Embed Size (px)
Citation preview

What Determines the Miscibility of Ionic Liquids with Water?Identification of the Underlying Factors toEnable a Straightforward Prediction
Marco Klahn,* Claudia Stuber, Abirami Seduraman, and Ping WuInstitute of High Performance Computing, 1 Fusionopolis Way, #16-16, Connexis,Singapore 138632, Republic of Singapore
ReceiVed: January 4, 2010; ReVised Manuscript ReceiVed: January 22, 2010
Whether an ionic liquid (IL) is water-miscible or immiscible depends on the particular ions that constitute it.We propose an explanation, based on molecular simulations, how ions determine the miscibility of ILs andsuggest a straightforward and computationally inexpensive method to predict the miscibility of arbitrary newILs. The influence of ions on the solvation of water is analyzed by comparing molecular dynamics simulationsof water in 9 different ILs with varying cation and anion constituents. The solvation of water in ILs is foundto depend primarily on the electrostatic water-ion interaction strength, which, in turn, is determined mainlyby two factors: primarily, by the size of the ions and secondarily by the amount of charge on the ion surfacethat is coordinated with water. It is demonstrated that large ions lead to weaker interactions with water, dueto the involved delocalization of the ion charge. A large charge on the ion surface, which is determined bythe chemical structure of the ion, strengthens water-ion interactions. We observe that whenever the interactionstrength of water with ions exceeds a certain threshold, an IL becomes water-miscible. On the basis of thesefindings, a simple equation is derived that estimates the water-ion interaction strength. With this equation itis possible to predict most of the observed water-miscibilities of a sample of 83 ILs correctly. A linear increaseof the water saturation concentration with the estimated water-ion interaction strength is observed in water-immiscible ILs, which can be utilized to predict the water concentration in new ILs.
1. Introduction
Ionic liquids (ILs) are molten salts with melting pointsbelow 100 °C, and in many cases below room temperature.Typical cations are based on an aromatic heterocycliccompound in the center, e.g., imidazole or guanidinium, towhich alkyl chains of varying length are attached. The largesize of the cations, the resulting delocalization of theirpositive charge, and their asymmetric structure impedecrystallization, which decreases the melting point. Anionsare usually compact and inorganic, and are often fluoridesor contain fluoroalkyls. ILs exhibit high viscosity, hamperedself-diffusion, and negligible volatility due to strong interionicinteractions. ILs are also characterized by a high ionconductivity and thermal and chemical stability. Thesecharacteristics, together with the possibility to adjust manyproperties of ILs by an independent variation of cations andanions, have brought massive attention to ILs. As a result, abroad variety of potential applications have been proposed.1–4
The study of the behavior of ILs in the presence of water isof fundamental interest, since contact with ubiquitous water canbe hardly avoided. It has been observed that some ILs are water-miscible, while other ILs are water-immiscible, even thoughtheir chemical structures might differ only slightly. Even in thecase of immiscibility, ILs are generally hygroscopic and capableof solvating substantial amounts of water from air humidity orother accessible sources of water. The amount of water thatwater-immiscible ILs solvate at thermal equilibrium has beenmeasured by several groups.5–13 The presence of water in ILs
modifies the characteristics of ILs in many ways: fundamentalproperties such as density, viscosity, and heat capacity aresubstantially affected.14–17 Furthermore, the solvation propertiesof ILs are profoundly altered, where water can act either as acosolvent to increase the solubility of polar compounds, e.g.,alcohols, or as an antisolvent to prevent the solvation of variousgases and nonpolar compounds.13,14,18–21 Moreover, water affectsthe catalytic abilities of ILs by changing reaction barriers,reaction energies and thus also the selectivity of catalyzedreactions.22 Essentially, given the enormous impact of water onIL properties, the variation of the water concentration in ILsprovides an additional powerful parameter, with which ILs canbe customized.
Also, the solubility of ILs in water is of great importance.ILs are usually considered as a potentially green alternative forvarious conventional molecular liquids. Since the volatility ofILs is negligible, the risk of air-pollution is minimal. However,the solvation of ions in water, especially when water-miscibleILs are used, might pose the risk of water pollution and needsto be addressed. Indeed, it has been found that the solvation ofions from water-immiscible ILs in water is not negligible, eventhough their solubility was found to be 1-4 orders of magnitudesmaller than the solubility of water in ILs.7,9–13,15,23–27 Once theseions are solvated in water, living organisms might be affecteddue to the adsorption of the cationic lipophilic alkyl chains tothe cell membrane.28,29 The toxicity of various ions to aquaticorganisms and human cells has been studied by several groups,where also ion modifications with reduced toxicity weresuggested (see refs 10 and 30 and references therein). Knowl-edge of the mutual solubility of ILs and water is also essentialfor the use of ILs in the treatment of wastewater, which is
* To whom correspondence should be addressed. Phone: (65) 6419 1468.Fax: (65) 6463 2536. E-mail: [email protected] and [email protected].
J. Phys. Chem. B 2010, 114, 2856–28682856
10.1021/jp1000557 2010 American Chemical SocietyPublished on Web 02/10/2010

considered to be an important possible application of ILs. WhenILs with suitable solvation properties are used, metal ions aswell as organic compounds can be extracted from wastewater.6,31
The ions of water-immiscible ILs need to exhibit a minimalsolubility in water to be suitable for this and many otherapplications.
Despite the fundamental importance of the above-mentionedproperties of ILs, the connection between the ion constituentsof the IL and the solubility of water in ILs has not beenunderstood. However, some observations have been made: ithas been pointed out by several authors that the influence ofthe anions on the water solubility is larger than the influence ofthe cations (e.g., ref 8). Interactions of water with ions can beobserved with IR spectroscopic methods, and it was found thatwater interactions with anions were stronger than with cations.32
Therefore, the identity of the anion often determines the water-miscibility of an IL, where some anions, e.g., halides, nitrate,and methyl sulfonate, strongly facilitate miscibility while otheranions, e.g., PF6 and bis(trifluoromethylsulfonyl)imide (Tf2N),usually prevent the mixing with water. Cations still influencethe solubility of water as well, even though to a lesser degree:it has been observed that an elongation of cation alkyl chainsreduce the solubility of water (see, e.g., ref 18). Furthermore, ithas been found that the central cation group plays a role aswell. Pyridinium- and pyrrolidinium-based cations impede thesolubility of water, compared to imidazolium-based cations, andthe solubility was hampered most, when piperidinium-basedcations were used.7 Regarding the solvation of ions in water, ithas been found that their solubility depends mostly on theirhydrophobicity, where large ions tend to be less soluble inwater.33
Molecular simulation techniques have been amply appliedby numerous groups to study ILs and a broad variety of theirproperties. These simulations are also an appropriate tool tostudy the solvation of water in ILs (for an overview, see, e.g.,the recent review from Maginn in ref 34). Mixtures of ILs andwater were first investigated computationally by Hanke et al.with molecular dynamics (MD) simulations, where the coordi-nation structure of water with ions was analyzed.35 Thearrangement of water molecules in imidazolium-based ILs hasbeen studied in various subsequent works by the same groupand others. It has been observed that water molecules stronglyassociated with anions. Furthermore, water molecules in the ILwere found to be isolated, while dimers and clusters of waterwere observed only at very high water concentrations in water-miscible ILs.36–38 In the case of nanostructured ILs, where longalkyl chains aggregate to form nonpolar domains (see, e.g., ref39), water was found to fluctuate within the polar domains thatare formed by the anions and polar groups of the cations.40,41
IL-water interfaces of miscible and immiscible ILs have beenstudied by Wipff and co-workers, where ion orientations andelectrostatic potentials at the interface were analyzed.40,42,43 Thesolubility of water in ILs was first studied by Lynden-Bell etal., where the excess chemical potential of water in dimeth-ylimidazolium chloride was determined with thermodynamicintegration to be 29 kJ/mol.44 Since the chemical potential hasnot been measured for this system, a confirmation of this valuehas not been possible so far. The excess chemical potential ofwater in 1-butyl-3-methylimidazolium hexafluorophosphate wasderived with free energy perturbation (FEP),45 where theresulting 23 kJ/mol somewhat overestimated the measuredchemical potential of 16 kJ/mol, according to Anthony et al.23
A more accurate value of 19 kJ/mol for the same system was
presented in ref 46 by applying Monte Carlo particle insertionas well as expanded ensemble simulations. The value for thechemical potential was derived from their published Henry’slaw constants. Molar enthalpy and entropy were calculated inthe same work, and a reasonable agreement with measured datawas found. The chemical potential was also calculated for1-hexyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imidewith Monte Carlo simulations.47 The saturation concentrationof water in this IL was found to be in good agreement with theexperiment. In addition, excess molar volumes and enthalpy ofmixing, which are quite difficult to derive from simulations,were calculated for a few ILs.36,47,48
The prediction of the water-miscibility or water saturationconcentration of new ILs has not been possible so far withoutemploying computationally expensive methods. The possibilityof prediction, however, would facilitate the optimization of ILs,considering the virtually infinite possibilities to modify a givenIL and the efforts to synthesize a new version. Therefore, theaim of the present work is to study how the choice of ionsinfluences the solvation of water and how the water-miscibilityof an IL can be estimated with a minimal computational effort.We start with MD simulations of water-miscible and water-immiscible ILs that are brought into contact with a waterreservoir, to verify if the simulations are able to reproduce theactually observed miscibilities. After confirming the suitabilityof the computational model, we proceed to analyze the solvationof water in 9 different ILs with varying cations and anions.Through comparison we identify the two main factors thatdetermine the solvation of water in ILs. These findings are usedin the final step to derive a simple model that estimates thewater-ion interaction strength in arbitrary ILs. These water-ioninteraction strengths are correlated with observed water misci-bilities of a large sample of ILs. The found correlation suggeststhat the water-miscibility of new ILs as well as their watersaturation concentration in case of immiscibility can be predictedwith the proposed model.
2. Methodology
2.1. Simulated ILs. The following ILs, which contain cationsbased on imidazole and guanidinium, complemented with nitrate(NO3), tetrafluoroborate (BF4) and hexafluorophosphate anions(PF6), were simulated in this work to study the influence ofions on the solvation of water: 1-butyl-3-methylimidazoliumpaired with nitrate (BMIM-NO3), tetrafluoroborate (BMIM-BF4),and hexafluorophosphate (BMIM-PF6) as well as 1-methoxy-ethyl-3-methylimidazolium tetrafluoroborate (MOEMIM-BF4),acyclic butylpentamethylguanidinium paired with tetrafluorobo-rate (BAGUA-BF4) and nitrate (BAGUA-NO3) and cyclicbutyltrimethylguanidinium nitrate (BCGUA-NO3). These ILswere complemented with cyclic tetramethylguanidinium nitrate(MCGUA-NO3) and cyclic heptyltrimethylguanidinium nitrate(HCGUA-NO3) to analyze the influence of a varying alkyl chainlength on the solvation of water. The structures of the simulatedions are displayed in Figure 1. Only the abbreviations of theILs will be used in the following as well as the abbreviationCGUA, which designates all ILs that contain cyclic guanidiniumgroups, i.e., MCGUA, BCGUA, and HCGUA.
2.2. Applied Force Field. An empirical molecular mechan-ical (MM) force field was used for all MD simulations tocalculate the potential energy of the simulated liquids, usingthe following standard functional form:
Determining the Miscibility of Ionic Liquids with Water J. Phys. Chem. B, Vol. 114, No. 8, 2010 2857

The first three terms in eq 1 are harmonic potentials thatdescribe vibrations along bonds, angles, and out-of-planevibrations, respectively, and are applied to covalently bondedatoms. The fourth term describes the torsion of molecular groupsalong a covalent bond and comprises together with the first threeterms the so-called bonded interactions. These interactions wereused whenever atoms were connected via three or less covalentbonds with each other. The fifth term is the electrostaticCoulomb potential and the last term is a short-range Lennard-Jones potential that includes Pauli repulsion and van der Waalsinteractions. The last two terms comprise the nonbondedinteractions and were applied to atom pairs that are connectedvia 4 or more covalent bonds or that are not connected witheach other. In the case of a separation of four bonds, a dihedralpotential as well as the nonbonded potentials were used, wherethe latter were scaled down with a factor of 0.5. A more detaileddescription of eq 1 and the used symbols is given elsewhere.49
Guanidinium-based cations and nitrate were simulated withthe force field that we developed in our previous work.49 For
imidazolium-based cations as well as for tetrafluoroborate andhexafluorophosphate, a force field based on the work from Lopeset al. was used.50 The water solute was represented in the initialIL-water mixing simulations by the TIP3P model51 and theTIP5P model.52,53 The charge distribution of TIP3P water isrepresented by three point charges at the atom positions. InTIP5P, two dummy atoms are attached to the oxygen atom, towhich the charge of the oxygen atom is transferred to mimicthe two electron lone pairs. All subsequent MD simulationsrelied solely on the TIP5P model, which turned out to providean improved description of IL-water interactions compared tothe TIP3P model, as demonstrated in section 3.1.
We like to emphasize that, for all simulated ILs, the chargedistribution in the actual liquid phase was used to parametrizethe Coulomb potential in eq 1. These charge distributions werealready published for the guanidinium-based ILs, except forBAGUA-BF4.49,54 For this IL and all ILs that contain imidazole,the liquid phase charge distributions were derived in this workand used instead of the previously published charge distributionof isolated ions. Additionally, the charge distribution of water,when solvated in the simulated ILs, was derived as well andused for the force field parametrization.
2.3. Derivation of Liquid Phase Charge Distributions. InILs, ions are subjected to interionic electron charge transfer andinternal charge polarization. Therefore, the charge distributionof an isolated ion or ion pair, which is the model that is usuallyexclusively considered in most force field parametrizations, arenot necessarily realistic. We demonstrated that energetic anddynamic properties, in contrast to structural properties, aresensitive to charge reorganization in the liquid phase.49,55 Wefollow in this work the approach that was applied to our previous
Figure 1. Liquid phase partial charges for five of the ILs that were simulated in this work. Partial charges of other simulated ILs are given inFigure 4 of ref 49 and Figure 2 of ref 54. These partial charges reproduce the electrostatic potential of the ions (ESP charges) in the actual liquidphase, derived with a QM/MM approach. For chemically equivalent atoms, only the average charge is given. Partial charges are given as fractionsof the elementary charge.
EpotMM ) ∑
i,j
bonds
kij(lij - l0,ij)2+ ∑
i,j,k
angles
kijk(�ijk - �0,ijk)2+
∑i,j,k,l
improper
kijkl(θijkl - θ0,ijkl)2 +
∑i,j,k,l
torsions
Eijkl(1 + cos(nijklφijkl - δ0,ijkl) +
∑i,j
qiqje2
4πε0rij+ 4εij[(σij
rij)12
- (σij
rij)6] (1)
2858 J. Phys. Chem. B, Vol. 114, No. 8, 2010 Klahn et al.

IL simulations to find atomic partial charges and derived liquidphase charge distributions by employing a combined quantummechanical/molecular mechanical approach (QM/MM).
A sample of six different ion clusters, containing sixcoordinated ion pairs each, was described with density functionaltheory (DFT) to assess charge transfer. Furthermore, a sampleof 12 different single cations and anions, respectively, wastreated with DFT to determine the intramolecular polarization.All samples were taken from equilibrated structures of the ILthat contained 500 ion pairs in total. The B3LYP hybridfunctional and 6-31+G(d) basis set were used, as implementedin the Gaussian 03 code.56–58 All the remaining non-DFT ionsin the system were treated with the previously described MMforce field, using the MD code GROMACS.59 The QM/MMinterface, which is a part of GROMACS, allows chargepolarization of the embedded DFT fragment by the electrostaticfield that is generated by the external MM fragment. Thepotential energy of these QM/MM systems was minimized.Atomic partial charges for each DFT fragment from the resultingstructures were derived. These partial charges were based onthe DFT fragment induced electrostatic potential (ESP charges),according to the CHELPG scheme.60 The charge transfer wasestimated by calculating the average total ion charge of the ionsin the sample of ion clusters. The average charge distributionof the ions in the liquid was then determined by averaging theatomic ESP charges over the sample of single DFT-ions. Thesecharges were scaled with the previously determined average totalion charge, resulting in the average liquid phase chargedistribution of the IL. These charge distributions were used toparametrize the Coulomb potential in eq 1. Details of thisprocedure are described in ref 49. It should be kept in mindthat partial charges are in principle not well-defined propertiesand that the method chosen to calculate them should be basedon their intended purpose. For MD simulations, a realisticrepresentation of electrostatic properties is essential, especiallyclose to the van der Waals surface of ions, which covers themost likely distances between ions. The method described abovewas chosen according to these requirements.
The liquid phase charge distribution, i.e., ESP partial charges,of the ILs BAGUA-NO3, MCGUA-NO3, and BCGUA-NO3 aregiven in ref 49 and of HCGUA-NO3 in ref 54. All other liquidphase charge distributions are shown in Figure 1. It was foundthat the internal polarization of BMIM cations in liquid phasewas small compared to guanidinium-based cations,49 which ledto a charge distribution that is similar to previously publishedvacuum charge distributions.61,62 The average total ion chargesin various simulated ILs is summarized in Table 1. Electroncharge is transferred from anions to cations. The amountincreases in the case of anions in the order of PF6 < BF4 < NO3,which agrees with the expected anion electrophilicities. In thecase of cations, the amount of transferred charge increased inthe order of BCGUA < BMIM < BAGUA. The strongelectrophilicity of BAGUA cations can be explained with thelarge positive partial charge of around +0.5 e on the centralcarbon of the guanidinium group, to which the anion chargewas found to be transferred.49 BMIM cations were slightly moreelectrophilic than CGUA cations, which might be due to themore emphasized localization of positive charge on the ringcompound.
For BMIM-PF6 total ion charges smaller than one wereproposed by other groups. A charge transfer of 0.1 e wasestimated in ref 61, based on the charge distribution of anisolated ion pair. In that case, however, charge transfer is likelyto be exaggerated due to the lack of surrounding counterions
that could balance the strong pull of charge that is exerted bya single counterion (see also ref 63). A charge transfer of 0.1and 0.2 e in this IL was later used in simulations to ensure water-immiscibility of the IL and to fit self-diffusion coefficients tomeasured values, respectively.42,43,64 Such an adjustment of thetotal ion charge to compensate inevitable inaccuracies of MMforce fields can be advantageous in many cases. However, sucha large charge transfer was not actually observed for this IL inthe present work. The charge transfer in some other ILs wasquite substantial on the other hand, with values up to 0.15 e forBAGUA-BF4 for instance, which justifies our approach to derivecharge distributions from the liquid phase. In any event, thederived partial charges together with the other force fieldparameters were used in simulations to determine IL densities,which are given in Table 1 together with their volume. Thecalculated densities of guanidinium-based ILs are in goodagreement with measured densities, and they are in satisfactoryagreement for ILs that contain BMIM cations.
The internal polarization of single water molecules solvatedin different ILs was derived with the QM/MM approachdescribed above. Water molecules turned out to be morepolarized in ILs than in water, due to strong interactions withdirectly coordinated ions. The variation of the polarization indifferent ILs is very small. Dipole moments of water rangedfrom 2.50 D in BAGUA-BF4 to 2.59 D in BMIM-NO3,compared to 2.35 D in TIP3P and 2.29 D in TIP5P. Thesecharge distributions of solvated water were used to parametrizethe Coulomb potential in eq 1.
2.4. Specification of MD Simulations. 2.4.1. Mixing of ILswith Water. All MD simulations, except those for the calculationof chemical potentials, were performed with the GROMACSsoftware package.59 For IL-water mixing simulations, a cubicbox with 500 ion pairs of BMIM-PF6, BMIM-BF4, and BMIM-NO3, respectively, were placed next to a similarly sized boxthat was filled with 5435 water molecules. These systemscontained in total around 30 000 atoms. All three mixingsimulations were set up twice: one that contained TIP3P waterand another that contained TIP5P water. Full periodic boundaryconditions were used, thereby generating two IL-water inter-faces, where IL and water were in direct contact with each other.Figure 2a illustrates the setup of these simulations. Furthermore,all hydrogen atoms were treated explicitly, and no degrees offreedom were constrained during the simulations, except thoseof the dummy atoms in the TIP5P model. Nonbonded interac-tions were calculated explicitly up to 15 Å, beyond which fast
TABLE 1: Average Total Ion Charge in the Liquid Phaseas Derived from QM/MM Simulations, Volume, and MassDensity of Simulated ILs
IL |q|a (e) V (cm3/mol) F (g/cm3)
BMIM-NO3 0.90 178.9 1.13 (1.16b)BMIM-BF4 0.95 196.9 1.15 (1.20c)BMIM-PF6 0.97 217.5 1.31 (1.36d)MOEMIM-BF4 0.95 185.7 1.23BAGUA-NO3 0.88 209.9 1.18 (1.17e)BAGUA-BF4 0.85 230.7 1.18BAGUA-ClO4 0.91 221.6f 1.29f (1.28e)MCGUA-NO3 0.92 149.6 1.36 (1.36e)BCGUA-NO3 0.92 190.3 1.29 (1.26e)HCGUA-NO3 0.92 232.1 1.24BCGUA-ClO4 0.94 203.6f 1.39f (1.37e)
a Same average total ion charges were assumed for ILs that differonly by the length of the cation alkyl chain. b Taken from ref 82.c Taken from ref 16. d Taken from ref 83. e Taken from ref 69.f Taken from ref 49.
Determining the Miscibility of Ionic Liquids with Water J. Phys. Chem. B, Vol. 114, No. 8, 2010 2859

particle-mesh Ewald electrostatics and dispersion energy cor-rections were used instead.65–67 These simulations were per-formed in the isothermal-isobaric (NPT) ensemble, wheretemperature and pressure were adjusted with a Berendsenthermostat.68 The coupling time constants for thermostat andbarostat were 0.1 and 1 ps, respectively. Compressibility andtarget pressure of the barostat were set to 4.5 × 10-5 bar-1
and 1 bar, respectively. A center of mass translation during thesimulation was prevented by applying correcting forces. Theintegration step size of all MD simulations was 1 fs.
Atom positions in the IL and water phases were initializedaccording to equilibrated structures of the pure liquids. Thederivation of these equilibrated structures is explained in thenext section. Close contacts at the IL-water interfaces wereremoved by minimizing the potential energy of the system witha steepest gradient method. The resulting structures were usedto initialize MD simulations of 3 ns length, at a temperature of300 K. MD simulations of systems that contained TIP5P waterwere continued for another 3 ns, resulting in total trajectorylengths of 6 ns.
2.4.2. Water SolWated in ILs Close to Infinite Dilution. Inthe first step 500 ion pairs of BMIM-NO3, BMIM-BF4, BMIM-
PF6, MOEMIM-BF4, BAGUA-BF4, BAGUA-NO3, BCGUA-NO3, MCGUA-NO3, and HCGUA-NO3 were simulated. Theinitial atom positions were generated by filling a cubic box withcopies of one ion pair. The dimension of the box was chosenso that the IL is diluted by about a factor of 10, compared tomeasured densities. Close contacts between atoms were removedby potential energy minimizations. MD simulations in the NPTensemble were started from the resulting structures, using thesame simulation settings as described above. During the first400 ps the temperature of the systems was linearly increasedto 600 K, after which the simulations were continued at thistemperature for another 400 ps. Finally, the temperature wasdecreased linearly to the target temperature of 300 K duringthe next 200 ps. This initial heating as well as the initially lowdensity of the ILs enabled a strong diffusion of the ions duringthe simulations. Therefore, the risk that the comparably im-mobile IL ions got trapped in local high energy minima of thepotential energy surface was substantially mitigated. Subse-quently, the MD simulations were continued for 2 ns at 300 K,where the last 500 ps were used for the data analysis. BAGUA-NO3 and MCGUA-NO3 exhibit melting points above roomtemperature and therefore represent in the simulations essentiallysupercooled liquids.69 These nine pure ILs served as referencesystems for simulations of ILs that contained water.
In these equilibrated structures of pure IL, 10 ion pairs werereplaced with 10 water molecules, respectively, to study thesolvation of water. The resulting water concentration corre-sponds to a mole fraction of water of 0.02, for which interactionsbetween water molecules could be neglected. All water mol-ecules were described with the TIP5P model. These IL-watersystems were equilibrated at 300 K for 1.5 ns, in which the last500 ps were used for analysis.
2.4.3. EWaluation of Excess Chemical Potentials of Waterin ILs. Excess chemical potentials of water in ILs were derivedwith the ENZYMIX module of the MD software MOLARIS,developed by Warshel and co-workers, which applies the FEPmethod.70 In this approach the interaction of the water solutewith the IL solvent is adiabatically switched off by slowlyreducing the involved Coulomb and Lennard-Jones pair poten-tials to zero. The following potential energy, Ei, was used forMD simulations:
The potential energy of the IL-water system, EIL+wat, isslowly transformed into the potential energy of the pure IL,EIL, through a coupling parameter, λi. This coupling parameteris stepwise increased from 0 to 1 during the MD simulation,thereby making the water molecule slowly invisible to the restof the system. When the potential energy is changed from Ei-1
to Ei, the excess chemical potential difference between the twostates, ∆µex,i, is given by the Zwanzig equation:
In eq 3, T is the temperature and kB is the Boltzmann constant.Equation 3 involves an average over the structure sample thatis generated with the MD simulation at step i, using the potentialEi. Equation 3 is only valid for small adiabatic changes of theenergy. After the simulation, all small chemical potentialchanges are added to yield the total excess chemical potential,µex, of the solute:
Figure 2. van der Waals representation of cations (blue), anions (green)and water molecules (red) of IL-water mixing simulations. ILs areseparated from water by two interfaces, because of periodic boundaryconditions, that are located in the figure to the left and right of the ILphase. Hydrogen atoms are not displayed for clarity. (a) Initializationof the MD simulation with a complete phase separation of BMIM-PF6
on the left-hand side and water on the right-hand side. (b) Equilibratedstructure of water-immiscible BMIM-PF6 after 6 ns of MD simulationat room temperature. Ions are transparent to highlight the position ofwater in the IL-rich phase. (c) Structure of water-miscible BMIM-BF4
after 6 ns of MD simulation, where the mixing of the two phases wasstill in progress. MD simulations reproduced the expected water-miscibilities of ILs.
Ei ) (1 - λi)EIL+wat + λiEIL (2)
∆µex,i ) -kBT ln⟨e-(Ei-Ei-1)/kBT)⟩MD with Ei(3)
2860 J. Phys. Chem. B, Vol. 114, No. 8, 2010 Klahn et al.
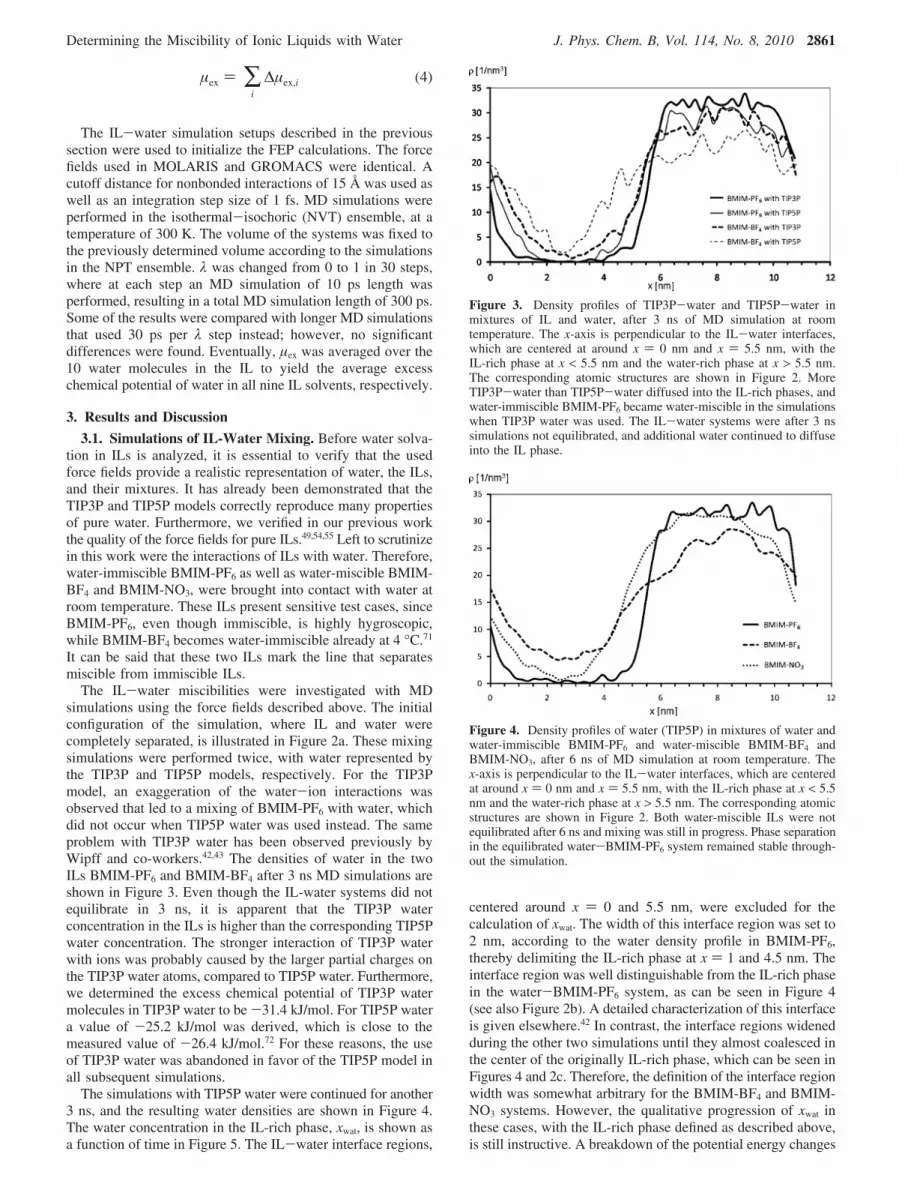
The IL-water simulation setups described in the previoussection were used to initialize the FEP calculations. The forcefields used in MOLARIS and GROMACS were identical. Acutoff distance for nonbonded interactions of 15 Å was used aswell as an integration step size of 1 fs. MD simulations wereperformed in the isothermal-isochoric (NVT) ensemble, at atemperature of 300 K. The volume of the systems was fixed tothe previously determined volume according to the simulationsin the NPT ensemble. λ was changed from 0 to 1 in 30 steps,where at each step an MD simulation of 10 ps length wasperformed, resulting in a total MD simulation length of 300 ps.Some of the results were compared with longer MD simulationsthat used 30 ps per λ step instead; however, no significantdifferences were found. Eventually, µex was averaged over the10 water molecules in the IL to yield the average excesschemical potential of water in all nine IL solvents, respectively.
3. Results and Discussion
3.1. Simulations of IL-Water Mixing. Before water solva-tion in ILs is analyzed, it is essential to verify that the usedforce fields provide a realistic representation of water, the ILs,and their mixtures. It has already been demonstrated that theTIP3P and TIP5P models correctly reproduce many propertiesof pure water. Furthermore, we verified in our previous workthe quality of the force fields for pure ILs.49,54,55 Left to scrutinizein this work were the interactions of ILs with water. Therefore,water-immiscible BMIM-PF6 as well as water-miscible BMIM-BF4 and BMIM-NO3, were brought into contact with water atroom temperature. These ILs present sensitive test cases, sinceBMIM-PF6, even though immiscible, is highly hygroscopic,while BMIM-BF4 becomes water-immiscible already at 4 °C.71
It can be said that these two ILs mark the line that separatesmiscible from immiscible ILs.
The IL-water miscibilities were investigated with MDsimulations using the force fields described above. The initialconfiguration of the simulation, where IL and water werecompletely separated, is illustrated in Figure 2a. These mixingsimulations were performed twice, with water represented bythe TIP3P and TIP5P models, respectively. For the TIP3Pmodel, an exaggeration of the water-ion interactions wasobserved that led to a mixing of BMIM-PF6 with water, whichdid not occur when TIP5P water was used instead. The sameproblem with TIP3P water has been observed previously byWipff and co-workers.42,43 The densities of water in the twoILs BMIM-PF6 and BMIM-BF4 after 3 ns MD simulations areshown in Figure 3. Even though the IL-water systems did notequilibrate in 3 ns, it is apparent that the TIP3P waterconcentration in the ILs is higher than the corresponding TIP5Pwater concentration. The stronger interaction of TIP3P waterwith ions was probably caused by the larger partial charges onthe TIP3P water atoms, compared to TIP5P water. Furthermore,we determined the excess chemical potential of TIP3P watermolecules in TIP3P water to be -31.4 kJ/mol. For TIP5P watera value of -25.2 kJ/mol was derived, which is close to themeasured value of -26.4 kJ/mol.72 For these reasons, the useof TIP3P water was abandoned in favor of the TIP5P model inall subsequent simulations.
The simulations with TIP5P water were continued for another3 ns, and the resulting water densities are shown in Figure 4.The water concentration in the IL-rich phase, xwat, is shown asa function of time in Figure 5. The IL-water interface regions,
centered around x ) 0 and 5.5 nm, were excluded for thecalculation of xwat. The width of this interface region was set to2 nm, according to the water density profile in BMIM-PF6,thereby delimiting the IL-rich phase at x ) 1 and 4.5 nm. Theinterface region was well distinguishable from the IL-rich phasein the water-BMIM-PF6 system, as can be seen in Figure 4(see also Figure 2b). A detailed characterization of this interfaceis given elsewhere.42 In contrast, the interface regions widenedduring the other two simulations until they almost coalesced inthe center of the originally IL-rich phase, which can be seen inFigures 4 and 2c. Therefore, the definition of the interface regionwidth was somewhat arbitrary for the BMIM-BF4 and BMIM-NO3 systems. However, the qualitative progression of xwat inthese cases, with the IL-rich phase defined as described above,is still instructive. A breakdown of the potential energy changes
µex ) ∑i
∆µex,i (4)
Figure 3. Density profiles of TIP3P-water and TIP5P-water inmixtures of IL and water, after 3 ns of MD simulation at roomtemperature. The x-axis is perpendicular to the IL-water interfaces,which are centered at around x ) 0 nm and x ) 5.5 nm, with theIL-rich phase at x < 5.5 nm and the water-rich phase at x > 5.5 nm.The corresponding atomic structures are shown in Figure 2. MoreTIP3P-water than TIP5P-water diffused into the IL-rich phases, andwater-immiscible BMIM-PF6 became water-miscible in the simulationswhen TIP3P water was used. The IL-water systems were after 3 nssimulations not equilibrated, and additional water continued to diffuseinto the IL phase.
Figure 4. Density profiles of water (TIP5P) in mixtures of water andwater-immiscible BMIM-PF6 and water-miscible BMIM-BF4 andBMIM-NO3, after 6 ns of MD simulation at room temperature. Thex-axis is perpendicular to the IL-water interfaces, which are centeredat around x ) 0 nm and x ) 5.5 nm, with the IL-rich phase at x < 5.5nm and the water-rich phase at x > 5.5 nm. The corresponding atomicstructures are shown in Figure 2. Both water-miscible ILs were notequilibrated after 6 ns and mixing was still in progress. Phase separationin the equilibrated water-BMIM-PF6 system remained stable through-out the simulation.
Determining the Miscibility of Ionic Liquids with Water J. Phys. Chem. B, Vol. 114, No. 8, 2010 2861

due to water-IL mixing is shown and described in Figure S1,Supporting Information.
The progression of xwat, according to Figure 5, suggeststhat the MD simulation of 6 ns length was insufficient toequilibrate the BMIM-BF4 and BMIM-NO3 systems and that,at the end of the simulation, mixing with water was still inprogress. While BMIM-BF4 mixed readily with water at thebeginning of the simulation, the influx of water into BMIM-NO3 was initially slow but eventually proceeded to a very highinflux rate toward the end of the simulation. The influx of waterinto BMIM-NO3 might have been initially impeded by the strongcounterion attraction in this IL (see also Figure S1). On thecontrary, the water-BMIM-PF6 system equilibrated after about4 ns of MD simulation. This is indicated by the convergenceof xwat toward a water saturation concentration in the IL-richphase of xwat ) 0.2 mol fractions of water. Measured values ofxwat in BMIM-PF6 range from 0.16 to 0.30, where an averagevalue of xwa ) 0.24 can be derived from these measurements,which agrees well with the value derived from our simulation.7,43
Despite of the chemical similarities of the three simulated ILsthat involve identical cations, simulations correctly reproducedthe water-immiscibility of BMIM-PF6 and its water saturationconcentration. Furthermore, the simulations also suggest themiscibility of BMIM-BF4 and BMIM-NO3 with water, inaccordance with measurements.
These results demonstrate that the applied force fields arecapable of providing a realistic model to describe IL-waterinteractions. Generally, miscibility and water saturation con-centration in ILs depend on the free energy changes that areinvolved when water is transferred from the water-phase intothe IL. By reproducing the measured excess chemical potentialof water in BMIM-PF6, as demonstrated in the next section,the quality of the force field was confirmed additionally. Withthese encouraging results in mind, we confidently proceeded toanalyze the interactions of water with various different ILs toidentify the factors that determine water-miscibility of ILs.
3.2. Analysis of Water-IL Interactions. The energetics ofsingle water molecules solvated in nine different ILs, close tothe limit of infinite dilution, was analyzed. The averagewater-ion interaction strength in these ILs, i.e., the sum of
Coulomb and van der Waals interactions between water andsurrounding ions, are shown in Figure 6. The correspondingpotential energy terms were derived from MD simulations ofthe ILs, in which single water molecules were solvated, asdescribed in section 2.4.2. Furthermore, the average excesschemical potential of water in ILs, µex, was derived. The methoddescribed in section 2.4.3 was applied to evaluate these chemicalpotentials, which also takes into account the reorganization ofthe IL-solvent upon solvation of water as well as entropic effects.These results are also displayed in Figure 6.
The obtained interaction energies of water with anions werea factor of 1.4-3.8 larger than with cations, which is in linewith numerous previous observations. The water-anion interac-tion strength increased in the order of PF6 < BF4 < NO3, whichis consistent with the findings from the mixing simulations (seealso Figure S1) as well as with IR measurements in whichanion-water interaction strengths were measured.73 Thewater-cation interaction strength increased in the order ofAGUA < CGUA < MOEMIM < BMIM and HCGUA <BCGUA < MCGUA. The latter relation means that thewater-cation interaction strength diminished with increasingalkyl chain length of the cation, which is confirmed bymeasurements as well.18 The interaction energy of water with aparticular ion was barely influenced by the identity of thecounterions.
Values of the calculated chemical potentials of water variedfrom -17 kJ/mol in BAGUA-BF4 to -26 kJ/mol in MCGUA-NO3 and follow the trend of the water-ion interaction energies,Ewat–IL ) Ewat-cat + Ewat-an. Regarding the three ILs that involveBMIM, water exhibits a smaller chemical potential, |µex|, inwater-immiscible BMIM-PF6 than in the other two water-miscible ILs, as expected. For BMIM-NO3, which is known tomix with water more readily than BMIM-BF4, the largestchemical potential was derived. A comparison with experimentaldata is possible in the case of BMIM-PF6, for which Henry’slaw constant of water vapor, H, was measured by Anthony etal.23 H can be converted to µex, in the limit of infinite dilutionof the solute, via H ) FRTeµex/RT, where F is the density of the
Figure 5. Change of water concentration in water-immiscible BMIM-PF6 and water-miscible BMIM-BF4 and BMIM-NO3, during 6 ns ofMD simulation. Concentrations are given as mole fractions of water.The water concentration converged to xwat ) 0.2 in BMIM-PF6, whichis close to the measured value of 0.24. The water concentration in theother two nonequilibrated systems did not converge after 6 ns, indicatingthat mixing is still in progress.
Figure 6. Interaction energies of water with cations, Ewat-cat, andanions, Ewat-an, as well as the excess chemical potential, µex, of waterin nine different simulated ILs. The numbers at the end of the upperbars are the total interaction energies of water with the IL, i.e., Ewat-cat
+ Ewat-an, respectively. The abbreviations of the ILs are explained insection 2.1. Values of µex follow the trend of Ewat-cat + Ewat-an.
2862 J. Phys. Chem. B, Vol. 114, No. 8, 2010 Klahn et al.

solvent. The resulting measured chemical potential is -16.2 kJ/mol. We obtained in our calculations a value of µex ) -18.3kJ/mol in this IL, which is in satisfactory agreement with themeasurement. Furthermore, the Henry’s law constants of twooctyl-substituted imidazolium-based ILs, namely C8MIM-BF4
and C8MIM-PF6, were measured as well in the same work.23
After conversion to |µex| it can be seen that in C8MIM-PF6 thechemical potential of water was 2.9 kJ/mol lower than inC8MIM-BF4. This value can be compared to the chemicalpotential difference of water in our simulated systems BMIM-PF6 and BMIM-BF4. The small contribution of the alkyl chainsto µex should basically cancel out in the chemical potentialdifference. The calculated value of ∆µex ) 2.1 kJ/mol compareswell with the measured value and provides a further confirmationof the applied computational approach.
When water molecules are solvated by the IL, the formationof solute cages in the solvent requires energy that is madeavailable by favorable interactions of water with surroundingions. Both energy contributions, as well as entropic changes ofthe solute and solvent, were taken into account when the excesschemical potentials were evaluated. The small values of µex,compared to values of Ewat-IL, indicate that a substantial amountof energy was used to form the water-accommodating cavitiesin ILs. Considering the strong electrostatic attraction of coun-terions in ILs, this is not surprising. Also a considerable entropypenalty due to the reduced mobility of water, caused by a tightcoordination to ions, might be involved. In any event, despiteof the substantial cavity formation energy, the qualitative trendof µex is basically determined by the largest of its contributions,Ewat-IL, according to the results in Figure 6. That, in turn, meansthat the interaction strength of water with ions is the main factorthat determines the solubility of water in ILs and therebywater-IL miscibility. This hypothesis will be confirmed laterin section 3.4.2 with the help of experimental data. A compu-tational analysis of the cage formation energy and entropiceffects, and how both are influenced by particular ions, deservespecial attention, but that is beyond the scope of the presentwork.
An inspection of the qualitative trend of Ewat-IL implies acorrelation with the ion size. In the following we use theMcGowan method rather than a more sophisticated computa-tional method to estimate ion volumes, because its simplicitywill ease the application of the empirical equation that will bederived in section 3.4.1.74 McGowan volumes are simplycalculated by adding the element specific volumes of all atomsin the compound, which are listed in ref 74, and by subsequentlysubtracting 6.56 cm3/mol for each present covalent bond, whilebond orders are ignored. This method was originally developedfor neutral molecules; however, its application can be extendedto ions, where volume changes due to additional or missingelectrons are taken into account.75 Here, cations were treated
in this particular case as neutral molecules, since the volumereduction due to the missing electron is negligible compared tothe total cation size. The volumes of the smaller anions weretaken from ref 75. The resulting McGowan volumes for cationsand anions are listed in Table 2.
The water-ion interaction energies, Ewat-cat and Ewat-an, arecompared with the corresponding ion volumes, Vcat and Van, inFigure 7. A correlation of ion volume and interaction strengthwith water is apparent, i.e., water favors interactions with smallions. The strongest interaction energies with values between40 and 43 kJ/mol were obtained for the smallest ion, NO3. Thesevalues decreased substantially to values between 28 and 33 kJ/mol in the case of BF4 and PF6. Interactions of water withcations, generally weaker than with anions, were strongest whensmall BMIM and MOEMIM cations were involved, with valuesbetween 16 and 19 kJ/mol. The interaction with the larger cationCGUA decreased from 14 to 11 kJ/mol due to an elongation ofthe alkyl chain. Finally, the weakest interaction was obtainedwhen BAGUA was involved, the largest ion in the simulations,with values between 9 and 11 kJ/mol. The stronger interactionwith small ions can be understood readily by considering thedominant Coulomb interactions between a water molecule andan ion. These Coulomb interactions are weak, when the averagedistance between the dipole of the water and the ion charge isincreased, which is the case for large ions. In other words, waterprefers to interact with ions that provide a localized charge thatcan be brought into close contact with the water molecule.
According to Figure 7, the ion size seems to be the primefactor that determines the interaction strength with water. Acloser look, however, reveals that another important factor isinvolved as well. In BMIM-BF4 and MOEMIM-BF4, forinstance, the interaction of water with BF4 was 4-5 kJ/molstronger than with PF6 in BMIM-PF6, even though both anionsare of similar size. That the interaction of water is indeedstronger with BF4 than with PF6 has been confirmed inmeasurements before.76 Inspecting the contributions to Ewat-an,
according to the simulations, the stronger interaction of waterwith BF4 was simply caused by the larger partial charges onthe fluorine atoms. These atoms exhibit an atomic partial chargeof -0.5 e each, compared to -0.4 e in the case of PF6, as can
TABLE 2: 2. McGowan Volume of Cations and Anions, andNumber of Water-Cation and Water-Anion Close Contactsin Simulated ILs
IL Vcat (cm3/mol) Van (cm3/mol) Nwat-cat Nwat-an
BMIM-NO3 125.9 25.8 3.9 1.7MCGUA-NO3 118.0 25.8 4.9 1.5BCGUA-NO3 160.3 25.8 4.8 1.3HCGUA-NO3 202.6 25.8 5.0 1.4BMIM-BF4 125.9 46.9 3.7 1.8MOEMIM-BF4 117.7 46.9 3.4 1.9BAGUA-NO3 179.6 25.8 5.2 1.4BMIM-PF6 125.9 48.3 3.2 2.2BAGUA-BF4 179.6 46.9 4.5 1.6
Figure 7. Comparison of the interaction strength of water with ions,Ewat-cat and Ewat-an, in nine different ILs, with the size of the involvedions, Vcat and Van. Ion volumes were estimated with the McGowanmethod.74 The influence of the counterion on the water-ion interactionstrength modified the values of Ewat-ion slightly, therefore some ionsare represented by more than one data point in the plot. Generally, theinteraction strength increases with the reciprocal ion volume.
Determining the Miscibility of Ionic Liquids with Water J. Phys. Chem. B, Vol. 114, No. 8, 2010 2863

be seen in Figure 1. These large partial charges on the surfaceof BF4 led to stronger interactions with the water dipole. Sincethe ion sizes are similar in this case, the different water-miscibilities of these ILs, in fact, can be attributed to the differentanion surface charges. In this context, we also like to remindthat all partial charges that were used in the simulations of thiswork were based on the IL charge distribution of the actualliquid phase. The two main factors that determine water-ioninteraction strength are summarized in Figure 8.
The findings of this section also explain why water preferablyinteracts with anions in ILs. In most ILs that have beensynthesized, anions are substantially smaller than cations.Furthermore, anions exhibit larger surface charges than cations.While on fluorine and oxygen atoms of anions typical surfacecharges vary between -0.4 and -0.8 e, the surface charges oncation hydrogen atoms vary between 0.1 and 0.2 e. Both, thesmall size and the large surface charge, result in strong attractionbetween water and anions. Therefore, it is plausible that thewater-miscibility of ILs is determined more by anions thancations, assuming that the solubility of water is mainlydetermined by the strength of water-ion interactions.
The findings of this section also constitute the basis withwhich the approach to predict water-miscibility of ILs will bederived in section 3.4. Before we turn to this approach, thealternative notion that the internal IL structure could determinewater solubility will be scrutinized in the next section.
3.3. Impact of the IL Structure on the Solvation of Water.3.3.1. Influence of the Coordination Structure of Water andIons. It was demonstrated in the last section that the magnitudeof the ion surface charge, together with the ion size, determinesthe water-ion interaction strength. The influence of the size ofthe ion surface area that is in direct contact with water was notconsidered so far. The size of this surface area is proportionalto the number of hydrogen bonds between the ion and water.Hydrogen bonds basically connect water oxygen atoms andcation hydrogen atoms as well as water hydrogen atoms andanion hydrogen acceptor atoms, i.e., oxygen and fluorine atoms.Since the covalent contribution to these hydrogen bonds is verysmall in ILs, compared to electrostatic attraction, we prefer touse in the following the more general expression “close contact”instead of hydrogen bond.
The number of close contacts between ions and water werequantified with radial distribution functions (RDFs) that werederived from the MD simulations. The RDFs for pairs of wateroxygen atoms and cation hydrogen atoms, gwat-cat, as well as forpairs of water hydrogen atoms and anion oxygen/fluorine atoms,gwat-an, were calculated. From these RDFs, the number of closecontacts, Nwat-cat and Nwat-an, can be derived as a function of themaximum distance up to which atom pairs are still considered toform a close contact, rmax, using the following equation:
Here, Fion designates the cation hydrogen atom density or anionoxygen/fluorine atom density, respectively. The resulting valuesfor N, in the case of rmax ) 3 Å, are given in Table 2.
A comparison of values for Nwat-cat and Nwat-an with thecorresponding water-ion interaction strengths, Ewat-cat andEwat-an in Figure 6, shows that a large number of water-ionclose contacts is related to weak water-ion interactions. Forinstance, in BMIM-NO3, Nwat-cat and Ewat-cat are 3.9 and -19kJ/mol, respectively, while in BAGUA-NO3 Nwat-cat and Ewat-cat
are 5.2 and -9 kJ/mol. This seemingly surprising result, inwhich a larger number of water-cation close contacts led toweaker interactions, can be simply explained by the largernumber of hydrogen atoms in BAGUA cations. This cationcontains 24 hydrogen atoms that can coordinate with anionoxygen atoms, compared to only 15 hydrogen atoms in BMIM.The larger number of hydrogen atoms translates to a largerhydrogen atom density in BAGUA-NO3, which increases thenumber of water-cation close contacts. A larger number ofhydrogen atoms per cation, on the other hand, also correspondsto a larger cation size, which leads to weaker interactions withwater, as explained in the previous section. That means that itwas indeed the cation size and not the number of close contactsthat determined the interaction strength with water. The findingsfor other cations as well as for anions were similar. Alsointeresting is a comparison of the interaction strength of waterwith anions in BMIM-BF4 and in BMIM-PF6. In the case ofBMIM-BF4, Nwat-an and Ewat-an are 1.8 and -32 kJ/mol,respectively, while in the case of BMIM-PF6, Nwat-an and Ewat-an
are 2.2 and -28 kJ/mol. The difference in Ewat-an is comparablysmall, because of the similar anion volume. The smaller partialcharge on the PF6 fluorine atoms resulted in a somewhat weakerinteraction with water, as explained before. This was notcompensated by the larger number of close contacts. Therefore,the ion volume and surface charge are by far more importantin determining the water-ion interaction strength than thestructural details of the particular water-ion coordination thatis realized in the IL.
3.3.2. Influence of Preformed CaWities. It has been suggestedthat interstices or preformed cavities in the IL solvent couldfacilitate the accommodation of small molecules and therebyimprove the solvation of water.45 For instance, the solubility ofwater in BMIM-PF6 could be lower than in BMIM-BF4, becausethe slightly larger PF6 anions would fill more interstices in theIL. Preformed cavities in ILs have indeed already been observedin simulations; however, their influence on IL properties remainsunclear.55,77 Preformed cavities that are sufficiently large toaccommodate water were located and quantified in the threeILs BMIM-PF6, BMIM-BF4, and BMIM-NO3 to scrutinize theirrelevance for the solvation of water. A sample of 10 structuresfrom MD simulations of the pure ILs was generated for eachIL, respectively. Subsequently, the simulation boxes that contained
Figure 8. Two main factors that determine the interaction strength ofwater with ions. (a) Water-ion interaction strength is primarilydetermined by the ion size: Water interaction with small ions is strongerthan with large ions, because the charge is more localized, i.e., theaverage distance between the electric water dipole and the ion monopoleis small. (b) Water-ion interaction strength is secondarily determinedby the surface charge of the ion: Water interacts favorably with ionsthat exhibit large surface charges, i.e., large partial charges on atomsthat are in direct contact with water, than with ions that exhibit smallsurface charges. The surface charge of an ion is determined by itschemical structure.
Nwat-ion ) 4πFion ∫0
rmax gwat-ion(r)r2dr (5)
2864 J. Phys. Chem. B, Vol. 114, No. 8, 2010 Klahn et al.

these structures were filled with small adjoining cubes of 2 Å edgesize. All cubes that overlapped with at least one of the atoms inthe IL, which were represented by spheres with atom-specific vander Waals radii, were removed. The volume of the remaining cubeswas added to give the percentage of unoccupied space in the ILs.Hence, small isolated cavities with a volume below 8 Å3 wereexcluded. Since cavities of that size are too small to be occupiedby water, they are not relevant for this analysis.
The largest amount of unoccupied space was obtained forBMIM-PF6, where 1.05% of the total volume remained unoc-cupied, followed by BMIM-BF4 with a value of 0.80% andBMIM-NO3, with a value of 0.45%. With a water volume of18 cm3/mol, these preformed cavities in BMIM-PF6 couldaccommodate up to 0.11 mol fractions of water and in the caseof BMIM-NO3 only up to 0.05 mol fractions. Since the solubilityof water is the lowest in BMIM-PF6 and largest in BMIM-NO3,the results of this analysis do not support the idea that preformedcavities are important for the solubility of water. Furthermore,the amount of unoccupied space seems to be insufficient to storelarger concentrations of water. On the basis of these findings,we did not consider cavities as an essential ingredient for theprediction of water-miscibilities of ILs. These results are alsoin line with a simulation work, in which cavities were not foundto cause the high solubility of CO2 in ILs.78
3.4. Prediction of Water-IL Miscibility and Water Satu-ration Concentration. 3.4.1. Estimation of the Water-IonInteraction Strength. According to the results in this work, thesolvation of water in ILs mainly depends on the water-ioninteraction strength. The interaction strength in turn dependson the size of the ions and the ion surface charge that is indirect contact with water, as explained before. A model toestimate water-ion interaction strengths, which is based on theseassumptions, is introduced in this section. An equation is derivedfrom this model, which can be used to predict the miscibilityof new ILs with water, while only minimal computational effortis required for its application.
The proposed water-ion interaction model is schematicallyshown in Figure 9a. One water molecule is coordinated to onecation and one anion. Ions are described as spheres with avolume that is estimated according to the McGowan method.Furthermore, the electron charge density of the ions is assumedto be constant within the sphere and can thus be represented bya single point charge in the center of the sphere. A part of thision charge is transferred to the ion surface, by placing a secondpoint charge on the part of the ion surface that is closest to thewater molecule. The sum of the ion center charge and the surfacecharge is +1 e for the cation and -1 e for the anion. Threepoint charges are placed at the atom positions of the watermolecule. A Coulomb potential is used to calculate the interac-tion energy of the three water point charges with the two pointcharges of the cation and anion, respectively. This model takesinto account the size of the ions and the magnitude of the surfacecharge that is in direct contact with the water molecule, asdemanded by the previous analysis.
Details of the described model, together with geometricspecifications, are shown in Figure 9b. The oxygen atom ofwater carries the charge -Q and the two water hydrogen atomscarry the charge +Q/2. The two surface charges are called qcat
and qan, leaving the charges 1 - qcat and -1 - qan in the centerof the cation and anion, respectively. The geometry of the watermolecule is specified by the oxygen-hydrogen bond length, d,the water bond angle, �, as well as by the van der Waals radiiof the oxygen atom and the hydrogen atoms, RO and RH. Theradii of the ion spheres, rcat and ran, can be derived from their
McGowan volume via r ) (3V/4π)1/3. The distance betweenthe water oxygen atom and the cation surface charge, qcat, aswell as the distance between the water hydrogen atoms and theanion surface charge, qan, are called p. With these specifications,the water-ion interaction strength, Ewi, can be calculated witha Coulomb sum, with the help of some simple geometricconsiderations, as shown in Figure 9b:
Figure 9. (a) Scheme of model to estimate the interaction strength ofwater with ILs. A water molecule is coordinated to one cation and oneanion. Both ions are treated as spheres with a volume according to theMcGowan method. Point charges are placed in the center of the spheres,with charges of +1 and -1 for cations and anions, respectively. Apart of these charges is transferred to the ion surface that is in directcontact with the water molecule. These surface charges are designatedas qcat and qan. (b) Details of the water-IL interaction model withgeometric specifications. Shown are the ion centered charges, 1 - qcat
and -1 - qan, the two ion surface charges, qcat and qan, the charge ofthe water oxygen atom, -Q, and the charge of the two water hydrogenatoms, +Q/2. Also shown are the hydrogen-oxygen bond length ofwater, d, and the water bond angle, �. The distance between wateroxygen atom and cation surface charge as well as the distance betweenwater hydrogen atom and anion surface charge is called p. The distancebetween the cation center charge and the water oxygen atom is thesum of cation radius, rcat, and the van der Waals radius of the oxygenatom, RO. Similarly, the distance between the anion center charge andthe water hydrogen atom is the sum of the anion radius, ran, and thevan der Waals radius of the hydrogen atom, RH.
4πε0
e2Ewi )
-Q(1 - qcat)
RO + rcat+
2
Q2
(1 - qcat)
�(d sin�2 )2
+ (d cos�2+ RO + rcat)2
+-Qqcat
p+
2
Q2
qcat
�d2 + p2 + 2dp cos�2
+
-Q(-1 - qan)
d cos�2+ �(RH + ran)
2 - (d sin�2 )2
+ 2
Q2
(-1 - qan)
RH + ran+
-Qqan
d cos�2+ �p2 - (d sin
�2 )2
+ 2
Q2
qan
p(6)
Determining the Miscibility of Ionic Liquids with Water J. Phys. Chem. B, Vol. 114, No. 8, 2010 2865

The specifications for water, namely d ) 0.96 Å, � ) 104.5°,RO ) 1.4 Å, and RH ) 1.0 Å, were substituted into eq 6 in thenext step. The charge Q was set to 0.9, according to thepreviously determined dipole moment of water in ILs. The closecontact distance, p, was set to 2 Å, which is a typical value fora hydrogen bond length. This leaves only the ion sizes andsurface charges, rcat, ran, qcat, and qan as parameters in eq 6. Withthe approximations RO + rcat . d and RH + ran . d, thefollowing equation for Ewi (in kJ/mol) can be derived:
We would like to emphasize that all coefficients in eq 7 weredirectly derived from eq 6 with the parameter values describedabove, i.e., no other data was used and no fitting of thecoefficients in eq 7 was performed. While the first two terms ineq 7 estimate the interaction of water with the electric monopoleof the ions, the last two terms describe the interaction of waterwith the surface charges.
3.4.2. Application and Verification of the Ion-WaterInteraction Model. The water-ion interaction strength, accord-ing to eq 7, was estimated for a sample of ILs and comparedwith measured water saturation concentrations and water-miscibilities to verify the proposed water-ion interaction model.The sample includes 83 different ILs and involves 28 differentcations and 18 different anions.4,5,7,9,18,23,26,27,30,79,80 Cations inthe sample are based on either imidazolium, pyridinium,pyrrolidinium, or piperidinium, to which alkyl chains of varyinglength were attached, involving in some cases also fluoroalkyl,allyl, and methoxyehtyl groups. The diversity of anions in thesample is illustrated in Figure S2. The list of ILs can be foundin the Supporting Information in Table S1, together with theMcGowan volume and radius of the constituting ions. Also,the water-miscibility of these ILs, and in case of immiscibilitythe measured water saturation concentrations as well, are givenin Table S1. The McGowan radii of monatomic halide anionssomewhat overestimate the distance between ion and water.Therefore, in this special case, the anion radius, ran, was chosenso that ran + RH, i.e., the distance between the halide ion andthe water hydrogen atoms, reproduced the actually measureddistances according to ref 81.
Equation 7 contains 4 parameters, two of which are the ionradii that were readily estimated with the McGowan method.The choice of the two ion surface charges, qcat and qan, requiredsome more consideration. When a water molecule diffuses intothe IL, it is safe to assume that it strives to coordinate with thepart of the ion that exhibits the largest surface charge, in aneffort to optimize its solvation. The ion surface charges are thepartial charges on the atoms that are directly coordinated withwater. That means that qcat and qan should be equal to the largestpartial charge, carried by an ion atom that is capable ofcoordinating with water. While the value of this charge variesconsiderably from anion to anion, the value and variation ofthis charge is small in the case of cations, as can be seen, forexample, in Figure 1. Therefore, we were content to use anaverage surface charge of qcat ) 0.1 for all cations to simplifymatters. In the case of anions, where the interaction model isrequired to capture the anion specific variation of the surfacecharge, qan was derived directly from the anion charge distribution.
The charge distributions of all anions that are listed in TableS1, excluding monatomic halides, were determined with DFT,
using the functional B3LYP and the basis set 6-31+G(d). Thegeometries of the anions were optimized in vacuum, and fromthe resulting geometries the ESP charges were calculated usingthe CHELPG scheme. The obtained charges are shown in FigureS2. The largest negative partial charge of each anion is listedin Table S1 as qan. In the case of monatomic anions, the entireanion charge was placed into the anion center. Interesting arethe large negative partial charges on anion groups such as,-CO2, -SO3, and -SO2-, compared to the charges on the ofteninvolved fluoroalkyl groups. For example, in the case of thecommonly used Tf2N anions, water coordinates much morelikely with one of the SO2 groups, whose oxygen atoms exhibita partial charge of -0.56 e, rather than with one of the fluorineatoms that carry only small charges of -0.12 e. This assumptionhas been confirmed also by MD simulations of BMIM-Tf2N,in which water was indeed found close to the SO2 groups.40
With this in mind, it should be expected that only thenonfluoroalkyl groups in larger anions influence the solubilityof water in the IL.
In Figure 10a, measured water saturation concentrations, xwatexp,
were plotted against the corresponding values of Ewi, listed inTable S1, according to eq 7. Indeed, the water-miscibilities ofILs strongly correlated with the estimated water-ion interactionstrengths. According to a visual inspection of Figure 10a, the
Ewi ) -738( 1 - qcat
(rcat + 1.67)2+
1 + qan
(ran + 1.26)2) -
161qcat + 111qan (7)
Figure 10. Comparison of estimated water-ion interaction strengthsaccording to eq 7, Ewi, and measured water saturation concentrations,xwat
exp, given in mole fractions of water, for a sample of 83 ILs. Largevalues of Ewi correlate with high water concentrations in the IL, up toa point where mixing of the IL with water occurs. (a) ILs are categorizedby the value of Ewi to be in one of 3 groups: for Ewi > -117 kJ/molwater and IL are immiscible and xwat
exp increases linearly with Ewi. For-124 kJ/mol < Ewi < -117 kJ/mol the phase separation of IL and waterbecomes unstable. For Ewi < -124 kJ/mol water and IL are miscible.(b) Magnification of the region in Figure 10a in which IL and waterare immiscible, together with a linear regression line according to eq8.
2866 J. Phys. Chem. B, Vol. 114, No. 8, 2010 Klahn et al.

plot can be divided into three regions. For weak water-ioninteractions with Ewi > -117 kJ/mol, ILs and water areimmiscible. Furthermore, xwat
exp increases linearly with Ewi, whichis plausible, since stronger water-ion interactions should allowthe absorption of a larger quantity of water. For strongwater-ion interactions with Ewi < -124 kJ/mol, ILs and waterbecome miscible. At these interaction strengths, a sufficientamount of water is absorbed by the IL to overcome the interionicinteractions, resulting in a mixing of the two phases. For anarrow range of medium values of Ewi, roughly between -117and -124 kJ/mol, miscibility cannot be predicted. In this regionof the plot, the ILs are either miscible or they are immisciblebut exhibit a very high concentration of water. Only 12 ILs inthe sample were found to fall into this category. It is likely thatmiscibility in this region of the plot also depends on how thewater-IL equilibrium is established in the experiment. Someexperiments involve especially long equilibration times andintense stirring, enabling mixing of the two phases that wouldhave otherwise not occurred. For BMIM-PF6, a value of Ewi )-113 kJ/mol was calculated, which classifies this IL correctlyas immiscible. For BMIM-BF4 a value of Ewi ) -122 kJ/molwas determined, classifying the separation of water and ILphases as unstable. Since BMIM-BF4 is water-miscible at roomtemperature but immiscible at 4 °C, as mentioned before, thisclassification seems to be very reasonable. BMIM-NO3, with avalue of Ewi ) -136 kJ/mol, is correctly classified as a miscibleIL. The region of the plot in Figure 10a in which ILs areimmiscible is enlarged in Figure 10b. Using a linear regressionline, the water saturation concentration in immiscible ILs canbe estimated from Ewi with
Considering the large data set of ILs that encompassed a largevariety of different ions and keeping in mind the simplicity ofthe proposed water-ion interaction model and that no param-eters in eq 7 were fitted to measured data, the observedcorrelation in Figure 10 is remarkable. The obtained correlationconfirms that the ingredients of the interaction model, the ionsizes and surface charges, were indeed sufficient to predict thewater-miscibility of ILs. It furthermore demonstrates that thewater-ion interaction strength indeed determines the trend ofthe excess chemical potential of water in ILs.
Equations 7 and 8 can be used to predict the miscibility andwater saturation concentration of new ILs, without the need toperform computationally expensive molecular simulations. Infact, only the derivation of the anion surface charges, qan,requires a computational treatment. Fortunately, deriving ananion charge distribution with the method explained above iscomputationally very inexpensive and straightforward to carryout. For instance, the charges of all 15 anions together, shownin Figure S2, were calculated within 2 days, using a singleprocessor. Values of Ewi for new ILs need to be compared withthe data shown in Figure 10 to enable a categorization of theIL. The absolute value of Ewi is not meaningful by itself.Therefore, new values for qan should be calculated with the sameDFT functional, basis set, and ESP scheme to ensure a maximumcomparability of the derived Ewi with the data presented in thiswork.
4. Conclusion
It was demonstrated with molecular simulations that thewater-ion interaction strength determines qualitatively the trend
of the excess chemical potential of water in ILs and therebyalso the miscibility of ILs with water. This interaction strengthin turn is primarily determined by the size of the ions.Interactions of water with small ions are more favorable becauseof the localization of the ion charge. The second factor thatdetermines water-ion interactions is the magnitude of the chargeon the ion surface atom that is directly coordinated with water.This charge in turn depends on the chemical structure of theion. A strong internal polarization of the ion, which leads tolarger partial charges on its surface atoms, results in morefavorable interactions with water. This also implies, for example,that the water saturation concentration in ILs can be minimizedby using ions that are large and that exhibit minimal partialcharges on their surface.
On the basis of these findings, a water-ion interaction modelwas proposed to derive an equation that estimates the water-ioninteraction strength. The only parameters of this equation arethe volume and surface charge of the ions. The validity of themodel was demonstrated by classifying a large sample of ILscorrectly as being miscible or immiscible ILs, merely based ontheir estimated water-ion interaction strengths. Thereby, it isalso established that the solvation of water in ILs is primarilydetermined by electrostatics. Additionally, a linear relationshipbetween water-ion interaction strength and water saturationconcentration was found for immiscible ILs.
The proposed model can be applied with a minimum ofcomputational effort to predict the miscibility of a new IL: first,McGowan volumes of cations and anions are determined fromtables, and, second, the ion surface charge is determined fromthe ion charge distribution, using a computationally inexpensiveDFT calculation. With these parameters, the water-ion interac-tion strength is estimated by evaluation of eq 7. Miscibility ofthe IL at room temperature is predicted for values below -124kJ/mol, and immiscibility is predicted for values above -117kJ/mol. A clear distinction cannot be made by the model forvalues in between. If immiscibility is predicted, eq 8 can beused to estimate the water saturation concentration in the IL atthermal equilibrium.
A refined version of eq 7 might be able to improve the qualityof the predictions further. However, a verification of such animproved equation would be hampered by the limited accuracyof the experimental data, due to difficulties in establishing theequilibrium of the water-IL systems. A generalization of theproposed model to predict the miscibility of ILs with other smallpolar solutes such as CO2 would also be promising.
In any event, the proposed method to predict the water-miscibility of ILs is computationally inexpensive, especiallywhen compared to the time-consuming calculation of chemicalpotentials with molecular simulations. This model providesguidelines to aid the considerable efforts that are being madeto optimize water-related properties of ILs.
Acknowledgment. We gratefully acknowledge the provisionof computing facilities by the Institute of High PerformanceComputing (IHPC) and the financial support from the Agencyfor Science, Technology and Research (A*STAR) of Singapore.
Supporting Information Available: Complete ref 58; tablelisting McGowan volume and radius of cations and anions, anionsurface charge, estimated water-IL interaction energy, andmeasured water-miscibility for a sample of ILs (Table S1);potential energy changes upon mixing of initially separated ILsand water, after 6 ns MD simulations at room temperature(Figure S1); and atomic partial charges for the 15 anions thatare included in the list of ILs in Table S1, excluding halide
xwatexp ) -0.0189Ewi - 1.91, for Ewi > -117kJ/mol
(8)
Determining the Miscibility of Ionic Liquids with Water J. Phys. Chem. B, Vol. 114, No. 8, 2010 2867

anions (Figure S2). This material is available free of chargevia the Internet at http://pubs.acs.org.
References and Notes
(1) Welton, T. Chem. ReV. 1999, 99, 2071.(2) Rogers, R. D.; Seddon, K. R. Science 2003, 302, 792.(3) Seddon, K. R. Nat. Mater. 2003, 2, 363.(4) Wasserscheid, P. and Welton, T. Ionic Liquids in Synthesis; Wiley-
VCH, Verlag: Weinheim, 2003.(5) Bonhote, P.; Dias, A. P.; Papageorgiou, N.; Kalyanasundaram, K.;
Gratzel, M. Inorg. Chem. 1996, 35, 1168.(6) Huddleston, J. G.; Willauer, H. W.; Swatloski, R. P.; Visser, A. E.;
Rogers, R. D. Chem. Commun. 1998, 16, 1765.(7) Freire, M. G.; Neves, C. M. S. S.; Carvalho, P. J.; Gardas, R. L.;
Fernandes, A. M.; Marrucho, I. M.; Santos, L. M. N. B. F.; Coutinho, J. A. P.J. Phys. Chem. B 2007, 111, 13082.
(8) Freire, M. G.; Santos, L. M. N. B. F.; Fernandes, A. M.; Coutinho,J. A. P.; Marrucho, I. M. Fluid Phase Equilib. 2007, 261, 449.
(9) Freire, M. G.; Carvalho, P. J.; Gardas, R. L.; Marrucho, I. M.;Santos, L. M. N. B. F.; Coutinho, J. A. P. J. Phys. Chem. B 2008, 112,1604.
(10) Salminen, J.; Papaiconomou, N.; Kumar, R. A.; Lee, J.-M.; Kerr,J.; Newman, J. P.; Prausnitz, J. M. Fluid Phase Equilib. 2007, 261, 421.
(11) Kakiuchi, T.; Tsujioka, N.; Kurita, S.; Iwami, Y. Electrochem.Commun. 2003, 5.
(12) Nishi, N.; Kawakami, T.; Shigematsu, F.; Yamamoto, M.; Kakiuchi,T. Green Chem. 2006, 8, 349.
(13) Chapeaux, A.; Simoni, L. D.; Stadtherr, M. A.; Brennecke, J. F.J. Chem. Eng. Data 2007, 52, 2462.
(14) Seddon, K. R.; Stark, A.; Torres, M.-J. Pure Appl. Chem. 2000,72, 2275.
(15) Carda-Broch, S.; Berthod, A.; Armstrong, D. W. Anal. Bioanal.Chem. 2003, 375, 191.
(16) Rebelo, L. P. N.; Najdanovic-Visak, V.; Visak, Z. P.; Nunes daPonte, M.; Szydlowski, J.; Cerdeirina, C. A.; Troncoso, J.; Romanı, L.;Esperanca, J. M. S. S.; Guedes, H. J. R.; de Sousa, H. J. R. Green Chem.2004, 6, 369.
(17) Garcia-Miaja, G.; Troncoso, J.; Romanı, L. J. Chem. Thermodyn.2009, 41, 161.
(18) Huddleston, J. G.; Visser, A. E.; Reichert, W. R.; Willauer, H. D.;Broker, G. A.; Rogers, D. Green Chem. 2001, 3, 156.
(19) Najdanovic-Visak, V.; Rebelo, L. P. N.; da Ponte, M. N. GreenChem. 2005, 7, 443.
(20) Fu, D.; Sun, X.; Pu, J.; Zhao, S. J. Chem. Eng. Data 2006, 51,371.
(21) Chaumont, A.; Wipff, G. J. Mol. Liq. 2007, 131–132, 36.(22) van Rantwijk, F.; Sheldon, R. A. Chem. ReV. 2007, 107, 2757.(23) Anthony, J. L.; Maginn, E. J.; Brennecke, J. F. J. Phys. Chem. B
2001, 105, 10942.(24) Luo, H.; Dzielawa, J. A.; Bonnesen, P. V. Anal. Chem. 2004, 76,
2773.(25) Shvedene, N. V.; Borovskaya, S. V.; Sviridov, V. V.; Ismailova,
E. R.; Pletnev, I. V. Anal. Bioanal. Chem. 2005, 381, 427.(26) Papaiconomou, N.; Yakelis, N.; Salminen, J.; Bergman, R.;
Prausnitz, J. M. J. Chem. Eng. Data 2006, 51, 1389.(27) Papaiconomou, N.; Salminen, J.; Lee, J. M.; Prausnitz, J. M.
J. Chem. Eng. Data 2007, 52, 833.(28) Gathergood, N.; Garcia, M. T.; Scammells, P. J. Green Chem. 2004,
6, 166.(29) Garcia, M. T.; Gathergood, N.; Scammells, P. J. Green Chem. 2005,
7, 9.(30) Toh, S. L. I.; McFarlane, J.; Tsouris, C.; DePaoli, D. W.; Luo, H.;
Dai, S. SolVent Extr. Ion Exch. 2006, 24, 33.(31) Dietz, M. L.; Dzielawa, J. A.; Laszak, I.; Young, B. A.; Jensen,
M. P. Green Chem. 2003, 5, 682.(32) (a) Vianello, R.; Kovacevic, B.; Ambrozic, G.; Mavri, J.; Maksic,
Z. B. Chem. Phys. Lett. 2004, 400, 117. (b) Tran, C. D.; Lacerda, S. H. D.;Oliveira, D. Appl. Spectrosc. 2003, 57, 152.
(33) Ranke, J.; Othman, A.; Fan, P.; Muller, A. Int. J. Mol. Sci. 2009,10, 1271.
(34) Maginn, E. J. J. Phys.: Condens. Matter 2009, 21, 373101.(35) Hanke, C. G.; Atamas, N. A.; Lynden-Bell, R. M. Green Chem.
2002, 4, 107.(36) Hanke, C. G.; Lynden-Bell, R. M. J. Phys. Chem. B 2003, 107,
10873.(37) Annapureddy, H. V. R.; Hu, Z.; Xia, J.; Margulis, C. J. J. Phys.
Chem. B 2008, 112, 1770.
(38) Moreno, M.; Castiglione, F.; Mele, A.; Pasqui, C.; Raos, G. J. Phys.Chem. B 2008, 112, 7826.
(39) Jiang, W.; Wang, Y. T.; Voth, G. A. J. Phys. Chem. B 2007, 111,4812.
(40) Sieffert, N.; Wipff, G. J. Phys. Chem. B 2006, 110, 13076.(41) Padua, A. A. H.; Gomes, M. F. C.; Lopes, J. N. C. Acc. Chem.
Res. 2007, 40, 1087.(42) Chaumont, A.; Schurhammer, R.; Wipff, G. J. Phys. Chem. B 2005,
109, 18964.(43) Chevrot, G.; Schurhammer, R.; Wipff, G. Phys. Chem. Chem. Phys.
2006, 8, 4166.(44) Lynden-Bell, R. M.; Atamas, N. A.; Vasilyuk, A.; Hanke, C. G.
Mol. Phys. 2002, 100, 3225.(45) Deschamps, J.; Costa Gomes, M. F.; Padua, A. A. H. ChemPhy-
sChem 2004, 5, 1049.(46) Shah, J. K.; Maginn, E. J. J. Phys. Chem. B 2005, 109, 10395.(47) Shi, W.; Maginn, E. J. J. Phys. Chem. B 2008, 112, 2045.(48) Kelkar, M. S.; Shi, W.; Maginn, E. J. Ind. Eng. Chem. Res. 2008,
47, 9115.(49) Klahn, M.; Seduraman, A.; Wu, P. J. Phys. Chem. B 2008, 112,
10989.(50) Lopes, J. N. C.; Padua, A. A. H.; Shimizu, K. J. Phys. Chem. B
2008, 112, 5039.(51) Jorgensen, W. L.; Chandrasekhar, J.; Madura, J. D.; Impey, R. W.;
Klein, M. L. J. Chem. Phys. 1983, 79, 926.(52) Mahoney, M. W.; Jorgensen, W. L. J. Chem. Phys. 2000, 112, 8910.(53) Mahoney, M. W.; Jorgensen, W. L. J. Chem. Phys. 2001, 114.(54) Seduraman, A.; Klahn, M.; Wu, P. Calphad 2009, 33, 605.(55) Klahn, M.; Seduraman, A.; Wu, P. J. Phys. Chem. B 2008, 112,
13849.(56) Becke, A. D. J. Chem. Phys. 1993, 98, 5648.(57) Lee, C. T.; Yang, W. T.; Parr, R. G. Phys. ReV. B 1988, 37, 785.(58) Frisch, M. J. et al. Gaussian 03, ReVision C.02; Gaussian, Inc.:
Wallingford, CT, 2004. (Full reference is given in the Supporting Informa-tion.)
(59) van der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A. E.;Berendsen, H. J. C. J. Comput. Chem. 2005, 26, 1701.
(60) Breneman, C. M.; Wiberg, K. B. J. Comput. Chem. 1990, 11, 361.(61) Morrow, T. I.; Maginn, E. J. J. Phys. Chem. B 2002, 106, 12807.(62) Lopes, J. N. C.; Deschamps, J.; Padua, A. A. H. J. Phys. Chem. B
2004, 108, 2038.(63) Klahn, M.; Braun-Sand, S.; Rosta, E.; Warshel, A. J. Phys. Chem.
B 2005, 109, 15645.(64) Bhargava, B. L.; Balasubramanian, S. J. Chem. Phys. 2007, 127,
114510.(65) Allen, M. P.; Tildesley, D. J. Computer Simulations of Liquids;
Oxford Science Publications: Oxford, 1987.(66) Darden, T.; York, D.; Pedersen, L. J. Chem. Phys. 1993, 98, 10089.(67) Essmann, U.; Perera, L.; Berkowitz, M. L.; Darden, T.; Lee, H.;
Pedersen, L. G. J. Chem. Phys. 1995, 103, 8577.(68) Berendsen, H. J. C.; Postma, J. P. M.; DiNola, A.; Haak, J. R.
J. Chem. Phys. 1984, 81, 3684.(69) Gao, Y.; Arritt, S. W.; Twamley, B.; Shreeve, J. M. Inorg. Chem.
2005, 44, 1704.(70) Lee, F. S.; Chu, Z. T.; Warshel, A. J. Comput. Chem. 1993, 14,
161.(71) Chiappe, C.; Pieraccini, D. J. Phys. Org. Chem. 2005, 18, 275.(72) Ben-Naim, A.; Marcus, Y. J. Chem. Phys. 1984, 81, 2016.(73) Cammarata, L.; Kazarian, S. G.; Salter, P. A.; Welton, T. Phys.
Chem. Chem. Phys. 2001, 3, 5192.(74) McGowan, J. C. J. Appl. Chem. Biotech. 1978, 28, 599.(75) Zhao, Y. H.; Abraham, M. H.; Zissimos, A. M. J. Chem. Inf.
Comput. Sci. 2003, 43, 1848.(76) Kaun, N.; Ayora-Canada, M. J.; Lendl, B. J. Phys. Chem. B 2007,
111, 4446.(77) Bresme, F.; Alejandre, J. J. Chem. Phys. 2003, 118, 4134.(78) Huang, X. H.; Margulis, C. J.; Li, Y. H.; Berne, B. J. J. Am. Chem.
Soc. 2005, 127, 17842.(79) Ionic Liquids Technologies GmbH Homepage. http://www.iolitec.de
(accessed Nov. 1, 2009).(80) Knozinger, H. AdVances in Catalysis; Elsevier, Inc.: Amsterdam,
Netherlands, 2006; Vol. 49.(81) Steiner, T. Acta Crystallogr. 1998, B54, 456.(82) Valderrama, J. O.; Zarricueta, K. Fluid Phase Equilib. 2009, 275,
145.(83) Gu, Z.; Brennecke, J. F. J. Chem. Eng. Data 2002, 47, 339.
JP1000557
2868 J. Phys. Chem. B, Vol. 114, No. 8, 2010 Klahn et al.





























