Embed Size (px)
Citation preview

THEORETICAL STUDIES ON THE SPECTROSCOPY
OF SOME INTRAGROUP IVA HETERONUCLEAR
DIATOMIC MOLECULES AND THEIR IONS
A
THESIS
SUBMITTED FOR THE DEGREE OF
DOCTOR OF PHILOSOPHY (SCIENCE)
OF
JADAVPUR UNIVERSITY
BY
ANUP PRAMANIK, M.Sc.
DEPARTMENT OF CHEMISTRY
PHYSICAL CHEMISTRY SECTION
JADAVPUR UNIVERSITY
KOLKATA – 700032
INDIA

CERTIFICATE FROM THE SUPERVISOR(S) This is to certify that the thesis entitled “THEORETICAL STUDIES ON THE SPECTROSCOPY OF SOME INTRAGROUP IVA HETERONUCLEAR DIATOMIC MOLECULES AND THEIR IONS” submitted by Sri / Smt. ANUP PRAMANIK, who got his/her name registered on 25.06.2007 for the award of Ph.D. (Science) degree of Jadavpur University, is based upon his own work under the supervision of PROF. DR. KALYAN KUMAR DAS and that neither this thesis nor any part of it has been submitted for either any degree / diploma or any other academic award anywhere before. (Signature of the Supervisor(s), date with official seal Prof. Dr. Kalyan Kumar Das, Department of Chemistry, Physical Chemistry Section, Jadavpur University, Kolkata – 700 032, India.

TO
MY FRIEND PHILOSOPHER AND GUIDE
“KOUSIK-UNCLE”

Acknowledgements The research work presented in the thesis has been performed in the Department of Chemistry,
Physical Chemistry Section, Jadavpur University since January, 2006. I would like to take the
opportunity to convey my thanks to the people whose constant help and encouragement have
finally laid me to complete the thesis.
I express my warmest gratitude to my supervisor, Prof. Dr. Kalyan Kumar Das for his kind
cooperation and thoughtful advices. What he has done for me is really beyond my expectation. In
each step I have learnt from him how to utilize the valuable times of our life properly. Great
scientific attitude as well as nice behavior of him is truly rememberable.
The financial support provided by CSIR, Govt. of India is gratefully acknowledged. Without
this it was impossible to carry out my research work, whatever I have done.
I am indebted to Prof. Dilip Kumar Bhattacharyya and Dr. Biplab Bhattacharjee for their
moral supports and valuable discussions. I am also thankful to the Head, other teaching and non-
teaching staffs of the department of Chemistry, Jadavpur University. Library and laboratory
facilities of this university are also gratefully acknowledged.
A lot of thanks to my lab-mates, Mr. Amartya Banerjee, Ms. Susmita Chakrabarti for their
ever helping hands and cooperation. The former guy deserves a speciality for his philosophical
sense and critical analysis, which helped me a lot during my research period.
My heartiest love and respect to my parents and other family members. Specially, my sincere
thanks to my mother, and my wife, Mitali. They have provided me continuous supports and all
kinds of facilities. I can’t make him dishonored by expressing only my thanks to Kousik-uncle
who induced the philosophy of science in my mind. It brings a great pleasure to me to dedicate
the thesis to him.
Date:
Department of chemistry, ANUP PRAMANIK
Physical Chemistry Section,
Jadavpur University,
Kolkata – 700032, India

Contents
Introduction 1
Plan of the thesis 4
1. A brief review of the electronic structure theory of atoms and
molecules
1.1 Introduction 7
1.2 The Schrdinger equation 8
1.3 The variational principle 9
1.4 The Hartree-Fock model 10
1.5 Basis sets 12
1.6 Relativistic effects 13
1.7 Electron correlation energy and post Hartree-Fock treatments 15
1.8 References 20
2. Brief review of the computational methodology: details of the
Configuration Interaction method
2.1 Introduction 22
2.2 Relativistic corrections
2.2.1 The Dirac equation 24
2.2.2 Effective core potential 25
2.2.3 Spin-orbit coupling 29
2.3 Computational methodology
2.3.1 Configuration selection technique 31
2.3.2 Role of unselected configurations 32
2.3.3 Spin-orbit interaction 34
2.3.4 Calculation of spectroscopic constants 36
2.3.5 Estimation of radiative lifetime 37
2.4 References 38
i

3. Electronic structure and spectroscopic properties of the SiC radical
3.1 Introduction 42
3.2 Computational details
3.2.1 RECPs and basis sets 44
3.2.2 SCF MOs and CI 44
3.2.3 Spin-orbit interaction 46
3.3 Results and discussion
3.3.1 Spectroscopic constants and potential energy curves of Λ-S states 46
3.3.2 Spectroscopic constants and potential energy curves of Ω states 56
3.3.3 Dipole moments and transition properties 59
3.4 Summary 62
3.5 References 64
4. Electronic structure and spectroscopic properties of SiC+ and SiC−
4.1 Introduction 66
4.2 Computational details
4.2.1 RECPs and basis sets 67
4.2.2 SCF MOs and CI 67
4.2.3 Spin-orbit interaction 69
4.3 Results and discussion
4.3.1 Spectroscopic constants and potential energy curves of Λ-S states
A. SiC+ 69
B. SiC− 75
4.3.2 Spectroscopic constants and potential energy curves of Ω states 81
4.3.3 Transition properties
A. SiC+ 83
B. SiC− 85
4.3.4 Dipole moments, ionization potentials, and electron affinities 86
4.4 Summary 89
4.5 References 91
5. Electronic structure and spectroscopic properties of SnC and SnC+
5.1 Introduction 93
ii

5.2 Computational details
5.2.1 RECPs and basis sets 95
5.2.2 SCF MOs and CI 95
5.2.3 Spin-orbit interaction 96
5.3 Results and discussion
5.3.1 Spectroscopic constants and potential energy curves of Λ-S states
A. SnC 97
B. SnC+ 105
5.3.2 Spectroscopic constants and potential energy curves of Ω states
A. SnC 110
B. SnC+ 113
5.3.3 Transition properties
A. SnC 117
B. SnC+ 120
5.3.4 Dipole moments and ionization energies 122
5.4 Summary 125
5.5 References 127
6. Electronic structure and spectroscopic properties of PbC and PbC+
6.1 Introduction 129
6.2 Computational details
6.2.1 RECPs and basis sets 130
6.2.2 SCF MOs and CI 130
6.2.3 Spin-orbit interaction 132
6.3 Results and discussion
6.3.1 Spectroscopic constants and potential energy curves of Λ-S states
A. PbC 132
B. PbC+ 138
6.3.2 Spectroscopic constants and potential energy curves of Ω states
A. PbC 142
B. PbC+ 147
6.3.3 Transition properties
A. PbC 151
B. PbC+ 154
iii

6.3.4 Dipole moments and ionization energies 157
6.3.5 Comparison of some spectroscopic properties of MC and MC+
(M = Si, Sn, Pb) 159
6.4 Summary 163
6.5 References 164
Conclusion 166
List of publications 169
iv

INTRODUCTION

The interpretation and understanding of every experimental finding requires the knowl-
edge of theoretical background. A large number of experimental results can be brought into
together by theoretical interpretation and suitable formulation. So, necessity of theoretical
research is urged by its own demand. A chemical problem can be solved theoretically by
proper use of physical laws and mathematical methods, often by the use of computer memory.
Large number of computational methods have been developed over the years for the com-
plete solution of chemical problems. Quantum mechanics is one such tool, which has been
developed enormously with the advancement of computer hardware and softwares. Modern
electronic structure theory, which is based on quantum mechanics, is capable of providing
reliable predictions of quantities of chemical interest. It is not surprising that, the variational
methods could be applied to systems as large as XeF6, azulene, and guanine-cytosine base
pair. Now a days, with the help of computation, a large number of organic molecules, such
as protein, DNA, RNA etc. are designed theoretically. Such attempts are very much helpful
to the experimentalists to reach the goal of real synthesis with prior experiences of chemical
hazards. Moreover, where we are bound to our experimental limit, theoretical investigation
is the only tool to interpret the natural observations. Thus structural chemistry, which is
based on spectroscopic measurement, is equally balanced by experimental results as well as
theoretical predictions.
The space trajectory and other dynamical properties of macroscopic objects can be well
studied by classical mechanics.1 However, classical mechanics fails in the domain of submi-
croscopic world of atom and its constituents. In the beginning of the nineteenth century,
Planck’s idea of quantization was brought into a new field of mechanics mainly by Heisenberg
and Schrodinger. The new mechanics, revealed as quantum mechanics2−10, is the the suc-
cessful treatment to describe the structural and dynamic properties of subatomic particles.
Now, if the velocity of the object is comparable to that of light, one must use the relativistic
mechanics of Einstein which takes into consideration of variation of mass with velocity. So,
subatomic particles of low mass and having very high velocity, comparable to that of light,
need to use of relativistic quantum mechanics, derived by Dirac. This uses the modified
Hamiltonian, containing various relativistic correction terms including mass-velocity, spin-
orbit, Darwin correction and Breit interaction. Thus, depending upon the mass and velocity,
the dynamics of a particle is governed by suitable mechanics and consequently it requires a
proper mathematical treatment.
The behavior of electrons in atoms or molecules are described by quantum mechanics.
1

Their space trajectories are described on the basis of probabilistic interpretation, accord-
ing to which the stationary sates of them are fitted with time independent Schrodinger
equations. On solving these quantum mechanical equations, which are second order differ-
ential in nature, one may get the electronic structure of atoms and molecules. By using
Born-Oppenheimer approximation, in which electronic motions are treated separately from
nuclear motion, the electronic Hamiltonian can be resolved and consequently the solution of
it gives the structural aspects and spectroscopic information of a molecule11−16. The task is
no longer a simple one, specially for molecules having heavier atoms, there involves a large
scale relativistic effects in hamiltonian and hence proper treatment is essential for that.17−19
A number of algorithms have been developed for this purpose over the past few decades
with the improvement of enormous computing facilities. Many of them give the results with
reasonably good accuracy. Large efforts are required for a bit of improvement of computed
result. Moreover, an enormous volume of computation may be necessary for this purpose.
So, there are always limitations in the accuracy. Parallel efforts are also being given for the
development of computing facilities. Thus, the real challenge is to exploit these developments
and carry out theoretical research to reach the stage more close to reality.
2

References
1 H. Goldstein, Classical Mechanics, Addition-Wesley, Reading, Mass., 1950.
2 L. Pauling, E.B. Wilson, Introduction to Quantum Mechanics, McGraw-Hill, 1935.
3 H. Eyring, J. Walter, G.E. Kimball, Quantum Chemistry, Wiley, New York, 1944.
4 L.I. Schiff, Quantum Mechanics, McGraw-Hill, New York, 1968.
5 J.P. Lowe, Quantum Chemistry, Academic Press, New York, 1978.
6 D.A. McQuarrie, Quantum Chemistry, University Science, Mill Valley, Calif. 1983.
7 P.W. Atkins, Molecular Quantum Mechanics, Oxford University Press, New York, 1983.
8 A. Hinchliffe, Computational Quantum Chemistry, Wiley, New York, 1988.
9 F.L. Pillar, Elementary Quantum Chemistry, McGraw-Hill, New York, 1990.
10 I.N. Levine, Quantum Chemistry, Printice-Hall, N.J., 1991.
11 R.G. Parr, Quantum theory of Molecular Electronic Structure, Benjamin, New York, 1963.
12 J.A. Pople, D.L. Beveridge, Approximate Molecular Orbital Theory, McGraw-Hill,
New York, 1970.
13 J.N. Murrell, A.J. Harget, Semiempirical Self-Consistent-Field Molecular Orbital
Theories of Molecules, Wiley-Interscience, New York, 1971.
14 R.S. Mulliken, W.C. Ermler, Diatomic Molecules, Academic Press, New York, 1977.
15 R.S. Mulliken, W.C. Ermler, Polyatomic Molecules, Academic Press, New York, 1981.
16 W.J. Hehre, L. Radom, P.V.R. Schleyer, and J.A. Pople, Ab Initio Molecular Orbital
Theory, Wiley-Interscience, New York, 1986.
17 P. Pyykko, Relativistic Theory of Atoms and Molecules, Springer-Verlag, Berlin and
New York, 1986.
18 K. Balasubramanian, Relativistic Effects in Chemistry Part A. Theory and Techniques,
Wiley-Interscience, New York, 1997.
19 K. Balasubramanian, Relativistic Effects in Chemistry Part B. Applications to molecules
and Clusters, Wiley-Interscience, New York, 1997.
3

PLAN OF THESIS

The aim of Quantum Chemistry is to have some information about chemical bonds. En-
ergetic information of chemical bonds involving permutation of all elements in the entire
periodic table have been collected over the years by many experimental scientists. Besides
their applications, the simple diatomic molecules draw special interest in contributing the in-
formation about their bond length, bond energy etc. Intragroup IVA heteronuclear diatomic
molecules have generated a special interest in recent years because of their possible applica-
tions in catalysis, sensor films and mostly, they are the building blocks of cluster materials
which have interesting solid state properties. Inspite of the facilities of many modern sophis-
ticated instruments like high resolution spectrophotometer, laser vaporization, supersonic
jet expansion, matrix isolation etc. this type of molecules are rarely studied in experi-
ment because of the difficulty of their isolation in gas phase. Even for the simplest of them
(SiC), ab initio calculations were performed before the experimental detection. Very recently
seven of the intragroup IVA diatomics have been energetically characterized by Knudesen
effusion mass spectroscopic (KEMS) technique. To verify the available experimental data
and to predict the spectroscopic characteristics it is common practice to use quantum me-
chanical techniques like configuration interaction (CI), complete active space self consistent
field (CASSCF), couple-cluster, many-body perturbation theories, density functional theo-
ries (DFT) etc. The present thesis aims to study the electronic structure and spectroscopic
properties of intragroup IVA heteronuclear diatomic molecules, specially the carbides of Si,
Sn, Pb, and some of their ions.
The multireference singles and doubles configuration interaction (MRDCI) calculations
have been performed in the present thesis using relativistic effective core potentials and
suitable Gaussian basis functions for the participating atoms. Potential energy curves of
some low-lying Λ-S as well as Ω states of the molecules and ions are constructed from the
estimated full-CI energies. Many avoided crossing interactions have been properly studied
by analyzing the CI state functions. Spectroscopic constants like re, ωe, and Te values are
calculated by fitting the potential energy curves. The variation of dipole moment functions
of some low-lying states and transition moment functions involving ground and some of the
excited states are followed against bond distances and subsequently radiative lifetimes of
few low-lying states are computed for neutral as well as the ionic species. Vertical ionization
energies (VIE) of the neutral species are reported. Electron affinity of SiC have also been
verified from the MRDCI studies of SiC and its anion.
Chapter 1 gives an overview of the basic quantum mechanics like time dependent
4

Schrodinger equation, variational principle, Hartree-Fock model, basis sets, relativistic effect,
electron correlation and post Hartree-Fock methods like MCSCF, CI etc.
Chapter 2 describes the computational methodologies which are used in the calculations
throughout the thesis. For many electron atoms, it is not possible to carry out all electron CI
calculations. So the effective core potential method is used. The details of MRDCI method
are discussed in this chapter. The configuration selection technique is also discussed in this
chapter along with the corrections due to the unselected configurations. The method of
including the spin-orbit coupling at the CI level is mentioned. The methods of estimation
of the spectroscopic constants and radiative lifetimes are also described here.
Chapter 3 deals with the results obtained from the calculations of SiC. Potential energy
curves and spectroscopic constants of a large number of Λ-S states of singlet, triplet, and
quintet spin multiplicities are reported and compared with the existing data. The ground-
state dissociation energy of the species is computed and verified with the experimental
results. E3Π is found to be an important one which have not been studied before. The
important transitions like A–X, B–X, C–X, D–X, E–X etc. are studied, at the same time
radiative lifetimes of some excited states are also reported. Dipole moments of some low-
lying states are computed. Finally, the effects of spin-orbit coupling on the spectroscopic
properties of SiC are discussed in the chapter.
Chapter 4 describes the effects of removal and addition of an electron to the neutral
silicon carbide using MRDCI methodologies. Ionization energies and electron affinities of
SiC are reported in this chapter. Spectroscopic aspects of the SiC+ and SiC− ions are
studied in detail. The ground states of SiC+ and SiC− are 4Σ− and 2Σ+, respectively. Thus,
the quartet-quartet transitions for SiC+ and the doublet-doublet transitions for SiC− are
of special interest. No experimental data are known, but a very few theoretical results are
available for comparison. The spectroscopic constants of low-lying states of both the species
up to an energy level of 6 eV are reported.
Chapter 5 contains the results of the electronic structure and spectroscopic properties
of SnC and SnC+. Spectroscopic constants and some other properties of these species, at
both Λ–S and Ω levels, are reported. Because of the heavier mass of Sn, the Ω states have
become more important as compared to those of SiC. Hence, the spin-forbidden transitions
are given a special attention. Radiative lifetimes of some low-lying states of these species
have been reported in this chapter.
5

In Chapter 6, we have discussed electronic structure and spectroscopic properties of
PbC and PbC+. Lead is the heaviest element of group IVA and consequently the spin-orbit
coupling has been found to be the most prominent. Hence, spectroscopic properties of the
Ω states are thoroughly studied. This chapter includes dipole and transition dipole moment
functions of some low-lying states. Many spin-forbidden transitions are computed, and a
comparison of the spectroscopic properties of all three carbides and their monopositive ions
has also been made in the last part of this chapter.
6

CHAPTER – 1
A BRIEF REVIEW OF THE ELECTRONIC
STRUCTURE THEORY OF ATOMS AND
MOLECULES

1.1. Introduction
As we have mentioned earlier, behaviors of electrons in an atom or molecule are described
by stationary state wave functions as given in time independent Schrodinger equations. To
have solutions of such equations is always a difficult task. If we neglect all the relativistic
effects and consider the electrons to be moving in a fixed nuclear framework as in the Born-
Oppenheimer approximation, the problem becomes much easier to deal with. But still it
is a formidable task to solve these equations because of the involvement of a large number
of inter-electronic interaction terms in the Hamoltonian. Approximate methods have been
developed over the years to solve the non-relativistic Schrodinger equation for determining
the electronic structure of the molecular systems as accurately as possible. As an approxima-
tion, firstly these two body terms are converted into separate one-electron potentials. This
transforms the many body problem into the effective one-body problems which is popularly
known as independent particle model. The model gives the best possible solution in which
the wave functions are represented by the antisymmetrized product of one-electron functions,
commonly called orbitals. Next, for the construction of the single configuration state, it has
to satisfy two criteria; the wave function would have minimum energy in its neighborhood,
and the orbitals must have maximum overlap. The minimum energy criterion is fulfilled by
Hartree-Fock model, based on the variational principle. On the other hand, the maximum
overlap criterion with the exact wave function leads to the Brueckner approximation.1 The
last one is not practicable to implement as it requires the knowledge of exact wave func-
tions, while it is easier to implement the Hartree-Fock theory in practice. The minimum
energy wave function of a given class can be obtained from the variational principle which
can be applied in quantum chemistry. There are several post-Hartree-Fock methodologies
which may then be applied for estimating the electron correlation missing in the Hartree-
Fock approximation. However, it requires rigorous mathematical calculations and numerical
methods. The basic principles and techniques, which are used in the electronic structure
theory of atoms and molecules, are briefly discussed in the following sections.
7

1.2. The Schrodinger equation
In quantum mechanics, dynamics of a system is described by the time dependent Schrodinger
equation
Hψ= ih(∂ψ/∂t), (1.1)
where H is the Hamiltonian operator consisting of the kinetic and potential energy operators
of the system and ψ is called state function which is the function of space coordinate (r) and
time (t).
Now, the state of a many-body system (say, consisting n number of electrons) is given by
the wave function
ψ=ψ(r1, r2, r3.....rn; t)
and the probability density is written as
P (r1, r2, r3.....rn; t)= |ψ(r1, r2, r3, .....rn; t) |2 .
The equation (1.1) shows how the wave function evolves in time. Now, the time-independent
Hamiltonian operator of the n-electron system in the absence of any external field but only
with the Coulomb interactions among the electrons can be written as follows
H=− h2
2meΣi(∂
2/∂x2i+∂
2/∂y2i +∂2/∂z2
i ) +ΣijQiQj
|ri − rj |. (1.2)
Since H does not contain time explicitly, one can apply the method of separation of variables.
Time dependent and time independent part of the wave function can be separated for the
stationary state problem,
ψ(r1, r2....., rn; t)=Ψ(r1, r2....., rn; t)e−iEt/h. (1.3)
This gives rise to time-independent Schrodinger equation,
HΨ=EΨ. (1.4)
There are 3n coordinates in the wave function Ψ for n electron system. In addition to
the spatial coordinates, if we consider spin coordinates into account the total number of
coordinates become 4n, where the spin is restricted to the value ±12. In the relativistic
8

treatment, the spin of the electrons appears naturally, and it is sometimes considered as an
intrinsic angular momentum of the particle.
The symmetry restriction is to be imposed on to the wave functions. The only acceptable
solutions of the equation (1.4) are those with appropriate symmetry on the application of
two particle permutation operator. For electronic systems, the wave functions must be
antisymmetric with respect to interchange of the coordinates of any pair of electrons. Time
independent Schrodinger equation will provide many solutions for stationary states. The
lowest energy state is obviously the ground state.
Schrodinger equations are simplified for stationary state problems by using Born-Oppenhei-
mer approximation,2 in which the nuclear coordinates are kept frozen and the electronic part
is solved at a fixed nuclear geometry.
Hel(r;R)Ψel(r;R)=Eel(R)Ψel(r;R) (1.5)
1.3. The variational principle
The Schrodinger equation cannot be solved exactly for many electron systems because
the variables are not separable. Many approximate methods are employed for getting the
solutions. The variational principle3 provides one such approximate technique to solve the
time-independent Schrodinger equation HΨk=EΨk, say for the k-th stationary state. In the
linear variational principle, a trial wave function (Ψk) is expanded as a linear combination
of basis functions χi
Ψk =Σiciχi, (1.6)
where ci denotes the expansion coefficients. If the functions χi form a complete set, one
obtains the true wave function of the system. However, truncation is needed for practical
purpose.
The energy functional is given as
Ek=〈Ψk | H | Ψk〉/ 〈Ψk|Ψk〉
=ΣijcicjHij/Σijcicj〈χi|χj〉. (1.7)
The linear variational principle ensures that the trial energy calculated above is always higher
9

than the true energy of the system. In other words, there is an upper bound to the electronic
energy. The energy functional, Ek is minimized with respect to all ci parameters. One gets
the exact energy if the trial function is the exact solution to the Schrodinger equation i.e.
Ψk. The above mentioned first-order variational conditions give the secular equations in the
matrix form as
Hc=Ec. (1.8)
From the computational view point, one must construct the Hamiltonian matrix element for
a given basis set used to expand the wave function in the form of the equation (1.6). The
diagonalization of the the H-matrix has to be done next to get eigenvalues and eigenfunctions.
The matrix elements may be computed by different semiemperical or abinitio methods.
1.4. The Hartree-Fock model
Electronic motions in atoms or molecules are correlated mainly because of the Coulombic
interactions among the electrons. Equation (1.2) suggests that the molecular Hamiltonian is
independent of three or higher body interaction terms. However, the two-electron interaction
terms are the most important and difficult to compute in the electronic structure theory.
Three or higher body interactions are approximated to zero.
In the independent particle model, one may write the wave function in terms of product
of orbitals, which are functions of both space and spin-coordinates of electrons.
Ψ(r1, r2, r3....) =Φ1(r1)Φ2(r2)Φ3(r3)..... (1.9)
Here the electron-electron repulsion term is taken into consideration in an indirect manner.
Each electron has been considered to be moving in the mean-field of the remaining (n-1)
electrons. This gives the following set of one-electron equations
hiΦi(ri)=εiΦi(ri), i=1,2,3...n (1.10)
where hi is an effective one-electron operator for the i-th electron which includes the mean-
field interaction with other electrons. The sum of all orbital energies (εi) differs from the
total energy of the system. A self-consistent field (SCF) method is employed to solve these
one-electron equations (1.10) iteratively.5,6
10

One must employ the antisymmetry requirement for the many electron wave function in
the next step to ensure the incorporation of Pauli exclusion principle. The antisymmetry
requirement is fulfilled if the product of one-electron functions is written in the form of a
Slater determinant.
Ψ(r1, r2, r3....)=|Φ 1(r1)Φ2(r2)Φ3(r3)....Φn(rn)| (1.11)
The use of complete set of orbitals gives rise to a complete set of determinants those span the
full space of the antisymmetric many-electron wave functions. Such an independent particle
model is known as the Hartree-Fock (HF) model.
Two rows of the determinant will be identical if two electrons possess the same coordinates
and hence the probability of such event is zero. So, in the Hartree-Fock model, electrons
must have different coordinates. The determinantal form of the wave function leads to a
certain correlation between their positions and movements (Fermi Correlation) for electrons
with the same spin. Moreover, the orbitals in the determinant must be linearly independent
and orthonormal. Instead of solving one 3n-dimensional equation, one has to solve n 3-
dimensional differential equations7 in the Hartree-Fock approximation. The one-electron
Fock operator (hi) has the following form:
hiΦi=Ti + VN + VC + VX ,
where
Ti =−12h2( ∂2
∂x2 + ∂2
∂y2+ ∂2
∂z2)Φi,
VN =−Σα( Zα
|r−rα|)Φi,
VC =Σj
∫dr2(
Φ∗j (r2)Φj(r2)Φi(r1)
r12),
VX =−Σj
∫dr2(
φ∗j (r2)φj(r1)Φi(r2)
r12). (1.12)
Ti is the kinetic energy, VN corresponds to all electron-nuclei attraction, VC is the Coulomb
interaction and VX is the exchange interaction. This last term VX is the outcome of the
antisymmetry requirement and has no classical analogue.
The integration of the spin-components must be considered in addition to the spatial
coordinates as each electron is associated with spin. The spin-orthogonality has a major role
in deciding the zero or nonzero value of the integrals. The Coulombic interaction between
11

electrons will always occur, but the exchange interaction has non-zero values only between
electrons of the same spin.
Although the Hartree-Fock equations are one electron equations, the Fock operator (hi)
itself is a function of all other orbitals in the system. Iterative methods are employed to solve
the equations. At first, one has to guess the trial orbitals which are used to compute the Fock
operator, and one-electron equations are then solved. A new set of orbitals is constructed
from the resulting orbitals and the iteration is continued. A convergence problem may be
encountered if the initial guess of trial orbitals is not good enough. Different numerical
techniques such as damping, scaling etc. are employed to achieve the convergence in those
situations. The SCF method has become a very important technique for modeling a variety
of many electron systems as it generates a number of symmetry adapted molecular orbitals
which are used as basis functions. At the lowest level, the closed-shell Hartree-Fock theory
gives good result for the ground state of molecules in the close vicinity at the equilibrium
configuration.
1.5. Basis sets
In the molecular orbital (MO) theory, the probability density for the electron in a molecule
is described by a set of MOs φi which are constructed from the set of atomic orbitals (AO)
of the constituent atoms in the molecule. The individual molecular orbital (φi) can be
expressed as linear combinations of a set of one-electron basis functions χj centered on
each atom
φi =∑nj=1 Cjiχj,
where Cji terms denote the expansion coefficients. To represent the MOs exactly, the basis
functions χj should form a complete set, hence an infinite number of basis functions is
required which is not possible in practice. So a finite number of basis functions is chosen
and their choice is important for the satisfactory representation of the molecular orbitals.
One may use Slater type orbitals (STO)8
χSTO =Yl,m(θ,Φ)e−αr (1.13)
as basis functions. However, these STOs are not often suitable for the numerical work.
12

Boys9 proposed another type of functions, namely, the Gaussian-type functions
glmn =N(x−xo)l(y−yo)m(z−zo)ne−α(r−ro)2 , (1.14)
where N is the normalization constant, l, m, n are positive integers and α is orbital exponent
which is also positive. The function glmn denotes s, p, d-type of Gaussian depending upon
the value of l+m+n=0, 1, 2, respectively.
The evaluation of various two-electron integrals in the Hamiltonian matrix elements is
the most difficult part in the MO calculation. Furthermore, the number of these integrals
increases rapidly with the number of basis functions. Wherever possible the symmetry of the
molecule may be used to reduce the number of integrals to a large extent. Instead of using a
single Gaussian function one can use a linear combination of a small number of Gaussians,
χj =∑i dijgi,
where gi s are Gaussians centered on the same atom and having the same l, m, n values as
one another with different α values. χj is called a contracted Gaussian function and gi s are
called primitive Gaussians, and dij terms are the suitable coefficients. The use of contracted
Gaussians instead of primitive Gaussians reduces the number of variational coefficients to
be determined. However, at the Hartree-Fock level, the number of MOs generated does not
pose much problem. But in the large scale post Hartree-Fock calculations such as MCSCF
and CI, the number of MOs can not be kept too large as it generates enormous number of
configurations for a given electronic state.
1.6. Relativistic effects
The velocity of light in classical mechanics is considered as infinite compared to that of
the object and the light does not interact with the object of measurement. Assuming the
velocity of light to be infinite, if the light is allowed to interact with the matter, one gets
the non-relativistic quantum mechanics through Heisenberg’s uncertainty principle . If both
the assumptions are relaxed, i.e., the velocity of light is finite relative to that of the object
and there exists an interaction with the object, one should include relativistic corrections.
In other words, if a particle moves at a velocity which is comparable with that of light, the
non-relativistic quantum mechanics is no longer accurate. Actually, the mass of the system
13

determines the extent of the relativistic correction necessary in the calculations. The energy
of the one-electron atom in the ground state with atomic number Z is -Z2/2 in atomic units.
The average velocity of the electron is of the order of Z which can be easily shown from virial
theorem. The velocity of light is about 137 in atomic unit which indicates that relativistic
effects can not be neglected for atoms of heavier masses.
Non-relativistic quantum mechanical methods are quite satisfactory for most of the molecu-
les having lighter atoms in the first and second row of the periodic table. But the situation
is not similar for molecules containing atoms of higher rows in the periodic table where the
relativistic effect comes into play. The inner electrons of heavy atoms attain faster speed
due to large nuclear charges, and the speed is comparable with that of light. As for exam-
ple the 1s electron of the Au atom acquires about 60% of the speed of light. As the core
electrons are subjected to larger nuclear charges, the relativistic effects10−14 are significantly
large for them. These core electrons in turn affect the valence space which is significant
for the chemical bonding. Therefore, the chemical bonding and spectroscopic properties of
these molecules are expected to change to a large extent due to the heavy nuclear masses.
Different types of relativistic corrections are made for heavy atoms and molecules. These
are mass-velocity correction, Darwin correction, spin-orbit correction, spin-spin interaction,
Breit interaction etc. The dominant part of the relativistic correction is the mass-velocity
correction which arises due to the variation in mass of the electron with its speed as it
compares the speed of light. The relativistic mass is written as
m= m0√1− v2
c2
.
The basic equation in the relativistic quantum mechanics is the Dirac equation
(−ih∂/∂t−V+cα · π+βmc2)Ψ=0, (1.15)
where π =−ih(∂/∂x, ∂/∂y, ∂/∂z),
α=
0 0 0 1
0 0 1 0
0 1 0 0
1 0 0 0
,
0 0 0 −i0 0 −i 0
0 −i 0 0
−i 0 0 0
,
0 0 1 0
0 0 0 −1
1 0 0 0
0 −1 0 0
,
14

β=
1 0 0 0
0 1 0 0
0 0 −1 0
0 0 0 −1
,
and Ψ=
Ψ1
Ψ2
Ψ3
Ψ4
.
For stationary states, time-independent one-electron Dirac equation takes the following form
(−V+cα · π+βmc2)Ψ=EΨ. (1.16)
Results of the solution of the Dirac equation for one-electron systems are in excellent agree-
ment with the experimental data but for many-electron systems, the applications of the
Dirac equation are not so simple. Many approximate schemes based on variation as well as
perturbation have been developed. In the next chapter of the thesis, we have reviewed the
method of computation for many electron atoms and molecules with the consideration of
relativistic effects.
1.7. Electron correlation energy and post Hartree-Focktreatments
Hartree-Fock wave functions are written as Slater determinants of one electron functions
under the independent particle model. The best possible determinant is chosen by using
variational methods. Therefore, it means that each electron experiences an effective mean
electrostatic field of all other electrons, but the motion and instantaneous positions of these
electrons are not explicitly correlated. The approximation is a crude one, though it works
well in some cases, especially for the ground state of the molecule. To obtain more accurate
results for studying the electronic structure and properties in the low-lying excited states
of the molecule, one must go beyond the Hartree-Fock approximation . Therefore, the
approximation by the mean effective field is not sufficient15,16 as the electron-correlation in
the post Hartree-Fock calculations becomes very important.
The Hartree-Fock model does not produce accurate results because of the inadequacy
15

of including correlations between the motions of electrons. The wave function written in
the single determinant form does not take into account of the electron correlation between
electrons of opposite spin. The correlation of the motions of electrons having the same spin
is partially, but not completely accounted for by virtue of the determinantal form of the
wave function. There are many qualitative deficiencies in the description of the electronic
structure of many electron system due to the omission of the correlation between electrons of
opposite spin. The closed-shell Hartree-Fock calculations do not describe the dissociation of
molecules correctly. The difference between the true non relativistic energy and the Hartree-
Fock energy is the measure of the correlation energy.
Eexact-EHF=Ecorrelation
Therefore, one has to achieve this amount of the correlation energy by some other means
for getting better results. The post Hartree-Fock methods are thus employed in quantum
chemistry so that the electron correlation17−19 which has been left out in the HF treatment
can be obtained. The methods like configuration interaction (CI), Many body perturbation
theory (MBPT), Coupled cluster (CC), density functional theory (DFT) are among the
post-Hartree Fock methods employed in quantum chemistry.
The CI method is one of the most useful methods which extend beyond the Hartree-Fock
model. In this method, the main concern is the choice of important configurations and
elimination of others at the optimum level so that the volume of the computations does not
increase very rapidly with the molecular size. Another requirement is the size consistency i.e.
the method must provide additive results when applied to an assembly of isolated molecules.
It is advantageous if the variational method can be applied as it ensures the upper boundness
of the total energy.
In order to incorporate the electron correlation within the variational principle, the wave
function is expressed as a linear combination of several Slater determinants, each of which
represents an individual electronic configuration. The variational method determines the
best possible combination. Multiconfiguration-SCF (MCSCF) method exploits this tech-
nique in which both expansion coefficients and orbitals forming the determinants are opti-
mized variationally while in the CI methodology, only CI coefficients are optimized. Variants
of MCSCF and CI methods are available and used to solve the actual problem depending
upon the computational capability and the desired accuracy. In recent years, many efficient
computational techniques are available in literature to tackle the problem in carrying out
16

MCSCF and CI calculations.
The lower energy molecular orbitals are generally occupied while the higher vacant ones
are virtual orbitals. One can generate antisymmetric many electron functions which have
different orbital occupancies. Each such many electron antisymmetrized function is a Slater
determinant or a linear combination of such determinants. As a result spin-adapted config-
uration state functions (CSF) can be formed.
The ground-state configuration is that distribution of electrons among the MOs which
possess the lowest energy. For even number of electrons, say, 2n, the single configuration
wave function is written as,
1Φ(0)G =A |φ1α(1)φ1β(2)........φnα(2n− 1)φnβ(2n)|; S=0, Ms=0.
For odd number of electrons (2n+1), the single configuration functions are,
2Φ(0)G =A |φ 1α(1)φ1β(2)........φnα(2n− 1)φnβ(2n)φpα(2n+ 1)|; S=1/2, Ms=1/2
and
2Φ(0)G =A |φ1α(1)φ1β(2)........φnα(2n− 1)φnβ(2n)φpβ(2n+ 1)|; S=1/2, Ms=-1/2.
The singly excited configurations are those distributions in which an electron has been pro-
moted from an occupied MO say, φk to a vacant MO φs. For 2n number of electrons
corresponding to singlet single-configuration wave function may be written as:
1Φ(1)k→s=
1√2[A |φ1α(1)φ1β(2).......φkα(2k − 1)φsβ(2k)........φnα(2n− 1)φnβ(2n)|
– A |φ1α(1)φ1β(2).......φkβ(2k − 1)φsα(2k)........φnα(2n− 1)φnβ(2n)|]; S=0, Ms=0.
Triplet state wave functions are,
3Φ(1)k→s=
1√2[A |φ1α(1)φ1β(2).......φkα(2k − 1)φsβ(2k)........φnα(2n− 1)φnβ(2n)|
+ A |φ1α(1)φ1β(2).......φkβ(2k − 1)φsα(2k)........φnα(2n− 1)φnβ(2n)|]; S=1, Ms=0
and
3Φ(1)k→s=A |φ1α(1).......φkα(2k − 1)φsα(2k)........φnβ(2n)|; S=1, Ms=1
3Φ(1)k→s=A |φ1α(1).......φkβ(2k − 1)φsβ(2k)........φnβ(2n)|; S=1, Ms=-1.
The doubly excited configurations are those distributions which are obtained by promoting
17

2 electrons from an occupied MO of 1Ψ(0)G to one vacant MO. The single-configuration wave
function is,
1Φ(2)k→s=A |φ1α(1)φ1β(2).......φsα(2k − 1)φsβ(2k)........φnα(2n− 1)φnβ(2n)|; S=0, Ms=0.
A linear combination of these CSF gives the CI wave function.
Ψ=∑iCiΦi (1.17)
The variation of coefficients Ci to minimize the energy functional leads to the determinantal
equation,
det(Hij-ESij)=0. (1.18)
It is important that only those CSF will contribute in the linear combination which have
the same angular momentum eigenvalues as that of the state Ψ or the CSF will have the
same symmetry properties (symmetry eigenvalue) as that of the state Ψ. The number of
configurations increases with the number of electrons and number of basis functions. For n
electrons and p number of basis functions, the number of CSF is roughly proportional to pn.
A CI calculation that includes all possible CSF with proper symmetry is a full CI calcula-
tion. Due to large number of CSF, full CI calculations are not possible to carry out except for
very small molecules with small basis set. There exist variants of CI calculations to choose
the proper configurations which will contribute largely to Ψ. It is, thus possible to perform
limited or truncated CI calculations.20 The simplest way of limiting the CI expansion is to
truncate the series in a given level of excitation. The truncated wave function can be written
as
Ψ=Φo+Σs>0CsΦs (1.19)
where Φ0 denotes the single determinant Hartree-Fock wave function while other determi-
nants are denoted by Φs. Ψ becomes Φ0, if no excitation is allowed, and one gets the HF
energy. The inclusion of all single excitations gives the CI wave function, ΨCIS as,
ΨCIS=C0Φ0 + Σocci Σvirt
a Cai Φa
i , (1.20)
where the excitation is indicated by i→a. If only single excitations are included it does not
improve the wave function or energy much. In the next step, the CI is limited with double
18

excitation only and the wave function may be written as,
ΨCID=C0Φ0 + ΣΣocci<j ΣΣvirt
a<b Cabij Φab
ij . (1.21)
If both single and double excitations are included in the next higher level of theory, it gives
the variational trial function as,
ΨCISD =C0Φ0+ Σocci Σvirt
a Cai Φa
i + ΣΣocci<j ΣΣvirt
a<b Cabij Φab
ij . (1.22)
These CI coefficients are optimized variationally. Multireference singles and doubles configu-
ration interaction (MRDCI) method, which includes the relativistic effects with and without
spin-orbit coupling, has been employed in the present work.
Size consistency is the modest requirement for a moderate system and all forms of the
truncated CI do not have this requirement. However, the full CI is size consistent, so also
for pair and coupled-pair theories. But the major disadvantage is that these pair theories
do not use variational principle. These are based on perturbative schemes, hence the total
energy obtained from these theories may be lower than the true energies. Sinanoglu and
Nesbet have introduced the independent electron-pair approximation (IEPA).15 These two
authors have used different terminology and formulations though the final results are same.
Sinanoglu termed his theory as Many-Electron Theory (MET) and Nesbet’s theory is called
Bethe-Goldstone Theory. Many body perturbation theory (MBPT) is also used for solving
infinite systems. For large systems, one needs a theory which is size consistent.
Molecular properties often may be expressed as derivatives of the total energy with respect
to parameters that correspond to perturbations of the system. The dipole moment of a
molecule is defined as the first derivative of the energy with respect to an external electric
field. The force constant for the molecular vibration is expressed as a second derivative
with respect to the displacement of nuclei in the molecule. Other electronic properties are
also defined in a similar way. It is, therefore, important to compute the potential energy
curves or surfaces of the ground and low-lying excited states which show the total energy as a
function of the coordinates of the nuclei. It is the most challenging task for both theoreticians
and experimentalists to construct such potential energy curves or surfaces. In the past few
decades, numerous working algorithms and computer codes13,14 have been developed for this
purpose.
19

1.8. References
1 R. Carbo, M. Klobukowski, Self-Consistent Field: Theory and Applications, Elsevier,
1990.
2 M. Born, J.R. Oppenheimer, Ann. Physik. 84, 457 (1927).
3 S.T. Epstein, The Variation Method in Quantum Chemistry, Academic Press, New York,
1974.
4 J.A. Pople, D.L. Beveridge, Approximate Molecular Orbital Theories, McGraw-Hill, New
York, 1970.
5 G.A. Segal, Ed. Semiempirical Methods of Electronic Structure Calculation, Part A and
B (vols. 7 and 8 of Modern Theoretical Chemistry, W. Miller et al. eds.), Plenum, New
York, 1977.
6 C.C.J. Roothaan, Rev. Mod. Phys. 23, 69 (1951).
7 B.A. Heβ, C.M. Marian, S.D. Peyerimhoff, Ab initio Calculation of spin-orbit Effects in
Molecules Including Electron Correlation.
8 J.C. Slater, Phys. Rev. 36, 57 (1930).
9 S.F. Boys, Proc. Roy. Soc. (London) A200, 542 (1950).
10 M. Krauss, W.J. Stevens, Annu. Rev. Phys. Chem. 35, 357 (1984).
11 P.A. Christiansen, W.C. Ermler, K.S. Pitzer, Annu. Rev. Phys. Chem. 36, 407 (1985).
12 K. Balasubramanian, K.S. Pitzer, Adv. Chem. Phys. 67, 287 (1987).
13 K. Balasubramanian, Relativistic Effects in Chemistry Part A. Theory and Techniques,
Wiley-Interscience, New York, p301, 1997.
14 K. Balasubramanian, Relativistic Effects in Chemistry Part B. Applications to Molecules
and Clusters, Wiley-Interscience, New York, p527, 1997.
15 A. Szabo, N.S. Ostland, Modern Quantum Chemistry, McGraw-Hill, New York, 1989.
16 S. Wilson, Electron Correlation in Molecules, Oxford University Press, New York, 1984.
17 H.F. Schaefer, Ed. Method of Electronic Structure Theory (vol.3 of Modern Theoretical
Chemistry, W. Miller et. al. eds.), Plenum, New York, 1977.
18 H.F. Schaefer, Ed. Applications of Electronic Structure Theory (vol.3 of Modern
20

Theoretical Chemistry, W. Miller et. al. eds.), Plenum, New York, 1977.
19 S.P. McGlynn, L.G. Vanquickenborne, M. Kinoshita, D.G. Carroll, Introduction to
Applied Quantum Chemistry, Holt, Rinehart and Winston Inc., New York, 1972.
20 W. Hehre, L. Radom, P.V.R. Schleyer, J.A. Pople, Ab initio Molecular Orbital Theory,
John Wiley and Sons, New York, 1986.
21

CHAPTER – 2
BRIEF REVIEW OF THE COMPUTATIONAL
METHODOLOGY: DETAILS OF THE
CONFIGURATION INTERACTION METHOD

2.1. Introduction
In the previous chapter we have mentioned that, if the velocity of light is assumed to be
infinite compared to that of a particle, one can use non-relativistic quantum mechanics to
describe its motion. The dynamics of the electrons in molecules consisting lighter atoms can
be treated accurately in non-relativistic quantum mechanics. But the treatment is somewhat
different for the molecules consisting of heavy atoms. The inner electrons in the heavy atoms
of the molecule attain a very high speed which cannot be neglected compared to that of light.
The non-relativistic quantum mechanics is no longer very accurate in this situation and
one should use the relativistic quantum mechanics.1−20 In general, non-relativistic quantum
mechanical methods based on ab initio techniques provide satisfactory results for most of the
molecules containing light elements in the first two rows of the periodic table. On the other
hand, relativistic effects become more important for molecules containing atoms of higher
rows in the periodic table. The difference is due to the fact that the inner electrons of very
heavy elements are subjected to large nuclear charges which increase their speed to such an
extent that is comparable with the speed of light.
Relativistic quantum mechanical treatment1,2 considers both finite velocity of light com-
pared to that of electrons and the interactions between them. The relativistic effect is
classified as mass-velocity correction, Darwin correction, spin-orbit interaction, spin-spin in-
teraction, Breit interaction etc.3 The mass-velocity correction on the kinetic energy of the
electron due to the variation of its mass with the velocity is the major part of the total rel-
ativistic correction. The inner s-orbitals, which are closest to the nucleus, experience higher
nuclear charge of the heavy atoms. Hence they contract because of the mass-velocity cor-
rection. This in turn, shrinks the outer s-orbitals due to orthogonality. The p-orbitals also
shrink due to the same reason, but to a lesser extent since the angular momentum allows
the electrons to keep away from the nucleus. If the coupling between the spin and orbital
angular momentum of the electron is strong, the spin-orbit correction is to be considered.
The correction becomes large for electronic states of molecules containing heavy atoms with
open-shell configurations. The electronic structure and spectroscopic properties may change
significantly because of the strong spin-orbit coupling. The two-electron counter part of the
spin-orbit interaction is known as Breit interaction. The Darwin correction is a characteristic
outcome of the Dirac’s relativistic equation, and as such it does not have any simple physical
significance. The spin-orbit interaction in the ground state of the gold atom is small but that
22

of lead is large because of open-shell configurations. The spin-orbit coupling not only splits
the electronic states into sub-states but also allows them to mix with electronic states, which
otherwise do not mix in the absence of the spin-orbit coupling. For example, the ground
state of the Pb atom in the absence of spin-orbit interaction is 3P, while its excited states
arising from the same electronic configuration are of 1D and 1S symmetries. The spin-orbit
coupling splits the 3P state into 3P0, 3P1, 3P2 components. The magnitude of this spin-orbit
splitting for Pb is as large as 1000 cm−1. Furthermore, 3P0 mixes with 1S0, similarly 3P2 and1D2 components mix together. The spin-orbit contamination is very large for heavy atoms
such as Pb, Pt, Au, etc. As a result, the spin-orbit interaction may change spectroscopic
properties of molecules containing heavy elements to a large extent. Sometimes a number of
important changes may take place in the potential energy surfaces (curves) e.g., appearance
of shoulders, barriers, double minima etc., due to some avoided curve-crossings. The higher
order interactions in the relativistic effect are generally ignored for the electronic and spec-
troscopic properties in the valence region. However, in case of fine structure calculations,
these smaller interactions become important and contribute significantly.
The relativistic effects can alter the nature of the chemical bonding in molecules containing
heavy atoms to a large extent. Some bonds may be weakened and some may be strengthened
depending upon the particular situation. The dissociation energies of heavy molecules are
found to change due to relativistic corrections. For instance, the dissociation energy of Au2 is
larger than that of Ag2 in contrary to the usual trend. This anomaly is due to the relativistic
contraction and stabilization of the 6s orbital of the gold atom. The well known lanthanide
contraction (i.e. the decrease of radii from La to Lu) is attributed to incomplete shielding of
the 4f shell. This effect is partly due to relativistic effects. Comparing the non-relativistic
and relativistic corrections, it has been found that a contribution4 of about 27% comes from
the relativistic effects in the form of lanthanide contraction.
All-electron molecular Dirac-Fock (DF) calculations are not easy to carry out as they in-
volve a large number of electrons. Moreover, additional integrals are generated due to each
molecular spinor having both large and small components. An enormous volume of compu-
tation is involved at the CI level because of the configurations generated from excitation of
valence electrons from shells with different angular momentum. An additional configuration
mixing takes place because of the relativistic interactions. The basis sets for the atoms in the
molecule must be sufficiently large so that the result of the CI calculation would be accurate
and acceptable for explaining the observed data. On the other hand, all-electron calcula-
23

tions become increasingly difficult with the increase in the size of the basis sets. Another
problem with all-electron DF calculations is the nature of the one-electron four-component
spinors. In general, these components are complex quantities. The behavior of these spinors
near the nucleus is difficult to describe by using the conventional basis functions. It has
been found5 that the large component of a molecular spinor having s or p1/2 population be-
haves like 1/rξ near a point nucleus, while the small component behaves like Z/r1+ξ, where
ξ=Z2α2/2. Moreover, the energy expectation values obtained from all-electron relativistic
calculations do not have the property of being upper bounds to the total energy. Hence,
there can be a variational collapse in attempting to use the variational principle. Ab initio
based all-electron calculations have been carried out by using the Dirac-Fock formalism for
heavy elements by Desclaus6 in 1973. An extensive configuration interaction is required for
this purpose. The relativistic effects on the orbital energies, and thus an excitation energies,
ionization potentials, and electron affinities have a direct influence on the chemically relevant
data. The relativistic effect may also change the bonding properties of the molecule as well.
2.2. Relativistic corrections
2.2.1 The Dirac equation
The relativistic quantum mechanical methods are based on Dirac equation which is an
analogue of Schrodinger equation. The Dirac Hamiltonian for a many-electron system can
be written as
HD=ΣihD(i) + Σi<j1rij
, (2.1)
where hD(i) is the one-electron Dirac Hamiltonian in the following form
hD(i)=αi· pi + βic2 − Z
ri. (2.2)
The wave function in the Dirac equation is a multi-component quantity, two components
describing the spin degrees of freedom of the electron, and other two components describing
the spin degrees of freedom for a charge-conjugated particle, loosely speaking, a positron.
In general, the Dirac equation has states of positive energy and infinite states of negative
energies, which are interpreted by Dirac as filled by an infinite number of electrons in the
ground state. Therefore, there exists four coupled first order differential equations for the
24

four components of the wave function.
The one-particle Dirac Hamiltonian involves 4 × 4 matrices instead of scalar functions
and differential operators. The solution is, therefore, a vector of four components, which are
called spinors.
Ψnkm = 1r
(Pnk(r) χkm(θ, φ)
iQnk(r) χ−km(θ, φ)
), (2.3)
where
χkm(θ, φ) = Σσ=± 12C(l
12 j; m− σ, σ)Y m−σ
λ (θ, φ)Φσ12
,
Y m−σλ is a spherical harmonics, Φ
1212
= α =
(1
0
)and Φ
− 12
12
= β =
(0
1
)are Pauli spinors,
and C(l12 j; m−σ, σ) are Clebsch-Gordan coefficients, k is the relativistic quantum number
k = (j + 12) for j = (l − 1
2)
−(j + 12) for j = (l + 1
2),
and λ is defined as
λ = k for j = (l − 12)
−(k + 1) for j = (l + 12).
The Pnk and Qnk are the large and small components, respectively. Thus for the central
force field V(r), the coupled differential equation of Dirac can be represented as
dPnkdr
+ kPnkr−(
2α
+α[V (r)−εnk])Qnk=0 (2.4)
dQnkdr−kQnk
r+α[V (r)−εnk]Pnk=0. (2.5)
In the non-relativistic limit (c →∞), the above coupled equation becomes the Schrodinger
equation if the small components Qnk are neglected. The small components are responsible
for the relativistic effects and make a significant contribution in the core region, while the
effect of these components in the valence region can be ignored.11−20
2.2.2 Effective core potential
A reliable pseudo potential method, known as relativistic effective potential method
25

(ECP) is used to perform relativistic quantum mechanical calculation. The basic assumption
used in the ECP method is the frozen core approximation which is nothing but the core-
valence separability. In the ECP method, the interaction of the valence electrons with the
core electrons are represented by effective potentials or pseudo potentials thereby reducing
the number of electrons in the calculation. These effective potentials may be relativistic or
non-relativistic depending on the nature of the wave function from which they are generated.
The calculations may be carried out semiempirically or by the ab initio methodologies.
In general, effective potentials replace the valence orbitals with pseudo-orbitals which
are smooth and nodeless in the core region but approximately resemble the true valence
orbitals at large radii. Therefore, one freezes not only the core orbitals but also that fraction
of valence electron density responsible for the inner oscillatory behavior. Once a nodeless
orbital has been generated, the one-electron atomic Fock equation is inverted to produce a
local operator which represents the core-valence interaction. Based on Phillips and Kleinman
transformation21, many studies have been made to construct effective potentials.
In the ECP method, the explicit core-valence orthogonality constraints are replaced by a
modified valence Hamiltonian. If the potential generated by core electrons is written as Vc,
the one-electron valence wave equation takes the following form.
(h+Vc)φv = Evφv (2.6)
Phillips and Kleinman21 suggested that φv can be written as
φv = χv−Σc〈χv|φc〉φc (2.7)
so that φv is orthogonal to φc.
Substituting φv into the one-electron eigenfunction,
(h+Vc+VEP )χv = Evχv, (2.8)
where VEP = Σc(E−Ec)|φc〉〈φc| is referred to as the Phillips-Kleinman pseudo-potential, χv
is known as the pseudo-orbital, and |φc〉〈φc| is the projection operator of the core orbitals.
When VEP is obtained from the non-relativistic atomic wave function, it will become non-
relativistic model potential. The details of these aspects can be found in the review of Krauss
and Stevens.13
Ab initio based relativistic effective core potentials (RECP) have been derived from the
26

numerical DF calculations of the atoms.22 As already mentioned, the solution of the DF
equation is a set of four component spinors. After partitioning the spinors as core and
valence, the overall many-electron relativistic wave function for a single configuration can be
written as
Ψ = A[(ψc1 ψc2 · · · ψcm)(ψv1 ψ
v2 · · · ψvn)], (2.9)
where A is the antisymmetrizer, ψc1 · · · ψcm and ψv1 · · · ψvn are core and valence orbitals,
respectively with m and n being the number of core and valence electrons. The total energy
is partitioned into core, valence, and core-valence interaction energies.
ET = Ec+Ev+Ecv (2.10)
One can show that
Ev + Ecv = 〈ψRv |Hrelv |ψRv 〉
where,
Hrelv = ΣihD(i) + Σc(Jc(i)−Kc(i)) + Σi<j
1rij
,
i and j indices refer to valence electrons. The core and valence orbital sets are assumed to
be orthogonal. The DF equation for a single electron is then given by
[hD+Σc(Jc−Kc)]ψv = εvψv+Σcψcεcv, (2.11)
where εcvs are the off-diagonal Lagrange multipliers
εcv = 〈ψv|hD + Σc(Jc −Kc)|ψc〉.
Defining the core-projection operator and the pseudo-orbital in the same way as done in
the Phillips-Kleinman method for relativistic spinor wave functions, one obtains relativistic
pseudo-orbitals and RECP.
χRv = ψRv + ΣcacψRc ,
ψRv = (1− P )χRv
P = Σc|ψc〉〈ψc|
27

V PK = −PHrelv P+PHrel
v P+εvP , (2.12)
where,
(Hrelv + V PK)χRv = εvχ
Rv
(hD + U core)χRv = εvχv
U core = Σc(Jc−Kc)+VPK , (2.13)
which is the relativistic effective potential of a 4 × 4 matrix that operates on the nodeless
four-component spinor χRv . The small components in the valence region are neglected. One
can use the non-relativistic kinetic energy operator along with relativistic large components
in an equation from which valence-level relativistic core potentials are generated.
Now, one-electron radial equation becomes
(−12∇2 − Z
r+ UEP )χ′v = εvχ
′v,
where χ′v is a two-component pseudo-function having a large radial components. For many
electron system, the equation becomes
(−12∇2−Z
r+UEP+Wvv′)χ
′v = εvχ
′v, (2.14)
where Wvv′ represents Coulomb and exchange potential involving pseudo-spinor χ′v and all
other pseudo-spinors. The RECP can be expressed by introducing the lj-dependent radial
potential URECPlj as
URECP = Σ∞l=0 Σ|l+ 1
2|
j=|l− 12| Σ
jm=−j U
RECPlj (r)|ljm〉〈ljm|. (2.15)
The projection operator |ljm〉〈ljm| is comprised of Pauli two-component spinors. The sum-
mation over l to ∞ is impractical to carry out, it requires potentials of all excited states
of the atom. A good approximation is to stop the summation at the maximum l (=L) and
maximum j (=J) values. After modification, the equation can be written in the following
form
URECP = URECPLJ (r)+ΣL−1
l=0 Σ|l+ 1
2|
j=|l− 12| Σ
jm=−j(U
RECPlj −URECP
LJ )|ljm〉〈ljm|. (2.16)
Relativistic calculations (single configuration SCF) on several diatomic molecules like
Au2, PbS, PbSe+ etc. have been carried out by Pitzer and his group.22−26 However, it has
28

now become possible to perform calculations at the multiconfiguration SCF (MCSCF) and
configuration interaction (CI) levels using the RECP of the constituent atoms.
The potentials generated in the above method are numerical potentials. However, Gaus-
sian analytic fit of these potentials is more desirable and useful. The Gaussian expansion of
these numerical potentials are proposed by Kahn, Baybutt, and Trular27 as
URECPLJ (r)− URECP
lj (r) = r−2ΣNi=0Cir
niexp(−αir2),
where Ci, ni, and αi are chosen such that best fitted results are obtained.
The RECP can be averaged with respect to spin. The averaged RECP (ARECP) takes
the following form.
UARECP (r) = UARECPL (r) + ΣL
l=0 Σlm=−l[U
ARECPl (r)− UARECP
L (r)]|lm〉〈lm|,
(2.17)
where
UARECPL = 1
(2l+1)[lURECP
l,l− 12
(r) + (l + 1)URECPl,l+ 1
2
(r)].
The advantages with ARECP are as follows:
a) These potentials can be used in standard molecular calculations which are based on
the Λ–S coupling.
b) These ARECP potentials may be interpreted as containing relativistic effects present
in the Dirac Hamiltonian except the spin-orbit coupling.
c) These potentials resemble non-relativistic effective potentials, and can be introduced
into the CI calculations.
2.2.3 Spin-orbit coupling
The spin-orbit operator has been defined by Hafner and Schwarz5 as the difference of
(l + 12) and (l − 1
2) relativistic effective potentials.
HSO = ΣL−1l=1 ∆URECP
l (r)[
l(2l+1)
Σl+ 1
2
m=−l− 12
l, l + 12,m〉〈l, l + 1
2,m
− l+12l+1
Σl− 1
2
m=−l− 12
l, l−12,m〉〈l, l−1
2,m
](2.18)
29

where, ∆URECPl (r) = URECP
l,l+ 12
(r) − URECPl,l− 1
2
(r). These spin-orbit operators and ARECP are
used in molecular calculations.
Hay and co-workers28−34 have published Gaussian fits of RECP without spin-orbit cou-
pling for all elements in the periodic table. Gaussian analytic fits of ARECP and spin-orbit
operators for Li to Ar have been derived by Pacios and Christiansen.35 Similarly, for other
elements these numbers are computed by other authors.36−38 MCSCF and CI calculations of
heavy molecules are now easy to carry out because of the availability of these potentials in
literature.
A large number of post Hartree-Fock calculations (like CAS-MCSCF, variants of CI)
on homonuclear and heteronuclear heavy diatomic molecules have been made by several
authors.17,18,39 These calculations demonstrate the requirement to consider the relativistic
effects including the spin-orbit coupling. From the results of SCF, MCSCF, and CI calcu-
lations, it has been found that the relativistic bond contractions for heavy molecules are
substantial. For molecules like AuH and AgH, the bond lengths contract by about 0.25 A
and 0.08 A, respectively.40−42 Christiansen and Pitzer43 have performed MCSCF calculations
on T l2 and T l+2 in the Ω–Ω coupling scheme.
2.3. Computational methodology
In the present thesis, a series of CI calculations on SiC, SnC, PbC, and some of their
ions have been performed. For such species, RECPs are required to be included in the
calculations. However, we have used either semi-core or full-core RECPs depending upon
the nature of the problem. Optimized Gaussian atomic orbital basis sets compatible with the
RECPs are employed for this purpose. These RECPs and basis sets are available in literature.
In some cases additional polarization and diffuse functions are included in the basis set.
The present CI calculations are based on multireference singles and doubles configuration
interaction (MRDCI).
In the first step, we perform a series of self-consistent-field (SCF) calculations for a partic-
ular molecular symmetry at each internuclear bond distance from 2-3 a0 upto the dissociation
limit, say ∼15 a0. The choice of the molecular state for this purpose would be such that, SCF
calculations converge at each bond length, and it would generate reasonably good optimized
SCF-MOs which can be used as one electron basis functions for the subsequent MRDCI
30

calculations. As it is not possible to work with the actual symmetry C∞v of the heteronu-
clear diatomic molecules, calculations are performed in the C2v subgroup. At first, the CI
calculations are done without considering spin-orbit coupling. The spin-orbit coupling is
introduced in the second step. A brief description of the MRDCI method is given in the
following section.
2.3.1 Configuration selection technique
It is well known that the dimension of a full-CI calculation increases enormously with the
number of the basis functions. For many electron problem it is impossible to carry out full CI
calculations. So, a simple straightforward approach to the attainment of multiconfiguration
wave functions is not practicable. An alternate approach to the CI treatment lies in the
proper selection of important configurations which are to be considered explicitly in a given
secular equation. It is possible to successfully implement this technique without losing much
accuracy.
The objective of the configuration-selection technique is to select those configurations
which contribute significantly to the total electronic wave function. The relatively less im-
portant configurations are also identified. Another important aspect of this method is to
predict accurately the amount of error occurred in omitting the configurations which are
not selected in the process. The contribution in energy for the unselected configurations
can be estimated. A set of dominant or main configurations (referred to as reference con-
figurations), φn must be chosen, and it forms the basis for the selection of the remaining
configurations. We must generate all configurations from the reference set by single and
double excitations. The energy lowering because of the inclusion of a test configuration, say
φt in the reference set φm, is obtained from the solution of the secular equation which is
larger than the previous by one.
Alternatively, one can estimate this energy lowering perturbatively using the expression44
∆E = Em+H2mt
(Em−Et) . (2.19)
The equation (2.19) has been used for the first time by Whitten and Hackmeyer45 as a
configuration selection method in non-relativistic CI calculations. Many selection techniques
based on the above-mentioned equation have been developed and used successfully by Bender
and Davidson,46 and extensively by Buenker and Peyerimhoff.47−54 The perturbative method
31

has the advantage of involving less computation, while the variational method can check the
energy-lowering simultaneously for all roots of that symmetry. We have used variational
methods of Buenker and co-workers for our computational purpose. It is also necessary to
obtain an accurate prediction of the total energy contribution of those configurations which
are not chosen for the final CI calculations.
Therefore, each test configuration φt is tested against every member of the main reference
set φm. The configuration φt, for which ∆E exceeds some cut-off or threshold value (T),
is selected for the final CI calculations. The choice of main configurations and the estimation
of the effect of adding more configurations to the initial set are to be made quite accurately.
It is important to make sure that the set of main configurations is sufficiently large to allow
for a realistic representation of all states. The desired number of roots is obtained from each
of the secular equations involving φm and the test configuration. The energy-lowerings are
computed separately for each of the roots. The maximal energy-lowerings for all the roots
are compared with some threshold value for the purpose of determining whether the test
species should be included in the final CI. The choice of the magnitude of the threshold is
also important in this regard. A smaller threshold will be required to solve larger number of
configurations in the final CI. Generally, threshold is kept in the order of microhartree.
2.3.2 Role of unselected configurations
Once the selection criterion is fixed, one must be able to accurately compute the effect
of those configurations which are not included in the final CI calculations. It is, therefore,
required to estimate the threshold as a parameter in the final results by means of some
reliable method. Davidson’s55,56 suggestion is to compare the sum of the lowering of the
neglected test species with the total energy change effected by the CI in order to get at least
a semiquantitative estimate of the consequence of employing a nonzero threshold at different
nuclear geometries.
The method for extrapolating the CI energies to zero-threshold can be formulated as
follows. Let the total configuration-set in the CI space at zero-threshold is partitioned into
three subsets φm, φs, and φr which are main, selected, and rejected configurations,
respectively. The zero order wave function ψ and that obtained in the final truncated CI
for a given threshold can be defined as
ψ = ΣmCmφm, (2.20)
32

ψ(T ) = ΣmCm(T )φm+ΣsCs(T )φs. (2.21)
The wave function ψ ≡ ψ(0) for T=0 is obtained as
ψ(T=0) = ΣmCm(0)φm+ΣsCs(0)φs+ΣrCr(0)φr. (2.22)
The φr configurations are weakly interacting when T is quite small, and φm is sufficiently
representative. With some approximations, the energy-lowering ∆Er by adding φr to the
original set of main configurations φm can be expressed as
∆Er = 〈ψrHψr〉 -〈ψHψ〉
≈ 2ReCr(0)〈ψHφr〉+Cr(0)2〈φrHφr〉−〈ψHψ〉 (2.23)
with ψr ≈ ψ + Cr(0)ψr.
Estimating Cr(0) from the first order perturbation theory, the equation simplifies as
∆Er ≈ Cr(0)〈ψHφr〉.
Total energy E ≡ E(0) can be obtained as
E = 〈ψHψ〉 ≈ E(T )+ΣrCr(0)〈ψ(T )Hφr〉 (2.24)
Therefore, employing a small value of T and a sufficiently representative set of main config-
urations φm, it is possible to estimate the CI energy at T=0.
As we are using variational principle in determining the energy roots and wave functions,
it is assured that the CI energy (E) decreases as T lowers. But E(T → 0) does not remain
constant as T changes. A general expression of E (T → 0) can be written with a scaling
factor λ
Eλ(T → 0) = E(T )+λΣr∆Er(T ), (2.25)
where λ is an arbitrary constant which is chosen such that Eλ(T → 0) is the most slowly
varying function of T. The λ factor takes care of the internal coupling and relaxation pro-
cesses in the CI performed with T=0. The secular equations are then solved for a series of
T. The [E(r) +λΣr∆Er(T )] curves are constructed for a given root at various values of λ as
a function of T. The optimum λ value for each root has been estimated.
33

The extrapolated energy is expected to be same, provided an appropriate set of reference
configurations is chosen. It is well known that in the full-CI treatment, the nature of the
molecular orbital basis set does not have much importance. Once the CI energy extrapolated
to zero threshold is obtained, one can use Davidson’s correction to estimate the contribution
due to higher order excitations. The estimated full-CI energy may be expressed as
E(full-CI) = E(T=0)+(1−ΣmC2m)(E(m)−E(T = 0)), (2.26)
provided the contribution of the reference species in the total CI expansion is above 90% i.e,
ΣC2m is more than 0.9. This has been extended later for a multireference case by Peyerimhoff
and Buenker.57 The correction has been shown to be good for the lack of size retentivity
of the energy with single and double excitations.58,59 The Table Direct-CI version of the
MRDCI codes of the Kerbs and Buenker60 are used in some cases. All properties other
than energy may be evaluated from the wave functions corresponding to the MRDCI space.
Results are found to be quite satisfactory for one-electron properties like dipole moments,
quadrupole moments, transition moments between various electronic states.
2.3.3 Spin-orbit interaction
Once the CI energies and wave functions of different Λ–S states are obtained, the next
step is to include the spin-orbit effects at the CI level of treatment. MRDCI wave functions
are employed as basis for the representation of the relativistic Hamiltonian including the
spin-orbit coupling. The spin-orbit interaction may be introduced in different ways.
In one way, the selected spatial configurations of different types for a given spatial sym-
metry are multiplied with spin functions which transform according to irreducible repre-
sentations of the C2v double group. In the actual computations, heteronuclear diatomic
molecules are always considered to be in the C2v subgroup. The correlation with the ac-
tual symmetry (C∞v) group is done afterward. The C2v double group for even number of
electrons consists of four irreducible representations namely, A1, A2, B1, and B2 of which
B1 and B2 are degenerate. For odd number of electrons, the irreducible representations in
C2v double group are either E1 or E2 which are degenerate. The resulting products of the
spatial configurations and spin functions, are grouped according to the symmetry. The spin-
orbit matrix elements between different configuration state functions (CSFs) are calculated.
These results are combined with the previous results for the spin-independent operator to
34

form a Hamiltonian matrix representation for each of the irreducible representations in the
C2v double group. The resulting secular equations are four to six times larger than the origi-
nal Λ–S CI treatments. However, the diagonalization can be done in relatively few iterations
by using Davidson’s algorithm61 because of the availability of very good starting vectors
from the spin-independent calculations. The resulting energies are used directly without
adding perturbation correction analogous to those employed for the Λ–S CI treatment. For
small number of active electrons, this procedure is found to be accurate. However, for large
number of electrons one can add the spin-orbit interaction in a different way.
In the alternative way, the appropriately estimated full-CI energies from the Λ–S CI treat-
ment are placed in the diagonals of the CI matrix. The off-diagonal matrix elements are
obtained by employing pairs of selected CI wave functions with Ms=S and applying spin-
projection techniques and the Wigner-Eckart theorem.62−65 It is now required to diagonalize
small CI matrices, called super-CI corresponding to A1, A2, and B1 (or B2), or E1 (or E2)
representations of the C2v and C′2v double group depending upon the number of electrons.
The dimensions of the secular equations in the super-CI treatment depend on the number
of roots of the Λ–S states involved in the spin-orbit interaction. Such a two-step method
is mostly used in the calculations carried out in the present thesis. This two-step method
for the inclusion of the spin-orbit interaction has become useful, especially, to analyze the
Ω-components in terms of different Λ–S eigenfunctions. However, the disadvantage of this
simpler two-step method of introducing the spin-orbit coupling is that the spin-orbit correc-
tion is not treated at the same level as the non-relativistic terms.
The spin wave functions corresponding to odd number of electrons belong to E represen-
tation of C′2v double point group. The character table for C′2v double point group66 is given
below.
C′2v E C2(z) σxz σyz R Operator Spin function
A1 1 1 1 1 1 z αβ - βα
A2 1 1 -1 -1 1 lz, sz αβ+βα
B1 1 -1 1 -1 1 x, ly, sy αα+ββ
B2 1 -1 -1 1 1 y, lx, sx αα - ββ
E 2 0 0 0 -2 α, β
The symmetry adapted spin wave functions66 for even number of electrons with fixed S and
35

MS values and those transforming according to irreducible representations of C2v point group
may be tabulated as follows.
S MS | S, MS〉 | S, |MS|, R〉0 0 αβ A1(0)=|0〉1 1 αα B1(1)= 1√
2(|+1〉+|-1〉)
-1 ββ B2(1)= 1√2(|+1〉 - |-1〉)
0 1√2P [αβ] A2(0)=|0〉
2 2 αααα A1(2)= 1√2(|+2〉+|-2〉)
-2 ββββ A2(2)= 1√2(|+2〉 - |-2〉)
1 1√4P [αααβ] B1(1)= 1√
2(|+1〉 - |-1〉)
-1 1√4P [βββα] B2(1)= 1√
2(|+1〉+|-1〉)
0 1√6P [ααββ] A1(0)=|0〉
3 3 αααααα B1(3)= 1√2(|+3〉+|-3〉)
-3 ββββββ B2(3)= 1√2(|+3〉 - |-3〉)
2 1√6P [αααααβ] A1(2)= 1√
2(|+2〉 - |-2〉)
-2 1√6P [βββββα] A2(2)= 1√
2(|+2〉+|-2〉)
1 1√15P [ααααββ] B1(1)= 1√
2(|+1〉+|-1〉)
-1 1√15P [ββββαα] B2(1)= 1√
2(|+1〉 - |-1〉)
0 1√20P [αααβββ] A2(0)=|0〉
Where P [α...β] denotes a sum of all possible permutations of α and β spins. In the fourth
column a short | MS〉 notation is used for the | S, MS〉 spin-functions from column 3.
2.3.4 Calculation of spectroscopic constants
CI energies of various Λ–S electronic states of the molecule are estimated from the short
range of internuclear distance, 2.5-3.0 a0 to the long range, say 15.0 or 20.0 a0 depending
upon the system. Around the equilibrium bond length, small grids (typically 0.05 to 0.1 a0)
are chosen, while in the dissociation limit of the molecule, the larger grids are used for
calculations. Spin-orbit corrections are carried out at each of these bond distances. MRDCI
calculations are capable of giving a reliable description of the entire potential energy curve
all the way from a short bond distance to the dissociation limit. The computed bound
36

state potential energy curves are fitted into polynomials for substitution in Schrodinger
equation for nuclear motion. One dimensional nuclear Schrodinger equations are then solved
numerically by using Neumerov-Cooley method67,68 to obtain the desired vibrational energies
and wave functions.
2.3.5 Estimation of radiative lifetime
The electric dipole transition moments between two states involved in the transition, are
calculated as a function of internuclear separation of the diatomic species. Knowing these
transition moments, the Einstein’s spontaneous emission coefficients Av′v′′ (sec−1) between
different vibrational levels (designated as v′) of the upper electronic states and those (desig-
nated as v′′) of the lower electronic state are obtained from the following standard formula
Av′v′′ = ge′e′′ 2.1419× 1010(∆E)3Sv′v′′ ,
where ∆E (transition energy) is in a.u., and
Sv′v′′ = 〈χv′′(r)Be′e′′(r)χv′(r)〉 2
is obtained by using a polynomial fit to the discrete data for the electronic transition moment
Be′e′′ and the vibrational wave functions χv(r) generated for the respective pairs of electronic
states. The factor ge′e′′ is derived from the fact that the transition moment used in the
calculation is only for one component of a given degenerate system. The radiative lifetime
(τv′) of the vibrational level (v′) of the upper electronic state is obtained as
τv′ =(Σv′′Av′v′′
)−1, (2.27)
where the sum runs over all vibrational levels which can be reached among the lower-lying
electronic states. The oscillator strength (f) for a given absorption process may also be
obtained from the following expression
fv′v′′ = 23g′e′e′′∆ESv′v′′ ,
where g′e′e′′ is the degeneracy factor.
37

2.4. References
1 K. Balasubramanian, Relativistic Effects in Chemistry Part A. Theory & Techniques,
Wiley-Interscience, New York, p 301, 1997.
2 K. Balasubramanian, Relativistic Effects in Chemistry Part B. Applications to Molecules
& Clusters, Wiley-Interscience, New York, p 527, 1997.
3 H.A. Bethe, E.E. Salpeter, Quantum Theory of One and Two Electron Atoms, Plenum,
New York, 1977.
4 P.S. Bagus, Y. Lee, K.S. Pitzer, Chem. Phys. Lett. 33, 408 (1975).
5 P. Hafner, W.H.E. Schwarz, Chem. Phys. Lett. 65, 537 (1979).
6 J.P. Desclaux, At. Data Nucl. Data 12, 311 (1973).
7 P. Pyykko, Adv. Quantum Chem. 11, 353 (1978).
8 K.S. Pitzer, Acc. Chem. Res. 12, 271 (1979).
9 P. Pyykko, J.P. Desclaux, Acc. Chem. Res. 12, 276 (1979).
10 P.A. Christiansen, W.C. Ermler, K.S. Pitzer, Annu. Rev. Phys. Chem. 36, 407 (1985).
11 K. Balasubramanian, K.S. Pitzer, Adv. Chem. Phys. 67, 287 (1987).
12 P. Pyykko, Relativistic Theory of Atoms and Molecules, Springer-Verlag: Berlin, New
Work, 1986.
13 M. Krauss, W.J. Stevens, Annu. Rev. Phys. Chem. 35, 357 (1984).
14 G.L. Malli, Ed.; Relativistic Effects in Atoms, Molecules, and Solids, Plenum, New York,
1982.
15 P. Pyykko, Ed.; Proceedings of the symposium on relativistic effects in quantum chemistry,
Int. J. Quantum Chem. 25, (1984).
16 K. Balasubramanian, J. Phys. Chem. 939, 6585 (1989).
17 K. Balasubramanian, Chem. Rev. 89, 1801 (1989).
18 K. Balasubramanian, Chem. Rev. 90, 93 (1990).
19 P. Pyykko, Chem. Rev. 88, 563 (1988).
20 W.C. Ermler, R.B. Ross, P.A. Christiansen, Adv. Quantum Chem. 19, 139 (1988).
21 J.C. Phillips, L. Kleinman, Phys. Rev. 116, 287 (1959).
38

22 Y.S. Lee, W.C. Ermler, K.S. Pitzer,J. Chem. Phys. 15, 5861 (1977).
23 W.C. Ermler, Y.S. Lee, K.S. Pitzer, N.W. Winter, J. Chem. Phys. 69, 976 (1978).
24 Y.S. Lee, W.C. Ermler, K.S. Pitzer, A.D. McLean, J. Chem. Phys. 70, 288 (1979).
25 W.C. Ermler, Y.S. Lee, K.S. Pitzer, J. Chem. Phys. 70, 293 (1979).
26 Y.S. Lee, W.C. Ermler, K.S. Pitzer, J. Chem. Phys. 73, 360 (1980).
27 L. Kahn, P. Baybutt, D.G. Truhlar, J. Chem. Phys. 65, 3826 (1976).
28 J.S. Cohen, W.R. Wadt, P.J. Hay, J. Chem. Phys. 71, 2955 (1979).
29 P.J. Hay, W.P. Wadt, L.R. Kahn, R.C. Raffenetti, D.H. Phillips, J. Chem. Phys 71, 1767
(1979).
30 P.J. Hay, W.R. Wadt, J. Chem. Phys. 82, 270 (1985).
31 W.R. Wadt, P.J. Hay, J. Chem. Phys. 82, 284 (1985).
32 P.J. Hay, W.R. Wadt, J. Chem. Phys. 82, 299 (1985).
33 P.J. Hay, W.R. Wadt, L. Kahn, J. Chem. Phys. 68, 3059 (1978).
34 L.R. Kahn, P.J. Hay, R.D. Cowan, J. Chem. Phys. 68, 2836 (1979).
35 L.F. Pacios, P.A. Christiansen, J. Chem. Phys. 82, 2664 (1985).
36 M.M. Hurley, L.F. Pacios, P.A. Christiansen, R.B. Ross, W.C. Ermler, J. Chem. Phys.
84, 6840 (1986).
37 L.A. LaJohn, P.A. Christiansen, R.B. Ross, T. Atashroo, W.C. Ermler, J. Chem. Phys.
87, 2812 (1987).
38 R.B. Ross, J.M. Powers, T. Atashroo, W.C. Ermler, L.A. LaJohn, P.A. Christiansen,
J. Chem. Phys. 93, 6654 (1990).
39 T. Ziegler, J.G. Snjders, E.J. Baerends, J. Chem. Phys. 74, 1271 (1981).
40 Y.S. Lee, A.D. McLean, J. Chem. Phys. 76, 735 (1982).
41 A.D. McLean, Y.S. Lee, Stud. Phys. Theor. Chem. 21, 219 (1982).
42 R.B. Ross, W.C. Ermler, J. Phys. Chem. 89, 5202 (1985).
43 P.A. Christiansen, K.S. Pitzer, J. Chem. Phys. 74, 1162 (1981).
44 J.C. Slater, Quantum Theory of Atomic Structure, Vol.1, McGraw-Hill, New York, 1960.
45 J.L. Whitten, M. Hackmeyer, J. Chem. Phys. 51, 5584 (1969).
46 C.E. Bender, E.R. Davidson, Phys. Rev. 183, 23 (1929).
39

47 R.J. Buenker, S.D. Peyerimhoff, Theor. Chim. Acta 35, 33 (1974).
48 R.J. Buenker, S.D. Peyerimhoff, Theor. Chim. Acta 39, 217 (1975).
49 R.J. Buenker, Int. J. Quantum. Chem. 29, 435 (1986).
50 R.J. Buenker, S.D. Peyerimhoff, W. Butcher, Mol. Phys. 35, 771 (1978).
51 R.J. Buenker, In Proceeding of the Workshop on Quantum Chemistry and Molecular
Physics, Burton, P. Ed.; University of Wollongong, Wollongong, Australia, 1980.
52 R.J. Buenker, In Studies in Physical Theoretical Chemistry, Carbo, R., Ed.; Elsevier,
Amsterdam; Vol. 21 (Current Aspects of Quantum Chemistry), 1981.
53 R.J. Buenker, R.A. Phillips, J. Mol. Struct. (THEOCHEM) 123, 291 (1985).
54 R.J. Buenker, S.D. Peyerimhoff, in New horizons of Quantum Chemistry eds., Lowdin,
P.O., and Pullman, B., (Reidel, Dordrecht, 1983) p. 183.
55 E.R. Davidson, In The World of Quantum Chemistry; R. Daudel, B. Pullman, Reidel:
Dordrecht (1974).
56 S.R. Langhoff, E.R. Davidson, Int. J. Quantum Chem. 8, 61 (1974).
57 S.D. Peyerimhoff, R.J. Buenker, Chem. Phys. 42, 167 (1979).
58 J. Paldus, P.E. Warmer, F. Vissar, A. Van der Avoird, Proceedings of the 5th Seminar on
Computational Methods in Quantum Chemistry, Groningen, The Netherlands (1983)
p. 13.
59 D.B. Knowles, J.R. Alvarez-Collado, G. Hirsch, R.J. Buenker, J. Chem. Phys. 95, 585
(1990).
60 S. Kerbs, R.J. Buenker, J. Chem. Phys. 103, 5613 (1995).
61 E.R. Davidson, J. Comput. Phys. 17, 87 (1975).
62 M. Tinkham, Group Theory and Quantum Mechanics, McGraw-Hill, New York (1964)
p. 131.
63 H.P. Liebermann, I. Boustani, S.N. Rai, A.B. Alekseyev, G. Hirsch, R.J. Buenker, Chem.
Phys. Letts. 214, 381 (1993).
64 I. Boustani, S.N. Rai, H.-P. Liebermann, A.B. Alekseyev, G. Hirsch, R.J. Buenker, Chem.
Phys. 177, 45 (1993).
65 A.B. Alekseyev, H.-P. Liebermann, I. Boustani, G. Hirsch, R.J. Buenker, Chem. Phys.
40

173, 333 (1993).
66 A.B. Alekseyev, H.-P. Liebermann, R.J. Buenker, In Recent Advances in Computational
Chemistry, K. Hirao, Y. Ishikawa, Ed.; 70-71; Vol. 5 (Recent Advances in Relativistic
Molecular Theory).
67 J. Cooley, Math. Comput. 15, 363 (1961).
68 M. Peric, R. Runau, J. Romelt, S.D. Peyerimhoff, R.J. Buenker, J. Mol. Spectrosc. 78,
309 (1979).
41

CHAPTER – 3
ELECTRONIC STRUCTURE AND
SPECTROSCOPIC PROPERTIES OF THE SiC
RADICAL

3.1. Introduction
The simple diatomic SiC radical is known to be an important component of the carbon
star. It is also present in interstellar regions of space.1 Though astrophysically important,
the spectroscopy of this radical was not observed in laboratory for a long time. The major
difficulty to study the silicon-carbon compounds was that it needed a very high temperature
to vaporize these elements. Bondybey2 and Michalopoulos et al.3 attempted experiments
using laser vaporization of silicon carbide rod. But the detection of the SiC radical was
unsuccessful. Although Si2 and C2 are spectroscopically well known species, the spectro-
scopic identification of SiC has been made much later. However, ab initio calculations of
SiC are performed before the experimental detection. Lutz and Ryan4 have performed the
configuration interaction (CI) calculations and found that the ground state of the mixed first
row-second row diatomic is 3Π. Bruna et al.5 have also carried out large scale CI calculations
for the potential curves of the isovalent series of diatomic species, CN+, Si2, SiC, CP+, and
SiN+ in their low-lying states. The results of these calculations for the SiC radical have
agreed well with those of the previous calculations.4 Rohlfing and Martin6 have studied the
structure and spectroscopic properties of the isovalent diatomic molecules such as C2, Si2,
and SiC. These authors have used Moller-Plesset perturbation theory based on UHF refer-
ence function as well as externally contracted CI based on a multireference function of the
complete-active-space type for determining the spectroscopic constants of a few low-lying
states. Meanwhile, low-lying electronic states of SiC− and electron affinity of SiC have been
studied by Anglada et al.7 from large scale CI calculations. Dohman et al.8 have made
a comparison among various isoelectronic radicals possessing eight valence electrons. The
CASSCF and contracted CI calculations have been performed by Larsson9 to study the po-
tential curves of X3Π, B3Σ+, and C3Π states of the SiC molecule. The author has predicted
transitions between the ground state and the B and C states to occur in the wave length
range 4000-6000 A. Bauschlicher and Langhoff10 have performed CI calculations at various
levels of electron correlation to compute the spectroscopic constants of X3Π and A3Σ− states
of SiC. Their best estimates of re, ωe, and De for the ground state of the radical were 1.719 A,
962 cm−1, and 4.4 eV, respectively.
The SiC radical was first observed11 by high-resolution Fourier transform emission spec-
troscopy from a composite wall hollow cathode. The 0-0 band of the d1Σ+-b1Π system of
SiC has been observed near 6100 cm−1. This has been confirmed by ab initio calculations
42

performed at different level of accuracy. Molecular constants of several low-lying states,
namely X3Π, A3Σ−, a1Σ+, b1Π, c1∆, and d1Σ+ of SiC are predicted from these calculations.
The results are found to be comparable with the predictions of Bauschlicher and Langhoff.10
The ground state of SiC has been characterized from the microwave transition observed by
Cernicharo et al.12 The A3Σ−-X3Π system of SiC is analogous to the Ballik-Ramsay system
of C2. Brazier et al.13 have observed the 0-0 band of this system in emission near 4500 cm−1
and the reported the bond lengths are 1.81356 and 1.72187 A for A3Σ− and X3Π states, re-
spectively. The A-X band was found to be weak because of the difficulty in making the SiC
radical. Multireference CI calculations have been performed by Langhoff and Bauschlicher14
to study the A3Σ−-X3Π infrared transition in the radical. The 0-0 band of the A-X transi-
tion has also been reassigned in another theoretical study.15 The dipole moment functions
of A3Σ− and X3Π, and the transition moment functions as well as radiative lifetimes of the
A-X transition have also been reported. Martin et al.16 have computed three lowest states,
X3Π, A3Σ−, and a1Σ+ of SiC using augmented coupled cluster methods and different basis
sets. Thermochemistry of the radical has also been reported by these authors. Butenhoff
and Rohlfing17 have studied the C3Π-X3Π band system of the jet-cooled SiC radical using
a laser induced fluorescence (LIF) spectroscopy. The vibrational energies and rotational
constants for the lowest few vibrational levels of both C3Π and X3Π states of SiC have been
determined by these authors. Almost at the same time, Ebben et al.18 have measured seven
rovibronic bands belonging to the C3Π-X3Π transition in SiC produced by the laser vapor-
ization in combination with supersonic cooling. The radiative lifetimes of the C3Π state
were found to vary from 2886 ns to 499 ns in the lowest seven vibrational levels. Singles and
doubles CI calculations from single SCF configuration have been carried out by McLean et
al.19 on a series of diatomic species including SiC and SiC−. Some spectroscopic information
of X3Π, A3Σ−, b1Π, and c1∆ states of SiC and the 2Π state of SiC− have been reported.
The millimeter-wave rotational spectra with hyperfine structure of two stable isotopes with
nuclear spin, namely 29SiC and Si13C, were detected in the ground state.20,21 The vertical
and adiabatic ionization energies and electron affinities of SinC and SinO (n=1-3) molecules
have been reported by Boldyrev et al.22 from large-scale ab initio calculations at different
levels of correlation.
Grutter et al.23 have identified the electronic absorption spectra of SiC− and SiC in 5K
neon matrices using mass-selected deposition. The neutralization of the anion leads to the
observation of a new band system B3Σ+←X3Π of SiC in addition to the known systems,
43

namely A3Σ−←X3Π and C3Π←X3Π. The B-X band has the origin at 11 749 cm−1, while
the ωe value for the B3Σ+ state of SiC in neon matrix has been reported to be 1178 cm−1.
However, no gas phase data is available for this state. Recently24, the infrared emission
spectrum of the A3Σ−-X3Π electronic transition of the SiC radical in the gas phase has been
observed using a high resolution Fourier transform spectrometer. Three bands, 0-1, 0-0, and
1-0 of this system are found in 2770, 3723, and 4578 cm−1, respectively.
In this chapter we report potential energy curves of 32 Λ–S states of singlet, triplet, and
quintet spin multiplicities. Spectroscopic constants (re, Te, and ωe) of 23 states within 6 eV
are determined42 and compared with the existing data. Effects of the spin-coupling on the
spectroscopic properties of these states have been studied. Potential energy curves of several
low-lying Ω states of SiC have also been constructed. The radiative lifetimes of some of the
excited states have been predicted.
3.2. Computational details
3.2.1 RECPs and basis sets
The RECPs of Pacios and Christiansen25 are used to replace the 1s22s22p6 core electrons
of the Si atom and 3s23p2 valence electrons are kept available for the CI calculations. For
the carbon atom, the RECPs of the same authors have been employed to replace inner 1s2
electrons. The total number of active electrons in the CI space is, therefore, eight. The 4s4p
Gaussian basis sets of Pacios and Christiansen25 for Si are augmented with some diffuse and
polarization functions. Three s functions (ξs = 0.04525, 0.02715, and 0.0163 a−20 ), two p
functions (ξp = 0.06911 and 0.02499 a−20 ), five d functions (ξd = 4.04168, 1.46155, 0.52852,
0.19112, and 0.06911 a−20 ), and two f functions (ξf = 0.19112 and 0.06911 a−2
0 ) are added.26
The first two sets of d functions are contracted using coefficients of 0.054268 and 0.06973.
Similarly, the two f functions are contracted using the coefficients of 0.29301 and 0.536102.
The final basis set for Si used in the present CI calculations is (7s6p5d2f/7s6p4d1f). For the
carbon atom, the (4s4p) basis set of Pacios and Christiansen25 has been enhanced by adding
two sets of d functions of exponents 1.2 and 0.35 a−20 .
3.2.2 SCF MOs and CI
At each internuclear distance of SiC, we have performed self-consistent-field (SCF) calcula-
44
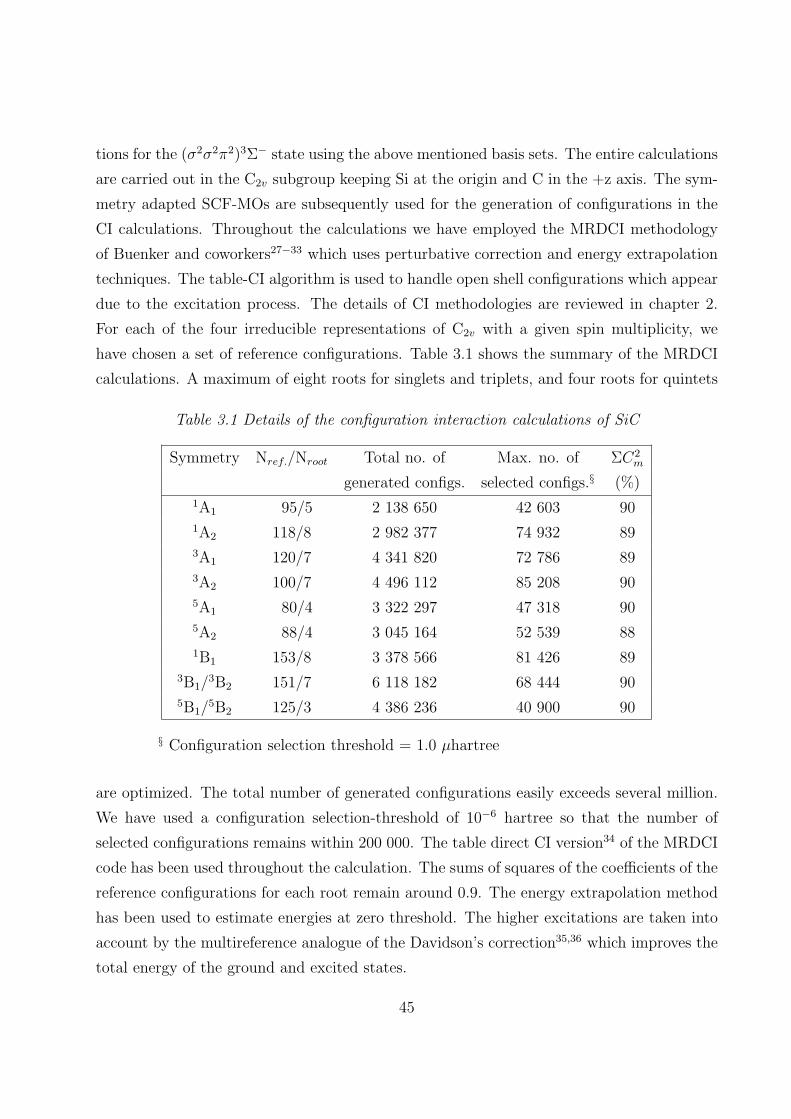
tions for the (σ2σ2π2)3Σ− state using the above mentioned basis sets. The entire calculations
are carried out in the C2v subgroup keeping Si at the origin and C in the +z axis. The sym-
metry adapted SCF-MOs are subsequently used for the generation of configurations in the
CI calculations. Throughout the calculations we have employed the MRDCI methodology
of Buenker and coworkers27−33 which uses perturbative correction and energy extrapolation
techniques. The table-CI algorithm is used to handle open shell configurations which appear
due to the excitation process. The details of CI methodologies are reviewed in chapter 2.
For each of the four irreducible representations of C2v with a given spin multiplicity, we
have chosen a set of reference configurations. Table 3.1 shows the summary of the MRDCI
calculations. A maximum of eight roots for singlets and triplets, and four roots for quintets
Table 3.1 Details of the configuration interaction calculations of SiC
Symmetry Nref./Nroot Total no. of Max. no. of ΣC2m
generated configs. selected configs.§ (%)1A1 95/5 2 138 650 42 603 901A2 118/8 2 982 377 74 932 893A1 120/7 4 341 820 72 786 893A2 100/7 4 496 112 85 208 905A1 80/4 3 322 297 47 318 905A2 88/4 3 045 164 52 539 881B1 153/8 3 378 566 81 426 89
3B1/3B2 151/7 6 118 182 68 444 905B1/5B2 125/3 4 386 236 40 900 90
§ Configuration selection threshold = 1.0 µhartree
are optimized. The total number of generated configurations easily exceeds several million.
We have used a configuration selection-threshold of 10−6 hartree so that the number of
selected configurations remains within 200 000. The table direct CI version34 of the MRDCI
code has been used throughout the calculation. The sums of squares of the coefficients of the
reference configurations for each root remain around 0.9. The energy extrapolation method
has been used to estimate energies at zero threshold. The higher excitations are taken into
account by the multireference analogue of the Davidson’s correction35,36 which improves the
total energy of the ground and excited states.
45

3.2.3 Spin-orbit interaction
The spin-orbit interaction is included in the calculation by two-step variational calculations.37
We have allowed all the spin components of low-lying Λ-S states of SiC to interact. The
spin-orbit operators, which are compatible with the RECPs are taken from Pacios and
Christiansen.25 The sizes of the secular equations of A1, A2, and B1/B2 blocks in the C22v
double group are 41×41, 41×41, and 40×40, respectively for some selected number of roots
of Λ–S symmetries of the molecule. The details of the spin-orbit CI calculations are already
discussed.
3.3. Results and discussion
3.3.1 Spectroscopic constants and potential energy curves of Λ–S states
The ground states of both C and Si belong to the 3Pg symmetry. Table 3.2 shows dissoci-
ation correlation between the Λ–S states and atomic states leading to lowest five asymptotes.
The lowest dissociation limit, Si(3Pg)+C(3Pg) of SiC correlates with Σ+(2), Σ−, Π(2), and
∆ symmetries, a total of 18 Λ-S states. The second dissociation limit, Si(1Dg)+C(3Pg) cor-
relates with triplets, namely 3Σ+, 3Σ−(2), 3Π(3), 3∆(2), and 3Φ. The relative energy of this
limit computed from the CI calculation of the SiC molecule at a very large bond distance is
about 7000 cm−1 which is 800-900 cm−1 higher than the J-averaged observed value.38 The
Table 3.2 Dissociation correlation between the molecular and atomic states of SiC
Λ-S states Atomic states Relative energy / cm−1
Si + C Expt.a Calc.1Σ+(2), 1Σ−, 1Π(2), 1∆, 3Pg + 3Pg 0 03Σ+(2), 3Σ−, 3Π(2), 3∆,5Σ+(2), 5Σ−, 5Π(2), 5∆3Σ+, 3Σ−(2), 3Π(3), 3∆(2), 3Φ 1Dg + 3Pg 6124 70003Σ+, 3Σ−(2), 3Π(3), 3∆(2), 3Φ 3Pg + 1Dg 10 1583Σ−, 3Π 1Sg + 3Pg 15 2191Σ+(3), 1Σ−(2), 1Π(4), 1∆(3), 1Dg + 1Dg 16 282 18 8001Φ(2), 1Γ
a Averaged over J; Ref. 38
46

next two dissociation limits also correlate only with the excited triplets of SiC. The combi-
nation of Si(1Dg) and C(1Dg) generates a set of 15 excited singlet states of SiC. The relative
energy of these states at the dissociation limit is about 16 300 cm−1.
Potential energy curves of singlet, triplet, and quintet states of SiC without spin-orbit
coupling are shown in Figs. 3.1a-c. All the states correlating with the lowest dissociation
limit and a few excited states dissociating into higher asymptotes are studied here. The
computed spectroscopic constants (re, ωe, Te) of 23 Λ–S states are tabulated in Table 3.3.
The computed bond length of the SiC radical in the ground state (X3Π) is 1.74 A which
is nearly 0.02 A longer than the observed data. The ground-state bond length obtained
in earlier calculations at various level of theories varies between 1.721 and 1.75 A. The
vibrational frequency (ωe) of the ground state obtained in the present calculation is 930 cm−1
which is about 35 cm−1 smaller than the observed value. A comparison of the present data
with other theoretical results is shown in Table 3.3. The ground state of the SiC radical
is dominated by the σ21σ2π
31(80%) configuration at re (Table 3.4). The σ1 MO is mainly a
bonding combination of s and pz orbitals of Si and C atoms, while σ2 has the antibonding
character and is mostly localized on the silicon atom. The π1 MO is a strongly bonding,
comprising the px/y orbitals of the two atoms. The ground-state dissociation energy (De) of
SiC computed here is 4.05 eV, which is somewhat lower than the thermochemical D00 value
of 4.64 eV.39,40 The MRD-CI calculations41 with DZP+bond functions have reported a De
value of 4.3 eV.
The A3Σ− state is the first excited state of SiC. Its computed transition energy (Te) is
3985 cm−1 which compares well with the experimental or other theoretical data shown in
Table 3.3. The observed 0-0 band of the A3Σ−-X3Π system has been found near 4500 cm−1
from which a T00 value of 4578 cm−1 is obtained.13 However, Langhoff and Bauschlicher14
suggested an alternative assignment of the band to a 1-0 transition. These authors have
established the 0-0 band at 3700+200 cm−1 from the theoretical calculations using atomic
natural orbitals. The equilibrium bond length of the A3Σ− state estimated here is about
1.82 A which agrees well with the available values as listed in Table 3.3. The ωe value of this
state, obtained in the present calculations, is 857 cm−1 which is very close to the values found
in other calculations.14,15 Around the potential minimum, the A3Σ− state is dominated by
the σ21σ
22π
21(88%) configuration in which the π1 MO has a similar bonding characteristic as
in the ground state.
47
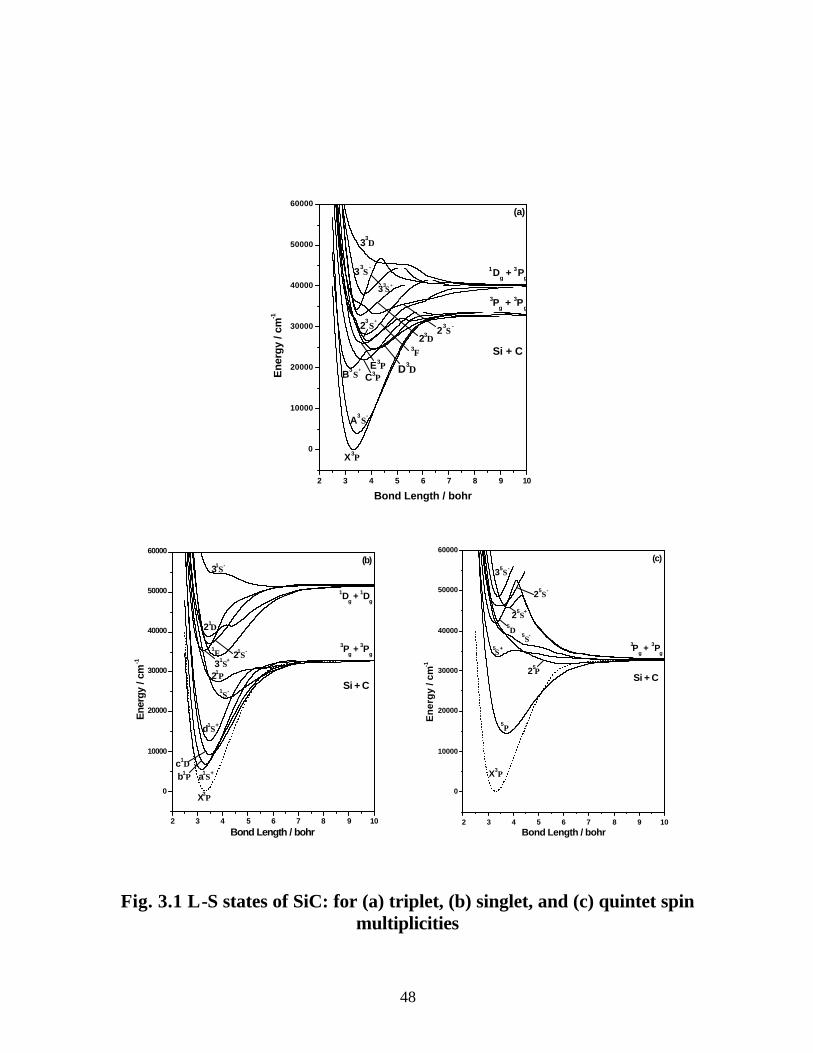
48
2 3 4 5 6 7 8 9 10
0
10000
20000
30000
40000
50000
60000
Si + C
(a)
3ΦE3Π
1Dg + 3P
g
3Pg + 3P
g
33Σ
-
23Σ
-
A3Σ
-
33∆
C3Π
X3Π
23∆
33Σ
+
23Σ
+
D3∆B3Σ
+Ene
rgy
/ cm
-1
Bond Length / bohr
2 3 4 5 6 7 8 9 10
0
10000
20000
30000
40000
50000
60000
Si + C
(b)
1Φ
1Dg + 1D
g
31Σ-
X3Π
21Π
b1Π
21Σ-
1Σ-
21∆
31Σ+
3Pg + 3P
g
c1∆
d1Σ+
a1Σ+
Ene
rgy
/ cm
-1
Bond Length / bohr
2 3 4 5 6 7 8 9 10
0
10000
20000
30000
40000
50000
60000
Si + C
(c)
3Pg + 3P
g
X3Π
25Π
5Π
35Σ-
25Σ-
5Σ-
5∆
25Σ+
5Σ+
En
erg
y / c
m-1
Bond Length / bohr
Fig. 3.1 Λ-S states of SiC: for (a) triplet, (b) singlet, and (c) quintet spin
multiplicities
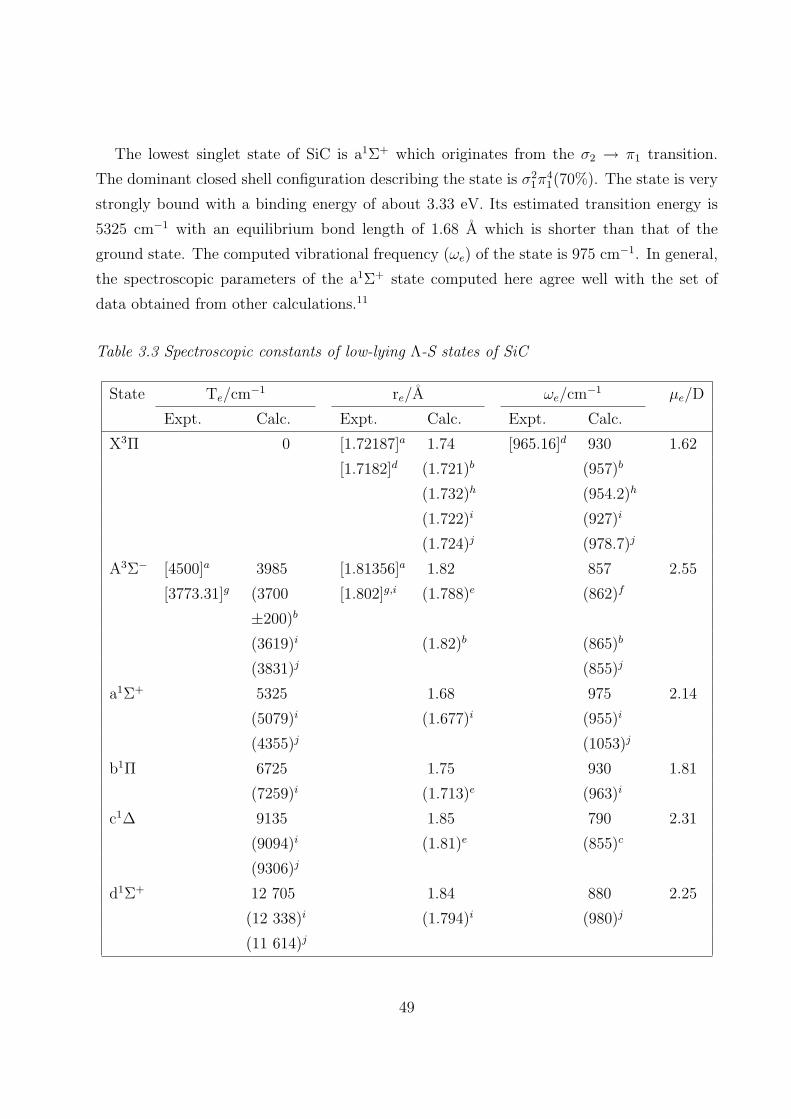
The lowest singlet state of SiC is a1Σ+ which originates from the σ2 → π1 transition.
The dominant closed shell configuration describing the state is σ21π
41(70%). The state is very
strongly bound with a binding energy of about 3.33 eV. Its estimated transition energy is
5325 cm−1 with an equilibrium bond length of 1.68 A which is shorter than that of the
ground state. The computed vibrational frequency (ωe) of the state is 975 cm−1. In general,
the spectroscopic parameters of the a1Σ+ state computed here agree well with the set of
data obtained from other calculations.11
Table 3.3 Spectroscopic constants of low-lying Λ-S states of SiC
State Te/cm−1 re/A ωe/cm−1 µe/D
Expt. Calc. Expt. Calc. Expt. Calc.
X3Π 0 [1.72187]a 1.74 [965.16]d 930 1.62
[1.7182]d (1.721)b (957)b
(1.732)h (954.2)h
(1.722)i (927)i
(1.724)j (978.7)j
A3Σ− [4500]a 3985 [1.81356]a 1.82 857 2.55
[3773.31]g (3700 [1.802]g,i (1.788)e (862)f
±200)b
(3619)i (1.82)b (865)b
(3831)j (855)j
a1Σ+ 5325 1.68 975 2.14
(5079)i (1.677)i (955)i
(4355)j (1053)j
b1Π 6725 1.75 930 1.81
(7259)i (1.713)e (963)i
c1∆ 9135 1.85 790 2.31
(9094)i (1.81)e (855)c
(9306)j
d1Σ+ 12 705 1.84 880 2.25
(12 338)i (1.794)i (980)j
(11 614)j
49
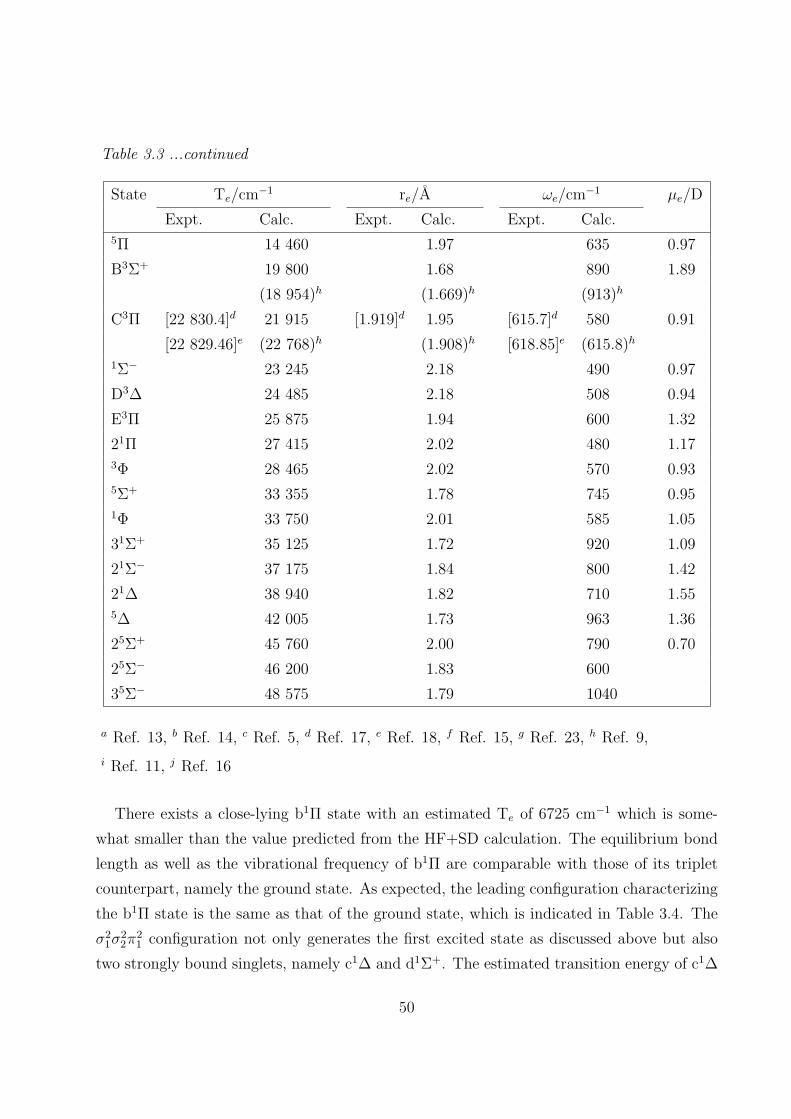
Table 3.3 ...continued
State Te/cm−1 re/A ωe/cm−1 µe/D
Expt. Calc. Expt. Calc. Expt. Calc.5Π 14 460 1.97 635 0.97
B3Σ+ 19 800 1.68 890 1.89
(18 954)h (1.669)h (913)h
C3Π [22 830.4]d 21 915 [1.919]d 1.95 [615.7]d 580 0.91
[22 829.46]e (22 768)h (1.908)h [618.85]e (615.8)h
1Σ− 23 245 2.18 490 0.97
D3∆ 24 485 2.18 508 0.94
E3Π 25 875 1.94 600 1.32
21Π 27 415 2.02 480 1.173Φ 28 465 2.02 570 0.935Σ+ 33 355 1.78 745 0.951Φ 33 750 2.01 585 1.05
31Σ+ 35 125 1.72 920 1.09
21Σ− 37 175 1.84 800 1.42
21∆ 38 940 1.82 710 1.555∆ 42 005 1.73 963 1.36
25Σ+ 45 760 2.00 790 0.70
25Σ− 46 200 1.83 600
35Σ− 48 575 1.79 1040
a Ref. 13, b Ref. 14, c Ref. 5, d Ref. 17, e Ref. 18, f Ref. 15, g Ref. 23, h Ref. 9,
i Ref. 11, j Ref. 16
There exists a close-lying b1Π state with an estimated Te of 6725 cm−1 which is some-
what smaller than the value predicted from the HF+SD calculation. The equilibrium bond
length as well as the vibrational frequency of b1Π are comparable with those of its triplet
counterpart, namely the ground state. As expected, the leading configuration characterizing
the b1Π state is the same as that of the ground state, which is indicated in Table 3.4. The
σ21σ
22π
21 configuration not only generates the first excited state as discussed above but also
two strongly bound singlets, namely c1∆ and d1Σ+. The estimated transition energy of c1∆
50

is 9135 cm−1 which is comparable with the value reported in the previous calculations.11,16
However, the calculation of Bruna et al.5 predicted its transition energy above 10 500 cm−1.
The ωe value of the c1∆ state estimated here is 790 cm−1 which is 65 cm−1 smaller than the
value reported in earlier calculations. The d1Σ+ state is the second root of the 1Σ+ symme-
try. At the equilibrium bond length, the state is dominated by an open shell configuration,
σ21σ
22π
21(60%), while there is at least 17% contribution of a closed shell configuration, σ2
1π41.
The computed transition energy of this state (Te=12 705 cm−1) agrees well, while the re
value reported here is 0.05 A longer than the value reported earlier.11 The vibrational fre-
quency of d1Σ+ at re is about 880 cm−1 which is 100 cm−1 smaller than the value estimated
in the earlier study.15
There is a low-lying 5Π state which has not been identified before. The state originates
from a σ21σ2π
21π2 (82%) configuration in which π2 is mostly antibonding MO comprising
the px/y orbitals of both Si and C. The computed transition energy of the state is about
14 460 cm−1 with ωe=635 cm−1 and re=1.97 A. The state is also strongly bound with an
estimated binding energy of 2.22 eV. Although 5Π is not a very important state from the
spectroscopic point of view, its spin components with Ω=0+, 0−, 1, 2, and 3 may influence
the lower states to some extent. Several other excited quintet bound states of the SiC radical
lie above 30 000 cm−1.
The next two important states of SiC are B3Σ+ and C3Π. The B state is the lowest root
of the 3Σ+ symmetry. Ab initio based CASSCF-CCI calculations of Larsson9 have predicted
the B3Σ+ state to lie around 18 954 cm−1 with a bond length of 1.67 A. In the present
calculation, the estimated Te of this state is about 19 800 cm−1. The computed bond length
is 0.01 A longer, while the calculated vibrational frequency of this state agrees well with
the value reported in the earlier calculation of Larsson.9 However, spectroscopic constants of
the B3Σ+ state obtained from both the theoretical calculations disagree with those derived
from the absorption spectrum of the B3Σ+←X3Π transition in a 5K neon matrix.23 The
origin of the observed band has been reported to be at 11 749 cm−1. The observed ωe of
1178 cm−1 also disagrees completely with the present value of 890 cm−1. The spectrum of
the B3Σ+-X3Π transition of SiC may need further experimental study. However, it is quite
clear that there is no other lower state except the ground one to which the transition from
B3Σ+ may take place. Theoretically, the B-X transition is expected to carry a reasonably
large intensity. Around the potential minimum, the B3Σ+ state is characterized by two
important configurations, σ1σ2π41 (45%) and σ2
1π31π2(30%) as shown in Table 3.4.
51

Table 3.4 Composition of Λ-S states of SiC at equilibrium bond length
State Configuration (% contribution)
X3Π σ21σ2π
31(80), σ1σ
22π
31(3)
A3Σ− σ21σ
22π
21(88)
a1Σ+ σ21π
41(70), σ2
2π41(2), σ2
1σ22π
21(6), σ1σ2π
31π2(5)
b1Π σ21σ2π
31(81), σ2
1σ2π1π22(2)
c1∆ σ21σ
22π
21(85)
d1Σ+ σ21σ
22π
21(60), σ2
1π41(16), σ2
2π41(3)
5Π σ21σ2π
21π2(82), σ2
1σ2π1π22(2), σ1σ
22π1π
22(2)
B3Σ+ σ1σ2π41(45), σ2
1π31π2(30), σ1σ2π
31π2(4), σ1σ2π
21π
22(2)
C3Π σ21σ2π
21π2(76), σ2
1σ2π31(3), σ1σ
22π
31(3)
1Σ− σ21σ
22π1π2(84)
D3∆ σ21σ
22π1π2(84)
E3Π σ21σ2π
21π2(78), σ1σ
22π
31(5)
21Π σ21σ2π
21π2(82), σ1σ
22π
31(2)
3Φ σ21σ2π
21π2(86)
5Σ+ σ1σ2π31π2(66), σ2
1π21π
22(13), σ1σ2π1π
32(4), σ1σ2π
21π
22(3), σ2
2π21π
22(2)
1Φ σ21σ2π
21π2(83)
31Σ+ σ1σ2π41(37), σ2
1π31π2(23), σ1σ2π
31π2(6), σ2
2π41(6), σ2
1π41(3)
21Σ− σ21π
31π2(78), σ2
2π31π2(4)
21∆ σ21π
31π2(78)
5∆ σ1σ2π31π2(82)
25Σ+ σ1σ2π31π2(42), σ2
1π21π
22(37)
25Σ− σ21σ2σ3π
21(47), σ1σ2π
31π2(18), σ2
1σ2σ5π21(10)
35Σ− σ21σ2σ3π
21(37), σ1σ2π
31π2(25), σ2
1σ2σ4π21(10)
The C3Π state is the second root of the ground-state symmetry and it originates predom-
inantly from the same configuration, σ21σ2π
21π2 that generates the lowest 5Π. This configu-
ration also generates eight more states of Π and Φ symmetries. The transition energy (Te)
of C3Π calculated here is about 21 915 cm−1 which is somewhat smaller than the observed
value17 of 22 830.4(9) cm−1 determined from the C3Π–X3Π band system of the SiC radical
produced by laser vaporization. In another experimental study18, analysis of seven rovibronic
52

bands involving the C3Π(v′=0-6)–X3Π(v′′=0) transition have resulted Te and ωe values of
22 829.46 cm−1 and 618.85 cm−1, respectively. The Si-C bond in the C3Π state is nearly
0.21 A longer than that in the ground state. However, the calculated re is 0.03 A longer than
the experimentally determined value of 1.919 A. The fitted vibrational frequency is about
580 cm−1, as compared to the observed value of 615.7(8) cm−1. The molecular constants
computed by Larsson9 are in better agreement with the observed data as shown in Table 3.3.
No other state beyond C3Π has been identified so far. We have predicted 1Σ− and 3∆ states
with their potential minima located almost at the same bond length of 2.18 A. The estimated
transition energies of 1Σ− and 3∆ states are 23 245 and 24 485 cm−1, respectively. Potential
energy curves of these states are a bit shallow compared to those of the other low-lying
states already discussed. This has been reflected in their smaller ωe values. Both the states
originate from the same configuration, σ21σ
22π1π2 with the same dominance at equilibrium.
We may therefore, expect a 3∆-X3Π transition to take place around 24 500 cm−1. Although
such a transition has not yet been observed, we have labeled 3∆ as the D state because it is
next to C3Π. There is another excited 3Π, denoted as E3Π which lies just above D3∆. The
computed transition energy of the E3Π state is about 25 875 cm−1. The state originates from
σ21σ2π
21π2(78%) which has also created the lower lying C3Π and 5Π states. The re and ωe
values of the E3Π state are very similar to those of C3Π. The E3Π state, however, dissociates
into the higher asymptote. The E-X transition is expected to be strong one, though no such
transition is experimentally reported so far.
The second root of 1Π, which is also characterized by the σ21σ2π
21π2 configuration, is weakly
bound. The potential minimum of the 21Π state is located around the bond length of 2.02 A
with an estimated transition energy of 27 415 cm−1 at equilibrium. Both the lowest singlet
and triplet Φ states are strongly bound and have similar spectroscopic properties. The energy
separation between them is about 5300 cm−1 with the 3Φ state lying lower. These states are
also predominantly characterized by the same configuration that generates 5Π, C3Π, E3Π,
and 21Π. The 3Φ–X3Π transition may be of some interest from the spectroscopic point of
view through their dipolar components only.
Potential energy curves of three lowest states of the 3Σ+ symmetry undergo avoided cross-
ings as seen in Fig. 3.1a. Analyzing the compositions of these states in the CI calculations, it
has been found that three important configurations, namely σ1σ2π41, σ2
1π31π2, and σ2
1σ22π1π2
mix up. An avoided curve crossing between the first and second root of 3Σ+ takes place
around 3.9 a0, while another crossing is noted between the second and third root in the
53

range 3.2-3.4 a0. The contributions of important configurations of all three 3Σ+ states over
a certain range of bond distances have been displayed in Table 3.5.
Two excited 3Σ− states, namely 23Σ− and 33Σ− also undergo an avoided crossing around
the bond length of 3.6 a0. This is reflected in the potential energy curves of these states
shown in Fig. 3.1a. The diabatic coupling is considerably large. In the bond length region
below 3.6 a0, the lower root is dominated by σ21π
31π2, while in the other region it is mainly
characterized by the σ21σ
22π1π2 configuration. The diabatic curve of the 23Σ− state is pre-
dicted to have a minimum around 2.2 A with an estimated transition energy of 33 100 cm−1.
We also expect the diabatic curve of 33Σ− to have a potential minimum around 1.8 A with
a predicted transition energy of about 36 200 cm−1.
Table 3.5 Compositions of the lowest three roots of 3Σ+ of SiC in the avoided crossing region
r/a0 B3Σ+ 23Σ+ 33Σ+
2.7 σ1σ2π41(70), σ2
1π31π2(8) σ2
1π31π2(69), σ1σ2π
41(9), σ1σ2π
31π2(80), σ1σ2π
41(6)
σ22π
31π2(4)
2.9 σ1σ2π41(63), σ2
1π31π2(13), σ2
1π31π2(64), σ1σ2π
41(14) σ2
1σ22π1π2(82)
σ1σ2π31π2(6)
3.1 σ1σ2π41(52), σ2
1π31π2(22), σ2
1π31π2(54), σ1σ2π
41(23) σ2
1σ22π1π2(82)
σ1σ2π31π2(6)
3.2 σ1σ2π41(45), σ2
1π31π2(29), σ2
1π31π2(45), σ1σ2π
41(27) σ2
1σ22π1π2(80), σ2
1π31π2(4)
σ1σ2π31π2(4)
3.5 σ21π
31π2(53), σ1σ2π
41(20) σ2
1σ22π1π2(80) σ1σ2π
41(45), σ2
1π31π2(25)
3.6 σ21π
31π2(57), σ1σ2π
41(13) σ2
1σ22π1π2(80) σ1σ2π
41(51), σ2
1π31π2(19)
3.8 σ21π
31π2(49), σ2
1σ22π1π2(27) σ2
1σ22π1π2(60), σ2
1π31π2(17) σ1σ2π
41(54), σ2
1π31π2(11)
3.9 σ21σ
22π1π2(55), σ2
1π31π2(26) σ2
1π31π2(41), σ2
1σ22π1π2(30) σ1σ2π
41(53), σ2
1π31π2(8),
σ1σ2π31π2(8)
4.2 σ21σ
22π1π2(74), σ2
1π31π2(11) σ2
1π31π2(59), σ2
1σ22π1π2(11) σ1σ2π
41(38), σ1σ2π
31π2(12),
σ21σ2σ6π
21(7), σ1σ2π
21π
22(5)
Both the 5Σ+ states reported here are weakly bound and dissociate into the lowest
limit through different channel. The potential minimum of 5Σ+ is located at 1.78 A with
ωe=745 cm−1 and Te=33 355 cm−1. Around the equilibrium bond length, the 5Σ+ state is
54

characterized predominantly by σ1σ2π31π2, while another configuration, σ2
1π21π
22 dominates at
the longer bond distances. This has resulted a predissociation of the 5Σ+ state through a
barrier of height 0.22 eV. A strong avoided crossing between the potential curves of the two
roots of 5Σ+ has created an apparent minimum in the potential curve of 25Σ+. Since the
coupling between these two roots is very strong, we have fitted the adiabatic curve and the
minimum is located at 2.0 A with ωe=790 cm−1 and Te=45 760 cm−1. The nature of the
potential energy curve of 25Σ+ reveals that there is a potential barrier of height 0.4 eV at
4.4 a0 beyond which the state dissociates through a repulsive path.
Three more excited singlets, namely 31Σ+, 21Σ−, and 21∆ of the SiC radical are strongly
bound. They correlate with the higher asymptote, Si(1Dg)+C(1Dg) as shown in Table 3.2.
The predicted transition energy of the 31Σ+ state is about 35 125 cm−1 at re=1.72 A.
The estimated vibrational frequency of the state is close to that of a1Σ+. A transition
of the type 31Σ+–a1Σ+ is expected to occur around 29 000 cm−1 with a large transition
probability. Both 21Σ− and 21∆ states, in the Franck-Condon region, are characterized
mainly by the σ21π
31π2 configuration. The potential energy curve of 21∆ shows an avoided
crossing near the bond distance of 4.0 a0. The estimated res of 21Σ− and 21∆ are 1.84
and 1.82 A, respectively. Analyzing the compositions of these states above 4.0 a0, a strong
mixing with other configurations is predicted. The vibrational frequency of the 21∆ state is
nearly 100 cm−1 smaller than that of 21Σ−. The computed transition energies of these two
excited singlets lie in the range 37 000-39 000 cm−1.
The lowest 5∆ state has a deep potential minimum at re=1.73 A with Te=42 005 cm−1
and it is mainly described by the σ1σ2π31π2 (82%) configuration. Potential energy curves in
Fig. 3.1c show that there is an avoided crossing around 4.1 a0 with a repulsive curve of an
excited 5∆ which is dominated by the σ21σ2σ6π1π2 configuration. This has resulted a predis-
sociation of 5∆ to the lowest dissociation limit through a large barrier of 1.3 eV. However,
the fitted potential curve in the Franck-Condon region has predicted ωe=963 cm−1. The
second and third root of 5Σ− have bound potential energy curves. The compositions of these
roots at different bond distances reveal that there is a strong mixing between (σ1σ2π31π2)5Σ−
and (σ21σ2σ3π
21)5Σ− in the range 3.4-3.5 a0. As a result, the potential minimum of 25Σ− is
shallow, while the adiabatic curve of 35Σ− is very sharp with a large ωe of 1040 cm−1. The
estimated re of the 35Σ− state is about 1.79 A, while the minimum of the adiabatic curve of
25Σ− is located around the bond length of 1.83 A with ωe=600 cm−1.
55

3.3.2 Spectroscopic constants and potential energy curves of Ω states
The spin-orbit coupling splits the ground-state dissociation limit, Si(3Pg)+C(3Pg) into
nine very closely spaced asymptotes as shown in Table 3.6. The largest splitting is only
267 cm−1 as obtained from the observed atomic spectral data.38 These asymptotes correlate
with 50 Ω states of the SiC radical. In the present calculations, we allow all these states to
Table 3.6 Dissociation correlation between Ω and atomic states of SiC
Ω States† Atomic states Relative energy/
Si + C cm−1
0+ 3P0 + 3P0 0
0−, 1 3P0 + 3P1 16
0+, 1, 2 3P0 + 3P2 43
0−, 1 3P1 + 3P0 77
0+(2), 0−, 1(2), 2 3P1 + 3P1 94
0+, 0−(2), 1(3), 2(2), 3 3P1 + 3P2 121
0+, 1, 2 3P2 + 3P0 223
0+, 0−(2), 1(3), 2(2), 3 3P2 + 3P1 240
0+(3), 0−(2), 1(4), 2(3), 3(2), 4 3P2 + 3P2 267
† Values in parenthesis are the corresponding number of states
interact. Potential energy curves of a few low-lying states of 0+, 0−, 1, 2 and 3 symmetries
of SiC are shown in Figs. 3.2a-d. These show several number of curve crossing phenomena.
Since both the atoms are light, the spin-orbit effects are expectedly small. The potential
energy curves show no great changes and the avoided crossings are very sharp. The ground
state of SiC splits in an inverted order with X3Π2 being the lowest spin-orbit component.
All four components of X3Π lie within 100 cm−1. The computed spectroscopic constants of
the low-lying Ω states up to 22 000 cm−1 of energy are shown in Table 3.7. Obviously, the
spectroscopic constants are obtained by fitting the diabatic curves. The spin-orbit mixing
does not change the spectroscopic constants much. The two components of A3Σ− are almost
inseparable. The transition energies are increased only by 70-75 cm−1 due to the spin-orbit
coupling. The diabatic curve of A3Σ−0+ cuts the similar component of a1Σ+ just beyond the
equilibrium point of the latter (3.2 a0). Thus, the potential energy curves of second and
56
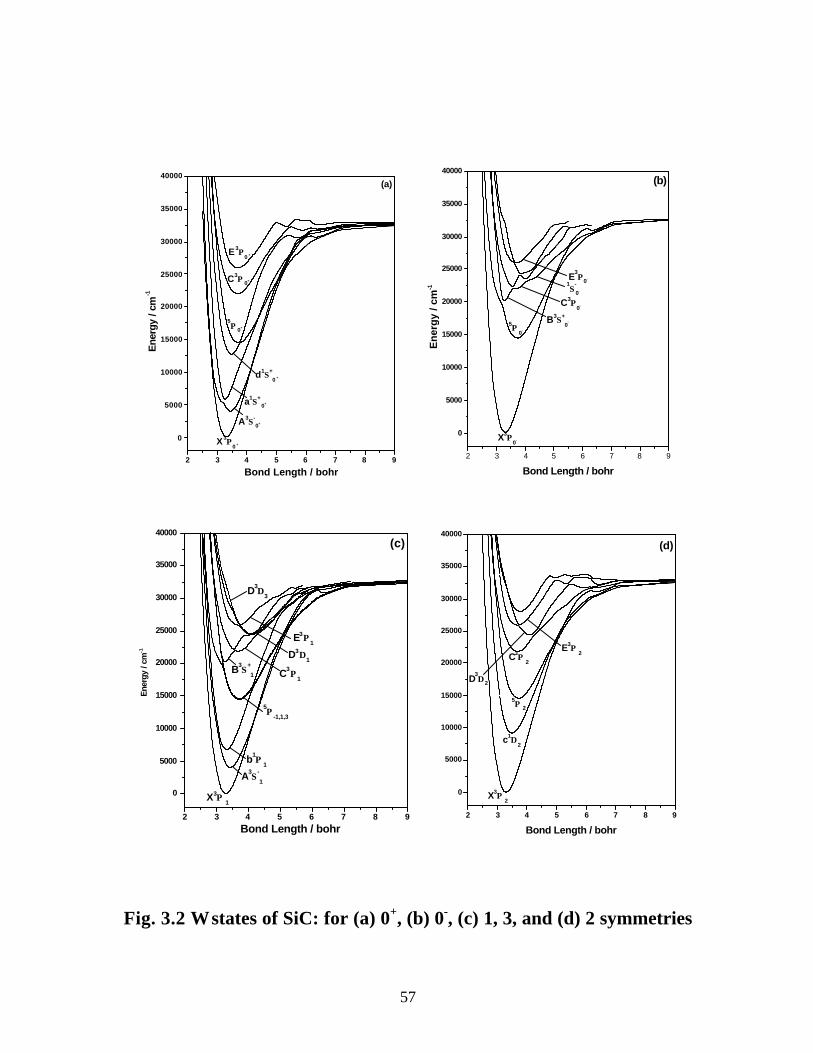
57
2 3 4 5 6 7 8 9
0
5000
10000
15000
20000
25000
30000
35000
40000(a)
E3Π0 +
C3Π0+
5Π0+
d1Σ+
0 +
a1Σ+0+
A3Σ-0+
X3Π0 +
Ene
rgy
/ cm
-1
Bond Length / bohr
2 3 4 5 6 7 8 9
0
5000
10000
15000
20000
25000
30000
35000
40000
1Σ
-
0-
E3Π0-
C3Π0-
B3Σ+
0-
(b)
5Π0-
X3Π0-
En
erg
y / c
m-1
Bond Length / bohr
2 3 4 5 6 7 8 9
0
5000
10000
15000
20000
25000
30000
35000
40000
D3∆ 1
D3∆
3
Bond Length / bohr
Ene
rgy
/ cm
-1
E3Π1
C3Π
1B3
Σ+
1
5Π
-1,1,3
b1Π
1
A3Σ
-
1
X3Π 1
(c)
2 3 4 5 6 7 8 9
0
5000
10000
15000
20000
25000
30000
35000
40000
E3Π 2
D3∆ 2
C3Π 2
5Π 2
c1∆ 2
X3Π 2
(d)
Bond Length / bohr
Fig. 3.2 Ω states of SiC: for (a) 0+, (b) 0-, (c) 1, 3, and (d) 2 symmetries

third root of 0+ look somewhat different as compared to the corresponding Λ-S states. All
the singlet components, a1Σ+0+ , b1Π1, c1∆2, and d1Σ+
0+ remain almost unchanged in their
spectroscopic properties. The spin-orbit components of 5Π split in a regular order with a
maximum separation of 135 cm−1. The components of C3Π, however, split in an inverted
order as displayed in Table 3.7. Spectroscopic properties of the 0− and 1 components of
B3Σ+ are nearly the same.
Table 3.7 Spectroscopic constants of low-lying Ω states of SiC
State Te/cm−1 re/A ωe/cm−1
X3Π2 0 1.74 930
(0)a
X3Π1 60 1.74 925
(37.3)a
X3Π0− 95 1.74 933
(74.6)a
X3Π0+ 100 1.74 933
(74.6)a
A3Σ−1 4055 1.82 854
A3Σ−0+ 4060 1.82 855
a1Σ+0+ 5370 1.68 975
b1Π1 6770 1.76 931
c1∆2 9185 1.85 800
d1Σ+0+ 12 745 1.84 882
5Π−1 14 450 1.97 6255Π0− 14 480 1.97 6255Π0+ 14 485 1.97 6255Π1 14 515 1.97 6255Π2 14 550 1.97 6255Π3 14 585 1.97 625
B3Σ+1 19 880 1.69 871
B3Σ+0− 19 885 1.69 871
58

Table 3.7 ...continued
State Te/cm−1 re/A ωe/cm−1
C3Π2 21 935 1.95 581
C3Π1 21 965 1.95 581
C3Π0− 21 995 1.95 581
C3Π0+ 22 000 1.95 582
a Ref. 16
3.3.3 Dipole moments and transition properties
In Table 3.3, we have reported the computed dipole moments (µe) of most of the low-
lying states at their equilibrium bond lengths. The ground-state µe is estimated to be about
1.62 D with Si+C− polarity. The first excited state (A3Σ−) has a larger µe value of 2.55 D.
All other excited states have the same sense of polarity as that of the ground state. The
variation of the dipole moment for the low-lying states of SiC as a function of bond distance
are plotted in Fig. 3.3a. The ground-state dipole moment function has a maximum at 4.6 a0.
After a short increase upto 3.2 a0, the dipole moment function of A3Σ− smoothly decreases
with increase in bond length. The situation is same for c1∆, while those of a1Σ+, b1Π, and
c1∆ have another similar type of variation as shown in the Fig. 3.3a. At very short bond
distance (2.5 a0), C3Π has the highest dipole moment of 1.17 ea0, but it suddenly falls with
increase in bond length. Dipole moment function of 5Π looks similar to that of the ground
state. Strikingly, all the functions tend to zero at very long bond distances.
Several electric dipole allowed transitions involving the low-lying singlets and triplets of
SiC are studied here. Transition probabilities of six triplet-triplet transitions, each of which
has the ground state as the lowest one, have been computed. Transition dipole moments
of these transitions as a function of bond distance are shown in Fig. 3.3b. The transition-
moment curve of A3Σ−-X3Π is smoothly decreasing to zero at the longer bond distance. In
the Franck-Condon region, the magnitude of the transition dipole moment of A-X is about
0.3 ea0. The computed partial radiative lifetime for this transition at v′=0 is about 125 µs and
it decreases considerably with the higher v′. Table 3.8 lists the partial radiative lifetimes for
eleven transitions at the lowest three vibrational levels. The B3Σ+-X3Π transition moments
in the Franck-Condon region are at least 10 times smaller than those of A-X. But the
computed lifetime for the B-X transition is lower due to larger energy differences. As
59
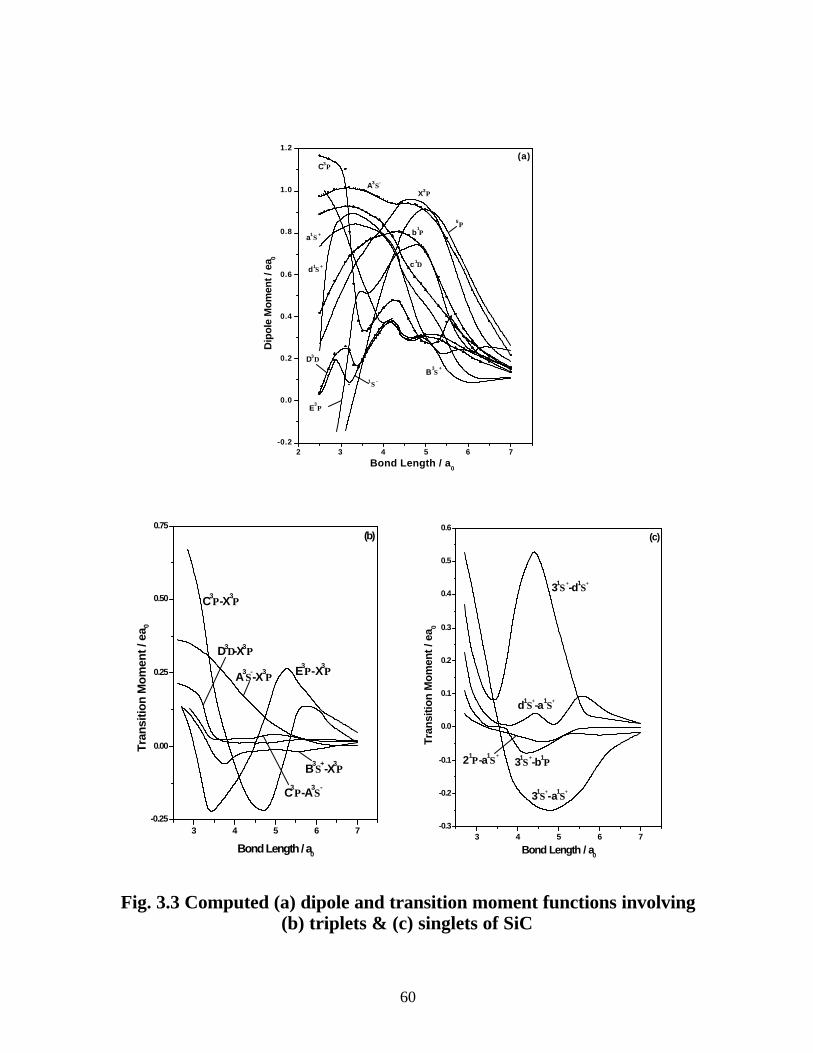
60
2 3 4 5 6 7-0.2
0.0
0.2
0.4
0.6
0.8
1.0
1.2(a)
Bond Length / a0
E3Π
D3∆
1Σ -
C3Π
B3Σ
+
5Π
d1Σ + c1∆
b1Π
a1Σ +
A3Σ-
X3Π
Dip
ole
Mom
ent /
ea 0
3 4 5 6 7-0.25
0.00
0.25
0.50
0.75(b)
C3Π-A3
Σ-
E3Π-X3
ΠA3Σ
--X3Π
B3Σ
+-X3Π
C3Π-X3
Π
D3∆-X3Π
Tran
sitio
n M
omen
t / e
a 0
Bond Length / a0
3 4 5 6 7-0.3
-0.2
-0.1
0.0
0.1
0.2
0.3
0.4
0.5
0.6(c)
31Σ+-d1Σ+
31Σ+-b1Π
d1Σ+-a1Σ+
21Π-a1Σ+
31Σ+-a1Σ+
Tran
sitio
n M
omen
t / e
a 0
Bond Length / a0
Fig. 3.3 Computed (a) dipole and transition moment functions involving (b) triplets & (c) singlets of SiC
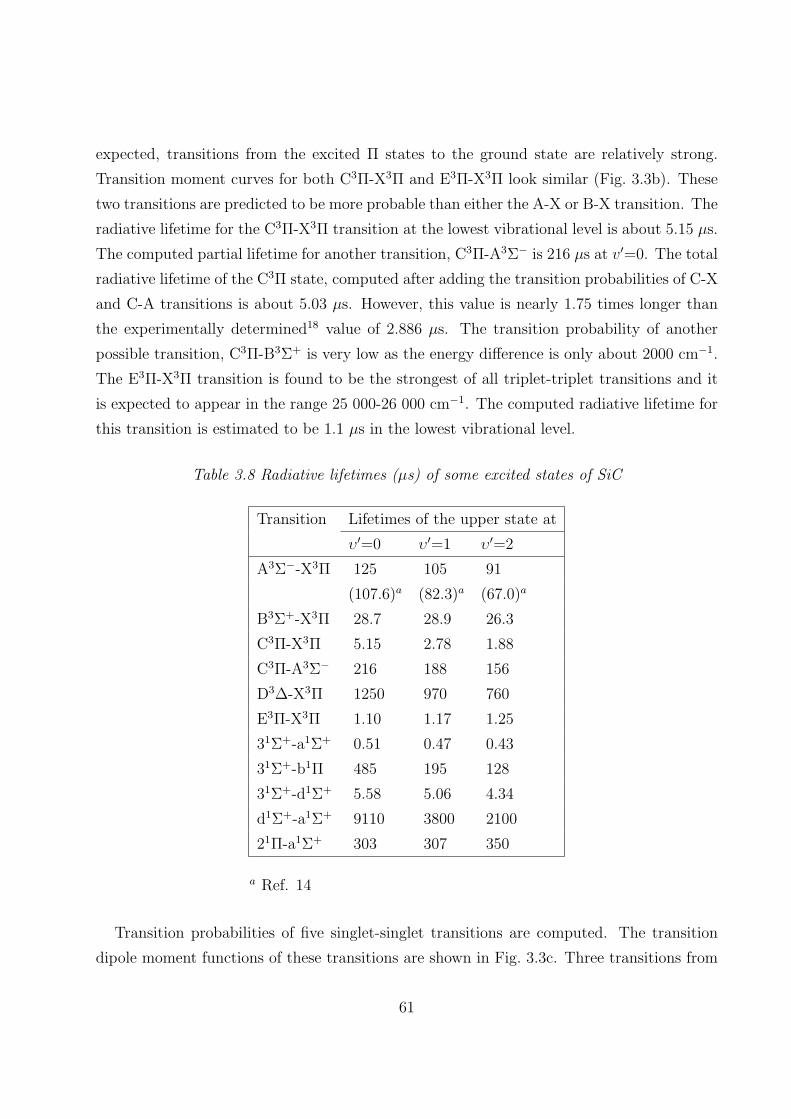
expected, transitions from the excited Π states to the ground state are relatively strong.
Transition moment curves for both C3Π-X3Π and E3Π-X3Π look similar (Fig. 3.3b). These
two transitions are predicted to be more probable than either the A-X or B-X transition. The
radiative lifetime for the C3Π-X3Π transition at the lowest vibrational level is about 5.15 µs.
The computed partial lifetime for another transition, C3Π-A3Σ− is 216 µs at v′=0. The total
radiative lifetime of the C3Π state, computed after adding the transition probabilities of C-X
and C-A transitions is about 5.03 µs. However, this value is nearly 1.75 times longer than
the experimentally determined18 value of 2.886 µs. The transition probability of another
possible transition, C3Π-B3Σ+ is very low as the energy difference is only about 2000 cm−1.
The E3Π-X3Π transition is found to be the strongest of all triplet-triplet transitions and it
is expected to appear in the range 25 000-26 000 cm−1. The computed radiative lifetime for
this transition is estimated to be 1.1 µs in the lowest vibrational level.
Table 3.8 Radiative lifetimes (µs) of some excited states of SiC
Transition Lifetimes of the upper state at
υ′=0 υ′=1 υ′=2
A3Σ−-X3Π 125 105 91
(107.6)a (82.3)a (67.0)a
B3Σ+-X3Π 28.7 28.9 26.3
C3Π-X3Π 5.15 2.78 1.88
C3Π-A3Σ− 216 188 156
D3∆-X3Π 1250 970 760
E3Π-X3Π 1.10 1.17 1.25
31Σ+-a1Σ+ 0.51 0.47 0.43
31Σ+-b1Π 485 195 128
31Σ+-d1Σ+ 5.58 5.06 4.34
d1Σ+-a1Σ+ 9110 3800 2100
21Π-a1Σ+ 303 307 350
a Ref. 14
Transition probabilities of five singlet-singlet transitions are computed. The transition
dipole moment functions of these transitions are shown in Fig. 3.3c. Three transitions from
61

the 31Σ+ state, namely 31Σ+-a1Σ+, 31Σ+-b1Π, and 31Σ+-d1Σ+ are studied. The 31Σ+-
a1Σ+ transition has the largest transition probability. The computed radiative lifetime for
this transition is about 510 ns at v′=0. Adding the transition probabilities of these three
transitions, the total radiative lifetime of the 31Σ+ state is estimated to be 467 ns at the
lowest vibrational level. However, no transition from the 31Σ+ state of SiC is experimentally
known yet. Among the other transitions reported here, the 21Π-a1Σ+ transition is predicted
to be stronger than d1Σ+-a1Σ+. The computed lifetime of the former transition is about
300 µs.
3.4. Summary
Low-lying electronic states of the SiC radical have been studied by using ab initio based
MRDCI calculations which include pseudo potentials of both Si and C atoms. We have
compared the computed spectroscopic constants with the observed and previously calculated
data. At least 23 states of Λ-S symmetries have been reported within 6 eV of energy. Besides
the ground state, the triplets such as A3Σ−, B3Σ+, and C3Π are experimentally well studied.
Four singlets, a1Σ+, b1Π, c1∆, and d1Σ+ are located in between 5000 and 13 000 cm−1. The
lowest bound state of the quintet spin multiplicity is 5Π which is located around 14 460 cm−1.
Potential energy curves of all the low-lying states are constructed. The excited E3Π state
is not previously known. We have predicted that the transition energy of the E3Π state
is about 25 875 cm−1. Its re and ωe are 1.94 A and 600 cm−1, respectively. A number of
singlet and quintet states exist in between 30 000 and 50 000 cm−1. Some of these singlet
and quintet states are strongly bound. The ground-state dipole moment (µe) is calculated
to be 1.62 D with a Si+C− polarity. All the other excited states have the same sense of
polarity. The largest dipole moment is reported for the A3Σ− state. The spin-orbit effects
on the low-lying states of SiC are not significant to change their spectroscopic properties.
The largest splitting among the four spin components of X3Π is only 100 cm−1 with X3Π2
being the lowest one. The two components of both A3Σ− and B3Σ+ are inseparable. Several
electric dipole allowed triplet-triplet and singlet-singlet transitions at the Λ-S level have
been studied. The strongest triplet-triplet transition is predicted to be E3Π-X3Π in the
range 25 000-26 000 cm−1. The computed radiative lifetime of this transition at v′=0 is
1.1 µs. However, such a transition has not been observed yet. The computed radiative
lifetime of C3Π is 5.03 µs compared to the experimentally determined value of 2.886 µs.
62

The lifetimes for A-X and B-X transitions are predicted to be 125 and 28.7 µs, respectively.
Of three transitions from the excited 31Σ+ state, the 31Σ+-a1Σ+ transition has the largest
transition probability and it has the shortest lifetime (τ=510 ns at v′=0) among all the
transitions studied here.
63

3.5 References
1 A. Suzuki, Prog. Theor. Phys. 62, 936 (1979).
2 V.E. Bondybey, J. Phys. Chem. 86, 3396 (1982).
3 D.L. Michalopoulos, M.E. Geusic, P.R.R. Langridge-Smith, R.E. Smalley, J. Chem. Phys.
80, 3556 (1984).
4 B.L. Lutz, J.A. Ryan, Astrophys. J. 194, 753 (1974).
5 P.J. Bruna, S.D. Peyerimhoff, R.J. Buenker, J. Chem. Phys. 72, 5437 (1980).
6 C.M. Rohlfing, R.L. Martin, J. Phys. Chem. 90, 2043 (1986).
7 J. Anglada, P.J. Bruna, S.D. Peyerimhoff, R.J. Buenker, Mol. Phys. 16, 2469 (1983).
8 H. Dohman, P.J. Bruna, S.D. Peyerimhoff, R.J. Buenker, Mol. Phys. 51, 1109 (1984).
9 M. Larsson, J. Phys. B: At. Mol. Phys. 19, L261 (1986).
10 C. Bauschlicher Jr., S.R. Langhoff, J. Chem. Phys. 87, 2919 (1987).
11 P.F. Bernath, S.A. Rogers, L.C. O′Brien, C.R. Brazier, Phys. Rev. Lett. 60, 197 (1988).
12 J. Cernicharo, C.A. Gottlieb, M. Guelin, P. Thaddeus, J.M. Vrtilek, Astrophys. J. Lett.
341, L25 (1989).
13 C.R. Brazier, L.C. O′Brien, P.F. Bernath, J. Chem. Phys. 91, 7384 (1989).
14 S.R. Langhoff, C. Bauschlicher Jr., J. Chem. Phys. 93, 42 (1990).
15 F.L. Sefyani, J. Schamps, Astrophys. J. 434, 816 (1994).
16 J.M.L. Martin, J.P. Francois, R. Gijbels, J. Chem. Phys. 92, 6655 (1990).
17 T.J. Butenhoff, E.A. Rohlfing, J. Chem. Phys. 95, 3939 (1991).
18 M. Ebben, M. Drabbels, J.J. ter Meulen, J. Chem. Phys. 45, 2292 (1991).
19 A.D. McLean, B. Liu, G.S. Chandler, J. Chem. Phys. 97, 8459 (1992).
20 R. Mollaaghababa, C.A. Gottlieb, J.M. Vrtilek, P. Thaddens, Astrophys. J. Lett. 352,
L21 (1990).
21 R. Mollaaghababa, C.A. Gottlieb, J.M. Vrtilek, P. Thaddens, J. Chem. Phys. 98, 968
(1993).
22 A.I. Boldyrev, J. Simons, V.G. Zahrzewski, W. Von Niessen, J. Phys. Chem. 98, 1427
(1994).
64

23 M. Grutter, P. Freivogel, J.P. Mair, J. Phys. Chem. A 101, 275 (1997).
24 M.N. Deo, K.J. Kawaguchi, Mol. Spectrosc. 228, 76 (2004).
25 L.F. Pacios, P.A. Christiansen, J. Chem. Phys. 82, 2664 (1985).
26 J.M.O. Matos, V. Kello, B.O. Ross, A.J. Sadlej, J. Chem. Phys. 89, 423 (1988).
27 R.J. Buenker, S.D. Peyerimhoff, Theo. Chim. Acta 35, 33 (1974).
28 R.J. Buenker, S.D. Peyerimhoff, Theo. Chim. Acta 39, 217 (1975).
29 R.J. Buenker, Int. J. Quantum Chem. 29, 435 (1986).
30 R.J. Buenker, in: P. Burton (Ed.), Proc. Workshop on Quantum Chemistry and Molecular
Physics in Wollongong, Wollongong, Australia, 1980.
31 R.J. Buenker, in: R. Carbo (Ed.), Studies in Physical and Theoretical Chemistry, vol. 21,
Current Aspects of Quantum Chemistry, Elsevier, Amsterdam, p.17, 1982.
32 R.J. Buenker, S.D. Peyerimhoff, W. Butscher, Mol. Phys. 35, 771 (1978).
33 R.J. Buenker, R.A. Philips, J. Mol. Struct. (Theochem) 123, 291 (1985).
34 S. Krebs, R.J. Buenker, J. Chem. Phys. 103, 5613 (1995).
35 E.R. Davidson, in: R. Daudel, B. Pullman (Eds.), The World of Quantum Chemistry,
Reidel, Dordrecht, The Netherland, 1974.
36 G. Hirsch, P.J. Bruna, S.D. Peyerimhoff, R.J. Buenker, Chem. Phys. Lett. 52, 442
(1977).
37 A.B. Alekseyev, R.J. Buenker, H.-P. Lieberman, G. Hirsch, J. Chem. Phys. 100, 2989
(1994).
38 C.E. Moore, Tables of Atomic Energy Levels: vols. I–III, U.S. National Bureau of
Standards: Washington, DC, 1971.
39 J. Drowart, G. De Maria, M.G. Inghram, J. Chem. Phys. 29, 1015 (1958).
40 G. Verhaegen, F.E. Stafford, J. Drowart, J. Chem. Phys. 40, 1622 (1964).
41 P.J. Bruna, C. Petrongolo, R.J. Buenker, S.D. Peyerimhoff, J. Chem. Phys. 74, 4611
(1981).
42 A. Pramanik, K.K. Das, J. Mol. Spectrosc. 224, 13 (2007).
65

CHAPTER – 4
ELECTRONIC STRUCTURE AND
SPECTROSCOPIC PROPERTIES OF SiC+ AND SiC-

4.1. Introduction
In chapter 3 we have discussed the astrophysical importance of the small molecules of
silicon and carbon. The simple SiC radical has been identified in interstellar clouds and
stellar atmospheres.1−2 Although in recent years the radical has been widely studied both
experimentally as well as theoretically3−11, data on the electronic states of the positive and
negative ions of SiC are not much available in the literature. No photoelectron spectrum of
the SiC radical has been reported as yet. First theoretical calculations of SiC+ have been
carried out by Bruna et al.9 using ab initio MRDCI method. Potential energy curves of
12 Λ-S states and spectroscopic constants of six bound states of SiC+ have been reported
by these authors. Ionization potentials and dissociation energies have also been determined.
They have also calculated potential energy curves of the isovalent Si+2 ion. Boldyrev et al.10
have computed ionization energies (vertical as well as adiabatic) and electron affinities of a
series of SinC and SinO (n=1-3) molecules using four different abinitio based methods. The
optimized bond lengths and vibrational frequencies of the SiC+ ion in its 4Σ+, 2Πi, and 2Σ+
states have been estimated.
The negative ion, SiC− is isovalent with C−2 for which the electronic spectrum is known
in detail both experimentally and theoretically. Moreover, the ionic species, C− and Si− are
known to exist in different electronic states. It is therefore, expected to have many stable
low-lying electronic states of SiC−. Grutter et al.12 identified SiC− in 5K neon matrices for
the first time using mass selected deposition. Two transitions, namely A2Π-X2Σ+ and B2Σ+-
X2Σ+ of SiC− with the absorption band origins at 3538 and 21 683 cm−1, respectively were
observed. Spectroscopic constants of X2Σ+, A2Π, and B2Σ+ states were determined from
the experimental data. The detachment of electrons from the SiC− ion in the B2Σ+ state
embedded in the neon matrix was established to take place between 3.5 and 4.0 eV by means
of wavelength-selected irradiation. Anglada et al.13 carried out the first theoretical study
on the low-lying electronic states of SiC− using multireference CI method. These authors
reported two bound states, X2Σ+ and A2Π in contrast to the results for isovalent C−2 in
which a third species like 2Σ+u is known. The calculated electron affinity of SiC obtained by
these authors has been found to be 1.98 eV. Singles and doubles CI calculations at the CISD
and CISD+Q levels from single SCF configuration were performed by McLean et al.14 on a
number of diatomic species including SiC and SiC−. Spectroscopic aspects of the 2Π state of
SiC− were also reported by these authors. Hunsicker and Jones15 made a density functional
66

study with simulated anealing on SixCy and SixC−y species with x+y ≤ 8. The ground-state
bond length of SiC− has been found to be 1.677 A. Recently, Cai and Francois16 have studied
potential energy curves and spectroscopic constants of the X2Σ+, A2Π, and B2Σ+ states of
SiC− using internally contracted multireference CI calculations. They have also computed
adiabatic and vertical electron affinities of SiC to the A2Π and B2Σ+ states of SiC−, and the
electronic transition moment functions for both B2Σ+-X2Σ+ and B2Σ+-A2Π transitions.
In this chapter, we report the electronic states and spectroscopic properties of SiC+ and
SiC− from a large scale MRDCI calculation. Potential energy curves of 14 low-lying Λ-S
states of the cation and 21 Λ-S states of the anion are constructed from the computed CI
energies. Spectroscopic constants of all these states are estimated by fitting the potential
curves. Effects of the spin-orbit coupling on the spectroscopic properties of the Ω states
of SiC+ are studied. Transition moments of the electric dipole allowed and spin-forbidden
transitions are computed. Subsequently, the partial radiative lifetimes for some of these
transitions are also estimated. The computed vertical as well as adiabatic ionization energies
and electron affinities of SiC are reported and compared with the available data.
4.2. Computational details
4.2.1 RECPs and basis sets
The whole calculations of SiC+ are performed using similar pseudo potentials and basis
sets of Pacios and Christiansen17,18 for Si and C as those used in case of SiC (sec. 3.2.1). This
is also important to determine the ionization potential of SiC. In case of SiC−, the RECPs
are same but an additional set of p polarization functions of an exponent of 0.034 a−20 and
another set of d polarization functions of an exponent of 0.15 a−20 are included in the basis
set of carbon. This essentially improves the correlation due to one extra electron. In order
to calculate the electron affinity of SiC, we have performed the ground state CI calculation
of SiC with these additional basis sets.
4.2.2 SCF MOs and CI
Self-consistent-field (SCF) calculations have been performed for the (σπ2)4Σ− state of
SiC+ and (σπ4)2Σ+ state of SiC− at different internuclear distances in the range 2.5-20 a0
using the AREP and basis sets mentioned above. The calculations are carried out in the
67
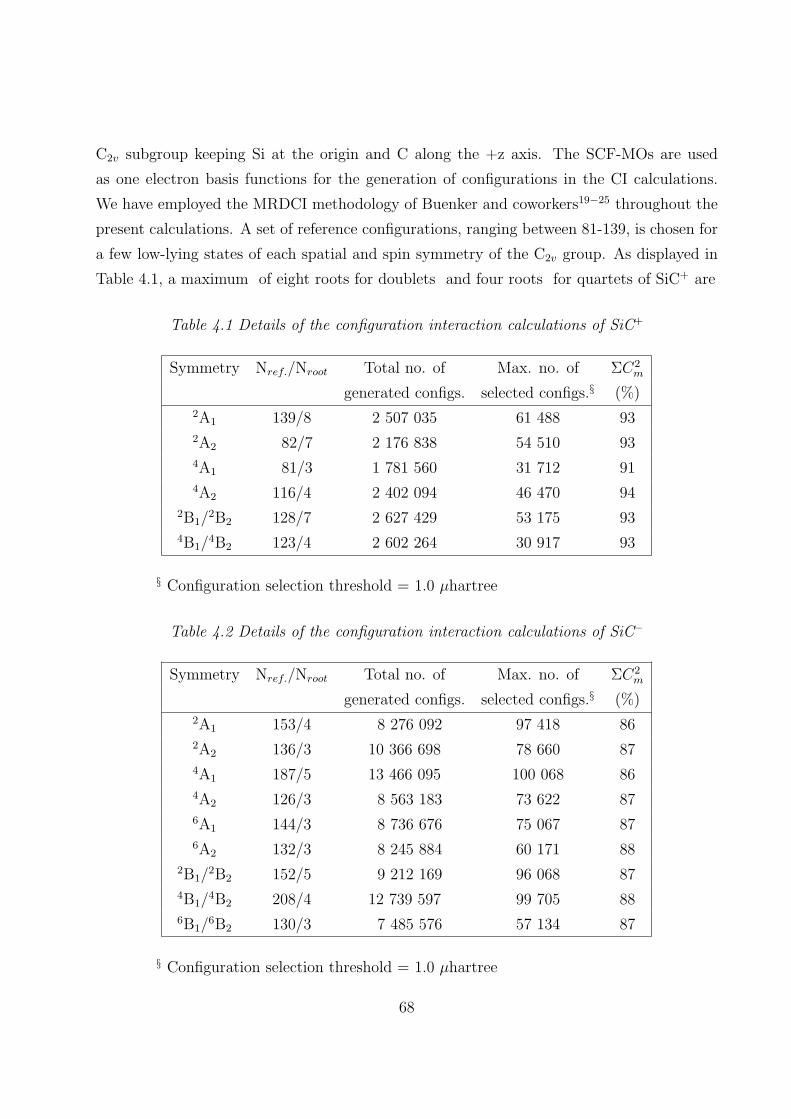
C2v subgroup keeping Si at the origin and C along the +z axis. The SCF-MOs are used
as one electron basis functions for the generation of configurations in the CI calculations.
We have employed the MRDCI methodology of Buenker and coworkers19−25 throughout the
present calculations. A set of reference configurations, ranging between 81-139, is chosen for
a few low-lying states of each spatial and spin symmetry of the C2v group. As displayed in
Table 4.1, a maximum of eight roots for doublets and four roots for quartets of SiC+ are
Table 4.1 Details of the configuration interaction calculations of SiC+
Symmetry Nref./Nroot Total no. of Max. no. of ΣC2m
generated configs. selected configs.§ (%)2A1 139/8 2 507 035 61 488 932A2 82/7 2 176 838 54 510 934A1 81/3 1 781 560 31 712 914A2 116/4 2 402 094 46 470 94
2B1/2B2 128/7 2 627 429 53 175 934B1/4B2 123/4 2 602 264 30 917 93
§ Configuration selection threshold = 1.0 µhartree
Table 4.2 Details of the configuration interaction calculations of SiC−
Symmetry Nref./Nroot Total no. of Max. no. of ΣC2m
generated configs. selected configs.§ (%)2A1 153/4 8 276 092 97 418 862A2 136/3 10 366 698 78 660 874A1 187/5 13 466 095 100 068 864A2 126/3 8 563 183 73 622 876A1 144/3 8 736 676 75 067 876A2 132/3 8 245 884 60 171 88
2B1/2B2 152/5 9 212 169 96 068 874B1/4B2 208/4 12 739 597 99 705 886B1/6B2 130/3 7 485 576 57 134 87
§ Configuration selection threshold = 1.0 µhartree
68

optimized. All single and double excitations are carried out from the reference configurations
generating a large number of configurations of the order of 2-3 millions. In case of SiC−, 4-5
roots are calculated for doublets and quartets, while the number of lowest roots optimized
for sextets is three. Table 4.2 shows that, the similar type of configuration interactions from
130-208 reference configurations of SiC− generate 7-12 million of configurations. However, a
configuration selection with a threshold of 1.0 µhartree has been made to reduce the number
of selected configurations for the CI below 200 000. The table direct CI version26 of the
MRDCI codes has been used here. The sums of the squares of the CI coefficients remain
around 0.90 for both the species. The energy extrapolation method has been used to estimate
CI energies at zero threshold. The multireference analogue of the Davidson’s correction27,28
takes care of the higher excitations. The full CI energies of the lowest few roots in each
symmetry for both SiC+ and SiC− are thus estimated.
4.2.3 Spin-orbit interaction
The spin-orbit coupling has been included by two-step variational calculations. The spin-
independent CI wave functions are multiplied with appropriate spin functions which trans-
form according to the symmetry. The diagonals of the spin-included Hamiltonian matrix
consist of the estimated full CI energies and the off-diagonals are the spin-orbit matrix ele-
ments which are calculated by the Wigner-Eckart theorem and spin-projection method. The
1/2, 3/2, and 5/2 states of SiC+ are obtained in two degenerate representations, E1 and E2
of C22v group. Potential energy curves of the low-lying bound states are fitted to estimate
the spectroscopic constants (re, Te, ωe), vibrational energies, and vibrational wave functions.
In the subsequent calculations, transition dipole moments of important dipole allowed and
spin-forbidden transitions and their partial radiative lifetimes are computed.
4.3. Results and discussion
4.3.1 Spectroscopic constants and potential energy curves of Λ–S states
A. SiC+
Six doublets and six quartets of Σ+, Σ−(2), Π(2), and ∆ symmetries of SiC+ correlate
with the lowest dissociation limit, Si+(2Pu)+C(3Pg). The second asymptote comprising the
ground-state Si+ and the first excited state (1Dg) of the carbon atom correlates with nine
69
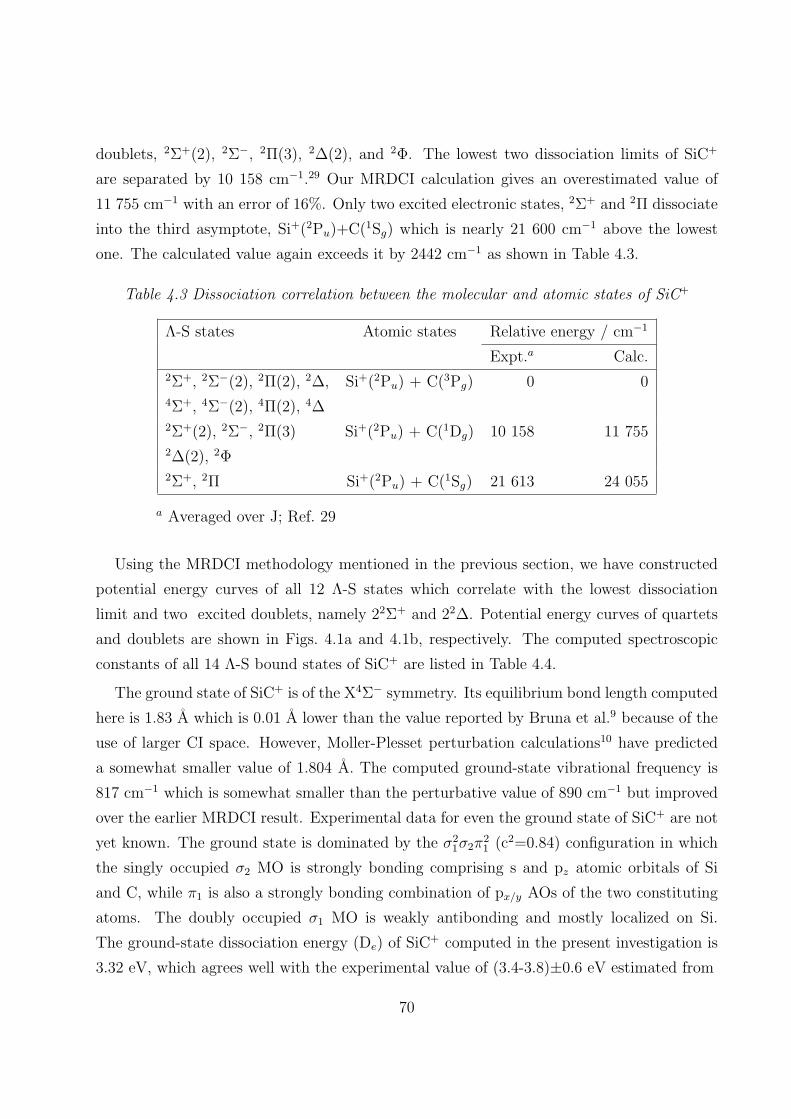
doublets, 2Σ+(2), 2Σ−, 2Π(3), 2∆(2), and 2Φ. The lowest two dissociation limits of SiC+
are separated by 10 158 cm−1.29 Our MRDCI calculation gives an overestimated value of
11 755 cm−1 with an error of 16%. Only two excited electronic states, 2Σ+ and 2Π dissociate
into the third asymptote, Si+(2Pu)+C(1Sg) which is nearly 21 600 cm−1 above the lowest
one. The calculated value again exceeds it by 2442 cm−1 as shown in Table 4.3.
Table 4.3 Dissociation correlation between the molecular and atomic states of SiC+
Λ-S states Atomic states Relative energy / cm−1
Expt.a Calc.2Σ+, 2Σ−(2), 2Π(2), 2∆, Si+(2Pu) + C(3Pg) 0 04Σ+, 4Σ−(2), 4Π(2), 4∆2Σ+(2), 2Σ−, 2Π(3) Si+(2Pu) + C(1Dg) 10 158 11 7552∆(2), 2Φ2Σ+, 2Π Si+(2Pu) + C(1Sg) 21 613 24 055
a Averaged over J; Ref. 29
Using the MRDCI methodology mentioned in the previous section, we have constructed
potential energy curves of all 12 Λ-S states which correlate with the lowest dissociation
limit and two excited doublets, namely 22Σ+ and 22∆. Potential energy curves of quartets
and doublets are shown in Figs. 4.1a and 4.1b, respectively. The computed spectroscopic
constants of all 14 Λ-S bound states of SiC+ are listed in Table 4.4.
The ground state of SiC+ is of the X4Σ− symmetry. Its equilibrium bond length computed
here is 1.83 A which is 0.01 A lower than the value reported by Bruna et al.9 because of the
use of larger CI space. However, Moller-Plesset perturbation calculations10 have predicted
a somewhat smaller value of 1.804 A. The computed ground-state vibrational frequency is
817 cm−1 which is somewhat smaller than the perturbative value of 890 cm−1 but improved
over the earlier MRDCI result. Experimental data for even the ground state of SiC+ are not
yet known. The ground state is dominated by the σ21σ2π
21 (c2=0.84) configuration in which
the singly occupied σ2 MO is strongly bonding comprising s and pz atomic orbitals of Si
and C, while π1 is also a strongly bonding combination of px/y AOs of the two constituting
atoms. The doubly occupied σ1 MO is weakly antibonding and mostly localized on Si.
The ground-state dissociation energy (De) of SiC+ computed in the present investigation is
3.32 eV, which agrees well with the experimental value of (3.4-3.8)±0.6 eV estimated from
70
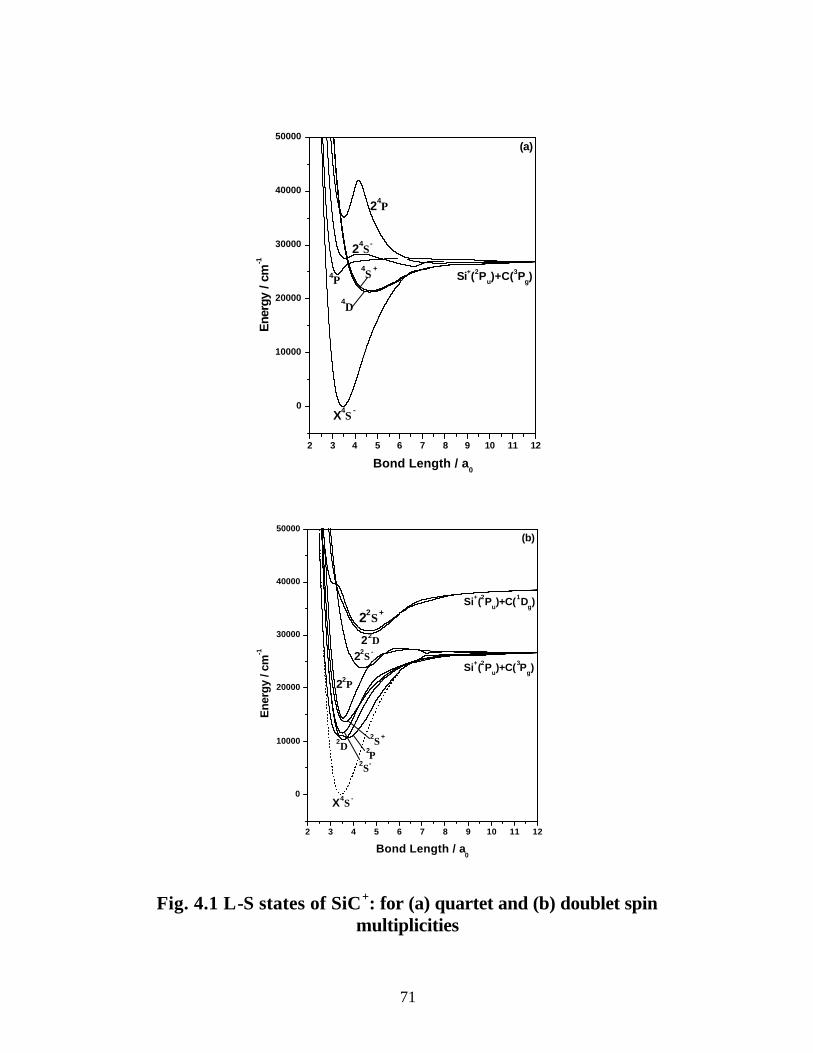
71
2 3 4 5 6 7 8 9 10 11 12
0
10000
20000
30000
40000
50000(a)
Si+(2Pu)+C(3Pg)4Σ
+
4∆
24Π
4Π
24Σ
-
X4Σ
-
Ene
rgy
/ cm
-1
Bond Length / a0
2 3 4 5 6 7 8 9 10 11 12
0
10000
20000
30000
40000
50000
Si+(2Pu)+C(1D
g)
(b)
22Σ+
22∆
Si+(2Pu)+C(3P
g)
22Σ -
22Π
2Σ+
2Σ-
2Π2∆
X4Σ -
Ene
rgy
/ cm
-1
Bond Length / a0
Fig. 4.1 Λ-S states of SiC+: for (a) quartet and (b) doublet spin multiplicities
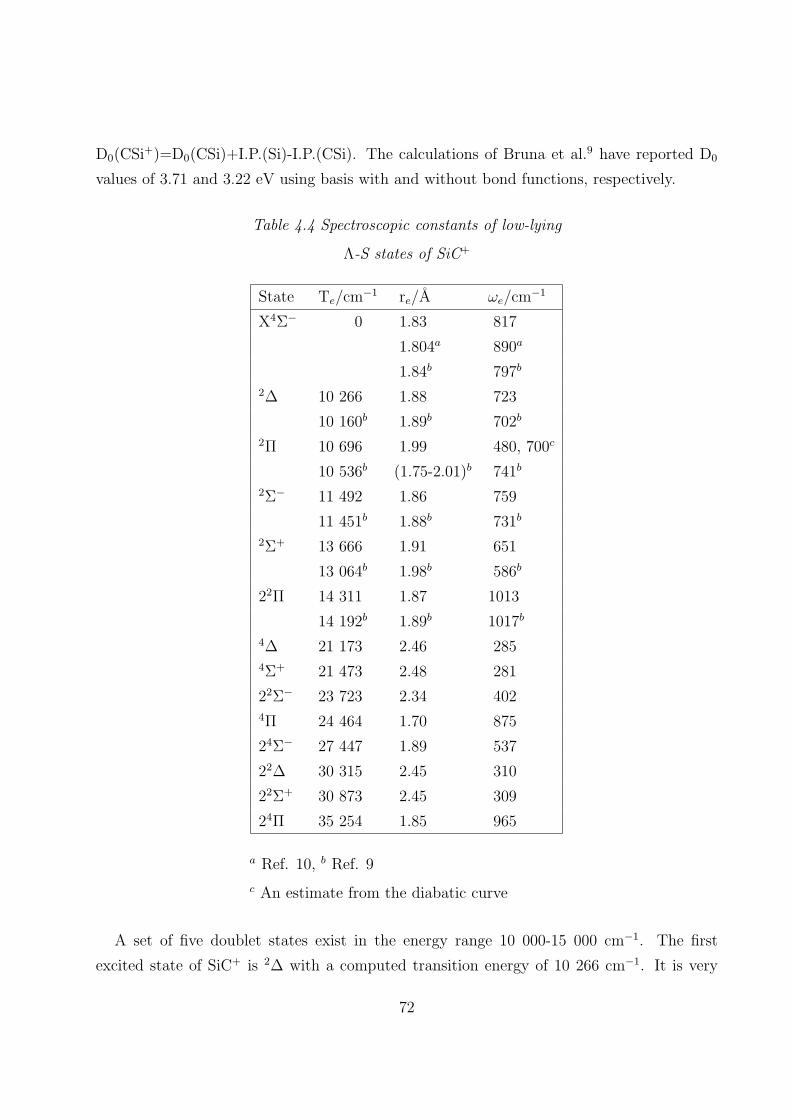
D0(CSi+)=D0(CSi)+I.P.(Si)-I.P.(CSi). The calculations of Bruna et al.9 have reported D0
values of 3.71 and 3.22 eV using basis with and without bond functions, respectively.
Table 4.4 Spectroscopic constants of low-lying
Λ-S states of SiC+
State Te/cm−1 re/A ωe/cm−1
X4Σ− 0 1.83 817
1.804a 890a
1.84b 797b
2∆ 10 266 1.88 723
10 160b 1.89b 702b
2Π 10 696 1.99 480, 700c
10 536b (1.75-2.01)b 741b
2Σ− 11 492 1.86 759
11 451b 1.88b 731b
2Σ+ 13 666 1.91 651
13 064b 1.98b 586b
22Π 14 311 1.87 1013
14 192b 1.89b 1017b
4∆ 21 173 2.46 2854Σ+ 21 473 2.48 281
22Σ− 23 723 2.34 4024Π 24 464 1.70 875
24Σ− 27 447 1.89 537
22∆ 30 315 2.45 310
22Σ+ 30 873 2.45 309
24Π 35 254 1.85 965
a Ref. 10, b Ref. 9
c An estimate from the diabatic curve
A set of five doublet states exist in the energy range 10 000-15 000 cm−1. The first
excited state of SiC+ is 2∆ with a computed transition energy of 10 266 cm−1. It is very
72

strongly bound with a binding energy of about 2.05 eV. The computed re and ωe of the 2∆
state match well with the earlier results of Bruna et al.9 At equilibrium, the state is mainly
characterized by the same dominant configuration as that of the ground state. However, two
other configurations, σ21σ2π1π2 (c2=0.07) and σ1σ
22π
21 (c2=0.04) also contribute to 2∆. The
two roots of 2Π interact strongly, as is evident from the avoided crossing between the two
potential curves in Fig. 4.1b. The curve-crossing point is located around 3.47 a0. Analysing
the CI wave functions for these two roots of 2Π symmetry, it is revealed that at bond lengths
shorter than 3.47 a0, the lower root is dominated by σ21π
31 and σ1σ2π
31 configurations, while
the higher one is described mainly by σ21σ
22π1. The situation is reversed after the avoided
crossing. In Table 4.5, we have tabulated the important configurations of these two roots at
six representative bond distances. The adiabatic potential energy curves of both 2Π and
Table 4.5 Composition (% contribution) of the lowest two roots of 2Π state of SiC+
in the bond length range 2.8-4.0 a0
r(a0) 2Π 22Π
2.8 σ21π
31(42), σ1σ2π
31(32), σ1σ2π
21π2(11) σ2
1σ22π1(73), σ1σ2π
31(8)
3.1 σ21π
31(43), σ1σ2π
31(25), σ1σ2π
21π2(12) σ2
1σ22π1(80), σ1σ2π
31(4)
3.3 σ21π
31(42), σ1σ2π
31(17), σ2
1σ22π1(12), σ2
1σ22π1(72), σ1σ2π
31(7)
σ1σ2π21π2(11)
3.5 σ21σ
22π1(50), σ2
1π31(24), σ1σ2π
21π2(6), σ2
1σ22π1(33), σ2
1π31(21), σ1σ2π
31(15),
σ1σ2π31(5) σ1σ2π
21π2(9), σ2
1π21π2(6)
3.7 σ21σ
22π1(74), σ2
1π31(8) σ2
1π31(35), σ1σ2π
31(15), σ1σ2π
21π2(12),
σ21π
21π2(11), σ2
1σ22π1(9)
4.0 σ21σ
22π1(79), σ2
1π31(4) σ2
1π31(37), σ2
1π21π2(18), σ1σ2π
21π2(13),
σ1σ2π31(11), σ2
1σ22π1(3)
22Π are fitted for estimating the spectroscopic constants. The potential energy curve of2Π is seen to have a double well. Although the inner well is not prominent, the minimum
lies around 1.77 A and does not hold any vibrational levels. On the other hand, the long-
distant minimum is found to be near 1.99 A. The corresponding Te and ωe are computed
to be 10 696 cm−1 and 480 cm−1, respectively. The vibrational frequency for the 2Π state
reported by Bruna et al.9 is 741 cm−1 which may be due to the fitting of the diabatic curve.
73

The corresponding ωe of the diabatic curve in our calculations is estimated to be about
700 cm−1. The interaction between the 2Π and 22Π states causes the 22Π state to have a
large ωe value of 1013 cm−1, which compares well to the previously calculated value. The
potential minimum of 22Π is located around 1.87 A. The energy gap between the minima of
the two roots of 2Π is about 3600 cm−1.
Potential minima of the next two doublets belong to 2Σ− and 2Σ+ symmetries. Both
states are dominated by the same configuration, σ21σ2π
21, as that in the ground state and like
2∆ state, two other excited configurations namely, σ1σ22π
21 and σ2
1σ2π1π2 contribute to a small
extent (Table 4.6). It may be noted that all four states of SiC+, X4Σ−, 2∆, 2Σ−, and 2Σ+ lie
within 14 000 cm−1. The computed Te of the 2Σ− state is about 11 492 cm−1 with re=1.86 A
and ωe=759 cm−1. The separation between 2Σ+ and 2Σ− is computed to be 2174 cm−1
which is larger than the value of 1613 cm−1 predicted in the earlier CI calculations.9 The
bond length of the 2Σ+ state is predicted to be shortened and the vibrational frequency is
predicted to be increased, relative to the previous calculation.
Table 4.6 Composition of Λ-S states of SiC+ at equilibrium bond length
State Configuration (% contribution)
X4Σ− σ21σ2π
21(84)
2∆ σ21σ2π
21(72), σ2
1σ2π1π2(7)), σ1σ22π
21(4)
2Π σ21σ
22π1(75), σ2
1π31(8)
2Σ− σ21σ2π
21(70), σ1σ
22π
21(7), σ2
1σ2π1π2(6))2Σ+ σ2
1σ2π21(65), σ2
1σ2π1π2(11), σ1σ22π
21(5), σ1σ
22π1π2(4))
22Π σ21π
31(28), σ2
1σ22π1(20), σ1σ2π
31(16), σ1σ2π
21π2(10), σ2
1π21π2(10)
4∆ σ21σ2π1π2(72), σ2
1σ3π1π2(8), σ1σ22π1π2(6)
4Σ+ σ21σ2π1π2(72), σ2
1σ3π1π2(8), σ1σ22π1π2(6)
22Σ− σ21σ2π1π2(64), σ2
1σ3π1π2(10), σ21σ2π
21(7)
4Π σ1σ2π31(52), σ1σ2π
21π2(33), σ2
1π21π2(5)
24Σ− σ1σ22π
21(32), σ1σ
22π1π2(25), σ2
1σ2π1π2(23), σ21σ2π
21(6)
22∆ σ21σ2π1π2(65), σ1σ
22π1π2(8), σ2
1σ3π1π2(7), σ21σ2π
21(4)
22Σ+ σ21σ2π1π2(67), σ1σ
22π1π2(9), σ2
1σ3π1π2(6), σ21σ2π
21(3)
24Π σ1σ2π21π2(30), σ2
1π21π2(28), σ1σ2π
31(26)
The next two quartet states, 4∆ and 4Σ+ have transition energies of 21 173 and 21 473 cm−1,
74

respectively. The potential energy curves (Fig. 4.1a) of these two states have similar char-
acteristics. Both of them dissociate into the lowest limit and are characterized by σ21σ2π1π2
(c2=0.72), σ21σ3π1π2 (c2=0.08), and σ1σ
22π1π2 (c2=0.06) configurations. The re and ωe of
these two states are very similar and their potential minima are shallow and shifted towards
the longer bond distance region. Neither of these two states is important from the spectro-
scopic point of view as their transitions to the ground state are forbidden. The 22Σ− state,
which originates from the same σ21σ2π1π2 configuration as the previous two quartets, also has
a shallow minimum at re=2.34 A with a smaller ωe of 402 cm−1. The computed transition
energy of the 22Σ− state at re is about 23 723 cm−1. The lowest two roots of 4Π also interact
strongly. The avoided crossing between the two 4Π roots has made the potential minimum
of the lower 4Π state very sharp but with a small binding energy of 0.29 eV. As a result of
the strong perturbation, the 4Π state has a shorter bond length (re=1.70 A) and larger ωe
(=875 cm−1). The upper state, 24Π has a deeper potential well with ωe=965 cm−1 and the
minimum is shifted at 1.85 A around which the avoided crossing takes place. Energetically,
the potential minimum of 24Π lies more than 10 000 cm−1 above 4Π. The analysis of CI
wave functions reveal that the electronic configurations, which contribute in the region of the
avoided crossing, are σ1σ2π31, σ1σ2π
21π2, σ2
1π21π2 and the compositions of both the 4Π states
are complex in nature. Two more excited doublets, 22∆ and 22Σ+ look very similar to the
corresponding lowest two quartet state. Potential energy curves and even the compositions
of these doublet states resemble those of the quartets. The computed transition energies of
22∆ and 22Σ+ states are estimated to be 30 315 and 30 873 cm−1, respectively with almost
the same re and ωes.
B. SiC−
Doublets, quartets, and sextets of Σ+ and Π symmetries of SiC− correlate with the lowest
asymptote, Si−(4Su)+C(3Pg). The second dissociation limit, Si(3Pg)+C−(4Su), which lies
only 0.12 eV away, correlates with another six excited Λ-S states of the same symmetries as
the previous one. A set of doublets and quartets of Σ+(2), Σ−, Π(3), ∆(2), and Φ symmetries
dissociate into Si−(2Du)+C(3Pg) with the observed atomic splitting (2Du-4Su) of 6952 cm−1
31 for Si−. The dissociation correlation between the atomic states and the corresponding
molecular states is shown in the Table 4.7. Potential energy curves of 21 Λ-S states of
doublet, quartet, and sextet spin multiplicities are shown in Figs. 4.2a-c, respectively, while
their spectroscopic constants (Te, re, ωe) are given in Table 4.8.
75
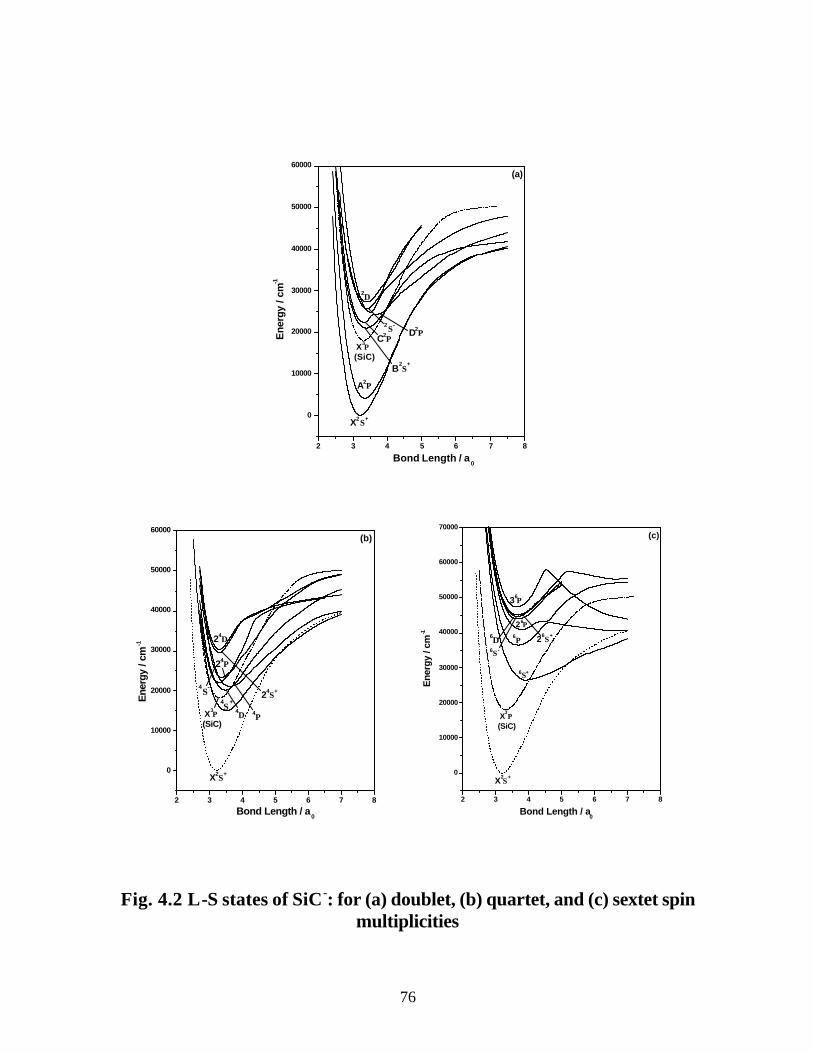
76
2 3 4 5 6 7 8
0
10000
20000
30000
40000
50000
60000(a)
2∆
2Σ-
D2ΠC2Π
B2Σ
+
X3Π
(SiC)
A2Π
X2Σ+
Ene
rgy
/ cm
-1
Bond Length / a0
2 3 4 5 6 7 8
0
10000
20000
30000
40000
50000
60000(b)
4Σ
-24Σ+
24∆
24Π
X3Π
(SiC)
X2Σ+
4Π4∆
4Σ
+
Ene
rgy
/ cm
-1
Bond Length / a0
2 3 4 5 6 7 8
0
10000
20000
30000
40000
50000
60000
70000(c)
26Σ
+
36Π
6Σ-
26Π6∆ 6
Π
X3Π
(SiC)
X2Σ
+
6Σ+
Bond Length / a0
Ene
rgy
/ cm
-1
Fig. 4.2 Λ-S states of SiC-: for (a) doublet, (b) quartet, and (c) sextet spin
multiplicities

Table 4.7 Dissociation correlation between the molecular and atomic states of SiC−
Λ-S states Atomic states Relative energya / cm−1
2Σ+, 2Π, 4Σ+, 4Π, 6Σ+, 6Π Si−(4Su) + C(3Pg) 02Σ+, 2Π, 4Σ+, 4Π, 6Σ+, 6Π Si(3Pg) + C−(4Su) 9682Σ+(2), 2Σ−, 2Π(3), 2∆(2), 2Φ, Si−(2Du) + C(3Pg) 69524Σ+(2), 4Σ−, 4Π(3), 4∆(2), 4Φ
a Averaged over J; Ref. 29, 31
The bond length of SiC− in the ground state (X2Σ+) is 1.69 A which compares well
with the previously calculated values. The CMRCI calculations of Cai and Francois16 at
various level predicted the ground-state bond length in the range 1.6794-1.7286 A. Their
best estimated ωe value of 1026.3 cm−1 agrees well with the present value of 1056 cm−1. The
second order Moller-Plesset perturbation (MP2) level calculations10 have resulted in a longer
bond length and a larger frequency for the ground state of SiC−. Experimental molecular
constants of SiC− in the ground state are not yet known. The ground state is dominated
by a pair of configurations, σ21σ2π
41 (c2=0.34) and σ1σ
22π
41(c2=0.28). The σ1 MO is strongly
antibonding comprising s and pz AOs of both Si and C atoms, while σ2 mainly consists of
their bonding combination. The π1 MO is a strongly bonding combination of px/y AOs of
the two constituting atoms. The computed ground-state dissociation energy (De) of SiC− is
about 5.14 eV which compares well with the previous value of 5.29 eV predicted from the
CMRCI calculations.16
The first excited state of the anion is A2Π, which lies close to the ground state. Its
computed transition energy is larger than the experimental value of 3556 cm−1 by 330 cm−1
only. Although the experimental bond length of A2Π is not known, the present re value
of 1.77 A supports the earlier theoretical prediction. The observed A2Π←X2Σ+ electronic
transition has a band origin at 3538 A in 5K neon matrices using mass selected deposition.12
The reported ωe of A2Π matches well with the value obtained in the present study (Table 4.8).
The state is dominated by σ21σ
22π
31 with three other open-shell configurations contributing
to a smaller extent as shown in Table 4.9. The next two quartets, 4Σ+ and 4∆, which
originate from the same set of configurations, are strongly bound. The 4∆ state has a
bond length shorter than 4Σ+ by about 0.05 A. The two states are separated by about
4500 cm−1 with their ωes lying in the range 810-850 cm−1. Both the quartets are dominated
77
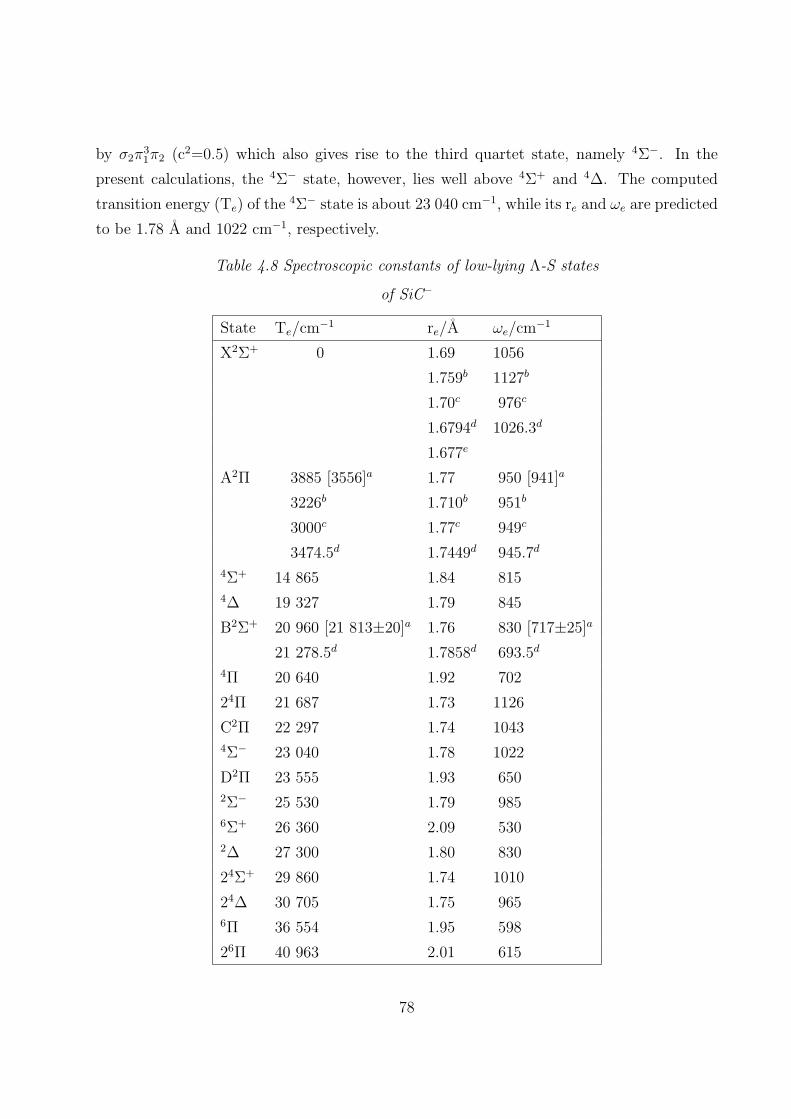
by σ2π31π2 (c2=0.5) which also gives rise to the third quartet state, namely 4Σ−. In the
present calculations, the 4Σ− state, however, lies well above 4Σ+ and 4∆. The computed
transition energy (Te) of the 4Σ− state is about 23 040 cm−1, while its re and ωe are predicted
to be 1.78 A and 1022 cm−1, respectively.
Table 4.8 Spectroscopic constants of low-lying Λ-S states
of SiC−
State Te/cm−1 re/A ωe/cm−1
X2Σ+ 0 1.69 1056
1.759b 1127b
1.70c 976c
1.6794d 1026.3d
1.677e
A2Π 3885 [3556]a 1.77 950 [941]a
3226b 1.710b 951b
3000c 1.77c 949c
3474.5d 1.7449d 945.7d
4Σ+ 14 865 1.84 8154∆ 19 327 1.79 845
B2Σ+ 20 960 [21 813±20]a 1.76 830 [717±25]a
21 278.5d 1.7858d 693.5d
4Π 20 640 1.92 702
24Π 21 687 1.73 1126
C2Π 22 297 1.74 10434Σ− 23 040 1.78 1022
D2Π 23 555 1.93 6502Σ− 25 530 1.79 9856Σ+ 26 360 2.09 5302∆ 27 300 1.80 830
24Σ+ 29 860 1.74 1010
24∆ 30 705 1.75 9656Π 36 554 1.95 598
26Π 40 963 2.01 615
78

Table 4.8 ...continued
State Te/cm−1 re/A ωe/cm−1
6∆ 44 015 1.95 6306Σ− 44 680 1.94 695
26Σ+ 45 090 1.95 625
36Π 47 380 1.93 745
a Ref. 12, b Ref. 10, c Ref. 13, d Ref. 16, e Ref. 15
The B2Σ+ state of SiC− is the most important one from the spectroscopic point of view,
as the B2Σ+←X2Σ+ transition has been observed in absorption with a band origin around
21 680 cm−1. The computed transition energy of B2Σ+ is 20 960 cm−1 with a bond length
of 1.76 A. The present ωe of this state is at least 100 cm−1 larger than the value determined
from the absorption study in neon matrix. Like the lowest two doublets, the B2Σ+ state of
SiC− has a multiconfiguration character with a dominant configuration, σ2π31π2 (c2=0.44).
Two roots of 4Π are closely spaced with a separation of about 1000 cm−1. The lowest 4Π state
has a longer equilibrium bond length and a smaller ωe than those of the higher root. The
avoided crossing between 4Π and 24Π curves takes place around 1.78 A. Both the 4Π states
originate from the same dominant configuration, ...π21π2. However, the greater complexity
of the 24Π state may be noted from the composition given in Table 4.9. The next two 2Π
states, designated as C and D, are located just above 4Π with a gap of 1250 cm−1 in between
them. The avoided crossing in the potential curves of C2Π and D2Π is found to be around
3.5 a0. The estimated equilibrium bond length of the higher energy state, D2Π, is about
0.2 A longer than that of C2Π. The vibrational frequency of C2Π is comparable to that of the
ground state. A strong transition, C2Π←X2Σ+ is expected to take place around 22 300 cm−1.
Another transition, D2Π-X2Σ+ may not be so strong as the Franck-Condon overlap factor
is small due to a longer re of the excited state than the ground state. Around the potential
minimum, the C2Π state is composed of a number of open shell configurations. The dominant
configuration, σ21σ2σ4π
31 has only 23% contribution. The D2Π state is, however, less complex
with a dominant configuration of σ21σ
22π
21π2(c2=0.62). Two more doublets, 2Σ− and 2∆
are predicted to be strongly bound. Their computed transition energies are 25 530 and
27 300 cm−1, respectively. Although the equilibrium bond lengths of these states are almost
same, their ωes differ significantly. These two states show multiconfiguration character.
79

Table 4.9 Composition of Λ-S states of SiC− at equilibrium bond length
State Configuration (% contribution)
X2Σ+ σ21σ2π
41(34), σ1σ
22π
41(28), σ2
1σ2π31π2(8), σ1σ
22π
31π2(2)
A2Π σ21σ
22π
31(58), σ2
1σ2σ5π31(9), σ2
1σ22π
21π2(9) σ2
1σ2σ7π31(3),
4Σ+ σ21σ2π
31π2(50), σ1σ
22π
31π2(23), σ2
1σ2π21π
22(4)
4∆ σ21σ2π
31π2(50), σ1σ
22π
31π2(23), σ2
1σ2π21π
22(4)
B2Σ+ σ21σ2π
31π2(44), σ1σ
22π
31π2(15), σ1σ
22π
41(7), σ2
1σ2π41(5), σ2
1σ2π21π
22(3)
4Π σ21σ
22π
21π2(72), σ2
1σ2σ4π21π2(10)
24Π σ21σ
22π
21π2(35), σ2
1σ2σ4π31(14), σ1σ
22σ4π
31(10), σ2
1σ2σ3π31(8), σ1σ
22σ3π
31(7)
C2Π σ21σ2σ4π
31(23), σ1σ
22σ4π
31(18), σ2
1σ2σ3π31(15), σ1σ
22σ3π
31(11),
σ21σ2σ6π
31(5), σ1σ
22σ6π
31(3), σ2
1σ22π
31(2)
4Σ− σ21σ2π
31π2(43), σ1σ
22π
31π2(25), σ2
1σ22σ3π
21(3), σ2
1σ22σ4π
21(3)
D2Π σ21σ
22π
21π2(62), σ2
1σ2σ4π21π2(8), σ2
1σ2σ3π31(4), σ1σ
22σ3π
31(2)
2Σ− σ21σ
22σ4π
21(39), σ2
1σ22σ3π
21(33), σ2
1σ22σ6π
21(8), σ2
1σ2σ3σ4π21(2), σ2
1σ2σ4σ6π21(2)
6Σ+ σ21σ2π
21π
22(69), σ1σ
22π
21π
22(13), σ2
1σ5π21π
22(2)
2∆ σ21σ2π
31π2(47), σ1σ
22π
31π2(25)
24Σ+ σ21σ2π
31π3(46), σ1σ
22π
31π3(31)
24∆ σ21σ2π
31π3(44), σ1σ
22π
31π3(29), σ2
1σ2π31π2(2)
6Π σ21σ2σ3π
21π2(51), σ1σ
22σ3π
21π2(15), σ2
1σ2σ6π21π2(9), σ2
1σ2σ4π21π2(5)
26Π σ21σ2σ4π
21π2(31), σ2
1σ2σ5π21π2(20), σ2
1σ2σ6π21π2(9), σ2
1σ2σ3π21π2(7),
σ1σ22σ4π
21π2(5), σ1σ
22σ5π
21π2(5), σ1σ
22σ6π
21π2(2), σ1σ
22σ3π
21π2(2)
6∆ σ21σ2π
21π2π3(64), σ1σ
22π
21π2π3(18), σ1σ
22π1π
22π3(2)
6Σ− σ21σ2π
21π2π3(64), σ1σ
22π
21π2π3(18), σ1σ
22π1π
22π3(2)
26Σ+ σ21σ2π
21π2π3(62), σ1σ
22π
21π2π3(16), σ2
1σ2π21π
23(2)
36Π σ21σ2σ5π
21π2(33), σ2
1σ2σ6π21π2(20), σ1σ
22σ5π
21π2(9), σ1σ
22σ6π
21π2(7),
σ21σ2σ3π
21π2(4), σ2
1σ2σ4π21π2(4)
The potential minima of two closely spaced excited quartets, 24Σ+ and 24∆ are located
around 29 900 and 30 700 cm−1, respectively, and their re and ωe values are comparable.
Both the states are characterized by two almost equally dominant configurations, σ21σ2π
31π3
and σ1σ22π
31π3, in which the excited π3 MO is primarily a localized Si(px/y) orbital. At least
seven bound sextets of the SiC− anion have been predicted for the first time in the present
80

study. The lowest sextet, 6Σ+ is mainly characterized by σ21σ2π
21π
22. Its potential energy
curve looks flat in the longer bond length region (Fig. 4.2c). At equilibrium, the bond length
of SiC− in the 6Σ+ state is estimated to be 2.09 A with a vibrational frequency of 530 cm−1.
Spectroscopic features of the remaining sextets lying in the energy range 36 500-47 500 cm−1
are more or less similar. At the longer bond distances, three higher roots of 6Π show avoided
crossings as seen in Fig. 4.2c.
4.3.2 Spectroscopic constants and potential energy curves of Ω states
In the presence of the spin-orbit coupling all 14 Λ-S states of SiC+ are allowed to interact.
The lowest dissociation limit, Si+(2Pu)+C(3Pg) splits into six closely spaced asymptotes
with a maximum separation of only 331 cm−1. The effects of the spin-orbit coupling on the
spectroscopic constants are, therefore, expected to be small. Six asymptotes correlate with
27 Ω states of SiC+. Potential energy curves of some of the low-lying 1/2, 3/2, and 5/2
states are shown in Figs. 4.3a-c, while Table 4.10 displays the spectroscopic constants of the
Table 4.10 Spectroscopic constants of low-
lying Ω states of SiC+
State Te(cm−1) re(A) ωe(cm−1)
X4Σ−1/2 0 1.827 825
X4Σ−3/2 0 1.827 8252∆5/2 10 278 1.878 7282∆3/2 10 365 1.876 7202Π1/2 10 681 1.995 4852Π3/2 10 735 1.995 4762Σ−1/2 11 498 1.855 7602Σ+
1/2 13 675 1.911 652
22Π3/2 14 313 1.874 999
22Π1/2 14 343 1.872 10044Π5/2 24 440 1.697 8794Π3/2 24 458 1.698 8754Π1/2(I) 24 478 1.699 8774Π1/2(II) 24 494 1.698 867
81
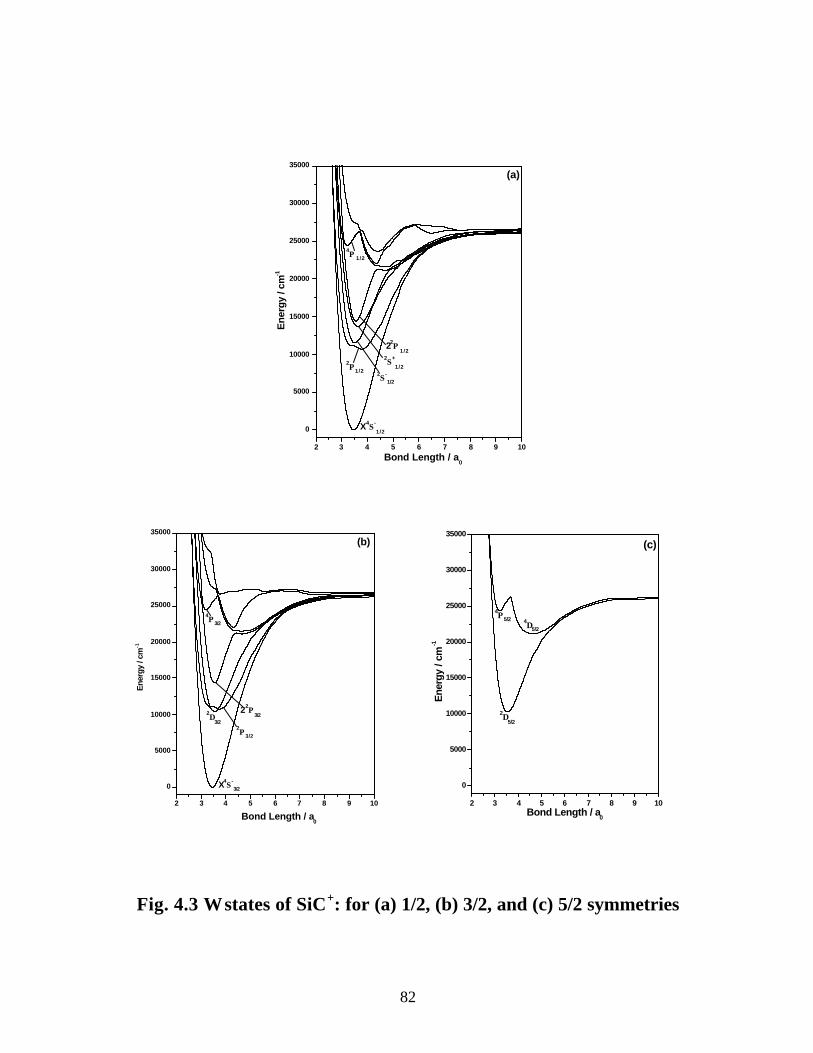
82
2 3 4 5 6 7 8 9 10
0
5000
10000
15000
20000
25000
30000
35000(a)
4Π1/2
22Π1/2
2Σ+
1/22Π1/2 2Σ-
1/2
X4Σ-
1/2
Ene
rgy
/ cm
-1
Bond Length / a0
2 3 4 5 6 7 8 9 10
0
5000
10000
15000
20000
25000
30000
35000
Ene
rgy
/ cm
-1
(b)
4Π3/2
22Π
3/22∆
3/22Π3/2
X4Σ
-3/2
Bond Length / a0
2 3 4 5 6 7 8 9 10
0
5000
10000
15000
20000
25000
30000
35000
(c)
4∆5/2
4Π5/2
2∆5/2
Ene
rgy
/ cm
-1
Bond Length / a0
Fig. 4.3 Ω states of SiC+: for (a) 1/2, (b) 3/2, and (c) 5/2 symmetries

bound states of SiC+. There is no splitting between the components of the ground state,
while the spin-orbit splitting between the components of 2∆ is only 87 cm−1 with no sig-
nificant changes in re and ωe values. The two components of the lowest 2Π state split in
a regular order with a separation of 54 cm−1. The spectroscopic parameters and potential
energy curves of 2Σ−1/2 and 2Σ+1/2 do not change much due to the spin-orbit coupling. The
spin components of the adiabatic 22Π state have similar potential curves with a splitting
of 30 cm−1 only. We have also predicted spectroscopic constants of four spin components
of 4Π with a maximum separation of 54 cm−1. It should be mentioned here that, for the
neutral species also (discussed in chapter 3) the spin-orbit mixing is very poor and almost
insignificant. For this reason no such effort is taken for SiC−.
4.3.3 Transition properties
A. SiC+
Transition probabilities of three electric dipole transitions, 4Π-X4Σ−, 24Π-X4Σ−, and
24Σ−-X4Σ− are large for SiC+. Fig. 4.4a shows the transition moments for these transitions
as a function of the bond length. Table 4.11 reports the computed radiative lifetimes for
these transitions. Because of the avoided crossings, the potential well of the 4Π state holds
only two vibrational levels. The computed radiative lifetime for the 4Π-X4Σ− transition at
the lowest two vibrational levels are 6.06 and 9.72 µs, respectively. The present calculations
predict that the 24Π-X4Σ− transition is more probable and the estimated lifetimes at the
Table 4.11 Radiative lifetime (s) of some excited states of SiC+
Transition Partial lifetime of the upper state ata
υ′=0 υ′=1 υ′=24Π-X4Σ− 6.06(-6) 9.72(-6)
24Π-X4Σ− 110(-9) 114(-9) 117(-9)
24Σ−-X4Σ− 4.31(-6) 1.80(-6)4Π1/2-X4Σ−3/2 0.39(-4) 1.48(-4)4Π1/2-X4Σ−1/2 1.32(-4) 3.94(-4)4Π3/2-X4Σ−1/2 1.29(-4) 3.76(-4)4Π5/2-X4Σ−3/2 1.29(-4) 3.50(-4)
a Values in parenthesis are power to base 10
83
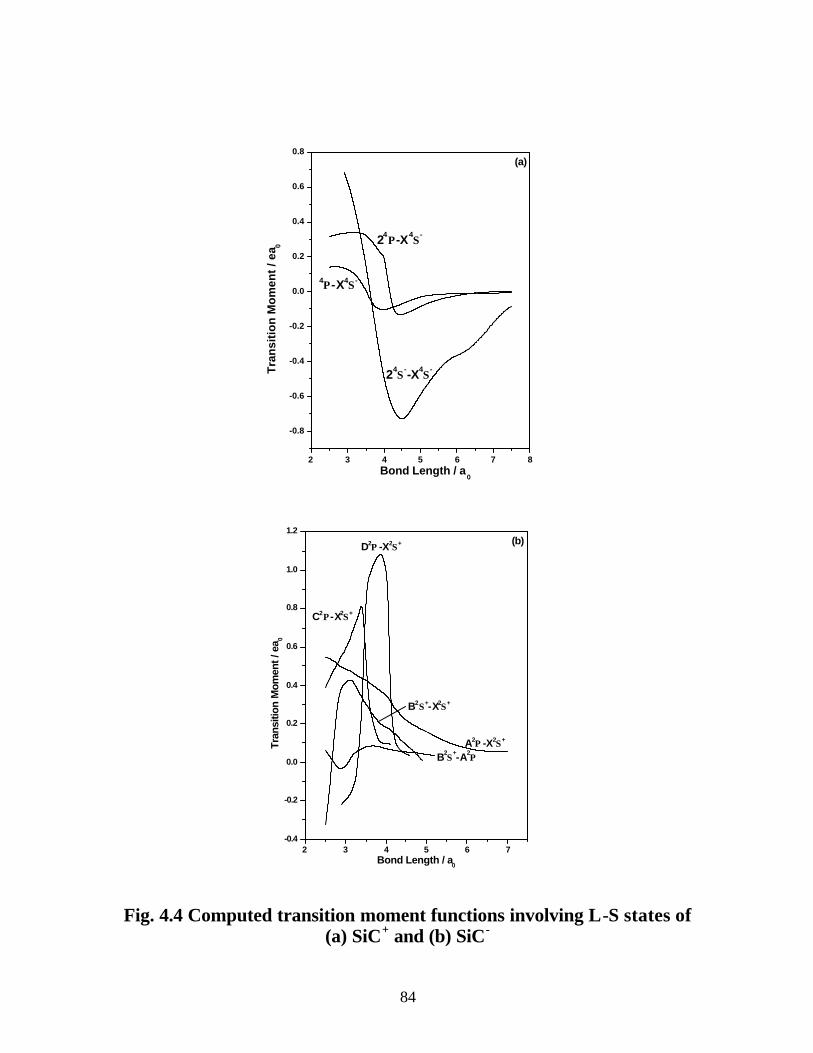
84
2 3 4 5 6 7 8
-0.8
-0.6
-0.4
-0.2
0.0
0.2
0.4
0.6
0.8(a)
4Π-X4Σ-
24Π-X4
Σ-
24Σ
--X4Σ
-
Tra
nsi
tio
n M
om
ent
/ ea 0
Bond Length / a0
2 3 4 5 6 7-0.4
-0.2
0.0
0.2
0.4
0.6
0.8
1.0
1.2(b)D2Π-X2Σ+
B2Σ+-X2Σ+
B2Σ
+-A2Π
A2Π-X2Σ+
C2Π-X2Σ+
Tran
sitio
n M
omen
t / e
a 0
Bond Length / a0
Fig. 4.4 Computed transition moment functions involving Λ-S states of (a) SiC+ and (b) SiC-

lowest few vibrational levels are of the order of one hundred nanoseconds. The radiative
lifetime for the 24Σ−-X4Σ− transition at v′=0 is 4.31 µs, while it is lowered to 1.8 µs in the
next vibrational level. With the inclusion of the spin-orbit coupling four transitions involving
quartet states have partial radiative lifetimes of the order of a few hundred microseconds
(Table 4.11). All the spin-forbidden transitions such as 22Π1/2-X4Σ−1/2, 22Π3/2-X4Σ−1/2 etc. are
very weak and are not expected to have sufficient intensities to be observed experimentally.
B. SiC−
Five doublet-doublet transitions are found to be significant for the anion. The computed
transition moments for these transitions are shown in Fig. 4.4b as a function of the bond
length. Transition moments of the A2Π-X2Σ+ transition decrease monotonically along the
bond distance, while other transition-moment curves look differently. In Table 4.12, we have
reported the computed radiative lifetimes for these transitions in the lowest four vibrational
levels. The predicted lifetime for the A-X transition is about 63 µs at v′=0, which reduces
further with v′. Transition probabilities of three transitions, B2Σ+-X2Σ+, C2Π-X2Σ+, and
D2Π-X2Σ+ are significantly high. The radiative lifetime for the C-X transition at v′=0 is
less than hundred nanoseconds, while for B-X and D-X, the computed lifetimes are 475
and 173 ns, respectively. However, the contracted CI calculations of Cai and Francois16
predicted somewhat smaller radiative lifetimes for the B2Σ+ state of the anion. The B2Σ+-
A2Π transition is also expected to be highly probable and the calculated lifetime for this
transition at v′=0 is 28 µs compared to the previously estimated value of 10 µs.
Table 4.12 Radiative lifetime (s) of some excited Λ-S states of SiC−
Transition Partial lifetime of the upper state ata
υ′=0 υ′=1 υ′=2 υ′=3
A2Π-X2Σ+ 6.30(-5) 5.10(-5) 4.30(-5) 3.80(-5)
B2Σ+-X2Σ+ 4.75(-7) 4.88(-7) 5.10(-7) 5.37(-7)
[3.06(-7)]b [3.11(-7)]b [3.17(-7)]b [3.24(-7)]b
C2Π-X2Σ+ 8.10(-8) 7.80(-8) 7.50(-8) 7.20(-8)
D2Π-X2Σ+ 1.73(-7) 3.32(-7) 1.75(-6) 2.69(-6)
B2Σ+-A2Π 2.80(-5) 2.70(-5) 2.60(-5) 2.50(-5)
[1.00(-5)]b [1.10(-5)]b [1.60(-5)]b [2.10(-5)]b
a Values in parenthesis are power to base 10, b Ref. 16
85
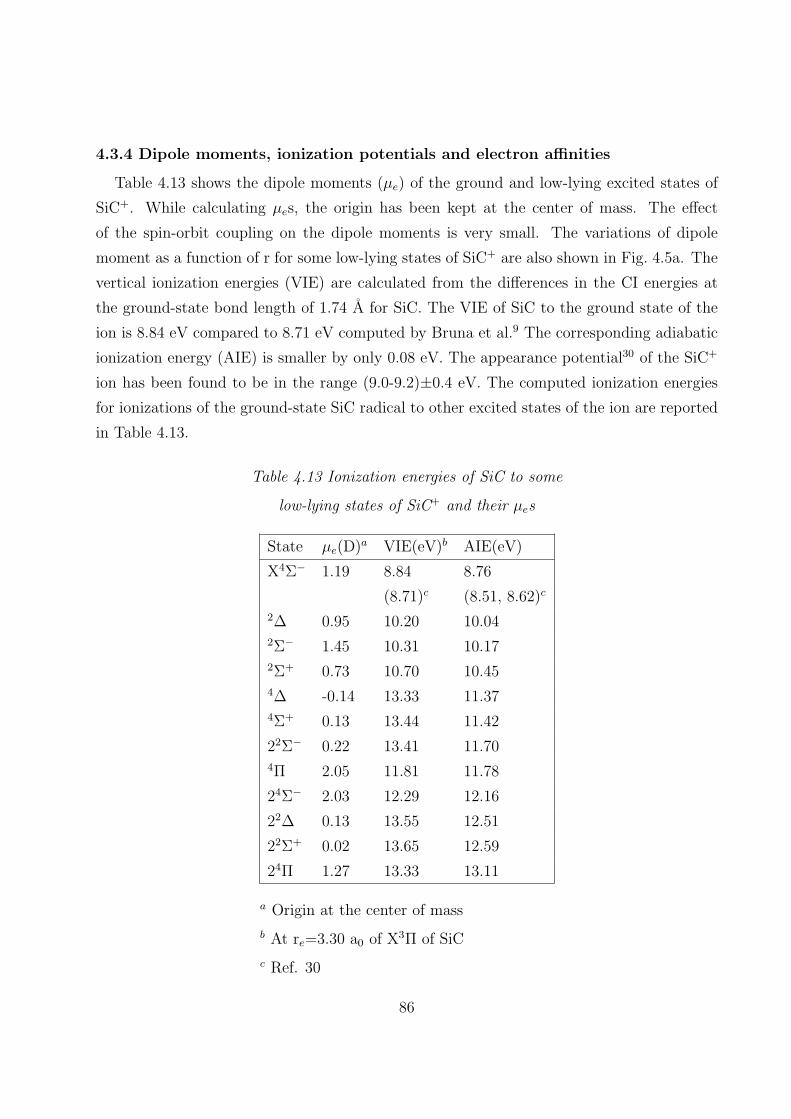
4.3.4 Dipole moments, ionization potentials and electron affinities
Table 4.13 shows the dipole moments (µe) of the ground and low-lying excited states of
SiC+. While calculating µes, the origin has been kept at the center of mass. The effect
of the spin-orbit coupling on the dipole moments is very small. The variations of dipole
moment as a function of r for some low-lying states of SiC+ are also shown in Fig. 4.5a. The
vertical ionization energies (VIE) are calculated from the differences in the CI energies at
the ground-state bond length of 1.74 A for SiC. The VIE of SiC to the ground state of the
ion is 8.84 eV compared to 8.71 eV computed by Bruna et al.9 The corresponding adiabatic
ionization energy (AIE) is smaller by only 0.08 eV. The appearance potential30 of the SiC+
ion has been found to be in the range (9.0-9.2)±0.4 eV. The computed ionization energies
for ionizations of the ground-state SiC radical to other excited states of the ion are reported
in Table 4.13.
Table 4.13 Ionization energies of SiC to some
low-lying states of SiC+ and their µes
State µe(D)a VIE(eV)b AIE(eV)
X4Σ− 1.19 8.84 8.76
(8.71)c (8.51, 8.62)c
2∆ 0.95 10.20 10.042Σ− 1.45 10.31 10.172Σ+ 0.73 10.70 10.454∆ -0.14 13.33 11.374Σ+ 0.13 13.44 11.42
22Σ− 0.22 13.41 11.704Π 2.05 11.81 11.78
24Σ− 2.03 12.29 12.16
22∆ 0.13 13.55 12.51
22Σ+ 0.02 13.65 12.59
24Π 1.27 13.33 13.11
a Origin at the center of mass
b At re=3.30 a0 of X3Π of SiC
c Ref. 30
86
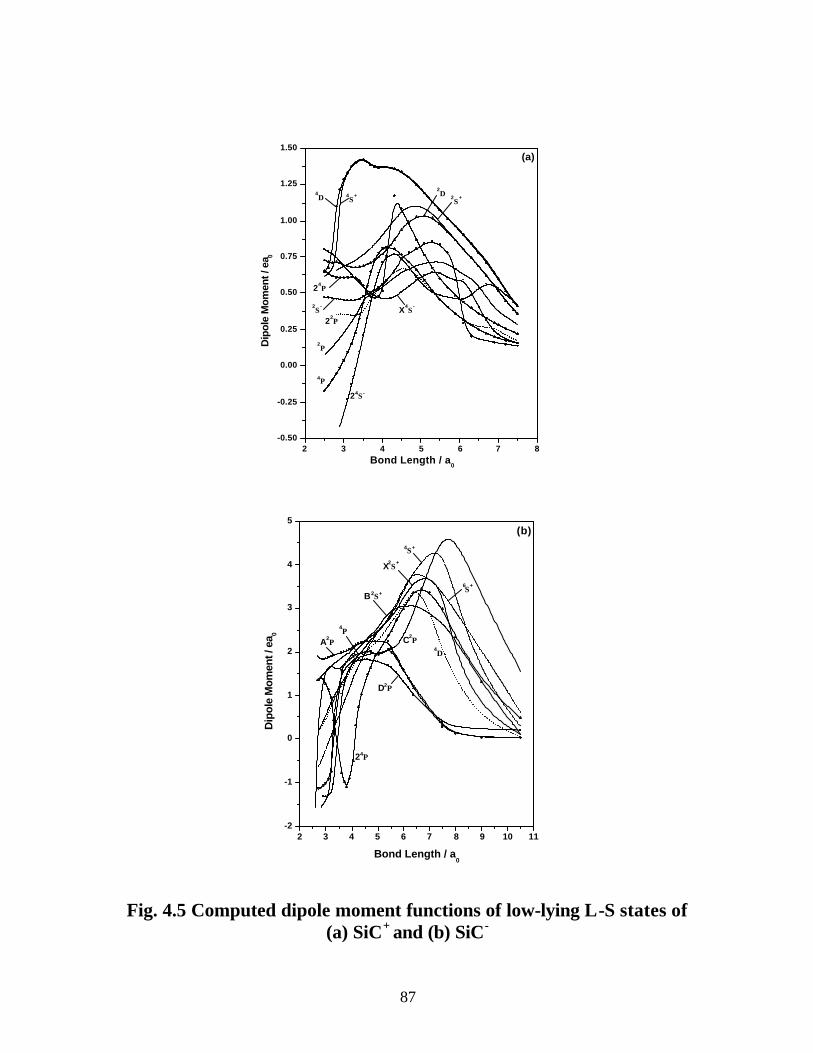
87
2 3 4 5 6 7 8-0.50
-0.25
0.00
0.25
0.50
0.75
1.00
1.25
1.50(a)
22Π
2Π
2Σ
-
2Σ
+
2∆
24Π
4Π
24Σ-
4Σ
+4∆
X4Σ
-
Dip
ole
Mom
ent /
ea 0
Bond Length / a0
2 3 4 5 6 7 8 9 10 11-2
-1
0
1
2
3
4
5(b)
24Π
4Π
6Σ
+
4∆
4Σ+
D2Π
C2ΠA2
Π
B2Σ+
X2Σ
+
Dip
ole
Mom
ent /
ea 0
Bond Length / a0
Fig. 4.5 Computed dipole moment functions of low-lying Λ-S states of (a) SiC+ and (b) SiC-

Dipole moments (µe) of the ground and low-lying states of SiC− are given in Table 4.14.
These have been calculated by keeping the origin at the center of mass. The ground-state
dipole moment of the anion is reported here as 1.95 D, while it has the highest dipole moment
in its 4Σ− state. Fig. 4.5b shows how the dipole moment functions for few low-lying states
of SiC− vary with bond distances. The vertical electron affinities (EAvert) are calculated
from the differences in the CI energies at the ground-state bond length of 1.74 A for SiC at
the same level of calculation. The EAvert of SiC to the ground state of the anion is 2.24 eV,
which is smaller than the MP2 calculated data10 by 0.8 eV. A better agreement is noted
for the corresponding adiabatic electron affinities (EAad). The electron affinities of other
excited states of SiC− are also reported in Table 4.14. Another form of electron affinity may
be defined as the vertical detachment energy (EAV DE) of the anion. EAV DE=E(neutral at
optimized anion geometry)- E(optimized anion). The calculated EAV DE of SiC− is found to
Table 4.14 Electron affinities of SiC to some
low-lying states of SiC− and their µes
State µe(D)a EAad(eV) EAvert(eV)b
X2Σ+ 1.95 2.28 2.24
(2.25)c (2.32)c
(1.98)d
A2Π 2.41 1.79 1.79
(1.71)c (1.73)c
4Σ+ 0.24 0.44 0.354∆ 0.29 -0.11 -0.14
B2Σ+ 1.63 -0.27 -0.274Π 1.41 -0.28 -0.44
24Π 1.86 -0.345 -0.53
C2Π 2.46 -0.49 -0.494Σ− 3.17 -0.58 -0.59
D2Π 2.55 -0.64 -0.84
a Origin at the center of mass
b At re=3.30 a0 of X3Π of SiC
c Ref. 10, d Ref. 13
88

be about 2.33 eV. The computed electron affinities follow the desired trend: EAvert < EAad
< EAV DE.
4.4. Summary
The electronic states of the SiC+ and SiC− ions have been investigated using ab initio
based MRDCI calculations which include pseudo potentials and the compatible Gaussian
basis functions of both Si and C atoms. There are at least 14 bound states of SiC+ and
21 electronic states of SiC− within 6 eV of energy.32,33
The spectroscopic constants of the ground state (X4Σ−) of SiC+ are improved over the
previous calculations of Bruna et al.9 The computed dissociation energy (D0) of SiC+ is
3.32 eV which matches well with the observed value. The strong interaction between the
lowest two 2Π states has created a shallow asymmetric double well in the potential energy
curve of 2Π. Consequently, the adiabatic potential curve of 22Π has a deeper potential well
with ωe=1013 cm−1. The energy separation between these two roots is about 3600 cm−1. Like
the doublets, the lowest two 4Π states undergo avoided crossing. Their strong interaction has
reduced the binding energy of the 4Π state, while the potential energy curve of 24Π shows
a sharp minimum. The spin-orbit interaction has almost no influence on the spectroscopic
properties of SiC+. The 24Π-X4Σ− transition is predicted to be the strongest transition
with a partial radiative lifetimes of about hundred nanoseconds at the lowest few vibrational
levels. Two other transitions, 4Π-X4Σ− and 24Σ−-X4Σ− are also expected to have sufficient
intensities for experimental observation. Transition dipole moments of none of the spin-
forbidden transitions involving quartet and doublet spin states are significant. All such
transitions are predicted to be very weak. The computed vertical and adiabatic ionization
energies for the ionization to the ground-state SiC+ ion compare well with the available data.
Only three states of the anion, namely X2Σ+, A2Π, and B2Σ+ have been studied before.
The computed ground-state dissociation energy of SiC− is 5.14 eV which compares well with
the previous results. Two new doublet Π states, denoted as C and D, are important from
the spectroscopic point of view. The re and ωe values of these two states differ significantly.
A number of hitherto unknown quartets and sextets of SiC− are predicted. The lowest
bound state of the quartet spin multiplicity is 4Σ+, which lies nearly 15 000 cm−1 above
the ground state. Like the doublets, spectroscopic features of the two closely spaced 4Π and
89

24Π states are quite different. The 4Π state has a longer equilibrium bond distance than the
higher root. Potential energy curve of the lowest sextet, 6Σ+ looks flat in the longer bond
length region. Three bound 6Π states exist within a gap of 11 000 cm−1. A2Π-X2Σ+ and
B2Σ+-A2Π transitions are not predicted to be very strong. Their partial radiative lifetimes
are estimated to be less than hundred microseconds. Three strong transitions, B-X, C-X,
and D-X are expected to be observed in the range 21 000-24 000 cm−1. The C-X and D-X
transitions are predicted for the first time in the present study. The radiative lifetimes for
these two transitions at v′=0 are expected to be around 80 and 170 ns, respectively. Vertical,
and adiabatic electron affinities of SiC to the ground and nine low-lying states of SiC− along
with their dipole moments are also computed.
90

4.5. References
1 A. Suzuki, Prog. Theor. Phys. 62, 936 (1979).
2 J. Cernicharo, C.A. Gottlieb, M. Guelin, P. Thaddeus, J.M. Vrtilek, Astrophys. J. Lett.
341, L25 (1989).
3 B.L. Lutz, J.A. Ryan, Astrophys. J. 194, 753 (1974).
4 P.J. Bruna, S.D. Peyerimhoff, R.J. Buenker, J. Chem. Phys. 72, 5437 (1980).
5 C.M. Rohlfing, R.L. Martin, J. Phys. Chem. 90, 2043 (1986).
6 H. Dohman, P.J. Bruna, S.D. Peyrimhoff, R.J. Buenker, Mol. Phys. 51, 1109 (1984).
7 M. Larsson, J. Phys. B: At. Mol. Phys. 19, L261 (1986).
8 P.F. Bernath, S.A. Rogers, L.C. O′Brien, C.R. Brazier, Phys. Rev. Lett. 60, 197 (1988).
9 P.J. Bruna, C. Petrongolo, R.J. Buenker, S.D. Peyerimhoff, J. Chem. Phys. 74, 4611
(1981).
10 A.I. Boldyrev, J. Simons, V.G. Zahrzewski, W. Von Niessen, J. Phys. Chem. 98, 1427
(1994).
11 A. Pramanik, K.K. Das, J. Mol. Spectrosc. 244, 13 (2007).
12 M. Grutter, P. Freivogel, J.P. Maier, J. Phys. Chem. 101, 275 (1997).
13 J. Anglada, P.J. Bruna, S.D. Peyerimhoff, R.J. Buenker, J. Phys. B: At. Mol. Phys. 16,
2469 (1983).
14 A.D. McLean, B. Liu, G.S. Chaudler, J. Chem. Phys. 97, 8459 (1992).
15 S. Hunsicker, R.O. Jones, J. Chem. Phys. 105, 5048 (1996).
16 J.-L. Cai, J.P. Francois, J. Phys. Chem. A 103, 1007 (1999).
17 L.F. Pacios, P.A. Christiansen, J. Chem. Phys. 82, 2664 (1985).
18 J.M.O. Matos, V. Kello, B.O. Roos, A. J. Sadlej, J. Chem. Phys. 89, 423 (1988).
19 R.J. Buenker, S.D. Peyerimhoff, Theo. Chim. Acta 35, 33 (1974).
20 R.J. Buenker, S.D. Peyerimhoff, Theo. Chim. Acta 39, 217 (1975).
21 R.J. Buenker, S.D. Peyerimhoff, W. Butscher, Mol. Phys. 35, 771 (1975).
22 R.J. Buenker, Int. J. Quant. Chem. 29, 435 (1986).
23 R.J. Buenker, in: P. Burton (Ed.), Proc. Workshop on Quantum Chemistry and Molecular
91

Physics in Wollongong, Wollongong, Australia, 1980.
24 R.J. Buenker, in: R.Carbo (Ed.), Studies in Physical and Theoretical Chemistry, Current
Aspects of Quantum Chemistry, vol. 21, Elsevier, Amsterdam, p.17, 1982.
25 R.J. Buenker, R.A. Phillips, J. Mol. Struc. (Theochem) 123, 291 (1985).
26 S. Krebs, R.J. Buenker, J. Chem. Phys. 103, 5613 (1995).
27 E.R. Davidson, in: R. Daudel, B. Pullman (Eds.), The World of Quantum Chemistry,
Reidel, Dordrecht, The Netherlands, 1974.
28 G. Hirsch, P.J. Bruna, S.D. Peyerimhoff, R.J. Buenker, Chem. Phys. lett. 52, 442 (1977).
29 C.E. Moore, Tables of Atomic Energy Levels: vols. I-III, US National Bureau of Standards,
Washington, DC, 1971.
30 J. Drowart, C. De Maria, M.G. Inghram, J. Chem. Phys. 29, 1015 (1958).
31 M.W. Chase Jr., C.A. Davis, J.R. Downey Jr., D.J. Frurip, R.A. McDonald, A.N. Syverud
JANAF Thermochemical Tables, 3rd ed, American Chemical Society and the American
Institute of Physics (for National Bureau of Standards), New York, 1986.
32 A. Pramanik, S. Chakrabarti, K.K. Das, Chem. Phys. Lett. 450, 221 (2008).
33 A. Pramanik, A. Banerjee, K.K. Das, Chem. Phys. Lett. 468, 124 (2009).
92

CHAPTER – 5
ELECTRONIC STRUCTURE AND
SPECTROSCOPIC PROPERTIES OF SnC AND SnC+

5.1. Introduction
Molecules of group 14 elements have drawn a special attention in recent years due to
their possible applications in catalysis, sensor films and new cluster materials.1,2 Most of
the intragroup 14 heteronuclear diatomic molecules such as SiC, GeC, SnC, GeSi etc. were
energetically characterized by using high temperature Knudsen effusion mass spectroscopic
experiments.3,4 Since the simple diatomic molecules are the building unit of cluster materials,
the knowledge of the chemical bonds in different electronic states of the molecules is essential.
In chapter 3 we have introduced the diatomic carbide SiC and in the subsequent chapter,
the spectroscopic properties of its cation and anion have been discussed. In this section, we
report the spectral behavior of SnC and its monopositive ion.
Goodfriend5 used empirical relationships for calculating vibrational constants of diatomic
molecules of group 14 elements. The predicted vibrational constant of SnC was reported
to be 1021 cm−1. The upper limit to the atomization enthalpy for SnC was determined as
452±14 kJ mol−1 from the Knudsen effusion mass spectroscopic method.6 The bond length
of SnC was estimated to be 1.97±0.08 A which is in consistent with the the trends in C2,
SiC and GeC bond lengths. In analogy with the SiC radical, it was assumed that the ground
state of SnC would be 3Π. Schmude Jr. and Gingerich6 also predicted excited states and
respective energies used for SnC as 3Σ (3300 cm−1), 1Σ (7200 cm−1), 1Π (7400 cm−1) and1∆ (7300 cm−1) based on theoretical work of Martin et al.7 for SiC and by Shim et al.8 for
GeC.
The structural parameters, optical constants and enthalpy of formation for cubic GeC and
SnC alloys were computed by Pandey et al.9 using methods based on a generalised gradient
approximation (GGA) to the density functional theory (DFT). Benzair et al.10 reported a
theoretical study of the ground-state and electronic properties of group 14 Zink-blende-like
GeC, SnC, and SiC compounds employing full-potential linearized augmented plane wave
(FP-LAPW) approach within the DFT in the local spin density approximation (LSDA)
including GGA. It was suggested from the distribution of the valence charge density that
the bond in GeC and SnC are more ionic than that in SiC. Another study was carried
out to see the effects of different forms of the correlation energy functional.11 Khenata et
al.12 made a complete analysis of the structural and electronic properties of GeC, SnC and
GeSn using FP-LAPW method. Very recently Li and Wang13 carried out DFT calculations
of the tin-doped carbon clusters SnCn/SnC+n /SnC−n (n=1-10) using B3LYP method with
93

TZP+ basis set. All neutral SnCn (n=1-10) clusters have Sn-terminated linear equilibrium
structures. The ground-state symmetry of SnC is 3Π, while that of other n-odd membered
clusters is 3Σ. Except for SnC2 and SnC10, the ground states of n-even numbered clusters
belong to 1Σ. They also predicted 2Π as the electronic ground state for linear SnC+n and
SnC−n , except for SnC/SnC+/SnC−, SnC2/SnC+2 , SnC+
4 , SnC+6 , and SnC10/SnC+
10. SnC+
has a 4Σ ground state with re=2.115 A as predicted by these authors.13 Another excited 2Π
state was proposed in the same calculation having a relative energy of 25 kJ/mol.
Extensive theoretical and experimental research about the ground and excited states
of homonuclear diatomic molecules of group 14 elements like C2, Si2, Ge2, Sn2, and Pb2
have been carried out successfully in past decades. But similar studies of the intragroup
14 diatomic molecules are not many, which may be due to the experimental difficulties in
isolating such heterodiatomic single molecules in the gas phase. Most of the previous studies
on SnC were performed in solid phase. Large scale MRDCI studies of Pramanik et al.14,15
on SiC and SiC+ have proposed a number of spectroscopically important states as well
as radiative lifetimes in the excited states. Although almost insignificant, they also have
performed spin-orbit interactions among the low-lying states of both the species. No such
experimental/theretical studies have so far been carried out on the SnC or SnC+ in the gas
phase. Spectroscopic constants and potential energy curves of the ground and low-lying
excited states are not known yet.
In this chapter we have presented an extensive theoretical study on SnC and SnC+ using
multireference singles and doubles configuration interaction (MRDCI) calculations. Potential
energy curves of a large number of electronic states have been constructed. Effects of spin-
orbit coupling on the spectroscopic constants of low-lying states are investigated. Transition
probabilities of some electric dipole allowed and spin-forbidden transitions are calculated.
Hence the radiative lifetimes of excited states are estimated from MRDCI wave functions.
At the same time we have computed the vertical and adiabatic ionization potential of SnC.
Dipole moment functions of some few states as well as transition moments involving ground
and low-lying excited states of the neutral and the cationic species are also investigated in
the present studies.
94

5.2. Computational details
5.2.1 RECPs and basis sets
The full core RECPs of Sn have been taken from LaJohn et al.16 in which 5s25p2 electrons
of the atom are kept in the valence space, while the remaining inner electrons are described
by means of pseudo potentials. Therefore, the number of active electrons for Sn is 4 only. For
the carbon atom, the RECPs of Pacios and Christiansen17, which include 2s22p2 electrons
in the valence space have been used. The total number of electrons used for generating the
configuration space is 8. The 3s3p4d primitive Gaussian basis set for Sn is taken from LaJohn
et al.16 The first two d functions are contracted with coefficients 0.333845 and 0.474286. The
4s4p basis function of Pacios and Christiansen17 for the carbon atom have been augmented
with two sets of d functions of exponents 1.2 and 0.35 a−20 .
5.2.2 SCF MOs and CI
SCF calculations have been carried out for the (σ2σ2π2)3Σ− state of SnC and (σ2σπ2)4Σ−
state of SnC+ at different internuclear distances in the range 3.0-15.0 a0 using the above
mentioned RECPs and basis sets. It generates reasonably good optimized symmetry adopted
molecular orbitals which are used for MRDCI calculations. In both cases the molecule/ion
has been placed along the +z axis with Sn at the origin. The computations are carried out
in the C2v symmetry. The preliminary studies show that the 4d10 electrons of Sn do not
participate in the formation of low-lying electronic states of the molecule like SnC. Therefore,
in the present calculations, 4d10 electrons are kept within the full core RECP. The d electrons
do not affect the structural and spectroscopic features to a large extent.
In the next step, the MRDCI methodology of Buenker and coworkers18−24 have been
employed. For a given spin and spatial symmetry, a set of reference configurations is chosen as
shown in Table 5.2 and 5.2, respectively. All possible single and double excitations generate
a large number of configurations. A configuration-selection threshold of 0.5 µhartree is used
to make the total number of selected configurations below 200 000. The sums of the square
of coefficients of the reference configurations for lowest few roots remain in the range 0.90-
0.94. The details of the CI calculations for SnC and SnC+ are tabulated in Table 5.1 and
Table 5.2, respectively. As mentioned previously, Davidson correction25,26 has been made
and which ultimately leads to estimated full-CI energies of different states of SnC and SnC+.
95
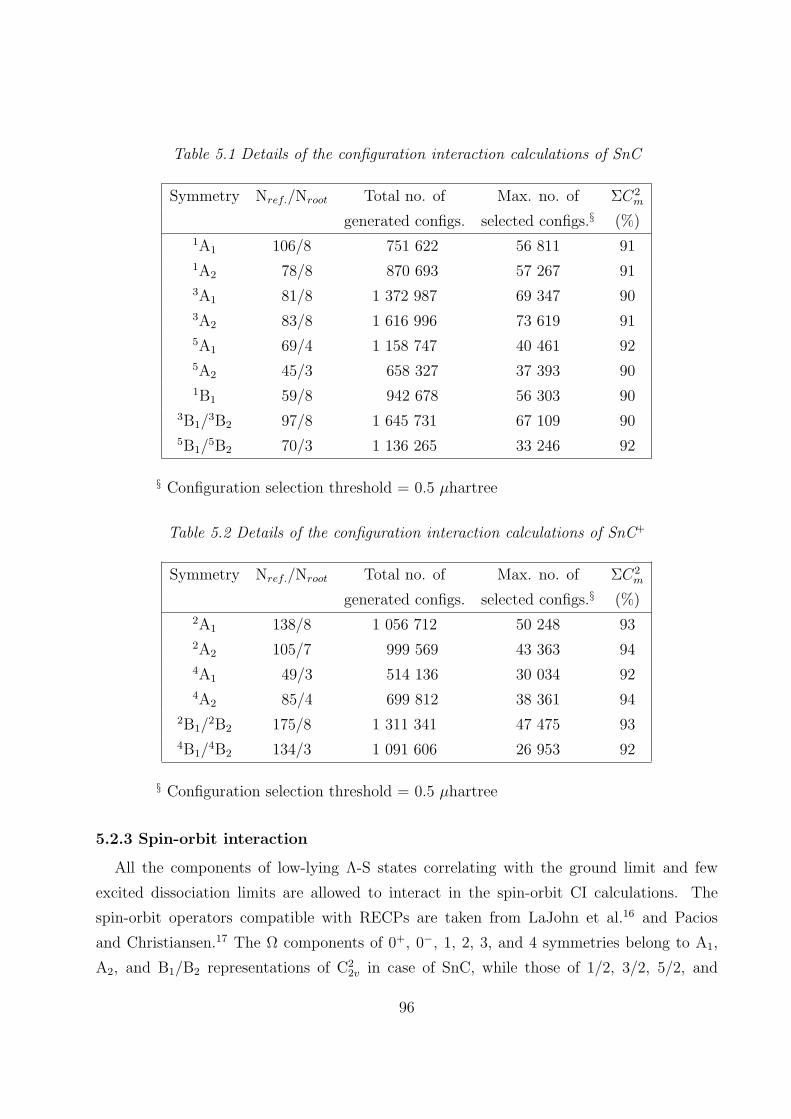
Table 5.1 Details of the configuration interaction calculations of SnC
Symmetry Nref./Nroot Total no. of Max. no. of ΣC2m
generated configs. selected configs.§ (%)1A1 106/8 751 622 56 811 911A2 78/8 870 693 57 267 913A1 81/8 1 372 987 69 347 903A2 83/8 1 616 996 73 619 915A1 69/4 1 158 747 40 461 925A2 45/3 658 327 37 393 901B1 59/8 942 678 56 303 90
3B1/3B2 97/8 1 645 731 67 109 905B1/5B2 70/3 1 136 265 33 246 92
§ Configuration selection threshold = 0.5 µhartree
Table 5.2 Details of the configuration interaction calculations of SnC+
Symmetry Nref./Nroot Total no. of Max. no. of ΣC2m
generated configs. selected configs.§ (%)2A1 138/8 1 056 712 50 248 932A2 105/7 999 569 43 363 944A1 49/3 514 136 30 034 924A2 85/4 699 812 38 361 94
2B1/2B2 175/8 1 311 341 47 475 934B1/4B2 134/3 1 091 606 26 953 92
§ Configuration selection threshold = 0.5 µhartree
5.2.3 Spin-orbit interaction
All the components of low-lying Λ-S states correlating with the ground limit and few
excited dissociation limits are allowed to interact in the spin-orbit CI calculations. The
spin-orbit operators compatible with RECPs are taken from LaJohn et al.16 and Pacios
and Christiansen.17 The Ω components of 0+, 0−, 1, 2, 3, and 4 symmetries belong to A1,
A2, and B1/B2 representations of C22v in case of SnC, while those of 1/2, 3/2, 5/2, and
96

7/2 symmetries belong to E1/E2 representations for SnC+. The dimensions of the secular
equations of A1, A2, and B1 blocks are 49, 48, 48 respectively for some selective number of
roots of Λ-S symmetries of the SnC molecule. On the other hand E1/E2 blocks of SnC+ have
the dimension of 57.
Potential energy curves for both spin-independent and spin-included low-lying states of
SnC/SnC+ are constructed. Spectroscopic constants are then determined by fitting these
potential energy curves. Transition dipole moments for the pair of vibrational functions in a
particular transition are computed. Einstein spontaneous emission coefficients and transition
probabilities are then calculated.
5.3. Results and discussion
5.3.1 Spectroscopic constants and potential energy curves of Λ–S states
A. SnC
The ground state of both Sn and C belong to 3Pg and 18 Λ-S states of SnC correlate with
them. The interaction between the first excited state (1Dg) of Sn and the ground state of C
results in a set of nine excited triplets. Similarly, the next dissociation limit, Sn(3Pg)+C(1Dg)
which lies 10 000 cm−1 above the lowest limit, correlates with another set of nine triplets of
same symmetry as the previous one. As shown in Table 5.3, a set of fifteen singlets correlates
Table 5.3 Dissociation correlation between the molecular and atomic states of SnC
Λ-S states Atomic states Relative energy / cm−1
Sn + C Expt.a Calc.1Σ+(2), 1Σ−, 1Π(2), 1∆, 3Pg + 3Pg 0 03Σ+(2), 3Σ−, 3Π(2), 3∆,5Σ+(2), 5Σ−, 5Π(2), 5∆3Σ+, 3Σ−(2), 3Π(3), 3∆(2), 3Φ 1Dg + 3Pg 5764 73963Σ+, 3Σ−(2), 3Π(3), 3∆(2), 3Φ 3Pg + 1Dg 10 159 11 6663Σ−, 3Π 1Sg + 3Pg 14 3141Σ+(3), 1Σ−(2), 1Π(4), 1∆(3), 1Dg + 1Dg 15 923 19 1451Φ(2), 1Γ
a Ref. 27
97

with the limit at 15 923 cm−1 comprising 1Dg states of both the atoms. The MRDCI
estimated value, however, overestimates it by about 3200 cm−1.
The computed potential energy curves of a large number of triplet, singlet, and quintet
states of SnC without spin-orbit coupling are shown in Figs. 5.1a-c. Spectroscopic constants
of 31 bound states of SnC are given in Table 5.4. Like the two lighter carbides, namely
SiC and GeC, the ground state of SnC is X3Π. The computed ground-state bond length of
the molecule is 2.023 A with a vibrational frequency of 646 cm−1. Although the molecular
constants of the ground state of SnC are not known, the computed equilibrium bond length
follows the desired trend: re(SiC) < re(GeC) < re(SnC). The ground state is dominated by
σ21σ2π
31 (73%) at re. The σ1 MO is a strongly a bonding combination of the s orbitals of both
Sn and C, while σ2 is weakly antibonding and mainly localized on the Sn atom. The π1 MO
is strongly bonding comprising the px/y orbitals of the two atoms. The computed ground-
state dissociation energy of SnC is about 3.06 eV. Neither any experimental study such as
Knudsen effusion mass spectrometric nor any theoretical calculation have been reported so
far for SnC. The present De value is smaller than that of either SiC or GeC which is expected
due to the heavier mass of SnC.
Analogous to SiC and GeC, the first excited state of SnC is predicted to be 3Σ−, which
is designated as A3Σ−. As expected, the state is slightly more stabilized compared to that
of the lighter carbides. The computed transition energy (Te) of the state is 3775 cm−1 and
the potential minimum is shifted towards the longer bond distance by about 0.09 A from
the ground-state re. The predicted vibrational frequency is 590 cm−1. Though not observed
as yet, the A3Σ−←X3Π transition in SnC is predicted to take place around 3800 cm−1.
Comparing the previous results of SiC, it is predicted that the calculated transition energy
of A3Σ− is underestimated by about 500 cm−1. In an analogous situation, the observed
0-0 band of the A-X system in SiC has been found to be near 4500 cm−1. The CI wave
functions show that the A3Σ− state of SnC is characterized mainly by σ21σ
22π
21 with c2=0.84
at re, where the MOs have similar bonding characteristics as the ground state. The state
is reasonably strongly bound with a binding energy of 2.6 eV estimated from the MRDCI
study.
The σ2→π1 excitation in the ground state generates the lowest singlet state belonging
to the 1Σ+ symmetry. The contribution of the closed shell leading configuration, σ21π
41 at
equilibrium is about 65%. We designate the state as a1Σ+ whose estimated transition energy
is 6505 cm−1. Its equilibrium bond length (1.942 A) is shorter than that of the ground state,
98
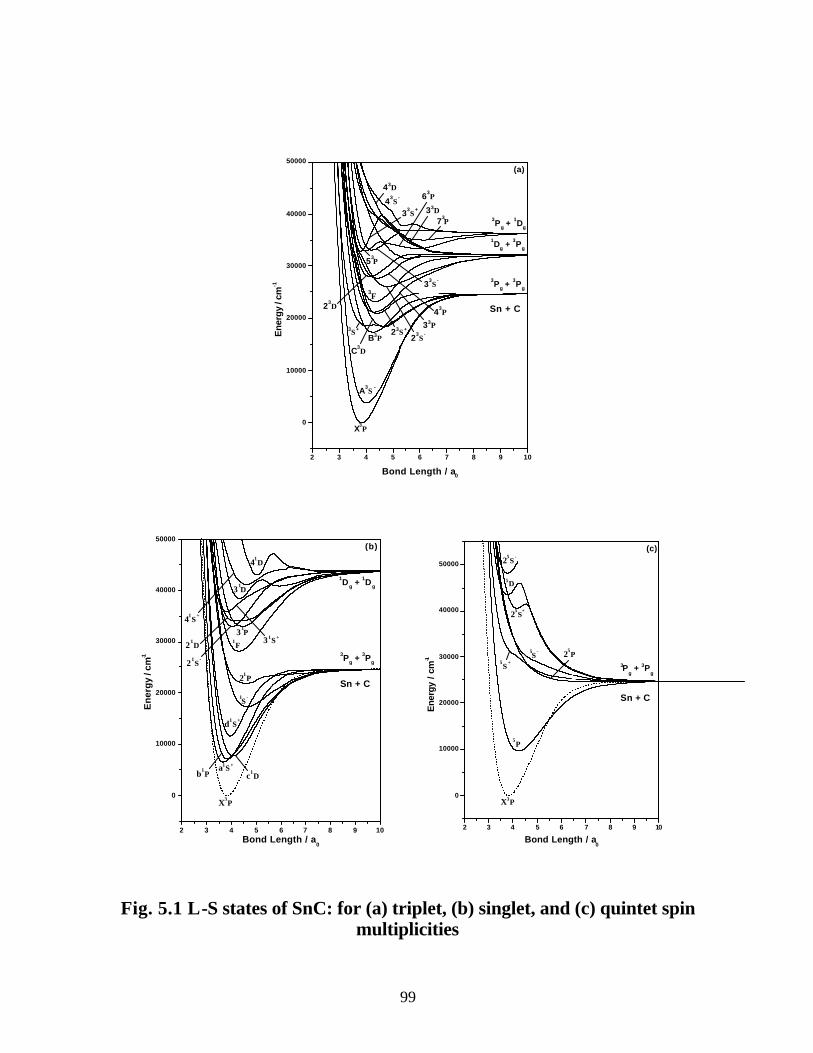
99
2 3 4 5 6 7 8 9 10
0
10000
20000
30000
40000
50000(a)
43∆
73Π
63Π
33∆43
Σ-
3Pg + 1Dg
1Dg + 3Pg
3Pg + 3Pg
Sn + C
33Σ
-
33Σ
+
53Π
23∆
43Π
23Σ
-
3Φ
33Π23Σ+
C3∆
3Σ+
B3Π
A3Σ
-
X3Π
Ene
rgy
/ cm
-1
Bond Length / a0
2 3 4 5 6 7 8 9 10
0
10000
20000
30000
40000
50000(b)
41∆
41Σ
+
31∆
21∆
1Dg + 1Dg
3Pg + 3Pg
Sn + C
31Σ+31
Π
21Σ
-
1Φ
21Π
1Σ
-
d1Σ
+
c1∆b1
Πa1Σ+
X3Π
En
erg
y / c
m-1
Bond Length / a0
2 3 4 5 6 7 8 9 10
0
10000
20000
30000
40000
50000
(c)
3Pg + 3P
g
Sn + C
25Σ
-
5∆
25Σ
+
5Σ
- 25Π
5Σ
+
5Π
X3Π
Ene
rgy
/ cm
-1
Bond Length / a0
Fig. 5.1 Λ-S states of SnC: for (a) triplet, (b) singlet, and (c) quintet spin
multiplicities
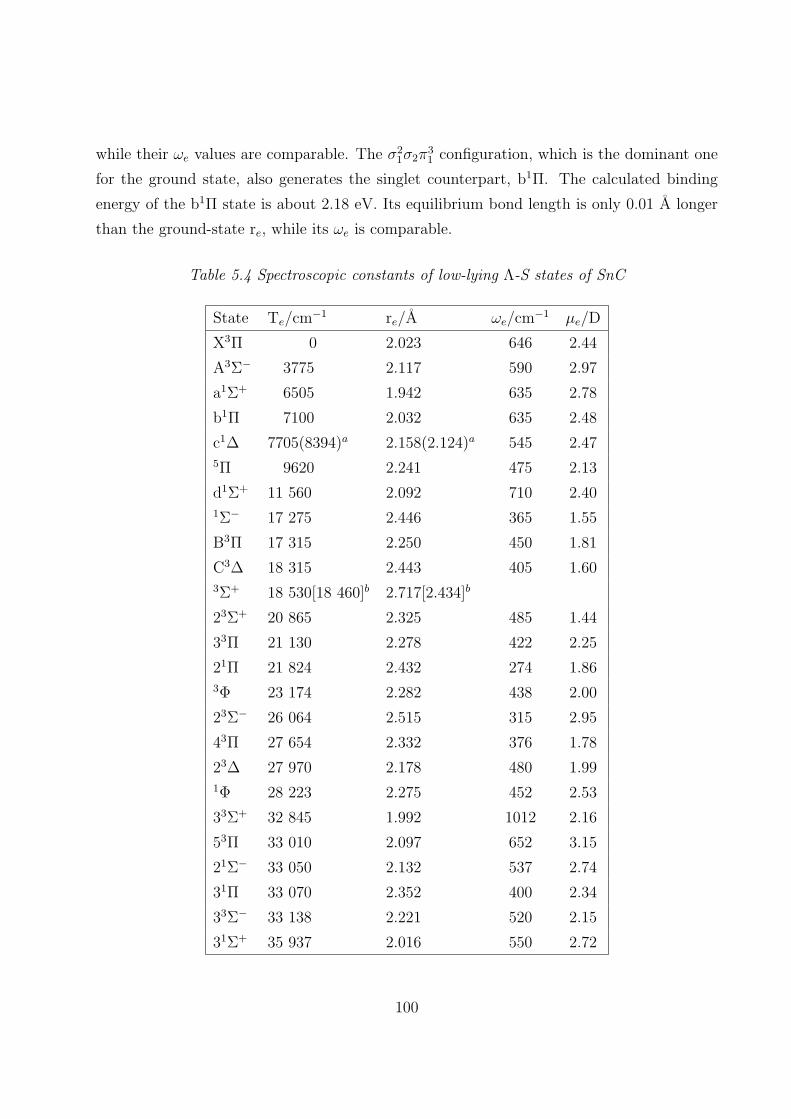
while their ωe values are comparable. The σ21σ2π
31 configuration, which is the dominant one
for the ground state, also generates the singlet counterpart, b1Π. The calculated binding
energy of the b1Π state is about 2.18 eV. Its equilibrium bond length is only 0.01 A longer
than the ground-state re, while its ωe is comparable.
Table 5.4 Spectroscopic constants of low-lying Λ-S states of SnC
State Te/cm−1 re/A ωe/cm−1 µe/D
X3Π 0 2.023 646 2.44
A3Σ− 3775 2.117 590 2.97
a1Σ+ 6505 1.942 635 2.78
b1Π 7100 2.032 635 2.48
c1∆ 7705(8394)a 2.158(2.124)a 545 2.475Π 9620 2.241 475 2.13
d1Σ+ 11 560 2.092 710 2.401Σ− 17 275 2.446 365 1.55
B3Π 17 315 2.250 450 1.81
C3∆ 18 315 2.443 405 1.603Σ+ 18 530[18 460]b 2.717[2.434]b
23Σ+ 20 865 2.325 485 1.44
33Π 21 130 2.278 422 2.25
21Π 21 824 2.432 274 1.863Φ 23 174 2.282 438 2.00
23Σ− 26 064 2.515 315 2.95
43Π 27 654 2.332 376 1.78
23∆ 27 970 2.178 480 1.991Φ 28 223 2.275 452 2.53
33Σ+ 32 845 1.992 1012 2.16
53Π 33 010 2.097 652 3.15
21Σ− 33 050 2.132 537 2.74
31Π 33 070 2.352 400 2.34
33Σ− 33 138 2.221 520 2.15
31Σ+ 35 937 2.016 550 2.72
100
Table 5.4 ...continued
State Te/cm−1 re/A ωe/cm−1 µe/D
31∆ 38 478 2.285 576 3.44
25Σ+ 40 639 2.208 556 1.46
41Σ+ 41 225 2.426 390 3.87
41∆ 43 051 2.660 693 -0.435∆ 43 529 2.000 651 2.34
25Σ− 48 191 2.000 659 2.46
a Ref. 13, b Second minimum
Besides A3Σ−, the σ21σ
22π
21 configuration generates two strongly bound singlets, c1∆ and
d1Σ+. Although none of these states of SnC is experimentally known yet, we have designated
them according to the labels given in SiC.14 The c1∆ state is energetically more stable than
d1Σ+. The estimated Te of c1∆ is about 7705 cm−1, which is somewhat smaller than the value
predicted by Li and Wang13 from B3LYP/TZVP+ calculations. The re and ωe of the state
predicted here are 2.158 A and 545 cm−1, respectively. The previously calculated re of the 1∆
state of SnC was reported to be 2.124 A. The d1Σ+ state, which lies 11 560 cm−1 above the
ground state, is strongly bound. Analyzing the CI wave functions at different bond distances
it is found that the d1Σ+ state strongly interacts with the lower state, a1Σ+. At equilibrium,
there is at least 19% contribution of the closed shell configuration σ21π
41 (Table 5.5). As a
result of the strong interaction, the vibrational frequency of d1Σ+ is predicted to be larger
than that of both the ground and a1Σ+ state. The estimated equilibrium bond length in the
d1Σ+ state is 2.092 A.
The lowest quintet state, 5Π originates from a σ21σ2π
21π2(83%) configuration in which π2
mainly consists of antibonding combination of px/y orbitals of both the atoms. The computed
transition energy of this strongly bound state is about 9620 cm−1 with ωe=475 cm−1 and
re=2.241 A. The binding energy of 5Π is predicted to be 1.84 eV. Although such a low-lying
quintet state may not have much influence on the spectroscopy of the molecule, the spin
components may result in many spin-forbidden transitions. The next important state of
SnC is B3Π which is the second root of the ground-state symmetry. It is predominantly
described by σ21σ2π
21π2, the same one that generates the lowest 5Π state. It may be noted
that this configuration yields another eight states of Π and Φ symmetries. The B3Π-X3Π
101

transition is predicted to take place around 17 300 cm−1. The computed re of the B3Π state is
at least 0.2 A longer than the ground-state equilibrium bond length. As a result, the Franck-
Condon overlap factor for the B-X transition should be much less than what is expected.
The fitted vibrational frequency of the B3Π state is 450 cm−1. In an analogous situation
such a transition is experimentally observed around 21 915 cm−1 for the SiC molecule. In
the present calculations, we have predicted 1Σ− and 3∆ states with their potential minima
located almost at the same bond length around 2.445 A. The former is more stable than
the latter by about 1040 cm−1. Potential energy curves of both the states are shallow
compared to those of other low-lying states. However, a weak 3∆-X3Π transition may take
place around 18 315 cm−1. Though not observed, the 3∆ state is labeled as C because it
lies next to B3Π. 1Σ− and C3∆ states arise from the configurations, σ21σ
22π1π2(77%) and
σ21σ
22π
21(76%), respectively.
Table 5.5 Composition of Λ-S states of SnC at equilibrium bond length
State Configuration (% contribution)
X3Π σ21σ2π
31(73), σ2
1σ2π21π2(5), σ2
1σ2π1π22(5), σ1σ
22π
31(3)
A3Σ− σ21σ
22π
21(84), σ2
1σ22π
22(2)
a1Σ+ σ21π
41(65), σ2
1σ22π
21(7), σ2
1π21π
22(6), σ1σ2π
31π2(4)
b1Π σ21σ2π
31(78), σ2
1σ2π21π2(5), σ2
1σ2π1π22(2)
c1∆ σ21σ
22π
21(79), σ2
1σ22π
22(5)
5Π σ21σ2π
21π2(83)
d1Σ+ σ21σ
22π
21(52), σ2
1π41(19), σ2
1π21π
22(5), σ2
1σ22π
22(3), σ2
2π41(2)
B3Π σ21σ2π
21π2(77), σ2
1σ2π31(6)
1Σ− σ21σ
22π1π2(77), σ2
1σ2σ3π1π2(7)
C3∆ σ21σ
22π
21(76), σ2
1σ2σ3π1π2(7)3Σ+ σ2
1π31π2(63), σ2
1σ22π1π2(8), σ1σ2π
41(5), σ2
1π1π32(4), σ1σ2π
21π
22(3)
23Σ+ σ21π
31π2(41), σ2
1σ22π1π2(29), σ2
1π1π32(7), σ2
1π21π
22(4)
33Π σ21σ2π
21π2(82)
21Π σ21σ2π
21π2(77), σ2
1σ2π1π22(3)
3Φ σ21σ2π
21π2(84)
23Σ− σ21σ
22π1π2(42), σ2
1σ22π
21(21), σ2
1σ2σ3π21(8), σ2
1π31π2(6), σ2
1σ2σ3π1π2(5)
43Π σ21σ2π
21π2(80)
102

Table 5.5 ...continued
State Configuration (% contribution)
23∆ σ21π
31π2(74), σ2
1σ22π1π2(5), σ2
1π1π32(3)
1Φ σ21σ2π
21π2(83)
33Σ+ σ1σ2π41(48), σ2
1π31π2(17), σ1σ2π
31π2(7), σ2
1σ22π1π2(5), σ1σ2π
21π
22(5)
21Σ− σ21π
31π2(80)
53Π σ21σ2π
21π2(41), σ1σ
22π
31(34), σ2
1σ2π32(2), σ1σ
22π
21π2(6)
31Π σ21σ2π
21π2(80)
33Σ− σ21π
31π2(62), σ2
1σ22π1π2(12), σ2
1σ2σ3π21(5)
31Σ+ σ1σ2π41(24), σ2
1π31π2(16), σ2
1π21π
22(13), σ1σ2π
31π2(10), σ2
1π41(5), σ1σ2π
21π
22(3)
31∆ σ21π
31π2(37), σ2
1σ22π1π2(34), σ2
1σ2σ3π21(8), σ2
1σ22π
22(2)
25Σ+ σ1σ2π31π2(53), σ2
1π21π
22(19), σ1σ2π1π
32(5), σ1σ2π
21π
22(5)
41Σ+ σ21σ
22π1π2(43), σ2
1σ22π
21(26), σ2
1σ2σ3π21(10)
41∆ σ21π
31π2(52), σ2
1π21π
22(17), σ2
1σ22π
21(8), σ2
1σ22π1π2(2)
5∆ σ1σ2π31π2(80), σ1σ2π
21π
22(4), σ1σ2π1π
32(3)
25Σ− σ21σ2σ3π
21(77), σ2
1σ2σ3π1π2(3), σ21σ2σ6π
21(3)
The potential energy curve of the 3Σ+ state of SnC shows a very shallow double minima
at 4.0 and 4.6 a0 (Fig. 5.1a) as a result of an avoided crossing between two close-lying roots
of 3Σ+ symmetry. The barrier height is estimated to be only 200 cm−1. Transition energies
at the two minima are located at 18 530 and 18 460 cm−1, respectively. The CI coefficients
show that the state at the shorter-distant minimum is characterized by σ21π
31π2, while at
the longer distant minimum, it is described by σ21σ
22π1π2. In Table 5.5, we have only given
the detailed composition of the 3Σ+ state at the short distant minimum. A minimum is
created in the potential energy curve of the second root of 3Σ+ due to avoided crossing. It
lies 20 865 cm−1 above the ground state. We have designated it as 23Σ+ and the fitted re is
about 2.325 A with ωe=485 cm−1. As expected, the composition of 23Σ+ at the potential
minimum consists of two important configurations, namely σ21π
31π2 and σ2
1σ22π1π2. A second
avoided crossing takes place between the second and third root of 3Σ+ in the range 3.4-3.8 a0.
The potential minimum in the adiabatic curve of 33Σ+ is predicted to be at 1.992 A, where
σ1σ2π41 and σ2
1π31π2 configurations dominate. The estimated transition energy of the 33Σ+
is 32 845 cm−1 with a large vibrational frequency of 1012 cm−1. The 33Σ+ state potential
103

curve predissociates into Sn(1Dg)+C(3Pg) through a barrier of about 0.85 eV as a result of
an avoided crossing with a repulsive state.
Three excited 3Π states, 33Π, 43Π and 53Π are found to be weakly bound and dissociate
into Sn(1Dg)+C(3Pg). They originate mainly from the π1→π2 excitation of the ground state.
At equilibrium, there is a strong contribution of the σ1σ22π
31 configuration in the 53Π state.
On the contrary, 33Π and 43Π states are relatively pure. Fig. 5.1a shows a low barrier of
only 0.2 eV in the potential energy curve of 53Π. Transitions from these excited 3Π states
to the ground state are expected to take place in the range 21 000-33 000 cm−1, though
none of them is experimentally observed. The higher excited state, 53Π has bond length and
vibrational frequency, comparable to those of the ground state.
The 21Π state, which lies just above 33Π, is also characterized predominantly by σ21σ2π
21π2.
The state is weakly bound with a much longer equilibrium bond length. Both 3Φ and 1Φ
state of SnC are bound and originate from σ21σ2π
21π2 configuration. The computed re and ωe
of these two states are very similar. However, the 3Φ state dissociates into Sn(1Dg)+C(3Pg),
while the singlet counterpart correlates with a higher asymptote. The triplet state is more
stable than the singlet one by about 5050 cm−1. The potential minima of the second and
third root of 3Σ− are separated by 7075 cm−1. The lower state, 23Σ− has a longer bond
length of 2.515 A, while for the higher state the Sn-C bond is relatively short (re=2.221 A).
Fig. 5.1a shows that the predissociation of the state to Sn(1Dg)+C(3Pg) may take place with
an estimated barrier of 0.4 eV. This is due to another curve crossing with a repulsive state
of the same symmetry.
The lowest 5Σ+ state is not bound and dissociates into the lowest limit. The nature of
the potential energy curves of 5Σ+ and its higher root, 25Σ+ show a very strong mixing.
As a result, a potential well is created at r=2.21 A in the potential curve of 25Σ+ with a
small barrier. It predissociates rapidly into Sn(3Pg)+C(3Pg). Two configuration, namely
σ1σ2π31π2 and σ2
1π21π
22 mainly dominate in the lowest two 5Σ+ states. The composition of
25Σ+ at 2.208 A is given in Table 5.5. Two other quintets, 5∆ and 25Σ− have potential
minima around 2.0 A having almost same vibrational frequencies. As shown in Fig. 5.1c,
the 5∆ state predissociates almost in the similar way as in 25Σ+, but with a larger barrier.
Of the remaining excited triplets, 23∆ is bound with a transition energy of 27 970 cm−1 at
equilibrium.
At least six more excited singlets which dissociate into Sn(1Dg)+C(1Dg) are bound. The
104
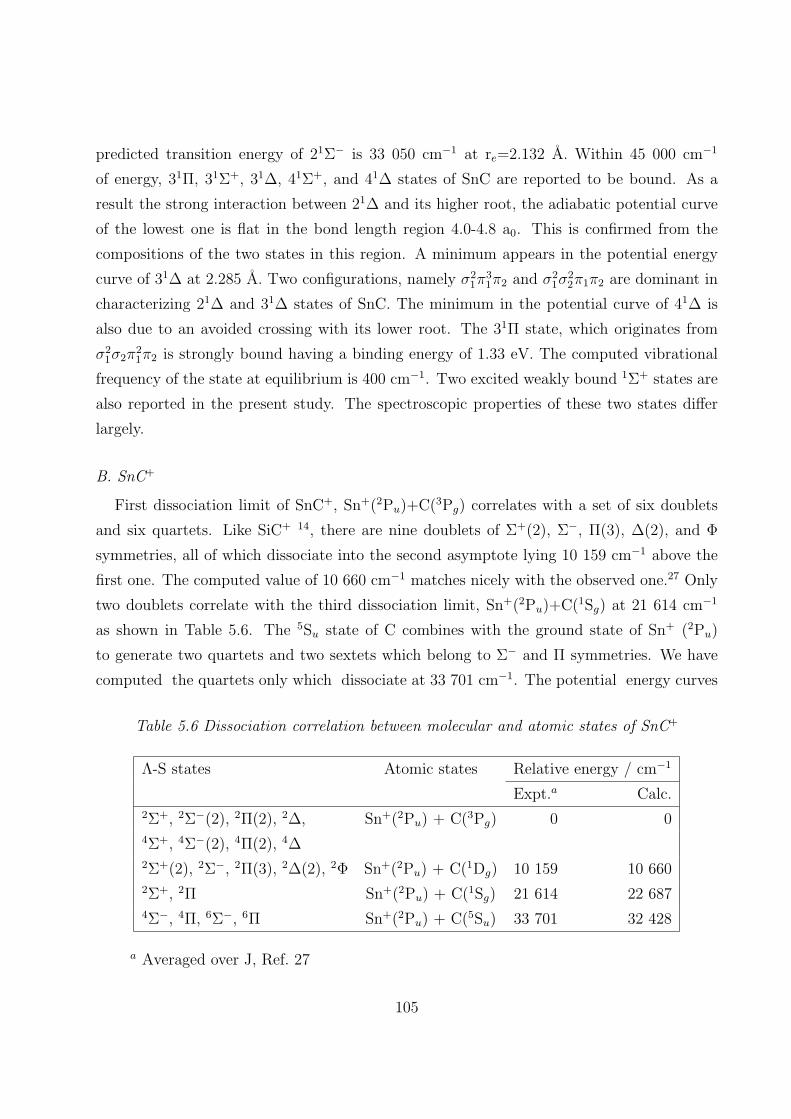
predicted transition energy of 21Σ− is 33 050 cm−1 at re=2.132 A. Within 45 000 cm−1
of energy, 31Π, 31Σ+, 31∆, 41Σ+, and 41∆ states of SnC are reported to be bound. As a
result the strong interaction between 21∆ and its higher root, the adiabatic potential curve
of the lowest one is flat in the bond length region 4.0-4.8 a0. This is confirmed from the
compositions of the two states in this region. A minimum appears in the potential energy
curve of 31∆ at 2.285 A. Two configurations, namely σ21π
31π2 and σ2
1σ22π1π2 are dominant in
characterizing 21∆ and 31∆ states of SnC. The minimum in the potential curve of 41∆ is
also due to an avoided crossing with its lower root. The 31Π state, which originates from
σ21σ2π
21π2 is strongly bound having a binding energy of 1.33 eV. The computed vibrational
frequency of the state at equilibrium is 400 cm−1. Two excited weakly bound 1Σ+ states are
also reported in the present study. The spectroscopic properties of these two states differ
largely.
B. SnC+
First dissociation limit of SnC+, Sn+(2Pu)+C(3Pg) correlates with a set of six doublets
and six quartets. Like SiC+ 14, there are nine doublets of Σ+(2), Σ−, Π(3), ∆(2), and Φ
symmetries, all of which dissociate into the second asymptote lying 10 159 cm−1 above the
first one. The computed value of 10 660 cm−1 matches nicely with the observed one.27 Only
two doublets correlate with the third dissociation limit, Sn+(2Pu)+C(1Sg) at 21 614 cm−1
as shown in Table 5.6. The 5Su state of C combines with the ground state of Sn+ (2Pu)
to generate two quartets and two sextets which belong to Σ− and Π symmetries. We have
computed the quartets only which dissociate at 33 701 cm−1. The potential energy curves
Table 5.6 Dissociation correlation between molecular and atomic states of SnC+
Λ-S states Atomic states Relative energy / cm−1
Expt.a Calc.2Σ+, 2Σ−(2), 2Π(2), 2∆, Sn+(2Pu) + C(3Pg) 0 04Σ+, 4Σ−(2), 4Π(2), 4∆2Σ+(2), 2Σ−, 2Π(3), 2∆(2), 2Φ Sn+(2Pu) + C(1Dg) 10 159 10 6602Σ+, 2Π Sn+(2Pu) + C(1Sg) 21 614 22 6874Σ−, 4Π, 6Σ−, 6Π Sn+(2Pu) + C(5Su) 33 701 32 428
a Averaged over J, Ref. 27
105
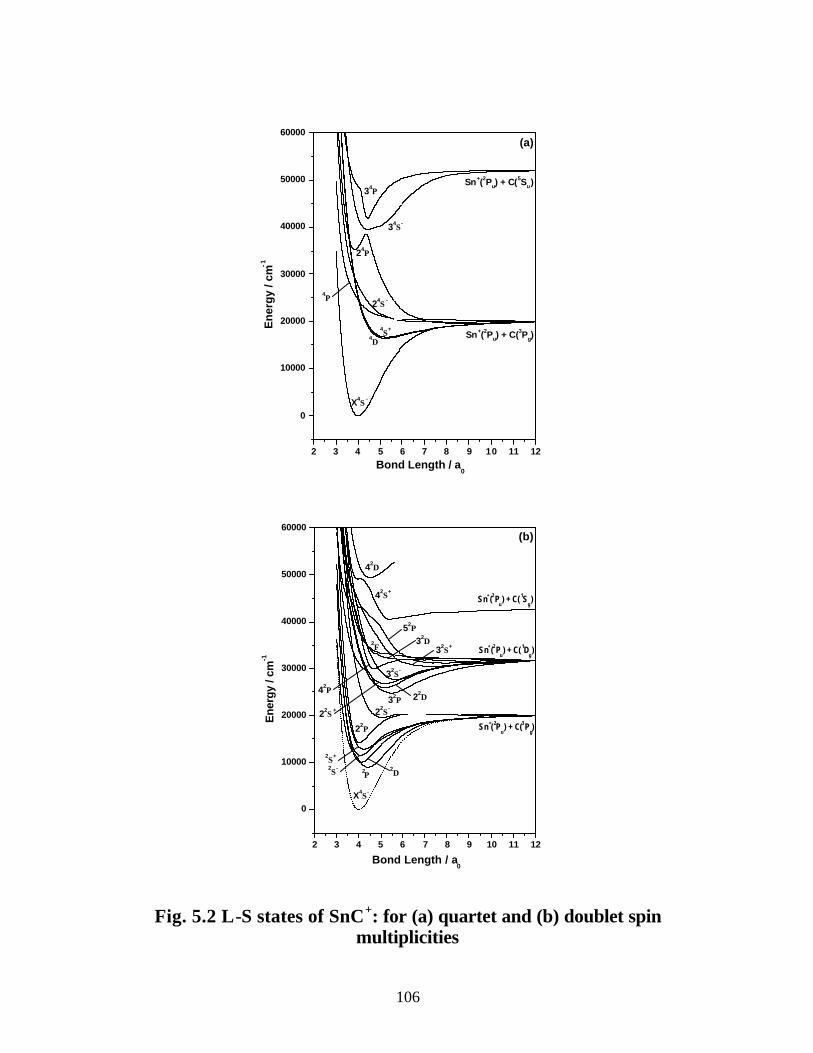
106
2 3 4 5 6 7 8 9 10 11 12
0
10000
20000
30000
40000
50000
60000(a)
Sn+(2Pu) + C(5Su)
Sn+(2Pu) + C(3Pg)
34Π
34Σ-
24Π
24Σ -4Π
4Σ+
4∆
X4Σ -
En
erg
y / c
m-1
Bond Length / a0
2 3 4 5 6 7 8 9 10 11 12
0
10000
20000
30000
40000
50000
60000(b)
52Π
42∆
Sn+(2Pu) + C( 1S
g)
Sn+(2Pu) + C( 1D
g)
Sn+(2Pu) + C(3P
g)
42Σ+
2Φ32∆
32Σ+
32Σ-
22Σ +
22∆42Π
32Π22Σ-
22Π
2Σ+
2Σ- 2∆2Π
X4Σ-
Ene
rgy
/ cm
-1
Bond Length / a0
Fig. 5.2 Λ-S states of SnC+: for (a) quartet and (b) doublet spin multiplicities
are plotted in Figs. 5.2a, b, while the spectroscopic constants of the bound states are tabu-
lated in Table 5.7.
Table 5.7 Spectroscopic constants of low-lying
Λ-S states of SnC+
State Te/cm−1 re/A ωe/cm−1
X4Σ− 0 2.112 591
2.115a
2Π 8965 2.330 423
8744a
2∆ 10 032 2.173 4852Σ− 11 459 2.156 4952Σ+ 12 797 2.233 410
22Π 14 184 2.132 6454∆ 16 366 2.773 2164Σ+ 16 622 2.795 207
22Σ− 19 546 2.664 269
32Π 24 714 2.889 251
22∆ 25 864 2.733 271
22Σ+ 26 723 2.738 258
32Σ− 27 551 3.011 226
42Π 29 941 2.488 226
24Π 35 330 2.032 785
34Σ− 39 614 2.345 395
42∆ 49 440 2.384 391
a Ref. 13
Like the lighter homologue, the ground state of SnC+ has a 4Σ− symmetry with an
equilibrium bond length of 2.112 A. The ionization involves the removal of a π1 bonding
electron from SnC. Our calculated re agrees well to the earlier value calculated by Li et
al.13 The estimated vibrational frequency of the ground state is 591 cm−1. The σ21σ2π
21
configuration dominates throughout the potential energy curve of the ground state, where
107

σ1 is mostly antibonding comprising s orbitals of Sn and C, σ2 is a bonding MO involving s
and pz orbitals of the constituting atoms, and π1 is weakly bonding, centering on C atom.
As a periodic trend the dissociation energy of the ground state is reduced from 3.32 eV for
SiC+ to 2.47 eV for SnC+.
Two strongly interacting 2Π states create an adiabatic potential minimum at 2.33 A with
a transition energy of 8965 cm−1. At equilibrium, it has been characterized by σ21σ
22π1(79%)
with 4% contribution from σ21π
31. The equilibrium vibrational frequency of the state is
computed here as 423 cm−1. The second 2Π state has a very sharp potential well with
re=2.132 A and ωe=645 cm−1. It is situated at 5219 cm−1 above 2Π. The composition of
22Π is multiconfigurational in nature, σ21π
31 being the highest contributing configuration with
c2=0.27. As shown in Table 5.8, σ21σ
22π1 also makes a large contribution (c2=0.24) to it.
Table 5.8 Composition of Λ-S states of SnC+ at equilibrium bond length
State Configuration (% contribution)
X4Σ− σ21σ2π
21(84)
2Π σ21σ
22π1(79), σ2
1π31(4)
2∆ σ21σ2π
21(69), σ2
1σ2π1π2(12)), σ1σ22π
21(3), σ1σ
22π1π2(3)
2Σ− σ21σ2π
21(71), σ2
1σ2π1π2(8), σ1σ22π
21(5), σ1σ
22π1π2(3)
2Σ+ σ21σ2π
21(58), σ2
1σ2π1π2(19), σ1σ22π1π2(4), σ1σ
22π
21(3)
22Π σ21π
31(27), σ2
1σ22π1(24), σ2
1π21π2(13), σ1σ2π
21π2(11), σ1σ2π
31(10)
4∆ σ21σ2π1π2(58), σ2
1σ3π1π2(28)4Σ+ σ2
1σ2π1π2(58), σ21σ3π1π2(28)
22Σ− σ21σ2π1π2(47), σ2
1σ2π21(24), σ2
1σ3π1π2(8), σ1σ22π1π2(4)
32Π σ21σ2σ3π2(38), σ2
1σ22π2(16), σ2
1σ23π2(14), σ2
1π21π2(10),
σ21σ2σ3π1(2), σ2
1σ22π1(2)
22∆ σ21σ2π1π2(55), σ2
1σ3π1π2(14), σ21σ2π
21(13), σ1σ
22π1π2(4)
22Σ+ σ21σ2π1π2(60), σ2
1σ3π1π2(15), σ21σ2π
21(6), σ1σ
22π1π2(5)
32Σ− σ21σ3π1π2(40), σ2
1σ2π1π2(36), σ21σ2π
21(6), σ2
1σ3π21(4)
42Π σ21π
21π2(52), σ1σ2π
21π2(16), σ2
1σ22π2(9), σ2
1π31(6)
24Π σ1σ2π31(37), σ1σ2π
21π2(31), σ2
1π21π2(16)
34Σ− σ1σ22π
21(32), σ2
1σ3π21(26), σ1σ2σ3π
21(12), σ1σ
22π
21(8)
42∆ σ1σ22π
21(26), σ1σ
22π1π2(19), σ2
1σ3π21(15), σ1σ2σ3π
21(9),
σ21σ2π1π2(6), σ1σ2σ3π1π2(4)
108

The generating configuration of the ground state, σ21σ2π
21 also gives rise to a set of three
doublets namely, 2∆, 2Σ−, and 2Σ+ having bond length of about 2.20 A. The estimated ωe
of these states vary between 410 and 495 cm−1. However, these are spectroscopically less
important as the relative population in the lowest doublet (2Π) is expectedly low. But after
including the spin-orbit coupling, the corresponding Ω components may be of great interest,
from the spectroscopic point of view. One more 2Σ− state having very shallow potential
well holding three vibrational levels also dissociates into the first dissociation limit. In the
longer bond length region (> 5 a0), it strongly interacts with the lower 2Σ−. Te of the state
is estimated to be 19 546 cm−1.
A set of closely lying 4∆ and 4Σ+ arises at around 16 500 cm−1 with considerably large
bond distances of 2.773 and 2.795 A, respectively. Their potential energy curves look flat-
tened with small binding energy of 0.44 eV only. Both the states are generated from an open
shell σ2π1π2 configuration. However, due to symmetry forbidness they are spectroscopically
unimportant. Unlike SiC+, the next 4Π and 24Σ− states of SnC+ are repulsive in nature.
It may be mentioned here that SiC+ showed two transitions namely, 4Π–X4Σ− and 24Σ−–
X4Σ− with band origins at 24 464 and 27 447 cm−1, respectively. Although no experimental
data is available, they have been predicted to have high intensities. In case of SnC+, both
the transitions are absent. The 24Π–X4Σ− transition is expected for SnC+ also, with an
excitation energy of 35 330 cm−1. The computed lifetime in the ground vibrational level of
24Π is reported to be 84 ns. This state strongly couples with another root of 4Π which is
strongly repulsive. Thus, the 24Π state predissociates through this repulsive channel crossing
a potential barrier of 0.41 eV.
All the bound doublets dissociating into second asymptote have bond length ≥2.73 A and
their vibrational frequencies are within 275 cm−1. The only exception is for 42Π. A relatively
shorter bond length (2.488 A) is due to the avoided crossing between the third and fourth
roots of 2Π. The state is about 21 000 cm−1 above the lowest doublet, 2Π. At equilibrium,
42Π is dominated by σ21π
21π2 (52%), while the composition of 32Π is complicated. The
potential energy curves of 32Σ+, 32∆, 2Φ, and 52Π are repulsive in nature. We also predict
two more excited bound states, 34Σ− and 42∆ at 39 614 and 49 440 cm−1, respectively. The
first one dissociates into Sn+(2Pu)+C(5Su), while the dissociation of 42∆ is not confirmed.
The equilibrium composition of 34Σ− is multiconfigurational in nature (Table 5.8), however,
the absorption may be confirmed by the experiment at 252 nm.
109
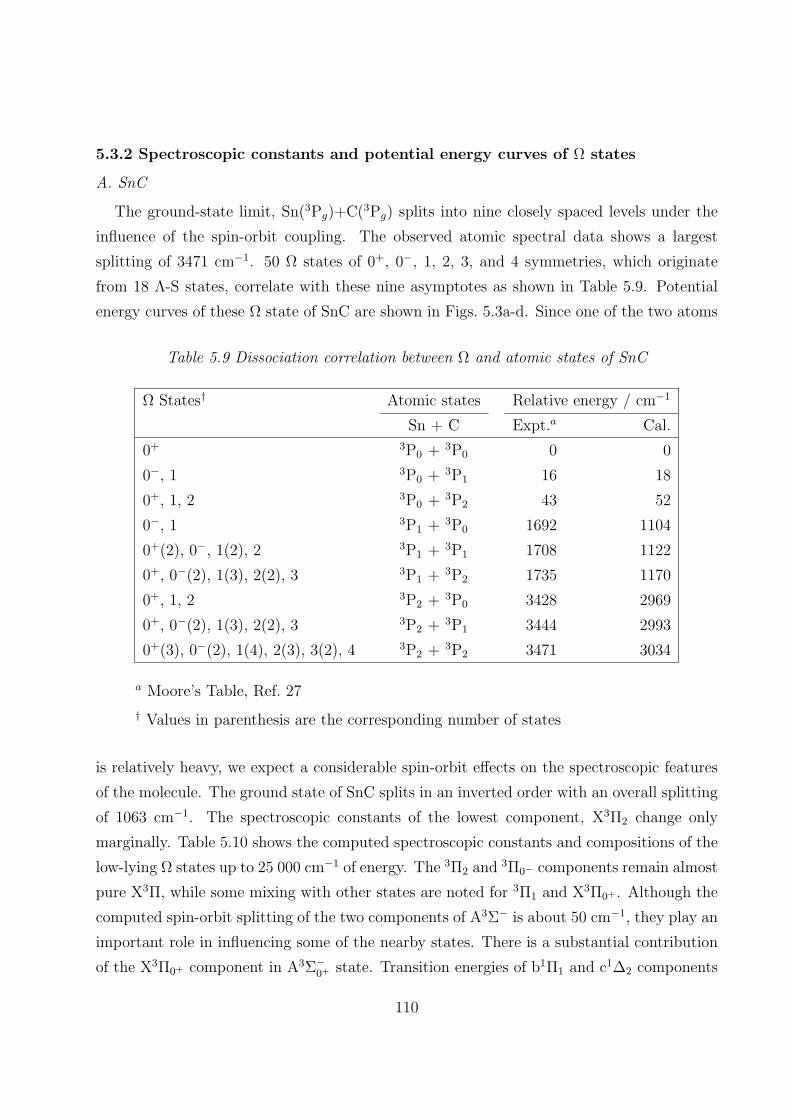
5.3.2 Spectroscopic constants and potential energy curves of Ω states
A. SnC
The ground-state limit, Sn(3Pg)+C(3Pg) splits into nine closely spaced levels under the
influence of the spin-orbit coupling. The observed atomic spectral data shows a largest
splitting of 3471 cm−1. 50 Ω states of 0+, 0−, 1, 2, 3, and 4 symmetries, which originate
from 18 Λ-S states, correlate with these nine asymptotes as shown in Table 5.9. Potential
energy curves of these Ω state of SnC are shown in Figs. 5.3a-d. Since one of the two atoms
Table 5.9 Dissociation correlation between Ω and atomic states of SnC
Ω States† Atomic states Relative energy / cm−1
Sn + C Expt.a Cal.
0+ 3P0 + 3P0 0 0
0−, 1 3P0 + 3P1 16 18
0+, 1, 2 3P0 + 3P2 43 52
0−, 1 3P1 + 3P0 1692 1104
0+(2), 0−, 1(2), 2 3P1 + 3P1 1708 1122
0+, 0−(2), 1(3), 2(2), 3 3P1 + 3P2 1735 1170
0+, 1, 2 3P2 + 3P0 3428 2969
0+, 0−(2), 1(3), 2(2), 3 3P2 + 3P1 3444 2993
0+(3), 0−(2), 1(4), 2(3), 3(2), 4 3P2 + 3P2 3471 3034
a Moore’s Table, Ref. 27
† Values in parenthesis are the corresponding number of states
is relatively heavy, we expect a considerable spin-orbit effects on the spectroscopic features
of the molecule. The ground state of SnC splits in an inverted order with an overall splitting
of 1063 cm−1. The spectroscopic constants of the lowest component, X3Π2 change only
marginally. Table 5.10 shows the computed spectroscopic constants and compositions of the
low-lying Ω states up to 25 000 cm−1 of energy. The 3Π2 and 3Π0− components remain almost
pure X3Π, while some mixing with other states are noted for 3Π1 and X3Π0+ . Although the
computed spin-orbit splitting of the two components of A3Σ− is about 50 cm−1, they play an
important role in influencing some of the nearby states. There is a substantial contribution
of the X3Π0+ component in A3Σ−0+ state. Transition energies of b1Π1 and c1∆2 components
110
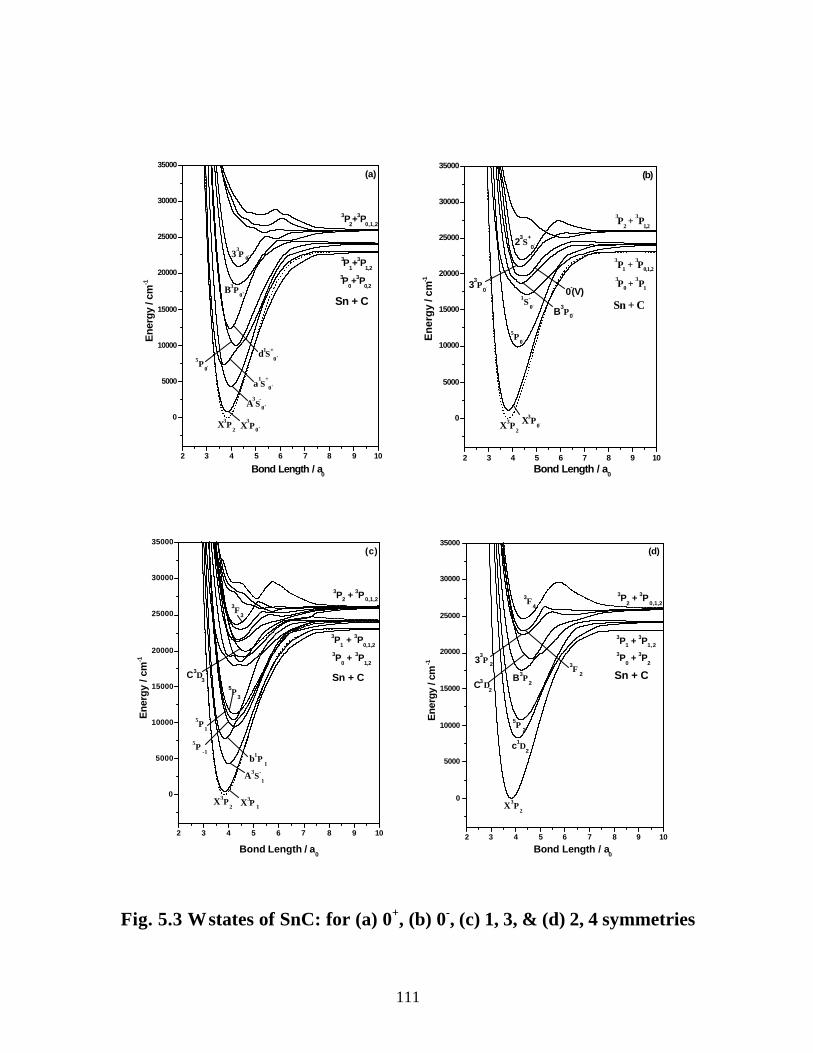
111
2 3 4 5 6 7 8 9 10
0
5000
10000
15000
20000
25000
30000
35000(a)
3P2+3P
0,1,2
3P1+3P
1,2
3P0+3P
0,2
Sn + C
33Π0+
B3Π0+
d1Σ+
0+5Π
0+
a1Σ
+
0+
A3Σ
-
0+
X3Π0+X3Π
2
E
nerg
y / c
m-1
Bond Length / a0
2 3 4 5 6 7 8 9 10
0
5000
10000
15000
20000
25000
30000
35000
0-(V)
(b)
23Σ
+
0-
33Π
0-
B3Π0-
3P2 + 3P
1,2
3P1 + 3P0,1,2
Sn + C
3P0 + 3P1
1Σ-
0-
5Π0-
X3Π0-X3Π
2
En
erg
y / c
m-1
Bond Length / a0
2 3 4 5 6 7 8 9 10
0
5000
10000
15000
20000
25000
30000
35000(c)
3Φ3
C3∆3
5Π3
3P2 + 3P0,1,2
3P1 + 3P
0,1,2
3P0 + 3P
1,2
Sn + C
5Π1
5Π-1
b1Π1
A3Σ-
1
X3Π1X3Π
2
Ene
rgy
/ cm
-1
Bond Length / a0
2 3 4 5 6 7 8 9 10
0
5000
10000
15000
20000
25000
30000
35000(d)
3Φ4
3P2 + 3P
0,1,2
3P1 + 3P1,2
3P0 + 3P
2
Sn + C3Φ
2
33Π2
C3∆2
B3Π2
5Π2
c1∆2
X3Π2
Ene
rgy
/ cm
-1
Bond Length / a0
Fig. 5.3 Ω states of SnC: for (a) 0+, (b) 0-, (c) 1, 3, & (d) 2, 4 symmetries
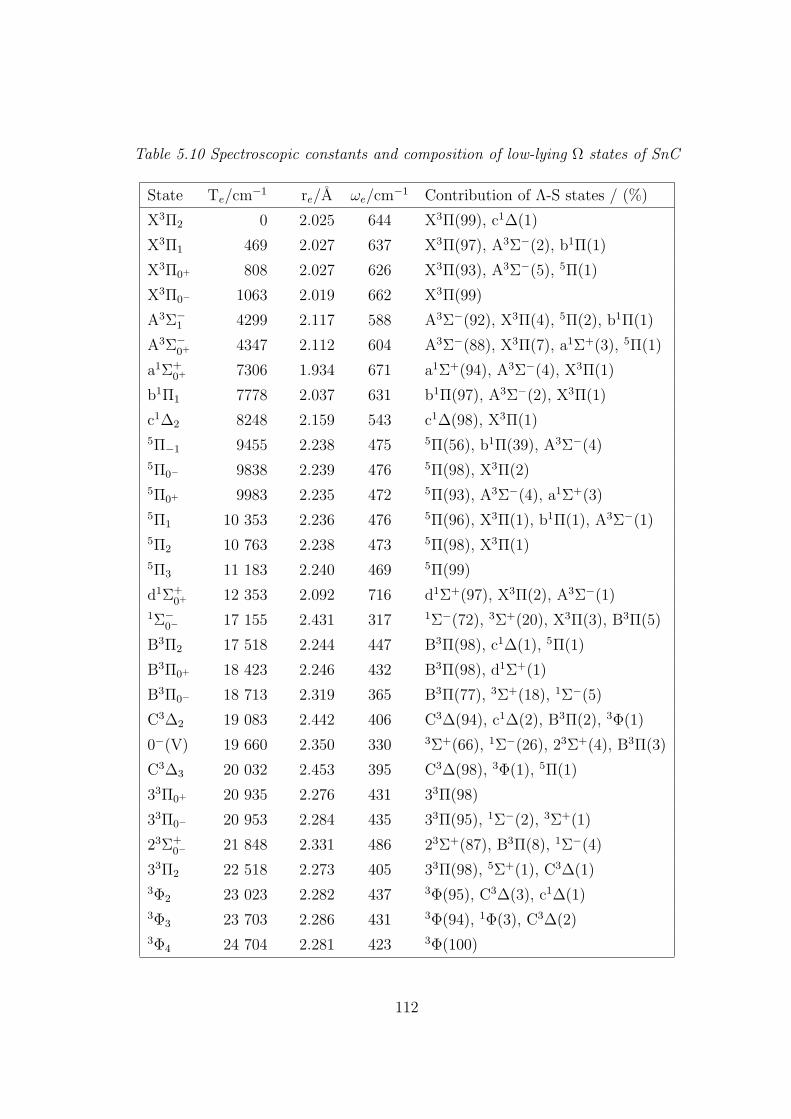
Table 5.10 Spectroscopic constants and composition of low-lying Ω states of SnC
State Te/cm−1 re/A ωe/cm−1 Contribution of Λ-S states / (%)
X3Π2 0 2.025 644 X3Π(99), c1∆(1)
X3Π1 469 2.027 637 X3Π(97), A3Σ−(2), b1Π(1)
X3Π0+ 808 2.027 626 X3Π(93), A3Σ−(5), 5Π(1)
X3Π0− 1063 2.019 662 X3Π(99)
A3Σ−1 4299 2.117 588 A3Σ−(92), X3Π(4), 5Π(2), b1Π(1)
A3Σ−0+ 4347 2.112 604 A3Σ−(88), X3Π(7), a1Σ+(3), 5Π(1)
a1Σ+0+ 7306 1.934 671 a1Σ+(94), A3Σ−(4), X3Π(1)
b1Π1 7778 2.037 631 b1Π(97), A3Σ−(2), X3Π(1)
c1∆2 8248 2.159 543 c1∆(98), X3Π(1)5Π−1 9455 2.238 475 5Π(56), b1Π(39), A3Σ−(4)5Π0− 9838 2.239 476 5Π(98), X3Π(2)5Π0+ 9983 2.235 472 5Π(93), A3Σ−(4), a1Σ+(3)5Π1 10 353 2.236 476 5Π(96), X3Π(1), b1Π(1), A3Σ−(1)5Π2 10 763 2.238 473 5Π(98), X3Π(1)5Π3 11 183 2.240 469 5Π(99)
d1Σ+0+ 12 353 2.092 716 d1Σ+(97), X3Π(2), A3Σ−(1)
1Σ−0− 17 155 2.431 317 1Σ−(72), 3Σ+(20), X3Π(3), B3Π(5)
B3Π2 17 518 2.244 447 B3Π(98), c1∆(1), 5Π(1)
B3Π0+ 18 423 2.246 432 B3Π(98), d1Σ+(1)
B3Π0− 18 713 2.319 365 B3Π(77), 3Σ+(18), 1Σ−(5)
C3∆2 19 083 2.442 406 C3∆(94), c1∆(2), B3Π(2), 3Φ(1)
0−(V) 19 660 2.350 330 3Σ+(66), 1Σ−(26), 23Σ+(4), B3Π(3)
C3∆3 20 032 2.453 395 C3∆(98), 3Φ(1), 5Π(1)
33Π0+ 20 935 2.276 431 33Π(98)
33Π0− 20 953 2.284 435 33Π(95), 1Σ−(2), 3Σ+(1)
23Σ+0− 21 848 2.331 486 23Σ+(87), B3Π(8), 1Σ−(4)
33Π2 22 518 2.273 405 33Π(98), 5Σ+(1), C3∆(1)3Φ2 23 023 2.282 437 3Φ(95), C3∆(3), c1∆(1)3Φ3 23 703 2.286 431 3Φ(94), 1Φ(3), C3∆(2)3Φ4 24 704 2.281 423 3Φ(100)
112

are increased by 500-700 cm−1 due to the spin-orbit coupling, while other spectroscopic
constants remain unchanged.
Six spin components of 5Π split in a regular order with 5Π−1 lying at the lowest. The
overall spin-orbit splitting is about 1728 cm−1. However, the equilibrium bond lengths and
vibrational frequencies of these components vary only slightly. The wave function of 5Π−1
at equilibrium shows about 39% contribution from 1Π (Table 5.10). Potential energy curves
of 5Π−1 and 5Π1 components undergo avoided curve crossings with that of b1Π1 due to their
larger equilibrium bond lengths. Except for 5Π0− , the spectroscopic parameters of other five
components of 5Π (reported in Table 5.10) are obtained from their respective diabatic curves.
The transition energy of d1Σ+0+ is increased by about 800 cm−1 due to the spin-orbit coupling.
Among the four components of the excited B3Π state, B3Π2 and B3Π0+ components have
distinct potential minima, while other two components involve several avoided crossings
making them difficult to fit for the calculations of the spectroscopic constants. However, the
calculations show that the spin-orbit splitting of the components of B3Π takes place in the
inverted order. The magnitude of splitting between the 0+ and 2 components of it is about
900 cm−1. The changes in the other spectroscopic constants are not significant. It should be
mentioned here that, while fitting the diabatic curve of B3Π0− , which has contribution from
B3Π by more than 75% at re (=2.319 A), gives an estimated value of Te=18 713 cm−1. The
relatively longer bond length of it is due to significant mixing with the similar components
of 3Σ+ and 1Σ−. Thus the overall splitting among the components of B3Π exceeds 1000
cm−1. Unlike B3Π, the spin components of the next higher root split in a regular order. The
potential energy curves of 33Π0+ , 33Π0− , and 33Π2 are fitted adiabatically. As the remaining
component of 1 undergoes strong spin mixing with the nearby components, the minimum
of it could not be located. The present results show that the separation between 0+ and
2 components is about 1585 cm−1. Like all other spin mixed states, re and ωe of these
components do not differ much. Spectroscopic constants of C3∆2, C3∆3, 3Φ2, 3Φ3, and 3Φ4
components are also reported in Table 5.10.
B. SnC+
After spin-orbit coupling the ground state dissociation limit splits into six sublevels. As
the spin-orbit splitting of 3P state of C is only 43 cm−1, the spin-orbit effect due to C is
negligible. Thus, the overall splitting among the Ω components of the SnC+ ion (4294 cm−1)
resembles the separation between 2P1/2 and 2P3/2 states of Sn+(Fig. 5.4). However, the
113
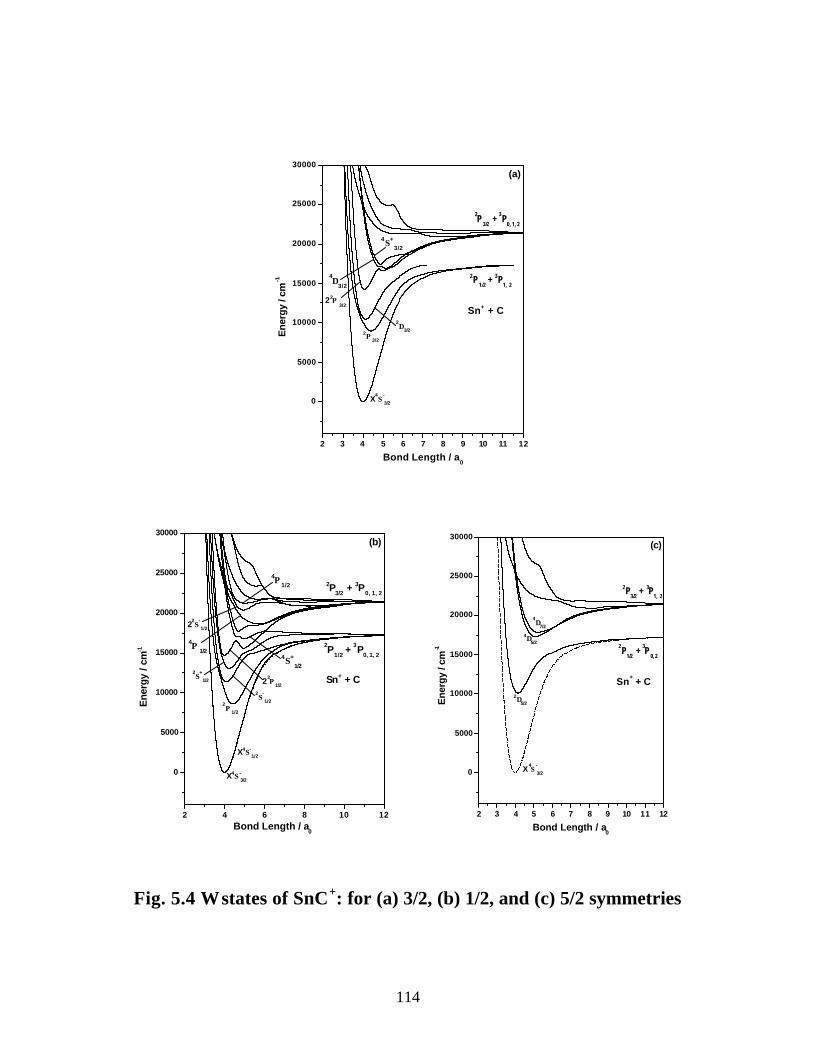
114
2 3 4 5 6 7 8 9 10 11 12
0
5000
10000
15000
20000
25000
30000
Sn+ + C
(a)
4∆
3/2
4Σ+
3/2
2P
3/2 +
3P
0, 1, 2
2P1/2
+ 3P1, 2
22Π3/2
2∆
3/22Π
3/2
X4Σ
-
3/2
Ene
rgy
/ cm
-1
Bond Length / a0
2 4 6 8 10 12
0
5000
10000
15000
20000
25000
30000
Sn+ + C
(b)
4Π1/2
22Σ
-
1/2
4Π1/2
4Σ+1/2
2P3/2
+ 3P0, 1, 2
2P1/2
+ 3P0, 1, 2
22Π
1/2
2Σ
+
1/2
2Σ
-
1/22Π
1/2
X4Σ-1/2
X4Σ -3/2
Ene
rgy
/ cm
-1
Bond Length / a0
2 3 4 5 6 7 8 9 10 11 12
0
5000
10000
15000
20000
25000
30000
Sn+ + C
(c)
2P3/2
+ 3P1, 2
2P
1/2 +
3P
0, 2
4∆
7/2
4∆5/2
2∆5/2
X4Σ
-
3/2
Ene
rgy
/ cm
-1
Bond Length / a0
Fig. 5.4 Ω states of SnC+: for (a) 3/2, (b) 1/2, and (c) 5/2 symmetries

calculated value of 4230 cm−1 (Table 5.11) compares it almost accurately. The ground state
splits into two almost inseparable Ω components, X4Σ−1/2 and X4Σ−3/2 with a separation of
only 21 cm−1 with similar re and ωe values. Two components of 2Π are shifted downwards
by 375 and 31 cm−1, respectively. The spin-orbit interaction does not change their re and ωe
much. 4∆ contributes to both of them by 1-2%. 2Π3/2 mixes with the similar component of2∆ by an amount of 8%. However, the spin-orbit coupling allows the 2Π–X4Σ− transition to
take place through their respective dipolar components. The predicted lifetime in the υ′=0
state of 2Π3/2 is of the order of 400 µs.
Table 5.11 Dissociation correlation between Ω and atomic states of SnC+
Ω states Atomic states Relative energy / cm−1
Sn+ + C Expt.a Cal.
1/2 2P1/2+3P0 0 0
1/2(2), 3/2 2P1/2+3P1 16 25
1/2(2), 3/2(2), 5/2 2P1/2+3P2 43 55
1/2, 3/2 2P3/2+3P0 4251 4185
1/2(3), 3/2(2), 5/2 2P3/2+3P1 4267 4205
1/2(4), 3/2(3), 5/2(2), 7/2 2P3/2+3P2 4294 4230
a Moore’s Table, Ref. 27
Ω=3/2 and 5/2 components of 2∆ split in an inverted order with a separation of 363 cm−1.
The re of 2∆5/2 is increased by 0.018 A, while its ωe is reduced by 20 cm−1. The contribution
of 4∆ in the 2∆5/2 state is 1% only. On the other hand, 2∆3/2 is strongly perturbed by the
component of 2Π. Its equilibrium bond length and vibrational frequency are changed only
marginally. Spin-orbit interactions do not change the spectroscopic constants of 2Σ−1/2 by
a considerable amount, only Te is increased by 134 cm−1. But in the longer bond length
region it is strongly perturbed by 4∆1/2. The next root of Ω=1/2 is dominated by 2Σ+. The
state is shifted upward by 237 cm−1, while re and ωe remain almost unchanged. However,
it interacts with the component of 2Σ− to a large extent and dissociates into the third
asymptote (Fig. 5.4b). Unlike 2Π, 22Π splits in an inverted pattern placing 22Π3/2 at least
400 cm−1 below 22Π1/2. Both the states mix to some extent with the similar components
of 4∆, 4Π, and 4Σ+. The extent of mixing is maximum at around 4.6 a0 which leads the
dissociation of both the states following complicated adiabatic pathway. We have fitted
115
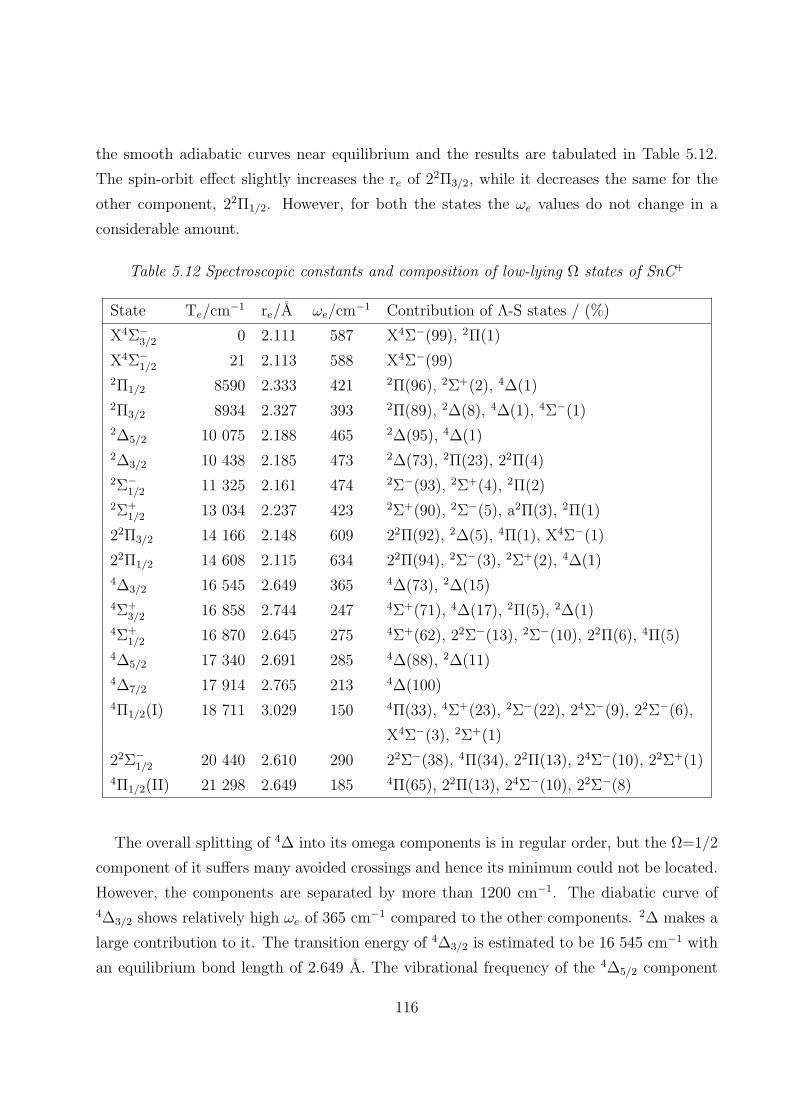
the smooth adiabatic curves near equilibrium and the results are tabulated in Table 5.12.
The spin-orbit effect slightly increases the re of 22Π3/2, while it decreases the same for the
other component, 22Π1/2. However, for both the states the ωe values do not change in a
considerable amount.
Table 5.12 Spectroscopic constants and composition of low-lying Ω states of SnC+
State Te/cm−1 re/A ωe/cm−1 Contribution of Λ-S states / (%)
X4Σ−3/2 0 2.111 587 X4Σ−(99), 2Π(1)
X4Σ−1/2 21 2.113 588 X4Σ−(99)2Π1/2 8590 2.333 421 2Π(96), 2Σ+(2), 4∆(1)2Π3/2 8934 2.327 393 2Π(89), 2∆(8), 4∆(1), 4Σ−(1)2∆5/2 10 075 2.188 465 2∆(95), 4∆(1)2∆3/2 10 438 2.185 473 2∆(73), 2Π(23), 22Π(4)2Σ−1/2 11 325 2.161 474 2Σ−(93), 2Σ+(4), 2Π(2)2Σ+
1/2 13 034 2.237 423 2Σ+(90), 2Σ−(5), a2Π(3), 2Π(1)
22Π3/2 14 166 2.148 609 22Π(92), 2∆(5), 4Π(1), X4Σ−(1)
22Π1/2 14 608 2.115 634 22Π(94), 2Σ−(3), 2Σ+(2), 4∆(1)4∆3/2 16 545 2.649 365 4∆(73), 2∆(15)4Σ+
3/2 16 858 2.744 247 4Σ+(71), 4∆(17), 2Π(5), 2∆(1)4Σ+
1/2 16 870 2.645 275 4Σ+(62), 22Σ−(13), 2Σ−(10), 22Π(6), 4Π(5)4∆5/2 17 340 2.691 285 4∆(88), 2∆(11)4∆7/2 17 914 2.765 213 4∆(100)4Π1/2(I) 18 711 3.029 150 4Π(33), 4Σ+(23), 2Σ−(22), 24Σ−(9), 22Σ−(6),
X4Σ−(3), 2Σ+(1)
22Σ−1/2 20 440 2.610 290 22Σ−(38), 4Π(34), 22Π(13), 24Σ−(10), 22Σ+(1)4Π1/2(II) 21 298 2.649 185 4Π(65), 22Π(13), 24Σ−(10), 22Σ−(8)
The overall splitting of 4∆ into its omega components is in regular order, but the Ω=1/2
component of it suffers many avoided crossings and hence its minimum could not be located.
However, the components are separated by more than 1200 cm−1. The diabatic curve of4∆3/2 shows relatively high ωe of 365 cm−1 compared to the other components. 2∆ makes a
large contribution to it. The transition energy of 4∆3/2 is estimated to be 16 545 cm−1 with
an equilibrium bond length of 2.649 A. The vibrational frequency of the 4∆5/2 component
116

is also somewhat larger while the re is shortened by 0.08 A because of spin-orbit mixing.
The 4∆7/2 remains almost pure with almost no changes in the spectroscopic constants. Two
components of 4Σ+ are almost inseparable. Both the components are energetically shifted
upward by more than 200 cm−1. Their potential minima are separated by 0.099 A, both of
which are lower than that of 4Σ+, however. 4∆ makes a large contribution (17%) to 4Σ+3/2,
while the components of lowest two roots of 2Σ− contribute to 4Σ+1/2 by more than 20%. The
potential minima of three more components with Ω=1/2 are located at 18 711, 20 440, and
21 298 cm−1, respectively. Two of them are dominated by 4Π. It may be mentioned here that
in the absence of any spin-orbit coupling 4Π was purely repulsive (see Fig. 5.2a). For example,
the eighth root of 1/2 is mainly characterized by 4Π(33%), 4Σ+(23%), and 2Σ−(22%) at
equilibrium. The shallow potential well has a characteristic frequency of 150 cm−1 only. A
large bond distance of 3.029 A makes the state spectroscopically insignificant. The next
root of 1/2 is characterized by 22Σ− (38%) with more than 30% contribution from 4Π. The
highest contribution of 4Π (c2=0.65) is noted at the tenth root of 1/2. The minimum of the
state is located at 2.649 A with a vibrational frequency of 185 cm−1. The above two states
are also spectroscopically unimportant due to very low Franck-Condon overlap factors.
5.3.3 Transition properties
A. SnC
As the ground state of SnC belongs to 3Π, there are several dipole allowed triplet-triplet
transitions. In addition, three transitions involving low-lying singlets are also studied here.
Transition probabilities of these transitions are calculated from transition moment data.
Fig. 5.5a shows transition moment curves of all nine transitions as a function of the bond
distance. For all the curves, transition moments smoothly decrease to zero at the longer
Sn-C bond distance. The partial radiative lifetimes at the lower few vibrational levels for
all nine transitions are tabulated in Table 5.13. As seen in Fig. 5.5a, transition moment of
A-X transition monotonically decreases with the increase in bond length. Its value around
the equilibrium bond length of the upper state is nearly 0.25 ea0. The computed lifetime
for the A-X transition at υ′=0 is about 220 µs, which decreases with υ′ due to longer re of
the A3Σ− compared to that of the ground state. Although transition dipole moments and
the energy differences for the B3Π-X3Π transition are sufficiently large, the Franck-Condon
overlap factors are small. As a result, the B-X transition is expected to be weak. The partial
lifetime at v′=0 for this transition is 6.5 ms. Four transitions from the excited triplets,
117
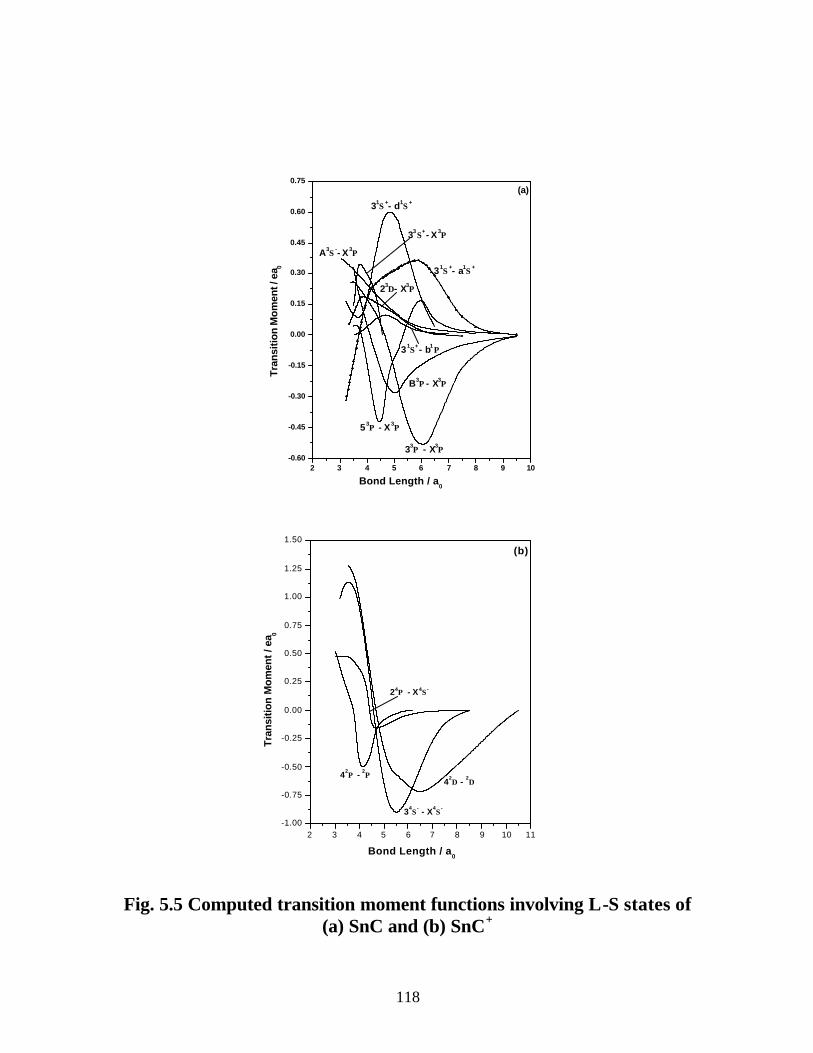
118
2 3 4 5 6 7 8 9 10-0.60
-0.45
-0.30
-0.15
0.00
0.15
0.30
0.45
0.60
0.75(a)
31Σ+- b1Π
31Σ+- a1Σ+
31Σ+- d1Σ+
33Σ+- X3Π
23∆- X3Π
53Π - X3Π
33Π - X3Π
B3Π- X3Π
A3Σ -- X3Π
Tran
sitio
n M
omen
t / e
a 0
Bond Length / a0
2 3 4 5 6 7 8 9 10 11-1.00
-0.75
-0.50
-0.25
0.00
0.25
0.50
0.75
1.00
1.25
1.50(b)
24Π - X4Σ-
42Π - 2Π
34Σ
- - X4Σ
-
42∆ - 2
∆
Tran
sitio
n M
omen
t / e
a 0
Bond Length / a0
Fig. 5.5 Computed transition moment functions involving Λ-S states of (a) SnC and (b) SnC+
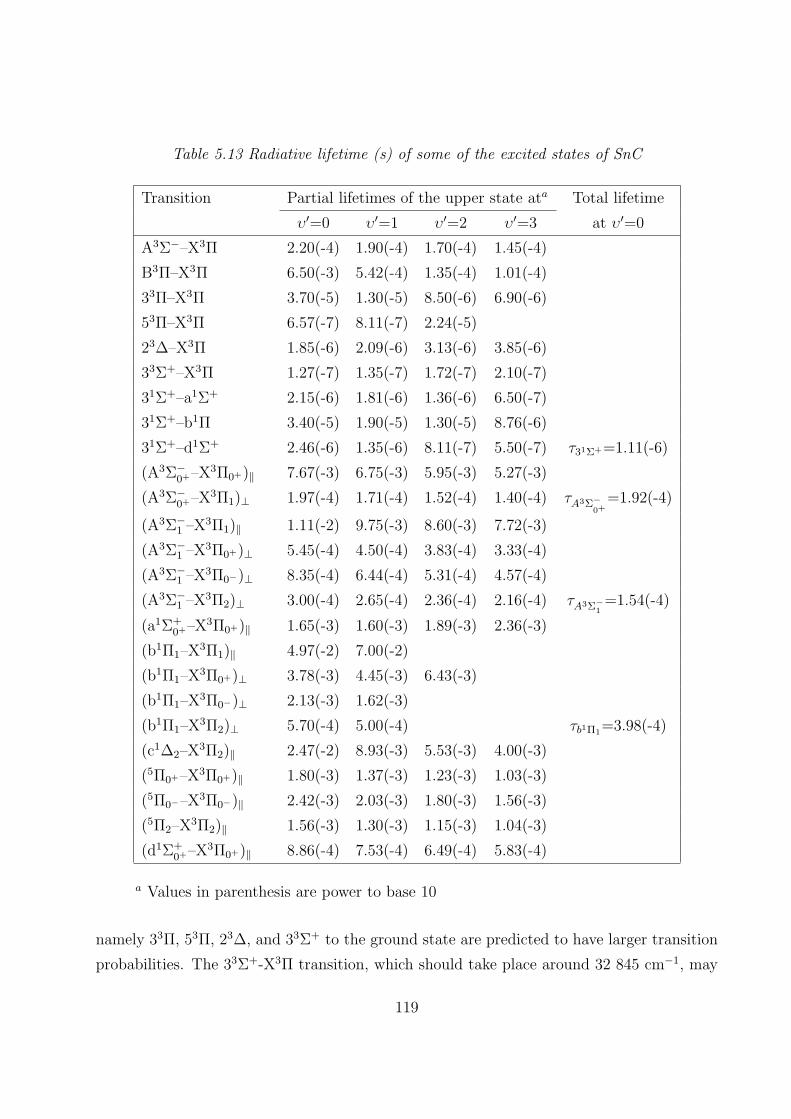
Table 5.13 Radiative lifetime (s) of some of the excited states of SnC
Transition Partial lifetimes of the upper state ata Total lifetime
υ′=0 υ′=1 υ′=2 υ′=3 at υ′=0
A3Σ−–X3Π 2.20(-4) 1.90(-4) 1.70(-4) 1.45(-4)
B3Π–X3Π 6.50(-3) 5.42(-4) 1.35(-4) 1.01(-4)
33Π–X3Π 3.70(-5) 1.30(-5) 8.50(-6) 6.90(-6)
53Π–X3Π 6.57(-7) 8.11(-7) 2.24(-5)
23∆–X3Π 1.85(-6) 2.09(-6) 3.13(-6) 3.85(-6)
33Σ+–X3Π 1.27(-7) 1.35(-7) 1.72(-7) 2.10(-7)
31Σ+–a1Σ+ 2.15(-6) 1.81(-6) 1.36(-6) 6.50(-7)
31Σ+–b1Π 3.40(-5) 1.90(-5) 1.30(-5) 8.76(-6)
31Σ+–d1Σ+ 2.46(-6) 1.35(-6) 8.11(-7) 5.50(-7) τ31Σ+=1.11(-6)
(A3Σ−0+–X3Π0+)‖ 7.67(-3) 6.75(-3) 5.95(-3) 5.27(-3)
(A3Σ−0+–X3Π1)⊥ 1.97(-4) 1.71(-4) 1.52(-4) 1.40(-4) τA3Σ−0+
=1.92(-4)
(A3Σ−1 –X3Π1)‖ 1.11(-2) 9.75(-3) 8.60(-3) 7.72(-3)
(A3Σ−1 –X3Π0+)⊥ 5.45(-4) 4.50(-4) 3.83(-4) 3.33(-4)
(A3Σ−1 –X3Π0−)⊥ 8.35(-4) 6.44(-4) 5.31(-4) 4.57(-4)
(A3Σ−1 –X3Π2)⊥ 3.00(-4) 2.65(-4) 2.36(-4) 2.16(-4) τA3Σ−1=1.54(-4)
(a1Σ+0+–X3Π0+)‖ 1.65(-3) 1.60(-3) 1.89(-3) 2.36(-3)
(b1Π1–X3Π1)‖ 4.97(-2) 7.00(-2)
(b1Π1–X3Π0+)⊥ 3.78(-3) 4.45(-3) 6.43(-3)
(b1Π1–X3Π0−)⊥ 2.13(-3) 1.62(-3)
(b1Π1–X3Π2)⊥ 5.70(-4) 5.00(-4) τb1Π1=3.98(-4)
(c1∆2–X3Π2)‖ 2.47(-2) 8.93(-3) 5.53(-3) 4.00(-3)
(5Π0+–X3Π0+)‖ 1.80(-3) 1.37(-3) 1.23(-3) 1.03(-3)
(5Π0−–X3Π0−)‖ 2.42(-3) 2.03(-3) 1.80(-3) 1.56(-3)
(5Π2–X3Π2)‖ 1.56(-3) 1.30(-3) 1.15(-3) 1.04(-3)
(d1Σ+0+–X3Π0+)‖ 8.86(-4) 7.53(-4) 6.49(-4) 5.83(-4)
a Values in parenthesis are power to base 10
namely 33Π, 53Π, 23∆, and 33Σ+ to the ground state are predicted to have larger transition
probabilities. The 33Σ+-X3Π transition, which should take place around 32 845 cm−1, may
119

be quite strong. The estimated radiative lifetime for this transition at the lowest vibrational
level is about 127 ns which increases with υ′. The 53Π-X3Π transition is found to be more
probable than the transition from either B3Π or 33Π state. The lifetime of the 23∆-X3Π
transition is of the order of microsecond. Transition probabilities of three singlet-singlet
transitions involving the excited 31Σ+ state are studied here. The computed transition
dipole moment functions of 31Σ+-a1Σ+, 31Σ+-d1Σ+, and 31Σ+-b1Π transitions are similar
in nature. First two transitions are more probable than the third one. The partial lifetime
for these three transitions are estimated to be 2.15, 2.46, and 34 µs, respectively at v′=0.
Summing up the transition probabilities, the total radiative lifetime of 31Σ+ is found to be
1.11 µs at the lowest vibrational level.
Transition probabilities of many transitions involving the X3Π2, X3Π1, and X3Π0+ com-
ponents of SnC are calculated. The A3Σ−1 -X3Π0+ transition is predicted to have larger
transition probabilities than A3Σ−0+-X3Π0+ . The computed partial lifetime for 0+-0+ tran-
sition is 7.67 ms, while for the 1-0+ transition it is less than a millisecond. The lifetimes
of A3Σ−1 –X3Π0− and A3Σ−1 –X3Π2 transitions at υ′=0 are found to be 0.84 and 0.30 ms,
respectively. Several weak spin-forbidden transitions are also reported in Table 5.13. At
the lowest vibrational level, the computed partial lifetimes of a1Σ+0+ and d1Σ+
0+ are 1.65
and 0.89 ms, respectively. Another important transition b1Π1-X3Π0+ is expected to carry
sufficient intensity. However, b1Π1-X3Π1 and b1Π1-X3Π2 transitions are predicted to have
larger transition probabilities. The spin-forbidden transitions originating from different spin
components of 5Π are weak and the partial radiative lifetimes of some of these transitions
are listed in Table 5.13.
B. SnC+
As mentioned earlier, only two spin allowed transitions from the ground state of SnC+
are expected to occur. The transition moment functions for both 24Π–X4Σ− and 34Σ−–
X4Σ− decrease sharply with the bond distance in the Franck-Condon region. The MRDCI
calculations predict a lifetime of 84 ns in the υ′=0 level of 24Π. The lifetime suddenly
increases for the next vibrational level as shown in Table 5.14. 34Σ−–X4Σ− transition is also
expected to have sufficient intensity. The radiative lifetime of the upper state (34Σ−) is 256
ns in its lowest vibrational level. It is much lower (111 ns) in its υ′=1 level. However, the
lifetime value increases for the next vibrational level possibly due to the avoided crossing
interaction between 34Σ− and its neighboring root. Two doublet-doublet transitions namely,
120

42Π–2Π and 42∆–2∆ also have comparatively high transition moments as shown in Fig. 5.5b.
Although the lowest doublet (2Π) is situated above the ground state by more than 1 eV,
transitions from these two states to 2Π and 2∆ are expected to occur due to large energy
gaps and high Franck-Condon factors. The total radiative lifetime in the ground vibrational
level of 42∆ is about 135 ns. 42Π state has a radiative lifetime of the order of 2-3 µs.
Table 5.14 Radiative lifetime (s) of some excited states of SnC+
Transition Partial lifetime of the upper state ata Total lifetime
υ′=0 υ′=1 υ′=2 υ′=3 at υ′=0
24Π–X4Σ− 8.37(-8) 2.07(-7) 2.15(-7) 5.48(-7)
34Σ−–X4Σ− 2.56(-7) 1.11(-7) 9.23(-7) 8.43(-7)
42Π–2Π 2.52(-6) 2.10(-6) 2.26(-6) 2.25(-6)
42∆–2∆ 2.14(-7) 9.66(-7) 8.10(-7) 8.00(-7)
42∆–2Π 3.63(-7) 3.86(-7) 4.06(-7) 4.21(-7) τ42∆=1.35(-7)
(2Π3/2–X4Σ−3/2)‖ 4.16(-4) 4.72(-4) 5.43(-4) 6.31(-4)
(2∆3/2–X4Σ−3/2)‖ 1.14(-3) 1.26(-3) 1.49(-3) 3.41(-3)
(2∆3/2–X4Σ−1/2)⊥ 6.01(-3) 6.02(-3) 6.25(-3) 7.28(-3) τ2∆3/2=9.58(-4)
(2∆5/2–X4Σ−3/2)⊥ 1.92(-3) 1.94(-3) 1.97(-3) 1.99(-3)
(2Σ−1/2–X4Σ−1/2)‖ 3.01(-3) 1.65(-3) 1.95(-3) 2.11(-3)
(2Σ−1/2–X4Σ−3/2)⊥ 4.32(-3) 3.79(-3) 3.84(-3) 5.30(-3) τ2Σ−1/2
=1.77(-3)
(2Σ+1/2–X4Σ−1/2)⊥ 4.68(-5) 4.77(-5) 6.59(-5) 1.34(-4)
(22Π1/2–X4Σ−1/2)‖ 8.51(-4) 7.80(-4) 8.42(-4)
(22Π3/2–X4Σ−3/2)‖ 1.72(-2) 2.83(-2) 4.20(-2) 4.65(-2)
(22Π3/2–X4Σ−1/2)⊥ 1.02(-3) 1.38(-3) 4.05(-3) τ22Π3/2=9.63(-4)
a Values in parenthesis are power to base 10
Many spin forbidden transitions through the respective dipolar components are reported
in Table 5.14. Their transition moment values are within 0.16 ea0. The parallel component
of 2Σ+1/2–X4Σ−1/2 has the highest transition moment value of 0.158 ea0 at 4.95 a0. Due to low
Franck-Condon overlap factor the computed lifetime is of the order of one tenth of a second.
Inspite of lower transition moment lifetime of the corresponding perpendicular transition is
around 50 µs at the lower vibrational level. Transitions involving the components of 2∆,
121

2Π, 2Σ− and the corresponding ground-state component are studied here. Of these, the
spin forbidden transitions like, (2Π3/2–X4Σ−3/2)‖ is of great interest. The computed radiative
lifetime of the 2Π3/2 state at υ′=0 is 416 µs. The value increases monotonically with the
increase in the vibrational quantum number. The 22Π1/2 state has a lifetime of the order of
850 µs for the parallel component of the 22Π1/2–X4Σ−1/2 transition. The transition lifetime
for the Ω=3/2 component of 22Π is much higher (963 µs).
5.3.4 Dipole moments and ionization energies
The computed dipole moment of SnC is reported to be 2.44 D in its ground state. It
is much higher in A3Σ− with the same sense of polarity (Sn+C−). Table 5.4 shows the
equilibrium dipole moments (µe) of other excited states. The Spin-orbit coupling decreases
the ground-state dipole moment by 0.02 D for X3Π2 component. But it increases to some
extent for other three components as shown in Table 5.16. Both the components of A3Σ−
have slightly lesser dipole moment than the originating states. The effect is insignificant for
the next three states, but in case of 5Π−1 it is more prominent and µe is increased by 0.22 D.
The variation of dipole moment functions for some low-lying states of SnC are shown in
Fig. 5.6a. All the curves smoothly converge to zero dipole moment at longer bond distances.
The ground-state dipole moment of SnC+ is computed here as -0.847 D. As the dipole
moment is origin dependent, the origin is kept at the center of mass of SnC+. However, the
variation of dipole moments with the bond distance for some low-lying Λ-S states of the ion
are shown in Fig. 5.6b. The values shown in Fig. 5.6b are obtained keeping Sn at the origin.
Dipole moment functions of most of the states pass through maxima. The 4∆ and 4Σ+
states of SnC+ have very high maximum values of 1.237 and 1.232 ea0, respectively, both
at 3.9 a0. In Table 5.15 we have tabulated the equilibrium dipole moments of few low-lying
states following the same convention as that of the ground state. The ground state and a
few low-lying doublets have opposite sense of polarity. Spin-orbit effect does not change the
ground-state dipole moment to a large amount, but it affects those of the excited states.2Π1/2 has at least 0.10 D less dipole moment than its originating Λ-S state. The effect is
more prominent for the two spin components of 2∆. Comparatively large dipole moment of2∆3/2 may be due to mixing of it with the similar component of 2Π.
We have calculated the vertical ionization energies (VIE) of SnC from the difference in
estimated CI energies of the ground-state neutral molecule and the cation at the ground
state as well as some low-lying excited states. The estimated CI energies are taken from
122
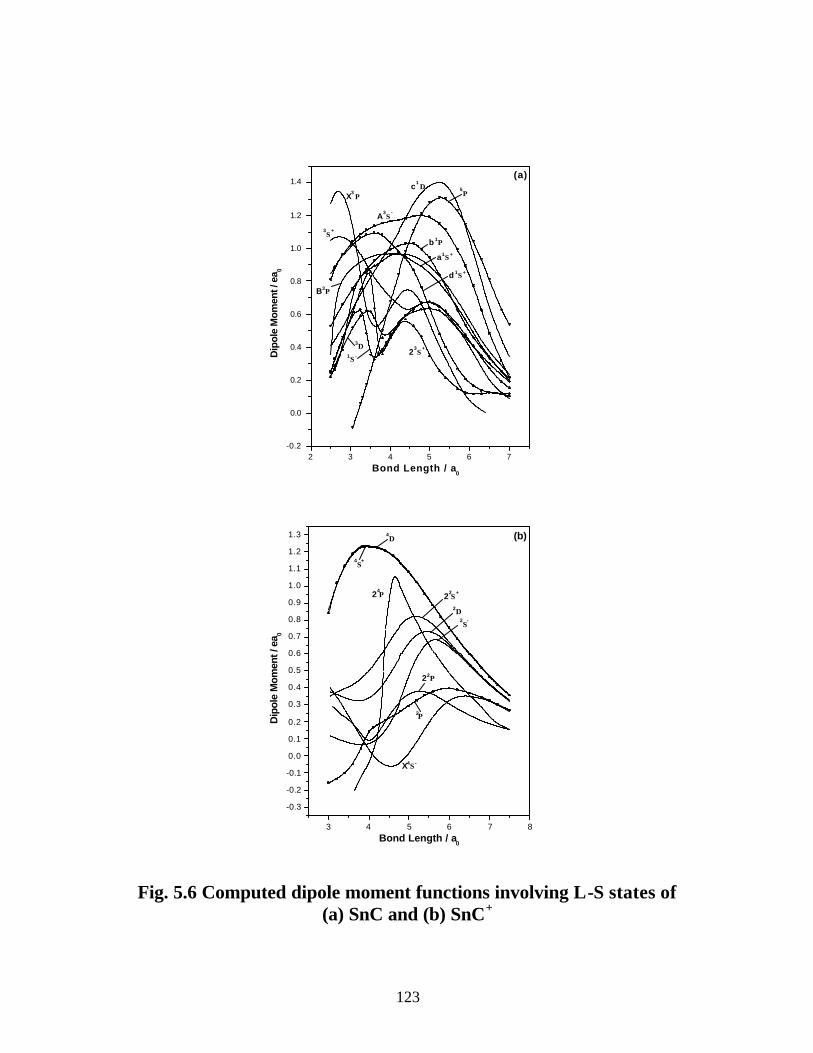
123
2 3 4 5 6 7-0.2
0.0
0.2
0.4
0.6
0.8
1.0
1.2
1.4
B3Π
(a)
b 1Π
1Σ -
d 1Σ+
a1Σ+
c1
∆
23Σ
+
3Σ
+
3∆
A3Σ
-
5ΠX
3Π
Dip
ole
Mom
ent /
ea 0
Bond Length / a0
3 4 5 6 7 8
-0.3
-0.2
-0.1
0.0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
1.1
1.2
1.3 (b)
22Π
2Π
2Σ
-
2∆
22Σ
+24Π
4∆
4Σ
+
X4Σ-
Dip
ole
Mom
ent /
ea 0
Bond Length / a0
Fig. 5.6 Computed dipole moment functions involving Λ-S states of (a) SnC and (b) SnC+
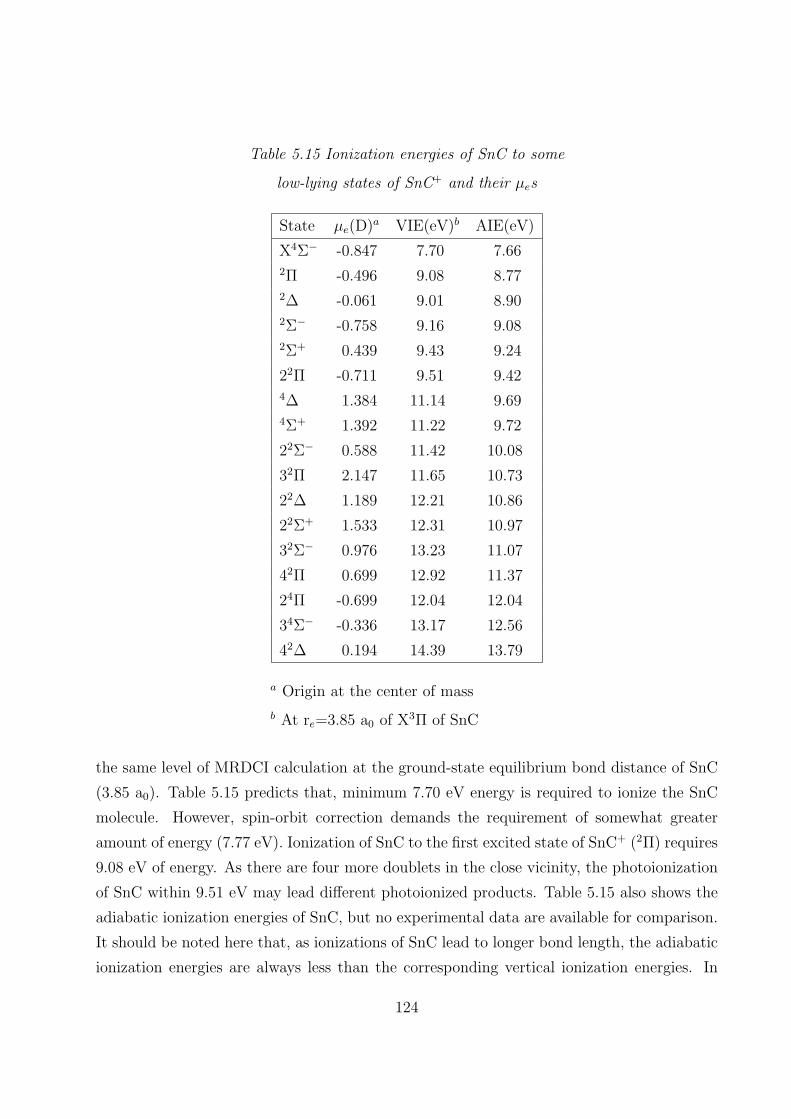
Table 5.15 Ionization energies of SnC to some
low-lying states of SnC+ and their µes
State µe(D)a VIE(eV)b AIE(eV)
X4Σ− -0.847 7.70 7.662Π -0.496 9.08 8.772∆ -0.061 9.01 8.902Σ− -0.758 9.16 9.082Σ+ 0.439 9.43 9.24
22Π -0.711 9.51 9.424∆ 1.384 11.14 9.694Σ+ 1.392 11.22 9.72
22Σ− 0.588 11.42 10.08
32Π 2.147 11.65 10.73
22∆ 1.189 12.21 10.86
22Σ+ 1.533 12.31 10.97
32Σ− 0.976 13.23 11.07
42Π 0.699 12.92 11.37
24Π -0.699 12.04 12.04
34Σ− -0.336 13.17 12.56
42∆ 0.194 14.39 13.79
a Origin at the center of mass
b At re=3.85 a0 of X3Π of SnC
the same level of MRDCI calculation at the ground-state equilibrium bond distance of SnC
(3.85 a0). Table 5.15 predicts that, minimum 7.70 eV energy is required to ionize the SnC
molecule. However, spin-orbit correction demands the requirement of somewhat greater
amount of energy (7.77 eV). Ionization of SnC to the first excited state of SnC+ (2Π) requires
9.08 eV of energy. As there are four more doublets in the close vicinity, the photoionization
of SnC within 9.51 eV may lead different photoionized products. Table 5.15 also shows the
adiabatic ionization energies of SnC, but no experimental data are available for comparison.
It should be noted here that, as ionizations of SnC lead to longer bond length, the adiabatic
ionization energies are always less than the corresponding vertical ionization energies. In
124
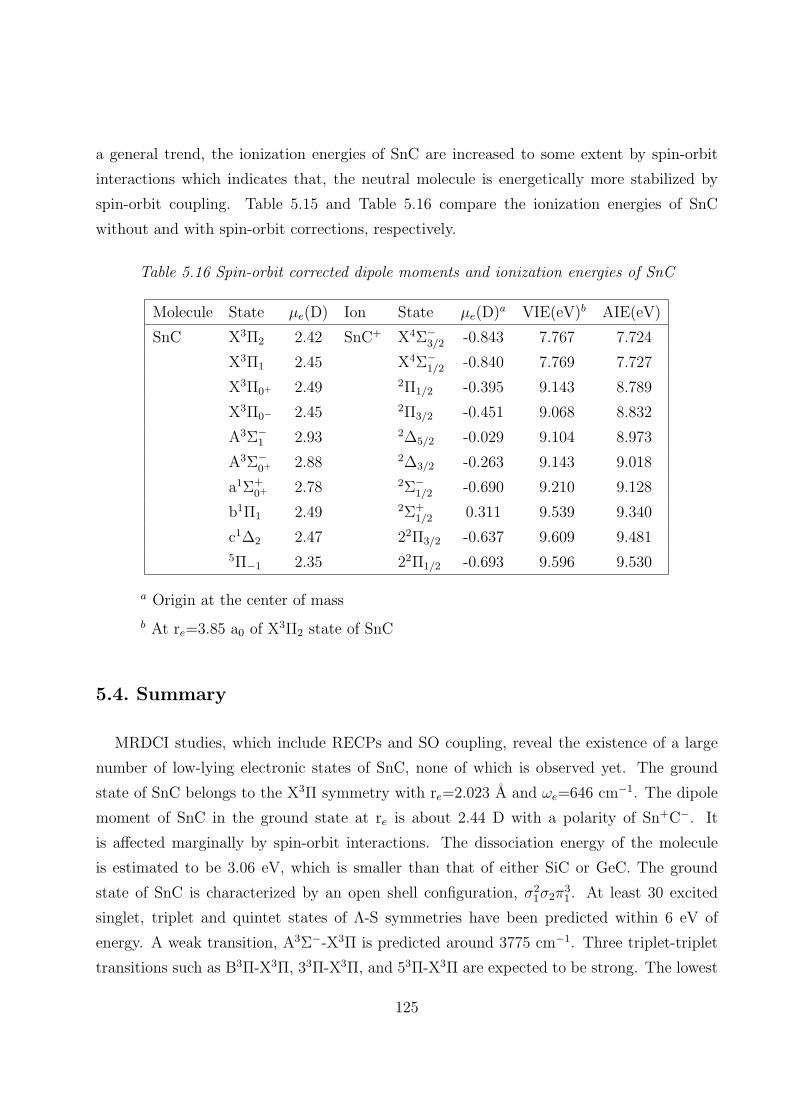
a general trend, the ionization energies of SnC are increased to some extent by spin-orbit
interactions which indicates that, the neutral molecule is energetically more stabilized by
spin-orbit coupling. Table 5.15 and Table 5.16 compare the ionization energies of SnC
without and with spin-orbit corrections, respectively.
Table 5.16 Spin-orbit corrected dipole moments and ionization energies of SnC
Molecule State µe(D) Ion State µe(D)a VIE(eV)b AIE(eV)
SnC X3Π2 2.42 SnC+ X4Σ−3/2 -0.843 7.767 7.724
X3Π1 2.45 X4Σ−1/2 -0.840 7.769 7.727
X3Π0+ 2.49 2Π1/2 -0.395 9.143 8.789
X3Π0− 2.45 2Π3/2 -0.451 9.068 8.832
A3Σ−1 2.93 2∆5/2 -0.029 9.104 8.973
A3Σ−0+ 2.88 2∆3/2 -0.263 9.143 9.018
a1Σ+0+ 2.78 2Σ−1/2 -0.690 9.210 9.128
b1Π1 2.49 2Σ+1/2 0.311 9.539 9.340
c1∆2 2.47 22Π3/2 -0.637 9.609 9.4815Π−1 2.35 22Π1/2 -0.693 9.596 9.530
a Origin at the center of mass
b At re=3.85 a0 of X3Π2 state of SnC
5.4. Summary
MRDCI studies, which include RECPs and SO coupling, reveal the existence of a large
number of low-lying electronic states of SnC, none of which is observed yet. The ground
state of SnC belongs to the X3Π symmetry with re=2.023 A and ωe=646 cm−1. The dipole
moment of SnC in the ground state at re is about 2.44 D with a polarity of Sn+C−. It
is affected marginally by spin-orbit interactions. The dissociation energy of the molecule
is estimated to be 3.06 eV, which is smaller than that of either SiC or GeC. The ground
state of SnC is characterized by an open shell configuration, σ21σ2π
31. At least 30 excited
singlet, triplet and quintet states of Λ-S symmetries have been predicted within 6 eV of
energy. A weak transition, A3Σ−-X3Π is predicted around 3775 cm−1. Three triplet-triplet
transitions such as B3Π-X3Π, 33Π-X3Π, and 53Π-X3Π are expected to be strong. The lowest
125

quintet state, 5Π is strongly bound. A strong mixing between the lowest two 3Σ+ states has
created a double minima in the potential energy curve of the lowest root. The extent of the
spin-orbit coupling is significantly large due to the presence of the heavier Sn atom. The
largest splitting among the components of X3Π, which split in an inverted order, is more
than 1000 cm−1. However, the spin-orbit splitting between 0+ and 1 components of A3Σ− is
expected to be small. Many spin-forbidden transitions such as a1Σ+0+–X3Π0+ , d1Σ+
0+–X3Π0+ ,
b1Π1–X3Π0+ etc. are studied, the partial lifetime of which are in the millisecond order and
they are predicted to be weak.
The removal of a π electron from SnC generates the cation SnC+ which has 4Σ− ground-
state like the other homologous cations like SiC+, GeC+. It requires at least 7.77 eV of energy.
The computed dissociation energy of the ground-state cation is 2.47 eV which follows the
trend SiC+> GeC+> SnC+. The ground-state bond length and vibrational frequency of
SnC+ are estimated to be 2.112 A and 591 cm−1, respectively which are not much affected
by spin-orbit effects. Two important transitions from the ground state are expected to be
occurred within 4.91 eV. Excited 24Π and 34Σ− states have the radiative lifetimes of 84
and 256 ns, respectively at υ′=0. Total radiative lifetime of the lowest vibrational state
of 42∆ is also of the order of hundred nanosecond. Several spin-forbidden transitions are
also expected for SnC+. The perpendicular component of the 2Σ+1/2–X4Σ−1/2 transition has
a lifetime of 50 µs in the lowest few vibrational levels. The ground-state dipole moment of
the ion is computed here as -0.847 D.
126

5.5. References
1 R. Roncka, J. Tolle, C. Cook, A.V.G. Chizmeshya and J. Kouvetakis, V. D’Costa,
J. Menendez, Z.D. Chen, and S. Zollner, Appl. Phys. Lett. 86, 191912 (2005).
2 J. Tottle, A.V.G. Chizmeshya, Y.-Y. Fang, J. Kouvetakis, V. D’Costa, C.-W. Hu,
J. Menendez and T.S.T. Tsong, Appl. Phys. Lett. 89, 231924 (2006).
3 G. Gigli, G.M. Eloni, M. Carrozzino, J. Chem. Phys. 122, 14303 (2005).
4 A. Ciccioli, G. Gigli, G. Meloni, E. Testani, J. Chem. Phys. 127, 54303 (2007).
5 P.L. Goodfriend, Canad. J. Phys. 45, 3425 (1967).
6 R.W. Schmude Jr., K.A. Gingerich, J. Chem. Phys. 109, 3069 (1998).
7 J.M.L. Martin, J.P. Francois, R. Gijbels, J. Chem. Phys. 92, 6655 (1990).
8 I. Shim, M. Sai Baba, K.A. Gingerich, J. Phys. Chem. 102, 10763 (1998).
9 R. Pandey, M. Rerat, C, Darrigan, M. Causa, J. Appl. Phys. 88, 6462 (2000).
10 A. Benzair, B. Bouhafs, C. Mathien, H. Aourag, Phys. Lett. A 282, 299 (2001).
11 A. Benzair, H. Aourag, Phys. Stat. Solidi B 231, 411 (2002).
12 R. Khenata, H. Baltache, M. Sahnun, M. Driz, M. Rerat, B. Abbar, Physica B 336, 321
(2003).
13 G. Li, C. Wang, J. Mol. Struct. (THEOCHEM) 824, 48 (2007).
14 A. Pramanik, K.K. Das, J. Mol. Spectrosc. 244, 13 (2007).
15 A. Pramanik, S. Chakrabarti, K.K. Das, Chem. Phys. Lett. 450, 221 (2008).
16 L.A. LaJohn, P.A. Christiansen, R.B. Ross, T. Atastroo, W.C. Ermler, J. Chem. Phys.
87, 2812 (1987).
17 L.F. Pacios, P.A. Christiansen, J. Chem. Phys. 82, 2664 (1985).
18 R.J. Buenker, S.D. Peyerimhoff, Theor. Chim. Acta 35, 33 (1974).
19 R.J. Buenker, S.D. Peyerimhoff, Theor. Chim. Acta 39, 217 (1975).
20 R.J. Buenker, S.D. Peyerimhoff, W. Butscher, Mol. Phys. 35, 771 (1978).
21 R.J. Buenker, Int. J. Quantum Chem. 29, 435 (1986).
22 R.J. Buenker, in: P. Burton (Ed.), Proc. Workshop on Quantum Chemistry and Molecular
Physics in Wollongong, Wollongong, Australia, 1980.
127

23 R.J. Buenker, in Studies in Physical and Theoretical Chemistry, R. Carbo, Ed.; Elsevier:
Amsterdam, The Natherlands, Vol. 21 (Current Aspects of Quantum Chemistry), 1981.
24 R.J. Buenker, R.A. Phillips, J. Mol. Struct. (THEOCHEM) 123, 291 (1985).
25 E.R. Davidson, in: R. Daudel, B. Pullman (Eds.), The World of Quantum Chemistry,
Reidel, Dordrecht, The Netherlands, 1974.
26 G. Hirsch, P.J. Bruna, S.D. Peyreimhoff, R.J. Buenker, Chem. Phys. Lett. 52, 442 (1977).
27 C.E. Moore, Tables of Atomic Energy Levels, vols. I-III, US National Bureau of Standards,
Washington, DC, 1971.
28 Electronic Spectrum of SnC: A Theoretical Study,
A. Pramanik, K.K. Das (communicated).
29 Theoretical Investigation of Electronic States of SnC+,
A. Pramanik, K.K. Das (to be communicated).
128

CHAPTER – 6
ELECTRONIC STRUCTURE AND
SPECTROSCOPIC PROPERTIES OF PbC AND PbC+

6.1. Introduction
Energetic information of chemical bond involving permutation of all elements in the en-
tire periodic table have been collected over the years.1 Besides their applications, the simple
diatomic molecules draw special interest in contributing the information about their bond
length, bond energy etc. As mentioned in the earlier chapters, intragroup 14 heteronuclear
diatomic molecules have generated a special interest in recent years because of their possible
applications in catalysis, sensor films and mostly, they are the building blocks of cluster
materials.2−4 Seven out of ten intragroup 14 diatomics have been energetically character-
ized by Knudesen effusion mass spectroscopic technique.5,6 Ciccioli et al.6 have investigated
the thermodynamic properties of diatomics containing lead. They have also predicted the
unknown dissociation energies of SiSn and PbC.
The number of electrons and the relativistic effect are quite high, so theoretical calcula-
tions on the molecules containing lead are very difficult task. RECP based DFT studies on
the lead-doped carbon clusters, PbCn/PbC+n /PbC−n (n=1-10) have been carried out using
B3LYP method with both CEP-31G and TZP+ basis sets by Li et al.7 Their studies include
the structure, stability, ionization potentials (IPs), electron affinities (EAs), and fragmenta-
tion energies of the PbCn/PbC+n /PbC−n (n=1-10) clusters. They predicted the ground state
of neutral PbC molecule as 3Π with an equilibrium bond length of 2.063 A. Two excited
states, 5Π and 1∆ at the energies 21.6 and 22.9 kcal/mole, respectively are also reported.
However, Ciccioli et al.6 have performed back to back electronic structure calculations in
the CCSD(T) level of theory together with small core relativistic pseudopotential (aug-cc-
pVTZ) on intragroup 14 diatomics along with PbC to determine the molecular constants
(re, ωe), necessary for their data analysis. They also computed adiabatic ionization energies
(AIEs), adiabatic electron affinities (AEAs), term energies, and dissociation energies (D00).
According to their prediction, the D00 value for PbC is about 1.93 eV. The 4H0 value for
the reaction PbC(g) = Pb(g) + C(g) is 248.1 kJmole−1 as predicted by Ciccioli et al.6
Contradictorily, they predicted the ground state of PbC as X3Σ− with re and ωe, 2.191 A
and 565 cm−1, respectively.
On the other hand, ionization of the neutral carbide leads to the 4Σ− ground-state of the
ionic species, as predicted in a recent theoretical study of SiC+.13 The B3LYP/DFT studies7
also report the ground state of PbC+ as 4Σ with ...π2 valence electronic configuration. Its
equilibrium bond length is predicted to be 2.179 A which is longer than that of the neutral
129

molecule by 0.096 A. They have also predicted an excited 2∆ state with re=2.191 A for
PbC+.
The diatomic carbides of third to fifth row elements are extensively studied by many
authors.8−12 But similar results of PbC are not available in literature. This is for the first
time, we have carried out a large scale MRDCI study using RECP to study electronic
structure and spectroscopic properties of the ground as well as low-lying excited states of
PbC and PbC+ within 6 eV of energy. Spin-orbit interaction has also been incorporated to
show the changes in the potential energy curves and spectroscopic parameters in comparison
to their corresponding Λ-S states. Transition properties of some excited states with different
spin multiplicities have been computed. Their radiative lifetimes have also been predicted by
calculating the Einstein’s spontaneous emission coefficients. An attempt has been made to
determine the dipole moment of the neutral as well as cationic species. Ionization energies
for the ionization to the low-lying states of the cation from the ground state have been
calculated.
6.2. Computational details
6.2.1 RECPs and basis sets
In the present study, the RECPs of Ross et al.14 replace the inner electrons of Pb, leaving
the remaining 6s26p2 electrons in the valence space. The valence (3s3p4d) Gaussian basis set
from the same reference14 is used for the calculation. For C atom, the RECPs of Pacios and
Christiansen15, which retain the outer 2s22p2 electrons in the valence space, are employed.
The optimized (4s4p) Gaussian basis set compatible with the above mentioned RECPs is
augmented with two sets of d functions of exponents 1.2 and 0.35 a−20 , respectively. The
total number of active electrons in the valence space is 8.
6.2.2 SCF MOs and CI
All the calculations are performed in the C2v subgroup of the main group C∞v, placing Pb
at origin and choosing z axis as the molecular axis. SCF calculations for the 3Σ−(σ21σ
22π
21)
state of PbC and 2Σ+(σ21σ2π
21) state of PbC+ are performed at different internuclear distances
between 3 to 15 a0. This generates 64 symmetry adapted molecular orbitals. Out of these
64 MOs, 2 MOs of very high energy are discarded, while the remaining 62 MOs are treated
130
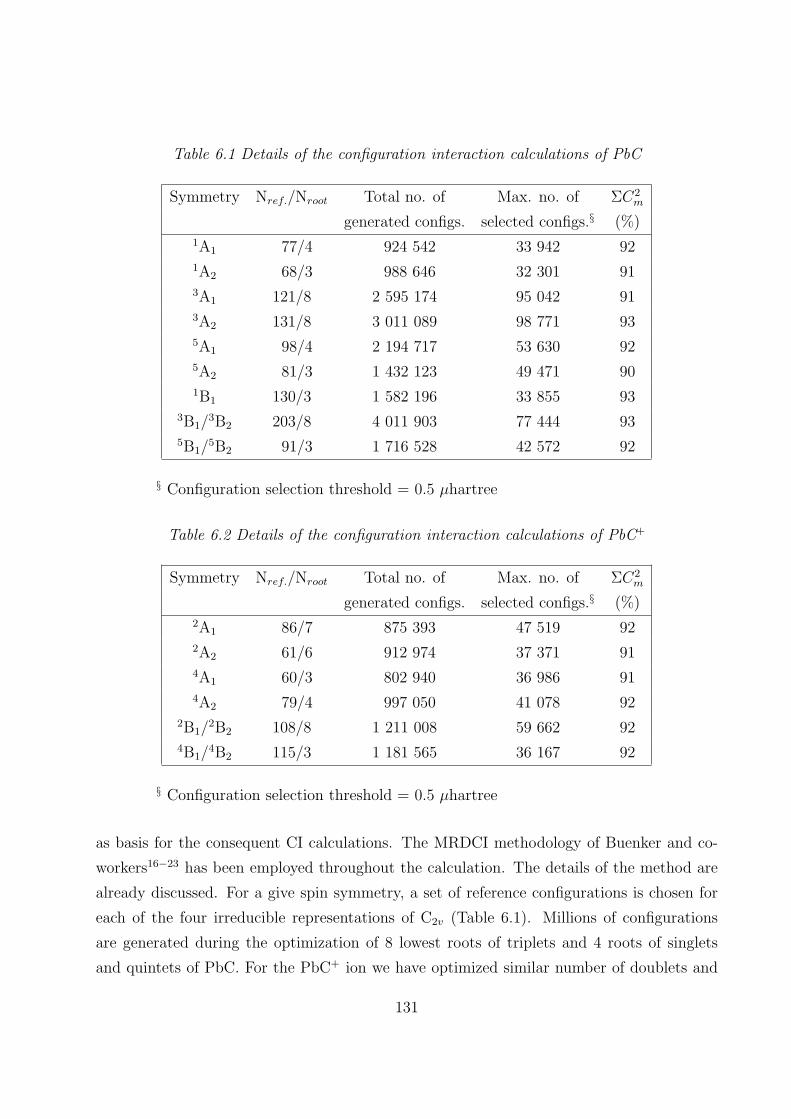
Table 6.1 Details of the configuration interaction calculations of PbC
Symmetry Nref./Nroot Total no. of Max. no. of ΣC2m
generated configs. selected configs.§ (%)1A1 77/4 924 542 33 942 921A2 68/3 988 646 32 301 913A1 121/8 2 595 174 95 042 913A2 131/8 3 011 089 98 771 935A1 98/4 2 194 717 53 630 925A2 81/3 1 432 123 49 471 901B1 130/3 1 582 196 33 855 93
3B1/3B2 203/8 4 011 903 77 444 935B1/5B2 91/3 1 716 528 42 572 92
§ Configuration selection threshold = 0.5 µhartree
Table 6.2 Details of the configuration interaction calculations of PbC+
Symmetry Nref./Nroot Total no. of Max. no. of ΣC2m
generated configs. selected configs.§ (%)2A1 86/7 875 393 47 519 922A2 61/6 912 974 37 371 914A1 60/3 802 940 36 986 914A2 79/4 997 050 41 078 92
2B1/2B2 108/8 1 211 008 59 662 924B1/4B2 115/3 1 181 565 36 167 92
§ Configuration selection threshold = 0.5 µhartree
as basis for the consequent CI calculations. The MRDCI methodology of Buenker and co-
workers16−23 has been employed throughout the calculation. The details of the method are
already discussed. For a give spin symmetry, a set of reference configurations is chosen for
each of the four irreducible representations of C2v (Table 6.1). Millions of configurations
are generated during the optimization of 8 lowest roots of triplets and 4 roots of singlets
and quintets of PbC. For the PbC+ ion we have optimized similar number of doublets and
131

quartets (see Table 6.2). A configuration selection threshold of 0.5 µhartree has been used
such that sum of the square of the CI coefficients becomes more or less 0.90. The higher order
excitations from the reference configurations are taken care by the multireference analogue
of Davidson’s correction.24,25
4.2.3 Spin-orbit interaction
In the subsequent steps, we have introduced the spin-orbit interaction through the spin-
orbit operators of Pb and C atoms. All the low-lying Λ-S states are allowed to interact.
In the C22v double group, the resulting Ω states of PbC belong to A1, A2, and B1/B2 and
those of PbC+ correspond to E1 and E2, respectively. The details are discussed in earlier
chapter.26
Spectroscopic constants (re, Te, ωe) are obtained by fitting PECs constructed from the
CI energies. The vibrational energies and wave functions are obtained from the numerical
solutions of one dimensional nuclear Schrodinger equation. Transition dipole moments in-
volving different Λ-S and Ω states of spin and/or dipole allowed transitions are computed.
These also give the estimate of the radiative lifetimes of the excited states.
6.3. Results and discussion
6.3.1 Spectroscopic constants and potential energy curves of Λ–S states
A. PbC
Eighteen electronic states of Σ+(2),Σ−,Π(2), and ∆ symmetries of singlet, triplet and
quintet spin multiplicities correlate with the lowest dissociation limit, Pb(3Pg)+C(3Pg). In
the atomic level, the first excited state (1Dg) of Pb and that of C differ in their relative energy
by 0.2 eV only. Thus, the two limits, Pb(3Pg)+C(1Dg) and Pb(1Dg)+C(3Pg) are expected
to be very closed. Both of them correlate with the triplets of Σ+,Σ−(2),Π(3),∆(2), and
Φ symmetries. Two sets of 3Σ− and 3Π states dissociate into the fourth and fifth limits
(Table 6.3) at 19 761 and 21 614 cm−1, respectively above the first dissociation limit. The
first excited states (1Dg) of both Pb and C combine to form 15 singlets of different symmetries
which lie at 21 911 cm−1 above the lowest limit. However, the calculated value of 18 710
cm−1 is underestimated by 3200 cm−1 from the experimental observation.27
132
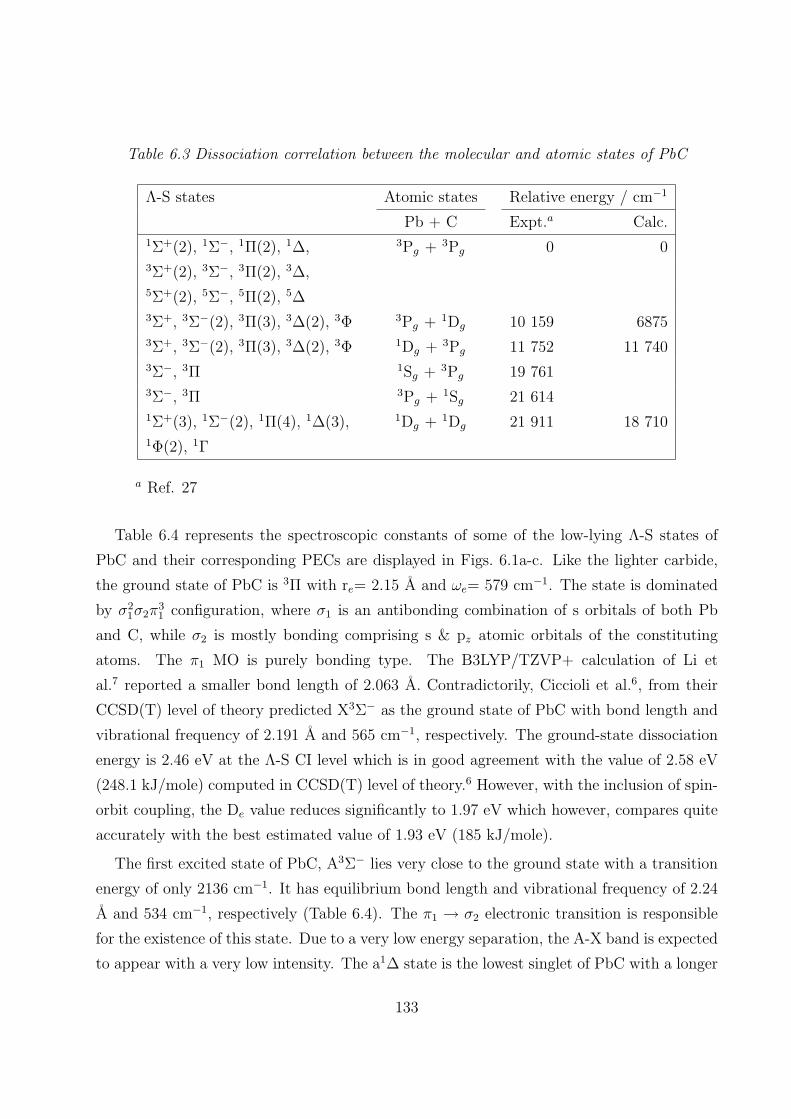
Table 6.3 Dissociation correlation between the molecular and atomic states of PbC
Λ-S states Atomic states Relative energy / cm−1
Pb + C Expt.a Calc.1Σ+(2), 1Σ−, 1Π(2), 1∆, 3Pg + 3Pg 0 03Σ+(2), 3Σ−, 3Π(2), 3∆,5Σ+(2), 5Σ−, 5Π(2), 5∆3Σ+, 3Σ−(2), 3Π(3), 3∆(2), 3Φ 3Pg + 1Dg 10 159 68753Σ+, 3Σ−(2), 3Π(3), 3∆(2), 3Φ 1Dg + 3Pg 11 752 11 7403Σ−, 3Π 1Sg + 3Pg 19 7613Σ−, 3Π 3Pg + 1Sg 21 6141Σ+(3), 1Σ−(2), 1Π(4), 1∆(3), 1Dg + 1Dg 21 911 18 7101Φ(2), 1Γ
a Ref. 27
Table 6.4 represents the spectroscopic constants of some of the low-lying Λ-S states of
PbC and their corresponding PECs are displayed in Figs. 6.1a-c. Like the lighter carbide,
the ground state of PbC is 3Π with re= 2.15 A and ωe= 579 cm−1. The state is dominated
by σ21σ2π
31 configuration, where σ1 is an antibonding combination of s orbitals of both Pb
and C, while σ2 is mostly bonding comprising s & pz atomic orbitals of the constituting
atoms. The π1 MO is purely bonding type. The B3LYP/TZVP+ calculation of Li et
al.7 reported a smaller bond length of 2.063 A. Contradictorily, Ciccioli et al.6, from their
CCSD(T) level of theory predicted X3Σ− as the ground state of PbC with bond length and
vibrational frequency of 2.191 A and 565 cm−1, respectively. The ground-state dissociation
energy is 2.46 eV at the Λ-S CI level which is in good agreement with the value of 2.58 eV
(248.1 kJ/mole) computed in CCSD(T) level of theory.6 However, with the inclusion of spin-
orbit coupling, the De value reduces significantly to 1.97 eV which however, compares quite
accurately with the best estimated value of 1.93 eV (185 kJ/mole).
The first excited state of PbC, A3Σ− lies very close to the ground state with a transition
energy of only 2136 cm−1. It has equilibrium bond length and vibrational frequency of 2.24
A and 534 cm−1, respectively (Table 6.4). The π1 → σ2 electronic transition is responsible
for the existence of this state. Due to a very low energy separation, the A-X band is expected
to appear with a very low intensity. The a1∆ state is the lowest singlet of PbC with a longer
133
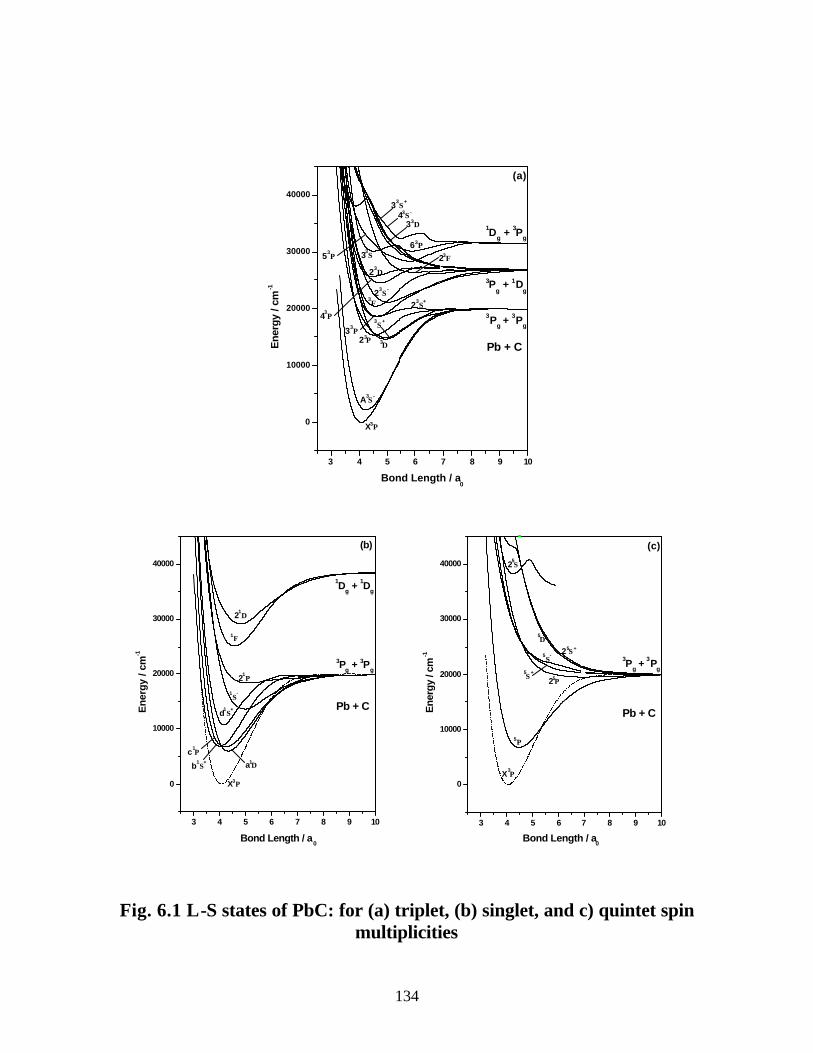
134
3 4 5 6 7 8 9 10
0
10000
20000
30000
40000
(a)
3Pg + 1D
g
1Dg + 3P
g
3Pg + 3P
g
Pb + C
33Σ
+
43Σ-
33∆
63Π53
Π 23Φ33Σ
-
23∆
43Π
23Σ
-
3Φ
33Π
23Σ
+
3Σ
+
23Π 3∆
A3Σ
-
X3Π
Ene
rgy
/ cm
-1
Bond Length / a0
3 4 5 6 7 8 9 10
0
10000
20000
30000
40000
(b)
1Dg + 1D
g
3Pg + 3P
g
Pb + C
21∆
1Φ
21Π
1Σ
-
d1Σ
+
c1Π
b1Σ
+ a1∆
X3Π
Ene
rgy
/ cm
-1
Bond Length / a0
3 4 5 6 7 8 9 10
0
10000
20000
30000
40000
(c)
3Pg + 3P
g
Pb + C
25Σ
-
25Σ+
5∆
5Σ
-
5Σ
+
25Π
5Π
X3Π
Ene
rgy
/ cm
-1
Bond Length / a0
Fig. 6.1 Λ-S states of PbC: for (a) triplet, (b) singlet, and c) quintet spin
multiplicities
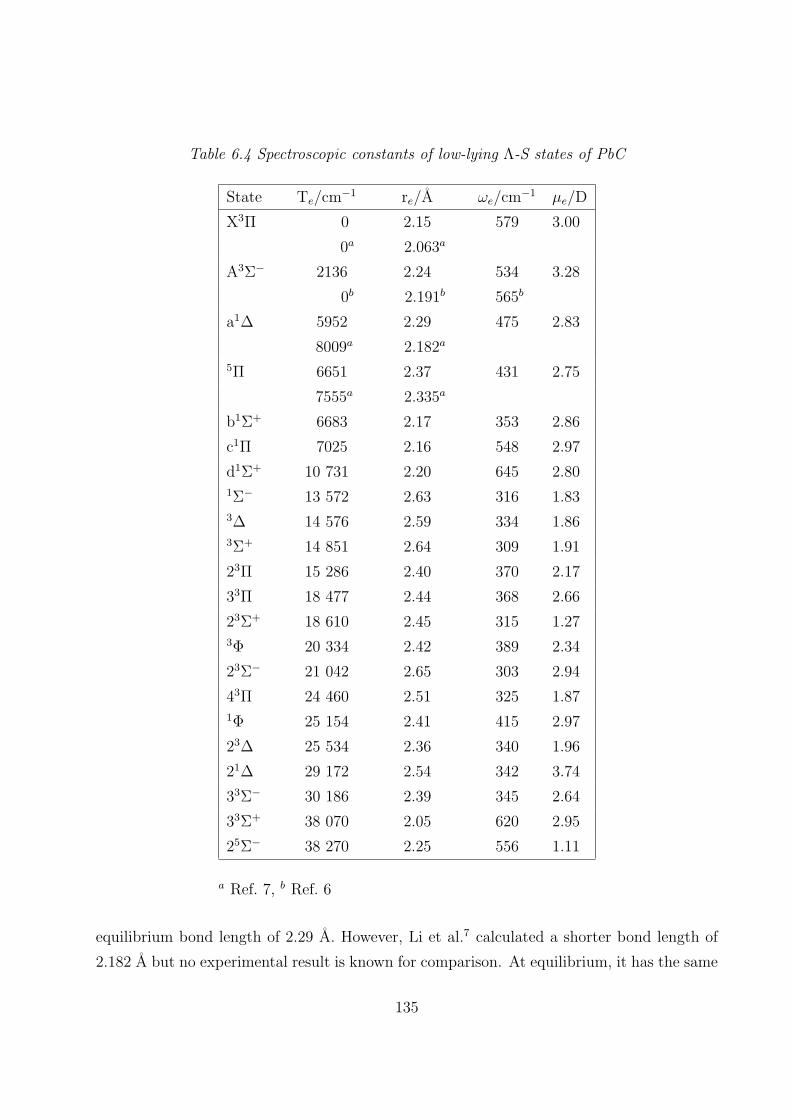
Table 6.4 Spectroscopic constants of low-lying Λ-S states of PbC
State Te/cm−1 re/A ωe/cm−1 µe/D
X3Π 0 2.15 579 3.00
0a 2.063a
A3Σ− 2136 2.24 534 3.28
0b 2.191b 565b
a1∆ 5952 2.29 475 2.83
8009a 2.182a
5Π 6651 2.37 431 2.75
7555a 2.335a
b1Σ+ 6683 2.17 353 2.86
c1Π 7025 2.16 548 2.97
d1Σ+ 10 731 2.20 645 2.801Σ− 13 572 2.63 316 1.833∆ 14 576 2.59 334 1.863Σ+ 14 851 2.64 309 1.91
23Π 15 286 2.40 370 2.17
33Π 18 477 2.44 368 2.66
23Σ+ 18 610 2.45 315 1.273Φ 20 334 2.42 389 2.34
23Σ− 21 042 2.65 303 2.94
43Π 24 460 2.51 325 1.871Φ 25 154 2.41 415 2.97
23∆ 25 534 2.36 340 1.96
21∆ 29 172 2.54 342 3.74
33Σ− 30 186 2.39 345 2.64
33Σ+ 38 070 2.05 620 2.95
25Σ− 38 270 2.25 556 1.11
a Ref. 7, b Ref. 6
equilibrium bond length of 2.29 A. However, Li et al.7 calculated a shorter bond length of
2.182 A but no experimental result is known for comparison. At equilibrium, it has the same
135

dominant configuration, σ21σ
22π
21 (77%) as that of A3Σ−. The longer bond length may be
attributed to the removal of an electron from strongly bonding π1 to the weaker bonding σ2.
The computed vibrational frequency of the state is 475 cm−1.
A strongly bound quintet is located around 6650 cm−1 above the ground state having a
longer bond length of 2.37 A. The state originates from the π1 → π2 electronic transition.
The antibonding character of π2 changes the bonding character of the molecule in this state.
As shown in Table 6.5, the same configuration (σ21σ2π
21π2) generates three more excited 3Π
and a pair of 1,3Φ states. It may be mentioned here that this configuration gives rise to four
more Π states, all of which are not computed here.
The lowest 1Σ+ state, designated as b, interacts strongly with the next root of the same
symmetry, namely d1Σ+. As a result a shallow double minima appear in the potential
energy curve of b1Σ+ (Fig. 6.1b). The energy barrier between the two minima of b1Σ+ is
extremely low and thus the adiabatic potential well on fitting gives an estimated re= 2.17 A
and ωe=353 cm−1. The ωe obtained by fitting the adiabatic curve of d1Σ+ is 645 cm−1.
The longer magnitude of ωe for this state is due to avoided crossing with its lower root.
However, at the potential minima, both the states b and d are described by two dominating
configurations, σ21σ
22π
21 and σ2
1π41 as shown in Table 6.5. The ground-state configuration
dominates in c1Π state having a similar bond length and vibrational frequency as those of
the ground state. The first dissociation limit correlates with two more singlets namely, 1Σ−
and 21Π. The 1Σ− state has a longer bond length and low binding energy of 0.78 eV, while
the 21Π state is repulsive.
Since the ground state of PbC is of 3Π symmetry and there exists another close lying 3Σ−,
the excited 3Π, 3Σ+, 3Σ−, and 3∆ states are spectroscopically important. The potential
minima of the lowest 3∆ and 3Σ+ states are at longer bond distances and thus, Franck-
Condon overlap factors are quite low for them. The second and third roots of 3Π can be
identified in emission spectroscopy at around 15 300 and 18 500 cm−1, respectively. The 33Π
state is more strongly bound than 23Π as evident from Fig. 6.1a. However, they dissociate
into two different asymptotes.
The 23Σ+ state interacts with the lowest root of 3Σ+. The state is spectroscopically less
important due to small Franck-Condon overlap factor. A 3Φ state is generated dominantly
by σ21σ2π
21π2 (80%) configuration with a binding energy of 0.794 eV. In the longer bond
length region (> 5.5 a0) it interacts with a higher root of 3Φ which is repulsive in nature.
136

Table 6.5 Composition of Λ-S states of PbC at equilibrium bond length
State Configuration (% contribution)
X3Π σ21σ2π
31(71), σ2
1σ2π21π2(7), σ2
1σ2π1π22(6)
A3Σ− σ21σ
22π
21(83)
a1∆ σ21σ
22π
21(77), σ2
1σ22π1π2(5), σ2
1σ22π
22(4)
5Π σ21σ2π
21π2(84)
b1Σ+ σ21σ
22π
21(42), σ2
1π41(33), σ2
1π21π
22(3)
c1Π σ21σ2π
31(76), σ2
1σ2π1π22(6), σ2
1σ2π21π2(4)
d1Σ+ σ21π
41(38), σ2
1σ22π
21(31), σ2
1π21π
22(8), σ2
1π31π2(4)
1Σ− σ21σ
22π1π2(63), σ2
1σ2σ4π1π2(15), σ21σ2σ7π1π2(6), σ2
1σ2σ3π1π2(3)3∆ σ2
1σ22π1π2(62), σ2
1σ2σ4π1π2(15), σ21σ2σ7π1π2(6), σ2
1σ2σ3π1π2(3)3Σ+ σ2
1σ22π1π2(54), σ2
1σ2σ4π1π2(15), σ21σ2σ7π1π2(6), σ2
1π31π2(5),
σ21π
21π
22(4), σ2
1σ2σ3π1π2(3)
23Π σ21σ2π
21π2(74), σ2
1σ2π31(4), σ2
1σ2π1π22(2)
33Π σ21σ2π
21π2(82), σ2
1σ2π31(2)
23Σ+ σ21π
31π2(47), σ2
1σ22π1π2(23), σ2
1π21π
22(8), σ2
1π1π32(6)
3Φ σ21σ2π
21π2(82)
23Σ− σ21σ
22π1π2(34), σ2
1σ22π
21(28), σ2
1σ2σ5π21(6), σ2
1π31π2(5)
43Π σ21σ2π
21π2(68), σ2
1σ2π1π22(7), σ2
1σ4π1π22(3), σ2
1σ4π21π2(2)
1Φ σ21σ2π
21π2(85)
23∆ σ21π
31π2(73), σ2
1π21π
22(5), σ2
1π1π32(4)
21∆ σ21σ
22π
21(37), σ2
1σ22π1π2(23), σ2
1π31π2(16), σ2
1π21π
22(3), σ2
1σ2σ4π1π2(3)
33Σ− σ21π
31π2(63), σ2
1π21π
22(8), σ2
1σ22π1π2(6)
33Σ+ σ1σ2π41(56), σ1σ2π
31π2(9), σ1σ2π
21π
22(6), σ2
1π31π2(5)
25Σ− σ21σ2σ3π
21(75), σ2
1σ2σ4π21(5), σ2
1σ2σ3π1π2(4)
Another 3Σ− state also exists at 21 042 cm−1 having bond length at least 0.50 A greater
than that of the ground state. Beyond the potential minimum, the state is flattened and
thus it has a low vibrational frequency of 303 cm−1. Transition to this state is also less
probable because of poor overlap region with the ground state. Among the bound 3Π states,
binding energy is the least for 43Π. It has a relatively longer re and hence spectroscopically
unimportant. However, it interacts with fifth and sixth root of the same symmetry in the
137

longer bond length region. Above 25 000 cm−1, there are two bound singlets designated as 1Φ
and 21∆, both of which dissociate into sixth asymptote, Pb(1Dg)+C(1Dg). Their potential
minima have been located at 2.41 and 2.54 A, respectively.
In the Λ-S level, 23∆ and 33Σ− are very important states of PbC from the spectroscopic
point of view. They are weakly bound with binding energies 0.19 and 0.14 eV, respectively.
Both the states are characterized by the σ21π
31π2 configuration, the origin of which is the
σ2 → π2 electronic transition. Like two other lighter carbides (SiC and SnC), the 33Σ+ state
of PbC arises dominantly from σ1σ2π41 configuration (c2=0.56). It is bound with an energy
barrier of 0.21 eV only. However, 33Σ+ may be isolated in the emission band at around
38 000 cm−1. All the quintets except 5Π and 25Σ− are repulsive. The latter one undergoes
predissociation at around 5.0 a0 through a potential barrier of 0.33 eV.
B. PbC+
The first dissociation limit of PbC+, comprising Pb+(2Pu) and C(3Pg), correlates with a
set of six doublets and six quartets of Σ+, Σ−(2), Π(2), and ∆ symmetries. The ground
state of Pb+ (2Pu) and the first excited state of C (1Dg) combine to form nine states of
Σ+(2), Σ−, Π(3), ∆(2), and Φ symmetries. All these states dissociate into the second
asymptote lying 11 782 cm−1 above the first one. The atomic spectral data27, however,
shows that the calculation is overestimated by about 1700 cm−1 with an error of 16%. Only
two doublets namely, 42Σ+ and 62Π correlate with the third asymptote, Pb+(2Pu) and C(1Sg)
which lies 22 782 cm−1 above the ground limit. Here also our estimated value exceeds the
experimental value, although by 5% only. Atomic combination of Pb+(2Pu)+C(5Su) around
Table 6.6 Dissociation correlation between the molecular and atomic states of PbC+
Λ-S states Atomic states Relative energy / cm−1
Expt.a Calc.2Σ+, 2Σ−(2), 2Π(2), 2∆, Pb+(2Pu) + C(3Pg) 0 04Σ+, 4Σ−(2), 4Π(2), 4∆2Σ+(2), 2Σ−, 2Π(3), 2∆(2), 2Φ Pb+(2Pu) + C(1Dg) 10 159 11 7822Σ+, 2Π Pb+(2Pu) + C(1Sg) 21 614 22 7824Σ−, 4Π, 6Σ−, 6Π Pb+(2Pu) + C(5Su) 33 701 33 580
a Averaged over J, Ref. 27
138
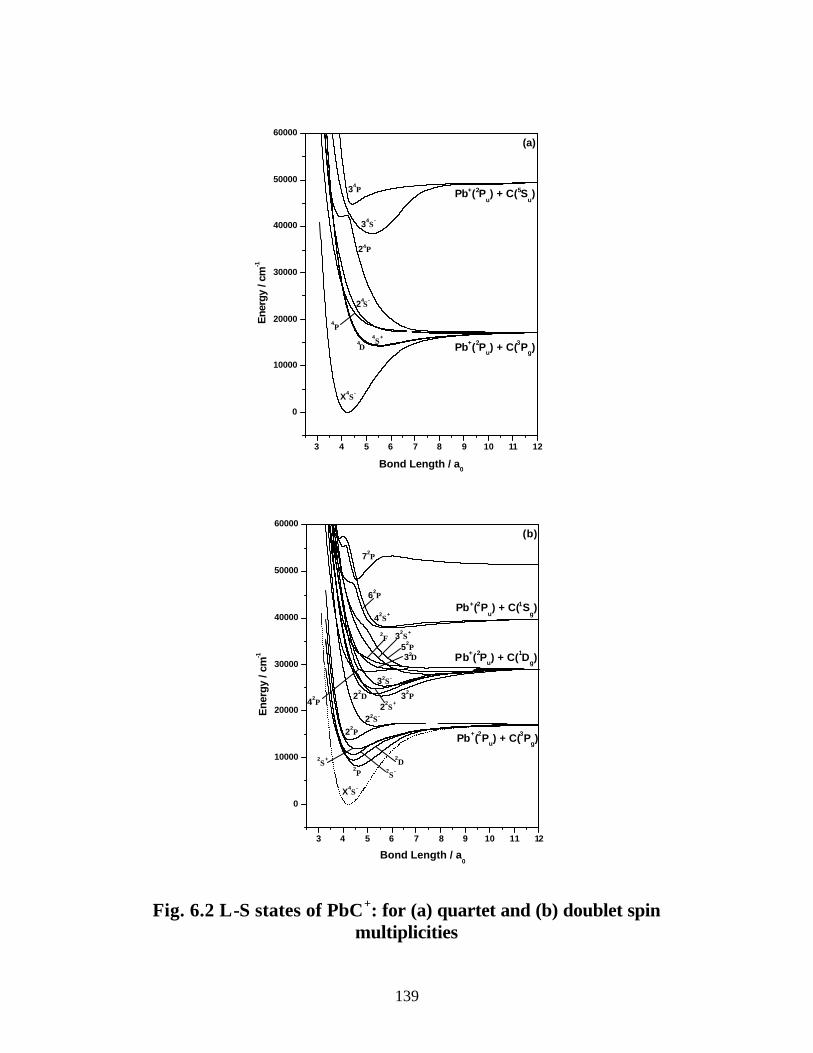
139
3 4 5 6 7 8 9 10 11 12
0
10000
20000
30000
40000
50000
60000(a)
Pb+(2Pu) + C(5S
u)
Pb+(2Pu) + C(3P
g)
34Π
34Σ
-
24Π
24Σ
-
4Π
4Σ
+4∆
X4Σ
-
Ene
rgy
/ cm
-1
Bond Length / a0
3 4 5 6 7 8 9 10 11 12
0
10000
20000
30000
40000
50000
60000(b)
Pb+(2Pu) + C(1S
g)
Pb+(2Pu) + C(1D
g)
Pb+(2Pu) + C(3P
g)
42Σ
+
32Σ
+
72Π
62Π
52Π
2Φ
32∆
42Π
32Σ-
22Σ
+22
∆ 32Π
22Σ-
22Π
2Σ
+
2Σ
-
2∆
2Π
X4Σ
-
Ene
rgy
/ cm
-1
Bond Length / a0
Fig. 6.2 Λ-S states of PbC+: for (a) quartet and (b) doublet spin multiplicities
33 580 cm−1 correlates with two quartet states, 34Σ− and 34Π. The MRDCI calculated value
agrees quite accurately with the experimental observation (Table 6.6). Potential energy
curves of most of the quartets and doublets are displayed in Figs. 6.2a and b, respectively.
The removal of one electron from the π1 bonding orbital of the ground-state PbC makes
the bond weaker in PbC+. The ground state X4Σ− has a longer bond length of 2.24 A. The
state has a multi configuration character with the dominant configuration σ21σ2π
21(58%). The
equilibrium vibrational frequency of the state is reported to be 543 cm−1 (Table 6.7), while
its dissociation energy is predicted to be 2.12 eV. No experimental data is available for
comparison.
Table 6.7 Spectroscopic constants of low-lying
Λ-S states of PbC+
State Te/cm−1 re/A ωe/cm−1
X4Σ− 0 2.24 543
2.179a
2Π 8183 2.44 3922∆ 9475 2.33 416
4582a 2.191a
2Σ− 10 665 2.33 3902Σ+ 11 856 2.41 356
22Π 13 767 2.26 4764∆ 14 176 2.94 1954Σ+ 14 392 2.95 191
22Σ− 16 712 2.82 249
32Π 23 148 2.95 264
22∆ 23 595 2.80 285
22Σ+ 24 685 2.83 255
32Σ− 25 278 3.04 201
42Π 28 390 2.65 206
34Σ− 38 540 2.77 315
24Π 42 151 2.09 582
a Ref. 7
140

The lowest two roots of 2Π interact strongly minimizing the lower one. Thus unlike SiC+
but like SnC+, the first excited state of PbC+ is 2Π having a longer bond length of 2.44 A and
adiabatic transition energy of 8183 cm−1. The upper 2Π state is comparatively loosely bound
and it has comparable bond length as that of the ground state. After the incorporation of
the spin-orbit interaction this state may be spectroscopically important. The lowest 2Π is
characterized by σ21σ
22π1(58%), while the upper one is dominated by σ2
1π31(45%) with some
other open shell configurations as given in Table 6.8. In between these two states there
exist three doublets, 2∆, 2Σ−, and 2Σ+ all of which originate from the same dominant
configuration as the ground state. The 22Σ− state with a longer bond length of 2.82 A exists
at around 16 700 cm−1.
Table 6.8 Composition of Λ-S states of PbC+ at equilibrium bond length
State Configuration (% contribution)
X4Σ− σ21σ2π
21(58), σ2
1σ2π1π2(12), σ1σ22π
21(9), σ1σ
22π1π2(4)
2Π σ21σ
22π1(58), σ2
1σ22π2(11), σ2
1σ2σ4π1(7), σ21σ2σ4π2(7)
2∆ σ21σ2π
21(70), σ2
1σ2π1π2(9)), σ1σ22π
21(5), σ2
1σ2π22(3)
2Σ− σ21σ2π
21(59), σ2
1σ2π1π2(14), σ1σ22π
21(4)
2Σ+ σ21σ2π
21(60), σ2
1σ2π1π2(11), σ21σ2π
22(6)
22Π σ21π
31(45), σ2
1π21π2(7), σ1σ2π
31(7), σ2
1σ22π1(5),
σ1σ2π21π2(4), σ2
1π1π22(4)
4∆ σ21σ2π1π2(82), σ2
1σ2π1π5(4)4Σ+ σ2
1σ2π1π2(82), σ21σ2π1π5(4)
22Σ− σ21σ2π1π2(47), σ2
1σ3π22(12), σ2
1σ3π1π2(9), σ21σ2π
22(8),
σ21σ2π
21(5)
32Π σ21σ
22π1(56), σ1σ
22π2(17), σ2
1π1π22(6), σ2
1σ2σ3π2(5)
22∆ σ21σ2π1π2(71), σ2
1σ2π22(3), σ2
1σ3π22(3), σ2
1σ2π21(2)
22Σ+ σ21σ2π1π2(76), σ2
1σ2π21(4)
32Σ− σ21σ2π1π2(42), σ2
1σ2π21(15), σ2
1σ2π22(15), σ2
1σ3π22(8)
42Π σ21π
21π2(38), σ2
1π1π22(26), σ2
1π31(8)
34Σ− σ21σ3π1π2(19), σ2
1σ3π22(12), σ2
1σ2π22(8), σ2
1σ3π21(5)
24Π σ1σ2π31(48), σ2
1π31(8), σ1σ2π
21π2(5), σ1σ2π1π
22(5)
A one electron transition, π1 → π2 gives rise to 4∆ and 4Σ+ states with comparable re
141

and ωe values. Both the states are weakly bound with a maximum of 10 vibrational levels.
The remaining quartets which dissociate into the first dissociation limit, are repulsive in
nature. The only exception is for 24Π which has been isolated with a single vibrational level.
Unlike the lighter carbide ions such as SiC+, SnC+, transition to this state is not expected
to occur because of its shorter bond length and low binding property. At equilibrium, 24Π
is characterized mainly by σ1σ2π31 with three other configurations as shown Table 6.8. An
excited 4Σ− is located with a vibrational frequency of 315 cm−1. The calculated equilibrium
bond length of the state is large enough (2.77 A), yet an emission band may be expected at
around 38 500 cm−1.
Eleven doublets correlate with second and third dissociation limits. Of these seven states
have been isolated as bound or quasi-bound in nature. Most of them are dominated by
an open shell ...π1π2 configuration. Their equilibrium bond lengths are above 2.65 A and
vibrational frequencies vary between 200 and 285 cm−1. The remaining four states are
repulsive, one of which has been assigned as 2Φ.
6.3.2 Spectroscopic constants and potential energy curves of Ω states
A. PbC
The spin-orbit splitting is quite strong for PbC because of the heavy atom, Pb. The lowest
dissociation limit, Pb(3Pg)+C(3Pg) under the spin-orbit coupling splits into ten asymptotes.
The dissociation correlation between the Ω states of PbC and the corresponding atomic
combinations are shown in Table 6.9. There are 53 omega states which are allowed to mix
in the spin-orbit CI calculations. The spin-orbit splitting of the components of 3P0,1,2 of the
carbon atom is only 43 cm−1 and thus the states coming from them are almost inseparable.
However, the low-lying 0+, 0−, 1, 2, 3, and 4 components are plotted in Figs. 6.3a-d.
Spectroscopic constants of the bound Ω states are also shown in Table 6.10.
Because of the spin-orbit coupling, there are extensive changes in the spectroscopic prop-
erties of the low-lying Ω states of PbC. The ground state splits into four Ω components
having a very large energy separation. Both the X3Π and A3Σ− states contribute largely to
the lowest component of 0+. Its re is intermediate between those of the most contributing
states, while ωe has been lowered to 470 cm−1. 5Π0+ also significantly contribute to the low-
est 0+ component. The first Ω=2 state has been assigned to X3Π2 which is slightly mixed
up with the similar components of a1∆2 and 5Π2. However, the spectroscopic properties of
this state are not changed too much from its originating Λ-S state. The lowest Ω=1 state is
142

located 230 cm−1 above the ground state. It has a significant contribution from X3Π, A3Σ−
as well as 5Π. The re and ωe are completely different as compared to those of the contributing
Λ-S states. The situations are similar for the second and third roots of 1 also. The spin-orbit
coupling affects the X3Π0− to a large extent. The computed transition energy of this state
is 3295 cm−1. The re and ωe are also altered by 0.035 A and 74 cm−1, respectively.
Table 6.9 Dissociation correlation between Ω and atomic states of PbC
Ω States† Atomic states Relative energy / cm−1
Pb + C Expt.a Cal.
0+ 3P0 + 3P0 0 0
0−, 1 3P0 + 3P1 16 20
0+, 1, 2 3P0 + 3P2 43 55
0−, 1 3P1 + 3P0 7819 3540
0+(2), 0−, 1(2), 2 3P1 + 3P1 7835 3565
0+, 0−(2), 1(3), 2(2), 3 3P1 + 3P2 7862 3590
0+, 1, 2 3P0 + 1D2 10 193 7025
0+, 1, 2 3P2 + 3P0 10 650 10 390
0+, 0−(2), 1(3), 2(2), 3 3P2 + 3P1 10 666 10 405
0+(3), 0−(2), 1(4), 2(3), 3(2), 4 3P2 + 3P2 10 693 10 440
a Moore’s Table, Ref. 27
† Values in parenthesis are the corresponding number of states
At equilibrium, 1(II) is composed of almost 30% of each of X3Π, A3Σ−, and 5Π states. The
minimum has been located at 4002 cm−1 with re and ωe, 2.258 A and 460 cm−1, respectively.
Like the ground-state, second root of 0+ is also contributed almost equally by X3Π and
A3Σ−. The computed re of the state is shorter than that of 0+(I), and ωe is more close to
that of X3Π. The X3Π state splits in an inverted order with Ω=2 lying lowest, while A3Σ−
splits in a regular pattern. Thus, 0+(II) has a longer contribution of the X3Π state. Due
to the spin-orbit mixing, the a1∆2 is energetically destabilized by an amount 1492 cm−1.
Its re is increased by 0.021 A, while ωe is decreased by 12 cm−1 only. Inspite of significant
mixing with other states, potential energy curves of all the five spin components of 5Π are
fitted. The spin-orbit components are separated in a regular pattern. The third root of 1 is
dominated by the 5Π−1 component. However, A3Σ−1 contributes in it by more than 30%,
143
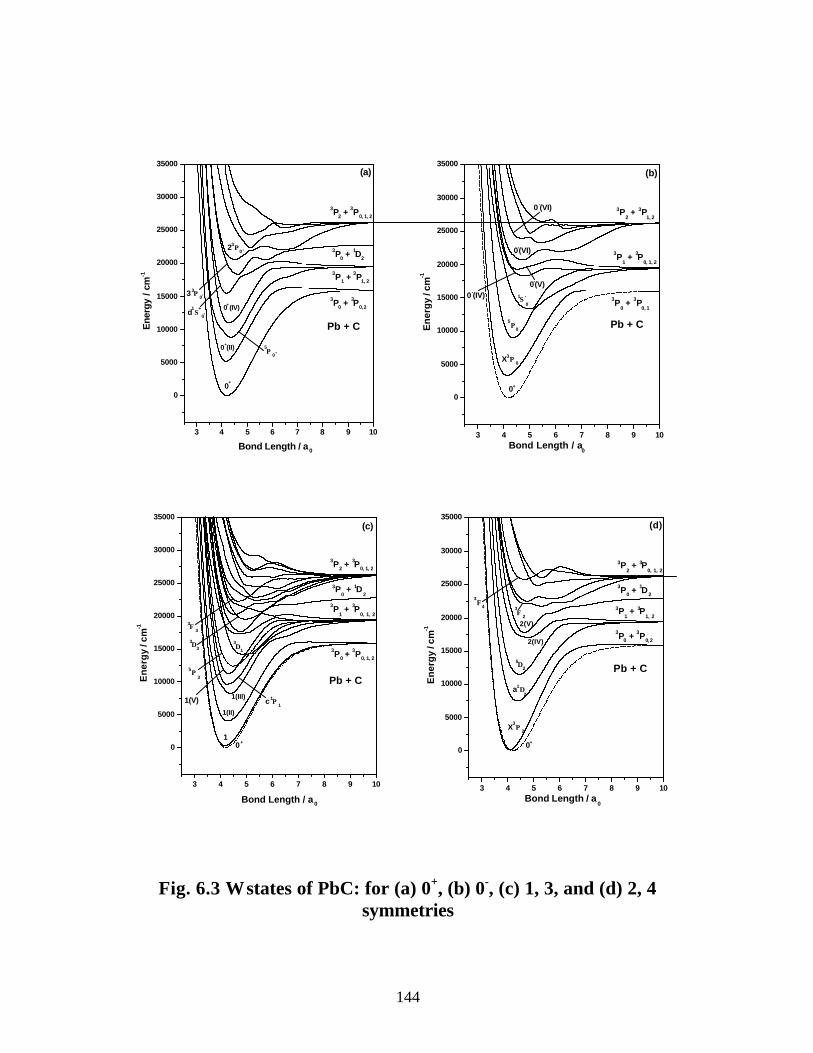
144
3 4 5 6 7 8 9 10
0
5000
10000
15000
20000
25000
30000
35000
33Π0
+
(a)
23Π0+
d1Σ
+
0+
0+(IV)
5Π
0+
0+(II)
0+
3P2 + 3P0, 1, 2
3P0 + 1D2
3P1 + 3P1, 2
3P0 + 3P0, 2
Pb + C
E
nerg
y / c
m-1
Bond Length / a0
3 4 5 6 7 8 9 10
0
5000
10000
15000
20000
25000
30000
35000
0 -(VI)
0-(VI)
0-(V)0 -(IV)
(b)
3P2 + 3P
1, 2
3P1 + 3P
0, 1, 2
3P0 + 3P
0, 1
Pb + C
1Σ -
0-
5Π
0-
X3Π0-
0+
En
erg
y / c
m-1
Bond Length / a0
3 4 5 6 7 8 9 10
0
5000
10000
15000
20000
25000
30000
35000
1(V)
(c)
3Φ3
3∆3
5Π3
3P2 + 3P0, 1, 2
3P0 + 1D
2
3P1 + 3P
0, 1, 2
3P0 + 3P0, 1, 2
Pb + C
3∆
1
c1Π1
1(III)
1(II)
10+
En
erg
y / c
m-1
Bond Length / a 0
3 4 5 6 7 8 9 10
0
5000
10000
15000
20000
25000
30000
35000
3Φ
2
(d)
3Φ
4
3P2 + 3P
0, 1, 2
3P0 + 1D2
3P1 + 3P
1, 2
3P0 + 3P0, 2
Pb + C
2(V)
2(IV)
5∆2
a1∆2
X3Π
2
0+
En
erg
y / c
m-1
Bond Length / a0
Fig. 6.3 Ω states of PbC: for (a) 0+, (b) 0-, (c) 1, 3, and (d) 2, 4 symmetries
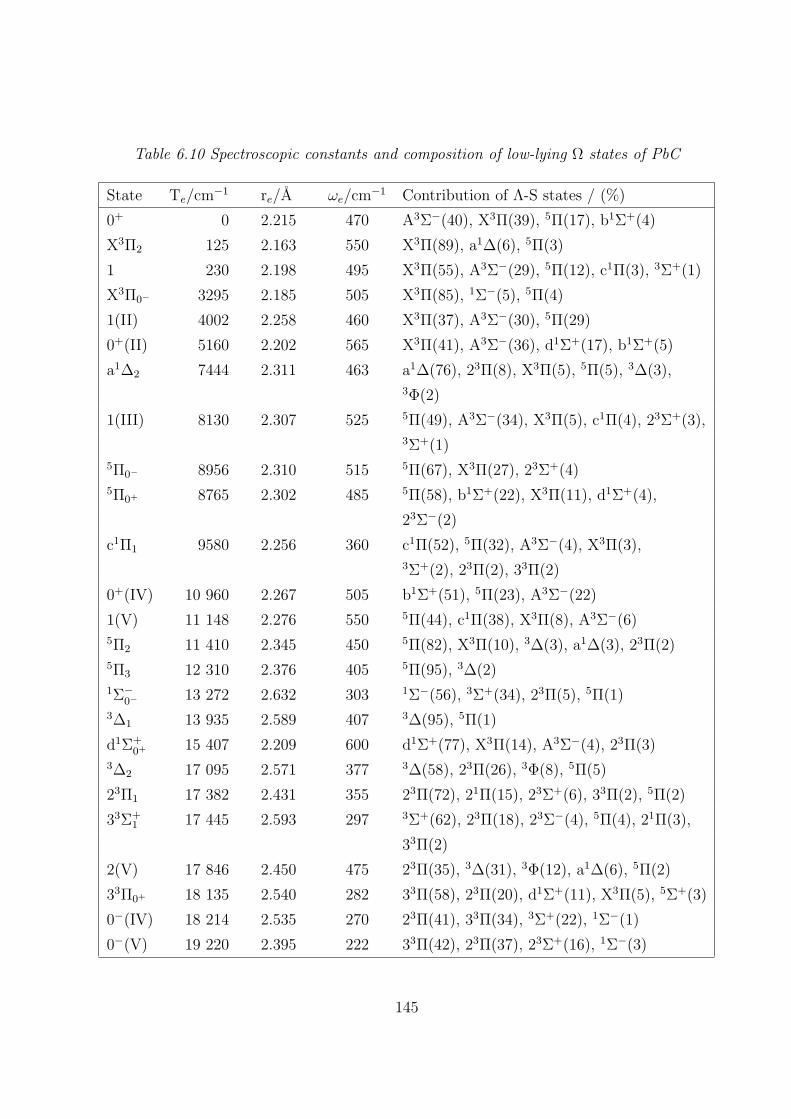
Table 6.10 Spectroscopic constants and composition of low-lying Ω states of PbC
State Te/cm−1 re/A ωe/cm−1 Contribution of Λ-S states / (%)
0+ 0 2.215 470 A3Σ−(40), X3Π(39), 5Π(17), b1Σ+(4)
X3Π2 125 2.163 550 X3Π(89), a1∆(6), 5Π(3)
1 230 2.198 495 X3Π(55), A3Σ−(29), 5Π(12), c1Π(3), 3Σ+(1)
X3Π0− 3295 2.185 505 X3Π(85), 1Σ−(5), 5Π(4)
1(II) 4002 2.258 460 X3Π(37), A3Σ−(30), 5Π(29)
0+(II) 5160 2.202 565 X3Π(41), A3Σ−(36), d1Σ+(17), b1Σ+(5)
a1∆2 7444 2.311 463 a1∆(76), 23Π(8), X3Π(5), 5Π(5), 3∆(3),3Φ(2)
1(III) 8130 2.307 525 5Π(49), A3Σ−(34), X3Π(5), c1Π(4), 23Σ+(3),3Σ+(1)
5Π0− 8956 2.310 515 5Π(67), X3Π(27), 23Σ+(4)5Π0+ 8765 2.302 485 5Π(58), b1Σ+(22), X3Π(11), d1Σ+(4),
23Σ−(2)
c1Π1 9580 2.256 360 c1Π(52), 5Π(32), A3Σ−(4), X3Π(3),3Σ+(2), 23Π(2), 33Π(2)
0+(IV) 10 960 2.267 505 b1Σ+(51), 5Π(23), A3Σ−(22)
1(V) 11 148 2.276 550 5Π(44), c1Π(38), X3Π(8), A3Σ−(6)5Π2 11 410 2.345 450 5Π(82), X3Π(10), 3∆(3), a1∆(3), 23Π(2)5Π3 12 310 2.376 405 5Π(95), 3∆(2)1Σ−0− 13 272 2.632 303 1Σ−(56), 3Σ+(34), 23Π(5), 5Π(1)3∆1 13 935 2.589 407 3∆(95), 5Π(1)
d1Σ+0+ 15 407 2.209 600 d1Σ+(77), X3Π(14), A3Σ−(4), 23Π(3)
3∆2 17 095 2.571 377 3∆(58), 23Π(26), 3Φ(8), 5Π(5)
23Π1 17 382 2.431 355 23Π(72), 21Π(15), 23Σ+(6), 33Π(2), 5Π(2)
33Σ+1 17 445 2.593 297 3Σ+(62), 23Π(18), 23Σ−(4), 5Π(4), 21Π(3),
33Π(2)
2(V) 17 846 2.450 475 23Π(35), 3∆(31), 3Φ(12), a1∆(6), 5Π(2)
33Π0+ 18 135 2.540 282 33Π(58), 23Π(20), d1Σ+(11), X3Π(5), 5Σ+(3)
0−(IV) 18 214 2.535 270 23Π(41), 33Π(34), 3Σ+(22), 1Σ−(1)
0−(V) 19 220 2.395 222 33Π(42), 23Π(37), 23Σ+(16), 1Σ−(3)
145
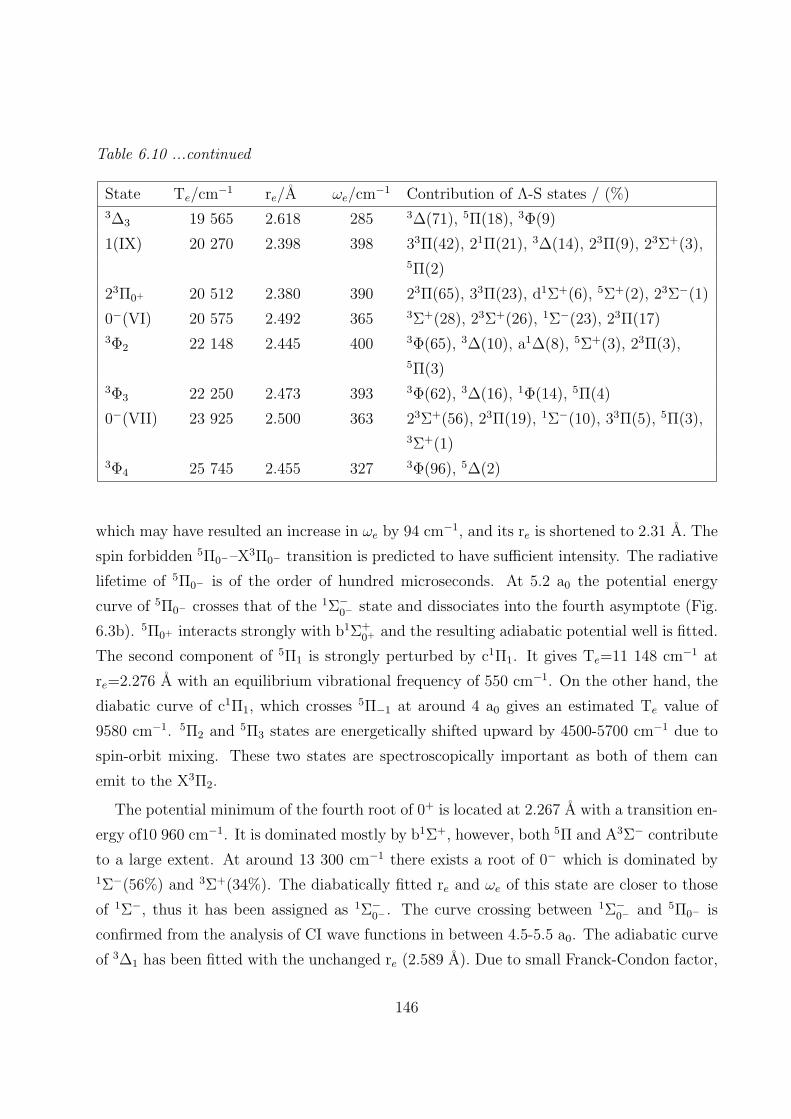
Table 6.10 ...continued
State Te/cm−1 re/A ωe/cm−1 Contribution of Λ-S states / (%)3∆3 19 565 2.618 285 3∆(71), 5Π(18), 3Φ(9)
1(IX) 20 270 2.398 398 33Π(42), 21Π(21), 3∆(14), 23Π(9), 23Σ+(3),5Π(2)
23Π0+ 20 512 2.380 390 23Π(65), 33Π(23), d1Σ+(6), 5Σ+(2), 23Σ−(1)
0−(VI) 20 575 2.492 365 3Σ+(28), 23Σ+(26), 1Σ−(23), 23Π(17)3Φ2 22 148 2.445 400 3Φ(65), 3∆(10), a1∆(8), 5Σ+(3), 23Π(3),
5Π(3)3Φ3 22 250 2.473 393 3Φ(62), 3∆(16), 1Φ(14), 5Π(4)
0−(VII) 23 925 2.500 363 23Σ+(56), 23Π(19), 1Σ−(10), 33Π(5), 5Π(3),3Σ+(1)
3Φ4 25 745 2.455 327 3Φ(96), 5∆(2)
which may have resulted an increase in ωe by 94 cm−1, and its re is shortened to 2.31 A. The
spin forbidden 5Π0−–X3Π0− transition is predicted to have sufficient intensity. The radiative
lifetime of 5Π0− is of the order of hundred microseconds. At 5.2 a0 the potential energy
curve of 5Π0− crosses that of the 1Σ−0− state and dissociates into the fourth asymptote (Fig.
6.3b). 5Π0+ interacts strongly with b1Σ+0+ and the resulting adiabatic potential well is fitted.
The second component of 5Π1 is strongly perturbed by c1Π1. It gives Te=11 148 cm−1 at
re=2.276 A with an equilibrium vibrational frequency of 550 cm−1. On the other hand, the
diabatic curve of c1Π1, which crosses 5Π−1 at around 4 a0 gives an estimated Te value of
9580 cm−1. 5Π2 and 5Π3 states are energetically shifted upward by 4500-5700 cm−1 due to
spin-orbit mixing. These two states are spectroscopically important as both of them can
emit to the X3Π2.
The potential minimum of the fourth root of 0+ is located at 2.267 A with a transition en-
ergy of10 960 cm−1. It is dominated mostly by b1Σ+, however, both 5Π and A3Σ− contribute
to a large extent. At around 13 300 cm−1 there exists a root of 0− which is dominated by1Σ−(56%) and 3Σ+(34%). The diabatically fitted re and ωe of this state are closer to those
of 1Σ−, thus it has been assigned as 1Σ−0− . The curve crossing between 1Σ−0− and 5Π0− is
confirmed from the analysis of CI wave functions in between 4.5-5.5 a0. The adiabatic curve
of 3∆1 has been fitted with the unchanged re (2.589 A). Due to small Franck-Condon factor,
146

the transition from this state is not expected to occur. A dipole allowed 0+-0+ transition
has been located at around 15 400 cm−1. The upper state is labeled as d1Σ+0+ with the
computed re and ωe of 2.209 A and 600 cm−1, respectively. It has significant mixing with
X3Π0+ . In the longer bond length region it interacts strongly with the 0+ components of 23Π
and 33Π. As a result of strong spin-orbit interaction the fourth root of Ω=2 looks flattened
in the equilibrium region. The diabatically fitted curve shows a potential minimum at 2.571
A with a transition energy of 17 095 cm−1. As 3∆ makes the major contribution (58%)
to it, it has been named as 3∆2. The next root of 2 has been fitted adiabatically. The
estimated Te of the state is reported to be 17 864 cm−1 at 2.450 A with ωe=475 cm−1. The3∆3 component is perturbed by two other components, namely 5Π3 and 3Φ3. The transition
energy of the state is computed to be 19 565 cm−1 which is 5000 cm−1 away from the 3∆
state itself. The ωe of the state is lowered by 49 cm−1. However, because of the smaller
Franck-Condon factor transitions from this state with 4Ω=0, 1 are expected to be weak.
Near 17 400 cm−1, two roots of Ω=1 cross each other at the bond distance of 4.8 a0.
Analysis of the wave functions reveals that they originate from 23Π and 3Σ+, respectively.
Fitting both the curves diabatically, the spectroscopic constants are displayed in Table 6.10.
All the four spin components of 23Π show potential minima at 17 382 17 864, 18 214, and
20 512 cm−1, respectively. The excited 33Π split in a regular order. A strong spin-orbit
mixing is indicated in the composition shown in Table 6.10. The 3Π2 does not have clear
potential minimum because of several avoided curve crossings around 22 000 cm−1 in the
4.8-5.6 a0 bond distance region. The Ω components of 3Φ mix up with these of 3∆ around
22 000 cm−1. The 3Φ4 state is almost pure, though its Te is increased by more than 5000
cm−1.
B. PbC+
The ground term of the ion Pb+ (2P) splits into J=1/2 and J=3/2 with a separation of
14 081 cm−1, while that of C (3P) splits into three sub-levels with a maximum separation
of 43 cm−1 only. Consequently, the ground-state dissociation limit of the molecular ion,
Pb+(2P)+C(3P) correlates with six sub levels with a maximum separation of 14 124 cm−1
as shown in Table 6.11. A set of two 1/2, two 3/2, and a 5/2 dissociates in the limit
Pb+(2P1/2)+C(1D2). Thus a total of 32 Ω states of 1/2, 3/2, 5/2, and 7/2 symmetries
correlate with three sets of sub-levels within 14 124 cm−1 of energy. The potential energy
curves and their spectroscopic constants are given in Figs. 6.4a-c and Table 6.12, respectively.
147
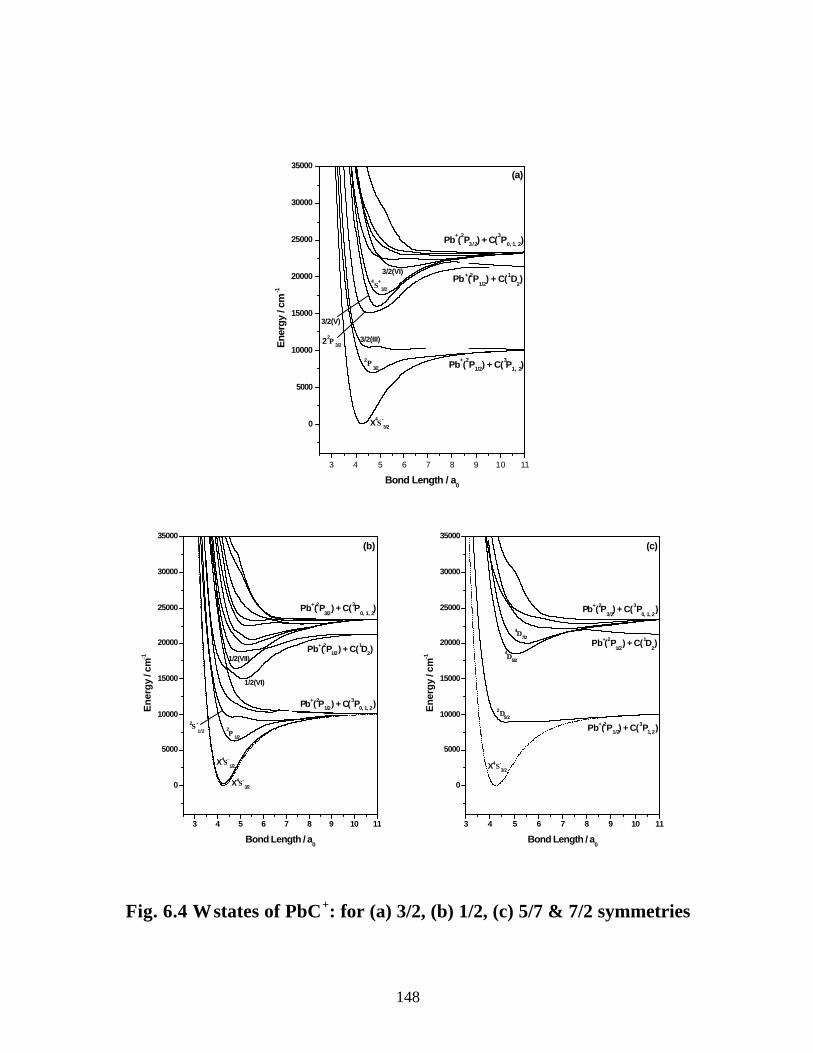
148
3 4 5 6 7 8 9 10 11
0
5000
10000
15000
20000
25000
30000
35000
3/2(VI)
22Π3/2
(a)
Pb+(2P3/2) + C(
3P0, 1, 2)
Pb+(2P1/2
) + C(1D2)
Pb+(2P1/2) + C(
3P1, 2)
4Σ
+
3/2
3/2(V)
3/2(III)
2Π3/2
X4Σ
-
3/2
Ene
rgy
/ cm
-1
Bond Length / a0
3 4 5 6 7 8 9 10 11
0
5000
10000
15000
20000
25000
30000
35000
1/2(VII)
(b)
Pb+(2P1/2) + C(1D2)
Pb+(2P3/2
) + C(3P0, 1, 2
)
Pb+(2P1/2) + C(3P0, 1, 2)
1/2(VI)
2Σ -
1/2 2Π
1/2
X4Σ-1/2
X4Σ-3/2
Ene
rgy
/ cm
-1
Bond Length / a0
3 4 5 6 7 8 9 10 11
0
5000
10000
15000
20000
25000
30000
35000(c)
Pb+(2P3/2
) + C(3P0, 1, 2
)
Pb+(2P1/2
) + C(1D2)
Pb+(2P1/2) + C(3P1, 2)
4∆7/2
4∆
5/2
2∆5/2
X4Σ-3/2
Ene
rgy
/ cm
-1
Bond Length / a0
Fig. 6.4 Ω states of PbC+: for (a) 3/2, (b) 1/2, (c) 5/7 & 7/2 symmetries

The bond length of ground-state spin component, X4Σ−3/2 is increased by 0.01 A, while its ωe
is lowered by 38 cm−1 due to the spin-orbit interaction. The spin-orbit zero-field splitting is
computed to be 286 cm−1. The composition of both the ground-state components are given
in Table 6.12.
Table 6.11 Dissociation correlation between Ω and atomic states of PbC+
Ω states Atomic states Relative energy / cm−1
Pb+ + C Expt.a Cal.
1/2 2P1/2+3P0 0 0
1/2(2), 3/2 2P1/2+3P1 16 23
1/2(2), 3/2(2), 5/2 2P1/2+3P2 43 54
1/2(2), 3/2(2), 5/2 2P1/2+1D2 10 193 11 360
1/2, 3/2 2P3/2+3P0 14 081 13 446
1/2(3), 3/2(2), 5/2 2P3/2+3P1 14 097 13 466
1/2(4), 3/2(3), 5/2(2), 7/2 2P3/2+3P2 14 124 13 496
a Moore’s Table, Ref. 27
The two components (1/2 and 3/2) of the first excited 2Π state are separated by about
623 cm−1. Their equilibrium bond lengths are elongated by 0.03-0.06 A, while their ωes are
reduced by about 75 cm−1 as a result of the spin-orbit mixing. Table 6.12 shows that 2Π1/2
mixes with the components of 4∆, 4Σ−, while the other component mixes strongly with2∆3/2, 4∆3/2, and 4Σ−3/2. The spin-orbit mixing permits the X4Σ− →2 Π transition through
their respective dipolar components. The Ω=3/2 component of 2∆ is very strongly perturbed
by the similar components of first and second root of 2Π. In absorption spectroscopy, the2∆3/2 state may be isolated at 10 430 cm−1. The situation is more complex for 2∆5/2. The
mixing contribution of 4∆ results its quasi bound nature with a very small potential barrier.
Due to its low binding energy, the emission from this state is not expected. The estimated
re and ωe of 2∆5/2 are 2.459 A and 249 cm−1, respectively.
The third root of 1/2 is dominated by 2Σ−(69%). Because of the interaction with the
Ω=1/2 component of repulsive 4Π, this state is also very poorly bound and consequently no
transition to this state is expected to be observed. The 2Σ+1/2 and 4Σ+
1/2 components also
contribute to this by 8% and 7%, respectively. Fourth and fifth root of 1/2 are repulsive in
149
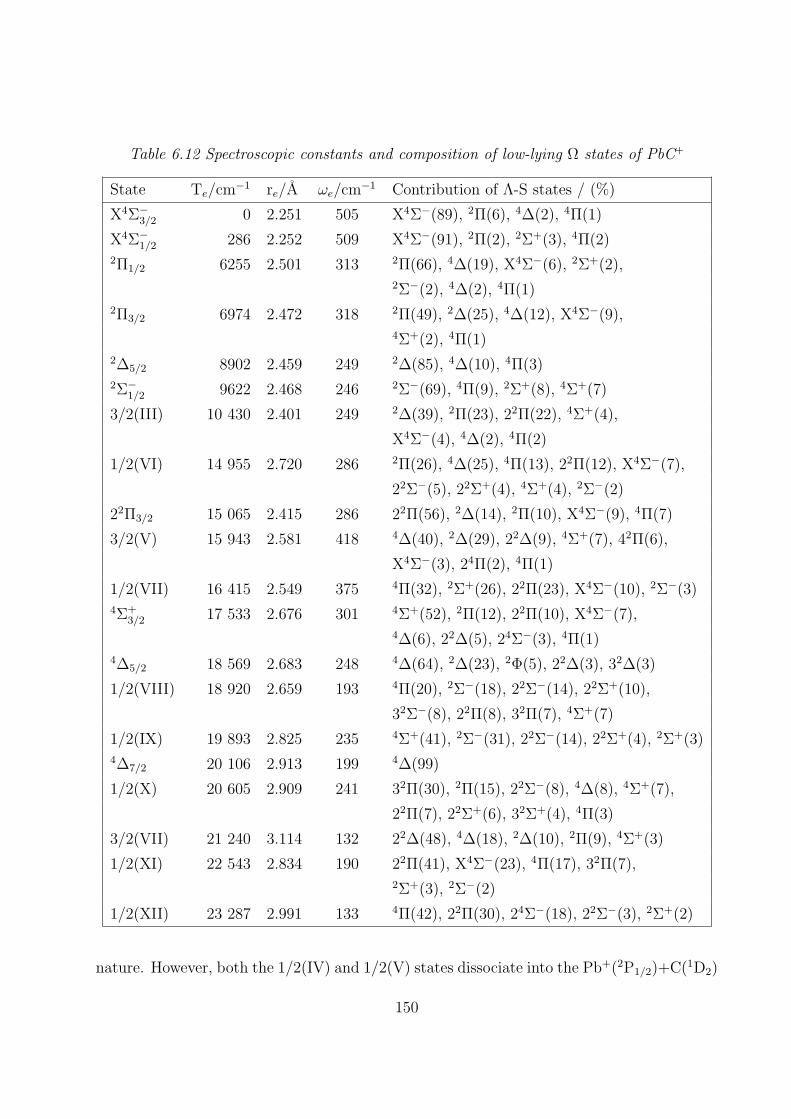
Table 6.12 Spectroscopic constants and composition of low-lying Ω states of PbC+
State Te/cm−1 re/A ωe/cm−1 Contribution of Λ-S states / (%)
X4Σ−3/2 0 2.251 505 X4Σ−(89), 2Π(6), 4∆(2), 4Π(1)
X4Σ−1/2 286 2.252 509 X4Σ−(91), 2Π(2), 2Σ+(3), 4Π(2)2Π1/2 6255 2.501 313 2Π(66), 4∆(19), X4Σ−(6), 2Σ+(2),
2Σ−(2), 4∆(2), 4Π(1)2Π3/2 6974 2.472 318 2Π(49), 2∆(25), 4∆(12), X4Σ−(9),
4Σ+(2), 4Π(1)2∆5/2 8902 2.459 249 2∆(85), 4∆(10), 4Π(3)2Σ−1/2 9622 2.468 246 2Σ−(69), 4Π(9), 2Σ+(8), 4Σ+(7)
3/2(III) 10 430 2.401 249 2∆(39), 2Π(23), 22Π(22), 4Σ+(4),
X4Σ−(4), 4∆(2), 4Π(2)
1/2(VI) 14 955 2.720 286 2Π(26), 4∆(25), 4Π(13), 22Π(12), X4Σ−(7),
22Σ−(5), 22Σ+(4), 4Σ+(4), 2Σ−(2)
22Π3/2 15 065 2.415 286 22Π(56), 2∆(14), 2Π(10), X4Σ−(9), 4Π(7)
3/2(V) 15 943 2.581 418 4∆(40), 2∆(29), 22∆(9), 4Σ+(7), 42Π(6),
X4Σ−(3), 24Π(2), 4Π(1)
1/2(VII) 16 415 2.549 375 4Π(32), 2Σ+(26), 22Π(23), X4Σ−(10), 2Σ−(3)4Σ+
3/2 17 533 2.676 301 4Σ+(52), 2Π(12), 22Π(10), X4Σ−(7),4∆(6), 22∆(5), 24Σ−(3), 4Π(1)
4∆5/2 18 569 2.683 248 4∆(64), 2∆(23), 2Φ(5), 22∆(3), 32∆(3)
1/2(VIII) 18 920 2.659 193 4Π(20), 2Σ−(18), 22Σ−(14), 22Σ+(10),
32Σ−(8), 22Π(8), 32Π(7), 4Σ+(7)
1/2(IX) 19 893 2.825 235 4Σ+(41), 2Σ−(31), 22Σ−(14), 22Σ+(4), 2Σ+(3)4∆7/2 20 106 2.913 199 4∆(99)
1/2(X) 20 605 2.909 241 32Π(30), 2Π(15), 22Σ−(8), 4∆(8), 4Σ+(7),
22Π(7), 22Σ+(6), 32Σ+(4), 4Π(3)
3/2(VII) 21 240 3.114 132 22∆(48), 4∆(18), 2∆(10), 2Π(9), 4Σ+(3)
1/2(XI) 22 543 2.834 190 22Π(41), X4Σ−(23), 4Π(17), 32Π(7),2Σ+(3), 2Σ−(2)
1/2(XII) 23 287 2.991 133 4Π(42), 22Π(30), 24Σ−(18), 22Σ−(3), 2Σ+(2)
nature. However, both the 1/2(IV) and 1/2(V) states dissociate into the Pb+(2P1/2)+C(1D2)
150

limit with a maximum contribution coming from 4Π. The equilibrium bond length of the
1/2(VI) state of PbC+ is 2.720 A and it is spectroscopically not significant due to poor
Franck-Condon overlap factor. Its potential minimum is located at 14 955 cm−1.
Above 15 000 cm−1, the purity of both the spin components Ω=1/2 and 3/2 is almost
destroyed. There occur several number of avoided curve crossing phenomena. Strong spin-
orbit interactions result enormous changes in the spectroscopic properties. The 22Π3/2 state
has been identified with re= 2.415 A and ωe = 286 cm−1. The X4Σ−3/2 → 22Π3/2 transition is
expected to occur with a band origin at 15 065 cm−1. The influence of the 3/2 components
of 2∆, 4Σ− and 4Π changes re and ωe to a large extent. The 11th root of 1/2 is characterized
by 22Π with a contribution of 41%. The adiabatic curve shows a potential minimum at
2.834 A with Te=22 543 cm−1 and a low vibrational frequency of 190 cm−1. The fifth root
of 3/2 at equilibrium is dominated by 4∆ (40%) with a transition energy of 15 943 cm−1.
However, 2∆ state also contributes largely. The equilibrium vibrational frequency of the
state is comparatively large (418 cm−1). Potential energy curve of 4∆1/2 suffers a large
number of avoided crossing and hence could not be fitted. But the other two components,4∆5/2 and 4∆7/2 are fitted. The 4∆5/2 state mixes with the components of 2∆ and 2Φ, while
the 4∆7/2 is almost pure. However, the four components of 4∆ split in a regular pattern.
The two components of 4Σ+ are shifted upward by 3141 and 5501 cm−1, respectively, by
spin-orbit effect. They split in an inverted manner, re of both the states are shortened, while
their vibrational frequencies are increased to a considerable amount. The adiabatic potential
minimum of tenth root of 1/2 is also complex with maximum contribution coming from 32Π
(30%). Very long bond distance makes it spectroscopically unimportant. A very shallow
minimum with a low vibrational frequency of 132 cm−1 is located at 21 240 cm−1 and the
state is assigned to be Ω=3/2 having 48% contribution from 22∆. Spectroscopic constants
of several other low-lying Ω states along with the equilibrium compositions are tabulated in
Table 6.12.
6.3.3 Transition properties
A. PbC
Many spin forbidden and dipole allowed transitions are studied for the neutral PbC
molecule. Transition moment functions for some selected transitions are plotted against
internuclear distance in Fig. 6.5a. The radiative lifetimes (partial as well as total) are re-
ported in Table 6.13. The 33Σ−–A3Σ− transition is predicted to be the strongest one. The
151
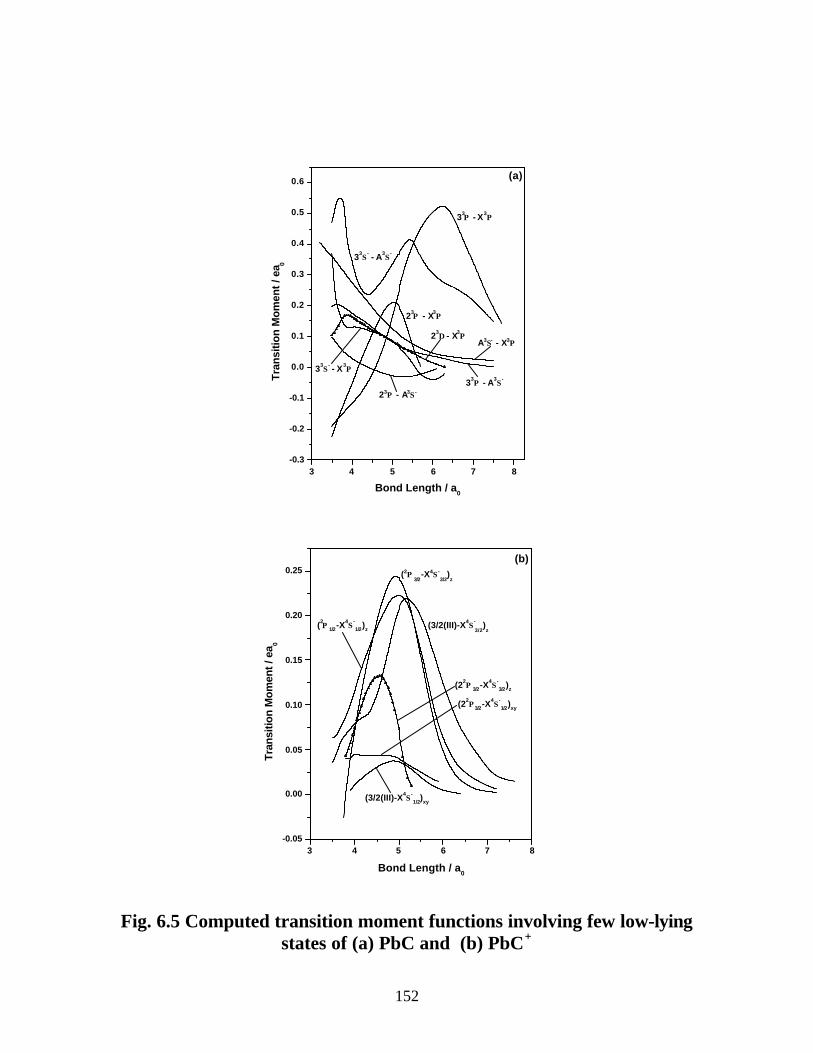
152
3 4 5 6 7 8-0.3
-0.2
-0.1
0.0
0.1
0.2
0.3
0.4
0.5
0.6 (a)
A3Σ- - X3Π
33Σ
- - X3Π
23Π - A3Σ-
23∆ - X3
Π
33Π - A3
Σ-
33Σ
- - A3Σ
-
33Π - X3
Π
23Π - X3
Π
Tran
sitio
n M
omen
t / e
a 0
Bond Length / a0
3 4 5 6 7 8-0.05
0.00
0.05
0.10
0.15
0.20
0.25(b)
(22Π3/2-X4
Σ-
1/2)xy
(22Π3/2-X4
Σ-
3/2)z
(3/2(III)-X4Σ
-
1/2)xy
(2Π1/2-X4
Σ-
1/2)z
(2Π3/2
-X4Σ-
3/2)
z
(3/2(III)-X4Σ
-
3/2)
z
Tran
sitio
n M
omen
t / e
a 0
Bond Length / a0
Fig. 6.5 Computed transition moment functions involving few low-lying states of (a) PbC and (b) PbC+

corresponding transition moment curve has a minimum in Franck-Condon region. Total
radiative lifetime in the υ′=0 state of 33Σ− of PbC is about 320 ns. 33Σ+–X3Π is another
strong transition for PbC. The partial radiative lifetime in the ground vibrational level of
33Σ+ is 0.84 µs, but it becomes almost double in the next higher vibrational level. Both
the transition moment curves of 23Π–X3Π and 33Π–X3Π pass through maximum. In the
Franck-Condon region, the former transition has a greater transition moment and hence a
shorter radiative lifetime which again increases monotonically with the higher vibrational
levels. The situation is opposite for the transition from A3Σ− state. In that case, transition
to the upper 3Π is much more probable than that to the lower one. The 33Π has shorter total
radiative lifetime than that of 23Π. The transition moment of 23∆–X3Π has a maximum
at 3.9 a0 and the curve then slowly converge to zero. The partial radiative lifetime for
A–X transition is estimated to be of the millisecond order. The dipole allowed transitions
such as (0+(IV)–0+(I))‖, (1(V)–X3Π1)‖, (5Π2–X3Π2)‖, (d1Σ+0+–X3Π1)⊥ etc. have radiative
lifetime less than hundred microsecond. Transitions involving the components of 5Π and the
corresponding ground state components are highly probable due to strong spin-orbit mixing.
The 5Π2 and 1(V) states have the total radiative lifetimes of 58 and 43 µs, respectively.
A strong transition is indicated at around 15 400 cm−1, the upper state of which is the
component of d1Σ+. As the 0− component of the ground state is shifted upward by more
than 3000 cm−1, transitions from this state are relatively weaker. However, 5Π0− has a total
lifetime of 136 µs. The predicted lifetime in 5Π3 state is also in the order of few millisecond.
The 5Π3–X3Π2 transition is expected to appear at 12 310 cm−1.
Table 6.13 Radiative lifetime (s) of some of the excited states of PbC
Transition Partial lifetimes of the upper state ata Total lifetime
υ′=0 υ′=1 υ′=2 υ′=3 υ′=4 at υ′=0
A3Σ−–X3Π 1.40(-3) 9.97(-4) 8.08(-4) 6.65(-4) 5.60(-4)
23Π–X3Π 1.66(-5) 1.67(-5) 1.69(-5) 1.89(-5) 2.77(-5)
23Π–A3Σ− 3.10(-3) 1.78(-3) 1.31(-3) 1.11(-3) 0.91(-3) τ23Π=1.65(-5)
33Π–X3Π 9.97(-5) 3.18(-5) 2.08(-5) 2.24(-5)
33Π–A3Σ− 1.04(-5) 1.03(-5) 1.02(-5) 1.02(-5) 1.01(-5) τ33Π=9.41(-6)
23∆–X3Π 2.33(-6) 2.80(-6) 3.65(-6) 4.14(-6) 4.06(-6)
33Σ−–X3Π 1.73(-6) 1.91(-6) 2.30(-6)
153
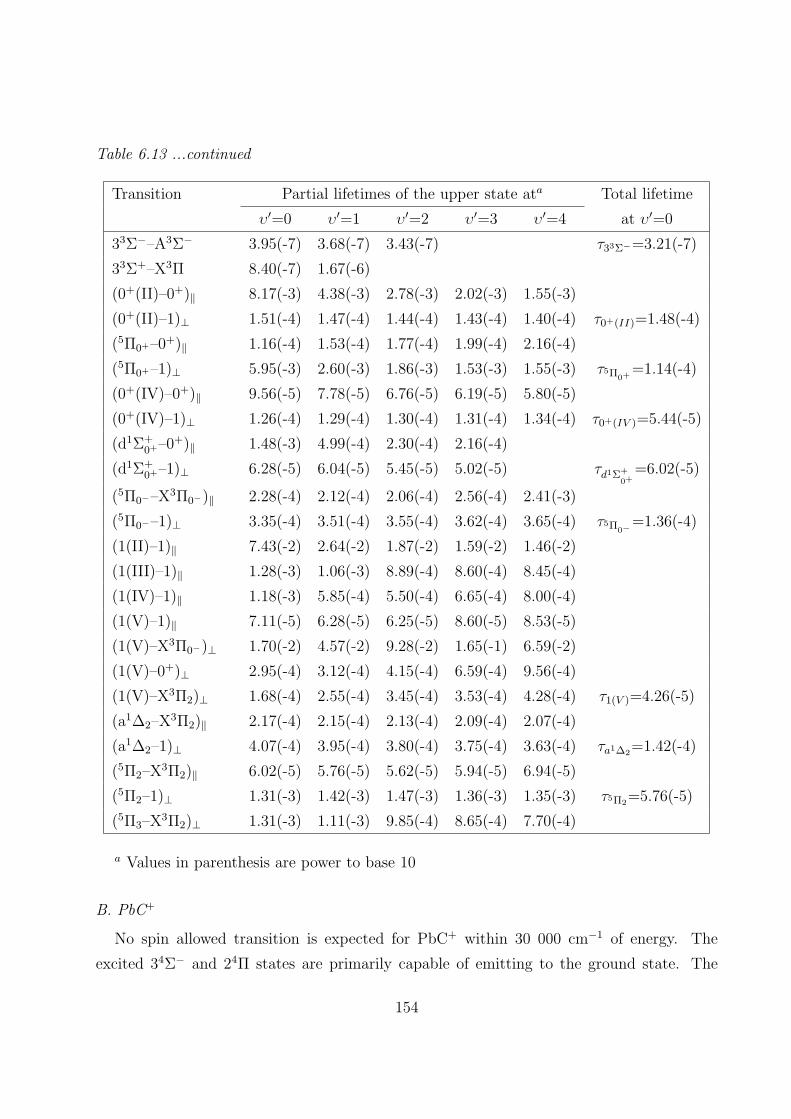
Table 6.13 ...continued
Transition Partial lifetimes of the upper state ata Total lifetime
υ′=0 υ′=1 υ′=2 υ′=3 υ′=4 at υ′=0
33Σ−–A3Σ− 3.95(-7) 3.68(-7) 3.43(-7) τ33Σ−=3.21(-7)
33Σ+–X3Π 8.40(-7) 1.67(-6)
(0+(II)–0+)‖ 8.17(-3) 4.38(-3) 2.78(-3) 2.02(-3) 1.55(-3)
(0+(II)–1)⊥ 1.51(-4) 1.47(-4) 1.44(-4) 1.43(-4) 1.40(-4) τ0+(II)=1.48(-4)
(5Π0+–0+)‖ 1.16(-4) 1.53(-4) 1.77(-4) 1.99(-4) 2.16(-4)
(5Π0+–1)⊥ 5.95(-3) 2.60(-3) 1.86(-3) 1.53(-3) 1.55(-3) τ5Π0+=1.14(-4)
(0+(IV)–0+)‖ 9.56(-5) 7.78(-5) 6.76(-5) 6.19(-5) 5.80(-5)
(0+(IV)–1)⊥ 1.26(-4) 1.29(-4) 1.30(-4) 1.31(-4) 1.34(-4) τ0+(IV )=5.44(-5)
(d1Σ+0+–0+)‖ 1.48(-3) 4.99(-4) 2.30(-4) 2.16(-4)
(d1Σ+0+–1)⊥ 6.28(-5) 6.04(-5) 5.45(-5) 5.02(-5) τd1Σ+
0+=6.02(-5)
(5Π0−–X3Π0−)‖ 2.28(-4) 2.12(-4) 2.06(-4) 2.56(-4) 2.41(-3)
(5Π0−–1)⊥ 3.35(-4) 3.51(-4) 3.55(-4) 3.62(-4) 3.65(-4) τ5Π0−=1.36(-4)
(1(II)–1)‖ 7.43(-2) 2.64(-2) 1.87(-2) 1.59(-2) 1.46(-2)
(1(III)–1)‖ 1.28(-3) 1.06(-3) 8.89(-4) 8.60(-4) 8.45(-4)
(1(IV)–1)‖ 1.18(-3) 5.85(-4) 5.50(-4) 6.65(-4) 8.00(-4)
(1(V)–1)‖ 7.11(-5) 6.28(-5) 6.25(-5) 8.60(-5) 8.53(-5)
(1(V)–X3Π0−)⊥ 1.70(-2) 4.57(-2) 9.28(-2) 1.65(-1) 6.59(-2)
(1(V)–0+)⊥ 2.95(-4) 3.12(-4) 4.15(-4) 6.59(-4) 9.56(-4)
(1(V)–X3Π2)⊥ 1.68(-4) 2.55(-4) 3.45(-4) 3.53(-4) 4.28(-4) τ1(V )=4.26(-5)
(a1∆2–X3Π2)‖ 2.17(-4) 2.15(-4) 2.13(-4) 2.09(-4) 2.07(-4)
(a1∆2–1)⊥ 4.07(-4) 3.95(-4) 3.80(-4) 3.75(-4) 3.63(-4) τa1∆2=1.42(-4)
(5Π2–X3Π2)‖ 6.02(-5) 5.76(-5) 5.62(-5) 5.94(-5) 6.94(-5)
(5Π2–1)⊥ 1.31(-3) 1.42(-3) 1.47(-3) 1.36(-3) 1.35(-3) τ5Π2=5.76(-5)
(5Π3–X3Π2)⊥ 1.31(-3) 1.11(-3) 9.85(-4) 8.65(-4) 7.70(-4)
a Values in parenthesis are power to base 10
B. PbC+
No spin allowed transition is expected for PbC+ within 30 000 cm−1 of energy. The
excited 34Σ− and 24Π states are primarily capable of emitting to the ground state. The
154

24Π state predissociates, while 34Σ− has a large bond distance. The computed radiative
lifetime for the 34Σ−–X4Σ− transition is 25 µs at υ′=0. As Table 6.14 shows, the lifetime
decreases rapidly with the increase in vibrational quantum number. A similar kind of tran-
sition was also predicted for SnC+ (section 5.3.3) with a radiative lifetime of the order of
few nanosecond. Six dipole allowed transitions along with partial radiative lifetimes of the
upper states of PbC+ are displayed in Table 6.14. The parallel component of 22Π3/2–X4Σ−3/2
has been predicted to be the strongest one. We have plotted the transition moment val-
ues for parallel and perpendicular components of six transitions in Fig. 6.5b. The parallel
component transition moments look similar; all of them have a highest peak near the Franck-
Condon overlap zone and subsequently they tend to zero value as the internuclear distance
increases. In general, parallel transitions involving 3/2-3/2 components are stronger than
the perpendicular transitions involving the corresponding 3/2-1/2 components. Radiative
lifetimes in the 22Π3/2 state for the (22Π3/2–X4Σ−1/2)⊥ transition is roughly 10 times larger
than that for (22Π3/2–X4Σ−3/2)‖. This is because of the fact that in the Franck-Condon region
the transition moment values for the parallel component are almost three times than those
of the perpendicular one. The transition moment value for the parallel component of the
22Π3/2–X4Σ−3/2 transition is maximum (0.245 ea0) at r= 4.9 a0. The radiative lifetime in the2Π3/2 state is about 60 µs at υ′=0. Although a low-lying 2∆5/2 has been isolated around
8900 cm−1, the radiative lifetime in that state could not be measured because of its very low
binding property.
Table 6.14 Radiative lifetime (s) of some of the excited states of PbC+
Transition Partial lifetime of the upper state ata Total lifetime
υ′=0 υ′=1 υ′=2 υ′=3 at υ′=0
34Σ−–X4Σ− 2.50(-5) 3.10(-6) 8.08(-7) 3.33(-7)
(2Π1/2–X4Σ−1/2)‖ 1.61(-4) 1.82(-4) 1.92(-4) 1.96(-4)
(2Π3/2–X4Σ−3/2)‖ 6.25(-5) 7.30(-5) 7.97(-5) 9.70(-5)
(3/2(III)–X4Σ−3/2)‖ 4.37(-5)
(3/2(III)–X4Σ−1/2)⊥ 7.19(-4) τ3/2(III)=4.12(-5)
(22Π3/2–X4Σ−3/2)‖ 1.15(-5) 1.30(-5)
(22Π3/2–X4Σ−1/2)⊥ 1.08(-4) 1.09(-4) τ22Π3/2=1.04(-5)
a Values in parenthesis are power to base 10
155
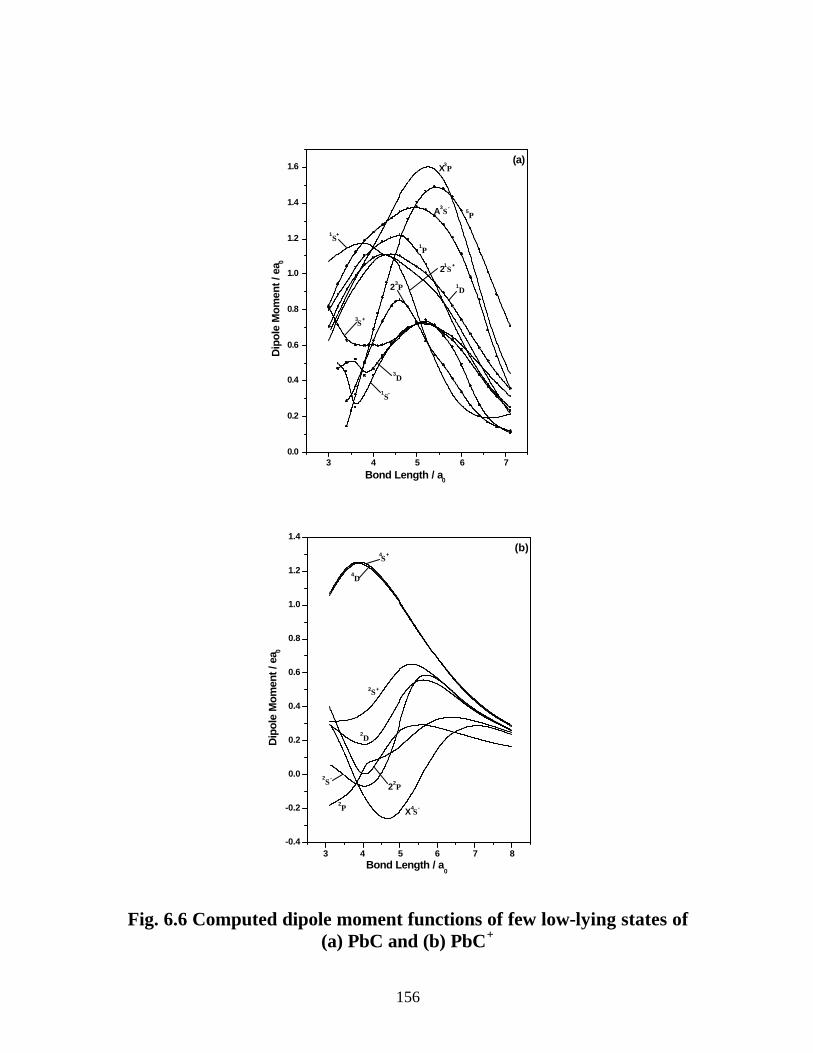
156
3 4 5 6 70.0
0.2
0.4
0.6
0.8
1.0
1.2
1.4
1.6(a)
3∆
23Π
1Σ
-
3Σ
+
1∆
21Σ
+
1Σ
+
1Π
A3Σ
-5Π
X3Π
Dip
ole
Mom
ent /
ea 0
Bond Length / a0
3 4 5 6 7 8-0.4
-0.2
0.0
0.2
0.4
0.6
0.8
1.0
1.2
1.4(b)
2Σ+
2∆
2Σ
-
22Π
2Π
4Σ
+
4∆
X4Σ
-
Dip
ole
Mom
ent /
ea 0
Bond Length / a0
Fig. 6.6 Computed dipole moment functions of few low-lying states of (a) PbC and (b) PbC+

6.3.4 Dipole moments and ionization energies
The computed dipole moment as a function of bond length for different Λ-S states of
PbC are plotted in Fig. 6.6a. Their corresponding equilibrium values (µe) are also listed in
Table 6.4. The computed µe varies from 1.83 to 3.74 D for the low-lying Λ-S states. Fig. 6.6a
shows that dipole moment tends to zero at longer bond distances as expected for the neutral
molecules. The ground state dipole moment of PbC is reported to be 3.0 D with Pb+C−
polarity. The π1 → σ2 transition not only increases the bond length but also increases the
charge separation as σ2 is more localized on C atom. Consequently, the molecule is more
polar in its A3Σ− state with a greater dipole moment of 3.28 D. All the other excited states
except 21∆ have smaller dipole moment than the ground state. The 21∆ state has the highest
dipole moment of 3.74 D due to very long bond length of 2.54 A. Inspite of their longer re,
the states like 1Σ−, 3∆, 3Σ+ etc., which arise from π1 → π2 electronic transition, have the
dipole moments less than 2 D. The reason behind it is the localization of antibonding π2
more on Pb and thus a π back donation from C→Pb decreases the µes.
Fig. 6.6b represents the dipole moment functions of several low-lying states of PbC+. The
numerical values are computed keeping Pb at the origin of the Cartesian coordinate system.
The corresponding dipole moment function of the ground state (X4Σ−) passes through a
minimum, while those of 4∆, and 4Σ+ pass through maximum. Rest of the curves look
similar; they have a minimum followed by a maximum point, and all the curves converge to
zero value at very large bond distances. In Table 6.15, we have presented the equilibrium
dipole moments of some of the low-lying states of PbC+. Here the values have been calculated
by keeping the origin at the the center of mass of the ion. The ground-state dipole moment is
predicted to be -1.11 D, while that of the first excited state is -0.36 D. Inspite of an increase
in bond length the decrease in µe may be attributed to the electronic transfer from more
polarized π1 bonding MO to less polarized σ2 bonding orbital. The dipole moment of 4∆
and 4Σ+ are extremely large in consistent with the lighter homologue.13
Table 6.15 gives the calculated values of vertical as well as adiabatic ionization energies
to the low-lying states of the cation in the Λ-S level, whereas in Table 6.16 we have given
the same values after the spin-orbit correction. Ionization of PbC to the ground-state PbC+
requires the energy of 7.26 eV which is approximately three times the ground-state dissoci-
ation energy of PbC at the Λ-S level. As the cation has to some extent longer bond length,
the adiabatic energy separation between these two states is somewhat lass than the VIE.
With the inclusion of spin-orbit interaction the VIE is changed to be 7.40 eV. However, no
157
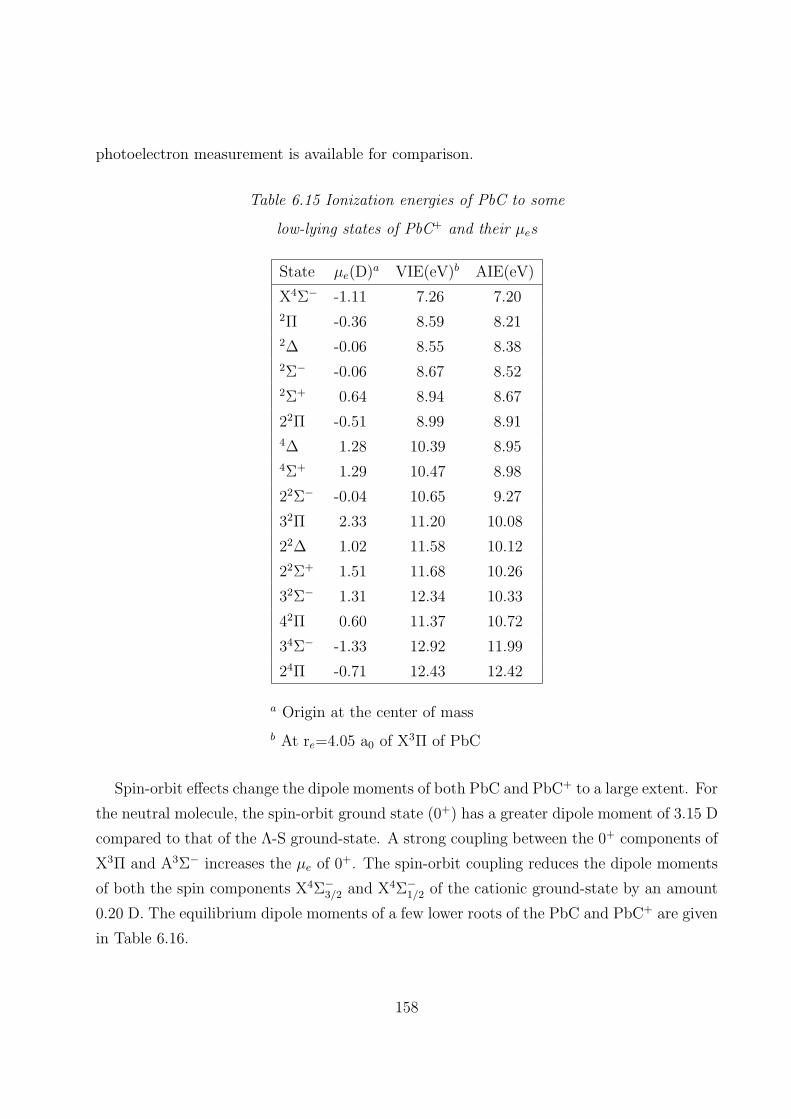
photoelectron measurement is available for comparison.
Table 6.15 Ionization energies of PbC to some
low-lying states of PbC+ and their µes
State µe(D)a VIE(eV)b AIE(eV)
X4Σ− -1.11 7.26 7.202Π -0.36 8.59 8.212∆ -0.06 8.55 8.382Σ− -0.06 8.67 8.522Σ+ 0.64 8.94 8.67
22Π -0.51 8.99 8.914∆ 1.28 10.39 8.954Σ+ 1.29 10.47 8.98
22Σ− -0.04 10.65 9.27
32Π 2.33 11.20 10.08
22∆ 1.02 11.58 10.12
22Σ+ 1.51 11.68 10.26
32Σ− 1.31 12.34 10.33
42Π 0.60 11.37 10.72
34Σ− -1.33 12.92 11.99
24Π -0.71 12.43 12.42
a Origin at the center of mass
b At re=4.05 a0 of X3Π of PbC
Spin-orbit effects change the dipole moments of both PbC and PbC+ to a large extent. For
the neutral molecule, the spin-orbit ground state (0+) has a greater dipole moment of 3.15 D
compared to that of the Λ-S ground-state. A strong coupling between the 0+ components of
X3Π and A3Σ− increases the µe of 0+. The spin-orbit coupling reduces the dipole moments
of both the spin components X4Σ−3/2 and X4Σ−1/2 of the cationic ground-state by an amount
0.20 D. The equilibrium dipole moments of a few lower roots of the PbC and PbC+ are given
in Table 6.16.
158
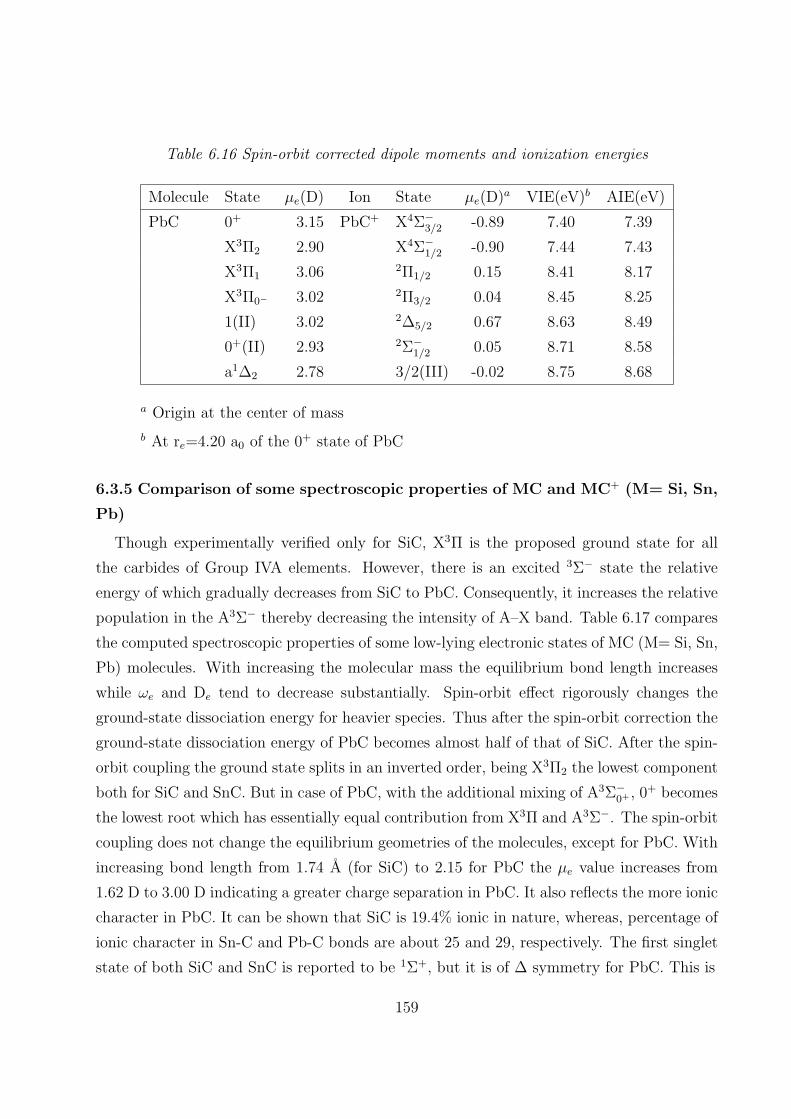
Table 6.16 Spin-orbit corrected dipole moments and ionization energies
Molecule State µe(D) Ion State µe(D)a VIE(eV)b AIE(eV)
PbC 0+ 3.15 PbC+ X4Σ−3/2 -0.89 7.40 7.39
X3Π2 2.90 X4Σ−1/2 -0.90 7.44 7.43
X3Π1 3.06 2Π1/2 0.15 8.41 8.17
X3Π0− 3.02 2Π3/2 0.04 8.45 8.25
1(II) 3.02 2∆5/2 0.67 8.63 8.49
0+(II) 2.93 2Σ−1/2 0.05 8.71 8.58
a1∆2 2.78 3/2(III) -0.02 8.75 8.68
a Origin at the center of mass
b At re=4.20 a0 of the 0+ state of PbC
6.3.5 Comparison of some spectroscopic properties of MC and MC+ (M= Si, Sn,
Pb)
Though experimentally verified only for SiC, X3Π is the proposed ground state for all
the carbides of Group IVA elements. However, there is an excited 3Σ− state the relative
energy of which gradually decreases from SiC to PbC. Consequently, it increases the relative
population in the A3Σ− thereby decreasing the intensity of A–X band. Table 6.17 compares
the computed spectroscopic properties of some low-lying electronic states of MC (M= Si, Sn,
Pb) molecules. With increasing the molecular mass the equilibrium bond length increases
while ωe and De tend to decrease substantially. Spin-orbit effect rigorously changes the
ground-state dissociation energy for heavier species. Thus after the spin-orbit correction the
ground-state dissociation energy of PbC becomes almost half of that of SiC. After the spin-
orbit coupling the ground state splits in an inverted order, being X3Π2 the lowest component
both for SiC and SnC. But in case of PbC, with the additional mixing of A3Σ−0+ , 0+ becomes
the lowest root which has essentially equal contribution from X3Π and A3Σ−. The spin-orbit
coupling does not change the equilibrium geometries of the molecules, except for PbC. With
increasing bond length from 1.74 A (for SiC) to 2.15 for PbC the µe value increases from
1.62 D to 3.00 D indicating a greater charge separation in PbC. It also reflects the more ionic
character in PbC. It can be shown that SiC is 19.4% ionic in nature, whereas, percentage of
ionic character in Sn-C and Pb-C bonds are about 25 and 29, respectively. The first singlet
state of both SiC and SnC is reported to be 1Σ+, but it is of ∆ symmetry for PbC. This is
159
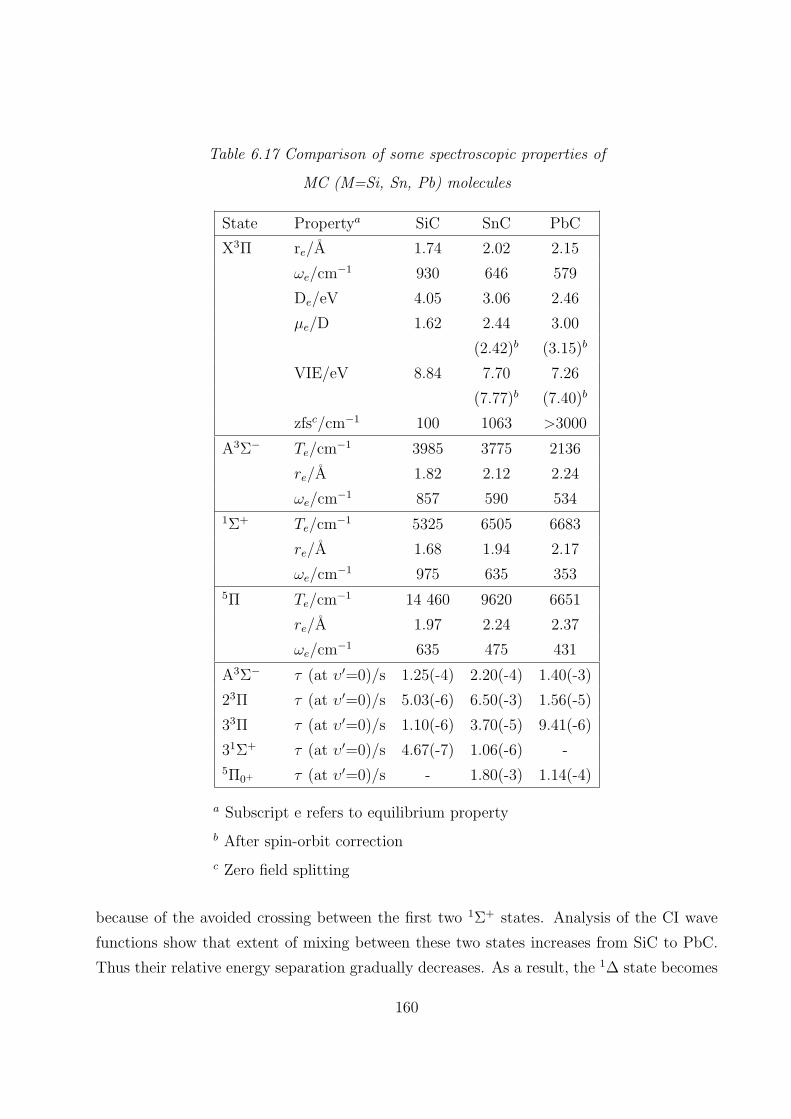
Table 6.17 Comparison of some spectroscopic properties of
MC (M=Si, Sn, Pb) molecules
State Propertya SiC SnC PbC
X3Π re/A 1.74 2.02 2.15
ωe/cm−1 930 646 579
De/eV 4.05 3.06 2.46
µe/D 1.62 2.44 3.00
(2.42)b (3.15)b
VIE/eV 8.84 7.70 7.26
(7.77)b (7.40)b
zfsc/cm−1 100 1063 >3000
A3Σ− Te/cm−1 3985 3775 2136
re/A 1.82 2.12 2.24
ωe/cm−1 857 590 5341Σ+ Te/cm−1 5325 6505 6683
re/A 1.68 1.94 2.17
ωe/cm−1 975 635 3535Π Te/cm−1 14 460 9620 6651
re/A 1.97 2.24 2.37
ωe/cm−1 635 475 431
A3Σ− τ (at υ′=0)/s 1.25(-4) 2.20(-4) 1.40(-3)
23Π τ (at υ′=0)/s 5.03(-6) 6.50(-3) 1.56(-5)
33Π τ (at υ′=0)/s 1.10(-6) 3.70(-5) 9.41(-6)
31Σ+ τ (at υ′=0)/s 4.67(-7) 1.06(-6) -5Π0+ τ (at υ′=0)/s - 1.80(-3) 1.14(-4)
a Subscript e refers to equilibrium property
b After spin-orbit correction
c Zero field splitting
because of the avoided crossing between the first two 1Σ+ states. Analysis of the CI wave
functions show that extent of mixing between these two states increases from SiC to PbC.
Thus their relative energy separation gradually decreases. As a result, the 1∆ state becomes
160
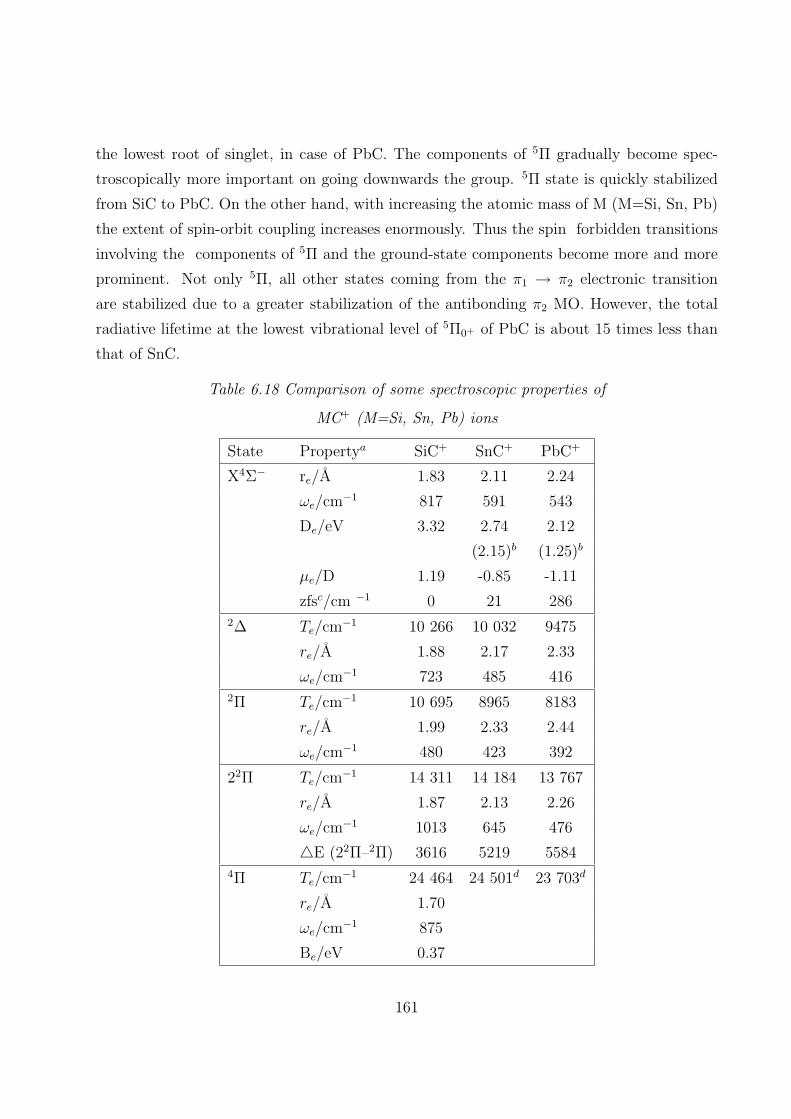
the lowest root of singlet, in case of PbC. The components of 5Π gradually become spec-
troscopically more important on going downwards the group. 5Π state is quickly stabilized
from SiC to PbC. On the other hand, with increasing the atomic mass of M (M=Si, Sn, Pb)
the extent of spin-orbit coupling increases enormously. Thus the spin forbidden transitions
involving the components of 5Π and the ground-state components become more and more
prominent. Not only 5Π, all other states coming from the π1 → π2 electronic transition
are stabilized due to a greater stabilization of the antibonding π2 MO. However, the total
radiative lifetime at the lowest vibrational level of 5Π0+ of PbC is about 15 times less than
that of SnC.
Table 6.18 Comparison of some spectroscopic properties of
MC+ (M=Si, Sn, Pb) ions
State Propertya SiC+ SnC+ PbC+
X4Σ− re/A 1.83 2.11 2.24
ωe/cm−1 817 591 543
De/eV 3.32 2.74 2.12
(2.15)b (1.25)b
µe/D 1.19 -0.85 -1.11
zfsc/cm −1 0 21 2862∆ Te/cm−1 10 266 10 032 9475
re/A 1.88 2.17 2.33
ωe/cm−1 723 485 4162Π Te/cm−1 10 695 8965 8183
re/A 1.99 2.33 2.44
ωe/cm−1 480 423 392
22Π Te/cm−1 14 311 14 184 13 767
re/A 1.87 2.13 2.26
ωe/cm−1 1013 645 476
4E (22Π–2Π) 3616 5219 55844Π Te/cm−1 24 464 24 501d 23 703d
re/A 1.70
ωe/cm−1 875
Be/eV 0.37
161
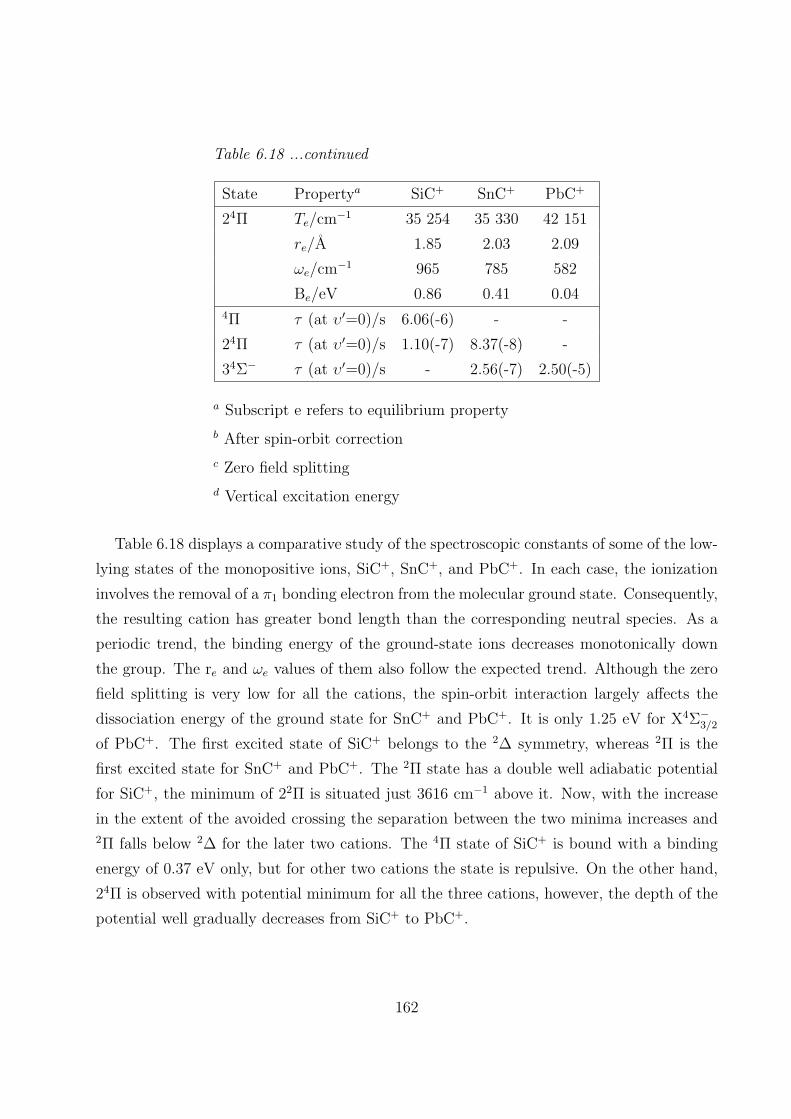
Table 6.18 ...continued
State Propertya SiC+ SnC+ PbC+
24Π Te/cm−1 35 254 35 330 42 151
re/A 1.85 2.03 2.09
ωe/cm−1 965 785 582
Be/eV 0.86 0.41 0.044Π τ (at υ′=0)/s 6.06(-6) - -
24Π τ (at υ′=0)/s 1.10(-7) 8.37(-8) -
34Σ− τ (at υ′=0)/s - 2.56(-7) 2.50(-5)
a Subscript e refers to equilibrium property
b After spin-orbit correction
c Zero field splitting
d Vertical excitation energy
Table 6.18 displays a comparative study of the spectroscopic constants of some of the low-
lying states of the monopositive ions, SiC+, SnC+, and PbC+. In each case, the ionization
involves the removal of a π1 bonding electron from the molecular ground state. Consequently,
the resulting cation has greater bond length than the corresponding neutral species. As a
periodic trend, the binding energy of the ground-state ions decreases monotonically down
the group. The re and ωe values of them also follow the expected trend. Although the zero
field splitting is very low for all the cations, the spin-orbit interaction largely affects the
dissociation energy of the ground state for SnC+ and PbC+. It is only 1.25 eV for X4Σ−3/2
of PbC+. The first excited state of SiC+ belongs to the 2∆ symmetry, whereas 2Π is the
first excited state for SnC+ and PbC+. The 2Π state has a double well adiabatic potential
for SiC+, the minimum of 22Π is situated just 3616 cm−1 above it. Now, with the increase
in the extent of the avoided crossing the separation between the two minima increases and2Π falls below 2∆ for the later two cations. The 4Π state of SiC+ is bound with a binding
energy of 0.37 eV only, but for other two cations the state is repulsive. On the other hand,
24Π is observed with potential minimum for all the three cations, however, the depth of the
potential well gradually decreases from SiC+ to PbC+.
162

6.4. Summary
Using relativistically corrected pseudo core potential, abinitio based MRDCI calculations
have been performed on the heaviest diatomic carbide of the group IVA and its monopositive
ion.28 Potential energy curves along with the spectroscopic properties of low-lying Λ-S states
are reported for the first time. The neutral PbC has X3Π ground-state and A3Σ− is lying
only 2136 cm−1 above it. On the other hand, due to the removal of one π1 electron from the
ground-state PbC, the resulting cation has 0.09 A longer bond length in its X4Σ− ground-
state. The spectroscopic constants of the states of PbC and PbC+ are largely affected by
spin-orbit interaction. The ground state of PbC splits into four Ω components with a large
equilibrium separation. Unlike other carbides in group IVA, it has the ground state of 0+
symmetry. Total spin-orbit splitting in the first dissociation limit matches well with the
experimental observation of 10 693 cm−1. The spin-orbit interaction splits Pb+(2P)+C(3P)
into six sub-levels which are overall 14 124 cm−1 apart. The computed spin-orbit corrected
IP of Pb is in good agreement with the experimental observation of 7.415 eV.
Several spin allowed or spin forbidden transitions are reported for PbC. Transitions to
23Π, 33Π, 23∆, and 33Σ− are of great interest. Dipole allowed transition lifetimes of the 5Π
components are noticeable, many of them are less than 100 µs. d1Σ+0+ also has total radiative
lifetime of 60 µs at its lowest vibrational level. Only one spin allowed transition is expected
for PbC+, but some dipole allowed transitions are important to be observed. Total radiative
lifetime at υ′=0 state of 2Π3/2 and 22Π3/2 are about 60 and 10 µs, respectively. Dipole and
transition dipole moment functions of several states of PbC and PbC+ are presented. The
equilibrium dipole moment of the ground-state PbC is predicted to be 3.00 D. The dipole
moments of some of the low-lying states of PbC+ are also reported. The spin-orbit coupling
increases the ground-state equilibrium dipole moment of PbC which is reported to be 3.15 D.
On the other hand similar effect reduces the µe of PbC+ from 1.11 D to 0.89 D. Though these
are origin dependent, they may be useful for the analysis of microwave spectra. The vertical
and adiabatic ionization energies of PbC to many low-lying states of PbC+ are reported, but
no experimental result is found for comparison. Inclusion of the spin-orbit effect predicts
the requirement of 7.40 eV of energy to ionize the molecule PbC.
163

6.5. References
1 K.P. Huber and G. Herzberg, Molecular Spectra and Molecular Structure IV: Constants
of Diatomic Molecules, Van Nistrand Reinhold, New York, 1979.
2 A. Druzhinin, I. OstrovSkii, and I. Kogut, Mater. Sci. Semicond. Process. 9, 853 (2006).
3 J. Tolle, R. Roucka, V. D’costa, J. Menendez, A. Chizmeshya, and J. Kouvetakis, Mater.
Res. Sco. Symp. Proc. 891, 579 (2006).
4 J. Tolle, A.V.G. Chizmeshya, Y.-Y. Fang, J. Kouvetakis, V.R. D’costa, C.-W. Hu,
J. Menendez, and I.S.T. Tsong, Appl. Phys. Lett. 89, 231924 (2006).
5 G. Gigli, G. Meloni, M. Carrozzino, J. Chem. Phys. 122, 14303 (2005).
6 A. Ciccioli, G. Gigli, G. Meloni, E. Testani, J. Chem. Phys. 127, 54303 (2007).
7 G. Li, X. Xing, and Z. Tang, J. Chem. Phys. 118, 6884 (2003).
8 P.A. Denis and K. Balasubramanian, J. Chem. Phys. 123, 54318 (2005).
9 B. Suo and K. Balasubramanian, J. Chem. Phys. 126, 224305 (2007).
10 L.T. Ueno, L.R. Martin, A.D. Pino Jr., F.R. Ornellas, F.B.S. Machado, Chem. Phys.
Lett. 432, 11 (2006).
11 A. Pramanik, K.K. Das, J. Mol. Spectrosc. 244, 13 (2007).
12 Electronic Spectrum of SnC: A Theoretical Study,
A. Pramanik, K.K. Das (communicated).
13 A. Pramanik, S. Chakrabarti, K.K. Das, Chem. Phys. Lett. 450, 221 (2008)
14 R.B. Ross, W.C. Ermler, P.A. Christiansen, J. Chem. Phys. 93, 6654 (1990).
15 L.F. Pacios, P.A. Christiansen, J. Chem. Phys. 82, 2664 (1985).
16 R.J. Buenker, S.D. Peyerimhoff, Theo. Chim. Acta 35, 33 (1974).
17 R.J. Buenker, S.D. Peyerimhoff, Theo. Chim. Acta 39, 217 (1975).
18 R.J. Buenker, Int. J. Quantum Chem. 29, 435 (1986).
19 R.J. Buenker, in: P. Burton (Ed.), Proc. Workshop on Quantum Chemistry and
Molecular Physics in Wollongong, Wollongong, Australia, 1980.
20 R.J. Buenker, in: R. Carbo (Ed.), Studies in Physical and Theoretical Chemistry, vol. 21,
Current Aspects of Quantum Chemistry, Elsevier, Amsterdam, p.17, 1982.
164

21 R.J. Buenker, S.D. Peyerimhoff, W. Butscher, Mol. Phys. 35, 771 (1978).
22 R.J. Buenker, R.A. Philips, J. Mol. Struct. (Theochem) 123, 291 (1985).
23 S. Krebs, R.J. Buenker, J. Chem. Phys. 103, 5613 (1995).
24 E.R. Davidson, in: R. Daudel, B. Pullman (Eds.), The World of Quantum Chemistry,
Reidel, Dordrecht, The Netherland, 1974.
25 G. Hirsch, P.J. Bruna, S.D. Peyerimhoff, R.J. Buenker, Chem. Phys. Lett. 52, 442
(1977).
26 A.B. Alekseyev, R.J. Buenker, H.-P. Lieberman, G. Hirsch, J. Chem. Phys. 100, 2989
(1994).
27 C.E. Moore, Tables of Atomic Energy Levels, vols. I–III, U.S. National Bureau of
Standards: Washington, DC, 1971.
28 Electronic Structure and Spectroscopic Properties of PbC and PbC+: an MRDCI Study,
A. Pramanik, K.K. Das (to be communicated).
165

CONCLUSION

The structural and spectroscopic aspects of intragroup IVA heteronuclear diatomic molec-
ules like SiC, SnC, PbC and some of their monopositive and mononegative ions have been
studied in the present thesis. It deals with the analysis of the results of abinitio based quan-
tum mechanical calculations on the above species. A high-level multireference singles and
doubles configuration interaction method, which incorporates relativistically corrected effec-
tive core potentials and spin-orbit coupling, has been employed throughout the calculations.
The diabatic as well as adiabatic potential energy curves are constructed for the ground and
low-lying electronic states. These provide different types of spin-forbidden and spin-allowed
transitions which are possible within experimental ranges. The dipole and transition dipole
moment functions are also constructed for these molecules and ions. The calculated ioniza-
tion energies and electron affinities match well with the available experimental and other
theoretical data.
Of all the species studied here, experimental results of SiC are available only. Although
SiC radical was first studied theoretically, later on many experimental spectroscopic studies
on the ground state (X3Π) as well as a few triplets like A3Σ−, B3Σ+, C3Π etc. were reported.
The calculated spectroscopic constants of several low-lying states of SiC agree well with the
experimental results. Besides the triplets, several bound states of singlet and quintet spin
multiplicities are computed here. The lowest quintet bound state 5Π is located around
14 460 cm−1. An excited 3Π namely E3Π is isolated for the first time in our calculation
with Te=25 875 cm−1. The ground-state dipole moment is calculated to be 1.62 D with a
Si+C− polarity. The molecule has the highest dipole moment of 2.55 D in its A3Σ− state.
The sense of polarity is same for other excited states also. The spin-orbit coupling is almost
insignificant for SiC. Due to the spin-orbit interaction, the largest splitting among the four
spin components of X3Π is only 100 cm−1. Many spin allowed transitions are studied for
the radical. The observed E–X transition is predicted to be the strongest one. A radiative
lifetime of 1.1 µs is estimated for this transition. 31Σ+–a1Σ+ is also an important transition
for SiC. It has a very short lifetime of the order of 450 ns in its lowest vibrational level.
Unlike the neutral species, a very few experimental data are available for the unicharged
ions of SiC. The computed ground state of SiC+ is X4Σ− while its dissociation energy of
3.32 eV matches well with the experimental data. An asymmetric double well is observed in
the potential energy curve of the lowest doublet state of SiC+ (2Π). Like the doublets, the
lowest two 4Π states undergo avoided crossing. Their strong interaction reduces the binding
energy of 4Π. On the other hand, 24Π has a sharp potential minimum. The potential energy
166

curves of SiC− are also of great interest. Only the ground (X2Σ+) and two other excited
states of the anion were studied before. We have computed two new doublets, namely C2Π
and D2Π which are spectroscopically important. A number of hitherto unknown quartets
and sextets of SiC− are also predicted. Like the SiC radical, spin-orbit interactions have
almost no influence on the spectroscopic properties of the ions. The 24Π-X4Σ− transition is
predicted to be the strongest one for SiC+ with a partial radiative lifetimes of about hundred
nanoseconds at the lowest few vibrational levels. In case of SiC−, A2Π–X2Σ+ and B2Σ+-A2Π
transitions are not predicted to be strong, but three strong transitions, B-X, C-X, and D-X
are expected to be observed in the range 21 000-24 000 cm−1. The last two transitions are
predicted for the first time in the present study. The present thesis also computes the dipole
moments of the ions, vertical and adiabatic ionization energies and electron affinities of SiC
which compare well with the available data.
The spectroscopic properties of SnC and SnC+ are not well known in literature. In
the present thesis, the CI calculations have been performed on these two species. The
ground state of SnC belongs to the X3Π symmetry with re=2.023 A and ωe=646 cm−1. It is
characterized by an open shell configuration, σ21σ2π
31. At least 30 excited singlet, triplet and
quintet states of Λ-S symmetries have been predicted within 6 eV of energy. The ground-
state dissociation energy of the molecule is estimated to be 3.06 eV which is reduced to
2.87 by spin-orbit interaction. The computed dipole moment of SnC in the ground state
is about 2.44 D with Sn+C− polarity. Three triplet-triplet transitions such as B3Π-X3Π,
33Π-X3Π, and 53Π-X3Π are expected to be strong for SnC. Besides these, transitions to
excited 31Σ+ from the lower singlets are expected to have enough intensities. Total radiative
lifetime of 31Σ+ in the lowest vibrational level is found to be 1.05 µs. Ionizing the molecule
by the energy of the order of 7.70 eV it reaches to 4Σ− ground-state of SnC+. Besides the
ground state, potential energy curves of 16 more excited states of doublet and quartet spin
multiplicities are constructed in the present thesis. Spectroscopic constants are not largely
affected by the spin-orbit coupling, but several spin forbidden transitions like 2Π3/2–X4Σ−3/2,
22Π1/2–X4Σ−1/2, 2Σ+1/2–X4Σ−1/2 etc. are expected to have significant intensities.
The spectral behaviors of the heaviest carbide of group IVA are somewhat different from
the lighter species due to a strong spin-orbit coupling. The ground state of PbC is computed
here as X3Π which is in analogy with the lighter carbides of group IVA and some other
intragroup IVA diatomics. The first excited state of the molecule (A3Σ−) lies only 2136
cm−1 above the ground state. The ground-state dissociation energy of PbC is reported to be
167

1.97 eV which matches well with the previously estimated value. The adiabatic separation
among the spin-orbit components of the ground state exceeds 3000 cm−1. The overall split-
ting in the dissociation limit agrees well with the experimental observation in the atomic
level. The computed ionization energy of Pb is also in good agreement to the experimentally
observed value. Many dipole allowed transitions of PbC are predicted for the first time.
Transitions involving the components of 5Π, d1Σ+, a1∆ etc. and the corresponding ground-
state components are found to be important. The PbC+ ion has X4Σ− ground-state with
excited 2Π and 2∆. Total radiative lifetime of the 22Π3/2 state of PbC+ at the ground vibra-
tional level is of the order of 10 µs. The spin-orbit corrected ground-state dipole moments
of PbC and PbC+ are 3.15 and -0.89 D, respectively. With the inclusion of the spin-orbit
interaction the vertical ionization potential of PbC is reported to be 7.40 eV.
Throughout the theses we have presented a detailed structural and spectroscopic infor-
mation of intragroup IVA heteronuclear diatomics from the state-of-the-art ab initio based
CI calculations. Emphasis has been given on the carbides of group IVA and some of their
monopositive and mononegative ions. We hope these spectroscopic data would be very
helpful to the experimentalist in future.
168

List of Publications
∗1. Electronic States and Spectroscopic Properties of SiTe and SiTe+.
Surya Chattopadhyaya, Anup Pramanik, Amartya Banerjee, and Kalyan Kumar Das,
J. Phys. Chem. A 110, 12303 (2006).
∗2. Ab initio configuration interaction study of the low-lying electronic states of InF.
Amartya Banerjee, Anup Pramanik, Kalyan Kumar Das,
Chem. Phys. Lett. 429, 62 (2006).
∗3. B2Σ+-X2Σ+ and C2Π-X2Σ+ transitions in InF++: A configuration interaction study.
Amartya Banerjee, Anup Pramanik, Kalyan Kumar Das,
Chem. Phys. Lett. 435, 208 (2007).
4. The electronic spectrum of the SiC radical: A theoretical study.
Anup Pramanik, Kalyan Kumar Das, J. Mol. Spectrosc. 244, 13 (2007).
5. Theoretical studies of the electronic spectrum of SiC+.
Anup Pramanik, Susmita Chakrabarti, Kalyan Kumar Das,
Chem. Phys. Lett. 450, 221 (2008).
∗6. MDRCI studies on the electronic states of InBr and InBr+.
Amartya Banerjee, Anup Pramanik, Susmita Chakrabarti, Kalyan Kumar Das,
J. Mol. Struc. (THEOCHEM) 893, 37 (2009).
7. Electronic states of SiC−: A theoretical study.
Anup Pramanik, Amartya Banerjee, Kalyan Kumar Das,
Chem. Phys. Lett. 468, 124 (2009).
8. Electronic spectrum of SnC: A theoretical study.
A. Pramanik, K.K. Das (communicated).
9. Theoretical investigation of electronic states of SnC+.
A. Pramanik, K.K. Das (to be communicated).
10. Electronic structure and spectroscopic properties of PbC and PbC+: an MRDCI study.
A. Pramanik, A. Banerjee, K.K. Das (to be communicated).
∗Papers are not included in the thesis
169


![Synthesis, Characterization and Crystal Structure of a Novel Three-Dimensional Supramolecular Framework Containing Ni···π Interaction Heteronuclear Complex: {[Cu(4(5)-Meim)4][Ni(CN)4] · H2O}n](https://img.pdfslide.net/doc/110x75/635011bf3be08ca4ad0c192d/synthesis-characterization-and-crystal-structure-of-a-novel-three-dimensional-supramolecular.jpg)


























