Embed Size (px)
Citation preview

C H A P T E R T W E N T Y - F O U R
☆
*
{
M
IS
ThisapprchapAmstDe BKluyvVU U
ethods
SN 0
A Practical Guide to Genome-Scale
Metabolic Models and Their
Analysis☆
Filipe Santos,*,† Joost Boele,* and Bas Teusink*,†
Contents
1. In
chaoacterserdoelerniv
in
076
troduction
pter and Chapter 4 in Section 7 are focused on large-scale metabolic model reconstrhes and viewpoints of the authors differ, and the editors would advise readers to cotogether for a broader understanding of the methodologies and issues within this fieldam Institute for Molecules, Medicines and Systems/NISB, VU Universityelaan 1085, Amsterdam, The NetherlandsCentre for Genomics of Industrial Fermentations/Netherlands Consortium Systeersity Amsterdam, De Boelelaan 1085, Amsterdam, The Netherlands
Enzymology, Volume 500 # 2011
-6879, DOI: 10.1016/B978-0-12-385118-5.00024-4 All rig
uctnsi.Am
ms
Else
hts
510
2. G
enome-Scale Metabolic Models: Their Place in the Spectrum ofModeling Options
5142
.1. B ottom-up (kinetic) models versus genome-scalemetabolic models
5142
.2. T op-down (biostatistical) models versus genome-scalemetabolic models
5183. T
he Art of Making Genome-Scale Metabolic Models 5203
.1. G enerating a draft reconstruction based on thegenome sequence
5203
.2. Id entify and resolve errors, gaps, and inconsistenciesin the network
5213
.3. D efine external metabolites and the biomass equation 5213
.4. V alidate the model with additional experiments 5234. A
pplications of Genome-Scale Metabolic Models 5234
.1. F lux balance analysis: The work horse of constraintbased modeling
5244
.2. F BA predicts rates only through yield maximization 5254
.3. U sing constraint-based modeling for discovery andinterpretation: Sensitivity analysis
5274
.4. F inal remarks 528Refe
rences 528ion. Theder both
sterdam,
Biology,
vier Inc.
reserved.
509

510 Filipe Santos et al.
Abstract
Genome-scale metabolic reconstructions and their analysis with constraint-
based modeling techniques have gained enormous momentum. It is a natural
next step after sequencing of a genome, as a technique that links top-down
systems biology analyses at genome scale with bottom-up systems biology
modeling scrutiny. This chapter aims at (systems) biologists that have an
interest in, but no extensive knowledge of, applying genome-scale metabolic
reconstruction and modeling to their organism. Rather than being comprehen-
sive—excellent and extensive reviews exist on every aspect of this field—we
give a rather personal account on our experience with the process of recon-
struction and modeling. First, we place genome-scale metabolic models in the
spectrum of modeling approaches, and rather extensively discuss, for nonex-
perts, the central concept in constraint-based modeling: the solution space that
is bounded through constraints on fluxes. We subsequently provide an over-
view of the different steps involved in metabolic reconstruction and modeling,
pointing to aspects that we found difficult, important, not well enough
addressed in the current reviews, or any combination thereof. In this way, we
hope that this chapter serves as a practical guide through the field.
1. Introduction
Today’s interest in systems biology is largely fuelled by high-throughputtechniques that generate large amounts of data. There is a general consensusthat functional genomics has enormous potential in the life sciences, in partic-ular, in biotechnology and medicine. How to use these technologies mostefficiently, for fundamental understanding, biomarker discovery, or concretebiotech applications, is an area of active research. It is clear that the volume andcomplexity of the data are becoming too large to copewith by biologists alone,especially when the latter are poorly trained in advanced mathematics andcomputation (which is still largely the case). So there is an understandable needfrom the biologist’s perspective for help in mining, interpreting, and using thedatasets that they collect. Such activities require modeling of one form or theother (Ideker and Lauffenburger, 2003).
Biostatistics and bioinformatics offer help in the analysis of genome-scaledata sets, but they often rely on purely mathematical and statistical analysis.Although extremely useful, it ignoreswhat is often referred to as “legacy data,”that is, the large body of biological knowledge that is often scattered inliterature and therefore poorly accessible. Moreover, many of the techniqueswere not designed to incorporate a priori knowledge, even if it is available (Liaoet al., 2003). “Bottom-up” systems biologists, however, construct detailedmechanistic models that aim at a fundamental understanding of systems behav-ior (Bruggeman and Westerhoff, 2007). To achieve this, they start from themolecular properties of biological components and their interactions in

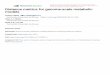
BOX 24.1 “Beginner’s Kit” for Genome-Scale Metabolic Model Reconstructionand Their Analysis
The first step of model development is generally obtaining a draft model.
Multiple tools can be used for this with different input demands. These can be
from only an assembled genome (The SEED) to the combination of multiple
genome sequences and respective curated models (AUTOGRAPH). Again
depending on the algorithms employed in the initial reconstruction, the list of
gene-reaction associations needs to be checked for consistency, gaps, and
errors, and also attached to the list of external metabolites and the biomass
equation. The latter two generally are obtained either from literature or from
experimental data. This first (almost) “growing”model then enters the refine-
ment stage in which the outputs of a succession of constraint-based analysis
techniques available within various software packages (see Table 24.1) are
compared with both literature and experimental data. For more detail, please
see the main text and/or references to external resources (Fig. 24.1).
(draft)
Model
Available modelsGenome
Model simulations
Literature andexperimental data
Modelreconstruction
Assessmentof model
performance
Manualcuration
Figure 24.1 Overview of draft genome-scale metabolic model generation and itera-tive refinement cycle.
Genome-Scale Metabolic Models 511
biological networks, be it metabolic, signaling, or gene regulatory, amongstothers. As will be briefly discussed below, both approaches have their limita-tions. Using genome-scale reconstructions, and their corresponding models,may be considered as a “middle-out” approach, as they combine -omics datawith more traditional modeling strategies. This approach is the focus of thischapter, in particular, its application to metabolic networks.
All aspects of genome-scale metabolic models have been extensivelyreviewed in recent years (Feist and Palsson, 2010; Feist et al., 2009; Hyduke

Table 24.1 Selected list of external resources useful for the generation of genome-scale metabolic model and their analysis
Tool/data source URL/reference Description
AUTOGRAPH www.
biomedcentral.
com/1471-2105/
7/296
AUTOGRAPH is a semi-automatic
approach to accelerate the process
of genome-scale metabolic
network reconstruction by taking
full advantage of already manually
curated networks
BiGG bigg.ucsd.edu BiGG is a knowledge base of
biochemically, genetically, and
genomically structured genome-
scale metabolic network
reconstructions
BioCyc biocyc.org BioCyc is a collection of >1000
pathway/genome databases. Each
database in the BioCyc collection
describes the genome and
metabolic pathways of a single
organism
BioMet
Toolbox
129.16.106.142 The BioMet ToolBox is a web-based
resource for analysis of high-
throughput data, together with
methods for flux analysis
(fluxomics) and integration of
transcriptome data exploiting the
capabilities of metabolic networks
described in genome scale models
BRENDA www.brenda-
enzymes.org
BRENDA is the main collection of
enzyme functional data available to
the scientific community.
COBRA gcrg.ucsd.edu/
Downloads/
Cobra_Toolbox
The COBRA Toolbox for Matlab
includes implementations of many
of the commonly used forms of
constraint-based analysis such as
FBA, gene deletions, flux
variability analysis, sampling, and
batch simulations together with
tools to read in and manipulate
constraint-based models
KEGG www.genome.jp/
kegg/
KEGG (Kyoto Encyclopedia of
Genes and Genomes) is a
bioinformatics resource, popular
for its visualization capabilities,
that links genomes to pathways
512 Filipe Santos et al.

Table 24.1 (Continued)
Tool/data source URL/reference Description
OptFlux www.optflux.org OptFlux incorporates strain
optimization tasks and also allows
the use of stoichiometric metabolic
models for (i) phenotype
simulation of both wild-type and
mutant organisms, (ii) metabolic
flux analysis, and (iii) pathway
analysis through the calculation of
elementary flux modes
Pathway Tools bioinformatics.ai.sri.
com/ptools
Pathway Tools is a comprehensive
symbolic systems biology software
system that supports several use
cases in bioinformatics and systems
biology
PubMed www.ncbi.nlm.nih.
gov/pubmed
PubMed is a service of the U.S.
National Library of Medicine that
includes over 19 million citations
from MEDLINE and other life
science journals for biomedical
articles back to the 1950s
The SEED www.theseed.org TheModel SEED automates as much
as possible the development of
genome-scale metabolic models
requiring from the user only an
assembled genome sequence
YANAsquare yana.bioapps.
biozentrum.uni-
wuerzburg.de
YANA is a user friendly software
package for the analysis of
metabolic networks
INSD http://www.insdc.
org/
The International Nucleotide
Sequence Databases (INSD)
consists of an international
collaboration between GenBank,
DDBJ, and ENA resulting in the
largest sequence repository
Genome-Scale Metabolic Models 513
and Palsson, 2010; Liu et al., 2010; Oberhardt et al., 2009; Orth et al., 2010;Teusink and Smid, 2006). In this chapter, therefore, we will try to guide thereader, whom we envisage is a biologist relatively new to systems biology ingeneral, and to genome-scale metabolic models, in particular, through thevarious steps of the modeling process. The account on the many steps

514 Filipe Santos et al.
involved is necessarily not comprehensive, and rather personal, based on ourexperience of the process. It will hopefully help to understand the basicconcepts and rationales, while providing many references to other resourceswhere more detailed information can be pursued. We also provide a sort of“beginner’s kit” of software that we found useful at different stages in thereconstruction and modeling process (see Box 24.1).
2. Genome-Scale Metabolic Models: Their Place
in the Spectrum of Modeling Options
As seen above, two opposing strategies have been generally employedto model biological systems. The so-called bottom-up approach focusesmostly on the very detailed description of the different components of thesystem, while the “top-down” approach looks for (uneducated) correlationsbetween different variables of the system. Genome-scale metabolic modelscan be seen as a “middle-out” strategy because they first delimit thegenomic pool of the system, mainly through bioinformatic approaches,and from there model the potential interactions of these components.
It should be clear that all modeling strategies are good for specific tasksbut have limited capabilities for others, and so it is important to understandthe possibilities, limitations, and underlying assumptions before embarkingon any of them. The direct comparison of genome-scale metabolic modelswith other modeling approaches places them in the spectrum of modelingoptions and helps understand their value.
2.1. Bottom-up (kinetic) models versus genome-scalemetabolic models
The structure of a kinetic model is actually quite simple. Suppose, we have asimple system with two branches (Fig. 24.2).
We often assume an environment in which S is some infinite sourcewith a constant concentration (often referred to as an external metabolite;Heinrich and Schuster, 1996; Schuster et al., 2000), for example, glucose inthe bloodstream or a nutrient in a chemostat. Similarly, P1 and P2, theproducts of this pathway, have constant concentrations and are also externalmetabolites. X is an internal metabolite, the concentration of whichdepends on the rates of production and consumption. In mathematicalterms, this can be described as:
dX
dt¼ v1 � v2 � v3 ð24:1Þ

SV1
V2
V3
X
P1
P2
Figure 24.2 Simple metabolic network consisting of three reactions, three externalmetabolites (S, P1, and P2) that act as source or sink, and one internal metabolite (X).
Genome-Scale Metabolic Models 515
The rates of the enzymes, vi, are a function (ƒ) of the kinetic parametersp (e.g., Michaelis–Menten constants Vmax and Km values) and concentra-tions of the metabolites, in this case:
vi ¼ ƒ p;X ; S;P1;P2ð Þ ð24:2Þ
Thus, Eq. (24.1) is a differential equation, the solution of which is afunction that describes the time course of the concentration of X, that is,
X tð Þ ¼ ƒ X0;p; S; P1; P2ð Þ ð24:3Þ
The X0 indicates that the actual behavior is dependent on the initialconcentration of X. As the rates depend on p, S, P1, and P2, so does X.IfX is computed at each time point, Eq. (24.2) can be solved to find the ratesof all the enzymes in time, that is, vi(t). Thus, such kinetic models can beconstructed and validated through time-series of metabolites and fluxmeasurements.
Detailed kinetic models can be used to explore the dynamics of a system,as well as the control structure of a pathway (Fell, 1997). Once a model isavailable, parameters and conditions (such as the concentration of S in ourexample) can be altered to study biologically relevant properties, such asbistabilities (Veening et al., 2008), oscillations (Goldbeter, 1997), androbustness or homeostasis (Kitano, 2002, 2007), just to mention a few. Ina practical sense, this understanding makes it possible to predict which stepsto enhance, or which feedbacks to remove, for a particular modification ofthe pathway’s behavior (e.g., enhanced production of a valuable product orimproving a diseased state). Even though there are examples of successfulapplications of these types of kinetic models (Bakker et al., 2010; Hoefnagelet al., 2002), they face some serious limitations.
First, kinetic parameters p of all enzymes in a pathway are very rarelyavailable, although databases such as BRENDA (Barthelmes et al., 2007)and Sabio-RK (Rojas et al., 2007) are a tremendous help. However, thesekinetic parameters are often measured at different or nonphysiological

516 Filipe Santos et al.
conditions, such as at the optimal pH of the enzymes, not the physiologi-cal pH. Also, it remains quite controversial to what extent the in vitrokinetics reflects the in vivo kinetics (Teusink et al., 2000). An alternative tothe in vitro kinetic parameter determinations is to estimate “in vivo kineticparameters” from time courses of metabolites after short timescale pertur-bations of the metabolic network (Mashego et al., 2007; Theobald et al.,1993; Visser et al., 2004). Parameter estimation is a field by itself and notan easy task. We refer to a recent review on the topic (Banga and Balsa-Canto, 2008).
Second, these models necessarily represent relatively small, isolatedmetabolic pathways. As these pathways are embedded in a large metabolicnetwork, the boundary conditions, that is, the exchanges of informationwith the rest of the system, become critically important in the success ofthe model predictions. It was shown that including the boundaries explic-itly, even with uncertain parameter values, can improve significantly thepredictive power of the model outcome (Liebermeister et al., 2005).Several approaches, in fact, have been described that take the uncertaintyin kinetic parameters explicitly into account, generating an ensemble ofmodel outcomes that can be inspected for robust and more uncertainmodel predictions (Liebermeister and Klipp, 2005, 2006; Steuer et al.,2006; Wang and Hatzimanikatis, 2006). Eventually such new approaches,together with further developments in metabolomics research and com-puting power, may substantially increase the size of systems for whichkinetic models can be constructed by reverse engineering ( Jamshidi andPalsson, 2010; Resendis-Antonio, 2009). However, at the moment theseapproaches scale poorly, and genome sizes are still a long way ahead.
Genome-scale metabolic models, however, do cover the total meta-bolic potential that is encoded in the genome of an organism, but thiscomes at a cost. They share with kinetic models the structure, that is, thestoichiometry of all reactions, but they leave out (almost) all of the kineticdetails. This is possible because many pathways, such as the one depictedin Fig. 24.2, when analyzed over a sufficiently long time will reach asteady state, that is, a state in which all internal metabolites will bebalanced by the producing and consuming reactions. In our example,this means that, in steady state:
dX
dt¼ v1 � v2 � v3 ¼ 0 ð24:4Þ
Equation (24.4) basically states that the concentration of X is constant intime, as it is balanced by its production and consumption rate. When thekinetics of the reactions are known, we can compute the steady stateconcentration of X, and the steady-state rates through the enzymes, whichwe then, in steady state, call fluxes:

Genome-Scale Metabolic Models 517
X t ! 1ð Þ ¼ Xss ¼ ƒ p; S;P1; P2ð Þ ð24:5aÞVi;ss ¼ ƒ Xss; p; S; P1; P2ð Þ ð24:5bÞ
Equation (24.4) leads to a set of linear equations that link the rates viwitheach other, which does not require the parameters p, S, P1, and P2.However, to truly predict the rates vi or the steady-state metabolite concen-tration from the conditions defined by S, P1, and P2, one does need thiskinetic information, as is specified in Eqs. (24.5a) and (24.5b). Via Eq. (24.4),we only look at steady-state stoichiometric interrelationships between(steady-state) fluxes, nothing more. This difference with kinetic modelingis absolutely crucial for understanding the limitations of the analyses ofgenome-scale metabolic models discussed later on. Imposing steady-staterelationships between rates (referred to as mass-balance constraints (Priceet al., 2004), for obvious reasons) constrains the possible states the system canbe in only with respect to fluxes through the network, also referred to as thesolution space (illustrated in Fig. 24.3).
The other type of constraints used in genome-scale metabolic modelingis called the capacity constraint. Such constraints also have a link withkinetics, as they represent any limitation that can be imposed on individualrates. Such can be measured Vmax values that form an upper limit through
v1
v2
v3
v1
v2
v3A B
Figure 24.3 Illustration of the concept of solution space, and how mass-balance andcapacity constraints result in a bounded space of possible flux states. (A) The steady-state solution space is two-dimensional (a plane), because there are three unknowns(three fluxes) and one equation: the mass balance of X, that is, Eq. (24.4). This planestretches out to all directions but is depicted only in the positive quadrant for compari-son with Fig. 24.3B. The arrows represent “extreme pathways,” basis vectors that spanthe solution space, see Papin et al. (2004) for a review on the topic. (B) Specific capacityconstraints on the fluxes v1, v2, and v3 turn the plane into a bounded space within whichall feasible flux states should lie. For each flux, we have set capacity constraints,0 � vi � vi,max.

518 Filipe Santos et al.
the reaction catalyzed by the enzyme. More often, it contains a constrainton the directionality of a reaction, that is, the assessment whether a reactioncan work in both directions under the physiological range of metaboliteconcentrations that is assumed (or sometimes measured) in the organismunder study (Hoppe et al., 2007; Kummel et al., 2006a,b). Mass balance andcapacity constraints together limit the solution space of all possible fluxdistributions (Fig. 24.3).
The analysis of genome-scale metabolic models, applying mass-balanceand capacity constraints, is collectively named constraint-based modeling.Note that a detailed kinetic model will predict a specific steady state withfluxes that lie inside the solution space of the corresponding stoichiometricnetwork (Teusink and Smid, 2006). Leaving out the kinetic details inconstraint-based modeling comes at the cost of being able to predict thespace of feasible states but not the specific state (unless specific assumptionsare made that will be outlined later). Also information about the sensitivityof the steady state to parameters changes, as well as the trajectory how thisstate is reached, is not available through stoichiometry alone.
2.2. Top-down (biostatistical) models versus genome-scalemetabolic models
Despite the -omics data explosion, it is clear that for true systems-levelunderstanding, the abundance of relevant data, unfortunately, is matched byan abundance of less relevant data. This has to do with the inductive andopen-ended nature of the typical -omics experiment. There is nothingwrong with this (well, a little perhaps; Lipton, 2005), and especially whendone in a systematic way with good experimental design, there are goodmethods to turn data into knowledge (Kell, 2004; van der Werf, 2005).Statistics-based data analysis often gives qualitative interaction networks, orcandidate components (genes, transcription factors, metabolites) that arescored to be related to the phenomenon under study. For many applica-tions, this may be sufficient because these leads can be followed up byvalidation experiments. Also, the -omics data can simply be used fordiagnostic purposes both in red and in white biotechnology, for example,to indicate a particular limitation or stress during a fermentation process(Knijnenburg et al., 2007; Tai et al., 2005), or to predict likely outcomes ofcancer treatment (Kim and Paik, 2010), just to mention a few of the many,many applications of recent years.
Attempts to enhance the purely statistical analyses by integrating differ-ent data sets and a priori knowledge lie within the realm of integrativebioinformatics. We will not make an attempt to give a full account onthat field but focus on where metabolic information was, or can be, used forintegration. The construction of genome-scale models starts from thegenome: using bioinformatic approaches, the sequenced genome is scanned

Genome-Scale Metabolic Models 519
for regions that encode proteins with enzymatic activity (Francke et al.,2005; Reed et al., 2006; Thiele and Palsson, 2010). Based on the presence ofsuch genes, and supplemented with known biochemical and physiologicalinformation, putative metabolic pathways can be inferred. Thus, genome-scale metabolic models do not only contain reaction information (as wasused above in comparison with kinetic models) but also the often many-to-man relationships between genes, proteins, and reactions (Reed et al.,2006). The mapping of genes to proteins and to reactions allows forintegration of these levels. Transcriptome data (gene level) can be mappedon metabolic maps in this way, providing a visual, metabolic context for theinterpretation of the transcriptome data (Gehlenborg et al., 2010; Konoet al., 2009). By visual inspection, this may give leads to parts ofthe metabolic network that are regulated. For instance, the need for CO2
in fermentations of Lactobacillus plantarum was identified in this way (Stevenset al., 2008).
A computational counterpart of visual inspection was developed asreporter metabolites for proteome or transcriptome data (Patil andNielsen, 2005). In these analyses, the differential expression of reactionsthat produce or consume a particular metabolite is scored for each metabo-lite and compared to an expected score (based on chance alone). Thosemetabolites, whose surrounding reactions change significantly, are thenreporters for metabolic effects. CO2 would be such a reporter metabolitein the L. plantarum example above. Reporter reactions are computed in asimilar vein from metabolome data (Cakir et al., 2006). Recently, a toolboxwas developed that allows these types of analyses, called the BioMet Tool-box (Cvijovic et al., 2010).
Also, the metabolic association of genes, via their mapping to thereaction network, has been used for functional association tests. Thus,genes close to each other on the metabolic map were found to have astronger correlation in gene expression. Using constraint-based modeling,via the so-called flux coupling analysis (Burgard et al., 2004), this relation-ship was resolved with a much higher functional resolution (Notebaart et al.,2008, 2009). The key to this higher resolution is the appreciation that tworeactions in series in a linear pathway are stoichiometrically fully coupled,whereas two reactions diverging from the same metabolite (such as reactions2 and 3 in Fig. 24.2) are uncoupled, that is, they can carry flux in steady stateindependently from each other (Notebaart et al., 2008).
Thus, genome-scale metabolic models provide a “context for content”(Palsson, 2004) and are very useful in -omics data integration. Reconstruc-tions of other networks, such as transcription regulation networks (Herrgardet al., 2004), signal transduction networks (Hyduke and Palsson, 2010),or the translation machinery (Thiele et al., 2009), allow for furtherextension of top-down analyses in which biological data are integrated ina systematic way.

520 Filipe Santos et al.
3. The Art of Making Genome-Scale
Metabolic Models
The step-by-step instructions for making a genome-scale reconstruc-tion have been published recently (Feist et al., 2009; Thiele and Palsson,2010), and so we will forego a detailed description of these steps in favor ofplacing emphasis on specific issues that we have experienced to be critical,difficult, easily overlooked, or a combination of these. The steps toward areconstruction are (see Feist et al., 2009; Thiele and Palsson, 2010, withmany illustrations or a flow chart in Francke et al., 2005; Henry et al., 2010):
1. generating a draft reconstruction based on the genome sequence2. identify and resolve errors, gaps, and inconsistencies in the network3. define external metabolites and the biomass equation4. validate the model with additional experiments
3.1. Generating a draft reconstruction based on thegenome sequence
Tools and methods exist that take either a sequence or an annotated genomeand produce a first draft of a metabolic reconstruction automatically. One suchsystem is Pathway Tools (Karp et al., 2010), which underlies the BioCyccollections including the famous EcoCyc database (Keseler et al., 2009).It takes annotated genomes and identifies the metabolic genes based on ECcode andnamematching.PathwayTools also includes algorithms for automaticgap filling. In our experience, the information in this resource iswonderful, butthe automatically generated output of Pathway Tools results in a rather frag-mented set of pathways that need extensive curation (Teusink et al., 2005b).Also, the system is not explicitly designed for modeling, but rather, for makingan inventory (“encyclopedia”) of metabolic pathways, and their regulation.
The SEED, a pipeline system for metabolic reconstruction, has beenrecently made publicly available (Henry et al., 2010). This system uses asinput a sequenced genome and produces an SBML (Hucka et al., 2003)model that is able to produce biomass (Feist and Palsson, 2010), one of thehallmarks of a completed reconstruction. However, we agree with Feist (Feistet al., 2009), and have stressed ourselves (Francke et al., 2005), that extensivemanual curation cannot be fully replacedby automatedmethods.This is simplybecause there are too many important organism-specific choices that confertheir own idiosyncrasies and set them apart from others. Expert domainknowledge about the organism under study is required to make these choices.
With this in mind, we ourselves have developed an automated methodthat uses orthology prediction between genes of a query genome and genes

Genome-Scale Metabolic Models 521
from already existing genome-scale metabolic models, to copy the gene–protein–reaction associations (Notebaart et al., 2006). The philosophy ofthis AUTOGRAPH method is that information of the manual curationprocess of the existing genome-scale metabolic models is being reused asmuch as possible. Issues resolved duringmanual curation relate to (i) reactionstoichiometries of specific reactions not found in the databases (or withwrong stoichiometries, still not uncommon); (ii) choices in cofactor usage;and (iii) annotations of genes on the basis of gap filling, extensive bioinfor-matic analyses, or experimental results that have not found their way intothe databases (yet). It does not remove the need for manual curation, but itclearly accelerates the curation process, in our experience. We expect thesame to be true for the SEED pipeline, and the two methods are to a largeextent complementary.
3.2. Identify and resolve errors, gaps, and inconsistenciesin the network
After the initial draft of the model, manual curation involves going throughthe draft gene–protein–reaction associations to check for omissions, wrongassignments and gaps, and inconsistencies (for a review on gap filling, seeBreitling et al., 2008). One insightful inconsistency that we have found(Francke et al., 2005) is the annotation of metA in L. plantarum to homo-serine succinyl CoA transferase, while this organism lacks a TCA cycle andhence cannot make succinyl CoA. All databases nevertheless agree on thisfunction for metA, presumably because it is a member of a distinct ortho-logous group that includes the Escherichia coli enzyme, which does takesuccinyl CoA. However, the protein from B. subtilis, also in this ortholo-gous cluster, has been experimentally shown to take acetyl CoA, butsomehow this result did not find its way into BRENDA or any otherdatabase. So, sequence similarity, even one-to-one orthology, is not aguarantee for function transfer. Interestingly, the model SEED has keptthe succinyl CoA-dependent annotation and hence, the gap, but allowsbiomass production through methionine transport, even though domainexperts will know that L. plantarum grows without methionine (Teusinket al., 2005a). This teaches us that it is both wise and necessary to not alwaystrust the databases (when tested on students, it was striking how much theytrust information from the internet).
3.3. Define external metabolites and the biomass equation
For a reconstruction to become a model, there are a number of additionalsteps that involve a minimum of experimental data without which themodel is of poor predictive value. They come in three flavors:

522 Filipe Santos et al.
(i) Biomass composition: biomass composition must be defined, ideally forthe different experimental conditions under study. The SupplementaryMaterial in Oliveira et al. (2005), on the biomass of L. lactis, gives anextensive and useful account of the data and computation that isrequired. Protein, RNA, and lipid content change with growth rateand may affect the model simulations.
(ii) Data on the bioenergetics: this pertains to information on stoichiometry ofpumps and the respiratory chain (P/O ratio), and on maintenanceenergy and ATP requirement for growth, often referred to as YATP
(Tempest and Neijssel, 1984). These data can be estimated by carefullymonitoring product formation and biomass yield under a sufficientnumber of substrate/growth rate regimes; for excellent reviews on thetechniques involved, see vanGulik and Heijnen (1995), Vanrolleghemet al. (1996), and Vanrolleghem and Heijnen (1998). Surprisingly, onlyvery recently these parameters were carefully estimated for E. coli(Taymaz-Nikerel et al., 2010). But there is an important assumptionhere: measurement and inclusion of energetic parameters are based onfull coupling between ATP production by catabolism and ATP utili-zation through anabolic processes. Without this assumption, we can-not do the calculations, and the predictions will be imprecise (Teusinket al., 2006).
(iii) Essential and possible nutrients: growth requirement data (e.g., on spe-cific auxotrophies for vitamins and amino acids, or possible carbon,nitrogen and sulfur sources) are used to decide on the biologicalrelevance of a gap, for example, if a vitamin is not essential, then agap in the vitamin biosynthesis pathways may be benign (Teusink et al.,2005b). These data can also be used to determine the potential externalmetabolites, that is, sources for the network, and the necessary trans-port systems to take them up.
Although it is not very often pointed out, sinks are equally important assources, as they also require transport reactions, even for simple diffusionover the membrane. Easily overlooked sinks include by-products of bio-synthesis pathways (e.g., folate biosynthesis produces glycolaldehyde), ordiffusible substrates such as water and CO2.
Because genome-scale models are not exclusively bottom-up but largelybased on genome content, they tend to contain more (futile) cycles, parallelroutes, and dead-end pathways which a biochemist or biochemical engineerwould never put into his or her bottom-up model. These cycles arenevertheless potentially there in the network (Pinchuk et al., 2010;Teusink et al., 2006) and are either thermodynamically infeasible (substratecycles; Beard et al., 2002) or probably, and hopefully, regulated in such away that they tend not to run in circles too much (in the case of futile cycles;if they do, however, we have uncoupling of ATP production and

Genome-Scale Metabolic Models 523
consumption by growth, and our bioenergetic parameters go down thedrain). The dead-end pathways point to missing biochemical knowledge(or to reductive evolution, see e.g., Hols et al., 2005) and can therefore bevaluable leads to new discoveries, especially in combination with the dataon substrate use and product formation. Metabolic reconstructions and theirmodeling therefore not only rely on gene annotation, but it can also help infunctional annotation by identifying functions that have to be there.
3.4. Validate the model with additional experiments
Once the model can form biomass by providing all the necessary compo-nents that form the biomass, validation of the model requires comparisonwith additional experimental data. One such set of data are phenotypes ofdeletion strains; comprehensive sets are unfortunately usually not availablebut for model organisms such as E. coli and yeast. Their metabolic recon-structions have been extensively tested against the deletion strain pheno-types, with relatively high success rates in the order of 80–90% (Covert et al.,2004; Kuepfer et al., 2005). A more accessible set of data that can be used isphysiological data on growth rates, yields, substrate utilization, and productformation rates. Especially chemostat data are useful, as they are the bestexperimental setup for establishing growth in steady state. The modelshould be compatible with the measured fluxes, or, at a higher level ofvalidation, should be able to predict some of the fluxes as a function of someinput fluxes (note that we cannot predict all fluxes de novo, only the steady-state dependencies between fluxes, Eq. (24.4)). Oberhardt (Oberhardt et al.,2009) gives a good overview of the different models that exist and the extentto which they have been validated through experimental data.
4. Applications of Genome-Scale Metabolic
Models
For an increasing number of microorganisms, a genome-scale meta-bolic model is available at the quality level that we have discussed above(Oberhardt et al., 2009; Reed et al., 2006). The group of Palsson developeda resource for such metabolic models (see Schellenberger et al., 2010). Thesemodels can be used for a number of purposes, see a recent account on theapplications of genome-scale metabolic models (Oberhardt et al., 2009).A large set of different methods, constraint-based modeling techniques, havebeen developed in the past years to accommodate these goals. An excellentoverview of these techniques is given in Price et al. (2004). In brief, successfuluse of genome-scale metabolic models have ranged from:

524 Filipe Santos et al.
(i) exploration of gene lethality (Blank et al., 2005; Covert et al., 2004)and/or synthetic lethality (Costanzo et al., 2010)
(ii) definition of metabolic context for integrative bioinformatics(Kharchenko et al., 2004; Notebaart et al., 2008; Patil and Nielsen,2005)
(iii) the study of pathway evolution (Pal et al., 2006; Papp et al., 2004)(iv) prediction of metabolic engineering strategies (Burgard et al., 2003;
Bro and Nielsen, 2004, Park et al., 2010)(v) prediction of adaptive evolution outcomes (Fong et al., 2003, 2005;
Ibarra et al., 2002; Teusink et al., 2009)(vi) interpretation of fermentation data (Goffin et al., 2010; Teusink et al.,
2006)
4.1. Flux balance analysis: The work horse of constraintbased modeling
In many of the applications of genome-scale models, flux balance analysis(FBA) is used, and this is by far the most popular constraint-based modelingtechnique (Gianchandani et al., 2010). For a more extensive primer on FBA,see Orth et al. (2010). FBA uses optimization of a certain objective functionto find a subset of optimal states in the large solution space of possible statesthat is shaped by the mass balance and capacity constraints (see above; Orthet al., 2010; Price et al., 2004). We purposely state subset of optimal states,because the flux value of the objective function is unique, but the pathwaydistributions that give this optimal flux value, may not. There may be manydifferent ways to Rome. If one is interested in the flux distributions, notonly in the optimal value of the objective function, one needs to do a fluxvariability analysis (FVA) to check the uniqueness of the flux distributions(Mahadevan and Schilling, 2003). FVA maximizes and minimizes eachreaction rate in the network at the optimal flux value: the resultant rangein fluxes gives an indication how flexible the network is in reaching theoptimum.
FBA basically tries to find a state (or set of states) that maximizes (orminimizes) some desired flux or linear combination of fluxes. The approachis an optimization technique, and hence it implies that the modeled system(e.g., organism) behaves optimally (by evolution). This may limit theunderstanding of how microorganisms behave in experiments, as theymay not have had the time to adapt to the environment.
There are three ways to deal with the optimality issue. First, we canallow cells to adapt to their (constant) environment by laboratory evolutionexperiments (see application (v)). On the positive side, FBA offers anopportunity to predict evolutionary engineering outcomes, which may beguided by in silico analysis of specific deletions to enhance productivity

Genome-Scale Metabolic Models 525
(Burgard et al., 2003; Cvijovic et al., 2010). It also provides optimal yieldswith which experimentally obtained yields can be benchmarked.
Second, alternatives to FBA have been proposed to circumvent the needfor evolution, notably MOMA (minimization of metabolic adjustments;Segre et al., 2002). MOMA requires a well-defined reference state and thentries to predict the response of the network to a perturbation (change inexternal conditions, deletion of a reaction) by assuming that the network isrobust. Technically, it tries to minimize the (Euclidian) distance betweenthe reference flux distribution and the new solution space (which haschanged by the perturbation). In our experience, the major problem withMOMA is that it weighs all fluxes equally with respect to biologicalrelevance, which is not very likely. Moreover, the response is dominatedby high fluxes. Thus, MOMA predictions in our hands do not produceproper specific predictions of fluxes, but it has been useful to see if a certainperturbation is likely to cause severe growth problems even though theoptimal FBA solution seems fine (e.g., see Teusink et al., 2009).
Third, the optimality issue can just be ignored, simply because one doesnot care about a specific, quantitative prediction. For example, one maywant to know if the model can produce biomass, or some compound, notnecessarily how much. This goes for (synthetic) lethality predictions, andpredictions of evolution of metabolic networks.
4.2. FBA predicts rates only through yield maximization
If one does want to optimize to obtain specific flux distributions, however,one requires a sensible objective function, and here some confusion andcontroversy have arisen in the literature. In a recent study, different objectivefunctions were tested for the extent to which they could predict actual fluxstates under different conditions (Schuetz et al., 2007). This study demon-strated that different objective functions were needed to describe the fluxstates under different conditions. Notably, under energy (or carbon) limita-tion, optimization of biomass yield appeared to be the best objective function.This is in line with earlier studies in which the biomass formation functionwas taken as objective to predict functional states (Edwards et al., 2001).
However, many microorganisms display overflow metabolism, which isa wasteful lifestyle in terms of ATP generation and consequently, biomassyield. Yet it is observed even in glucose-limited chemostats above a certaincritical dilution rate, such as ethanol fermentation in Saccharomyces cerevisiae(van Dijken et al., 1993), acetate formation in E. coli (Vemuri et al., 2006), orlactate formation in lactic acid bacteria (Thomas et al., 1979). This behaviorcannot be predicted by FBA, because it predicts rates through optimalyields. It could only be described by including additional capacity con-straints on the oxidative phosphorylation pathways in the correspondingmetabolic networks (Famili et al., 2003; Varma and Palsson, 1994).

526 Filipe Santos et al.
It is important to fully appreciate the point that FBA predicts ratesthrough optimal yields, and not rates directly. If we take our examplefrom Fig. 24.2, and suppose we want to maximize the production rate ofP1 from S, we may formulate the FBA problem as:
max v2given :
mass balance constraint :
v1 � v2 � v3 ¼ 0
capacity constraints :
0 � v1 � 10
�1 � v2 � 10 � v3 � 1
8>>>>>><>>>>>>:
ð24:6Þ
The solution is easy in this case: v1 should be 10, v2 should be 10, and v3should be 0. But note that the rate of v2 is fully dictated by the constraint onv1. We could write the rate of v2 as:
v2 ¼ v1� � �YP1;S ð24:7Þ
where YP1,S is the yield of P1 on S (1 in this case). As v1 is fixed by thecapacity constraint, the only way to maximize v2 is to maximize the yield;this is also fixed in this simple case, but this is not so in larger systems whilethe capacity constraint is always required to bound the problem. Forlarger, real genome-scale models, this argument therefore remains(Schuster et al., 2007): as the solution space is bounded by an input flux,FBA finds an optimal objective flux by optimizing the yield on theincoming substrate. Hence, if in a model there are two options to makeATP from glucose, fermentation (low ATP yield) and respiration (highATP yield), optimization of ATP production rate will necessarily beachieved by respiration. Therefore, when applying biomass optimizationin FBA, the underlying biological assumption is that biomass yield maximi-zation was the strategy through which the organism has reached its fitness.It is clear that there are also other strategies that lead to fitness, and hence,the validity of FBA, using biomass yield as objective function, is organismand condition specific (Fong et al., 2003; Ibarra et al., 2002; Schuetz et al.,2007).
Understanding the basic assumptions of FBA, we can turn the problemaround and change the conditions in such a way that organisms are likely togrow efficiently, thereby increasing the predictive power of FBA. Growthyield maximization on poor substrates must have been the growth strategyunder the conditions where FBA predictions were successful (Fong et al.,2003; Teusink et al., 2009). Growth yield maximization is not the best

Genome-Scale Metabolic Models 527
strategy under high glucose concentrations and explains why adaptation onglucose led the cells away from the line of optimality predicted by FBA(Ibarra et al., 2002).
4.3. Using constraint-based modeling for discovery andinterpretation: Sensitivity analysis
Even under conditions where FBA is not predictive, such as high glucoseconcentrations, FBA can be very useful when combined with experimentaldata on input and output fluxes (which could not be predicted by FBA). Byusing those measured fluxes as constraints, there are a number of interestingthings one can do. We will discuss two of them.
First, we can set the measured flux data and then perform FVA to assesswhich parts of the network flux distribution is resolved by the measuredfluxes and which are not. One basically maximizes and minimizes eachreaction in the network, given the measured values as constraints (note weused FVA before to test the uniqueness of an FBA solution: in that case,fluxes were minimized and maximized with the optimal objective value asconstraint). It thus gives a range of reaction flux values for each reaction: thelarger the range, the poorer do the measured fluxes predict this flux. Thisanalysis could be considered as a genome-scale analogue to the well-knownmetabolic flux analysis (Maertens and Vanrolleghem, 2010), but applicableto large, underdetermined, systems. Alternatively, the ranges of fluxesobtained this way may include products of fermentation that were notmeasured but may be required to obtain the proper mass (or redox)balances. In this way, we were pointed at degradation products of aminoacids in a recent retentostat study (Goffin et al., 2010).
Second, growth rate optimization under measured flux constraints willindicate which medium compounds could further contribute to growth.This can be done through sensitivity analysis, which comes for free duringthe optimization procedure. These sensitivity coefficients are called reducedcosts, and they quantify to what extent the objective flux could be improvedby changing a capacity constraint. For example, the reduced cost of reactionv1 in the FBA problem of Eq. (24.6) is 1. We demonstrated the use ofreduced costs by analysis of growth limitations of L. plantarum on a mediumcontaining 3 carbon sources and 18 amino acids (Teusink et al., 2006).It turned out that some amino acids contributed to growth via ATPproduction, even though the mechanism for this ATP production was notimmediately obvious. In this way, we discovered a previously unknowntranshydrogenase activity in the degradation of branched chain amino acids.The latter is a good example of how genome-scale metabolic models canlead to the discovery of new metabolic capacities. In a later study, thisapproach was used to understand the puzzling observation that some aminoacids were being produced under conditions of extremely slow growth

528 Filipe Santos et al.
(Goffin et al., 2010). We can safely say that without the model, we wouldnot have been able to solve these puzzles.
4.4. Final remarks
There is a general consensus that functional genomics has enormous poten-tial in metabolic engineering and biotechnology. Systems biology, as a newinterdisciplinary branch of science, is rapidly gaining momentum. Thefunctional genomics toolbox has allowed a global view and thereby forcedmany (molecular) biologists to focus more on the system’s behavior than onthe behavior of one of its single components. Systems biology aims atunderstanding and ultimately predicting such system level behavior interms of the underlying molecular components and their interactions. Ithas model building at its centre: models to integrate data, models tointerpret the data, and models to make predictions. It is our view thatsuch models will play a central role in the advancement of biology in thiscentury, simply because we cannot grasp the complexity of biologicalsystems by intuition alone.
Genome-scale metabolic models are a first approach to combine bot-tom-up modeling with top-down, genomics data-driven modeling. How-ever, this can be used for exploration, for testing scenarios, for scanningconditions, and for eliminating impossibilities. However, these models canbe used for interpretation and integration of high-throughput data. Butthese models are a middle-out approach, and probably only a temporaryacceptance of our limitations. The challenge for the future will be to bringthese models to life, that is, to make them dynamic and put them underphysicochemical constraints and ultimately make them subject to ourmanipulation by understanding (exactly) how the control over flux andmetabolite levels is distributed. Only then can we truly claim to be able torationally engineer strains via computer-aided design.
REFERENCES
Bakker, B. M., et al. (2010). Systems biology from micro-organisms to human metabolicdiseases: The role of detailed kinetic models. Biochem. Soc. Trans. 38, 1294–1301.
Banga, J. R., and Balsa-Canto, E. (2008). Parameter estimation and optimal experimentaldesign. Essays Biochem. 45, 195–209.
Barthelmes, J., et al. (2007). BRENDA, AMENDA and FRENDA: The enzyme informa-tion system in 2007. Nucleic Acids Res. 35, D511–D514.
Beard, D. A., et al. (2002). Energy balance for analysis of complex metabolic networks.Biophys. J. 83, 79–86.
Blank, L. M., et al. (2005). Large-scale 13C-flux analysis reveals mechanistic principles ofmetabolic network robustness to null mutations in yeast. Genome Biol. 6, R49.
Breitling, R., et al. (2008). New surveyor tools for charting microbial metabolic maps. Nat.Rev. Microbiol. 6, 156–161.

Genome-Scale Metabolic Models 529
Bro, C., and Nielsen, J. (2004). Impact of ‘ome’ analyses on inverse metabolic engineering.Metab. Eng. 6, 204–211.
Bruggeman, F. J., and Westerhoff, H. V. (2007). The nature of systems biology. TrendsMicrobiol. 15, 45–50.
Burgard, A. P., et al. (2003). Optknock: A bilevel programming framework for identifyinggene knockout strategies for microbial strain optimization. Biotechnol. Bioeng. 84, 647–657.
Burgard, A. P., et al. (2004). Flux coupling analysis of genome-scale metabolic networkreconstructions. Genome Res. 14, 301–312.
Cakir, T., et al. (2006). Integration of metabolome data with metabolic networks revealsreporter reactions. Mol. Syst. Biol. 2, 50.
Costanzo, M., et al. (2010). The genetic landscape of a cell. Science 327, 425–431.Covert, M. W., et al. (2004). Integrating high-throughput and computational data elucidates
bacterial networks. Nature 429, 92–96.Cvijovic, M., et al. (2010). BioMet Toolbox: Genome-wide analysis of metabolism. Nucleic
Acids Res. 38(Suppl.), W144–W149.Edwards, J. S., et al. (2001). In silico predictions of Escherichia coli metabolic capabilities are
consistent with experimental data. Nat. Biotechnol. 19, 125–130.Famili, I., et al. (2003). Saccharomyces cerevisiae phenotypes can be predicted by using
constraint-based analysis of a genome-scale reconstructed metabolic network. Proc. Natl.Acad. Sci. USA 100, 13134–13139.
Feist, A. M., and Palsson, B. O. (2010). The biomass objective function. Curr. Opin.Microbiol. 13, 344–349.
Feist, A. M., et al. (2009). Reconstruction of biochemical networks in microorganisms. Nat.Rev. Microbiol. 7, 129–143.
Fell, D. (1997). Understanding the Control of Metabolism. Portland Press, London.Fong, S. S., et al. (2003). Description and interpretation of adaptive evolution of Escherichia
coli K-12 MG1655 by using a genome-scale in silico metabolic model. J. Bacteriol. 185,6400–6408.
Fong, S. S., et al. (2005). Parallel adaptive evolution cultures of Escherichia coli lead toconvergent growth phenotypes with different gene expression states. Genome Res. 15,1365–1372.
Francke, C., et al. (2005). Reconstructing the metabolic network of a bacterium from itsgenome. Trends Microbiol. 13, 550–558.
Gehlenborg, N., et al. (2010). Visualization of omics data for systems biology. Nat. Methods7, S56–S68.
Gianchandani, E. P., et al. (2010). The application of flux balance analysis in systems biology.Wiley Interdiscip. Rev. Syst. Biol. Med. 2, 372–382.
Goffin, P., et al. (2010). Understanding the physiology of Lactobacillus plantarum at zerogrowth. Mol. Syst. Biol. 6, 413.
Goldbeter, A. (1997). Biochemical Oscillations and Cellular Rhythms—The MolecularBases of Periodic and Chaotic Behaviour. Cambridge University Press, Cambridge,United Kingdom.
Heinrich, R., and Schuster, S. (1996). The Regulation of Cellular Systems. Chapman &Hall, New York.
Henry, C. S., et al. (2010). High-throughput generation, optimization and analysis ofgenome-scale metabolic models. Nat. Biotechnol. 28, 977–982.
Herrgard, M. J., et al. (2004). Reconstruction of microbial transcriptional regulatory net-works. Curr. Opin. Biotechnol. 15, 70–77.
Hoefnagel, M. H., et al. (2002). Metabolic engineering of lactic acid bacteria, the combinedapproach: Kinetic modelling, metabolic control and experimental analysis. Microbiology148, 1003–1013.
Hols, P., et al. (2005). New insights in the molecular biology and physiology of Streptococ-cus thermophilus revealed by comparative genomics. FEMS Microbiol. Rev. 29, 435–463.

530 Filipe Santos et al.
Hoppe, A., et al. (2007). Including metabolite concentrations into flux balance analysis:Thermodynamic realizability as a constraint on flux distributions in metabolic networks.BMC Syst. Biol. 1, 23.
Hucka, M., et al. (2003). The systems biology markup language (SBML): A medium forrepresentation and exchange of biochemical network models. Bioinformatics 19, 524–531.
Hyduke, D. R., and Palsson, B. O. (2010). Towards genome-scale signalling-networkreconstructions. Nat. Rev. Genet. 11, 297–307.
Ibarra, R. U., et al. (2002). Escherichia coli K-12 undergoes adaptive evolution to achieve insilico predicted optimal growth. Nature 420, 186–189.
Ideker, T., and Lauffenburger, D. (2003). Building with a scaffold: Emerging strategies forhigh- to low-level cellular modelling. Trends Biotechnol. 21, 255–262.
Jamshidi, N., and Palsson, B. O. (2010). Mass action stoichiometric simulation models:Incorporating kinetics and regulation into stoichiometric models. Biophys. J. 98,175–185.
Karp, P. D., et al. (2010). Pathway Tools version 13.0: Integrated software for pathway/genome informatics and systems biology. Brief. Bioinform. 11, 40–79.
Kell, D. B. (2004). Metabolomics and systems biology: Making sense of the soup. Curr.Opin. Microbiol. 7, 296–307.
Keseler, I. M., et al. (2009). EcoCyc: A comprehensive view of Escherichia coli biology.Nucleic Acids Res. 37, D464–D470.
Kharchenko, P., et al. (2004). Filling gaps in a metabolic network using expression informa-tion. Bioinformatics 20(Suppl. 1), I178–I185.
Kim, C., and Paik, S. (2010). Gene-expression-based prognostic assays for breast cancer.Nat. Rev. Clin. Oncol. 7, 340–347.
Kitano, H. (2002). Systems biology: A brief overview. Science 295, 1662–1664.Kitano, H. (2007). Towards a theory of biological robustness. Mol. Syst. Biol. 3, 137.Knijnenburg, T. A., et al. (2007). Exploiting combinatorial cultivation conditions to infer
transcriptional regulation. BMC Genomics 8, 25.Kono, N., et al. (2009). Pathway projector: Web-based zoomable pathway browser using
KEGG atlas and Google Maps API. PLoS One 4, e7710.Kuepfer, L., et al. (2005). Metabolic functions of duplicate genes in Saccharomyces cerevi-
siae. Genome Res. 15, 1421–1430.Kummel, A., et al. (2006a). Putative regulatory sites unraveled by network-embedded
thermodynamic analysis of metabolome data. Mol. Syst. Biol. 2, 0034.Kummel, A., et al. (2006b). Systematic assignment of thermodynamic constraints in
metabolic network models. BMC Bioinformatics 7, 512.Liao, J. C., et al. (2003). Network component analysis: Reconstruction of regulatory signals
in biological systems. Proc. Natl. Acad. Sci. USA 100, 15522–15527.Liebermeister, W., and Klipp, E. (2005). Biochemical networks with uncertain parameters.
IEE Proc. Syst. Biol. 152, 97–107.Liebermeister, W., and Klipp, E. (2006). Bringing metabolic networks to life: Convenience
rate law and thermodynamic constraints. Theor. Biol. Med. Model. 3, 41.Liebermeister, W., et al. (2005). Biochemical network models simplified by balanced
truncation. FEBS J. 272, 4034–4043.Lipton, P. (2005). Testing hypotheses: Prediction and prejudice. Science 307, 219–221.Liu, L., et al. (2010). Use of genome-scale metabolic models for understanding microbial
physiology. FEBS Lett. 584, 2556–2564.Maertens, J., and Vanrolleghem, P. A. (2010). Modelling with a view to target identification
in metabolic engineering: A critical evaluation of the available tools. Biotechnol. Prog. 26,313–331.
Mahadevan, R., and Schilling, C. H. (2003). The effects of alternate optimal solutions inconstraint-based genome-scale metabolic models. Metab. Eng. 5, 264–276.

Genome-Scale Metabolic Models 531
Mashego, M. R., et al. (2007). Metabolome dynamic responses of Saccharomyces cerevisiaeto simultaneous rapid perturbations in external electron acceptor and electron donor.FEMS Yeast Res. 7, 48–66.
Notebaart, R. A., et al. (2006). Accelerating the reconstruction of genome-scale metabolicnetworks. BMC Bioinformatics 7, 296.
Notebaart, R. A., et al. (2008). Co-regulation of metabolic genes is better explained by fluxcoupling than network distance. PLoS Comput. Biol. 4, e26.
Notebaart, R. A., et al. (2009). Asymmetric relationships between proteins shape genomeevolution. Genome Biol. 10, R19.
Oberhardt, M. A., et al. (2009). Applications of genome-scale metabolic reconstructions.Mol. Syst. Biol. 5, 320.
Oliveira, A. P., et al. (2005). Modelling Lactococcus lactis using a genome-scale flux model.BMC Microbiol. 5, 39.
Orth, J. D., et al. (2010). What is flux balance analysis? Nat. Biotechnol. 28, 245–248.Pal, C., et al. (2006). Chance and necessity in the evolution of minimal metabolic networks.
Nature 440, 667–670.Palsson, B. (2004). Two-dimensional annotation of genomes. Nat. Biotechnol. 22,
1218–1219.Papin, J. A., et al. (2004). Comparison of network-based pathway analysis methods. Trends
Biotechnol. 22, 400–405.Papp, B., et al. (2004). Metabolic network analysis of the causes and evolution of enzyme
dispensability in yeast. Nature 429, 661–664.Park, J. H., et al. (2010). Fed-batch culture of Escherichia coli for L-valine production based
on in silico flux response analysis. Biotechnol Bioeng. 108, 934–946.Patil, K. R., and Nielsen, J. (2005). Uncovering transcriptional regulation of metabolism by
using metabolic network topology. Proc. Natl. Acad. Sci. USA 102, 2685–2689.Pinchuk, G. E., et al. (2010). Constraint-based model of Shewanella oneidensis MR-1
metabolism: A tool for data analysis and hypothesis generation. PLoS Comput. Biol. 6,e1000822.
Price, N. D., et al. (2004). Genome-scale models of microbial cells: Evaluating the con-sequences of constraints. Nat. Rev. Microbiol. 2, 886–897.
Reed, J. L., et al. (2006). Towards multidimensional genome annotation.Nat. Rev. Genet. 7,130–141.
Resendis-Antonio, O. (2009). Filling kinetic gaps: Dynamic modelling of metabolism wheredetailed kinetic information is lacking. PLoS One 4, e4967.
Rojas, I., et al. (2007). Storing and annotating of kinetic data. In Silico Biol. 7, S37–S44.Schellenberger, J., et al. (2010). BiGG: A Biochemical Genetic and Genomic knowledgebase
of large scale metabolic reconstructions. BMC Bioinformatics 11, 213.Schuetz, R., et al. (2007). Systematic evaluation of objective functions for predicting
intracellular fluxes in Escherichia coli. Mol. Syst. Biol. 3, 119.Schuster, S., et al. (2000). A general definition of metabolic pathways useful for systematic
organization and analysis of complex metabolic networks. Nat. Biotechnol. 18, 326–332.Schuster, S., et al. (2007). Is maximization of molar yield in metabolic networks favoured by
evolution? J. Theor. Biol. 252, 497–504.Segre, D., et al. (2002). Analysis of optimality in natural and perturbed metabolic networks.
Proc. Natl. Acad. Sci. USA 99, 15112–15117.Steuer, R., et al. (2006). Structural kinetic modelling of metabolic networks. Proc. Natl. Acad.
Sci. USA 103, 11868–11873.Stevens, M. J., et al. (2008). Improvement of Lactobacillus plantarum aerobic growth as
directed by comprehensive transcriptome analysis. Appl. Environ. Microbiol. 74,4776–4778.
Tai, S. L., et al. (2005). Two-dimensional transcriptome analysis in chemostat cultures.Combinatorial effects of oxygen availability and macronutrient limitation in Saccharo-myces cerevisiae. J. Biol. Chem. 280, 437–447.

532 Filipe Santos et al.
Taymaz-Nikerel, H., et al. (2010). Genome-derived minimal metabolic models for Escher-ichia coli MG1655 with estimated in vivo respiratory ATP stoichiometry. Biotechnol.Bioeng. 107, 369–381.
Tempest, D.W., and Neijssel, O. M. (1984). The status of YATP and maintenance energy asbiologically interpretable phenomena. Annu. Rev. Microbiol. 38, 459–486.
Teusink, B., and Smid, E. J. (2006). Modelling strategies for the industrial exploitation oflactic acid bacteria. Nat. Rev. Microbiol. 4, 46–56.
Teusink, B., et al. (2000). Can yeast glycolysis be understood in terms of in vitro kinetics ofthe constituent enzymes? Testing biochemistry. Eur. J. Biochem. 267, 5313–5329.
Teusink, B., et al. (2005). In silico reconstruction of the metabolic pathways of Lactobacillusplantarum: Comparing predictions of nutrient requirements with those from growthexperiments. Appl. Environ. Microbiol. 71, 7253–7262.
Teusink, B., et al. (2006). Analysis of growth of Lactobacillus plantarumWCFS1 on a complexmedium using a genome-scale metabolic model. J. Biol. Chem. 281, 40041–40048.
Teusink, B., et al. (2009). Understanding the adaptive growth strategy of L. plantarum by insilico optimisation. PLoS Comput. Biol. 5, e1000410.
Theobald, U., et al. (1993). In vivo analysis of glucose-induced fast changes in yeast adeninenucleotide pool applying a rapid sampling technique. Anal. Biochem. 214, 31–37.
Thiele, I., and Palsson, B. O. (2010). A protocol for generating a high-quality genome-scalemetabolic reconstruction. Nat. Protoc. 5, 93–121.
Thiele, I., et al. (2009). Genome-scale reconstruction of Escherichia coli’s transcriptionaland translational machinery: A knowledge base, its mathematical formulation, and itsfunctional characterization. PLoS Comput. Biol. 5, e1000312.
Thomas, T. D., et al. (1979). Change from homo- to heterolactic fermentation by Strepto-coccus lactis resulting from glucose limitation in anaerobic chemostat cultures. J. Bacteriol.138, 109–117.
van der Werf, M. J. (2005). Towards replacing closed with open target selection strategies.Trends Biotechnol. 23, 11–16.
van Dijken, J. P., et al. (1993). Kinetics of growth and sugar consumption in yeasts. AntonieVan Leeuwenhoek 63, 343–352.
vanGulik, W. M., and Heijnen, J. J. (1995). A metabolic network stoichiometry analysis ofmicrobial growth and product formation. Biotechnol. Bioeng. 48, 681–698.
Vanrolleghem, P. A., and Heijnen, J. J. (1998). A structured approach for selection amongcandidate metabolic network models and estimation of unknown stoichiometric coeffi-cients. Biotechnol. Bioeng. 58, 133–138.
Vanrolleghem, P. A., et al. (1996). Validation of a metabolic network for Saccharomycescerevisiae using mixed substrate studies. Biotechnol. Prog. 12, 434–448.
Varma, A., and Palsson, B. O. (1994). Stoichiometric flux balance models quantitativelypredict growth and metabolic by-product secretion in wild-type Escherichia coli W3110.Appl. Environ. Microbiol. 60, 3724–3731.
Veening, J. W., et al. (2008). Bistability, epigenetics, and bet-hedging in bacteria. Annu. Rev.Microbiol. 62, 193–210.
Vemuri, G. N., et al. (2006). Overflow metabolism in Escherichia coli during steady-stategrowth: Transcriptional regulation and effect of the redox ratio. Appl. Environ. Microbiol.72, 3653–3661.
Visser, D., et al. (2004). Analysis of in vivo kinetics of glycolysis in aerobic Saccharomycescerevisiae by application of glucose and ethanol pulses. Biotechnol. Bioeng. 88, 157–167.
Wang, L., and Hatzimanikatis, V. (2006). Metabolic engineering under uncertainty. I:Framework development. Metab. Eng. 8, 133–141.