Embed Size (px)
Citation preview

Eur. J . Biochem. 195,465-475 (1991)
001429569100056Z
FEBS 1991
Tumour-necrosis-factor-mediated cytotoxicity is correlated with phospholipase-A, activity, but not with arachidonic acid release per se Philip SUFFYS, Rudi BEYAERT, Dirk DE VALCK, Bart VANHAESEBROECK, Frans VAN ROY and Walter FIERS
Laboratory of Molecular Biology, State University of Gent, Belgium
(Received April 9/September 24, 1990) - EJB 90 0390
L929, a murine fibrosarcoma cell line highly sensitive to the anti-proliferative and cytotoxic action of tumour necrosis factor (TNF), was used as a target cell in our studies. We [Suffys et al. (1987) Biochem. Biophys. Res. Commun. 149, 735 - 7431, as well as others, have previously provided evidence that a phospholipase (PL), most probably a PL-A,-type enzyme, is likely to be involved in TNF-mediated cell killing. We now further document this conclusion and provide suggestive evidence that the enzyme activity specifically involved in TNF cytotoxicity differs from activities associated with the eventual cell death process itself or with non-toxic serum treatment. We also show that the 5,8,11,14-icosatetraenoic acid (arachidonic acid, A4Ach) released by PL, and possibly metabolized, is unlikely to be a key mediator of the TNF-mediated cytotoxicity. These conclusions are based on the following experimental findings.
1. TNF treatment of cells, prelabelled for 24 h with [jH]A4Ach or [14C]A3Ach (A3Ach = 5,8,11- icosatrienoic acid) resulted in an early, time-dependent and concentration-dependent release of radioactivity in the supernatant preceding actual cell death. The extent of this response was moderate, albeit reproducible and significant. Analysis of the total lipid fraction from cells plus supernatant revealed that only release of arachidonic acid from phospholipids, but not its metabolization was induced by TNF. However, the release of less unsaturated fatty acids, such as linoleic acid (Lin) or palmitic acid (Pam), was not affected during the first hours after TNF addition.
2. An L929 subclone, selected for resistance to TNF toxicity, was found to be defective in TNF-induced A4Ach liberation.
3. Interleukin-1 (IL1) was not cytotoxic for L929 and did not induce release of A4Ach. 4. Release of A4Ach was not restricted to TNF; the addition of serum to the cells also induced release of fatty
acids into the medium. In this case, however, there was no specificity, as all fatty acids tested, including Lin and Pam, were released.
5. Inhibition of PL-A, activity by appropriate drugs markedly diminished TNF-induced A4Ach release and resulted also in a strong decrease in TNF-induced cytotoxicity.
6. Other drugs, including serine protease inhibitors, which strongly inhibit TNF-induced cytotoxicity, also decreased the TNF-induced A,Ach release, whereas LiCl potentiated both TNF-mediated effects.
7. Protection of cells against TNF toxicity by means of various inhibitors was not counteracted by addition of exogenous fatty acids, including A,Ach.
8. We could not detect an increase in the amount of free inositol or any inositol phosphate after TNF treatment of cells, prelabelled with my~-[~H]inositol. This indicates that the phosphatidylinositol-specific PL-C is not involved in the TNF-induced fatty acid release.
9. Neither were we able to observe a decrease in fatty acid incorporation into phospholipids during TNF treatment. This excludes a decrease in acyltransferase activity as the reason for increased levels of free fatty acid.
The above observations strongly suggest that an activated PL-A, is involved in TNF-mediated cell killing. The limited extent of TNF-specific fatty acid release, the relative specificity for A4Ach of this early response which is not shared by serum treatment, and the characteristic drug-resistance profile of this activity, all argue for the involvement of a particular PL-A, species. Moreover, our data also suggest that A4Ach itself and its metabolites do not play a key role in TNF cytotoxicity.
Correspondence to W. Fiers, Laboratorium voor Moleculaire Bio- logic, K . L. Ledeganckstraat 35, B-9000 Gent, Belgium
Abbreviations. AcPheONap, N-acetyl-DL-phenylalanine-b-naph- thy1 ester; BSA, bovine serum albumin; A,Ach, 5,8,1l-icosatrienoic acid; A,Ach, arachidonic acid, 5,8,11,14-icosatetraenoic acid; DMEM, Dulbecco’s modified Eagle’s medium; EM-BSA, fatty-acid- free bovine serum albumin; FCS, foetal calf serum; ILI, interleukin- 1 ; Lin, cis,cis-9,12-octadecadienoic acid (linoleic acid); MEFs, murine
embryonic fibroblast-like cells; MEM, minimal essential medium; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; NaCl/Pi, phosphate-buffered saline; NCS, newborn calf serum; NDGA, nordihydroguaiaretic acid ; Pam, hexadecanoic acid (palmitic acid); PL, phospholipase; PtdIns, phosphatidylinositol; TNF, tu- mour necrosis factor; TosArgOMe, W-p-tosyl-L-arginine methyl es- ter; TosLysCH,Cl, W-p-tosyl-L-lysine chloromethane.
Enzymes. Phospholipase Az (EC 3.1.1.4); phospholipase C (EC 3.1.4.3).

466
The name tuinour necrosis factor (TNF) was chosen be- cause of the in vivo antitumour activities of this cytokine [l]. TNF also exerts selective cytotoxic effects (growth inhibition and/or cytolysis) on tumour cells in vitro [2]. TNF-encoding nucleotide sequences of various species were cloned and ex- pressed by several groups [3] and the resulting pure proteins are now available in large quantities.
There are numerous reports on metabolic changes induced by TNF in different cell types (reviewed in [4]), but the exact biochemical steps between the first contact of TNF with its receptor on a susceptible target cell and the final death of the cell remain obscure. There is experimental evidence that binding of TNF to a high-affinity receptor [5] is followed by internalization of the complex [6, 71. TNF could then induce a second messenger and become metabolized [8, 91. Although the nature of a second mediator following TNF binding and internalization is not known, we and others have suggested the involvement of a G-protein [lo- 121, while there are con- tradictory results concerning the possible involvement of CAMP in TNF cytotoxicity [I31 (and A. Hepburn, unpub- lished results). GTP-binding proteins are known to transduce activation signals from ligand-receptor complexes to various enzymes (reviewed in [14]). Phospholipases (PLs) are examples of such enzymes and their activation leads to liberation of fatty acids from membrane phospholipids. Treatment of sensi- tive cells with TNF may result in (a) activation of PL-A2 [15, 161, (b) increase in free 5,8,11,14-icosatetraenoic acid (arachidonic acid, d,Ach) in the medium [16, 171 and increase in production of d,Ach metabolites [18-231.
It is well known that polyunsaturated fatty acids can be selectively cytotoxic to some tumour cells [24]. This is presum- ably due to oxygen radicals formed upon metabolization of the fatty acids [25]. The observation that some scavengers of oxygen radicals may protect cells from TNF cytotoxicity [26, 271 and the recent finding that manganese superoxide dismu- tase may be part of an 'inherent protection mechanism' of at least some cells against TNF [28,29], could mean that oxygen radicals as well as their potential source, viz. unsaturated fatty acids, may play an essential role in TNF cytotoxicity.
It was our goal in the present study to determine the relevance for TNF-induced cytotoxicity of PL activity, of release of d4Ach and of polyunsaturated fatty acids.
MATERIALS AND METHODS
Cell culttires
L929, a murine fibrosarcoma cell line (obtained from the Rega Institute, Leuven, Belgium) and L929(r)2, a TNF-resist- ant, but not TNF-producing cell clone [23] (and our unpub- lished results), were grown in Dulbecco's modified Eagle's medium (DMEM ; Gibco Bio-Cult, Paisley, UK) supplement- ed with 10% heat-inactivated (30 min, 57°C) newborn calf serum (NCS; Gibco Bio-Cult). Murine embryonic fibroblast- like cells (MEFs) were prepared as described previously [30]. HeLa D98/AH2, a human cervix carcinoma cell line (from the Imperial Cancer Research Fund, London, UK), and FS4, a human foreskin fibroblast cell line (from Dr J. Viltek, New York University Medical Center, USA), were grown in DMEM supplemented with 10% foetal calf serum (FCS; Gibco Bio-Cult). MCF-7(AZ), a huinan breast carcinoma cell line (from Dr M. Mareel, Universitair Ziekenhuis, Gent, Belgium) was cultured in minimal essential medium (MEM; Gibco Bio-Cult) supplemented with 5% FCS, nonessential amino acids and 6 ng bovine insulin/ml. WEHI 164 clone 13,
a murine fibrosarcoma cell line which is highly TNF-sensitive (from Dr T. Espevik, University of Trondheim, Norway), was cultured in RPMI 1640 supplemented with 10% FCS. All media were also supplemented with 100 U penicillin/ml and 0.1 mg streptomycin/ml.
Monokine preparations
Throughout this study we used recombinant human TNF, synthesized in Escherichia coli and purified to at least 99% purity [31]. The preparation had a specific activity of 20- 30 MU/mg protein (units as previously defined in a cytotoxicity assay on L929 cells in the presence of 1 pg/ml actinomycin D ; Sigma Chemical Co., St. Louis, MO, USA [32]) contained less than 3 3 ng endotoxinjmg protein, and was stored in aliquots in phosphate-buffered saline (NaCl/Pi) at - 70 'C. Recombinant human interleukin-1P (1L1-/?) was a generous gift from Dr A. Shaw (Glaxo Institute for Molecular Biology, Geneva, Switzerland) and had a specific activity of 270 MU/mg protein (units as previously defined in a cytotoxicity induction assay with PC60 cells [33]).
Materials
The following chemicals were used without further purifi- cation: quinacrine (Sigma); tetracaine (Sigma); chloroquine (Sigma); hydrocortisone hemisuccinate (Diosynth. Oss, The Net herlands) ; dexamethasone (Sigma), p - bromophenacyl bromide (Sigma), dimethylsulfoxide (Me2SO; E. Merck, Darmstadt, FRG); nordihydroguaiaretic acid (NDGA; Sigma); P-diethylaminoethyl diphenylpropylacetate hydro- chloride (SKF 525-A; a gift from Smith Kline Beckman Cor- poration, Philadelphia, PA, USA); indomethacin (Sigma): LiCl (E. Merck); N"-p-tosyl-L-arginine methyl ester (Tos- ArgOMe; Sigma) ; N-acetyl-oL-phenylalanine-fl-naphthyl es- ter (AcPheONap; Sigma); N"-p-tosyl-L-lysine chloromethane (TosLysCH2C1; Sigma); EGTA (Sigma); verapamil (Sigma).
All chemicals were dissolved in medium, except for tetracaine, p-bromophenacyl bromide, indomethacin and verapamil (in ethanol), and for NDGA and AcPheONap (in Me2SO).
Human serum and insulin were from Sigma; murine epi- dermal growth factor was from Boehringer, Mannheim, FRG; human platelet-derived growth factor (AA and BB forms) was a gift from Dr C.-H. Heldin (Ludwig Institute for Cancer Research, Uppsala, Sweden) ; human transforming growth factor PI was obtained from Paise1 (Frankfurt, FRG).
Fatty acid release
For kinetic studies, cells were grown in six-well plates (10 cm2/dish) in 5% C 0 2 at 37'C until near confluency. Me- dium was removed and 1 ml fresh medium, containing either 18.5 kBq [5,6,8,9,11,12,14,1 5-3H]arachidonic acid (d ,Ach ; 7.46 TBq/mmol) or 18.5 kBq [l-'4C]linoleic acid (Lin; 2.06 GBq/mmol) or 18.5 kBq [l-'4C]palmitic acid (Pam; 2.18 GBq/mmol) or 18.5 kBq [2-'4C]5,8,11-icosatrienoic acid (d,Ach; 1.76 GBq/mmol) was added. All radioactively labelled fatty acids were purchased from Amersham Inter- national (Amersham, UK). The cells were further incubated for 24 h until confluency; at that time they had taken up 92.4 A 4.2% [3H]d4A~h, 96.3 5.6% ['4C]Lin, 85.7 2.0% [14C]Pam and 96.9 & 7.3% ['4C]d,Ach. respectively. In the case of d,Ach, the bulk of the fatty acid (> 80%) was incor-

467
porated in the phospholipid fraction, while only about 10% was located in neutral lipids. We could not detect any label associated with the protein fraction (data not shown). Then the cells were washed three times with conditioned medium. This was followed by addition of 4.5 ml conditioned medium containing 5 mg/ml fatty-acid-free bovine serum albumin (EM-BSA; Sigma), and 2 h later by 0.5 ml of the appropriate stimulant. At indicated times after stimulation, 0.3 ml super- natant was cleared from cell material by centrifugation, and radioactivity in the medium was measured by scintillation counting after addition of 10 vol. Triton X-lOO/scintillation liquid (1 :2, by vol.).
The effect of drugs on A4Ach release was monitored as follows. Cells were grown in 24-well plates (2 cm2/well) in 5% C 0 2 at 37°C until near confluency. Then the medium was removed and 0.2 ml fresh medium containing 3.7 kBq [3H]A4Ach was added, after which cells were further incubated for 24 h until confluency. The cells were washed three times with conditioned medium and 0.45 ml of conditioned medium containing 5 mg/ml EM-BSA plus the proper drug was added. After 1 h, 0.05 ml of either control or TNF-containing me- dium was added; after the indicated time, the amount of radioactivity released in 0.3 ml supernatant was measured as described above.
Acyltransferase activity
Cells were grown in six-well plates in 5% C 0 2 at 37°C until confluency. The medium was removed and 1 ml of fresh control or TNF-containing medium was added. At different times after addition of TNF, 8.25 kBq [3H]A4Ach or 3.7 kBq ['4C]A3Ach were added for 1 h. Subsequently, cells were scraped from the plates and frozen at - 20 "C until analysis.
Lipid extraction and analysis
To separate fatty acids from phospholipids, 2 vol. ice-cold acidified acetone was added to the aqueous sample and the resulting protein pellet was removed. Then 2 vol. chloroform was added and, after vigorous shaking, the lower chloroform/ acetone/lipid phase was removed and evaporated. The dry residue was redissolved in 0.05 ml chloroform/methanol(9: 1 , by vol.) and spotted on thin-layer silica gel chromatography plates (E. Merck). The latter were developed in a solvent system containing petroleum ether/diethyl ether/acetic acid (80 : 20 : 1, by vol.).
Separation of metabolites of A4Ach was accomplished on thin-layer silica gel plates with a pre-concentration zone (Whatman Chemical Separation, Clifton, NJ, USA) and de- veloped in chloroform/methanol/acetic acid/water (90: 8 : 1 : 0.8, by vol.). Radioactive spots were visualized and localized by fluorography [34], cut from the plates for liquid scintillation counting or quantified by absorbance measurement of the autoradiography spots by a Gemini gel analysis system (Joyce Loebl, Gateshead, UK).
PtdIns-specific PL-C
Cells were grown in six-well plates. One day before confluency, 37 kBq my0-[2-~H]inositol (740 GBq/mmol; Amersham International) was added in 1 ml fresh medium/ dish; 24 h later, the cells were washed three times with con- ditioned medium and 4.5 ml conditioned medium was added; 2 h later, 0.5 ml TNF-containing medium or control medium were added. At indicated times, the amount of Ins released in
the supernatant was measured by scintillation counting. In another experiment, free Ins and Ins phosphates were mea- sured by adding, at indicated times, an equal volume of ice- cold 10% trichloroacetic acid (E. Merck) to the incubation mixture containing 10 mM LiC1. The precipitates were re- moved and radioactivity in the supernatant was measured by scintillation counting.
TNF-mediated cytotoxicity assays
The effect of various inhibitors on the cytotoxicity of TNF was measured as previously described [35]. Briefly, cells were seeded in microtiter plates at a concentration of 5 x 103/0.1 ml medium in each well. One day later, various concentrations of drugs were applied, followed 2 h later by a serial dilution of TNF (100-0.75 Ujml). Three days after TNF addition, cell numbers were measured either by fixing and staining with crystal-violet-containing solution [35], or by a modified colorimetric assay using a tetrazolium salt [36].
The cytolytic activity of TNF was measured on a confluent monolayer in the presence or absence of actinomycin D. L929 cells were seeded at a concentration of 3 x 104/0.1 ml medium in each well. One day later, serial dilutions of TNF were added, either 20-0.12 U/ml together with actinomycin D, or 4000- 0.25 U/ml in the absence of the drug. Potential inhibitors were added 2 h before TNF addition. Cell survival was measured, as described above, at 18 h after addition of TNF, for exper- iments with actinomycin D or at 24 - 36 h without.
For the experiments in which we tested the effect of fatty acids on the inhibition of TNF cytotoxicity by various drugs, A4Ach (Sigma), A3Ach (Sigma) or methyl arachidonate (Nu Check Prep, Elysian, MS, USA) were administered 1 h before drug addition; TNF was added 1 h later. These experiments were set up with or without supplementation of the medium with 5 mg EM-BSA/ml.
Uvidine release assay
Cells were cultured as described above for the kinetic stud- ies of fatty acid release. When nearly confluent, supernatant was removed and 1 ml fresh medium, containing 185 kBq [5,6- 3H]uridine (1.67 TBq/mmol; Amersham International), was added and incorporation allowed to proceed for 24 h. Then the cells were washed, incubated with or without TNF, and, at the indicated time points, the amount of radioactivity re- leased in the supernatant was measured as described above.
RESULTS
TNF induces the release of arachidonic acid in various cell lines sensitive to T N F cytotoxicity
The effect of TNF on the metabolism of [3H]A4Ach- labelled L929 cells was studied. As shown in Fig. 1, the release of A,Ach in the supernatant became evident as early as 2- 3 h after TNF addition and continued to increase during the time of observation. Fatty acid release preceded cell lysis, as measured by uridine release. The latter started to occur only about 6 h after TNF treatment. Most cells were dead 24 h after treatment. It may be noted that the use of fresh medium for washing and TNF stimulation procedures resulted in a spontaneous increase in A4Ach release in the control cultures without TNF. This could be avoided by the use of conditioned, instead of fresh medium. The increase in A4Ach release is clearly dependent on the concentration of TNF applied

468
- 9 - s ...- : a - 2 7 - : 0 6 -
IJ 5 -
6 1 -
0 3 - Y IJ 2 -
.- '0 1 .-
- I
1 - 0 1 2 3 4 5 6 7 8
time (h)
Fig. 1. Aruchidmic w i d releuse ut various times after the sturt of the TNF treatment us cornpured with cell survival. 10 cm2 of LY2Y cells, prelabelled with 18.5 kBq ['H]A,Ach or with 18.5 kBq [3H]uridine, were washed and further treated as described in Materials and Methods. Release of A4Ach ( 0 ) was compared with that of uridine (0). For each time point, the radioactivity released into the super- natant of cells treated with T N F (5000 Ujml) was diminished by the radioactivity in the supernatant of cells without TNF. This value was then expressed as a percentage of the total radioactivity incorporated. After 10 h, 4.8 f 0.2% of the d,Ach and 6.9 k 0.5% of the uridine were spontaneously released. Each value represents the mean ( f SD) of an experiment carried out in triplicate
0-
10,
r
I T
- 10
33
1
- 1(
3 TNF concentration (U/ml)
330
I 100
3300 33000
Fig. 2. Release of urachidonic acid,from cells treated \rich increasing umounts of TNF. 10 cm2 of L92V cells were prelabelled with 3.7 kBq [3H]d,Ach and further treated as described in Materials and Methods. The amount of radioactivity released after 5-h treatment is expressed as a percentage of total cell-associated 3H before treatment with TNF. Each value represents the mean (f SD) of an cxperiment carried out in triplicate
Tdble 1, TNF-induced rc,leuse of aruchidonic acid from different cell types The prelabelling and incubation procedures were as described in Fig. 1. The released [3H]d,Ach is expressed as a percentage of the total previously incorporated in the cells. Each value is the mean (f SD) of three experimental values after a 5-h treatment. The significance (P) of difference between values obtained without and with T N F (5000 Ujml) was calculated according to the student's t-test (ns indicates not significant)
Cell line Susceptibility t o T N F [3H]A,Ach release cytotoxicity _ _
-TNF + T N F P
% total ~~ ~~
LY2Y + + 9.3 0.3 11.5 +_ 0.4 < 0.01 LY2V" + + 2.9 & 0.1 4.3 f 0.1 < 0.001 L929(r)2" - 3.2 k 0.3 3.3 & 0.4 ns MEF - 4.2 f 0.4 4.2 0.3 ns WEHI 164 clone 13 + + + 2.1 fO.2 16.6 f 0.4 < 0.001 MCF-7(AZ) + + 5.1 f 0.3 1.2 f 0.2 < 0.01 HcLa DVt''AH2 + + 1.7 0.3 2.9 k 0.6 < 0.05 FS4 - 13.1 F 0.7 13.1 f 1.0 ns
* Culturcs tcsted in conditioned medium instead of fresh medium.
(Fig. 2), and led in some experiments to liberation of up to 10% of the total incorporated label at 5 h after TNF addition. At that time, no cell lysis was yet detectable.
treatment. The untransformed FS4 cells, on which TNF has a mitogenic effect under certain conditions [37, 381, did not release A4Ach upon TNF treatment.
The nature of the radioactivity released w o n TNF treat-
TNF-induced rcleusr oj'rxracliidonic acid is not restricted to LY2Y cclls
The effect of TNF on A4Ach metabolism was also tested in several other murine and human cell lines (Table 1). TNF- resistant L929(r)2 and MEF did not release increased amounts of A4Ach upon treatment with TNF for up to 24 h. This contrasts with the considerable TNF-induced A,Ach release by WEHI 364 clone 13 cells, which are very sensitive to TNF. MCF-7(AZ) and HeLa D98jAH2 are human cell lines killed by TNF treatment and both released more A4Ach upon TNF
ment of [3H]d4Ach-labelled cells was further analysed by TLC. The results showed that most of the radioactivity re- leased was associated with A,Ach itself, although about 20% of the 3H in the supernatant corresponded to a single, lower band (data not shown). Analysis of the complete incubation mixture (cells plus supernatant) also revealed the same two bands, different from phospholipid and neutral lipid. TNF increased only the amount of free d4Ach; the amount of the slower-moving 'side-product' did not change (Fig. 3). A4Ach was released from the phospholipid fraction; the amount of radioactivity associated with neutral lipid was not affected by

469
TNF. Analysis by TLC of the commercial A4Ach preparation before use in metabolic experiments also revealed a small amount of a contaminating product, presumably identical to the aforementioned 'side-product'.
NL-( 8.9 f. 0.1)
AA-( 5.4 f. 0.2)
SP-( 2.8 f. 0.3)
PL-(80.0 f. 0.3)
( 8.9 f. 0.2)
( 9.2 2 0.5)
( 2.6 f. 0.1 )
(76.2 -t 0.8 )
0-
- + Fig. 3. Analysis of [ Hlarachidonic-acid-derived radioactivity. L929 cells prelabclled with [3H]d4Ach were incubated for 5 b in the presence (+)or absence (-) of 5000 U TNF/ml. Extraction ofcells plus medium and separation procedures are described in Materials and Methods. 0, origin; AA, arachidonic acid (idcntified on the basis of a corre- sponding marker); SP, unknown side-product; PL, phospholipids; NL, neutral lipids. The percentage of the total radioactivity per lane associated with each spot is given between brackets. Each value rep- resents the mean (* SD) of a single experiment carried out in duplicate
TNF induces the release of several polyunsaturuted,fatty acids both before and during cell killing
L929 cells, prelabelled with radioactive fatty acid for 24 h, were treated with TNF, after which the liberation of fatty acid into the medium was monitored. The induction of fatty acid release was not limited to A,Ach, as we also observed an increased release of another polyunsaturated fatty acid, viz. A,Ach. Both the kinetics and the amount of the TNF-induced release were comparable to that of A,Ach (Fig. 4A and B). Furthermore, it is clear from the data in Fig. 4 (C and D) that, before the occurrence of any cell killing which starts at 6 - 8 h after TNF addition (Fig. l), TNF was unable to increase specifically the amount of free Pam or Lin relative to the controls. After longer TNF treatment periods, cell death oc- curred, and both Pam and Lin were released above control levels. After 15 h of TNF treatment, TNF-induced A4Ach release was 6-fold above control levels, Lin release 5.5-fold and Pam release 3.1-fold. To exclude the possibility that fatty acid release was solely due to a non-specific release of free fatty acid during cell lysis at all times, we separated fatty acids from phospholipids in the complete incubation mixture (cells plus supernatant) of prelabelled cells by use of TLC. After TNF treatments for up to 6 h, A,Ach was released, but not Pam or Lin; longer incubation, however, resulted in an in- crease of the other fatty acids as well (data not shown).
Induction of fa t ty acid release is not restricted to TNF
Although TNF and IL1 share many activities on a variety of cells [39], IL1 is not toxic for L929 cells (results not shown), nor could it stimulate A,Ach release from these cells (Fig. 5). On the other hand, NCS, which is not toxic for L929 cells, did induce A,Ach release, to a much higher level than even TNF did (Figs 4A and 5). Human serum was also active and its effect was even more pronounced than that of NCS. We did some preliminary experiments to find out what component in the serum might be responsible for this stimulation, but neither inurine epidermal growth factor (10 ngiml), nor hu-
I c 1
I
" 0 1 2 3 4 5 6 1 2 3 1 5 6 Time after addition of stimulant (h)
Fig. 4. Comparison of TNF-induced and serum-induced fatty acid release from L929 cells. 10 cmz of cells were prelabelled with 18.5 kBq [3H]d4Ach (A), ['4C]d3Ach (B), ['4C]Pam (C) or [14C]Lin (D) and further treated as described in Materials and Methods. The values in the absence (0) or presence of 5000 U TNF/ml ( 0 ) or 10% NCS ( A ) represent the amount of radioactivity released in the supernatant, expressed as a percentage of the total label taken up by the cells during the prelabelling period. Each value represents the mean (f SD) of a single experiment carried out in triplicate
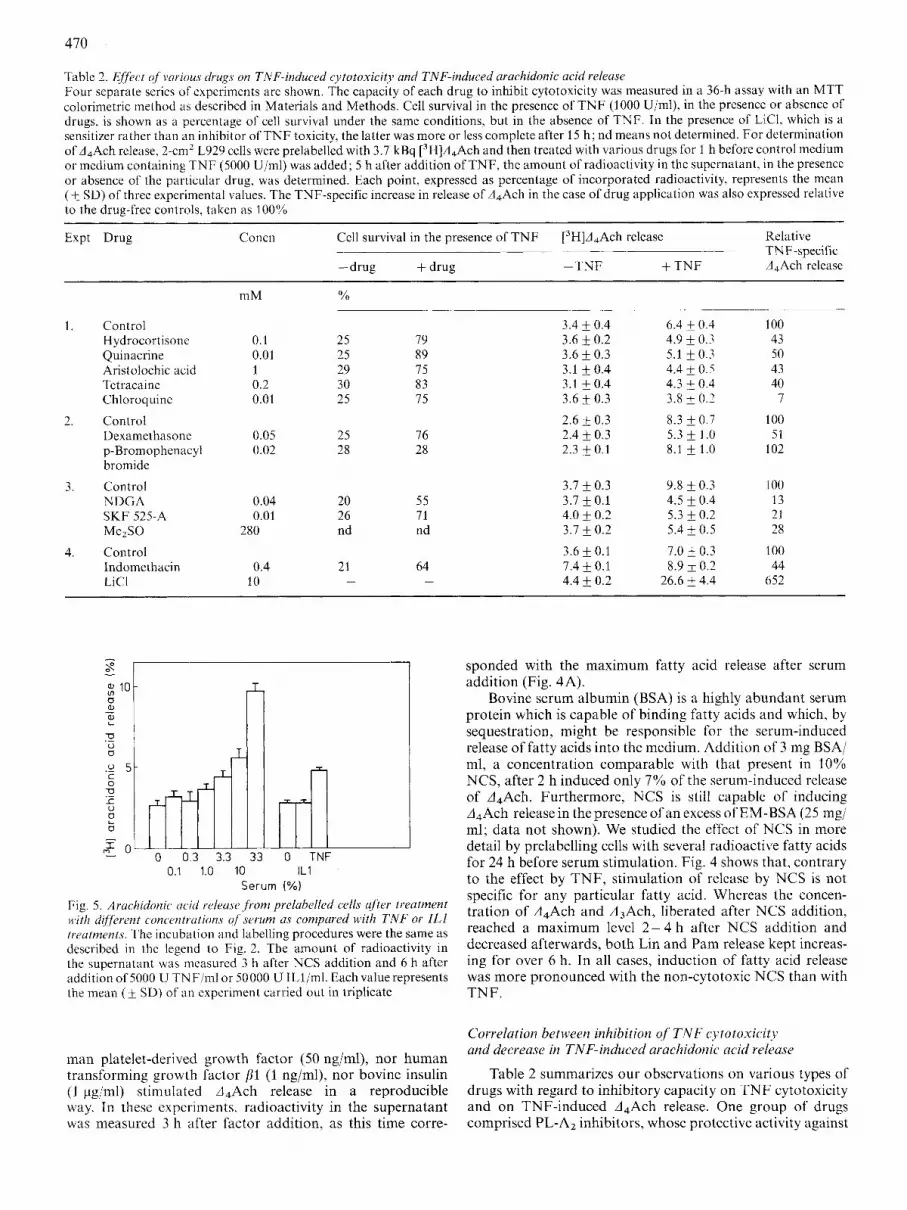
Table 2. Elfect of wrious drugs on TNF-induced cytotoxicity and TNF-induced arachidonic acid releusr Four separate series of experiments are shown. The capacity of each drug to inhibit cytotoxicity was measured in a 36-h assay with an MTT colorimetric method as described in Materials and Methods. Cell survival in the presence of T N F (3000 Ujml), in thc presence or absence of drugs, is shown as a percentage of cell survival under the same conditions, but in the absence of TNF. In the presence of LICI, which is a sensiti~er rather than an inhibitor of T N F toxicity, the latter was more or less complete after 15 h ; nd means not determined. For determination of d,Ach release, 2-cmZ L929 cells were prelabelled with 3.7 kBq [3H]A,Ach and then treated with various drugs for 1 h before control medium or medium containing T N F (5000 Ujml) was added; 5 h after addition of TNF, the amount of radioactivity in the supernatant, in the presence or absence of the particular drug, was determined. Each point, expressed as percentage of incorporated radioactivity, represents the mean ( jz SD) of three experimental values. The TNF-specific increase in release of A,Ach in the case of drug application was also expressed relative to the drug-free controls, taken as 100%
Expt Drug Relative TNF-specific A,Ach release
Concn Cell survival in the presence of T N F [3H]d4Ach release ___ ~ ~ _ _
-drug + drug -TNF + T N F
mM
3.4 + 0.4 3.6 & 0.2 3.6 f 0.3 3.1 & 0.4 3.1 +0.4 3.6 0.3
2.6 & 0.3 2.4 f 0.3 2.3 & 0.1
6.4 & 0.4 4.9 f 0.7 5.1 f 0.3 4.4 f 0.5 4.3 & 0.4 3.8 + 0.2
8.3 f 0.7 5.3 f 1 .0 8.1 f 1.0
100 43 50 43 40
7
100 51
L 02
1. Control H ydrocortisonc Quinacrine Aristolochic acid Tetracaine Chloroquinc
Dexamethasone p-Bromophenacyl bromide
3. Control NDGA
MezSO
4. Control Indomethacin LiCl
2. Control
SKF 525-A
0.1 0.01 1 0.2 0.01
25 25 29 30 25
79 89 75 83 75
0.05 0.02
25 28
76 28
3.7 f 0.3 3.7 * 0.1 4.0 F 0.2 3.1 f 0.2
3.6 f 0.1 7.4 f 0.1 4.4 & 0.2
9.8 f 0.3 4.5 f 0.4 5.3 * 0.2 5.4 f 0.5
7.0 & 0.3 8.9 * 0.2
26.6 + 4.4
I00 13 22 28
I00 44
652
0.04 0.01
280
20 26 nd
55 71 nd
64 -
0.4 10
21 -
sponded with the maximum fatty acid release after serum addition (Fig. 4A).
Bovine serum albumin (BSA) is a highly abundant serum protein which is capable of binding fatty acids and which, by sequestration, might be responsible for the serum-induced release of fatty acids into the medium. Addition of 3 mg BSA/ ml, a concentration comparable with that present in 10% NCS, after 2 h induced only 7% of the serum-induced release of A,Ach. Furthermore, NCS is still capable of inducing A,Ach release in the presence of an excess of EM-BSA (25 mgi ml; data not shown). We studied the effect of NCS in more detail by prelabelling cells with several radioactive fatty acids for 24 h before serum stimulation. Fig. 4 shows that, contrary to the effect by TNF, stimulation of release by NCS is not specific for any particular fatty acid. Whereas the concen- tration of A,Ach and A,Ach, liberated after NCS addition, reached a maximum level 2-4 h after NCS addition and decreased afterwards, both Lin and Pam release kept increas- ing for over 6 h. In all cases, induction of fatty acid release was more pronounced with the non-cytotoxic NCS than with TNF.
0.1 1.0 10 IL1 Serum (%)
Fig. 5. Arucliidonic w i d rrlruse ,from prelahelled cells after treutrnent ii>ith different concentrutions of serum as compared with TNF or ILI freutmmts. The incubation and labelling procedures were the same as described in the legend to Fig. 2. The amount of radioactivity in the supernatant was measured 3 h after NCS addition and 6 h after addition of 5000 U T N F/ml or 50000 U ILl/ml. Each value represents the mean (F SD) of an experiment carried out in triplicate
Correlation between inhibition of TNF cytotoxicity and decrease in TNF-induced arachidonic acid release
Table 2 summarizes our observations on various types of drugs with regard to inhibitory capacity on TNF cytotoxicity and on TNF-induced A,Ach release. One group of drugs comprised PL-A2 inhibitors, whose protective activity against
man platelet-derived growth factor (50 ngiml), nor human transforming growth factor fll (1 ngiml), nor bovine insulin (1 pglml) stimulated A,Ach release in a reproducible way. In these cxpcriments. radioactivity in the supernatant was measured 3 h after factor addition, as this time corre-

471
TNF cytotoxicity has been described [lo, 18, 26, 27, 40, 411. Hydrocortisone and dexamethasone are both corticoids which supposedly inhibit PLs by induction of synthesis or release of a cellular inhibitor of the enzyme, although other mechanisms of inhibition of PL-A2 have been described [42]. Quinacrine [43] and the alkaloid aristolochic acid [44] both inhibit PL-A2 directly, whereas the local anaesthetic tetracaine [45] and the anti-malarial agent chloroquine (461 are also supposed to de- crease PL-A2 activity. We found that they all counteracted both TNF-mediated cytotoxicity and TNF-induced A4Ach release. in contrast, p-bromophenacyl bromide, an irreversible inhibitor of some, but not of all types of PL-A2 [47], neither protected cells against the toxicity of TNF (see also [26]) nor influenced TNF-specific A4Ach release.
A second group of inhibitors comprised drugs we orig- inally used because they have been described to affect activities different from PL activity. NDGA inhibits lipoxygenases, whereas SKF 525-A blocks cytochrome P450 activity. Both drugs inhibited TNF-induced cytotoxicity, albeit the underly- ing mechanism of this activity is unclear; both drugs also reduced the TNF-induced A4Ach release. Similar experiments also showed that Me2S0, a scavenger of oxygen radicals which protects against TNF cytotoxicity [26], strongly affected A,Ach release. Indomethacin is an inhibitor of cyclo- oxygenase, but high concentrations may also reduce PL ac- tivity [48] and do protect against TNF cytotoxicity [26]. In the present experiments, indomethdcin at high concentration appeared to inhibit partly the TNF-induced release of fatty acids, although it also increased the background level of re- leased fatty acid. Finally, LiC1, which enhances TNF cyto- toxicity considerably [49], also strongly increased the TNF- induced A4Ach release.
interestingly, inhibitors of serine proteases, known to pro- tect cells from TNF cytotoxicity [35], also reduced the A4Ach release. Fig. 6A shows that the reversible inhibitors TosArgOMe and AcPheONap, as well as the irreversible in- hibitor TosLysCH2Cl almost completely blocked TNF-in- duced release of A4Ach.
Furthermore, we tested drugs that influence extracellular calcium levels and entry into the cells. EGTA chelates extra- cellular calcium, whereas verapamil blocks the voltage-depen- dent calcium channel, and both are described to decrease PL- A2 activity [50]. EGTA and verapamil were found to inhibit TNF-induced cytotoxicity (67% and 68% cell survival, respec- tively, under the conditions described in Table 2, whereas control survival with TNF in the absence of drug was 25%) as well as A4Ach release (Fig. 6B).
Exogenous addition of polyunsaturated f a t t y acids does not counteract protection by PL inhibitors against TNF cytotoxicity
Neither A4Ach nor A,Ach were able to restore TNF cytotoxicity in cells treated with PL inhibitors, in either a 72- h or a 36-h assay (Fig. 7). In the absence of these inhibitors, the fatty acids had no effect at all on cell survival or on TNF cytotoxicity. Comparable experiments with 200 pM methyl arachidonate, which may penetrate the cell membranes better, gave similar results (data not shown).
TNF does not induce a Ptdlns-specfic PL-C
We measured the activity of this PL-C in two different ways. First, L929 cells prelabelled for 24 h with myo- [3H]inositol, were treated with TNF for increasing time periods, after which the radioactivity in the supernatant was
A
B
L
0 u 0 u .- g 5
; -0 f u .-
I n
0
Fig. 6. Protease inhibitors and culcium levels influence TNF-mediated avachidonic acid release. The experimental conditions were the same as described in the legend to Table 2. The values represent the percentage amount of radioactivity released in the supernatant in the absence (open columns) or presence (dashed columns) of 5000 U TNF/ml. Each value, expressed as a percentage of incorporated radioactivity, represents the mean ( 5 SD) of an experiment carried out in triplicate, as obtained in the absence or presence of either protease inhibitors (A) or drugs affecting calcium uptake (B)
determined. Released radioactivity gradually increased with time, but there was no difference between control and TNF- treated cultures (Fig. 8A). As this method assays PL activity simply by measuring the amount of free Ins [51], we had to consider the possibility that the cellular InsP(s) formed after cleavage of PtdInsP(s) was not, or only partially, converted to free Ins and/or that the Ins was reincorporated into membrane phospholipids. To this end, we measured free Ins together with InsP(s) in the presence of 10 mM LiCl, the latter being used to prevent dephosphorylation. An increase in acid-sol- uble radioactivity, as a function of time, was not observed in the control nor in the TNF-treated cultures (Fig. 8 B).

T I
Fig. 7. ~ X O p n O i 4 , \ . l j ~ tickkd pol~i4nsatiirated,fatty acids do not restore smsitivity to TNF c!;toio.uicity itz wlls treated with PL inhibitors. L929 cells were seeded in microliter plates and incubatcd with TNF, PL inhibitors and polyunsaturated fatty acids ill the presence of medium supplemented with 5 mg:ml EM-BSA, as described in Materials and Methods. Data are shown for cell survival in the presence of quina- crine. chloroquine or hydrocortisone in a 36-h assay with 1000 U TNF/ml (A and C) or in a 72-11 y with 10 U TNF/ml (B and D). Each value is the m a n (k SD) of three values obtained either in the absence of additives (open columns) or in the presence of PL inhibitor (shaded columns) or in thc prcscncc of PL inhibitor plus either 100 pM A,Ach (A atid B) or 100 pM A,Ach (C and D) (filled columns). Thc survival values are expressed as a percentage of a control cell culturc, incubated undcr idcritical conditions, but in the absence of T N F
TNF does not off2Jc.t winmrporation of'pol~vzmsutcircitc~1,f~ittl. u i d s into phospholipids
Fatty acids are incorporated into phospholipids by acyltransferases. We determined the effect of T N F on this enzymatic activity as described in Materials and Methods. After 1 h, 39% of the ['H]d,Ach and 42% of the ['4C]A3Ach was taken up bq the cells. We could not, however. observe a difference in incorporation rate of fatty acids into phos- pholipids between TNF-treated and untreated cultures up to 6 h after T N F stimulation (data not shown).
DISCUSSION
Only cells sensitive to T N F cytotoxicity showed TNF- enhanced arachidonic acid (A,Ach) release in our experiments (Table l), which was both a time-dependent and TNF-concen- tration-dependcnt process (Figs I and 2). The absence of TNF-induced A4Ach release in the resistant L929(r)s cells is not due to the absence of T N F receptors (our unpublished results). Furthermore. growth stimulation of FS4 cells by T N F has been described (37, 3x1. Under our assay conditions for
, I 1 2 3 4 5
time after addition of TNF ( h )
Fig. 8. TNFdoes not activate u PtdIns- specific^ PL-C. I 0 cm' of L929 cells were labelled with [3H]Ins and further treated as described in Materials and Methods. The cells were either unstimulated ( C ) or stimulated with 5000 U TNF/ml (0) . The data show Ins release in the medium in the absence of LiCl (A) or amounts 01' frcc Ins plus InsP(s) in the cell extracts in the presence of LiCl (B)
fatty acid release, neither the proliferation rate of the FS4 cells nor the release of d4Ach increased after treatment with TNF. However, an increase in fatty acid release from cells susceptible to growth stimulation by T N F [18, 521, and even its necessity for this stimulation [53], have been reported. On the other hand, T N F growth-enhancing activity is stimulated by cor- ticoids [38, 411, which are known to inhibit A4Ach release at least in some cells. Hence, the role of A4Ach in the growth stimulatory activity of T N F is still uncertain and contro- versial.
Analysis of the labelled material from L929 cells, pre- labelled with A4Ach and treated with TNF, revealed that, although a second radioactive band different from neutral lipids was observed, only the amount of free A4Ach was specifically enhanced by T N F treatment. Together with our earlier data [26], this shows that metabolization of A4Ach in L929 cells, if any, is in no way related to T N F cytotoxicity. The non-involvement of A4Ach metabolites in T N F cyto- toxicity has also been reported previously [lo, 18, 531.
We further studied the effect of T N F addition to cells prelabelled with fatty acids other than A,Ach for two reasons: first, to obtain an internal control for TNF-induced A4Ach release; second, to obtain an idea about the specificity of the fatty acid release system. Early after T N F addition, and before the start of cell lysis, A4Ach and A,Ach release was specific for T N F action (Figs 1 and 4). Later on, and more particularly subsequent to cell death, this specificity was lost. We have no experimental data to explain the inolccular basis of the latter phenomenon, but it has been shown that TNF, albeit at very high concentrations, increases the permeability of liposomes [54], and that TNF-induced loss of cell viability is ac- companied by lysis of the plasma membrane [55]. Further- more, it is known that PL activity is affected by structural perturbations of the cell membrane [56]. In any case, the

413
early release of A,Ach and A3Ach precedes, and possibly even induces, the structural alterations of the membrane, leading to subsequent non-specific release of fatty acids. Hence we focused on the involvement of A4Ach release in the mechanism of TNF cytotoxicity at times preceding these non-specific events.
The way TNF induces A,Ach release strongly resembles the formation of A4Ach (Fig. 4). Both highly unsaturated fat- ty acids are mainly incorporated in the 2-position of phospholipids. The differential subcellular localization of phospholipids and PLs, or the activation of a PL with a defined specificity [57], could explain both the relative selec- tivity of early TNF-induced fatty acid release and the limited extent of the specific early TNF response observed. We also have to keep in mind that mammalian cells can enzymatically convert A,Ach to A,Ach. As we could not separate these two fatty acids with the chromatographic system used, we cannot rigorously exclude that the increase in amount of radioactivity in the supernatant of the cells prelabelled with [14C]A3Ach was due to A,Ach formed by enzymatic dehydrogenation.
As the release of polyunsaturated fatty acids seems to be coupled to sensitivity to TNF cytotoxicity, various inhibitors of TNF cytotoxicity were tested in this respect. As several inhibitors may exert multiple or poorly defined effects, the data obtained should be interpreted with care. Nonetheless, we indeed found a close positive correlation between anti- cytotoxic effect of the drugs applied and their ability to prevent TNF-induced A,Ach release (Table 2). Somewhat surprising was the capacity of some drugs, which are normally not used in studies on PLs, to inhibit TNF-induced A,Ach release. For example, protection by Me,SO, NDGA or SKF 525A was possibly mediated, directly or indirectly, by inhibition of the TNF-induced PL activation. Conversely, p-bromophenacyl bromide influenced neither TNF-mediated cytotoxicity nor TNF-induced A4Ach release. This drug has been reported to inhibit a secreted macrophage PL-A2 species, but not a membrane-associated one [43]. Our result with indomethacin, which at high concentration affects enzymes other than cyclooxygenase, was even more surprising: this drug enhanced the background [3H]A4Ach release, which might possibly be explained by an effect on a PL (or even an acyltransferase), different from the one that becomes activated by TNF, while the latter enzyme might still be inhibited. The monokine IL1 has been reported to share with TNF the ability to stimulate PL activity in some cell systems, i.e. chondrocytes and synovial fibroblasts [15 - 17, 201. 1L1 treatment was not toxic to the L929 cells (data not shown) and did not increase fatty acid release (Fig. 5).
The data presented here suggest that cytotoxicity in L929 may be associated with or even due to release of fatty acids. However, addition of serum to L929 cells, without being cytotoxic, induced fatty acid release to an extent far exceeding the one induced by TNF (Figs 4 and 5). We do not know what component or combination of factors in the serum is responsible for this stimulation. Some growth factors are known to induce A4Ach release in various cell types [58 - 601. But epidermal and platelet-derived growth factors, trans- forming growth factor Pl and insulin all failed to induce fatty acid release in our system. Mitogenic responses to growth factors are, however, complex processes which sometimes need cooperation of different factors [61]. It is striking that, in contrast to TNF treatment, there was less or virtually no specificity regarding the type of fatty acid released after stimu- lation of L929 cells with serum. Hence, both TNF and serum activate (a) PL(s), but the phenomena associated with the
former reaction are cytotoxic, whereas those associated with the latter are not, and the specificities of the enzymes activated are clearly different.
The fact that both cytotoxic and non-toxic treatments induce the release of A4Ach in the same cell line has important implications regarding the importance of fatty acid release for TNF cytotoxicity. Based on the experiments so far described, one might conclude that activation of at least some PL-A2 species is indeed required for TNF action, but also that the released A4Ach is not per se sufficient to induce growth inhi- bition and/or cell killing. Instead, one should give due con- sideration to the possible importance of the nature of the PL substrate, and possibly even the cellular localization of the fatty acid released, as these can determine the effect on the cell metabolism [57, 621.
A necessity for A4Ach itself for TNF cytotoxicity is also unlikely by the observation that exogenously added fatty acid is unable to restore sensitivity to TNF toxicity of cells treated with inhibitors of PL (Fig. 7). However, this failure might be due to different effects on cells by exogenously added vs endogenously released fatty acid [63]. At this point, we may mention the recent observation that exogenously added A,Ach inhibits the TNF-induced growth stimulation of hu- man fibroblasts [52]. At higher concentrations, its combi- nation with TNF even becomes cytotoxic. This seemingly contrasts with our results; it documents, however, the com- plexity of cytotoxicity induced by TNF. Furthermore, data suggesting the existence of different mechanisms of cytotoxicity in different cell types have been reported [64].
Furthermore, we attempted to identify the nature of the PL involved in the TNF action. It seems likely, based on our experiments with inhibitors of A4Ach release, that this effect is due to a PL-A,-type activity, although the inhibition of another type of PL by the drugs used cannot be rigorously excluded. Both A,Ach and A3Ach are mainly incorporated in the 2-position of phospholipids. The amount of free fatty acid is generally regulated either by increased liberation from phospholipids by (a) PL-A2; or by production of diacylglycerol by (a) PL-C, followed by further cleavage by diacylglycerol lipase and monoacylglycerol lipase; or by de- crease in fatty acid (re)incorporation by an acyltransferase [57]. We could exclude both an increase in the activity of PtdIns-specific PL-C (Fig. 8) [26] and a decrease in acyltransferase activity (data not shown). So far, we cannot exclude the involvement of other less characterized PLs. One example is increased diacylglycerol production by a PL-C, non-selective for PtdIns in 1L1-treated human T cells [65]. However, TNF was reportedly unable to activate this particu- lar PL [65]. PL-A2 activities are heterogeneous in several as- pects: (a) occurrence in various tissues and cell types; (b) subcellular localization; (c) activation route; (d) pH optimum; (e) substrate specificity; and (0 calcium requirements [56, 57, 66, 671. The results on the involvement of calcium on TNF cytotoxicity are complex [68], but our preliminary data (Fig. 6B) seem to indicate that both TNF cytotoxicity and PL activity are correlated with the availability of calcium.
We have suggested previously a connection between two important steps of the TNF-induced cytotoxicity cascade, namely a protease activity and a PL activity [35]. This working model of the mode of action is now further supported by our observation that TNF-induced A,Ach release is strongly decreased in the presence of (protecting) protease inhibitors (Fig. 6A). The requirement of a protease for the direct or indirect modulation of PL-A2 or one of its regulatory proteins is the simplest explanation for these observations. Further-

474
more, protease activity seems to be required also for TNF- induced synthesis of platelet-activating factor in endothelial cells [69]. Whatever pathway is used to activate PL activity, it is also likely that TNF action is linked to a G-protein, as pertussis toxin has been shown to inhibit TNF cytotoxicity [lo- 12,261. Moreover, LiCl which, among other effects, may positively influence at least some G-proteins [70], strongly enhanced cytotoxicity by TNF on various transformed cells, both in vitro and in vivo [49], and in fact also enhanced TNF- induced d,Ach release (Table 2).
Hence we may conclude that activation of a PL, most probably a particular PL-A2 species, is required for TNF cytotoxicity. But the product of the enzymatic action, i.e. the A4Ach released, does not seem to play a key role in causing the cytotoxicity, a t least not in L929 cells. The other reaction product, lysophosphatidyl ester, does not accumulate and also is unlikely to be an essential mediator (our unpublished re- sults). Other explanations remain: for example, a peculiar PL may specifically degrade a particular and essential phospholipid-type of substrate. Evidently, this problem re- quires further study and attempts are in progress to charac- terize in more detail the PL-A2 activated by TNF.
We thank Dr A. Shaw for gifts of ILI, Dr C.-H. Heldin for his gift of plaielei-derived growth factor, M. Woutcrs for the preparation of MEFs, F. Boeykens for assistance with the chromatography analy- ses, F. Van Houtte for technical assistance and W. Drijvers for artistic contribution. P.S., R . B . and D.D. thank the Znstituut voor WetenschuppeLijk Ondcwork in N<jvdieid en Lundbouw for a fellow- ship. F.V.R. is a Senior Research Associate. and B.V. a Research Assistant with the Nutionad Fonds voor Wetenschappelljk Onderzoek. The research project was supported by grants from the Fonds voor Geneeskundig Wc~ten.c-chuppel<jk Onderzoek, the Nationaul Sti- muleringsprugrumiria ~001' F~m~hnen tee l Onderzoek inzuke Bio- \vc~tensc~happen, ihe Algcwierie Spaur- en Lijfrentekas and the Sportvereniging regen Kunker.
REFERENCES 1 .
2.
3.
4
5
6 7
8.
9.
10.
11 .
12.
13. 14.
Carswell, E. A,. Old, L. J., Kassel, R. L., Green, S., Fiore, N. & Williamson, B. (1975) Proc. Nut1 Acud. Sci. U S A 72, 3666- 3670.
Helson, L., Grcen, S.. Carswell, E. & Old. L. J. (1975) Nuture
Fiers, W., Brouckaert, P., Devos, R., Fransen, L., Leroux-Roels, G., Remaut, E., Suffys, P., Tavernier, J., Van der Heyden, J. & Van Roy, F. (1986) Cold Spring Harbor Symp. Quunt. B id . 51,
Bonavida. B. & Granger, G. (eds) (1990) Tumor necrosis.fuctor: structure, niechuni.sm of crction, role in disease and therapy, Karger, Basel.
Baglioni, C., McCandless, S., Tavernier, J. & Fiers, W. (1985) J . Bid. Cl1en.1. 260, 13 395 - 3 3 397.
Kull, F. C. & Cuatrecasas. P. (2981) Cancer Rrs. 41,4885-4890. Mossclmans, R., Hepburn, A., Dumont, J. E., Fiers, W. &
Tsujimoto. M.. Yip, Y. K. & VilEek, J. (1985) Proc. Nut1 Acud.
Ohsawa, F. & Natori. S. (1988) J . Biochem. (Tolcyo) 103, 730-
Hepburn, A,, Boeynaems. J . M., Fiers, W. & Dumont, J. E. (1987)
Imamura, K.. Shcrman, M. L., Spriggs, D. & Kufe, D. (1988) J .
Beyaert. R., Suffys. P., Van Roy, F. & Fiers, W. (1988) Arch. Inr.
Chun, M. & Hofl'niann. M. K. (1987) Lymph. Res. 6, 161 - 167. Neer, 15. J. & Clapham, D. E. (1988) Nutuw 333, 129 - 134.
258,731-732.
587-595.
Galand, P. (1988) J . Immuno(. 141, 3096-3100.
Sci. (JSA 82. 7626 - 7630.
734.
Biochem. Bioplij..r. Res. Commun. 149, 81 5 - 822.
Riol. C'liem. 263. 10247- 10253.
PIi~:riol. Hiocliini. 96. B 136.
15. Suffys, P., Van Roy, F. & Ficrs, W. (1988) FEBS Lett. 232, 24-
16. Godfrey, R. W., Johnson, W. J., Newman. T. & Hoffstein, S. T.
17. Godfrey. R. W.. Johnson. W. J. & Hoffsiein, S. T. (1987) Biocheni.
18. Neale, M. L., Fiera, R. A. & Matthews, N. (1988) Immunology
19. Dayer, J. M., Beutler, B. & Cerami, A. (1985) J . Exp. Med. 162,
20. Bachwich, P. R., Chensuc, S. W., Larrick, J. W. & Kunkel, S. L. (1986) Biochem. Biop1z.y.~. Res. Comnzun. 136, 94- 101.
21. Kawakami, M., Ishibashi, S., Ogawa, H., Murase, T., Takaku, F. & Shibata, S. (1986) Biochem. Biophys. Res. Coinniun. 141,
22. Roubin, R., Elsas, P. P., Fiers, W. & Desscin, A. J. (1987) Clin.
23. Huber, M., Beutler, B. & Keppler, D. (1988) Eur. J . Zmmunol. 18,
24. Begin, M. E., Ells, G. & Das, U. N. (1985) Antjcuncer Res. 6,
25. Flohk, L., Beckmann, R., Giertz, H. & Loschen, G. (1985) in Oxidative stress (Sies, H., ed.) pp. 403-435, Academic Press, New York.
26. Suffys, P., Beyaert, R., Van Roy, F. & Fiers. W. (1987) Biochem. Biophys. Res. Comniun. 149, 735 - 743.
27. Matthews, N., Neale, M. L., Jackson, S. K. & Stark, 3 . M. (1987) Immunology 62, 153 - 155.
28. Wong, G. H. W. & Goeddel, D. V. (1988) Scicvce 242,941 -944. 29. Wong. G. H. W., Elwell, J. H., Oberley. L. W. & Goeddel, D. V.
30. Suffys, P., Beyaert, R., Van Roy, F. & Fiers, W. (1989) Anticuncer Res. 9, 167 - 172.
31. Tavernier, J., Fransen, L., Marmenout, A,, Van der Heyden, J., Muller, R., Ruysschaert, M. R.. Van Vlict, A,, Rauden, R. & Fiers, W. (1987) in Molecular Cloning und Anu1vsi.r o j Lymphokines (Webb, D. R. & Goeddel, D. V., eds) pp. 181 - 198, Academic Press, Orlando FL.
32. Ostrove, J. & Gifford, G. (1979) Ploc. Soc. E x p . Biol. Med. 160,
33. Erard, F., Corthesy, P., Smith, K. A., Fiers, W., Conzelmann,
34. Bonncr, W. M. & Stedman, J . D. (1978) Anal. Biochem. 89, 247 - 256.
35. Suffys, P., Beyaert, R., Van Roy, F. & Fiers, W. (1988) Eur. J . Biochem. 178, 257-265.
36. Tada, H., Shiho, O., Kuroshima, K., Koyama, M. & Tsukamoto, K . (1986) J . Immunol. Method~y 93, 157-165.
37. Fiers, W., Brouckaert, P., Guisez, Y.. Remaut, E., Van Roy, F., Devos, R., Fransen, I.., Leroux-Rods. G., Marmenout, A., Tavernier, J. & Van dcr Heyden. J. (1986) in The biology of the interferon system 1985 (Schellekens, H. & Stewart 11, W. E., eds) pp. 241 - 248, Elsevier Science Publishers. Amsterdam, New York, Oxford.
38. Kohase, M., Henriksen-DeStefano, D., Sehgal, P. B. & Viltek. J. (1987) J . Cell. Physiol. 132, 271 -278.
39. Fiers, W., Brouckaert, P., Devos, R., Fransen. L., Haegeman, G., Leroux-Roels, G., Marmenout, A,, Remaut, E., Suffys, P.. Tavernier, J., Van der Heyden, J. &Van Roy, F. (1987) in The biology of the interferon system I986 (Cantell, K. & Schellekens, H., eds)pp. 205-216, Martinus NijhoffPublishers, Dordrecht, Boston, Lancaster.
40. Kull, F. C. (1988) Biochem. Biopliys. Rcjs. Commuiz. 1.53, 402- 409.
41. Tsujimoto, M. & Adachi, H. (1988) J . Biochem. (Tokyo) 103. 393 - 395.
42. Hirata, F., Schiffmann, E., Venkatasubramanian, K.. Salomon. D. & Axelrod, J. (1980) Proc. Nut1 Acud. Sci. U S A 77, 2533- 2536.
43. Lapetina, E. G., Billah, M. M. & Cuairecasas, P. (1981) J . Biol. Chem. 256, 5037 - 5040.
28.
(1988) Prostaglandins 35, 107-134.
Biophys. Res. Commun. 142, 235 - 241.
64, 81 -85.
2163 -2168.
482-487.
Exp. Immunol. 70,484-490.
2085 -2088.
291 -296.
(1989) Cell.58, 923-931.
354-358.
A. & Nabholz, M. (1984) J . EX^. Med. 160, 584-599.

475
44. Vishwanath, B. S., Appu-Rao, A. G. & Gowda, T. V . (1987)
45. Kunze, H., Nahas, N., Traynor, J . R. & Wurl, M. (1976) Biochim.
46. Vadas, P., Stefanski, E. & Pruzanski, W. (1986) Agents Actians
47. Lister, M. D., Glaser, K . B., Ulevitch, R. J. & Dennis, E. A.
48. Kaplan, L., Weiss, J. & Elsbach, P. (1978) Proc. Nut1 Acud. Sci.
49. Beyaert, R., Vanhaesebroeck, B., Suffys, P., Van Roy, F. & Fiers,
50. Satoh, H., Suzuki, J. & Satoh, S. (1985) Biochem. Biophys. Kes.
51. Murayama, T. & Ui, M. (1985) J . Biol. Chem. 260,7226-7233. 52. Hori, T., Kashiyama, S., Hayakawa, M., Shibamoto, S.,
Tsujimoto, M., Oku, N. & Ito, F. (1989) Exp. Cdl Res. 185,
53. Palombella, V. J. & Viltek, J. (1989) J . Biol. Chem. 264, 18128- 18136.
54. Oku, N., Araki, R., Araki, H., Shibamoto, S., Ito, F., Nishihara, T. & Tsujimoto, M. (1987) J . Biochem. (Tokyo) 1112, 1303- 1310.
55. Scanlon, M., Laster, S. M., Wood, J. G. & Gooding, L. R. (1989) Proc. Natl Acud. Sci. USA 86, 182 - 186.
Toxicon 25,939-946.
Biophys. Acta 441, 93 - 102.
l 9> 194-202.
(1989) J . Biol. Chem. 264, 8520-8528.
USA 75,2955-2958.
W. (1989) Proc. Natl Acad. Sci. USA 86, 9494-9498.
Commun. 126,464-470.
41 -49.
56. Van den Bosch, H. (1980) Biochim. Biophys. Acta 604, 191 -246. 57. Irvinc, R. F. (1982) Biochem. J . 204, 3-16. 58. Jamieson, G. A. & Villereal, M. L. (1 987) Arch. Biochem. Biopkys.
59. Levine, L., Xiao, D., Worth, N. & Lilley, W. E. (1985) Biochem.
60. Margolis, B. L., Bonventre, J. V., Kremer, S. G. & Kudlow, J . E.
61. Rozengurt, E. (1986) Science 234, 161 -166. 62. Capriotti, A. M., Furth, E. E., Arrasmith, M. E. & Laposata, M.
(1988) J . Biol. Chem. 263, 10029-10034. 63. Metz, S. A. (1988) Prostug. Leuk. Ess. Fatty Acids 32, 187-202. 64. Reid, T. R., Torti, F. M. & Ringold, G. M. (1989) J . Biol. Chem.
65. Rosoff, P. M., Savage, N. & Dinarello, C. A. (1988) CeN54,73 -
66. Balsinde, J., Diez, E., Schiiller, A. & Mollinedo, F. (1988) J . B id .
67. Hsueh, W., Desai, U., Gonzalez-Crussi, F., Lamb, R. & Chu, A.
68. Schiitze, S., Scheurich, P., Pfizenmaier, K. & Kronke, M. (1989)
69. Camussi, G., Bussolino, F., Salvidio, G. & Baglioni, C. (1987) J .
70. Volontk, C. (1988) Neurosci. Lett. 87, 127-132.
252,478-486.
Biophys. Res. Commun. 130, 110 - 1 17.
(1988) Biochem. J . 249, 587-592.
264,4583-4589.
81.
Chem. 263, 1929- 1936.
(1981) Nature 290, 710-713.
J . Biol. Chem. 264, 3562-3567.
EX^. Med. 166, 1390-3404.





























