Embed Size (px)
Citation preview

THE ROLE OF APOLIPOPROTEIN E IN RECOVERY FROM TRAUMATIC BRAIN
INJURY AND DEVELOPMENT OF CHIMERA: A NOVEL CLOSED-HEAD IMPACT
MODEL OF ENGINEERED ROTATIONAL ACCELERATION
by
Dhananjay Rajaram Namjoshi
M.Sc., The University of British Columbia, 2008
M.Pharm., The University of Mumbai, 2003
B.Pharm., The University of Mumbai, 1999
A THESIS SUBMITTED IN PARTIAL FULFILMENT OF THE REQUIREMENTS FOR THE DEGREE OF
DOCTOR OF PHILOSOPHY
in
The Faculty of Graduate and Postdoctoral Studies
(Neuroscience)
THE UNIVERSITY OF BRITISH COLUMBIA
(Vancouver)
February 2015
© Dhananjay Rajaram Namjoshi, 2015

Abstract
Traumatic brain injury (TBI) is a “silent epidemic” that currently lacks any effective
treatment. While a major health care problem in itself, TBI also increases Alzheimer’s
disease (AD) risk and leads to the deposition of neurofibrillary tangles and amyloid
deposits similar to those found in AD. Agonists of Liver X receptors (LXRs), which
regulate the expression of many genes involved in lipid homeostasis and inflammation,
improve cognition and reduce neuropathology in AD mice. One pathway by which LXR
agonists exert their beneficial effects is through ATP-binding cassette transporter A1-
mediated lipid transport onto apolipoprotein E (apoE). In the first part of this thesis, I
show that a short-term treatment with synthetic LXR agonist GW3965 improves post-
injury outcomes in mice subjected to closed-head, mild, repetitive weight drop TBI
(mrTBI). My results suggest that both apoE-dependent and apoE-independent
pathways contribute to the ability of GW3965 to promote recovery from mrTBI. While
many drugs have shown promising outcomes in preclinical TBI models, clinical drug
trials for TBI so far have failed, suggesting that the translational potential of TBI models
may require further improvement. As most human TBIs result from impact to an intact
skull, closed head injury (CHI) rodent models are highly relevant. Traditional CHI
models like weight drop however suffer from large experimental variability that may be
due to poor control over biomechanical inputs. To address this caveat we developed a
novel CHI model called CHIMERA (Closed-Head Impact Model of Engineered
Rotational Acceleration) that fully integrates biomechanical, behavioral, and
neuropathological analyses. CHIMERA is distinct from existing neurotrauma model
systems in that it uses a completely non-surgical procedure to precisely deliver impacts
ii

of prescribed dynamic characteristics to a closed skull while enabling kinematic analysis
of unconstrained head movement. Here I show that repeated TBI in mice using
CHIMERA mimics many features of the human TBI including neurological, motor, and
cognitive deficits along with persistent neuroinflammation and diffuse axonal injury, and
increased endogenous tau phosphorylation up to 14 days with a reliable biomechanical
response of the head. This makes CHIMERA well suited to investigate the
pathophysiology of TBI and for drug development programs.
iii

Preface
Chapter 1.
Portions of the introductory text are reproduced from the published review article:
Namjoshi DR, Good C, Cheng WH, Panenka W, Richards D, Cripton PA, Wellington
CL (2013) Towards clinical management of traumatic brain injury: a review of models
and mechanisms from a biomechanical perspective. Disease Models & Mechanisms 6:
1325-1338.
Contributions:
Conception and outline of the article were designed by Dhananjay R. Namjoshi and
Cheryl L. Wellingtion. All authors contributed towards the preparation of the manuscript
with major contribution from Dhananjay R. Namjoshi.
Portions of the introductory text are reproduced from the published review article:
Namjoshi D, Stukas S, Wellington CL (2011) ABCA1, apoE and apoA-I as potential
therapeutic targets for treating Alzheimer’s disease. Neurodegenerative Disease
Management 1: 245-259.
Contributions:
Conception and outline of the article were designed by Dhananjay R. Namjoshi, Sophie
S. Stukas and Cheryl L. Wellingtion. Manuscript was prepared by Dhananjay R.
Namjoshi, Sophie Stukas and Cheryl L. Wellington with equal contribution from
Dhananjay Namjoshi and Sophie Stukas.
iv

Chapter 2.
A version is published in:
Namjoshi DR, Martin G, Donkin J, Wilkinson A, Stukas S, Fan J, Carr M, Tabarestani
S, Wuerth K, Hancock REW, Wellington CL (2013) The Liver X receptor agonist
GW3965 improves recovery from mild repetitive traumatic brain injury in mice partly
thorough apolipoprotein E. PLoS ONE 8(1): e53529.
All the animal experiments were carried out at UBC. All the biochemical and histology
experiments, except the cytokine assays described in this chapter were carried out in
the Wellington Laboratory at UBC. Cytokine assays were carried out by Kelli Wuerth in
the Hancock Laboratory at UBC.
Contributions:
All the experiments were designed by Dhananjay R Namjoshi and Cheryl L. Wellington.
James Donkin contributed towards the initial stages of experimental design. The
majority of experiments and data analyses were performed by Dhananjay R. Namjoshi.
Support for histological experiments and analysis was provided by Georgina Martin,
Michael Carr, and Sepediah Tabarestani and that for biochemical assays was provided
by Anna Wilkinson, Sophie Stukas, and Jianjia Fan. Manuscript was written by
Dhananjay R. Namjoshi and Cheryl L. Wellington.
v

Chapter 3.
A version is published in:
Namjoshi DR, Cheng WH, McInnes K, Martens KM, Carr M, Wilkinson A, Fan J,
Roberts J, Hayat A, Cripton PA, Wellington CL (2014) Merging Pathology with
Biomechanics using CHIMERA (Closed-Head Impact Model of Engineered Rotational
Acceleration): A Novel, Surgery-Free Model of Traumatic Brain Injury. Molecular
Neurodegeneration 9:55.
CHIMERA impactor was constructed by Kurt McInnes in the Cripton Laboratory at UBC.
All the animal experiments described in this Chapter were carried out at UBC. All the
biochemical and histology experiments described in this Chapter were carried out in the
Wellington Laboratory at UBC.
Contributions:
The CHIMERA impactor design was conceived by Dhananjay R. Namjoshi, Wai Hang
Cheng, Kurt McInnes, Peter A. Cripton, and Cheryl L. Wellington. Dhananjay R.
Namjoshi and Wai Hang Cheng designed and conducted experiments and analyzed
data. Support for head injury procedure was provided by Michael Carr and Kris M.
Martens. Behavioral assays were conducted by Dhananjay R. Namjoshi, Wai Hang
Cheng and Kris M. Martens. Head kinematics analysis was performed by Wai Hang
Cheng. Kurt McInnes and Peter A. Cripton provided expert advice on kinematic
analysis. Biochemical assays were carried out by Anna Wilkinson and Jianjia Fan.
Histology experiments were conducted by Dhananjay R. Namjoshi and Wai Hang
vi

Cheng. Support for histology was provided by Michael Carr, Jerome Roberts and Arooj
Hayat. Manuscript was prepared by Dhananjay R. Namjoshi, Wai Hang Cheng, and
Cheryl L. Wellington.
Study Approval by UBC Ethics Boards:
All the animal experiments described in this thesis were conducted according to the
protocols approved by the UBC Animal Care Committee as follows:
Protocol # A07-0706: ABCA1 and apoE function in traumatic brain injury.
Protocol # A11-0225: ABCA1 and apoE function in traumatic brain injury (New
Protocol).
The candidate’s animal care and training certification record:
3937-09: Online animal care training program
RBH-661-09: Rodent biology and husbandry
RA-343-09: Rodent anesthesia
RSHX-268-09: Rodent surgery
vii

Table of Contents
Abstract ............................................................................................................................ii
Preface ............................................................................................................................iv
Table of Contents .......................................................................................................... viii
List of Tables ..................................................................................................................xv
List of Figures ................................................................................................................ xvi
List of Abbreviations ...................................................................................................... xix
Acknowledgements ..................................................................................................... xxiii
Dedication .................................................................................................................... xxv
Chapter 1: Introduction .................................................................................................... 1
1.1 Traumatic Brain Injury (TBI): Definition ............................................................. 1
1.2 TBI: Epidemiology ............................................................................................. 4
1.3 TBI: Classification ............................................................................................. 7
1.3.1 TBI Classification Based on Clinical Severity .......................................... 7
1.3.2 TBI Classification Based on Pathophysiology ....................................... 10
1.3.3 TBI Classification Based on Physical Mechanism ................................. 11
1.4 TBI: Biomechanical Principles ........................................................................ 12
1.4.1 Head Motion during Impact TBI ............................................................. 15
1.4.2 Brain Motion during TBI ......................................................................... 17
1.4.3 The Clinical Picture: Human Tolerance and Related Biomechanical
Studies of Human TBI .................................................................................... 19
1.5 TBI: Pathophysiology ...................................................................................... 22
1.5.1 Primary Brain Injury ............................................................................... 23
viii

1.5.1.1 Focal Brain Injury ...................................................................... 24
1.5.1.1.1 Cerebral Contusion and Laceration .............................. 25
1.5.1.1.2 Intracranial Hemorrhage ............................................... 27
1.5.1.2 Diffuse Brain Injury .................................................................... 30
1.5.1.2.1 Diffuse Axonal Injury ..................................................... 30
1.5.1.2.2 Diffuse Vascular Injury .................................................. 34
1.5.2 Secondary Brain Injury .......................................................................... 34
1.5.2.1 Cerebrovascular Response to TBI ............................................ 35
1.5.2.2 Neurometabolic Changes Following TBI ................................... 36
1.5.2.3 Neuroinflammation Following TBI .............................................. 37
1.6 TBI, Alzheimer Disease, and Apolipoprotein E ............................................... 38
1.6.1 TBI and Alzheimer Disease ................................................................... 38
1.6.2 Alzheimer’s Disease: Pathology ............................................................ 39
1.6.2.1 Amyloid Beta Formation in the CNS .......................................... 40
1.6.2.2 Tauopathy in Alzheimer Disease ............................................... 42
1.6.3 TBI and Alzheimer’s Pathology: Similarities and Differences ................ 45
1.6.3.1 Amyloid Pathology after Moderate to Severe Single TBI ........... 45
1.6.3.2 Consequences of Repetitive TBI: Chronic Traumatic
Encephalopathy ..................................................................................... 50
1.6.4 Apolipoprotein E (ApoE) at the Nexus of TBI and AD Pathology ........... 55
1.6.4.1 ApoE Synthesis in the CNS ....................................................... 56
1.6.4.2 Liver X Receptors (LXR) ........................................................... 58
1.6.4.3 LXR: Molecular Mechanism of Action ........................................ 59
ix

1.6.4.4 LXR: Regulation of Cholesterol Metabolism .............................. 60
1.6.4.5 ABCA1: The Principal LXR Target ............................................ 62
1.6.4.6 The Role of LXRs in the CNS .................................................... 67
1.6.4.8 ApoE and Amyloid Beta (Aβ) Metabolism ................................. 75
1.6.4.8.1 ApoE and Aβ Interaction ............................................... 76
1.6.4.8.2 ApoE and Enzymatic Degradation of Aβ ....................... 77
1.6.4.8.3 Cellular Uptake and Degradation of Aβ ......................... 78
1.6.4.8.4 Aβ Clearance through the Cerebrovasculature ............. 79
1.6.4.9 ApoE, Synaptic Function, and Synaptogenesis ......................... 82
1.6.4.10 Role of ApoE in Inflammation .................................................. 84
1.6.5 ApoE and TBI ........................................................................................ 85
1.7 TBI: An Overview of Animal Models................................................................ 87
1.7.1 Open Head Injury Models ...................................................................... 89
1.7.1.1 Fluid Percussion (FP) ................................................................ 90
1.7.1.2 Controlled Cortical Impact (CCI) ................................................ 91
1.7.2 Closed Head Injury Models.................................................................... 92
1.7.2.1 CHI by Impact ............................................................................ 93
1.7.2.2 Non-Impact CHI by Blast Waves ............................................... 96
1.7.2.3 Non-Impact CHI by Inertial Loading .......................................... 97
1.7.3 Challenges Associated With Current TBI Models .................................. 98
1.8 Pharmacological and Non-pharmacological Management of TBI: An Overview
of Randomized Clinical Trials ............................................................................. 103
1.9 Study Rationale, Hypothesis, and Objectives ............................................... 106
x

Chapter 2: The Liver X Receptor Agonist GW3965 Improves Recovery from Mild
Repetitive Traumatic Brain Injury in Mice Partly Through Apolipoprotein E ................ 110
2.1 Summary ...................................................................................................... 110
2.2 Introduction ................................................................................................... 111
2.3 Materials and Methods .................................................................................. 115
2.3.1 Animals ................................................................................................ 115
2.3.2 Mild Repetitive Traumatic Brain Injury (mrTBI) .................................... 115
2.3.3 GW3965 Treatment ............................................................................. 119
2.3.4 Cognitive Assessment by Novel Object Recognition ........................... 120
2.3.5 Motor Function Assessment by Accelerating Rotarod ......................... 122
2.3.6 Biochemical Analyses .......................................................................... 122
2.3.6.1 Tissue Collection ..................................................................... 123
2.3.6.2 Protein Extraction .................................................................... 123
2.3.6.3 Western Blot Analyses ............................................................ 124
2.3.6.4 Quantitative Assessment of Endogenous Aβ by ELISA .......... 125
2.3.6.5 Quantitative Assessment of IL-6, TNFα, and MCP-1 by ELISA
............................................................................................................. 125
2.3.7 Histology .............................................................................................. 126
2.3.7.1 Assessment of Axonal Injury by Silver Staining ....................... 126
2.3.7.2 Assessment of Microglial Activation by Iba1
Immunohistochemistry ......................................................................... 127
2.3.8 Statistical Analyses .............................................................................. 128
2.4 Results .......................................................................................................... 129
xi

2.4.1 ApoE is Required for GW3965 to Improve Cognitive Function After
mrTBI ............................................................................................................ 129
2.4.2 Spontaneous Recovery of Motor Dysfunction After mrTBI is Unaffected
by GW3965 .................................................................................................. 133
2.4.3 GW3965 Prevents mrTBI-Induced Elevation of Endogenous Aβ Levels
..................................................................................................................... 135
2.4.4 GW3965 Enhances ABCA1 Induction Aafter mrTBI: ........................... 139
2.4.5 ApoE is Required for GW3965-Mediated Suppression of Axonal Damage
After mrTBI ................................................................................................... 142
2.4.6 Weight Drop TBI Model Produces Negligible Neuroinflammation........ 148
2.4.7 Retrospective Power Analysis ............................................................. 152
2.5 Discussion .................................................................................................... 154
Chapter 3: Merging Pathology and Biomechanics Using CHIMERA: A Novel, Surgery-
Free, Closed-Head Impact Model of Engineered Rotational Acceleration................... 161
3.1 Summary ...................................................................................................... 161
3.2 Introduction ................................................................................................... 162
3.3 Materials and Methods .................................................................................. 165
3.3.1 CHIMERA Impactor ............................................................................. 165
3.3.1.1 Animal Holding Platform .......................................................... 165
3.3.1.2 Pneumatic Impactor System.................................................... 166
3.3.1.3 Impact Piston and Barrel System ............................................ 166
3.3.1.4 CHIMERA Calibration .............................................................. 168
3.3.2 CHIMERA Repetitive TBI (rTBI) Procedure ......................................... 170
xii

3.3.3 High-Speed Videography and Kinematic Analyses ............................. 172
3.3.4 Behavioral Analyses ............................................................................ 174
3.3.5 Tissue Collection and Processing ........................................................ 178
3.3.6 Iba1 Immunohistochemistry and Silver Staining .................................. 179
3.3.7 Biochemical Analyses .......................................................................... 180
3.3.7.1 Tissue Processing ................................................................... 180
3.3.7.2 Assessment of TNF-α and IL-1β by ELISA .............................. 180
3.3.7.3 Quantitative Assessment of Phosphorylated and Total Tau by
Simple Western Analysis ..................................................................... 180
3.3.8 Statistical Analyses .............................................................................. 182
3.4 Results .......................................................................................................... 183
3.4.1 Head Kinematics Following CHIMERA rTBI ........................................ 183
3.4.2 CHIMERA rTBI Induces Behavioral Deficits ........................................ 189
3.4.3 CHIMERA rTBI Induces Widespread Diffuse Axonal Injury ................. 193
3.4.4 CHIMERA rTBI Induces Widespread Microglial Activation .................. 196
3.4.5 CHIMERA rTBI Increases Proinflammatory Cytokine Levels .............. 202
3.4.6 CHIMERA rTBI Increases Endogenous Tau Phosphorylation ............. 203
3.5 Discussion .................................................................................................... 205
Chapter 4: Conclusions and Future Directions ............................................................ 213
4.1 Chapter 2: Conclusions ................................................................................. 214
4.2 Chapter 2: Study Limitations and Future Directions ...................................... 219
4.3 Chapter 3: Conclusions ................................................................................. 223
4.4 Chapter 3: Study Limitations and Future Directions ...................................... 227
xiii

Bibliography ................................................................................................................ 230
xiv

List of Tables
Table 1.1 TBI incidence (in %) by cause across the globe. ............................................. 6
Table 1.2: Glasgow coma scale. ..................................................................................... 8
Table 1.3: North American classification of TBI according to severity of clinical signs. ... 8
Table 1.4: TBI classification according to European Federation of Neurological Societies
guidelines. ..................................................................................................................... 10
Table 1.5: Differences in the pathological features of CTE and AD. ............................. 53
Table 1.6: Pathological and clinical stages of CTE. ...................................................... 54
Table 1.7: Summary of physical and physiological properties of apoE alleles. ............. 75
Table 2.1: Details of primary antibodies used for Western blotting. ............................ 124
Table 2.2: Details of primary and secondary antibodies used for cytokine ELISA. ...... 126
Table 2.3: Summary of retrospective power calculations ............................................ 154
Table 3.1: Neurological severity score (NSS) tasks. ................................................... 178
Table 3.2: Details of monoclonal antibodies for probing p-tau and total tau. ............... 181
Table 3.3: Summary of peak values of head kinematic parameters. ........................... 187
Table 3.4: Comparison of head kinematic parameters between rodent TBI models and
human TBI. .................................................................................................................. 188
Table 4.1: Comparison of weight drop-rTBI and CHIMERA-rTBI models……………...226
xv

List of Figures
Figure 1.1: Annual incidence of TBI in people under 40 years of age ............................. 5
Figure 1.2: Biomechanics of head impact and brain tissue deformation. ...................... 14
Figure 1.3: Wayne State Tolerance Curve (WSTC). ..................................................... 20
Figure 1.4: Primary TBI. ................................................................................................ 23
Figure 1.5: Proposed molecular mechanism of diffuse axonal injury and generation of
Aβ. ................................................................................................................................. 33
Figure 1.6: Amyloidogenic and anti-amyloidogenic processing of APP. ........................ 42
Figure 1.7: Regulation of ApoE Lipidation by LXR. ....................................................... 62
Figure 1.8: ApoE structure, polymorphism and domain interaction. .............................. 74
Figure 1.9: Animal models of TBI. ................................................................................. 89
Figure 1.10: Fluid percussion injury model. ................................................................... 91
Figure 1.11: Controlled cortical impact model. .............................................................. 92
Figure 1.12: Blast Wave TBI Model. .............................................................................. 97
Figure 1.13: Variability in input parameters and outcomes in weight-drop TBI studies
reported in the literature. ............................................................................................. 100
Figure 2.1: Weight drop CHI model and impact location. ............................................ 118
Figure 2.2: Average animal body weight and drop height across all study groups. ..... 119
Figure 2.3: Novel object recognition phases. .............................................................. 122
Figure 2.4: Summary of mrTBI, Gw3965 treatment and post-injury outcomes. ........... 128
Figure 2.5: ApoE is required for GW3965 to improve NOR performance after mrTBI. 131
Figure 2.6: NOR performance was not affected by motor impairment. ....................... 132
xvi

Figure 2.7: mrTBI-induced motor impairment recovers spontaneously independent of
GW3965 and apoE. ..................................................................................................... 134
Figure 2.8: GW3965 prevents mrTBI-induced accumulation of endogenous Aβ in WT
and apoE-/- mice. ........................................................................................................ 137
Figure 2.9: APP and APP-CTFα levels remain unchanged following mrTBI. .............. 138
Figure 2. 10: GW3965 augments ABCA1 levels in WT and apoE-/- mice following
mrTBI. ......................................................................................................................... 140
Figure 2.11: ApoE and LDLR levels are unaffected by mrTBI or GW3965. ................. 141
Figure 2.12: Loss of apoE exacerbates axonal injury after mrTBI. .............................. 143
Figure 2.13: mrTBI leads to mild axonal damage in WT mice. .................................... 144
Figure 2.14: Loss of apoE exacerbates axonal damage after mrTBI. ......................... 145
Figure 2.15: ApoE is required for GW3965 to suppress axonal damage after mrTBI. . 147
Figure 2.16: mrTBI does not induce microglial activation in the cortex. ...................... 149
Figure 2.17: mrTBI induces negligible hippocampal microglial activation.................... 150
Figure 2.18: Pronounced microglial activation is localized only around contused areas.
.................................................................................................................................... 151
Figure 2.19: Pronounced microglial activation is localized only around contused areas.
.................................................................................................................................... 152
Figure 3.1: CHIMERA impactor and impact pistons. ................................................... 168
Figure 3.2: Piston energy-air pressure calibration curve. ............................................ 169
Figure 3.3: Mouse head and impact position. .............................................................. 171
Figure 3.4: External markers used for mouse head tracking. ...................................... 174
Figure 3.5: Open field thigmotaxis. .............................................................................. 177
xvii

Figure 3.6: CHIMERA-rTBI procedure and post-rTBI endpoints ................................. 182
Figure 3.7: Head kinematics following CHIMERA rTBI. ............................................... 185
Figure 3.8: CHIMERA allows unrestricted head motion during TBI. ............................ 186
Figure 3.9: CHIMERA rTBI induces behavioral deficits. .............................................. 192
Figure 3.10: CHIMERA rTBI does not significantly affect general mobility. ................. 193
Figure 3.11: CHIMERA rTBI induces diffuse axonal injury. ......................................... 195
Figure 3.12: CHIMERA rTBI induces sustained axonal injury ..................................... 196
Figure 3.13: CHIMERA rTBI induces widespread microglial activation. ...................... 199
Figure 3.14: Quantitative analysis of microglial response to rTBI. ............................... 201
Figure 3.15: CHIMERA rTBI increases proinflammatory cytokine levels. .................... 202
Figure 3.16: CHIMERA rTBI increases endogenous tau phosphorylation. .................. 204
xviii

List of Abbreviations
ABCA1: Adenosine triphosphate binding cassette transporter A1
Aβ: Amyloid beta peptide
ACRM: American Congress of Rehabilitation Medicine
AICD: Amyloid intracellular domain
ApoE/apoE: Apolipoprotein E
ApoA-I/apoA-I: Apolipoprotein A-I
APP: Amyloid precursor protein
AD: Alzheimer disease
ANOVA: Analysis of variance
BASE-1: Beta site APP cleaving enzyme-1
BBB: Blood-brain barrier
CAA: Cerebral amyloid angiopathy
CCI: Controlled cortical impact
CDC: Centers for disease control and prevention
CG: Center of gravity
CHI: Closed-head injury
CHIMERA: Closed-head impact model of engineered rotational acceleration
CNS: Central nervous system
CSF: Cerebrospinal fluid
CT: Computed tomography
CTE: Chronic traumatic encephalopathy
CTF: APP C-terminal fragment
xix

DAI: Diffuse axonal injury
DI: Discrimination index
DMSO: Dimethyl sulfoxide
DTI: Diffuse tensor imaging
DVI: Diffuse vascular injury
EDH: Epidural hemorrhage/hematoma
EFNS: European federation of neurological societies
FP: Fluid percussion
GCS: Glasgow coma scale
HDL: High-density lipoprotein
HIC: Head injury criterion
ICH: Intracranial hemorrhage/hematoma
ICP: Intracranial pressure
IDE: Insulin degrading enzyme
IL-1β: Interleukin 1 beta
IL-6: Interleukin 6
i.p.: Intraperitoneal
ISF: Interstitial fluid
LDL: Low-density lipoprotein
LDLR: LDL receptor
LRP1: LDLR related protein 1
LOC: Loss of consciousness
LRR: Loss of righting reflex
xx

LTP: Long-term potentiation
LXR: Liver X receptor
LXRE: LXR response element
MAPT: Microtubule associated protein tau
MCP-1: Monocyte chemotactic protein-1
MRI: Magnetic resonance imaging
mTBI: Mild traumatic brain injury
mrTBI: Mild, repetitive traumatic brain injury
MTBR: Microtubule binding repeat
MVA: Motor vehicle accident
NEP: Neprilysin
NFL: National football league
NFT: Neurofibrillary tangles
NMDA: N-methyl-d-aspartate
NOR: Novel object recognition
NSS: Neurological severity score
OHI: Open head injury
PBS: Phosphate-buffered saline
PS-1/2: Presenilin 1/2
PTA: Post-traumatic amnesia
RCT: Reverse cholesterol transport
RIPA: Radioimmunoprecipitation assay
rTBI: Repetitive traumatic brain injury
xxi

RXR: Retinoid X receptor
TBI: Traumatic brain injury
SDS: Sodium dodecyl sulfate
s.c.: Subcutaneous
SCI: Spinal cord injury
SDH: Subdural hemorrhage/hematoma
TNF-α: Tumor necrosis factor alpha
VLDL: Very low-density lipoprotein
WHO: World health organization
WSTC: Wayne state tolerance curve
WT: Wild-type (C57Bl/6) mice
xxii

Acknowledgements
As I come to this milestone in the pursuit of knowledge, I cannot help but reflect back
and acknowledge all those who have contributed towards completion of this work in one
or the other way.
First of all, I offer my deepest gratitude to Almighty GOD for showering ITS infinite
bounties, graces and mercies on me and giving me strength in the most difficult times.
Without ITS wishes and blessings, this work could have remained a dream only.
I would like to dedicate this work to my parents for their unconditional love, constant
encouragement, to my wife Archana for her immense love, patience, and unstinting
support, who stood with me in every good and bad moment and to my parents-in-law for
their constant motivation and support.
I express my deepest gratitude for my mentor Dr. Cheryl Wellington. Thank you Cheryl
for your guidance, support, constant encouragement, constructive criticism, and
valuable suggestions throughout my work. It has been an enriching experience and
privilege working in your laboratory.
I sincerely thank my supervisory committee members, Drs. Brian MacVicar, Wolfram
Tetzlaff, and Gordon Francis for providing critical guidance throughout this work.
xxiii

I thank all the current and past Wellington Lab members, especially, Anna Wilkinson,
Jeniffer Chan, Dr. James Donkin, Dr. Veronica Hirsh-Reinshagen, Dr. Kris Martens, Dr.
Jianjia Fan, Dr. Sophie Stukas, Tom Cheng, Mike Carr, Georgina Martin, and Arooj
Hayat for all the help and support throughout this work. Special thanks to Tom for all the
intellectual talks and exchange of ideas and who has a lion’s share in the development
of CHIMERA.
Special thanks to Dr. Peter Cripton and Kurt McInnes for helping me understand the
biomechanics of traumatic brain injury as well as for the wonderful collaboration that
resulted in the development of CHIMERA.
I express my gratitude towards Alzheimer Society of Canada and its patrons for
supporting me through Alzheimer Society Research Program doctoral award. This work
was also supported by the grants from Canadian Institutes of Health Research to Dr.
Cheryl Wellington.
I am grateful to Child and Family Research Institute, Centre for Disease Modeling, as
well as Djavad Mowafaghian Centre for Brain Health for allowing me to use their core
facilities and space to conduct this research work.
Last but not the least I would like to acknowledge the unsung heroes of this work, the
laboratory mice without whom this work would not be possible.
xxiv

Dedication
To my parents and parents-in-law.
To my wife, Archana
xxv

Chapter 1: Introduction
1.1 Traumatic Brain Injury (TBI): Definition
Traumatic brain injury (TBI) occurs when an external mechanical force traumatically
injures the brain resulting in altered mental state. TBI is a type of acquired brain injury
that is caused by events after birth as opposed to genetic or congenital defects. As
discussed further in this Chapter, TBI pathology is heterogeneous and each TBI is
unique and hence defining TBI has been challenging. The definition as well as the
classification of TBI has evolved through several attempts by different groups. As
discussed further about 75% of the human TBI are mild and therefore in most of the
attempts of defining TBI have considered only mild TBI (mTBI). The most commonly
used outcomes defining mTBI are Glasgow Coma Scale (GCS) score, loss of
consciousness (LOC) and post-traumatic amnesia (PTA). For example, the American
Congress of Rehabilitation Medicine (ACRM) defined mTBI as “traumatically induced
physiological disruption of brain function as manifested by at least one of the following:
1) any period of LOC, 2) any loss of memory events immediately before or after the
accident, 3) any alteration in mental state (e.g., dizziness, disorientation, or confusion)
at the time of the accident, and 4) focal neurological deficits which may or may not be
transient; but where the severity of injury does not exceed the following: LOC ≤ 30 min,
GCS score of 13-15 after 30 min, and PTA < 24 h” (Kay et al., 1993). The U.S. Centers
for Disease Control and Prevention (CDC) proposed a general definition of mTBI as “ an
injury to the head as a result of blunt trauma or acceleration forces that result in: 1)
transient confusion, disorientation, or impaired consciousness or 2) dysfunction of
memory around the time of injury or 3) LOC < 30 min or 4) neurological or
1

neuropsychological dysfunction such as seizures following injury (Gerberding and
Binder, 2003). The CDC also recommends developing case-specific definition of mTBI
based on this general definition. Later, the World Health Organization (WHO)
Collaborating Centre for Neurotrauma Task Force on Mild Traumatic Brain Injury
published another definition for mTBI as “an acute brain injury resulting from mechanical
energy to the head from external physical forces” manifesting into confusion or
disorientation, LOC for 30 min or PTA for < 24 h and GCS score of 13-15 after 30 min
(Carroll et al., 2004). The WHO definition further emphasizes that mTBI manifestations
must not be due to drugs, alcohol, or injuries caused by other mechanisms, e.g.,
systemic injuries, facial injuries or penetrating craniocerebral injury.
Thus, the ACRM, CDC, and WHO definitions address only the mild spectrum of TBI and
not moderate and severe TBIs, including penetrating brain injuries. Moreover, these
definitions consider only neurological signs while ignoring any evidence of the resulting
brain pathology.
Most recently, the Demographics and Clinical Assessment Working Group of the
International and Interagency Initiative toward Common Data Elements for Research on
Traumatic Brain Injury and Psychological Health proposed a broader definition of TBI as
“an alteration in brain function, or other evidence of brain pathology caused by an
external force” (Menon et al., 2010).
2

The alteration of brain function is assessed by clinical examination and includes any
one of the following: 1) any period of LOC, 2) any loss of memory immediately before
(retrograde amnesia) or after (PTA) TBI, 3) neurological deficits including weakness,
loss of balance, sensory loss, aphasia (problem with speaking, listening, reading and
writing), and paralysis, and 4) any alteration in mental state (confusion, disorientation) at
the time of injury (Menon et al., 2010). With the advances in modern imaging techniques
such as Magnetic Resonance Imaging (MRI), Diffusion Tensor Imaging (DTI) and
Computed Tomography (CT), it is now possible to visualize brain damage in some
cases. The neurological examination of TBI can therefore be supplemented with these
techniques (in addition to potential biomarkers of great interest for future use) to obtain
evidence of brain pathology. Lastly, TBI is caused by an external mechanical force,
which distinguishes TBI from other types of acquired brain injuries such as
cerebrovascular accident (stroke), brain tumors, brain infection, and brain injury caused
by substance abuse. The external mechanical force can be an impact (e.g., a moving
object striking stationary head such as during an assault or a moving head striking the
stationary object such as during a fall) or non-impact (e.g., rapid head movement during
whiplash or head exposed to blast waves) or a penetrating force (e.g., a bullet
penetrating the skull).
The new definition also encourages using the more appropriate term “traumatic brain
injury” rather than “head injury”, which may be limited to injury to the face and the scalp
and may not result in neurological deficits.
3

1.2 TBI: Epidemiology
TBI is a leading cause of death and disability throughout the world. The global annual
incidence of TBI is estimated to be ~200 per 100,000 people (Reilly, 2007). The annual
incidence of TBI in the North America is greater than the combined incidence of breast
cancer, HIV/AIDS, multiple sclerosis, and spinal cord injuries (Figure 1.1A). In the
United States, the overall incidence of TBI is estimated to be 538 per 100,000
population, which represents at least 1.7 million new cases per year since 2003
(Gerberding and Binder, 2003; Langlois et al., 2006; Faul et al., 2010). Annually
approximately 50,000 Canadians sustain a TBI with more than 11,000 deaths occurring
each year (Brain Injury Society of Brain Injury Society of Toronto, 2014). The rate of TBI
is reportedly lower in Europe (235 per 100,000) and in Australia (322 per 100,000)
(Cassidy et al., 2004; Tagliaferri et al., 2006) although the emerging evidence indicates
the otherwise (see below).
Estimating the true burden of TBI is challenging due to several factors. Most of the
epidemiological studies report data based on hospitalization and government records
that lack systematic epidemiological monitoring. Compounding this is the growing
awareness that more than 75% of TBI are mild (mTBI, a term synonymous to
concussion) that do not necessarily need hospitalization and therefore are not always
reported. As will be noted below, underdiagnosis of mTBI may nonetheless pose
challenges to interpretation of potential long-term consequences. Finally, most of the
epidemiological studies are retrospective, include small patient number and suffer from
recall bias. These limitations have been highlighted by a recent study in which a
4

substantially higher TBI incidence (749 per 100,000) was observed than previously
appreciated (Feigin et al., 2013). In this study, the authors collected data in an urban
and rural New Zealand population by both prospective and retrospective surveillance
systems. This study suggested that TBI incidence may be far greater than reported in
early epidemiological studies that mostly used retrospective data.
Furthermore, although most of the epidemiological data on TBI comes from the
developed countries, it is estimated that the incidence of TBI in developing countries is
rising and posing a significant health problem (Figure 1.1B). With the increasing
worldwide incidence rate as well as high number undiagnosed/underdiagnosed cases, it
is not surprising that TBI is often called a “silent epidemic”.
Figure 1.1: Annual incidence of TBI in people under 40 years of age (A) TBI has higher incidence rate compared to the combined incidence of other major health
issues including multiple sclerosis (MS), spinal cord injury (SCI), HIV/AIDS and breast cancer
combined. (B) Estimated annual incidence of TBI across the globe. For data sources, please
refer Table 1.1.
5
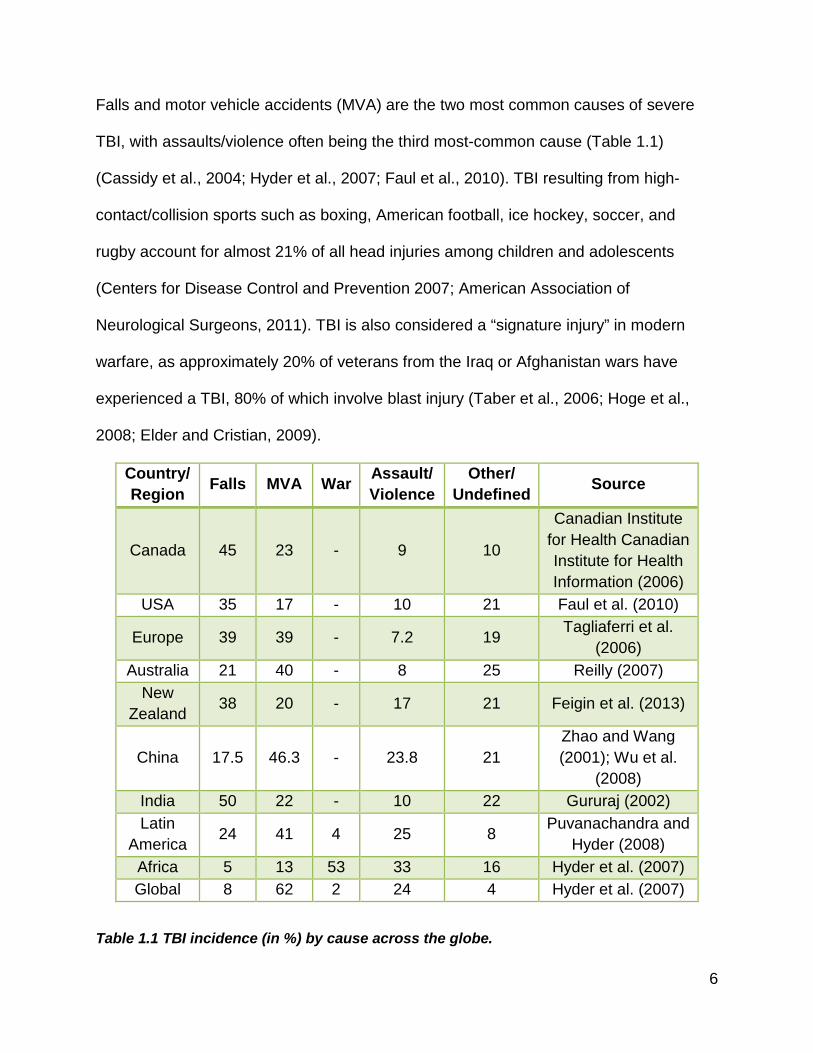
Falls and motor vehicle accidents (MVA) are the two most common causes of severe
TBI, with assaults/violence often being the third most-common cause (Table 1.1)
(Cassidy et al., 2004; Hyder et al., 2007; Faul et al., 2010). TBI resulting from high-
contact/collision sports such as boxing, American football, ice hockey, soccer, and
rugby account for almost 21% of all head injuries among children and adolescents
(Centers for Disease Control and Prevention 2007; American Association of
Neurological Surgeons, 2011). TBI is also considered a “signature injury” in modern
warfare, as approximately 20% of veterans from the Iraq or Afghanistan wars have
experienced a TBI, 80% of which involve blast injury (Taber et al., 2006; Hoge et al.,
2008; Elder and Cristian, 2009).
Country/ Region Falls MVA War Assault/
Violence Other/
Undefined Source
Canada 45 23 - 9 10
Canadian Institute for Health Canadian Institute for Health Information (2006)
USA 35 17 - 10 21 Faul et al. (2010)
Europe 39 39 - 7.2 19 Tagliaferri et al. (2006)
Australia 21 40 - 8 25 Reilly (2007) New
Zealand 38 20 - 17 21 Feigin et al. (2013)
China 17.5 46.3 - 23.8 21 Zhao and Wang (2001); Wu et al.
(2008) India 50 22 - 10 22 Gururaj (2002) Latin
America 24 41 4 25 8 Puvanachandra and Hyder (2008)
Africa 5 13 53 33 16 Hyder et al. (2007) Global 8 62 2 24 4 Hyder et al. (2007)
Table 1.1 TBI incidence (in %) by cause across the globe.
6

MVA: motor vehicle accident.
1.3 TBI: Classification
Traditionally TBI is classified based on three systems: 1. clinical severity, 2.
pathophysiology and 3. physical mechanism (Saatman et al., 2008).
1.3.1 TBI Classification Based on Clinical Severity
Conventional clinical TBI taxonomy divides severity of injury into three categories; mild,
moderate and severe. The most commonly accepted method to determine injury
severity is the GCS score (Table 1.2), which measures the patient’s level of
consciousness based on verbal, motor, and eye opening responses after injury. A
patient with a GCS score of 3-8 (out of 15) is considered to have sustained a severe
traumatic brain injury, 9-12 is moderate, and >12 mild (Table 1.3) (Teasdale and
Jennett, 1974). While the GCS is useful for the clinical management of TBI, it does not
provide specific information about the neuropathological mechanisms involved. The
GCS is also less useful in pediatric TBI. The prognostic ability of this system thus is
limited and as such other descriptors have been added. In the most recent iteration of
the widely adopted Departments of Defense and Veteran Affairs classification of
severity of brain injury, mTBI is further denoted by PTA lasting < 24 h, and a LOC of <
30 min. Similarly, to meet moderate TBI criteria, GCS must be between 9-12, PTA must
not exceed one week, and LOC must not last longer than 24 h (U.S Departments of
Defense and Veterans United Sates Departments of Defense and Veterans Affairs,
2008) (Table 1.3).
7

Score Eye Opening Response Verbal Response Motor Response
6 N/A N/A Obeys commands for movement
5 N/A Oriented Purposeful movements to painful stimulus
4 Spontaneous Confused, but able to answer questions
Withdraws in response to pain
3 To verbal stimuli Inappropriate words
Flexion in response to pain (decorticate posturing)
2 To pain only (not applied to face)
Incomprehensible speech
Extension in response to pain (decerebrate posturing)
1 No response No response No response
Table 1.2: Glasgow coma scale.
Glasgow coma scale determines a patient’s neurological state based on eye opening,
verbal and motor response. Adapted from (Teasdale and Jennett, 1974).
Injury Severity GCS Score (Out of 15)* LOC Duration PTA Duration Mild > 12 < 30 min < 24 h
Moderate 9-12 30 min to 24 h 1 to 7 days Severe 3-8 > 24 h > 7 days
Table 1.3: North American classification of TBI according to severity of clinical signs.
* Best available score within 24 h of injury. GCS: Glasgow Coma Score, LOC: Loss of
Consciousness, PTA: Posttraumatic Amnesia.
The European Federation of Neurological Societies (EFNS) guidelines have taken a
slightly different approach and categorized TBI into four levels of severity: mild,
moderate, severe, and critical (Table 1.4) (Vos et al., 2002). The heterogeneity inherent
8

to mTBI has been addressed with various refinements to this category. For example,
the EFNS guidelines further classify mTBI into four categories, 0, 1, 2, and 3. In addition
to the parameters used by the North American guidelines (i.e., GCS score, duration of
LOC and PTA), the EFNS guidelines also include risk factors for intracranial
complications in the grading of mTBI severity. The risk factors for intracranial
complications include unclear or ambiguous accident history, continued PTA (GCS
verbal score of 4), retrograde amnesia for > 30 min, trauma above clavicles including
clinical signs of skull fracture, severe headache, vomiting, focal neurological deficits,
seizure, age of < 2 or > 60 years, coagulation disorders, and alcohol/drug intoxication
(Vos et al., 2002). In this system, the subcategories of mTBI dictate post-TBI
management. Thus, a person with mTBI category 0 is considered to have no TBI and is
immediately discharged, mTBI category 1 recommends CT scanning while CT scanning
is mandatory for persons with mTBI categories 2 and 3 (Vos et al., 2002). The finer
distinction of mTBI may be of questionable value since in a recent major revision of
evidence-based guidelines for evaluation and management of sports-related
concussions, endorsed by multiple sporting bodies and physician groups, these finer
distinctions have now been dropped due to a lack of prognostic utility (Giza et al., 2013).
9

TBI Class GCS Score
LOC Duration
PTA Duration
Risk Factors for Intracranial
Complications Mild GCS = 13-15 mild Category 0 15 No No Not present mild Category 1 15 < 30 min < 1 h Not present mild Category 2 GCS = 15 and risk factors present mild Category 3 13-14 < 30 min < 1 h With or without risk factors Moderate GCS = 9-12 Severe GCS ≤ 8
Critical GCS = 3-4 with loss of pupillary reactions and absent or decerebrate motor reactions
Table 1.4: TBI classification according to European Federation of Neurological Societies guidelines.
GCS: Glasgow Coma Score, LOC: Loss of Consciousness, PTA: Posttraumatic
Amnesia. Adapted from (Vos et al., 2002).
1.3.2 TBI Classification Based on Pathophysiology
The action of external forces on the head leads to a cascade of pathological processes.
These processes are broadly classified into primary and secondary brain injuries
(discussed in section 1.5). Primary brain injury occurs at the time of mechanical loading
on the head leading to structural damage to the brain parenchyma and
cerebrovasculature. Primary injury results in laceration or contusion of the cortical
surface, diffuse axonal injury (DAI), and hematoma. Damage caused by primary injury is
considered to be irreversible and may not respond to any pharmacological intervention.
Primary brain injury initiates a plethora of secondary processes that result in complex
cellular, inflammatory, neurochemical, and metabolic alterations (McIntosh et al., 1996;
Blumbergs, 1997; Davis, 2000; Giza and Hovda, 2001; Werner and Engelhard, 2007;
10

McAllister, 2011). The secondary injury pathways may be more amenable to
pharmacological treatment.
1.3.3 TBI Classification Based on Physical Mechanism
TBI is caused by external mechanical forces, which can be static or dynamic. Static
stress (e.g., a head caught between elevator doors) does not cause head acceleration
and does not lead to brain injury unless the crushing forces exceed the threshold
required for skull fracture causing substantial tissue damage (Gennarelli, 1993). The
majority of TBIs result from dynamic forces including contact (impact) or non-contact
(inertial) forces. Dynamic stresses induce rapid acceleration and/or deceleration of the
head followed by distribution of inertial forces to the underlying brain tissue causing
tissue damage. Contact injuries are induced by a moving object striking a stationary
head or a moving head striking a stationary object and usually involve intense
mechanical loading of short duration (Davis, 2000). The basis of classifying TBI on the
basis of physical mechanism lies in the observed relation between the type of
mechanical force and the resulting neuropathological characteristics. Thus, brain
injuries as a result of impact forces are thought to induce localized (focal) damage
including contusions, skull fracture, and epidural hematoma while inertial forces tend to
cause more widespread (diffuse) brain damage such as diffuse axonal injury and
subdural hematoma (McLean and Anderson, 1997; King, 2000; Saatman et al., 2008).
11

1.4 TBI: Biomechanical Principles
Biomechanics involves the study of the motion (such as head acceleration) of a person,
animal or anthropomorphic test device (crash test device) and mechanical loads
sustained or applied (such as head impact forces). The work of Gurdjian and Lissner
(Gurdjian and Lissner, 1945, 1946, 1947; Gurdjian et al., 1947; Gurdjian and Webster,
1947) and Holbourn (Holbourn, 1943, 1945) pioneered the biomechanical evaluation of
TBI to understand the relationship between mechanical loading (force/stress) and the
physical response of the head and brain (deformation/strain) and resulting pathology.
Besides understanding the mechanism of brain injury, biomechanical studies also help
to predict head injury tolerance levels, which are used for developing and standardizing
vehicle safety parameters as well as in helmet design to provide improved protection for
the head during impact (Versace; Henn, 1998; Eppinger et al., 2000). Although, the
mechanisms by which mechanical forces induce brain injury are a subject of
considerable debate (Hardy et al., 1994; Drew and Drew, 2004), brain deformation or
strain resulting from external mechanical loading (i.e., force/stress) is the currently
accepted biomechanical theory of TBI (McLean and Anderson, 1997; King et al., 2003;
King et al., 2004; LaPlaca et al., 2007). Brain deformation increases with increasing
stress in a non-linear fashion (LaPlaca et al., 2007). In a head impact, dynamic
mechanical forces act on the skull and brain to cause both linear and rotational
movement of the head and skull. This in turn leads to deformation and structural
damage of brain and cerebrovascular tissues and triggers many secondary injury
pathways.
12

Human impact TBI can occur under many conditions. One example involves the slowly
moving or stationary head being impacted by a rapidly-moving object, such as when a
vehicle strikes a pedestrian’s head. Another example involves a head moving at a high
rate of speed impacting a stationary object, such as when a hockey player slides head
first into the boards. These impacts cause intense mechanical loading that lasts for only
a fraction of a second (< 50 ms) (Gurdjian et al., 1966; Ono et al., 1980; Pellman et al.,
2003b) and causes pressure gradients and mechanical strain (i.e., local areas of
stretching or compression of the brain tissue) to be induced within the brain tissue as
shown in Figure 1.2 (Meaney and Smith, 2011). Head impact can result in pure linear
(straight line) motion of the head or combined linear and rotational motion in response
to impact. Combined linear and rotational motion is more common than pure linear
motion because the head is coupled to the body by the neck and so almost any impact
to the head results in a combination of linear and rotational motion of the head due to
restraint forces on the head applied by the neck (Greaves et al., 2009). In pure linear
motion of the head, the pressure gradients and tissue strains described above will
occur. In combined rotational and translational motion of the head the pressure
gradients and tissue strains described above will have much larger tissue strains, which
arise from the rotation of the head, superimposed on the pressure gradient-related
strains (Meaney and Smith, 2011).
13

Figure 1.2: Biomechanics of head impact and brain tissue deformation.
(A) Impact to the back of the skull with the skull moving downwards (red arrow) causes
momentary skull deformation (red outline). (B) The skull stops suddenly (in ~ 50 ms)
and the momentum of the brain keeps it moving causing relative motion of the brain with
respect to the skull (orange outline). (C) This motion of the brain sets up positive
pressure at the impact site (i.e., coup) and negative pressure opposite the coup site at
the contrecoup site. Reproduced with permission from Namjoshi et al. (2013a)
Impact is by far the most common cause of TBi in civilian populations. In the military,
however, improvised explosive devices (IEDs) can lead to blast forces capable of
causing TBI. In contrast to impact TBI, pure blast TBI is non-contact and involves
dynamic forces with very short durations on the order of a few microseconds (µs)
(Goldstein et al., 2012). Other non-contact TBI mechanisms are hypothesized to occur
as a result of “inertial” loading of the head where torso motion, for example when a
football player’s torso is impacted by another player during a tackle or during “shaken
baby” syndrome, causes the head to move even when no direct impact forces are
subjected to it. This mechanism of TBI is a matter of some controversy and many
investigators have concluded that this does not occur in real world human TBI (Lau et
al., 1989; McLean, 1995; Yoganandan et al., 2009; Meaney and Smith, 2011; Wright et
14

al., 2013). The duration of inertial head loading in these situations varies from 50 to 200
ms (Gennarelli, 1993). Most epidemiological studies also conclude that these injuries
cannot feasibly occur in adult humans (Lau et al., 1989; McLean, 1995).
TBI can also result from static or near static loads that essentially crush the brain and
skull resulting in direct compression of the brain or contusion injury via bone fragments
(Denny-Brown and Russell, 1940; Lopez-Guerrero et al., 2012; Mattei et al., 2012). In
these crushing or nutcracker injuries, the head is generally not subjected to rapid linear
or rotational movements that occur during impact injuries.
1.4.1 Head Motion during Impact TBI
Head/skull acceleration that occurs during a head impact can be described using three-
dimensional linear (translational) and rotational (angular) accelerations. Linear
acceleration is defined as the change in velocity over a given time through translational
coordinates of the head’s center of gravity (CG) e.g., x-y-z or its resultant, and is usually
expressed in units of “g” (one g is the acceleration due to gravity on earth) or m/s2.
Rotational acceleration is the change in rotational velocity of the head over a given time
and is expressed in units of radians/s2 (rad/s2) or degrees/s2 (°/s2). One revolution is
equal to 360° or 2π rad. Acceleration can be measured using devices called
accelerometers.
The relative amounts of linear and rotational head acceleration that result from a
particular head impact depend on several factors including the type of impact force, the
15

direction of the force, the location of force on the skull, and the material properties of the
skull and brain. An impulsive contact force applied to the head is a vector with
magnitude and direction. A force that passes through the CG of the head (i.e., aligned
with the maxilla) will initiate primarily linear motion of the head during the impact. A
force that does not pass through the CG (e.g., an impact to the high forehead) will
produce an impulsive moment (conceptually a “twisting force”) about the CG and will
initiate both linear and rotational acceleration.
There is considerable debate about whether linear (Gurdjian et al., 1955; Haddad et al.,
1955; Gurdjian et al., 1961) or rotational (Holbourn, 1943; Gennarelli et al., 1981;
Gennarelli and Thibault, 1982; Gennarelli et al., 1982) acceleration is a better predictor
of brain injury. Advocates of rotational acceleration argue that pure linear impact is rare
in the clinical setting and angular acceleration is the principle mechanism underlying
brain injury (Holbourn, 1943; Hardy et al., 1994). Notably, the Head Injury Criterion
(HIC), which is a currently incorporated in vehicle safety standards around the world,
takes only linear acceleration into account. The HIC is calculated as a function of the
acceleration magnitude and duration of acceleration so that high accelerations acting for
long time intervals result in high HIC while lower acceleration values or shorter
exposure times result in lower HIC. In the context of automotive safety testing, the HIC
has been credited with considerably reducing the incidence of MVA-related head
injuries for over three decades (King et al., 2004). Moreover, recent studies by King and
colleagues contend that helmets significantly reduce linear acceleration without
changing rotational acceleration (King et al., 2003), leading these authors to propose
16

that the response of the brain itself (i.e., deformation of the structures of the brain) to
mechanical loading may be a better predictor of brain injury than linear or rotational
acceleration of the skull (Hardy et al., 2001; King et al., 2003; King et al., 2004; King et
al., 2011). Many authors have proposed metrics that are a combination of both linear
and rotational acceleration (and other factors such as HIC and impact force location on
the skull) (Gurdjian, 1975; Ono et al., 1980; Pellman et al., 2003b; Greenwald et al.,
2008; Rowson and Duma, 2013) as the most predictive mechanisms of brain injury.
1.4.2 Brain Motion during TBI
Like most soft tissue, the brain has viscoelastic properties with non-linear mechanical
stress-strain responses (LaPlaca et al., 2007). Importantly, it is shear strain, rather than
tissue compression or pressure gradients, which is believed to be the major mechanism
underlying most concussion pathology (Meaney and Smith, 2011). Brain motion inside
the skull during impact has been studied using a variety of techniques. In the earlier
studies, brain motion during impact was directly recorded through a cranial window
created by surgically replacing the skull cap of Macaque monkeys with a cap made up
of acrylic (called the “Lucite Calvarium”) (Shelden et al., 1944; Pudenz and Shelden,
1946) or polycarbonate (the “Lexan Calvarium”) (Ommaya et al., 1969; Gosch et al.,
1970). Brain motion following sub-concussive impacts in the frontal, temporal, and
parietal regions was recorded using high-speed film. In these studies, it was found that
the regardless the direction of impact, the principal brain displacement was in parieto-
occipital region with minimum displacement in the frontal region, which is likely due to
the anterior cranial fossa that restricts movement of frontal lobes. This study showed
17

relative movements of brain and the skull, concluding that the relative displacement of
the brain depended on the degree of skull movement. Thus, an immobile skull following
impact caused little to no brain displacement while a freely moving skull resulted in
considerable brain movement. With the advances in high-speed X-ray videography in
1960’s it was possible to record brain movement with minimal surgical manipulation.
Using intravascular contrast media and lead targets, brain motion was captured after
impact with high speed X-ray videography in dogs (Hodgson et al., 1966) and primates
(Shatsky et al., 1974). Hardy and colleagues improved on this technique by using
neutral density particles to record brain displacement in human cadavers (Hardy et al.,
2001; Hardy et al., 2007). The studies in human cadavers reported brain displacement
of ± 5 mm relative to the skull (King et al., 2011). These studies also reported a lag in
the brain displacement relative to the motion of the skull. Moreover, these studies also
show that the radio-opaque particles return to their original position following
displacement suggesting elastic properties of the brain tissue, which may be important
for studying brain displacement over repeat impacts. The effects of post-mortem
changes in the brain tissue on the brain displacement however are not known. Recently
brain motion was studied in human volunteers using tagged MRI. Bayly and colleagues
conducted a series of studies in which the head of human volunteers was subjected to
deceleration with occipital impact of 2-3 g (Bayly et al., 2005) or angular acceleration of
250-300 rad/s2 (Sabet et al., 2008) and the resulting strains were assessed using
tagged MRI (tagged MRI enables tracking the motions of tissue by tagging specific
tissue points with a sequence of radio-frequency pulses before imaging) although
without isolating relative movements of brain and the skull. By tracking the points along
18

tag lines imposed on MRI images, the authors reported that mechanical inputs that were
10-15% of those experienced by soccer players resulted in strains in the range of 0.02-
0.06 (a 0.05 strain corresponds to a 5% change in the dimension of local tissue).
1.4.3 The Clinical Picture: Human Tolerance and Related Biomechanical Studies
of Human TBI
The traumatic injury threshold for humans has been investigated using animal models,
physical brain surrogate models, analytical models and finite element models and
studies in athletes (football and hockey) who experience frequent head impacts and
concussion.
Using animal models in the 1950s, researchers at the Wayne State University
demonstrated that the duration of intracranial pressure was an important exposure
variable in injury tolerance; higher exposure generated a more severe injury in a shorter
amount of time (Lissner et al., 1960). Increasingly sophisticated instrumentation allowed
head acceleration to be measured at the occiput (posterior part of the skull) along with
changes in intracranial pressure in response to forehead impacts for whole and partial
cadavers on automotive instrument panels, windshields, and non-yielding surfaces
(Evans et al., 1958; Lissner et al., 1960). These experiments led to the preliminary
Wayne State Tolerance Curve (WSTC) for head injury. The initial tolerance curve
predicted whether head injury would occur as a function of the head impact duration
and the average linear acceleration measured at the occiput. This initial curve was
further refined through additional cadaver testing and in human volunteers (Eiband,
19

1959; Gurdjian et al., 1961; Patrick et al., 1963). The revised WSTC, as shown in Figure
1.3, assumed that the underlying experimental impacts that caused a linear skull
fracture also caused a moderate to severe concussion (Gurdjian et al., 1966). The first
experimental and biomechanics-based, quantitative human brain injury criterion, was
based on linear head acceleration. Rotational based human injury criteria would not
come until later (Newman, 2002). The WSTC data provides the basis for several widely
used injury metrics currently in use such as the Gadd Severity Index (GSI) (Gadd,
1966), and the HIC (Versace, 1971).
Figure 1.3: Wayne State Tolerance Curve (WSTC).
The WSTC describes the relationship between linear head acceleration, duration of
acceleration, and onset of concussion. The WSTC suggests that the head can
withstand very high acceleration for a very short duration. Conversely, any increase in
20

the duration of impact for the same intensity of acceleration is likely to cause head
injury. Reproduced with permission from Namjoshi et al. (2013a).
Several groups have generated head injury risk curves using logistic regression models
based on linear acceleration as a measure of exposure (Prasad and Mertz, 1985; Hertz,
1993; Kuppa, 2004). Prasad and Mertz suggest that a HIC15 (measurement of impact
over 15 ms) value of 700 represents a less than 5% risk of life-threatening brain injury.
More recently, Zhang et al. (2004) used a validated finite element human head model
and predicted the maximum resultant linear acceleration at the CG of the head to be 66,
82, and 106 g for a 25%, 50%, and 80% probability of mTBI, respectively, whereas the
maximum resultant rotational accelerations for a 25%, 50%, and 80% probability of
sustaining a mTBI are estimated at 4600, 5900, and 7900 rad/s2, respectively. Funk et
al. (2007) estimated a 10% risk of mTBI at 165 g, a HIC of 400, and an angular head
acceleration of 9000 rad/s2.
To put injury threshold experiments into context with mTBI, many groups have directly
measured or reconstructed concussive impacts during sporting events. Using helmets
fitted with triaxial accelerometers, Pellman et al. (2003b) reconstructed impacts
involving NFL players where players sustained concussions or significant head impacts
and determined that peak head accelerations in concussed players averaged 98 g and
in uninjured players averaged 60 g. The lowest measured acceleration where a player
sustained a concussion was 48 g. The peak angular acceleration in concussed and
uninjured players averaged 6432 rad/s2 and 4235 rad/s2, respectively. The lowest
angular acceleration where a player sustained a concussion was 2615 rad/s2. These
21

data are consistent with the predictions made from finite element modeling (FEM)
studies.
Researchers at Virginia Tech used instrumented helmets (helmets designed with
embedded accelerometers) to record tens of thousands of head impacts in football
players including 57 diagnosed concussions. Linear acceleration of 171 g, 192 g, and
214 g were identified to result in a 25%, 50%, and 75% risk of mTBI (Rowson and
Duma, 2011). Rotational accelerations of 5821 rad/s2, 6383 rad/s2, and 6945 rad/s2
were associated with a 25%, 50%, and 75% chance of mTBI (Rowson et al., 2012).
The most recent study investigated mTBI injury risk as a function of linear acceleration
alone, rotational acceleration alone and a combination of both linear and rotational
acceleration using logistic regression (Rowson and Duma, 2013). All three models were
found to be good predictors of mTBI outcome. Although the combined model is
preferred, it was statistically equivalent to the model using linear acceleration alone.
1.5 TBI: Pathophysiology
The TBI event can be divided into four stages: 1) mechanical forces (usually dynamic),
act on the skull and brain to cause 2) head motion (rotation and/or translation of the
head and skull) and 3) brain motion/deformation causing structural damage of brain and
vascular tissues (primary injury), which leads to 4) a delayed biological response of the
brain (i.e., deleterious biochemical and cellular processes that cause many TBI patients
to gradually deteriorate in the hours and days after injury) referred to as secondary
injury.
22

1.5.1 Primary Brain Injury
The primary brain injury is caused by mechanical forces resulting into structural damage
to the brain and the cerebrovasculature. The primary brain injury can be focal or diffuse
(Figure 1.4). Most clinical TBIs result in both focal and diffuse brain damage.
Figure 1.4: Primary TBI. Primary TBI results from structural damage caused to the brain parenchyma and
cerebrovasculature from mechanical forces. Focal injury results in contusion, laceration
and/or vascular injury
Primary TBI
Diffuse
Diffuse axonal injury
Diffuse vascular
injury
Focal
Skull Fracture Contusion Laceration Vascular
injury
Subdural hemorrhage
Epidural hemorrhage
Intracerebral hemorrhage
Intraventricular hemorrhage
23

1.5.1.1 Focal Brain Injury
Focal brain injury is caused with or without skull fracture by impact forces such as a
moving object striking against a stationary head (e.g., during physical assault) or a
moving head striking against a stationary object (e.g., during fall). Focal injuries are
thought to account for about two-thirds of TBI-related deaths (Davis, 2000). Whether or
not the head is stationary may result in characteristic focal damage. Thus, a moving
object striking a stationary head initiates head acceleration resulting in focal injury at the
side of the impact called “coup injury”. On the other hand, a moving head striking
stationary object results in sudden head deceleration and increases the probability of
focal damage to the side opposite to the impact site called “contrecoup injury” (Dawson
et al., 1980). Counterintuitively, the contrecoup injuries tend to be more severe than
coup injuries as reported in several clinical observations (Dawson et al., 1980). The
mechanisms of coup-contrecoup injuries are not well understood and are subject to
controversies and as such several theories are proposed. According to the positive
pressure theory, as the skull moves before impact, the brain lags and is pressed
against the lagging surface of the skull (i.e., side opposite to the forthcoming impact
site) (Lindenberg and Freytag, 1960; Drew and Drew, 2004). Also, as the brain lags, the
cerebrospinal fluid (CSF) is displaced in the space in the coup site created by the
lagging brain. The impact generates pressure waves that further increase compression
of the brain against the lagging surface of the skull. Accumulation of CSF near the
impact site further cushions against the coup injury causing more contrecoup damage.
The negative pressure theory (cavitation theory) argues that upon impact the head
suddenly stops while the brain still continues to move towards the impact site (Russel,
24

1932; Drew and Drew, 2004). This creates negative pressure (cavitation) at the
contrecoup site pulling brain towards it. Dawson et al proposed the angular
acceleration theory which is similar to positive pressure theory (Dawson et al., 1980).
This theory is based on the principle of rotation of a tethered object around its center;
the more distant the object is from the center of rotation or more massive the object is
than to which it is tethered, slower will it move. By applying this principle, when the
accelerating head strikes a stationary object, the brain lags resulting injury at the
lagging side (i.e., contrecoup side). Using the principles of Newtonian mechanics and
based on the difference between the relative densities of the brain (0.96 g/L) and the
CSF (1 g/L) Drew and Drew recently proposed an alternate theory. According to this
theory, when the moving head suddenly stops by impact, the CSF being denser than
the brain, continues to move in the direction of the head movement and accumulates at
the site of the impact pushing the brain towards the contrecoup side (Drew and Drew,
2004). This initial displacement of the brain results in more severe contrecoup injury
compared to the coup injury.
1.5.1.1.1 Cerebral Contusion and Laceration
The most common result of coup-contrecoup injury is cerebral contusion, which is
observed in about 20-30% of severe TBI (Khoshyomn and Tranmer, 2004). Cerebral
contusion usually involves the cortex and is associated with damage to the gyri and
small blood vessels often resulting in microhemorrhages (Yokobori and Bullock, 2013).
The pia mater remains intact in contusion differentiating it from laceration in which the
pia mater is torn (see below). The most common areas susceptible to contusion include
25

the orbitofrontal cortex, anterior temporal lobes, and posterior portion of superior
temporal gyrus and are thought to be due to the irregular surface of the bony floor
formed by the frontal and middle cranial fossa on which these structures lie
(Khoshyomn and Tranmer, 2004). The acute phase of contusion is characterized by
inflammatory cascades including vascular migration of polymorphonuclear leukocytes
seen within 24 h of injury, microglial activation seen over 3-5 days leading to release of
proinflammatory cytokines such as tumor necrosis factor–α (TNF-α) and interleukin-1β
(IL-1β) (Holmin et al., 1998; Holmin and Hojeberg, 2004; Clausen et al., 2007),
apoptosis and necrosis (Raghupathi, 2004). This contributes to damage of the blood
brain barrier (BBB) and vasogenic edema resulting in swelling of the areas adjacent to
the contusion site, which is seen in about 30% of all contusions (Khoshyomn and
Tranmer, 2004). Extensive contusions may lead to subdural hematoma and together
they are called a “burst lobe” (Gennarelli and Graham, 2005). In addition to cortical
contusions, shearing forces (often in the sagittal plane) within the white matter can lead
to gliding contusions that occur in the subcortical regions causing damage at the gray-
white matter junction. Gliding contusions tend to be bilateral and are often associated
with DAI caused by differential motion of subcortical structures and acute subdural
hematomas caused by tearing of parasagittal veins (Adams et al., 1986; Sganzerla et
al., 1989).
Laceration is the most severe form of primary TBI resulting in disruption of the brain
parenchyma and involves torn pia-arachnoid membranes. Laceration requires greater
mechanical force compared to cerebral contusions. Penetrating brain injury or
26

depressing skull fractures lead to direct laceration, while parenchymal disruption
secondary to tissue deformation caused by mechanical stress cause indirect lacerations
(Blumbergs, 1997).
1.5.1.1.2 Intracranial Hemorrhage
Intracranial bleeding is a life-threatening complication of TBI and is the most common
cause of TBI fatalities. Intracranial bleeding starts at the time of trauma and can expand
to hematoma in 30-60% of patients with post-TBI coma (Gennarelli and Graham, 2005).
Intracranial bleeding is categorized into four types: epidural hematoma, subdural
hematoma, subarachnoid hemorrhage and intracerebral hemorrhage.
Epidural hematoma/hemorrhage (EDH) is accumulation of blood in the epidural space
(i.e., between the skull and dura) caused by rupture of interposed meningeal vessels
due to shearing stress. Usually EDH occurs due to coup injury and results in separation
of the dura from periosteum. About two-thirds of EDHs are associated with skull fracture
(McKissock et al., 1960). EDH most commonly occurs in the temporoparietal region and
usually involves damage to the middle meningeal artery (Blumbergs, 1997). The
incidence of EDH is about 2-4% of overall TBI and 15% of severe TBI (Bullock et al.,
2006). Compared to adults, EDH is less common in pediatric TBI as the skull is more
pliable reducing occurrence of skull fracture, the dura is firmly attached to the skull and
the meningeal vessels are not yet embedded in the developing skull of infants (Yokobori
and Bullock, 2013). In many cases, patients with EDH have a brief LOC, followed by a
lucid interval and may deteriorate if the symptoms are not readily recognized/treated
27

(‘talk and die’ phenomenon). The common symptoms of EDH include headache,
nausea/vomiting, seizures, and focal neurological deficits including aphagia, weakness,
and numbness. EDH treated with neurosurgical intervention have better prognosis.
Subdural hematoma/hemorrhage (SDH) is bleeding between the dura and arachnoid
membrane resulting from the rupture of bridging veins that cross the subdural space
connecting the cerebral surface with the superior sagittal sinus. SDH occurs in about
5% of TBI and is associated with 65% of cases with prolonged LOC. While EDH are
localized, SDH tend to spread as the fluid can freely move within the subdural space
(Yokobori and Bullock, 2013). SDH is one of the classical triads of shaken baby
syndrome, the other two being retinal hemorrhage and cerebral edema. SDH are
classified into acute, subacute, and chronic depending on the duration of onset and
composition (clotted blood, fluid blood or mixture of clotted and fluid blood) (Blumbergs,
1997; Gennarelli and Graham, 2005). Acute SDH is composed of clotted blood and
forms within first 48 h of trauma and usually is caused by intense mechanical forces.
Acute SDH can be associated with cerebral contusion/laceration and intracerebral
hemorrhage together forming ‘burst lobes’ that cause delayed neurological deterioration
(Blumbergs, 1997). Acute SDH is life-threatening with 30 to 90% mortality rate unless
the patients are operated within the ‘golden hours’ (usually within 4 h) of injury (Seelig et
al., 1981). The poor neurological outcome following acute SDH is associated with
increased intracranial pressure (ICP) that decreases cerebral perfusion pressure (CPP),
which in turn leads to cerebral ischemia (Graham et al., 1978). Subacute SDH is
composed of both clotted and fluid blood that develops over 3-21 days while chronic
28

SDH is composed of fluid blood and develops for > 21 days. Chronic SDH is common in
elderly and alcoholic patients who are more prone to cerebral atrophy and/or
coagulopathy (Blumbergs, 1997). Subacute and chronic SDH have better prognosis
than acute SDH.
Subarachnoid hemorrhage (SAH) is bleeding between the arachnoid membrane and
pia mater and is the most common intracranial hemorrhage. SAH is observed in 40-65%
of moderate-severe TBI and is an independent predictor of poor post-TBI prognosis
(Eisenberg et al., 1990; Kakarieka et al., 1994; Taneda et al., 1996; Servadei et al.,
2002). In most cases, SAH is mild but the severity of SAH increases when associated
with cerebral contusions/lacerations (Blumbergs, 1997).
Intracerebral hemorrhage (ICH) is a hematoma 2 cm or greater in size, not in contact
with the surface of brain, that is caused by rupture of intraparenchymal blood vessels at
the time of impact. ICH is found in 15-20% of autopsy cases of severe TBI (Blumbergs,
1997; Yokobori and Bullock, 2013). Rupture of multiple vessels result in multiple ICH of
which about 28% are associated with SDH and 10% with EDH (Soloniuk et al., 1986).
Small ICH can occur at multiple sites including corpus callosum, the walls of third
ventricle, and basal ganglia. Intraventricular hemorrhage is thought to be caused by
deformation and rupture of subependymal veins due to sudden dilation of ventricular
system during mechanical impact (Zuccarello et al., 1981) or extension of hematomas in
the adjacent regions (Fujitsu et al., 1988).
29

1.5.1.2 Diffuse Brain Injury
1.5.1.2.1 Diffuse Axonal Injury
Diffuse axonal injury (DAI) is the widespread damage to the white matter and is the
most common, most important, and yet the most difficult-to-diagnose TBI pathology.
DAI is the major cause of TBI-induced unconsciousness/coma and vegetative state and
associated with long-term disabilities (Gusmao and Pittella, 2003; Fork et al., 2005).
Although the diagnosis of DAI with traditional imaging techniques such as CT and MRI
has been challenging, recent advances in MRI techniques, such as DTI and
susceptibility weighed imaging may enable the clinicians to capture this ‘stealth
pathology’ even in mild cases (Hunter et al., 2012). DAI was first described by Strich as
“diffuse degeneration of the white matter” observed in post-TBI human brain autopsies
(Strich, 1956).
The principal biomechanical mechanism of DAI is thought to be rapid inertial loading
(e.g., car crash) that induces dynamic shear, tensile and compressive stain in the brain
tissue (Gennarelli et al., 1982; Smith and Meaney, 2000; Smith et al., 2003a). The
axonal damage during TBI is affected by the unique material properties, size, and mass
of the brain tissue, the presence of intracranial folds of dura mater (e.g., falx cerebri)
and the rate and magnitude of strain (Smith and Meaney, 2000; Smith et al., 2003a).
Several studies have shown that nervous tissue is a nonlinear, viscoelastic material
(Donnelly and Medige, 1997; Darvish and Crandall, 2001; Takhounts et al., 2003). In
addition, brain is highly inhomogeneous and anisotropic, thus showing regional
differences in the biomechanical response to the mechanical loading. Thus, the gray
30

matter is isotropic while highly-organized white matter shows anisotropy with the
greatest anisotropy in the corpus callosum (Prange et al., 2000; Prange and Margulies,
2002). When axons are subjected to slowly applied external load (e.g., during normal
head rotation), they behave like a compliant and ductile material and return to their
original geometry after removal of the stress (Franze et al., 2009; van Dommelen et al.,
2009). On the other hand, under rapid and severe external loading (e.g., during car
crash), axons behave like a stiffer and brittle material resulting structural failure. This
property of axons along with their high anisotropic nature is thought to make white
matter more vulnerable to the mechanical trauma (Geddes et al., 1997; Geddes et al.,
2000). The direction of inertial loading and presence of intracranial dural folds my also
affect DAI characteristics. For example, mechanical loading in the temporal region
causes head rotation in the coronal plane. During such rotation, the falx cerebri (the
dural fold that descends vertically between the two cerebral hemispheres) impedes
motion of the following cerebral hemisphere while the leading cerebral hemisphere
continues moving along the direction of motion. This creates excessive shearing forces
along the midline between the two hemispheres. This, along with highly anisotropic
nature of the corpus callosum, may lead to greater axonal damage in this region (Smith
and Meaney, 2000; Smith et al., 2003a).
In extreme but rare TBI cases mechanical forces can cause direct axonal breakage,
called ‘primary axotomy’. In most cases, however axonal pathology develops over time
following mechanical insult (Buki and Povlishock, 2006). Smith and colleagues
conducted a series of elegant studies to understand the mechanism of axonal injury at
31

the cellular level. Using an in vitro axonal stretch injury model, Smith and colleagues
showed that immediately following mechanical loading the axons become temporarily
undulated at multiple sites, which is attributed to partial breakage of microtubules
leading to cytoskeletal damage (Smith et al., 1999a; Tang-Schomer et al., 2010; Tang-
Schomer et al., 2012). The initial axonal stretch results in massive Na+ influx through
mechanosensitive voltage-gated Na+ channels causing membrane depolarization.
Increased intracellular Na+ levels trigger Ca2+ influx, principally via opening of voltage-
gated calcium channels and to a modest level, by reversal of Na+-Ca2+ exchanger (Wolf
et al., 2001; Iwata et al., 2004). The abnormal increase in intracellular Ca2+ leads to
calpain-mediated structural proteolysis causing delayed damage to the axonal
cytoskeleton and ion channels that ensues over days to weeks (Buki et al., 1999a; Buki
et al., 1999b; Iwata et al., 2004; Huh et al., 2006; McGinn et al., 2009). Structural
damage to the axonal cytoskeleton along multiple sites leads to disruption of axonal
transport and subsequent accumulation of axonal proteins causing characteristic axonal
swellings (axonal varicosities or beads) (Tang-Schomer et al., 2012). Two important
proteins that accumulate at the site of axonal swellings are amyloid precursor protein
(APP) and neurofilament (NF), which are transported by fast and slow transport
mechanisms, respectively, and as such are the most commonly-used diagnostic
markers of DAI (Gentleman et al., 1993; Grady et al., 1993; Sherriff et al., 1994a;
McKenzie et al., 1996; Pierce et al., 1996). Accumulation of APP in axonal varicosities
is thought to act as a nidus for the enhanced amyloid beta (Aβ) production, which has
an important implication in the increased risk of Alzheimer disease in TBI survivors
(discussed further). The proposed molecular mechanism of DAI and subsequent Aβ
32
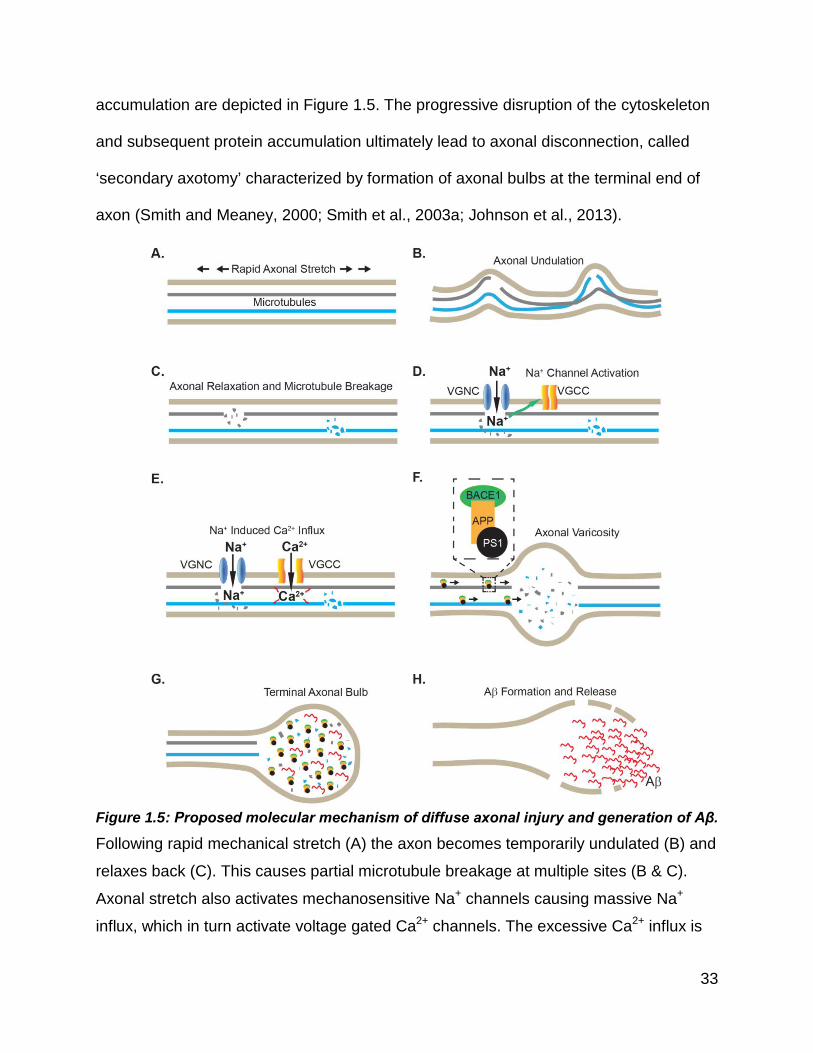
accumulation are depicted in Figure 1.5. The progressive disruption of the cytoskeleton
and subsequent protein accumulation ultimately lead to axonal disconnection, called
‘secondary axotomy’ characterized by formation of axonal bulbs at the terminal end of
axon (Smith and Meaney, 2000; Smith et al., 2003a; Johnson et al., 2013).
Figure 1.5: Proposed molecular mechanism of diffuse axonal injury and generation of Aβ.
Following rapid mechanical stretch (A) the axon becomes temporarily undulated (B) and
relaxes back (C). This causes partial microtubule breakage at multiple sites (B & C).
Axonal stretch also activates mechanosensitive Na+ channels causing massive Na+
influx, which in turn activate voltage gated Ca2+ channels. The excessive Ca2+ influx is
33

thought to exacerbate calpain-mediated disruption of the cytoskeleton (red arrows in E).
Microtubule interrupts protein transport along the axon causing accumulation of
proteins, including APP along with components of APP-cleaving enzymes, BACE1 and
PS1 resulting in axonal varicosities (F). The progressive disruption of cytoskeleton
results in axonal disconnection with formation of axonal bulb (G). The cleavage of APP
accumulated in the axonal varicosities/bulbs by BACE1 and PS1 is thought to lead to a
burst of Aβ production (H). APP: Amyloid Precursor Protein, BACE1: Beta Site APP
Cleaving Enzyme 1, PS1: Presenilin-1, VGCC: Voltage-Gated Ca2+ Channel, VGNC:
Voltage-Gated Na+ Channel. Adapted from Tang-Schomer et al. (2012) and Johnson et
al. (2010)
1.5.1.2.2 Diffuse Vascular Injury
Diffuse vascular injury (DVI) is characterized by multiple macroscopic and/or
microscopic hemorrhagic brain lesions (Tomlinson, 1970; Adams, 1980). Diffuse
vascular injury is almost always fatal with short survival duration (< 24 h) post-trauma
(Pittella and Gusmao, 2003). DVI and DAI are suggested to be associated with each
other and share the same biomechanical mechanism (Pittella and Gusmao, 2003). One
major difference between the two pathologies however, is that while DAI can occur at
low acceleration, DVI is almost exclusively seen in motor vehicle accidents that involve
high-rate inertial forces (Gennarelli and Thibault, 1982; Pittella and Gusmao, 2003).
1.5.2 Secondary Brain Injury
The primary TBI triggers downstream cellular and biochemical cascades that ensue for
days to months, called secondary brain injury, which lead to long-term disabilities. In
most cases the damage caused by the primary brain injury is irreparable however
secondary brain injury is amenable to therapeutic and supportive interventions (Park et
34

al., 2008). The consequences of secondary injury include disruption of
cerebrovasculature including damage to the BBB, alteration in the cerebral blood flow
(CBF), edema, neurometabolic changes including disruption of brain energy
metabolism, inflammation, free radical formation, necrosis and apoptosis. While a
complete discussion of secondary injury pathways is beyond the scope of this thesis,
key secondary injury mechanisms are briefly discussed below.
1.5.2.1 Cerebrovascular Response to TBI
Brain injury causes changes in cerebrovascular response through alteration in BBB
function and CBF that lead to edema, increased intracranial pressure (ICP), cerebral
ischemia and hypoxia. The BBB consists of specialized endothelial cells that form tight
junctions with each other and are surrounded by the basal lamina matrix, pericytes and
astrocyte end feet. This gliovascular unit tightly regulates transport of metabolites,
nutrients and xenobiotics between the brain and the periphery (Abbott et al., 2006). TBI
is often accompanied by disruption of BBB via a variety of mechanisms including,
increased BBB permeability and proinflammatory cytokine activity, loss and/or
redistribution of tight junction proteins and changes in the activity of BBB transporters
(Zink et al., 2010). Changes in BBB function in response to injury are thought to
influence the evolution of injury and response to therapeutic interventions. A
compromised BBB allows entry of plasma-derived, osmotically-active molecules such
as albumin and fibrinogen into the brain interstitial space leading to development of
vasogenic edema (Chodobski et al., 2011).
35

The brain receives about 20% of the cardiac output. CBF is tightly controlled through
constant modulation of vascular resistance and BBB (Zink et al., 2010). TBI decreases
CBF as shown by CT and PET scanning (Bouma et al., 1992; Scalfani et al., 2012). The
reduction in CBF is thought to occur due to dysfunction of both macro- and
microvasculature leading to focal or in severe cases global ischemia (Bouzat et al.,
2013).
1.5.2.2 Neurometabolic Changes Following TBI
TBI leads to perturbations of cell membrane leading to redistribution of ions and
neurotransmitters. During the acute phase of injury, stretching of axonal membrane
opens voltage-dependent K+ channels leading to massive K+ efflux. This has been
shown with increased extracellular K+ concentration measured by in vivo microdialysis
in various animal models of TBI (Takahashi et al., 1981; Hubschmann and Kornhauser,
1983; Katayama et al., 1990). The duration of K+ increase is shown to be dependent on
the injury dose with shorter duration following mTBI whereas moderate-severe TBI
cause prolonged K+ increase (Katayama et al., 1990). The former is associated with
increased neuronal firing while the latter coincides with indiscriminate glutamate release
(Katayama et al., 1990). In addition to K+, excessive glutamate also leads to
uncontrolled shifts in Na+ and Ca2+ homeostasis (Bullock, 1994; Bullock et al., 1998). To
compensate for these ionic alterations, the ion pumps, especially Na+/K+-ATPase go
into the overdrive mode further increasing metabolic demand setting a vicious cycle of
CBF-metabolism uncoupling to the cell (Werner and Engelhard, 2007).
36

1.5.2.3 Neuroinflammation Following TBI
Neuroinflammation is an important secondary injury mechanism that contributes to
ongoing impairment following TBI. Microglia, the resident macrophages in the central
nervous system (CNS) act as the “first responders” mediating the immune response to
the brain injury. Microglial activation has been demonstrated as early as 72 hours after
human TBI and can persist for years after injury (Engel et al., 2000; Beschorner et al.,
2002; Gentleman et al., 2004; Ramlackhansingh et al., 2011). This activation profile is
also mirrored in animal models of TBI (Csuka et al., 2000; Koshinaga et al., 2000;
Maeda et al., 2007). Studies have shown that under physiological and pathological
conditions microglia assume different activation states that have distinct morphological
characteristics (Boche et al., 2013). At resting state, microglia show ramified
morphology characterized by short, fine processes and are thought to constantly
monitor their local environment. In response to an acute insult, such as trauma, resting
microglia are activated with dramatic changes in morphology and behavior. The cells
retract their processes and assume amoeboid morphology resembling macrophages
that proliferate and migrate towards the site of injury (Loane and Byrnes, 2010; Boche
et al., 2013). In certain chronic infections, microglia can assume rod morphology
characterized by markedly elongated nuclei, scanty cytoplasm and few processes.
Recently, rod microglia were observed in the brain regions that have an associated
sensory sensitivity following diffuse brain injury in rats (Ziebell et al., 2012) although
their significance in TBI pathology is currently unknown. Classically activated microglia,
also called M1 microglia release proinflammatory mediators, including cytokines,
chemokines, nitric oxide and superoxide free radicals that contribute to neuronal
37

dysfunction and cell death in response to injury (Loane and Byrnes, 2010). Both human
and animal studies have shown rapid elevation of proinflammatory cytokine IL-1β within
hours following TBI (Woodroofe et al., 1991; Fan et al., 1995; Winter et al., 2002), which
activates proinflammatory pathways such as TNF-α (Rothwell, 2003) through interleukin
1 receptor type 1 expressed in microglia and neurons (Pinteaux et al., 2002; Lu et al.,
2005). Recent studies have identified an alternatively activated state of microglia, called
M2 microglia (Colton, 2009; Kigerl et al., 2009) that are considered to have anti-
inflammatory actions and secrete anti-inflammatory cytokines including IL-10 and
neurotrophic factors such as nerve growth factor and transforming growth factor β
following injury (Csuka et al., 1999).
1.6 TBI, Alzheimer Disease, and Apolipoprotein E
1.6.1 TBI and Alzheimer Disease
Research over last 30 years indicates an increased risk of dementia, particularly of
Alzheimer’s disease (AD) in TBI survivors (Van Den Heuvel et al., 2007; May et al.,
2011). Moreover, besides age, antecedent TBI is now considered as one of the most
important environmental risk factors for dementia. Several epidemiological studies have
indicated increased dementia risk in TBI victims (French et al., 1985; Mortimer et al.,
1985; Mortimer et al., 1991; van Duijn et al., 1992; Rasmusson et al., 1995; Guo et al.,
2000; Plassman et al., 2000; Fleminger et al., 2003; Barnes et al., 2014), although this
association is not always observed (Katzman et al., 1989; Li et al., 1992; Fratiglioni et
al., 1993). One potential reason for the disagreement between the epidemiological
studies is that the data collection in most of these studies was retrospective, based on
38

low sample number and thus may have suffered from recall bias (May et al., 2011).
Nonetheless, a high-powered EURODEM meta-analysis pooled the findings of 11 case-
control studies and reported a significant relative risk of 1.82 (95% CI: 1.26-2.67) for
head trauma associated with AD (Mortimer et al., 1991). Based on the available
epidemiological data, the Committee on Gulf War and Health: Brain Injury in Veterans
and Long-Term Health Outcomes recently concluded that “there is sufficient evidence of
association between moderate to severe TBI and AD, limited/suggestive evidence of
association between mild TBI with LOC and AD and inadequate evidence of association
between mild TBI without LOC and AD” (Rutherford et al., 2008). TBI and AD show
striking similarities in their pathologies, which may shed light into their association. In
the following sub-sections I have discussed similarities and differences between TBI
and AD pathologies.
1.6.2 Alzheimer’s Disease: Pathology
AD is defined by two neuropathological hallmarks: 1) extraneuronal amyloid plaques
mainly composed by aggregated amyloid beta (Aβ) peptides and 2) intraneuronal
neurofibrillary tangles (NFT) formed by hyperphosphorylated tau protein. Central to the
AD pathology is the widely accepted, yet controversial amyloid cascade hypothesis
according to which the deposition of Aβ in the brain parenchyma is a crucial step that
triggers formation of intraneuronal tau aggregates and ultimately leads to AD (Hardy
and Higgins, 1992; Selkoe, 1994; Hardy and Selkoe, 2002; Karran et al., 2011). The
amyloid cascade hypothesis was developed on the basis of the discovery of autosomal
dominant mutations in APP, the parent protein of Aβ peptides and Presenilin-1 (PS1)
39

and Presinilin-2 (PS2), the components of γ-secretase (an enzyme involved in cleavage
of APP that leads to Aβ generation) that cause Familial/Early Onset AD (EOAD)
(Chartier-Harlin et al., 1991; Goate et al., 1991; Citron et al., 1992; Levy-Lahad et al.,
1995; Rogaev et al., 1995; Scheuner et al., 1996). On the other hand, apolipoprotein E
(apoE), specifically the apoE ε4 allele is an established genetic risk factor for the
sporadic or Late Onset AD (LOAD) and is thought to affect several aspects of AD
pathology, including Aβ aggregation and clearance, tau phosphorylation, inflammation,
neuronal repair and synaptic plasticity (discussed further).
1.6.2.1 Amyloid Beta Formation in the CNS
Aβ peptides are physiologically produced by the sequential enzymatic processing of the
highly-conserved type I transmembrane glycoprotein, APP. APP is a single
transmembrane domain protein that consists of a large extracellular domain, a
hydrophobic transmembrane domain and a short intracellular C terminal tail (Suh and
Checler, 2002). The juxtamembrane region of APP contains the Aβ sequence
composed of 43 amino acids (amino acids 597-639 of APP). APP is highly expressed in
neurons (Slunt et al., 1994; Lorent et al., 1995), undergoes fast anterograde axonal
transport (Koo et al., 1990; Buxbaum et al., 1998) and is processed via two mutually-
exclusive pathways: anti-amyloidogenic and amyloidogenic (Figure 1.6). The majority of
APP is processed through the anti-amyloidogenic pathway that involves enzymatic
cleavage, first by α-secretase followed by γ-secretase (Suh and Checler, 2002). Alpha-
secretase, a membrane bound, zinc metalloproteinase of the ADAM family, first cleaves
the APP within the Aβ sequence between residues Lys16 and Leu17 releasing a large
40

secreted α APP (sAPPα) domain and a truncated, 83-residue carboxyl terminal
fragment (CTFα or C83) that lacks the N-terminal portion of Aβ (Naslund et al., 1994). In
the amyloidogenic pathway instead of α-secretase APP is cleaved by β-secretase,
which is the rate-limiting step in the formation of Aβ species (Vassar, 2004). The
neuronal form of β-secretase, also known as β-Site APP Cleaving Enzyme 1 (BASE1),
a membrane-bound aspartyl protease cleaves the extracellular domain of APP
generating sAPPβ and a 99-residue CTFβ or C99 that contains the N-terminal domain
of Aβ. Both CTFα and CTFβ are substrates of γ-secretase, which is a complex of four
subunits: PS-1, PS-2, nicastrin, APH-1 and PEN-2 (Iwatsubo, 2004). Gamma-secretase
cleaves CTFα in the transmembrane domain releasing a 3 kDa, N-terminal-truncated
Aβ peptide, called P3 in the extracellular fluid and a cytosolic amyloid intracellular
domain (AICD). Cleavage of CTFβ by γ-secretase results in the release of 4 kDa Aβ in
the extracellular fluid and cytosolic AICD. Gamma-secretase can cut CTFβ at multiple
sites; first in the ε site generating 48 or 49-residue Aβ and subsequently every 3-4
amino acid residues generating 39-43 residue Aβ species (Kakuda et al., 2006; Selkoe
and Wolfe, 2007). The longer residue Aβ species tend to aggregate more readily and
are toxic. Under physiological conditions, γ-secretase cleavage generates less-toxic
Aβ40 and more-toxic and fibrillogenic Aβ42 species that account for 90% and <10% of
secreted Aβ species, respectively (Thinakaran and Koo, 2008). While Aβ42 is the major
component of amyloid plaques, Aβ40 accounts for most of the amyloid deposited in
cerebral vessels, which is called cerebral amyloid angiopathy (CAA) and is observed in
80% of AD patients (Biffi and Greenberg, 2011; Masters and Selkoe, 2012).
41

Figure 1.6: Amyloidogenic and anti-amyloidogenic processing of APP. The anti-amyloidogenic processing of APP involves sequential cleavage of APP by first
α-secretase that generates sAPPα and CTFα followed by cleavage of CTFα by γ-
secretase generating AICD and truncated Aβ fragment, P3. The amyloidogenic pathway
involves cleavage of APP by first β-secretase that generates CTFβ and sAPPβ followed
by cleavage of CTFβ generating Aβ and AICD.
1.6.2.2 Tauopathy in Alzheimer Disease
The second pathological hallmark of AD is intraneuronal neurofibrillary tangles (NFT),
which are composed of abnormally hyperphosphorylated tau (p-tau) protein that forms
paired helical filaments (PHF) (Iqbal et al., 2010). According to the amyloid hypothesis,
while amyloid plaque formation is the initiating event of AD pathology, formation of NFT
is a downstream event and executes neuronal death (Hardy and Selkoe, 2002; Karran
42

et al., 2011). Tau is the major microtubule associated protein (MAP), expressed almost
exclusively in neurons and is mostly localized in axons (Binder et al., 1985).
Microtubules are non-covalent cytoskeletal polymers composed of α- and β-tubulins that
play an important role in trafficking of organelles and other substances along axons
(Conde and Caceres, 2009). Humans express six tau isoforms including three of each
of three-repeat (0N3R, 1N3R, and 2N3R) and four-repeat (0N4R, 1N4R, and 2N4R) tau,
which are generated by alternative splicing of the MAP tau (MAPT) gene on
chromosome 17q21.31 (Neve et al., 1986; Goedert et al., 1989; Andreadis et al., 1992).
In contrast to humans, the adult mouse brain expresses only four-repeat isoforms
(0N4R, 1N4R, and 2N4R) (Gotz et al., 2010). The two important known functions of tau
are 1) facilitation of microtubule assembly and 2) microtubule stabilization by reducing
microtubule dynamics through its interaction with tubulin (Weingarten et al., 1975; Bré
and Karsenti, 1990). The interaction of tau with tubulin occurs at the C-terminus of tau
protein that contains microtubule binding repeat (MTBR) domain composed of three or
four tandem repeat motifs (Lee et al., 1989). The binding of tau to tubulin is regulated
and is inversely correlated to the degree of tau phosphorylation, especially at Ser262
and Thr231 residues within and flanking the MTBR domain (Wang et al., 2007). Thus,
phosphorylation of Ser262 within the flanking region of MTBR significantly reduces
binding of tau with microtubules (Biernat et al., 1993). Phosphorylated tau under
physiological conditions contains 2-3 moles of phosphate per tau molecule, while
hyperphosphorylated tau associated with AD is defined to contain > 6 moles of
phosphate per tau molecule (Iqbal et al., 2010). Abnormally p-tau is incapable of binding
to tubulin, thus remains free and polymerizes into PHF, which negatively affects
43

microtubule assembly and stabilization (Alonso et al., 2006). While tau can be
phosphorylated by a variety of kinases (e.g., glycogen synthase kinase, cdk5, MAP
kinase, JNK, and cdc2), glycogen synthase kinase 3β (GSK3β) plays the most
important role in regulating tau phosphorylation under physiological and pathological
conditions (Avila et al., 2004). The principle enzyme that controls tau dephosphorylation
is protein phosphatase 2A (PP2A). The phosphorylation state of tau is regulated by the
balance between the activities of protein kinases and phosphatases. For example,
PP2A levels in AD brains are significantly downregulated (Sontag et al., 2004a; Sontag
et al., 2004b). On the other hand, Pei et al found approximately 50% increase although
without any increase in the activity of GSK3β levels in AD brains (Pei et al., 1997).
While Aβ plaque pathology is seen exclusively in AD, tauopathy besides AD is also
associated with other neurodegenerative disorders such as Frontotemporal Dementia
and Parkinson’s Disease linked to chromosome 17, Pick’s Disease, Corticobasal
Degeneration, Progressive Supranuclear Palsy, Argyrophilic Grain Disease,
Amyotrophic Lateral Sclerosis and Parkinsonism-Dementia Complex (Spillantini and
Goedert, 2013) and Chronic Traumatic Encephalopathy (CTE) (McKee et al., 2009). A
distinct feature of tauopathy seen in AD and CTE compared to other tauopathy-related
neurodegenerative disorders is that the NFT in AD and CTE are composed of all six
abnormally p-tau isoforms (Goedert et al., 1992; Schmidt et al., 2001). On the other
hand, the tauopathy in other neurodegenerative disorders involves abnormal
hyperphosphorylation of either four-repeat (e.g., Progressive Supranuclear Palsy,
Corticobasal Degeneration and Argyrophilic Grain Disease) or three-repeat (e.g., Pick’s
44

Disease) tau (Flament et al., 1991; Ksiezak-Reding et al., 1994; Delacourte et al., 1996;
Spillantini et al., 1997; Tolnay et al., 2002).
1.6.3 TBI and Alzheimer’s Pathology: Similarities and Differences
1.6.3.1 Amyloid Pathology after Moderate to Severe Single TBI
One of the first studies linking TBI to dementia was by Rudelli et al who reported
classical AD pathology seen in the brain autopsy of a 38-year old man who died 16
years following recovery after a single severe head trauma (Rudelli et al., 1982). In
subsequent brain autopsy studies it was reported that about 30% of fatal TBI cases had
cortical Aβ deposition, strikingly similar to AD plaques (Roberts et al., 1991; Roberts et
al., 1994; Gentleman et al., 1997). Moreover, the Aβ deposits seen in severe TBI cases
were predominantly composed of insoluble Aβ42 (Gentleman et al., 1997; DeKosky et
al., 2007). There are however, important differences in the plaque pathology found in
the severe head trauma cases and AD. First, Aβ deposition in these acute TBI cases
was observed irrespective of the patient’s age, i.e., even in children, which is in contrast
to AD in which the plaque pathology developed in advanced age. Second, severe head
trauma triggered rapid Aβ deposition, seen as early as 2 h of TBI, as opposed to AD
pathology that develops over years (Ikonomovic et al., 2004). Third, TBI-associated Aβ
deposits are not restricted to the gray matter but have also been reported in the white
matter (Smith et al., 2003b). Finally, the plaques formed following TBI tend to be diffuse
compared to solid, dense-core plaques seen in AD brains. In contrast to the above
studies, Adle-Biasette et al found no evidence of Aβ pathology in 23 cases of age
45

between 17 and 63 years and 17 cases of age between 69 and 79 years who died
between 0-76 days after head trauma (Adle-Biassette et al., 1996).
The mechanisms of rapid Aβ deposition following TBI are unclear. A potential source of
Aβ is postulated to be the axonal swellings (varicosities) formed by DAI (Johnson et al.,
2010). In vitro studies have shown that axonal stretch at high shear rate and intensity
induces microtubule breakage at multiple sites along the length of the axon resulting in
interruption of axonal transport of various proteins, including APP (as discussed in
section 1.5.1.2.1). Intra-axonal accumulation of APP following TBI has been reported in
humans (Gentleman et al., 1993; Sherriff et al., 1994a; Gorrie et al., 2002) and animal
models (Lewen et al., 1995; Pierce et al., 1996; Smith et al., 1999b). Interestingly, the
components of APP cleaving enzymes, BASE1 and PS1 have also been shown to co-
accumulate along with APP in the damaged axons in humans (Chen et al., 2004; Uryu
et al., 2007) and animals (Chen et al., 2004). The axonal transport of these proteins is
shown to be mediated by the axonal transport protein, kinesin-I (Kamal et al., 2000) and
APP is thought to act as kinesin-I receptor facilitating transport of BACE-1 and PS1
(Kamal et al., 2001). Based on these observations it is proposed that the co-
accumulation of APP, BASE-1 and PS1 within the axonal varicosities form a nidus for
Aβ production (Johnson et al., 2010). Supporting this hypothesis, intra-axonal
accumulation of Aβ has been reported following TBI (Chen et al., 2004; Uryu et al.,
2007; Chen et al., 2009). Thus, DAI seems to create a unique environment bringing
together all the machinery necessary for Aβ production (i.e., APP, BACE-1 and PS1),
which is thought to lead to a burst of Aβ production that can deposit as amyloid plaques
46

(Johnson et al., 2010). To counteract the increased Aβ production, the traumatized
brain may also upregulate Aβ clearance mechanisms. For example, Chen et al reported
increased intra-axonal accumulation of neprilysin (NEP), a major Aβ degrading enzyme,
following TBI (Chen et al., 2009). In support of this observation, Johnson et al reported
that a genetic polymorphism in NEP that resulted into longer GT repeats in the enzyme
was associated with increased risk of Aβ deposition after TBI while shorter GT repeats
NEP decreased the incidence of Aβ deposition (Johnson et al., 2009). Thus, a balance
between Aβ production and clearance after TBI may be crucial in preventing Aβ –
related pathologies in TBI. In this regard, apoE may also play an important role as it is
shown to modulate Aβ clearance mechanisms (discussed further).
While the majority of clinical studies indicate rapid plaque pathology in the acute phase
of TBI, the Aβ plaque dynamics in the long-term TBI survivors seems controversial.
Chen and colleagues studied 23 cases with post-TBI survival up to 3 years (mean
survival 245 days) and found widespread axonal pathology along with intra-axonal
accumulation of APP, BASE1, PS1 and Aβ over the long term. The authors also
reported NEP immunoreactivity in axons for up to 8 month post-injury. Surprisingly, the
authors did not find any evidence of Aβ plaques in the same cases (Chen et al., 2009).
The authors suggested that while Aβ plaques are seen in short-term post-TBI survivors,
plaques may undergo NEP-mediated regression over months-to-years post-injury. In
contrast to the above study, in a recent study by the same group, widespread tau and
amyloid pathology was found in 39 single moderate to severe TBI cases with survival up
to 47 years (Johnson et al., 2012). It should be noted however that Chen et al have not
47

reported on any tauopathy as reported by Johnson et al in the absence of Aβ
deposition. This may be important for two reasons. First, while amyloid hypothesis is
widely accepted as the basis of AD pathology, it is not without controversies. For
example, few studies have found no correlation between amyloid plaque load and the
degree of cognitive impairment, i.e., some subjects with substantial amyloid plaques do
not exhibit clinical signs of AD (Armstrong, 1994; Giannakopoulos et al., 2003). Second,
in most of the studies mentioned above, Aβ plaque deposits were found only in one-
third of the cases. Thus, as suggested for AD pathogenesis (Hardy and Selkoe, 2002;
Karran et al., 2011), while Aβ plaque pathology may be a triggering event, tauopathy
may be the executioner of the synaptic and neuronal loss and long-term cognitive
deficits in TBI survivors. This is underlined by tauopathy which is the principal pathology
of CTE, a progressive neurodegenerative disorder thought to be caused by repeated
head trauma (see the next section). Moreover, in a recent study it was shown that
inhibition of APP processing by treating 3XTg-AD mice with a γ-secretase inhibitor
blocked Aβ accumulation without affecting tauopathy following controlled cortical impact
injury suggesting that Aβ accumulation and tauopathy in TBI may be independent
pathways (Tran et al., 2011a).
While the data from post-mortem brain analyses indicate increased Aβ production
following TBI, Aβ and tau levels have also been measured in CSF and recently in brain
interstitial fluid (ISF) of TBI patients and have revealed intriguing changes in Aβ
dynamics. For example, two studies reported an initial increase the CSF Aβ42 levels
peaking at ~ 5 days after severe TBI (GCS <8) followed by decline over several weeks
48

(Raby et al., 1998; Olsson et al., 2004). In both these studies, CSF tau levels were not
measured. In contrast to these findings, Franz et al found a significant decrease in CSF
Aβ42 measured over 1-30 days in severe TBI cases (Franz et al., 2003). Tau levels in
the same patients were significantly increased peaking between 5-15 days followed by
a decline over 30 days (Franz et al., 2003). Marklund et al assessed total tau and Aβ42
levels in brain ISF collected by real-time in situ microdialysis from eight patients with
either focal/mixed TBI or DAI over 8 days following TBI and found a non-significant
increase in Aβ42 levels in patients with DAI compared to patients with focal/mixed TBI.
Brody et al measured Aβ levels in ISF sample obtained from 18 TBI patients from 12-48
h and observed some very intriguing trends in Aβ dynamics (Brody et al., 2008). The
authors found that the ISF Aβ levels correlated positively with the patient’s neurological
status as determined by GCS such that ISF Aβ levels declined in patients with worse
neurological status, increased as the patient’s neurological status improved and
remained stable in clinically stable patients (Brody et al., 2008). In their follow-up study,
Brody and colleagues found > 3-fold increase in ISF tau levels at 1-12 h followed by a
decline over 61-72 h following TBI (Magnoni et al., 2012). The authors further found that
the high initial tau levels were correlated with worse neurological outcomes and
inversely correlated with initial low ISF Aβ levels. Based on these surprising findings,
the authors hypothesized that low Aβ and high tau levels during the acute post-TBI
phase may be associated with reduced synaptic activity (Magnoni and Brody, 2010) as
ISF Aβ levels are directly regulated by synaptic activity (Cirrito et al., 2005). Brain ISF
Aβ dynamics was also replicated in a controlled cortical impact (CCI) model in AD as
well as wild-type mice wherein both human as well as murine ISF Aβ levels were
49

significantly reduced following TBI (Schwetye et al., 2010). The authors further found
that in concordance with ISF, Aβ levels in phosphate buffered saline (PBS)-soluble
hippocampal and cortical lysates were also reduced. The pathophysiological
implications of differential Aβ dynamics following brain trauma in the short-term vs long-
term effects of TBI are unclear and require further investigation.
1.6.3.2 Consequences of Repetitive TBI: Chronic Traumatic Encephalopathy
While the data on long-term consequences of moderate to severe single TBI is limited, it
is long known that mild, repetitive head trauma can increase the risk of late-life
dementia. In 1928, Harrison Martland first described “Punch Drunk” syndrome among
boxers who sustained multiple head blows with early symptoms characterized by gait
deficits with slow muscular movements, hand tremors and speech hesitancy (Martland,
1928). In the later stages, the fighters with severe head blows suffered from
Parkinsonian-like facial tremors, and vertigo ultimately resulting in mental deterioration.
Millispaugh coined the term ‘dementia pugilistica’ (pugilist: a professional boxer) to the
symptoms described by Martland in boxers (Millspaugh, 1937). The first detailed
pathological features of dementia pugilistica were described by Brandenburg and
Hallervorden (Brandenburg and Hallervorden, 1954) and later by Corsellis et al
(Corsellis et al., 1973). While the earlier studies on the aftermath of repetitive head
impacts were focused on boxing, studies in the last decade showed that the clinical
symptoms and neuropathology described as dementia pugilistica were also seen in
other situations that involved increased risk of repetitive head impacts including contact
sports such as American football, wrestling, hockey and association football (soccer)
50

(Cantu, 2007; McKee et al., 2009) and in military veterans exposed to blast injuries
(Omalu et al., 2011; Goldstein et al., 2012). Thus the currently-accepted term for
dementia pugilistica is chronic traumatic encephalopathy (CTE).
Presently no biomarkers are available for the diagnosis of CTE. The current
neuropathological data comes from the post-mortem analysis of athlete brains, while the
clinical features of CTE are mostly based on the interviews of friends and family
members. The clinical manifestation of CTE occurs several years after exposure to
repetitive TBI and involves cognitive impairments, neuropsychiatric alteration, and motor
deficits that are thought to occur over three stages (Chin et al., 2011). Stage 1 is
characterized by affective disturbances and psychotic symptoms (paranoia, agitation)
and changes in mood and behavior (depression, aggression or violence, irritability,
suicidal tendency). Stage 2 involves social instability and memory deficits, with initial
symptoms of Parkinsonism. In the last stage the patient manifests general cognitive
dysfunction and progression of dementia, speech or gait deficits or full-blown
Parkinsonism.
The macroscopic neuropathology of CTE is characterized by marked atrophy of
cerebral regions (frontal and temporal regions) (Neubuerger et al., 1959; Corsellis et al.,
1973; McKee et al., 2009) and general atrophy of the cerebellum (Williams and
Tannenberg, 1996). Another gross neuropathological feature of CTE is cavum septum
pellucidum (separation of leaflets of septum pellucidum) with a fenestrated septum
(Omalu et al., 2006; McKee et al., 2009; Stern et al., 2011; McKee et al., 2013).
51

Although many of the clinical features of CTE overlap with those of AD, the microscopic
pathology of CTE shows some features that distinguish CTE from AD (Table 1.5). Thus,
the primary histological feature of CTE is extensive deposition of p-tau as NFT such that
CTE is now often considered as a tauopathy (McKee et al., 2009; McKee et al., 2013).
A second distinctive histopathologic feature of CTE is presence of TAR DNA-binding
protein 43 (TDP-43) with TDP-43 positive neurites in the cortex, brain stem, and
temporal lobe seen in early stages and in frontal subcortical white matter and fornix,
brainstem and medial temporal lobes in later stages (McKee et al., 2010; McKee et al.,
2013). TDP-43 pathology is seen in about 80% of CTE cases (Baugh et al., 2014) as
opposed to a lower incidence (25-30%) in AD (Wilson et al., 2011). Lastly, while amyloid
deposition is the neuropathological signature of AD, the presence of Aβ deposition is an
inconsistent feature of CTE and is reported in only 25-50% of CTE cases (McKee et al.,
2009; McKee et al., 2013).
52

Chronic Traumatic Encephalopathy Alzheimer Disease Prominent presence of tau tangles in the astrocytes
Astrocyte tau tangles absent
Perivascular deposition of NFT Perivascular NFT absent Prominent NFT deposition at depths of cerebral sulci
No NFT deposition at depths of cerebral sulci
Prominent subpial astrocytic tangles Absence of subpial astrocytic tangles
Periventricular astrocytic tangles Absence of periventricular astrocytic tangles
Focal NFT seen in the frontal lobe cerebral cortex in early stages
NFT seen in entorhinal cortex, amygdala and hippocampus
Patchy and irregular NFT distribution in advanced stages
Uniform NFT distribution in advanced stages
Tau pathology seen in white matter Virtual absence of tau pathology in white matter
Amyloid deposits seen only in 25-50% of CTE cases
Amyloid deposits is the signature pathology
High incidence (~ 80%) of TDP-43 pathology
TDP-43 pathology seen in 25-30% of cases
Table 1.5: Differences in the pathological features of CTE and AD. Adapted from (McKee et al., 2013).
Recently McKee and colleagues proposed four stages of CTE neuropathology based on
the severity of pathological and clinical features recorded from 68 cases with confirmed
CTE (McKee et al., 2013). The four stages of CTE are briefly described in the Table 1.6.
53

CTE Stage
Neuropathological Features Associated Clinical Features
I
• Mild lateral ventricle enlargement seen in some cases
• Focal perivascular NFT at depth of cortical sulci
• Headache • Loss of attention and
concentration
II
• Mild enlargement of frontal horn of lateral and 3rd ventricles
• Small cavum septum pellucidum • NFT in superficial cortical layers
adjacent to focal epicenters and in nucleus basalis of Meynert and locus ceruleus
• Depression and mood swings • Explosivity • Headache • Loss of attention and
concentration • Short-term memory loss
III
• Mild cerebral atrophy, ventricular dilation, septal abnormalities, a sharply concave contour of 3rd ventricle, depigmentation of locus ceruleus and substantia nigra
• Dense p-tau pathology in hippocampus, entorhinal cortex, amygdala and widespread in cortex, diencephalon, brainstem and spinal cord
• Cognitive impairment • Memory loss • Executive dysfunction • Depression • Explosivity • Loss of attention and
concentration • Visuospatial abnormalities
IV
• Atrophy of medial temporal lobe, cerebrum, hypothalamus, thalamus, mammillary body, septal abnormalities, ventricular dilation and pallor of locus ceruleus and substantial nigra
• Widespread p-tau pathology including white matter
• Prominent neuronal loss and gliosis in cortex
• Hippocampal sclerosis
• Dementia with profound short-term memory loss
• Executive dysfunction • Loss of attention and
concentration • Explosivity and aggression • Paranoia, depression, impulsivity • Visuospatial abnormalities
Table 1.6: Pathological and clinical stages of CTE.
Adapted from McKee et al. (2013).
54

1.6.4 Apolipoprotein E (ApoE) at the Nexus of TBI and AD Pathology
Familial or early onset AD is caused by mutations in APP, PS1 and PS2 genes and
represents less than 10% of total AD cases. The majority of the AD cases has no known
causative genetic mutations and is thus called sporadic or late onset AD (LOAD). While
the exact cause of LOAD is currently unknown it is believed that the development and
progression of AD is affected by multiple environmental and genetic risk factors. For
example, advanced aging is considered the greatest environmental risk factor. On the
other hand, apolipoprotein E (apoE), particularly the ε4 (apoE4) allele is the most
established genetic risk factor for LOAD (Corder et al., 1993; Saunders et al., 1993).
Interestingly, apoE4 is also thought to affect recovery from TBI (discussed further).
The CNS is the most lipid-rich tissue in the body, containing over 25% of total body
cholesterol in only 2% of total body weight (Dietschy and Turley, 2001; Vance et al.,
2005). Almost all of the CNS cholesterol is in the unesterified form and about 70% of
this form is present in oligodendrocytes that make myelin sheaths, which accounts for ~
50% of the white matter (Bjorkhem and Meaney, 2004). Being insoluble in water,
cholesterol is transported on lipoprotein particles that consist of apolipoproteins that
surround, stabilize, and solubilize their fatty cargo. The CNS lipid metabolism has some
unique features that separate it from the peripheral lipid metabolism. For example,
because of the presence of the BBB, there is virtually no exchange of cholesterol and
lipoproteins between the CNS and periphery and virtually all cholesterol is synthesized
in situ (Dietschy and Turley, 2001). This observation is supported by many studies that
showed virtual absence of radioactivity in the CNS following peripheral administration of
55

radiolabeled cholesterol (Chobanian and Hollander, 1962; Plotz et al., 1968; Meaney et
al., 2001). All brain cells synthesize cholesterol during embryonic development, but
mature neurons do not produce sufficient cholesterol to meet their lifelong ongoing
requirements for membrane synthesis and repair (Dietschy and Turley, 2004). Adult
neurons therefore depend upon lipids transported on glial-derived lipoproteins for
optimal survival and maintenance. The CNS lipoprotein metabolism is based entirely
upon particles that resemble plasma high-density lipoproteins (HDL) in density, size,
and composition (LaDu et al., 2000b). A major difference between brain and plasma
HDL is that apoE is the major apoprotein in brain HDL whereas apoA-I is the primary
apoprotein of plasma HDL (LaDu et al., 2000b).
1.6.4.1 ApoE Synthesis in the CNS
Under physiological conditions, apoE in the CNS is synthesized by primarily by
astrocytes (DeMattos et al., 2001) and to a lesser extent by microglia (Gong et al.,
2002). ApoE mRNA expression has been reported in cortical and hippocampal neurons
but not in the cerebellar neurons (Xu et al., 1999). Some studies suggest that neurons
can express apoE under certain conditions like excitotoxic stress (Xu et al., 2006) and is
controlled by astrocytes through Erk kinase pathway (Harris et al., 2004). Compared to
the peripheral lipoprotein synthesis and metabolism, little is known about CNS
lipoprotein metabolism. In the periphery, liver secretes lipid-poor apoA-I containing
lipoprotein particles that acquire free cholesterol and phospholipid from the peripheral
tissue (Yu et al., 2011). This initial transfer of free cholesterol and phospholipid is
mediated by the membrane-bound cholesterol transporter, Adenosine Triphosphate
56

Binding Cassette A1 (ABCA1) (Oram and Vaughan, 2000). ABCA1-mediated transport
of cholesterol is the critical and rate-limiting step in the biogenesis of HDL (Bodzioch et
al., 1999; Brooks-Wilson et al., 1999). The partially-lipidated apoA-I particles further
acquire more lipids from another ABC transporter, ABCG1 forming nascent HDL3
(Nakamura et al., 2004; Kennedy et al., 2005). The HDL3 particles are matured by
further acquiring phospholipids, free cholesterol, and triglycerides via lecithin-cholesterol
acyltransferase (LCAT), an enzyme that esterifies free cholesterol resulting in spherical
HDL2.
Based on the pathway of peripheral HDL biosynthesis, it is proposed that glia secrete
apoE-containing lipoproteins as lipid-poor nascent discoid particles that are composed
of phospholipids and free cholesterol but lack a cholesterol ester/triglyceride core (LaDu
et al., 1998; Fagan et al., 1999). The nascent apoE particles are remodeled into mature
lipoproteins in the brain parenchyma by acquiring cholesterol and phospholipids (LaDu
et al., 1998; Koch et al., 2001). We and others have shown that the cholesterol
transporter, ABCA1 is expressed by both glia and neurons (Wellington et al., 2002;
Hirsch-Reinshagen et al., 2004; Tachikawa et al., 2005) and is required for the lipidation
of apoE in the brain (Hirsch-Reinshagen et al., 2004; Wahrle et al., 2004). The ABCA1-
mediated lipidation of apoE is regulated by nuclear transcription factors, liver X
receptors (LXR).
57

1.6.4.2 Liver X Receptors (LXR)
LXR have two isoforms in mammals: LXRα and LXRβ, which share 80% sequence
homology and have differential expression patterns (Wojcicka et al., 2007). LXRα is
expressed principally in the liver, intestine, kidney, spleen, and adipose tissue (Auboeuf
et al., 1997) whereas LXRβ is ubiquitously expressed albeit at lower levels (Song et al.,
1994). LXRs were initially identified as orphan receptors, i.e., there were no known
endogenous ligands at the time of their discovery. Later studies identified oxysterols as
their endogenous ligands (Janowski et al., 1996; Janowski et al., 1999). Oxysterols are
monooxygeneted derivatives of cholesterol that are generated as intermediates in the
cholesterol biosynthetic pathway [24(S),25-epoxycholesterol], steroid hormone
biosynthesis [22(R)-hydroxycholesterol], and cholesterol metabolism pathways [24(S)-
hydroxycholesterol, 25-hydroxycholesterol, and 27-hydroxycholesterol] (Schroepfer,
2000). 24(S)-hydroxycholesterol (cerebrosterol) is the principal cholesterol metabolite
and mechanism through which cholesterol is removed from the CNS (Lund et al., 1999)
whereas 27-hydroxycholesterol is the most abundant cholesterol metabolite in the
periphery (Wojcicka et al., 2007). Most oxysterols have similar affinity towards both LXR
isoforms. Various synthetic LXR agonists have been developed; the two most
commonly used LXR agonists in experimental studies are T0901317 and GW3965.
T0901317 activates both LXR isoforms with almost equal affinity (Schultz et al., 2000)
as well as pregnane X receptor (Mitro et al., 2007) and farnesoid X receptor (Houck et
al., 2004). GW3965 (2-[3-[3-[[2-chloro-3-(trifluoromethyl)phenyl]methyl-[2,2-
di(phenyl)ethyl]amino]propoxy]phenyl]acetic acid) is a potent, orally-active synthetic
LXR agonist with slightly higher affinity for LXRβ (EC50: 30 nM) than LXRα (EC50: 190
58

nM) (Collins et al., 2002). Unlike T0901317, GW3965 has higher selectivity for LXR over
other nuclear receptors (Collins et al., 2002). The natural LXR antagonists include
geranylgeranylpyrophosphate (Forman et al., 1997), polyunsaturated fatty acids
(arachidonic acid, eicosapentaenoic acid, docosahexaenoic acid and linoleic acid) (Ou
et al., 2001; Yoshikawa et al., 2002) and 5α,6α-epoxycholesterol-3-sulfonate (Song et
al., 2001).
1.6.4.3 LXR: Molecular Mechanism of Action
LXRs regulate gene expression by forming obligate heterodimers with another nuclear
receptor, the retinoid X receptor (RXR). The LXR-RXR complex can regulate gene
expression via two mechanisms: direct activation and transrepression (Gabbi et al.,
2014). In the absence of agonist, the transcriptional activity of LXR/RXR is suppressed
by corepressors [e.g., Nuclear Receptor Corepressor (NCoR), Silencing Mediator of
Retinoic Acid and Thyroid Hormone Receptor (SMRT)] (Chen and Evans, 1995; Horlein
et al., 1995). During direct activation, agonist binding induces conformational change
in the LXR causing dissociation of the corepressor from the LXR/RXR dimer and results
in moderate level of transcription. Full transcriptional activity is gained by recruitment of
coactivators [e.g., Peroxisome Proliferator Activated Receptor-γ (PPARγ) coativator-1a
(PGC-1a), Steroid Receptor Coactivator-1 (SRC-1), Activating Signal Cointegrator-2
(ASC-2)] (Oberkofler et al., 2003; Huuskonen et al., 2004; Lee et al., 2008). The
activated LXR/RXR binds to the LXR-response element (LXRE) that consists of a direct
repeat of the core sequence 50-AGGTCA-30 separated by 4 nucleotides (DR4) of the
target gene (Wiebel and Gustafsson, 1997). LXRs, particularly LXRβ inhibit transcription
59

of NF-κB-regulated proinflammatory genes that lack LXRE by an alternate mechanism
called transrepression. In this case, upon agonist binding LXRβ undergoes a specific
SUMOylation by SUMO-2/3 that promotes interaction with the corepressor, NCoR and
its dissociation from the LXR, which in turn blocks transcription of proinflammatory
genes (Ghisletti et al., 2007; Venteclef et al., 2010).
1.6.4.4 LXR: Regulation of Cholesterol Metabolism
Much of the current understanding of the physiological role of LXRs comes from the
studies of their actions in peripheral tissues. It is now well established that LXRs act as
the “master regulators” of cholesterol metabolism. LXRs act as cholesterol sensors and
regulate intracellular cholesterol levels via three processes: 1) by promoting the
transport of excessive cholesterol in the peripheral tissues to the liver for excretion via
process called reverse cholesterol transport (RCT), 2) by inhibiting intestinal cholesterol
absorption and, to a lesser extent, 3) by inhibiting cellular cholesterol synthesis
(Wojcicka et al., 2007). LXRs regulate the RCT by controlling the expression of several
target genes. Excessive intracellular cholesterol leads to accumulation of oxysterols,
which activate LXRs that in turn increase expression of their principle target proteins,
ABCA1 and ABCG1, thus leading to enhanced intracellular cholesterol efflux (Figure
1.7). This pathway has been demonstrated in a wide variety of cell types (Costet et al.,
2000; Venkateswaran et al., 2000; Kennedy et al., 2001). Both ABCA1 and ABCG1 are
highly expressed in macrophages and prevent excessive accumulation of intracellular
cholesterol and formation of foam cells (Oram and Lawn, 2001). The cholesterol
pumped out by ABC transporters is accepted by apoA-I, the principle apolipoprotein in
60

peripheral HDL, which transports cholesterol to liver for excretion. In addition to ABC
transporters, LXRs also increase the expression of Niemann-Pick C1 (NPC1) and
Niemann-Pick C (NPC2), which are intracellular cholesterol carriers that redistribute
cholesterol from the endosomal compartment to the plasma membrane (Rigamonti et
al., 2005). LXRs also increase expression of ABCG5 and ABCG8 in hepatocytes that
transport cholesterol to bile canaliculi for its ultimate secretion into bile (Yu et al., 2002;
Yu et al., 2003). Finally, in rats and mice synthetic LXR agonists upregulate cholesterol
7α-hydroxylase (CYP7a), a rate limiting enzyme that converts cholesterol to bile acids
(Lehmann et al., 1997; Menke et al., 2002). This mechanism is unique in rats and mice
as the human CYP7a gene lacks an LXRE and therefore cannot be upregulated by LXR
agonists (Chiang et al., 2001; Goodwin et al., 2003) and may partly explain the
susceptibility of humans to dietary-induced hypercholesterolemia (Menke et al., 2002).
In addition to induction of RCT, LXR agonists have also shown to reduce intestinal
cholesterol absorption by upregulating ABCG5 and ABCG8 expression in the
enterocytes that increase cholesterol recycling into the intestinal lumen (Repa et al.,
2002; Yu et al., 2003).
61

Figure 1.7: Regulation of ApoE Lipidation by LXR. (1) In the absence of agonist LXR/RXR are bound to corepressors (CoR) which inhibit
their transcription activity. (2) Agonist (A) binding induces dissociation of corepressors
and association with coactivators (CoA) turning on transcriptional activity of LXR/RXR.
(3) Activated LXR activate the membrane-bound cholesterol transporter, ABCA1 that
pumps free cholesterol and phospholipids out of the cell. (4) Unlipidated ApoE act as
cholesterol acceptor and mature to lipidated apoE.
1.6.4.5 ABCA1: The Principal LXR Target
The principal target gene controlled by LXRs is ABCA1, a 2261 amino acid integral
membrane protein. ABCA1 is almost ubiquitously expressed with high levels of
expression in the macrophages, liver, intestine, heart, adrenal gland, endothelial cells,
testes, and in placental trophoblasts (Langmann et al., 1999; Lawn et al., 2001; Liao et
62

al., 2002; Wellington et al., 2002). High ABCA1 expression levels have been also
reported in mouse (Wellington et al., 2002) and rat (Fukumoto et al., 2002) brains with
highest levels in neuronal layer of the cerebellum. ABCA1 belongs to subfamily A of
large superfamily of ABC transporters. Although exact domain structure and topology of
ABCA1 are not fully elucidated, several models have been predicted based on
hydropathy analysis (analysis of hydrophobic and hydrophilic amino acids) that predict
ABCA1 structure (Schmitz and Langmann, 2001; Cavelier et al., 2006). According to
these models, ABCA1 is a full ABC transporter and consists of two transmembrane
domains, each consisting of six membrane-spanning helices followed by two
cytoplasmic nucleotide (ATP) binding domains. In addition ABCA1 also has a regulatory
domain between the two halves, a feature unique to ABCA1 among other ABC
transporters. The ATP-binding domains contain two conserved peptide motifs, Walker A
and Walker B and a family signature region. In addition, ABCA1 has two large
extracellular domains linked with two disulfide bonds and are essential for apoA-I
binding and HDL formation (Tanaka et al., 2001; Nagao et al., 2012).
Cellular expression of ABCA1 is tightly regulated at both the transcriptional and post-
transcriptional levels and its regulation is integrated with mechanisms that sense
intracellular cholesterol content (Tall, 2008). ABCA1 transcription is controlled
primarily by members of the nuclear receptor family of ligand-activated transcriptional
factors, specifically LXR-RXRs and PPARs (Schmitz and Langmann, 2005). ABCA1
expression is increased by natural oxysterols along with 9-cis retinoic acid, an RXR
agonist, as shown in various cell lines including macrophages, liver, intestine, and
63

neuronal cells in vitro (Costet et al., 2000; Schwartz et al., 2000; Kennedy et al., 2001;
Murthy et al., 2002; Koldamova et al., 2003). In addition, PPARγ agonists also induce
ABCA1 and associated apolipoprotein-mediated lipid efflux, although this induction is
likely secondary to activation of LXRs by activated PPARγ (Chawla et al., 2001; Ogata
et al., 2009). The in vivo efficacy and relevance of PPARγ-mediated activation of
ABCA1 via LXRs remains to be determined.
Cellular levels of ABCA1 are also controlled by a fine balance of post-transcriptional
modifications that either stabilize or degrade the ABCA1 protein, which has a half-life
of less than 1h (Oram et al., 2000; Wang and Tall, 2003). For example, interaction of
apoA-I with ABCA1 stabilizes ABCA1 protein (Wang et al., 2000; Wang et al., 2003).
ABCA1 contains a PEST sequence – proline (P), glutamate (E), serine (S) and
threonine (T) - in the cytoplasmic loop of ABCA1, the protein kinase A-dependent
phosphorylation of which induces degradation of ABCA1 by calpain (Martinez et al.,
2003; Wang et al., 2003). Interaction of ABCA1 with apoA-I prevents the
phosphorylation of this PEST sequence, thus stabilizing ABCA1. ABCA1 also
undergoes ubiquitin-proteosome-mediated proteolytic degradation (Feng and Tabas,
2002). Recently, a small non-coding microRNA, miR-33, has been shown to play an
important role in ABCA1 expression (Moore et al., 2010). MiR-33 inhibits ABCA1
expression in mouse hepatocytes and macrophages, blocking cholesterol efflux and
reducing plasma HDL levels. Conversely, inhibiting miR-33 by anti-sense
oligonucleotides increases ABCA1 expression and promotes lipid efflux both in vitro in
murine macrophages as well as in human THP-1, IMR-90 and HepG2 cell lines and in
64

vivo (Moore et al., 2010; Rayner et al., 2010). Whether CNS ABCA1 levels can also be
regulated by microRNAs remains an important question to address.
ABCA1 plays the pivotal role in controlling apoA-I and apoE metabolism. The critical
role of ABCA1 in HDL metabolism was first highlighted by loss-of-function mutations of
chromosome 9q31 leading to defective ABCA1 that cause Tangier disease, which is
characterized by extremely low plasma HDL levels (Hobbs and Rader, 1999; Rader and
deGoma, 2012). ABCA1 mediates transport of free cholesterol and phoshpolipids to
lipid-free apoA-I generating pre-β HDL, a rate-limiting step in HDL biogenesis (Oram
and Vaughan, 2000). Although the role of ABCA1 in HDL biogenesis is well established,
the exact mechanism of lipid transport to apoA-I by ABCA1 is still unclear and as such
various models have been proposed. Vedhachalam et al. (2007) proposed a three step
model of ABCA1-mediated lipidation of apoA-I in murine macrophages: in the first step,
apoA-I directly binds to ABCA1 causing ABCA1 upregulation that results in enhanced
translocation of phospholipids to the outer leaflet of the cell membrane. The net
increase and reduction in phospholipid content leads to later compression of
phospholipids in the outer half and expansion of phospholipids in the cytoplasmic leaflet
of the lipid bilayer, respectively. This asymmetric packing in the phospholipids causes
strain in the local cell membrane region. In the second step, the cell membrane relieves
the localized strain by bulging into the extracellular space forming an exovesiculated
domain to which apoA-I binds with high affinity. In the final step, binding of apoA-I
initiates spontaneous microsolubilization of the membrane phospholipids and
65

cholesterol and creation of discoidal HDL particles containing two, three or four apoA-I
molecules/particle.
A second model proposes that apoA-I binds to ABCA1 at the plasma membrane
followed by internalization of apoA-I-ABCA1 complex and subsequent delivery to the
late endosomes where apoA-I acquires lipids (Takahashi and Smith, 1999; Chen et al.,
2001). The lipidated apoA-I is then resecreted from the cell by exocytosis.
Using state of the art single-molecule fluorescence tracking and diffusion analysis
technique in HeLa cell line, Nagata et al recently proposed a novel mechanism of apoA-
I lipidation by ABCA1 (Nagata et al., 2013). According to this model, ABCA1 monomers
freely diffuse and translocate lipids on the plasma membrane, even in the absence of
apoA-I in an ATP-dependent manner. Upon lipid acquisition, presumably in its
extracellular domain or near its vicinity in the plasma membrane, ABCA1 monomer
undergoes conformational changes and forms dimers. The dimers are stabilized to the
plasma membrane by their interaction with the membrane-skeleton actin filaments.
Lipid-free apoA-I directly binds to the extracellular domains of ABCA1 dimers but not the
monomers and accepts lipids reserved by ABCA1. Upon lipid transfer to apoA-I, the
ABCA1 dimers dissociate into monomers, which are released from actin filament and
resumes its function to translocate lipids in an ATP-dependent manner.
Although the above models of apoA-I lipidation have been proposed based on the
studies in macrophages, it is proposed that ABCA1 may mediate apoE lipidation in the
CNS through similar mechanisms (Koldamova et al., 2010).
66

1.6.4.6 The Role of LXRs in the CNS
Although LXR function in the CNS is poorly understood, it is conceivable that like in the
periphery, LXRs regulate cholesterol metabolism in the CNS. The net movement of
cholesterol out of the brain is largely controlled by its conversion to the principal
metabolite, 24(S)-hydroxycholesterol (Dietschy and Turley, 2004). The second route of
intracellular cholesterol removal is via ABCA1 and apoE-mediated transport. Both
24(S)-hydroxycholesterol and synthetic LXR agonists increase ABCA1 expression in
neurons and astrocytes in vitro as well as in vivo along with increased cholesterol efflux
(Whitney et al., 2002; Liang et al., 2004; Abildayeva et al., 2006). Similarly, LXRs also
increase apoE expression in astrocytes in vivo (Liang et al., 2004; Abildayeva et al.,
2006). Cholesterol carried on apoE in the brain is thought to play minor role in the net
cholesterol removal from the CNS but may be rather important in redistribution of
cholesterol required for synaptogenesis, especially during development and following
recovery from brain injury.
As discussed further apoE plays crucial role in Aβ clearance and thus in turn in AD
pathophysiology. It is thus imperative to determine whether enhancing apoE function
through LXR activation may be beneficial in AD. Indeed, the in vivo activation of LXRs
has shown to decrease brain Aβ levels as well as improve cognition in mouse models of
AD. Koldamova et al reported decreased soluble Aβ40 and Aβ42 levels following a six-
day treatment with T0901317 in four-month old APP-23 mice (Koldamova et al., 2005b).
These findings were replicated by Riddell et al in another AD mouse model, Tg2527 in
67

which a six-day treatment with T0901317 in five-month old mice selectively lowered
Aβ42 but not Aβ40 in the hippocampus and reversed contextual memory deficits
(Riddell et al., 2007). It should be noted that in the above two studies the LXR agonist
response was assessed in young mice (4-5 months) and not in old mice (> 10 months)
that that develop extensive Aβ deposits. Jiang et al treated one-year old Tg2527 mice
with another LXR agonist, GW3965 for four months and observed ~50% reduction in
Aβ40 and Aβ42 levels with corresponding ~67% reduction in the plaque load (Jiang et
al., 2008a). The authors also reported significant decrease in the number of plaque-
associated activated microglia as well as IL-6 mRNA levels in GW3965-treated mice,
supporting anti-inflammatory effects of LXRs in the brain (Zelcer et al., 2007). Recently
Fitz et al treated 9 months old APP-23 mice with T0901317 for four months and
reported a significant decrease in the amyloid plaque load, soluble and insoluble Aβ40
and Aβ42 as well as A11-positive Aβ oligomers in cortex and hippocampus along with
reversal of Morris water memory deficits (Fitz et al., 2010). In all of the studies
mentioned above, treatment with LXR agonists did not affect APP processing, indicating
that LXR activation may affect Aβ metabolism but not its synthesis.
The beneficial effects of LXRs in Aβ metabolism are dependent on ABCA1 as shown by
several studies. We and other have shown that deletion of ABCA1 in mice results in
poorly-lipidated apoE and reduced apoE levels (Hirsch-Reinshagen et al., 2004; Wahrle
et al., 2004), which is associated with increased amyloid burden in several mouse
models of AD (Hirsch-Reinshagen et al., 2005; Koldamova et al., 2005a; Wahrle et al.,
2005). Moreover, in a recent study, lack of even one copy of ABCA1 resulted increased
68

amyloid deposition and exacerbated memory deficits in AD mice expressing human
apoE4 but not in mice expressing human apoE3 (Fitz et al., 2012). Conversely,
selective overexpression of ABCA1 in PDAPP mice has been shown to increase apoE
lipidation and prevent amyloid deposition (Wahrle et al., 2008). We also showed that
ABCA1 is required for the beneficial effects of GW3965 in AD mice in increasing CSF
apoE levels, reducing amyloid burden, and restoring novel object recognition (NOR)
memory, suggesting that lipidated apoE particles may underlie the neuroprotective
effects of LXR agonists (Donkin et al., 2010).
While the in vivo studies indicate beneficial effects of LXR activation in AD mice, the
results from in vitro studies are conflicting. Fukumoto et al showed increased secreted
levels of both Aβ40 and Aβ42 from neuroblastoma cell lines treated with T0901317, an
effect antagonized by blocking ABCA1 expression by RNAi (Fukumoto et al., 2002). On
the other hand, in two other studies treatment with the same LXR agonist resulted in
decreased Aβ secretion from neuroblastoma (Sun et al., 2003) and neuroglioma
(Koldamova et al., 2005b) cell lines. The reason for this discrepancy is not clear.
The role of LXRs in the CNS is further highlighted by gross anatomical abnormalities as
well as excessive lipid deposits in the brain and spinal cord seen in LXR knock-out
mice. Specifically, in LXRα/β double knockout mice, Wang et al reported closed lateral
ventricles that were lined with lipid-laden cells, enlargement of blood vessels particularly
in the substantia nigra pars reticulata and globus pallidus, excessive lipid deposits in the
brain parenchyma, neuronal loss, astrocyte proliferation and myelin sheath defects
69

(Wang et al., 2002). The astrocytes surrounding blood vessels also accumulated
excessive lipids in aged LXRα/β null mice (Wang et al., 2002). Recently, Anderson et al
reported that deletion of LXRβ in mice resulted in spinal-cord specific deficits, including
motor neuron loss, axonal atrophy, astrogliosis and lipid accumulation, and impaired
motor coordination; some of these features akin to that of amyotrophic lateral sclerosis
(Andersson et al., 2005).
1.6.4.7 ApoE Structure and Polymorphisms and their Significance in AD
Pathology
ApoE is a 299 amino acid glycoprotein with the molecular mass of 34 kDa and is
composed of N- and C- terminal domains that are connected by a flexible hinge domain
such that each domain can fold independently (Figure 1.8 A) (Aggerbeck et al., 1988;
Wetterau et al., 1988). The N-terminal domain is an elongated four-helix bundle and
contains receptor-binding site (22 kDa, amino acid residues 136-150) located in helix 4
(Wilson et al., 1991). The C-terminal domain contains an amphipathic α-helical lipid-
binding site (10kDa, amino acid residues 244-272) that can switch between lipid-bound
and lipid-free states (Hatters et al., 2006; Zhong and Weisgraber, 2009). The receptor-
binding site is rich in lysine and arginine residues that are thought to interact with the
ligand-binding domain of the apoE receptors, i.e. members of the LDL receptor family
(Hatters et al., 2006). Lipid-free apoE has poor affinity for binding to LDL receptor
(LDLR), while lipid-bound apoE binds with LDLR with high affinity.
70

Apolipoprotein E is encoded by the APOE gene located on chromosome 19, which is
highly polymorphic. In humans, APOE gene has three alleles, ε2, ε3, and ε4 that
encode three apoE isoforms, apoE2, apoE3 and apoE4, respectively (Weisgraber,
1994; Mahley, 1998). This apoE polymorphism leads to expression of six phenotypes:
E2/E2, E3/E3, E4/E4, E2/E3, E2/E4 and E3/E4. The three apoE isoforms differ by
single amino acid changes at residues 112 and 158: apoE 2 (Cys112, Cys158), apoE3
(Cys112, Arg158) and apoE4 (Arg112, Arg158) (Figure 1.8 B).
The single amino acid change has profound influence on the physical and physiological
properties as well as stability of the three alleles (Table 1.7), which may help to
understand their role in pathological conditions (Weisgraber, 1994). In apoE3 and
apoE4, the Arg at position 158 forms a salt bridge with Asp154. In apoE2 the Cys at 158
cannot form such bridge with Asp154. Instead, Asp154 forms salt bridge with Arg150
which lies in the receptor binding domain of apoE2, causing conformational changes
and interfering with receptor binding. As a result, apoE2 poorly binds with LDLR, while
apoE3 and apoE4 have ~50-fold higher affinity for LDLR (Weisgraber et al., 1982).
Two physical properties of apoE4, viz. domain interaction and poor stability have
been thought to alter its function and contribute to increased AD risk. X-ray
crystallography, site-directed mutagenesis, and fluorescent resonance energy transfer
studies have revealed that the amino acid at position 112 is the key to domain
interaction. Thus, Arg112 in apoE4 induces conformational changes in Arg61 causing
its side chain away from the four-helix bundle promoting interaction with Glu255 in the
71

C-terminal domain (Figure 1.8 C) (Dong et al., 1994; Dong and Weisgraber, 1996;
Hatters et al., 2005). On the other hand Cys at position 112 in apoE2 and apoE3 tucks
Arg61 side chain into the two-helix bundles preventing its interaction with Glu255
(Figure 1.8 C). All non-human species have threonine instead of arginine at the position
61, which does not engage in domain interaction. The mouse apoE, for example is
structurally similar to human apoE4 (Arg112, Arg158) (Figure 1.8 B) but functionally
behaves like apoE3 due to the absence of domain interaction (Figure 1.8 C).
Interestingly, replacing Thr61 with Arg in mouse apoE gene introduces domain
interaction causing the mutated apoE behave like human apoE4 resulting in lower apoE
levels (Raffai et al., 2001) along with cognitive deficits (Adeosun et al., 2014).
It is hypothesized that domain interaction alters apoE4 properties in two ways that may
contribute to the negative effects of apoE4 in AD. First, domain interaction is thought to
make the C-terminus of apoE4 more susceptible to cleavage by an unidentified
neuronal protease yielding C-terminal truncated apoE fractions that are thought to be
neurotoxic (Zhong and Weisgraber, 2009). This is supported by the observation that
neuron-specific expression of apoE4 in mice leads to intraneuronal accumulation of C-
terminal truncated apoE fragments, neuronal loss, increased tau phosphorylation, and
cognitive deficits (Harris et al., 2003; Brecht et al., 2004). Moreover, truncated apoE4
fragments have been reported in AD brains (Huang et al., 2001). Second, domain
interaction is also thought to influence the binding preference of apoE to lipid particles;
apoE2 and apoE3 preferentially bind to HDL while apoE4 is principally associated with
VLDL and LDL (Weisgraber, 1990; Dong and Weisgraber, 1996). The preference of
72

apoE4 to triglyceride-rich LDL and VLDL is thought to make it inefficient in lipid transport
required for synaptogenesis and neuronal repair (Zhong and Weisgraber, 2009).
The other physical property of apoE that is postulated to contribute to its influence in AD
pathology is the differences in the stability of the three isoforms. The N-terminal
domain of apoE2 is the most stable, that of apoE4 the least stable while apoE3 stability
is in between the two (Mahley and Huang, 2006; Zhong and Weisgraber, 2009). As a
result, apoE4 acquires a unique molten globule state in which the four-helix bundle is
partially unfolded exposing the proteolytic cleavage site (Morrow et al., 2002). A likely
effect of molten globule formation is apoE4-mediated potentiation of Aβ42-induced
lysosomal leakage and apoptosis, which has been shown in Neuro-2a cells transfected
with apoE4 (Ji et al., 2002). It is proposed that the acidic pH inside lysosomes favors
molten globule state of apoE4 that has increased affinity to phospholipids and
membranes, which in turn can result in destabilization of the lysosomal membrane and
ultimately lysosomal leakage (Mahley and Huang, 2006).
73

Figure 1.8: ApoE structure, polymorphism and domain interaction. (A) Schematic of the basic structure of apoE depicting the receptor and ligand binding
regions. (B) Human apoE isoforms with amino acids at key positions. Mouse apoE is
shown at the bottom for comparison. (C) Domain interaction in apoE4. In human apoE2
and 3 (blue) Cys112 in the N terminal domain changes conformation of Arg61 such that
it cannot engage in domain interaction with Glu225 in the C terminus. Mouse apoE
(yellow) although has arginine at position 112, the Thr61 cannot engage in domain
interaction with Glu225. In human apoE4, Arg112 changes conformation of Arg61 such
that it engages in domain interaction with Glu225.
74

Parameter ApoE2 ApoE3 ApoE4 General Allelic Frequency (%) 8.4 77.9 13.7
Frequency in AD Population (%)
3.9 59.4 36.7
Amino Acid at Position 112 Cys Cys Arg
Amino Acid at Position 158 Cys Arg Arg
Stability Highest Intermediate Least Folding Intermediates No Yes Yes
Domain Interaction No No Yes
Resistance to Molten Globule Formation
Highest Intermediate Least
LDLR Binding Affinity
Poor (associated with type III
hyperlipoproteinemia)
Bind with ~ 50-fold greater affinity than apoE2
Lipoprotein Binding Preference
Preferential binding to HDL Preferential
binding to LDL and VLDL
Table 1.7: Summary of physical and physiological properties of apoE alleles.
1.6.4.8 ApoE and Amyloid Beta (Aβ) Metabolism
The genetic association of apoE with AD pathologies stems from its well-documented
role in Aβ metabolism including clearance and BBB integrity. Data from these studies
indicate that the allele, lipidation status, and levels of apoE influence three aspects of
Aβ and amyloid metabolism: interaction with Aβ, Aβ degradation and Aβ clearance via
cerebrovasculature. While Aβ is continually generated at a rate of 7.6%/h, it is also
efficiently cleared from the CNS at an equivalent rate of 8.3%/h (Bateman et al., 2006).
75

Thus, the net steady state level of Aβ represents a delicate balance between its
formation and degradation. Aβ is cleared through three major pathways: 1) through
intracellular and extracellular enzymatic degradation including lysosomal degradation,
2) along interstitial fluid drainage pathways, and 3) through direct transport across
cerebrovasculature.
1.6.4.8.1 ApoE and Aβ Interaction
The first clues on the association between apoE and Aβ came from apoE
immunoreactivity found to be co-localized with parenchymal and cerebrovascular
amyloid deposits in AD patients (Namba et al., 1991; Wisniewski and Frangione, 1992)
and in AD mouse models (Burns et al., 2003). The lipid binding site, especially residues
244-272 located in the C terminal domain of apoE interact with residues 12-28 of Aβ
(Strittmatter et al., 1993). A later epitope mapping study revealed that residues 144-148
located in the receptor binding site on the N-terminal domain of apoE can also interact
with residues 13-17 of Aβ (Winkler et al., 1999). Several in vitro studies have shown that
apoE can bind to Aβ in an isoform-specific manner with apoE3 forming a more stable
complex with Aβ than apoE4 (LaDu et al., 1994; Tokuda et al., 2000; Petrlova et al.,
2011; Tai et al., 2013). It should, however, be noted that the findings of these studies
are influenced not only by the apoE isoform but also by its lipidation status and
experimental conditions. For example, purified and lipid-free recombinant apoE4 was
found to bind with Aβ with faster kinetics than apoE3 in vitro (Strittmatter et al., 1993).
Contrary to this finding, Tokuda et al reported that delipidation of apoE caused a 5-10-
fold decrease in Aβ binding without any influence of isoform (Tokuda et al., 2000). On
76

the other hand studies using HEK23 cell-secreted or native plasma apoE showed that
apoE3 formed more abundant sodium dodecyl sulfate (SDS)-stable complexes with Aβ
than apoE4 (LaDu et al., 1994; LaDu et al., 1995; Tokuda et al., 2000). To address this
discrepancy, LaDu et al compared Aβ binding with purified, lipid-free apoE vs native,
lipidated apoE and reported that delipidation of apoE during purification process causes
conformational changes in apoE resulting in diminished interactions between apoE3
and Aβ (LaDu et al., 1995).
1.6.4.8.2 ApoE and Enzymatic Degradation of Aβ
Several metalloproteases can degrade monomeric Aβ, the most important of which are
neprilysin (NEP) and insulin degrading enzyme (IDE) (Saido and Leissring, 2012). NEP,
a type-II membrane-associated zinc metalloprotease is the most potent Aβ degrading
enzyme in the CNS (Shirotani et al., 2001) and can degrade both monomeric and
synthetic oligomeric Aβ (Kanemitsu et al., 2003). NEP is highly expressed in microglia
(Iwata et al., 2000). Partial loss or decreased activity of NEP induced by intracerebral
injection of a NEP inhibitor in rats (Iwata et al., 2000) or in NEP knock-out mice (Iwata et
al., 2001; Rogers et al., 2002) increases Aβ levels by approximately two-fold, especially
in the hippocampus, suggesting important role of NEP in Aβ catabolism. Cortical and
hippocampal expression of NEP and activity decreases with aging in both rodents (Apelt
et al., 2003) and humans with AD pathology (Yasojima et al., 2001b; Yasojima et al.,
2001a; Russo et al., 2005; Wang et al., 2005) as well as in the cerebrovasculature in
AD cases with CAA (Carpentier et al., 2002; Miners et al., 2006). Another important
enzyme involved in Aβ catabolism is IDE, which is both membrane-bound as well as
77

secreted by microglia and mediates extracellular degradation exclusively of monomeric
Aβ (Qiu et al., 1998; Vekrellis et al., 2000). The Aβ degrading activity (Perez et al.,
2000) as well as expression levels (Cook et al., 2003) of IDE are significantly decreased
in AD compared to control cases. Complete IDE knockdown causes > 50% reduction in
Aβ degradation in mice suggesting an important role of IDE in regulation of Aβ levels
(Farris et al., 2003; Miller et al., 2003). Conversely, overexpression of NEP or IDE in AD
mice significantly decreases Aβ levels and retards plaque pathology suggesting that
these two enzymes may be potential therapeutic targets (Leissring et al., 2003).
ApoE facilitates both intracellular and extracellular proteolytic degradation of Aβ in vitro
(Jiang et al., 2008a). Further, the ability of apoE to facilitate enzymatic degradation of
Aβ is dependent on the lipidation status of apoE; poorly-lipidated apoE is inefficient
while lipidated apoE generated through LXR activation is efficient in stimulating
proteolytic degradation (Jiang et al., 2008a). Moreover, loss of ABCA1 impairs while
enhancement of ABCA1 expression through LXR activation promotes the ability of
microglia to degrade Aβ. This study also showed that human apoE2 promotes maximal
Aβ degradation while apoE4 is the least efficient, consistent with the isoform-dependent
effect of apoE in human AD pathology.
1.6.4.8.3 Cellular Uptake and Degradation of Aβ
Another route of Aβ degradation is through lipoprotein receptor-mediated uptake and
subsequent lysosomal degradation by the brain parenchyma and cerebrovasculature
(Kanekiyo et al.; Bu, 2009). The major lipoprotein receptors involved in Aβ uptake are
78

low density lipoprotein (LDL) receptor and LDL receptor related protein 1 (LRP1), which
are expressed in microglia, astrocytes, neurons, and smooth muscle cells at the BBB
and cerebral arteries (Fan et al., 2001). Aβ binds indirectly to LDLR and LRP1 through a
chaperone, mainly apoE, which can affect Aβ clearance. Recent in vitro studies have
shown that Aβ can also directly bind to both LDLR (Basak et al., 2012) as well as LRP1
(Deane et al., 2004b) indicating an apoE-independent receptor-mediated clearance
pathway. Overexpression of LDLR enhances cellular uptake of Aβ in vitro (Basak et al.,
2012) and reduces aggregation as well as enhances clearance of Aβ in APP/PS1
transgenic mice (Kim et al., 2009), suggesting LDLR modulation as a therapeutic target
for AD. Following endocytosis, the majority of endocytosed Aβ is transferred through
early and late endosomes before degradation in the lysosomes (Hu et al., 2009; Li et
al., 2012), while a small fraction is recycled through Ras-related protein, Rab-11-
mediated mechanism (Li et al., 2012). These studies have also shown that increasing
intracellular Aβ concentration through inhibition of Aβ degradation by lysosome
inhibitors or by excessive transport of Aβ to lysosomes by Rab-5 and Rab-7 can be
detrimental due to enhanced Aβ aggregation (Hu et al., 2009; Li et al., 2012) indicating
importance of lysosomal degradation in intracellular Aβ degradation.
1.6.4.8.4 Aβ Clearance through the Cerebrovasculature
Aβ is also cleared by the cerebrovasculature through two important routes. The first
clearance route is through the perivascular drainage pathway in which brain solutes,
including Aβ and ISF diffuse along the basement membrane between smooth muscle
cells of cerebral arterioles and ultimately drain into the CSF and lymphatic system
79

(Weller et al., 2008). The movement of Aβ and ISF along the perivascular space is in
the opposite direction of the blood flow and is thought to be driven by the blood vessel
pulsations (Schley et al., 2006). Aging as well as pathologies such as CAA and
cardiovascular disease can stiffen the vessel walls, reducing vessel pulsation and
slowing Aβ and ISF movement along the perivascular space, ultimately promoting Aβ
deposition within the capillary walls (Weller et al., 2002; Schley et al., 2006). The
second vascular clearance route involves receptor-mediated trafficking of Aβ from the
brain parenchyma into the peripheral circulation across the BBB. The two most
important receptors of this system are LRP1 and receptor for advanced glycation end
products (RAGE), which are shown to have opposite actions on Aβ clearance across
BBB (Deane et al., 2004a). Thus, while LRP1 mediates efflux of soluble Aβ40 into the
peripheral circulation preventing Aβ accumulation in the brain, RAGE can mediate influx
of Aβ from plasma to brain ISF and promote Aβ accumulation (Deane et al., 2003;
Deane et al., 2004a; Zlokovic, 2004). Elegant studies by the Zolkovic group suggest that
LRP1-mediated Aβ clearance across BBB may be more rapid (~ 6 fold faster) than ISF
flow (Bell et al., 2007) and may account for about 10-15% of the total Aβ clearance
(Shibata et al., 2000).
While lipidated apoE enhances enzymatic degradation of Aβ, the data on its effect on
Aβ transport across the BBB is conflicting. ApoE was initially postulated to enhance Aβ
clearance across the BBB through its interaction with Aβ as well as its ability to be
exported to the peripheral circulation via LRP1 in vascular endothelial cells (Deane et
al., 2004b). However, using in situ, real-time microdialysis, Bell et al reported that
80

association of Aβ40 with lipid-poor human apoE3 decreased while lipidated apoE
virtually blocked Aβ transport across the BBB in mice (Bell et al., 2007). The findings of
Bell et al were replicated in a later study where lipidation of all three apoE isoforms
significantly reduced the transport of apoE as well as apoE:Aβ complexes across the
BBB (Deane et al., 2008). In the same study Deane et al also showed that apoE, in fact
disrupts the transport of Aβ from brain ISF in an isoform-specific manner (apoE4 >
apoE3 > apoE2). Thus, binding of apoE to Aβ redirected the rapid export of free Aβ40
and Aβ42 from LRP1 to the VLDL receptor, which in comparison to LRP1, has a much
slower interaction with apoE-Aβ complex, resulting in 15- and 9-fold retention of Aβ40
and Aβ42, respectively (Deane et al., 2008). Along similar lines, apoE deficiency leads
to reduction of fibrillar Aβ deposits in AD mice (Irizarry et al., 2000) which may be due in
part to by reduced Aβ retention as suggested by the above microdialysis studies.
Although current evidence indicates that apoE interacts with Aβ in an isoform-specific
manner, its influence on Aβ clearance has been challenged by a recent study by
Holtzman and coworkers. In this study, Verghese et al mixed astrocyte-derived, human
CSF-derived or reconstituted lipidated apoE with cell-derived Aβ using a physiologically-
relevant apoE:Aβ ratio of 50-150:1 and found negligible levels of apoE-Aβ complexes
(Verghese et al., 2013). Moreover, the authors reported that apoE and Aβ competed for
LRP1-dependent cellular uptake by astrocytes. These data indicate that there is yet
much to learn about the relationship between apoE and Aβ.
81

Besides its influence on Aβ, apoE is also shown to serve other important role in the
CNS including synaptic function and synaptogenesis and inflammation.
1.6.4.9 ApoE, Synaptic Function, and Synaptogenesis
Neuronal membranes are rich in lipids, which are crucial in neuronal homeostasis and
function as well as synaptogenesis. Since adult neurons are almost entirely dependent
on external source for their lipid demands it is conceivable that apoE plays a crucial role
in the normal neuronal function including synaptic activity and synaptogenesis. In
support of this hypothesis, apoE modulates axonal growth in vitro. Incubation of cultured
fetal rabbit dorsal root ganglion cells with β-VLDL, which is rich in apoE promoted
neurite outgrowth and branching via LDLR-mediated mechanism (Handelmann et al.,
1992). Intriguingly, incubation with purified, lipid free apoE along with β-VLDL or
cholesterol resulted in increased neurite outgrowth but decreased branching
(Handelmann et al., 1992). Later studies showed that the neurotrophic effects of apoE
were isoform dependent; apoE3 promoted whereas apoE4 reduced neurite extension
and branching through an LDLR-mediated mechanism (Nathan et al., 1994; Holtzman
et al., 1995; Fagan et al., 1996; Nathan et al., 2002). The isoform-specific effect of apoE
was further demonstrated in murine neuroblastoma cells stably transfected with human
apoE3 and apoE4 wherein apoE3-secreting cells promoted neurite extension and
branching while apoE4 suppressed this effect (Bellosta et al., 1995). The inhibitory
effect of apoE4 on the neurite outgrowth was shown to be associated with apoE4-
mediated depolymerization of microtubules (Nathan et al., 1995).
82

In addition to synaptic remodeling, apoE is also thought to (in)directly affect learning
and memory by modulating synaptic plasticity. For example, apoE-deficient mice exhibit
spatial memory deficits as assessed by poor Morris water maze performance (Gordon
et al., 1995) and display impaired long-term potentiation (LTP) in hippocampal CA1
neurons (Valastro et al., 2001). Moreover, recent studies indicate that apoE can alter
synaptic plasticity in isoform-dependent manner. For example, LTP in the dentate gyrus
neurons is significantly impaired in an apoE isoform-dependent manner with apoE4 >
apoE2 and apoE-/- > apoE3 and wild-type mice (Trommer et al., 2004). Intriguingly,
opposite results were reported when LTP was assessed in the Schaffer collateral
synapses in the CA1 area with greatest LTP enhancement in apoE4 mice and least
amount of LTP induction in apoE2 mice (Korwek et al., 2009). ApoE isoform effects on
changes in LTP seems to be age-dependent with LTP enhancement seen in younger
apoE4 mice that disappears in old apoE4 mice, while LTP enhancement is unaltered in
the mice expressing apoE3 (Kitamura et al., 2004). The exact molecular mechanisms of
isoform-dependent effects of apoE on LTP are not fully understood. Reelin, an
endogenous ligand that regulates neuronal migration during embryonic development, as
well as neurotransmission in the adult brain enhances LTP through clustering of the two
apoE receptors, apoE receptor-2 (ApoER2) and VLDL receptors activating Src family
non-receptor tyrosine kinases, which in turn phosphorylate NMDA receptors, the key
receptors mediating LTP (Herz and Chen, 2006). Recently, apoE4 was shown to
selectively impair Reelin-mediated LTP by reducing surface expression of ApoER2 (by
locking ApoER2 in the intracellular compartment), thus interfering with Reelin-
dependent phosphorylation and activation of NMDA receptors (Chen et al., 2010). The
83

isoform-dependent effects on LTP are also reflected in hippocampal-dependent spatial
memory wherein female apoE4 mice show worse memory deficits compared to apoE3
mice (Grootendorst et al., 2005).
1.6.4.10 Role of ApoE in Inflammation
Studies in apoE-deficient mice suggest that apoE may have immunomodulatory roles
both in the periphery and in the CNS. ApoE-deficient mice show impaired immunity after
bacterial challenge with Listeria monocytogenes (Roselaar and Daugherty, 1998),
increased susceptibility to endotoxemia after intravenous lipopolysaccharide (LPS)
administration and inoculation with Klebsiella pneumonia (de Bont et al., 1999) and
increased systemic inflammatory response and higher mortality following LPS injection
(Van Oosten et al., 2001). Further, apoE modulates inflammation in an isoform-
dependent manner. Macrophages expressing apoE3 show lowest LPS-induced
expression of TNF-α and IL-6 compared to apoE4 and apoE2 (Tsoi et al., 2007). Similar
results were obtained in cultured primary microglia wherein apoE stimulated secretion of
proinflammatory cytokines such as TNF-α, IL-1β as well as prostaglandin E2 in an
isoform-specific manner with apoE4 > apoE3 > apoE2 (Chen et al., 2005; Maezawa et
al., 2006). The isoform-specific modulation of inflammation has been also studied in
apoE target replacement mice wherein apoE4 mice had significantly elevated TNF-α
and IL6 levels in brain as well as in the periphery compared to apoE3 mice (Lynch et al.,
2003). Moreover, brain autopsies revealed increased numbers of scattered microglia
and microglial activation in AD patients carrying the apoE4 allele (Egensperger et al.,
1998).
84

1.6.5 ApoE and TBI
Given the role of apoE in the lipid metabolism, synaptic function, inflammation, Aβ
metabolism, and possible in tau metabolism as well as the pathophysiological
similarities between TBI and AD, it is conceivable that apoE may modulate quite a few
aspects of TBI pathophysiology. While the role of apoE in AD pathology is well
established, its role in TBI is yet elusive. Two studies in apoE-deficient mice subjected
to closed head injury (CHI) give us a glimpse of the possible contribution of apoE in TBI
pathophysiology. In these studies, apoE-deficient mice showed greater cerebral edema
along with increased TNF-α mRNA levels (Lynch et al., 2002) as well as had greater
motor deficits, impaired spatial memory and overt neuronal death compared to wild-type
mice after CHI (Chen et al., 1997).
CNS injury leads to the release of large amounts of lipids from the degenerating axonal
membranes and myelin. Since CNS lipid metabolism is almost entirely based on apoE,
it is possible that apoE dynamics may change in response to TBI. In humans for
example, TBI results in a striking 14-fold selective reduction in CSF apoE-containing
lipoproteins from 2 to 5 days following injury and a 2- and 4-fold increase in total and
free cholesterol (Kay et al., 2003), reflecting a pronounced uptake of apoE particles by
the injured brain. Although data on the dynamics of apoE synthesis and utilization are
not yet available from human subjects, studies in animal models support a role for
increased synthesis of apoE in the CNS in the hours to weeks after many types of CNS
injury including TBI and spinal cord injury (SCI) (Poirier et al., 1991; Poirier et al.,
85

1993b; White et al., 2001a; Seitz et al., 2003; Iwata et al., 2005). For example, lesions
to the entorhinal cortex have been shown to induce coordinated increases in apoE
mRNA and its receptor, LDLR in parallel to clearance of lipid and membrane debris
supporting the idea that apoE may accept cholesterol and lipids released from the
damaged neuronal membranes and transport them back to the neurons undergoing
reinnervation and taken up through LDLR (Poirier et al., 1991; Poirier et al., 1993a;
White et al., 2001a). Iwata et al reported a biphasic upregulation of apoE and another
CNS lipoprotein, apoJ following fluid percussion injury in rats (Iwata et al., 2005). In this
model the initial peak appears at 2 days followed by return of apoE to baseline. ApoE
levels then again gradually increased over 6 months post-injury. Intriguingly, the authors
reported an uncoupling between the apoE protein and mRNA levels with mRNA levels
showing a delayed and sustained increase following an increase in brain apoE protein
levels, suggesting that apoE may enter into the injured brain by transfer through the
BBB or CSF (Iwata et al., 2005). ApoE isoform may also influence synaptic plasticity
after brain injury as apoE4 mice exhibit impaired synaptic plasticity compared to apoE3
mice following entorhinal cortex lesions (White et al., 2001b).
Epidemiological studies indicate that apoE may influence TBI outcomes in an isoform-
dependent manner although this relation is not as clear as in AD. Many studies have
reported poor post-TBI outcomes in apoE4 carriers compared to non-carriers (Sorbi et
al., 1995; Teasdale et al., 1997; Lichtman et al., 2000; Fleminger and Ponsford, 2005;
Smith et al., 2006) while others have found no such association (Jiang et al., 2008b;
Hiekkanen et al., 2009; Pruthi et al., 2010). These conflicting data may have resulted
86

from various confounding factors such as short follow-up times after TBI and a large
diversity of the subject population with respect to age, gender, and injury severity.
Nonetheless, a recent meta-analysis of 14 studies with a total of 2527 subjects
concluded that apoE4 does not affect initial injury severity but rather compromises
recovery at 6 months post-injury (Zhou et al., 2008). Moreover, accumulating evidence
suggests that age at injury may alter the association of apoE4 with TBI outcome. For
example, in a study of mTBI in pediatric subjects it was observed that the apoE4-
carriers were significantly more likely to have a worse GCS score compared to non-
carriers, yet performed equivalently, if not better, than non-apoE4 carriers on
neuropsychological tests over 12 months of follow-up (Moran et al., 2009). Another
recent study of CTE cases, which are also highly representative of brain injuries
experienced in late adolescence and early adulthood, also reported that the association
of apoE4 with worse outcomes is weaker than originally anticipated (Katsnelson, 2011).
1.7 TBI: An Overview of Animal Models
Since no single animal model can replicate the entire spectrum of human TBI
pathophysiology, several large and small animal models have been developed to mimic
particular pathophysiological aspects. Large animal models (e.g., non-human primates,
pigs, dogs, sheep, cats) are useful for investigating questions related to the response of
the gyrencephalic brain to injury, and may be particularly applicable for advanced
preclinical evaluation of therapeutic agents before introduction into humans (Kwon et
al., 2010). However, the high cost of large animal models and considerable ethical
concerns associated with their use limits their wide adoption into preclinical studies. In
87

contrast, rodent (e.g., rat and mouse) models have emerged as the most commonly
used animal models in TBI research, as they are widely available, cost-effective,
amenable to many behavioral, physiological and drug discovery evaluations, and,
particularly for mice, offer a wide variety of genetically modified strains.
Animal TBI models can be broadly classified into open head and closed head injury
models (Figure 1.9). The following section briefly summarizes important animal TBI
models along with their key advantages and disadvantages.
88

Figure 1.9: Animal models of TBI.
1.7.1 Open Head Injury Models
In open head injury (OHI) models, the mechanical force is applied directly to the dura,
which is exposed by a craniotomy and as such results in almost no head movement.
Animal Models of TBI
Open Head Injury
Fluid Percussion
Controlled-Cortical Impact
Penetrating Ballistic-Like Brain Injury
Needle Stab
Closed Head Injury
Impact-Acceleration (e.g., weight-drop)
Inertial Acceleration
Blast
89

The two most popular OHI models are fluid percussion (FP) and controlled cortical
impact (CCI).
1.7.1.1 Fluid Percussion (FP)
FP models generate brain injury by rapidly injecting fluid onto the intact dura through a
craniotomy, typically made laterally over the parietal cortex (lateral fluid percussion) or
medially over the sagittal suture midway between the bregma and lambda (midline fluid
percussion) (Figure 1.10) (Gurdjian et al., 1966; Thompson et al., 2005; Alder et al.,
2011). Fluid propulsion is driven by dropping a pendulum onto a fluid reservoir, and
pulse pressure, load duration and pulse velocity are reported. The pressure pulse of the
fluid can be varied to produce more or less severe injury, thus enabling FP models to
mimic a variety of human TBI injuries. FP induces mixed injury including petechial and
SAH, vascular damage at the gray/white matter interface, DAI, focal necrosis, and cell
loss (Povlishock and Kontos, 1985; McIntosh et al., 1987; Dixon et al., 1988; McIntosh
et al., 1989; Wang et al., 1997; Thompson et al., 2005). The biomechanical studies of
FP models have mainly concentrated on characterizing the input parameters such
volume loading, pressure peaks, and rate of fluid flow (Stalhammar et al., 1987). High-
speed X-ray imaging of the fluid pulse in rats (Dixon et al., 1988) and ferrets (Lighthall et
al., 1989) shows that the complex movement of fluid in the epidural space induces
gross movement of brain tissue. Data on the biomechanical response of the brain tissue
to the fluid movement, however is limited to a physical model (Thibault et al., 1992).
Although very widely used, particularly for rats, FP exhibits a high mortality rate due to
apnea and considerable variability of outcomes among different laboratories (Cernak,
90

2005; Marklund and Hillered, 2011). This variability is hypothesized to be due in part to
the surgical precision required to generate reproducible craniotomy position and angle
as well as minimal control over injury parameter (i.e., the height of the pendulum). As a
result, use of FP models often requires extensive operator training.
Figure 1.10: Fluid percussion injury model.
The injury is caused by a rapid pulse of saline caused by a pendulum striking the saline
reservoir. The inset shows movement of fluid in the epidural space. Reproduced with
permission from Macmillan Publishers Ltd: [Nature Reviews Neuroscience] (Xiong et al.,
2013).
1.7.1.2 Controlled Cortical Impact (CCI)
CCI models use a weight drop device (Feeney et al., 1981), a pneumatic (Dixon et al.,
1991; Smith et al., 1995), an electromechanic (Onyszchuk et al., 2007) or, most
recently, an electromagnetic (Brody et al., 2007) piston to deliver precisely controllable
tissue deformation to an exposed dura (Figure 1.11). This method induces primarily
focal damage with extensive cortical loss, hippocampal and thalamic damage,
intracranial hemorrhage, edema, and increased ICP (Saatman et al., 2006; Marklund
91

and Hillered, 2011), and is therefore used primarily to mimic severe TBI with frank
tissue destruction. The injury severity and pathology are affected by several variables
including craniotomy placement (midline vs lateral vs frontal), the impactor tip
configuration (size and shape), impact velocity and depth, and whether or not the
craniotomy is resealed after impact, which can be adjusted to produce various degrees
of injury with excellent precision and reproducibility. CCI models do not suffer from
“rebound injury” typically seen in weight-drop models (see below). On the other hand,
CCI models tend to induce severe injury destroying large cortical areas, which is
observed only in severe human TBI (Marklund and Hillered, 2011). Moreover, the
cortical damage during impact is difficult to visualize if the craniotomy is barely larger
than the impactor size, which may cause variation in the injury severity.
Figure 1.11: Controlled cortical impact model. The injury is caused by a piston compressing the exposed dura. The inset shows
direction of compression of the brain tissue. Reproduced with permission from
Macmillan Publishers Ltd: [Nature Reviews Neuroscience] (Xiong et al., 2013).
1.7.2 Closed Head Injury Models
92

In closed head injury (CHI) models, the injury is induced through the intact skull by
impact (e.g., dropping a weight on the intact skull or striking the intact skull with a
piston), non-impact (e.g., blast) or inertial loading (by rapid rotation of head in sagittal,
coronal or oblique planes). CHI models are characterized by varying degrees of head
acceleration.
1.7.2.1 CHI by Impact
In impact CHI, the mechanical force is delivered to the intact skull through an impact
that is delivered by various methods. The most common and simplest method is by
dropping a weight of known mass from a pre-determined height that falls under gravity
over the animal’s head (Marmarou et al., 1994; Chen et al., 1996; Tang et al., 1997;
Beni-Adani et al., 2001; DeFord et al., 2002; Pan et al., 2003; Zohar et al., 2003; Ucar et
al., 2006; Flierl et al., 2009; Zohar et al., 2011). Injury severity can be adjusted by
varying the mass of the drop weight or drop height. Impact is also delivered using a
pneumatically or electromagnetically-driven piston. Injury severity is usually controlled
by adjusting piston speed. In another method, Kilbourne et al used a steel ball that was
rolled down on a metal ramp to deliver impact (Kilbourne et al., 2009). The impact force
can be directed to the vertex (Marmarou et al., 1994; Tang et al., 1997; DeFord et al.,
2002; Zohar et al., 2003; Creeley et al., 2004; Wang et al., 2006), the dorso-lateral
surface (Laurer et al., 2001; Shitaka et al., 2011), lateral side (Viano et al., 2009, 2012;
Ren et al., 2013) or to the frontal side (Kilbourne et al., 2009) of the skull. The animal’s
skull is usually surgically exposed (but without any craniotomy) to direct impact on a
particular location on the skull, although in recent models (including the one developed
93

by us and described in the Chapter 3 of this thesis) can deliver impact to the head
without any surgery (see below).
Various support systems have been used to stabilize the animal’s head during impact
that also affect the degree of skull movement after impact and injury characteristics. For
example, the rodent’s head is supported on a thick block of foam or gel that allows
partial head acceleration causing moderate to severe brain injury with DAI as revealed
by the APP histochemistry (Foda and Marmarou, 1994; Marmarou et al., 1994; Viano et
al., 2012). An important variable is the spring constant of the foam used to support the
head that can influence the head movement after impact. Unfortunately, physical
characteristics of the foam, including the spring constant are rarely reported,
challenging the replication of the model across laboratories. In another configuration,
the animal head is held in a stereotaxic frame and undergoes the least amount of
movement and show mild APP staining (Laurer et al., 2001; Shitaka et al., 2011).
Recently, Whalen and colleagues described a new murine impact CHI model
characterized by a completely unrestrained head movement (Khuman et al., 2011;
Meehan et al., 2012). In this model, isoflurane-anesthetized mice were supported on a
piece of Kimwipes®, manually grasped by the tail and placed under a vertical guide
tube. The impact was delivered without surgically exposing the animal skull, between
coronal and lambdoid sutures with a 53 g metal bolt. Upon impact the mouse head tore
the Kimwipes support with free rotation in the anterior-posterior plane. This method
however, resulted in a moderately high post-CHI mortality rate (~ 20%). In a recent
variant of the ‘Kimwipe model’ the animal’s body was supported on a piece of aluminum
94

foil suspended over an empty case with a thick foam pad at the base of the case (Kane
et al., 2012). Upon impact, the aluminum foil completely tears off allowing unrestricted
head and body movement of the animal as it falls into the cage. These authors reported
repeatable, mild, concussive injury and suggested that the technique could be used for
repeated impacts. Ren and colleagues recently developed a ‘hit and run’ TBI model in
which the anesthetized mouse was hung head up vertically from its incisor teeth by a
mounted metal ring and impacted by a pneumatically-driven piston over the lateral side
of the head (Ren et al., 2013). Following impact the mouse fell onto a soft pad. The
authors reported that driving piston at 4.8 m/s resulted in mild injury characterized by
diffuse cortical astrogliosis, while impacting head at 5.2 m/s resulted in moderate injury
characterized by frank tissue disruption or cavitation.
Advantages of CHI models include the concordance of the mechanism with the vast
majority of human TBI that occur without skull fracture and, relative to FP and CCI,
employ less-invasive and simpler surgical methods that allow rapid behavioral
assessment of injury severity. Weight drop models, however, pose a number of very
significant limitations. For example, high-speed videography analysis shows that many
weight drop models deliver a primary and a rebound impact to the head. As well, weight
drop models are limited in that velocity of head impact and head displacement cannot
be varied independently as they can be when using an actuator. In addition, appropriate
release of head constraint to allow human-like head kinematics is challenging with
weight drop models and actuators that move downwards to create impact. A principal
caveat of most current CHI animal models is that both the input parameters (e.g.,
95

mechanical loading, method of mechanical input, response of animal’s head to
mechanical loading) as well as the cognitive, histological, and biochemical endpoints
used can vary considerably among different laboratories, which has challenged the
reproducibility of results and thus, the translational potential of these models. Much
more needs to be learned about how these CHI models alter animal head kinematics
(e.g., calculation of linear vs. angular acceleration, quantification of head acceleration),
how head kinematics relate to injury severity, how brain movement relates to head
movement in the species used, and to functional, molecular, and histological outcomes.
1.7.2.2 Non-Impact CHI by Blast Waves
Improvised explosive devices are a major source of TBI in military personnel. Blasts can
induce TBI through several mechanisms including the blast wave itself,
acceleration/deceleration forces, and particles that impact the head during the blast. To
simulate non-impact, blast TBI conditions in animals, the animal is secured at one end
of a long shock tube and is subjected to blast waves generated by compressed air or
gas or explosives (Figure 1.12) (Rubovitch et al., 2011; Goldstein et al., 2012; Rafaels
et al., 2012). The animal body is usually protected while the head is exposed to the
blast waves. These injuries typically present with diffuse brain edema accompanied by
hyperemia and delayed vasospasm. Again, the degree of head motion after blast
appears to have a significant effect on behavioral and neuropathological outcomes as
stabilizing the rodent head during blast was recently reported to be neuroprotective
(Goldstein et al., 2012).
96

Figure 1.12: Blast Wave TBI Model.
An animal is secured at one end of the shock tube. Blast waves are generated by
compressed air or explosives at the other end of the tube which travel toward the animal
causing head trauma. Reproduced with permission from Macmillan Publishers Ltd:
[Nature Reviews Neuroscience] (Xiong et al., 2013).
1.7.2.3 Non-Impact CHI by Inertial Loading
Many laboratories have developed inertial loading CHI models in which rotational force
causes rapid acceleration of the animal head followed by a longer deceleration phase.
These models have been developed for larger animals, including non-human primates
(Gennarelli et al., 1981; Gennarelli et al., 1982), pigs (Smith et al., 1997; Smith et al.,
2000; Browne et al., 2011), and rabbits (Gutierrez et al., 2001; Runnerstam et al., 2001;
Hamberger et al., 2003; Krave et al., 2005; Krave et al., 2011), as well as for small
animals such as rats (Davidsson and Risling, 2011). In these models, the animal head
is secured to a mechanical system that consists of a snout clamp or a skull fixation plate.
Linear motion induced by a piston is converted to rotational motion of the device, which
in turn results in a rotational acceleration of the head, along either the coronal
(Gennarelli et al., 1982; Smith et al., 1997; Smith et al., 2000), sagittal (Gennarelli et al.,
1982; Gutierrez et al., 2001; Runnerstam et al., 2001; Davidsson and Risling, 2011;
97

Krave et al., 2011), axial (Smith et al., 2000; Browne et al., 2011), lateral or oblique
(head turned 30° to right or left side and moving anterior or posterior with the center of
rotation in the lower cervical spine) planes (Gennarelli et al., 1982). The severity of
trauma can be adjusted by varying the angle of rotation or pulse duration. Studies using
these models have shown that post-traumatic neurological status, such as coma, is
related to the energy and form of rotation induced (Gennarelli et al., 1981; Gennarelli et
al., 1982; Browne et al., 2011; Krave et al., 2011). These models have also shown that
rotational forces can induce pathologies including SAH, increased ICP and elevated
serum levels of an astrocyte-specific protein, S100B (Runnerstam et al., 2001;
Davidsson and Risling, 2011). A distinctive neuropathological feature of these models is
the DAI, as evident by the formation of axonal bulbs and accumulation of APP (Smith et
al., 2000; Hamberger et al., 2003; Davidsson and Risling, 2011). Other microscopic
pathologies include gliosis (Smith et al., 1997; Gutierrez et al., 2001), neuronal death
(Smith et al., 1997; Runnerstam et al., 2001), and accumulation of phosphorylated
heavy neurofilament subunits at neuronal cell bodies (Smith et al., 1997; Smith et al.,
2000; Hamberger et al., 2003).
1.7.3 Challenges Associated With Current TBI Models
The choice of the most appropriate preclinical TBI model to use depends on what
factors of human injury need to be modeled. Compared to CHI models the input
parameters of FP and CCI models are well-controlled and thus induce highly
reproducible injuries but have some disadvantages. First, these models represent only a
small minority (0.8-3%) of human injuries that involve penetration of dura (Masson et al.,
98

2001; Myburgh et al., 2008; Wu et al., 2008). Second, these models, especially CCI,
tend to induce severe injury through destruction of large cortical areas, which is typically
observed only in severe human TBI (Marklund and Hillered, 2011). Third, these
methods often require a precisely positioned craniotomy that can reduce secondary
brain swelling by mimicking decompressive craniotomy often used to lower ICP
(Zweckberger et al., 2006; De Bonis et al., 2010). Fourth, these methods of injury cause
almost no head movement, which is rarely observed in human TBI.
CHI models, that apply impulsive (impact) loads to the head, by contrast, are
increasingly attractive for modeling the majority (>95%) of human TBIs that occur
without skull fracture or penetration of brain tissue. However, most CHI model systems
have significant caveats with respect to their high incidence of skull fracture
(necessitating immediate euthanasia of the animal) and highly variable outcomes
between laboratories. We can gain further insights into the relative strengths and
weaknesses of the various rodent injury models if we interpret these models in the
context of what is known about the biomechanics of human, large animal, and rodent
TBI. The vast majority of rodent CHI studies however, lack systematic biomechanical
rigor, particularly for weight drop models where only gross mechanical input parameters
such as the mass of the weight and drop height are typically reported. The
reproducibility of these mechanical inputs and measures to decrease variability in
mechanical input are rarely stated, and the mechanical response of animal’s head to the
forces applied is even more rarely recorded. As a result, the outcomes of studies even
99

with similar reported mechanical inputs can vary from no detectable injury to death
(Figure 1.13).
Figure 1.13: Variability in input parameters and outcomes in weight-drop TBI studies reported in the literature.
The figure depicts variation in two most commonly reported input parameters, drop
height and drop mass (A) and associated theoretical values of input energy (B) used in
mouse weight drop impact TBI studies. Panel C shows variation in mortality rate
associated with input energy. Reproduced from Namjoshi et al. (2013a).
One potential source of variability especially relevant for CHI models is the relationship
of head kinematics (moment of the head) following impact to both behavioral and
pathological outcomes after injury. The head kinematics can vary depending on how the
head is supported during impact. A few recent studies have begun to report details of
head kinematics following impact CHI in rats ((Viano et al., 2009; Li et al., 2011; Viano
100

et al., 2012). For example, Li et al. (2011) recently studied the biomechanical
parameters of the weight drop-based Marmarou impact acceleration model (Marmarou
et al., 1994). Head acceleration, change in head velocity, linear and angular head
acceleration were measured and compared to 1) kinematic responses predicted by a
finite element model and 2) axonal damage as assessed by APP histochemistry. The
authors reported that the severity of DAI was related to the linear and angular response
of the rat head but, against expectation, not with the drop height. Viano and colleagues
developed a rat model of CHI with unrestricted head movement to mimic concussions
experienced by professional football players (Viano et al., 2009, 2012). The ability to
study the detailed biomechanical responses of the rats and to allow unrestricted motion
in response to impact and the proposed methods for scaling these responses to human
concussions offer new insights into factors that may increase the translational potential
of future CHI studies in rodents.
In animal models, classification of injury severity as mild, moderate or severe is often
determined on functional outcomes such as cognitive and motor impairment that
develop over hours to weeks. Importantly, these outcomes can vary widely among
different research groups. For example, motor impairment may (Tang et al., 1997;
DeFord et al., 2002; Andoh and Kuraishi, 2003; Creeley et al., 2004; Rubovitch et al.,
2011) or may not (Tsenter et al., 2008) be observed with “mild” CHI. The severity,
temporal response, and extent of secondary injury response of brain can also vary
among rodent species and also within different strains of the same species. Because
the severity of the primary injury at the time of impact is often unknown and may be
101

below the threshold of detection even when using methods such as MRI, understanding
how biomechanical data affect the functional and histological outcomes of rodent CHI
may offer an alternative approach to improve the predictive and translational power of
preclinical TBI research in rodents.
Economic, ethical and scientific drivers have shifted the focus of preclinical TBI studies
from large animals to mainly rodents, even though large animals are better mimics of
the size and anatomy of the human brain (Denny-Brown and Russell, 1940; Gennarelli
et al., 1981; Gennarelli et al., 1982; Duhaime, 2006). One important consideration for
selecting an appropriate rodent TBI model is that that the rodent and human brain
differs considerably with respect to skull anatomy, brain mass and size, craniospinal
angle, gray-to-white matter ratio and cortical folding, all of which influence the
biomechanical parameters of brain injury. For example, rotational loading cannot be
linearly scaled between human and rodent TBI (Finnie, 2001). Furthermore, the long
axis of the rodent brain and spinal cord are nearly linear, as opposed to perpendicular
axes of the human brain and spinal cord, which causes less rotational shearing in the
rodent brain and renders rodents less vulnerable to diffuse axonal damage. Although
the anatomical differences between the rodent and human brain will always pose a
caveat for preclinical research, rodent-based experiments are expected to form the
majority of mechanism of action and proof-of-concept studies that can then be further
validated in large animal species. This progression will provide the greatest adherence
to ethical principles and scientific feasibility.
102

A common question regarding rodent models of TBI is whether the response of the
lissencephalic rodent brain to impact, acceleration or blast injury is representative of
that of the gyrencephalic human brain. For example, cases of CTE, which occurs from
repeated concussions, commonly show more extensive pathology in sulcal depths than
elsewhere along the cortex, suggesting that shear strain may be maximal at the sulcal
depths (Smith et al., 2013). Further work is warranted to investigate the role that these
anatomical differences may play in the context of mTBI research. Even if the
lissencephalic vs. gyrencephalic differences are found to result in an inability to
reproduce anatomical injury patterns observed in humans, the vast potential of rodent
models for studies investigating secondary injury pathways and for drug discovery
research is a major incentive to continued use of these species.
1.8 Pharmacological and Non-pharmacological Management of TBI: An Overview
of Randomized Clinical Trials
A recent review of 100 randomized clinical trials (RCT) examined efficacy of
interdisciplinary interventions of 55 acute phase and 45 post-acute phase TBI trials (Lu
et al., 2012). Acute phase trials were defined as interventions that occurred within 24h
of TBI. These studies largely focused on patient stabilization and minimizing secondary
injury, and employed standard outcomes comprised of GCS scores and mortality. By
comparison, post-acute trials can be very heterogeneous in design. The post-acute
trials reviewed were initiated from days-to-years post-TBI and used a wide variety of
outcomes including cognitive, neuropsychological, and quality of life measures (Lu et
al., 2012).
103

In the acute phase, only 15 of 55 trials showed a positive treatment effect and, of these,
11 evaluated non-pharmacological interventions. Furthermore, many interventions that
were tested across multiple trials led to mixed results. For example, decompressive
craniotomy, hyperosmotic therapy and hypothermia have shown inconsistent effects,
with studies reporting positive (Cruz et al., 2001, 2002; Zhi et al., 2003; Jiang et al.,
2005; Qiu et al., 2005; Jiang et al., 2006; Qiu et al., 2007), negative (Cooper et al.,
2011), or no effect (Smith et al., 1986; Marion et al., 1997; Shiozaki et al., 2001; Clifton
et al., 2002; Lu et al., 2003; Cooper et al., 2004; Harris et al., 2009; Clifton et al., 2011)
on post-TBI outcomes. A single trial of hyperventilation to reduce ICP also revealed an
adverse effect (Muizelaar et al., 1991). Other non-pharmacological interventions that
have shown benefit include early nutritional supplementation with zinc within 48 hours
or total parenteral nutrition within 72 hours post-TBI (Young et al., 1996). Lastly, a pre-
hospital rapid sequence intubation trial also yielded positive results (Bernard et al.,
2010).
The 32 acute phase pharmacological trials reviewed were grouped according to the
targeted treatment mechanism. Anti-excitotoxic agents, insulin, magnesium,
corticosteroids, and targeting lipid peroxidation or free radical damage either failed to
show a positive treatment effect or led to adverse effects. Importantly, only 4 of 32
pharmacological acute phase RCTs for TBI showed positive treatment effects, including
the psychostimulant, methylphenidate (Ritalin) (Moein et al., 2006), 2 RCTs for
104

progesterone (Wright et al., 2007; Xiao et al., 2008) and 1 RCT for the calcium channel
blocker, nimodipine (Harders et al., 1996).
Of the 45 post-acute phase RCTs reviewed, 24 evaluated cognitive rehabilitation
approaches and of these 22 showed positive treatment effects. The approaches used
included comprehensive interdisciplinary rehabilitation, cognitive/academic exercises
and communication skill training, compensatory techniques and computer assisted
training, educational intervention, psychotherapy, and behavior modification. Six of 8
trials using physical rehabilitation also showed a positive treatment effects, and nutrition
and acupuncture were found to be beneficial in the single trials conducted thus far.
Potential TBI pharmacotherapies were tested in 11 post-acute RCTs, with positive
treatment effects reported in 6 studies, including methylphenidate (Whyte et al., 2004;
Willmott and Ponsford, 2009), CDP-choline (Calatayud Maldonado et al., 1991), and a
vitamin B6 analog, pyritinol (Kitamura, 1981). On the other hand, a single trial of
phenytoin and carbamazepine was negative (Smith et al., 1994), while sertraline
(Novack et al., 2009; Banos et al., 2010), rivsatigmine (Tenovuo et al., 2009) and
modafinil (Jha et al., 2008) were found to have no significant treatment effects.
A number of RCTs in TBI have been completed in 2012-2013. Highlights include a
Phase III randomized trial of hypothermia in children, termed the “Cool Kids” study,
which recently reported that 48 h of hypothermia followed by rewarming does not
reduce mortality or positively affect recovery from pediatric brain injury (Adelson et al.,
2013). An intervention trial randomizing high-risk post-concussion syndrome patients to
105

an early visit to a physician similarly was negative, with similar outcomes to the
treatment as usual group (Matuseviciene et al., 2013). Chesnut et al recently reported
on their much anticipated trial of continuous invasive ICP monitoring following severe
TBI, showing no advantage over imaging and clinical examination alone (Chesnut et al.,
2012). Citocoline, which demonstrated promise in earlier trials as a neuroprotective
agent, failed in a subsequent Phase III trial (Zafonte et al., 2012). A trial of 30
hyperbaric oxygen therapy sessions, done in military personnel with mTBI, did not
demonstrate a significant effect (Wolf et al., 2012). Finally, in perhaps the most
influential trial of 2012, amantadine, given to TBI patients who were minimally conscious
or in a vegetative state 4 to 16 weeks following TBI, showed a highly significant effect
on accelerating the pace of functional recovery (Giacino et al., 2012).
The emerging consensus from these 100 RCTs conducted on TBI subjects is that early
intervention in the acute phase and comprehensive rehabilitation approaches in the
post-acute phase provide the most beneficial outcomes. What is far less clear is
whether a deeper understanding of the pathways involved in TBI pathogenesis may
eventually offer effective pharmacologic therapies. A critical requirement for any drug
discovery program is the availability of preclinical models that are as biofidelic to the
human condition as possible. With respect to TBI, however, a working knowledge about
neurophysiology and biomechanics is necessary to select the most appropriate model
system to address the experimental question being considered.
1.9 Study Rationale, Hypothesis, and Objectives
106

As discussed above, repair of damaged CNS critically depends on the lipid transport
mediated by brain lipoproteins. Lipid-laden brain lipoproteins aid in neuronal membrane
repair and synaptogenesis and stimulate the degradation of Aβ peptides that
accumulate after TBI. Genetic and pharmacological methods to increase the cholesterol
and phospholipid carried on brain lipoproteins improve cognitive function and reduce
neuropathology in AD mice. However, it is not known whether similar strategies may
also be beneficial for acute CNS injury.
We and others have previously demonstrated that synthetic LXR agonists effectively
induce ABCA1 expression, increase apoE lipidation, reduce Aβ levels, lower
inflammation and restore cognitive function in AD mouse models. Given that TBI
pathology shares common features with that of AD pathology including Aβ deposition
and NFT formation as well as neuroinflammation, it is of considerable interest to
determine whether LXR agonists may also have potential therapeutic utility for TBI. The
importance of addressing this question is two-fold. First, LXR treatment may minimize
neuronal damage and promote recovery in the acute phase after injury by reducing
inflammation and promoting apoE-mediated neuronal repair processes. Second, by
facilitating Aβ clearance in the first weeks after injury, LXR treatment may also lessen
the increased long-term risk of AD decades later.
Accordingly I hypothesized that the synthetic liver X receptor agonist, GW3965 will
promote neuronal recovery following closed-head mild, repetitive traumatic brain
injury in mice via apoE-mediated mechanisms.
107

Thus, the first objective of this thesis was to study therapeutic utility of the non-
steroidal LXR agonist, GW3965 in mild, repetitive closed head TBI in adult male mice
and whether the hypothesized beneficial effects of GW3965 were mediated through an
apoE-based mechanism. I tested this hypothesis using a widely-used weight drop CHI
model due to its several advantages over OHI models discussed previously. The
experimental details of the first objectives are presented in Chapter 2 of this thesis.
While weight drop TBI is one of the most popular rodent CHI model, it suffers from
several caveats like skull fractures and considerable experimental variation across
cognitive, histological, and biochemical outcome measures. This can be due to poor
control over the input parameters used to induce injury (e.g., mechanical loading,
method of mechanical input, response of the animal’s head to mechanical loading). In
addition, surgical manipulations required to localize the impact on the exposed skull
necessitate the use analgesic cocktails, sedatives and long-term exposure to
anesthetic. For example, in the weight drop CHI model used in the first objective, ethical
constraints necessitated us to use a non-steroidal anti-inflammatory drug (NSAID),
meloxicam for post-TBI pain suppression as well as ketamine-xylazine cocktail to keep
the animal sedated during the CHI procedure. These agents can prolong the post-
anesthetic recovery interfering with the immediate post-TBI assessment of neurological
outcomes. In addition, NSAIDs can suppress post-TBI inflammation, while, ketamine, a
NMDA receptor blocker, can suppress post-TBI glutamate-induced excitotoxicity,
interfering with the secondary injury pathways. Thus, it was essential to develop a
108

neurotrauma model that would mitigate these limitations and thus increase its
translational potential. While the second objective of this thesis was not driven by a
specific hypothesis, it was derived from the caveats encountered in the first study
objective.
Accordingly, the second objective of this thesis was to develop and a CHI model that:
1) offers tight control over injury parameters such as impact location and impact energy,
2) allows unrestricted head movement after impact mimicking most of the human TBI
conditions, 3) allows CHI without any surgical manipulation thus reducing anesthetic
analgesic and sedative requirement, 4) allows assessment of head kinematics during
and following impact, and 5) allows immediate post-TBI assessment of neurological
outcomes with minimal interference from anesthetics and sedatives. I further
characterized acute post-TBI outcomes in mice using this new CHI model.
The experimental details of the second objective are presented in Chapter 3 of this
thesis.
109

Chapter 2: The Liver X Receptor Agonist GW3965 Improves Recovery from Mild
Repetitive Traumatic Brain Injury in Mice Partly Through Apolipoprotein E
2.1 Summary
Traumatic brain injury (TBI) increases Alzheimer’s disease (AD) risk and leads to the
deposition of neurofibrillary tangles and amyloid deposits similar to those found in AD.
Agonists of Liver X receptors (LXRs), which regulate the expression of many genes
involved in lipid homeostasis and inflammation, improve cognition and reduce
neuropathology in AD mice. One pathway by which LXR agonists exert their beneficial
effects is through ABCA1-mediated lipid transport onto apolipoprotein E (apoE). To test
the therapeutic utility of this pathway for TBI, we subjected male wild-type (WT) and
apoE-/- mice to mild repetitive traumatic brain injury (mrTBI) followed by treatment with
vehicle or the LXR agonist GW3965 at 15 mg/kg/day. GW3965 treatment restored
impaired novel object recognition memory in WT but not apoE-/- mice. GW3965 did not
significantly enhance the spontaneous recovery of motor deficits observed in all groups.
Total soluble Aβ40 and Aβ42 levels were significantly elevated in WT and apoE-/- mice
after injury, a response that was suppressed by GW3965 in both genotypes. WT mice
showed mild but significant axonal damage at 2 d post-mrTBI, which was suppressed
by GW3965. In contrast, apoE-/- mice showed severe axonal damage from 2 to 14 d
after mrTBI that was unresponsive to GW3965. Because our mrTBI model does not
produce significant inflammation, the beneficial effects of GW3965 we observed are
unlikely to be related to reduced inflammation. Rather, our results suggest that both
110

apoE-dependent and apoE-independent pathways contribute to the ability of GW3965
to promote recovery from mrTBI.
2.2 Introduction
Several epidemiological studies suggest that TBI may increase the risk of dementia,
particularly AD, although this association is not always observed (May et al., 2011). TBI
has also been associated with an earlier onset of AD (May et al., 2011). Both
neurofibrillary tau tangles and amyloid plaques are found in post-mortem TBI brain
tissue. However, the widespread white matter involvement in TBI that is not present in
AD results in noteworthy differences in the pattern and distribution of their common
neuropathological features. For example, TBI neuropathology is largely that of tau
deposition, as only approximately 30% of TBI patients also contain Aβ deposits
(Roberts et al., 1994). Aβ plaques can appear within hours and remain detectable up to
47 years after a single moderate-severe TBI (Johnson et al., 2012). NFTs are also
detectable after a single TBI and are remarkably prominent in mild, repetitive,
concussive injuries (Johnson et al., 2012). Despite the low prevalence of amyloid
deposits in the post-mortem TBI brain, APP accumulation in damaged axons is a
striking histological hallmark of diffuse axonal injury (DAI) (Gentleman et al., 1993;
Sherriff et al., 1994b). Increased APP in the post-TBI brain is hypothesized to trigger a
burst of Aβ production that can deposit in amyloid plaques (Johnson et al., 2010).
Intriguingly, microdialysis experiments in brain-injured humans have demonstrated that
ISF Aβ levels correlate positively with the patient’s GCS score (Magnoni and Brody,
111

2010; Magnoni et al., 2012), suggesting that Aβ release is associated with recovery of
synaptic function, as has been demonstrated in animals (Schwetye et al., 2010).
Apolipoprotein E (apoE) is the major lipid carrier in the brain (LaDu et al., 2000a).
Brain injury increases apoE levels in order to scavenge lipids released by degenerating
neurons and myelin, which are later delivered to surviving neurons during reinnervation
and synaptogenesis (Poirier, 1994; Laskowitz et al., 1998). In humans, TBI results in a
14-fold selective reduction in CSF apoE levels by 2 to 5 days following injury and a 2- and
4-fold increase in total and free cholesterol, respectively (Kay et al., 2003), reflecting the
pronounced lipid mobilization, remodeling, and uptake of CSF apoE lipoproteins by the
injured brain. In animal models, apoE is induced within hours to weeks after TBI
followed by increased neuronal uptake of apoE-containing lipoproteins through the
LDLR (Poirier, 1994; Iwata et al., 2005). In mice, apoE deficiency compromises
recovery from acute neurological injuries, including TBI (Chen et al., 1997; Han and
Chung, 2000).
ApoE also regulates Aβ metabolism, a function that underlies its genetic association
with AD. The human apoE4 allele increases AD risk and hastens its onset (Corder et al.,
1993; Poirier et al., 1993b), whereas apoE2 delays onset and reduces AD risk (Corder et
al., 1994). Recent microdialysis studies demonstrated that apoE genotype modulates the
Aβ half-life in the brain ISF, with the rate of Aβ degradation accelerated by apoE2 (t1/2 =
0.56 h) and prolonged by apoE4 (t1/2 = 1.1 h) compared to apoE3 (t1/2 = 0.71 h)
(Castellano et al., 2011). Although AD risk and age of onset are indisputably modified
112

by apoE genotype, the relationship between apoE genotype and TBI outcome is
complex. Some studies report that apoE4 carriers have significantly poorer outcomes
compared to non-carriers (Teasdale et al., 1997; Lichtman et al., 2000; Smith et al.,
2006), while others find no association (Jiang et al., 2008b; Pruthi et al., 2010). Many
variables pose challenges to resolving these uncertainties, including relatively short
follow-up times after TBI and the diversity of subjects with respect to the age, gender,
and injury severity. Despite these challenges, a meta-analysis of 14 studies with 2,527
subjects concluded that apoE4 does not affect initial injury severity but rather may
compromise recovery at 6 months post-injury (Zhou et al., 2008).
Improving apoE function is thought to facilitate both neuronal repair and Aβ clearance
after TBI, thus potentially offering both acute and long-term benefits. One method to
enhance apoE function is to promote its lipidation by ABCA1, which is the rate-limiting
step in generating apoE-containing lipoprotein particles in the CNS. ABCA1 deficiency
leads to poorly-lipidated apoE in the CNS (Hirsch-Reinshagen et al., 2004; Wahrle et
al., 2004), and increases amyloid load in AD mice (Hirsch-Reinshagen et al., 2005;
Koldamova et al., 2005a; Wahrle et al., 2005). Conversely, overexpression of ABCA1 in
the CNS increases apoE lipidation and greatly decreases amyloid deposits in AD mice
(Wahrle et al., 2008). Transcription of ABCA1 and apoE is induced by agonists of LXRs,
which regulate many genes involved in lipid metabolism and inflammation (Jamroz-
Wisniewska et al., 2007; Wojcicka et al., 2007). Genetic deficiency of LXRs increases
amyloid burden in AD mice (Zelcer et al., 2007). Synthetic LXR agonists including
TO901317 and GW3965 cross the BBB, induce ABCA1 and apoE expression, improve
113

memory and reduce Aβ levels in AD mice (Jiang et al., 2008a; Donkin et al., 2010; Fitz
et al., 2010). Importantly, ABCA1 is required for several beneficial effects of GW3965 in
AD mice (Donkin et al., 2010), including increased CSF apoE levels, reduced amyloid
load, and improved memory. These observations provide a compelling rationale for
testing the therapeutic potential of LXR agonists for TBI. Indeed, TO901317 reduces Aβ
accumulation and promotes cognitive recovery in a CCI model of moderate-severe brain
injury in mice (Loane et al., 2011).
Approximately 80% of human TBI are mild injuries without skull fracture and loss of
consciousness (Decuypere and Klimo, 2012). It is increasingly appreciated that
repetitive, mild TBI, commonly experienced by athletes in high-contact sports may lead
to chronic traumatic encephalopathy (CTE), which is characterized by cognitive,
executive, and motor function disturbances, tau deposition and, in some cases, amyloid
deposits similar to those found in AD (Gavett et al., 2011).
To determine the therapeutic utility of LXR agonists for this type of brain injury, we
established a mouse model of mild repetitive TBI (mrTBI), wherein a gravity-driven
weight drop device was used to deliver two consecutive injuries 24h apart. Here we
report that GW3965 improves cognitive recovery and suppresses axonal damage in an
apoE-dependent manner. Surprisingly, apoE was not required for GW3965 to suppress
the transient increase in Aβ levels. Our results provide additional support for the
therapeutic potential of LXR agonists in TBI, and demonstrate that both apoE-
dependent and apoE-independent pathways contribute to their beneficial effects.
114

2.3 Materials and Methods
2.3.1 Animals
Male C57Bl/6 (WT) and apoE-/- (strain name: B6.129P2-Apoetm1Unc/J) mice were
obtained from Jackson Laboratories (Bar Harbor, ME). ApoE-/- mice are on C57Bl/6
background. Animals were housed with a reverse 12 h light - 12 h dark cycle for at least
10 days before TBI. The animals were fed with standard chow diet (PMI LabDiet® 5010,
containing 24% protein, 5.15 fat, and 0.03% cholesterol) and had free access to water.
All animal procedures were approved by the University of British Columbia Committee
on Animal Care (Protocol # A07-0706) and adhered to the Canadian Council for Animal
Care guidelines.
2.3.2 Mild Repetitive Traumatic Brain Injury (mrTBI)
WT (mean body weight: 29.6 ± 0.24 g) and apoE-/- (mean body weight: 30 ± 0.26 g)
mice at 4 months of age were subjected to two mild CHI spaced 24 h apart using a
gravity-driven weight-drop device obtained from Dr. Esther Shohami (The Hebrew
University of Jerusalem). The device consists of ~ 50 cm long graduated acrylic guide
tube held vertically over a metal plate (Figure 2.1A). A Teflon-tipped cone (tip diameter:
2 mm) is located at the base of the tube that can be freely raised inside the tube. A 95 g
brass weight is hung inside the tube with a thread. The height of weight is manually
adjusted by a thread holding lever. For inducing injury, the animal’s head is positioned
under the lower end of the guide tube and the Teflon-tipped cone is rested over the
exposed skull. In the original Shohami model, the animal’s head was supported by a
115

rigid Teflon disk. We modified this support by introducing ~ 3 mm thick computer mouse
pad between the animal’s head and the Teflon disk to reduce incidence of skull fracture
by partially allowing head movement following impact. This modification was made after
conducting a small pilot study with and without the mouse pad. The weight is raised to
the desired height and dropped over the Teflon-tipped cone, which directs the impact
force to a localized area of the animal skull.
All surgical procedures were conducted using aseptic technique. One day before the
TBI procedure, animals were weighed and the scalp was shaved under isoflurane
anesthesia (induction: 3-4%, maintenance: 1.5% - 2%). On the day of TBI surgery,
animals were anesthetized with isoflurane (induction: 3-4%, maintenance: 1.5% - 2%) in
oxygen (0.9 L/min). Lubricating eye ointment was applied to prevent corneal drying.
Animals were kept warm with an electric heating pad. The scalp was scrubbed three
times with 4% chlorhexidine solution and 70% ethanol. Ketamine (15 mg/kg) and
xylazine (1.75 mg/kg) mixture [subcutaneous (s.c.)], meloxicam (1 mg/kg, s.c.), and
bupivacaine (0.125 mg/kg, s.c., under the scalp) were administered for analgesia.
Sterile saline (1 ml/100 g body weight) was administered s.c. for hydration. The surgical
plane of anesthesia was confirmed with the absence of toe-pinch reflex and a midline
longitudinal incision was made in the scalp to expose the calvarium. After clearing the
parietal bone, a sterile piece of gauze (~ 3 mm x 5 mm), pre-moistened with
bupivacaine solution (2.5 mg/ml) was overlaid over the left parietal bone to minimize the
incidence of skull fracture (Figure 2.1 B). Anesthesia was temporarily discontinued (not
more than 30 s) and the animal was quickly moved under the weight-drop device. The
116

animal remained sedated with ketamine-xylazine during this temporary discontinuation
of anesthesia. The head was manually held in place such that the tip of the Teflon-
tipped cone rested over the piece of gauze in the left mid-parietal bone [approximately
between -1 and -3 mm from bregma and 1 mm left of the sagittal suture (Paxinos and
Franklin, 2001) ] (Figure 2.1 B and C). A 95 g brass weight was dropped from a pre-
determined height to result in injury across the left hemisphere. The precise drop height
(cm) of the weight was adjusted to be one-third the weight (g) of the animal (Mean drop
height: WT, 10 ± 0.08 cm; apoE-/-, 10.1± 0.09 cm). Mean body weight and drop height
did not defer significantly across the genotypes, time points, and treatment groups
(Figure 2.2). Anesthesia was reinstituted following injury. After removing the gauze and
confirming the absence of skull fracture, the incision was closed and the animals were
returned to their cages. Twenty-four hours after the first injury, the incision was
reopened under isoflurane anesthesia and a second identical TBI was induced as
described above. Mice suffering from skull fracture after injury were immediately
euthanized. Post-TBI recovery was monitored until animals moved freely and fed
normally. Sham controls received isoflurane anesthesia, pre-surgical analgesia, and
skin incision only. Mice within each group were randomly assigned to one of three post-
TBI assessment time points, viz., 2, 7, and 14 days after the second injury. The time of
the 2nd TBI was defined as time zero.
117

Figure 2.1: Weight drop CHI model and impact location. (A) Schematic of weight drop device and mouse head under the device. (B) Illustration
of surgical preparation for TBI showing an anesthetized mouse with the midline scalp
incision exposing the calvarium. A sterile piece of cotton gauze is overlaid across the
skull to help reduce incidence of skull fracture upon impact. The location of impact is
indicated by red dot. (C) Schematic diagram of murine skull showing approximate
location of the weight drop impact (red circle) on the left parietal bone. The dotted
rectangle represents cotton gauze.
118

Figure 2.2: Average animal body weight and drop height across all study groups.
To induce mrTBI, a 95 g brass weight was dropped on the animal’s skull using a height
(cm) adjusted to one third of the animal’s body weight (g) (see Methods and Materials).
The figure depicts body weight (A, B) and drop height (C, D) of WT and apoE-/- mice,
respectively, across post-mrTBI time points and treatment groups. Neither drop height
nor body weight differed significantly across genotypes, time points or treatment groups.
Data within each genotype were analyzed by two-way ANOVA followed by a Bonferroni
post-hoc test. Legend: V: untreated mice, open bars, G: GW3965-treated mice, black
bars.
2.3.3 GW3965 Treatment
Injured mice were randomized into treated (GW3965) and untreated (vehicle) groups
(WT mice: N=45/group, apoE-/- mice: N=30/group). Since, the animals do not become
fully ambulatory and start feeding for up to 2 h following TBI, the GW3965 treatment
was started with a single intraperitoneal (i.p.) bolus (20 mg/kg) 30 min after the 2nd
119

injury. GW3965 treatment was continued thereafter by feeding mice with standard
rodent chow (Research Diets Inc., New Brunswick, NJ) in which GW3965 was
compounded at 120 mg/kg until the end of the study, resulting in an average daily dose
of 15 mg/kg upon determination of average daily food intake. Because we had
previously demonstrated that GW3965 doses of 2.5 mg/kg/day and 33 mg/kg/day
provided cognitive and neuropathological benefits in APP/PS1 mice (Donkin et al.,
2010), we aimed to achieve a dose between these two extremes. A complete dose-
response study was beyond the scope of this thesis project. Because GW3965 has a
short plasma half-life of 2 h and high oral bioavailability (70%) (Collins et al., 2002)
delivering GW3965 in chow provides more constant plasma levels than by once-daily
oral gavage. Injured mice in the untreated group received a single i.p. bolus of
equivalent volume of the vehicle used for dissolving GW3965, dimethyl sulfoxide 30 min
after 2nd injury. These mice were thereafter fed standard rodent chow. Sham-operated
mice (WT mice: N=24; apoE-/- mice: N=18) received neither GW3965 nor vehicle and
were fed standard rodent chow. We have previously determined that GW3965 at a dose
of 33 mg/kg/day does not cause hepatotoxicity in mice (Donkin et al., 2010).
2.3.4 Cognitive Assessment by Novel Object Recognition
Medial temporal lobe function was assessed using the novel object recognition (NOR)
task performed at 2, 7, or 14 days following mrTBI (Ennaceur and Delacour, 1988). This
task was selected to facilitate comparisons to our previous NOR study of GW3965
efficacy in APP/PS1 mice (Donkin et al., 2010), and because fine motor impairments
observed after TBI may confound tasks such as Morris Water Maze. Each animal was
120

assessed with the NOR task at only one time point after sham or mrTBI surgery.
Twenty-four hours prior to the NOR task, mice were habituated to an empty open field
(14” x 24” x 14”) for 10 min in a brightly-lit room. The NOR task consisted of two phases:
training and testing. During training, mice were placed in the center of the open field
containing two identical objects spaced equidistant from the walls (Figure 2.3). The time
the animal explored each of the two objects during a 5 min epoch was quantified, with
exploring defined as sniffing, climbing on, or touching the object and at a body length or
less away from the object while oriented toward it. Trials were tracked using an
overhead digital camera and video tracking system (ANY-maze, Stoelting Co, Wood
Dale, IL). Between animals, the open field and objects were cleaned with 70% ethanol
to eliminate olfactory cues. Four hours after training, mice were tested by replacing one
of the identical objects with a novel object of similar size but different shape and color
(Figure 2.3). The side of the open field with the novel and familiar objects was
alternated for each mouse to avoid any side preference. Each animal was tracked for a
period of 5 min. Novel object recognition was quantified using the discrimination index
(DI) calculated as: DI = (TN – TF)/(TN + TF), where TN and TF represent exploration time
for the novel and familiar objects, respectively. DI values range from +1 to –1, where a
positive score indicates preference for the novel object, a negative score indicates
preference for the familiar object, and zero indicates no discrimination (Antunes and
Biala, 2012).
121

Figure 2.3: Novel object recognition phases.
During the training phase the mouse is placed in a rectangular box containing two
identical objects (indicated by yellow circles) and exploration time for each object is
recorded for 5 min. After a retention period of 4 h, the animals are re-introduced to the
same box with one familiar (yellow circle) and a novel object (red hexagon).
2.3.5 Motor Function Assessment by Accelerating Rotarod
Motor performance was evaluated using an accelerating rotarod (Model 7650, Ugo
Basile, Collegeville, PA). Mice were first trained for two days before the 1st TBI to walk
on accelerating rotarod (0 to 30 rpm in 210 s). The average of the last two trials of the
second training day was used to establish baseline failing latency. Unless the animal
was scheduled for sacrifice, rotarod latencies were repeatedly assessed at 1, 2, 7, and
14 d post-mrTBI. At each test day, every mouse underwent two accelerating speed
trials (0 to 30 rpm in 210 s) separated by an inter-trial interval of at least 15 min. The
animal was recorded as failing during the 210 s period if: (1) the animal fell completely
off the rotating rod before 210 s, or (2) the animal gripped the rod and made one
complete passive revolution without actively walking on the rod. The average failing
latency was calculated from the two trials for each animal.
2.3.6 Biochemical Analyses
122

2.3.6.1 Tissue Collection
Mice were anesthetized with an i.p. injection of 150 mg/kg ketamine (Bimeda-MTC) and
20 mg/kg xylazine (Bayer) and transcardially perfused with ice-cold phosphate-buffered
saline (PBS) containing heparin (0.5 units/mL) for 7 min. For biochemistry, brains were
longitudinally bisected and the ipsilateral half-brains were rapidly frozen over dry ice and
stored at -80°C until analysis (WT mice N: injured = 10/group/time point, sham = 6/time
point; apoE-/- mice N: injured = 5/group/time point, sham = 4/time point). For histology,
anesthetized mice were perfused with heparinized PBS and whole brains were
immersion-fixed in 10% neutral buffered formalin for 48 h followed by cryoprotection in
30% sucrose in PBS at 4°C for additional two days (N: injured = 5/treatment group/time
point/genotype, sham = 2/time point/genotype).
2.3.6.2 Protein Extraction
Frozen ipsilateral half-brains were thawed over ice and homogenized in 1.5 mL of ice-
cold RIPA lysis buffer (5mM EDTA, 50mM NaCl, 10mM sodium pyrophosphate, 50mM
NaF, and 1% NP40 alternative, pH: 7.4) containing complete protease inhibitor cocktail
(Roche Applied Science) with a Tissuemite homogenizer at full speed for 20 s. The
homogenate was sonicated at 20% output for 10 s followed by centrifugation at 4°C for
10 min at 9,000 rpm in a microcentrifuge (Eppendorf). The supernatant was separated
and used for analysis of ABCA1, apoE, APP, APP-CTF, LDLR, IL-6, TNF-α, MCP-1,
Aβ40, and Aβ42. Aliquots of all lysates were immediately frozen at –80°C until analysis.
Protein concentration was determined by DC Protein assay (Bio-Rad, Hercules, CA).
123
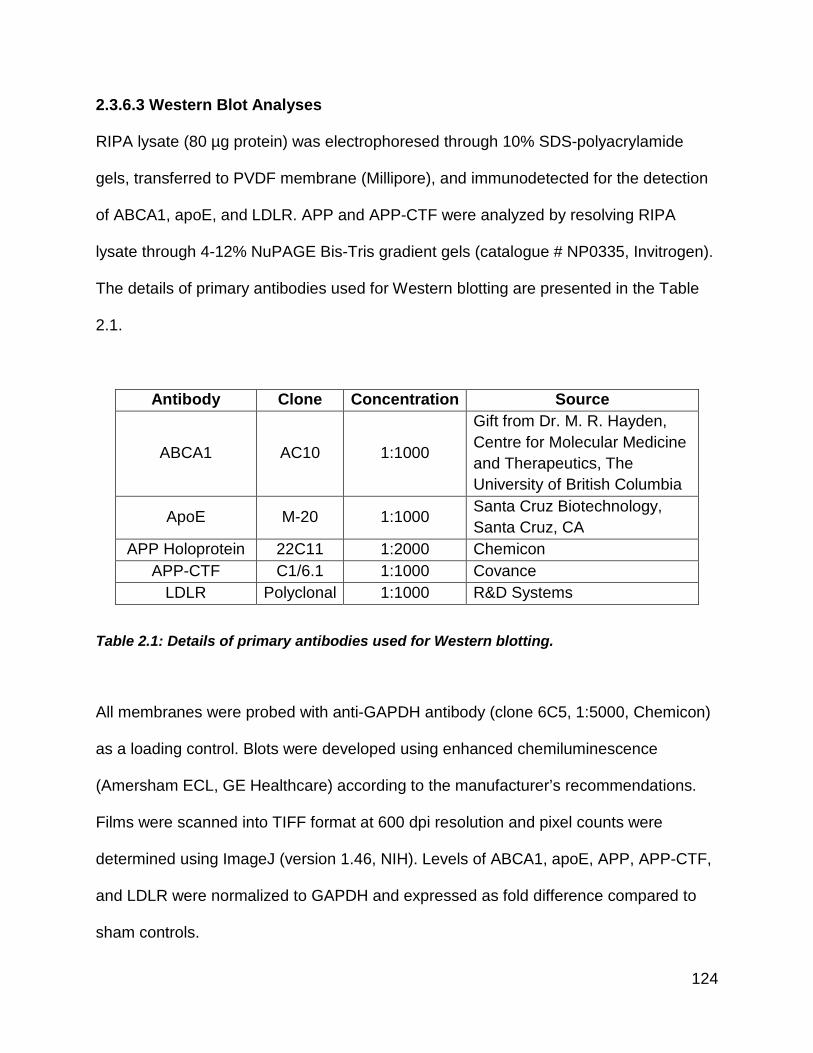
2.3.6.3 Western Blot Analyses
RIPA lysate (80 µg protein) was electrophoresed through 10% SDS-polyacrylamide
gels, transferred to PVDF membrane (Millipore), and immunodetected for the detection
of ABCA1, apoE, and LDLR. APP and APP-CTF were analyzed by resolving RIPA
lysate through 4-12% NuPAGE Bis-Tris gradient gels (catalogue # NP0335, Invitrogen).
The details of primary antibodies used for Western blotting are presented in the Table
2.1.
Antibody Clone Concentration Source
ABCA1 AC10 1:1000
Gift from Dr. M. R. Hayden, Centre for Molecular Medicine and Therapeutics, The University of British Columbia
ApoE M-20 1:1000 Santa Cruz Biotechnology, Santa Cruz, CA
APP Holoprotein 22C11 1:2000 Chemicon APP-CTF C1/6.1 1:1000 Covance
LDLR Polyclonal 1:1000 R&D Systems
Table 2.1: Details of primary antibodies used for Western blotting.
All membranes were probed with anti-GAPDH antibody (clone 6C5, 1:5000, Chemicon)
as a loading control. Blots were developed using enhanced chemiluminescence
(Amersham ECL, GE Healthcare) according to the manufacturer’s recommendations.
Films were scanned into TIFF format at 600 dpi resolution and pixel counts were
determined using ImageJ (version 1.46, NIH). Levels of ABCA1, apoE, APP, APP-CTF,
and LDLR were normalized to GAPDH and expressed as fold difference compared to
sham controls.
124

2.3.6.4 Quantitative Assessment of Endogenous Aβ by ELISA
Endogenous Aβ40 and Aβ42 levels in ipsilateral half-brain lysates were quantified by
ELISA (KMB3481 and KMB3431, Invitrogen) following the manufacturer’s instructions.
Levels of soluble Aβ40 and Aβ42 were normalized to total protein.
2.3.6.5 Quantitative Assessment of IL-6, TNFα, and MCP-1 by ELISA
IL-6, TNFα and monocyte chemotactic protein-1 (MCP-1) levels in ipsilateral half-brain
lysates were determined by ELISA. Briefly, 96-well plates were coated and incubated
overnight at 4°C with primary antibody against murine IL-6, TNFα, and MCP-1. Brain
lysates were incubated with primary antibody at room temperature for 1 h. Following
washing, samples were incubated with biotinylated secondary antibodies at room
temperature for 1h. All the primary and secondary antibodies were obtained from
eBioscience. The details of primary and secondary antibodies are presented in the
Table 2.2. Samples were developed with avidin-horseradish peroxidase (eBioscience)
and TMB substrate.
Cytokine Primary Antibody Secondary Antibody Clone Concentration Clone Concentration
IL-6 MP5-20F3 1:1000 MP5-32C11 1:1000 TNFα TN3-19.12 1:1000 Polyclonal 1:1000,
125

(Catalogue # 13-7341) MCP-1 4E2 1:1000 2H5 1:1000
Table 2.2: Details of primary and secondary antibodies used for cytokine ELISA.
2.3.7 Histology
Brains were cut into coronal sections (40 µm) on a cryotome and all sections starting
from the genu of corpus callosum (~1.1 mm anterior to bregma) to the posterior
hippocampus (~3.3 mm posterior to Bregma) were collected in 1X PBS with sodium
azide.
2.3.7.1 Assessment of Axonal Injury by Silver Staining
Three to four coronal brain sections, separated by 400 µm, from the bregma to the
posterior hippocampus, were stained using FD NeuroSilver Kit II (FD
NeuroTechnologies Inc, Columbia, MD) according to the manufacturer’s protocol, which
is based on the method originally described by Gallyas et al (1990). The manufacturer’s
protocol was further optimized with the following modifications: 1) Sections were stored
in 4% paraformaldehyde in saline-free phosphate buffer at 4°C for a minimum of 5 d
prior to staining. 2) Step 4 was decreased from 2x2 min incubations to 1x2 min
incubation to increase staining intensity. 3) Steps 3, 4, and 5 were performed with
vigorous shaking. From each section, 40X-magnified images of ipsilateral corpus
callosum, cingulum, external capsule, internal capsule, and optic tracts were captured
with a Zeiss Axio Imager A1 microscope attached to an Olympus DP72 camera and
digitized using cellSens® Standard digital imaging software (version 1.4.1, Build 8624,
OLYMPUS). A semiquantitative scale was developed to score staining intensity, ranging
126

from 0 (0 to <10% staining) to 3 (> 70% staining). The scale was based on the extent
and intensity of argyrophilic structures (cell bodies and axonal fibers) within an image
field. Every image was independently scored by two trained raters who were blinded to
the injury, genotype, and treatment status. Scores from the two raters were averaged. A
minimum of 3 sections per brain were scored and combined to define the average silver
stain score of each brain region. The silver staining was carried out on samples
containing 5 injured brains/genotype/time point/group and 2 sham brains/genotype/time
point.
2.3.7.2 Assessment of Microglial Activation by Iba1 Immunohistochemistry
Microglial activation induced by mrTBI was assessed with Iba1 immunohistochemistry.
Briefly, 3-4 coronal brain sections, separated by 400 µm, starting from the bregma to the
posterior hippocampus were washed in Tris-buffered saline (TBS), treated with 0.3%
hydrogen peroxide for 10 min and blocked with 3% normal goat serum (NGS) in TBS
containing 0.25% Triton-X (TBS-X) for 30 min. Sections were incubated overnight in
TBS-X containing anti-Iba1 polyclonal antibody (Catalogue # 019-19741, 1:1000, Wako
Chemicals, Richmond, VA) and 1% NGS. Following washing, sections were incubated
in biotinylated goat-anti-rabbit secondary antibody (1:1000 in TBS-X) for 1 h. Sections
were visualized with horseradish peroxidase (Vectastain Elite ABC Kit PK-6120, Vector
Laboratories (Canada) Inc, Burlington, ON) and DAB substrate. Sections were mounted
on Superfrost® Plus (Fisher Scientific) slides, dehydrated in ascending concentrations
(50, 70, 90, and 95%) of alcohol, cleared with xylene, and coverslipped in DPX
127
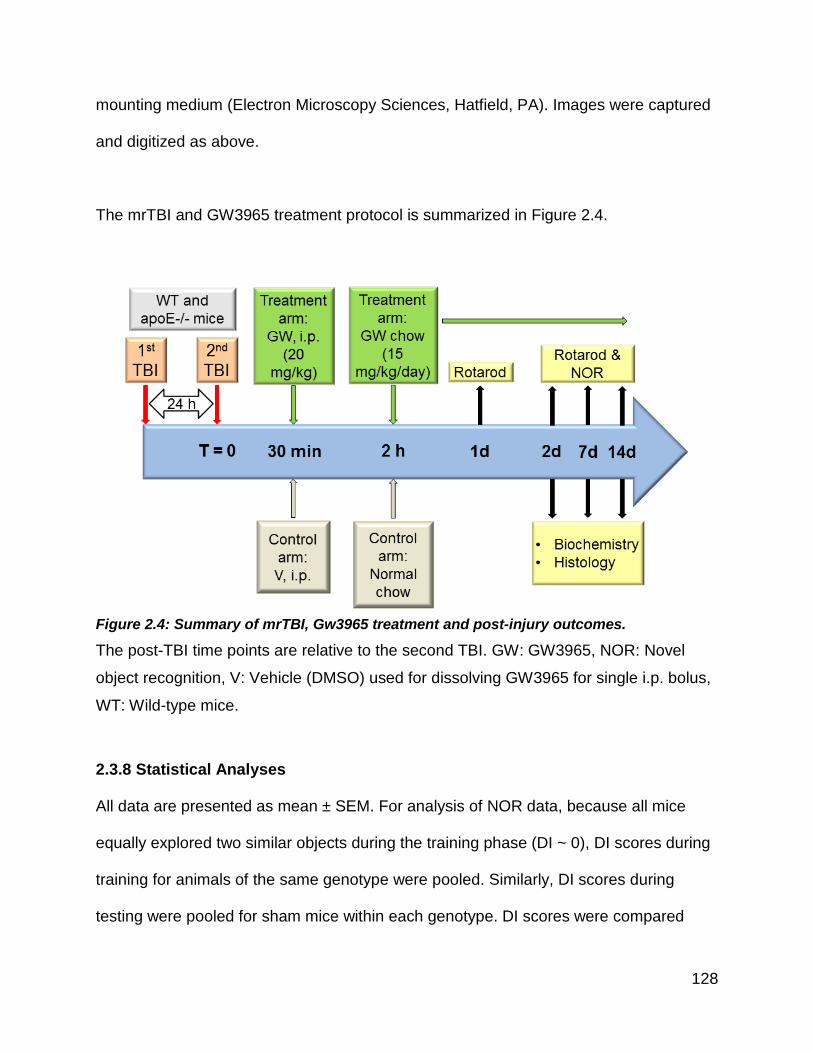
mounting medium (Electron Microscopy Sciences, Hatfield, PA). Images were captured
and digitized as above.
The mrTBI and GW3965 treatment protocol is summarized in Figure 2.4.
Figure 2.4: Summary of mrTBI, Gw3965 treatment and post-injury outcomes.
The post-TBI time points are relative to the second TBI. GW: GW3965, NOR: Novel
object recognition, V: Vehicle (DMSO) used for dissolving GW3965 for single i.p. bolus,
WT: Wild-type mice.
2.3.8 Statistical Analyses
All data are presented as mean ± SEM. For analysis of NOR data, because all mice
equally explored two similar objects during the training phase (DI ~ 0), DI scores during
training for animals of the same genotype were pooled. Similarly, DI scores during
testing were pooled for sham mice within each genotype. DI scores were compared
128

using two-way ANOVA followed by a Bonferroni post-hoc test. For rotarod analysis,
baseline latencies of mice from all time points within genotype and treatment groups
were pooled. Rotarod data were analyzed for time and treatment as well as time and
genotype effects using two-way repeated measures ANOVA followed by a Bonferroni
post-hoc test, as animals were tested repeatedly until sacrifice. For biochemical and
histological analyses, data from sham mice from all time points within each genotype
were pooled. Biochemistry and silver stain data were analyzed for time and treatment
as well as for time and genotype effects using two-way ANOVA followed by a Bonferroni
post-hoc test. A p value of < 0.05 was considered significant. All statistical analyses
were performed using GraphPad Prism (version 5.04, GraphPad Software Inc).
2.4 Results
2.4.1 ApoE is Required for GW3965 to Improve Cognitive Function After mrTBI
Non-spatial, working memory was assessed using the novel object recognition (NOR)
task at 2, 7, and 14 days following mrTBI. Discrimination between the novel and familiar
objects was determined using the discrimination index (DI) (Antunes and Biala, 2012).
During training, all animals displayed an equivalent time exploring each of the two
identical objects (DI ~ 0) (Figure 2.5) indicating that genotype, surgical procedures,
treatment, and time did not affect baseline exploratory behavior. Sham-operated WT
(Figure 2.5 A, gray bar, p < 0.001 compared to training, two-way ANOVA) and apoE-/-
(Figure 2.5 B, gray bar, p < 0.01 compared to training, two-way ANOVA) mice retained
preference to novelty as indicated by positive DI values. Untreated WT (Figure 2.5 A,
open bars) and apoE-/- (Figure 2.5 B, open bars) failed to discriminate between the
129

novel and familiar objects (p > 0.05 compared to training, two-way ANOVA) at all post-
mrTBI time points. Although GW3965-treated WT mice showed impaired memory at 2d
post-mrTBI (Figure 2.5 A, black bars, p > 0.05 compared to training, two-way ANOVA)
object recognition was restored by day 7 and 14 post-mrTBI (p < 0.001 compared to
training, two-way ANOVA). Finally, GW3965 treatment failed to improve NOR
performance in apoE-/- mice at any post-mrTBI time points (Figure 2.5 B, black bars, p
> 0.05 compared to training, two-way ANOVA). Gross motor impairment can affect NOR
performance. To rule out this possibility, we assessed the spontaneous activity of the
animal in the open field by comparing the total distance covered by the animal in the 5
min epochs of training and testing phases. We found that the total path length covered
by sham and injured WT as well as apoE-/- mice were not significantly different
indicating no gross motor impairment (Figure 2.6, p > 0.05, two-way ANOVA).
130

Figure 2.5: ApoE is required for GW3965 to improve NOR performance after mrTBI.
NOR memory was evaluated in untreated (V) and GW3965-treated (G) WT (A) and
apoE -/- (B) mice at 2, 7, and 14 days post-mrTBI. Bars represent discrimination index
(DI) scores. Cohorts were: sham (WT, n=23, pooled; apoE-/-, n=18, pooled) and 2 day
(WT: untreated, n=18, GW3965-treated, n= 15; apoE-/-: untreated, n=10, GW3965-
treated, n=10), 7 day (WT: untreated, n=13, GW3965-treated, n=10; apoE-/-: untreated,
n=11, GW3965-treated, n=11), and 14 day (WT: untreated, n=14, GW3965-treated,
131

n=14; apoE-/-: untreated, n=9, GW3965-treated, n=9) post-mrTBI. DI scores were
analyzed using two-way ANOVA and Bonferroni post hoc test. **: p<0.01, ***: p<0.001,
###: p<0.001.
Figure 2.6: NOR performance was not affected by motor impairment.
To assess whether NOR performance was affected by motor impairment; the total path
length (m) covered by WT and apoE-/- mice during testing and training was measured.
(A, B), total path length covered by WT mice during training and testing, respectively.
(C, D), total path length covered by apoE-/- mice during training and testing,
respectively. The path lengths covered by WT and apoE-/- mice were not significantly
different from sham animals during testing, indicating that NOR was not affected by
motor impairment. *: p<0.05 and **: p<0.01. Data were analyzed by two-way ANOVA
followed by a Bonferroni post hoc test. Legend: V- untreated mice, open bars, G-
GW3965-treated mice, black bars.
132

2.4.2 Spontaneous Recovery of Motor Dysfunction After mrTBI is Unaffected by
GW3965
We next assessed post-mrTBI fine motor functions with accelerating rotarod. mrTBI
significantly impaired the motor performance of WT (Figure 2.7 A, p < 0.05, two-way
repeated measures ANOVA) and apoE-/- (Figure 2.7 B, p < 0.05, two-way repeated
measures ANOVA) mice at 1, 2, and 7 d post-injury as indicated by reduced failing
latencies on an accelerating rotarod, compared to the baseline performance. By 14 d,
however, motor performance had fully recovered in WT and apoE-/- animals (Figure 2.7
A and B, p > 0.05, two-way repeated measures ANOVA). GW3965 treatment of WT or
apoE-/- mice did not significantly reduce the severity of motor impairment at any time
point, nor did it accelerate the rate of spontaneous recovery in either genotype (Figure
2.7 A and B, curly brackets). In contrast to GW3965, loss of apoE-/- led to significantly
more severe motor impairment compared to WT animals at 1 and 2 d post-mrTBI in
both untreated and treated groups (Figure 2.7 C and D, p < 0.05, two-way repeated
measures ANOVA).
133

Figure 2.7: mrTBI-induced motor impairment recovers spontaneously independent of GW3965 and apoE.
Motor performance of WT (n=38) and apoE-/- (n=30) mice was evaluated by the
accelerating rotarod task (0 to 30 rpm in 210 s). (A) Rotarod latencies of untreated (V,
open squares) and GW3965-treated (G, black filled squares) WT mice before and
following mrTBI. (B) Rotarod latencies of untreated (V, open circles) and GW3965-
treated (G, black filled circles) apoE-/- mice before and following mrTBI. Asterisks in A
and B denote significant differences between baseline and post-mrTBI latencies within
each group. For both genotypes, the treatment effect was not significant at any post-TBI
time point (curly brackets). (C) Rotarod latencies of untreated WT (open squares) and
apoE-/- (open circles) mice before and following mrTBI. (D) Rotarod latencies of
GW3965-treated WT (black filled squares) and apoE-/- (black filled circles) mice before
and following mrTBI. # and § in C and D denote significant differences between the
latencies of WT and apoE-/- mice at the respective time points. Cohorts were: Untreated
WT mice: (1 d=34, 2 d=34, 7 d=24; 14 d=14); GW3965-treated WT mice: (1 d=35, 2
d=35, 7 d=25, 14 d=14); untreated apoE-/- mice: (1 d=24, 2 d=24, 7 d=20, 14 d=9), and
134

GW3965-treated apoE-/- mice: (1 d=27, 2 d=22, 7 d=20, 14 d=9). Data were analyzed
using two-way repeated measures ANOVA followed by a Bonferroni post hoc test. *:
p<0.05, **: p<0.01, ***: p<0.001, ****: p<0.0001, #: p<0.05, ##: p<0.01, §§: p<0.01.
2.4.3 GW3965 Prevents mrTBI-Induced Elevation of Endogenous Aβ Levels
We determined levels of soluble endogenous Aβ40 and Aβ42 species from ipsilateral
half brains of WT and apoE-/- mice at 2, 7, and 14 d after mrTBI. Compared to sham
controls, injured WT mice showed a sustained ~1.5-fold increase in Aβ40 (Figure 2.8 A,
open bars, p < 0.05, two-way ANOVA), and a transient ~1.7 fold increase in Aβ42 that
returned to baseline by 14 d post-injury (Figure 2.8 B, open bars, p < 0.05, two-way
ANOVA). In contrast to untreated controls, GW3965 suppressed the increase of both
Aβ40 and Aβ42 in WT mice (Figure 2.8 A and B, black bars, p > 0.05, two-way ANOVA)
as compared to the sham. Similarly, apoE-/- mice exhibited a sustained ~1.5-fold
increase in Aβ40 (Figure 2.8 C, open bars, p < 0.05, two-way ANOVA), and a transient
~1.4-fold increase in Aβ42 after mrTBI that returned to baseline by 14 d post-injury
(Figure 2.8 D, open bars, p < 0.05, two-way ANOVA). Surprisingly, GW3965 effectively
suppressed the increase in both Aβ40 and Aβ42 in apoE-/- mice (Figure 2.8 C and D,
black bars, p > 0.05, two-way ANOVA) as compared to the sham. The GW3965
treatment effect was significant in both, WT and apoE-/- mice for Aβ40 (Figure 2.8 A
and C, p < 0.05, two-way ANOVA), but only in WT mice for Aβ42 (Figure 2.8 B, p <
0.05, two-way ANOVA). Furthermore, compared to WT mice, loss of apoE did not lead
to significantly greater accumulation of Aβ species, with the single exception of Aβ40
levels 7 d post-mrTBI in both vehicle (Figure 2.8 A and C, #, p < 0.05, two-way ANOVA)
and GW3965-treated groups (Figure 2.8 A and C, §, p < 0.05, two-way ANOVA). APP
135

holoprotein and APP-CTFα levels remained unchanged in both untreated and treated
WT and apoE-/- mice following mrTBI (Figure 2.9, p > 0.05, two-way ANOVA),
indicating that increased APP processing does not account for the changes in Aβ levels.
136

Figure 2.8: GW3965 prevents mrTBI-induced accumulation of endogenous Aβ in WT and apoE-/- mice.
Total, soluble murine Aβ40 and Aβ42 levels were measured from ipsilateral half brains of
WT (A, B) and apoE-/- (C, D) mice, respectively, at 2, 7, and 14 d post-mrTBI. Data
from sham animals within each genotype were pooled (grey bars). Numbers inside the
bars indicate sample size. Asterisks above individual bars indicate significant difference
compared to the respective sham levels. *: p<0.05, **: p<0.01, ***: p<0.001, ****:
p<0.0001. # indicates a significant difference (p < 0.05) between Aβ40 levels of
untreated WT and apoE-/- brains at 7 d post-mrTBI. § indicates a significant difference
(p < 0.05) between Aβ40 levels of GW3965-treated WT and apoE-/- brains at 7 d post-
mrTBI. Data were analyzed by two-way ANOVA followed by a Bonferroni post hoc test.
137

Legend: S: sham-operated mice, gray bars, V: untreated mice, open bars, G: GW3965-
treated mice, black bars.
Figure 2.9: APP and APP-CTFα levels remain unchanged following mrTBI.
APP holoprotein (A, C) and APP-CTFα (B, D) protein levels in ipsilateral half brains
were analyzed by Western blotting, with representative blots shown for WT (A, B) and
apoE-/- (C, D) mice. Data are expressed as fold difference relative to sham values. Data
from sham animals within each genotype were pooled (grey bars). Numbers inside bars
138

indicate sample size. Data were analyzed by two-way ANOVA followed by a Bonferroni
post hoc test. Legend: S: sham-operated mice, gray bars, V: untreated mice, open bars,
G: GW3965-treated mice, black bars.
2.4.4 GW3965 Enhances ABCA1 Induction Aafter mrTBI:
To determine whether lipid mobilization pathways are induced in our mrTBI model, we
determined the levels of ABCA1, apoE, and LDLR in treated and untreated WT and
apoE-/- mice. Compared to sham controls, untreated WT brains showed a transient 1.6-
fold increase in ABCA1 levels on post-mrTBI day 7 (Figure 2.10 A, open bars, p < 0.01,
two-way ANOVA) that returned to baseline by 14 d. This finding is consistent to the
findings reported in a FP injury model (Cartagena et al., 2008) and suggests that
endogenous LXR agonists may be transiently induced after TBI. As ABCA1 is a
sensitive LXR target, GW3965 augmented this response, leading to a 1.5-2.4-fold
increase in ABCA1 protein levels over the time points examined (Figure 2.10 A, black
bars, p < 0.05, two-way ANOVA). These results are consistent with our previous
observations of ABCA1 upregulation in GW3965-treated APP/PS1 mice (Donkin et al.,
2010). In contrast to WT animals, untreated apoE-/- mice did not show a transient
elevation of ABCA1 after mrTBI (Figure 2.10 B, open bars, p > 0.05, two-way ANOVA),
suggesting that the endogenous signals that lead to ABCA1 induction after TBI may be
compromised in the absence of apoE. Nevertheless, similar to the WT mice GW3965
treatment significantly increased ABCA1 levels in injured apoE-/- mice (Figure 2.10 B,
black bars, p < 0.05, two-way ANOVA), suggesting that GW3965 may bypass apoE and
directly increase ABCA1 expression after mrTBI. Neither mrTBI nor GW3965 treatment
led to significant changes in apoE levels in WT mice (Figure 2.11 A, p > 0.05, two-way
139

ANOVA), a result consistent with the relative insensitivity of apoE compared to ABCA1
as a LXR target (Koldamova et al., 2005b; Zelcer et al., 2007; Donkin et al., 2010).
Similar to apoE, LDLR levels remained unaffected after mrTBI in both treated and
untreated WT (Figure 2.11 B) and apoE-/- (Figure 2.11 C) mice (p > 0.05, two-way
ANOVA).
Figure 2. 10: GW3965 augments ABCA1 levels in WT and apoE-/- mice following mrTBI.
ABCA1 protein levels were determined in ipsilateral half brains of WT (A) and apoE-/-
(B) mice following mrTBI using Western blots, with representative blots shown below
the graphs. Data are expressed as fold difference normalized to sham values. Data
from sham animals within each genotype were pooled (grey bars). Numbers inside the
bars indicate sample size. Asterisks above individual bars indicate significant difference
compared to the respective sham levels. *: p<0.05, **: p<0.01, ***: p<0.001, ****:
p<0.0001. Data were analyzed by two-way ANOVA followed by a Bonferroni post hoc
test. Legend: S: sham V: untreated mice, open bars, G: GW3965-treated mice, black
bars.
140

Figure 2.11: ApoE and LDLR levels are unaffected by mrTBI or GW3965.
Levels of apoE and LDLR protein in WT mice (A, B) and LDLR protein in apoE-/- mice
(C) were determined in ipsilateral half brains following mrTBI using Western blotting,
with representative blots shown on the right. Data are expressed as fold difference
normalized to sham values. Data from sham animals within each genotype were pooled.
Numbers inside bars indicate sample size. Data were analyzed by two-way ANOVA
followed by Bonferroni post hoc test. Legend: S: sham-operated mice, gray bars, V:
untreated mice, G: GW3965-treated mice.
141

2.4.5 ApoE is Required for GW3965-Mediated Suppression of Axonal Damage
After mrTBI
Histological assessment of axonal damage using silver staining revealed argyrophilic
structures in cell bodies and axons in several ipsilateral white matter regions, including
the corpus callosum, cingulum, external and internal capsules, and optic tracts, with
strikingly greater accumulation observed in apoE-/- mice (Figures 2.12, 2.13, and 2.14
arrows). Notably, intense argyrophilia was observed in the bilateral optic tracts of WT
and apoE-/- mice, suggesting the involvement of strong coup-contrecoup forces in our
model. In comparison, very mild silver staining was observed in grey matter regions
including the sensory-motor cortex directly under the impact site as well as the piriform
cortex, farthest from the impact site (not shown).
142

Figure 2.12: Loss of apoE exacerbates axonal injury after mrTBI.
Axonal damage following mrTBI was assessed with silver staining (arrows). The left
panel depicts representative images of silver-stained coronal sections at approximately
-1.58 mm from bregma (Paxinos and Franklin, 2001) of untreated WT (A) and apoE-/-
(B) mouse brains harvested at 7 d post-mrTBI. White matter areas with prominent silver
staining are indicated by the black squares. Injury location is indicated by the
arrowhead. The right panel (C-L) depicts representative 40x-magnified images of silver
staining in five white matter areas in the brains of untreated WT (C-G) and apoE-/- (H-L) mice harvested at 7 d post-mrTBI.
143

Figure 2.13: mrTBI leads to mild axonal damage in WT mice.
Axonal damage following mrTBI was assessed with silver staining. The far left column
depicts representative images of silver-stained coronal sections at approximately -1.82
mm from bregma (Paxinos and Franklin, 2001) of sham (A), untreated (B), and
GW3965-treated (C) WT brains harvested at 2 d post-mrTBI. Locations of the white
matter areas where silver staining was most prominent are indicated by black squares.
Injury location is indicated by the arrowhead. Columns on the right depict representative
40X-magnified images of silver staining in five white matter areas in sham-operated (D-H), untreated TBI (I-M), and GW3965-treated (N-R) brains of WT mice harvested at 2 d
post-mrTBI.
144

Figure 2.14: Loss of apoE exacerbates axonal damage after mrTBI.
Axonal damage following mrTBI was assessed with silver staining (arrows). The far left
column depicts representative images of silver-stained coronal sections at
approximately -1.82 mm from bregma (Paxinos and Franklin, 2001) of sham (A), untreated (B), and GW3965-treated (C) apoE-/- brains harvested at 7 d post-mrTBI.
Locations of the white matter areas where silver staining was most prominent are
indicated by black squares. Injury location is indicated by the arrowhead. The columns
on the right depict representative 40X-magnified images of silver staining in five white
matter areas in sham-operated (D-H), untreated (I-M), and GW3965-treated (N-R) brains of apoE-/- mice harvested at 7 d post-mrTBI.
145

In untreated WT mice, axonal damage was significantly elevated in the corpus
callosum, cingulum, external capsule, and internal capsule at 2 d post mrTBI as
revealed by the semiquantitative analysis of silver-stained images (Figure 2.15 A-D,
open bars, p < 0.05, two-way ANOVA). In this group, silver staining intensity returned to
baseline by 7 d, suggesting that endogenous neuronal repair pathways were efficiently
activated in these regions. More robust and sustained axonal damage was observed in
the optic tracts of untreated WT mice (Figure 2.15 E, open bars, p < 0.05, two-way
ANOVA). In WT mice, GW3965 effectively suppressed axonal damage in all regions
examined (Figure 2.15 A-E, black bars, p < 0.05, two-way ANOVA). Untreated apoE-/-
mice showed extensive axonal damage that peaked at 7 d post-injury and remained
elevated at 14 d in the corpus callosum, external and internal capsules, and optic tract
(Figure 2.15 F-J, open bars, p < 0.05, two-way ANOVA). In contrast to WT mice,
GW3965 failed to suppress axonal damage in apoE-/- animals (Figure 2.15 F-J, black
bars, p < 0.05, two-way ANOVA). In all regions examined, axonal damage was
significantly more extensive in apoE-/- mice compared to WT controls (Figure 2.15 F-J,
p < 0.05, two-way ANOVA; # and § denote comparison of WT and apoE-/- mice within
untreated and treated groups, respectively).
146

Figure 2.15: ApoE is required for GW3965 to suppress axonal damage after mrTBI.
Silver-stained images of white matter areas in WT and apoE-/- brains were analyzed
semiquantitatively using an arbitrary silver staining scale extending from 0 (<10%
argyrophilic structures covering the image field) to 3 (>70% argyrophilic structures
covering the image field). The bar graphs represent mean ± SEM silver stain intensity
score (arbitrary value) of WT (A-E) and apoE-/- (F-J) brains in the corpus callosum (A,
147

F), cingulum (B, G), external capsule (C, H), internal capsule (D, I), and optic tracts (E, J). Sample sizes were: sham, n=6/genotype (pooled); untreated mrTBI, n=5/time
point/genotype; GW3965-treated mrTBI, n=5/time point/genotype. Asterisks above
individual bars indicate significant differences compared to the respective sham values
(grey bars). *: p<0.05, **: p<0.01, ***: p<0.001, ****: p<0.0001. ### (p<0.001) and ####
(p<0.0001) represent significant differences in silver stain scores between untreated WT
and apoE-/- mice. § (p<0.05), §§ (p<0.01), §§§ (p<0.001), and §§§§ (p<0.0001)
represent significant differences in silver stain scores between GW3965-treated WT and
apoE-/- mice. Data were analyzed by two-way ANOVA followed by a Bonferroni post
hoc test. Legend: V- untreated mice, open bars, G- GW3965-treated mice, black bars.
2.4.6 Weight Drop TBI Model Produces Negligible Neuroinflammation
To investigate the extent to which our mrTBI model induces inflammation, we examined
post-injury microglial activation using Iba1 immunohistochemistry and measured IL-6,
TNFα, and MCP-1 levels in ipsilateral half brains. Examination of coronal sections
revealed uniform distribution of Iba1-immunoreactive microglia with negligible changes
in the immunoreactivity in the injured WT and apoE-/- mice in either the ipsilateral cortex
(Figure 2.16) or hippocampus (Figure 2.17) compared to sham controls. Further,
GW3965 treatment did not reduce Iba1 staining below baseline (Figure 2.16 and 2.17).
148
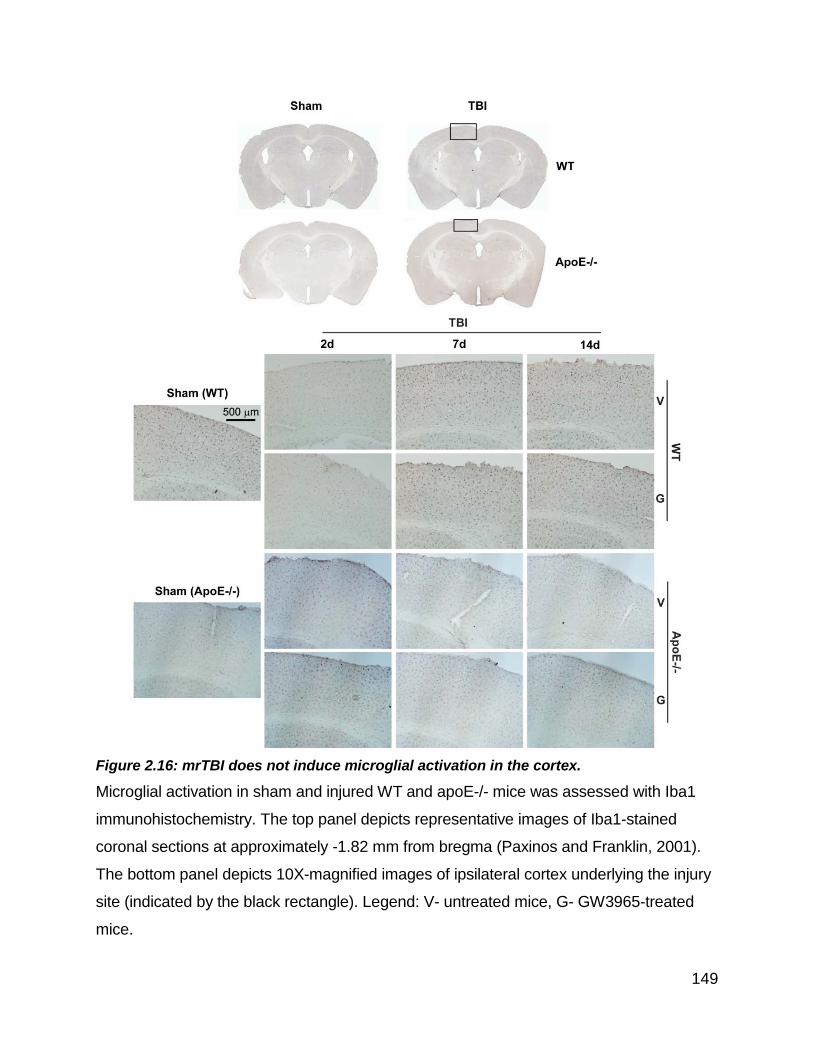
Figure 2.16: mrTBI does not induce microglial activation in the cortex.
Microglial activation in sham and injured WT and apoE-/- mice was assessed with Iba1
immunohistochemistry. The top panel depicts representative images of Iba1-stained
coronal sections at approximately -1.82 mm from bregma (Paxinos and Franklin, 2001).
The bottom panel depicts 10X-magnified images of ipsilateral cortex underlying the injury
site (indicated by the black rectangle). Legend: V- untreated mice, G- GW3965-treated
mice.
149

Figure 2.17: mrTBI induces negligible hippocampal microglial activation.
Microglial activation in sham and injured WT and apoE-/- hippocampi was assessed
with Iba1 immunohistochemistry. The top panel depicts representative images of Iba1-
stained coronal sections at approximately -1.82 mm from bregma (Paxinos and
Franklin, 2001). The bottom panel depicts 10X-magnified images ipsilateral
150

hippocampus (indicated by black rectangle). Legend: V- untreated mice, G- GW3965-
treated mice.
Although no animal in this study had any obvious skull fracture, approximately 4-8% of
mice showed evidence of microcontusions in the cortex directly under the impact site
(Figure 2.18, arrows). Increased Iba1 immunoreactivity was clearly associated with
these localized areas of more severe injury (Figure 2.18).
Figure 2.18: Pronounced microglial activation is localized only around contused areas.
In this study, approximately 4-8% of brains subjected to mrTBI showed micro contusions
in the absence of gross skull fracture. The left panel shows a Iba1-stained coronal section
at approximately -1.82 mm from bregma (Paxinos and Franklin, 2001) with a micro
contusion in the cortex below impact site (black square). The right panel shows
representative 10X-magnified images of Iba1-stained untreated WT and apoE-/- contused
cortices. Microcontusions are denoted by black arrows. Note the pronounced localized
activation of microglia around the contusion.
Consistent with this model inducing negligible microglial activation, IL-6 levels in the
ipsilateral brains were unchanged in both WT and apoE-/- mice, with or without
151

GW3965 (Figure 2.19, p > 0.05, two-way ANOVA). TNFα and MCP-1 levels were below
the detection limit (not shown).
Figure 2.19: Pronounced microglial activation is localized only around contused areas.
Interleukin- 6 (IL-6) levels were measured from ipsilateral half brains of WT and apoE-/- mice harvested at 2, 7, and 14 d post-mrTBI. Data from sham animals within each
genotype were pooled. Cohorts were: sham (WT, n=9, pooled; apoE-/-, n=10, pooled), 2
day (WT: untreated, n=5, GW3965-treated, n=5 ; apoE-/-: untreated, n=5, GW3965-
treated, n=5), 7 day (WT: untreated, n=4, GW3965-treated, n=4; apoE-/-: untreated, n=6,
GW3965-treated, n=6), and 14 day (WT: untreated, n=5, GW3965-treated, n=5; apoE-/-:
untreated, n=5, GW3965-treated, n=5) post-mrTBI. Data were analyzed by two-way
ANOVA followed by a Bonferroni post hoc test. Legend: Sham WT: open bars, sham
apoE-/-: grey bars, vehicle-treated WT: open striped bars, vehicle-treated apoE-/-: grey
striped bars, GW3965-treated WT: open stippled bars, GW3965-treated apoE-/-: grey
stippled bars.
2.4.7 Retrospective Power Analysis
In this study we did not perform prospective power calculations to determine the sample
size. A retrospective power analysis was thus carried out based on the collected data
152

using an online post-hoc power calculator (http://clincalc.com/stats/power.aspx). For
power calculations it was assumed that the post-TBI outcomes in apoE-/- mice will be
worse compared to those in the WT mice, hence any significant change in the WT mice
will likely indicate similar or more change in apoE-/- mice. In this study we there were
four factors that could influence the study outcome: genotype, injury status, treatment
and time point. Of these we hypothesized that the smallest effect will be of TBI and TBI
x treatment interaction in WT mice and thus a significant difference for these
interactions will likely indicate significant results for the other group comparisons. Based
on these assumptions the retrospective power analysis was carried for TBI effect and
treatment x TBI effect at 7 d time point.
The retrospective power analysis revealed that the study was adequately powered to
detect TBI effect in WT mice and was underpowered to detect treatment x TBI effect
although all the observed changes were statistically significant.
The results of power calculations for various post-TBI outcomes are summarized in the
Table 2.3.
Post-TBI Outcome
Time Point
TBI Effect Treatment x TBI Effect Observed Change
(%) N Power
Observed Change
(%) N Power
NOR 7d - - - 47 (Observed) 13 5.4
153

Rotarod WT 7d 10 24 73.7% 10
(Estimated) 24 9.6% (4.8%
change) Rotarod ApoE-/- 7d 16 20 95.5% 16
(Estimated) 20 5.4%
ABCA1 (WT) 7d 55 8 96.6% 71 8 25.8%
ABCA1 (ApoE-/-) 7d 26 5 29.4 68 5 63
Abeta 7d 15 12 99.1 15 8 95 Silver Stain
(WT-CC) 2d 100 5 83.4 50 5 93.8
Silver Stain
(ApoE-/-CC)
2d 107 2 84.3 50 5 4.7
Table 2.3: Summary of retrospective power calculations
The table summarizes results of retrospective power calculations for various post-TBI
outcomes that were assessed in the present study. All the observed changes reported
in this table were statistically significant.
2.5 Discussion
The goal of this study was to evaluate the ability of GW3965 to promote recovery in a
model of mrTBI specifically designed to mimic repeated concussion. We found that
therapeutic administration of GW3965 improved NOR performance, suppressed Aβ
accumulation, and reduced axonal damage after mrTBI. Loss of apoE exacerbated the
severity of motor impairment and axonal damage and eliminated the ability of GW3965
to restore NOR performance and to promote axonal recovery. These results are
consistent with the role of apoE in neuronal repair and synaptic restoration. ApoE levels
did not change after TBI or after GW3965 treatment, which suggests that injury severity
154

was not sufficient to elevate apoE as well as reflects the poor sensitivity of apoE as an
LXR target compared to ABCA1 (Donkin et al., 2010). However, it is possible that apoE
may show localized upregulation in regions with more severe damage where microglial
activation is pronounced. Moreover, it is also possible that GW3965-mediated lipidation
of the available apoE mass may have been sufficient to promote post-TBI recovery in
our model. Although we did not assess post-TBI lipidation status of apoE with or without
GW3965 treatment in the present study, future studies will test whether ABCA1-
mediated lipidation of apoE contributes to the beneficial effects of GW3965 after mrTBI.
We used a commonly-used weight drop model to induce CHI in mice. The rationale for
using this model was less invasive surgical manipulations including craniotomy that is
required for FPI and CCI models, and relative simplicity compared to FPI and CCI
model, and high throughput. In addition, being a CHI model, it is clinically more relevant
as about 90% of human TBI do not involve penetration of dura. Using the weight drop
device we established a TBI protocol wherein every mouse received two CHI spaced at
24 h. In our initial pilot studies, we found that for mice weighing 25 to 32 g, a drop height
(in cm) equal to the one third the body weight (in g) resulted in mild TBI with low (<
10%) incidence of skull fracture.
Surprisingly, apoE was not required for GW3965 to suppress the transient increase in
Aβ levels induced in our model. Further studies will be required to characterize these
apoE-independent pathways that promote Aβ clearance after TBI. This will be an
important endeavor, as axonal APP accumulation is a hallmark of TBI (Gentleman et al.,
155

1993; Sherriff et al., 1994b). Theoretically, the Aβ produced after TBI could trigger Aβ-
dependent toxic pathways that exacerbate damage (Johnson et al., 2010). In support of
this γ-secretase inhibitors, which block Aβ production, reduce cognitive and pathological
changes following CCI in mice (Loane et al., 2009). However, the relationship between
Aβ levels and TBI recovery is complex. In WT and AD mice, PBS-soluble Aβ levels in
brain ISF microdialysates decrease by up to 50% within 90 min after CCI with impact
depths ranging from 1-2 mm (Schwetye et al., 2010). Intriguingly, Schwetye et al. (2010)
found no evidence of transient increases in Aβ levels after CCI, which differs from our
findings and those from other investigators using relatively crude tissue lysates (see
references in the Schwetey et al. (Schwetye et al., 2010) paper/discussion). Intra-axonal
Aβ accumulation is observed in 3XTg-AD and APP/PS1 mice from 1-24 h after
moderate-severe CCI (Tran et al., 2011b; Tran et al., 2011c), again suggesting that
distinct pools of Aβ may differ in their responses after injury. Future studies are needed
to clarify the factors that influence Aβ dynamics after TBI, including apoE.
Although our mrTBI model clearly leads to behavioral deficits and axonal damage, it is
not severe enough to trigger significant inflammation except in localized regions near
microcontusions. Importantly, inclusion or exclusion of mice with microcontusions did
not influence the group outcomes for NOR, rotarod, and silver stain data. These results
suggest that Iba1-positive staining of reactive microglia may be a potent secondary
effect of structural brain damage, although it is possible that the time points examined in
this study may have missed a peak of more global inflammation in our model.
156

Interest in understanding the pathophysiology of mild closed-head TBI has led to the
development of various impact and blast models. However, there is considerable
variation in outcomes. For example, many groups have reported mild cognitive deficits
without motor impairment in weight drop-based mild TBI mouse models (Tang et al.,
1997; DeFord et al., 2002; Pan et al., 2003; Creeley et al., 2004; Zohar et al., 2011). In
contrast, the Shohami group observed motor deficits in both severe and mild TBI, but
found that severe injury was required for cognitive impairment (Tsenter et al., 2008).
Using an electromagnetically-controlled piston to produce mrTBI, Shitaka et al observed
cognitive deficits, white matter damage, and robust microglial activation without
significant structural damage or APP immunoreactivity (Shitaka et al., 2011). Our mrTBI
model also shows sustained cognitive and transient motor deficits, axonal damage, and
transient Aβ accumulation. The most noteworthy difference between our model and that
of Shitaka et al is that we observed very little microglial activation, suggesting that
marked inflammation is not required to lead to behavioral and axonal changes. Our
model also does not lead to significant increases in apoE or LDLR levels, which have
been observed in models of moderate-severe TBI (Poirier, 1994; Iwata et al., 2005;
Petit-Turcotte et al., 2007; Loane et al., 2011).
We and others have demonstrated that synthetic LXR agonists effectively enhance the
ability of lipidated apoE to reduce Aβ levels and restore cognitive function in AD mice
(Riddell et al., 2007; Donkin et al., 2010). Given that TBI also involves altered apoE and
Aβ metabolism, it is of considerable interest to determine whether LXR agonists also
have potential therapeutic benefits for TBI. The importance of addressing this question
157

is two-fold. First, LXR treatment may minimize neuronal damage and promote acute
recovery by reducing inflammation and promoting neuronal repair. Second, by
facilitating Aβ clearance in the first weeks after injury, LXR treatment may also reduce
the increased long-term risk of AD years or decades later. Although current LXR
agonists have safety issues such as hypertriglyceridemia and hepatic steatosis that
preclude their present clinical use (Grefhorst et al., 2002; Groot et al., 2005), ongoing
drug discovery efforts may lead to a safe and effective compound. Furthermore, the
metabolic risks of LXR agonists may be clinically tolerable if short-term treatment could
improve functional recovery as well as decrease long-term AD risk. Building on the
findings of Loane and colleagues (Loane et al., 2011), our study provides additional
support for the potential of LXR agonists to treat mrTBI.
Our study also illustrates that the mechanisms by which LXR agonists promote recovery
after TBI may not directly correspond to their effects in AD models. For example,
outcomes such as cognitive recovery and axonal damage are apoE-dependent, but
surprisingly, Aβ clearance is independent of apoE in our model. Additionally, because
our model produces negligible inflammation, the beneficial effects of LXR agonists on
TBI recovery may be independent from their established anti-inflammatory effects.
Future investigations will be designed to identify the LXR targets operative in our mrTBI
model, assess their efficacy to reduce tau pathology, and evaluate their therapeutic
utility in models of moderate-severe TBI.
158

One caveat of this study was the lack of prospective power analysis to determine
adequate sample size. We therefore performed retrospective power calculations and
found that the study was adequately powered to capture TBI effect. It should however
be noted that the use retrospective power calculations is controversial (Thomas, 1997).
For example, retrospective power analysis is generally carried out only for the negative
study results and as such will produce a low post-hoc power result. This may be
misinterpreted as the study having inadequate power, which is reflected by the low
power observed in the present study to detect TBI x treatment effect, although the
interaction was significant. Based on the results of the present studies future studies will
be designed to include prospective power analysis.
A caveat of the weight drop TBI model used in this study is the absence of a systematic
biomechanical assessment of injury. Although input parameters such as weight mass
and drop height are reported in most CHI studies including ours, the reproducibility of
these mechanical inputs and the response of the animal’s head to the forces applied are
not well understood. Li and colleagues recently studied the biomechanical parameters
of the weight-drop based Marmarou rat TBI model (Marmarou et al., 1994) and found
that DAI severity was related to the linear and angular response of the rat head but not
with the drop height (Li et al., 2011). The importance of free head acceleration after
application of mechanical forces was further underlined by a recent study by Goldstein
and colleagues who demonstrated that a single blast injury in mice can generate
significantly impaired memory performance, long-term potentiation, and axonal
conductance accompanied by tau pathology, myelinated axonopathy,
159

microvasculopathy, and neuroinflammation (Goldstein et al., 2012). Intriguingly,
immobilizing the animal’s head to prevent blast-induced head oscillations prevented
memory deficits (Goldstein et al., 2012). It is possible that the very mild pathology in our
model is due to relatively little head movement after impact.
The caveats associated with the weight drop model inspired us to develop a better TBI
model which can allow unrestricted head movement and integrate biomechanics with
TBI outcomes. In the next chapter of this thesis I have described the novel rodent
neurotrauma model called CHIMERA (Closed-Head Impact Model of Engineered
Rotational Acceleration) that we developed in collaboration with the biomechanical
engineers at the International Collaboration on Repair Disorders (ICORD), The
University of British Columbia.
160

Chapter 3: Merging Pathology and Biomechanics Using CHIMERA: A Novel,
Surgery-Free, Closed-Head Impact Model of Engineered Rotational Acceleration
3.1 Summary
Traumatic brain injury (TBI) is a major health care concern that currently lacks any
effective treatment. Despite promising outcomes from many preclinical studies, clinical
evaluations have failed to identify effective pharmacological therapies, suggesting that
the translational potential of preclinical models may require improvement. Rodents
continue to be the most widely used species for preclinical TBI research. As most
human TBIs result from impact to an intact skull, closed head injury (CHI) models are
highly relevant, however, traditional CHI models suffer from extensive experimental
variability that may be due to poor control over biomechanical inputs. Here we describe
a novel CHI model called CHIMERA (Closed-Head Impact Model of Engineered
Rotational Acceleration) that fully integrates biomechanical, behavioral, and
neuropathological analyses. CHIMERA is distinct from existing neurotrauma model
systems in that it uses a completely non-surgical procedure to precisely deliver impacts
of prescribed dynamic characteristics to a closed skull while enabling kinematic analysis
of unconstrained head movement. In this study we characterized head kinematics as
well as functional, neuropathological, and biochemical outcomes up to 14d following
repeated TBI (rTBI) in adult C57BL/6 mice using CHIMERA. Head kinematic analysis
showed excellent repeatability over two closed head impacts separated at 24h. Injured
mice showed significantly prolonged loss of righting reflex and displayed neurological,
motor, and cognitive deficits along with anxiety-like behavior. Repeated TBI led to
161

diffuse axonal injury with extensive microgliosis in white matter from 2-14d post-rTBI.
Injured mouse brains also showed significantly increased levels of TNF-α and IL-1β and
increased endogenous tau phosphorylation. Repeated TBI using CHIMERA mimics
many of the functional and pathological characteristics of human TBI with a reliable
biomechanical response of the head. This makes CHIMERA well suited to investigate
the pathophysiology of TBI and for drug development programs.
3.2 Introduction
Traumatic brain injury (TBI) is a leading worldwide cause of death and disability for
persons under 45 years of age with a cost to society of over 17 billion USD per year. In
the United States, the overall incidence of TBI is estimated to be 538 per 100,000
persons, which represents at least 1.7 million new cases per year since 2003
(Gerberding and Binder, 2003; Langlois et al., 2006; Faul et al., 2010). TBI incidence is
reportedly lower in Europe (235 per 100,000) and Australia (322 per 100,000) (Cassidy
et al., 2004; Tagliaferri et al., 2006) although recent epidemiological data suggests far
greater incidence (749 per 100,000) (Feigin et al., 2013). Mild TBI (mTBI), which
includes concussion, comprises over 75% of all TBIs (Gerberding and Binder, 2003).
The reported incidence of mTBI is rising and mTBI is increasingly recognized as an
injury for which medical attention should be sought.
Falls are the most prevalent cause of TBI, although motor vehicle accidents and
impacts against objects are also common causes (Cassidy et al., 2004; Faul et al.,
2010; Roozenbeek et al., 2013). TBI resulting from high-contact sports such as boxing,
162

American football, ice hockey, soccer, and rugby account for almost 21% of all head
injuries among children and adolescents, particularly for mTBI (Centers for Disease
Control and Prevention, 2007). In these situations, the skull experiences an impact
resulting in brain deformation and resulting injury that most often occurs without skull
fracture. TBI is also considered a “signature injury” in modern warfare, as approximately
20% of veterans from the Iraq or Afghanistan wars are reported to have experienced a
TBI, 80% of which involve both blunt impact and overpressure mechanisms (Taber et
al., 2006; Hoge et al., 2008; Elder and Cristian, 2009; Sevagan et al., 2013).
Furthermore, the growing awareness that mTBI may have long-lasting and severe
consequences (Gavett et al., 2011; Jordan, 2013; Rusnak, 2013; Smith et al., 2013)
highlights the urgency to understand much more about the acute and long-term
consequences of brain injury.
The rapid accelerations and rotations during impact TBI lead to vigorous movement and
deformation of brain tissue within the skull that can result in contusions as the brain
contacts the interior of the bony skull. These inertial and contact forces directly affect
neurons, glia, and blood vessels producing a primary injury that initiates secondary
processes within hours to weeks after the initial injury (McIntosh et al., 1996;
Blumbergs, 1997; Davis, 2000; Giza and Hovda, 2001; Werner and Engelhard, 2007;
McAllister, 2011). These secondary changes lead to a plethora of events including
edema, raised ICP, impaired CBF, increased BBB permeability, inflammation, axonal
injury, calcium influx, elevated oxidative stress, free radical-mediated damage,
excitatory neurotransmitter release, and cell death (McIntosh et al., 1996; Blumbergs,
163

1997; Davis, 2000; Giza and Hovda, 2001; Werner and Engelhard, 2007; McAllister,
2011). Although very few treatment options are available for the primary injury,
secondary injury pathways are potentially modifiable (Graham et al., 2000). An
increasingly wide variety of experimental animal models are therefore being developed
to characterize secondary injury processes and for the evaluation of candidate
therapeutic approaches.
No single animal model can replicate the entire spectrum of human TBI
pathophysiology; therefore, several large and small animal models have been
developed to mimic particular aspects. Popular rodent models include OHI models
including FP and CCI systems, and CHI models that use either gravity or mechanical
methods to impact the intact skull (reviewed in Chapter 1). Although FP and CCI models
employ highly reproducible mechanical inputs and can mimic many pathological
features of human TBI, the prominent tissue destruction and lack of head movement
decreases the resemblance to the majority of human injuries that occur due to impact
and/or acceleration on an intact skull. In contrast, CHI models employ methods that do
not generally cause overt brain tissue loss and can also allow rapid behavioral
assessment of injury severity. As such, CHI models are considered by some to better
mimic the majority of human TBI. However, a major limitation of most current CHI
animal models is that the input parameters used to induce injury (e.g., mechanical
loading, method of mechanical input, response of the animal’s head to mechanical
loading) are often poorly controlled and this can lead to considerable experimental
164

variation across cognitive, histological, and biochemical outcome measures (Namjoshi
et al., 2013a).
Here we report a novel neurotrauma model called CHIMERA (Closed-Head Impact
Model of Engineered Rotational Acceleration). CHIMERA was developed to address the
absence of a simple and reliable model of rodent CHI that mimics the majority of human
TBI cases. CHIMERA is distinct from existing neurotrauma model systems in that it is
completely surgery-free and fully integrates biomechanical, behavioral, and
neuropathological analyses after delivering impacts of defined energy to a closed skull
with unconstrained head motion after impact. We show that that repeated TBI (rTBI) in
mice using CHIMERA reliably induces motor deficits and anxiety-like behavior and
leads to diffuse axonal injury (DAI) with extensive white matter inflammation and
increased phosphorylation of endogenous tau.
3.3 Materials and Methods
3.3.1 CHIMERA Impactor
The CHIMERA impactor consists of an aluminum frame that supports an animal holding
platform above a pneumatic system platform (Figure 3.1 A).
3.3.1.1 Animal Holding Platform
The animal holding platform is composed of a fixed head plate that supports the
animal’s head in a supine position and a body plate that positions and secures the
animal’s torso. The head plate has a hole through which the tip of the impactor piston
165

projects to impact the animal’s head. A cushion made from closed-cell foam surrounds
the hole to minimize rebound impact when the animal’s head falls back on the head
plate following impacted by the piston. Two perpendicular lines across the piston hole
act as crosshairs for aligning the animal’s head over the hole. The body plate holds a
restraint system consisting of an animal bed of closed-cell foam contoured to the shape
of the animal’s body and two Velcro straps. The animal holding platform is attached to
the frame by hinges and its angle of inclination can be adjusted. In this study, the angle
was set to approximately 32° such that the frontal and parietal bones laid flat over the
hole in the head plate, thus delivering impact to the dorsal cortical region.
3.3.1.2 Pneumatic Impactor System
The pneumatic impactor system includes a 1.9 L accumulator air tank (Model F9124,
Firestone Industrial Products, Indianapolis, IN), pressure regulator (Model 700-BF,
ControlAir Inc., Amherst, NH), a digital pressure gauge (Model DPG409-150G, Omega,
Laval, QC), a two-way solenoid valve (Model 21HN2KY110, Granzow Inc., Charlotte,
NC), and a trigger button. The pressure regulator and digital pressure gauge allow
precise adjustment of air pressure with minimum increments of 0.1 psi (0.69 kPa),
enabling accurate delivery of piston velocity and impact energy (Figure 3.2).
3.3.1.3 Impact Piston and Barrel System
Impact is induced with a free-floating metal impact piston with a body diameter of 15
mm and a tip diameter of 5 mm whose trajectory is constrained to linear motion by a
stainless steel barrel (internal diameter: 16 mm, external diameter: 20 mm). Four
166

different masses of impact pistons are available with the current system: 50 g, 75 g and
100 g pistons machined out of a chrome-coated steel rod, and a 25 g piston machined
out of an aluminum rod, to allow impact velocity or impact energy to be independently
adjusted (Figure 3.1 B). Only the piston tip (length: 2 cm, diameter: 0.5 cm) extrudes
from the barrel. The barrel design includes a series of holes drilled along the length to
vent air as the piston moves towards the open end. The piston is accelerated by a
controlled pulse of compressed air along the length of the barrel until it clears the
venting holes. The barrel can be mounted in either a vertical or horizontal configuration
relative to the animal holding platform. In the vertical configuration, the barrel is located
below the supine animal’s head such that the impact cylinder strikes the head to initiate
flexion. In this configuration the animal holding platform can be inclined relative to the
head plate such that the frontal and parietal bones lie flat over the hole in the head
plate, thus delivering impact to the dorsal cortical region. Alternatively, the animal
holding platform can be adjusted to lie parallel to the head plate, such that the impact is
directed towards the interparietal and occipital bones. In the lateral configuration the
barrel is mounted orthogonally relative to the head plate and the body is plate is inclined
to keep the head flat over the hole. In this configuration, the piston travels horizontally to
impact the left side of the head and initiate lateral and twisting head movements.
167

Figure 3.1: CHIMERA impactor and impact pistons.
(A) CHIMERA with various parts labeled as follows: 1. head plate, 2. body plate, 3.
animal bed, 4. Velcro straps, 5. air tank, 6. air pressure regulator, 7. digital pressure
gauge, 8. two-way solenoid valve, 9 vertical piston barrel. B) Impact pistons. CHIMERA
impactor can use impact pistons of four different masses, a 25 g piston constructed of
aluminum and 50 g, 75 g and 100 g pistons constructed of chrome-coated steel.
3.3.1.4 CHIMERA Calibration
The impactor was first calibrated by determining the air pressure-piston velocity
relationship by measuring the exit velocity of the piston at various air pressures (0.5, 1,
1.5, 2, 3, 5, 7, 10, 15, 20, 30, 50, 70, 100, and 130 psi). The velocity was measured in
triplicate at each pressure value for all four piston masses in both horizontal and vertical
orientations. Each impact event was recorded by a high-speed video camera at 10,000
fps. Video motion analysis software (TEMA Motion, Image Systems AB, Sweden) was
used to track the trajectory of a specific point on the impact piston. The calibration
curves for all four piston masses showed an inflection point between 10 and 15 psi such
168

that they were concave upwards before this point and concave downwards after the
point. The cause of this is unknown, but it is believed to be due to the interaction
between friction, drag, and piston velocity. Because of this effect, two 2nd-order
polynomial curves were used to fit the data. An R2 value of > 0.995 was found for all
curves. A representative calibration curve of impact energy for the 50 g piston at air
pressure values between 0 and 12 psi is shown in the Figure 3.2. Using these curves,
the desired impact velocity or energy can be independently interpolated. By choosing
the appropriate combination of piston mass and air pressure, impacts of input energy
ranging from 0.01 J to 18 J can be precisely generated.
Figure 3.2: Piston energy-air pressure calibration curve.
Air pressure-energy calibration curve was obtained by driving a 50 g piston at
increasing air pressure values and calculating the resultant impact energy. The graph
depicts energy measurements in triplicates for each air pressure value.
All experiments reported in the present Chapter were performed using a 50 g piston and
a vertically configured barrel. We initially performed a pilot experiment using three
169

impact energies (0.4, 0.5 and 0.6 J) to determine the maximum allowable impact energy
without leading to animal mortality. In the experiments described in this Chapter we
used impact energy of 0.5 J with the corresponding piston velocity of 4.5 m/s.
3.3.2 CHIMERA Repetitive TBI (rTBI) Procedure
All animal procedures were approved by the University of British Columbia Committee
on Animal Care (protocol # A11-0225) and were carried out in strict accordance with the
Canadian Council on Animal Care guidelines. Male C57Bl/6 mice (mean ± SD body
weight 34 ± 4 g) at 4-5 months of age were housed with a reversed 12 h light-12 h dark
cycle for at least 10 days and were subjected to repetitive TBI (rTBI) using CHIMERA
impactor as follows.
The animal was first anesthetized in an anesthesia induction chamber with 5%
isoflurane in oxygen (0.9 L/min). Once the animal lost righting reflex, it was quickly
transferred to the CHIMERA device and kept in a prone position on the animal bed. The
anesthesia was maintained with 2.5-3% isoflurane. Lubricating eye ointment was
applied to prevent corneal drying. Meloxicam (1 mg/kg) and saline (1 mL/100 g body
weight) were administered by subcutaneous injections for pain control and hydration,
respectively. The animal was then reoriented in a supine position in the animal bed. The
body plate was inclined such that the top of the animal’s head laid flat over the piston
hole in the head plate and the head was aligned using crosshairs such that the piston
struck the vertex of the head covering a 5 mm area surrounding the bregma (Figure 3.3
B and C). The animal’s body was secured with two Velcro straps (Figure 3.3 A).
170

Figure 3.3: Mouse head and impact position.
(A) Close-up view of animal strapped on the holding platform. (B) Location of impact
relative to the mouse head and brain in sagittal plane. P: impact piston. (C) Estimated
location of impact on the mouse skull (left) and brain (right) based on mouse brain atlas
(Paxinos and Franklin, 2001). The area covered by the impactor tip is shown by black
circle.
Impact was induced by pressing a trigger button that fires the piston and when
connected, simultaneously activates a high-speed camera to record the resulting head
trajectory. Isoflurane delivery was immediately stopped following the impact, the animal
was transferred to recovery cage and continuously monitored until fully ambulatory.
Twenty-four hours after the first impact, a second identical impact (including same drug
regimen) was delivered. Sham animals underwent all of these procedures except for the
171

impact. Isoflurane exposure time was measured for each impact from the start of
isoflurane delivery till isoflurane discontinuation. Approximately ~ 4 % of animals
displayed hind limb paralysis that lasted for several hours after TBI and were
euthanized. These animals were excluded from the data analysis. Sham-operated and
rTBI mice were randomly assigned to one of three post-TBI assessment time points,
viz., 2, 7, and 14 days after the second injury. The time of the 2nd TBI was defined as
time zero.
3.3.3 High-Speed Videography and Kinematic Analyses
For kinematic analysis, a cohort of 8 mice was subjected to rTBI and impact events
were recorded at 5,000 frames per second using a high-speed video camera (Q-PRI,
AOS Technologies, Switzerland). Head motion was tracked using two markers, one
being non-toxic paint applied on lateral side of head to mark the cheek area. Because
the skin is loose over the bony skull, we also marked the position of the maxilla by
wrapping dental floss positioned just caudal to the upper incisors around the animal’s
snout (Figure 3.4). Videos were analyzed using ProAnalyst Professional motion analysis
software (v 1.5.6.8, Xcitex Inc., Woburn, MA) as follows. The origin was set using the
paint marker in the Cartesian coordinates (Figure 3.4 A). The X and Y coordinates of
the position of each marker were tracked on a frame-by-frame basis and were
processed with a 400-Hz low-pass Butterworth filter to mitigate the noise in the recorded
measurements. Linear kinematic parameters were assessed from trajectories obtained
by tracking the paint mark, which was closest to the CG of the animal’s head. Velocities
and accelerations were determined by discrete differentiation of the marker position
172

data. Resultant linear velocity and acceleration were calculated as the magnitude of
their respective X and Y components using the following equations.
Linear Head Displacement = �𝑋𝑋𝑋𝑋𝑋𝑋𝑋𝑋2 + 𝑌𝑌𝑋𝑋𝑋𝑋𝑋𝑋2
Where, Xpos and Ypos represent X and Y positions of the paint mark from the origin,
respectively.
Linear Head Veloxity = �Xvel2 + Yvel2
Where, Xvel and Yvel represent velocities at X and Y positions, respectively.
Linear Head Acceleration = �Xacc2 + Yacc2
Where, Xacc and Yacc represent acceleration at X and Y positions, respectively.
Angular rotation of the head during TBI was determined by the angle of the line joining
the dental floss and the paint mark with the horizon (Figure 3.4 B). Immediately after
video capture, the dental floss was removed. The kinetic energy (KE) transferred from
the piston to the head was determined by KE = 0.5 x Me X ΔV2 (Viano et al., 2012),
where Me is the effective mass and approximated by head mass measured from the 8
mice (3.4 g) and ΔV is change in head velocity. A scaling factor λ [λ = (mass of human
brain / mass of mouse brain)1/3 = 13.8] was used to estimate the human head-
equivalent kinematic parameters from the animal data (Viano et al., 2009).
173

Figure 3.4: External markers used for mouse head tracking.
(A) Head movement was tracked using two markers: a dental floss (red arrow in the first
image) wrapped around the maxilla and a non-toxic paint (yellow arrow in the first
image) applied at the lateral size of the head. Head trajectories and linear kinematic
parameters were obtained by tracking the paint (yellow arrow) marker. Two dots
separated by 10 mm were marked on the CHIMERA frame and were used to calibrate
the distance. (B) Angle of head deflection and angular kinematic parameters were
obtained by determining the angle of the line joining the two tracking markers with the
horizon.
3.3.4 Behavioral Analyses
Duration of loss of righting reflex (LRR) was calculated as the time interval from
isoflurane discontinuation to the first sign of righting after each impact. Neurological
impairment was assessed using the neurological severity score (NSS, Table 3.1) (Flierl
et al., 2009) determined at 1 h and at 1, 2, and 7 d after the second TBI. Motor
performance was evaluated at 1, 2, 7, and 14 d after the second TBI using an
accelerating rotarod as described in Chapter 2. Open field activity was assessed at 1, 7
174

and 14 d after the second TBI using a Plexiglas box (14” x 24” x 14”). The floor of the
box was virtually divided into 60 equal squares using an overhead digital camera and
video tracking software (ANY-maze, v. 4.99, Stoelting Co, Wood Dale, IL). The field was
further subdivided into a peripheral zone along the walls of the open field consisting of
28 squares that surrounded a central zone consisting of 32 squares (Figure 3.5). The
animal was placed in the center of the box and spontaneous activity was recorded over
a 10 min epoch, including quantification of the total distance traveled, number of line
crossings, and immobile time. The thigmotaxis index (TI) was calculated as: TI = (TP-
TC)/(TP+TC) where TP and TC represent time spent in the peripheral and central zones,
respectively. Working memory was assessed by the passive avoidance task from 7 to
10 days after second TBI. Passive avoidance testing was conducted in a device that
consisted of two adjoining compartments, one illuminated (20.3 x 15.9 x 21.3 cm) and
one darkened (20.3 x 15.9 x 21.3 cm), divided by a guillotine-style door (Med
Associates Inc., St. Albans, VT). The floor of the compartments consisted of steel rods
capable of delivering an electric foot-shock. The electric shock was delivered by a
programmable animal shocker (Med Associates Inc.). Each session consisted of placing
mice into the illuminated compartment and using a timer to record the latency of the
mice to cross into the darkened compartment. On day 7 after the second TBI (training)
mice received an electric foot-shock (0.3 mA, 2 s) as soon as they crossed from the
illuminated into the darkened compartment. Following foot-shock, mice were removed
from the apparatus and returned to their home cage. On days 8-10 after the second TBI
mice were tested for memory retention. The latency for the mice to cross into the
darkened compartment was recorded. No shock was delivered during testing. Mice that
175

did not cross over into the darkened compartment were allowed to remain in the
illuminated compartment for the full 5 min and assigned a latency of 300 s. Non-working
memory was assessed with the Barnes maze from 8-13 d post-rTBI. Barnes maze
testing was conducted on a grey circular platform (91 cm diameter) with 20 circular
holes (5 cm diameter; Stoelting Co) at its periphery located in a 3.0 x 3.6 m room. An
escape box was positioned beneath one of the holes. Extra-maze visual cues consisted
of black and white images (cross, lightning bolt, smiley face) printed on glossy white
paper and placed on the walls surrounding the maze. Other visual cues included a cart
with a laptop and the experimenter. For motivational purposes, mice were subjected to
mild food deprivation (1 g of food per day) from days 8-13 after the second TBI and
maintained at 90% of free feeding body weight. During testing mice were exposed to
aversive stimuli in the form of two lights (100 W) positioned at either side of the maze
and a buzzer (2.9 kHz, 65 dB) which hung 10 cm above the maze. On day 8 mice
completed one 5-min habituation trial to become familiar with the maze environment
and to practice descending into the escape box. On days 9-13 mice were tested for
memory retention. Mice were given four trials per day (15-min inter-trial interval) for five
days. For each trial the animal was placed in a black plastic start tube (7 cm diameter,
12 cm height) at the center of the maze. After 10 s, the start tube was raised and the
buzzer was turned on to start the trial. Mice that were unable to locate the escape hole
in 90 s were gently guided to it. Mice remained in the escape box for 10 s before being
returned to their home cage. Mice were tracked during each trial using a digital camera
placed 4 m above the center of the maze and ANY-maze tracking system.
176

Figure 3.5: Open field thigmotaxis. The floor of the open field was virtually divided into central (gray area) and peripheral
(white area) zones. Thigmotaxis was assessed by placing the mouse in the open field
and measuring time spent in central and peripheral zones.
Task Description Points
177

(success/failure)
Exit circle Ability to exit a circle of 30 cm diameter within 3 min 0/1
Monoparesis/hemiparesis Paresis of front and/or back limbs 0/1
Straight walk Alertness, initiative and motor ability to walk straight 0/1
Startle reflex Innate reflex; the mouse will bounce in response to a loud hand clap
0/1
Seeking behavior
Physiological behavior as a sign of 'interest' in the environment 0/1
Beam balancing
Ability to balance on a beam of 7 mm width for at least 10 s 0/1
Round stick balancing
Ability to balance on a round stick of 5 mm diameter for at least 10 s 0/1
3 cm wide beam walk
Ability to cross a 30-cm long beam of 3 cm width 0/1
2 cm wide beam walk
Same task, increased difficulty on a 2-cm wide beam 0/1
1 cm wide beam walk
Same task, increased difficulty on a 1-cm wide beam 0/1
Maximal Score 10
Table 3.1: Neurological severity score (NSS) tasks.
Post-TBI neurological impairment is assessed by NSS, which is a composite of ten
different tasks that mice perform. Failure in each task earns one point, while passing the
task gets no point. Total NSS is obtained by adding scores of individual tasks. Based on
the total NSS, injury can be classified as mild (NSS: < 5), moderate (NSS: 5-7) and
severe/fatal (NSS: 8-10). Adapted from (Flierl et al., 2009).
3.3.5 Tissue Collection and Processing
For histological analyses, mice were anesthetized with an i.p. injection of 150 mg/kg
ketamine (Zoetis) and 20 mg/kg xylazine (Bayer) at 2, 7, and 14 d post-rTBI, and brains
were collected from perfused animals as described in the Chapter 2 except that 4%
178

paraformaldehyde rather than 10% neutral buffered formalin was used to post-fix
hemisected brain tissue for histology. For biochemical analyses, brains were harvested
as described in the Chapter 2 at 6 and 12 d and at 2, 7, and 14 d post-rTBI,
longitudinally hemisected and rapidly frozen over dry ice and stored at -80°C until
analysis.
3.3.6 Iba1 Immunohistochemistry and Silver Staining
Microglial activation and axonal injury were assessed using Iba-1 immunohistochemistry
and silver staining, respectively as described in Chapter 2. Microglial activation was
quantified using fractal analysis using ImageJ (NIH). Three to four microglia were
randomly chosen from 40X-magnified Iba-1 stained images, converted to 8-bit images,
and thresholded. Thresholded images were converted to outline images and analyzed
using the FracLac plugin (Karperien, A., FracLac for ImageJ.
http://rsb.info.nih.gov/ij/plugins/fraclac/FLHelp/Introduction.htm. 1999-2013.). The box
counting method was used and the mean fractal dimension was analyzed. The number
of microglia in each white matter regions was quantified by scanning the entire area
using a Olympus BX61 microscope at 10X magnification with a ProScan motorized XY
stage (Prior Scientific Inc, Rockland, MA). The component images were stitched
together into a montage using ImagePro Plus image analysis software (Media
Cybernetics Inc., Rockville, MD). The number of Iba1-positive cells was manually
counted in the entire white matter track ROI. The area of ROI was measured by ImageJ
as pixels and scaled to mm2. Cell density was finally expressed as number of Iba-1
positive cells per mm2. Silver staining intensity was quantified using ImageJ (version
179

1.48, NIH) on 40X-magnified images. The images were thresholded and the region of
interest (ROI) was selected. The ratio of area of positive signal in ROI to total ROI area
was reported as percent positive.
3.3.7 Biochemical Analyses
3.3.7.1 Tissue Processing
For protein determination, half-brains were homogenized in RIPA lysis buffer as
described in Chapter 2.
3.3.7.2 Assessment of TNF-α and IL-1β by ELISA
Endogenous TNF-α and IL-1β protein levels in the half-brain homogenates were
quantified by commercial ELISA kits (BD Biosciences OptEIA 559603 and 555268,
respectively) following the manufacturer’s instructions.
3.3.7.3 Quantitative Assessment of Phosphorylated and Total Tau by Simple
Western Analysis
Phosphorylated and total tau were assessed using a completely automated capillary
electrophoresis-sized-based Simple Western system (O'Neill et al., 2006) using the
WES machine (ProteinSimple, San Jose, CA). Simple Western is a gel-free, blot-free,
capillary-based, automated protein immunodetection system that automates all the
steps following sample preparation including sample loading, size-based protein
separation, immunoprobing, washing, detection, and data analysis. All procedures were
performed with manufacturer’s reagents according to the user manual. Briefly, 5 µl of
180

RIPA lysate (2 µg of protein) was mixed with 1.2 µl of 5x fluorescent master mix and
heated at 95°C for 5 min. The samples, blocking reagent, wash buffer, primary
antibodies, secondary antibodies, and chemiluminescent substrate were dispensed into
designated wells in the manufacturer provided microplate. Following plate loading the
separation and immunodetection were performed automatically using default settings.
Data were analyzed with Compass software (ProteinSimple). All tau antibodies were
kind gifts from Dr. Peter Davies (Albert Einstein College of Medicine, Manhasset, NY,
USA). The details of tau antibodies are presented in the Table 3.2. GAPDH (clone 6C5,
1:25000, Chemicon) was used as a loading control. Levels of p-tau and total tau were
normalized to GAPDH. Levels of phosphorylated tau were expressed as fold difference
compared to sham controls at the respective time points.
Antibody Tau Form Epitope(s) Concentration RZ3 p-Tau Thr231 1:25
PHF1 p-Tau Ser396 & Ser404 1:25
CP13 p-Tau Ser202 & Thr404 1:25
DA9 Total Tau 102 to 140 1:5000
Table 3.2: Details of monoclonal antibodies for probing p-tau and total tau.
The rTBI protocol and post-rTBI end points are summarized in Figure 3.6.
181

Figure 3.6: CHIMERA-rTBI procedure and post-rTBI endpoints
The figure indicates time line for rTBI/sham procedure and behavioral, biochemical and
histological end points at various post-rTBI time points.
BM: Barnes maze, Iba-1: Iba-1 immunohistochemistry, NSS: neurological severity score,
OF: open field behavior, PA: passive avoidance, RR: rotarod, SS: silver stain
3.3.8 Statistical Analyses
The head kinematics data and graphs are presented as mean ± 95% CI. Behavioral
data and graphs are presented as mean ± SEM. All other data and graphs are
presented as mean ± SD unless otherwise specified. NSS, LRR, thigmotaxis, and
rotarod data were analyzed using repeated measures two-way ANOVA followed by the
Holm-Sidak post-hoc test, as animals were tested repeatedly until sacrifice. Passive
avoidance and Barnes maze data were analyzed by repeated measures two-way
ANOVA. Iba-1 and silver staining data were analyzed by two-way ANOVA followed by
Tukey’s post-hoc test. For all the above statistical analyses, a p value of <0.05 was
considered significant. Tau phosphorylation and cytokine protein expression at each
post-rTBI time point was compared to the respective sham values by t test followed by
182

Bonferroni correction of multiple comparison, with p value set to <0.01 (5 comparisons)
for detecting statistical significance. Statistical analyses of behavioral data were
performed using SigmaPlot (version 12.5, Systat Software Inc.). Statistical analyses for
rest of the data were performed using GraphPad Prism (version 6.04, GraphPad
Software Inc).
3.4 Results
3.4.1 Head Kinematics Following CHIMERA rTBI
Analysis of high-speed videography (5000 fps) was used to assess the biomechanical
responses of the head in a group of 8 mice during CHIMERA rTBI at impact energy of
0.5 J. Peak kinematic parameters are depicted in Figure 3.7 and Table 3.3. Following a
vertical upward impact of a pneumatically-accelerated piston onto an intact skull of a
supine mouse, the head follows a looped trajectory in the sagittal plane (Figures 3.7 A
and 3.8). The average head trajectories following two TBIs were highly consistent
(Figure 3.7 A). The head traveled a peak linear displacement of 49.6 ± 3.5 mm in 15.7 ±
2.4 ms (Figure 3.7 B) and a peak angular rotation of 2.6 ± 0.28 rad in 24.8 ± 3.1 ms
(Figure 3.7 C). The peak linear velocity was 6.6 ± 0.8 m/s at 3.4 ± 1.0 ms (Figure 3.7 D),
and the peak angular velocity was 305.8 ± 73.7 rad/s at 2.8 ± 1.9 ms following initial
impactor contact (Figure 3.7 F). The head experienced large linear and angular
accelerations following impact, achieving a peak linear acceleration of 385.3 ± 52 g in
1.5 ± 0.3 ms (Figure 3.7 E) and peak angular acceleration of 253.6 ± 69.0 krad/s2 in 0.8
± 1.1 ms (Figure 3.7 G). As the head was stationary before impact, the change in head
velocity (ΔV) equals peak head velocity and was found to be 6.6 m/s. The energy
183

transferred from the piston to the head was 0.07 J, which equals to about 86% loss of
the input energy (0.5 J).
184
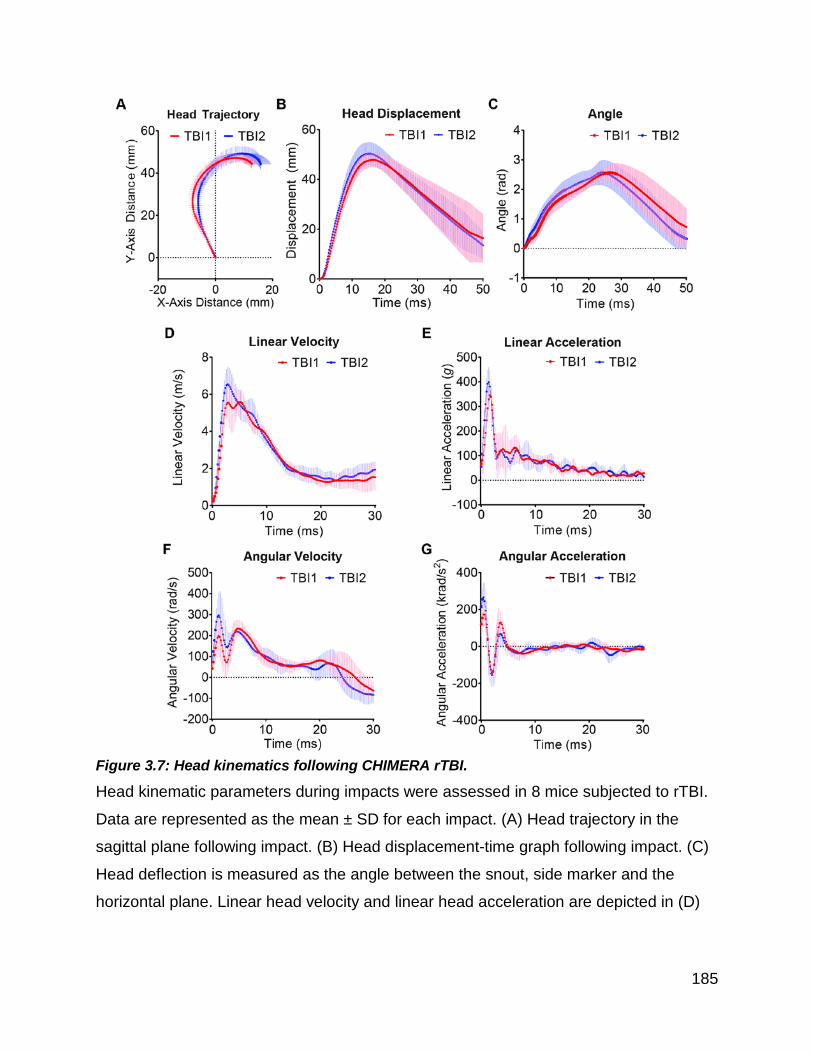
Figure 3.7: Head kinematics following CHIMERA rTBI.
Head kinematic parameters during impacts were assessed in 8 mice subjected to rTBI.
Data are represented as the mean ± SD for each impact. (A) Head trajectory in the
sagittal plane following impact. (B) Head displacement-time graph following impact. (C)
Head deflection is measured as the angle between the snout, side marker and the
horizontal plane. Linear head velocity and linear head acceleration are depicted in (D)
185

and (E), respectively. (F) and (G) show angular head velocity and angular acceleration,
respectively. All graphs are presented as mean ± SD.
Figure 3.8: CHIMERA allows unrestricted head motion during TBI.
Before impact, the mouse head was freely supported on a foam pad in the supine
position. Velcro straps were applied to the torso. Impact from the piston deflects the
head, which then subsequently returns to its original position on the foam pad. The
images were taken at 5,000 fps, at an angle perpendicular to the direction of impact and
along the mouse sagittal plane. Each image shown was 12 ms apart.
186

Kinematic Parameter 1st TBI (mean ± SD)
2nd TBI (mean ± SD)
Average of Two TBI % CV
Head displacement (mm) 48.4 ± 2.5 50.8 ± 4.1 49.6 8 Linear velocity (m/s) 6.3 ± 0.93 6.9 ± 0.94 6.6 5 Linear acceleration (g) 362.6 ± 42 407.6 ± 57.3 385.3 13 Angle of head deflection (rad) 2.6 ± 0.24 2.6 ± 0.31 2.6 9 Angular velocity (rad/s) 273.3 ± 82.9 338.4 ± 85.7 305.8 20 Angular acceleration (krad/s2) 216.2 ± 77.2 291 ± 69.7 253.6 21
Table 3.3: Summary of peak values of head kinematic parameters.
The table depicts average peak values of head kinematic parameters for rTBI from 8
rTBI mice. The coefficient of variation (CV) was calculated as the average of day 1 and
day 2 peak values from all available recordings.
Using the equal stress/equal velocity approach (Holbourn, 1943; Gutierrez et al., 2001;
Viano et al., 2009) to scale our murine kinematic data to human-equivalent values, ΔV
was found to be comparable to National Football League (NFL) values and higher than
Olympic boxing values, whereas scaled linear and angular velocity and acceleration
parameters were lower than NFL values but comparable to Olympic boxing values
Table 3.4.
Study TBI Type Species / Model
Head Kinematic Parameters (Scaled to Human) HIC15
187

ΔV (m/s)
Angular Velocity (rad/s)
Acceleration Linear
(g) Angular (krad/s2)
CHIMERA Impact-acceleration Mouse 6.6 22.6 27.9 1.4 40
Goldstein et al. (2012) Blast Mouse NA 36.2 NA 5.0 NA
Xiao-Sheng et al. (2000)
Non-impact rotation Rat NA 72.9 14.2 16.9 NA
Fijalkowski et al. (2007)
Non-impact rotation Rat NA NA NA 3.0 NA
Viano et al. (2009)
Impact-acceleration Rat NA NA 93.5 NA NA
Li et al. (2010)
Non-impact rotation Rat NA NA 14.7 1.1 NA
Wang et al. (2010)
Non-impact rotation Rat NA 6.4 NA 1.1 NA
Davidsson and Risling (2011)
Non-impact rotation Rat NA NA 32.8-
68.6 2.5-17.4 NA
Li et al. (2011) Weight drop Rat NA 4.3-5.7 60.5-
82.5 1.4-1.5 NA
Pellman et al. (2003a), Pellman et al. (2003b)
NFL concussion Human NA 34.8 97.8 6.4 250
Viano et al. (2005), Peng et al. (2013)
Olympic boxing – hook punch
Hybrid III dummy 3.1 29.3 71.2 9.3 79
Olympic boxing – uppercut
Hybrid III dummy 2.8 17.5 24.1 3.2 17
MVA involving pedestrian head impact (50% probability resulting in moderate injury)
Hybrid III dummy
and FEM NA NA 116 11.4 825
Table 3.4: Comparison of head kinematic parameters between rodent TBI models and human TBI.
The table compares most commonly-reported head kinematic parameters between
different TBI models in mice and rats. All the kinematic parameters are scaled to human
188

values according to the equal velocity/equal stress approach. According to this
approach, ΔV is unscaled, linear acceleration and angular velocity are scaled by 1 scale
factor (λ), and angular acceleration is scaled by λ2. The λ values for mice and rats were
13.8 and 11.0, respectively. National Football League (NFL) concussion values and
data from punches by Olympic boxers are included for comparison. Head Injury
Criterion for 15 ms (HIC15) is included to compare the likelihood of head injury. HIC15
for the CHIMERA was calculated based on the definition:
HIC = max
12
5.2
12)()(1 2
1
−
− ∫ ttdttatt
t
t
where linear acceleration (a) is in g and scaled by λ, time (t) is in seconds and scaled by
1/λ, and t1 and t2 are determined to give maximum value to the HIC function such that t2
- t1 = 15 ms.
FEM: Finite element model
NA: Data not available/not applicable
HIC15: Head Injury Criterion for 15 ms
ΔV: Change in velocity
MVA: Motor vehicle accidents
3.4.2 CHIMERA rTBI Induces Behavioral Deficits
Loss of righting reflex (LRR) in animals after TBI is considered analogous to loss of
consciousness in humans after TBI and can be considered as a behavioral indicator of
injury severity (Dewitt et al., 2013). Mice subjected to CHIMERA rTBI showed
significantly increased LRR duration compared to sham animals (Figure 3.9 A; TBI
effect: F(1, 65) = 59.61, p < 0.001). LRR duration was consistent between the first and
189

second impacts (Figure 3.9 A). We further assessed injury severity using the NSS,
which is a composite of ten tasks that assess motor reflexes, alertness, and
physiological behavior (Flierl et al., 2009). The NSS of injured animals was significantly
higher than sham mice from 1 h to 7d post-procedure (Figure 3.9 B, TBI effect: F(1,
191) = 44.12, p < 0.001). In injured mice, the NSS score showed maximum deficits at
1h post-procedure (p < 0.001) followed by steady spontaneous improvement over 1-7d,
albeit remaining significantly higher than sham animals at each post-rTBI time point
(Figure 3.9 B, p < 0.001). Similarly, rTBI significantly impaired motor performance from
1-7d post-injury as indicated by reduced fall latencies on an accelerating rotarod
compared to sham controls (Figure 3.9 C, TBI effect: F(1, 220) = 11.99, p < 0.001). Fall
latencies showed both time effects (F(4, 220) = 13.70, p < 0.001) and TBI x Time
interaction (F(4, 220) = 11.22, p < 0.001). Motor deficits in injured mice peaked at 1d (p
< 0.001) and returned to baseline conditions by 14d post-injury (p = 0.22), whereas
sham mice did not show any motor deficit (p > 0.82). Injured mice showed anxiety-like
behavior as indicated by significantly increased thigmotaxis in an open field test (Figure
3.9 D, TBI effect: F(1, 53) = 12.30, p < 0.001). The thigmotactic behavior of both groups
significantly declined over time in similar trend (Figure 3.9 D, Time effect: F(2, 53) =
5.45, p = 0.007; TBI x Time interaction insignificant). Open field thigmotaxis was not
affected by gross motor activity as no significant differences in total distance traveled or
time immobile were observed between injured and sham-operated mice (Figure 3.10).
Repeated TBI also induced working memory impairment as indicated by decreased
latencies to enter the darkened compartment on the passive avoidance task (Figure 3.9
E, TBI effect: F(1, 28) = 4.6, p = 0.041). For all mice, a main effect of Day indicated that
190

mouse behavior changed over the training and testing sessions evaluated in this study
(F(3, 84) = 58.55, p < 0.001). For the main effect of Day, pairwise comparisons
indicated that mice entered the darkened compartment significantly faster on Day 7 (p <
0.001) than on Days 8–10 (p > 0.05) signifying that all mice remembered the shock to
some degree (Figure 3.9 E). Injured mice also showed spatial reference memory
impairment as indicated by increased latencies to locate the escape hole on the Barnes
maze (Figure 3.9 F, TBI effect: F(1, 28) = 6.27, p = 0.018). Under our experimental
conditions, cognitive performance did not spontaneously resolve by the end of our
testing period.
191
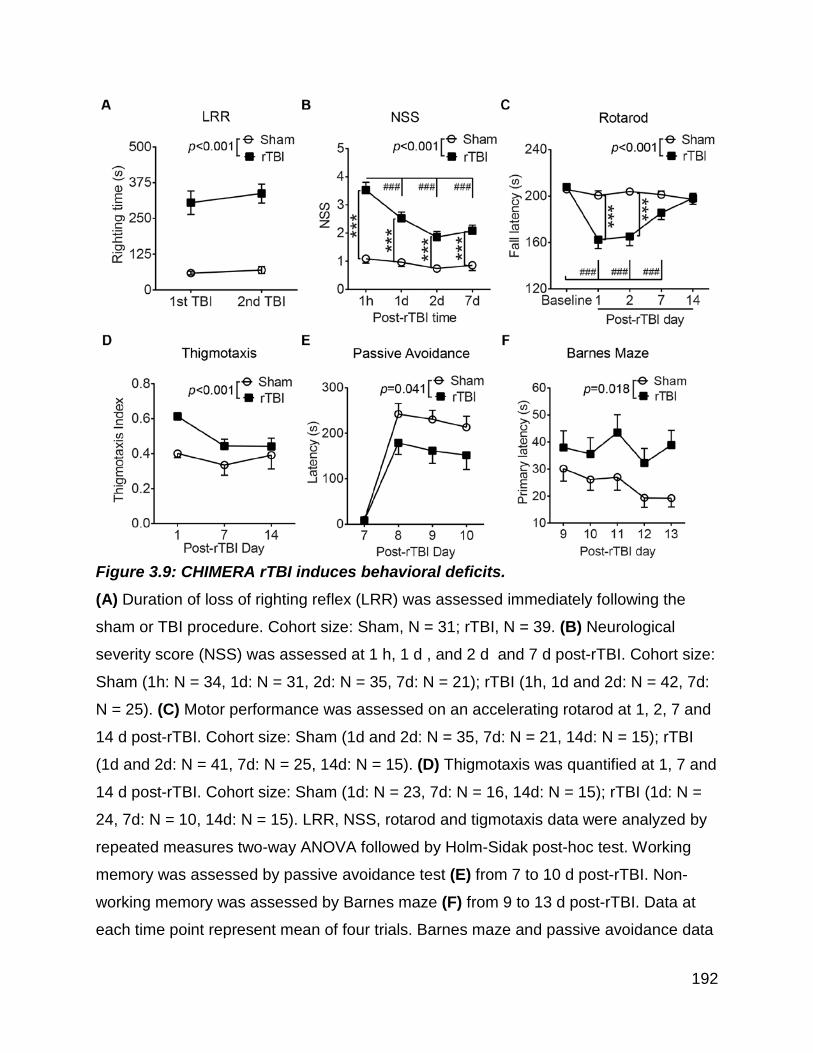
Figure 3.9: CHIMERA rTBI induces behavioral deficits. (A) Duration of loss of righting reflex (LRR) was assessed immediately following the
sham or TBI procedure. Cohort size: Sham, N = 31; rTBI, N = 39. (B) Neurological
severity score (NSS) was assessed at 1 h, 1 d , and 2 d and 7 d post-rTBI. Cohort size:
Sham (1h: N = 34, 1d: N = 31, 2d: N = 35, 7d: N = 21); rTBI (1h, 1d and 2d: N = 42, 7d:
N = 25). (C) Motor performance was assessed on an accelerating rotarod at 1, 2, 7 and
14 d post-rTBI. Cohort size: Sham (1d and 2d: N = 35, 7d: N = 21, 14d: N = 15); rTBI
(1d and 2d: N = 41, 7d: N = 25, 14d: N = 15). (D) Thigmotaxis was quantified at 1, 7 and
14 d post-rTBI. Cohort size: Sham (1d: N = 23, 7d: N = 16, 14d: N = 15); rTBI (1d: N =
24, 7d: N = 10, 14d: N = 15). LRR, NSS, rotarod and tigmotaxis data were analyzed by
repeated measures two-way ANOVA followed by Holm-Sidak post-hoc test. Working
memory was assessed by passive avoidance test (E) from 7 to 10 d post-rTBI. Non-
working memory was assessed by Barnes maze (F) from 9 to 13 d post-rTBI. Data at
each time point represent mean of four trials. Barnes maze and passive avoidance data
192

(Cohort size: sham, N = 15; rTBI, N = 15) were analyzed by repeated measures two-
way ANOVA. For all graphs, * indicates significant rTBI effect within time point and #
indicates significant time effect within each group. *, **, *** and ***** indicate p<0.05,
p<0.01, p<0.001, and p<0.0001, respectively. #, ### and #### indicate p<0.05,
p<0.001, and p<0.0001, respectively.
Figure 3.10: CHIMERA rTBI does not significantly affect general mobility. General mobility was tested by the open field test at 1, 7 and 14 d post-injury. There
was no rTBI effect at any time point for the total distance travelled (A) and time spent
immobile (B). Data are presented as the mean ± SEM and analyzed by repeated
measures two-way ANOVA followed by Holm-Sidak post-hoc test. In all graphs, *** and
# indicate significant time effect for rTBI (p<0.001) and sham (p<0.05) mice,
respectively.
3.4.3 CHIMERA rTBI Induces Widespread Diffuse Axonal Injury
Silver staining was used to assess post-rTBI axonal damage at 2, 7, and 14 d (Figure
3.11). rTBI brains revealed widespread axonal injury, as indicated by intense punctate
and fiber-associated argyrophilic structures in white matter tracts including the olfactory
nerve layer of the olfactory bulb, corpus callosum, and optic tracts (Figure 3.11 A and
B). Axonal injury was observed at both coup (corpus callosum) and contra-coup (optic
193

tract) regions, indicating a diffuse injury pattern. High-magnification of the affected areas
at 100X revealed numerous axonal varicosities (Figure 3.11 C), which is a characteristic
histological feature of human axonal pathology after TBI (Johnson et al., 2013).
Quantitative analysis revealed significant silver uptake in the injured olfactory nerve
layer (TBI effect: F(1, 27) = 16.89, p < 0.0001), which was maximum at 2d (p < 0.001)
and returned to sham levels over 7-14d (Figure 3.12 A). On the other hand, rTBI
induced persistent silver stain uptake in the corpus callosum (Figure 3.12 B, TBI effect:
F(1, 37) = 41.54, p < 0.0001; Time effect insignificant) (Figure 3.12 C). In optic tract,
silver uptake was most intense (Figures 3.12 B and C TBI effect: F(1, 36) = 107.4) and
there was a significant time and injury interaction (TBI x Time interaction: F(2, 36) =
11.66), indicating persistent increase in axonal degeneration in contrecoup regions.
194

Figure 3.11: CHIMERA rTBI induces diffuse axonal injury.
Axonal degeneration was assessed by silver staining at 2, 7 and 14 d post-rTBI. (A) Coronal sections showing white matter areas including olfactory nerve layer, corpus
195

callosum and optic tract with prominent silver staining indicated by black rectangles. (B) Representative 40X-magnified images of the same brain regions in sham-operated
(upper panel) or rTBI-induced (lower panel) animals. (C) 100X-magnified images of the
same brain regions in rTBI-induced animals. Axonal varicosities are indicated by
arrows.
Figure 3.12: CHIMERA rTBI induces sustained axonal injury
Silver stained images were quantified by calculating % of region of interest (ROI) in the
white matter tract area stained with silver stain. The bars indicate mean ± SEM percent
of ROI showing positive signal in sham and rTBI-induced animals in (A) olfactory nerve
layer, (B) corpus callosum and (C) optic tract. Data were analyzed using two-way
ANOVA followed Tukey post-hoc test. For all graphs * indicates rTBI effect within each
time point and # indicates time effect within each group. For all graphs, *, **, *** and ****
indicate p<0.05, p<0.01, p<0.001, and p<0.0001, respectively and ### and ####
indicate p<0.001, p<0.0001, respectively.
3.4.4 CHIMERA rTBI Induces Widespread Microglial Activation
Using Iba-1 immunohistochemistry, we observed significantly increased activated
microglia throughout several white matter tracts including the olfactory nerve layer,
corpus callosum, optic tracts, and brachium of superior colliculus of injured brains
compared to the sham controls as assessed using both fractal analysis and microglial
196

density (Figures 3.13 and 3.14). Quantification of microglial morphology by fractal
analysis revealed that microglia in sham animals displayed highly ramified and
extensively branched processes that are characteristic of the resting state (Figure 3.13
C, upper row, Figure 3.14 A-D, open bars). By contrast, microglia in the corpus
callosum, brachium of superior colliculus, and olfactory nerve layer of injured animals
had predominantly hypertrophic to bushy morphology with primary branches only,
whereas those in the optic tract showed amoeboid morphology characteristic of highly
activated microglia (Figure 3.13 C, lower row, Figure 3.14 A-D, black bars). Quantitative
analysis showed significant and persistent microglial activation in the injured olfactory
nerve layer (TBI effect: F(1, 35) = 13.64, p = 0.0008), optic tract (TBI effect: F(1, 37) =
9.77, p = 0.0034), corpus callosum (TBI effect: F(1, 38) = 29.51, p < 0.0001), and
brachium of superior colliculus (TBI effect: F(1, 38) = 24.5, p < 0.0001) as soon as 2d
(Figure 3.14 A-D).
In addition to changes in microglial morphology, we observed significant increases in
the number of microglia in the same white matter regions including the olfactory nerve
layer (Figure 3.14 E, TBI effect: F(1, 16) = 21.53, p = 0.0003), optic tract (Figure 3.14 G,
TBI effect: F(1, 16) = 90.30, p = 0.0001), corpus callosum (Figure 3.14 F, TBI effect:
F(1, 19) = 22.25, p = 0.0002) and brachium of superior colliculus (Figure 3.14 H, TBI
effect: F(1, 18) = 34.85, p < 0.0001), indicating that injury induced proliferation or
recruitment of immune cells. In olfactory bulb (Time effect insignificant), corpus
callosum (Time effect insignificant) and optic tract, microglia cell number was
persistently increased from 2d up to 14d (p < 0.05). In the brachium of superior
197

colliculus, a delayed but persistent increase in the Iba-1 positive cell number was
observed from 7d to 14d (p < 0.01).
198

Figure 3.13: CHIMERA rTBI induces widespread microglial activation.
Microglial activation was assessed using Iba-1 immunohistochemistry at 2 d post-rTBI.
(A) Representative images of Iba-1 stained coronal sections of olfactory bulb and brain.
Areas with prominent microglial activation are indicated by black rectangles. (B) Representative 40X-magnified images of olfactory never layer, corpus callosum, optic
199

tract, and brachium of superior colliculus showing resting microglia in sham brains
(upper row) and activated microglia in injured brains (lower row). (C) Representative
100X-magnified images showing the morphology of Iba-1-stained resting microglia in
sham (upper row) and activated microglia in rTBI (lower row) brains.
200

Figure 3.14: Quantitative analysis of microglial response to rTBI.
Bar graphs in the left column (A-D) indicate mean ± SD fractal dimension for microglial
morphology in (A) olfactory nerve layer, (B) corpus callosum, (C) brachium of superior
201

colliculus, and (D) optic tract. Bar graphs in the right column (E-H) show mean ± SD
number of Iba-1 positive cells per mm2 in the same white matter regions. Data were
analyzed by two-way ANOVA followed by a Tukey post-hoc test. Numbers inside the
bars indicate sample size. For all graphs, * indicates a significant rTBI effect within a
particular time point while # indicates a significant time effect within rTBI group. *: p <
0.05, **: p < 0.01, ***: p < 0.001, ****: p < 0.0001. ##: p < 0.01, ####: p < 0.0001.
3.4.5 CHIMERA rTBI Increases Proinflammatory Cytokine Levels
In addition to the microglial response, we also measured protein levels of the
proinflammatory cytokines TNFα and IL-1β in the half brain homogenates. Protein levels
TNFα (Figure 3.15 A) and IL-1β (Figure 3.15 B) were significantly higher at 2 d post-TBI
compared to the respective sham levels (p<0.01, t tests with Bonferroni corrections).
Figure 3.15: CHIMERA rTBI increases proinflammatory cytokine levels.
Values are expressed as mean % fold change in TNFα (A) and IL-1β (B) levels in rTBI
brain lysates compared to cytokine levels in the sham brain lysates at respective time
points. For both graphs, ** indicates p<0.01 in comparison of rTBI vs sham, using t-test
with Bonferroni correction of multiple comparisons (p = 0.05/5 = 0.01).
202

3.4.6 CHIMERA rTBI Increases Endogenous Tau Phosphorylation
We next assessed the phosphorylation levels of endogenous murine tau using three
antibodies directed against different phosphorylation sites, namely CP13 (pSer202),
RZ3 (pThr231), and PHF1 (pSer396 and pSer404). In addition, total murine tau levels
were determined by the antibody DA9. Western blot analysis showed significantly
increased phosphorylation of all the probed epitopes in rTBI brain lysates at 6 h, 12 h
and 2d compared to the respective sham brain lysates (Figure 3.16 A-C and G-I,
p<0.01, t tests with Bonferroni correction). The change in tau phosphorylation was due
to a significant increase in ratio of phosphorylated tau:total tau, but not as a result of
change in total tau level (Figure 3.16 D-F and G-I, p<0.01, t tests with Bonferroni
corrections).
203

Figure 3.16: CHIMERA rTBI increases endogenous tau phosphorylation.
The graphs in the left column (A-C) depict fold change in endogenous phosphorylated
tau levels in rTBI half-brain homogenates compared to the sham brains using antibodies
CP13 (pSer202 and pThr205), RZ3 (pThr231) and PHF1 (pSer396 and pSer404),
respectively. Graphs in the middle column (D-F) depict quantitation of phosphorylated
tau as a proportion of total tau (DA9). Representative immunoblots of phosphorylated
and corresponding total tau are depicted in the right column (G-I). Arrows on the left of
the blots indicate position of 66 kDa marker. Data are presented as the mean ± SEM
204

fold change in rTBI compared to the respective shams at each time point. For all
graphs, *, ** and *** indicate p<0.01 for the comparison of rTBI vs respective sham
values, using multiple t-tests with Bonferroni correction (p = 0.05/5 = 0.01).
3.5 Discussion
The major goal of this study was to develop a simple, reliable model of murine CHI that
replicates fundamental aspects of human impact TBI through precise delivery of known
biomechanical inputs. CHIMERA fulfills these criteria and offers several key advantages
over comparable rodent TBI models.
The first and foremost advantage is CHIMERA allows completely surgery-free induction
of TBI eliminating the need of longer anesthesia duration as well as sedatives such as
ketamine and xylazine (that were used in the weight drop model), as well as learning
complex surgery such as craniotomy that is required for FP and CCI models. Almost all
of the currently available TBI models require some surgery to deliver the mechanical
force to either exposed skull (e.g. in weight drop TBI) or to the dura (as in FP and CCI
models) that increases the learning curve of the operator. For example, a novice
learning FP injury may struggle at the beginning to perform the craniotomy without
disrupting the dura as well as gluing the hub for attaching fluid injection tube without
introducing the glue into the craniotomy site (Alder et al., 2011). Moreover, head
securing method such as a stereotaxic setup used for CCI model and surgery
themselves increase the duration of injury procedure. For example, the duration
required for weight drop injury model discussed in Chapter 2 is ~ 20 min. In contrast,
using CHIMERA a fully-trained individual can finish the whole procedure in ~ 4 and ~ 8
205

minutes with and without high speed videography, respectively. Shorter injury procedure
also reduces the duration of anesthesia exposure. Duration of anesthesia exposure is
important as general anesthetic may influence injury outcomes. For example, isoflurane,
the most commonly used general anesthetic for rodent surgery is shown to exert
neuroprotective actions in TBI (Statler et al., 2000; Statler et al., 2006).
A second advantage of CHIMERA is that it allows unrestricted head movement after
impact replicating clinically encountered head impact situations as opposed to other TBI
models (e.g. weight drop) in which head support systems restrict head movement to
varying degree (Namjoshi et al., 2013a). As discussed in Chapter 1, head acceleration,
rather the impact itself is thought to induce more brain damage. In support of this,
CHIMERA-rTBI induces robust and sustained axonal damage and significant
neuroinflammation compared to the weight drop rTBI as discussed in Chapter 2.
Third advantage is the very high reproducibility of mechanical inputs (i.e. piston velocity
and impact energy) as exemplified by high R2 values (~ 0.99) of the piston velocity vs
air pressure calibration curve (Figure 3.2). This reflected in the high reproducibility in the
head kinematic parameters with a reasonable cohort size (N = 8). By fine adjustment in
the air pressure, CHIMERA allows precise control over mechanical input. Moreover, the
biomechanical inputs can be delivered by changing variety of parameters (impact
energy, location, and direction) that make CHIMERA a highly versatile model to study
TBI under a variety of mechanical input conditions. Thus, by choosing appropriate
combination of piston mass (25 g, 50 g, 75 g or 100 g) and air pressure, CHIMERA can
206

deliver impacts over a wide energy range from 0.01 J to 19 J. The impact can be
delivered either in vertical direction that results in head acceleration in the sagittal plane
or in the horizontal direction resulting head movement in an oblique plane. By adjusting
the angle between body and the head, the impact can be delivered at the vertex or on
the posterior aspect of the skull. By using the horizontal configuration, the impact can be
delivered to the lateral side of the head.
Lastly, the build-in mechanism of CHIMERA allows simultaneous high-speed video
recording that can be used to measure head kinematics following impact. This is very
important for the validation of impact procedures.
In this study, a 50 g piston delivered an input impact with a kinetic energy of 0.5 J.
Calibration curves show that the 50 g piston has an energy range of 0.01 J to 14.0 J
(useful range for mouse TBI is 0.1 J to 1 J) in minimum steps of 0.01 J, with highly
reproducible performance (Figure 3.2). Adjusting piston mass and dimensions will allow
other biomechanical parameters to be controlled, so that it may be possible to
experimentally model the biomechanical conditions observed under different types of
human impact TBI.
High-speed video integrated into CHIMERA enables measurement of mouse head
kinematic parameters that can be scaled to humans. The most-commonly used method
for scaling kinematic parameters between humans and animals is based on the equal
stress/equal velocity approach (Holbourn, 1943; Gutierrez et al., 2001; Viano et al.,
207

2009). For this certain assumptions are made such as: 1) neurons, blood and CSF all
have about the same density as water, 2) brain tissue is extremely incompressible, 3)
brain tissue has small modulus of rigidity and thus it is extremely easy to change shape
of the brain, and 4) human and animal brains have similar density and geometry, are
elastic and are isotropic (i.e. uniform in all directions) (Holbourn, 1943). According to
this principle, velocity does not scale and is the same for human and animal data. We
used a scaling factor λ [λ = (mass of human brain / mass of mouse brain)1/3 = 13.8] to
estimate the human head-equivalent kinematic parameters from our animal data.
Although this scaling approach has been widely used in the study of impact
biomechanics (Holbourn, 1943; Gutierrez et al., 2001; Viano et al., 2009), and has been
applied to a rat model (Viano et al., 2009), it is important to be cautious in its
extrapolation as the human and rodent brains differ in geometry, white : grey matter
ratio, ventricular volume and position, and cortical folding. All these factors render the
“scaling” of rodent data to human to be an approximation at best. Under our
experimental conditions, behavioral and neuropathological changes reliably occurred at
lower scaled values of all kinematic parameters (except for impact duration) than those
reported for NFL concussions (Pellman et al., 2003a; Pellman et al., 2003b). Because
clinical mTBI can occur under many different circumstances (e.g., falls, passengers and
pedestrians in motor vehicle accidents, non-NFL sports), an important area for future
research will be to determine how various impact characteristics lead to functional and
biochemical changes. The precision and flexibility CHIMERA offers with respect to
impact parameters will help to refine the relationship between impact characteristics and
physiological outcomes.
208

An additional advantage of CHIMERA is that it overcomes much of the variability
observed with most closed-head injury models. Typical weight-drop models (Marmarou
et al., 1994; Flierl et al., 2009) have poor control over biomechanical input parameters,
such as friction and air resistance inside the guide tube that may contribute to variable
outcomes, high incidence of skull fractures, and limited dynamic range, thereby posing
challenges in comparing results from different laboratories (Namjoshi et al., 2013a).
Head movement during many CHI impact models is often at least partially restricted by
anchoring the head within a stereotaxic frame or by resting the animal on a foam
support. In a recent modification, Kane et al supported mice on a piece of aluminum foil
that ruptures upon impact and leads to a 180° rotation of the animal (Kane et al., 2012).
While this modification allows unrestricted head movement, it still includes possible
sources of variability including stretching of the aluminum foil before yielding to the force
generated by the weight-drop and less reliable and adjustable positioning of impact
location compared to CHIMERA.
Loss of consciousness for <30 min is one of the clinical criteria for mTBI (Carroll et al.,
2004) and the analogous measure in mice is LRR. Interestingly, a second TBI occurring
24h after the first impact did not prolong LRR time. Post-concussion patients may also
show balance difficulties or postural instability up to several days (Guskiewicz, 2003;
Blume et al., 2011; Putukian, 2011), as well as mood changes such as irritability or
anxiety (Blume et al., 2011; Daneshvar et al., 2011). Though general locomotor
activities were not severely affected, CHIMERA TBI resulted in deficits in fine motor
209

coordination and neurological performance. Similar changes have also been reported
by other groups (Tsenter et al., 2008; Mouzon et al., 2012). CHIMERA also increased
thigmotaxis in TBI animals, suggesting an anxiety-like behavior (Simon et al., 1994).
Our model also revealed deficits in both working and non-working memory as assessed
by passive avoidance test and Barnes maze, respectively.
Human and experimental TBI induces rapid neuroinflammatory responses as
demonstrated by changes in cytokine levels (e.g., IL-1β and IL-6) and microglial
activation (Taupin et al., 1993; Shohami et al., 1997; Rooker et al., 2006; Shein et al.,
2007; Shitaka et al., 2011; Perez-Polo et al., 2013). Under our conditions, rTBI led to
elevated IL-1β and TNF-α levels at 2 d after injury, which was accompanied by
histological evidence of microglial activation. Because our animal ethics committee
required the use of meloxicam for pre-emptive pain control, it is possible that an
inflammatory response occurring during the first few hours after impact (Taupin et al.,
1993; Shohami et al., 1997; Shein et al., 2007) was suppressed (Engelhardt, 1996). Iba-
1-positive activated microglia were particularly evident along white matter tracts
throughout the brain, whereas grey matter was essentially spared. Microglial activation
as assessed by fractal analysis and cell density was significant at 2d and persistent until
14d. However, as Iba-1 does not distinguish the source of immune cells, the increase in
cell number in this study may be due to proliferation of resident immune cells in the
brain or recruitment of immune cells from the periphery, or both.
210

Diffuse axonal injury (DAI) is one of the characteristic pathologies of TBI (Adams et al.,
1989; Johnson et al., 2013). Using silver staining, we observed increased argyrophilic
fibers and punctate structures in several white matter tracts across the brain, suggesting
a diffuse pattern of damaged axons. Axonal varicosities, a classical feature of DAI in
humans, were also present (Johnson et al., 2013). Interestingly, both white matter areas
that were close to (e.g., corpus callosum) and distant from (e.g., optic tract and olfactory
nerve layer) the impact site were affected, suggesting coup and contra-coup injuries are
present in our model. Affected white matter areas, including the corpus callosum, optic
tract, and the olfactory system, have been reported in other CHI models that induce
impact at the superior side of skull (Foda and Marmarou, 1994; Creeley et al., 2004;
Shitaka et al., 2011; Namjoshi et al., 2013b). Several white matter areas, including
olfactory nerve layer, corpus callosum and optic tract showed both increased
argyrophilic staining and microglial activation, suggesting a possible relationship
between axonal damage and neuroinflammation, in agreement with previous reports
(Ciallella et al., 2002; Shitaka et al., 2011; Lin and Wen, 2013). Interestingly, axonal
injury in corpus callosum and optic tract continued to increase over 14 days while
microglial activation resolved in all white matter tract areas over 2-7 days. On the other
hand, axonal injury was resolved in the olfactory neuronal layer within 7 days
suggesting efficient neural repair/neuroregeneration in this white matter region. Future
studies will characterize the dynamics of post-rTBI axonal damage over long-term follow
up (up to 6 months).
211

Hyperphosphorylation of the cytoskeletal protein tau is a pathological event observed in
many neurodegenerative diseases including AD (Ballatore et al., 2007) and CTE
(McKee et al., 2013). In our model, we demonstrated that tau hyperphosphorylation is
an early event after rTBI in wild-type mice, again in agreement with other models of CHI
(Genis et al., 2000; Goldstein et al., 2012). It should, however be noted that post-TBI
changes in phosphorylation of murine tau does not predict whether human tau will show
similar dynamics. Further experiments using transgenic human tau mice will be required
to investigate the influence of rTBI on tau deposition.
Future studies will be conducted to characterize the relationships between kinematics
and the resulting behavioral and neuropathological responses across a variety of impact
parameters. The significant advantages CHIMERA offers over comparable rodent TBI
models are expected to facilitate the acquisition of preclinical data with improved
relevance to human TBI, thereby accelerating the pace of successful research to
understand the mechanisms of TBI and to develop effective therapeutic approaches for
this devastating condition.
212

Chapter 4: Conclusions and Future Directions
The experiments described in Chapter 2 of this thesis were designed to assess the role
of apoE in the recovery from TBI. As discussed in the introductory chapter, TBI, while a
pathological entity in itself, is also suggested to increase risk of dementia in TBI
survivors. Although the cause and effect mechanisms linking these two pathological
conditions are still unclear, several observations common to both diseases may help to
understand the relationship between the two. For example, post-mortem analyses of
traumatized human brains have revealed formation of diffuse Aβ deposits and NFTs,
which are the two pathological hallmarks of AD. Neurons are rich in lipids, which play
crucial role in neuronal function as well as synaptogenesis. Thus it is conceivable that
the brain lipid metabolism, which is based on apoE may serve important roles in
neuronal recovery following TBI. While the apoE4 isoform is the best established
genetic risk factor for AD, the association between apoE and TBI is still unclear.
Epidemiological data suggests that apoE may influence post-TBI recovery. The role of
apoE in AD pathogenesis stems from its role in Aβ metabolism, inflammation, and
synaptic remodeling. We and other groups have previously shown that ABCA1-
mediated lipidation of apoE is crucial for its role in Aβ clearance. Thus enhancing apoE
lipidation and function may be beneficial in AD pathology which is reflected by the
beneficial effects of LXR agonists on facilitating Aβ clearance and improving cognitive
function in AD mouse model.
213

Based on these observations we hypothesized that LXR agonists may have improved
recovery from TBI through apoE-based mechanisms. As described in Chapter 2 of this
thesis, we tested this hypothesis by treating wild-type and apoE-/- mice that were
subjected to mrTBI using popular weight drop method with a synthetic LXR agonist
GW3965.
4.1 Chapter 2: Conclusions
The first objective of this thesis was to test the therapeutic utility of synthetic LXR
agonist, GW3965 in mild, repetitive TBI (mrTBI), which is described in Chapter 2. I
found that GW3965 treatment improves NOR performance as well as promotes axonal
recovery following mrTBI and these outcomes require apoE. I further found that
GW3965 can also suppress the elevation of Aβ following TBI, although surprisingly this
outcome was apoE-independent. The overall results from this study suggest that
synthetic LXR agonists may be a promising pharmacological treatment for TBI.
Moreover, we observed treatment effects on the above-mentioned the post-TBI deficits
as early as two days following GW3965 administration, suggesting that LXR agonists
may be beneficial in the management of acute post-TBI phase.
We found that mrTBI induced significant NOR memory deficits in untreated WT as well
as apoE-/- mice that persisted from 2 to 14 days. While GW3965 treatment restored
NOR memory deficits in WT animals, it failed to show this beneficial effect in apoE-/-
mice. This indicates that apoE is required for the beneficial effect of GW3965 in post-
TBI cognitive deficits. Previously thought to assess pure working memory, NOR task is
214

now suggested to test episodic memory of the animal (Ennaceur, 2010). Although the
exact neural circuits for NOR task are not fully understood, studies indicate that
ventromedial prefrontal cortex (Akirav and Maroun, 2006) and hippocampus (Broadbent
et al., 2010) are essential for object recognition.
We tested TBI-induced motor deficits on an accelerating rotarod. The rationale for using
accelerating vs constant speed rotarod was that in the former method, the accelerating
rod constantly challenges the animal requiring motor coordination and planning while
the later may suffer from testing endurance rather than motor coordination (Deacon,
2013). Our TBI model induced impairment in finer motor tasks, such as motor
coordination and planning. While the exact mechanism for spontaneous recovery of
motor function observed in this study is presently unknown, it is possible that with the
repetitive rotarod testing after TBI we may have inadvertently introduced motor training
akin to post-TBI rehabilitation program in humans. It is well known that repeated motor
tasks promote development of spinal neural circuits, called central pattern generators
that when activated can produce rhythmic motor patterns such as walking in the
absence of sensory or descending inputs from brain (Marder and Bucher, 2001). The
above-mentioned possibility is further supported by a recent study, in which a 30 min
daily bilateral movement training in mice using the rotarod for 33 days improved motor
function and promoted axonal remodeling and rewiring of the corticospinal tract (the
major motor pathway that controls fine movements and posture) after unilateral
contusion of the motor cortex (Nakagawa et al., 2013). The authors further showed that
rotarod training increased neural activity in the spinal cord laminae VII, VIII and X. The
215

commissural interneurons located in these laminae of the rodent spinal cord are
involved in generating locomotor-like activity (Kjaerulff and Kiehn, 1996; Kiehn and
Kjaerulff, 1998). We further observed that loss of apoE further exacerbated the
magnitude of motor impairment following mrTBI. Intriguingly, GW3965 treatment neither
affected the severity of motor deficits nor the kinetics of spontaneous recovery. Future
studies will be necessary to assess specific motor pathways affected in our TBI model,
changes in the plasticity and remodeling of these neurons and role of apoE in these
processes.
Untreated WT brains showed a transient increase in ABCA1, the principal LXR target
protein that peaked at 7 days and dropped back to sham levels by day 14. It is
hypothesized that the damaged neurons release lipids that are taken up by the viable
cells as shown by increased levels of LDLR, a major apoE receptor with no change in
ABCA1 in first 3 days after CCI injury (Loane et al., 2011). In the same model, ABCA1
levels peaked at 7 days with corresponding decrease in LDLR (Loane et al., 2011). In
our study, while the LDLR levels were not affected at any post-TBI time point, the
ABCA1 dynamics was similar to that reported in the CCI model, i.e. with no change at 2
day post-TBI and peaking by day 7. This delayed increase ABCA1 may indicate
cholesterol efflux by the viable cells that can be used for synaptic repair and remodeling
during post-TBI recovery. Interestingly, unlike WT mice we did not see any changes in
ABCA1 levels in untreated apoE-/- mice following TBI. This suggests that the
endogenous signals leading to upregulation of ABCA1 might be compromised by the
absence of apoE. Tall and coworkers have shown that interaction of ABCA1 with apoA-
216

I, the peripheral counterpart of apoE, at the cell membrane inhibits calpain-mediated
degradation of ABCA1 that results in increase in the both cell surface expression and
activity of ABCA1 (Martinez et al., 2003). Extending this concept, it is also possible that
ABCA1 turnover is increased in the CNS leading to its rapid degradation in the absence
of apoE (Krimbou et al., 2004). As expected, GW3965 treatment caused sustained
increase in ABCA1 in both WT and apoE-/- mice.
We further found that neither TBI alone nor GW3965 treatment had any effect on apoE
levels in WT mice. This result is consistent with the relative insensitivity of apoE
compared to ABCA1 as a LXR target (Donkin et al., 2010). Since we determined apoE
levels in the whole half-brain extracts, it is possible that regional changes in the apoE
levels, if any may have been missed. While we did not assess the lipidation status of
apoE, it is also possible that even though the total apoE pool does not change per say
the ABCA1-mediated lipidation of the available apoE may have been sufficient to
promote recovery mechanisms after mild injury. More studies can be done to assess
whether the same holds true in case of moderate-to-severe TBI. Moreover, the
contribution of ABCA1 in LXR agonist-mediated recovery may be more important as
recently shown by our group that ABCA1 is required for the beneficial effects of
GW3965 in AD mice (Donkin et al., 2010). This hypothesis can be tested in future
studies using ABCA1-/- mice.
Surprisingly, contrary to our hypothesis GW3965 treatment blocked Aβ elevation even
in the absence of apoE. While, it is generally agreed that apoE modulates Aβ clearance
217

through enzymatic degradation as well as clearance through cerebrovasculature recent
data suggests that the interaction of apoE with Aβ and its influence on Aβ metabolism is
complex than thought (see section 1.6.4.7). Our observation indicates that LXR
activation may engage compensatory mechanisms in the absence of apoE to enhance
Aβ clearance. A potential candidate for such compensatory mechanism is apoA-I, the
peripheral counterpart of apoE. Although apoA-I is not synthesized in the brain, it is
present in the CSF at relatively high levels of 0.1%-0.5% relative to plasma levels,
suggesting that it crosses BBB (Stukas et al., 2014). A recent study from our lab has
shown that apoA-I levels remain significantly higher in the CNS than in plasma in
ABCA1-/- mice and that 8-week treatment of AD mice with GW3965 causes a dramatic
increase in the cerebral apoA-I levels (Stukas et al., 2011). In the present study we did
not observe any changes in brain as well as CSF apoA-I levels (data not shown) either
following TBI or with GW3965 treatment, which may be due to short duration of
treatment compared to long-term treatment (Stukas et al., 2011). Nonetheless, recent
studies suggest apoA-I as another apolipoprotein that may play important role in injured
brain for several reasons. First, AD mice that lack apoA-I show increased
cerebrovascular amyloid deposits and impaired spatial learning and memory function
(Lefterov et al., 2010) whereas overexpression of human apoA-I protects age-
associated cognitive deficits and lowers neuroinflammation and CAA (Lewis et al.,
2010). Interestingly, deletion or overexpression of apoA-I seem to affect Aβ and amyloid
deposits only in the cerebrovasculature and not in the brain parenchyma (Lefterov et al.,
2010; Lewis et al., 2010) suggesting that role of apoA-I may be more prominent in the
maintenance of the cerebrovasculature (Stukas et al., 2014). Second, in vitro studies
218

have shown that apoA-I binds to Aβ (Koldamova et al., 2001), prevents Aβ aggregation
and reduces Aβ-induced cytotoxicity, oxidative stress, and neurodegeneration (Lefterov
et al., 2010; Lewis et al., 2010). Lastly, serum apoA-I levels were recently shown to be
elevated in patients with mTBI and assessment serum apoA-I levels in combination with
S100B, an astrocyte-specific protein has been proposed to be a useful biomarker for
classification of mTBI (Bazarian et al., 2013). ApoA-I therefore may have more
important role in the CNS function and lipid metabolism than it is appreciated (Stukas et
al., 2014). Thus, future studies will be designed to get a deeper insight into the role of
apoA-I in the brain, which may offer novel approaches to promote neuronal recovery
after TBI.
4.2 Chapter 2: Study Limitations and Future Directions
While the results from this study indicate that synthetic LXR agonists may offer
beneficial therapeutic effects in TBI partly through apoE-mediated mechanism there are
several limitations to this study that may help guide future studies.
Although the LXR stimulation offers beneficial effects in mrTBI, current LXR agonists
have safety issues such as hypertriglyceridemia and hepatic steatosis (Grefhorst et al.,
2002; Groot et al., 2005) that has virtually precluded the development of LXR agonists
for chronic diseases. To date, only one Phase I study of a synthetic LXR agonist, LXR-
623, in humans has been reported (Katz et al., 2009). In this study, the safety,
pharmacokinetic and pharmacodynamic profile of LXR-623, was studied in healthy
human volunteers after a single ascending dose. Although the authors reported a
219

favorable safety profile with dose-dependent increase in ABCA1 and ABCG1
expression, further development of this compound was discontinued due to the
development of adverse CNS events at the highest doses used (150 and 300 mg),
including confusion, forgetfulness, decreased comprehension, lightheadedness,
drowsiness, paresthesias, palpitations, decreased concentration, altered time
perception and paranoid ideation (Katz et al., 2009). It is therefore of interest to develop
strategies that can retain the beneficial effects of LXR activation and circumvent their
unwanted side effects. For example, development of LXRβ-selective agonists is an
active area of research, as LXRα underlies much of the undesired lipogenic effects
(Quinet et al., 2006). Another promising approach is to develop tissue-specific LXR
agonists or antagonists, as the magnitude of induction of LXR response genes varies
considerably among tissues (Brunham et al., 2006). As shown in this study, the
beneficial effects of LXR agonist were seen even in the acute post-TBI phase, which
suggests that these agents may be useful for short-term rather than chronic treatment,
which could improve functional recovery as well as decrease long-term AD risk. While
studying long-term effects of LXR treatment was beyond the scope of this thesis project,
future studies could be designed for this. Finally, less potent compounds or compounds
with LXR-agonist like activity may offer another approach, as proton pump inhibitors
have some LXR activity in murine primary astrocytes and human glial-derived cell lines
(Cronican et al., 2010).
Our study suggests that apoE is not necessary for LXR-mediated suppression of
endogenous Aβ. While the amyloid plaques are one of the pathological hallmarks of AD,
220

the overall influence of Aβ in TBI pathology is questionable as amyloid deposits are
found in only 30% of the clinical TBI cases. On the other hand, tau
hyperphosphorylation is common neuropathology of TBI, which suggests that the
cascade of events that occur following TBI may lead directly to tau pathogenesis
independent of Aβ. This hypothesis is supported by a recent study where tau pathology
still occurs when Aβ production is blocked (Tran et al., 2011a). Although in the present
study we did not undertake systematic examination tauopathy using specific anti-tau
antibodies, the possibility of tauopathy may come from indirect evidence. For example,
silver uptake is associated with both 3-repeat and 4-repeat tau deposits (Uchihara et al.,
2005). Silver uptake into damaged axons in white matter tracts is by far the most
sensitive histological marker of injury in our weight drop mrTBI model as it is detectable
even in the absence of marked microglial activation or cell death. This suggests that
these white matter tracts may be sites where tau pathology begins after mrTBI. This
possibility is further supported by robust silver uptake along with increased tau
phosphorylation observed in CHIMERA-TBI (discussed in the next section). Moreover,
apoE-/- mice clearly show profoundly greater and prolonged silver staining of damaged
axons, suggesting that apoE’s role in repair of damaged neuronal membranes may
(in)directly influence pathways that lead to tau phosphorylation. Supporting this
hypothesis is our observation that promoting apoE function with GW3965 suppresses
silver uptake in WT but not in apoE-/- mice. The influence of apoE on tau
phosphorylation is unclear although the current evidence suggests that apoE may inhibit
tau phosphorylation by inhibition of tau kinases, including GSK3β (Ohkubo et al., 2003;
221

Hoe et al., 2006). Thus, it will be now important to determine the interaction between
murine as well as human apoE on tau phosphorylation in mrTBI.
Last but not least, although we used the widely-used weight drop model in this study,
this model has several caveats, including poor control over mechanical parameters and
variable injury outcomes (discussed in Chapter 1). Although currently underappreciated,
rigorous biomechanical validation of TBI models is essential to minimize variations in
the outcomes and increase their translational potential (Namjoshi et al., 2013a). As
described in the Discussion section of Chapter 2, we did not conduct a systematic
biomechanical evaluation of the weight drop model. This strongly motivated us to
develop a novel TBI model that can mitigate the caveats associated with the weight
drop model, as well as allow us to study injury biomechanics. Accordingly in
collaboration with the biomechanical engineers at the University of British Columbia, we
developed a novel neurotrauma model called CHIMERA that uses completely non-
surgical procedure to induce closed head injury in mice. In the Chapter 3 of this thesis, I
have discussed the biomechanical, functional as well as neuropathological
characterization of CHIMERA.
As will be discussed in the next section of this Chapter, we now have a novel, simple
yet highly versatile TBI model that can reliably mimic some neuropathological features
of human TBI with high experimental reproducibility. Based on the results discussed in
this thesis, we believe that CHIMERA will prove a highly useful and biofidelic model for
conducting future studies including those proposed above.
222

4.3 Chapter 3: Conclusions
The study described in Chapter 3 was not driven by a specific hypothesis but rather
describes characterization of a new, simple, and reliable model of murine CHI,
CHIMERA that replicates fundamental aspects of human impact TBI through precise
delivery of known biomechanical inputs.
CHIMERA is completely nonsurgical and requires only isoflurane anesthesia, therefore
enabling immediate neurological severity assessments using LRR and NSS measures.
Being nonsurgical, CHIMERA is ideal for studies investigating multiple impacts as well
the long-term consequences of impact TBI. These advantages overcome many
limitations of surgically-induced TBI models, including longer anesthetic exposure as
well as inclusion of opioid analgesics (e.g., buprenorphine) and sedatives like xylazine
that can interfere with rapid neurological assessment, low throughput, extensive
operator training, and limited suitability for studies involving repetitive TBI or long-term
TBI outcomes. CHIMERA produces injury using a simple, reliable and semi-automated
procedure that requires < 10 min per animal to produce defined injury. As the
biomechanical input parameters are highly adjustable, i.e., across impact energy,
velocity, and direction, CHIMERA offers a wide dynamic range of precisely controllable
inputs to reproduce the conditions that occur in human impact TBI. Importantly, the
kinematic analyses facilitated by CHIMERA enable head motion parameters to be
integrated with behavioral and neuropathological outcome measures, potentially
enabling improved relevance to human TBI. CHIMERA produces diffuse injury
223

characterized by activation of inflammatory reactions, axonal damage, and tau
phosphorylation, replicating many aspects of human impact TBI without overt focal
damage. Taken together, these attributes make CHIMERA a valuable new model to
investigate the mechanisms of TBI and for use in preclinical drug discovery and
development programs.
We found several differences in the neuropathological outcomes of CHIMERA-rTBI
compared to the weight drop rTBI. Thus, silver stain data revealed that the axonal injury
in CHIMERA-rTBI was different than that in weight drop rTBI in the terms of extent,
intensity and dynamics. In the weight drop rTBI, for example the axonal injury was
restricted in the coup and contrecoup white matter tracts below the impact site. In the
CHIMERA-rTBI in addition to the coup-contrecoup spread, we found significant axonal
pathology at the site distant from the impact site, such as olfactory nerve layer.
Qualitative comparison revealed robust axonal pathology in CHIMERA-rTBI compared
to mild axonal injury in weight drop rTBI. Lastly, while the axonal pathology in weight
drop rTBI resolved over 7-14 days, it continued to increase over the same duration in
CHIMERA-rTBI suggesting ongoing secondary injury pathways that may have
overwhelmed endogenous recovery mechanisms. CHIMERA-rTBI induced robust
microglial activation in the white matter tracts along with moderate increase in the
cytokine levels. On the other hand both microglial activation and elevation of cytokine
levels were absent in weight drop rTBI. There are several possibilities for these
differences. For example the mechanical inputs as well as head support system used in
these two models were different that may have affected head kinematics. For example,
224

while we did not systematically assessed head kinematics in the weight drop rTBI
model, a preliminary high-speed video analysis revealed almost no head moment but a
crush type of injury following weight drop (data not presented). On the other hand
CHIMERA-rTBI allows free head movement that translates into robust, widespread and
sustained axonal injury coupled with significant microglial activation and elevated
cytokine levels. Moreover, we cannot rule out influence of longer duration of anesthesia
as well as effects of ketamine, xylazine, and bupivacaine on the secondary injury
pathways in weight drop rTBI. Lastly, while CHIMERA-rTBI induced a transient increase
in phosphorylation of endogenous tau, tau phosphorylation in weight drop rTBI was
variable (data not shown).
Comparisons of weight drop rTBI and CHIMERA-rTBI models are summarized in the
Table 4.1
225

Weight Drop rTBI CHIMERA rTBI TBI Procedure: TBI Procedure: Surgery required Surgery-free Total duration of isoflurane anesthesia: ~ 20 min
Total duration of isoflurane anesthesia: ~ 4 min (without videography) and ~ 8 min (with videography)
Local anesthetic used: Bupivacaine No local anesthetic required Analgesics used: Meloxicam Analgesics used: Meloxicam Sedatives used: Ketamine and xylazine No sedatives required Total duration of injury procedure: ~ 20 min/animal
Total duration of injury procedure: 4-8 min/animal
Duration of recovery from anesthesia: 60-90 min
Duration of recovery from anesthesia: 6-10 min
Mortality rate: 10-15 % Mortality rate: 3-4 % Input Parameters and Head Kinematics: Input Parameters and Head Kinematics: Drop height is controlled, however friction and air resistance can have unpredictable influence on the velocity of gravity-driven mass
Input parameters are precisely controlled. Piston velocity is maintained throughout the piston travel by constant air pressure.
The head support system allows virtually no head movement following impact
The head support system allows free head movement following impact
Head requires to be held manually under the guide tube that can influence head kinematics following impact
Head does not require manual holding
High-speed videography requires manual trigger and cannot be easily integrated into the injury procedure
High-speed videography is triggered by CHIMERA impactor and is easily integrated into the injury procedure
Proposed Injury Mechanism Proposed Injury Mechanism Impact-head crush/compression Impact-head acceleration Behavioral Outcomes Behavioral Outcomes Injury induced cognitive deficits from 2-14 d as assessed by NOR
Injury induced cognitive deficits from 2-14 days as assessed by passive avoidance test and Barnes maze
Injury induced motor deficits from 1-7 d with spontaneous recovery by 14 d.
Injury induced motor deficits from 1-7 d with spontaneous recovery by 14 d.
Neuropathological Outcomes Neuropathological Outcomes Axonal injury restricted to coup-contrecoup white matter under the impact site
Widespread axonal injury including coup-contrecoup white matter as well as site distant from the impact (olfactory bulb)
Mild axonal injury Robust axonal injury Axonal injury in most of white matter resolves over 7-14 days
Axonal injury continues to increase over 2-14 days
No microglial activation Robust microglial activation Biochemical Outcomes Biochemical Outcomes No change in the proinflammatory cytokines Moderate and transient increase in the
proinflammatory cytokine levels Variable changes in phosphorylation of endogenous tau
Transient but significant increase in endogenous tau phosphorylation
Table 4.1: Comparison of weight drop-rTBI and CHIMERA-rTBI models.
226

4.4 Chapter 3: Study Limitations and Future Directions
While CHIMERA offers several advantages over traditional TBI models, we also
acknowledge several limitations of the present study, which stem principally from the
very nascent nature of the model. An interesting aspect of this study was although
several behavioral, inflammatory, and tau responses mostly returned to baseline by 14
days post-injury using the injury parameters used in this study, cognitive performance
and axonal pathology remained robust. Understanding the temporal response of the
various behavioral, biochemical and neuropathological outcomes used in this study
under a variety of injury conditions is now a very important future topic. Thus, the injury
characteristics using CHIMERA can be varied by independently adjusting various
parameters (e.g., impact energy, number of impacts, duration between successive
impacts, impact location and impact direction). Such assessment was beyond the scope
of this thesis.
While CHIMERA-TBI allows free head movement following TBI, high speed videos
revealed that the impact induced significant hyperflexion of the animal’s neck. It is
possible the frontal Velcro straps used to restrain the animal body may lead to
significant strain in the neck during such hyperflexion that can result in the spinal cord
injury. Indeed during the pilot study, post-mortem examination revealed that
approximately 30% of mice injured by CHIMERA develop visible contusions of the
cervical and thoracic spinal cord and these animals have significantly worse motor
behavior. Moreover the most common reason for the post-TBI mortality (~ 3.75%)
227

associated with CHIMERA was the development of hind limb paralysis. While seemingly
a caveat of the model, this can be exploited to develop a neurotrauma model that
induces either TBI alone or TBI along with spinal cord injury (SCI). Accordingly, future
studies are designed to control neck hyperflexion. One way to achieve this is by
gradually slowing head acceleration after peak acceleration phase (that is thought to
cause TBI) using soft foam. The utility of such model is further highlighted by recent
data indicating that TBI is a common co-occurring injury with SCI (Macciocchi et al.,
2008). Moreover, up to 30% of neurotrauma cases may also have some degree of
combined TBI/SCI, which can go undiagnosed and untreated relative to the most
obvious injury (Povolny and Kaplan, 1993). To our knowledge, there is only one animal
model of combined TBI/SCI that uses a double surgical method to induce separate TBI
and SCI (Inoue et al., 2013). By controlling neck flexion, we will be in a unique position
to study either TBI or combined TBI and SCI without any surgical manipulations in a
single model.
A major limitation of the present study is short post-TBI follow-up time. While inclusion
of longer post-injury time points was beyond the scope of this thesis, ongoing and future
studies are designed to address this. While thigmotaxis indicates only anxiety-like
behavior, more appropriate tests such as elevated plus maze will be used in future to
assess post-TBI anxiety.
Ongoing and future studies are also designed to undertake a detailed injury
characterization of CHIMERA to test influence of various input parameters such as full
228

dynamic range of CHIMERA, comparison of vertical vs lateral head impact and varying
the number or intervals of repetitive impacts. As mentioned previously, the wide impact
energy range and relatively simple design makes CHIMERA easily adaptable for
studying TBI in large rodents such as rats, which have been the traditional rodent model
of choice for most TBI studies due to their superior behavioral repertoire and larger
brains for imaging. Future studies are planned to compare injury responses in mice and
rats using CHIMERA.
Notwithstanding these limitations, the present study indicates that CHIMERA will be a
valuable neurotrauma model for conducting both the mechanistic as well as
pharmacological studies.
Taken together, the first study described in this thesis shows that the synthetic LXR
agonists may be further developed as a potential treatment for mild TBI. While this
study indicates that the beneficial effects of LXR in TBI are mediated partially through
apoE-dependent mechanisms there are several unanswered questions related to the
mechanism. We believe that these questions can be addressed in future studies using a
highly innovative neurotrauma model, CHIMERA described in the second study of this
thesis.
229

Bibliography
Abbott NJ, Ronnback L, Hansson E (2006) Astrocyte-endothelial interactions at the
blood-brain barrier. Nat Rev Neurosci 7:41-53.
Abildayeva K, Jansen PJ, Hirsch-Reinshagen V, Bloks VW, Bakker AHF, Ramaekers
FCS, de Vante J, Groen AK, Wellington CL, Kuipers F, Mulder M (2006) 24(S)-
hydroxycholesterol participates in a liver X recpetor-controlled pathway in astrocytes
that regulates apolipoprotein E-mediated cholesterol efflux. J Biol Chem 281:12799-
12808.
Adams JH (1980) Brain damage in fatal non-missile head injury in man. Amsterdam:
Elsevier Publishing company.
Adams JH, Doyle D, Graham DI, Lawrence AE, McLellan DR (1986) Gliding contusions
in nonmissile head injury in humans. Arch Pathol Lab Med 110:485-488.
Adams JH, Doyle D, Ford I, Gennarelli TA, Graham DI, McLellan DR (1989) Diffuse
axonal injury in head injury: definition, diagnosis and grading. Histopathology 15:49-59.
Adelson PD, Wisniewski SR, Beca J, Brown SD, Bell M, Muizelaar JP, Okada P, Beers
SR, Balasubramani GK, Hirtz D (2013) Comparison of hypothermia and normothermia
after severe traumatic brain injury in children (Cool Kids): a phase 3, randomised
controlled trial. Lancet Neurol 12:546-553.
230

Adeosun SO, Hou X, Zheng B, Stockmeier C, Ou X, Paul I, Mosley T, Weisgraber K,
Wang JM (2014) Cognitive deficits and disruption of neurogenesis in a mouse model of
apolipoprotein E4 domain interaction. J Biol Chem 289:2946-2959.
Adle-Biassette H, Duyckaerts C, Wasowicz M, He Y, Fornes P, Foncin JF, Lecomte D,
Hauw JJ (1996) Beta AP deposition and head trauma. Neurobiol Aging 17:415-419.
Aggerbeck LP, Wetterau JR, Weisgraber KH, Wu CS, Lindgren FT (1988) Human
apolipoprotein E3 in aqueous solution. II. Properties of the amino- and carboxyl-terminal
domains. J Biol Chem 263:6249-6258.
Akirav I, Maroun M (2006) Ventromedial prefrontal cortex is obligatory for consolidation
and reconsolidation of object recognition memory. Cereb Cortex 16:1759-1765.
Alder J, Fujioka W, Lifshitz J, Crockett DP, Thakker-Varia S (2011) Lateral fluid
percussion: model of traumatic brain injury in mice. J Vis Exp:e3063.
Alonso AdC, Li B, Grundke-Iqbal I, Iqbal K (2006) Polymerization of
hyperphosphorylated tau into filaments eliminates its inhibitory activity. Proc Natl Acad
Sci U S A 103:8864-8869.
Andersson S, Gustafsson N, Warner M, Gustafsson JA (2005) Inactivation of liver X
receptor beta leads to adult-onset motor neuron degeneration in male mice. Proc Natl
Acad Sci U S A 102:3857-3862.
231

Andoh T, Kuraishi Y (2003) Nitric oxide enhances substance P-induced itch-associated
responses in mice. Br J Pharmacol 138:202-208.
Andreadis A, Brown WM, Kosik KS (1992) Structure and novel exons of the human tau
gene. Biochemistry 31:10626-10633.
Antunes M, Biala G (2012) The novel object recognition memory: neurobiology, test
procedure, and its modifications. Cogn Process 13:93-110.
Apelt J, Ach K, Schliebs R (2003) Aging-related down-regulation of neprilysin, a putative
beta-amyloid-degrading enzyme, in transgenic Tg2576 Alzheimer-like mouse brain is
accompanied by an astroglial upregulation in the vicinity of beta-amyloid plaques.
Neurosci Lett 339:183-186.
Armstrong RA (1994) beta-Amyloid (A beta) deposition in elderly non-demented
patients and patients with Alzheimer's disease. Neurosci Lett 178:59-62.
Auboeuf D, Rieusset J, Fajas L, Vallier P, Frering V, Riou JP, Staels B, Auwerx J,
Laville M, Vidal H (1997) Tissue distribution and quantification of the expression of
mRNAs of peroxisome proliferator-activated receptors and liver X receptor-alpha in
humans: no alteration in adipose tissue of obese and NIDDM patients. Diabetes
46:1319-1327.
232

Avila J, Lucas JJ, Perez M, Hernandez F (2004) Role of tau protein in both physiological
and pathological conditions. Physiol Rev 84:361-384.
Ballatore C, Lee VM, Trojanowski JQ (2007) Tau-mediated neurodegeneration in
Alzheimer's disease and related disorders. Nat Rev Neurosci 8:663-672.
Banos JH, Novack TA, Brunner R, Renfroe S, Lin HY, Meythaler J (2010) Impact of
early administration of sertraline on cognitive and behavioral recovery in the first year
after moderate to severe traumatic brain injury. J Head Trauma Rehabil 25:357-361.
Barnes DE, Kaup A, Kirby KA, Byers AL, Diaz-Arrastia R, Yaffe K (2014) Traumatic
brain injury and risk of dementia in older veterans. Neurology 83:312-319.
Basak JM, Verghese PB, Yoon H, Kim J, Holtzman DM (2012) Low-density lipoprotein
receptor represents an apolipoprotein E-independent pathway of Abeta uptake and
degradation by astrocytes. J Biol Chem 287:13959-13971.
Bateman RJ, Munsell LY, Morris JC, Swarm R, Yarasheski KE, Holtzman DM (2006)
Human amyloid-beta synthesis and clearance rates as measured in cerebrospinal fluid
in vivo. Nat Med 12:856-861.
Baugh CM, Robbins CA, Stern RA, McKee AC (2014) Current understanding of chronic
traumatic encephalopathy. Curr Treat Options Neurol 16:306.
233

Bayly PV, Cohen TS, Leister EP, Ajo D, Leuthardt EC, Genin GM (2005) Deformation of
the human brain induced by mild acceleration. J Neurotrauma 22:845-856.
Bazarian JJ, Blyth BJ, He H, Mookerjee S, Jones C, Kiechle K, Moynihan R, Wojcik SM,
Grant WD, Secreti LM, Triner W, Moscati R, Leinhart A, Ellis GL, Khan J (2013)
Classification accuracy of serum Apo A-I and S100B for the diagnosis of mild traumatic
brain injury and prediction of abnormal initial head computed tomography scan. J
Neurotrauma 30:1747-1754.
Bell RD, Sagare AP, Friedman AE, Bedi GS, Holtzman DM, Deane R, Zlokovic BV
(2007) Transport pathways for clearance of human Alzheimer's amyloid beta-peptide
and apolipoproteins E and J in the mouse central nervous system. J Cereb Blood Flow
Metab 27:909-918.
Bellosta S, Nathan BP, Orth M, Dong LM, Mahley RW, Pitas RE (1995) Stable
expression and secretion of apolipoproteins E3 and E4 in mouse neuroblastoma cells
produces differential effects on neurite outgrowth. J Biol Chem 270:27063-27071.
Beni-Adani L, Gozes I, Cohen Y, Assaf Y, Steingart RA, Brenneman DE, Eizenberg O,
Trembolver V, Shohami E (2001) A peptide derived from activity-dependent
neuroprotective protein (ADNP) ameliorates injury response in closed head injury in
mice. J Pharmacol Exp Ther 296:57-63.
234

Bernard SA, Nguyen V, Cameron P, Masci K, Fitzgerald M, Cooper DJ, Walker T, Std
BP, Myles P, Murray L, David, Taylor, Smith K, Patrick I, Edington J, Bacon A,
Rosenfeld JV, Judson R (2010) Prehospital rapid sequence intubation improves
functional outcome for patients with severe traumatic brain injury: a randomized
controlled trial. Ann Surg 252:959-965.
Beschorner R, Nguyen TD, Gozalan F, Pedal I, Mattern R, Schluesener HJ, Meyermann
R, Schwab JM (2002) CD14 expression by activated parenchymal
microglia/macrophages and infiltrating monocytes following human traumatic brain
injury. Acta Neuropathol 103:541-549.
Biernat J, Gustke N, Drewes G, Mandelkow E, Mandelkow E (1993) Phosphorylation of
Ser262 strongly reduces binding of tau to microtubules: Distinction between PHF-like
immunoreactivity and microtubule binding. Neuron 11:153-163.
Biffi A, Greenberg SM (2011) Cerebral amyloid angiopathy: a systematic review. J Clin
Neurol 7:1-9.
Binder LI, Frankfurter A, Rebhun LI (1985) The distribution of tau in the mammalian
central nervous system. J Cell Biol 101:1371-1378.
Bjorkhem I, Meaney S (2004) Brain cholesterol: long secret life behind a barrier.
Arterioscler Thromb Vasc Biol 24:806-815.
235

Blumbergs PC (1997) Pathology. In: Head Injury - Pathophysiology and Management of
Severe Closed Head Injury (Reilly P, Bullock R, eds), pp 39-70. London: Chapman and
Hall.
Blume HK, Lucas S, Bell KR (2011) Subacute concussion-related symptoms in youth.
Phys Med Rehabil Clin N Am 22:665-681, viii-ix.
Boche D, Perry VH, Nicoll JA (2013) Review: activation patterns of microglia and their
identification in the human brain. Neuropathol Appl Neurobiol 39:3-18.
Bodzioch M, Ors¢ E, Klucken J, Langmann T, B”ttcher A, Diederich W, Drobnik W,
Barlage S, B �chler C, Porsch-™zc �r�mez M, Kam
Rothe G, Aslanidis C, Lackner KJ, Schmitz G (1999) The gene encoding ATP-binding
cassette transporter 1 is mutated in Tangier Disease. Nat Genet 22:347-351.
Bouma GJ, Muizelaar JP, Bandoh K, Marmarou A (1992) Blood pressure and
intracranial pressure-volume dynamics in severe head injury: relationship with cerebral
blood flow. J Neurosurg 77:15-19.
Bouzat P, Sala N, Payen JF, Oddo M (2013) Beyond intracranial pressure: optimization
of cerebral blood flow, oxygen, and substrate delivery after traumatic brain injury. Ann
Intensive Care 3:23.
236

Brain Injury Society of Toronto (2014) Brain injury facts and figure.
http://www.bist.ca/brain-injury-fact-figures. Accessed on May 22, 2014.
Brandenburg W, Hallervorden J (1954) Dementia pugilistica with anatomical findings.
Virchows Arch 325:680-709.
Bré MH, Karsenti E (1990) Effects of brain microtubule-associated proteins on
microtubule dynamics and the nucleating activity of centrosomes. Cell Motil
Cytoskeleton 15:88-98.
Brecht WJ, Harris FM, Chang S, Tesseur I, Yu GQ, Xu Q, Dee Fish J, Wyss-Coray T,
Buttini M, Mucke L, Mahley RW, Huang Y (2004) Neuron-specific apolipoprotein e4
proteolysis is associated with increased tau phosphorylation in brains of transgenic
mice. J Neurosci 24:2527-2534.
Broadbent NJ, Gaskin S, Squire LR, Clark RE (2010) Object recognition memory and
the rodent hippocampus. Learn Mem 17:5-11.
Brody DL, Magnoni S, Schwetey KE, Spinner ML, Esparza TJ, Stocchetti N, Zipfel GJ,
Holtzman DM (2008) Amyloid-β dynamics correlate with neurological status in the
injured human brain. Science 321:1221-1224.
Brody DL, Mac Donald C, Kessens CC, Yuede C, Parsadanian M, Spinner M, Kim E,
Schwetye KE, Holtzman DM, Bayly PV (2007) Electromagnetic controlled cortical
237

impact device for precise, graded experimental traumatic brain injury. J Neurotrauma
24:657-673.
Brooks-Wilson A et al. (1999) Mutations in ABC1 in Tangier disease and familial high-
density lipoprotein deficiency. Nat Genet 22:336-345.
Browne KD, Chen XH, Meaney DF, Smith DH (2011) Mild traumatic brain injury and
diffuse axonal injury in swine. J Neurotrauma 28:1747-1755.
Brunham LR, Kruit JK, Pape TD, Parks JS, Kuipers F, Hayden MR (2006) Tissue-
specific induction of intestinal ABCA1 expression with a liver X receptor agonist raises
plasma HDL cholesterol levels. Circ Res 99:672-674.
Bu G (2009) Apolipoprotein E and its receptors in Alzheimer's disease: pathways,
pathogenesis and therapy. Nat Rev Neurosci 10:333-344.
Buki A, Povlishock JT (2006) All roads lead to disconnection?--Traumatic axonal injury
revisited. Acta Neurochir (Wien) 148:181-193; discussion 193-184.
Buki A, Koizumi H, Povlishock JT (1999a) Moderate posttraumatic hypothermia
decreases early calpain-mediated proteolysis and concomitant cytoskeletal compromise
in traumatic axonal injury. Exp Neurol 159:319-328.
238

Buki A, Siman R, Trojanowski JQ, Povlishock JT (1999b) The role of calpain-mediated
spectrin proteolysis in traumatically induced axonal injury. J Neuropathol Exp Neurol
58:365-375.
Bullock MR, Chesnut R, Ghajar J, Gordon D, Hartl R, Newell DW, Servadei F, Walters
BC, Wilberger JE (2006) Surgical management of acute epidural hematomas.
Neurosurgery 58:S7-15; discussion Si-iv.
Bullock R (1994) Excitatory amino acids following brain injury. J Neurosurg 80:595-596.
Bullock R, Zauner A, Woodward JJ, Myseros J, Choi SC, Ward JD, Marmarou A, Young
HF (1998) Factors affecting excitatory amino acid release following severe human head
injury. J Neurosurg 89:507-518.
Burns MP, Noble WJ, Olm V, Gaynor K, Casey E, LaFrancois J, Wang L, Duff K (2003)
Co-localization of cholesterol, apolipoprotein E and fibrillar Abeta in amyloid plaques.
Brain Res Mol Brain Res 110:119-125.
Buxbaum JD, Thinakaran G, Koliatsos V, O'Callahan J, Slunt HH, Price DL, Sisodia SS
(1998) Alzheimer amyloid protein precursor in the rat hippocampus: transport and
processing through the perforant path. J Neurosci 18:9629-9637.
Calatayud Maldonado V, Calatayud Perez JB, Aso Escario J (1991) Effects of CDP-
choline on the recovery of patients with head injury. J Neurol Sci 103 Suppl:S15-18.
239

Canadian Institute for Health Information (2006) Head Injuries in Canada: A Decade of
Change (1994-1995 to 2003-2004). In: Analysis in Brief: Canadian Institute for Health
Information.
Cantu RC (2007) Chronic traumatic encephalopathy in the National Football League.
Neurosurgery 61:223-225.
Carpentier M, Robitaille Y, DesGroseillers L, Boileau G, Marcinkiewicz M (2002)
Declining expression of neprilysin in Alzheimer disease vasculature: possible
involvement in cerebral amyloid angiopathy. J Neuropathol Exp Neurol 61:849-856.
Carroll LJ, Cassidy JD, Holm L, Kraus J, Coronado VG (2004) Methodological issues
and research recommendations for mild traumatic brain injury: the WHO Collaborating
Centre Task Force on Mild Traumatic Brain Injury. J Rehabil Med:113-125.
Cartagena CM, Ahmed F, Burns MP, Pajoohesh-Ganji A, Pak DT, Faden AI, Rebeck
GW (2008) Cortical injury increases cholesterol 24S hydroxylase (Cyp46) levels in the
rat brain. J Neurotrauma 25:1087-1098.
Cassidy JD, Carroll LJ, Peloso PM, Borg J, von Holst H, Holm L, Kraus J, Coronado VG
(2004) Incidence, risk factors and prevention of mild traumatic brain injury: results of the
WHO Collaborating Centre Task Force on Mild Traumatic Brain Injury. J Rehabil
Med:28-60.
240

Castellano JM, Kim J, Stewart FR, Jiang H, DeMattos RB, Patterson BW, Fagan AM,
Morris JC, Mawuenyega KG, Cruchaga C, Goate AM, Bales KR, Paul SM, Bateman RJ,
Holtzman DM (2011) Human apoE isoforms differentially regulate brain amyloid-beta
peptide clearance. Sci Transl Med 3:89ra57.
Cavelier C, Lorenzi I, Rohrer L, von Eckardstein A (2006) Lipid efflux by the ATP-
binding cassette transporters ABCA1 and ABCG1. Biochim Biophys Acta 1761:655-666.
Centers for Disease Control and Prevention (2007) Nonfatal traumatic brain injuries
from sports and recreation activities--United States, 2001-2009. MMWR Morb Mortal
Wkly Rep 56:733-737.
Cernak I (2005) Animal models of head trauma. NeuroRx 2:410-422.
Chartier-Harlin M-C, Crawford F, Houlden H, Warren A, Hughes D, Fidani L, Goate A,
Rossor M, Roques P, Hardy J, Mullan M (1991) Early-onset Alzheimer's disease caused
by mutations at codon 717 of the [beta]-amyloid precursor protein gene. Nature
353:844-846.
Chawla A, Boisvert WA, Lee CH, Laffitte BA, Barak Y, Joseph SB, Liao D, Nagy P,
Edwards PA, Curtiss LK, Evans RM, Tontonoz P (2001) A PPAR g -LXR-ABCA1
pathway in macrophages is involved in cholesterol efflux and atherogenesis. Molec Cell
7:161-171.
241

Chen JD, Evans RM (1995) A transcriptional co-repressor that interacts with nuclear
hormone receptors. Nature 377:454-457.
Chen S, Averett NT, Manelli A, Ladu MJ, May W, Ard MD (2005) Isoform-specific
effects of apolipoprotein E on secretion of inflammatory mediators in adult rat microglia.
J Alzheimers Dis 7:25-35.
Chen W, Sun Y, Welch C, Gorelik A, Leventhal AR, Tabas I, Tall AR (2001) Preferential
ATP-binding cassette transporter A1-mediated cholesterol efflux from late
endosomes/lysosomes. J Biol Chem 276:43564-43569.
Chen XH, Johnson VE, Uryu K, Trojanowski JQ, Smith DH (2009) A lack of amyloid á
plaques despite persistent accumulation of amyloid á in axons of long-term survivors of
traumatic brain injury. Brain Pathol 19:214-223.
Chen XH, Siman R, Iwata A, Meaney DF, Trojanowski JQ, Smith DH (2004) Long-term
accumulation of amyloid-beta, beta-secretase, presenilin-1, and caspase-3 in damaged
axons following brain trauma. Am J Pathol 165:357-371.
Chen Y, Lomnitski L, Michaelson DM, Shohami E (1997) Motor and cognitive deficits in
apolipoprotein E-deficient mice after closed head injury. Neuroscience 80:1255-1262.
242

Chen Y, Durakoglugil MS, Xian X, Herz J (2010) ApoE4 reduces glutamate receptor
function and synaptic plasticity by selectively impairing ApoE receptor recycling. Proc
Natl Acad Sci U S A 107:12011-12016.
Chen Y, Constantini S, Trembovler V, Weinstock M, Shohami E (1996) An experimental
model of closed head injury in mice: Pathophysiology, histopathology, and cognitive
deficits. J Neurotrauma 13:557-568.
Chesnut RM, Temkin N, Carney N, Dikmen S, Rondina C, Videtta W, Petroni G, Lujan
S, Pridgeon J, Barber J, Machamer J, Chaddock K, Celix JM, Cherner M, Hendrix T
(2012) A Trial of Intracranial-Pressure Monitoring in Traumatic Brain Injury. New Eng J
Med 367:2471-2481.
Chiang JY, Kimmel R, Stroup D (2001) Regulation of cholesterol 7alpha-hydroxylase
gene (CYP7A1) transcription by the liver orphan receptor (LXRalpha). Gene 262:257-
265.
Chin LS, Toshkezi G, Cantu RC (2011) Traumatic encephalopathy related to sports
injury. US Neurology 7:33-36.
Chobanian AV, Hollander W (1962) Body cholesterol metabolism in man. I. The
equilibration of serum and tissue cholesterol. J Clin Invest 41:1732-1737.
243

Chodobski A, Zink BJ, Szmydynger-Chodobska J (2011) Blood-brain barrier
pathophysiology in traumatic brain injury. Transl Stroke Res 2:492-516.
Ciallella JR, Ikonomovic MD, Paljug WR, Wilbur YI, Dixon CE, Kochanek PM, Marion
DW, DeKosky ST (2002) Changes in expression of amyloid precursor protein and
interleukin-1beta after experimental traumatic brain injury in rats. J Neurotrauma
19:1555-1567.
Cirrito JR, Yamada KA, Finn MB, Sloviter RS, Bales KR, May PC, Schoepp DD, Paul
SM, Mennerick S, Holtzman DM (2005) Synaptic activity regulates interstitial fluid
amyloid-beta levels in vivo. Neuron 48:913-922.
Citron M, Oltersdorf T, Haass C, McConlogue L, Hung AY, Seubert P, Vigo-Pelfrey C,
Lieberburg I, Selkoe DJ (1992) Mutation of the beta-amyloid precursor protein in familial
Alzheimer's disease increases beta-protein production. Nature 360:672-674.
Clausen F, Lorant T, Lewen A, Hillered L (2007) T lymphocyte trafficking: a novel target
for neuroprotection in traumatic brain injury. J Neurotrauma 24:1295-1307.
Clifton GL, Miller ER, Choi SC, Levin HS, McCauley S, Smith KR, Jr., Muizelaar JP,
Marion DW, Luerssen TG (2002) Hypothermia on admission in patients with severe
brain injury. J Neurotrauma 19:293-301.
244

Clifton GL, Valadka A, Zygun D, Coffey CS, Drever P, Fourwinds S, Janis LS, Wilde E,
Taylor P, Harshman K, Conley A, Puccio A, Levin HS, McCauley SR, Bucholz RD,
Smith KR, Schmidt JH, Scott JN, Yonas H, Okonkwo DO (2011) Very early hypothermia
induction in patients with severe brain injury (the National Acute Brain Injury Study:
Hypothermia II): a randomised trial. Lancet Neurol 10:131-139.
Collins JL, Fivush AM, Watson MA, Galardi CM, Lewis MC, Moore LB, Parks DJ, Wilson
JG, Tippin TK, Binz JG, Plunket KD, Morgan DG, Beaudet EJ, Whitney KD, Kliewer SA,
Willson TM (2002) Identification of a nonsteroidal liver X receptor agonist through
parallel array synthesis of tertiary amines. J Med Chem 45:1963-1966.
Colton CA (2009) Heterogeneity of microglial activation in the innate immune response
in the brain. J Neuroimmune Pharmacol 4:399-418.
Conde C, Caceres A (2009) Microtubule assembly, organization and dynamics in axons
and dendrites. Nat Rev Neurosci 10:319-332.
Cook DG, Leverenz JB, McMillan PJ, Kulstad JJ, Ericksen S, Roth RA, Schellenberg
GD, Jin LW, Kovacina KS, Craft S (2003) Reduced hippocampal insulin-degrading
enzyme in late-onset Alzheimer's disease is associated with the apolipoprotein E-
epsilon4 allele. Am J Pathol 162:313-319.
Cooper DJ, Myles PS, McDermott FT, Murray LJ, Laidlaw J, Cooper G, Tremayne AB,
Bernard SS, Ponsford J (2004) Prehospital hypertonic saline resuscitation of patients
245

with hypotension and severe traumatic brain injury: a randomized controlled trial. J Am
Med Assoc 291:1350-1357.
Cooper DJ, Rosenfeld JV, Murray L, Arabi YM, Davies AR, D'Urso P, Kossmann T,
Ponsford J, Seppelt I, Reilly P, Wolfe R (2011) Decompressive craniectomy in diffuse
traumatic brain injury. N Engl J Med 364:1493-1502.
Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW,
Roses AD, Haines JL, Pericak-Vance MA (1993) Gene dose of apolipoprotein E type 4
allele and the risk of Alzheimer's disease in late onset families. Science 261:921-923.
Corder EH, Saunders AM, Risch NJ, Strittmatter WJ, Schmechel DE, Gaskell PC,
Rimmler JB, Locke PA, Conneally PM, Schmader KE, Small GW, Roses AD, Haines JL,
Pericak-Vance MA (1994) Protective effect of apolipoprotein E type 2 for late onset
Alzheimer disease. Nat Genet 7:180-184.
Corsellis JA, Bruton CJ, Freeman-Browne D (1973) The aftermath of boxing. Psychol
Med 3:270-303.
Costet P, Luo Y, Wang N, Tall AR (2000) Sterol-dependent transactivation of the ABC1
promoter by the liver X receptor/retinoid X receptor. J Biol Chem 275:28240-28245.
246

Creeley CE, Wozniak DF, Bayly PV, Olney JW, Lewis LM (2004) Multiple episodes of
mild traumatic brain injury result in impaired cognitive performance in mice. Acad Emerg
Med 11:809-819.
Cronican AA, Fitz NF, Pham T, Fogg A, Kifer B, Koldamova R, Lefterov I (2010) Proton
pump inhibitor lansoprazole is a nuclear liver X receptor agonist. Biochem Pharmacol
79:1310-1316.
Cruz J, Minoja G, Okuchi K (2001) Improving clinical outcomes from acute subdural
hematomas with the emergency preoperative administration of high doses of mannitol:
a randomized trial. Neurosurgery 49:864-871.
Cruz J, Minoja G, Okuchi K (2002) Major clinical and physiological benefits of early high
doses of mannitol for intraparenchymal temporal lobe hemorrhages with abnormal
pupillary widening: a randomized trial. Neurosurgery 51:628-637; discussion 637-628.
Csuka E, Morganti-Kossmann MC, Lenzlinger PM, Joller H, Trentz O, Kossmann T
(1999) IL-10 levels in cerebrospinal fluid and serum of patients with severe traumatic
brain injury: relationship to IL-6, TNF-α, TGF-β1 and blood–brain barrier function. J
Neuroimmunol 101:211-221.
Csuka E, Hans VH, Ammann E, Trentz O, Kossmann T, Morganti-Kossmann MC (2000)
Cell activation and inflammatory response following traumatic axonal injury in the rat.
Neuroreport 11:2587-2590.
247

Daneshvar DH, Riley DO, Nowinski CJ, McKee AC, Stern RA, Cantu RC (2011) Long-
term consequences: effects on normal development profile after concussion. Phys Med
Rehabil Clin N Am 22:683-700, ix.
Darvish KK, Crandall JR (2001) Nonlinear viscoelastic effects in oscillatory shear
deformation of brain tissue. Med Eng Phys 23:633-645.
Davidsson J, Risling M (2011) A new model to produce sagittal plane rotational induced
diffuse axonal injuries. Front Neurol 2:41.
Davis AE (2000) Mechanisms of traumatic brain injury: biomechanical, structural and
cellular considerations. Crit Care Nurs Q 23:1-13.
Dawson SL, Hirsch CS, Lucas FV, Sebek BA (1980) The contrecoup phenomenon:
Reappraisal of a classic problem. Hum Pathol 11:155-166.
De Bonis P, Pompucci A, Mangiola A, D'Alessandris QG, Rigante L, Anile C (2010)
Decompressive craniectomy for the treatment of traumatic brain injury: does an age limit
exist? J Neurosurg 112:1150-1153.
de Bont N, Netea MG, Demacker PNM, Verschueren I, Kullberg BJ, van Dijk KW, van
der Meer JWM, Stalenhoef AFH (1999) Apolipoprotein E knock-out mice are highly
susceptible to endotoxemia and Klebsiella pneumoniae infection. J Lipid Res 40:680-
685.
248

Deacon RMJ (2013) Measuring Motor Coordination in Mice. J Vis Exp:e2609.
Deane R, Wu Z, Zlokovic BV (2004a) RAGE (yin) versus LRP (yang) balance regulates
alzheimer amyloid beta-peptide clearance through transport across the blood-brain
barrier. Stroke 35:2628-2631.
Deane R, Sagare A, Hamm K, Parisi M, Lane S, Finn MB, Holtzman DM, Zlokovic BV
(2008) apoE isoform-specific disruption of amyloid beta peptide clearance from mouse
brain. J Clin Invest 118:4002-4013.
Deane R, Wu Z, Sagare A, Davis J, Du Yan S, Hamm K, Xu F, Parisi M, LaRue B, Hu
HW, Spijkers P, Guo H, Song X, Lenting PJ, Van Nostrand WE, Zlokovic BV (2004b)
LRP/Amyloid β-Peptide Interaction Mediates Differential Brain Efflux of Aβ Isoforms.
Neuron 43:333-344.
Deane R et al. (2003) RAGE mediates amyloid-beta peptide transport across the blood-
brain barrier and accumulation in brain. Nat Med 9:907-913.
Decuypere M, Klimo P, Jr. (2012) Spectrum of traumatic brain injury from mild to
severe. Surg Clin North Am 92:939-957, ix.
DeFord SM, Wilson MS, Rice AC, Clausen T, Rice LK, Barabnova A, Hamm RJ (2002)
Repeated mild brain injuries result in cognitive impairment in B6C3F1 mice. J
Neurotrauma 19:427-438.
249

DeKosky ST, Abrahamson EE, Ciallella JR, Paljug WR, Wisniewski SR, Clark RS,
Ikonomovic MD (2007) Association of increased cortical soluble abeta42 levels with
diffuse plaques after severe brain injury in humans. Arch Neurol 64:541-544.
Delacourte A, Robitaille Y, Sergeant N, Buee L, Hof PR, Wattez A, Laroche-Cholette A,
Mathieu J, Chagnon P, Gauvreau D (1996) Specific pathological Tau protein variants
characterize Pick's disease. J Neuropathol Exp Neurol 55:159-168.
DeMattos RB, Brendza RP, Heuser JE, Kierson M, Cirrito JR, Fryer J, Sullivan PM,
Fagan AM, Han X, Holtzman DA (2001) Purification and characterization of astrocyte-
secreted apolipoprotein E and J-containing lipoproteins from wild-type and human apoE
transgenic mice. Neurochem Int 39:415-425.
Denny-Brown D, Russell WR (1940) Experimental cerebral concussion. J Physiol
99:153.
Dewitt DS, Perez-Polo R, Hulsebosch CE, Dash PK, Robertson CS (2013) Challenges
in the development of rodent models of mild traumatic brain injury. J Neurotrauma
30:688-701.
Dietschy JM, Turley SD (2001) Cholesterol metabolism in the brain. Curr Opin Lipidol
12:105-112.
250

Dietschy JM, Turley SD (2004) Cholesterol metabolism in the central nervous system
during early development and in the mature animal. J Lipid Res 45:1375-1397.
Dixon CE, Lighthall JW, Anderson TE (1988) Physiologic, histopathologic, and
cineradiographic characterization of a new fluid-percussion model of experimental brain
injury in the rat. J Neurotrauma 5:91-104.
Dixon CE, Clifton GL, Lighthall JW, Yaghmai AA, Hayes RL (1991) A controlled cortical
impact model of traumatic brain injury in the rat. J Neurosci Methods 39:253-262.
Dong LM, Weisgraber KH (1996) Human apolipoprotein E4 domain interaction. Arginine
61 and glutamic acid 255 interact to direct the preference for very low density
lipoproteins. J Biol Chem 271:19053-19057.
Dong LM, Wilson C, Wardell MR, Simmons T, Mahley RW, Weisgraber KH, Agard DA
(1994) Human apolipoprotein E. Role of arginine 61 in mediating the lipoprotein
preferences of the E3 and E4 isoforms. J Biol Chem 269:22358-22365.
Donkin JJ, Stukas S, Hirsch-Reinshagen V, Namjoshi D, Wilkinson A, May S, Chan J,
Fan J, Collins J, Wellington CL (2010) ATP-binding cassette transporter A1 mediates
the beneficial effects of the liver-X-receptor agonist GW3965 on object recognition
memory and amyloid burden in APP/PS1 mice. J Biol Chem 285:34144-34154.
251

Donnelly BR, Medige J (1997) Shear properties of human brain tissue. J Biomech Eng
119:423-432.
Drew LB, Drew WE (2004) The contrecoup-coup phenomenon: a new understanding of
the mechanism of closed head injury. Neurocrit Care 1:385-390.
Duhaime AC (2006) Large animal models of traumatic injury to the immature brain. Dev
Neurosci 28:380-387.
Egensperger R, Kosel S, von Eitzen U, Graeber MB (1998) Microglial activation in
Alzheimer disease: Association with APOE genotype. Brain Pathol 8:439-447.
Eiband AM (1959) Human tolerance to rapidly applied accelerations: a summary of the
literature. In: NASA Memorandum. Cleveland, Ohio: NASA Lewis Research Center.
Eisenberg HM, Gary HE, Jr., Aldrich EF, Saydjari C, Turner B, Foulkes MA, Jane JA,
Marmarou A, Marshall LF, Young HF (1990) Initial CT findings in 753 patients with
severe head injury. A report from the NIH Traumatic Coma Data Bank. J Neurosurg
73:688-698.
Elder GA, Cristian A (2009) Blast-related mild traumatic brain injury: mechanisms of
injury and impact on clinical care. Mount Sinai J Med 76:111-118.
252

Engel S, Schluesener H, Mittelbronn M, Seid K, Adjodah D, Wehner HD, Meyermann R
(2000) Dynamics of microglial activation after human traumatic brain injury are revealed
by delayed expression of macrophage-related proteins MRP8 and MRP14. Acta
Neuropathol (Berl) 100:313-322.
Engelhardt G (1996) Pharmacology of meloxicam, a new non-steroidal anti-
inflammatory drug with an improved safety profile through preferential inhibition of COX-
2. Br J Rheumatol 35 Suppl 1:4-12.
Ennaceur A (2010) One-trial object recognition in rats and mice: methodological and
theoretical issues. Behav Brain Res 215:244-254.
Ennaceur A, Delacour J (1988) A new one-trial test for neurobiological studies of
memory in rats. 1: Behavioral data. Behav Brain Res 31:47-59.
Eppinger R, Sun E, Kuppa S, Saul R (2000) Suppliment: Development of Improved
Injury Criteria for the Assessment of Advanced Automotive Restraint Systems - II. In, pp
1-40: National Highway Traffic Safety Administration,
http://www.nhtsa.gov/DOT/NHTSA/NRD/Multimedia/PDFs/Crashworthiness/Air%20Bag
s/finalrule_all.pdf, Accessed on March 2012.
Evans FG, Lissner HR, Lebow M (1958) The relation of energy, velocity, and
acceleration to skull deformation and fracture. Surg Gynecol Obstet 107:593-601.
253

Fagan AM, Bu G, Sun Y, Daugherty A, Holtzman DM (1996) Apolipoprotein E-
containing high density lipoprotein promotes neurite outgrowth and is a ligand for the
low density lipoprotein receptor-related protein. J Biol Chem 271:30121-30125.
Fagan AM, Holtzman DM, Munson G, Mathur T, Schnieder D, Chang LK, Getz GS,
Reardon CA, Lukens J, Shah JA, Ladu MJ (1999) Unique lipoproteins secreted by
primary astrocytes from wild type, apoE (-/-), and human apoE transgenic mice. J Biol
Chem 274:30001-30004.
Fan L, Young PR, Barone FC, Feuerstein GZ, Smith DH, McIntosh TK (1995)
Experimental brain injury induces expression of interleukin-1 beta mRNA in the rat
brain. Brain Res Mol Brain Res 30:125-130.
Fan QW, Iosbe I, Asou H, Yanagisawa K, Michikawa M (2001) Expression and
regulation of apolipoprotein E receptors in the cells of the central nervous system in
culture: A review. J Am Aging Assoc 24:1-10.
Farris W, Mansourian S, Chang Y, Lindsley L, Eckman EA, Frosch MP, Eckman CB,
Tanzi RE, Selkoe DJ, Guenette S (2003) Insulin-degrading enzyme regulates the levels
of insulin, amyloid beta-protein, and the beta-amyloid precursor protein intracellular
domain in vivo. Proc Natl Acad Sci U S A 100:4162-4167.
Faul MM, Xu L, Wald MM, Coronado VG (2010) Traumatic brain injury in the united
states: emergency department visits, hospitalizations and death 2002-2006. In. Atlanta,
254

GA: Centers for Disease Control and Prevention, National Center for Injury Prevention
and Control.
Feeney DM, Boyeson MG, Linn RT, Murray HM, Dail WG (1981) Responses to cortical
injury: I. Methodology and local effects of contusions in the rat. Brain Res 211:67-77.
Feigin VL, Theadom A, Barker-Collo S, Starkey NJ, McPherson K, Kahan M, Dowell A,
Brown P, Parag V, Kydd R, Jones K, Jones A, Ameratunga S (2013) Incidence of
traumatic brain injury in New Zealand: a population-based study. Lancet Neurol 12:53-
64.
Feng B, Tabas I (2002) ABCA1-mediated cholesterol efflux is defective in free
cholesterol-loaded macrophages. J Biol Chem 277:43271-43280.
Fijalkowski RJ, Stemper BD, Pintar FA, Yoganandan N, Crowe MJ, Gennarelli TA
(2007) New rat model for diffuse brain injury using coronal plane angular acceleration. J
Neurotrauma 24:1387-1398.
Finnie J (2001) Animal models of traumatic brain injury: a review. Aust Vet J 79:628-
633.
Fitz NF, Cronican AA, Saleem M, Fauq AH, Chapman R, Lefterov I, Koldamova R
(2012) Abca1 deficiency affects Alzheimer's disease-like phenotype in human ApoE4
but not in ApoE3-targeted replacement mice. J Neurosci 32:13125-13136.
255

Fitz NF, Cronican A, Pham T, Fogg A, Fauq AH, Chapman R, Lefterov I, Koldamova R
(2010) Liver X receptor agonist treatment ameliorates amyloid pathology and memory
deficits caused by high-fat diet in APP23 mice. J Neurosci 30:6862-6872.
Flament S, Delacourte A, Verny M, Hauw JJ, Javoy-Agid F (1991) Abnormal Tau
proteins in progressive supranuclear palsy. Similarities and differences with the
neurofibrillary degeneration of the Alzheimer type. Acta Neuropathol 81:591-596.
Fleminger S, Ponsford J (2005) Long term outcome after traumatic brain injury. Br Med
J 331:1419-1420.
Fleminger S, Oliver DL, Lovestone S, Rabe-Hesketh S, Giora A (2003) Head injury as a
risk factor for Alzheimer's disease: the evidence 10 years on; a partial replication. J
Neurol Neurosurg Psychiatry 74:857-862.
Flierl MA, Stahel PF, Beauchamp KM, Morgan SJ, Smith WR, Shohami E (2009) Mouse
closed head injury model induced by a weight-drop device. Nat Protoc 4:1328-1337.
Foda MA, Marmarou A (1994) A new model of diffuse brain injury in rats. Part II:
Morphological characterization. J Neurosurg 80:301-313.
Fork M, Bartels C, Ebert AD, Grubich C, Synowitz H, Wallesch CW (2005)
Neuropsychological sequelae of diffuse traumatic brain injury. Brain Inj 19:101-108.
256

Forman BM, Ruan B, Chen J, Schroepfer GJ, Jr., Evans RM (1997) The orphan nuclear
receptor LXRalpha is positively and negatively regulated by distinct products of
mevalonate metabolism. Proc Natl Acad Sci U S A 94:10588-10593.
Franz G, Beer R, Kampfl A, Engelhardt K, Schmutzhard E, Ulmer H, Deisenhammer F
(2003) Amyloid beta 1-42 and tau in cerebrospinal fluid after severe traumatic brain
injury. Neurology 60:1457-1461.
Franze K, Reichenbach A, Käs J (2009) Biomechanics of the CNS. In:
Mechanosensitivity of the Nervous System (Kamkim A, Kiseleva I, eds), pp 173-213:
Springer Netherlands.
Fratiglioni L, Ahlbom A, Viitanen M, Winblad B (1993) Risk factors for late-onset
Alzheimer's disease: a population-based, case-control study. Ann Neurol 33:258-266.
French LR, Schuman LM, Mortimer JA, Hutton JT, Boatman RA, Christians B (1985) A
case-control study of dementia of the Alzheimer type. Am J Epidemiol 121:414-421.
Fujitsu K, Kuwabara T, Muramoto M, Hirata K, Mochimatsu Y (1988) Traumatic
Intraventricular Hemorrhage: Report of Twenty-six Cases and Consideration of the
Pathogenic Mechanism. Neurosurgery 23:423-430.
257

Fukumoto H, Deng A, Irizarry MC, Fitzgerald ML, Rebeck GW (2002) Induction of the
cholesterol transporter ABCA1 in central nervous system cells by liver X receptor
agonists increases secreted Abeta levels. J Biol Chem 277:48508-48513.
Funk JR, Duma SM, Manoogian SJ, Rowson S (2007) Biomechanical risk estimates for
mild traumatic brain injury. Annu Proc Assoc Adv Automot Med 51:343-361.
Gabbi C, Warner M, Gustafsson JA (2014) Action mechanisms of Liver X Receptors.
Biochem Biophys Res Commun 446:647-650.
Gadd GW (1966) Use of a Weighted-Impulse Criterion for Estimation Injury Hazard. In:
10th Stapp Car Crash Conf, pp 164-174: Society of Automotive Engineers.
Gallyas F, Guldner FH, Zoltay G, Wolff JR (1990) Golgi-like demonstration of "dark"
neurons with an argyrophil III method for experimental neuropathology. Acta
Neuropathol 79:620-628.
Gavett BE, Stern RA, McKee AC (2011) Chronic traumatic encephalopathy: a potential
late effect of sport-related concussive and subconcussive head trauma. Clin Sports Med
30:179-188.
Geddes JF, Whitwell HL, Graham DI (2000) Traumatic axonal injury: practical issues for
diagnosis in medicolegal cases. Neuropathol Appl Neurobiol 26:105-116.
258

Geddes JF, Vowles GH, Beer TW, Ellison DW (1997) The diagnosis of diffuse axonal
injury: implications for forensic practice. Neuropathol Appl Neurobiol 23:339-347.
Genis L, Chen Y, Shohami E, Michaelson DM (2000) Tau hyperphosphorylation in
apolipoprotein E-deficient and control mice after closed head injury. J Neurosci Res
15:559-564.
Gennarelli TA (1993) Mechanisms of brain injury. J Emerg Med 11 Suppl 1:5-11.
Gennarelli TA, Thibault LE (1982) Biomechanics of acute subdural hematoma. J
Trauma 22:680-686.
Gennarelli TA, Graham DI (2005) Neuropathology. In: Textbook of traumatic brain
injury, 1st Edition (Silver JM, McAllister TW, Yudofsky SC, eds), pp 27-50. Washington,
DC: American Psychiatric Publishing, Inc.
Gennarelli TA, Adams JH, Graham DI (1981) Acceleration induced head injury in the
monkey.I. The model, its mechanical and physiological correlates. Acta Neuropathol
Suppl 7:23-25.
Gennarelli TA, Thibault LE, Adams JH, Graham DI, Thompson CJ, Marcincin RP (1982)
Diffuse axonal injury and traumatic coma in the primate. Ann Neurol 12:564-574.
259

Gentleman SM, Nash MJ, Sweeting CJ, Graham DI, Roberts GW (1993) Beta-amyloid
precursor protein (beta APP) as a marker for axonal injury after head injury. Neurosci
Lett 160:139-144.
Gentleman SM, Leclercq PD, Moyes L, Graham DI, Smith C, Griffin WS, Nicoll JA
(2004) Long-term intracerebral inflammatory response after traumatic brain injury.
Forensic Sci Int 146:97-104.
Gentleman SM, Greenberg BD, Savage MJ, Noori M, Newman SJ, Roberts GW, Griffin
WS, Graham DI (1997) A beta 42 is the predominant form of amyloid beta-protein in the
brains of short-term survivors of head injury. Neuroreport 8:1519-1522.
Gerberding JL, Binder S (2003) The report to congress on mild traumatic brain injury in
the United States: Steps to prevent a serious public health problem. In, pp 1-45. Atlanta,
GA: Centers for Disease Control and Prevention.
Ghisletti S, Huang W, Ogawa S, Pascual G, Lin M-E, Willson TM, Rosenfeld MG, Glass
CK (2007) Parallel SUMOylation-Dependent Pathways Mediate Gene- and Signal-
Specific Transrepression by LXRs and PPARγ. Molecular Cell 25:57-70.
Giacino JT, Whyte J, Bagiella E, Kalmar K, Childs N, Khademi A, Eifert B, Long D, Katz
DI, Cho S, Yablon SA, Luther M, Hammond FM, Nordenbo A, Novak P, Mercer W,
Maurer-Karattup P, Sherer M (2012) Placebo-Controlled Trial of Amantadine for Severe
Traumatic Brain Injury. New England Journal of Medicine 366:819-826.
260

Giannakopoulos P, Herrmann FR, Bussiere T, Bouras C, Kovari E, Perl DP, Morrison
JH, Gold G, Hof PR (2003) Tangle and neuron numbers, but not amyloid load, predict
cognitive status in Alzheimer's disease. Neurology 60:1495-1500.
Giza CC, Hovda DA (2001) The Neurometabolic Cascade of Concussion. J Athl Train
36:228-235.
Giza CC, Kutcher JS, Ashwal S, Barth J, Getchius TS, Gioia GA, Gronseth GS,
Guskiewicz K, Mandel S, Manley G, McKeag DB, Thurman DJ, Zafonte R (2013)
Summary of evidence-based guideline update: Evaluation and management of
concussion in sports: Report of the Guideline Development Subcommittee of the
American Academy of Neurology. Neurology 80:2250-2257.
Goate A et al. (1991) Segregation of a missense mutation in the amyloid precursor
protein gene with familial Alzheimer's disease. Nature 349:704-706.
Goedert M, Spillantini MG, Cairns NJ, Crowther RA (1992) Tau proteins of Alzheimer
paired helical filaments: abnormal phosphorylation of all six brain isoforms. Neuron
8:159-168.
Goedert M, Spillantini MG, Jakes R, Rutherford D, Crowther RA (1989) Multiple
isoforms of human microtubule-associated protein tau: sequences and localization in
neurofibrillary tangles of Alzheimer's disease. Neuron 3:519-526.
261

Goldstein LE et al. (2012) Chronic traumatic encephalopathy in blast-exposed military
veterans and a blast neurotrauma mouse model. Sci Transl Med 4:143ra160.
Gong JS, Kobayashi H, Hayashi H, Zou K, Sawamura N, Fujita SC, Yanagisawas K,
Michikawa M (2002) Apolipoprotein E (ApoE) isoform-dependent lipid release from
astrocytes prepared from hyman ApoE3 and ApoE4 knock-in mice. J Biol Chem
277:29919-29926.
Goodwin B, Watson MA, Kim H, Miao J, Kemper JK, Kliewer SA (2003) Differential
regulation of rat and human CYP7A1 by the nuclear oxysterol receptor liver X receptor-
alpha. Mol Endocrinol 17:386-394.
Gordon I, Grauer E, Genis I, Sehayek E, Michaelson DM (1995) Memory deficits and
cholinergic impairments in apolipoprotein E-deficient mice. Neurosci Lett 199:1-4.
Gorrie C, Oakes S, Duflou J, Blumbergs P, Waite PM (2002) Axonal injury in children
after motor vehicle crashes: extent, distribution, and size of axonal swellings using beta-
APP immunohistochemistry. J Neurotrauma 19:1171-1182.
Gosch HH, Gooding E, Schneider RC (1970) The lexan calvarium for the study of
cerebral responses to acute trauma. J Trauma Acute Care Surg 10:370-385.
262

Gotz J, Gladbach A, Pennanen L, van Eersel J, Schild A, David D, Ittner LM (2010)
Animal models reveal role for tau phosphorylation in human disease. Biochim Biophys
Acta 1802:860-871.
Grady MS, McLaughlin MR, Christman CW, Valadka AB, Fligner CL, Povlishock JT
(1993) The use of antibodies targeted against the neurofilament subunits for the
detection of diffuse axonal injury in humans. J Neuropathol Exp Neurol 52:143-152.
Graham DI, Adams JH, Doyle D (1978) Ischaemic brain damage in fatal non-missile
head injuries. J Neurol Sci 39:213-234.
Graham DI, McIntosh TK, Maxwell WL, Nicoll JA (2000) Recent Advances in
Neurotrauma. J Neuropathol Exp Neurol 59:641-651.
Greaves LL, Van Toen C, Melnyk A, Koenig L, Zhu Q, Tredwell S, Mulpuri K, Cripton
PA (2009) Pediatric and adult three-dimensional cervical spine kinematics: effect of age
and sex through overall motion. Spine (Phila Pa 1976) 34:1650-1657.
Greenwald RM, Gwin JT, Chu JJ, Crisco JJ (2008) Head impact severity measures for
evaluating mild traumatic brain injury risk exposure. Neurosurgery 62:789-798;
discussion 798.
Grefhorst A, Elzinga BM, Voshol PJ, Bl”sch T, Kok T, Bloks VW, van der Sluijs FH,
Havekes LM, Romijn JA, Verkade HJ, Kuipers F (2002) Stimulation of lipogenesis by
263

pharmacological activation of the liver X receptor leads to production of large,
triglyceride-rich very low density lipoprotein particles. J Biol Chem 277:34182-34190.
Groot PH et al. (2005) Synthetic LXR agonists increase LDL in CETP species. J Lipid
Res 46:2182-2191.
Grootendorst J, Bour A, Vogel E, Kelche C, Sullivan PM, Dodart JC, Bales K, Mathis C
(2005) Human apoE targeted replacement mouse lines: h-apoE4 and h-apoE3 mice
differ on spatial memory performance and avoidance behavior. Behav Brain Res 159:1-
14.
Guo Z, Cupples LA, Kurz A, Auerbach SH, Volicer L, Chui H, Green RC, Sadovnick AD,
Duara R, DeCarli C, Johnson K, Go RC, Growdon JH, Haines JL, Kukull WA, Farrer LA
(2000) Head injury and the risk of AD in the MIRAGE study. Neurology 54:1316-1323.
Gurdjian ES (1975) Re-evaluation of the biomechanics of blunt impact injury of the
head. Surg Gynecol Obstet 140:845-850.
Gurdjian ES, Lissner HR (1945) Deformation of the skull in head injury; a study with the
stresscoat technique. Surg Gynecol Obstet 81:679-687.
Gurdjian ES, Lissner HR (1946) Study of deformations of the skull by the stresscoat
technique. Anat Rec 94:465.
264

Gurdjian ES, Webster JE (1947) The mechanism and management of injuries of the
head. J Am Med Assoc 134:1072-1077.
Gurdjian ES, Lissner HR (1947) Deformations of the skull in head injury as studied by
the stresscoat technic. Am J Surg 73:269-281.
Gurdjian ES, Lissner HR, Webster JE (1947) The mechanism of production of linear
skull fracture; further studies on deformation of the skull by the stresscoat technique.
Surg Gynecol Obstet 85:195-210.
Gurdjian ES, Webster JE, Lissner HR (1955) Observations on the mechanism of brain
concussion, contusion, and laceration. Surg Gynecol Obstet 101:680-690.
Gurdjian ES, Roberts VL, Thomas LM (1966) Tolerance curves of acceleration and
intracranial pressure and protective index in experimental head injury. J Trauma 6:600-
604.
Gurdjian ES, Lissner HR, Evans FG, Patrick LM, Hardy WG (1961) Intracranial pressure
and acceleration accompanying head impacts in human cadavers. Surg Gynecol Obstet
113:185-190.
Gururaj G (2002) Epidemiology of traumatic brain injuries: Indian scenario. Neurol Res
24:24-28.
265

Guskiewicz KM (2003) Assessment of postural stability following sport-related
concussion. Curr Sports Med Rep 2:24-30.
Gusmao SN, Pittella JE (2003) Acute subdural hematoma and diffuse axonal injury in
fatal road traffic accident victims: a clinico-pathological study of 15 patients. Arq
Neuropsiquiatr 61:746-750.
Gutierrez E, Huang Y, Haglid K, Bao F, Hansson HA, Hamberger A, Viano D (2001) A
new model for diffuse brain injury by rotational acceleration: I model, gross appearance,
and astrocytosis. J Neurotrauma 18:247-257.
Haddad BF, Lissner HR, Webster JE, Gurdjian ES (1955) Experimental concussion;
relation to acceleration to physiologic effect. Neurology 5:798-800.
Hamberger A, Huang YL, Zhu H, Bao F, Ding M, Blennow K, Olsson A, Hansson HA,
Viano D, Haglid KG (2003) Redistribution of neurofilaments and accumulation of beta-
amyloid protein after brain injury by rotational acceleration of the head. J Neurotrauma
20:169-178.
Han SH, Chung SY (2000) Marked hippocampal neuronal damage without motor
deficits after mild concussive-like brain injury in apolipoprotein E-deficient mice. Ann N
Y Acad Sci 903:357-365.
266

Handelmann GE, Boyles JK, Weisgraber KH, Mahley RW, Pitas RE (1992) Effects of
apolipoprotein E, beta-very low density lipoproteins, and cholesterol on the extension of
neurites by rabbit dorsal root ganglion neurons in vitro. J Lipid Res 33:1677-1688.
Harders A, Kakarieka A, Braakman R (1996) Traumatic subarachnoid hemorrhage and
its treatment with nimodipine. German tSAH Study Group. J Neurosurg 85:82-89.
Hardy J, Higgins G (1992) Alzheimer's disease: the amyloid cascade hypothesis.
Science 256:184-185.
Hardy J, Selkoe DJ (2002) The amyloid hypothesis of Alzheimer's disease: progress
and problems on the road to therapeutics. Science 297:353-356.
Hardy WN, Khalil TB, King AI (1994) Literature review of head injury biomechanics. Int
Journal Impact Eng 15:561-586.
Hardy WN, Foster CD, Mason MJ, Yang KH, King AI, Tashman S (2001) Investigation
of Head Injury Mechanisms Using Neutral Density Technology and High-Speed Biplanar
X-ray. Stapp Car Crash J 45:337-368.
Hardy WN, Mason MJ, Foster CD, Shah CS, Kopacz JM, Yang KH, King AI, Bishop J,
Bey M, Anderst W, Tashman S (2007) A study of the response of the human cadaver
head to impact. Stapp Car Crash J 51:17-80.
267

Harris FM, Tesseur I, Brecht WJ, Xu Q, Mullendorff K, Chang S, Wyss-Coray T, Mahley
RW, Huang Y (2004) Astroglial regulation of apolipoprotein E expression in neuronal
cells. Implications for Alzheimer's disease. J Biol Chem 279:3862-3868.
Harris FM, Brecht WJ, Xu Q, Tesseur I, Kekonius L, Wyss-Coray T, Fish JD, Masliah E,
Hopkins PC, Scearce-Levie K, Weisgraber KH, Mucke L, Mahley RW, Huang Y (2003)
Carboxyl-terminal-truncated apolipoprotein E4 causes Alzheimer's disease-like
neurodegeneration and behavioral deficits in transgenic mice. Proc Natl Acad Sci U S A
100:10966-10971.
Harris OA, Muh CR, Surles MC, Pan Y, Rozycki G, Macleod J, Easley K (2009) Discrete
cerebral hypothermia in the management of traumatic brain injury: a randomized
controlled trial. J Neurosurg 110:1256-1264.
Hatters DM, Peters-Libeu CA, Weisgraber KH (2006) Apolipoprotein E structure:
insights into function. Trends Biochem Sci 31:445-454.
Hatters DM, Budamagunta MS, Voss JC, Weisgraber KH (2005) Modulation of
apolipoprotein E structure by domain interaction: differences in lipid-bound and lipid-free
forms. J Biol Chem 280:34288-34295.
Henn H-W (1998) Crash Tests and the Head Injury Criterion. Teach Math Appl 17:162-
170.
268

Hertz E (1993) A Note on the Head Injury Criteria (HIC) as a predictor of the Risk of
Skull Fracture. In: Proceedings of the 37th Annual Conference of Association for the
Advancement of Automotive Medicine, pp 303-312. San Antonio, TX: Association for
the Advancement of Automotive Medicine.
Herz J, Chen Y (2006) Reelin, lipoprotein receptors and synaptic plasticity. Nat Rev
Neurosci 7:850-859.
Hiekkanen H, Kurki T, Brandstack N, Kairisto V, Tenovuo O (2009) Association of injury
severity, MRI-results and ApoE genotype with 1-year outcome in mainly mild TBI: a
preliminary study. Brain Inj 23:396-402.
Hirsch-Reinshagen V, Zhou S, Burgess BL, Bernier L, McIsaac SA, Chan JY, Tansley
GH, Cohn JS, Hayden MR, Wellington CL (2004) Deficiency of ABCA1 impairs
apolipoprotein E metabolism in brain. J Biol Chem 279:41197-41207.
Hirsch-Reinshagen V, Maia LF, Burgess BL, Blain JF, Naus KE, McIsaac SA, Parkinson
PF, Chan JY, Tansley GH, Hayden MR, Poirier J, Van Nostrand WE, Wellington CL
(2005) The absence of ABCA1 decreases soluble apoE levels but does not diminish
amyloid deposition in two murine models of Alzheimer's Disease. J Biol Chem
280:43243-43256.
Hobbs HH, Rader DJ (1999) ABC1: connecting yellow tonsils, neuropathy, and very low
HDL. J Clin Invest 104:1015-1017.
269

Hodgson VR, Gurdjian ES, Thomas LM (1966) Experimental skull deformation and
brain displacement demonstrated by flash x-ray technique. J Neurosurg 25:549-552.
Hoe HS, Freeman J, Rebeck GW (2006) Apolipoprotein E decreases tau kinases and
phospho-tau levels in primary neurons. Mol Neurodegener 1:18.
Hoge CW, McGurk D, Thomas JL, Cox AL, Engel CC, Castro CA (2008) Mild traumatic
brain injury in US soldiers returning from Iraq. New Eng J Med 358:453-463.
Holbourn AH (1943) Mechanics of head injury. The Lancet 2:438-441.
Holbourn AH (1945) Mechanics of brain injuries. Br Med Bull 3:147-149.
Holmin S, Hojeberg B (2004) In situ detection of intracerebral cytokine expression after
human brain contusion. Neurosci Lett 369:108-114.
Holmin S, Soderlund J, Biberfeld P, Mathiesen T (1998) Intracerebral inflammation after
human brain contusion. Neurosurgery 42:291-298; discussion 298-299.
Holtzman DM, Pitas RE, Kilbridge J, Nathan B, Mahley RW, Bu G, Schwartz AL (1995)
Low density lipoprotein receptor-related protein mediates apolipoprotein E-dependent
neurite outgrowth in a central nervous system-derived neuronal cell line. Proc Natl Acad
Sci U S A 92:9480-9484.
270

Horlein AJ, Naar AM, Heinzel T, Torchia J, Gloss B, Kurokawa R, Ryan A, Kamei Y,
Soderstrom M, Glass CK, Rosenfeld MG (1995) Ligand-independent repression by the
thyroid hormone receptor mediated by a nuclear receptor co-repressor. Nature 377:397-
404.
Houck KA, Borchert KM, Hepler CD, Thomas JS, Bramlett KS, Michael LF, Burris TP
(2004) T0901317 is a dual LXR/FXR agonist. Mol Genet Metab 83:184-187.
Hu X, Crick SL, Bu G, Frieden C, Pappu RV, Lee J-M (2009) Amyloid seeds formed by
cellular uptake, concentration, and aggregation of the amyloid-beta peptide. Proc Natl
Acad Sci U S A.
Huang Y, Liu XQ, Wyss-Coray T, Brecht WJ, Sanan DA, Mahley RW (2001)
Apolipoprotein E fragments present in Alzheimer's disease brains induce neurofibrillary
tangle-like intracellular inclusions in neurons. Proc Natl Acad Sci U S A 98:8838-8843.
Hubschmann OR, Kornhauser D (1983) Effects of intraparenchymal hemorrhage on
extracellular cortical potassium in experimental head trauma. J Neurosurg 59:289-293.
Huh JW, Franklin MA, Widing AG, Raghupathi R (2006) Regionally distinct patterns of
calpain activation and traumatic axonal injury following contusive brain injury in
immature rats. Dev Neurosci 28:466-476.
271

Hunter JV, Wilde EA, Tong KA, Holshouser BA (2012) Emerging imaging tools for use
with traumatic brain injury research. J Neurotrauma 29:654-671.
Huuskonen J, Fielding PE, Fielding CJ (2004) Role of p160 coactivator complex in the
activation of liver X receptor. Arterioscler Thromb Vasc Biol 24:703-708.
Hyder AA, Wunderlich CA, Puvanachandra P, Gururaj G, Kobusingye OC (2007) The
impact of traumatic brain injuries: a global perspective. NeuroRehabilitation 22:341-353.
Ikonomovic MD, Uryu K, Abrahamson EE, Ciallella JR, Trojanowski JQ, Lee VM, Clark
RS, Marion DW, Wisniewski SR, DeKosky ST (2004) Alzheimer's pathology in human
temporal cortex surgically excised after severe brain injury. Exp Neurol 190:192-203.
Inoue T, Lin A, Ma X, McKenna SL, Creasey GH, Manley GT, Ferguson AR, Bresnahan
JC, Beattie MS (2013) Combined SCI and TBI: recovery of forelimb function after
unilateral cervical spinal cord injury (SCI) is retarded by contralateral traumatic brain
injury (TBI), and ipsilateral TBI balances the effects of SCI on paw placement. Exp
Neurol 248:136-147.
Iqbal K, Liu F, Gong CX, Grundke-Iqbal I (2010) Tau in Alzheimer disease and related
tauopathies. Curr Alzheimer Res 7:656-664.
Irizarry MC, Cheung BS, Rebeck GW, Paul SM, Bales KR, Hyman BT (2000)
Apolipoprotein E affects the amount, form, and anatomical distribution of amyloid beta-
272

peptide deposition in homozygous APP(V717F) transgenic mice. Acta Neuropathol
100:451-458.
Iwata A, Browne KD, Chen XH, Yugushi T, Smith DH (2005) Traumatic brain injury
induces biphasic upregulation of ApoE and ApoJ protein in rats. J Neurosci Res 82:103-
114.
Iwata A, Stys PK, Wolf JA, Chen XH, Taylor AG, Meaney DF, Smith DH (2004)
Traumatic axonal injury induces proteolytic cleavage of the voltage-gated sodium
channels modulated by tetrodotoxin and protease inhibitors. J Neurosci 24:4605-4613.
Iwata N, Tsubuki S, Takaki Y, Shirotani K, Lu B, Gerard NP, Gerard C, Hama E, Lee H-
J, Saido TC (2001) Metabolic Regulation of Brain Aβ by Neprilysin. Science 292:1550-
1552.
Iwata N, Tsubuki S, Takaki Y, Watanabe K, Sekiguchi M, Hosoki E, Kawashima-
Morishima M, Lee HJ, Hama E, Sekine-Aizawa Y, Saido TC (2000) Identification of the
major Abeta1-42-degrading catabolic pathway in brain parenchyma: suppression leads
to biochemical and pathological deposition. Nat Med 6:143-150.
Iwatsubo T (2004) The gamma-secretase complex: machinery for intramembrane
proteolysis. Curr Opin Neurobiol 14:379-383.
273

Jamroz-Wisniewska A, W¢jcicka G, Horoszewicz K, Beltowski J (2007) Liver X
receptors (LXRs). Part II: Non-lipid effects, role in pathology, and therapeutic
implications. Postepy Hig Med Dosw 61:760-785.
Janowski BA, Willy PJ, Devi TR, Falck JR, Mangelsdorf DJ (1996) An oxysterol
signalling pathway mediated by the nuclear receptor LXR alpha. Nature 383:728-731.
Janowski BA, Grogan MJ, Jones SA, Wisely GB, Kliewer SA, Corey EJ, Mangelsdorf DJ
(1999) Structural requirements of ligands for the oxysterol liver X receptors LXRalpha
and LXRbeta. Proc Natl Acad Sci U S A 96:266-271.
Jha A, Weintraub A, Allshouse A, Morey C, Cusick C, Kittelson J, Harrison-Felix C,
Whiteneck G, Gerber D (2008) A randomized trial of modafinil for the treatment of
fatigue and excessive daytime sleepiness in individuals with chronic traumatic brain
injury. J Head Trauma Rehabil 23:52-63.
Ji ZS, Miranda RD, Newhouse YM, Weisgraber KH, Huang Y, Mahley RW (2002)
Apolipoprotein E4 potentiates amyloid beta peptide-induced lysosomal leakage and
apoptosis in neuronal cells. J Biol Chem 277:21821-21828.
Jiang JY, Xu W, Li WP, Gao GY, Bao YH, Liang YM, Luo QZ (2006) Effect of long-term
mild hypothermia or short-term mild hypothermia on outcome of patients with severe
traumatic brain injury. J Cereb Blood Flow Metab 26:771-776.
274

Jiang JY, Xu W, Li WP, Xu WH, Zhang J, Bao YH, Ying YH, Luo QZ (2005) Efficacy of
standard trauma craniectomy for refractory intracranial hypertension with severe
traumatic brain injury: a multicenter, prospective, randomized controlled study. J
Neurotrauma 22:623-628.
Jiang Q, Lee CY, Mandrekar S, Wilkinson B, Cramer P, Zelcer N, Mann K, Lamb B,
Willson TM, Collins JL, Richardson JC, Smith JD, Comery TA, Riddell D, Holtzman DM,
Tontonoz P, Landreth GE (2008a) ApoE promotes the proteolytic degradation of Abeta.
Neuron 58:681-693.
Jiang Y, Sun XC, Gui L, Tang WY, Zhen LP, Gu YJ, Wu HT (2008b) Lack of association
between apolipoprotein E promoters in epsilon-4 carriers and worsening on computed
tomography in early stage of traumatic brain injury. Acta Neurochir (Suppl) 105:233-
236.
Johnson VE, Stewart W, Smith DH (2010) Traumatic brain injury and amyloid-beta
pathology: a link to Alzheimer's disease? Nat Rev Neurosci 11:361-370.
Johnson VE, Stewart W, Smith DH (2012) Widespread tau and amyloid-beta pathology
many years after a single traumatic brain injury in humans. Brain Pathol 22:142-149.
Johnson VE, Stewart W, Smith DH (2013) Axonal pathology in traumatic brain injury.
Exp Neurol 246:35-43.
275

Johnson VE, Stewart W, Graham DI, Stewart JE, Praestgaard AH, Smith DH (2009) A
neprilysin polymorphism and amyloid-beta plaques after traumatic brain injury. J
Neurotrauma 26:1197-1202.
Jordan BD (2013) The clinical spectrum of sport-related traumatic brain injury. Nat Rev
Neurol 9:222-230.
Kakarieka A, Braakman R, Schakel EH (1994) Clinical significance of the finding of
subarachnoid blood on CT scan after head injury. Acta Neurochir (Wien) 129:1-5.
Kakuda N, Funamoto S, Yagishita S, Takami M, Osawa S, Dohmae N, Ihara Y (2006)
Equimolar production of amyloid beta-protein and amyloid precursor protein intracellular
domain from beta-carboxyl-terminal fragment by gamma-secretase. J Biol Chem
281:14776-14786.
Kamal A, Stokin GB, Yang Z, Xia CH, Goldstein LS (2000) Axonal transport of amyloid
precursor protein is mediated by direct binding to the kinesin light chain subunit of
kinesin-I. Neuron 28:449-459.
Kamal A, Almenar-Queralt A, LeBlanc JF, Roberts EA, Goldstein LS (2001) Kinesin-
mediated axonal transport of a membrane compartment containing beta-secretase and
presenilin-1 requires APP. Nature 414:643-648.
276

Kane MJ, Angoa-Perez M, Briggs DI, Viano DC, Kreipke CW, Kuhn DM (2012) A mouse
model of human repetitive mild traumatic brain injury. J Neurosci Methods 203:41-49.
Kanekiyo T, Xu H, Bu G ApoE and Aβ in Alzheimer’s Disease: Accidental Encounters or
Partners? Neuron 81:740-754.
Kanemitsu H, Tomiyama T, Mori H (2003) Human neprilysin is capable of degrading
amyloid beta peptide not only in the monomeric form but also the pathological
oligomeric form. Neurosci Lett 350:113-116.
Karran E, Mercken M, De Strooper B (2011) The amyloid cascade hypothesis for
Alzheimer's disease: an appraisal for the development of therapeutics. Nat Rev Drug
Discov 10:698-712.
Katayama Y, Becker DP, Tamura T, Hovda DA (1990) Massive increases in
extracellular potassium and the indiscriminate release of glutamate following concussive
brain injury. J Neurosurg 73:889-900.
Katsnelson A (2011) Gene tests for brain injury still far from the football field. Nat Med
17:638-638.
Katz A, Udata C, Ott E, Hickey L, Burczynski ME, Burghart P, Vesterqvist O, Meng X
(2009) Safety, pharmacokinetics and pharmacodynamics of single doses of LXR-623, a
novel liver-X-receptor agonist, in healthy participants. J Clin Pharmacol 49:643-649.
277

Katzman R, Aronson M, Fuld P, Kawas C, Brown T, Morgenstern H, Frishman W, Gidez
L, Eder H, Ooi WL (1989) Development of dementing illnesses in an 80-year-old
volunteer cohort. Ann Neurol 25:317-324.
Kay AD, Day SP, Kerr M, Nicoll JA, Packard CJ, Caslake MJ (2003) Remodeling of
cerebrospinal fluid lipoprotein particles after human traumatic brain injury. J
Neurotrauma 20:717-723.
Kay T, Harrington DE, Admas R, Anderson T, Berrol S, Cicerone K, Dahlberg C, Gerber
D, Goka R, Harley P, Hilt J, Horn L, Lehmkuhl D, Malec J (1993) Definition of mild
traumatic brain injury. In: J Head Trauma Rehabil, pp 86-87.
Kennedy MA, Venkateswaran A, Tarr PT, Xenarios I, Kudoh J, Shimizu N, Edwards PA
(2001) Characterization of the human ABCG1 gene: liver X receptor activates an
internal promoter that produces a novel transcript encoding an alternative form of the
protein. J Biol Chem 276:39438-39447.
Kennedy MA, Barrera GC, Nakamura K, Bald n A, Tarr P, Fishbein MC, Frank J,
Francone OL, Edwards PA (2005) ABCG1 has a critical role in mediating cholesterol
efflux to HDL and preventing cellular lipid accumulation. Cell Metabolism 1 121-131.
Khoshyomn S, Tranmer BI (2004) Diagnosis and management of pediatric closed head
injury. Semin Pediatr Surg 13:80-86.
278

Khuman J, Meehan WP, 3rd, Zhu X, Qiu J, Hoffmann U, Zhang J, Giovannone E, Lo
EH, Whalen MJ (2011) Tumor necrosis factor alpha and Fas receptor contribute to
cognitive deficits independent of cell death after concussive traumatic brain injury in
mice. J Cereb Blood Flow Metab 31:778-789.
Kiehn O, Kjaerulff O (1998) Distribution of central pattern generators for rhythmic motor
outputs in the spinal cord of limbed vertebrates. Ann N Y Acad Sci 860:110-129.
Kigerl KA, Gensel JC, Ankeny DP, Alexander JK, Donnelly DJ, Popovich PG (2009)
Identification of two distinct macrophage subsets with divergent effects causing either
neurotoxicity or regeneration in the injured mouse spinal cord. J Neurosci 29:13435-
13444.
Kilbourne M, Kuehn R, Tosun C, Caridi J, Keledjian K, Bochicchio G, Scalea T,
Gerzanich V, Simard JM (2009) Novel model of frontal impact closed head injury in the
rat. J Neurotrauma 26:2233-2243.
Kim J, Castellano JM, Jiang H, Basak JM, Parsadanian M, Pham V, Mason SM, Paul
SM, Holtzman DM (2009) Overexpression of low-density lipoprotein receptor in the
brain markedly inhibits amyloid deposition and increases extracellular A beta clearance.
Neuron 64:632-644.
King AI (2000) Fundamentals of impact biomechanics: Part I--Biomechanics of the
head, neck, and thorax. Annu Rev Biomed Eng 2:55-81.
279

King AI, Yang KH, Hardy WN (2011) Recent firsts in cadaveric impact biomechanics
research. Clin Anat 24:294-308.
King AI, Yang KH, Zhang L, Hardy W (2004) Is Rotational Acceleration More Injurious to
the Brain Than Linear Acceleration? In: Frontiers in Biomedical Engineering (Hwang
NHC, Woo SLY, eds), pp 135-147: Springer US.
King AI, Yang KH, Zhang L, Hardy W, Viano DC (2003) Is head injury caused by linear
or angular acceleration? In: Proceedings of the International Research Conference on
the Biomechanics of Impacts (IRCOBI), pp 1-12. Lisbon, Portugal.
Kitamura HW, Hamanaka H, Watanabe M, Wada K, Yamazaki C, Fujita SC, Manabe T,
Nukina N (2004) Age-dependent enhancement of hippocampal long-term potentiation in
knock-in mice expressing human apolipoprotein E4 instead of mouse apolipoprotein E.
Neurosci Lett 369:173-178.
Kitamura K (1981) Therapeutic effect of pyritinol on sequelae of head injuries. J Int Med
Res 9:215-221.
Kjaerulff O, Kiehn O (1996) Distribution of networks generating and coordinating
locomotor activity in the neonatal rat spinal cord in vitro: a lesion study. J Neurosci
16:5777-5794.
280

Koch S, Donarski N, Goetze K, Kreckel M, Sturernburg HJ, Buhmann C, Beisiegel U
(2001) Characterization of four lipoprotein classes in human cerebrospinal fluid. J Lipid
Res 42:1143-1151.
Koldamova R, Staufenbiel M, Lefterov I (2005a) Lack of ABCA1 considerably
decreased brain apoE level and increases amyloid deposition in APP23 mice. J Biol
Chem 280:43224-43235.
Koldamova R, Fitz NF, Lefterov I (2010) The role of ATP-binding cassette transporter
A1 in Alzheimer's disease and neurodegeneration. Biochim Biophys Acta 180:824-830.
Koldamova RP, Lefterov IM, Lefterova MI, Lazo JS (2001) Apolipoprotein A1 directly
interacts with amyloid precursor protein and inhibits A beta aggregation and toxicity.
Biochemistry 40:3553-3560.
Koldamova RP, Lefterov IM, Ikonomovic MD, Skoko J, Lefterov PI, Isanski BA,
DeKosky ST, Lazo JS (2003) 22R-hydroxycholesterol and 9-cis-retinoic acid induce
ATP-binding cassette transporter A1 expression and cholesterol efflux in brain cells and
decrease amyloid á secretion. J Biol Chem 278:13244-13256.
Koldamova RP, Lefterov IM, Staufenbiel M, Wolfe D, Huang S, Glorioso JC, Walter M,
Roth MG, Lazo JS (2005b) The liver X receptor ligand T0901317 decreases amyloid
beta production in vitro and in a mouse model of Alzheimer's disease. J Biol Chem
280:4079-4088.
281

Koo EH, Sisodia SS, Archer DR, Martin LJ, Weidemann A, Beyreuther K, Fischer P,
Masters CL, Price DL (1990) Precursor of amyloid protein in Alzheimer disease
undergoes fast anterograde axonal transport. Proc Natl Acad Sci U S A 87:1561-1565.
Korwek KM, Trotter JH, Ladu MJ, Sullivan PM, Weeber EJ (2009) ApoE isoform-
dependent changes in hippocampal synaptic function. Mol Neurodegener 4:21.
Koshinaga M, Katayama Y, Fukushima M, Oshima H, Suma T, Takahata T (2000)
Rapid and widespread microglial activation induced by traumatic brain injury in rat brain
slices. J Neurotrauma 17:185-192.
Krave U, Hojer S, Hansson HA (2005) Transient, powerful pressures are generated in
the brain by a rotational acceleration impulse to the head. Eur J Neurosci 21:2876-2882.
Krave U, Al-Olama M, Hansson HA (2011) Rotational acceleration closed head flexion
trauma generates more extensive diffuse brain injury than extension trauma. J
Neurotrauma 28:57-70.
Krimbou L, Denis B, Haidar M, Carrier M, Marcil M, Genest J, Jr. (2004) Molecular
interactions between apoE and ABCA1: impact on apoE lipidation. J Lipid Res 45:839-
848.
282

Ksiezak-Reding H, Morgan K, Mattiace LA, Davies P, Liu WK, Yen SH, Weidenheim K,
Dickson DW (1994) Ultrastructure and biochemical composition of paired helical
filaments in corticobasal degeneration. Am J Pathol 145:1496-1508.
Kuppa S (2004) Injury Criteria for Side Impact Dummies. In: National Transportation
Biomechanics Research Center.
Kwon BK, Hillyer J, Tetzlaff W (2010) Translational research in spinal cord injury: a
survey of opinion from the SCI community. J Neurotrauma 27:21-33.
LaDu MJ, Falduto MT, Manelli AM, Reardon CA, Getz GS, Frail DE (1994) Isoform-
specific binding of apolipoprotein E to beta-amyloid. J Biol Chem 269:23403-23406.
LaDu MJ, Pederson TM, Frail DE, Reardon CA, Getz GS, Falduto MT (1995)
Purification of apolipoprotein E attenuates isoform-specific binding to beta-amyloid. J
Biol Chem 270:9039-9042.
LaDu MJ, Gilligan SM, Lukens JR, Cabana VG, Reardon CA, Van Eldik LJ, Holtzman
DA (1998) Nascent astrocyte particles differ from lipoproteins in CSF. J Neurochem
70:2070-2081.
LaDu MJ, Reardon C, Van Eldik L, Fagan AM, Bu G, Holtzmann D, Getz GS (2000a)
Lipoproteins in the central nervous system. Ann New York Acad Sci 903:167-175.
283

LaDu MJ, Reardon C, Van Eldik L, Fagan AM, Bu G, Holtzman D, Getz GS (2000b)
Lipoproteins in the central nervous system. Ann N Y Acad Sci 903:167-175.
Langlois JA, Rutland-Brown W, Wald MM (2006) The Epidemiology and Impact of
Traumatic Brain Injury: A Brief Overview. J Head Trauma Rehabil 21:375-378.
Langmann T, Klucken J, Reil M, Liebisch G, Luciani M-Fo, Chimini G, Kaminski WE,
Schmitz G (1999) Molecular cloning of the human ATP-binding cassette transporter 1
(hABC1): Evidence for sterol-dependent regulation in macrophages. Biochem Biophys
Res Commun 257:29-33.
LaPlaca MC, Simon CM, Prado GR, Cullen DK (2007) CNS injury biomechanics and
experimental models. Prog Brain Res 161:13-26.
Laskowitz DT, Horsburgh K, Roses AD (1998) Apolipoprotein E and the CNS response
to injury. J Cereb Blood Flow Metab 18:465-471.
Lau IV, Viano DC, Gamero F (1989) Invalidity of speculated injury mechanism in
autopsy reports. Injury 20:16-21.
Laurer HL, Bareyre FM, Lee VM, Trojanowski JQ, Longhi L, Hoover R, Saatman KE,
Raghupathi R, Hoshino S, Grady MS, McIntosh TK (2001) Mild head injury increasing
the brain's vulnerability to a second concussive impact. J Neurosurg 95:859-870.
284

Lawn RM, Wade DP, Couse TL, Wilcox JN (2001) Localization of human ATP-Binding
cassette transporter 1 (ABC1) in normal and atherosclerotic tissues. Arterioscler
Thromb Vasc Biol 21:378-385.
Lee G, Neve RL, Kosik KS (1989) The microtubule binding domain of tau protein.
Neuron 2:1615-1624.
Lee Y-H, Jung MG, Kang HB, Choi K-C, Haam S, Jun W, Kim Y-J, Cho HY, Yoon H-G
(2008) Effect of anti-histone acetyltransferase activity from Rosa rugosa Thunb.
(Rosaceae) extracts on androgen receptor-mediated transcriptional regulation. J
Ethnopharmacol 118:412-417.
Lefterov I, Fitz NF, Cronican AA, Fogg A, Lefterov P, Kodali R, Wetze R, Koldamova R
(2010) Apolipoprotein A-I deficiency increases cerebral amyloid angiopathy and
cognitive deficits in APP/PS1E9 mice. J Biol Chem 285 36945-36957.
Lehmann JM, Kliewer SA, Moore LB, Smith-Oliver TA, Sundseth SS, Winegar DA,
Blanchard DE, Spencer TA, Wilson TM (1997) Activation of the nuclear receptor LXR by
oxysterols defines a new hormone response pathway. J Biol Chem 272:3137-3140.
Leissring MA, Farris W, Chang AY, Walsh DM, Wu X, Sun X, Frosch MP, Selkoe DJ
(2003) Enhanced proteolysis of beta-amyloid in APP transgenic mice prevents plaque
formation, secondary pathology, and premature death. Neuron 40:1087-1093.
285

Levy-Lahad E, Wasco W, Poorkaj P, Romano DM, Oshima J, Pettingell WH, Yu CE,
Jondro PD, Schmidt SD, Wang K, et al. (1995) Candidate gene for the chromosome 1
familial Alzheimer's disease locus. Science 269:973-977.
Lewen A, Li GL, Nilsson P, Olsson Y, Hillered L (1995) Traumatic brain injury in rat
produces changes of beta-amyloid precursor protein immunoreactivity. Neuroreport
6:357-360.
Lewis TL, Cao D, Lu H, Mans RA, Su YR, Jungbauer L, Linton MF, Fazio S, Ladu MJ, Li
L (2010) Overexpression of human apolipoprotein A-I preserves cognitive function and
attenuates neuroinflammation and cerebral amyloid angiopathy in a mouse model of
Alzheimer's Disease. J Biol Chem 285:36958-36968.
Li G, Shen YC, Li YT, Chen CH, Zhau YW, Silverman JM (1992) A case-control study of
Alzheimer's disease in China. Neurology 42:1481-1488.
Li J, Kanekiyo T, Shinohara M, Zhang Y, LaDu MJ, Xu H, Bu G (2012) Differential
regulation of amyloid-beta endocytic trafficking and lysosomal degradation by
apolipoprotein E isoforms. J Biol Chem 287:44593-44601.
Li XY, Li J, Feng DF, Gu L (2010) Diffuse axonal injury induced by simultaneous
moderate linear and angular head accelerations in rats. Neuroscience 169:357-369.
286

Li Y, Zhang L, Kallakuri S, Zhou R, Cavanaugh JM (2011) Quantitative relationship
between axonal injury and mechanical response in a rodent head impact acceleration
model. J Neurotrauma 28:1767-1782.
Liang Y, Lin S, Beyer TP, Zhang Y, Wu X, Bales KR, DeMattos RB, May PC, Li SD,
Jiang XC, Eacho PI, Cao G, Paul SM (2004) A liver X receptor and retinoid X receptor
heterodimer mediates apolipoprotein E expression, secretion and cholesterol
homeostasis in astrocytes. J Neurochem 88 623-634.
Liao H, Langmann T, Schmitz G, Zhu Y (2002) Native LDL upregulation of ATP-binding
cassette transporter-1 in human vascular endothelial cells. Arterioscler Thromb Vasc
Biol 22:127-132.
Lichtman SW, Seliger G, Tycko B, Marder K (2000) Apolipoprotein E and functional
recovery from brain injury following postacute rehabilitation. Neurology 55:1536-1539.
Lighthall JW, Dixon CE, Anderson TE (1989) Experimental models of brain injury. J
Neurotrauma 6:83-97.
Lin Y, Wen L (2013) Inflammatory response following diffuse axonal injury. Int J Med Sci
10:515-521.
Lindenberg R, Freytag E (1960) The mechanism of cerebral contusions. A pathologic-
anatomic study. Arch Pathol 69:440-469.
287

Lissner HR, Lebow M, Evans FG (1960) Experimental studies on the relation between
acceleration and intracranial pressure changes in man. Surg Gynecol Obstet 111:329-
338.
Loane DJ, Byrnes KR (2010) Role of microglia in neurotrauma. Neurotherapeutics
7:366-377.
Loane DJ, Pocivavasek A, Moussa CE, Thompson R, Matsuoka Y, Faden AI, Rebeck
GW, Burns MP (2009) Amyloid precursor protein secretases as therapeutic targets for
traumatic brain injury. Nat Med 15:377-379.
Loane DL, Washington PM, Vardanian L, Pocivavsek A, Hoe HS, Duff KE, Cernak I,
Rebek GW, Faden AI, Burns MP (2011) Modulation of ABCA1 by an LXR agonist
reduces beta-amyloid levels and improves outcome after traumatic brain injury. J
Neurotrauma 28:225-236.
Lopez-Guerrero AL, Martinez-Lage JF, Gonzalez-Tortosa J, Almagro MJ, Garcia-
Martinez S, Reyes SB (2012) Pediatric crushing head injury: biomechanics and clinical
features of an uncommon type of craniocerebral trauma. Childs Nerv Syst 28:2033-
2040.
Lorent K, Overbergh L, Moechars D, De Strooper B, Van Leuven F, Van den Berghe H
(1995) Expression in mouse embryos and in adult mouse brain of three members of the
amyloid precursor protein family, of the alpha-2-macroglobulin receptor/low density
288

lipoprotein receptor-related protein and of its ligands apolipoprotein E, lipoprotein lipase,
alpha-2-macroglobulin and the 40,000 molecular weight receptor-associated protein.
Neuroscience 65:1009-1025.
Lu J, Gary KW, Neimeier JP, Ward J, Lapane KL (2012) Randomized controlled trials in
adult traumatic brain injury. Brain Inj 26:1523-1548.
Lu KT, Wang YW, Yang JT, Yang YL, Chen HI (2005) Effect of interleukin-1 on
traumatic brain injury-induced damage to hippocampal neurons. J Neurotrauma 22:885-
895.
Lu LQ, Jiang JY, Yu MK, Hou LJ, Chen ZG, Zhang GJ, Zhu C (2003) Standard large
trauma craniotomy for severe traumatic brain injury. Chin J Traumatol 6:302-304.
Lund EG, Guileyardo JM, Russell DW (1999) cDNA cloning of cholesterol 24-
hydroxylase, a mediator of cholesterol homeostasis in the brain. Proc Natl Acad Sci U S
A 96:7238-7243.
Lynch JR, Pineda JA, Morgan D, Zhang L, Warner DS, Benveniste H, Laskowitz DT
(2002) Apolipoprotein E affects the central nervous system response to injury and the
development of cerebral edema. Ann Neurol 51:113-117.
289

Lynch JR, Tang W, Wang H, Vitek MP, Bennett ER, Sullivan PM, Warner DS, Laskowitz
DT (2003) APOE genotype and an ApoE-mimetic peptide modify the systemic and
central nervous system inflammatory response. J Biol Chem 278:48529-48533.
Macciocchi S, Seel RT, Thompson N, Byams R, Bowman B (2008) Spinal cord injury
and co-occurring traumatic brain injury: assessment and incidence. Arch Phys Med
Rehabil 89:1350-1357.
Maeda J, Higuchi M, Inaji M, Ji B, Haneda E, Okauchi T, Zhang MR, Suzuki K, Suhara
T (2007) Phase-dependent roles of reactive microglia and astrocytes in nervous system
injury as delineated by imaging of peripheral benzodiazepine receptor. Brain Res
1157:100-111.
Maezawa I, Nivison M, Montine KS, Maeda N, Montine TJ (2006) Neurotoxicity from
innate immune response is greatest with targeted replacement of E4 allele of
apolipoprotein E gene and is mediated by microglial p38MAPK. FASEB J 20:797-799.
Magnoni S, Brody DL (2010) New perspectives on amyloid-beta dynamics after acute
brain injury: moving between experimental approaches and studies in the human brain.
Arch Neurol 67:1068-1073.
Magnoni S, Esparza TJ, Conte V, Carbonara M, Carrabba G, Holtzman DM, Zipfel GJ,
Stocchetti N, Brody DL (2012) Tau elevations in the brain extracellular space correlate
290

with reduced amyloid-beta levels and predict adverse clinical outcomes after severe
traumatic brain injury. Brain 135:1268-1280.
Mahley RW (1998) Apolipoprotein E: cholesterol transport protein with expanding role in
cell biology. Science 240:622-630.
Mahley RW, Huang Y (2006) Apolipoprotein (apo) E4 and Alzheimer's disease: unique
conformational and biophysical properties of apoE4 can modulate neuropathology. Acta
Neurologica Scandinavica 114:8-14.
Marder E, Bucher D (2001) Central pattern generators and the control of rhythmic
movements. Current Biology 11:R986-R996.
Marion DW, Penrod LE, Kelsey SF, Obrist WD, Kochanek PM, Palmer AM, Wisniewski
SR, DeKosky ST (1997) Treatment of traumatic brain injury with moderate hypothermia.
N Engl J Med 336:540-546.
Marklund N, Hillered L (2011) Animal modelling of traumatic brain injury in preclinical
drug development: where do we go from here? Br J Pharmacol 164:1207-1229.
Marmarou A, Foda MA, van den Brink W, Campbell J, Kita H, Demetriadou K (1994) A
new model of diffuse brain injury in rats. Part I: Pathophysiology and biomechanics. J
Neurosurg 80:291-300.
291

Martinez LO, Agerholm-Larsen B, Wang N, Chen W, Tall AR (2003) Phosphorylation of
a Pest sequence in ABCA1 promotes calpain degradation and is reversed by apoA-I. J
Biol Chem 278:37368-37374.
Martland HS (1928) Punch drunk. J Am Med Assoc 91:1103-1107.
Masson F, Thicoipe M, Aye P, Mokni T, Senjean P, Schmitt V, Dessalles PH,
Cazaugade M, Labadens P (2001) Epidemiology of severe brain injuries: a prospective
population-based study. J Trauma 51:481-489.
Masters CL, Selkoe DJ (2012) Biochemistry of amyloid beta-protein and amyloid
deposits in Alzheimer disease. Cold Spring Harb Perspect Med 2:a006262.
Mattei TA, Bond BJ, Goulart CR, Sloffer CA, Morris MJ, Lin JJ (2012) Performance
analysis of the protective effects of bicycle helmets during impact and crush tests in
pediatric skull models. J Neurosurg Pediatr 10:490-497.
Matuseviciene G, Borg J, Stalnacke BM, Ulfarsson T, de Boussard C (2013) Early
intervention for patients at risk for persisting disability after mild traumatic brain injury: a
randomized, controlled study. Brain Inj 27:318-324.
May S, Martin G, Wellington C (2011) Brain injury and dementia: Is there a connection?
J Neurology Neurosci 2:1-7.
292

McAllister TW (2011) Neurobiological consequences of traumatic brain injury. Dialogues
Clin Neurosci 13:287-300.
McGinn MJ, Kelley BJ, Akinyi L, Oli MW, Liu MC, Hayes RL, Wang KK, Povlishock JT
(2009) Biochemical, structural, and biomarker evidence for calpain-mediated
cytoskeletal change after diffuse brain injury uncomplicated by contusion. J Neuropathol
Exp Neurol 68:241-249.
McIntosh TK, Noble L, Andrews B, Faden AI (1987) Traumatic brain injury in the rat:
characterization of a midline fluid-percussion model. Cent Nerv Syst Trauma 4:119-134.
McIntosh TK, Smith DH, Meaney DF, Kotapka MJ, Gennarelli TA, Graham DI (1996)
Neuropathological sequelae of traumatic brain injury: relationship to neurochemical and
biomechanical mechanisms. Lab Invest 74:315-342.
McIntosh TK, Vink R, Noble L, Yamakami I, Fernyak S, Soares H, Faden AL (1989)
Traumatic brain injury in the rat: characterization of a lateral fluid-percussion model.
Neuroscience 28:233-244.
McKee AC, Cantu RC, Nowinski CJ, Hedley-Whyte ET, Gavett BE, Busdon AE, Santini
VE, Lee HS, Kubilus CA, Stern RA (2009) Chronic traumatic encephalopathy in
athletes: Progressive tauopathy following repetitive head injury. J Neuropathol Exp
Neurol 68:709-735.
293

McKee AC, Gavett BE, Stern RA, Nowinski CJ, Cantu RC, Kowall NW, Perl DP, Hedley-
Whyte ET, Price B, Sullivan C, Morin P, Lee HS, Kubilus CA, Daneshvar DH, Wulff M,
Budson AE (2010) TDP-43 proteinopathy and motor neuron disease in chronic
traumatic encephalopathy. J Neuropathol Exp Neurol 69:918-929.
McKee AC et al. (2013) The spectrum of disease in chronic traumatic encephalopathy.
Brain 136:43-64.
McKenzie KJ, McLellan DR, Gentleman SM, Maxwell WL, Gennarelli TA, Graham DI
(1996) Is beta-APP a marker of axonal damage in short-surviving head injury? Acta
Neuropathol 92:608-613.
McKissock W, Taylor JC, Bloom WH, Till K (1960) Extradural haematoma. Observations
on 125 cases. Lancet 2:167–172.
McLean AJ (1995) Brain injury without head impact? J Neurotrauma 12:621-625.
McLean AJ, Anderson RWG (1997) Biomechanics of closed head injury. In: Head Injury
- Pathophysiology and Management of Severe Closed Head Injury (Reilly P, Bullock R,
eds), pp 25-37. London: Chapman and Hall.
Meaney DF, Smith DH (2011) Biomechanics of concussion. Clin Sports Med 30:19-31,
vii.
294

Meaney S, Hassan M, Sakinis A, Lutjohann D, von Bergmann K, Wennmalm U,
Diczfalusy U, Bjorkhem I (2001) Evidence that the major oxysterols in human circulation
originate from distinct pools of cholesterol: a stable isotope study. J Lipid Res 42:70-78.
Meehan WP, 3rd, Zhang J, Mannix R, Whalen MJ (2012) Increasing recovery time
between injuries improves cognitive outcome after repetitive mild concussive brain
injuries in mice. Neurosurgery 71:885-891.
Menke JG, Macnaul KL, Hayes NS, Baffic J, Chao YS, Elbrecht A, Kelly LJ, Lam MH,
Schmidt A, Sahoo S, Wang J, Wright SD, Xin P, Zhou G, Moller DE, Sparrow CP (2002)
A novel liver X receptor agonist establishes species differences in the regulation of
cholesterol 7alpha-hydroxylase (CYP7a). Endocrinology 143:2548-2558.
Menon DK, Schwab K, Wright DW, Maas AI (2010) Position statement: definition of
traumatic brain injury. Arch Phys Med Rehabil 91:1637-1640.
Miller BC, Eckman EA, Sambamurti K, Dobbs N, Chow KM, Eckman CB, Hersh LB,
Thiele DL (2003) Amyloid-β peptide levels in brain are inversely correlated with insulysin
activity levels in vivo. Proc Natl Acad Sci U S A 100:6221-6226.
Millspaugh JA (1937) Dementia pugilistica. US Naval Med Bull 35:297-303.
295

Miners JS, Van Helmond Z, Chalmers K, Wilcock G, Love S, Kehoe PG (2006)
Decreased expression and activity of neprilysin in Alzheimer disease are associated
with cerebral amyloid angiopathy. J Neuropathol Exp Neurol 65:1012-1021.
Mitro N, Vargas L, Romeo R, Koder A, Saez E (2007) T0901317 is a potent PXR ligand:
Implications for the biology ascribed to LXR. FEBS Lett 581:1721-1726.
Moein H, Khalili HA, Keramatian K (2006) Effect of methylphenidate on ICU and
hospital length of stay in patients with severe and moderate traumatic brain injury. Clin
Neurol Neurosurg 108:539-542.
Moore KJ, Rayner KJ, Suarez Y, Fernandez-Hernando C (2010) microRNAs and
cholesterol metabolism. Trends Endocrinol Metab 21:699-706.
Moran LM, Taylor HG, Ganesalingam K, Gastier-Foster JM, Frick J, Bangert B, Dietrich
A, Nuss KE, Rusin J, Wright M, Yeates KO (2009) Apolipoprotein E4 as a predictor of
outcomes in pediatric mild traumatic brain injury. J Neurotrauma 26:1489-1495.
Morrow JA, Hatters DM, Lu B, Höchtl P, Oberg KA, Rupp B, Weisgraber KH (2002)
Apolipoprotein E4 forms a molten globule: a potential basis for its association with
disease. J Biol Chem 277:50380-50385.
Mortimer JA, French LR, Hutton JT, Schuman LM (1985) Head injury as a risk factor for
Alzheimer's disease. Neurology 35:264-267.
296

Mortimer JA, van Duijn CM, Chandra V, Fratiglioni L, Graves AB, Heyman A, Jorm AF,
Kokmen E, Kondo K, Rocca WA (1991) Head trauma as a risk factor for Alzheimer's
disease: a collaborative re-analysis of case-control studies. EURODEM Risk Factors
Research Group. Int J Epidemiol 20 Suppl 2:S28-S35.
Mouzon B, Chaytow H, Crynen G, Bachmeier C, Stewart J, Mullan M, Stewart W,
Crawford F (2012) Repetitive mild traumatic brain injury in a mouse model produces
learning and memory deficits accompanied by histological changes. J Neurotrauma
29:2761-2773.
Muizelaar JP, Marmarou A, Ward JD, Kontos HA, Choi SC, Becker DP, Gruemer H,
Young HF (1991) Adverse effects of prolonged hyperventilation in patients with severe
head injury: a randomized clinical trial. J Neurosurg 75:731-739.
Murthy S, Born E, Mathur SN, Field FJ (2002) LXR/RXR activation enhances
basolateral efflux of cholesterol in CaCo-2 cells. J Lipid Res 43:1054-1064.
Myburgh JA, Cooper DJ, Finfer SR, Venkatesh B, Jones D, Higgins A, Bishop N, Higlett
T, Australian Traumatic Brain Injury Study Investigators for the Australian:New Zealand
Intensive Care Society Clinical Trials G (2008) Epidemiology and 12-month outcomes
from traumatic brain injury in Australia and New Zealand. J Trauma 64:854-862.
297

Nagao K, Takahashi K, Azuma Y, Takada M, Kimura Y, Matsuo M, Kioka N, Ueda K
(2012) ATP hydrolysis-dependent conformational changes in the extracellular domain of
ABCA1 are associated with apoA-I binding. J Lipid Res 53:126-136.
Nagata KO, Nakada C, Kasai RS, Kusumi A, Ueda K (2013) ABCA1 dimer–monomer
interconversion during HDL generation revealed by single-molecule imaging. Proc Natl
Acad Sci U S A 110:5034-5039.
Nakagawa H, Ueno M, Itokazu T, Yamashita T (2013) Bilateral movement training
promotes axonal remodeling of the corticospinal tract and recovery of motor function
following traumatic brain injury in mice. Cell Death Dis 4:e534.
Nakamura K, Kennedy MA, Bald n A, Bohanic DD, Lyons K, Edwards PA (2004)
Expression and regulation of multiple murine ATP-binding cassette transporter G1
mRNAs/isoforms that stimulate cellular cholesterol efflux to high density lipoprotein. J
Biol Chem 279 45980-45989.
Namba Y, Tomonaga M, Kawasaki H, Otomo E, Ikeda K (1991) Apolipoprotein E
immunoreactivity in cerebral amyloid deposits and neurofibrillary tangles in Alzheimer's
disease and kuru plaque amyloid in Creutzfeldt-Jakob disease. Brain Res 541:163-166.
Namjoshi DR, Good C, Cheng WH, Panenka W, Richards D, Cripton PA, Wellington CL
(2013a) Towards clinical management of traumatic brain injury: a review of models and
mechanisms from a biomechanical perspective. Dis Model Mech 6:1325-1338.
298

Namjoshi DR, Martin G, Donkin J, Wilkinson A, Stukas S, Fan J, Carr M, Tabarestani S,
Wuerth K, Hancock RE, Wellington CL (2013b) The liver X receptor agonist GW3965
improves recovery from mild repetitive traumatic brain injury in mice partly through
apolipoprotein E. PLoS One 8:e53529.
Naslund J, Jensen M, Tjernberg LO, Thyberg J, Terenius L, Nordstedt C (1994) The
metabolic pathway generating p3, an A beta-peptide fragment, is probably non-
amyloidogenic. Biochem Biophys Res Commun 204:780-787.
Nathan BP, Bellosta S, Sanan DA, Weisgraber KH, Mahley RW, Pitas RE (1994)
Differential effects of apolipoproteins E3 and E4 on neuronal growth in vitro. Science
264:850-852.
Nathan BP, Jiang Y, Wong GK, Shen F, Brewer GJ, Struble RG (2002) Apolipoprotein
E4 inhibits, and apolipoprotein E3 promotes neurite outgrowth in cultured adult mouse
cortical neurons through the low-density lipoprotein receptor-related protein. Brain Res
928:96-105.
Nathan BP, Chang KC, Bellosta S, Brisch E, Ge N, Mahley RW, Pitas RE (1995) The
inhibitory effect of apolipoprotein E4 on neurite outgrowth is associated with microtubule
depolymerization. J Biol Chem 270:19791-19799.
Neubuerger KT, Sinton DW, Denst J (1959) Cerebral atrophy associated with boxing.
AMA Arch Neurol Psychiatry 81:403-408.
299

Neve RL, Harris P, Kosik KS, Kurnit DM, Donlon TA (1986) Identification of cDNA
clones for the human microtubule-associated protein tau and chromosomal localization
of the genes for tau and microtubule-associated protein 2. Mol Brain Res 1:271-280.
Newman JA (2002) Biomechanics of Head Trauma: Head Protection. In: Accidental
Injury: Biomechanics and Prevention (Nahum AM, Melvin JW, eds), pp 303–323. New
York, USA: Springer.
Novack TA, Banos JH, Brunner R, Renfroe S, Meythaler JM (2009) Impact of early
administration of sertraline on depressive symptoms in the first year after traumatic
brain injury. J Neurotrauma 26:1921-1928.
O'Neill RA et al. (2006) Isoelectric focusing technology quantifies protein signaling in 25
cells. Proc Natl Acad Sci U S A 103:16153-16158.
Oberkofler H, Schraml E, Krempler F, Patsch W (2003) Potentiation of liver X receptor
transcriptional activity by peroxisome-proliferator-activated receptor gamma co-activator
1 alpha. Biochem J 371:89-96.
Ogata M, Tsujita M, Hossain M, Akita N, Gonzales FJ, Staels B, Suzuki S, Fukutomi T,
Kimura G, Yokoyama S (2009) On the mechanism for PPAR agonists to enhance
ABCA1 expression. Atherosclerosis 205:413-419.
300

Ohkubo N, Lee YD, Morishima A, Terashima T, Kikkawa S, Tohyama M, Sakanaka M,
Tanaka J, Maeda N, Vitek MP, Mitsuda N (2003) Apolipoprotein E and reelin ligands
modulate tau phosphorylation through an apolipoprotein E receptor/disabled-1/glycogen
synthase kinase-3beta cascade. FASEB J 17:295-297.
Olsson A, Csajbok L, Ost M, Hoglund K, Nylen K, Rosengren L, Nellgard B, Blennow K
(2004) Marked increase of beta-amyloid(1-42) and amyloid precursor protein in
ventricular cerebrospinal fluid after severe traumatic brain injury. J Neurol 251:870-876.
Omalu B, Hammers JL, Bailes J, Hamilton RL, Kamboh MI, Webster G, Fitzsimmons
RP (2011) Chronic traumatic encephalopathy in an Iraqi war veteran with posttraumatic
stress disorder who committed suicide. Neurosurg Focus 31:E3.
Omalu BI, DeKosky ST, Hamilton RL, Minster RL, Kamboh MI, Shakir AM, Wecht CH
(2006) Chronic traumatic encephalopathy in a national football league player: part II.
Neurosurgery 59:1086-1093.
Ommaya AK, Boretos JW, Beile EE (1969) The Lexan Calvarium: An Improved Method
for Direct Observation of the Brain. Journal of Neurosurgery 30:25-29.
Ono K, Kikuchi A, Nakamura M, Kobayashi H, Nakamura N (1980) Human Head
Tolerance to Sagittal Impact Reliable Estimation Deduced from Experimental Head
Injury Using Subhuman Primates and Human Cadaver Skulls. SAE Technical Paper
801303.
301

Onyszchuk G, Al-Hafez B, He Y-Y, Bilgen M, Berman NEJ, Brooks WM (2007) A mouse
model of sensorimotor controlled cortical impact: Characterization using longitudinal
magnetic resonance imaging, behavioral assessments and histology. J Neurosci
Methods 160:187-196.
Oram JF, Vaughan AM (2000) ABCA1-mediated transport of cellular cholesterol and
phosopholipids to HDL apoplipoproteins. Curr Opin Lipidol 11:253-260.
Oram JF, Lawn RM (2001) ABCA1. The gatekeeper for eliminating excess tissue
cholesterol. J Lipid Res 42:1173-1179.
Oram JF, Lawn RM, Garvin MR, Wade DP (2000) ABCA1 is the cAMP-inducible
apolipoprotein receptor that mediates cholesterol secretion from macrophages. J Biol
Chem 275:34508-34511.
Ou J, Tu H, Shan B, Luk A, DeBose-Boyd RA, Bashmakov Y, Goldstein JL, Brown MS
(2001) Unsaturated fatty acids inhibit transcription of the sterol regulatory element-
binding protein-1c (SREBP-1c) gene by antagonizing ligand-dependent activation of the
LXR. Proc Natl Acad Sci U S A 98:6027-6032.
Pan W, Kastin AJ, Rigai T, McLay R, Pick CG (2003) Increased hippocampal uptake of
tumor necrosis factor alpha and behavioral changes in mice. Exp Brain Res 149:195-
199.
302

Park E, Bell JD, Baker AJ (2008) Traumatic brain injury: can the consequences be
stopped? CMAJ 178:1163-1170.
Patrick LM, Lissner HR, Gurdjian ES (1963) Survival by design: Head protection. In:
Proc. 7th STAPP Car Crash Conference, pp 483-499. Warren dale, PA: Association for
the Advancement of Automotive Medicine.
Paxinos G, Franklin KBJ (2001) The Mouse Brain In Stereotaxic Coordinates. San
Diego: Elsevier/Academic Press.
Pei JJ, Tanaka T, Tung YC, Braak E, Iqbal K, Grundke-Iqbal I (1997) Distribution,
levels, and activity of glycogen synthase kinase-3 in the Alzheimer disease brain. J
Neuropathol Exp Neurol 56:70-78.
Pellman EJ, Viano DC, Tucker AM, Casson IR (2003a) Concussion in professional
football: location and direction of helmet impacts-Part 2. Neurosurgery 53:1328-1340;
discussion 1340-1321.
Pellman EJ, Viano DC, Tucker AM, Casson IR, Waeckerle JF (2003b) Concussion in
professional football: reconstruction of game impacts and injuries. Neurosurgery
53:799-812; discussion 812-794.
303

Peng Y, Yang J, Deck C, Otte D, Willinger R (2013) Development of head injury risk
functions based on real-world accident reconstruction. Int J Crashworthiness 19:105-
114.
Perez-Polo JR, Rea HC, Johnson KM, Parsley MA, Unabia GC, Xu G, Infante SK,
Dewitt DS, Hulsebosch CE (2013) Inflammatory consequences in a rodent model of
mild traumatic brain injury. J Neurotrauma 30:727-740.
Perez A, Morelli L, Cresto JC, Castano EM (2000) Degradation of soluble amyloid beta-
peptides 1-40, 1-42, and the Dutch variant 1-40Q by insulin degrading enzyme from
Alzheimer disease and control brains. Neurochem Res 25:247-255.
Petit-Turcotte C, Aumont N, Blain JF, Poirier J (2007) Apolipoprotein E receptors and
amyloid expression are modulated in an apolipoprotein E-dependent fashion in
response to hippocampal deafferentation in rodent. Neuroscience 150:58-63.
Petrlova J, Hong HS, Bricarello DA, Harishchandra G, Lorigan GA, Jin LW, Voss JC
(2011) A differential association of Apolipoprotein E isoforms with the amyloid-beta
oligomer in solution. Proteins 79:402-416.
Pierce JE, Trojanowski JQ, Graham DI, Smith DH, McIntosh TK (1996)
Immunohistochemical characterization of alterations in the distribution of amyloid
precursor proteins and beta-amyloid peptide after experimental brain injury in the rat. J
Neurosci 16:1083-1090.
304

Pinteaux E, Parker LC, Rothwell NJ, Luheshi GN (2002) Expression of interleukin-1
receptors and their role in interleukin-1 actions in murine microglial cells. J Neurochem
83:754-763.
Pittella JE, Gusmao SN (2003) Diffuse vascular injury in fatal road traffic accident
victims: its relationship to diffuse axonal injury. J Forensic Sci 48:626-630.
Plassman BL, Havlik RJ, Steffens DC, Helms MJ, Newman TN, Drosdick D, Phillips C,
Gau BA, Welsh-Bohmer KA, Burke JR, Guralnik JM, Breitner JC (2000) Documented
head injury in early adulthood and risk of Alzheimer's disease and other dementias.
Neurology 55:1158-1166.
Plotz EJ, Kabara JJ, Davis ME, LeRoy GV, Gould RG (1968) Studies on the synthesis
of cholesterol in the brain of the human fetus. Am J Obstet Gynecol 101:534-538.
Poirier J (1994) Apolipoprotein E in animal models of CNS injury and Alzheimer's
disease. Trends Neurosci 17:525-530.
Poirier J, Hess M, May PC, Finch CE (1991) Astrocytic apolipoprotein E mRNA and
GFAP mRNA in hippocampus after entorhinal cortex lesioning. Brain Res Mol Brain Res
11:97-106.
305

Poirier J, Baccichet A, Dea D, Gauthier S (1993a) Cholesterol synthesis and lipoprotein
reuptake during synaptic remodelling in hippocampus in adult rats. Neuroscience 55:81-
90.
Poirier J, Davignon J, Bouthillier D, Kogan S, Bertrand P, Gauthier S (1993b)
Apolipoprotein E polymorphism and Alzheimer's disease. The Lancet 342:697-699.
Povlishock JT, Kontos HA (1985) Continuing axonal and vascular change following
experimental brain trauma. Cent Nerv Syst Trauma 2:285-298.
Povolny MA, Kaplan SP (1993) Traumatic brain injury occuring with spinal cord injury:
Significance for rehabilitation. J Rehabil:23-37.
Prange MT, Margulies SS (2002) Regional, directional, and age-dependent properties
of the brain undergoing large deformation. J Biomech Eng 124:244-252.
Prange MT, Meaney DF, Margulies SS (2000) Defining brain mechanical properties:
effects of region, direction, and species. Stapp Car Crash J 44:205-213.
Prasad P, Mertz H (1985) The Position of the United States Delegation to the ISO
Working Group 6 on the Use of HIC in the Automotive Environment. SAE Technical
Paper 851246.
306

Prevention CfDCa (2007) Nonfatal traumatic brain injuries from sports and recreation
activities-United States, 2001-2005. MMWR Morb Mortal Wkly Rep 56:733-737.
Pruthi N, Chandramouli BA, Kuttappa TB, Rao SL, Subbakrishna DK, Abraham MP,
Mahadevan A, Shankar SK (2010) Apolipoprotein E polymorphism and outcome after
mild to moderate traumatic brain injury: a study of patient population in India. Neurol
India 58:264-269.
Pudenz RH, Shelden CH (1946) The lucite calvarium; a method for direct observation of
the brain; cranial trauma and brain movement. J Neurosurg 3:487-505.
Putukian M (2011) The acute symptoms of sport-related concussion: diagnosis and on-
field management. Clin Sports Med 30:49-61, viii.
Puvanachandra P, Hyder AA (2008) Traumatic brain injury in Latin America and the
Caribbean: a call for research. Salud Pública de México 50:s3-s5.
Qiu W, Zhang Y, Sheng H, Zhang J, Wang W, Liu W, Chen K, Zhou J, Xu Z (2007)
Effects of therapeutic mild hypothermia on patients with severe traumatic brain injury
after craniotomy. J Crit Care 22:229-235.
Qiu WQ, Walsh DM, Ye Z, Vekrellis K, Zhang J, Podlisny MB, Rosner MR, Safavi A,
Hersh LB, Selkoe DJ (1998) Insulin-degrading enzyme regulates extracellular levels of
amyloid beta-protein by degradation. J Biol Chem 273:32730-32738.
307

Qiu WS, Liu WG, Shen H, Wang WM, Hang ZL, Zhang Y, Jiang SJ, Yang XF (2005)
Therapeutic effect of mild hypothermia on severe traumatic head injury. Chin J
Traumatol 8:27-32.
Quinet EM, Savio DA, Halpern AR, Chen L, Schuster GU, Gustafsson JA, Basso MD,
Nambi P (2006) Liver X receptor (LXR)-beta regulation in LXRalpha-deficient mice:
implications for therapeutic targeting. Mol Pharmacol 70:1340-1349.
Raby CA, Morganti-Kossmann MC, Kossmann T, Stahel PF, Watson MD, Evans LM,
Mehta PD, Spiegel K, Kuo YM, Roher AE, Emmerling MR (1998) Traumatic brain injury
increases beta-amyloid peptide 1-42 in cerebrospinal fluid. J Neurochem 71:2505-2509.
Rader DJ, deGoma EM (2012) Approach to the Patient with Extremely Low HDL-
Cholesterol. J Clin Endocrinol Metab 97:3399-3407.
Rafaels KA, Bass CR, Panzer MB, Salzar RS, Woods WA, Feldman SH, Walilko T,
Kent RW, Capehart BP, Foster JB, Derkunt B, Toman A (2012) Brain injury risk from
primary blast. J Trauma Acute Care Surg 73:895-901.
Raffai RL, Dong LM, Farese RV, Jr., Weisgraber KH (2001) Introduction of human
apolipoprotein E4 "domain interaction" into mouse apolipoprotein E. Proc Natl Acad Sci
U S A 98:11587-11591.
308

Raghupathi R (2004) Cell death mechanisms following traumatic brain injury. Brain
Pathol 14:215-222.
Ramlackhansingh AF, Brooks DJ, Greenwood RJ, Bose SK, Turkheimer FE, Kinnunen
KM, Gentleman S, Heckemann RA, Gunanayagam K, Gelosa G, Sharp DJ (2011)
Inflammation after trauma: microglial activation and traumatic brain injury. Ann Neurol
70:374-383.
Rasmusson DX, Brandt J, Martin DB, Folstein MF (1995) Head injury as a risk factor in
Alzheimer's disease. Brain Inj 9:213-219.
Rayner KJ, Suarez Y, Davalos A, Parathath S, Fitzgerald ML, Tamehiro N, Fisher EA,
Moore KJ, Fernandez-Hernando C (2010) MiR-33 contributes to the regulation of
cholesterol homeostasis. Science 328:1570-1573.
Reilly P (2007) The impact of neurotrauma on society: an international perspective.
Prog Brain Res 161:3-9.
Ren Z, Iliff JJ, Yang L, Yang J, Chen X, Chen MJ, Giese RN, Wang B, Shi X,
Nedergaard M (2013) 'Hit & Run' model of closed-skull traumatic brain injury (TBI)
reveals complex patterns of post-traumatic AQP4 dysregulation. J Cereb Blood Flow
Metab 33:834-845.
309

Repa JJ, Berge KE, Pomajzl C, Richardson JA, Hobbs H, Mangelsdorf DJ (2002)
Regulation of ATP-binding cassette sterol transporters ABCG5 and ABCG8 by the liver
X receptors alpha and beta. J Biol Chem 277:18793-18800.
Riddell DR et al. (2007) The LXR agonist TO901317 selectively lowers hippocampal
Abeta42 and improves memory in the Tg2576 mouse model of Alzheimer's disease. Mol
Cell Neurosci 34:621-628.
Rigamonti E, Helin L, Lestavel S, Mutka AL, Lepore M, Fontaine C, Bouhlel MA, Bultel
S, Fruchart JC, Ikonen E, Clavey V, Staels B, Chinetti-Gbaguidi G (2005) Liver X
receptor activation controls intracellular cholesterol trafficking and esterification in
human macrophages. Circ Res 97 682-689.
Roberts GW, Gentleman SM, Lynch A, Graham DI (1991) beta A4 amyloid protein
deposition in brain after head trauma. The Lancet 338:1422-1423.
Roberts GW, Gentleman SM, Lynch A, Murray L, Landon M, Graham DI (1994) Beta
amyloid protein deposition in the brain after severe head injury: implications for the
pathogenesis of Alzheimer's disease. J Neurol Neurosurg Psychiatry 57:419-425.
Rogaev EI et al. (1995) Familial Alzheimer's disease in kindreds with missense
mutations in a gene on chromosome 1 related to the Alzheimer's disease type 3 gene.
Nature 376:775-778.
310

Rogers J, Strohmeyer R, Kovelowski CJ, Li R (2002) Microglia and inflammatory
mechanisms in the clearance of amyloid beta peptide. Glia 40:260-269.
Rooker S, Jander S, Van Reempts J, Stoll G, Jorens PG, Borgers M, Verlooy J (2006)
Spatiotemporal pattern of neuroinflammation after impact-acceleration closed head
injury in the rat. Mediators Inflamm 2006:90123.
Roozenbeek B, Maas AI, Menon DK (2013) Changing patterns in the epidemiology of
traumatic brain injury. Nat Rev Neurol 9:231-236.
Roselaar SE, Daugherty A (1998) Apolipoprotein E-deficient mice have impaired innate
immune responses to Listeria monocytogenes in vivo. J Lipid Res 39:1740-1743.
Rothwell N (2003) Interleukin-1 and neuronal injury: mechanisms, modification, and
therapeutic potential. Brain Behav Immun 17:152-157.
Rowson S, Duma SM (2011) Development of the STAR evaluation system for football
helmets: integrating player head impact exposure and risk of concussion. Ann Biomed
Eng 39:2130-2140.
Rowson S, Duma SM (2013) Brain Injury Prediction: Assessing the Combined
Probability of Concussion Using Linear and Rotational Head Acceleration. Ann Biomed
Eng.
311

Rowson S, Duma SM, Beckwith JG, Chu JJ, Greenwald RM, Crisco JJ, Brolinson PG,
Duhaime AC, McAllister TW, Maerlender AC (2012) Rotational head kinematics in
football impacts: an injury risk function for concussion. Ann Biomed Eng 40:1-13.
Rubovitch V, Ten-Bosch M, Zohar O, Harrison CR, Tempel-Brami C, Stein E, Hoffer BJ,
Balaban CD, Schreiber S, Chiu WT, Pick CG (2011) A mouse model of blast-induced
mild traumatic brain injury. Exp Neurol 232:280-289.
Rudelli R, Strom JO, Welch PT, Ambler MW (1982) Posttraumatic premature
Alzheimer's disease. Neuropathologic findings and pathogenetic considerations. Arch
Neurol 39:570-575.
Runnerstam M, Bao F, Huang Y, Shi J, Gutierrez E, Hamberger A, Hansson HA, Viano
D, Haglid K (2001) A new model for diffuse brain injury by rotational acceleration: II.
Effects on extracellular glutamate, intracranial pressure, and neuronal apoptosis. J
Neurotrauma 18:259-273.
Rusnak M (2013) Traumatic brain injury: Giving voice to a silent epidemic. Nat Rev
Neurol 9:186-187.
Russel WR (1932) Cerebral involvement in head injury: a study based on the
examination of two hundred cases. Brain 55:549-603.
312

Russo R, Borghi R, Markesbery W, Tabaton M, Piccini A (2005) Neprylisin decreases
uniformly in Alzheimer’s disease and in normal aging. FEBS Letters 579:6027-6030.
Rutherford GW, Bazarian JJ, Cernak I, Corrigan JD, Dikmen SS, Grady SM, Hesdorffer
DC, Kraus JF, Levin HS, Noble L, Potolicchio SJ, Rauch SL, Stiers W, Tamminga CA,
Temkin N, Weisskopf MG (2008) Gulf War and Health: Long-Term Consequences of
Traumatic Brain Injury: The National Academies Press.
Saatman KE, Feeko KJ, Pape RL, Raghupathi R (2006) Differential behavioral and
histopathological responses to graded cortical impact injury in mice. J Neurotrauma
23:1241-1253.
Saatman KE, Duhaime AC, Bullock R, Maas AI, Valadka A, Manley GT (2008)
Classification of traumatic brain injury for targeted therapies. J Neurotrauma 25:719-
738.
Sabet AA, Christoforou E, Zatlin B, Genin GM, Bayly PV (2008) Deformation of the
human brain induced by mild angular head acceleration. J Biomech 41:307-315.
Saido T, Leissring MA (2012) Proteolytic degradation of amyloid beta-protein. Cold
Spring Harb Perspect Med 2:a006379.
Saunders AM, Strittmatter WJ, Schmechel D, George-Hyslop PH, Pericak-Vance MA,
Joo SH, Rosi BL, Gusella JF, Crapper-MacLachlan DR, Alberts MJ (1993) Association
313

of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer's
disease. Neurology 43:1467-1472.
Scalfani MT, Dhar R, Zazulia AR, Videen TO, Diringer MN (2012) Effect of osmotic
agents on regional cerebral blood flow in traumatic brain injury. J Crit Care
27:526.e527-512.
Scheuner D et al. (1996) Secreted amyloid beta-protein similar to that in the senile
plaques of Alzheimer's disease is increased in vivo by the presenilin 1 and 2 and APP
mutations linked to familial Alzheimer's disease. Nat Med 2:864-870.
Schley D, Carare-Nnadi R, Please CP, Perry VH, Weller RO (2006) Mechanisms to
explain the reverse perivascular transport of solutes out of the brain. J Theor Biol
238:962-974.
Schmidt ML, Zhukareva V, Newell KL, Lee VM, Trojanowski JQ (2001) Tau isoform
profile and phosphorylation state in dementia pugilistica recapitulate Alzheimer's
disease. Acta Neuropathol 101:518-524.
Schmitz G, Langmann T (2001) Structure, function and regulation of the ABC1 gene
product. Curr Opin Lipidol 12:129-140.
Schmitz G, Langmann T (2005) Transcriptional regulatory networks in lipid metabolism
control ABCA1 expression. Biochim Biophys Acta 1735:1-19.
314

Schroepfer GJ, Jr. (2000) Oxysterols: modulators of cholesterol metabolism and other
processes. Physiol Rev 80:361-554.
Schultz JR, Tu H, Luk A, Repa JJ, Medina JC, Li L, Schwendner S, Wang S, Thoolen
M, Mangelsdorf DJ, Lustig KD, Shan B (2000) Role of LXRs in control of lipogenesis.
Genes & Development 14:2831-2838.
Schwartz K, Lawn RM, Wade DP (2000) ABC1 gene expression and ApoA-1-mediated
cholesterol efflux are regulated by LXR. Biochem Biophys Res Commun 274:794-802.
Schwetye KE, Cirrito JR, Esparza TJ, Mac Donald CL, Holtzman DM, Brody DL (2010)
Traumatic brain injury reduces soluble extracellular amyloid-β in mice: A
methodologically novel combined microdialysis-controlled cortical impact study.
Neurobiol Disease 40:555-564.
Seelig JM, Becker DP, Miller JD, Greenberg RP, Ward JD, Choi SC (1981) Traumatic
acute subdural hematoma: major mortality reduction in comatose patients treated within
four hours. N Engl J Med 304:1511-1518.
Seitz A, Kragol M, Aglow E, Showe L, Heber-Katz E (2003) Apolipoprotein E expression
after spinal cord injury in the mouse. J Neurosci Res.
Selkoe DJ (1994) Alzheimer's disease: a central role for amyloid. J Neuropathol Exp
Neurol 53:438-447.
315

Selkoe DJ, Wolfe MS (2007) Presenilin: Running with Scissors in the Membrane. Cell
131:215-221.
Servadei F, Murray GD, Teasdale GM, Dearden M, Iannotti F, Lapierre F, Maas AJ,
Karimi A, Ohman J, Persson L, Stocchetti N, Trojanowski T, Unterberg A (2002)
Traumatic subarachnoid hemorrhage: demographic and clinical study of 750 patients
from the European brain injury consortium survey of head injuries. Neurosurgery
50:261-267; discussion 267-269.
Sevagan G, Zhu F, Jiang B, Yang KH (2013) Numerical simulations of the occupant
head response in an infantry vehicle under blunt impact and blast loading conditions.
Proc Inst Mech Eng H 227:778-787.
Sganzerla EP, Tomei G, Rampini P, Ceretti L, Gaini SM, Giovanelli M, Villani R (1989)
A peculiar intracerebral hemorrhage: The gliding contusion, it s relationship to diffuse
brain damage. Neurosurgical Review 12:215-218.
Shatsky SA, Alter WA, Evans DE, Armbrustmacher VW, Clark G, Earle KM (1974)
Traumatic distortions of the primate head and chest: correlation of biomechanical,
radiological and pathological data. Proceedings: Stapp Car Crash Conference 18:-.
Shein NA, Doron H, Horowitz M, Trembovler V, Alexandrovich AG, Shohami E (2007)
Altered cytokine expression and sustained hypothermia following traumatic brain injury
in heat acclimated mice. Brain Res 1185:313-320.
316

Shelden CH, Pudenz RH, Restarski JS, Craig WM (1944) The Lucite Calvarium—A
Method for Direct Observation of the Brain. J Neurosurg 1:67-75.
Sherriff FE, Bridges LR, Sivaloganathan S (1994a) Early detection of axonal injury after
human head trauma using immunocytochemistry for beta-amyloid precursor protein.
Acta Neuropathol 87:55-62.
Sherriff FE, Bridges LR, Gentleman SM, Sivaloganathan S, Wilson S (1994b) Markers
of axonal injury in post mortem human brain. Acta Neuropathol (Berl) 88:433-439.
Shibata M, Yamada S, Kumar SR, Calero M, Bading J, Frangione B, Holtzman DM,
Miller CA, Strickland DK, Ghiso J, Zlokovic BV (2000) Clearance of Alzheimer's
amyloid-{beta}1-40 peptide from brain by LDL receptor-related protein-1 at the blood-
brain barrier. J Clin Invest 106:1489-1499.
Shiozaki T, Hayakata T, Taneda M, Nakajima Y, Hashiguchi N, Fujimi S, Nakamori Y,
Tanaka H, Shimazu T, Sugimoto H (2001) A multicenter prospective randomized
controlled trial of the efficacy of mild hypothermia for severely head injured patients with
low intracranial pressure. Mild Hypothermia Study Group in Japan. J Neurosurg 94:50-
54.
Shirotani K, Tsubuki S, Iwata N, Takaki Y, Harigaya W, Maruyama K, Kiryu-Seo S,
Kiyama H, Iwata H, Tomita T, Iwatsubo T, Saido TC (2001) Neprilysin degrades both
317

amyloid beta peptides 1-40 and 1-42 most rapidly and efficiently among thiorphan- and
phosphoramidon-sensitive endopeptidases. J Biol Chem 276:21895-21901.
Shitaka Y, Tran HT, Bennett RE, Sanchez L, Levy MA, Dikranian K, Brody DL (2011)
Repetitive closed-skull traumatic brain injury in mice causes persistent multifocal axonal
injury and microglial reactivity. J Neuropathol Exp Neurol 70:551-567.
Shohami E, Gallily R, Mechoulam R, Bass R, Ben-Hur T (1997) Cytokine production in
the brain following closed head injury: dexanabinol (HU-211) is a novel TNF-alpha
inhibitor and an effective neuroprotectant. J Neuroimmunol 72:169-177.
Simon P, Dupuis R, Costentin J (1994) Thigmotaxis as an index of anxiety in mice.
Influence of dopaminergic transmissions. Behav Brain Res 61:59-64.
Slunt HH, Thinakaran G, Von Koch C, Lo AC, Tanzi RE, Sisodia SS (1994) Expression
of a ubiquitous, cross-reactive homologue of the mouse beta-amyloid precursor protein
(APP). J Biol Chem 269:2637-2644.
Smith C, Graham DI, Murray LS, Stewart J, Nicoll JA (2006) Association of APOE e4
and cerebrovascular pathology in traumatic brain injury. J Neurol Neurosurg Psychiatry
77:363-366.
Smith DH, Meaney DF (2000) Axonal Damage in Traumatic Brain Injury. Neuroscientist
6:483-495.
318

Smith DH, Meaney DF, Shull WH (2003a) Diffuse axonal injury in head trauma. J Head
Trauma Rehabil 18:307-316.
Smith DH, Johnson VE, Stewart W (2013) Chronic neuropathologies of single and
repetitive TBI: substrates of dementia? Nat Rev Neurol 9:211-221.
Smith DH, Chen XH, Iwata A, Graham DI (2003b) Amyloid beta accumulation in axons
after traumatic brain injury in humans. J Neurosurg 98:1072-1077.
Smith DH, Wolf JA, Lusardi TA, Lee VM, Meaney DF (1999a) High tolerance and
delayed elastic response of cultured axons to dynamic stretch injury. J Neurosci
19:4263-4269.
Smith DH, Chen XH, Xu BN, McIntosh TK, Gennarelli TA, Meaney DF (1997)
Characterization of diffuse axonal pathology and selective hippocampal damage
following inertial brain trauma in the pig. J Neuropathol Exp Neurol 56:822-834.
Smith DH, Nonaka M, Miller R, Leoni M, Chen XH, Alsop D, Meaney DF (2000)
Immediate coma following inertial brain injury dependent on axonal damage in the
brainstem. J Neurosurg 93:315-322.
Smith DH, Soares HD, Pierce JS, Perlman KG, Saatman KE, Meaney DF, Dixon CE,
McIntosh TK (1995) A model of parasagittal controlled cortical impact in the mouse:
cognitive and histopathologic effects. J Neurotrauma 12:169-178.
319

Smith DH, Chen XH, Nonaka M, Trojanowski JQ, Lee VM, Saatman KE, Leoni MJ, Xu
BN, Wolf JA, Meaney DF (1999b) Accumulation of amyloid beta and tau and the
formation of neurofilament inclusions following diffuse brain injury in the pig. J
Neuropathol Exp Neurol 58:982-992.
Smith HP, Kelly DL, Jr., McWhorter JM, Armstrong D, Johnson R, Transou C, Howard
G (1986) Comparison of mannitol regimens in patients with severe head injury
undergoing intracranial monitoring. J Neurosurg 65:820-824.
Smith KR, Jr., Goulding PM, Wilderman D, Goldfader PR, Holterman-Hommes P, Wei F
(1994) Neurobehavioral effects of phenytoin and carbamazepine in patients recovering
from brain trauma: a comparative study. Arch Neurol 51:653-660.
Soloniuk D, Pitts LH, Lovely M, Bartkowski H (1986) Traumatic intracerebral
hematomas: timing of appearance and indications for operative removal. J Trauma
26:787-794.
Song C, Hiipakka RA, Liao S (2001) Auto-oxidized cholesterol sulfates are antagonistic
ligands of liver X receptors: implications for the development and treatment of
atherosclerosis. Steroids 66:473-479.
Song C, Kokontis JM, Hiipakka RA, Liao S (1994) Ubiquitous receptor: a receptor that
modulates gene activation by retinoic acid and thyroid hormone receptors. Proc Natl
Acad Sci U S A 91:10809-10813.
320

Sontag E, Hladik C, Montgomery L, Luangpirom A, Mudrak I, Ogris E, White CL, 3rd
(2004a) Downregulation of protein phosphatase 2A carboxyl methylation and
methyltransferase may contribute to Alzheimer disease pathogenesis. J Neuropathol
Exp Neurol 63:1080-1091.
Sontag E, Luangpirom A, Hladik C, Mudrak I, Ogris E, Speciale S, White CL, 3rd
(2004b) Altered expression levels of the protein phosphatase 2A ABalphaC enzyme are
associated with Alzheimer disease pathology. J Neuropathol Exp Neurol 63:287-301.
Sorbi S, Nacmias B, Piacentini S, Repice A, Latorraca S, Forleo P, Amaducci L (1995)
ApoE as a prognostic factor for post-traumatic coma. Nat Med 1:852.
Spillantini MG, Goedert M (2013) Tau pathology and neurodegeneration. Lancet Neurol
12:609-622.
Spillantini MG, Goedert M, Crowther RA, Murrell JR, Farlow MR, Ghetti B (1997)
Familial multiple system tauopathy with presenile dementia: a disease with abundant
neuronal and glial tau filaments. Proc Natl Acad Sci U S A 94:4113-4118.
Stalhammar D, Galinat BJ, Allen AM, Becker DP, Stonnington HH, Hayes RL (1987) A
new model of concussive brain injury in the cat produced by extradural fluid volume
loading: I. Biomechanical properties. Brain Inj 1:73-91.
321

Statler KD, Alexander H, Vagni V, Holubkov R, Dixon CE, Clark RS, Jenkins L,
Kochanek PM (2006) Isoflurane exerts neuroprotective actions at or near the time of
severe traumatic brain injury. Brain Res 1076:216-224.
Statler KD, Kochanek PM, Dixon CE, Alexander HL, Warner DS, Clark RS, Wisniewski
SR, Graham SH, Jenkins LW, Marion DW, Safar PJ (2000) Isoflurane improves long-
term neurologic outcome versus fentanyl after traumatic brain injury in rats. J
Neurotrauma 17:1179-1189.
Stern RA, Riley DO, Daneshvar DH, Nowinski CJ, Cantu RC, McKee AC (2011) Long-
term consequences of repetitive brain trauma: chronic traumatic encephalopathy.
PM&R 3:S460-S467.
Strich SJ (1956) Diffuse degeneration of the cerebral white matter in severe dementia
following head injury. J Neurol Neurosurg Psychiat 19:163-185.
Strittmatter WJ, Weisgraber KH, Huang DY, Dong LM, Salvesen GS, Pericak-Vance M,
Schmechel D, Saunders AM, Goldgaber D, Roses AD (1993) Binding of human
apolipoprotein E to synthetic amyloid beta peptide: isoform-specific effects and
implications for late-onset Alzheimer disease. Proc Natl Acad Sci U S A 90:8098-8102.
Stukas S, Robert J, Wellington CL (2014) High-density lipoproteins and cerebrovascular
integrity in Alzheimer's disease. Cell Metab 19:574-591.
322

Stukas S, May S, Wilkinson A, Chan J, Donkin J, Wellington CL (2011) The LXR
agonist GW3965 increases apoA-I protien levels in the central nervous system
independent of ABCA1. Biochim Biophys Acta doi:10.1016.
Suh YH, Checler F (2002) Amyloid precursor protein, presenilins, and alpha-synuclein:
molecular pathogenesis and pharmacological applications in Alzheimer's disease.
Pharmacol Rev 54:469-525.
Sun Y, Yao J, Kim T-W, Tall AR (2003) Expression of Liver X Receptor Target Genes
Decreases Cellular Amyloid β Peptide Secretion. J Biol Chem 278:27688-27694.
Surgeons AAoN (2011) Sports-related head injury. In.
Taber KH, Warden DL, Hurley RA (2006) Blast-related traumatic brain injury: what is
known? J Neuropsychiatry Clin Neurosci 18:141-145.
Tachikawa M, Watanabe M, Hori S, Fukaya M, Ohtsuki S, Asashima T, Terasaki T
(2005) Distinct spatio-temporal expression of ABCA and ABCG transporters in the
developing and adult mouse brain. J Neurochem 95 294-304.
Tagliaferri F, Compagnone C, Korsic M, Servadei F, Kraus J (2006) A systematic review
of brain injury epidemiology in Europe. Acta Neurochir (Wien) 148:255-268.
323

Tai LM, Bilousova T, Jungbauer L, Roeske SK, Youmans KL, Yu C, Poon WW,
Cornwell LB, Miller CA, Vinters HV, Van Eldik LJ, Fardo DW, Estus S, Bu G, Gylys KH,
Ladu MJ (2013) Levels of soluble apolipoprotein E/amyloid-beta (Abeta) complex are
reduced and oligomeric Abeta increased with APOE4 and Alzheimer disease in a
transgenic mouse model and human samples. J Biol Chem 288:5914-5926.
Takahashi H, Manaka S, Sano K (1981) Changes in extracellular potassium
concentration in cortex and brain stem during the acute phase of experimental closed
head injury. J Neurosurg 55:708-717.
Takahashi Y, Smith JD (1999) Cholesterol efflux to apolipoprotein A1 involves
endocytosis and resecretion in a calcium-dependent pathway. Proc Natl Acad Sci USA
96 11358-11363.
Takhounts EG, Crandall JR, Darvish K (2003) On the importance of nonlinearity of brain
tissue under large deformations. Stapp Car Crash J 47:79-92.
Tall AR (2008) Cholesterol efflux pathways and other potential mechanisms involved in
the athero-protective effect of high density lipoproteins. J Intern Med 263:256-273.
Tanaka AR, Ikeda Y, Abe-Dohmae S, Arakawa R, Sadanami K, Kidera A, Nakagawa S,
Nagase T, Aoki R, Kioka N, Amachi T, Yokoyama S, Ueda K (2001) Human ABCA1
Contains a Large Amino-Terminal Extracellular Domain Homologous to an Epitope of
Sjögren's Syndrome. Biochem Biophys Res Commun 283:1019-1025.
324

Taneda M, Kataoka K, Akai F, Asai T, Sakata I (1996) Traumatic subarachnoid
hemorrhage as a predictable indicator of delayed ischemic symptoms. J Neurosurg
84:762-768.
Tang-Schomer MD, Patel AR, Baas PW, Smith DH (2010) Mechanical breaking of
microtubules in axons during dynamic stretch injury underlies delayed elasticity,
microtubule disassembly, and axon degeneration. FASEB J 24:1401-1410.
Tang-Schomer MD, Johnson VE, Baas PW, Stewart W, Smith DH (2012) Partial
interruption of axonal transport due to microtubule breakage accounts for the formation
of periodic varicosities after traumatic axonal injury. Exp Neurol 233:364-372.
Tang YP, Noda Y, Hasegawa T, Nabeshima T (1997) A concussive-like brain injury
model in mice (I): impairment in learning and memory. J Neurotrauma 14:851-862.
Taupin V, Toulmond S, Serrano A, Benavides J, Zavala F (1993) Increase in IL-6, IL-1
and TNF levels in rat brain following traumatic lesion. Influence of pre- and post-
traumatic treatment with Ro5 4864, a peripheral-type (p site) benzodiazepine ligand. J
Neuroimmunol 42:177-185.
Teasdale G, Jennett B (1974) Assessment of coma and impaired consciousness: a
practical scale. The Lancet 304:81-84.
325

Teasdale GM, Nicoll JA, Murray G, Fiddes M (1997) Association of apolipoprotein E
polymorphism with outcome after head injury. The Lancet 350:1069-1071.
Tenovuo O, Alin J, Helenius H (2009) A randomized controlled trial of rivastigmine for
chronic sequels of traumatic brain injury-what it showed and taught? Brain Inj 23:548-
558.
Thibault LE, Meaney DF, Anderson BJ, Marmarou A (1992) Biomechanical aspects of a
fluid percussion model of brain injury. J Neurotrauma 9:311-322.
Thinakaran G, Koo EH (2008) Amyloid precursor protein trafficking, processing, and
function. J Biol Chem 283:29615-29619.
Thomas L (1997) Retrospective Power Analysis. Conserv Biol 11:276-280.
Thompson HJ, Lifshitz J, Marklund N, Grady MS, Graham DI, Hovda DA, McIntosh TK
(2005) Lateral fluid percussion brain injury: a 15-year review and evaluation. J
Neurotrauma 22:42-75.
Tokuda T, Calero M, Matsubara E, Vidal R, Kumar A, Permanne B, Zlokovic B, Smith
JD, Ladu MJ, Rostagno A, Ghiso J (2000) Lipidation of apolipoprotein E influences its
isoform-specific interaction with Alzheimer's amyloid beta peptides. Biochem J 348:359-
365.
326

Tolnay M, Sergeant N, Ghestem A, Chalbot S, De Vos RA, Jansen Steur EN, Probst A,
Delacourte A (2002) Argyrophilic grain disease and Alzheimer's disease are
distinguished by their different distribution of tau protein isoforms. Acta Neuropathol
104:425-434.
Tomlinson BE (1970) Brain-stem lesions after head injury. J Clin Pathol Suppl (R Coll
Pathol) 4:154-165.
Tran HT, LaFerla FM, Holtzman DM, Brody DL (2011a) Controlled cortical impact
traumatic brain injury in 3xTg-AD mice causes acute intra-axonal amyloid-á
accumulation and independently accelerates the development of tau abnormalities. J
Neurosci 31:9513-9525.
Tran HT, LaFerla FM, Holtzman DM, Brody DL (2011b) Controlled cortical impact
traumatic brain injury in 3xTg-AD mice causes acute intra-axonal amyloid-β
accumulation and independently accelerates the development of tau abnormalities. J
Neurosci 31:9513-9525.
Tran HT, Sanchez L, Esperza TJ, Brody DL (2011c) Distint temporal and anatomical
distributions of amyloid-β and tau abnormalities following controlled cortical impact in
transgenic mice. PLoS One 6:e2575.
327

Trommer BL, Shah C, Yun SH, Gamkrelidze G, Pasternak ES, Ye GL, Sotak M,
Sullivan PM, Pasternak JF, LaDu MJ (2004) ApoE isoform affects LTP in human
targeted replacement mice. Neuroreport 15:2655-2658.
Tsenter J, Beni-Adani L, Assaf Y, Alexandrovich AG, Trembovler V, Shohami E (2008)
Dynamic changes in the recovery after traumatic brain injury in mice: effect of injury
severity on T2-weighted MRI abnormalities, and motor and cognitive functions. J
Neurotrauma 25:324-333.
Tsoi LM, Wong KY, Liu YM, Ho YY (2007) Apoprotein E isoform-dependent expression
and secretion of pro-inflammatory cytokines TNF-alpha and IL-6 in macrophages. Arch
Biochem Biophys 460:33-40.
Ucar T, Tanriover G, Gurer I, Onal MZ, Kazan S (2006) Modified experimental mild
traumatic brain injury model. J Trauma 60:558-565.
Uchihara T, Shibuya K, Nakemura A, Yagishita S (2005) Silver stains distinguish tau-
positive structures in corticobasal degeneration/progressive supranuclear palso and in
Alzheimer's disease - comparison between Gallyas and Campbell-Switzer methods.
Acta Neuropathol 109:299-305.
United Sates Departments of Defense and Veterans Affairs (2008) Common definition
of TBI. In: U.S. Departments of Defense and Veterans Affairs.
328

Uryu K, Chen XH, Martinez D, Browne KD, Johnson VE, Graham DI, Lee VM,
Trojanowski JQ, Smith DH (2007) Multiple proteins implicated in neurodegenerative
diseases accumulate in axons after brain trauma in humans. Exp Neurol 208:185-192.
Valastro B, Ghribi O, Poirier J, Krzywkowski P, Massicotte G (2001) AMPA receptor
regulation and LTP in the hippocampus of young and aged apolipoprotein E-deficient
mice. Neurobiol Aging 22:9-15.
Van Den Heuvel C, Thornton E, Vink R (2007) Traumatic brain injury and Alzheimer's
Disease: a review. Prog Brain Res 161:297-310.
van Dommelen JAW, Hrapko M, Peters GWM (2009) Mechanical Properties of Brain
Tissue: Characterisation and Constitutive Modelling. In: Mechanosensitivity of the
Nervous System (Kamkim A, Kiseleva I, eds), pp 249-279: Springer Netherlands.
van Duijn CM, Tanja TA, Haaxma R, Schulte W, Saan RJ, Lameris AJ, Antonides-
Hendriks G, Hofman A (1992) Head trauma and the risk of Alzheimer's disease. Am J
Epidemiol 135:775-782.
Van Oosten M, Rensen PCN, Van Amersfoort ES, Van Eck M, Van Dam A-M, Brevé
JJP, Vogel T, Panet A, Van Berkel TJC, Kuiper J (2001) Apolipoprotein E protects
against bacterial lipopolysaccharide-induced lethality: a new therapeutic approach to
treat gram-negative sepsis. J Biol Chem 276:8820-8824.
329

Vance JE, Hayashi H, Karten B (2005) Cholesterol homeostasis in neurons and glial
cells. Semin Cell Dev Biol 16:193-212.
Vassar R (2004) BACE1: the beta-secretase enzyme in Alzheimer's disease. J Mol
Neurosci 23:105-114.
Vedhachalam C, Duong PT, Nickel M, Nguyen D, Dhanasekaran P, Saito H, Rothblat
GH, Lund-Katz S, Phillips MC (2007) Mechanism of ATP-binding cassette transporter
A1 (ABCA1)-mediated cellular lipid efflux to apolipoprotein A-I and formation of high
density lipoprotein particles. J Biol Chem 282:25123-25130.
Vekrellis K, Ye Z, Qiu WQ, Walsh D, Hartley D, Chesneau V, Rosner MR, Selkoe DJ
(2000) Neurons regulate extracellular levels of amyloid beta-protein via proteolysis by
insulin-degrading enzyme. J Neurosci 20:1657-1665.
Venkateswaran A, Repa JJ, Lobaccaro JM, Bronson A, Mangelsdorf DJ, Edwards PA
(2000) Human white/murine ABC8 mRNA levels are highly induced in lipid-loaded
macrophages. A transcriptional role for specific oxysterols. J Biol Chem 275:14700-
14707.
Venteclef N, Jakobsson T, Ehrlund A, Damdimopoulos A, Mikkonen L, Ellis E, Nilsson
L-M, Parini P, Jänne OA, Gustafsson J-Å, Steffensen KR, Treuter E (2010) GPS2-
dependent corepressor/SUMO pathways govern anti-inflammatory actions of LRH-1
and LXRβ in the hepatic acute phase response. Genes & Development 24:381-395.
330

Verghese PB, Castellano JM, Garai K, Wang Y, Jiang H, Shah A, Bu G, Frieden C,
Holtzman DM (2013) ApoE influences amyloid-beta (Abeta) clearance despite minimal
apoE/Abeta association in physiological conditions. Proc Natl Acad Sci U S A
110:E1807-1816.
Versace J National Highway Traffic Safety Adm. In: 15th Stapp Car Crash Conference,
SAE Paper #710881, pp 771-796. 1971.
Versace J (1971) A review of the severity index. In: Proc. of the 15th Stapp Car Crash
Conference, pp 771-796.
Viano DC, Hamberger A, Bolouri H, Saljo A (2009) Concussion in professional football:
animal model of brain injury--part 15. Neurosurgery 64:1162-1173; discussion 1173.
Viano DC, Hamberger A, Bolouri H, Saljo A (2012) Evaluation of three animal models
for concussion and serious brain injury. Ann Biomed Eng 40:213-226.
Viano DC, Casson IR, Pellman EJ, Bir CA, Zhang L, Sherman DC, Boitano MA (2005)
Concussion in professional football: comparison with boxing head impacts--part 10.
Neurosurgery 57:1154-1172; discussion 1154-1172.
Vos PE, Battistin L, Birbamer G, Gerstenbrand F, Potapov A, Prevec T, Stepan Ch A,
Traubner P, Twijnstra A, Vecsei L, von Wild K (2002) EFNS guideline on mild traumatic
brain injury: report of an EFNS task force. Eur J Neurol 9:207-219.
331

Wahrle S, Jiang H, Parsadanian M, Hartman RE, Bales KR, Paul SM, Holtzman DM
(2005) Deletion of Abca1 increases Abeta deposition in the PDAPP transgenic mouse
model of Alzheimer disease. J Biol Chem 280 43236-43242.
Wahrle SE, Jiang H, Parsadanian M, Legleiter J, Han X, Fryer JD, Kowalewski T,
Holtzman DM (2004) ABCA1 is required for normal CNS apoE levels and for lipidation
of astrocyte-secreted apoE. J Biol Chem 279 40987-40993.
Wahrle SE, Jiang H, Parsadanian M, Kim J, Li A, Knoten A, Jain S, Hirsch-Reinshagen
V, Wellington CL, Bales KR, Paul SM, Holtzman DM (2008) Overexpression of ABCA1
reduces amyloid deposition in the PDAPP mouse model of Alzheimer disease. J Clin
Invest 118:671-682.
Wang DS, Lipton RB, Katz MJ, Davies P, Buschke H, Kuslansky G, Verghese J,
Younkin SG, Eckman C, Dickson DW (2005) Decreased neprilysin immunoreactivity in
Alzheimer disease, but not in pathological aging. J Neuropathol Exp Neurol 64:378-385.
Wang H, Gao J, Lassiter T, McDonagh D, Sheng H, Warner D, Lynch J, Laskowitz D
(2006) Levetiracetam is neuroprotective in murine models of closed head injury and
subarachnoid hemorrhage. Neurocrit Care 5:71-78.
Wang HC, Duan ZX, Wu FF, Xie L, Zhang H, Ma YB (2010) A new rat model for diffuse
axonal injury using a combination of linear acceleration and angular acceleration. J
Neurotrauma 27:707-719.
332

Wang JZ, Grundke-Iqbal I, Iqbal K (2007) Kinases and phosphatases and tau sites
involved in Alzheimer neurofibrillary degeneration. Eur J Neurosci 25:59-68.
Wang L, Schuster GU, Hultenby K, Zhang Q, Andersson S, Gustafsson JA (2002) Liver
X receptors in the central nervous system: from lipid homeostasis to neuronal
degeneration. Proc Natl Acad Sci U S A 99:13878-13883.
Wang N, Tall AR (2003) Regulation and mechanisms of ATP-binding cassette
transporter A1-mediated cellular cholesterol efflux. Arterioscler Thromb Vasc Biol
23:1178-1184.
Wang N, Silver DL, Costet P, Tall AR (2000) Specific binding of ApoA-I, enhanced
cholesterol efflux, and altered plasma membrane morphology in cells expressing ABC1
[In Process Citation]. J Biol Chem 275:33053-33058.
Wang N, Chen W, Linsel-Nitschke P, Martinez LO, Agerholm-Larsen B, Silver DL, Tall
AR (2003) A PEST sequence in ABCA1 regulates degradation by calpain protease and
stabilization of ABCA1 by apoA-I. J Clin Invest 111:99-107.
Wang YJ, Shimura T, Kobayashi S, Teramoto A, Nakazawa S (1997) A lateral fluid
percussion model for the experimental severe brain injury and a morphological study in
the rats. Nihon Ika Daigaku Zasshi 64:172-175.
333

Weingarten MD, Lockwood AH, Hwo SY, Kirschner MW (1975) A protein factor
essential for microtubule assembly. Proc Natl Acad Sci U S A 72:1858-1862.
Weisgraber KH (1990) Apolipoprotein E distribution among human plasma lipoproteins:
role of the cysteine-arginine interchange at residue 112. J Lipid Res 31:1503-1511.
Weisgraber KH (1994) Apolipoprotein E: structure-function relationships. Adv Protein
Chem 45:249-302.
Weisgraber KH, Innerarity TL, Mahley RW (1982) Abnormal lipoprotein receptor-binding
activity of the human E apoprotein due to cysteine-arginine interchange at a single site.
J Biol Chem 257:2518-2521.
Weller RO, Yow HY, Preston SD, Mazanti I, Nicoll JA (2002) Cerebrovascular disease
is a major factor in the failure of elimination of Abeta from the aging human brain:
implications for therapy of Alzheimer's disease. Ann N Y Acad Sci 977:162-168.
Weller RO, Subash M, Preston SD, Mazanti I, Carare RO (2008) SYMPOSIUM:
Clearance of Aβ from the Brain in Alzheimer's Disease: Perivascular Drainage of
Amyloid-β Peptides from the Brain and Its Failure in Cerebral Amyloid Angiopathy and
Alzheimer's Disease. Brain Pathology 18:253-266.
Wellington CL, Walker EK, Suarez A, Kwok A, Bissada N, Singaraja R, Yang YZ, Zhang
LH, James E, Wilson JE, Francone O, McManus BM, Hayden MR (2002) ABCA1
334

mRNA and protein distribution patterns predict multiple different roles and levels of
regulation. Lab Invest 82:273-283.
Werner C, Engelhard K (2007) Pathophysiology of traumatic brain injury. Br J Anaesth
99:4-9.
Wetterau JR, Aggerbeck LP, Rall SC, Jr., Weisgraber KH (1988) Human apolipoprotein
E3 in aqueous solution. I. Evidence for two structural domains. J Biol Chem 263:6240-
6248.
White F, Nicoll JA, Horsburgh K (2001a) Alterations in apoE and apoJ in relation to
degeneration and regeneration in a mouse model of entorhinal cortex lesion. Exp
Neurol 169:307-318.
White F, Nicoll JA, Roses AD, Horsburgh K (2001b) Impaired neuronal plasticity in
transgenic mice expressing human apolipoprotein E4 compared to E3 in a model of
entorhinal cortex lesion. Neurobiol Dis 8:611-625.
Whitney KD, Watson MA, Collins JL, Benson WG, Stone TM, Numerick MJ, Tippin TK,
Wilson JG, Winegar DA, Kliewer SA (2002) Regulation of cholesterol homeostasis by
the liver X receptors in the central nervous system. Molec Endocrinol 16:1378-1385.
335

Whyte J, Hart T, Vaccaro M, Grieb-Neff P, Risser A, Polansky M, Coslett HB (2004)
Effects of methylphenidate on attention deficits after traumatic brain injury: a
multidimensional, randomized, controlled trial. Am J Phys Med Rehabil 83:401-420.
Wiebel FF, Gustafsson JA (1997) Heterodimeric interaction between retinoid X receptor
alpha and orphan nuclear receptor OR1 reveals dimerization-induced activation as a
novel mechanism of nuclear receptor activation. Mol Cell Biol 17:3977-3986.
Williams DJ, Tannenberg AE (1996) Dementia pugilistica in an alcoholic
achondroplastic dwarf. Pathology 28:102-104.
Willmott C, Ponsford J (2009) Efficacy of methylphenidate in the rehabilitation of
attention following traumatic brain injury: a randomised, crossover, double blind,
placebo controlled inpatient trial. J Neurol Neurosurg Psychiatry 80:552-557.
Wilson AC, Dugger BN, Dickson DW, Wang DS (2011) TDP-43 in aging and
Alzheimer's disease - a review. Int J Clin Exp Pathol 4:147-155.
Wilson C, Wardell MR, Weisgraber KH, Mahley RW, Agard DA (1991) Three-
dimensional structure of the LDL receptor-binding domain of human apolipoprotein E.
Science 252:1817-1822.
336

Winkler K, Scharnagl H, Tisljar U, Hoschutzky H, Friedrich I, Hoffmann MM, Huttinger
M, Wieland H, Marz W (1999) Competition of Abeta amyloid peptide and apolipoprotein
E for receptor-mediated endocytosis. J Lipid Res 40:447-455.
Winter CD, Iannotti F, Pringle AK, Trikkas C, Clough GF, Church MK (2002) A
microdialysis method for the recovery of IL-1beta, IL-6 and nerve growth factor from
human brain in vivo. J Neurosci Methods 119:45-50.
Wisniewski T, Frangione B (1992) Apolipoprotein E: A pathological chaperone protein in
patients with cerebral and systemic amyloid. Neurosci Lett 135:235-238.
Wojcicka G, Jamroz-Wisniewska A, Horoszewicz K, Beltowski J (2007) Liver X
receptors (LXRs). Part I: structure, function, regulation of activity, and role in lipid
metabolism. Postepy Hig Med Dosw (Online) 61:736-759.
Wolf G, Cifu D, Baugh L, Carne W, Profenna L (2012) The effect of hyperbaric oxygen
on symptoms after mild traumatic brain injury. J Neurotrauma 29:2606-2612.
Wolf JA, Stys PK, Lusardi T, Meaney D, Smith DH (2001) Traumatic axonal injury
induces calcium influx modulated by tetrodotoxin-sensitive sodium channels. J Neurosci
21:1923-1930.
Woodroofe MN, Sarna GS, Wadhwa M, Hayes GM, Loughlin AJ, Tinker A, Cuzner ML
(1991) Detection of interleukin-1 and interleukin-6 in adult rat brain, following
337

mechanical injury, by in vivo microdialysis: evidence of a role for microglia in cytokine
production. J Neuroimmunol 33:227-236.
Wright DW, Kellermann AL, Hertzberg VS, Clark PL, Frankel M, Goldstein FC,
Salomone JP, Dent LL, Harris OA, Ander DS, Lowery DW, Patel MM, Denson DD,
Gordon AB, Wald MM, Gupta S, Hoffman SW, Stein DG (2007) ProTECT: a
randomized clinical trial of progesterone for acute traumatic brain injury. Ann Emerg
Med 49:391-402.
Wright RM, Post A, Hoshizaki B, Ramesh KT (2013) A Multiscale Computational
Approach to Estimating Axonal Damage under Inertial Loading of the Head. J
Neurotrauma 30:102-118.
Wu X, Hu J, Zhuo L, Fu C, Hui G, Wang Y, Yang W, Teng L, Lu S, Xu G (2008)
Epidemiology of traumatic brain injury in eastern China, 2004: a prospective large case
study. J Trauma 64:1313-1319.
Xiao-Sheng H, Sheng-Yu Y, Xiang Z, Zhou F, Jian-ning Z (2000) Diffuse axonal injury
due to lateral head rotation in a rat model. J Neurosurg 93:626-633.
Xiao G, Wei J, Yan W, Wang W, Lu Z (2008) Improved outcomes from the
administration of progesterone for patients with acute severe traumatic brain injury: a
randomized controlled trial. Crit Care 12:R61.
338

Xiong Y, Mahmood A, Chopp M (2013) Animal models of traumatic brain injury. Nat Rev
Neurosci 14:128-142.
Xu PT, Gilbert JR, Qiu HL, Ervin J, Rothrock-Christian TR, Hulette C, Schmechel DE
(1999) Specific regional transcription of apolipoprotein E in human brain neurons. Am J
Pathol 154:601-611.
Xu Q, Bernardo A, Walker D, Kanegawa T, Mahley RW, Huang Y (2006) Profile and
regulation of apolipoprotein E (ApoE) expression in the CNS in mice with targeting of
green fluorescent protein gene to the ApoE locus. J Neurosci 26:4985-4994.
Yasojima K, McGeer EG, McGeer PL (2001a) Relationship between beta amyloid
peptide generating molecules and neprilysin in Alzheimer disease and normal brain.
Brain Res 919:115-121.
Yasojima K, Akiyama H, McGeer EG, McGeer PL (2001b) Reduced neprilysin in high
plaque areas of Alzheimer brain: a possible relationship to deficient degradation of β-
amyloid peptide. Neurosci Lett 297:97-100.
Yoganandan N, Gennarelli TA, Zhang J, Pintar FA, Takhounts E, Ridella SA (2009)
Association of contact loading in diffuse axonal injuries from motor vehicle crashes. J
Trauma 66:309-315.
339

Yokobori S, Bullock MR (2013) Pathobiology of primary traumatic brain injury. In: Brain
Injury Medicine: Principles and Practice, 2nd Edition (Zasler ND, Katz DI, Zafonte RD,
eds), pp 137-147. New York, NY: Demos Medical Publishing , LLC.
Yoshikawa T, Shimano H, Yahagi N, Ide T, Amemiya-Kudo M, Matsuzaka T, Nakakuki
M, Tomita S, Okazaki H, Tamura Y, Iizuka Y, Ohashi K, Takahashi A, Sone H, Osuga Ji
J, Gotoda T, Ishibashi S, Yamada N (2002) Polyunsaturated fatty acids suppress sterol
regulatory element-binding protein 1c promoter activity by inhibition of liver X receptor
(LXR) binding to LXR response elements. J Biol Chem 277:1705-1711.
Young B, Ott L, Kasarskis E, Rapp R, Moles K, Dempsey RJ, Tibbs PA, Kryscio R,
McClain C (1996) Zinc supplementation is associated with improved neurologic
recovery rate and visceral protein levels of patients with severe closed head injury. J
Neurotrauma 13:25-34.
Yu C, Youmans KL, LaDu MJ (2011) Proposed mechanism for lipoprotein remodelling
in the brain. Biochim Biophys Acta 1801:19-23.
Yu L, York J, von Bergmann K, Lutjohann D, Cohen JC, Hobbs HH (2003) Stimulation
of cholesterol excretion by the liver X receptor agonist required ATP-binding cassette
transporters G5 and G8. J Biol Chem 278:15565-15570.
340

Yu L, Li-Hawkins J, Hammer RE, Berge KE, Horton JD, Cohen JC, Hobbs H (2002)
Overexpression of ABCG5 and ABCG8 promotes biliary cholesterol secretion and
reduces fractional absorption of dietary cholesterol. J Clin Invest 110:671-680.
Zafonte RD, Bagiella E, Ansel BM, Novack TA, Friedewald WT, Hesdorffer DC,
Timmons SD, Jallo J, Eisenberg H, Hart T, Ricker JH, Diaz-Arrastia R, Merchant RE,
Temkin NR, Melton S, Dikmen SS (2012) Effect of citicoline on functional and cognitive
status among patients with traumatic brain injury: Citicoline Brain Injury Treatment Trial
(COBRIT). JAMA 308:1993-2000.
Zelcer N, Khanlou N, Clare R, Jiang Q, Reed-Geaghan EG, Landreth GE, Vinters HV,
Tontonoz P (2007) Attenuation of neuroinflammation and Alzheimer's disease pathology
by liver x receptors. Proc Natl Acad Sci U S A 104:10601-10606.
Zhang L, Yang KH, King AI (2004) A proposed injury threshold for mild traumatic brain
injury. J Biomech Eng 126:226-236.
Zhao YD, Wang W (2001) Neurosurgical trauma in People's Republic of China. World J
Surg 25:1202-1204.
Zhi D, Zhang S, Lin X (2003) Study on therapeutic mechanism and clinical effect of mild
hypothermia in patients with severe head injury. Surg Neurol 59:381-385.
341

Zhong N, Weisgraber KH (2009) Understanding the Association of Apolipoprotein E4
with Alzheimer Disease: Clues from Its Structure. J Biol Chem 284:6027-6031.
Zhou W, Xu D, Peng X, Zhang Q, Jia J, Crutcher KA (2008) Meta-analysis of APOE4
allele and outcomes after traumatic brain injury. J Neurotrauma 25:279-290.
Ziebell JM, Taylor SE, Cao T, Harrison JL, Lifshitz J (2012) Rod microglia: elongation,
alignment, and coupling to form trains across the somatosensory cortex after
experimental diffuse brain injury. J Neuroinflammation 9:247.
Zink BJ, Szmydynger-Chodobska J, Choobski A (2010) Emerging concepts in the
pathophysiology of trauamtic brain injury. Psychiatr Clin North Am 33:741-756.
Zlokovic BV (2004) Clearing amyloid through the blood–brain barrier. J Neurochem
89:807-811.
Zohar O, Rubovitch V, Milman A, Schreiber S, Pick CG (2011) Behavioral
consequences of minimal traumatic brain injury in mice. Acta Neurobiol Exp 71:36-45.
Zohar O, Schreiber S, Getslev V, Schwartz JP, Mullins PG, Pick CG (2003) Closed-
head minimal traumatic brain injury produces long-term cognitive deficits in mice.
Neuroscience 118:949-955.
342

Zuccarello M, Iavicoli R, Pardatscher K, Cervellini P, Fiore D, Mingrino S, Gerosa M
(1981) Posttraumatic intfaventricular haemorrhages. Acta Neurochirurgica 55:283-293.
Zweckberger K, Eros C, Zimmermann R, Kim SW, Engel D, Plesnila N (2006) Effect of
early and delayed decompressive craniectomy on secondary brain damage after
controlled cortical impact in mice. J Neurotrauma 23:1083-1093.
343





























