) neurons: A new tool for dissecting the molecular and cellular basis of LHRH physiology. Cell Mol Neurobiol. [1995;15:43-78.
324. Eraly SA, Mellon PL. Regulation of gonadotropin-releasing hormone transcription by protein kinase C is mediated by evolutionarily conserved, promoter-proximal elements. Mol Endocrinol. [1995;9:848-59.
325. Allen LH. Nutritional influences on linear growth: a general review. European journal of clinical nutrition. [1994 Feb;48 Suppl 1:S75-89.
326. Hileman SM, Pierroz DD, Flier JS. Leptin, nutrition, and reproduction: timing is everything. The Journal of clinical endocrinology and metabolism. [2000 Feb;85(2):804-7.

167
167
327. Temple JL, Rissman EF. Acute re-feeding reverses food restriction-induced hypothalamic-pituitary-gonadal axis deficits. Biology of reproduction. [2000 Dec;63(6):1721-6.
328. Tonkelaar ID, Seidell JC, van Noord PA, Baanders-van Halewijn EA, Jacobus JH, Bruning PF. Factors influencing waist/hip ratio in randomly selected pre- and post-menopausal women in the dom-project (preliminary results). International journal of obesity. [1989;13(6):817-24.
329. Andersson B, Mattsson LA, Hahn L, Marin P, Lapidus L, Holm G, et al. Estrogen replacement therapy decreases hyperandrogenicity and improves glucose homeostasis and plasma lipids in postmenopausal women with noninsulin-dependent diabetes mellitus. The Journal of clinical endocrinology and metabolism. [1997 Feb;82(2):638-43.
330. Santollo J, Eckel LA. Effect of a putative ERalpha antagonist, MPP, on food intake in cycling and ovariectomized rats. Physiology & behavior. [2009 May 25;97(2):193-8.
331. Silva AP, Cavadas C, Grouzmann E. Neuropeptide Y and its receptors as potential therapeutic drug targets. Clinica chimica acta; international journal of clinical chemistry. [2002 Dec;326(1-2):3-25.
332. Rogers NH, Witczak CA, Hirshman MF, Goodyear LJ, Greenberg AS. Estradiol stimulates Akt, AMP-activated protein kinase (AMPK) and TBC1D1/4, but not glucose uptake in rat soleus. Biochemical and biophysical research communications. [2009 May 15;382(4):646-50.
333. Simoncini T, Rabkin E, Liao JK. Molecular basis of cell membrane estrogen receptor interaction with phosphatidylinositol 3-kinase in endothelial cells. Arteriosclerosis, thrombosis, and vascular biology. [2003 Feb 1;23(2):198-203.
334. Mori H, Hanada R, Hanada T, Aki D, Mashima R, Nishinakamura H, et al. Socs3 deficiency in the brain elevates leptin sensitivity and confers resistance to diet-induced obesity. Nature medicine. [2004 Jul;10(7):739-43.
335. Bjorbaek C, El-Haschimi K, Frantz JD, Flier JS. The role of SOCS-3 in leptin signaling and leptin resistance. J Biol Chem. [1999 Oct 15;274(42):30059-65.
336. Zabolotny JM, Bence-Hanulec KK, Stricker-Krongrad A, Haj F, Wang Y, Minokoshi Y, et al. PTP1B regulates leptin signal transduction in vivo. Developmental cell. [2002 Apr;2(4):489-95.
337. Van Heek M, Compton DS, France CF, Tedesco RP, Fawzi AB, Graziano MP, et al. Diet-induced obese mice develop peripheral, but not central, resistance to leptin. The Journal of clinical investigation. [1997 Feb 1;99(3):385-90.
338. Fioramonti X, Contie S, Song Z, Routh VH, Lorsignol A, Penicaud L. Characterization of glucosensing neuron subpopulations in the arcuate nucleus:

168
168
integration in neuropeptide Y and pro-opio melanocortin networks? Diabetes. [2007 May;56(5):1219-27.
339. Fick LJ, Fick GH, Belsham DD. Rhythmic clock and neuropeptide gene expression in hypothalamic mHypoE-44 neurons. Molecular and cellular endocrinology. [Jul 29;323(2):298-306.
340. May T, Hauser H, Wirth D. Transcriptional control of SV40 T-antigen expression allows a complete reversion of immortalization. Nucleic acids research. [2004;32(18):5529-38.
341. Taguchi Y, Koslowski M, Bodenner DL. Binding of estrogen receptor with estrogen conjugated to bovine serum albumin (BSA). Nuclear receptor. [2004 Aug 19;2(1):5.
342. Watters JJ, Campbell JS, Cunningham MJ, Krebs EG, Dorsa DM. Rapid membrane effects of steroids in neuroblastoma cells: effects of estrogen on mitogen activated protein kinase signalling cascade and c-fos immediate early gene transcription. Endocrinology. [1997 Sep;138(9):4030-3.
343. Harrington WR, Kim SH, Funk CC, Madak-Erdogan Z, Schiff R, Katzenellenbogen JA, et al. Estrogen dendrimer conjugates that preferentially activate extranuclear, nongenomic versus genomic pathways of estrogen action. Molecular endocrinology (Baltimore, Md. [2006 Mar;20(3):491-502.
344. Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, McLauchlan H, et al. The selectivity of protein kinase inhibitors: a further update. The Biochemical journal. [2007 Dec 15;408(3):297-315.
345. Gao Q, Horvath TL. Cross-talk between estrogen and leptin signaling in the hypothalamus. American journal of physiology. [2008 May;294(5):E817-26.
346. Diano S, Kalra SP, Sakamoto H, Horvath TL. Leptin receptors in estrogen receptor-containing neurons of the female rat hypothalamus. Brain research. [1998 Nov 23;812(1-2):256-9.
347. Thorn SR, Meyer MJ, Van Amburgh ME, Boisclair YR. Effect of estrogen on leptin and expression of leptin receptor transcripts in prepubertal dairy heifers. Journal of dairy science. [2007 Aug;90(8):3742-50.





























