Embed Size (px)
DESCRIPTION
固 相 反 应 第一节 引言 第二节 固相反应机理 第三节 固相反应动力学 第四节 影响固相反应的因素. 第一节 引 言. 一、 固态反应特征 基于研究结果,泰曼认为: ( 1 )固态物质间的反应是 直接进行 的,气相或液相没有或不起重要作用。 ( 2 )固态反应开始温度 远低于 反应物的熔点或系统的低共熔温度,通常相当于一种反应物开始呈现显著扩散作用的温度,此温度称为泰曼温度或烧结温度。. 泰曼的观点长期以来一直为学术界所普遍接受。但随着生产和科学实验的进展,发现许多问题。 - PowerPoint PPT Presentation
Citation preview

固 相 反 应第一节 引言 第二节 固相反应机理 第三节 固相反应动力学 第四节 影响固相反应的因素

一、 固态反应特征 基于研究结果,泰曼认为:( 1 )固态物质间的反应是直接进行的,气相或液相
没有或不起重要作用。( 2 )固态反应开始温度远低于反应物的熔点或系统
的低共熔温度,通常相当于一种反应物开始呈现显著扩散作用的温度,此温度称为泰曼温度或烧结温度。
第一节 引 言

( 3 )当反应物之一存在有多晶转变时,则转变温度通常也是反应开始明显进行的温度,这一规律也称为海得华定律。
泰曼的观点长期以来一直为学术界所普遍接受。但随着生产和科学实验的进展,发现许多问题。 因此,金斯特林格等人提出,固态反应中,反应物可能转为气相或液相,然后通过颗粒外部扩散到另一固相的非接触表面上进行反应。指出了气相或液相也可能对固态反应过程起重要作用。并指出这些反应有如下一些共同的特点:

固相反应的共同特点:( 1 )固态反应一般包括相界面上的反应和物质迁移两个过程。( 2 )固态反应通常需在高温下进行。而且由于反应发生在非均相系统,因而传热和传质过程都对反应速度有重要影响。

二、原子迁移
1 、固态物质点的可动性2 、质点的迁移形式

第二节 固相反应机理
相界面上化学反应机理相界面上反应和离子扩散的关系中间产物和连续反应不同反应类型和机理

固态反应一般是由相界面上的化学反应和固相内的物质迁移两个过程构成。但不同类型的反应既表现出一些共性规律,也存在着差异和特点。
一 相界面上化学反应机理 傅梯格( Hlütting )研究了 ZnO和 Fe2O3 合成的反应过程。图 1 示出加热到不同温度的反应化合物,经迅速冷却后分别测定的物性变化结果。可把整个反应过程划分为六个阶段。
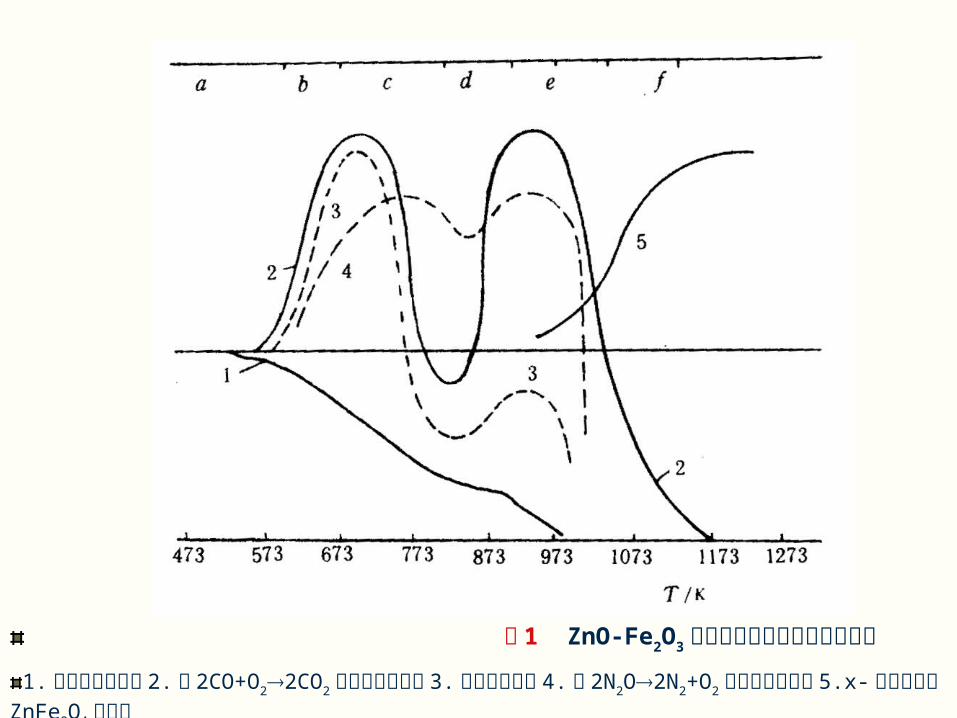
图 1 ZnO-Fe2O3 混合物加热过程中性质的变化
1. 对色剂的吸附性 2.对 2CO+O22CO2 反应的催化活性 3. 物系的吸湿性 4.对 2N2O2N2+O2 反应的催化活性 5.x- 射线图谱上 ZnFe2O4 的强度

( 1 )隐蔽期:约低于 300℃ 。( 2 )第一活化期:约在 300~400℃ 之间。( 3 )第一脱活期:约在 400~500℃ 之间。( 4 )二次活化期:约在 500~620℃ 之间。( 5 )二次脱活期或晶体形成期:约在620~750℃ 之间。( 6 )反应产物晶格校正期:约> 750℃ 。

当然,对不同反应系统,并不一定都划分成上述六个阶段。但都包括以下三个过程:( 1 )反应物之间的混合接触并产生表面效应;( 2 )化学反应和新相形成;( 3 )晶体成长和结构缺陷的校正。
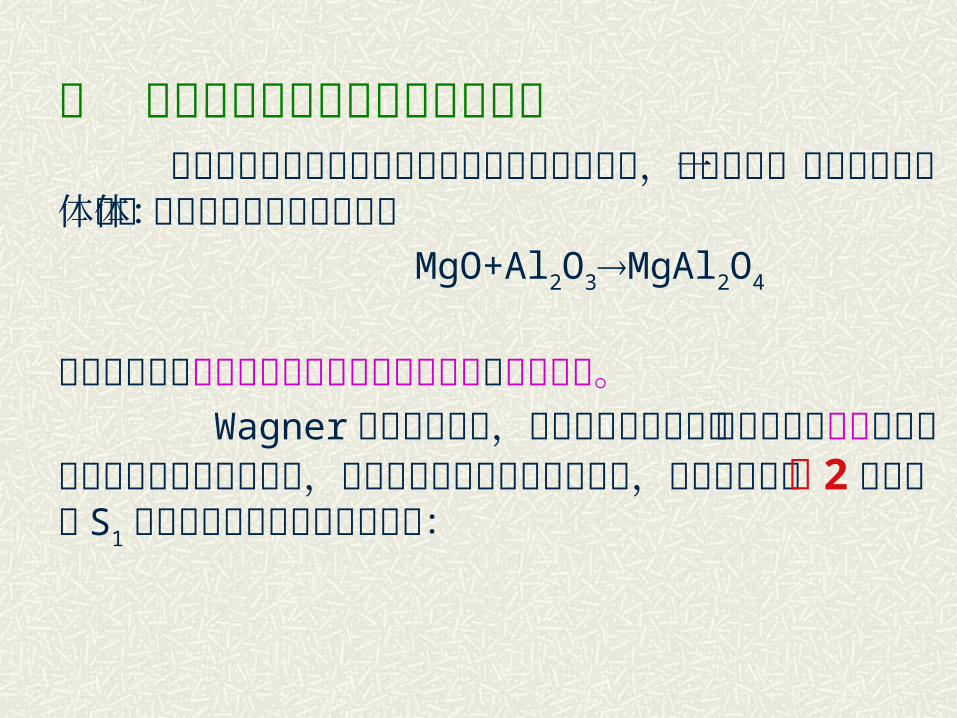
二 相界面上反应和离子扩散的关系 以尖晶石类三元化合物的生成反应为例进行讨论,尖晶石是一类重要的铁氧体晶体反应式可以下式为代表: MgO+Al2O3MgAl2O4
这种反应属于反应物通过固相产物层中扩散的加成反应。 Wagner 通过长期研究,提出尖晶石形成是由两种正离子逆向经过两种氧化物界面扩散所决定,氧离子则不参与扩散迁移过程,按此观点则在图 2 中在界面S1 上由于扩散过来必有如下反应:
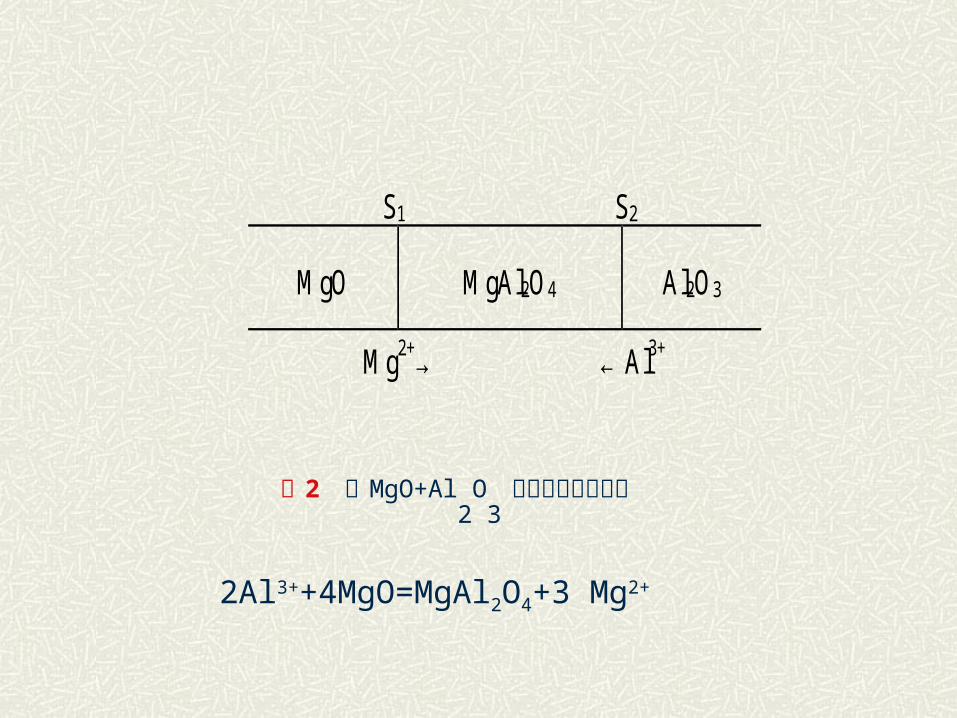
S1 S2
MgO MgAl2O4 Al2O3
Mg2+→ ← Al3+
图 2 由 MgO+Al2O
3形成尖晶石示意图
2Al3++4MgO=MgAl2O4+3 Mg2+

反应物的离子的扩散需要穿过相的界面以及穿过产物的物相。反应产物中间层形成之后,反应物离子在其中的扩散便成为这类尖晶石型反应的控制速度的因素。因为决定反应速度的是扩散的离子流,所以可以有: J 1/x dx/dt∝ ∝
对此式积分便得到抛物线增长定律。

三 中间产物和连续反应
在固态反应中,有时反应不是一步完成,而是经由
不同的中间产物才最终完成,这通常称为连续反应。
例如 :CaO和 SiO2 的反应,尽管配料的摩尔比为 1:1 ,
但反应首先形成 C2S, C3S 等中间产物,最终才转变
为 CS 。其反应顺序和量的变化如图 3 所示。
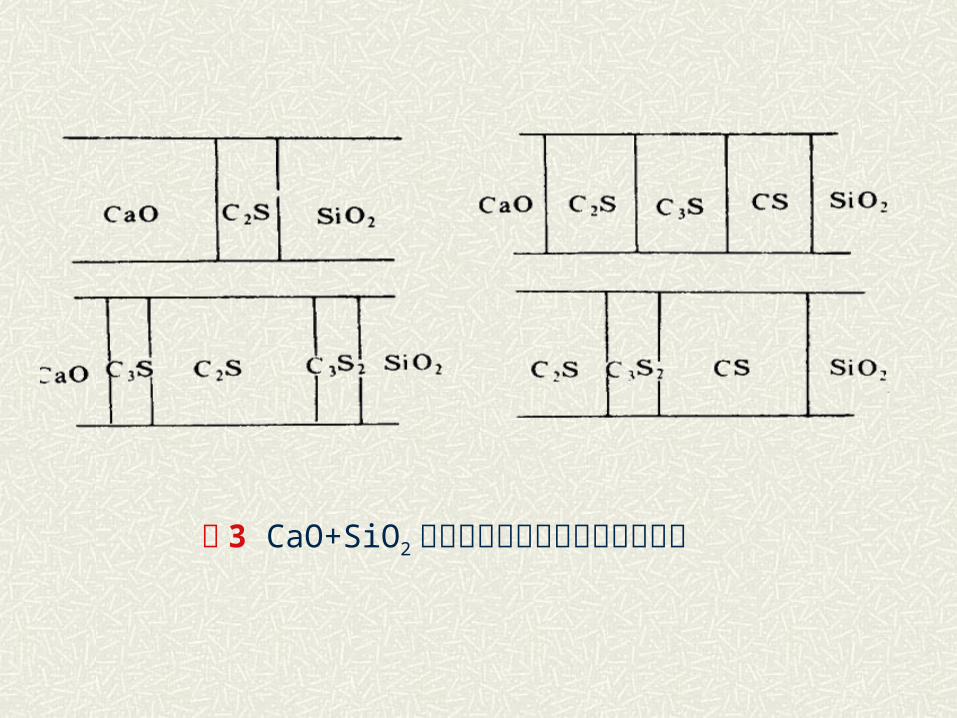
图 3 CaO+SiO2 反应形成多钙硅酸盐过程示意图

四 不同反应类型和机理1. 加成反应 一般形式为: A+B→C
当化合物 C 不溶于 A或 B 中任一相时,则在 A、B 两层间就形成产物层 C 。当 C与 A或 B 之间形成部分或完全互溶时,则在初始反应物中生成一个或两个新相。当 A与 B 形成成分连续变化的产物时,则在反应物间可能形成几个新相。作为这类反应的一个典型代表,是尖晶石生成反应:
AO+B2O3→AB2O4

2. 造膜反应 这类反应实际上也属于加成反应,但 A、 B 常是单质元素。若生成物 C 不溶于 A、 B 中任一相,或能以任意比例固溶,则产物中排列方式分别为 :
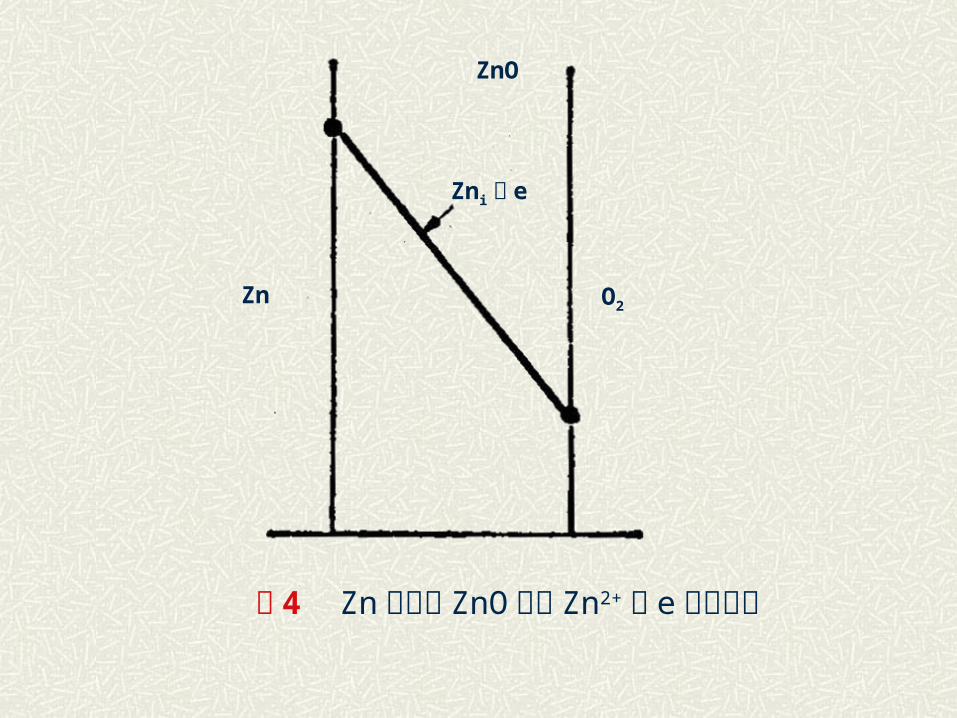
A│C│B, A( B )│ B及 A│B( A )。例如 : Zn+O2→ZnO
伴随上述反应进行,系统自由焓减少,即气相中 O2 的化学位 μa
与 Zn-ZnO 界面上平衡氧的化学位 μi 的差值是此反应的推动力。当氧化膜增厚速度由扩散控制时,上述氧的化学位降低将在氧化膜中完成,相关离子的浓度分布如图 4 所示。
ZnO
Zni或 e
ZnO2
图 4 Zn 氧化的 ZnO 层内 Zn2+及 e 浓度分布

3. 置换反应 置换反应是另一类重要的固态反应,其反应通式为 :
A+BC→AC+B; AB+CD→AD+BC;ABX+CB→CBX+AB
这时反应物必须在两种产物层中扩散才能使反应继续进行。并将形成种种反应物与生成物的排列情况。 产物层排列主要取决于反应物的扩散组元、产物与反应物的固溶性等。对于三组分以上的多元系统,则产物层的排列就更复杂。

4. 转变反应特点 :
( 1 )反应仅在一个固相内进行,反应物或生成物不必参与迁移;( 2 )反应通常是吸热的,在转变点附近会出现比热值异常增大。对于一级相变,熵变是不连续的;对于二级相变则是连续的。由此可见,传热对转变反应速度有着决定性影响。石英的多晶转变反应是硅酸盐工业种最常见的实例。
5. 热分解反应
这类反应常伴有较大的吸热效应,并在某一狭窄范围内迅速进行,所不同的是热分解反应伴有分解产物的扩散过程。

第三节 固相反应动力学
一般动力学关系化学动力学范围扩散动力学范围
一、一般动力学关系
整个过程的速度将由其中速度最慢的一
环控制。现以金属氧化反应 M +1/2O 2→
MO 为例(图 5 )说明之。
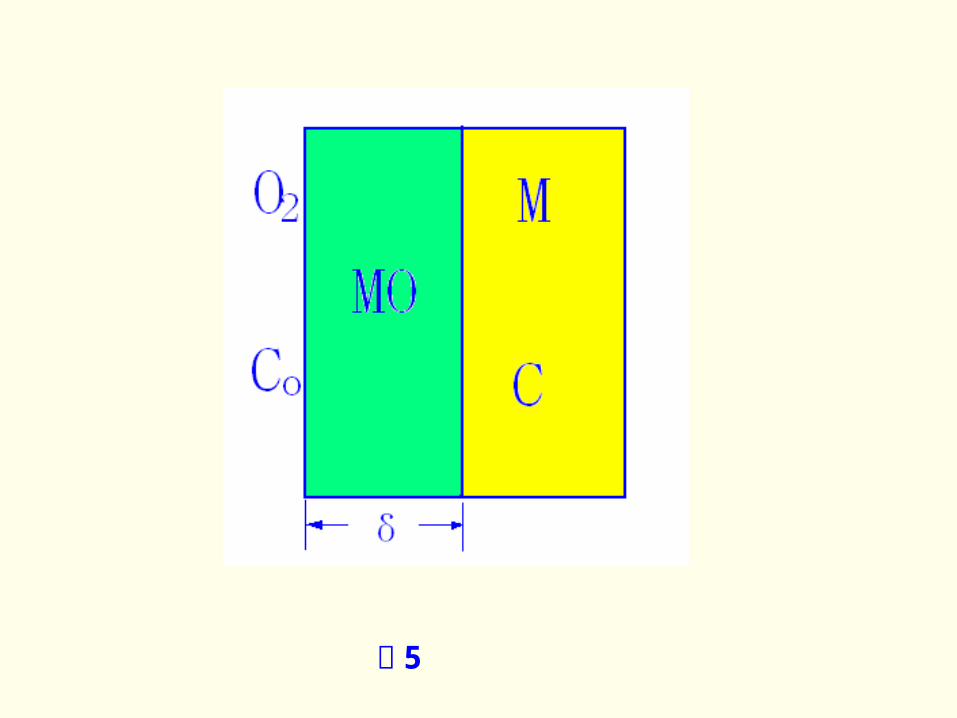
图 5
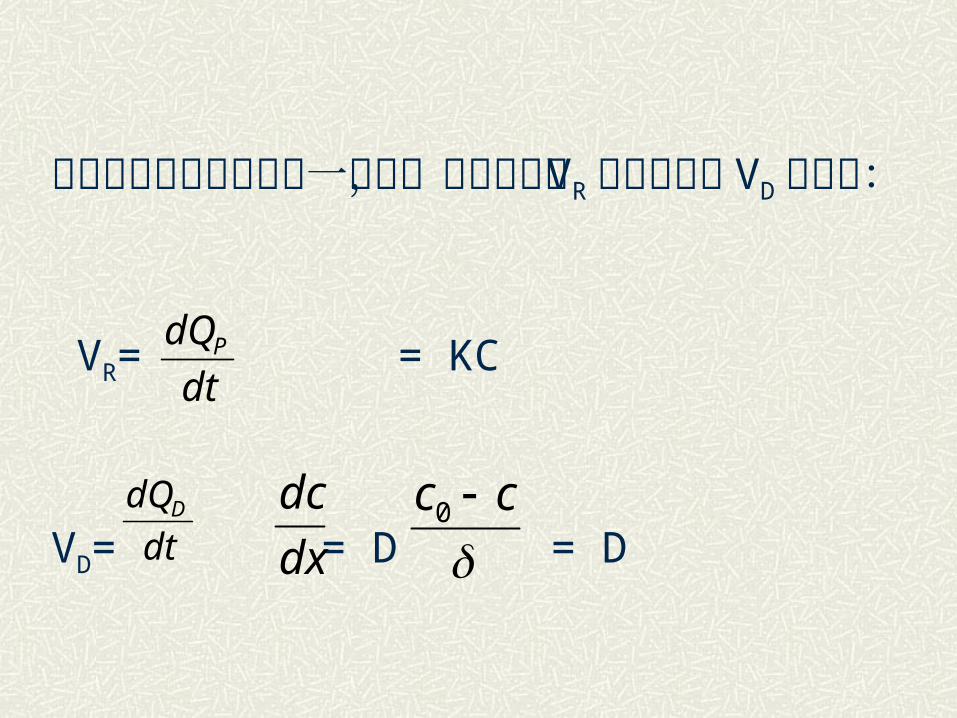
由化学动力学和菲克第一定律,其反应速度VR 和扩散速度 VD 分别为:
VR= = KC
VD= = D = D
dt
dQP
dt
dQD
dx
dc
cc 0
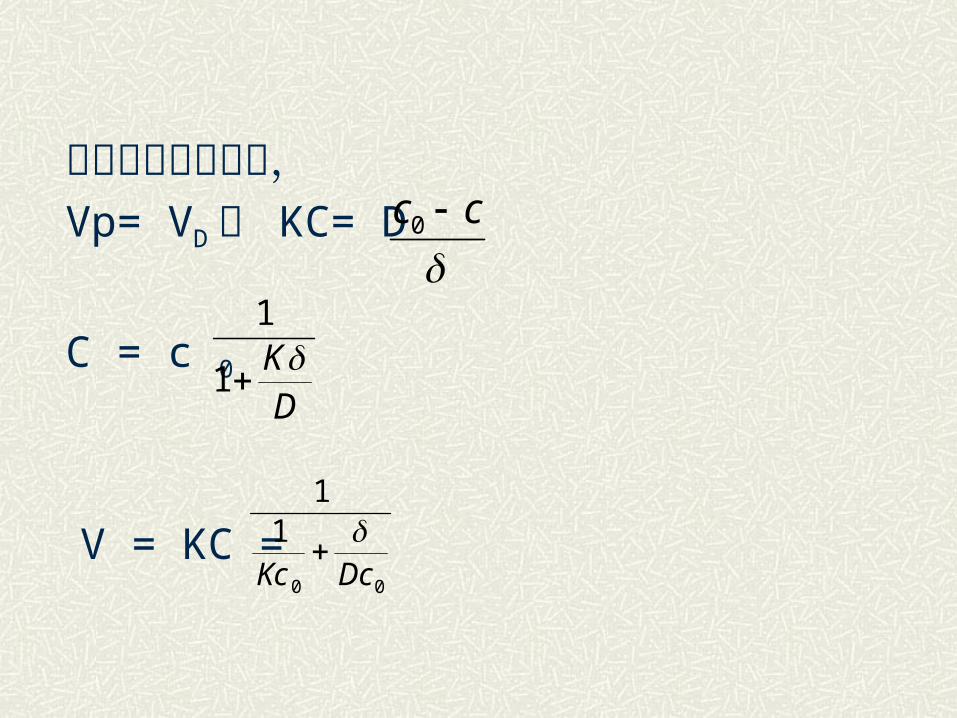
当过程达到平衡时, Vp= VD 或 KC= D
C = c 0
V = KC =
cc 0
DK1
1
00
11
DcKc
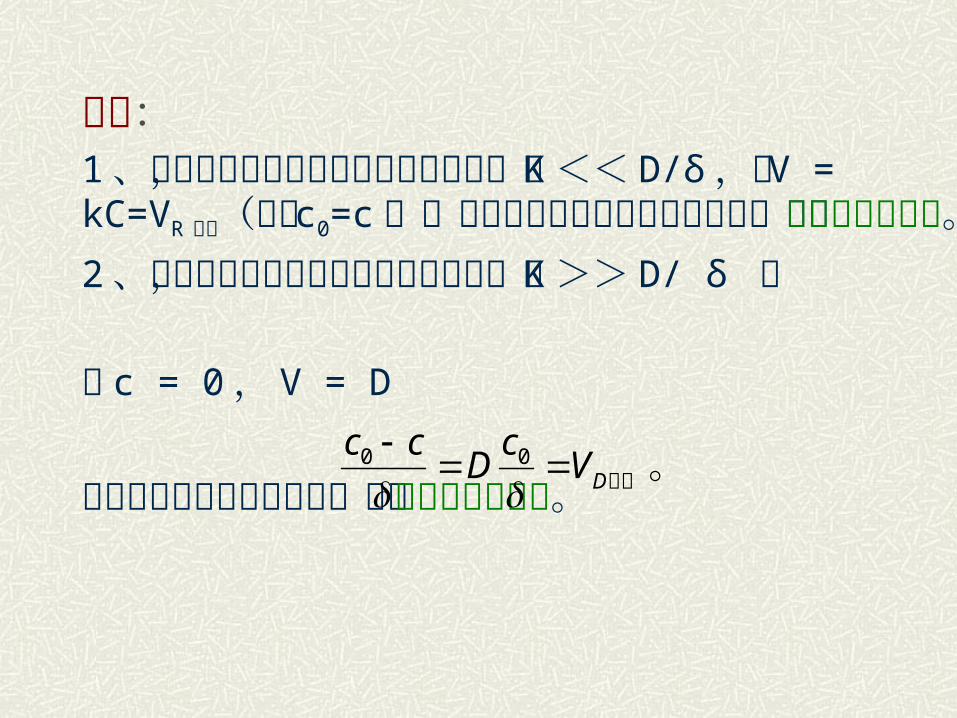
讨论:1 、当扩散速度远大于化学反应速度时,即 K <<D/δ ,则 V = kC=VR 最大(式中 c0=c ),说明化学反应速度控制此过程,称为化学动力学范围。2 、当扩散速度远小于化学反应速度时,即 K >> D/ δ ,
即 c = 0, V = D
说明扩散速度控制此过程,称为扩散动力学范围。
。最大DVc
Dcc
00
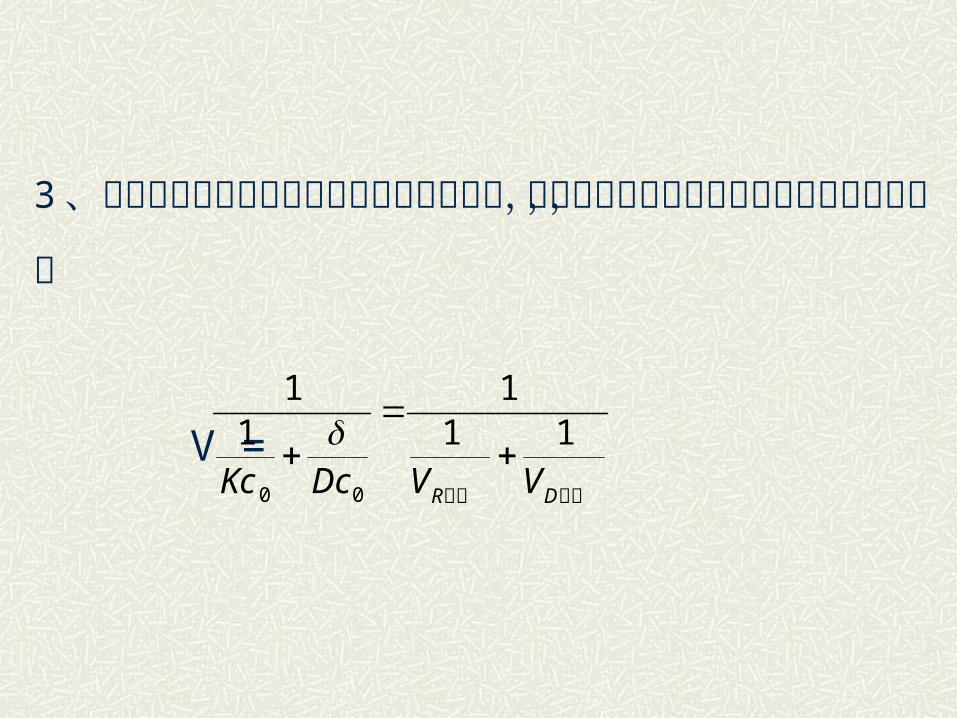
3 、当扩散速度远和化学反应速度可相比拟时,则过程
速度由上式确定,称为过渡范围,即
V =最大最大 DR VVDcKc11
11
1
00

二 化学动力学范围1. 此过程的特点是:反应物通过产物层的扩散速度远大于接触面上的化学反应速度。过程总的速度由化学反应速度所控制。2. 均相二元系统化学反应速度的表达式对于均相二元系统,化学反应速度的一般表达式是 V = KcA
mc Bn
对于反应过程中只有一个反应物的浓度是可变的,则上式可简化为: V = Kncn( n≠1, n> 0 )
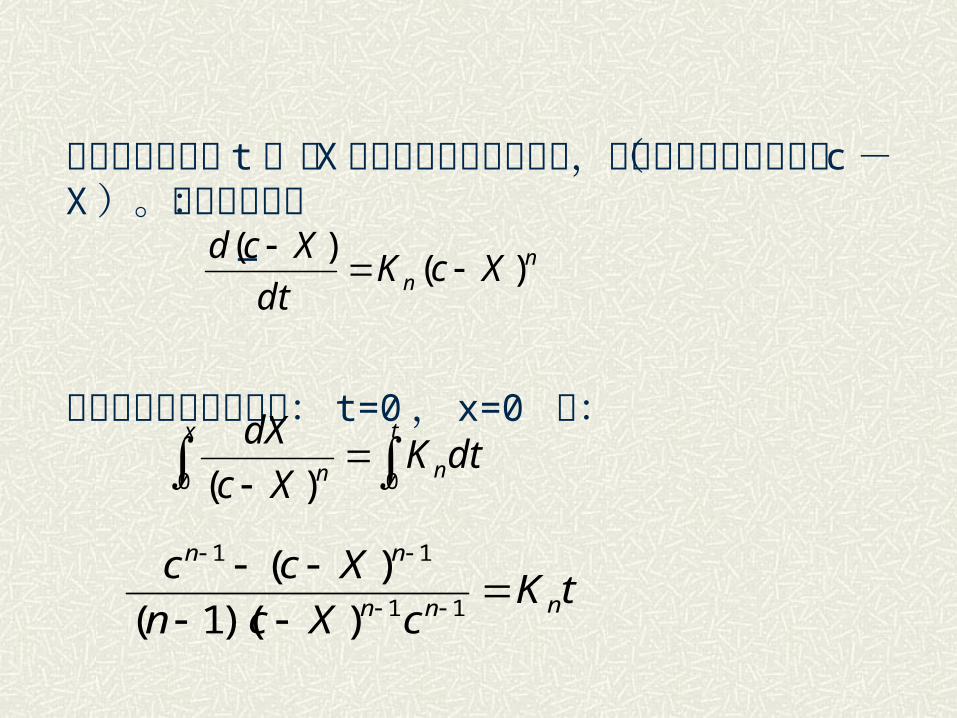
令经过任意时间 t ,有 X 部分反应物消耗于反应,而剩下的反应物量为( c- X )。上式可写成: —
积分并考虑到初始条件: t=0, x=0 得:
nn XcK
dt
Xcd)(
)(
dtKXc
dXx t
nn 0 0)(
tKcXcn
Xccnnn
nn
11
11
))(1(
)(
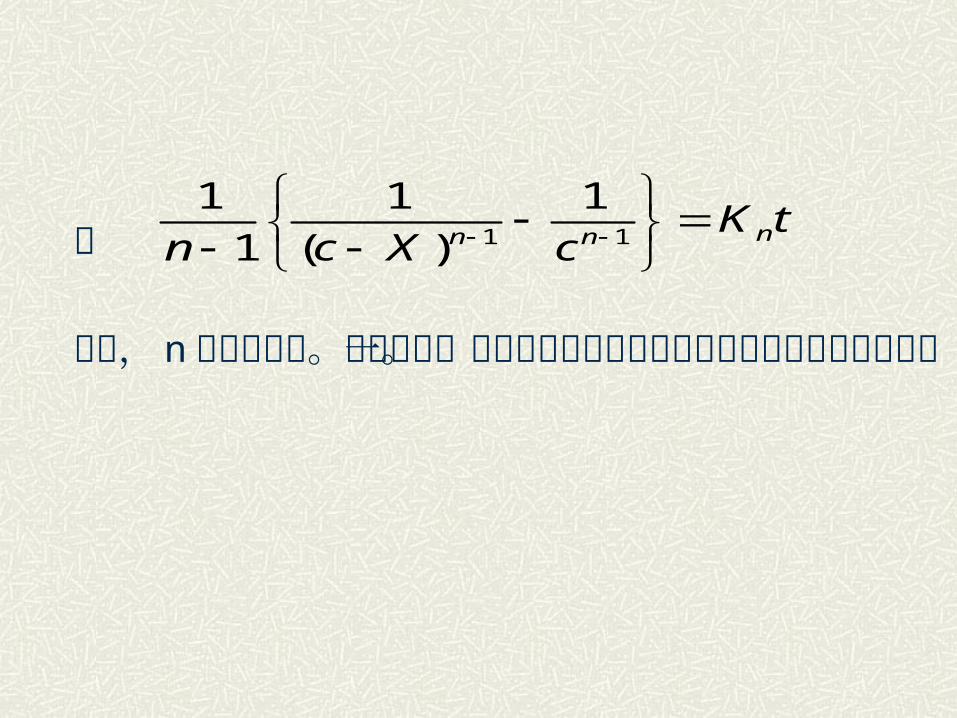
或
这里, n 是反应级数。故给出除一级以外的任意级数的这类反应的动力学积分式。
tKcXcn nnn
11
1
)(
1
1
1
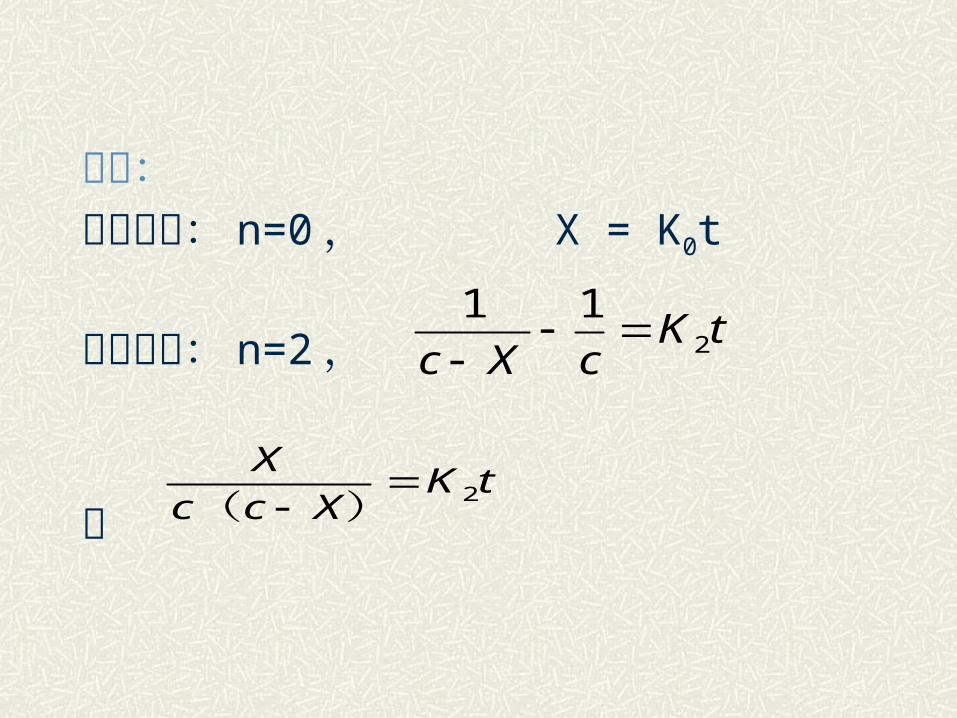
讨论:零级反应: n=0 , X = K0t
二级反应: n=2 ,
或
tKcXc 2
11
tKXcc
X2
)(
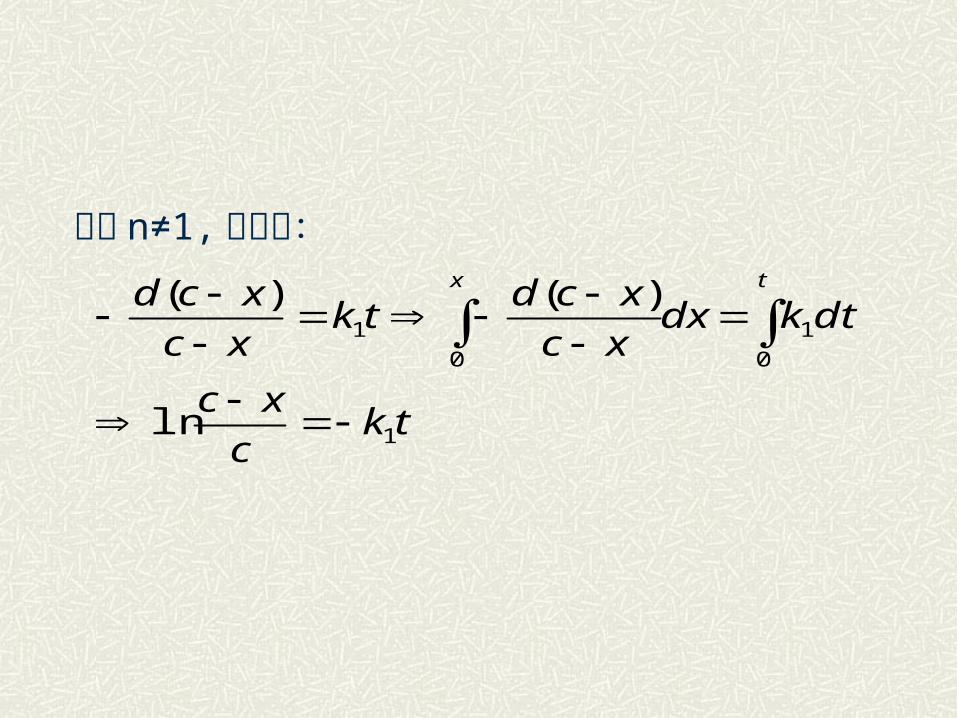
对于 n≠1, 可求得:
tkc
xc
dtkdxxc
xcdtk
xc
xcd tx
1
0
1
0
1
ln
)()(
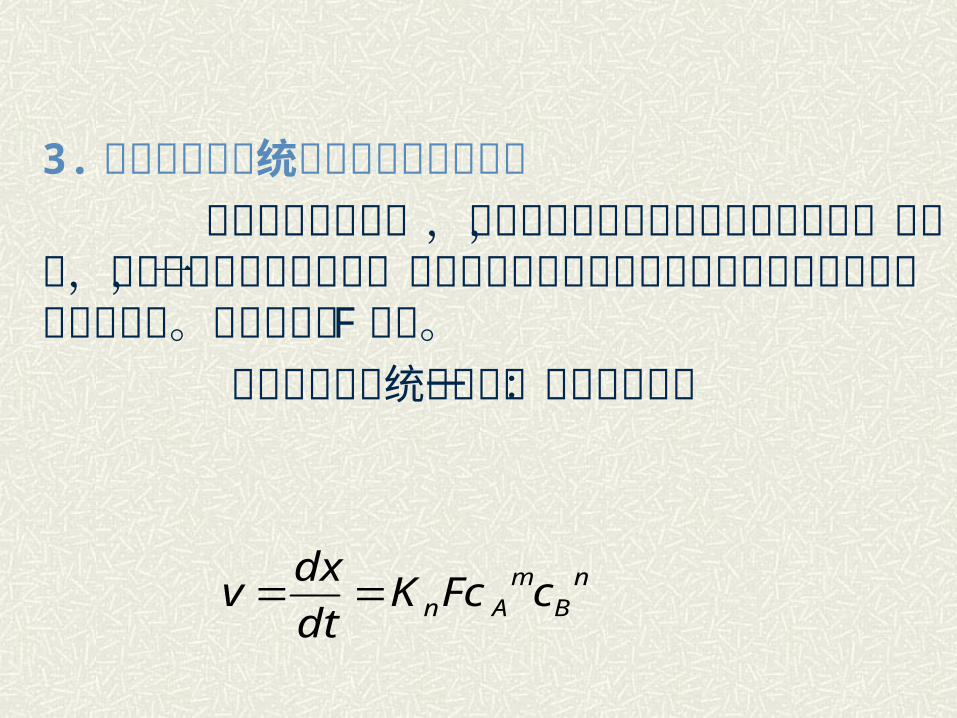
3. 非均相二元系统化学反应速度表达式 对于均相二元系统,计算过程中未考虑接触面积的影响,实际上,固相反应层非均相反应,接触面积在反应过程是影响固相反应速度的一个重要参数。接触面积用 F 表示。 非均相二元系统反应的一般速度方程:
nB
mAn cFcK
dt
dxv
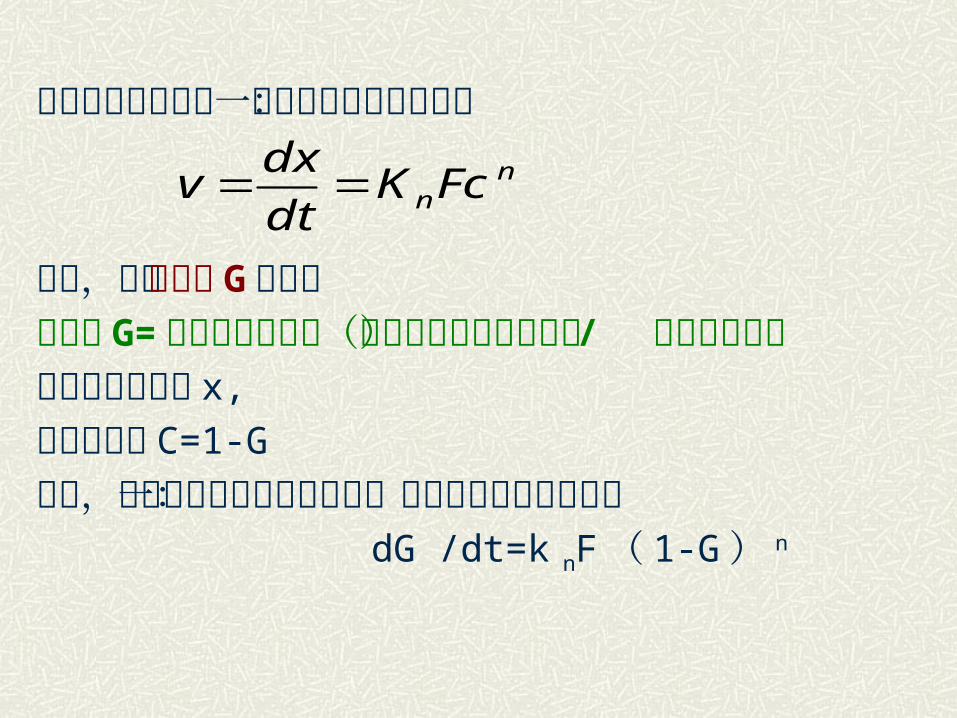
当反应过程中只有一个反应物浓度可变时:
下面,引入转化率 G 的概念转化率 G= 转化的反应物量(或消耗掉的反应物量) / 原始反应物量消耗掉的反应物 x,
反应物浓度 C=1-G
那么,二元系统非均相化学反应一般速度方程即可写成: dG /dt=k nF( 1-G) n
nnFcK
dt
dxv

4. 接触面积 F 的计算设反应物为球形颗粒,半径为 R0 ,经 t 时间反应后反应层消耗掉厚度为 x ,则经 t 时间的转化率为 G
G =
R0—x = R0 ( 1—G) 1/3 或 x = R0[1-( 1- G) 1/3]
相应于每个颗粒的反应表面积 F’ 与转化程度 G 的关系:
F’=4πR02( 1—G) 2/3
30
30
30 (
R
xRR )

5. 化学反应控制范围的动力学方程1 )一级反应 n=1
将上式反应级数 n 代入数值,就可求出不同级数反应的微积分形式:
d G/dt= K 4πR02 ( 1-G) 2/3 ( 1-G) 1
=K 1( 1-G) 5/3
nn
nn GGkGFk
dt
dG)1()1(R4)1( 3/22
O
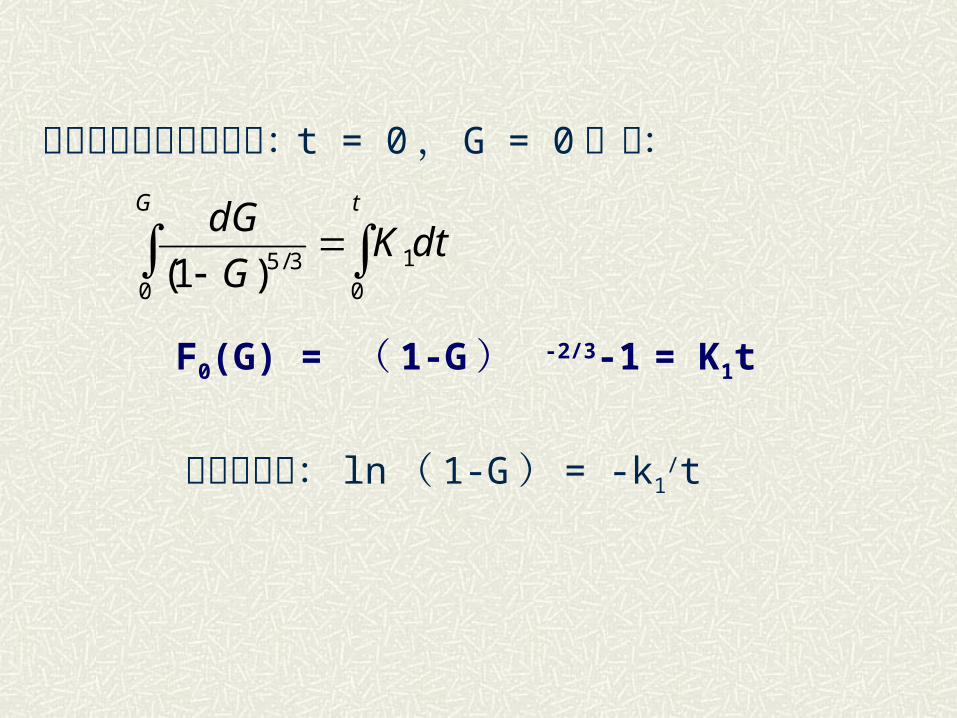
积分并考虑到初始条件: t = 0, G = 0 ,得 :
F0(G) = ( 1-G ) -2/3-1 = K1t
平板状颗粒: ln( 1-G) = -k1/t
tG
dtKG
dG
0
1
03/5)1(

6. 实验验证
如何验证上述动力学是正确的?如果我们能够使扩散阻力很小,这时扩散很快,反应为化学反应所控制。实验上常采取降低反应物颗粒度,再加入助熔剂来使反应
处于化学动力学范围。如 NaCO3: SiO2=1: 1 ,进行
固相反应,其动力学是化学反应控制的一级反应。

三 扩散动力学范围
1. 过程特点
扩散速度很慢,起控制作用,为整个固相反应中速度最慢的一步。
在多数情况下 , 扩散速度往往起控制作用。

2. 动力学方程
( 1 )抛物线型速度方程——平板模型
此方程可从平板扩散模型导出。如图 6 所示。
若化学反应速度远大于扩散速度,则过程由扩散控制。经 dt 时间,通过单位 AB 层迁移的 A 物质量为 dm ,浓度梯度为 dc/dx ,则按菲克定律有:
( 1 )dx
dcD
dt
dm
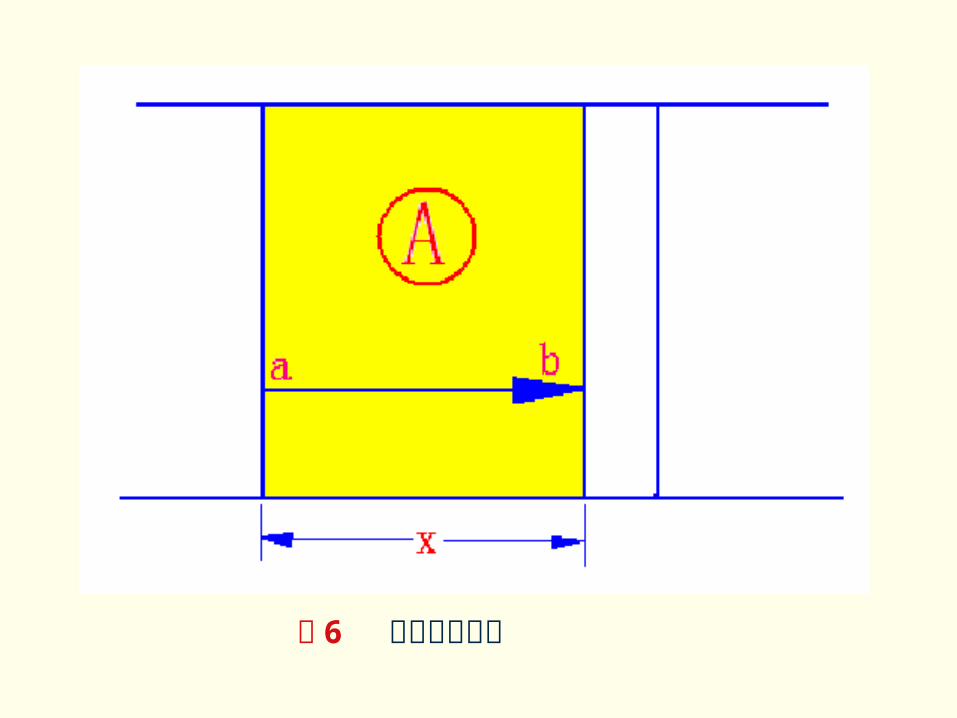
图 6 平板扩散模型
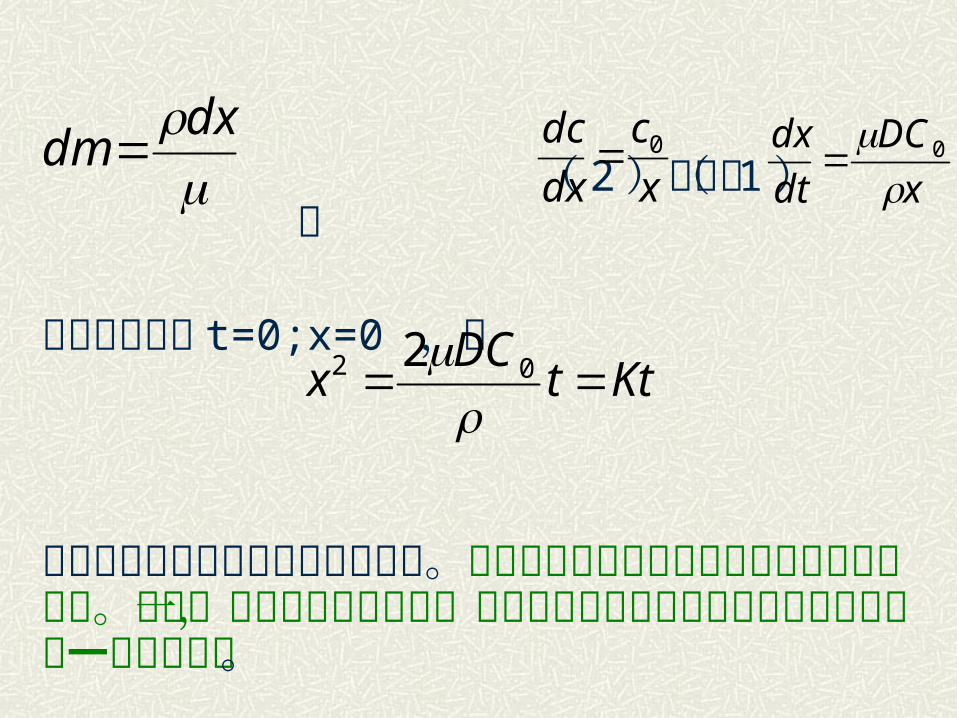
( 2 )代入( 1 ) 则
考虑边界条件 t=0;x=0 ,故
上式即为抛物线速度方程的积分式。说明反应产物层厚度与时间的平方根成比例。这是一个重要的基本关系,可以描述各种物理或化学的控制过程并有一定的精确度。
dx
dmx
c
dx
dc 0x
DC
dt
dx
0
KttDC
x
02 2
实验验证:
图 7 示出的金属镍氧化时的增重曲线就是一个例证。
局限性:
但是,由于采用的是平板模型,忽略了反应物间接触面积随时间变化的因素,使方程的准确度和适用性都受到局限。
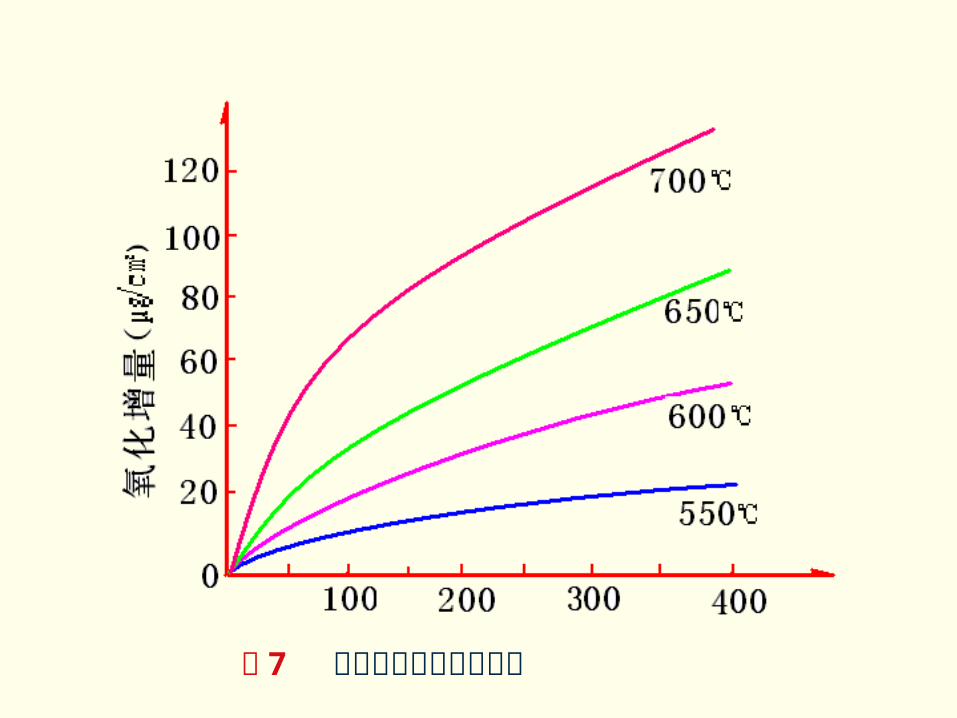
图 7 金属镍的氧化增重曲线

( 2 )杨德方程——球体模型 在材料生产中通常采用粉状物料作为原料,这时,在反应过
程中,颗粒间接触界面积是不断变化的。 为此,扬德在抛物线速度方程基础上采用了“球体模型”导
出了扩散控制的动力学关系。A.扬德假设: a )反应物是半径为 R 的等径球粒; b )反应物 A 是扩散相,即 A 成分总是包围着 B 的颗粒,而
且 A、 B 同产物 C 是完全接触的,反应自球表面向中心进行;
c) A 在产物层中的浓度是线性的,而扩散层截面积一定;
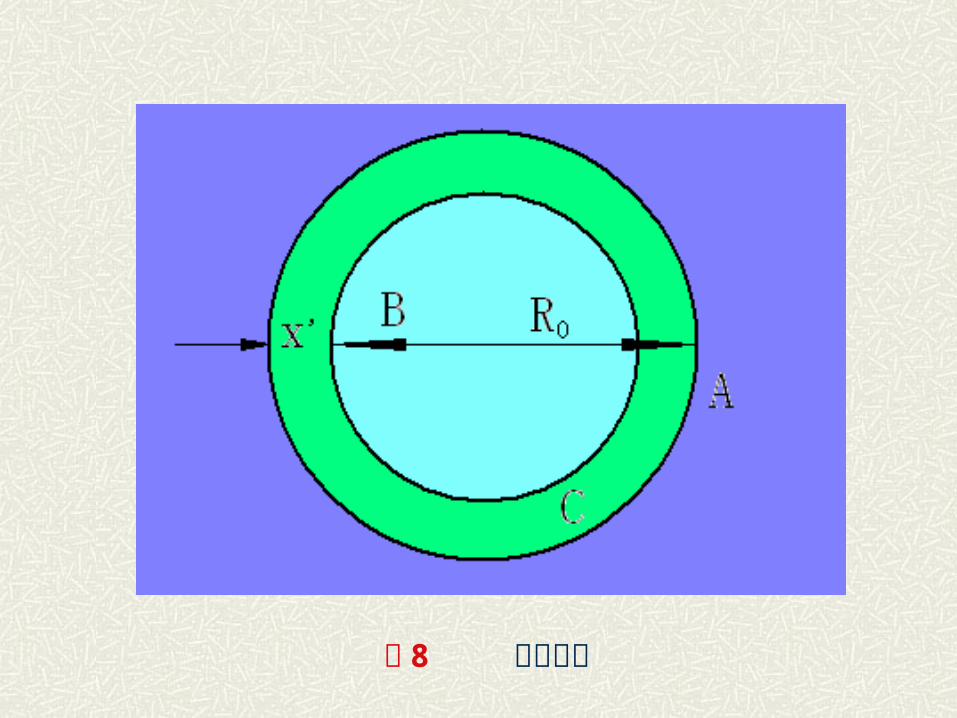
图 8 杨德模型
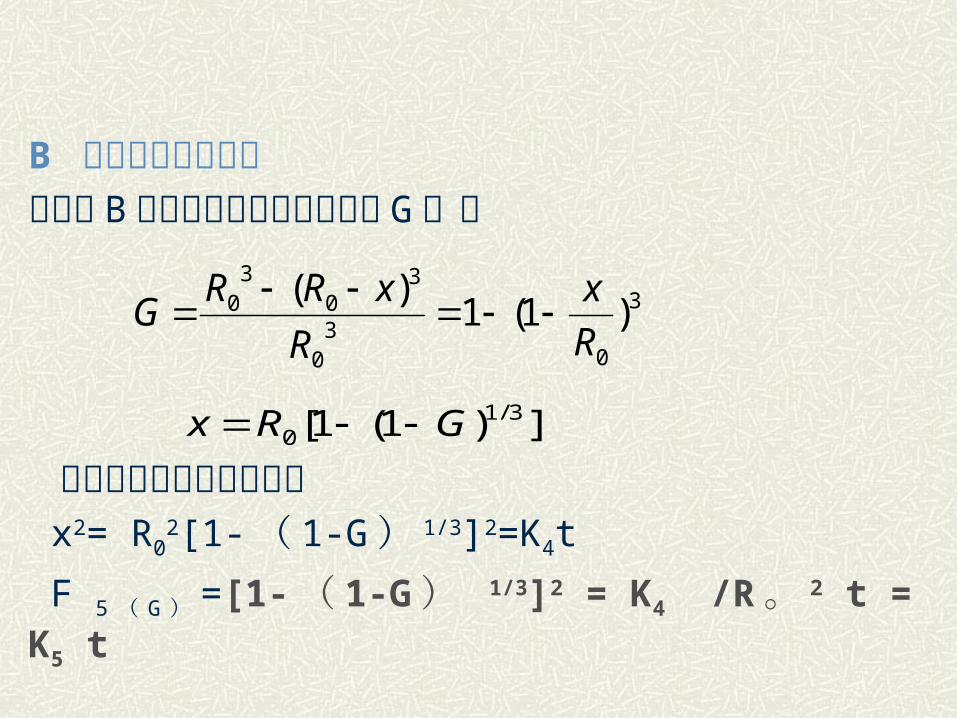
B 动力学方程的推导现令以 B 物质为基准的转化程度为 G ,则
代入抛物线速度方程式得 x2= R0
2[1-( 1-G) 1/3]2=K4t
F 5 ( G ) =[1-( 1-G ) 1/3]2 = K4 /R。 2 t = K5 t
3
03
0
30
30 )1(1
)(
R
x
R
xRRG
])1(1[ 3/10 GRx
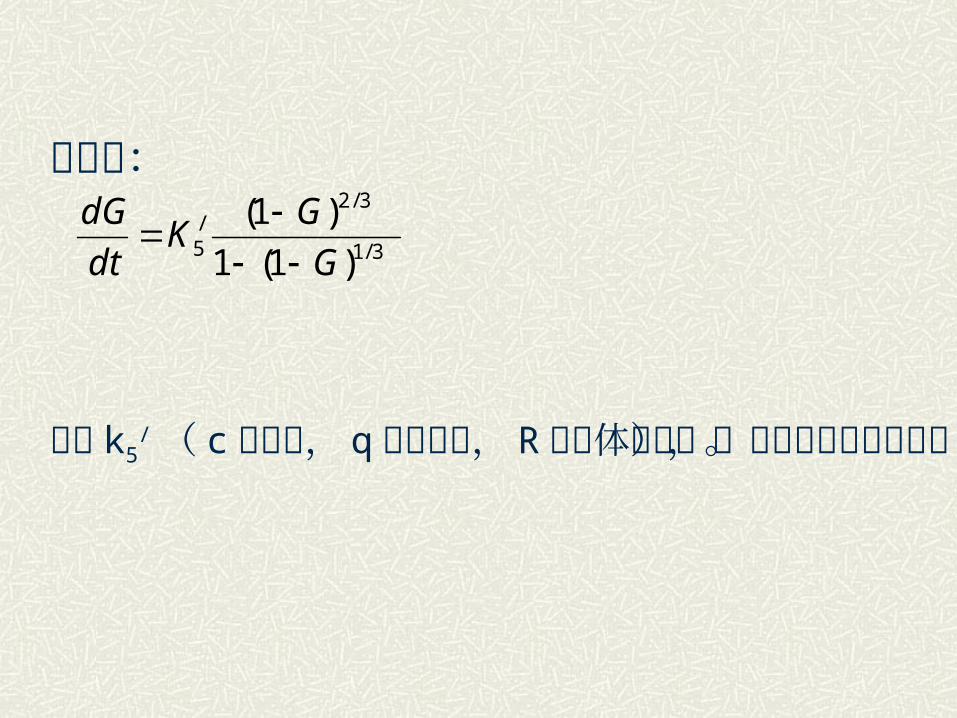
微分得:
其中 k5
/( c 是常数, q 是活化能, R 是气体常数),也称杨德速度常数。
3/1
3/2/5 )1(1
)1(
G
GK
dt
dG

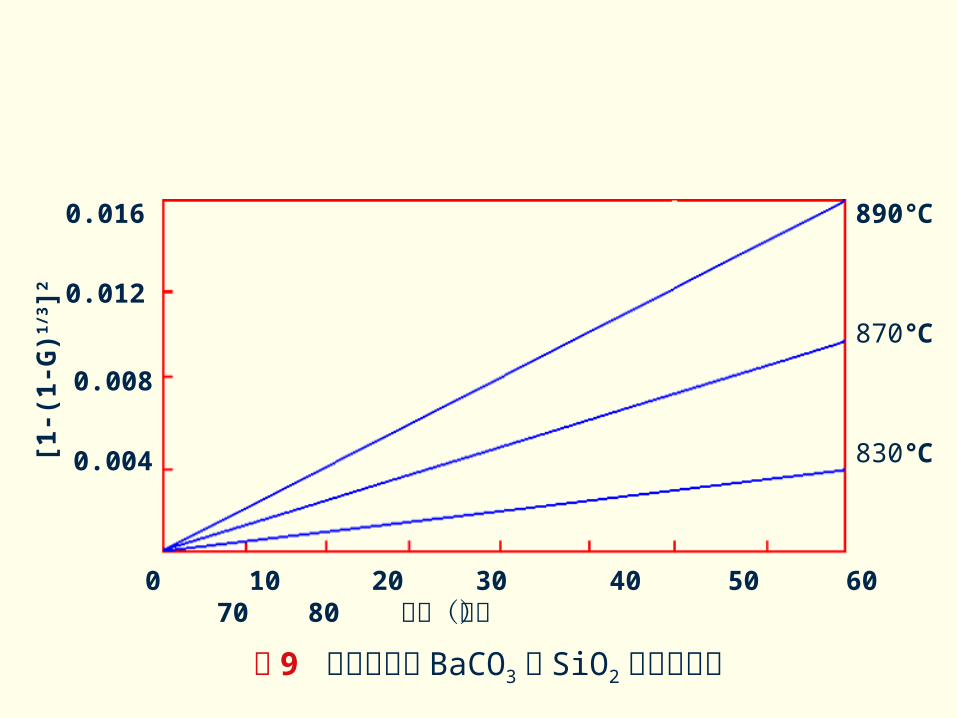
C )实验验证
对于反应 BaCO3 + SiO2→ BaSiO3+CO2 的实测结果
示于图 9 。由图可见,随着反应温度的升高,反应强度规律也提高了,但都很好地符合杨德方程。
0.016
0.012
0.008
0.004
0 10 20 30 40 50 60 70 80
[1-(
1-G
)1/3]2
时间(分)
890℃
870℃
830℃
图 9 不同温度下 BaCO3与 SiO2 的反应情况

D 、杨德方程的适用范围——反应初期、 G较小时
对碳酸盐和氧化物间的一系列反应进行实验研究,发现在反应初期都基本符合杨德方程式,而后偏差就愈来愈大。为什么会这样呢?
原因是杨德方程虽然采用了球体模型,在计算产物厚度时考虑了接触界面的变化,即利用反应前后球体之体积差算出产物层厚度 x 。但在将 x值代入抛物线方程时实质上又保留了扩散面积恒定的假设,这是导致其局限性的主要原因。

( 3 )金斯特林格方程——三维球体模型 金斯特林格采用了杨德的球状模形,但放弃了扩散截面不变的假设从而导出了更有普遍性的新动力学关系。A 金斯特林格假设: a )假设反应 A 是扩散相, B 是平均半径为 R 。的球形颗粒,反应沿 B 整个球表面同时进行,首先, A和 B 形成产物 AB ,厚度为 x, x随反应进行而增厚 b) A 扩散到 A-AB 界面的阻力远小于通过 AB 层的扩散阻力,则 A-AB 界面上 A 的浓度不变为 C 。,因扩散控制则 A在 B-
AB 界面上的浓度为 0
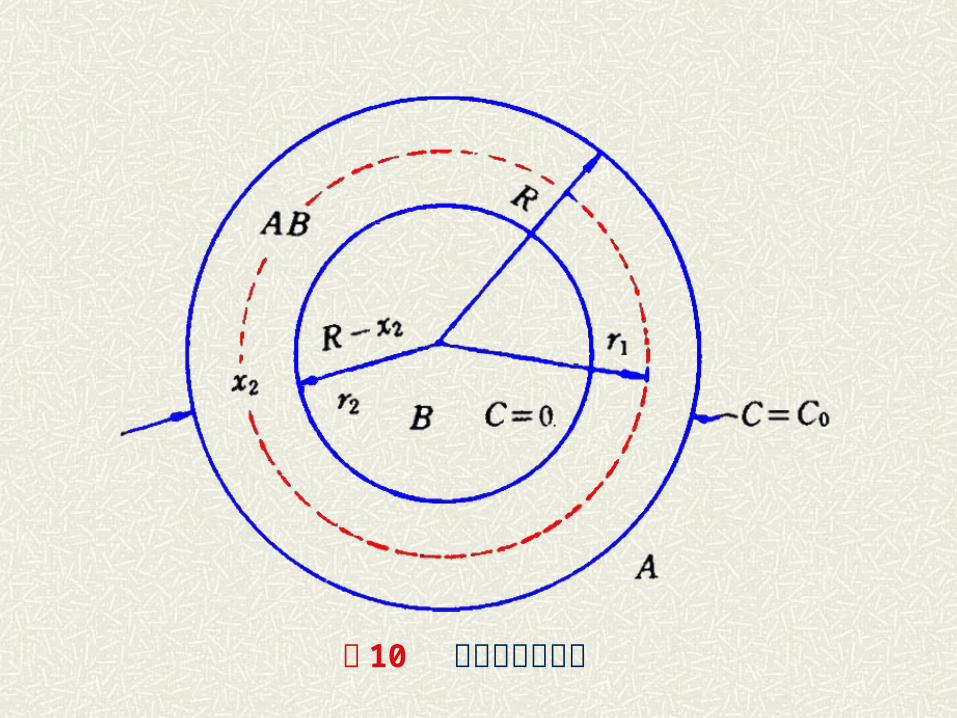
图 10 金斯特林格模型

B 方程推导 由于粒子是球形的,产物两侧界面 A 的浓度不变,故随产物层增厚, A 在层内的浓度分布是 r 和时间 t 函数,即过程是一个不稳定扩散问题,可以用球面坐标情况下的菲克扩散方程描述:
根据初始和边界条件 r = R t> 0, C(R0,t)= C 0
r = R0-x t> 0, C (R0-x,t) = 0
t=0 , x=0
]2
[),(
2
2
)(r
C
rr
CD
t
tr
xRrr
CD
dt
dx
0)(

式中 是比例常数,其中和分别是产物 AB 的比重和分子量, n 是反应的化学计量常数,即和一个 B 分子化合所需的A 的分子数, D 是 A 在 AB 中的扩散系数。
求解得:
F6(G)=1—2/3G—( 1-G) 2/3=K6t
许多试验研究表明,金斯特林格方程具有更好的普遍性。
n
3/1
3/1
3/1
3/1
6 )1(1
)1(
)1(1
)1(
G
GK
G
GK
dt
dGr

杨德方程与金斯特林格方程之比较 , 得:
按上式令 对 G 作图可得图 11曲线。可见,当 G 值较小即转化程度较低时,说明两方程是基本一致的,反之,随 G 值增加,两式偏差越来越大。 可见,杨德方程只是在转化程度较小时适用。当 G 值较大时, Kj将随 G 的增大而增大,而金斯特林格方程则在一定程度上克服了杨德方程的局限。
3/13/2
3/1
)1()1(
)1(
)(
)(
GGK
GK
dtdGdtdG
i
r
i
r
jr dt
dG
dt
dG)/()
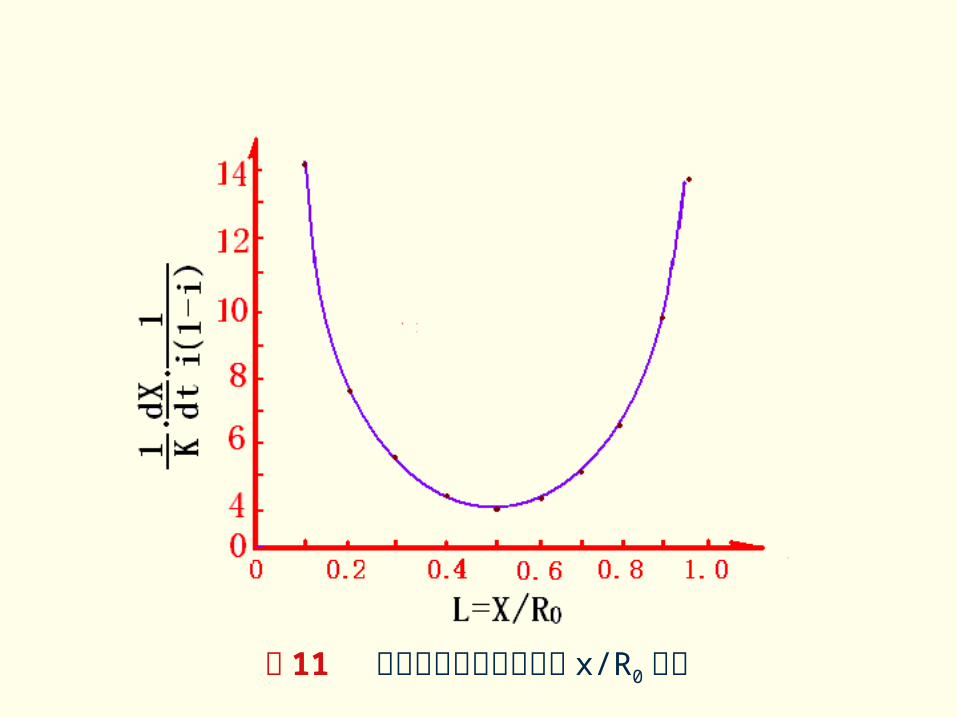
图 11 反应产物层增厚速率与 x/R0关系
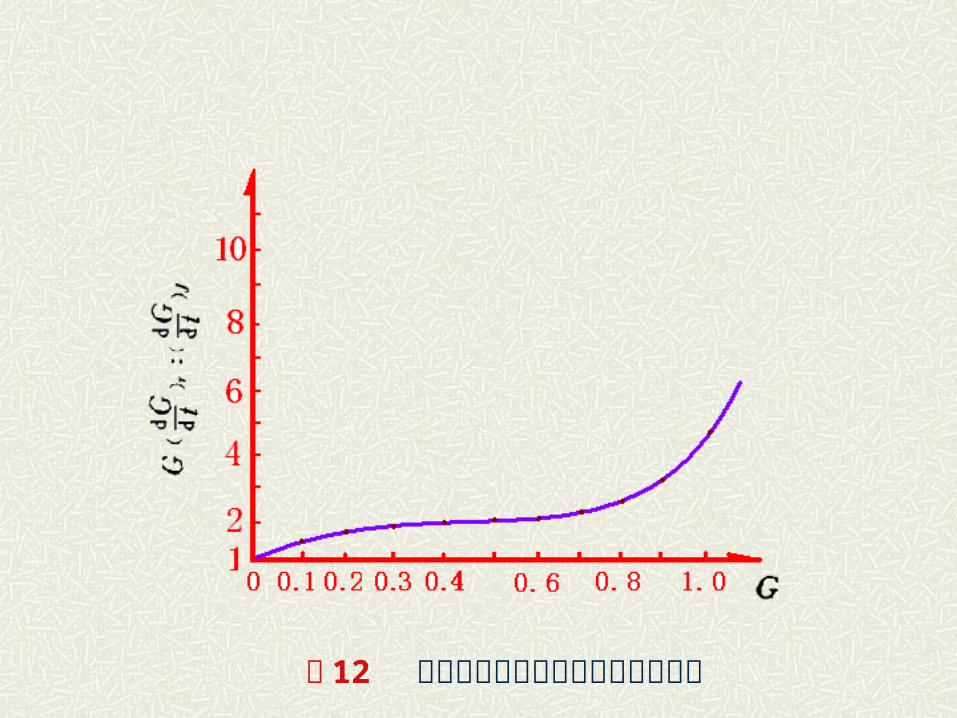
图 12 金斯特林格方程与杨德方程之比较

表 1 部分重要的固相反应动力学方程
控制 范围
反应类别 动力学方翟的积分式 A值 对应于图13的曲线
界 面
零级反应(对于球形颗粒) F0(G)=1一(1- -G) 1/ 3=K0t=0. 2063(t/ t 0.5) 0.2063 7
化学
零级反应(对于圆柱形颗粒) F1(G)=1一(1- -G) 1/ 2=K1t=0. 2929(t/ t 0.5) 0.2929 6
反应
零级反应(对于乎板试样) F2(G)= G) =K2t=0. 5000(t/ t 0.5) 0.5000 5
控制
一级反应(球形颗粒) F’ 3(G)=l n(1- -G) =-K3t=0. 6931(t/ t 0.5) 0.6931 9
扩 散
抛物线速度方程(干板试样) F4(G)= G2=K4’ t=0. 2500(t/ t 0.5) 0.2500 1
控 制
对圆柱形试样 F7(G)= (1- -G)l n(1-G)+G =K7t=0. 1534(t/ t 0.5) 0:1534 2
杨德方程(球形试样) F5(G)=[1一(1- -G) 1/ 3] 2=K5t=0. 0426(t/ t 0.5) 0.0426 3 范 围
金斯特林格方程(球形试样) F6(G)=1一 2/ 3G- (1- -G) 2/ 3=K6t=0. 0367(t/ t 0.5) 0.0367 4
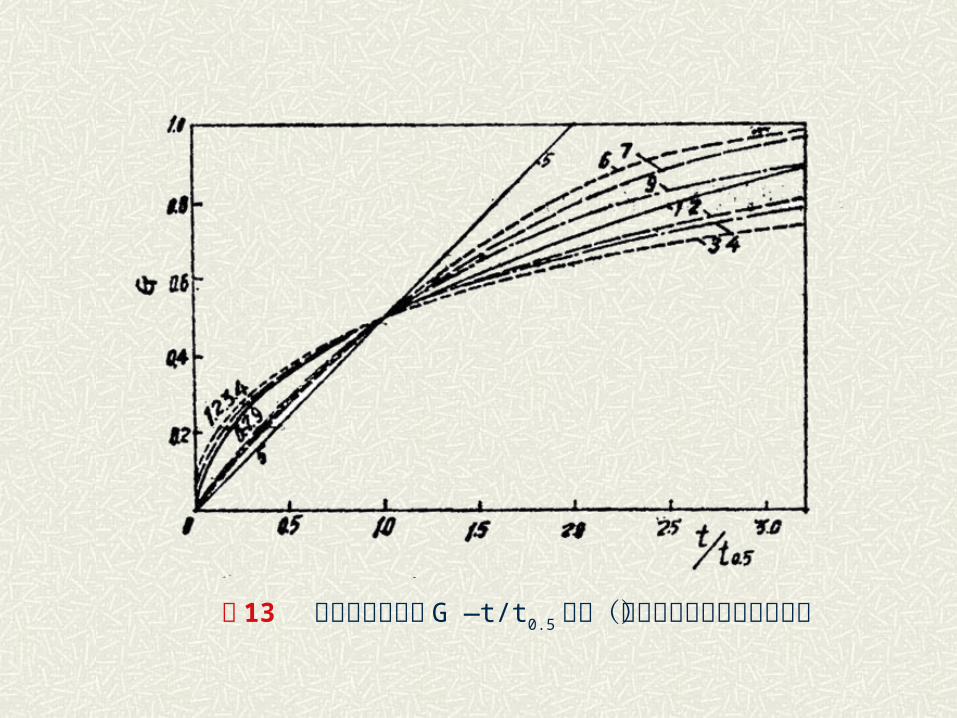
图 13 各种类型反应中 G —t/t0.5曲线(曲线序号对应的方程见表)

第四节 影响固相反应的因素
反应物化学组成的影响反应物颗粒及均匀性的影响反应温度的影响压力和气氛的影响反应物活性的影响
1 、反应物化学组成的影响 化学组成是影响固相反应的内因,是决定反应方向和速度的重要条件。 从热力学角度看,在一定温度、压力条件下,反应能进行的方向是自由焓减少(△ G
< 0 )的过程,而且的负值愈大,该过程的推动力也愈大,沿该方向反应的几率也大。

从结构角度看,反应物中质点间的作用键愈大,则 可 动 性 和 反 应 能 力 愈 小 , 反 之 亦 然 。 其次,在同一反应系统中,固相反应速度还与各反应物间的比例有关。如果颗粒相同的 A 和 B 反应生成物 AB ,若改变 A 与 B 比例会改变产物层温度、反应物表面积和扩散截面积的大小,从而影响反应速度。例如增加反应混合物中“遮盖”物的含量,则产物层厚度变薄,相应的反应速度也增加。
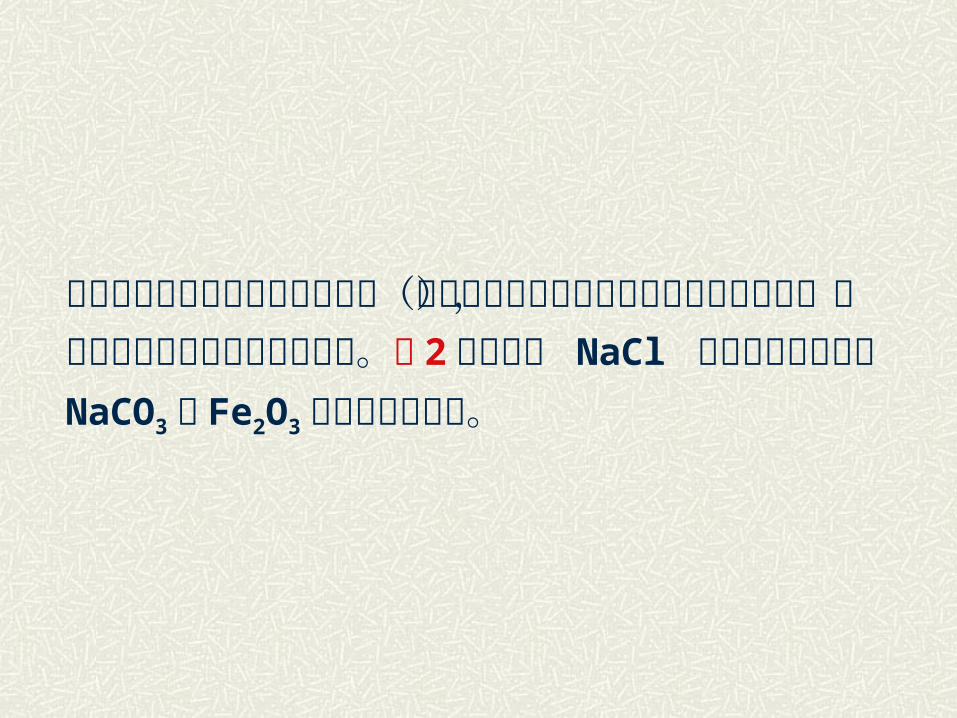
当反应混合物中加入少量矿化剂(也可能是由存在于原料中的杂质引起的),则常会对反应产生特殊的作用。表 2列出少量 NaCl 可使不同颗粒尺寸NaCO3与 Fe2O3 反应的加速作用。
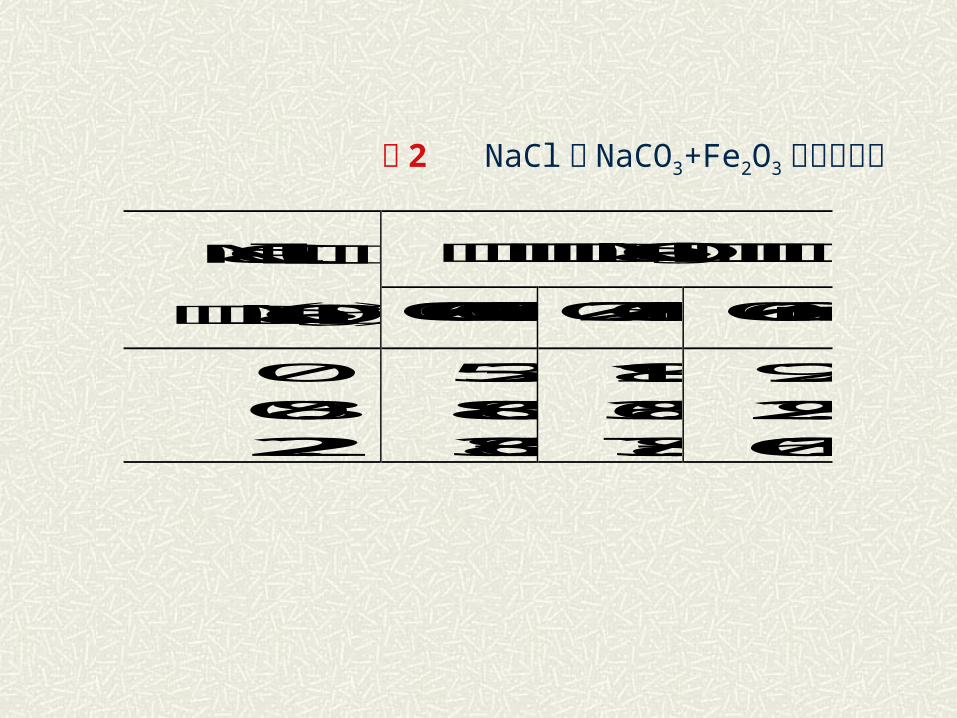
表 2 NaCl对 NaCO3+Fe2O3 反应的作用
不同颗粒尺寸的NaCO3转化率百分率 NaCl添加量
(相对于NaCO3的%) 0.06~0.088mm 0.27~0.35mm 0.6~2mm
0
0.8
2.2
53.2
88.6
38.6
18.9
36.8
73.8
9.2
22.9
60.1

2 、反应物颗粒及均匀性的影响 颗粒尺寸大小主要是通过以下途径对固相反应起影响的。1 )物料颗粒尺寸愈小,比表面积愈大,反应界面和扩散截面增加,反应产物层厚度减少,使反应速度增大。 图 14 表示出不同尺寸的 ZnO 和 Al2O3在 1200 ℃时形成速率的影响。
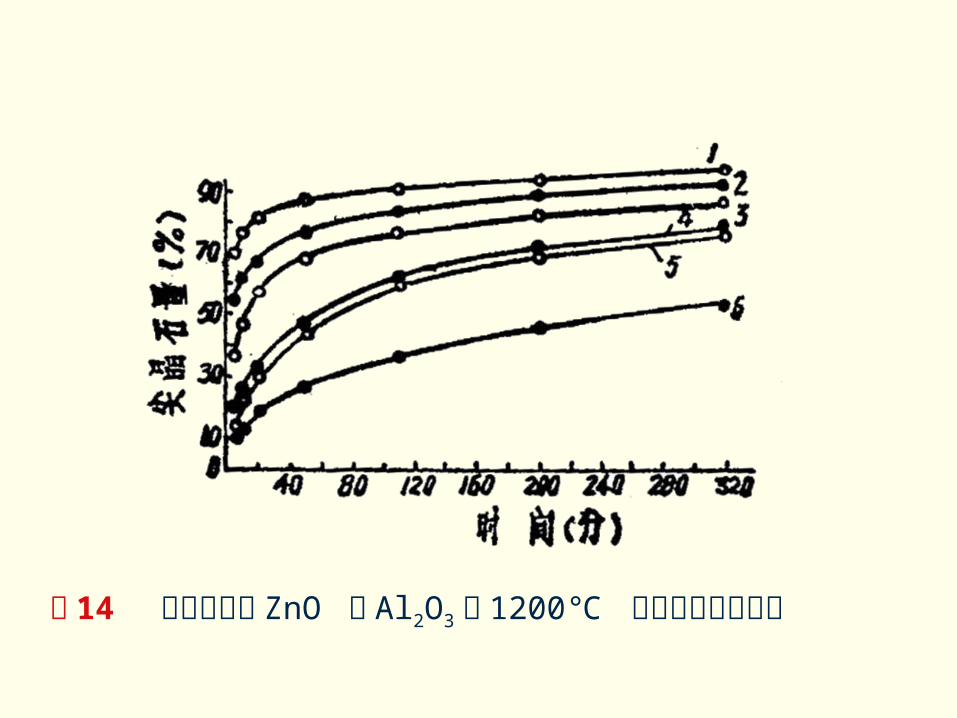
图 14 不同尺寸的 ZnO 和 Al2O3在 1200 ℃ 时形成速率的影响

2 )同一反应物系由于物料尺寸不同,反应速度可能会属于不同动力学范围控制。 例如 CaCO3与MoO3 反应,当取等分子比成分并在较高温度( 600℃ )下反应时,若 CaCO3颗粒大于MoO3 ,反应由扩散控制,反应速度主要由随 CaCO3
颗粒减少而加速。倘若 CaCO3与MoO3比值较大, CaCO3颗粒度小于 MoO3 时,由于产物层厚度减薄,扩散阻力很小,则反应将由 MoO3升华过程所控制,并随 MoO3粒径减少而加剧。

3 、反应温度的影响 温度是影响固相反应速度达到重要外部条件。一般随温度升高,质点热运动动能增大,反应能力和扩散能力增强。对于化学反应,因其速度常数 K =A 。 因此,温度对化学反应的加速作
用一般也比对扩散过程为大。
)exp(RT
Q

4 、压力和气氛的影响 对不同反应类型,压力的影响也不同。在两相间的反应中,增大压力有助于颗粒的接触面积,加速物质传递过程,使反应速度增加。但对于有液、气相参与达到反应中,扩散过程主要不是通过固体粒子的直接接触实现的。因此提高压力有时并不表现出积极作用,甚至会适得其反。

5 、反应物活性的影响 实践证明,同一物质处于不同结构状态时其反应活性差异甚大。一般说来,晶格能愈高、结构愈完整和稳定的,其反应活性也低。因此,对于难熔氧化物间的反应和烧结往往是困难的。为此通常采用具有高活性的活性固体作为原料。例如 Al2O3 + CoO → CoAl2O4 反应中,若分别采用轻烧 Al2O3 和较高温度煅烧制得的死烧 Al2O3
作原料,其反应速度相差近十倍,表明轻烧 Al2O3具有高得多的反应活性。
根据海德华定律,即物质在转变温度附近质点可动性显著增大、晶格松懈和活化的原理,工艺上可以利用多晶转变伴随的晶格重排来活化晶格;或是利用热分解反应和脱水反应形式具有较大比表面和晶格缺陷的初生态或无定形物质等措施提高反应活性。





























