Embed Size (px)
Citation preview

J. Chem. Soc., Faraday Trans. 2, 1989, 85(3), 163-176
A Phenomenological Theory of Long-range Hydrophobic Attraction Forces based on a
Square-gradient Variational Approach
Jan Christer Eriksson,* Stig Ljunggren and Per M. Claesson Department of Physical Chemistry, The Royal Institute of Technology,
S-1 00 44 Stockholm, Sweden
Through recent surface force measurements it has been convincingly demon- strated that strong and amazingly long-range, attractive interaction forces act between hydrophobic surfaces immersed in water. Upon separating two such surfaces from molecular contact a vapour/gas cavity normally forms. This is not the case, however, when gradually diminishing the surface separation. The hydrophobic attraction forces have been recorded in this latter, metastable regime.
A mean-field theory based on a square-gradient assumption is presented in this paper which is shown to account reasonably well for the surface forces found experimentally for two cylindrically shaped, hydrophobic sur- faces interacting in water. The order parameter/variational approach taken is closely related conceptually to earlier theories of repulsive hydration forces by Marcelja et al. and Cevc et a/. The present theory implies that rather minor, hydrogen-bond-propagated molecular ordering effects, in the contact layers of water molecules next to the hydrophobic surfaces and in the core of the thin water film, give rise to the attraction observed. However, it does not fully address the intriguing question as to how it comes about that the hydrophobic attraction forces extend over such a wide range as 70-90 nm. It merely points in the direction that surface-induced structural changes in the core of the thin water film (so far not captured by molecular dynamics simulations) which demand minimal free-energy expense may generate an interaction of a long-range nature.
It has become firmly established that a long-range attractive force acts between hydro- phobic surfaces immersed in water.’-’ The most recent investigations by Claesson and Christenson,’ Rabinovich a n d Derjaguin6 and Claesson and Christenson’ have shown that this attractive force is measurable for surfaces which are free of charges even a t separations of 70-90 nm, and that the long-range tail of the surface force curve decays exponentially. However, at distances less than ca. 10 nm, the hydrophobic attraction is further amplified. These observations are substantially different from those first reported by Israelachvili and Pashley’ and by Pashley et al.,’ who claimed an interaction range of 10-15 nm and a purely exponential force law.
The strength and decay rate of this hydrophobic interaction depend on the hydropho- bicity of the two approaching surfaces, but seemingly not in a straightforward fashion. New studies by Herder* and Christenson et al.9 indicate that the hydrocarbon chain packing density in the hydrophobic surfaces and the adsorption of small amounts of surfactant or alkylammonium ions may drastically reduce the range and magnitude of the attraction. Furthermore, it is worth noting that besides a van der Waals force, there seems to be no long-range attractive force acting between hydrophilic surfaces immersed in water. Instead, there is often a repulsive hydration force of shorter range.
163
Dow
nloa
ded
by U
nive
rsity
of
Illin
ois
at C
hica
go o
n 07
Mar
ch 2
013
Publ
ishe
d on
01
Janu
ary
1989
on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/F
2989
8500
163
View Article Online / Journal Homepage / Table of Contents for this issue

164 Long-range Hydrophobic Attraction
The origin of the hydrophobic attraction force as well as of the corresponding repulsive hydration force observed for hydrophilic surfaces has been a recurrent matter of discussion during recent years.””’ In 1976 Marcelja and Radic12 published a vari- ational mean-field theory which was based upon the idea that interaction forces of this kind are mediated by structural changes in the thin water film separating the two surfaces. As a general concept this idea dates back to a large number of investigations carried out over the years, especially by Derjaguin et aZ.13 However, in its original version, Marcelja’s theory did not yield a satisfactory agreement with experimental results on hydration forces and free energies, and hence modified theories were advanced later by Cevc et al.I4 and by Ostrowsky and Sornette.’’
A different line of thinking starts out from the observation that cavity formation occurs between strongly hydrophobic surfaces (contact angle >90”) when they are brought into molecular ont tact.^-^'-^^ In fact, for the systems studied by Claesson and Chr i~ tenson ,~ the thermodynamically stable state is a state with a vapour cavity between the surfaces whenever the separation is less than a few hundred nm. The reason why no cavities form until the surfaces are in molecular contact is that the activation energy for.cavity formation would otherwise be enormous. In this respect the capillary theory of cavity formationl8 is consistent with The hydrophobic attraction which is measured without any cavities present between the hydrophobic surfaces has accordingly been supposed to be related, in some way or other, to a precursor state of a vapour c a ~ i t y . ~ ’ ~
A third possible origin of the hydrophobic attraction, which has been suggested is that electrostatic forces might be involved. However, it follows from a comparison between theory and experiment that attractive, classical double-layer forces are incon- sistent with the measured attraction.’ Furthermore, charge-fluctuation forces are too weak to be able to account for the hydrophobic
In this unsatisfactory situation as regards the theory of the hydrophobic attraction, we have considered it worthwhile to turn back to the mean-field theory of the repulsive hydration forces in an attempt to modify this theory so as to cover explicitly also the case of the hydrophobic attraction. Choosing such an approach seems even more appropriate for hydrophobic than for hydrophilic surfaces because a thoroughly hydro- phobic, smooth solid surface is likely to be more homogeneous on the molecular scale than a hydrophilic surface containing a certain number of ionic or polar groups. Furthermore, it can be argued that a correct mean-field theory should be capable of accounting at least for the long-range attraction forces recorded for water film thickness larger than a few nm. This kind of theoretical approach involves only short-range contact forces which are propagated by structural and bonding effects and there is no need to invoke long-range forces of some more or less evasive nature acting across the entire width of the water film.
Ordering Effects due to the Hydrophobic Surfaces
Several years ago Forslind** suggested to one of the present authors that one important feature characterizing the molecular constitution of the water/air interface might be that fragments of oxygen double layers, as defined by the ice Ih lattice, tend to orientate along the plane of the interface and that this is the main cause of the comparatively low surface entropy of water.2’ The molecular-dynamics simulations made by Lee et al.24 of ST2 water molecules between hydrophobic walls, ca. 20 8, apart, substantiate this view, as do the more recent molecular-dynamics studies of water clusters performed by Brodskaya and R ~ s a n o v ~ ~ and by Matsumoto and K a t a ~ k a . ~ ~
Lee et al. discussed their results employing the ideal ice Ih lattice as the point of departure. For the water molecules which are bonded in the oxygen double layers
Dow
nloa
ded
by U
nive
rsity
of
Illin
ois
at C
hica
go o
n 07
Mar
ch 2
013
Publ
ishe
d on
01
Janu
ary
1989
on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/F
2989
8500
163
View Article Online

J. C. Eriksson, S. Ljunggren and P. M. Claesson 165
orientated at right angles to the c axes, the bisectors of the HOH angles are directed either with 8 = 55" or with 8 = 125" from the surface normal. Thus, they could account for the bimodal 8 distribution found for the water molecules in the vicinity of the walls by assuming a preferential orientation of portions of (distorted) oxygen double layers parallel with the interface. As regards the first monolayers of water molecules, immedi- ately adjacent to the two hydrophobic walls, these simulations indicated an even more planar arrangement with a 8 distribution peaked around 90", i.e. implying a clear trend towards a strictly tangential alignment. This kind of structural reorganization of the water between hydrophobic surfaces results in a larger average number of hydrogen bonds per water molecule as compared with a corresponding slab of bulk water, particularly close to the walls. Important issues in the present context are how far this effect actually extends and to what degree the ST2 water model is capable of reproducing the very delicate free-energy balance that is likely to control the ordering process. The simulations of Lee et al. show that bulk properties would be attained in the centre of a 20 A thick water slab. This result is at variance, however, with the recent surface force measurements which indicate a much larger range of surface interaction.
Our theoretical description of the hydrophobic attraction forces to be presented below is based upon the structural features just discussed even though the calculations involved do not a priori contain any explicit assumptions about the molecular structure of the thin water film. Hence, we hypothesize that, relatively speaking, there is a significant free-energy decrease associated with the development of the first layer of water molecules which is well bonded in lateral directions. As water is an associated liquid there is a tendency to avoid the rupture of hydrogen bonds. This causes the surface-induced structure to be propagated (with a certain decay rate) towards the centre of the water film, resulting in an enhanced average number of hydrogen bonds per water molecule. Hence, assuming short-range interaction forces only, there is a free-energy increase arising in the core of the thin film owing to the imposed hydrogen-bond formation or, more precisely, to the concomitant entropy decrease that is supposed to predominate. The final (inhomogeneous) water state in the film reflects a balance between the favourable molecular reorganization occurring in the first (contact) layers of water molecules and the induced, unfavourable hydrogen-bonding effects in the remainder of the film. We shall treat the corresponding free-energy changes making use of a parameter s(x), which may tentatively be thought of as the local, relative increase of the average number of hydrogen bonds per molecule in the film as compared with bulk water or, alternatively, as the excess number of hydrogen bonds crossing a geometrical surface located at x and orientated parallel with the hydrophobic walls.
In an important paper from 1977, Marcelja et aL2' discussed the foundations of the kind of approach further developed here. However, their work was primarily directed towards modelling the properties of thin water films between hydrophilic (lecithin) surfaces that are heterogeneous on the (water) molecular scale, yielding rapidly decaying structural effects. The interaction between small hydrophobic solute molecules was treated similarly, resulting in short-ranged, attractive free-energy changes.
Theoretical We consider a planar water film of thickness h formed between two hydrophobic surfaces, each of area A, immersed in pure water. The two hydrophobic surfaces are assumed to be ideal, i.e. of a solid-like nature and without any residual charges or adsorbed surface-active compounds and, in addition, atomically smooth. The x coordinate is along the direction perpendicular to the two surfaces which are located at x = *h/2.
The excess Helmholtz free energy of the water film, as compared with an equally thick film made up of the surrounding bulk water ( i .e . without any surface effects), is
Dow
nloa
ded
by U
nive
rsity
of
Illin
ois
at C
hica
go o
n 07
Mar
ch 2
013
Publ
ishe
d on
01
Janu
ary
1989
on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/F
2989
8500
163
View Article Online

166 Long-range Hydrophobic Attraction
denoted by F and the corresponding excess number of moles of water is n. These excess film properties correspond exactly with the Gibbs excesses commonly employed for single surfaces. The Kramers function (or grand s1 potential) per unit surface area is then defined by
where p is the chemical potential of the water component and r = n / A. For convenience, we will call y t h e j l m tension, although it should be realized that the exact mechanical significance of y may be somewhat difficult to assess since we are dealing with two approaching solid /liquid interfaces.*’ For a fully equilibrated water film y is minimal at constant T and p.
Let us now imagine that the final equilibrium state of the water film, which is an open system, is reached in a step-wise fashion. Starting from the bulk state, the first step would involve the formation of the hydrocarbon/water interfaces (with associated local density changes) while retaining the average spherical-symmetric orientation of all the water molecules in the film. The second step would imply a change of the packing and the average orientations of the water molecules in each of the first molecular layers next to the two hydrophobic surfaces to yield a denser state with an increased number of hydrogen bonds and a preference for tangential alignment of the HOH bisectors of the water molecules. The film tension value obtained after these two equilibrization steps is denoted by yo. The third step is a reorganization of the hydrogen bonding pattern throughout the core of the film, whereby the parameter s(x) becomes a function of x and the final equilibrium film tension value y is attained.
We shall assume that the film tension, y, of the planar water film, can be expressed as
in a formalism that closely resembles the so-called ‘square-gradient approximation’. Here, so is the relative increase of the hydrogen-bond order parameter in the first layers of water molecules associated with the third equilibration step mentioned above. The function g(so) is the corresponding contact contribution to y with the property that dg/dso < 0 since the contact with the hydrophobic surfaces tends to promote hydrogen- bond formation. The integral expression included in eqn (2) is intended to account for the disadvantages (in the 0-potential sense) associated with the imposed hydrogen-bond ordering in the core of the water film and of the terminating chains of hydrogen-bonded water molecules.
In addition, the form of eqn (2) can be motivated on mathematical grounds similarly to those often inferred to explain the square-gradient approximation approach. Accord- ingly, the second term in the integrand may be thought of as an expansion in powers of s’(x) and higher derivatives, truncated after the s’(x)’ term. The necessary invariance to the choice of the direction of positive x requires that there be only even powers of s’(x). A term in s”(x) would lead to the same integrated excess free energy as a term proportional to ~ ‘ ( x ) ~ , as may be seen from integrating by parts. The mathematical calculations below indicate that a term like s’(x)* is necessary if s(x) is to become equal to zero at x = 0 for an infinitely thick film (ie. at an infinite distance from the hydrophobic surfaces). In fact, if this kind of term were missing the resulting Euler variational equation would only have the solution s = constant. From a physical point of view, the term ;CQ’(X)~ is also most essential since it accounts for the energetic coupling between successive layers of water molecules. In the simple model treated here, it will be assumed that the coupling coefficient c3 can be considered as a constant.
Dow
nloa
ded
by U
nive
rsity
of
Illin
ois
at C
hica
go o
n 07
Mar
ch 2
013
Publ
ishe
d on
01
Janu
ary
1989
on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/F
2989
8500
163
View Article Online

J. C. Eriksson, S. Ljunggren and P. M. Claesson 167
Finally, as a first ansatz we suppose that fo(s) can be approximated with sufficient accuracy by a second-degree polynomial in s, at least for low degrees of ordering:?
f o ( d = C d X ) + c2[s(x)12. (3)
This expression is also justified a posteriori from the agreement in functional form between experimental surface force curves with the theoretical expression derived below. The equilibrium condition requiring y to be minimal can now be written as
Sy dfo d2s c3 2 = 0.
S s ( x ) - d s dx (4)
If dfo/ds as derived from eqn (3) is inserted in eqn (4) the following differential equation emerges
d2s 2c2 C1 s (x) = - dx2 c3 c3
which in the case of two hydrophobic surfaces should be solved with the boundary conditions
s ( - h / 2 ) = S( h / 2 ) = SO. (6)
The solution satisfying these boundary conditions is easily seen to be
C1 cosh (bx) 2c2 cosh ( b h / 2 )
s (x) = --+so (7)
where we have introduced b = ( ~ C ~ / C , ) ~ ” . If, however, s ( x ) is to vanish at x = 0 for large values of h ( i e . far from the surface) it follows that c, must be zero and that c2 and c3 must have the same (positive) sign in order to avoid an oscillating behaviour of the function s(x) . Hence we obtain
cosh (6x) cosh (bh/2) ’
s(x) = so
This solution corresponds to a minimum in the film tension y according to Legendre’s condition because c3 is always a positive constant. By inserting this expression for s(x) in eqn (2) we obtain the following expression for the overall film tension
11 / 2
[ cosh2 (bx) + sinh’ ( b x ) J dx c,s:b’
’= Yofg(so)+2 cash' ( b h / 2 )
= yo+ g(so) + c3s;b tanh ( b h / 2 ) .
However, y should be a minimum with respect to varying so as well, i.e.
As a first approximation it seems reasonable to set dg/dso = -a, where a is a positive constant, i.e. to assume a linear-contact free-energy response function g(so) = -aso, resulting in
so = ( a /2c3b) coth ( b h / 2 ) . (11)
i A much more complicated form of the function corresponding to is necessary in the square-gradient theory of a liquid/vapour interface where the density varies beween the liquid-phase value and the gas-phase value.
Dow
nloa
ded
by U
nive
rsity
of
Illin
ois
at C
hica
go o
n 07
Mar
ch 2
013
Publ
ishe
d on
01
Janu
ary
1989
on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/F
2989
8500
163
View Article Online

168 Long-range Hydrophobic Attraction
h l n m
Fig. 1. The attractive surface force, F / R , plotted us. the surface separation in the case of hydrocarbon (DD0A)-covered mica surfaces. The points denote experimental values measured by Claesson and Christenson: (-) eqn (23) inserting B = 0.6 mJ mP2 and b-' = 15.8 nm; (. . a )
eqn (24) (so = constant) with B = B and (- - -) eqn (26) (second-degree contact response function) with B + C = 0.6 mJ m-*, C = 0.2 B and b-' = 15.8 nm. The full-drawn straight line denoted by R+ D represents the surface force function valid between h = 20 nm and h = 60 nm, which was obtained by Rabinovich and Derjaguin' using hydrophobic silica filaments immersed in lop4 mol dm-3 KCl solution. The corresponding experimental 1 values are scattered around this
line wiihin a factor of ca. 3.
When this expression is inserted into eqn (8) and (9) we
cosh ( b x ) s(x) = (z)
2c,b sinh ( b h / 2 )
obtain the equations
(12 )
and
y = yo- (a2 ; /4c3b) coth ( b h / 2 ) ,
Yo0 = Yo - (a"4c3b)
( 1 3 )
(14)
respectively. For infinitely large h we obtain from eqn ( 1 3 ) that
implying that a2/8c ,b is the reduction of the interfacial tension between water and the hydrophobic surface caused by the imposed ordering effect of the surface.
According to the Derjaguin a p p r o ~ i m a t i o n ~ ~ eqn ( 1 3 ) and (14 ) yield the following expression for the normalized force, F / R, between cylindrically shaped hydrophobic surfaces with radii equal to R, as measured in the surface force apparatus:
Dow
nloa
ded
by U
nive
rsity
of
Illin
ois
at C
hica
go o
n 07
Mar
ch 2
013
Publ
ishe
d on
01
Janu
ary
1989
on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/F
2989
8500
163
View Article Online

J. C. Eriksson, S. Ljunggren and P. M. Claesson
-10
-10; - I
E z 9 2
rr, .-. v
-lo3
-10'
h / n m
10 20 50 60 70 80
. * - I i";
169
Fig, 2. The attractive surface force, F / R , plotted us. the surface separation in the case of fluorocarbon ( F-surfactant)-covered mica surfaces. The points mark the values measured by Claesson and Christenson: (-) eqn (23) with B = 0.9 mJ m-? and b-' = 15.8 nm, (. - a ) eqn (24) (so = constant) with B = B and (- - -) eqn ( 2 6 ) (second-degree contact response function)
with B + C = 0.9 mJ m-I, C = 0.2 B and b-' = 15.8 nm.
Since coth ( b h / 2 ) 2 1 this surface force is always negative, i.e. attractive, and rapidly increasing in magnitude when h diminishes, in line with the experimental results (cf: fig. 1 and 2).
The interaction (disjoining) pressure between two planar hydrophobic plates, on the other hand, has thz following functional form:
- (5) [coth2 ( b h / 2 ) - 11 aY ah P N e - p = - - - -
where pN is the pressure tensor component perpendicular to the plane of the film and p e the exterior pressure.
It is instructive to consider in more detail the mechanisms behind eqn (15). The contact term g ( so) = -as, in eqn (9) and the film core contribution term c,s$ tanh ( b h / 2 ) are competing. Inserting so as given by eqn ( l l ) , it is seen that
a'
2 c d y - yo = -as,+ c ,s ib tanh (F) =- coth (F) ( - 1 +:)
as agrees, of course, with eqn (13). This implies that the contact term (-1) always yields a negative contribution to y twice as large in magnitude as the positive contribution from the core term (+:).
Dow
nloa
ded
by U
nive
rsity
of
Illin
ois
at C
hica
go o
n 07
Mar
ch 2
013
Publ
ishe
d on
01
Janu
ary
1989
on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/F
2989
8500
163
View Article Online
170
E t 2
2
- v
-10 -5
--loo0 +
0
x/nm
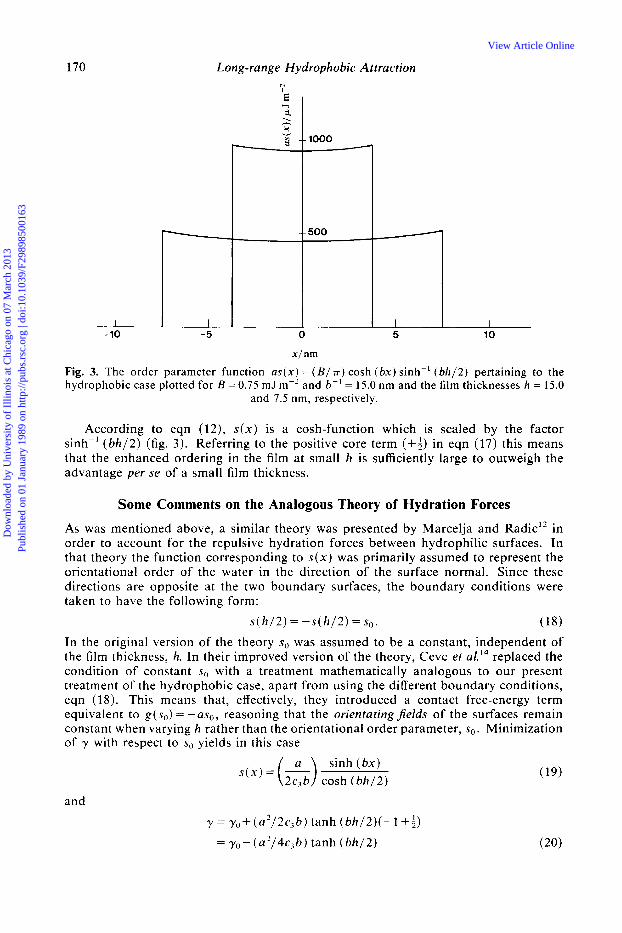
Fig. 3. The order parameter function as( x) = ( B / 7r) cosh ( b x ) sinh-’ ( b h / 2 ) pertaining to the hydrophobic case plotted for B = 0.75 mJ rnp2 and b-’ = 15.0 nm and the film thicknesses h = 15.0
and 7.5 nm, respectively.
According to eqn ( 1 2 ) , s(x) is a cosh-function which is scaled by the factor sinh-’ ( b h / 2 ) (fig. 3 ) . Referring to the positive core term (+$) in eqn ( 1 7 ) this means that the enhanced ordering in the film at small h is sufficiently large to outweigh the advantage per se of a small film thickness.
Some Comments on the Analogous Theory of Hydration Forces
As was mentioned above, a similar theory was presented by Marcelja and Radic’’ in order to account for the repulsive hydration forces between hydrophilic surfaces. In that theory the function corresponding to s(x) was primarily assumed to represent the orientational order of the water in the direction of the surface normal. Since these directions are opposite at the two boundary surfaces, the boundary conditions were taken to have the following form:
S ( h / 2 ) = - s ( h / 2 ) = SO. ( 1 8 ) In the original version of the theory so was assumed to be a constant, independent of the film thickness, h. In their improved version of the theory, Cevc et ~ 1 . ‘ ~ replaced the condition of constant so with a treatment mathematically analogous to our present treatment of the hydrophobic case, apart from using the different boundary conditions, eqn ( 1 8 ) . This means that, effectively, they introduced a contact free-energy term equivalent to g ( s o ) = -aso, reasoning that the orientating fields of the surfaces remain constant when varying h rather than the orientational order parameter, so. Minimization of y with respect to so yields in this case
sinh ( b x ) s(x) = (L)
2c,b cosh ( b h / 2 )
and
y = y o + ( a ’ / 2 c 3 h ) tanh ( b h / 2 ) ( - 1 + ; ) = yo- (a2 /4c ,b ) tanh ( b h / 2 )
Dow
nloa
ded
by U
nive
rsity
of
Illin
ois
at C
hica
go o
n 07
Mar
ch 2
013
Publ
ishe
d on
01
Janu
ary
1989
on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/F
2989
8500
163
View Article Online

J. C. Eriksson, S. Ljunggren and P. M. Claesson
c1 t I
171
Fig. 4. The order parameter function a s ( x > = ( B / 7 ~ ) sinh ( b x ) cosh-’ ( b h / 2 ) pertaining to the hydration force case plotted for B = 7.5 mJ rnP2 and b = 8.8 nm and the film thicknesses h = 8.8
and 4.4 nm, respectively.
whence
and, finally,
r a 2c3b
F I R = 2 7 ( y - 7tm) =- [ 1 -tanh (bh/2)]
which is the expression for the repulsive hydration force corresponding to eqn (15). Eqn (20) shows that the contact term ( - 1 ) is predominant in this case also. Thus,
the major cause of the hydration force would be the disturbance of the orientational order in each of the contact layers due to the approach of the other hydrophilic surface. This effect is only partially counteracted by the film core contribution term (+$).
The corresponding s(x) functions are displayed in fig. 4. These monotonic functions resemble only broadly the oscillatory polarization and dipole orientation profiles com- puted by Kjellander and Marcelja” for ST2 water between mineral surfaces ( h =z 29.3 A ) using the molecular-dynamics method, indicating that a mean-field theory 9f the present kind is somewhat unrealistic for water films with thicknesses <ca. 30 A, for which significant orientational ordering effects may be anticipated. Still, it may well be that it captures the chief mechanism leading to repulsive hydration forces, at least for atomically smooth and homogeneous polar surfaces.
Comparison with Experiment
For convenience we introduce the notation B = .rra2/2c3b so that eqn (15) can be rewritten as
-F /R=B[coth (bh/2)-11. (23) For sufficiently large h values, the right-hand side of this equation tends to 2 B exp ( - bh) , i.e. in this range we predict In ( - F / R ) to be a linear function of h. Provided this actually
Dow
nloa
ded
by U
nive
rsity
of
Illin
ois
at C
hica
go o
n 07
Mar
ch 2
013
Publ
ishe
d on
01
Janu
ary
1989
on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/F
2989
8500
163
View Article Online

172 Long-range Hydrophobic Attraction
turns out to be the case, the constants 6 and B are easily determined from the experimental In ( - F / R ) us. h plot.
Fig. 1 and 2 show the theoretical curves obtained in this way for mica substrates made hydrophobic with the double-chain cationics dimethyldioctadecylammonium (DDOA) bromide and N - ( a-trimethylammonioacety1)-0, 0'-bis-( 1 H,l H,2H,2H-per- fluorodecy1)-L-glutamate chloride (F-surfactant), respectively, together with the corre- sponding experimental points determined by Claesson and Chr i~ tenson .~ In fig. 1 we have also marked the linear log ( - F / R ) us. h function valid for h > 20 nm derived by Rabinovich and Derjaguin6 on the basis of their recent surface force measurements using silica filaments, made hydrophobic by reaction with dimethyldichlorosilane, immersed in mol cm-3 KCl solution. In general, the agreement is satisfactory except, perhaps, for surface separations < ca. 5 nm where, however, the experimental accuracy is worse. Accordingly, within the larger part of the entire range, the attractive surface force between hydrophobic surfaces immersed in water seems to be well described by eqn (23). For comparison we have included functions of the type
- F / R = i[ 1 - tanh (&h/2)] in fig. 1 and 2, where denotes the constant 2nc3s5b. These surface force functions were obtained by assuming the boundary conditions so = constant to be fulfilled. Obviously, the corresponding curves fail to reproduce the characteristic amplification of the attraction observed for small surface separations. In addition, we have explored the effect of including a quadratic term in the contact free-energy contribution, i e .
When C/2<< B, where C = n ~ a ~ / 2 ( c , b ) ~ , this assumption yields the following surface force equation:
For large values of h we obtain from eqn (26) that
-g(so) = as ,+ t s s i . (25)
- F / R ={B+iC[coth (6h/2) + l]}[coth (bh/2) - 13.
In ( - F / R ) = In [2( B + C ) ] - bh.
(26)
(27) It appears from fig. 1 and 2 that a somewhat improved agreement with the experimental data can be generated in this way at small film thicknesses. This supports the view that the effect causing the attractive surface force is of a cooperative nature.
It is interesting to note that the overall film tension change associated with the molecular reorganization can readily be estimated by means of eqn (13), from which we have that
(28) Since B is found to be somewhat below 1 mN m-' for the cases considered, it appears that the entire gain in film tension due to the molecular reorganization is quite small, ca. 0.1-1.0 mJ m-*, i.e. only ca. O. l - l .Oo/~ of the total film tension (ie. ca. 100 mJ m-2) for thicknesses between ca. 600 and 30 A, respectively.
According to the present theory, the strength of the hydrophobic attraction is geared by the interaction constant B = ~ a ' / J ( 8 ~ 2 ~ 3 ) . Thus, it would be enhanced by a strong free-energy response to so (i.e. to the degree of hydrogen bonding in the layers of water molecules next to the hydrophobic surfaces) and attenuated by strong responses to ordering and decoupling effects in the core of the film.
As regards the decay length 6-' = J( c3/2c2), a long-ranged attractive force would be favoured by a weak free-energy response to increasing the hydrogen-bond parameter s(x) (i.e. a small constant c2 and a strong tendency to avoid s(x)-gradients, i.e. a large value of c3).
Thus, this theory predicts ( i ) that the decay length b-' should be the same for all hydrophobic surfaces under identical solution conditions and (ii) that B should increase
70- y = ( B / ~ T ) coth (6h/2).
Dow
nloa
ded
by U
nive
rsity
of
Illin
ois
at C
hica
go o
n 07
Mar
ch 2
013
Publ
ishe
d on
01
Janu
ary
1989
on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/F
2989
8500
163
View Article Online

J. C. Eriksson, S. Ljunggren and P. M. Claesson 173
with the degree of hydrophobicity of the surfaces. Within the limits of error, this is, indeed, consistent with the experimental findings (fig. 1 and 2). Furthermore, our theory implies that if the molecular mechanism which propagates the (dynamic) water structure is disturbed by ions or other solutes, causing c3 to diminish but c2 to remain essentially unchanged, one would expect a shorter decay length and, simultanteously, a larger constant B. Our recent studies on the effect of adding tetrapentylammoniumbromide using fluorocarbon surfaces’ are in agreement with this, but further experiments are needed to check whether this effect is caused solely by the presence of the salt in the solution or if it is due in part also to ion adsorption on the surfaces, making them less hydrophobic.
Note also that some recent experiments show that the hydrophobic attraction depends strongly on the detailed state of the hydrophobic mica surface^.'^' Thus, surfaces which are free of residual charges but are less densely packed with hydrocarbon or fluorocarbon chains seem to generate much weaker attraction forces than those with thicker hydro- carbon or fluorocarbon layers. This might be attributed to the critical importance of a smooth and chemically homogeneous hydrophobic substrate surface which promotes the formation of the first contact layer of water molecules in such a way that the constant a (and hence B) becomes comparatively large.
The Effect of Curvature on the Interfacial Tension of Hydrophobic Solute Particles
The same interactibis which appear in thin planar films are presumably of a major importance also for the theory of the curvature dependence of hydrophobic par- ticle/water interfacial tensions. Marcelja et aL2’ have previously treated this problem, assuming, however, so to be independent of the radius R of the particle. Assuming, instead, that so varies with R, and carrying out a minimization of the overall excess free energy with respect to so, it is found that so is strongly dependent upon R, particularly for small values of R, and the following expressions for the interfacial tension 7 emerge:
B 1 n l + b R
j L j&+- - (sphere)
where K O and K , are modified Bessel functions of the second kind. These expressions are valid insofar as the same contact response function g(so) = -aso holds at all particle radii.
Both for the sphere and the cylinder 7 = qa+ B/ n at small values of R, i.e. when R<< b-’. However, the entire curvature effect is minute since B / T may amount to 0.2 mJ m-’, whereas the interfacial tension q is ca. 50 mJ m-2.
Discussion In this paper we have advanced a mean-field theory of the hydrophobic attraction which is based on the notion of enhanced hydrogen bonding for the water molecules close to hydrophobic surfaces. It is conceptually related to the theories of hydration forces proposed by Marcelja and Radic12 and Cevc et all4 According to our theory, an exponential surface force us. h relationship is predicted for large values of h, in good agreement with recent experimental results. ’-’”’ More importantly, the amplification effect observed for small values of h is also reproduced accurately, particularly if a second-degree contact free-energy response function is assumed.
Dow
nloa
ded
by U
nive
rsity
of
Illin
ois
at C
hica
go o
n 07
Mar
ch 2
013
Publ
ishe
d on
01
Janu
ary
1989
on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/F
2989
8500
163
View Article Online

174 Long-range Hydrophobic Attraction
0.1 1 0.08 1
h
0
0.06
0.04 i 0.02
v
20 40 60 80 100 h/nm
Fig. 5. The order parameter in the centre of the film s(x = 0) 1 ( B / m ) sinh-’ ( b h / 2 ) plotted us. the film thickness h, with B = 0.6 mJ m-2, b-’ = 15.8 nm and a = 65.0 mJ rn-’ [estimated from
ref. ( 2 4 ) inserting s(x = 0.6 nm) = 0.05 for h = 18.6 nm].
The ordering process modelled by this variational approach is supposed to be controlled by a delicate balance between the free-energy gain for the water molecules next to the hydrophobic walls as a result of the structural reorganization and an associated free-energy expenditure for the water molecules in the core of the thin film due to the imposed hydrogen-bond ordering. The molecular-dynamics simulations of Lee et ~ l . * ~ indicate a substantial orientational ordering effect only in the first few layers of water molecules next to the walls, and they also show a rapidly decaying excess of the average number of hydrogen bonds per water molecule when receding from the walls. It appears natural to associate the hydrophobic attraction with the latter property because, accord- ing to the simulations, the excess number of hydrogen bonds varies somewhat similarly as a cosh function. On closer inspection, however, one finds that full agreement is lacking between the ST2 M C simulations and the present investigation, since it follows from combining eqn (11) with the experimental surface force results that s(x) would decay much more slowly upon approaching the centre of the film than was actually found through the simulations ( c j fig. 3) .
The excess free energy per molecule related to the hydrophobic attraction is very small, typically only 10-3-10-5 k T 3 whereas the energy of a hydrogen bond is ca. 7 kT at room temperature. As to the magnitude of the effect, a similar picture emerges upon computing the order parameter in the centre of the film s(x = 0) = ( B / n a ) sinh-’ ( b h / 2 ) [eqn (12)] through estimating the constant a on basis of the simulation results of Lee et al. close to the hydrophobic walls (fig. 5). Accordingly, s(x = 0) would be ca. lo-’ for a 10 nm thick film and ca. for a 60 nm thick film. Toward this background one realizes that we are concerned with ordering effects which may not easily be captured by molecular-dynamics calculations for comparatively thick films using the ST2 water model. In particular, we might argue that only a water model that can correctly account for the phase transition from ice to liquid water would be worthwhile trying when it comes to modelling what has become known as ‘typical aqueous behaviour’, which implies that comparatively large structural reorganizations are possible at low free-energy costs. Like other similar models, the ST2 water model is clearly unsatisfactory in this respect.3‘
Dow
nloa
ded
by U
nive
rsity
of
Illin
ois
at C
hica
go o
n 07
Mar
ch 2
013
Publ
ishe
d on
01
Janu
ary
1989
on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/F
2989
8500
163
View Article Online

J. C. Eriksson, S. Ljunggren and P. M. Claesson 175
A complete molecular theory should, of course, also yield values of the interaction constant B as well as of the decay length b-' . Hence, it is evident that the theory presented in this paper is only a partial, mainly phenomenological description of the long-range and still rather puzzling hydrophobic attraction. In order to make further progress, additional experiments are called for, especially on the effects of varying the temperature, adding salts and other solutes and switching to other hydrogen-bonded liquids.
Finally, we should add a few comments on the alternative way proposed of accounting for the hydrophobic attraction which is based on the idea that a thin water film between hydrophobic walls is in some metastable precursor state to a vapour cavity. Somewhat vaguely, this proposition assumes that thermodynamically important density fluctuations are involved, resulting in progressively lower values of the film tension upon diminishing the film thickness. However, there is so far no experimental (or computational) evidence in favour of a significantly reduced density in the centre of thin water films and, furthermore, large fluctuations are anticipated to occur only close to a critical point. The vapour cavity formation seems rather to have the nature of a regular first-order transition, similar to the formation of a capillary condensate between wetting walls. Normally, it is hindered by a large free-energy barrier, except very close to molecular contact between two curved hydrophobic surfaces.I8 However, in a rather broad sense, we can reach agreement also with these preliminary notions by reasoning as follows. The surprisingly long range of the hydrophobic attraction would, according to the present theory, be due to a hydrogen-bond-mediated structural reorganization in the core of the water film that takes place at a minimal free-energy cost. Hence, considering a small column of water stretching between the two hydrophobic surfaces we will have to envisage that the water contained in this volume fluctuates in its structural state because the concomitant y changes are relatively minute. Such a dynamic ordering- disordering process would necessarily be associated with some density fluctuations, however minor. This points in the direction that we would need to know more about the molecular properties of liquid water and, in particular perhaps, about the conditions which favour the formation of elongated hydrogen-bond-connected clusters of water molecules, before a full understanding of the hydrophobic attraction is within reach.
We are most grateful to Hugo Christenson and Roland Kjellander for valuable comments on the present work.
References
1 J. N. Israelachvili and R. M. Pashley, Nature (London), 1982, 300, 341; J. Colloid Interface Sci., 1984, 98, 500.
2 R. M. Pashley, P. M. McGuiggan, B. W. Ninham and D. F. Evans, Science, 1985, 229, 1088. 3 P. M. Claesson, 77iesis (Stockholm, 1986). 4 P. M. Claesson, C . E. Blom, P. C . Herder and B. W. Ninham, J. Colloid Interface Sci., 1986, 114, 234. 5 H. K. Christenson, P. M. Claesson and P. M. Pashley, Proc. Indian Acad. Sci. (Chern. Sci.), 1987,98,379. 6 Ya. I . Rabinovich and B. V. Derjaguir., Colloids Sur-f, 1988, 30, 243. 7 P. M. Claesson and H. K. Christenson, J. Phj*s. Chem., 1988, 92, 1650. 8 P. C. Herder, J. Colloid Interface Sci., submitted. 9 H. K. Christenson, P. M. Claesson, P. C. Herder and J.: Berg, J. Phvs. Chem., in press.
10 Hj-dration Forces and Molecular Aspects of Solvation, Orenas, Sweden, 1984, ed. Bo Jonsson, Chem. Scr., 1985, 25.
1 1 J. Israelachvili, Acc. C'hern. Rex, 1987, 20, 415. 12 S. Marcelja and N. Radic, Chern. Phys. Lett., 1976, 42, 129. 13 B. V. Derjaguin and N. V. Churaev, Frog. SurJ Mernbr. Sci., 1981, 14, 69; Langrnuir, 1987, 3, 607. 14 G. Cevc, R. Podgornik and B. Zeks, Chern. Phys. Lett. 1982, 91, 193. 15 N. Ostrovsky and D. Sornette, Chem. Scr., 1985, 25, 108. 16 Ya. I . Rabinovich, B. V. Derjaguin and N. V. Churaev, Adc. Colloid Interface Sci., 1982, 16, 63. 17 V. V. Yaminsky, V. S. Yushchenko, E. A. Amefina and E. D. Shchukin, J. Colloid Inrefface Sci., 1983,
96, 301.
Dow
nloa
ded
by U
nive
rsity
of
Illin
ois
at C
hica
go o
n 07
Mar
ch 2
013
Publ
ishe
d on
01
Janu
ary
1989
on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/F
2989
8500
163
View Article Online

176 Long-range Hydrophobic Attraction
18 V. S. Yushchenko, V. V. Yaminsky and E. D. Shchukin, J. Colloid Interface Sci., 1983, 96 307. 19 H. K. Christenson and P. M. Claesson, Science, 1988, 239, 390. 20 P. Attard, R. Kjellander and D. J. Mitchell, Chern. Phys. Letr., 1987, 139, 219; P. Attard, R. Kjellander,
D. J. Mitchell and B. Jonsson, J. Chern fhys . , in press. 21 R. Kjellander and S. Marcelja, Chern. Phys. Lett., 1987, 142, 485. 22 E. Forslind, Acta folytechn. Scand., 1952, 115, 9. 23 K. Johansson and J. C. Eriksson, J. Colloid Interface Sci., 1974, 49, 469. 24 C. Y. Lee, J. A. McCammon and P. J . Rossky, J. Chern. Phys., 1984, 80, 4448. 25 E. N. Brodskaya and A. I . Rusanov, Mol. Phys., 1987, 62, 251. 26 M. Matsumoto and Y. Kataoka, J. Chern. Phys., 1988, 88, 3233. 27 S. Marcelja, D. J. Mitchell, B. W. Ninham and M. J. Sculley, J. Chern. Soc., Faraday Trans. 2, 1977,
28 J . C. Eriksson, Surj Sci., 1969, 14, 221. 29 B. V. Derjaguin, Kolloid Z., 1934, 69, 155. 30 R. Kjellander and S. Marcelja, Chern. Scr., 1985 25, 7 3 . 31 J . N. Israelachvili, Interrnolecular and Surface Forces (Academic Press, New York, 1985), pp. 99.
73, 630.
Paper 8/01628C; Received 25th April, 1988
Dow
nloa
ded
by U
nive
rsity
of
Illin
ois
at C
hica
go o
n 07
Mar
ch 2
013
Publ
ishe
d on
01
Janu
ary
1989
on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/F
2989
8500
163
View Article Online





























