Embed Size (px)
Citation preview

101
CHAPTER - 4
SYNTHESIS OF SUBSTITUTED CARBAZOLE AND
PYRAZOLES
This chapter is divided into two sections namely Section-A and
Section-B. Section-A deals with the synthesis of substituted carbazole
and pyrazoles. Section-B describes an alternate synthesis of
naratriptan drug using a new intermediate, N-benzyl-N-
methylethanesulfonamide.
SECTION-A
4A.1 INTRODUCTION:
Carbazole derivatives are well known for their pharmacological
activities [130]. These compounds have been reported to possess
diverse biological activity like antibacterial [131], antifungal [132],
anticancer and anti-HIV activities [133-134]. Pyrazole derivatives also
well established in the literature as important biologically active
heterocyclic compounds [135]. These compounds exhibit anti-
inflammatory [136], antipyretic [137], antimicrobial [138], antiviral
[139] antitumour [140] and antidepressant [141] etc. In this chapter
we synthesized and characterized some new sulfonamide carbazole
and pyrazole derivatives.
4A.2 LITERATURE SURVEY
Several synthetic methods have been reported in the literature
for carbazole and pyrazoles. Some of the synthetic methods are
discussed below.

102
4A.2.1 CARBAZOLE
Deoxygenation of o-nitrobiphenyl (113) to carbazole (114)
(Scheme- 4A.1) was first reported by Waterman and Viviens [142] by
using iron oxalate at 200 ºC. The widely accepted mechanism involves
exhaustive deoxygenation to singlet nitrene that undergoes a down
stream C-H insertion.
….. Scheme - 4A.1
Cadogan et. al. [143] reported a similar deoxygenative
cyclization of o-nitrobiphenyl (113) to carbazole (114) (Scheme-4A.2)
under reflux using triethylphosphite as solvent.
….. Scheme 4A.2
Smith and brown [144] reported the synthesis of carbazole (116)
(Scheme-4A.3) by thermal decomposition of o-azidobiphenyl (115) in
kerosene at 180 ºC. The reaction is believed to proceed via loss of
nitrogen gas forming nitrene, followed by cyclization to 9a-hydro-9H-
carbazole.

103
….. Scheme - 4A.3
In 1982, Tauber et. al. [145] reported the synthesis of carbazole
(114) (Scheme-4A.4) by the cyclization of 2,2-diaminobiphenyl (117).
This reaction relies on high temperature and acidic conditions
involving cyclization with the elimination of ammonia.
….. Scheme - 4A.4
The Fischer method [146] of indole synthesis by the indolization
of an arylhydrazone by treatment with an acid catalyst was applied to
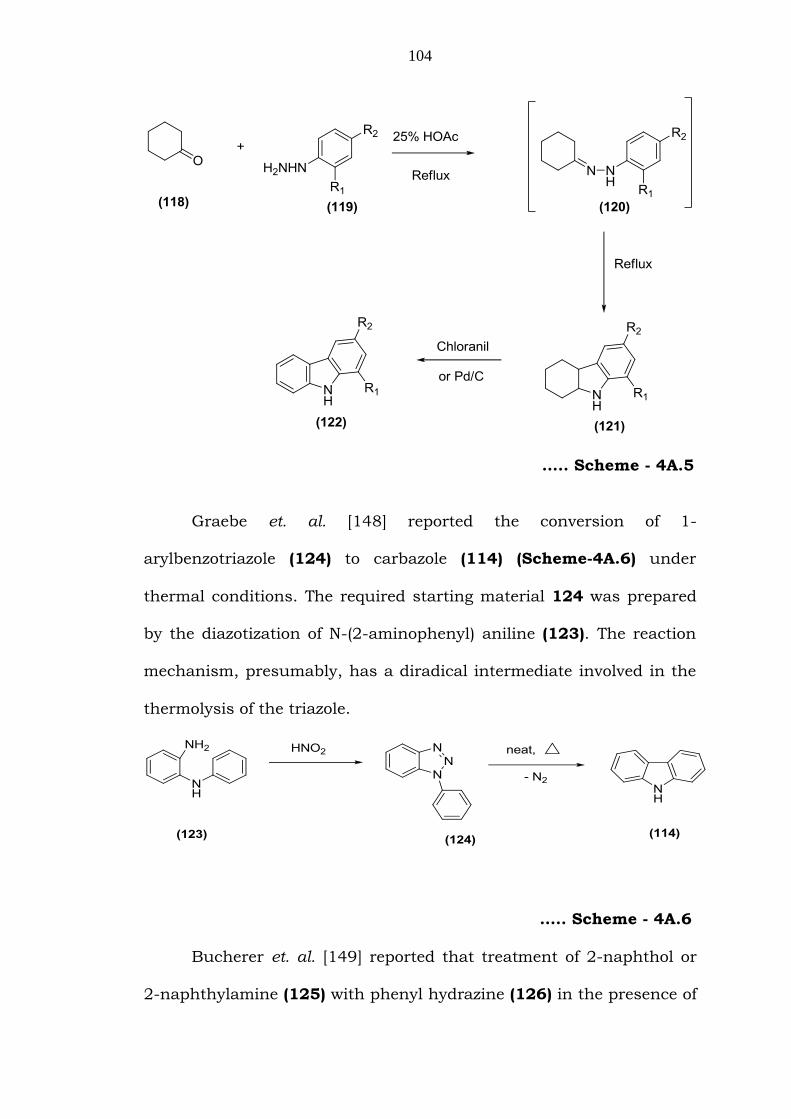
the synthesis of tetrahydrocarbazole by Borsche [147]. The
cyclohexanone phenylhydrazone (120) obtained by reacting
cyclohexanone (118) with substituted phenylhydrazine (119), on
indolization, furnished tetra hydrocarbazole (121) which on
dehydrogenation, afforded carbazole (122) (Scheme-4A.5). The
mechanism for the formation of tetrahydrocarbazole involves a
tautomeric equilibrium and formation of new C-C bond via [3,3]-
sigmatropic rearrangement followed by elimination of ammonia.
104
….. Scheme - 4A.5
Graebe et. al. [148] reported the conversion of 1-
arylbenzotriazole (124) to carbazole (114) (Scheme-4A.6) under
thermal conditions. The required starting material 124 was prepared
by the diazotization of N-(2-aminophenyl) aniline (123). The reaction
mechanism, presumably, has a diradical intermediate involved in the
thermolysis of the triazole.
….. Scheme - 4A.6
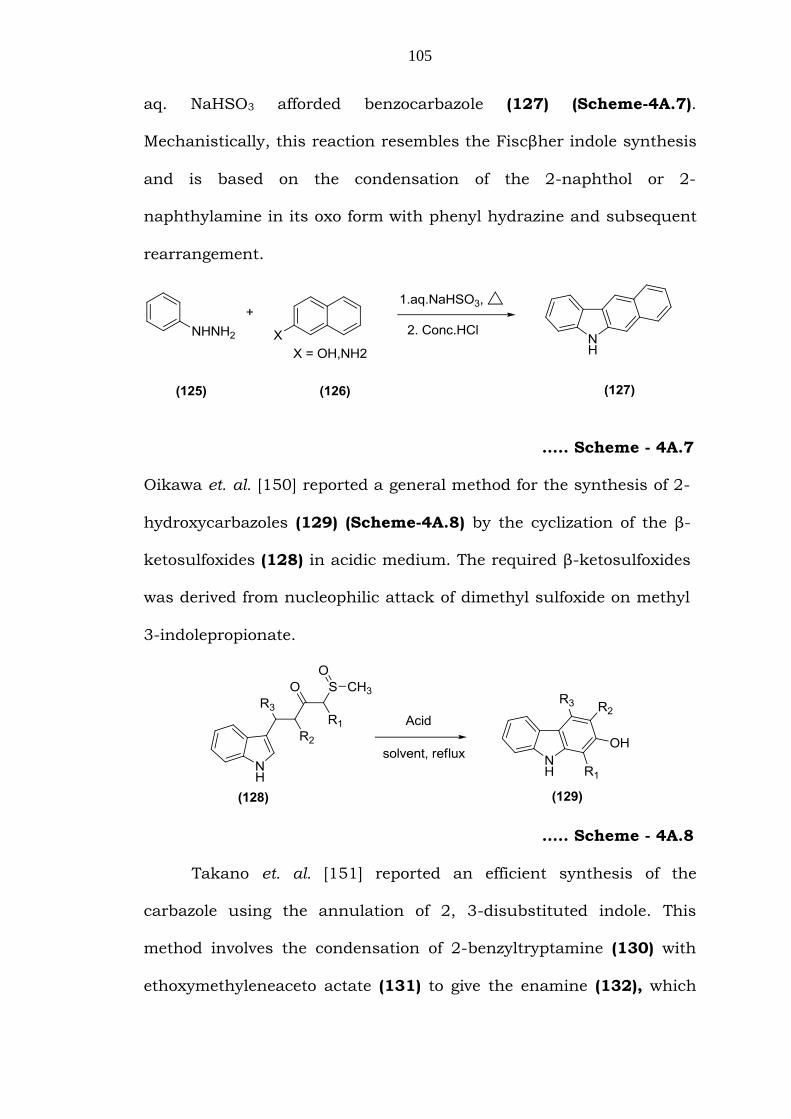
Bucherer et. al. [149] reported that treatment of 2-naphthol or
2-naphthylamine (125) with phenyl hydrazine (126) in the presence of
105
aq. NaHSO3 afforded benzocarbazole (127) (Scheme-4A.7).
Mechanistically, this reaction resembles the Fiscβher indole synthesis
and is based on the condensation of the 2-naphthol or 2-
naphthylamine in its oxo form with phenyl hydrazine and subsequent
rearrangement.
….. Scheme - 4A.7
Oikawa et. al. [150] reported a general method for the synthesis of 2-
hydroxycarbazoles (129) (Scheme-4A.8) by the cyclization of the β-
ketosulfoxides (128) in acidic medium. The required β-ketosulfoxides
was derived from nucleophilic attack of dimethyl sulfoxide on methyl
3-indolepropionate.
….. Scheme - 4A.8
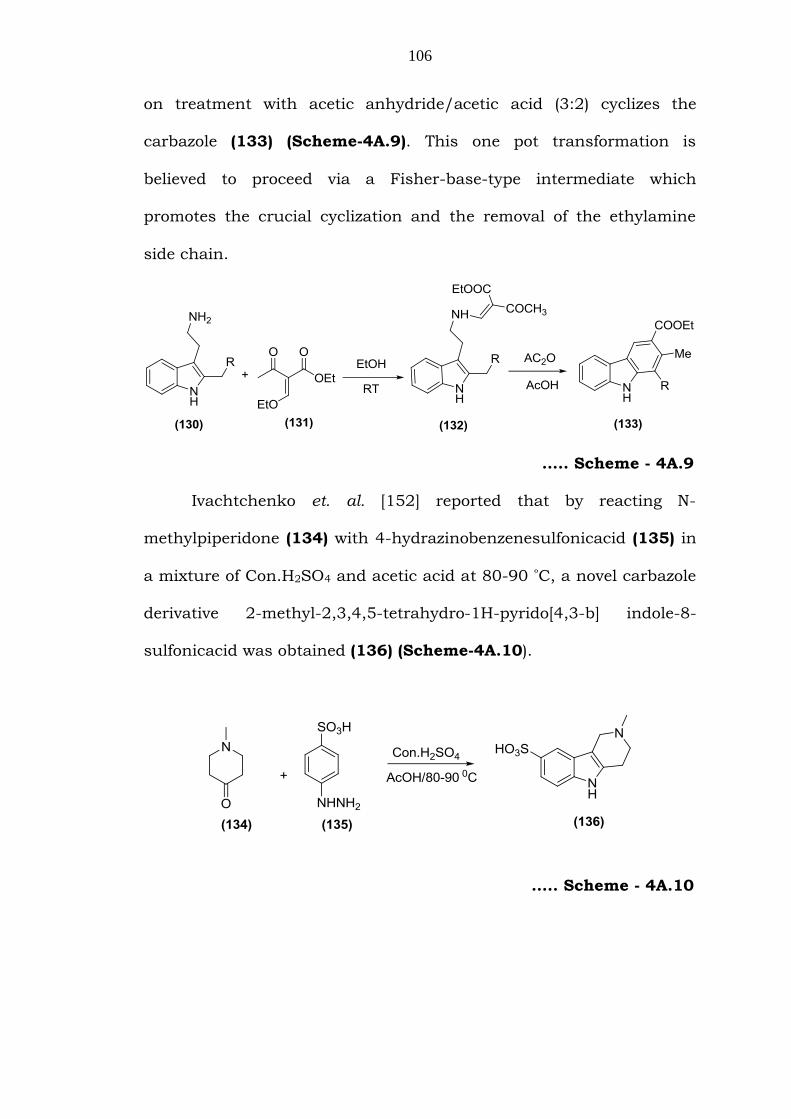
Takano et. al. [151] reported an efficient synthesis of the
carbazole using the annulation of 2, 3-disubstituted indole. This
method involves the condensation of 2-benzyltryptamine (130) with
ethoxymethyleneaceto actate (131) to give the enamine (132), which
106
on treatment with acetic anhydride/acetic acid (3:2) cyclizes the
carbazole (133) (Scheme-4A.9). This one pot transformation is
believed to proceed via a Fisher-base-type intermediate which
promotes the crucial cyclization and the removal of the ethylamine
side chain.
….. Scheme - 4A.9
Ivachtchenko et. al. [152] reported that by reacting N-
methylpiperidone (134) with 4-hydrazinobenzenesulfonicacid (135) in
a mixture of Con.H2SO4 and acetic acid at 80-90 ºC, a novel carbazole
derivative 2-methyl-2,3,4,5-tetrahydro-1H-pyrido[4,3-b] indole-8-
sulfonicacid was obtained (136) (Scheme-4A.10).
….. Scheme - 4A.10
107
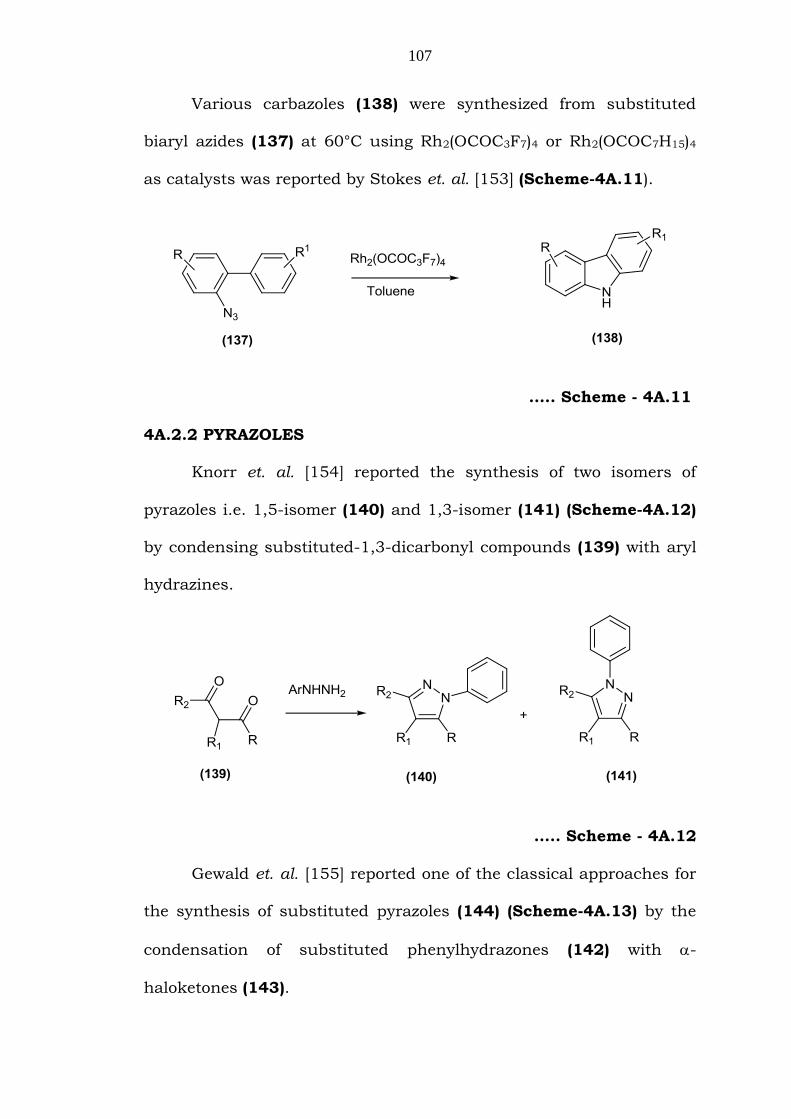
Various carbazoles (138) were synthesized from substituted
biaryl azides (137) at 60°C using Rh2(OCOC3F7)4 or Rh2(OCOC7H15)4
as catalysts was reported by Stokes et. al. [153] (Scheme-4A.11).
….. Scheme - 4A.11
4A.2.2 PYRAZOLES
Knorr et. al. [154] reported the synthesis of two isomers of
pyrazoles i.e. 1,5-isomer (140) and 1,3-isomer (141) (Scheme-4A.12)
by condensing substituted-1,3-dicarbonyl compounds (139) with aryl
hydrazines.
….. Scheme - 4A.12
Gewald et. al. [155] reported one of the classical approaches for
the synthesis of substituted pyrazoles (144) (Scheme-4A.13) by the
condensation of substituted phenylhydrazones (142) with -
haloketones (143).
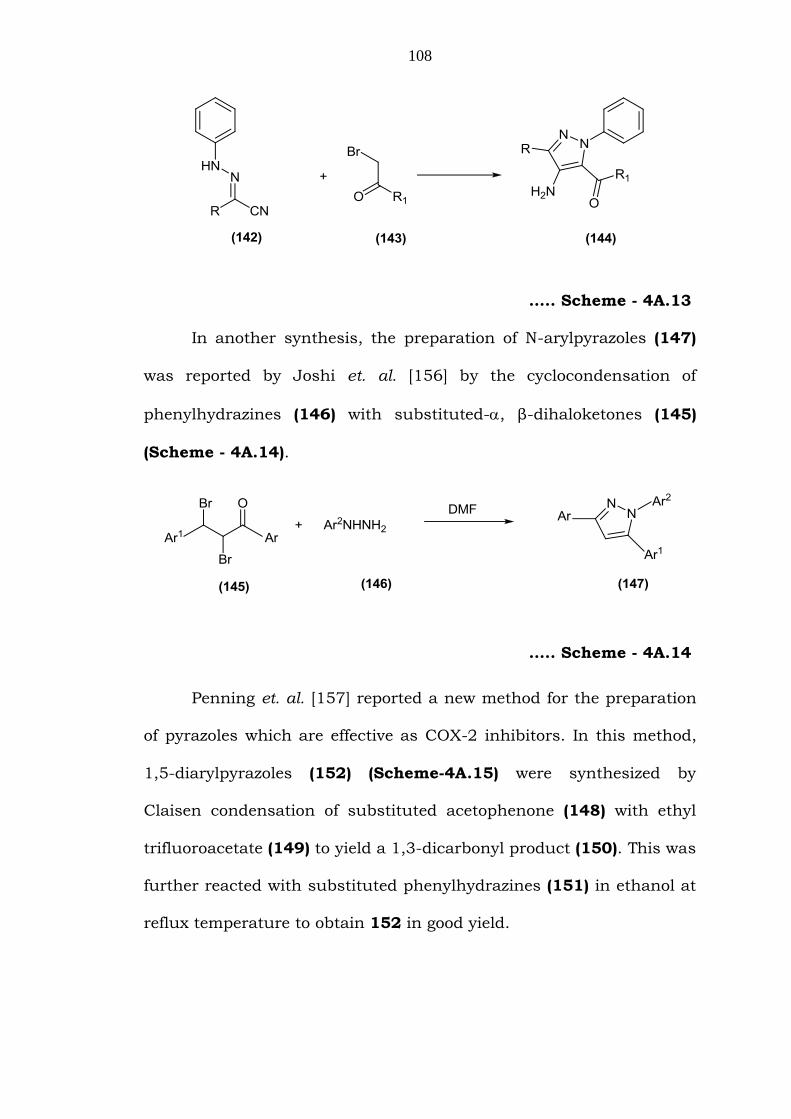
108
….. Scheme - 4A.13
In another synthesis, the preparation of N-arylpyrazoles (147)
was reported by Joshi et. al. [156] by the cyclocondensation of
phenylhydrazines (146) with substituted-, β-dihaloketones (145)
(Scheme - 4A.14).
….. Scheme - 4A.14
Penning et. al. [157] reported a new method for the preparation
of pyrazoles which are effective as COX-2 inhibitors. In this method,
1,5-diarylpyrazoles (152) (Scheme-4A.15) were synthesized by
Claisen condensation of substituted acetophenone (148) with ethyl
trifluoroacetate (149) to yield a 1,3-dicarbonyl product (150). This was
further reacted with substituted phenylhydrazines (151) in ethanol at
reflux temperature to obtain 152 in good yield.
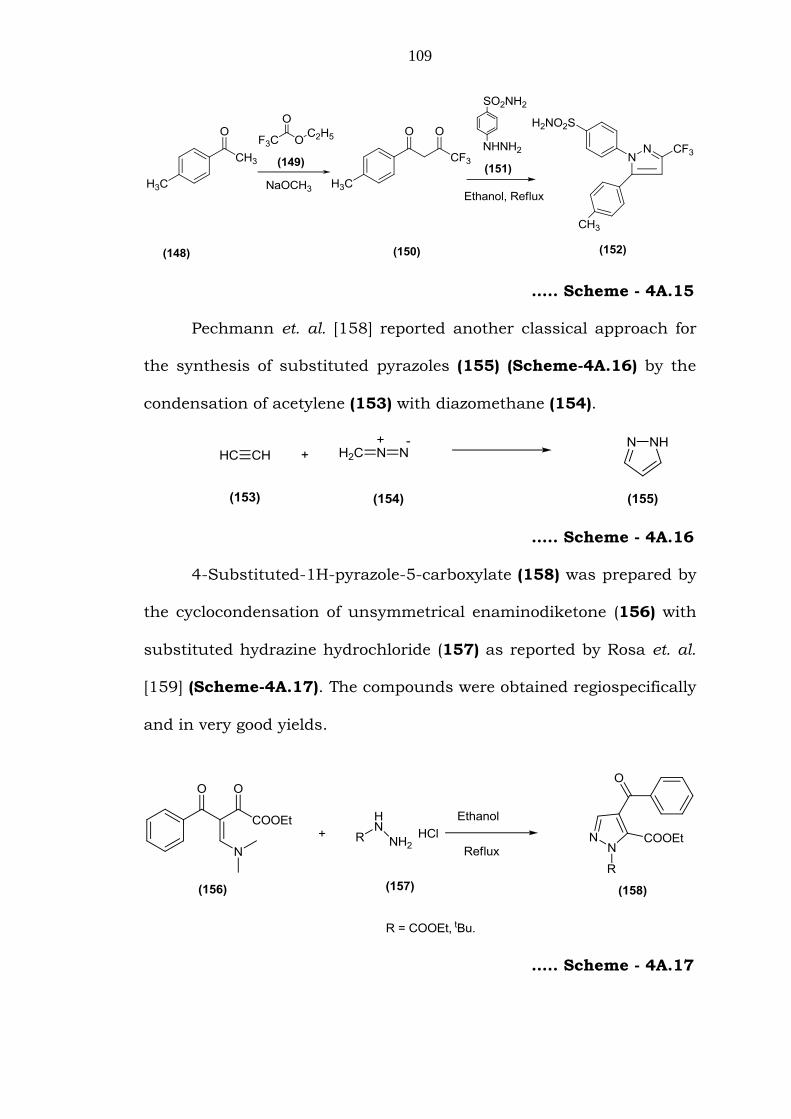
109
….. Scheme - 4A.15
Pechmann et. al. [158] reported another classical approach for
the synthesis of substituted pyrazoles (155) (Scheme-4A.16) by the
condensation of acetylene (153) with diazomethane (154).
….. Scheme - 4A.16
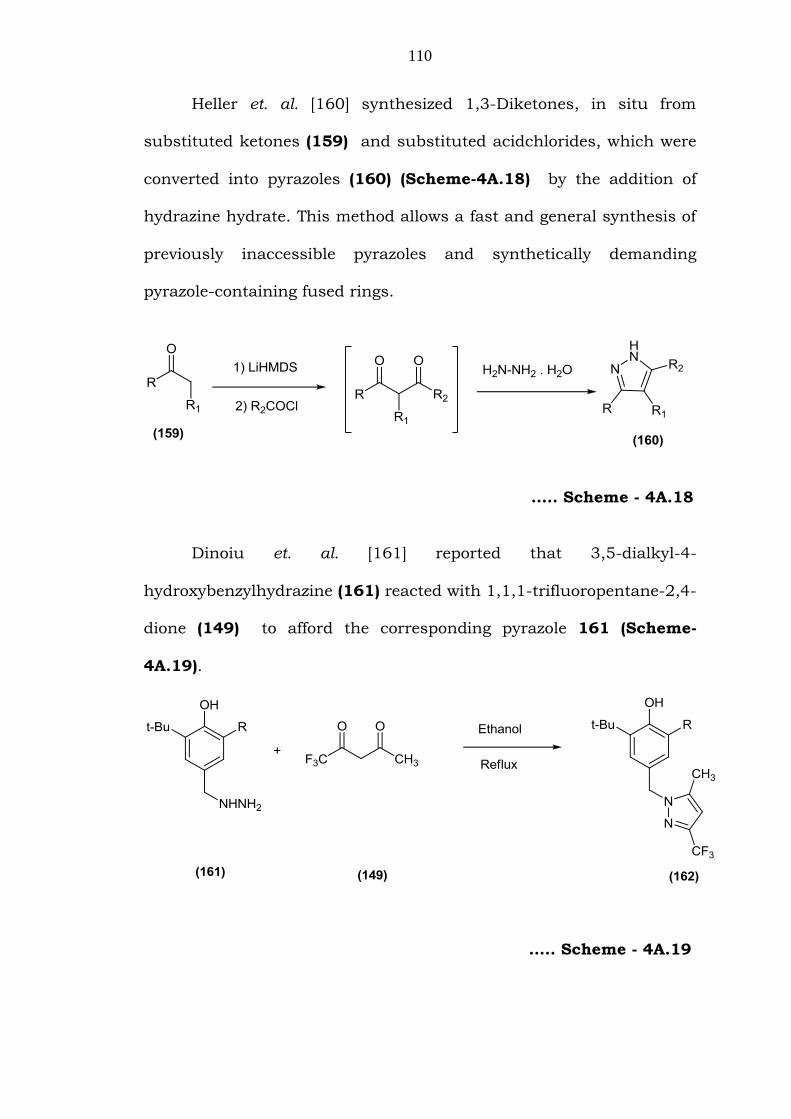
4-Substituted-1H-pyrazole-5-carboxylate (158) was prepared by
the cyclocondensation of unsymmetrical enaminodiketone (156) with
substituted hydrazine hydrochloride (157) as reported by Rosa et. al.
[159] (Scheme-4A.17). The compounds were obtained regiospecifically
and in very good yields.
….. Scheme - 4A.17
110
Heller et. al. [160] synthesized 1,3-Diketones, in situ from
substituted ketones (159) and substituted acidchlorides, which were
converted into pyrazoles (160) (Scheme-4A.18) by the addition of
hydrazine hydrate. This method allows a fast and general synthesis of
previously inaccessible pyrazoles and synthetically demanding
pyrazole-containing fused rings.
….. Scheme - 4A.18
Dinoiu et. al. [161] reported that 3,5-dialkyl-4-
hydroxybenzylhydrazine (161) reacted with 1,1,1-trifluoropentane-2,4-
dione (149) to afford the corresponding pyrazole 161 (Scheme-
4A.19).
….. Scheme - 4A.19

111
4A.3 PRESENT WORK
It is obvious from the references cited above number of
researchers have synthesized carbazole and pyrazole analogues which
are biologically active molecules. Hence, in this chapter the synthesis
of new pyrazole and carbazole derivatives are reported by combining
these ring compounds with methanesulfonamide functionality as
potentially biologically active compounds.
4A.4 RESULTS AND DISCUSSIONS
4A.4.1 CARBAZOLES
The required starting materials substituted hydrazines 163(a-c)
were prepared by using reported procedure [95] as depicted in
scheme- 4A.20. Thus, diazotization of 26a, 26d, and 26e resulting
the corresponding diazonium salts, which on reduction with stannous
chloride gave 163(a-c).
….. Scheme - 4A.20
2-(4-Hydrazinylphenyl)-N-methylethanesulfonamide hydrochloride
163c (i.e. 163, R=-NHCH3 n=2) was reacted with cyclohexanone (118)
(Scheme-4A.21) to give a new compound whose structure was
established by its spectral data as N-methyl-2-(2,3,4,9-tetrahydro-1H-
carbazol-6-yl)ethanesulfonamide 164c (i.e. 164, R=-NHCH3, n=2).

112
Thus, its IR spectrum in KBr (Fig.4A.1) showed a characteristic peak
at 3386 cm-1 which can be attributed to -NH group. The peaks at 1310
cm-1 and 1148 cm-1 indicate the presence of -SO2 group. Its 1H NMR
spectrum (CDCl3/TMS) (Fig.4A.2) showed multiplets at 1.92-2.75
with eight proton integration can be assigned to eight aliphatic
protons. The signals at 2.7 and 6.9 confirm the presence of –
NHCH3. Two triplets at 3.2 and 3.4 corresponding to the (-Ar-CH2-
CH2-SO2-). The peaks between 7.0-7.4 confirm the three aromatic
protons. Broard singlet at 7.8 can be assigned to –NH of carbazole
ring. The mass spectrum of 164c showed the molecular ion at m/z
293 (M++1) ion peak corresponding to a molecular mass of 292 which
further supports the assigned structure (Fig.4A.3). Its 13C NMR
(Fig.4A.4) showed the peaks at δ 21.03, 23.18, 23.25, 23.40, 29.02,
29.89, 52.01, 108.26, 110.90, 116.99, 121.01, 127.95, 128.16,
134.89 and 135.23.
….. Scheme - 4A.21
The above reaction of 163c with cyclohexanone has been found
to be a general one and has been extended to substituted hydrazines

113
(163a-b). The products 165a-b, obtained was assigned structures on
the basis of their spectral and analytical data.
Similarly, condensation of 163c (i.e. 163, R=-NHCH3 n=2) with N-
methyl-4-piperidone (134), followed by cyclisation in Con.HCl gave the
corresponding N-methyl-2-(2-methyl-2,3,4,5-tetrahydro-1H-pyrido
[4,3-b]indol-8-yl)ethanesulfonamide (165c) (i.e. 165, R=-NHCH3 n=2)
(Scheme-4A.22). Its IR spectrum in KBr (Fig.4A.5) showed
characteristic peak at 3436 cm-1, which can be attributed to -NH
group. Peaks at 1319 cm-1 and 1125 cm-1 indicates the presence of -
SO2 group. Its 1H NMR spectrum (DMSO-d6/TMS) (Fig.4A.6) showed
signal at 2.4 (3H) and 2.6 (3H) which can be attributed to methyl
protons of –NHCH3 and –NCH3 respectively. Two multiplets between
2.70-2.78 can be assigned to –CH2-CH2NCH3. Two triplets at 2.9-3.2
(4H) can be assigned to -Ar-CH2-CH2-SO2. The singlet at 3.4 (2H)
can be assigned to –CH2NCH3. The three aromatic protons and –
NHCH3 proton appeared between 6.8-7.1 as a multiplet. A singlet at
10.6 can be attributed to –NH proton of carbazole. The mass
spectrum (Fig.4A.7) of 165c showed a molecular ion at m/z 307. Its
13C NMR (Fig.4A.8) showed the peaks at δ 23.7, 29.0, 29.8, 45.8,
51.8, 52.4, 107.1, 111.1, 116.8, 121.2, 126.0, 128.4, 133.2 and
135.1.
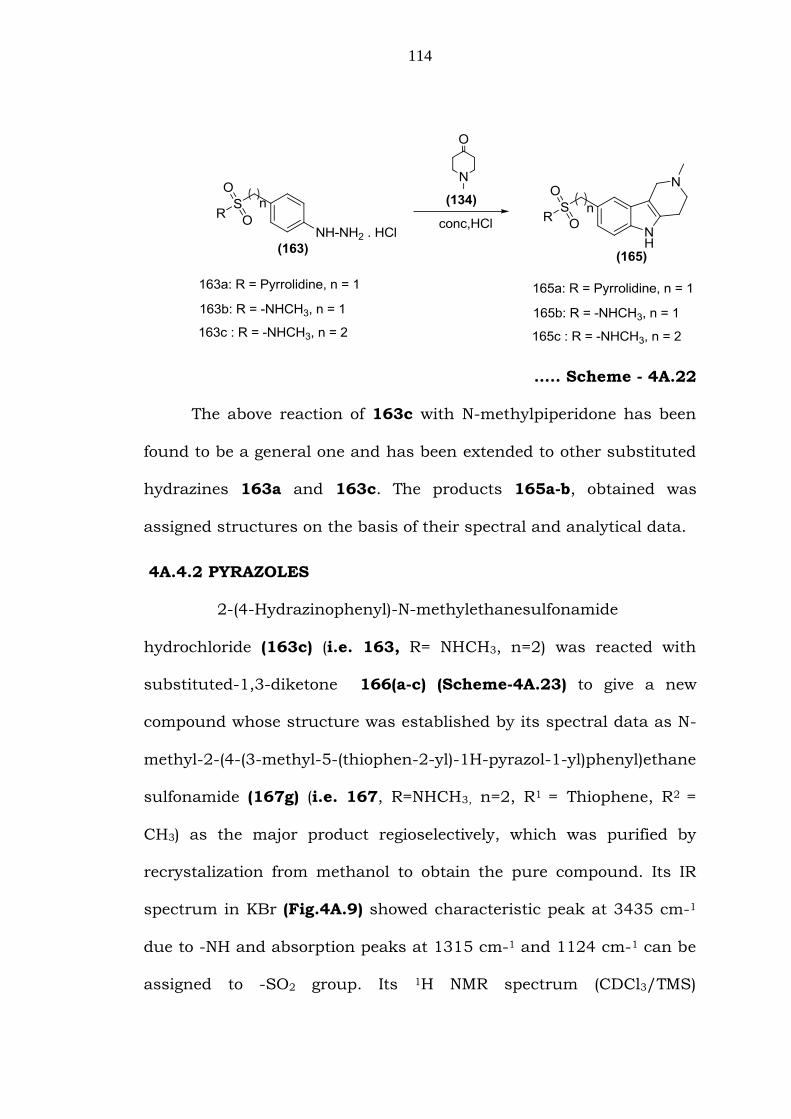
114
….. Scheme - 4A.22
The above reaction of 163c with N-methylpiperidone has been
found to be a general one and has been extended to other substituted
hydrazines 163a and 163c. The products 165a-b, obtained was
assigned structures on the basis of their spectral and analytical data.
4A.4.2 PYRAZOLES
2-(4-Hydrazinophenyl)-N-methylethanesulfonamide
hydrochloride (163c) (i.e. 163, R= NHCH3, n=2) was reacted with
substituted-1,3-diketone 166(a-c) (Scheme-4A.23) to give a new
compound whose structure was established by its spectral data as N-
methyl-2-(4-(3-methyl-5-(thiophen-2-yl)-1H-pyrazol-1-yl)phenyl)ethane
sulfonamide (167g) (i.e. 167, R=NHCH3, n=2, R1 = Thiophene, R2 =
CH3) as the major product regioselectively, which was purified by
recrystalization from methanol to obtain the pure compound. Its IR
spectrum in KBr (Fig.4A.9) showed characteristic peak at 3435 cm-1
due to -NH and absorption peaks at 1315 cm-1 and 1124 cm-1 can be
assigned to -SO2 group. Its 1H NMR spectrum (CDCl3/TMS)

115
(Fig.4A.10) showed three proton singlets at 2.35 due to the methyl
group of pyrazole and doublet at 2.75 is due to –NHCH3. Two
triplets at 3.25-3.45 can be assigned to (–Ph-CH2-CH2-SO2-). A
multpilet appeared at 4.0 is due to -NHCH3. A singlet at 6.35 can
be assigned to pyrazole-CH. The multiplet between 6.9-7.3 with
seven protons integration can be assigned to the four aryl protons and
three thiazole protons. The mass spectrum (Fig.4A.11) of 167g
showed the molecular ion at m/z 361. Its 13C NMR (DMSO-d6)
spectrum (Fig. 3.22) showed peaks at 13.16, 28.53, 28.73, 50.12,
107.29, 126.00, 127.24, 127.36, 127.55, 129.08, 130.74, 136.92,
137.91, 138.78 and 148.30.
….. Scheme - 4A.23
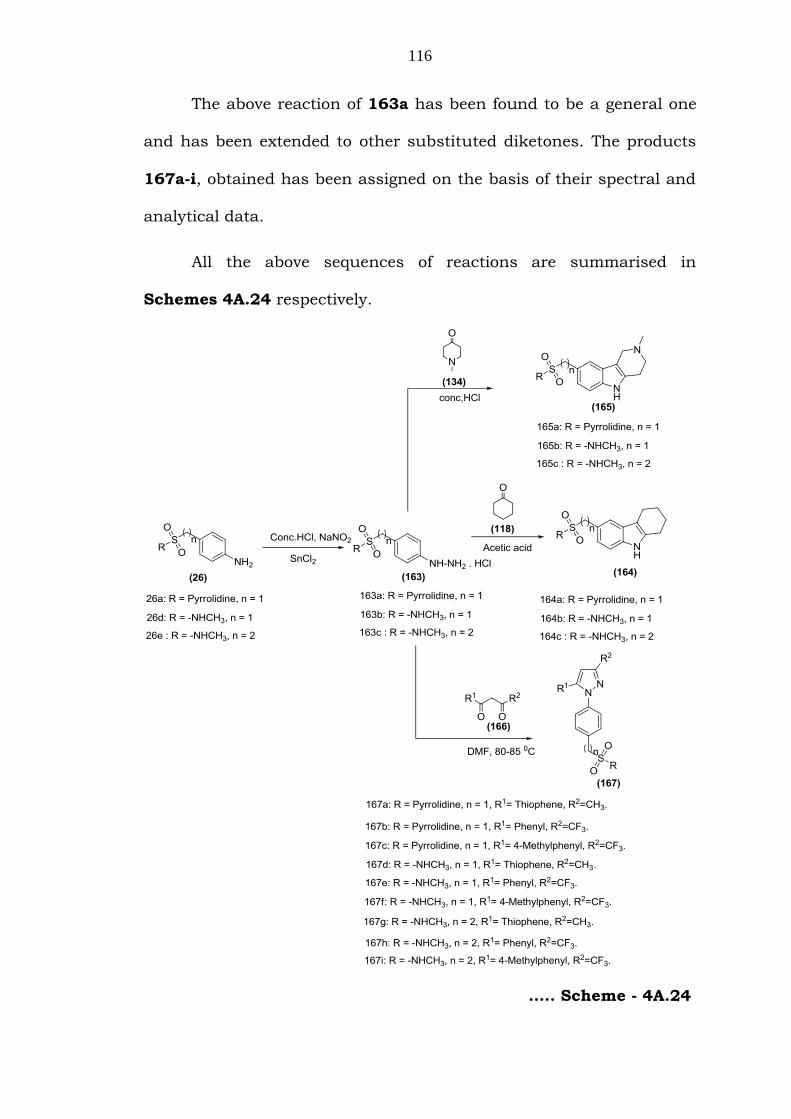
116
The above reaction of 163a has been found to be a general one
and has been extended to other substituted diketones. The products
167a-i, obtained has been assigned on the basis of their spectral and
analytical data.
All the above sequences of reactions are summarised in
Schemes 4A.24 respectively.
….. Scheme - 4A.24

117
4A.5. EXPERIMENTAL SECTION:
PREPARATION OF 164:
4A.5.1. GENERAL PROCEDURE FOR THE SYNTHESIS OF 164(a-c):
A mixture of 163(a-c) (0.011 mol), cyclohexanone (118) (0.011 mol),
sodium acetate (0.015 mol) and acetic acid (25 mL) was refluxed with
stirring for 5 hours. The solvent was distilled off under reduced
pressure. The residue was partitioned between water and ethyl
acetate. The organic layer was dried, concentrated and crystallized
from methanol (10 mL). The crude compound was re-crystallized from
a suitable solvent to obtain pure 164(a-c).
164a: R = pyrrolidine, n=1, Yield: 2.23 gm (64 %), M.R: ~250 ºC; IR
(KBr, cm-1) 3372 (-NH), 1300, 1144 (-SO2); 1H NMR (DMSO-d6/TMS) δ
1.80-1.90 (m, 8H, 2 x -CH2 pyrrolidine and 2 x -CH2 carbazole), 2.70
(m, 2H, -CH2 carbazole), 2.75 (m, 2H, -CH2 carbazole), 3.2 (m, 4H,
pyrrolidine), 4.3 (s, 2H, -SO2CH2), 7.0-7.4 (m, 3H, Ar-H), 7.8 (bs, 1H,
-NH indole, D2O exchangable), M++1: 318; Anal.Calcd for
(C17H22N2O2S) requires: C, 64.12; H, 6.96; N, 8.80. Found: C, 64.02;
H, 6.91; N, 8.80.
164b: R = -NHCH3, n=1, Yield: 1.8 gm (59 %), M.R: 213-215 ºC; IR
(KBr, cm-1) 3362 (-NH), 1139, 1120 (-SO2); 1H NMR (CDCl3/TMS) δ 1.9
(m, 4H, 2 x CH2 carbazole), 2.6 (d, 3H, -NHCH3), 2.70 (m, 2H, -CH2
carbazole), 2.75 (m, 2H, -CH2 carbazole) 3.8 (m, 1H, -NHCH3, D2O
exchangable), 4.3 (s, 2H, -SO2CH2), 7.0-7.4 (m, 3H, Ar-H), 7.8 (bs, 1H,
-NH indole, D20 exchangable), M++1: 278. Anal.Calcd for

118
(C14H18N2O2S) requires: C, 60.41; H, 6.52; N, 10.06. Found: C, 60.45;
H, 6.50; N, 10.16.
164c: R = -NHCH3, n=2, Yield: 2.0 gm (65 %), M.R: 154-157 °C; IR
(KBr, cm-1) 3386 (-NH),1310, 1144 (-SO2); 1H NMR (CDCl3/TMS): δ
1.92 (m, 4H, 2 x -CH2 carbazole), 2.6 (d, 3H, -NHCH3), 2.70 (m, 2H, -
CH2 carbazole), 2.75 (m, 2H, -CH2 carbazole) 3.2 (m, 2H,- SO2CH2),
3.4 (m, 2H, -Ar-CH2), 3.8 (m, 1H, -NHCH3, D2O exchangable), 7.0-7.4
(m, 3H, Ar-H), 7.8 (bs, 1H, -NH indole, D20 exchangable), M++1: 293.
Anal.Calcd for (C15H20N2O2S) requires: C, 61.62; H, 6.89; N, 9.58.
Found: C, 61.52; H, 6.81; N, 9.53.
4A.5.2. GENERAL PROCEDURE FOR THE SYNTHESIS OF 165(a-c):
163(a-c) (0.011 mol), was dissolved in water (47.4 mL) and N-
Methyl-4-piperidone (134) (0.011 mol) was added over a period of five
minutes. After the addition, the mixture was slowly heated to 50-55 ºC
and Con.HCl (7 mL) was added over period of 30 minutes. The
reaction mass was stirred overnight at 50-55 ºC, then cooled to 25-30
ºC and the pH adjusted to 12.0 with sodium hydroxide solution. The
slurry was allowed to stirr for 30 minutes and then cooled to 10-15 ºC,
filtered and washed with water. The crude compound was
recrystallized from a suitable solvent to get pure compound 165(a-c).
165a: R = pyrrolidine, n=1, Yield: 1.97 gm (54 %), M.R: 170-172 ºC; IR
(KBr, cm-1) 1330, 1120 (-SO2); 1H NMR (DMSO-d6/TMS) δ 1.8-1.9 (m,
4H, pyrrolidine), 2.4 (s, 3H, -NCH3), 2.6-2.9 (m, 4H, 2 x -CH2
carbazole) 3.2 (m, 4H, pyrrolidine) 3.40 (s, 2H, -CH2 carbazole), 4.3

119
(s, 2H, -SO2 CH2), 6.8-7.3 (m, 3H, Ar-H), 10.6 (br, s, -NH, D2O
exchangable); M++1: 334.43. Anal.Calcd for (C17H23N3O2S) requires: C,
61.23; H, 6.95; N, 12.60. Found: C, 61.13; H, 6.90; N, 12.58.
165b: R = -NHCH3, n=1, Yield: 1.57 gm (49 %), M.R: 142-145 ºC; IR
(KBr, cm-1) 3372 (-NH), 1275, 1118 (-SO2); 1H NMR (DMSO-d6/TMS): δ
2.4 (s, 3H, -NCH3), 2.6 (d, 3H, -NHCH3), 2.6-2.9 (m, 4H, 2 x -CH2
carbazole) 3.40 (s, 2H, -CH2 carbazole), 4.3 (s, 2H, -SO2CH2), 6.8-7.3
(m, 4H, Ar-H and –NHCH3), 10.6 (br, s, -NH, D2O exchangable); M++1:
318.43; Anal.Calcd for (C17H22N2O2S) requires: C, 64.12; H, 6.96; N,
8.80. Found: C, 64.02; H, 6.91; N, 8.80.
165c: R = -NHCH3, n=2, Yield: 2.45 gm (73 %), M.R: 158-160 °C; IR
(KBr, cm-1) 3436 (-NH), 1319, 1125 (-SO2); 1H NMR (DMSO-d6/TMS): δ
2.4 (s, 3H, -NCH3), 2.6 (d, 3H, -NHCH3), 2.6-2.9 (m, 4H, 2 x -CH2
carbazole) 3.0 (t, 2H, -Ar-CH2), 3.2 (t, 2H, -SO2CH2), 3.40 (s, 2H, -CH2
carbazole), 6.8-7.3 (m, 4H, Ar-H and –NHCH3), 10.6 (bs, -NH, D2O
exchangable); M++1: 318.43. Anal.Calcd for (C17H22N2O2S) requires: C,
64.12; H, 6.96; N, 8.80. Found: C, 64.02; H, 6.91; N, 8.80.
4A.5.3. GENERAL PROCEDURE FOR THE SYNTHESIS OF 167(a-c):
163(a-c) (0.01 mol) was added to a stirred solution of the 1-
(thiophen-2-yl) butane-1,3-dione 166(a-c) (0.01 mol) in N,N-
dimethylformamide (20 mL). The mixture was heated to 80-85 ºC and
stirred for 3 hours. After cooling to room temperature, the reaction
mixture was quenched into water (50 mL) and extracted into ethyl
acetate. The organic layer was washed with water and dried over anh.
MgSO4. The organic layer was filtered and concentrated under

120
reduced pressure to give a light brown solid. The crude product was
recrystallized from methanol to give pure 167(a-i).
167a: R = pyrrolidine, n=1, R1 = Thiophene, R2 = CH3, Yield: 2.48 gm
(64 %), M.R: 122-125 ºC; IR (KBr, cm-1) 1320, 1125 (-SO2); 1H NMR
(CDCl3): δ 1.70-1.90 (m, 4H, pyrrolidine), 2.30 (s, 3H, -CH3 pyrazole),
3.0-3.2 (m, 4H, pyrrolidine), 4.50 (s, 2H, -SO2CH2), 6.50 (s, 1H, CH
pyrazole), 6.90 (m, 1H, CH thiazole), 6.95 (m, 1H, CH thiazole), 7.30
(d, 4H, Ar-H J= 8.4), 7.40 (d, 4H, Ar-H J= 8.4), 7.50 (m, 1H, CH
thiazole); M++1: 386. Anal.Calcd for (C20H22N2O2S2) requires: C, 62.15;
H, 5.74; N, 7.25. Found: C, 62.10; H, 5.64; N, 7.20.
167b: R = pyrrolidine, n=1, R1 = Phenyl, R2 = CF3, Yield: 3.14 gm (73
%), M.R: 162-165 ºC; IR (KBr, cm-1) 1320, 1120 (-SO2); 1H NMR
(CDCl3): δ 1.70-1.90 (m, 4H, pyrrolidine), 3.0-3.2 (m, 4H,
pyrrolidine), 4.50 (s, 2H, -SO2CH2), 6.70 (s, 1H, CH pyrazole), 7.0 (m,
1H, -NHCH3), 7.20-7.60 (m, 9H, Ar-H); M++1: 435; Anal.Calcd for
(C22H21F3N2O2S) requires: C, 60.82; H, 4.87; N, 6.45. Found: C, 60.72;
H, 4.85; N, 6.35.
167c: R = pyrrolidine, n=1, R1 = 4-Methylphenyl, R2 = CF3, Yield: 4.20
gm (94 %), M.R: 147-149 ºC; IR (KBr, cm-1) 1317, 1124 (-SO2); 1H
NMR (DMSO-d6/TMS): δ 1.70-1.90 (m, 4H, pyrrolidine), 2.30 (s, 3H, -
CH3), 3.00-3.20 (m, 4H, pyrrolidine), 4.50 (s, 2H, -SO2CH2), 6.70 (s,
1H, CH pyrazole), 7.20-7.60 (m, 8H, Ar-H); M++1: 449; Anal.Calcd for
(C23H23F3N2O2S) requires: C, 61.59; H, 5.17; N, 6.25. Found: C, 61.49;
H, 5.10; N, 6.20.

121
167d: R = -NHCH3, n=1, R1 = Thiophene, R2 = CH3, Yield: 2.70 gm
(79 %), M.R: 150-152 ºC; IR (KBr, cm-1) 3345 (-NH), 1322, 1124 (-
SO2); 1H NMR (CDCl3/TMS) δ 2.30 (s, 3H, -CH3 pyrazole), 2.75 (d, 3H,
-NHCH3), 4.00 (m, 1H, -NHCH3, D2O exchangable), 4.40 (s, 2H, -
SO2CH2), 6.35 (s, 1H, CH pyrazole), 6.90 (m, 1H, CH thiazole), 6.95
(m, 1H, CH thiazole), 7.25-7.4 (m, 4H, Ar-H), 7.35 (s, 1H, CH thiazole);
M++1: 348; Anal.Calcd for (C16H17N3O2S2) requires: C, 55.31; H, 4.93;
N, 12.09. Found: C, 55.21; H, 4.96; N, 12.00.
167e: R = -NHCH3, n=1, R1 = Phenyl, R2 = CF3, Yield: 2.50 gm (64 %),
M.R: 181-183 ºC; IR (KBr, cm-1) 3347 (-NH), 1309, 1135 (-SO2); 1H
NMR (CDCl3/TMS): δ 2.75 (d, 3H, -NHCH3), 4.00 (m, 1H, -NHCH3, D2O
exchangable), 4.40 (s, 2H, -SO2CH2), 6.70 (s, 1H, CH pyrazole), 7.20-
7.60 (m, 9H, Ar-H); M++1: 396. Anal.Calcd for (C18H16F3N3O2S)
requires: C, 54.68; H, 4.08; 10.63. Found: C, 54.63; H, 4.04; 10.73.
167f: R = -NHCH3, n=1, R1 = 4-Methylphenyl, R2 = CF3, Yield: 3.19 gm
(79 %), M.R: 139-141 ºC; IR (KBr, cm-1) 3347 (-NH), 1332, 1126 (-
SO2); 1H NMR (CDCl3/TMS): δ 2.30 (s, 3H, -CH3), 2.75 (d, 3H, -
NHCH3), 4.40 (s, 2H, -Ar-CH2), 6.70 (s, 1H, CH pyrazole), 4.00 (q, 1H,
-NHCH3, D2O exchangable), 7.20-7.60 (m, 8H, Ar-H); M++1: 410.
Anal.Calcd for (C19H18F3N3O2S) requires: C, 55.74; H, 4.43; N, 10.26.
Found: C, 55.64; H, 4.33; N, 10.29.
167g: R = -NHCH3 n=2, R1 = Thiophene, R2 = CH3, Yield: 2.70 gm (75
%), M.R: 191-194 °C; IR (KBr, cm-1) 3347, 1280, 1120 ; 1H NMR
(CDCl3): 2.35 (s, 3H, -CH3 pyrazole), 2.75 (d, 3H, -NHCH3), 3.25 (t,
2H, -Ar- CH2), 3.45 (t, 2H, -SO2CH2), 4.00 (q, 1H, -NHCH3, D20

122
exchangable), 6.35 (s, 1H, CH pyrazole), 6.90 (dd, 1H, thiazole), 6.95
(dd, 1H, thiazole), 7.25-7.4 (m, 4H, Ar-H), 7.35 (s, 1H, thiazole); M++1:
362. Anal.Calcd for (C17H19N3O2S2) requires: C, 56.48; H, 5.30; N,
11.62. Found: C, 56.40; H, 5.20; N, 11.52.
167h: R = -NHCH3, n=2, R1 = Phenyl, R2 = CF3, Yield: 2.82 gm (70 %),
M.R: 174-176 °C; IR (KBr, cm-1) 3343, 1265, 1130; 1H NMR (CDCl3): δ
2.80 (d, 3H, -NHCH3), 3.20 (t, 2H, - Ar-CH2), 3.45 (t, 2H, -SO2CH2),
4.10 (q, 1H, -NHCH3, D2O exchangable), 6.70 (s, 1H, CH pyrazole),
7.20-7.60 (m, 9H, Ar-H); M++1: 410. Anal.Calcd for (C19H18F3N3O2S)
requires: C, 55.74; H, 4.43; N, 10.26. Found: C, 55.64; H, 4.40; N,
10.16.
167i: R = -NHCH3 n=2, R1 = 4-Methylphenyl, R2 = CF3, Yield: 3.77 gm
(90 %), M.R: 160-165 °C; IR (KBr, cm-1) 3337, 1270, 1117; 1H NMR
(DMSO-d6/TMS): δ 2.30 (s, 3H, -CH3), 2.60 (d, 3H, -NHCH3), 3.00 (t,
2H, -Ar- CH2), 3.30 (m, 2H, -SO2CH2), 6.90 (s, 1H, CH pyrazole), 7.00
(q, 1H, -NHCH3, D2O exchangable), 7.00 7.20-7.60 (m, 8H, Ar-H);
M++1: 424. Anal.Calcd for (C20H20F3N3O2S) requires: C, 56.73; H, 4.76;
N, 9.92. Found: C, 56.63; H, 4.72; N, 9.82.

123
CHAPTER-4
SYNTHESIS OF NARATRIPTAN USING N-BENZYL-N-
METHYL ETHENESULFONAMIDE
SECTION-B
4B.1 INTRODUCTION:
Triptans are a new class of compounds developed for the
treatment of migraine attacks [13]. The first one of this class,
sumatriptan [162] and the newer triptans are zolmitriptan [163],
naratriptan [164], rizatriptan [165], eletriptan [166], almotriptan [167]
and frovatriptan [168] display high antagonist activity mainly at the
serotonin 5-HT1B and 5-HT1D receptor subtypes. Among these
triptans, naratriptan is one of the important drugs for the treatment of
acute attacks of migraine exhibiting high affinity for 5-HT1D
receptors, a serotonin (5-hydroxytryotamine, 5-HT) receptor. This
chapter describes synthesis of naratriptan using new intermediate N-
benzyl-N-methylethenesulfonamide.
4B.2 LITERATURE SURVEY
Several methods for the synthesis of naratriptan have been
reported in the literature and few of them are discussed below.
Oxford et. al. [169] reported that 5-bromoindole (168), when
subjected to Heck coupling with N-methylethenesulfonamide (169)
gave 2-(1H-Indol-5-yl)-N-methylethenesulfonamide (170), which on
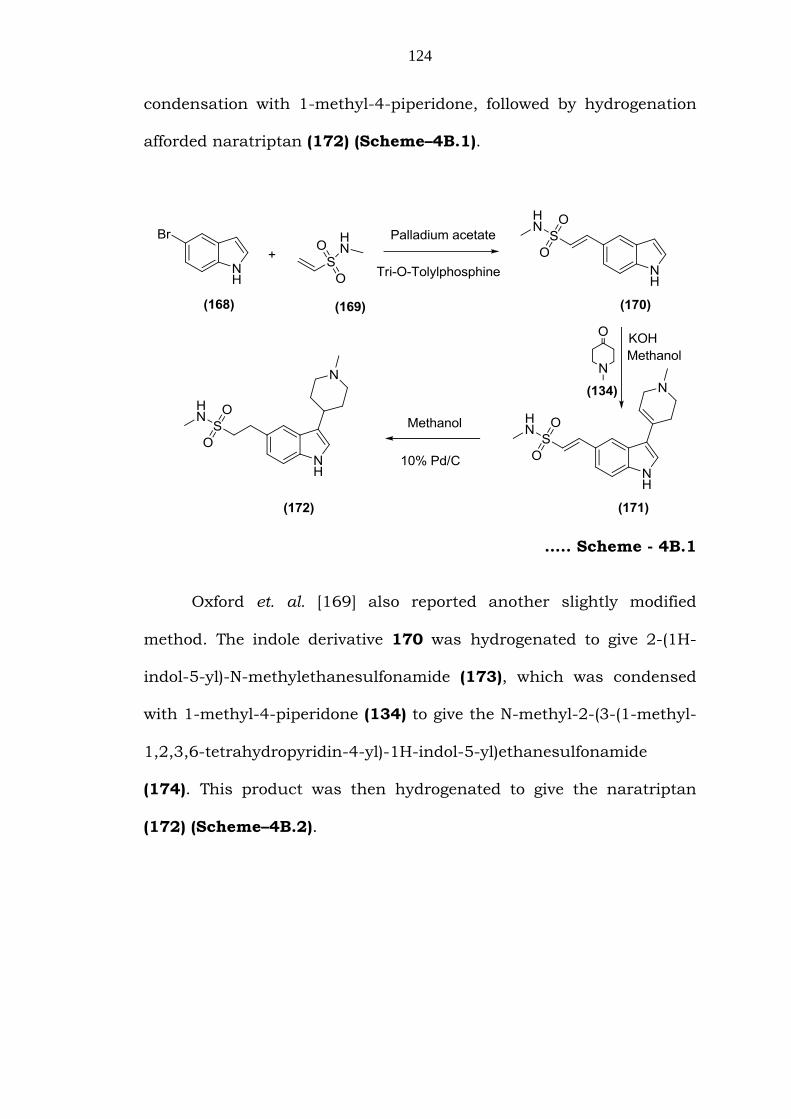
124
condensation with 1-methyl-4-piperidone, followed by hydrogenation
afforded naratriptan (172) (Scheme–4B.1).
….. Scheme - 4B.1
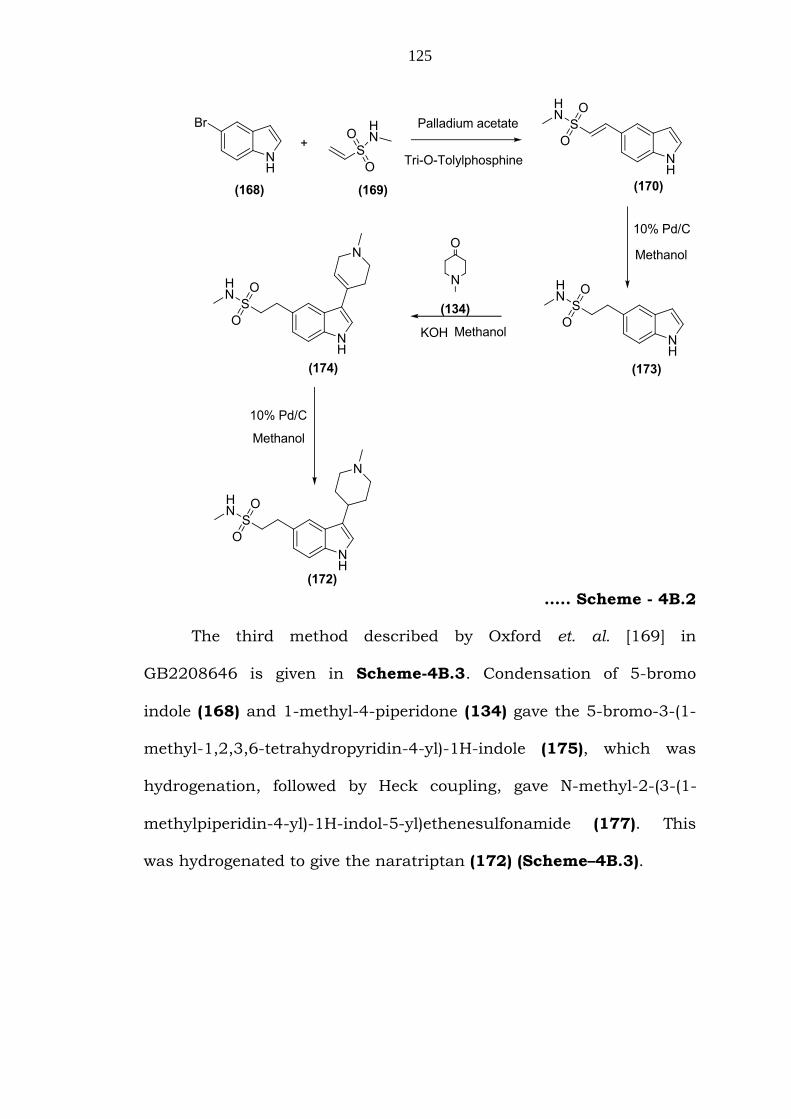
Oxford et. al. [169] also reported another slightly modified
method. The indole derivative 170 was hydrogenated to give 2-(1H-
indol-5-yl)-N-methylethanesulfonamide (173), which was condensed
with 1-methyl-4-piperidone (134) to give the N-methyl-2-(3-(1-methyl-
1,2,3,6-tetrahydropyridin-4-yl)-1H-indol-5-yl)ethanesulfonamide
(174). This product was then hydrogenated to give the naratriptan
(172) (Scheme–4B.2).
125
….. Scheme - 4B.2
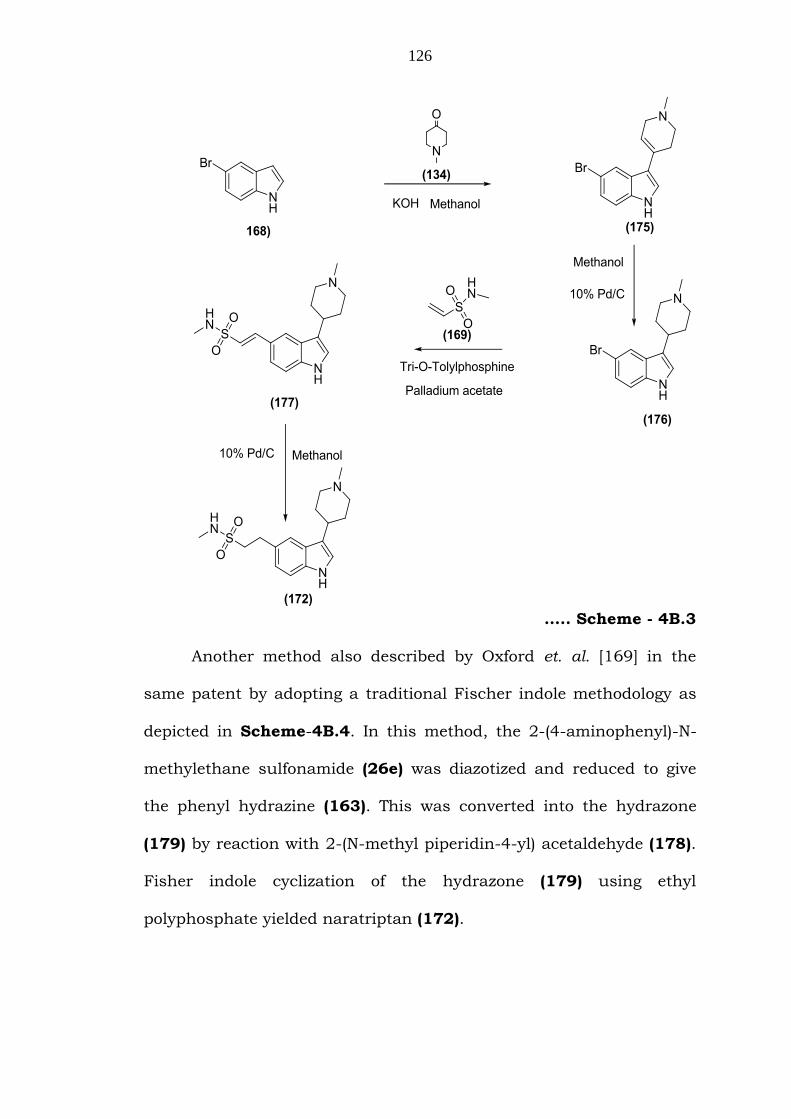
The third method described by Oxford et. al. [169] in
GB2208646 is given in Scheme-4B.3. Condensation of 5-bromo
indole (168) and 1-methyl-4-piperidone (134) gave the 5-bromo-3-(1-
methyl-1,2,3,6-tetrahydropyridin-4-yl)-1H-indole (175), which was
hydrogenation, followed by Heck coupling, gave N-methyl-2-(3-(1-
methylpiperidin-4-yl)-1H-indol-5-yl)ethenesulfonamide (177). This
was hydrogenated to give the naratriptan (172) (Scheme–4B.3).
126
….. Scheme - 4B.3
Another method also described by Oxford et. al. [169] in the
same patent by adopting a traditional Fischer indole methodology as
depicted in Scheme-4B.4. In this method, the 2-(4-aminophenyl)-N-
methylethane sulfonamide (26e) was diazotized and reduced to give
the phenyl hydrazine (163). This was converted into the hydrazone
(179) by reaction with 2-(N-methyl piperidin-4-yl) acetaldehyde (178).
Fisher indole cyclization of the hydrazone (179) using ethyl
polyphosphate yielded naratriptan (172).
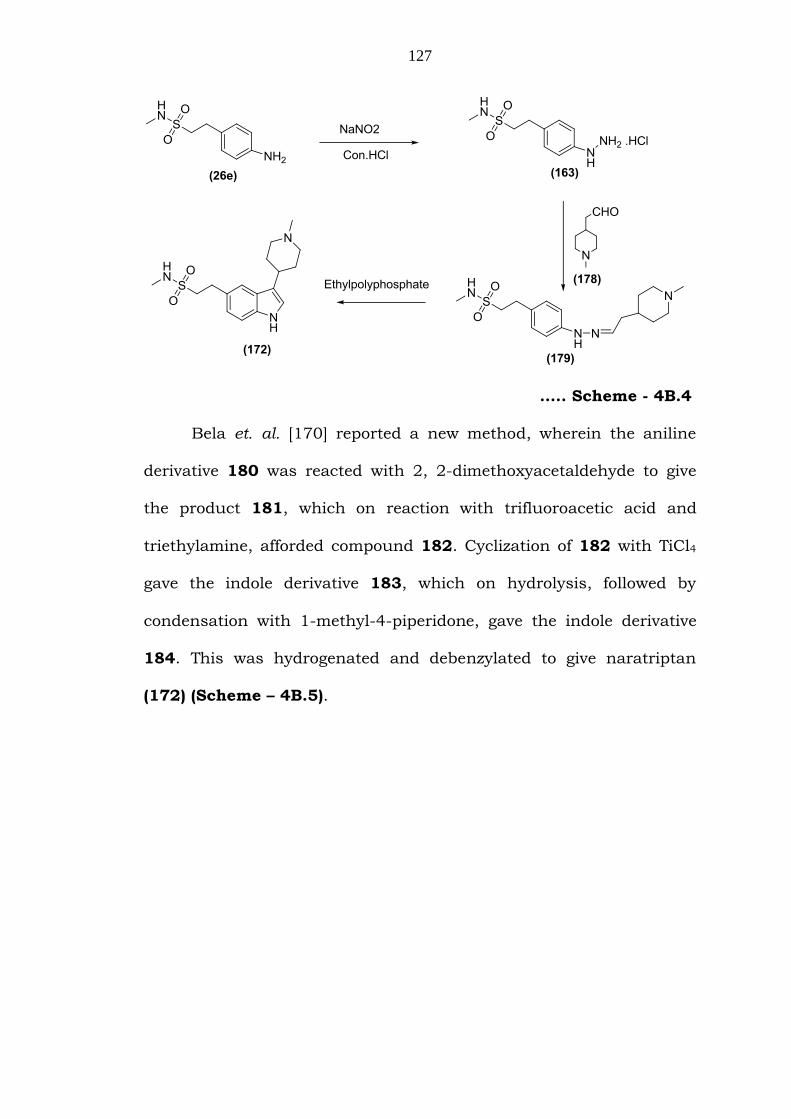
127
….. Scheme - 4B.4
Bela et. al. [170] reported a new method, wherein the aniline
derivative 180 was reacted with 2, 2-dimethoxyacetaldehyde to give
the product 181, which on reaction with trifluoroacetic acid and
triethylamine, afforded compound 182. Cyclization of 182 with TiCl4
gave the indole derivative 183, which on hydrolysis, followed by
condensation with 1-methyl-4-piperidone, gave the indole derivative
184. This was hydrogenated and debenzylated to give naratriptan
(172) (Scheme – 4B.5).
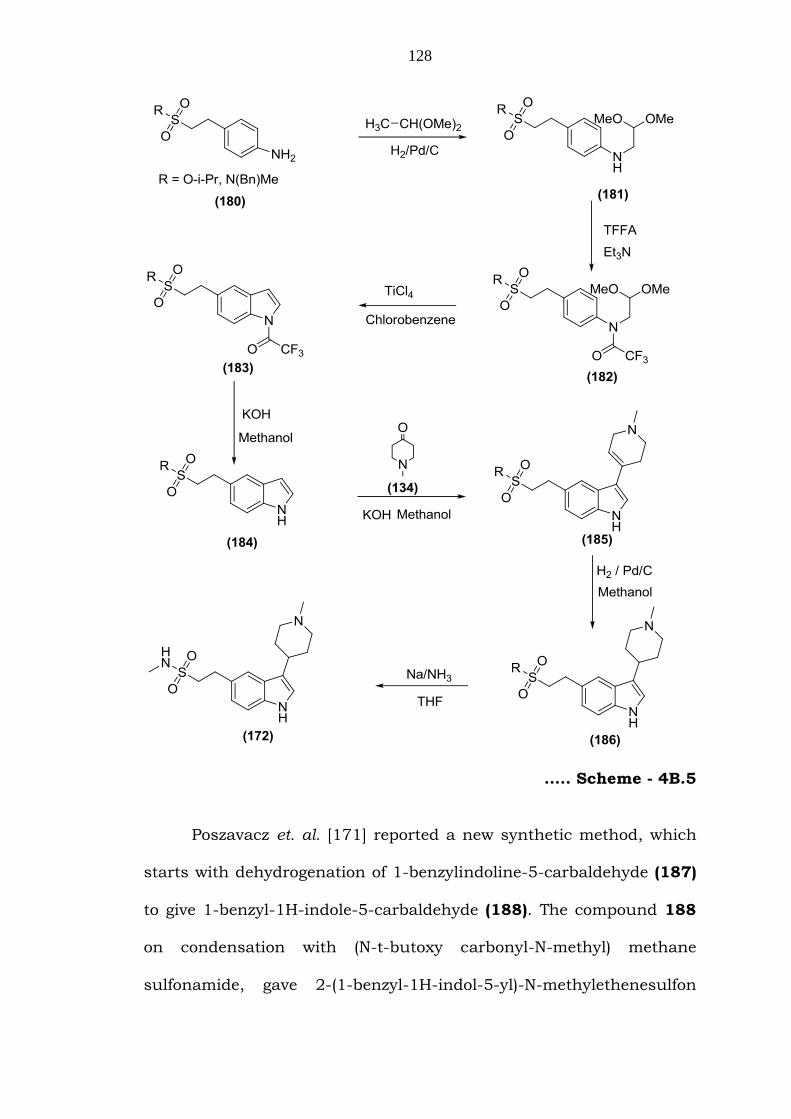
128
….. Scheme - 4B.5
Poszavacz et. al. [171] reported a new synthetic method, which
starts with dehydrogenation of 1-benzylindoline-5-carbaldehyde (187)
to give 1-benzyl-1H-indole-5-carbaldehyde (188). The compound 188
on condensation with (N-t-butoxy carbonyl-N-methyl) methane
sulfonamide, gave 2-(1-benzyl-1H-indol-5-yl)-N-methylethenesulfon
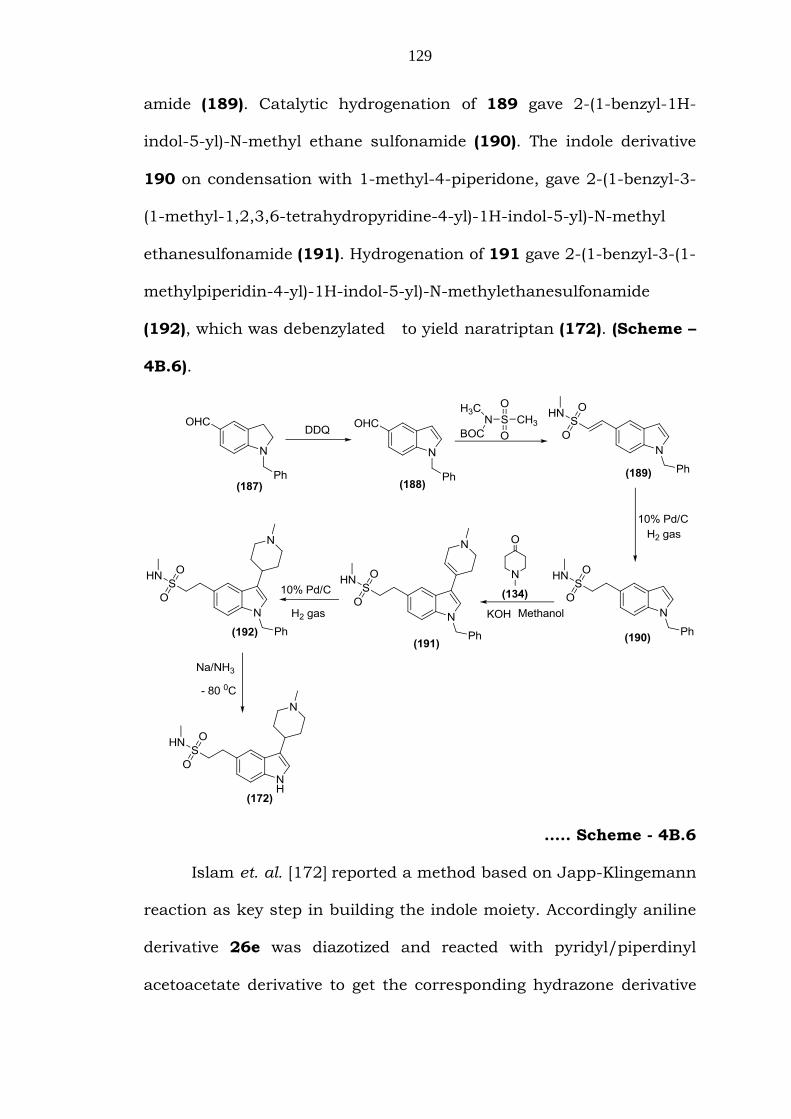
129
amide (189). Catalytic hydrogenation of 189 gave 2-(1-benzyl-1H-
indol-5-yl)-N-methyl ethane sulfonamide (190). The indole derivative
190 on condensation with 1-methyl-4-piperidone, gave 2-(1-benzyl-3-
(1-methyl-1,2,3,6-tetrahydropyridine-4-yl)-1H-indol-5-yl)-N-methyl
ethanesulfonamide (191). Hydrogenation of 191 gave 2-(1-benzyl-3-(1-
methylpiperidin-4-yl)-1H-indol-5-yl)-N-methylethanesulfonamide
(192), which was debenzylated to yield naratriptan (172). (Scheme –
4B.6).
….. Scheme - 4B.6
Islam et. al. [172] reported a method based on Japp-Klingemann
reaction as key step in building the indole moiety. Accordingly aniline
derivative 26e was diazotized and reacted with pyridyl/piperdinyl
acetoacetate derivative to get the corresponding hydrazone derivative
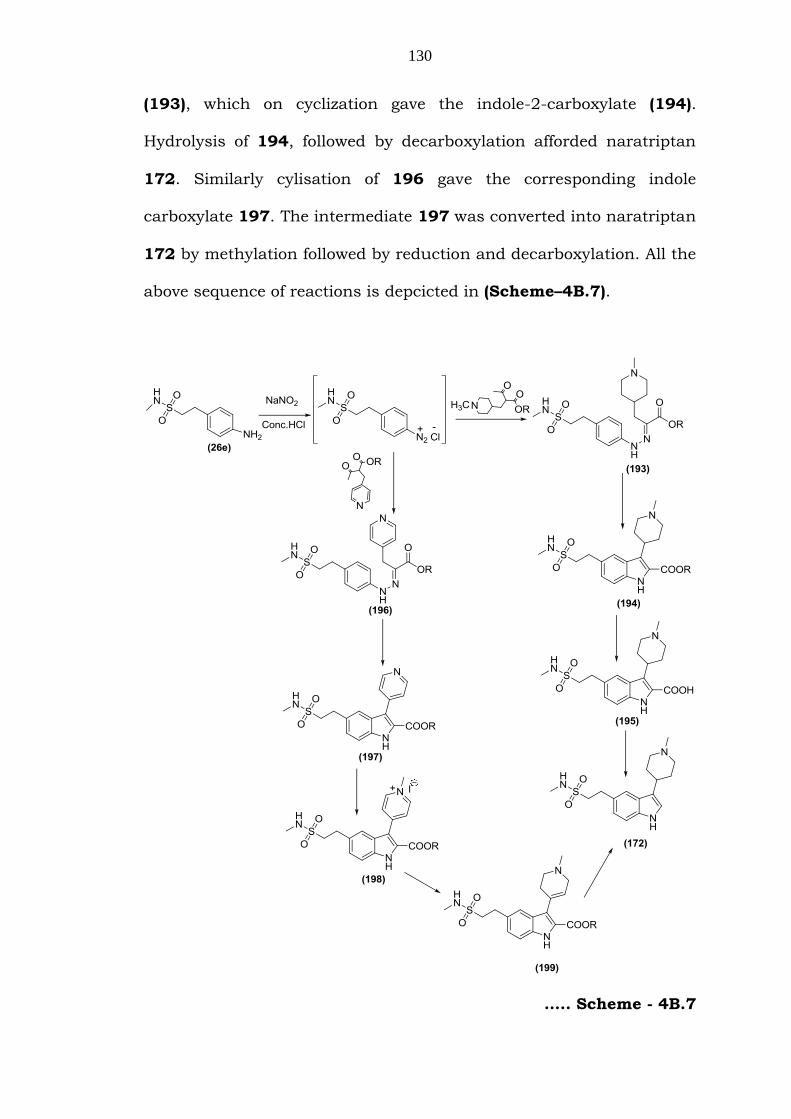
130
(193), which on cyclization gave the indole-2-carboxylate (194).
Hydrolysis of 194, followed by decarboxylation afforded naratriptan
172. Similarly cylisation of 196 gave the corresponding indole
carboxylate 197. The intermediate 197 was converted into naratriptan
172 by methylation followed by reduction and decarboxylation. All the
above sequence of reactions is depcicted in (Scheme–4B.7).
….. Scheme - 4B.7

131
4B.3 PRESENT WORK
Synthesis of a novel intermediate N-benzyl-N-methylethene
sulfonamide was achieved. 5-Bromo-3-(1-methyl-1,2,3,6-tetrahydro-
pyridin-4-yl)-1H-indole was reacted with N-methyl-N-benzyl
ethenesulfonamide using Heck coupling method. The resulting
intermediate was subjected to hydrogenation followed by
debenzylation to afford naratriptan in high purity. These results are
described below.
4B.4 RESULTS AND DISCUSSIONS
A new compound N-benzyl-N-methylethenesulfonamide (202)
was prepared by reaction of 2-chloroethanesulfonyl chloride (200)
with N-methylbenzylamine (201) in the presence of triethylamine at
-20 to -10 ºC (Scheme-4B.8). The progress of this reaction was
studied in various solvents such as dimethylformamide, diethylether,
diisopropylether, tetrahydrofuran, dichloromethane, ethylene
dichloride and chloroform using triethylamine as a base. Among all
the solvents, dichloromethane was found to be the best solvent to get
good yield. The product was characterized by its spectral data. Thus,
its IR spectrum (neat) (Fig.4B.1) showed peak at 1606 cm-1 due to
C=C and presence of the peaks at 1338 cm-1 and 1151 cm-1
conforming the -SO2 group. Its 1H NMR (CDCl3/TMS) (Fig.4B.2)
showed singlet at 2.6 due to the methyl group and 4.2 is due to –
CH2-Ph. A three proton multiplet at 6.0-6.5 can be assigned to (–
CH2=CH-SO2-) and multpilet at 7.3-7.6 due to five Ar-H. Its APCI

132
mass spectrum (Fig. 3.3) showed M++1 ion peak at 212 corresponding
to a molecular mass of 211 further confirms the structur 202.
….. Scheme - 4B.8
The compound 202 was reacted with 5-bromo-3-(1-methyl-
1,2,3,6-tetrahydro-pyridin-4-yl)-1H-indole (175) in the presence of
palladium acetate and tri-o-tolylphosphine using triethylamine as a
base at 100-110 ºC in dimethylformamide and obtained 2-(3-(1,2,3,6-
tetrahydro-1-methylpyridin-4-yl)-1H-indol-5-yl)vinylsulfonyl)-N-methyl
(phenyl)methan amine (203) (Scheme–4B.9). The product was
characterized by spectral methods. Thus, its IR spectrum (Fig. 4B.4)
in KBr showed a peak at 1607 cm-1 indicates the presence of C=C.
Peaks at 1331 cm-1 and 1142 cm-1 absorptions assignable to
asymmetric and symmetric stretching of –SO2 group. Its 1H-NMR
spectrum (DMSO-d6/TMS) (Fig.4B.5) showed signals at δ 2.25 (s, 3H,
-NCH3), 2.4-2.5 (m, 4H, Piperidine), 2.6 (s, 3H, -NCH3), 3.3 (m, 2H,
Piperdine), 4.2 (s, 2H, -NCH2Ar), 6.2 (s, 1H, Piperdine), 6.50 (d, 1H
J=15.2 Hz, -SO2CH=CH), 7.2-7.4 (m, 8H, Ar-H), 7.5 (d, 1H J=15.2 Hz,
CH=CH-Ar), 7.8 (s, 1H, CH indole), 11.0 (br, s, 1H, -NH indole D2O
exchangable). Its mass spectrum (Fig. 4B.6) showed its molecular ion
peak at m/z 422 corresponding to molecular mass of 421 when
recorded in the Q+1 mode.
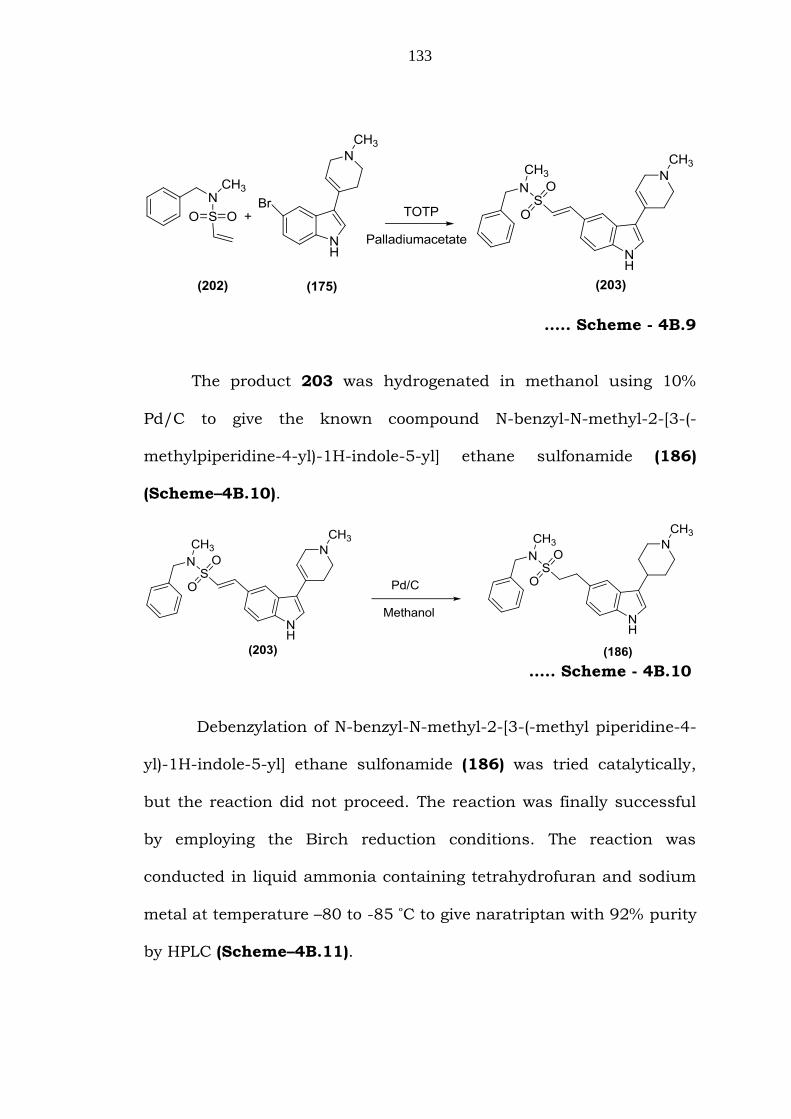
133
….. Scheme - 4B.9
The product 203 was hydrogenated in methanol using 10%
Pd/C to give the known coompound N-benzyl-N-methyl-2-[3-(-
methylpiperidine-4-yl)-1H-indole-5-yl] ethane sulfonamide (186)
(Scheme–4B.10).
….. Scheme - 4B.10
Debenzylation of N-benzyl-N-methyl-2-[3-(-methyl piperidine-4-
yl)-1H-indole-5-yl] ethane sulfonamide (186) was tried catalytically,
but the reaction did not proceed. The reaction was finally successful
by employing the Birch reduction conditions. The reaction was
conducted in liquid ammonia containing tetrahydrofuran and sodium
metal at temperature –80 to -85 ºC to give naratriptan with 92% purity
by HPLC (Scheme–4B.11).

134
….. Scheme - 4B.11
In order to get pure compound the crude product is to be
purified. Crystallisation in different solvents did not help. Hence other
methods of purification like salt formation were tried. Of the several
salts viz., hydrochloride, sulphate, succinate, maleate and oxalate ete.,
Among these oxalate salt was most favourable. Naratriptan oxalate
salt on basification yielded naratriptan base of 99.8% HPLC purity.
The product was characterised by its analytical and spectral data, IR
(Fig.4B.7), 1H NMR (Fig.4B.8), Mass (Fig.4B.9) and 13C NMR
(Fig.4B.10) and the product was identical, in all the respects when
compared with reference standard of naratriptan.
All the above sequences of reactions were summarized in
Schemes- 4B.12.
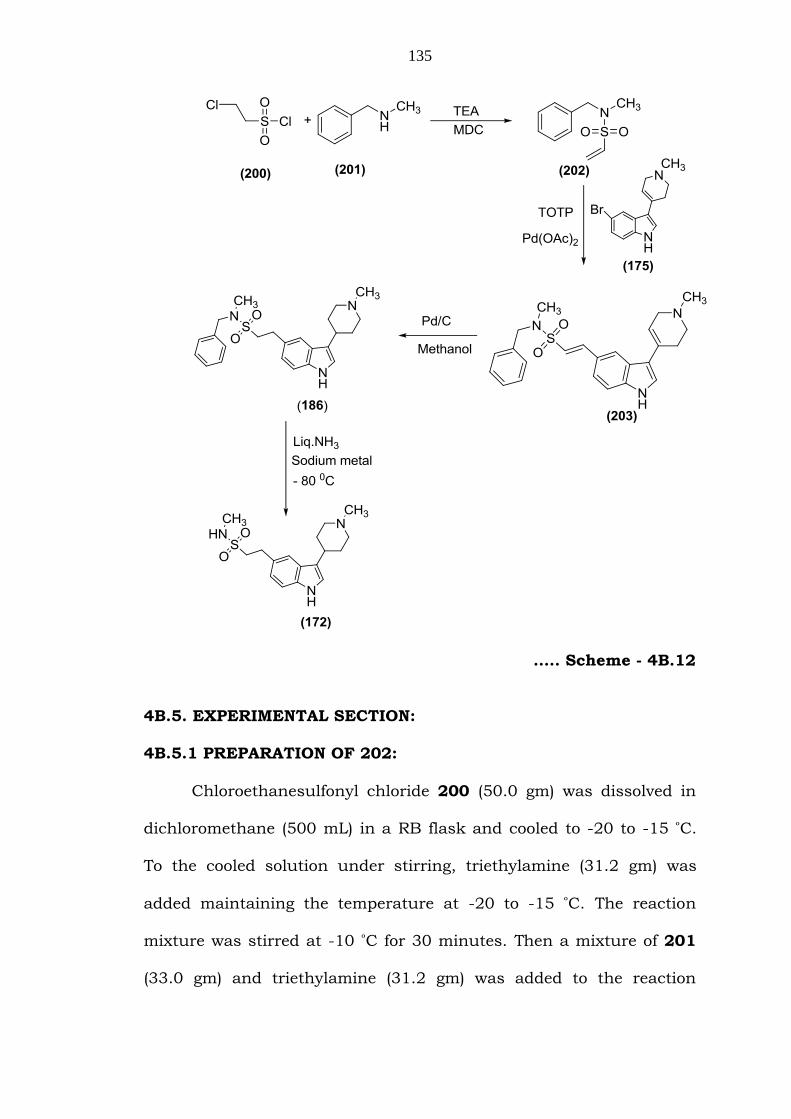
135
….. Scheme - 4B.12
4B.5. EXPERIMENTAL SECTION:
4B.5.1 PREPARATION OF 202:
Chloroethanesulfonyl chloride 200 (50.0 gm) was dissolved in
dichloromethane (500 mL) in a RB flask and cooled to -20 to -15 ºC.
To the cooled solution under stirring, triethylamine (31.2 gm) was
added maintaining the temperature at -20 to -15 ºC. The reaction
mixture was stirred at -10 ºC for 30 minutes. Then a mixture of 201
(33.0 gm) and triethylamine (31.2 gm) was added to the reaction

136
mixture at -10 ºC. After completion of the addition, the temperature of
the reaction was raised to 0 ºC and water (100 mL) was added. The
reaction mixture was stirred for 10 to 20 minutes and the organic
layer was separated, dried over anh.Na2SO4 and the solvent was
evaporated. The residue obtained was repeatedly triturated with
diethylether. The ether extract, on concentration, gave oil product (42
gm, 70%) of N-benzyl-N-methylethenesulfonamide (202) with 95%
purity (by GC). This was used as such in subsequent reactions.
IR (neat): 3030, 3059, 1606, 1338, 1152, 978, 779; 1H NMR:
(CDCl3/TMS) 2.69 (s, 3H, -NCH3), 4.25 (s, 2H, -CH2Ph), 6.02 (m, 1H,
CH2), 6.27 (m, 1H, CH2), 6.45 (m, 1H, CH), 7.31 (m, 5H, Ar-H); M+1:
212; Anal. Calcd. for (C10H13NO2S) requires: C, 56.85; H, 6.20; N,
6.63 Found: C, 56.79; H, 6.27; N, 6.60.
4B.5.2. PREPARATION OF 203:
In a round bottom flask fitted with a mechanical stirrer and
condenser, were charged N,N- dimethylformamide (100 mL) and 175
(25 gm), 202 (30 gm), triethylamine (32.1 gm), triorthotolylphosphine
(7.75 gm) and palladium acetate (0.42 gm). The reaction mixture was
gradually heated to 100-110 ºC under stirring and maintained for 8
hours. The reaction mass was cooled to 60 ºC and then filtered. The
filterate was diluted with water (300 mL) and extracted with ethyl
acetate (3x100 mL). The ethyl acetate layer was washed with water (75
mL) and dried over anh.Na2SO4, filtered and then concentrated. The
residue was stirred with methanol (100 mL), cooled to 0-5 ºC and solid

137
formed was filtered and dried to give 27.0 gm (75%) of the product. An
analytical sample was obtained after recrystalization from methanol
M.R: 205-210 ºC; IR (KBr): 3416, 3027, 2847, 2890, 1647,1607, 1494,
1445, 1337, 1148, 1073, 938, 846; 1H NMR: (DMSO-d6/TMS) δ 2.25
(s, 3H, -NCH3), 2.4-2.5 (m, 4H, Piperdine), 2.6 (s, 3H, -NCH3), 3.3 (m,
2H, Piperdine), 4.2 (s, 2H, -NCH2Ar), 6.2 (s, 1H, Piperdine), 6.50 (d,
1H J=15.2 Hz, -SO2CH=CH), 7.2-7.4 (m, 8H, Ar-H), 7.5 (d, 1H J=15.2
Hz, CH=CH-Ar), 7.8 (s, 1H, indole), 11.0 (br, s, 1H, -NH indole D2O
exchangable). M+1: 422. Anal. Calcd. for (C24H27N3O2S) requires: C,
68.38; H, 6.46; N, 9.97; Found: C, 68.30; H, 6.51; N, 9.92.
4B.5.3. PREPARATION OF 186:
A hydrogenation kettle was charged methanol (500 mL), 10%
Pd/C (20 gm) slurrey and 203 (30 gm) at room tempreature.
Hydrogenation was carried out at 6kg/cm2 pressure and continued for
20-24 hours till hydrogen gas absorption ceased. The reaction mass
was filtered to remove the catalyst and filterate was concentrated
under reduced pressure. The resulting residue was dissolved in
methanol (100 mL), cooled to 20-25 ºC and solid formed was filtered
and dried to give (14 gm, 47%) of product with m.p: 155-160 ºC. IR
(KBr): 3441, 3127, 2913, 1448, 1323, 1144, 987. 1H NMR: (DMSO-
d6/TMS) 2.0-2.1 (m, 4H, piperdine), 2.6 (s, 2H, -NCH3), 2.7 (s, 3H,-
NCH3), 3.02 (m, 3H, piperidine), 3.20 (m, 2H, -SO2CH2), 3.40 (m, 2H,
piperdine), 3.5 (m, 2H, -CH2-Ar), 4.1 (s, 2H, -CH2-Ar), 7.2-7.5 (m, 8H
Ar-H), 7.8 (s, 1H, CH indole), 10.90 (s, 1H, NH indole, D2O

138
exchangable). M+1: 425. Anal. Calcd. for (C24H31N3O2S) requires: C,
67.73; H, 7.34; N, 9.87; Found: C, 67.30; H, 7.51; N, 9.89.
4B.5.4. PREPARATION OF 172:
To a stirred solution of 186 (25 gm) in liquid ammonia (250 mL) and
THF (100 mL), was added sodium metal (18 gm) at -80 ºC over a
period of 30 min. The reaction mixture was stirred for another one
hour at -80 ºC. The reaction mass was treated with aqueous
ammonium chloride solution (25 gm in 75 mL water) at -80 ºC and
then allowed to warm to room temperature and extracted with
dichloromethane (3x150 mL). The organic layer was washed with
water (75 mL), dried over anh.Na2SO4, filtered and then concentrated.
The residue was stirred with methanol (100 mL), cooled to 0-5 ºC and
the solid was filtered and dried to give 17 gm (80%) of the crude
naratriptan base, M.R: 170-172 ºC.
Purification: In to a round bottomed flask fitted with a mechanical
stirrer and condenser was charged methanol (100 mL) and crude
naratriptan (25 gm). The reaction mixture was gradually heated to
about 45- 50 ºC under stirring and maintained for 30 min. Oxalic acid
(17 gm) was added at 40-45 ºC and stirred for another 30min. The
reaction mixture was then slowly cooled to 10-15 ºC and filtered to
give naratriptan oxalate with a purity of 99.5% (HPLC). The wet cake
of naratriptan oxalate was taken in to water (200 mL) and basified
with potassium carbonate to pH 9.5-10.0 and stirred for 20 minutes.
The solid was filtered, washed with water and dried to give 20 gm of
naratriptan with HPLC purity 99.8%. IR (KBr): 3441, 3127, 2913,

139
1448, 1323, 1144, 987. 1H NMR: (DMSO-d6/TMS) 2.09 (m, 4H,
piperidine), 2.62 (d, 3H, -NHCH3), 2.76 (s, 3H, -NCH3), 3.02 (m, 3H,
piperdine), 3.1 (m, 2H, -SO2CH2), 3.3 (m, 2H, piperidine), 3.4 (m, 2H,-
CH2-Ar), 7.0 (m, 2H, Ar-H) ), 7.1 (m, 1H, -NHCH3), 7.3 (d, 1H, Ar-H),
7.6 (s, 1H, CH indole), 10.70 (br, s, 1H, -NH indole D2O exchangable);
M+1: 336. Anal. Calcd. for (C17H25N3O2S) requires: C, 60.87; H, 7.51;
N, 12.53; Found: C, 60.85; H, 7.51; N, 12.50.






![Synthesis, crystal structures and properties of carbazole-based … · 2021. 1. 4. · 11 Synthesis, crystal structures and properties of carbazole-based [6]helicenes fused with an](https://img.pdfslide.net/doc/110x75/60fee8ef13348044ac53521d/synthesis-crystal-structures-and-properties-of-carbazole-based-2021-1-4-11.jpg)






















