Embed Size (px)
Citation preview

In Vivo �-Secretase 1 Inhibition Leads to Brain A� Loweringand Increased �-Secretase Processing of Amyloid PrecursorProtein without Effect on Neuregulin-1□S
Sethu Sankaranarayanan, Eric A. Price, Guoxin Wu, Ming-Chih Crouthamel, Xiao-Ping Shi,Katherine Tugusheva, Keala X. Tyler, Jason Kahana,1 Joan Ellis, Lixia Jin, Thomas Steele,Shawn Stachel, Craig Coburn, and Adam J. SimonDepartments of Alzheimer’s Research (S.Sa., E.A.P., G.W., M.-C.C., X.-P.S., K.T., K.X.T., J.K., A.J.S.), Drug Metabolism(J.E., L.J.), and Medicinal Chemistry (T.S., S.St., C.C.), Merck Research Laboratories, West Point, Pennsylvania
Received August 13, 2007; accepted December 20, 2007
ABSTRACT�-Secretase (BACE) cleavage of amyloid precursor protein(APP) is one of the first steps in the production of amyloid �peptide A�42, the putative neurotoxic species in Alzheimer’sdisease. Recent studies have shown that BACE1 knockdownleads to hypomyelination, putatively caused by a decline inneuregulin (NRG)-1 processing. In this study, we have tested apotent cell-permeable BACE1 inhibitor (IC50 � 30 nM) by ad-ministering it directly into the lateral ventricles of mice, express-ing human wild-type (WT)-APP, to determine the consequencesof BACE1 inhibition on brain APP and NRG-1 processing.BACE1 inhibition, in vivo, led to a significant dose- and time-dependent lowering of brain A�40 and A�42. BACE1 inhibitionalso led to a robust brain secreted (s)APP� lowering that wasaccompanied by an increase in brain sAPP� levels. Although an
increase in full-length NRG-1 levels was evident in 15-day-oldBACE1 homozygous knockout (KO) (�/�) mice, in agreementwith previous studies, this effect was also observed in 15-day-old heterozygous (�/�) mice, but it was not evident in 30-day-old and 2-year-old BACE1 KO (�/�) mice. Thus, BACE1knockdown led to a transient decrease in NRG-1 processing inmice. Pharmacological inhibition of BACE1 in adult mice, whichled to significant A� lowering, was without any significant effecton brain NRG-1 processing. Taken together, these results sug-gest that BACE1 is the major �-site cleavage enzyme for APPand that its inhibition can lower brain A� and redirect APPprocessing via the potentially nonamyloidogenic �-secretasepathway, without significantly altering NRG-1 processing.
Alzheimer’s disease (AD) is a neurodegenerative diseasecharacterized by the progressive accumulation of extracellu-lar amyloid plaques, intracellular neurofibrillary � tangles,and neuronal loss leading to memory deficits and dementia(Braak and Braak, 1991; Terry et al., 1991; Trojanowski etal., 1995; Naslund et al., 2000; Scheff and Price, 2003).�-Secretase (BACE) is a type I transmembrane aspartyl pro-tease that cleaves amyloid precursor protein (APP) to pro-duce the secreted N-terminal fragment sAPP� and a mem-
brane-anchored c-terminal fragment CTF� (for review, seeVassar, 2004). CTF� is further processed by the �-secretaseenzyme complex, leading to the production of amyloidogenic40 or 42 amino acid A� peptides. In contrast, APP processingby �-secretase enzyme followed by �-secretase cleavage re-sults in generation of secreted N-terminal fragment calledsAPP�, a putative neuroprotective factor (Furukawa et al.,1996), and a shorter “P3 fragment,” which is thought to benot amyloidogenic (Sambamurti et al., 2002).
BACE1-deficient mice generated from multiple groupswere viable, and they showed subtle alterations in behavioraland neurochemical phenotype (Roberds et al., 2001; Harrisonet al., 2003), but robust reduction in neuronal A� production(Cai et al., 2001; Luo et al., 2001) and amyloid plaque depo-sition (Ohno et al., 2004; Laird et al., 2005). In contrast, mice
1 Current affiliation: Department of Oncology, GlaxoSmithKline, Colle-geville, Pennsylvania.
Article, publication date, and citation information can be found athttp://jpet.aspetjournals.org.
doi:10.1124/jpet.107.130039.□S The online version of this article (available at http://jpet.aspetjournals.org)
contains supplemental material.
ABBREVIATIONS: AD, Alzheimer’s disease; BACE, �-secretase; APP, amyloid precursor protein; sAPP, soluble amyloid precursor protein; CTF,C-terminal fragment; A�, amyloid � peptide (those mentioned herein consist of 40 or 42 amino acids); KO, knockout; NRG, neuregulin-1; P,postnatal day; WT, wild type; ELISA, enzyme-linked immunosorbent assay; AP, alkaline phosphatase; mkd, milligrams per kilogram per day; PBS,phosphate-buffered saline; MBP, myelin basic protein; HSD, honestly significant difference; NTF, N-terminal fragment; P-gp, P-glycoprotein;ANOVA, analysis of variance.
0022-3565/08/3243-957–969$20.00THE JOURNAL OF PHARMACOLOGY AND EXPERIMENTAL THERAPEUTICS Vol. 324, No. 3Copyright © 2008 by The American Society for Pharmacology and Experimental Therapeutics 130039/3312711JPET 324:957–969, 2008 Printed in U.S.A.
957
http://jpet.aspetjournals.org/content/suppl/2007/12/26/jpet.107.130039.DC1Supplemental material to this article can be found at:
at ASPE
T Journals on June 12, 2018
jpet.aspetjournals.orgD
ownloaded from

overexpressing human BACE1 showed an increase in brainsAPP�, CTF�, and A� levels, suggesting enhanced amyloi-dogenic processing of APP and amyloid plaque deposition(Bodendorf et al., 2002; Mohajeri et al., 2004; Willem et al.,2004), whereas reduced plaque deposition has been observedin an independent mice line (Lee et al., 2005). In contrast, inmice overexpressing the �-secretase enzyme ADAM10, a re-duction in A� peptides and amyloid plaque deposition wasobserved previously (Postina et al., 2004). Thus, either inhi-bition of BACE1 enzymatic activity or an elevation of �-secre-tase activity can reduce brain A� production (Hardy andSelkoe, 2002).
Although there is evidence for early lethality (Dominguezet al., 2005) and age-dependent development of cognitivedeficits (Laird et al., 2005) in BACE1 KO mice, the overallbenign phenotype from multiple studies had supported theidea that BACE1 inhibition is a safe therapeutic target forA� lowering in human AD (Citron, 2004). However, recentstudies have shown that BACE1 KO mice show hypomyeli-nation and putative alteration in neuregulin (NRG)-1) pro-cessing, as evidenced by an increase in brain full-lengthNRG-1 levels (Willem et al., 2006). These deficits in NRG-1processing are thought to lead to defects in both peripheral(Willem et al., 2006) and central nervous system myelinationin young mice (Hu et al., 2006). Thus, a mechanism-basedeffect on peripheral or central myelination could potentiallyaffect BACE1 inhibitor development as an AD therapeutic.Although BACE1 inhibitors have shown acute in vivo efficacyto lower brain A� levels in murine models when adminis-tered peripherally (Chang et al., 2004; Stachel et al., 2006;Hussain et al., 2007) or via direct intracranial injection (Asaiet al., 2006; Nishitomi et al., 2006), their poor pharmacoki-netic properties have prevented effective evaluation of sub-chronic to chronic BACE1 inhibition on brain APP processingand NRG-1 processing in adult mice.
In a recent study, we have shown that small-moleculehydroxyethylamine dipeptide isosteres are very potent inhib-itors of BACE1 (IC50 � 15 nM) (Stachel et al., 2004; Pietraket al., 2005; Shi et al., 2005). In the present study, we dem-onstrate that direct infusion of this inhibitor, over 1 to 2weeks, into the lateral ventricles of brains of mice led to asustained reduction of brain A�40, A�42, and sAPP�, alongwith a corresponding elevation of sAPP�. Thus, the impact ofsubchronic to chronic BACE1 inhibition in mice on both APPprocessing and NRG-1 processing could be evaluated. Thequestions that are addressed in this study include whetherthere is a gene-dosage effect on NRG-1 processing in youngBACE1 KO mice; whether defects in NRG-1 processing areobserved in aged BACE1 KO mice similar to that in youngmice; and finally, whether in vivo BACE1 inhibition in adultmice leads to deficits in NRG-1 processing. We confirmedprevious findings that there is an increase in full-lengthNRG-1 protein levels in young postnatal day P15 BACE1 KO(�/�) mice compared with WT mice (Willem et al., 2006). InP15 BACE1 (�/�) mice, we observed an increase in full-length NRG-1 protein similar to (�/�) mice, suggesting alack of BACE1 gene dosage on NRG-1 processing. In contrast,P30 and 2-year-old BACE1 KO mice showed no significantdifferences in either the full-length or N-terminal fragmentof NRG-1 among the genotypes. These results indicate thatNRG-1 processing may decline rapidly with age in mice, andthey suggest an early developmental alteration in NRG-1
processing in BACE1 KO mice. Finally, we demonstrate thatBACE1 inhibition over 1 week was not accompanied by anyalterations in NRG-1 levels in adult mice. These data supportthe hypothesis that inhibition of BACE1 can potentially ame-liorate the amyloid burden in the brain of AD patients byboth decreasing A� production and shifting APP processing tothe �-secretase pathway, while not affecting NRG-1 processing.
Materials and MethodsAntibodies. A� N-terminal antibody 6E10 (Kim et al., 1988)
(catalog number SIG-39320; Covance Research Products, Princeton,NJ), which recognizes A� amino acids 3 to 8, was used as a captureantibody for A�40, A�42, and sAPP� sandwich ELISAs. Neoepitope-specific antibodies were used to detect A�40 and A�42 peptides,similar to that described previously (Ida et al., 1996). We raisedrabbit polyclonal antibodies to peptides encoding the sAPP�_KMneoepitope by using an artificial norleucine amino acid in place ofmethionine. This antibody was affinity purified, it showed neo-epitope specificity compared with the linear epitope around theBACE cleavage site, and it was used as a capture antibody for sAPP�ELISAs. The 22C11 antibody to amino acids 66 to 81 of APP Nterminus (MAB348; Millipore Bioscience Research Reagents, Te-mecula, CA) was used to detect both sAPP� and sAPP� in ELISAassays. Detection antibodies for ELISA assays were conjugated withalkaline phosphatase (AP) using the EZ-link maleimide-activatedAP kit (31493; Pierce Chemical, Rockford, IL) using standardprotocols.
Mice Models. The APP-YAC mice expressing the human WT-APP transgene under control of endogenous human APP promoterelements, in a yeast artificial chromosome vector, was used in thesestudies (Lamb et al., 1993). These mice show APP expression and A�levels that are �2 to 3-fold above endogenous murine levels, and theydisplay no amyloid plaques with age (Lamb et al., 1993) comparedwith other mutant-APP transgenic mice driven by strong promoterelements (Kawarabayashi et al., 2001). All in vivo inhibitor studieswere performed on 3- to 6-month-old mice. The APP-YAC miceshowed no age-dependent changes in soluble brain A� levels, andthey allowed efficient measurement of the human A� peptide frombrain homogenates. Mice lacking APP (Zheng et al., 1995) were usedto generate brain homogenates for running standard curves inELISA assays. The BACE1 knockout mice (Cai et al., 2001) wereobtained from Johns Hopkins University (Baltimore, MD), and theywere backcrossed to the C57BL/6 background.
Intracerebroventricular Administration of BACE Inhibitorin Mice. Intracerebroventricular infusions of compound were doneusing osmotic minipumps (1002, 1003D, 1007D; Alzet, Cupertino,CA) with capacity of �100 �l and pumping rates of 0.25 to 1 �l/h.BACE inhibitor Merck-3 (Stachel et al., 2004) was dissolved in 50%polyethylene glycol 300 and 50% dimethyl sulfoxide (D2650; Sigma-Aldrich, St. Louis, MO) at concentrations of 12.5 to 50 mg/ml todeliver doses ranging from 7.5 to 30 mg/kg/day (mkd) of compound.Osmotic minipumps were filled with compound and connected viapolyethylene tubing to customized infusion cannulas (�2.2 mm;Plastics One, Roanoke, VA), and they were primed overnight at 37°C.The next day, droplets were observed at the outlet of the cannula,indicating that the pumps were primed and exuding compound.APP-YAC mice were surgically prepared, and then they were placedin a mouse stereotaxic station (model 900; David Kopf Instruments,Tujunga, CA). Mice were anesthetized via a mouse mask using 2 to5% isoflurane. A midline incision was made in the scalp to expose theskull. The bregma was identified and coordinates AP, 1.2 mm; ML,�0.5 mm; and DV, �2.2 mm were used to target the left lateralventricle (Paxinos and Franklin, 2001). A trephine drill was used toremove �1 mm in diameter of bone centered on the marked coordi-nates to expose the brain. The osmotic minipump was placed withinan s.c. pocket under the skin on the back of the mouse. The attached
958 Sankaranarayanan et al.
at ASPE
T Journals on June 12, 2018
jpet.aspetjournals.orgD
ownloaded from

infusion cannula was stabilized in a holder, and then it was gentlyinserted into the brain until it was flush with the skull and stabilizedwith dental cement. The tubing was buried under the skin, takingcare not to introduce kinks, and the overlying skin was sealed withcyanoacrylate cement (Vetbond tissue adhesive; Henry Schein,Melville, NY). The mice were removed from the stereotaxic appara-tus, and they were allowed to recover over warm water circulatingblankets. Then, they were returned to their home cage until the endof the study. All animal procedures were done using sterile techniqueand procedures were fully approved by the Institutional Animal Careand Use Committee at Merck.
After the desired time of treatment with compounds, the micewere euthanized using a CO2 chamber. Blood was removed by car-diac puncture, collected in K2EDTA tubes, and centrifuged at 5000rpm for 10 min. Plasma was stored frozen at �80°C. Brains wereremoved and sectioned mid-sagittally. Each hemisphere wasweighed and stored at �80°C in polypropylene tubes until furtheranalysis.
Measurement of Brain A�40, A�42, sAPP�, and sAPP� bySandwich ELISA. Mouse brains were extracted in 0.2% diethyl-amine in 10� volume (w/v), heat-treated, and spun at 170,000g for90 min. Supernatant was collected and neutralized with 10% volume0.5M Tris-HCl, pH 6.8, and then it was either run in Western blotsor plated for ELISA for specific analytes. Brain A�40, A�42, andsAPP� analytes were captured in ELISA plates with the 6E10 anti-body, whereas the KM neoepitope antibody was the capture antibodyfor sAPP�. Black polystyrene Costar plates (Costar 3925; CorningLife Sciences, Acton, MA) were coated overnight with the 5 �g/mlcapture antibody, washed, and then blocked with 3% bovine serumalbumin in PBS. The plates were stored at 4°C until use. Onehundred microliters of sample was added per well in duplicate,followed by 50 �l of detection antibody conjugated with alkalinephosphatase (final IgG concentration at 0.1 �g/ml). Brain A�40 andA�42 were detected with C-terminal neoepitope antibodies, whereasN-terminal linear epitope antibody 22C11 was used to detect bothsAPP� and sAPP�. A�40 and A�42 standards (American PeptideCo., Inc., Berkeley, CA) and sAPP� and sAPP� peptide standards(S4316 and S9564; Sigma-Aldrich) were stored in frozen aliquots at�80°C, and they were used in standard curve dilutions in APP-KObrain matrix, prepared in an identical manner as the study samples.After overnight incubation at 4°C, plates were washed and developedusing alkaline phosphatase substrate (T2214; Applied Biosystems,Foster City, CA). Luminescence counts were measured using LJLAnalyst (Molecular Devices, Sunnyvale, CA).
Standard curves were fit using a third order spline fit, coefficientswere determined, and unknown sample counts were converted toactual analyte concentrations. All statistical analysis was performedon log-transformed data. The brain A�40 and A�42 assays had alower limit of sensitivity of 3 and 1.56 pM (�33 and �17 fmol/g afteradjusting for brain weight), respectively. Brain levels of A�40 werein the range of 750 to 1500 fmol/g, whereas brain A�42 was in therange of 100 to 400 fmol/g. Brain sAPP� and sAPP� assays had alower limit of sensitivity of �12.5 pM (�300 fmol/g adjusting forbrain weight). Brain sAPP� and sAPP� were in the range of 100 to8000 fmol/g. Because the standards showed lot-to-lot variability insignal and calculated amounts, these data were normalized to thevehicle group for analysis.
Western Blotting for Brain sAPP� and sAPP�. For Westernblots, 25 �l of mouse brain homogenate, as described above underMeasurement of Brain A�40, A�42, sAPP�, and sAPP� by SandwichELISA, was mixed with 25 �l of 2� sample loading buffer withreducing agent, it was boiled for 5 min, and then it was resolvedusing 10 to 20% Tris-Tricine SDS-polyacrylamide gel electrophoresisgels (Bio-Rad, Hercules, CA). Separated proteins were electro-phoretically transferred onto 0.2-�m nitrocellulose membranes,backed by 0.1-�m nitrocellulose. The membranes were boiled for 5min in PBS, and then they were blocked with Odyssey blocker(LI-COR, Lincoln, NE), followed by overnight incubation with pri-
mary antibodies 6E10 for sAPP� and rabbit KM-neoepitope antibodyfor sAPP�. Secondary antibodies IRDye goat anti-mouse 800 nm(Lonza Rockland, Inc., Rockland, MD) and Alexa Fluor goat anti-rabbit 680 nm (A21076; Invitrogen, Carlsbad, CA) were diluted1:2500 in Odyssey blocker with 0.1% Tween 20 for 1 h and washedwith PBS containing 0.1% Tween 20. Blots were then visualized andquantified using an Odyssey infrared scanner (LI-COR).
Western Blotting for Brain BACE1, Neuregulin, and MyelinBasic Protein. To quantify BACE1 and neuregulin, brain hemi-spheres were homogenized using a modified radioimmunoprecipita-tion assay buffer (1% Nonidet-40, 0.25% Na-deoxycholate, 150 mMNaCl, 1 mM EDTA, 1 mM phenylmethylsulfonyl fluoride, 1 �g/mleach of aprotinin, leupeptin, and pepstatin, 1 mM Na3VO4, 1 mMNaF in 50 mM Tris-HCl, pH 7.4). Ten microliters of brain homogenateswas mixed with 10 �l of 2� sample loading buffer, boiled for 5 min,and resolved using 4 to 20% gradient Tris-glycine SDS-polyacryl-amide gel electrophoresis gel (Invitrogen). Separated proteins wereelectrophoretically transferred onto 0.2-�m polyvinylidene difluoridemembranes. The membranes were blocked with Odyssey blocker(LI-COR), followed by overnight incubation with primary antibodies.Blots were incubated with rabbit polyclonal antibody to BACE1(EE17; Sigma-Aldrich) with or without mouse monoclonal antibodyto myelin basic protein (MBP) (smi94; Covance Research Products).A second set of blots was incubated with rabbit polyclonal antibodyto the N terminus of NRG-1 (sc28916; Santa Cruz Biotechnology,Inc., Santa Cruz, CA). Tubulin staining with mouse monoclonalantibody to �III-tubulin (MMS-435P; Covance Research Products)was used as a loading control. Secondary antibodies IRDye goatanti-mouse 800 nm (610-132-121; Lonza Rockland, Inc.) and AlexaFluor goat anti-rabbit 680 nm (catalog number A21109; Invitrogen)were diluted 1:2500 in Odyssey blocker with 0.1% Tween 20 for 1 hand washed with PBS containing 0.1% Tween 20. Blots were thenvisualized and quantified using an Odyssey infrared scanner(LI-COR).
Data Presentation and Statistical Analysis. Statistical anal-ysis was performed using JMP version 5.0 (SAS Institute, Cary, NC).Graphs and figures were prepared using JMP or Origin 7.5 (Origin-Lab Corp., Northampton, MA) and collated using Adobe Illustrator(Adobe Systems, Mountain View, CA).
Box and whisker plots were used to display data from individualanimal data using JMP. The box plots in red summarize the datafrom each treatment group. The ends of the box are the 25th and75th quantiles. The red line across the middle of the box indicates themedian. Each box has red lines or whiskers that extend from theends of the box to the highest and lowest magnitude data point. Thehorizontal green line represents the mean of the data for each group.
Average data were plotted as the mean � S.E.M. Statistical sig-nificance was denoted by �, p � 0.05, ��, p � 0.01, and ���, p � 0.001using Tukey-Kramer honestly significant difference (HSD) or Stu-dent’s t test.
ResultsIntracerebroventricular Administration of BACE1
Inhibitor Merck-3 Led to Robust Brain A�40 and A�42Lowering in Mice. We have reported previously thatMerck-3 is a potent inhibitor of in vitro BACE1 enzymeactivity, with an IC50 of �10 nM and reduced sAPP� produc-tion from cells, with an IC50 of �30 nM. Merck-3 was selec-tive for BACE1, and it had an in vitro IC50 value of �230 nMfor BACE2, �7 �M for cathepsin D, and �50 �M for renin,respectively (Stachel et al., 2004; Pietrak et al., 2005).Merck-3 showed a robust inhibition of secreted sAPP�, and itdisplayed a dose-dependent reduction of secreted A�40 inprimary brain slice cultures, with an IC50 of �20 nM (Sup-plemental Fig. 1). These results suggested that Merck-3 caninhibit endogenous BACE1 in mice brain slice cultures.
BACE1 Inhibition, A� Lowering, and Neuregulin-1 959
at ASPE
T Journals on June 12, 2018
jpet.aspetjournals.orgD
ownloaded from
Initially, Merck-3 was tested at a dose of 10 mpk via tailvein bolus administration in APP-YAC mice, and brain A�40levels were examined at 2 h after dosing. Merck-3 showed noeffect on brain A�40 via i.v. dosing (data not shown). Averageplasma concentrations of �1.3 � 1.2 �M Merck-3 wereachieved after 2 h (n � 3 separate experiments), whereasbrain concentrations were below the limit of detection (�90nM). Merck-3 had poor intrinsic permeability of �0.7 � 10�6
cm/s as measured in a renal epithelial cell line (LLC-PK1).Merck-3 was also a P-glycoprotein (P-gp) substrate becausebrain levels in mice lacking the p-glycoprotein mdr1A (Um-benhauer et al., 1997) were at least 4-fold greater than thelimit of detection. These results suggest that Merck-3 ispoorly brain penetrant; therefore, it is not efficacious in low-ering brain A�40 after i.v. administration.
We next tested whether brain A� can be lowered if Merck-3was administered directly into the lateral ventricles of thebrains of APP-YAC mice. We surgically implanted a cannulato deliver Merck-3 into the left lateral ventricle, by minipumpinfusion. Subcutaneous miniosmotic pumps (100 �l; 1 �l/hrate) were filled with Merck-3 at a concentration of 50 mg/ml,and they were connected via polyethylene tubing to a can-nula and primed overnight at 37°C. The pump deliveredvehicle or Merck-3 at a dose of �30 mkd into the left lateralventricles. After recovery, the mice were sent to their homecage; they were sacrificed 24 h later, and brains were har-vested. The mice tolerated the i.c.v. infusions well, and theyshowed normal feeding and exploratory behavior after recov-ery from anesthesia. Overall, survival was in the order of95% among all surgically implanted animals, and there wasno statistical difference between vehicle-treated and Merck-3-treated animals (vehicle, 53 of 55 and Merck-3, 68 of 71animals survived from n � 11 experiments; p � 0.7). Brainconcentrations of Merck-3 were in the range of 50 to 300 �M
in this study. This is probably due to the large dose of com-pound infused into the brain and the possibility of localizedprecipitation in the ventricle leading to very high brain ex-posures of Merck-3.
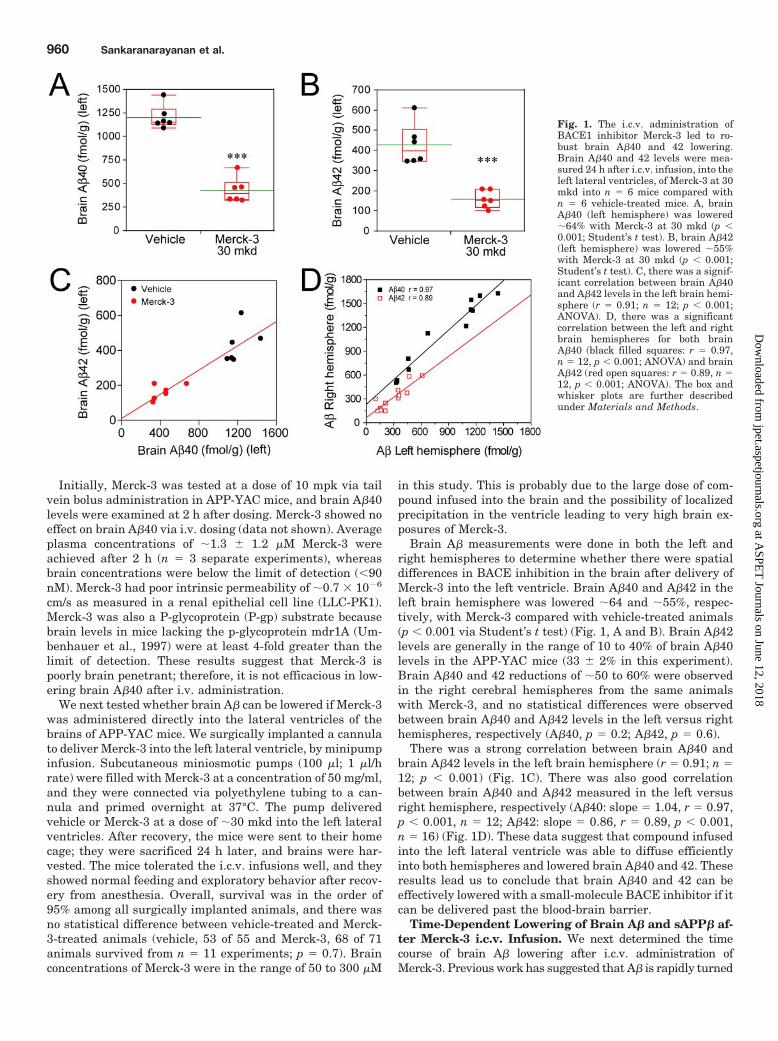
Brain A� measurements were done in both the left andright hemispheres to determine whether there were spatialdifferences in BACE inhibition in the brain after delivery ofMerck-3 into the left ventricle. Brain A�40 and A�42 in theleft brain hemisphere was lowered �64 and �55%, respec-tively, with Merck-3 compared with vehicle-treated animals(p � 0.001 via Student’s t test) (Fig. 1, A and B). Brain A�42levels are generally in the range of 10 to 40% of brain A�40levels in the APP-YAC mice (33 � 2% in this experiment).Brain A�40 and 42 reductions of �50 to 60% were observedin the right cerebral hemispheres from the same animalswith Merck-3, and no statistical differences were observedbetween brain A�40 and A�42 levels in the left versus righthemispheres, respectively (A�40, p � 0.2; A�42, p � 0.6).
There was a strong correlation between brain A�40 andbrain A�42 levels in the left brain hemisphere (r � 0.91; n �12; p � 0.001) (Fig. 1C). There was also good correlationbetween brain A�40 and A�42 measured in the left versusright hemisphere, respectively (A�40: slope � 1.04, r � 0.97,p � 0.001, n � 12; A�42: slope � 0.86, r � 0.89, p � 0.001,n � 16) (Fig. 1D). These data suggest that compound infusedinto the left lateral ventricle was able to diffuse efficientlyinto both hemispheres and lowered brain A�40 and 42. Theseresults lead us to conclude that brain A�40 and 42 can beeffectively lowered with a small-molecule BACE inhibitor if itcan be delivered past the blood-brain barrier.
Time-Dependent Lowering of Brain A� and sAPP� af-ter Merck-3 i.c.v. Infusion. We next determined the timecourse of brain A� lowering after i.c.v. administration ofMerck-3. Previous work has suggested that A� is rapidly turned
Fig. 1. The i.c.v. administration ofBACE1 inhibitor Merck-3 led to ro-bust brain A�40 and 42 lowering.Brain A�40 and 42 levels were mea-sured 24 h after i.c.v. infusion, into theleft lateral ventricles, of Merck-3 at 30mkd into n � 6 mice compared withn � 6 vehicle-treated mice. A, brainA�40 (left hemisphere) was lowered�64% with Merck-3 at 30 mkd (p �0.001; Student’s t test). B, brain A�42(left hemisphere) was lowered �55%with Merck-3 at 30 mkd (p � 0.001;Student’s t test). C, there was a signif-icant correlation between brain A�40and A�42 levels in the left brain hemi-sphere (r � 0.91; n � 12; p � 0.001;ANOVA). D, there was a significantcorrelation between the left and rightbrain hemispheres for both brainA�40 (black filled squares: r � 0.97,n � 12, p � 0.001; ANOVA) and brainA�42 (red open squares: r � 0.89, n �12, p � 0.001; ANOVA). The box andwhisker plots are further describedunder Materials and Methods.
960 Sankaranarayanan et al.
at ASPE
T Journals on June 12, 2018
jpet.aspetjournals.orgD
ownloaded from
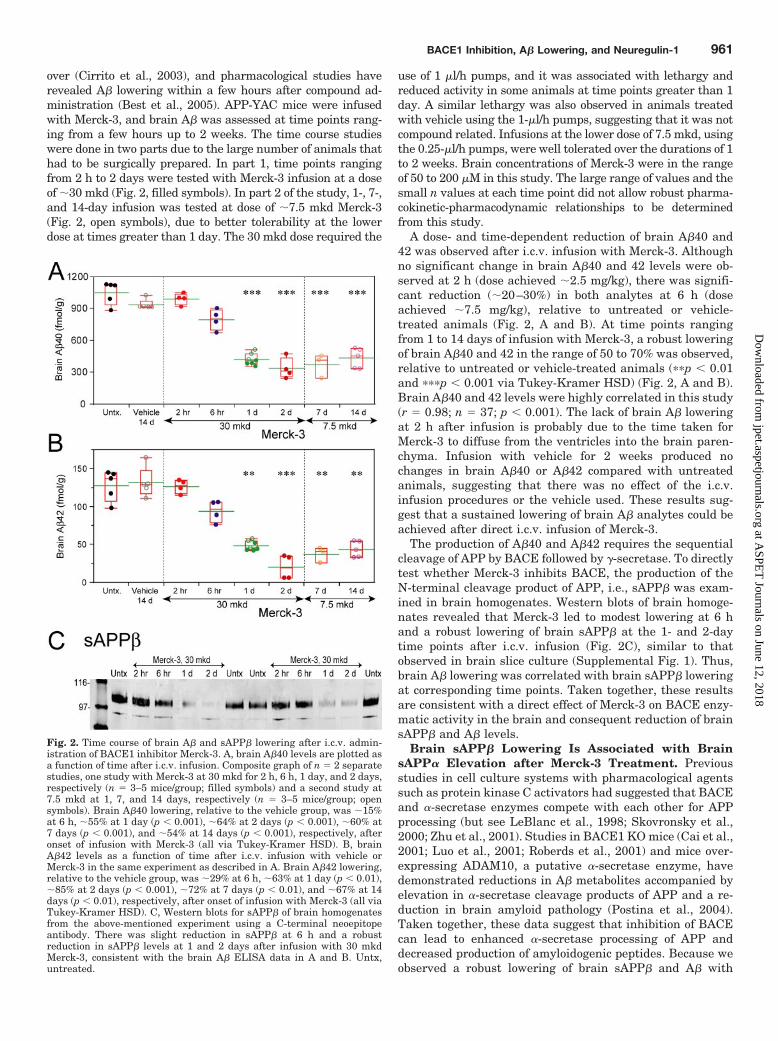
over (Cirrito et al., 2003), and pharmacological studies haverevealed A� lowering within a few hours after compound ad-ministration (Best et al., 2005). APP-YAC mice were infusedwith Merck-3, and brain A� was assessed at time points rang-ing from a few hours up to 2 weeks. The time course studieswere done in two parts due to the large number of animals thathad to be surgically prepared. In part 1, time points rangingfrom 2 h to 2 days were tested with Merck-3 infusion at a doseof �30 mkd (Fig. 2, filled symbols). In part 2 of the study, 1-, 7-,and 14-day infusion was tested at dose of �7.5 mkd Merck-3(Fig. 2, open symbols), due to better tolerability at the lowerdose at times greater than 1 day. The 30 mkd dose required the
use of 1 �l/h pumps, and it was associated with lethargy andreduced activity in some animals at time points greater than 1day. A similar lethargy was also observed in animals treatedwith vehicle using the 1-�l/h pumps, suggesting that it was notcompound related. Infusions at the lower dose of 7.5 mkd, usingthe 0.25-�l/h pumps, were well tolerated over the durations of 1to 2 weeks. Brain concentrations of Merck-3 were in the rangeof 50 to 200 �M in this study. The large range of values and thesmall n values at each time point did not allow robust pharma-cokinetic-pharmacodynamic relationships to be determinedfrom this study.
A dose- and time-dependent reduction of brain A�40 and42 was observed after i.c.v. infusion with Merck-3. Althoughno significant change in brain A�40 and 42 levels were ob-served at 2 h (dose achieved �2.5 mg/kg), there was signifi-cant reduction (�20–30%) in both analytes at 6 h (doseachieved �7.5 mg/kg), relative to untreated or vehicle-treated animals (Fig. 2, A and B). At time points rangingfrom 1 to 14 days of infusion with Merck-3, a robust loweringof brain A�40 and 42 in the range of 50 to 70% was observed,relative to untreated or vehicle-treated animals (��p � 0.01and ���p � 0.001 via Tukey-Kramer HSD) (Fig. 2, A and B).Brain A�40 and 42 levels were highly correlated in this study(r � 0.98; n � 37; p � 0.001). The lack of brain A� loweringat 2 h after infusion is probably due to the time taken forMerck-3 to diffuse from the ventricles into the brain paren-chyma. Infusion with vehicle for 2 weeks produced nochanges in brain A�40 or A�42 compared with untreatedanimals, suggesting that there was no effect of the i.c.v.infusion procedures or the vehicle used. These results sug-gest that a sustained lowering of brain A� analytes could beachieved after direct i.c.v. infusion of Merck-3.
The production of A�40 and A�42 requires the sequentialcleavage of APP by BACE followed by �-secretase. To directlytest whether Merck-3 inhibits BACE, the production of theN-terminal cleavage product of APP, i.e., sAPP� was exam-ined in brain homogenates. Western blots of brain homoge-nates revealed that Merck-3 led to modest lowering at 6 hand a robust lowering of brain sAPP� at the 1- and 2-daytime points after i.c.v. infusion (Fig. 2C), similar to thatobserved in brain slice culture (Supplemental Fig. 1). Thus,brain A� lowering was correlated with brain sAPP� loweringat corresponding time points. Taken together, these resultsare consistent with a direct effect of Merck-3 on BACE enzy-matic activity in the brain and consequent reduction of brainsAPP� and A� levels.
Brain sAPP� Lowering Is Associated with BrainsAPP� Elevation after Merck-3 Treatment. Previousstudies in cell culture systems with pharmacological agentssuch as protein kinase C activators had suggested that BACEand �-secretase enzymes compete with each other for APPprocessing (but see LeBlanc et al., 1998; Skovronsky et al.,2000; Zhu et al., 2001). Studies in BACE1 KO mice (Cai et al.,2001; Luo et al., 2001; Roberds et al., 2001) and mice over-expressing ADAM10, a putative �-secretase enzyme, havedemonstrated reductions in A� metabolites accompanied byelevation in �-secretase cleavage products of APP and a re-duction in brain amyloid pathology (Postina et al., 2004).Taken together, these data suggest that inhibition of BACEcan lead to enhanced �-secretase processing of APP anddecreased production of amyloidogenic peptides. Because weobserved a robust lowering of brain sAPP� and A� with
Fig. 2. Time course of brain A� and sAPP� lowering after i.c.v. admin-istration of BACE1 inhibitor Merck-3. A, brain A�40 levels are plotted asa function of time after i.c.v. infusion. Composite graph of n � 2 separatestudies, one study with Merck-3 at 30 mkd for 2 h, 6 h, 1 day, and 2 days,respectively (n � 3–5 mice/group; filled symbols) and a second study at7.5 mkd at 1, 7, and 14 days, respectively (n � 3–5 mice/group; opensymbols). Brain A�40 lowering, relative to the vehicle group, was �15%at 6 h, �55% at 1 day (p � 0.001), �64% at 2 days (p � 0.001), �60% at7 days (p � 0.001), and �54% at 14 days (p � 0.001), respectively, afteronset of infusion with Merck-3 (all via Tukey-Kramer HSD). B, brainA�42 levels as a function of time after i.c.v. infusion with vehicle orMerck-3 in the same experiment as described in A. Brain A�42 lowering,relative to the vehicle group, was �29% at 6 h, �63% at 1 day (p � 0.01),�85% at 2 days (p � 0.001), �72% at 7 days (p � 0.01), and �67% at 14days (p � 0.01), respectively, after onset of infusion with Merck-3 (all viaTukey-Kramer HSD). C, Western blots for sAPP� of brain homogenatesfrom the above-mentioned experiment using a C-terminal neoepitopeantibody. There was slight reduction in sAPP� at 6 h and a robustreduction in sAPP� levels at 1 and 2 days after infusion with 30 mkdMerck-3, consistent with the brain A� ELISA data in A and B. Untx,untreated.
BACE1 Inhibition, A� Lowering, and Neuregulin-1 961
at ASPE
T Journals on June 12, 2018
jpet.aspetjournals.orgD
ownloaded from
Merck-3, we examined whether direct inhibition of BACEwith Merck-3 also alters �-secretase processing of APP.
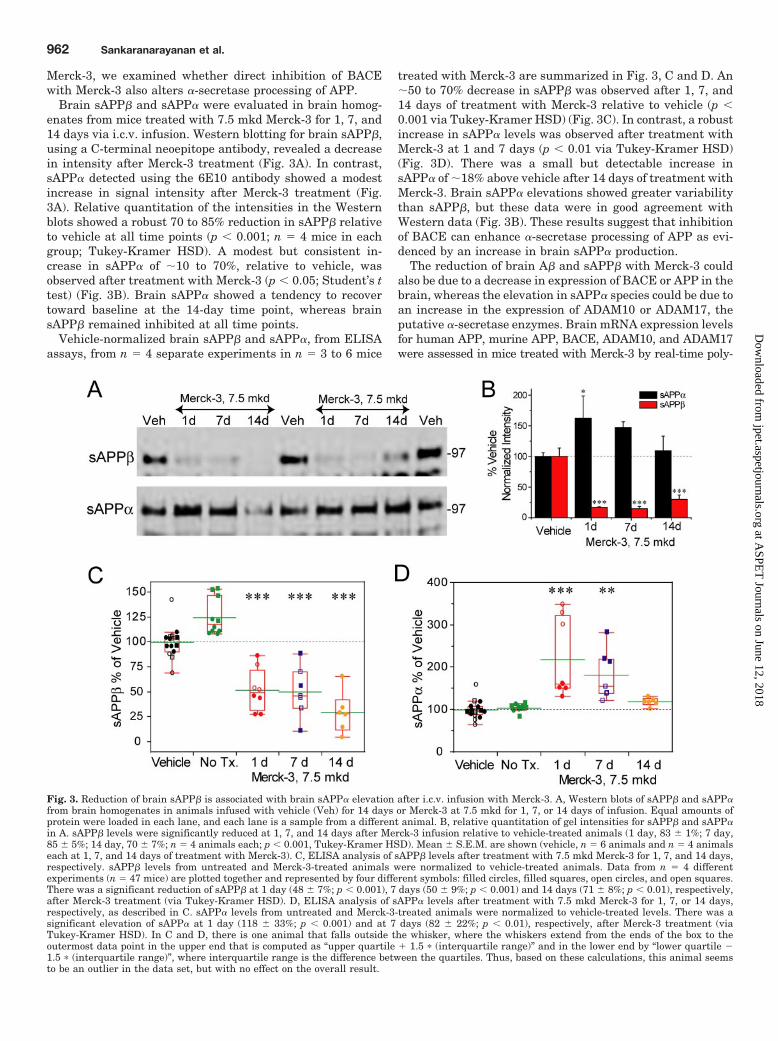
Brain sAPP� and sAPP� were evaluated in brain homog-enates from mice treated with 7.5 mkd Merck-3 for 1, 7, and14 days via i.c.v. infusion. Western blotting for brain sAPP�,using a C-terminal neoepitope antibody, revealed a decreasein intensity after Merck-3 treatment (Fig. 3A). In contrast,sAPP� detected using the 6E10 antibody showed a modestincrease in signal intensity after Merck-3 treatment (Fig.3A). Relative quantitation of the intensities in the Westernblots showed a robust 70 to 85% reduction in sAPP� relativeto vehicle at all time points (p � 0.001; n � 4 mice in eachgroup; Tukey-Kramer HSD). A modest but consistent in-crease in sAPP� of �10 to 70%, relative to vehicle, wasobserved after treatment with Merck-3 (p � 0.05; Student’s ttest) (Fig. 3B). Brain sAPP� showed a tendency to recovertoward baseline at the 14-day time point, whereas brainsAPP� remained inhibited at all time points.
Vehicle-normalized brain sAPP� and sAPP�, from ELISAassays, from n � 4 separate experiments in n � 3 to 6 mice
treated with Merck-3 are summarized in Fig. 3, C and D. An�50 to 70% decrease in sAPP� was observed after 1, 7, and14 days of treatment with Merck-3 relative to vehicle (p �0.001 via Tukey-Kramer HSD) (Fig. 3C). In contrast, a robustincrease in sAPP� levels was observed after treatment withMerck-3 at 1 and 7 days (p � 0.01 via Tukey-Kramer HSD)(Fig. 3D). There was a small but detectable increase insAPP� of �18% above vehicle after 14 days of treatment withMerck-3. Brain sAPP� elevations showed greater variabilitythan sAPP�, but these data were in good agreement withWestern data (Fig. 3B). These results suggest that inhibitionof BACE can enhance �-secretase processing of APP as evi-denced by an increase in brain sAPP� production.
The reduction of brain A� and sAPP� with Merck-3 couldalso be due to a decrease in expression of BACE or APP in thebrain, whereas the elevation in sAPP� species could be due toan increase in the expression of ADAM10 or ADAM17, theputative �-secretase enzymes. Brain mRNA expression levelsfor human APP, murine APP, BACE, ADAM10, and ADAM17were assessed in mice treated with Merck-3 by real-time poly-
Fig. 3. Reduction of brain sAPP� is associated with brain sAPP� elevation after i.c.v. infusion with Merck-3. A, Western blots of sAPP� and sAPP�from brain homogenates in animals infused with vehicle (Veh) for 14 days or Merck-3 at 7.5 mkd for 1, 7, or 14 days of infusion. Equal amounts ofprotein were loaded in each lane, and each lane is a sample from a different animal. B, relative quantitation of gel intensities for sAPP� and sAPP�in A. sAPP� levels were significantly reduced at 1, 7, and 14 days after Merck-3 infusion relative to vehicle-treated animals (1 day, 83 � 1%; 7 day,85 � 5%; 14 day, 70 � 7%; n � 4 animals each; p � 0.001, Tukey-Kramer HSD). Mean � S.E.M. are shown (vehicle, n � 6 animals and n � 4 animalseach at 1, 7, and 14 days of treatment with Merck-3). C, ELISA analysis of sAPP� levels after treatment with 7.5 mkd Merck-3 for 1, 7, and 14 days,respectively. sAPP� levels from untreated and Merck-3-treated animals were normalized to vehicle-treated animals. Data from n � 4 differentexperiments (n � 47 mice) are plotted together and represented by four different symbols: filled circles, filled squares, open circles, and open squares.There was a significant reduction of sAPP� at 1 day (48 � 7%; p � 0.001), 7 days (50 � 9%; p � 0.001) and 14 days (71 � 8%; p � 0.01), respectively,after Merck-3 treatment (via Tukey-Kramer HSD). D, ELISA analysis of sAPP� levels after treatment with 7.5 mkd Merck-3 for 1, 7, or 14 days,respectively, as described in C. sAPP� levels from untreated and Merck-3-treated animals were normalized to vehicle-treated levels. There was asignificant elevation of sAPP� at 1 day (118 � 33%; p � 0.001) and at 7 days (82 � 22%; p � 0.01), respectively, after Merck-3 treatment (viaTukey-Kramer HSD). In C and D, there is one animal that falls outside the whisker, where the whiskers extend from the ends of the box to theoutermost data point in the upper end that is computed as “upper quartile � 1.5 � (interquartile range)” and in the lower end by “lower quartile �1.5 � (interquartile range)”, where interquartile range is the difference between the quartiles. Thus, based on these calculations, this animal seemsto be an outlier in the data set, but with no effect on the overall result.
962 Sankaranarayanan et al.
at ASPE
T Journals on June 12, 2018
jpet.aspetjournals.orgD
ownloaded from

merase chain reaction (Supplemental Fig. 2). We observed atransient 1.5- to 2-fold increase in BACE1 and murine-APPmRNA levels at day 1 and 7, which declined back to levelssimilar to vehicle-treated animals, after Merck-3 treatment.There was no significant change in expression levels of human-APP, ADAM10, or ADAM17 at any of these time points. Thus,neither the brain A� nor sAPP� lowering or sAPP� elevation,with Merck-3 is due to a reduction in brain BACE or APPexpression or to an up-regulation of ADAM10 or ADAM17 lev-els, respectively. These results confirm that inhibition ofBACE1 leads to a decrease in brain sAPP� production and anincrease in brain sAPP� secretion.
Neuregulin 1 Full-Length Protein Is Elevated in P15Homozygote (�/�) and Heterozygote (�/�)-BACE1 KOmice compared with (�/�) Mice but Not in P30 or AgedHomozygote (�/�) Mice. Recent studies suggest thatNRG-1, a protein that mediates juxtacrine-signaling betweenaxons and Schwann cells and regulates axon myelination(Nave and Salzer, 2006), is a putative substrate for BACE1(Hu et al., 2006; Willem et al., 2006). A reduction in NRG-1processing was observed in young BACE1 KO mice (�/�), asevidenced by an increase in full-length NRG-1 protein levels(Willem et al., 2006) and a decrease in the N-terminal frag-ment of NRG-1 (Hu et al., 2006). BACE1 KO mice display areduction in myelin thickness in peripheral nerves (Hu et al.,2006; Willem et al., 2006) and in the central nervous system(Hu et al., 2006). Consequently, it has been suggested thatthe decline in myelination during development in BACE1 KOmice, may be due to a reduction in NRG-1 processing andraises questions regarding the in vivo consequences of pro-longed BACE1 inhibition on these parameters.
Before testing the effect of in vivo pharmacological inhibi-tion of BACE1 on NRG-1 processing, we examined the con-sequences of gene dosage of BACE1 on BACE1 protein andNRG-1 processing in young mice, and we also tested whetherthe defect in NRG-1 processing was persistent in agedBACE1 KO or restricted to an early developmental period.We observed a gene dosage-dependent decrease in BACE1protein in the BACE1 KO mice and cleavage products of APP(Supplemental Fig. 3). In the BACE1 KO (�/�) mice, BACE1protein was absent, whereas (�/�) mice show intermediatelevels compared with (�/�) mice. Likewise c99, the C-termi-nal fragment derived after BACE1 cleavage of APP, andsAPP�, the N-terminal fragment of APP, were absent in the(�/�) mice compared with the (�/�) mice, whereas (�/�)mice show diminished levels but closer to levels in (�/�)animals (Cai et al., 2001; Luo et al., 2001) (Supplemental Fig.3A). BACE1 KO (�/�) mice also show a complete lack ofendogenous murine A�40 production, whereas the BACE1(�/�) mice show �20% lower brain A�40 levels comparedwith the (�/�) mice (Supplemental Fig. 3B). Thus, an �50%drop in BACE1 protein levels only leads to �20% drop inbrain A�40 levels in 3- to 4-month-old BACE1 KO mice,suggesting a nonlinear relationship of BACE activity on invivo APP processing. Finally, an age-dependent decrease inboth BACE1 protein levels (similar to Willem et al., 2006)and ex vivo BACE1 activity was observed in APP-YAC mice(Supplemental Fig. 3, C and D). Although the age-dependentdecrease in BACE1 activity levels was correlated with adecline in BACE1 protein levels, BACE1 activity normalizedto BACE1 protein levels were similar among the differentages (data not shown). These results suggest an age-depen-
dent decrease in BACE1 protein levels and prompted us toexamine the effect of age on NRG-1 processing in BACE1 KOmice.
Previous studies have shown that full-length NRG-1 pro-tein levels were elevated in brains from BACE1 KO (�/�)mice compared with (�/�) animals from postnatal day 5 mice(Willem et al., 2006) and up to �2 months of age (Hu et al.,2006). Therefore, we first examined the gene dosage ofBACE1 protein in brains from young (15 and 30 days old; P15and P30) (�/�), (�/�), and (�/�) BACE1 KO mice and aged(2-year-old) (�/�) and (�/�) mice [unfortunately aged (�/�)mice were not available]. BACE1 KO (�/�) mice showed nodetectable BACE1 protein in brain extracts from all threeages (Fig. 4, A–F). BACE1 KO (�/�) mice showed elevatedintermediate levels of BACE1 protein, in the P15 and P30animals, compared with (�/�) and (�/�) mice (p � 0.01;Tukey-Kramer HSD) (Fig. 4, A–D).
Consistent with previous results, an increase in full-lengthNRG-1 protein of �130 kDa was observed in BACE1 KO(�/�) mice, compared with (�/�) in the P15 mice (p � 0.01;Tukey-Kramer HSD) (Fig. 4, A and B). In addition, theBACE1 (�/�) mice also displayed an increase in full-lengthNRG-1 protein levels, compared with WT (�/�) mice, verysimilar to or even greater than that of (�/�) mice (p � 0.01;Tukey-Kramer HSD). Although by eye one could think thereis a difference, there was no detectable difference in full-length NRG-1 protein levels between the P15 (�/�) and(�/�) BACE1 KO mice after quantitation. Next, we evalu-ated the levels of the N-terminal fragment of NRG-1 (NRG-NTF), which would result after putative cleavage by BACE1.The NRG-NTF in the P15 (�/�) mice, examined in the sameblots as the full-length protein by using an N-terminalNRG-1 antibody, showed a modest decline in signal com-pared with the BACE1 (�/�) and (�/�) mice (p � 0.05;Tukey-Kramer HSD). In contrast to the P15 mice, the P30mice showed no apparent difference in the levels of bothfull-length NRG-1 and NRG-NTF among the 3 genotypes ofBACE1 KO mice (Fig. 4, C and D). Finally, BACE1 (�/�) and(�/�) mice showed no detectable differences in NRG-1 andNRG-NTF levels in 2-year-old mice (Fig. 4, E and F).Whereas P15 BACE (�/�) and (�/�) animals showed anincrease in full-length NRG-1, contrary to our expectations,the N-terminal fragment of NRG-1 showed no decline com-pared with (�/�) animals as would be expected if NRG-1processing is altered in BACE1KO mice (Fig. 4B). In addi-tion, we observed no differences in the NRG-1 and NRG-NTFlevels in P30 and 2-year-old mice (Fig. 4, D and F). Finally,the lack of difference in NRG-1 or NRG-NTF levels betweenBACE1 (�/�) and (�/�) mice suggests a lack of BACE1 genedosage on NRG-1 processing (Fig. 4, B, D, and F).
MBP levels were modestly reduced in BACE1 (�/�) micecompared with (�/�) and (�/�) mice in P15 mice, but nosignificant differences were observed among the genotypes inthe P30 mice (Fig. 4, A–D). Finally, 2-year-old BACE1 (�/�)mice showed no detectable difference in MBP levels com-pared with (�/�) animals (Fig. 4, E and F). Thus, there wasno evidence for long-term effects on brain MBP levels in theBACE1 KO mice.
Taken together, these results suggest that mature andolder BACE1 KO animals do not show differences in brainNRG-1 processing like that observed in young P15 animals.Thus, BACE1-dependent processing of NRG-1 maybe more
BACE1 Inhibition, A� Lowering, and Neuregulin-1 963
at ASPE
T Journals on June 12, 2018
jpet.aspetjournals.orgD
ownloaded from
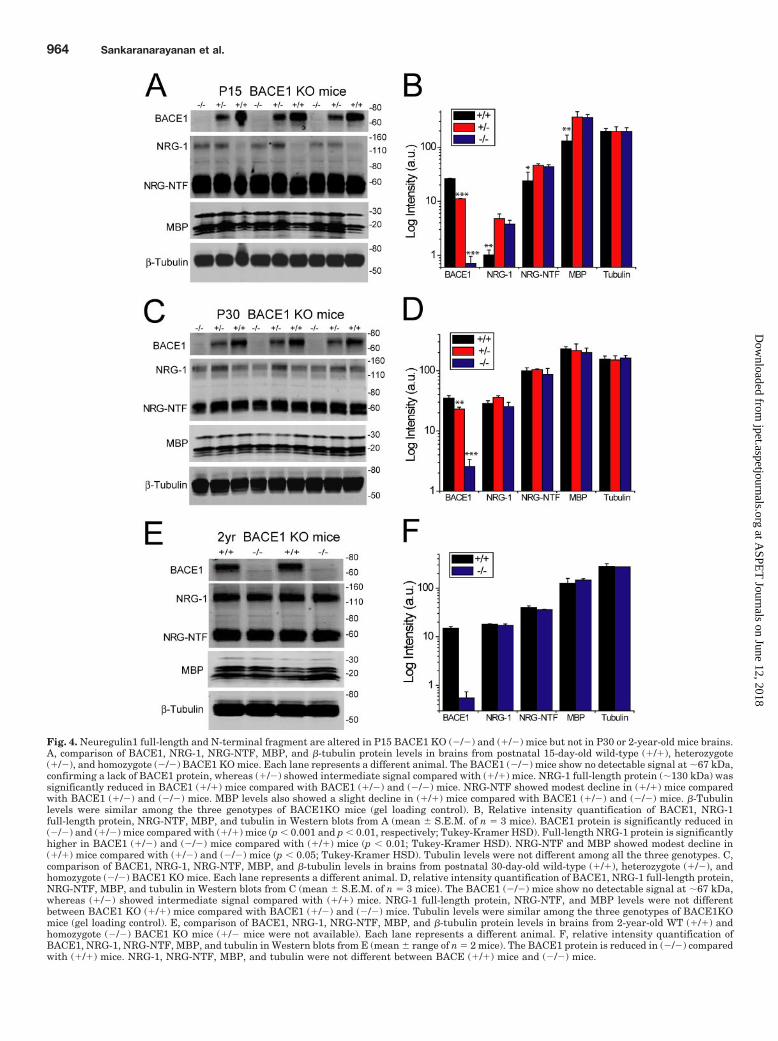
Fig. 4. Neuregulin1 full-length and N-terminal fragment are altered in P15 BACE1 KO (�/�) and (�/�) mice but not in P30 or 2-year-old mice brains.A, comparison of BACE1, NRG-1, NRG-NTF, MBP, and �-tubulin protein levels in brains from postnatal 15-day-old wild-type (�/�), heterozygote(�/�), and homozygote (�/�) BACE1 KO mice. Each lane represents a different animal. The BACE1 (�/�) mice show no detectable signal at �67 kDa,confirming a lack of BACE1 protein, whereas (�/�) showed intermediate signal compared with (�/�) mice. NRG-1 full-length protein (�130 kDa) wassignificantly reduced in BACE1 (�/�) mice compared with BACE1 (�/�) and (�/�) mice. NRG-NTF showed modest decline in (�/�) mice comparedwith BACE1 (�/�) and (�/�) mice. MBP levels also showed a slight decline in (�/�) mice compared with BACE1 (�/�) and (�/�) mice. �-Tubulinlevels were similar among the three genotypes of BACE1KO mice (gel loading control). B, Relative intensity quantification of BACE1, NRG-1full-length protein, NRG-NTF, MBP, and tubulin in Western blots from A (mean � S.E.M. of n � 3 mice). BACE1 protein is significantly reduced in(�/�) and (�/�) mice compared with (�/�) mice (p � 0.001 and p � 0.01, respectively; Tukey-Kramer HSD). Full-length NRG-1 protein is significantlyhigher in BACE1 (�/�) and (�/�) mice compared with (�/�) mice (p � 0.01; Tukey-Kramer HSD). NRG-NTF and MBP showed modest decline in(�/�) mice compared with (�/�) and (�/�) mice (p � 0.05; Tukey-Kramer HSD). Tubulin levels were not different among all the three genotypes. C,comparison of BACE1, NRG-1, NRG-NTF, MBP, and �-tubulin levels in brains from postnatal 30-day-old wild-type (�/�), heterozygote (�/�), andhomozygote (�/�) BACE1 KO mice. Each lane represents a different animal. D, relative intensity quantification of BACE1, NRG-1 full-length protein,NRG-NTF, MBP, and tubulin in Western blots from C (mean � S.E.M. of n � 3 mice). The BACE1 (�/�) mice show no detectable signal at �67 kDa,whereas (�/�) showed intermediate signal compared with (�/�) mice. NRG-1 full-length protein, NRG-NTF, and MBP levels were not differentbetween BACE1 KO (�/�) mice compared with BACE1 (�/�) and (�/�) mice. Tubulin levels were similar among the three genotypes of BACE1KOmice (gel loading control). E, comparison of BACE1, NRG-1, NRG-NTF, MBP, and �-tubulin protein levels in brains from 2-year-old WT (�/�) andhomozygote (�/�) BACE1 KO mice (�/� mice were not available). Each lane represents a different animal. F, relative intensity quantification ofBACE1, NRG-1, NRG-NTF, MBP, and tubulin in Western blots from E (mean � range of n � 2 mice). The BACE1 protein is reduced in (�/�) comparedwith (�/�) mice. NRG-1, NRG-NTF, MBP, and tubulin were not different between BACE (�/�) mice and (�/�) mice.
964 Sankaranarayanan et al.
at ASPE
T Journals on June 12, 2018
jpet.aspetjournals.orgD
ownloaded from
active in early development, and these effects may be com-pensated for with age in these mice. An alternate interpre-tation is that cleavage of NRG-1 is more robust in youngeranimals as evidenced by reduced levels of full-length NRG-1,whereas with increasing age NRG-1 cleavage declines tolevels where no significant differences are observed betweenthe BACE1 (�/�) and (�/�) animals.
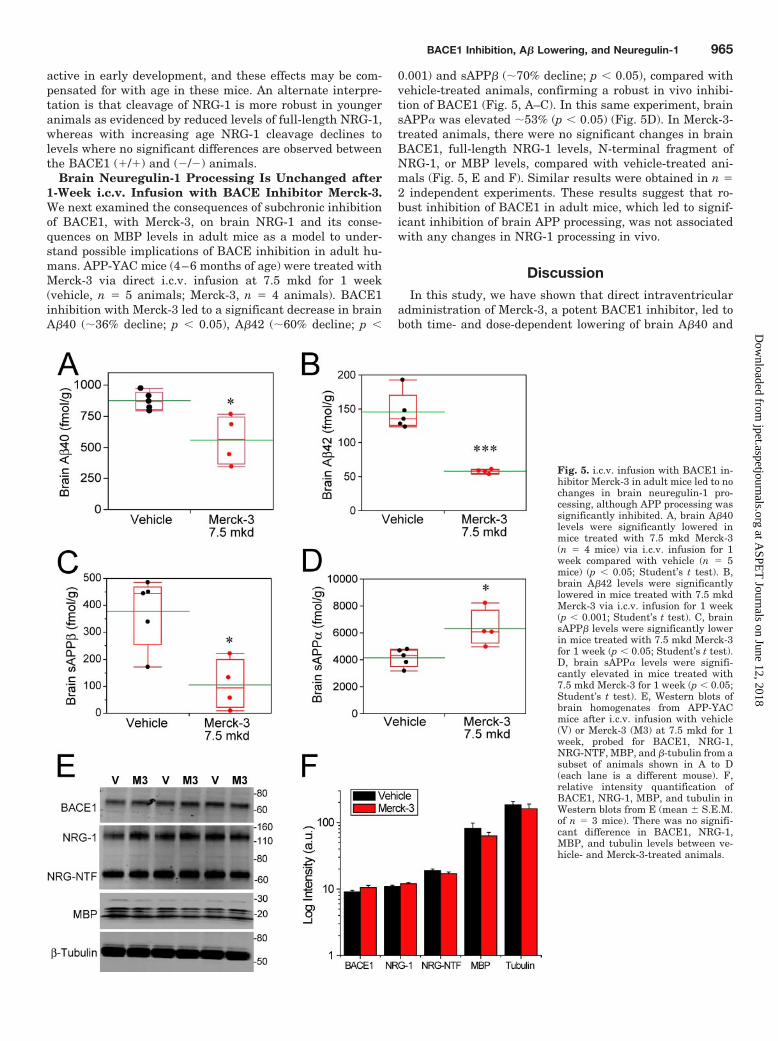
Brain Neuregulin-1 Processing Is Unchanged after1-Week i.c.v. Infusion with BACE Inhibitor Merck-3.We next examined the consequences of subchronic inhibitionof BACE1, with Merck-3, on brain NRG-1 and its conse-quences on MBP levels in adult mice as a model to under-stand possible implications of BACE inhibition in adult hu-mans. APP-YAC mice (4–6 months of age) were treated withMerck-3 via direct i.c.v. infusion at 7.5 mkd for 1 week(vehicle, n � 5 animals; Merck-3, n � 4 animals). BACE1inhibition with Merck-3 led to a significant decrease in brainA�40 (�36% decline; p � 0.05), A�42 (�60% decline; p �
0.001) and sAPP� (�70% decline; p � 0.05), compared withvehicle-treated animals, confirming a robust in vivo inhibi-tion of BACE1 (Fig. 5, A–C). In this same experiment, brainsAPP� was elevated �53% (p � 0.05) (Fig. 5D). In Merck-3-treated animals, there were no significant changes in brainBACE1, full-length NRG-1 levels, N-terminal fragment ofNRG-1, or MBP levels, compared with vehicle-treated ani-mals (Fig. 5, E and F). Similar results were obtained in n �2 independent experiments. These results suggest that ro-bust inhibition of BACE1 in adult mice, which led to signif-icant inhibition of brain APP processing, was not associatedwith any changes in NRG-1 processing in vivo.
DiscussionIn this study, we have shown that direct intraventricular
administration of Merck-3, a potent BACE1 inhibitor, led toboth time- and dose-dependent lowering of brain A�40 and
Fig. 5. i.c.v. infusion with BACE1 in-hibitor Merck-3 in adult mice led to nochanges in brain neuregulin-1 pro-cessing, although APP processing wassignificantly inhibited. A, brain A�40levels were significantly lowered inmice treated with 7.5 mkd Merck-3(n � 4 mice) via i.c.v. infusion for 1week compared with vehicle (n � 5mice) (p � 0.05; Student’s t test). B,brain A�42 levels were significantlylowered in mice treated with 7.5 mkdMerck-3 via i.c.v. infusion for 1 week(p � 0.001; Student’s t test). C, brainsAPP� levels were significantly lowerin mice treated with 7.5 mkd Merck-3for 1 week (p � 0.05; Student’s t test).D, brain sAPP� levels were signifi-cantly elevated in mice treated with7.5 mkd Merck-3 for 1 week (p � 0.05;Student’s t test). E, Western blots ofbrain homogenates from APP-YACmice after i.c.v. infusion with vehicle(V) or Merck-3 (M3) at 7.5 mkd for 1week, probed for BACE1, NRG-1,NRG-NTF, MBP, and �-tubulin from asubset of animals shown in A to D(each lane is a different mouse). F,relative intensity quantification ofBACE1, NRG-1, MBP, and tubulin inWestern blots from E (mean � S.E.M.of n � 3 mice). There was no signifi-cant difference in BACE1, NRG-1,MBP, and tubulin levels between ve-hicle- and Merck-3-treated animals.
BACE1 Inhibition, A� Lowering, and Neuregulin-1 965
at ASPE
T Journals on June 12, 2018
jpet.aspetjournals.orgD
ownloaded from

A�42 that could be sustained for up to 7 to 14 days oftreatment. The decline in brain A� species was correlatedwith a decrease in brain sAPP�, the N-terminal fragment ofAPP after BACE cleavage, thus confirming direct BACE1inhibition. Concurrently, BACE1 inhibition led to a modestincrease in brain sAPP�, confirming that BACE1 and�-secretase enzymes compete for cleavage of APP in vivo.Consistent with previous reports (Hu et al., 2006; Willem etal., 2006), an increase in full-length NRG-1 levels was ob-served in young P15 BACE1 KO (�/�) mice compared with(�/�) mice. Interestingly, we report first observations where(�/�) BACE1 KO mice displayed a similar increase in full-length NRG-1 like the (�/�) mice, and they lacked BACE1gene dosage on NRG-1, compared with (�/�) mice. In con-trast, there was a lack of a correlated decline in the N-terminal fragment of NRG-1 in the P15 BACE1 KO mice. Inaddition, 1-month-old P30 and 2-year-old BACE1 KO (�/�)mice and adult mice treated with a BACE1 inhibitor show nochange in steady-state levels of brain full-length NRG-1 orNRG-NTF. Thus, in vivo BACE1 inhibition with Merck-3 inadult animals, which led to a significant decrease in brain APPprocessing, produced no measurable effect on NRG-1 process-ing. Taken together, these results suggest that pharmacologicalinhibition of BACE1 in adult animals can significantly lowerbrain A� levels and increase �-secretase processing of APPwithout adversely affecting NRG-1 processing.
BACE1 is the major �-secretase in the brain that wasidentified and cloned using a variety of strategies (Hussain etal., 1999; Sinha et al., 1999; Vassar et al., 1999; Yan et al.,1999; Lin et al., 2000). Although BACE1 inhibitor develop-ment has been challenging due to the large size and presenceof numerous charged residues in the active site (Hong et al.,2000), numerous peptidomimetic BACE inhibitors have beendeveloped (Baxter and Reitz, 2005; Ghosh et al., 2005). Al-though the first potent BACE1 inhibitor (Statine-val) was apeptidic inhibitor with a noncleavable statine and valineresidue (IC50 �30 nM) (Sinha et al., 1999), the subsequentstatine-based inhibitors (Hong et al., 2000; Ghosh et al.,2001; Hu et al., 2004) had a variety of modifications, includ-ing hydroxyethylene isosteres (Hong et al., 2002; Hom et al.,2004), hydroxyethylamine isosteres (Tamamura et al., 2003;Stachel et al., 2004), hydroxymethylcarbonyl isosteres (Shutoet al., 2003; Kimura et al., 2004, 2005), and aminoethylenetetrahedral isosteres (Yang et al., 2006). Most of these tran-sition state inhibitors had in vitro activity on BACE1, butmany failed to work in cell culture systems or in vivo.
Merck-3, a hydoxyethylamine isostere, had an in vitroBACE1 IC50 of �15 nM, and it showed robust lowering ofsecreted A�40 from brain slice cultures (IC50 �30 nM). Whenadministered peripherally via i.v. injection, no brain A� low-ering was observed because it had poor apparent permeabil-ity and was a P-gp substrate. Central nervous system drugstypically require apparent permeabilities greater than 15 �10�6 cm/s and a P-gp transport ratio of �2.5 (Mahar Doan etal., 2002). When Merck-3 was introduced directly into the leftlateral ventricle, a robust lowering of brain A�40 and A�42 inboth brain hemispheres was observed. Merck-3 also led to atime- and dose-dependent lowering of brain sAPP�, A�40,and A�42. Although brain A�40 and A�42 showed good cor-relation within a brain hemisphere, there was also excellentcorrelation of A�40 and 42 levels between the left and rightbrain hemisphere. The robust brain A� lowering is not prob-
ably due to nonspecific cellular toxicity of the surgery or toMerck-3, because animals infused with vehicle for 14 daysshowed no A� lowering compared with untreated animals;there was no difference in survival or gross behavioralchanges between vehicle- and Merck-3-treated animals; amodest but consistent elevation of brain sAPP� levels wasobserved, indicating lack of toxicity; and finally, brain mRNAexpression studies suggest that brain A� lowering withMerck-3 is not due to a reduction of brain BACE or human-APP expression or to an up-regulation of ADAM10 orADAM17. Therefore, we conclude that if a potent BACE1inhibitor can be delivered past the blood-brain barrier, asustained lowering of brain A� species can be achieved.
Although Merck-3 is a potent inhibitor of BACE1, it canalso inhibit BACE2, with an IC50 value of �300 nM (Pietraket al., 2005). Thus, both BACE1 and BACE2 are probablyinhibited at the concentrations of Merck-3 achieved via i.c.v.infusion. Although, BACE1 knockdown in cell cultures (Basiet al., 2003; Kao et al., 2004) or in mice (Singer et al., 2005)can lower sAPP� production, BACE2 knockdown can actu-ally increase sAPP� and A� secretion, whereas simultaneousknockdown of BACE1 and BACE2 may produce no effects onsecreted A� (Basi et al., 2003). Thus, BACE isoforms maycompete for APP processing in some cell lines and reductionin cleavage at position 19 or 20 by BACE2 (Yan et al., 2001)could enhance cleavage at position 1 of A� sequence byBACE1 (Basi et al., 2003). Merck-3 led to robust lowering ofbrain sAPP� while producing a significant elevation of brainsAPP� levels, consistent with results from primary neuronsfrom the BACE1 KO mice (Cai et al., 2001), and Tg2576 micecrossed with the BACE1 KO mice (Ohno et al., 2004; Laird etal., 2005). These observations, together with the poor brainexpression of BACE2 (Bennett et al., 2000), strongly supportthe idea that BACE1 is the predominant �-site cleavageenzyme in brains of mice and that inhibition of BACE1 caneffectively lower brain A� production.
The reduction of brain sAPP� and associated elevation ofbrain sAPP� may have beneficial effects because sAPP� issuggested to have putative neuroprotective functions (Fu-rukawa et al., 1996). In previous studies, it has been shownthat activation of the M1-cholinergic receptors (Nitsch et al.,1992; Caccamo et al., 2006) and serotonin receptors (Nitschet al., 1996) led to increased �-secretase processing of APP,probably via protein kinase C activation (Skovronsky et al.,2000). Direct intracortical injection of PKC activators in miceshowed an acute lowering of brain A� and sAPP�, but nochange in sAPP� up to 6 h after injection (however, seeRossner et al., 2000; Savage et al., 1998). The rapid loweringof brain A� and sAPP� are consistent with the short half-lives of �1 to 2 h estimated for brain A� and c99 (Savage etal., 1998; Cirrito et al., 2003). Mice overexpressing the�-secretase enzyme ADAM10, crossed with the APPV717Itransgenic mice, showed reduced A� production and amyloidplaque burden. In contrast, coexpression of dominant-nega-tive mutant of ADAM10 with APPV717I enhanced amyloidpathology in double transgenic mice (Postina et al., 2004).Taken together, these results suggest that BACE1 and�-secretase enzymes can compete for APP processing andthat BACE1 inhibition can indirectly enhance �-secretaseprocessing of APP.
Our results are consistent with previous studies in whichintraperitoneal administration of a hydroxyethylene isostere
966 Sankaranarayanan et al.
at ASPE
T Journals on June 12, 2018
jpet.aspetjournals.orgD
ownloaded from

inhibitor conjugated to a peptide carrier (Chang et al., 2004)led to a dose- and time-dependent reduction of plasma andbrain A�40. Intrahippocampal injection of a different BACE1inhibitor (cell IC50 � 10 �M) showed in vivo lowering of brainA�40, A�42, and C99, but no change in C83 was observed inmice (Asai et al., 2006). More recently, a poorly brain pene-trant BACE1 inhibitor when coadministered with a P-gpinhibitor lowered both brain A�40 and A�42, and it wasaccompanied by an increase in brain sAPP� (Hussain et al.,2007). However, these previous studies have not addressedthe impact of BACE1 inhibition on brain NRG-1 processing.
Neuregulin 1 type III (NRG-1) is a double transmembraneprotein expressed on axonal membranes and cleaved by met-alloproteases. The N-terminal membrane-bound fragment ofNRG-1 can function as a juxtacrine-signaling molecule thatregulates axon myelination via activation of ErbB receptorson Schwann cells (Nave and Salzer 2006). Recent studieshave suggested that BACE1 regulates NRG-1 processing andsignaling during development and thereby affects early my-elination in the nervous system (Hu et al., 2006; Willem etal., 2006). BACE1 KO mice showed defects in myelination, asmeasured by an increase in G-ratio or myelin thickness in thesciatic nerves (Hu et al., 2006; Willem et al., 2006). Myelinthickness seems to increase gradually with age in BACE1 KOmice, change in G-ratio of 0.75 to 0.95 at P5 to P17 days to0.70 to 0.85 in the adult (6–8 weeks of age), whereas the WTmice exhibit G-ratio of 0.60 to 0.80 over this age (Willem etal., 2006). These results suggest that downstream signalingof NRG-1, after BACE1 cleavage, maybe a critical eventduring myelination in early development.
An increase in full-length NRG-1 levels in the brain wasobserved in young P5 mice (Willem et al., 2006), but thisstudy did not examine changes in the N-terminal fragment ofNRG-1. Proximal studies by Hu et al. (2006) showed anincrease in full-length NRG-1 levels in brain along with areduction in the N-terminal fragment of NRG-1 in young(P30–P60) BACE1 KO (�/�) mice. These results suggestedthat BACE1 knockdown maybe accompanied by a decrease inin vivo processing of NRG-1 in young mice. However, theseprevious studies did not examine the affect of BACE1 genedosage and older ages on NRG-1 processing.
The results in this study confirmed an increase in full-length NRG-1 protein in young P15 BACE1 KO (�/�) micecompared with WT mice, suggesting reduced NRG-1 process-ing. Although BACE1 protein levels were halved, an identicalincrease in full-length NRG-1, equivalent to that of BACE1(�/�) mice, was observed in BACE1 (�/�) mice comparedwith WT (�/�) mice. These results suggest a decrease inNRG-1 processing in both BACE1 (�/�) and (�/�) mice.These results are in contrast to the effect of BACE1 knock-down on APP processing, where BACE1 (�/�) mice showedonly �20% reduction in brain A�40 levels compared with(�/�) mice, whereas (�/�) mice showed an almost completereduction in brain A� levels (Supplemental Fig. 3). Thus,neither APP nor NRG-1 processing shows precise gene dos-age dependence with BACE1 knockdown, raising questionsregarding the exact physiological substrate(s) for BACE1 invivo. In contrast, BACE1-dependent processing of substratesmay not be solely regulated by absolute expression levels, butby differential colocalization with substrates and intracellu-lar localization similar to that suggested in other reports(Sinha and Lieberburg, 1999; Greenfield et al., 2000; Paster-
nak et al., 2004). We envision that future studies could ex-amine the affect of BACE1 gene dosage on myelination inparallel with NRG-1 and other substrates.
Although an increase in full-length NRG-1 was observed inP15 BACE1 (�/�) and (�/�) mice, we observed a modestdecline in the N-terminal fragment of NRG-1 in BACE1(�/�) mice compared with BACE1 (�/�) and (�/�) mice incontrast to a previous report (Hu et al., 2006). The possiblereasons for the differences between these studies are pres-ently unknown, but they could be due to difference in geneticbackground or difference in the locus where BACE1 wasknocked out in these mice. Mice used in our studies wererederived from that generated in Cai et al. (2001) and back-crossed to the C57BL/6 mice. Thus, although mice used in ourstudies and Hu et al. (2006) seem to be of the same origin,backcrossing to a pure genetic background may have led tothe differences observed.
BACE1 protein levels and activity are �2- to 3-fold higher(our results) and manyfold higher (Willem et al., 2006) inmice from ages of P0 to P15 compared with mice P30 andolder, thus motivating experiments to examine the age de-pendence of NRG-1 processing. Examination of NRG-1 pro-cessing in P30 BACE1 KO mice revealed no differences insteady-state levels of either full-length NRG-1 or NRG-NTFamong all three genotypes in our studies. Finally, 2-year-oldBACE1 KO (�/�) mice showed no change in full-lengthNRG-1 or NRG-NTF levels compared with WT mice. Ourresults are consistent with those of both Willem et al. (2006)and Hu et al. (2006) in that young BACE1 KO mice show anincrease in full-length NRG-1 compared with (�/�) mice.Although NRG-NTF was modestly elevated in (�/�) mice inHu et al. (2006), we observed a modest decline in NRG-NTFin P15 mice, and no differences were seen in P30 and 2-year-old mice, whereas Willem et al. (2006) did not examine NRG-NTF in their studies. Thus, our results point to a transienteffect of BACE1 knockdown on NRG-1 processing in mice,and they suggest differences in NRG-1 processing only inearly development. The age-dependent decrease in BACE1expression could explain the transient effect on NRG-1 pro-cessing, and it can also account for the lack of significantdifferences in NRG-1 and NRG-NTF between BACE1 KOand (�/�) mice at ages P30 and older. Thus, it seems thatwith age, NRG-1 processing declines in (�/�) mice to levelsequivalent to that in the BACE1 (�/�) mice, and this mightexplain the lack of differences in NRG-1 processing in olderages.
Finally, inhibition of BACE1 over the course of 1 week inadult APP-YAC mice, via direct i.c.v. infusion of Merck-3, didnot alter full-length NRG-1 or NRG-NTF levels or MBP lev-els compared with vehicle-treated mice, although a robustinhibition of APP processing and A� production was ob-served. These results suggest that acute inhibition of BACE1in adult mice is without significant impact on NRG-1 orNRG-NTF, although APP processing is significantly altered.The lack of an effect of Merck-3 on NRG-1 processing isconsistent with our observations that there was minimalimpact of BACE1 knockdown on NRG-1 processing in oldermice. However, this cannot be predicted a priori becausepharmacological inhibition with a compound can signifi-cantly change the steady-state processing of substrate as inthe case of APP. In conclusion, although we have consistentlyobserved that in vivo BACE1 inhibition leads to significant
BACE1 Inhibition, A� Lowering, and Neuregulin-1 967
at ASPE
T Journals on June 12, 2018
jpet.aspetjournals.orgD
ownloaded from

decline in brain sAPP� and A� production, steady-stateNRG-1 processing was unaffected. These results suggest thatBACE1 inhibition may be devoid of adverse effects due toaltered NRG-1 processing in adult humans. Recent studiesindicate that NRG-1 can directly affect glutamatergic andGABAergic synapse maturation and transmission in vivo (Liet al., 2007; Woo et al., 2007). Therefore, the interplay ofNRG-1 and BACE1 on synaptic function and the impact ofBACE1 inhibition should motivate further investigation.
In conclusion, our results support BACE1 inhibition as aviable target to lower brain A�40 and A�42 and to enhancethe nonamyloidogenic processing of APP via �-secretasecleavage without adversely affecting NRG-1 downstreamevents. In recent studies, there have been a number of prom-ising reports of acute lowering of brain A� with BACE inhib-itors after peripheral administration, but these inhibitorslack the properties required for sustained in vivo efficacy(Stachel et al., 2006; Hussain et al., 2007; Stanton et al.,2007). Further optimization of BACE inhibitors for improvedpotency, physical, and pharmacokinetic properties would al-low better understanding of the therapeutic effects and po-tential drawbacks of long-term BACE inhibition.
Acknowledgments
We thank Jennifer Shapiro and Mark Stiteler for help in theneuregulin experiments and Dr. Thomas William Mitchell, JimDestefano, Sandra Veggian, Jeanine Hange, and Ryan Desautels(Merck Research Laboratories, West Point Laboratory Animal Re-sources team) for technical support in the animal surgeries. Finally,we gratefully acknowledge the extensive comments of the reviewers.
ReferencesAsai M, Hattori C, Iwata N, Saido TC, Sasagawa N, Szabo B, Hashimoto Y, Ma-
ruyama K, Tanuma S, Kiso Y, et al. (2006) The novel beta-secretase inhibitorKMI-429 reduces amyloid beta peptide production in amyloid precursor proteintransgenic and wild-type mice. J Neurochem 96:533–540.
Basi G, Frigon N, Barbour R, Doan T, Gordon G, McConlogue L, Sinha S, and ZellerM (2003) Antagonistic effects of beta-site amyloid precursor protein-cleaving en-zymes 1 and 2 on �-amyloid peptide production in cells. J Biol Chem 278:31512–31520.
Baxter EW and Reitz AB (2005) BACE inhibitors for the treatment of Alzheimer’sdisease, in Annual Reports in Medicinal Chemistry, Vol. 40, pp. 35–48, Elsevier,New York.
Bennett BD, Babu-Khan S, Loeloff R, Louis JC, Curran E, Citron M, and Vassar R(2000) Expression analysis of BACE2 in brain and peripheral tissues. J Biol Chem275:20647–20651.
Best JD, Jay MT, Otu F, Ma J, Nadin A, Ellis S, Lewis HD, Pattison C, Reilly M,Harrison T, et al. (2005) Quantitative measurement of changes in amyloid-�(40) inthe rat brain and cerebrospinal fluid following treatment with the gamma-secretase inhibitor LY-411575 [N2-[(2S)-2-(3,5-difluorophenyl)-2-hydroxyeth-anoyl]-N1-[(7S)-5-methyl-6-oxo-6,7-dihydro-5H-dibenzo[b,d]azepin-7-yl]-L-alaninamide]. J Pharmacol Exp Ther 313:902–908.
Bodendorf U, Danner S, Fischer F, Stefani M, Sturchler-Pierrat C, Wiederhold KH,Staufenbiel M, and Paganetti P (2002) Expression of human beta-secretase in themouse brain increases the steady-state level of beta-amyloid. J Neurochem 80:799–806.
Braak H and Braak E (1991) Neuropathological staging of Alzheimer-relatedchanges. Acta Neuropathol 82:239–259.
Caccamo A, Oddo S, Billings LM, Green KN, Martinez-Coria H, Fisher A, andLaFerla FM (2006) M1 receptors play a central role in modulating AD-like pathol-ogy in transgenic mice. Neuron 49:671–682.
Cai H, Wang Y, McCarthy D, Wen H, Borchelt DR, Price DL, and Wong PC (2001)BACE1 is the major beta-secretase for generation of Abeta peptides by neurons.Nat Neurosci 4:233–234.
Chang WP, Koelsch G, Wong S, Downs D, Da H, Weerasena V, Gordon B, Devasam-udram T, Bilcer G, Ghosh AK, et al. (2004) In vivo inhibition of Abeta productionby memapsin 2 (beta-secretase) inhibitors. J Neurochem 89:1409–1416.
Cirrito JR, May PC, O’Dell MA, Taylor JW, Parsadanian M, Cramer JW, Audia JE,Nissen JS, Bales KR, Paul SM, et al. (2003) In vivo assessment of brain interstitialfluid with microdialysis reveals plaque-associated changes in amyloid-beta metab-olism and half-life. J Neurosci 23:8844–8853.
Citron M (2004) Strategies for disease modification in Alzheimer’s disease. Nat RevNeurosci 5:677–685.
Dominguez D, Tournoy J, Hartmann D, Huth T, Cryns K, Deforce S, Serneels L,Camacho IE, Marjaux E, Craessaerts K, et al. (2005) Phenotypic and biochemicalanalyses of BACE1- and BACE2-deficient mice. J Biol Chem 280:30797–30806.
Furukawa K, Sopher BL, Rydel RE, Begley JG, Pham DG, Martin GM, Fox M, and
Mattson MP (1996) Increased activity-regulating and neuroprotective efficacy ofalpha-secretase-derived secreted amyloid precursor protein conferred by a C-terminal heparin-binding domain. J Neurochem 67:1882–1896.
Ghosh AK, Bilcer G, Harwood C, Kawahama R, Shin D, Hussain KA, Hong L, LoyJA, Nguyen C, Koelsch G, et al. (2001) Structure-based design: potent inhibitors ofhuman brain memapsin 2 (beta-secretase). J Med Chem 44:2865–2868.
Ghosh AK, Kumaragurubaran N, and Tang J (2005) Recent developments of struc-ture based beta-secretase inhibitors forAlzheimer’s disease. Curr Top Med Chem5:1609–1622.
Greenfield JP, Gross RS, Gouras GK, and Xu H (2000) Cellular and molecular basisof beta-amyloid precursor protein metabolism. Front Biosci 5:D72–D83.
Hardy J and Selkoe DJ (2002) The amyloid hypothesis of Alzheimer’s disease:progress and problems on the road to therapeutics. Science 297:353–356.
Harrison SM, Harper AJ, Hawkins J, Duddy G, Grau E, Pugh PL, Winter PH,Shilliam CS, Hughes ZA, Dawson LA, et al. (2003) BACE1 (beta-secretase) trans-genic and knockout mice: identification of neurochemical deficits and behavioralchanges. Mol Cell Neurosci 24:646–655.
Hom RK, Gailunas AF, Mamo S, Fang LY, Tung JS, Walker DE, Davis D, ThorsettED, Jewett NE, Moon JB, et al. (2004) Design and synthesis of hydroxyethylene-based peptidomimetic inhibitors of human beta-secretase. J Med Chem 47:158–164.
Hong L, Koelsch G, Lin X, Wu S, Terzyan S, Ghosh AK, Zhang XC, and Tang J (2000)Structure of the protease domain of memapsin 2 (beta-secretase) complexed withinhibitor. Science 290:150–153.
Hong L, Turner RT III, Koelsch G, Shin D, Ghosh AK, and Tang J (2002) Crystalstructure of memapsin 2 (�-secretase) in complex with an inhibitor OM00–3.Biochemistry 41:10963–10967.
Hu B, Fan KY, Bridges K, Chopra R, Lovering F, Cole D, Zhou P, Ellingboe J, Jin G,Cowling R, et al. (2004) Synthesis and SAR of bis-statine based peptides as BACE1 inhibitors. Bioorg Med Chem Lett 14:3457–4560.
Hussain I, Hawkins J, Harrison D, Hille C, Wayne G, Cutler L, Buck T, Walter D,Demont E, Howes C, et al. (2007) Oral administration of a potent and selectivenon-peptidic BACE-1 inhibitor decreases beta-cleavage of amyloid precursor pro-tein and amyloid-beta production in vivo. J Neurochem 100:802–809.
Hussain I, Powell D, Howlett DR, Tew DG, Week TD, Chapman C, Gloger IS,Murphy KE, Southan CD, Ryan DM, et al. (1999) Identification of a novel asparticprotease (Asp 2) as beta-secretase. Mol Cell Neurosci 14:419–427.
Hu X, Hicks CW, He W, Wong P, Macklin WB, Trapp BD, and Yan R (2006) Bace1modulates myelination in the central and peripheral nervous system. Nat Neurosci9:1520–1525.
Ida N, Hartmann T, Pantel J, Schroder J, Zerfass R, Forstl H, Sandbrink R, MastersCL, and Beyreuther K (1996) Analysis of heterogeneous A4 peptides in humancerebrospinal fluid and blood by a newly developed sensitive Western blot assay.J Biol Chem 271:22908–22914.
Kao SC, Krichevsky AM, Kosik KS, and Tsai LH (2004) BACE1 suppression by RNAinterference in primary cortical neurons. J Biol Chem 279:1942–1949.
Kawarabayashi T, Younkin LH, Saido TC, Shoji M, Ashe KH, and Younkin SG (2001)Age-dependent changes in brain, CSF, and plasma amyloid (beta) protein in theTg2576 transgenic mouse model of Alzheimer’s disease. J Neurosci 21:372–381.
Kim KS, Miller DL, Sapienza VJ, Chen CJ, Bai C, Grundke-Iqbal I, Currie JR, andWisniewski HM (1988) Production and characterization of monoclonal antibodiesreactive to synthetic cerebrovascular amyloid peptide. Neurosci Res Commun2:121–130.
Kimura T, Shuto D, Hamada Y, Igawa N, Kasai S, Liu P, Hidaka K, Hamada T,Hayashi Y, and Kiso Y (2005) Design and synthesis of highly active Alzheimer’sbeta-secretase (BACE1) inhibitors, KMI-420 and KMI-429, with enhanced chemi-cal stability. Bioorg Med Chem Lett 15:211–215.
Kimura T, Shuto D, Kasai S, Liu P, Hidaka K, Hamada T, Hayashi Y, Hattori C, AsaiM, Kitazume S, et al. (2004) KMI-358 and KMI-370, highly potent and small-sizedBACE1 inhibitors containing phenylnorstatine. Bioorg Med Chem Lett 14:1527–1531.
Laird FM, Cai H, Savonenko AV, Farah MH, He K, Melnikova T, Wen H, Chiang HC,Xu G, Koliatsos VE, et al. (2005) BACE1, a major determinant of selective vulner-ability of the brain to amyloid-beta amyloidogenesis, is essential for cognitive,emotional, and synaptic functions. J Neurosci 25:11693–11709.
Lamb BT, Sisodia SS, Lawler AM, Slunt HH, Kitt CA, Kearns WG, Pearson PL, PriceDL, and Gearhart JD (1993) Introduction and expression of the 400 kilobaseamyloid precursor protein gene in transgenic mice. Nat Genet 5:22–30.
LeBlanc AC, Koutroumanis M, and Goodyer CG (1998) Protein kinase C activationincreases release of secreted amyloid precursor protein without decreasing Abetaproduction in human primary neuron cultures. J Neurosci 18:2907–2913.
Lee EB, Zhang B, Liu K, Greenbaum EA, Doms RW, Trojanowski JQ, and Lee VM(2005) BACE overexpression alters the subcellular processing of APP and inhibitsAbeta deposition in vivo. J Cell Biol 168:291–302.
Li B, Woo RS, Mei L, and Malinow R (2007) The neuregulin-1 receptor erbB4 controlsglutamatergic synapse maturation and plasticity. Neuron 54:583–597.
Lin X, Koelsch G, Wu S, Downs D, Dashti A, and Tang J (2000) Human asparticprotease memapsin 2 cleaves the beta-secretase site of beta-amyloid precursorprotein. Proc Natl Acad Sci U S A 97:1456–1460.
Luo Y, Bolon B, Kahn S, Bennett BD, Babu-Khan S, Denis P, Fan W, Kha H, ZhangJ, Gong Y, et al. (2001) Mice deficient in BACE1, the Alzheimer’s beta-secretase,have normal phenotype and abolished beta-amyloid generation. Nat Neurosci4:231–232.
Mahar Doan KM, Humphreys JE, Webster LO, Wring SA, Shampine LJ, Serabjit-Singh CJ, Adkison KK, and Polli JW (2002) Passive permeability and P-glycoprotein-mediated efflux differentiate central nervous system (CNS) and non-CNS marketed drugs. J Pharmacol Exp Ther 303:1029–1037.
Mohajeri MH, Saini KD, and Nitsch RM (2004) Transgenic BACE expression inmouse neurons accelerates amyloid plaque pathology. J Neural Transm 111:413–425.
968 Sankaranarayanan et al.
at ASPE
T Journals on June 12, 2018
jpet.aspetjournals.orgD
ownloaded from

Naslund J, Haroutunian V, Mohs R, Davis KL, Davies P, Greengard P, and BuxbaumJD (2000) Correlation between elevated levels of amyloid beta peptide in the brainand cognitive decline. JAMA 283:1571–1577.
Nave KA and Salzer JL (2006) Axonal regulation of myelination by neuregulin 1.Curr Opin Neurobiol 16:492–500.
Nishitomi K, Sakaguchi G, Horikoshi Y, Gray AJ, Maeda M, Hirata-Fukae C, BeckerAG, Hosono M, Sakaguchi I, Minami SS, et al. (2006) BACE1 inhibition reducesendogenous Abeta and alters APP processing in wild-type mice. J Neurochem99:1555–1563.
Nitsch RM, Deng M, Growdon JH, and Wurtman RJ (1996) Serotonin 5-HT2a and5-HT2c receptors stimulate amyloid precursor protein ectodomain secretion. J BiolChem 271:4188–4194.
Nitsch RM, Slack BE, Wurtman RJ, and Growdon JH (1992) Release of Alzheimeramyloid precursor derivatives stimulated by activation of muscarinic acetylcholinereceptors. Science 258:304–307.
Ohno M, Sametsky EA, Younkin LH, Oakley H, Younkin SG, Citron M, Vassar R,and Disterhoft JF (2004) BACE1 deficiency rescues memory deficits and cholin-ergic dysfunction in a mouse model of Alzheimer’s disease. Neuron 41:27–33.
Pasternak SH, Callahan JW, and Mahuran DJ (2004) The role of the endosomal/lysosomal system in amyloid-beta production and the pathophysiology of Alzhei-mer’s disease: reexamining the spatial paradox from a lysosomal perspective. JAlzheimers Dis 6:53–65.
Paxinos G and Franklin KBJ (2001) The Mouse Brain in Stereotaxic Coordinates, 2nded, Academic Press, San Diego.
Pietrak BL, Crouthamel MC, Tugusheva K, Lineberger JE, Xu M, DiMuzio JM,Steele T, Espeseth AS, Stachel SJ, Coburn CA, et al. (2005) Biochemical andcell-based assays for characterization of BACE-1 inhibitors. Anal Biochem 342:144–151.
Postina R, Schroeder A, Dewachter A, Bohl J, Schmitt U, Kojro E, Prinzen C, EndresK, Hiemke C, Blessing M, et al. (2004) A disintegrin-metalloproteinase preventsamyloid plaque formation and hippocampal defects in an Alzheimer disease mousemodel. J Clin Invest 113:1456–1464.
Roberds SL, Anderson J, Basi G, Bienkowski MJ, Branstetter DG, Chen KS, Freed-man SB, Frigon NL, Games D, Hu K, et al. (2001) BACE knockout mice are healthydespite lacking the primary beta-secretase activity in brain: implications for Alz-heimer’s disease therapeutics. Hum Mol Genet 10:1317–1324.
Rossner S, Beck M, Stahl T, Mendla K, Schliebs R, and Bigl V (2000) Constitutiveoveractivation of protein kinase C in guinea pig brain increases alpha-secretoryAPP processing without decreasing beta-amyloid generation. Eur J Neurosci 12:3191–3200.
Sambamurti K, Greig NH, and Lahiri DK (2002) Advances in the cellular andmolecular biology of the beta-amyloid protein in Alzheimer’s disease. Neuromo-lecular Med 1:1–31.
Savage MJ, Trusko SP, Howland DS, Pinsker LR, Mistretta S, Reaume AG, Green-berg BD, Siman R, and Scott RW (1998) Turnover of amyloid beta-protein in mousebrain and acute reduction of its level by phorbol ester. J Neurosci 18:1743–1752.
Scheff SW and Price DA (2003) Synaptic pathology in Alzheimer’s disease: a reviewof ultrastructural studies. Neurobiol Aging 24:1029–1046.
Shi XP, Tugusheva K, Bruce JE, Lucka A, Chen-Dodson E, Hu B, Wu GX, Price E,Register RB, Lineberger J, et al. (2005) Novel mutations introduced at the beta-site of amyloid beta protein precursor enhance the production of amyloid betapeptide by BACE1 in vitro and in cells. J Alzheimers Dis 7:139–148.
Shuto D, Kasai S, Kimura T, Liu P, Hidaka K, Hamada T, Shibakawa S, Hayashi Y,Hattori C, Szabo B, et al. (2003) KMI-008, a novel beta-secretase inhibitor con-taining a hydroxymethylcarbonyl isostere as a transition-state mimic: design andsynthesis of substrate-based octapeptides. Bioorg Med Chem Lett 13:4273–4276.
Singer O, Marr RA, Rockenstein E, Crews L, Coufal NG, Gage FH, Verma IM, andMasliah E (2005) Targeting BACE1 with siRNAs ameliorates Alzheimer diseaseneuropathology in a transgenic model. Nat Neurosci 8:1343–1349.
Sinha S, Anderson JP, Barbour R, Basi GS, Caccavello R, Davis D, Doan M, DoveyHF, Frigon N, Hong J, et al. (1999) Purification and cloning of amyloid precursorprotein beta-secretase from human brain. Nature 402:537–540.
Sinha S and Lieberburg I (1999) Cellular mechanisms of beta-amyloid productionand secretion. Proc Natl Acad Sci U S A 96:11049–11053.
Skovronsky DM, Moore DB, Milla ME, Doms RW, and Lee VM (2000) Protein kinase
C-dependent �-secretase competes with �-secretase for cleavage of amyloid-�precursor protein in the trans-Golgi network. J Biol Chem 275:2568–2575.
Stachel SJ, Coburn CA, Sankaranarayanan S, Price EA, Pietrak BL, Huang Q,Lineberger J, Espeseth AS, Jin L, Ellis J, et al. (2006) Macrocyclic inhibitors ofbeta-secretase: functional activity in an animal model. J Med Chem 49:6147–6150.
Stachel SJ, Coburn CA, Steele TG, Jones KG, Loutzenhiser EF, Gregro AR, Ra-japakse HA, Lai MT, Crouthamel MC, Xu M, et al. (2004) Structure-based designof potent and selective cell-permeable inhibitors of human beta-secretase (BACE-1). J Med Chem 47:6447–6450.
Stanton MG, Stauffer SR, Gregro AR, Steinbeiser M, Nantermet P, Sankaranaray-anan S, Price EA, Wu G, Crouthamel MC, Ellis J, et al. (2007) Discovery ofisonicotinamide derived beta-secretase inhibitors: in vivo reduction of beta-amyloid. J Med Chem 50:3431–3433.
Tamamura H, Kato T, Otaka A, and Fujii N (2003) Synthesis of potent �-secretaseinhibitors containing a hydroxyethylamine dipeptide isostere and their structure-activity relationship studies. Org Biomol Chem 1:2468–2473.
Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, andKatzman R (1991) Physical basis of cognitive alterations in Alzheimer’s disease:synapse loss is the major correlate of cognitive impairment. Ann Neurol 30:572–580.
Trojanowski JQ, Shin RW, Schmidt ML, and Lee VM (1995) Relationship betweenplaques, tangles, and dystrophic processes in Alzheimer’s disease. Neurobiol Aging16:335–340.
Umbenhauer DR, Lankas GR, Pippert TR, Wise LD, Cartwright ME, Hall SJ, andBeare CM (1997) Identification of a P-glycoprotein-deficient subpopulation in theCF-1 mouse strain using a restriction fragment length polymorphism. Toxicol ApplPharmacol 146:88–94.
Vassar R (2004) BACE1: the beta-secretase enzyme in Alzheimer’s disease. J MolNeurosci 23:105–114.
Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, Teplow DB,Ross S, Amarante P, Loeloff R, et al. (1999) Beta-Secretase cleavage of Alzheimer’samyloid precursor protein by the transmembrane aspartic protease BACE. Science286:735–741.
Willem M, Dewachter I, Smyth N, Van Dooren T, Borghgraef P, Haass C, and VanLeuven F (2004) �-Site amyloid precursor protein cleaving enzyme 1 increasesamyloid deposition in brain parenchyma but reduces cerebrovascular amyloidangiopathy in aging BACExAPP[V717I] double-transgenic mice. Am J Pathol165:1621–1631.
Willem M, Garratt AN, Novak B, Citron M, Kaufmann S, Rittger A, Destrooper B,Saftig P, Birchmeier C, and Haass C (2006) Control of peripheral nerve myelina-tion by the beta-secretase BACE1. Science 314:664–666.
Woo RS, Li XM, Tao Y, Carpenter-Hyland E, Huang YZ, Weber J, Neiswender H,Dong XP, Wu J, Gassmann M, et al. (2007) Neuregulin-1 enhances depolarization-induced GABA release. Neuron 54:599–610.
Yang W, Lu W, Lu Y, Zhong M, Sun J, Thomas AE, Wilkinson JM, Fucini RV, LamM, Randal M, et al. (2006) Aminoethylenes: a tetrahedral intermediate isostereyielding potent inhibitors of the aspartyl protease BACE-1. J Med Chem 49:839–842.
Yan R, Munzner JB, Shuck ME, and Bienkowski MJ (2001) BACE2 functions as analternative alpha-secretase in cells. J Biol Chem 276:34019–34027.
Yan RQ, Bienkowski MJ, Shuck ME, Miao H, Tory MC, Pauley AM, Brashier JR,Stratman NC, Mathews WR, Buhl AE, et al. (1999) Membrane-anchored aspartylprotease with Alzheimer’s disease beta-secretase activity. Nature 402:533–537.
Zheng H, Jiang M, Trumbauer ME, Sirinathsinghji DJ, Hopkins R, Smith DW,Heavens RP, Dawson GR, Boyce S, Conner MW, et al. (1995) beta-Amyloid pre-cursor protein-deficient mice show reactive gliosis and decreased locomotor activ-ity. Cell 81:525–531.
Zhu G, Wang D, Lin YH, McMahon T, Koo EH, and Messing RO (2001) Proteinkinase C epsilon suppresses Abeta production and promotes activation of alpha-secretase. Biochem Biophys Res Commun 285:997–1006.
Address correspondence to: Dr. Adam J. Simon, Department of Alzheimer’sResearch, WP 26A-2000, Merck Research Laboratories, 770, SumneytownPike, West Point, PA 19486. E-mail: [email protected]
BACE1 Inhibition, A� Lowering, and Neuregulin-1 969
at ASPE
T Journals on June 12, 2018
jpet.aspetjournals.orgD
ownloaded from






![Inhibition of the Otub1/c-Maf axis by the herbal acevaltrate ......that c-Maf confers to chemoresistance in MM [- 6]. Con sistently, downregulation of c-Maf by genetic inhibition leads](https://img.pdfslide.net/doc/110x75/613844900ad5d206764926ec/inhibition-of-the-otub1c-maf-axis-by-the-herbal-acevaltrate-that-c-maf.jpg)




![Inhibition of γ-Secretase Leads to an Increase in Presenilin-1 · defective 1 (APH1), and presenilin enhancer 2 (PEN2) [7]. γ-Secretase acts an aspartyl protease, which catalytic](https://img.pdfslide.net/doc/110x75/5fcf13aeec1c843f815764d3/inhibition-of-secretase-leads-to-an-increase-in-presenilin-1-defective-1-aph1.jpg)

















