Embed Size (px)
Citation preview

VOLUME 74, NUMBER 8 PH YS ICAL REVIEW LETTERS 20 FEBRUARY 1995
Quantum Monte Carlo Determination of Electronic and Structural Properties of Si„Clusters(n ~ 20)
Jeffrey C. Grossman' and Lubos Mitas'Department of Physics, University of Illinois at Urbana Cha-mpaign, Urbana, Illinois 61801
2National Center for Supercomputing Applications, University of Illinot sa't Urbana Cha-mpaign, Urbana, Illinois 61801(Received 18 July 1994)
Variational and fixed-node diffusion Monte Carlo methods are applied to study the structuraland valence electronic properties of Si„clusters. Binding energies for n ~ 7 agree within =4%with experiments and within =2% when the fixed-node error is decreased by using natural orbitals.For n ~ 9 we show that the local density approximation overbinds by =25%. We determinedunambiguously (i) the role of correlation in the energy ordering for different structures, including ournew lowest energy structure of Si2O, and (ii) a different ground state for Si~3 than the one predictedby the local density approximation.
PACS numbers: 36.40.—c, 71.45.Nt
The study of atomic clusters has become one of themost exciting areas of research because of the uniquecombination of molecular and condensed matter physicsand the possibility of studying the transition frommolecules to crystalline solids [1]. Recently, covalentlybonded semiconductor clusters (such as C, Si, and Ge)have been given much attention [2—4] as they have ratherdifferent properties than the bulk materials and formunique structures which may be useful in generatingnew materials with novel and unusual properties [4,5].In particular, Si„clusters have been studied by boththeoretical and experimental approaches [6—12], andfor small n ground state structures have been identified[13] and very recently confirmed experimentally [14].In spite of this effort, the situation is far from clear forlarger clusters; e.g. , a lower energy structure has justbeen found [15] for Si9. Except for the small clusterswhich were studied by quantum chemistry methods,most of the electronic structure calculations performed todate have used either empirical tight-binding models orlocal density approximation (LDA) approaches. Whilein most cases LDA provides excellent geometries andalso a correct picture of the energy differences betweenstructures with the same or similar number of atoms, onecan expect a significant overbinding; in addition, it isnot possible to assess the role of correlation energy instabilizing a particular structure. This uncertainty in thecorrelation effect description has led, for example, to acontroversy concerning the validity of LDA predictionsregarding the stable structure of Si f3 [11,16].
This Letter determines without ambiguity the impact ofcorrelation on the structural and electronic properties of Si„clusters using high-accuracy quantum Monte Carlo (QMC)methods. We find that the valence electron correlationenergy per atom varies significantly from one structure toanother even for larger clusters. We also evaluate the bind-ing energies, which differ from available experiment by2% to 5%, a significant improvement over the accuracy ofLDA. For Si20, a new elongated structure is proposed that
where f(R, t) = t/tr(R)P(R, t) and G(R, R', r) is a Greenfunction which is known in the r ~ 0 limit [21]. Wetreated the fermion antisymmetry problem by the com-monly used fixed-node approximation [21], which re-stricts the nodes of @(R,t) to be identical with the nodesof Pz. (R). The trial function used is a linear combinationof Slater determinants of single electron spin-up and spin-down orbitals times a correlation factor [22,23],
Pz. = g d„Dt Dl exp P u(r;t, r,t, r,,) (2)
we find to be about 4 eV lower in energy than the dodec-ahedron, and about 2 eV lower than previously proposedelongated structures [12,17,18]. The ability of the QMCmethod to study effects which are very sensitive to an accu-rate description of correlation is illustrated by calculatingthe negative ion for the closed shell Si]3 cluster, for whichwe find a different electronic ground state than LDA andevaluate the electron affinity to be 2.0(1) eV. Finally, wecontribute to the development of the QMC method by ad-dressing the fixed-node error as a significant source of thefew percent discrepancy between our calculated bindingenergies and experiment. We show that the use of natu-ral orbitals from correlated calculations instead of Hartree-Fock orbitals produces more accurate wave functions,decreasing the differences with experiment for the n =2, 3, 4 cases to 1%—2%. To our knowledge, these QMCresults represent the first ab initio many-body calculationsof valence electronic structure for molecular systems witha large number of valence electrons.
In our QMC approach [19],we treat the core electronswith nonlocal pseudopotentials generated within Hartree-Fock theory [20] and use variational Monte Carlo (VMC)calculations to optimize the trial function Pr(R), where Rdenotes coordinates of the valence electrons. In diffusionMonte Carlo (DMC) calculations, one simulates stochas-tically the imaginary time Schrodinger equation,
f(R, t + r) = G(R, R';r) f(R', t) dR',
0031-9007/95/74(8)/1323(4)$06. 00 1995 The American Physical Society 1323

VOLUME 74, NUMBER 8 PHYSICAL REVIEW LETTERS 20 FEBRUARY 1995
where I corresponds to the ionsions, i, g to the electrons,
are band r;I, r,I, r;, to the distances. The Slater determe erminantsare uilt from Hartree-Fock (HF) orbitals which arecomputed using the GAMESS pack . Pac age. arametrizationand optimization of u~r,d p
' of u(r;l, r,j, r;, ), which represents thee ectron-electron and electron-elect-e ec ron-ion correlations,are described elsewhere [24]. With VMC hin t e optimizedPr(R) gave 85%—90% of the DMC correlation energy forall sizes of studied clusters.
Since the calculation of forces is not developed forQMC, searching and optimization of the structures isperformed using LDA, which is well known to giveexcellent geometries. All structures are optimized within
c comparisonthe given symmetries in order to have a directwit previous calculations. A conjugate gradient as wellas quasi Newton minimization are used a d t, an a om positionsare relaxed until forces become less than 0.003 a.u.
n=2to 7—to 7.—For these smaller clusters, the groundstate structures were first determ d thRaghavachari and Rohlfing [13] and recently confirmedexperimentally through Raman spectroscopy [14], andthus provide an excellent test for the ~~MCTable I
r e ~~approach.a e ists our results for these cluste rs, as we as11
avai a e experimental data. We are encouraged by thelevel of accuracy of the QMC results when compared withexperiment [25] and point out that there is no decrease inaccuracy with increased cluster size for n = 4, 6, 7. Bycontrast, LDA overbinding is 15%—20% for these smallclusters, typical of errors found in LDA calculations [26).
n = 9 and 10.—For Si9 the distorted tricapped octahe-ricappe prism (seedron is compared with a distorted t d
Fig. 1) recently proposed by Ordej6n, Lebedenko, and
prism and the tetracapped octahedron. The total energieso t ese different geometries for each cluster size differed
y ess than 1 eV in previous calculations [13,15,27], and
(c)
(e)
n=l0
(h)
LDA DMC Expt.
TABLE I. BindinHF, LDA and D
'
g energies (eV/atom) calculated b th
FIG. l. Struuctures of calculated clusters for n ~ 9. Thatom ~C d(,) distorted tricapped octahedron; (b) the 9-atom (D
n . a e9-
distorted tricapped prism; (c) the 10-atom Te -atom (C3 ) tetracapped trigonal prism;
(e) the 13-atom (Ih) icosahedron; (f) the 13-atom C3, cappedtrigonal antiprism; (g) the 20-atom (Ih) dodecahedron; (h) the20-atom (C3,) new elongated structure.
S12 (D2h)Si, (C,.)Si4 (D~h)Sih (C2, )»7 (DSh)
Si9 (C,)S19 (Dh3)Sicko (Td)Ss&o (C3„)S43 (Ih)Si,3 (C3.)Si„(C, )
S120 (Ih)Si2o (Cq. )
1324
0.851.121.611.821.911.741.771.941.891.411.801.881.611.55
1.982.923.504.004.144.064.144.254.323.984.284.434.104.28
1.580(7)2.374(8)2.86(2)3.26(1)3.43(2)3.28(2)3.39(2)3.44(2)3.48(2)3.12(2)3.41(l)3.56(l)3.23(3)3.43(3)
1.61(4)2.45(6)3.01(6)3.42(4)3.60(4)
it has not been clear which structure was more stable.Our results confirm the LDA and empirical tight-bindingpredictions in both cases showing th d de istorte tricappedtrigonal prism and the tetracapped trigonal prism to be
n = 13.—Theslower in energy for Si9 and Si &o, respectivel .
e clusters are of interest because of the
lh) geometry, which, for example, has recently been foundto be a stable geometry for doped Al clusters [28]. How-ever a lowerower C3 symmetry structure was found by a sim-ulate annealing LDA search to be energeticall f-ica y more a-
e for Si by about 0.5 eV/atom [11]. This led tothe conjecture [16] that the electron correlation had not
VOLUME 74, NUMBER 8 PH YS ICAL REVIEW LETTERS 20 FEBRUARY 1995
been treated adequately and that proper calculations wouldshow the icosahedron to be more stable. We calculatedboth structures, and for completeness three different occu-pations of the highest orbitals of the icosahedron are con-sidered: Ag singlet, Gg singlet, and G triple. These pos-sibilities come from the fact that Si~3++ (lh) is a closed-shell molecule with the first two virtual orbitals (As and
Gs) very near in energy. The two open-shell states of Si~3with double occupancy of the fourfold degenerate Gg or-bital are essentially equal in energy, whereas the closed-shell Ag state is considerably higher, thus confirming theJahn-Teller instability of the icosahedron as found in LDA[11]. We find indeed that the icosahedral structure has alarger correlation energy. It is not nearly enough, how-ever, to overcome Coulomb and exchange contributions,and so the closed-shell C3 structure is substantially lowerin total energy.
n = 20.—In order to compare our results with recentcalculations [12], we first considered the perfect dodeca-hedron structure even though it is not stable against Jahn-Teller distortion. We then went further and focused ona new structure for Sicko which we have recently foundfrom a determination of a new class of stacked clusters[18]. Surprisingly, this new structure (shown in Fig. 1)is found to be significantly lower in total energy thanthe ring structure of Kaxiras and Jackson [12], and 4 eVlower in energy than the dodecahedron. Note that in thisstructure every atom except for the caps is fourfold coor-dinated, an important difference from the previously pro-posed threefold coordinated ring structures [12]. Our Si2o(C3 ) cluster can be further relaxed to lower symmetry,but the gain in total energy is less than 0.2 eV.
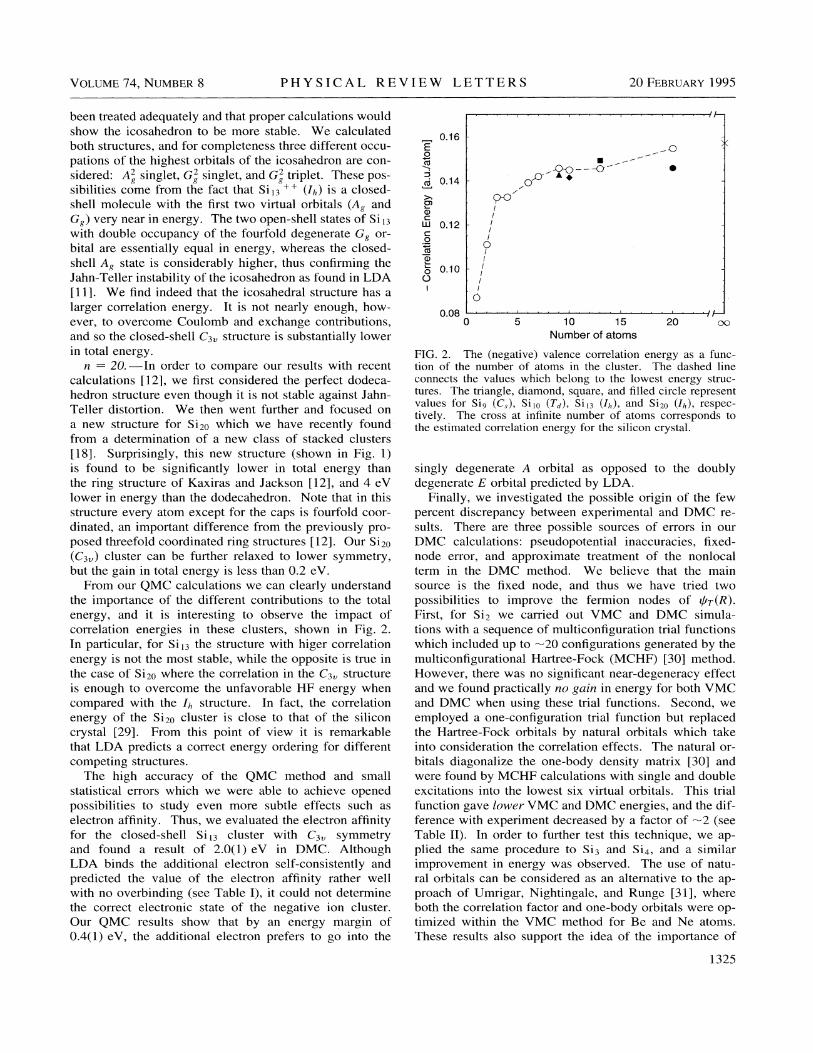
From our QMC calculations we can clearly understandthe importance of the different contributions to the totalenergy, and it is interesting to observe the impact ofcorrelation energies in these clusters, shown in Fig. 2.In particular, for Sii3 the structure with higer correlationenergy is not the most stable, whi1e the opposite is true inthe case of Si20 where the correlation in the C3 structureis enough to overcome the unfavorable HF energy whencompared with the I~ structure. In fact, the correlationenergy of the Si20 cluster is close to that of the siliconcrystal [29]. From this point of view it is remarkablethat LDA predicts a correct energy ordering for differentcompeting structures.
The high accuracy of the QMC method and smallstatistical errors which we were able to achieve openedpossibilities to study even more subtle effects such aselectron affinity. Thus, we evaluated the electron affinityfor the closed-shell Si » cluster with C3 symmetryand found a result of 2.0(l) eV in DMC. AlthoughLDA binds the additional electron self-consistently andpredicted the value of the electron affinity rather wellwith no overbinding (see Table I), it could not determinethe correct electronic state of the negative ion cluster.Our QMC results show that by an energy margin of0.4(1) eV, the additional electron prefers to go into the
0.16E0
gj 0.14
O
+
CDI0.12
C06$
LD
o 0.100
/
/
/
/
/0/
/
/
/
/
/
0.080 10 15
Number of atoms
I
20
FIG. 2. The (negative) valence correlation energy as a func-tion of the number of atoms in the cluster. The dashed lineconnects the values which belong to the lowest energy struc-tures. The triangle, diamond, square, and filled circle representvalues for Si9 (C, ), Si&o (Td), Si~3 (Ih), and Si20 (II, ), respec-tively. The cross at infinite number of atoms corresponds tothe estimated correlation energy for the silicon crystal.
singly degenerate A orbital as opposed to the doublydegenerate F. orbital predicted by LDA.
Finally, we investigated the possible origin of the fewpercent discrepancy between experimental and DMC re-sults. There are three possible sources of errors in ourDMC calculations: pseudopotential inaccuracies, fixed-node error, and approximate treatment of the nonlocalterm in the DMC method. We believe that the mainsource is the fixed node, and thus we have tried twopossibilities to improve the fermion nodes of Pr(R).First, for Siq we carried out VMC and DMC simula-tions with a sequence of multiconfiguration trial functionswhich included up to -20 configurations generated by themulticonfigurational Hartree-Fock (MCHF) [30] method.However, there was no significant near-degeneracy effectand we found practically no gain in energy for both VMCand DMC when using these trial functions. Second, weemployed a one-configuration trial function but replacedthe Hartree-Fock orbitals by natural orbitals which takeinto consideration the correlation effects. The natural or-bitals diagonalize the one-body density matrix [30] andwere found by MCHF calculations with single and doubleexcitations into the lowest six virtual orbitals. This trialfunction gave low er VMC and DMC energies, and the dif-ference with experiment decreased by a factor of —2 (seeTable II). In order to further test this technique, we ap-plied the same procedure to Si3 and Si4, and a similarimprovement in energy was observed. The use of natu-ral orbitals can be considered as an alternative to the ap-proach of Umrigar, Nightingale, and Runge [31], whereboth the correlation factor and one-body orbitals were op-timized within the VMC method for Be and Ne atoms.These results also support the idea of the importance of
1325

VOLUME 74, NUMBER 8 PHYS ICAL REVIEW LETTERS 20 FEBRUARY 1995
TABLE II. Comparison of binding energies (eV/atom) calcu-lated using Hartree-Fock (HF) and natural (NO) orbitals forsmall clusters.
Si2Si3Si4
DMC (HF)
1.580(7)2.374(8)2.86(2)
DMC (NO)
1.592(6)2.423(7)2.92(2)
Expt.
1.61(4)2.45(6)3.01(6)
*On leave from Institute of Physics, Dubravska cesta 9,84228 Bratislava, Slovakia.
[1] Physics and Chemistry of Small Clusters, edited byP. Jena, B.K. Rao, and S.N. Khanna (Plenum, New York,1987).
[2] L. A. Bloomfield, R. R. Freeman, and W. L. Brown, Phys.Rev. Lett. 54, 2246 (1985).
using one-body orbitals which are optimal in the presenceof correlation [31,32]. Moreover, this raises a new chal-lenge for QMC, namely the task of efficient optimizationof one-body orbitals for large systems.
In conclusion, we have carried out QMC calculationsof silicon clusters and evaluated valence correlation andbinding energies, as well as the electron affinity for onecase. Our calculations give an unambiguous picture ofthe correlation energy in silicon clusters and its role onthe energetical ordering of structures. The QMC bindingenergies agree with available experimental data withina few percent and provide a significant improvementover the mean-field results. For Si2O, we discovereda new elongated structure cluster that is energeticallymore favorable than the previously studied ones by=2 eV. In addition, for small clusters we have shownthat the use of natural orbitals from correlated calculationsinstead of Hartree-Fock ones gives a trial function whichsignificantly decreases the fixed-node error. %'e believethat these calculations demonstrate the feasibility andaccuracy of QMC for molecular systems with up to about100 valence electrons and open new perspectives forstudying even larger stystems in the near future.
We thank Shao-ping Tang for help in carrying out theLDA calculations and David Ceperley and Richard M.Martin for constant support and encouragement. We arealso grateful to Krishnan Raghavachari for -communicat-ing his results prior to publication and for pointing out ref-erences to experimental data, Arthur J. Freeman for read-ing the manuscript and for useful discussions, and BalajiVeeraraghavan for help with GAMEss calculations. Thiswork was supported by NSF Grants No. DMR91-17822and No. ASC 92-11123. The calculations have been per-formed on the NCSA HP 9000/715 cluster and the CrayY-MP.
[3]
[4][5]
[6]
[8]
[9]
[10]
[I I]
[12]
[13]
[14][15]
[16][17]
[18][19]
I2o]
[21]
[22]
[24]
[25]
[26]
[27]
[28]
[29]
[30]
[31]
[32]
M. F. Jarrold, U. Ray, and K. M. Creegan, J. Chem. Phys.93, 224 (1990).W. L. Brown et al. , Science 235, 860 (1987).S.N. Khanna and P. Jena, Phys. Rev. Lett. 69, 1664(1992).M. F. Jarrold, Science 252, 1085 (1991).P. Ballone et al. , Phys. Rev. Lett. 60, 271 (1988).N. Binggeli, J. L. Martins, and J. R. Chelikowsky, Phys.Rev. Lett. 6S, 2956 (1992).M. Menon and K. R. Subbaswamy, Phys. Rev. B 47,12 754 (1993).J.R. Chelikowsky and J.C. Phillips, Phys. Rev. Lett. 63,1653 (1989).U. Rothlisberger, W. Andreoni, and P. Giannozzi, J.Chem. Phys. 92, 1248 (1992).E. Kaxiras and K. Jackson, Phys. Rev. Lett. 71, 727(1993).K. Raghavachari and C. M. Rohlfing, J. Chem. Phys. 89,2219 (1988).E.C. Honea et al. , Nature (London) 366, 42 (1993).P. Ordejon, D. Lebedenko, and M. Menon, Phys. Rev. B50, 5645 (1994).J.C. Phillips, Phys. Rev. B 47, 14132 (1993).M. F. Jarrold and V. A. Constant, Phys. Rev. Lett. 67,2994 (1991).J.C. Grossman and L. Mitas (to be published).L. Mitas, E.L. Shirley, and D. M. Ceperley, J. Chem.Phys. 95, 3467 (1991).W. J. Stevens, H. Basch, and M. Krauss, J. Chem. Phys.Sl, 6026 (1984).P. J. Reynolds et al. , J. Chem. Phys. 77, 5593 (1982);J.W. Moskowitz et al. , ibid 77, 349 (19.82).K. E. Schmidt and J.W. Moskowitz, J. Chem. Phys. 93,4172 (1990).C. J. Umrigar, K. G. Wilson, and J.W. Wilkins, Phys.Rev. Lett. 60, 1719 (1988).L. Mitas, in Computer Simulation Studies in Condensed-Matter Physics V, edited by D. P. Landau, K. K. Mon, andH. B. Schuttler (Springer, Berlin, 1993).R. W. Schmude et al. (to be published); R. W. Schmude,Q. Ran, and K. A. Gingerich, J. Chem. Phys. 99, 7998(1993); Q. Ran et al. (to be published).G. S. Painter and F.W. Averill, Phys. Rev. B 26, 1781(1982).I. Lee, K. J. Chang, and Y. H. Lee, J. Phys. Condens.Matter 6, 741 (1993).X.G. Gong and V. Kumar, Phys. Rev. Lett. 70, 2078(1993).The Si crystal correlation energy was estimated fromprevious calculations: X.-P. Li, D. M. Ceperley, and R. M.Martin, Phys. Rev. B 44, 10929 (1991);and Ref. [32].Modern Quantum Chemistry, edited by A. Szabo andN. S. Ostlund (McGraw-Hill, New York, 1989).C. J. Umrigar, M. P. Nightingale, and K. J. Runge, J.Chem. Phys. 99, 2865 (1993).S. Fahy, X.W. Wang, and S.G. Louie, Phys. Rev. B 42,3503 (1990).
1326








![Second Slide%20 %20 Slide%20 Sharing%20 Made%20 Easy%20with%20the%20 Innovation%20 Second Slide%20 Service[1]](https://img.pdfslide.net/doc/110x75/55a267641a28abca6b8b47e1/second-slide20-20-slide20-sharing20-made20-easy20with20the20-innovation20-second-slide20-service1.jpg)











![Bi%20 Consulting%20 Govt%20 Overview%20 %20 July09[1]](https://img.pdfslide.net/doc/110x75/5481fe46b07959290c8b46b6/bi20-consulting20-govt20-overview20-20-july091.jpg)







