Embed Size (px)
Citation preview

a
2OT
Aslot
cc
Developmental Biology 239, 95–106 (2001)doi:10.1006/dbio.2001.0419, available online at http://www.idealibrary.com on
A Spontaneous Mouse Mutation, mesenchymaldysplasia (mes), Is Caused by a Deletionof the Most C-Terminal CytoplasmicDomain of patched (ptc)
Shigeru Makino,* ,§ Hiroshi Masuya,† Junko Ishijima,* Yukari Yada,* ,‡nd Toshihiko Shiroishi* ,§ ,1
*Mammalian Genetics Laboratory, National Institute of Genetics, Mishima, Shizuoka-ken411-8540, Japan; †Mouse Functional Genomics Group, RIKEN Genomic Science Center, Maedacho14, Totsukaku, Yokohamashi, Kanagawa-ken, 244-0804, Japan; ‡Ochanomizu University, 2-1-1htsuka, Bunkyoku, Tokyo, 112-8610, Japan; and §Department of Genetics, School of Life Science,he Graduate University for Advanced Studies, Yata, 1111, Shizuoka-ken, 411-8540, Japan
recessive mouse mutation, mesenchymal dysplasia (mes), which arose spontaneously on Chromosome 13, causes excesskin, increased body weight, and mild preaxial polydactyly. Fine gene mapping in this study indicated that mes is tightlyinked to patched (ptc) that encodes a transmembrane receptor protein for Shh. Molecular characterization of the ptc genef the mes mutant and an allelism test using a ptc knockout allele (ptc2) demonstrated that mes is caused by a deletion ofhe most C-terminal cytoplasmic domain of the ptc gene. Since mes homozygous embryos exhibit normal spinal cord
development as compared with ptc2 homozygotes, which die around 10 dpc with severe neural tube defects, the C-terminalytoplasmic domain lost in mes mutation is dispensable for inhibition of Shh signaling in early embryogenesis. However,ompound heterozygotes of ptc2 and mes alleles, which survive up to birth and die neonatally, had increased body weight
and exhibited abnormal anteroposterior axis formation of the limb buds. These findings indicate that Ptc is a negativeregulator of body weight and ectopic activation of Shh signaling in the anterior mesenchyme of the limb buds, and that theC-terminal cytoplasmic domain of Ptc is involved in its repressive action. © 2001 Academic Press
Key Words: patched; Sonic hedgehog; limb; mesenchymal dysplasia; mes; developing spinal cord; polydactyly; bodyweight.
eapmcIada
INTRODUCTION
In vertebrates, the secreted signaling molecule Sonichedgehog (Shh), which is a mouse homologue of the Dro-sophila segment polarity gene Hedgehog (Hh), has essentialroles in a variety of patterning events in developing em-bryos. Shh is produced in organizing centers, notochord,floor plate, and the zone of polarizing activity (ZPA) of limbbuds (Echelard et al., 1993; Riddle et al., 1993), and inepithelia at numerous sites, teeth, hair follicles, gut, andlung (Bitgood and McMahon, 1995).
Genetic data in Drosophila suggested that Hh inducestranscription of the downstream target genes by antagoniz-
1 To whom correspondence should be addressed. Fax: 81-559-81-
s6817. E-mail: [email protected].0012-1606/01 $35.00Copyright © 2001 by Academic PressAll rights of reproduction in any form reserved.
ing the activity of a segment polarity gene, patched (ptc)(Hooper, 1994; Ingham et al., 1991). It was shown that Ptc isa novel protein with multiple transmembrane domains andis present at the plasma membrane (Capdevila et al., 1994;Hooper and Scott, 1989; Nakano et al., 1989). Biochemicalstudies (Carpenter et al., 1998; Marigo et al., 1996b; Muronet al., 1999; Stone et al., 1996) indicated that Ptc constitutesreceptor complex for Hh together with another segment
olarity gene, Smoothened (Smo). Smo encodes a seven-passembrane protein with the characteristics of a G protein-
oupled receptor (Alcedo et al., 1996; van den Heuvel andngham, 1996). In the absence of Hh, Ptc represses thectivity of Smo, which constitutively activates the genesownstream of Hh signaling. Hh binding antagonizes thection of Ptc, and Smo becomes active and transduces Hh
ignaling to downstream genes.95

mpoo
tbmtgs
LIgfp
96 Makino et al.
In mice, the roles of Shh signaling have been studied withShh and ptc knockout mutants (Chiang et al., 1996; Good-rich et al., 1997). Mice homozygous for a Shh knockoutmutation lose ventral neural progenitor cells and showsevere morphological defects in the early stage of develop-ment of the lung, limbs, and other organs. Homozygotes ofthe ptc knockout mutation also exhibit severe neural tubedefects and die around day 10. On the other hand, heterozy-gotes of both mutations have almost normal phenotypes,except for slight enlargement of the body size in the ptc
utant. The properties of these null mutants of the Shh andtc genes have hampered the detailed experimental analysisf Shh signaling in later stages of development, especially inrganogenesis processes.In this study, we have analyzed a recessive mouse mu-
ant, mesenchymal dysplasia (mes) (Sweet et al., 1996),ecause it has the following interesting characteristics: (1)es exhibits pleiotropic phenotypes, some of which seem
o result from aberrant Shh signaling. These include over-rowth of mesenchyme-derived tissues, especially excesskin and increased musculature; mes also shows preaxial
polydactyly of all four feet, a shortened face, wide-set eyes,dome head, and a shortened kinky tail. (2) mes was mappedto the middle of Chromosome 13, and shown to be linked tothe ptc locus. These facts suggested that mes is a mutationof the ptc gene. A genetic study of the ptc gene of the mesmutant demonstrated that mes is indeed caused by a 32-bpdeletion in the coding region of the C-terminal cytoplasmicdomain of the ptc gene, which results in truncation of thePtc protein at the beginning of the cytoplasmic domain.
Recently, the function of the C-terminal cytoplasmicdomain of Drosophila Ptc was analyzed in wing disc, usingtruncated Ptc which had lost almost all of the C-terminalcytoplasmic domain (Johnson et al., 2000). That studyindicated that the C-terminal cytoplasmic domain has anessential role in blocking Hh signaling. In contrast to theobservations in Drosophila, the mes homozygotes do notexhibit gross abnormalities of the axis formation in thedeveloping spinal cord. This indicates that the C-terminalcytoplasmic domain of mouse Ptc is not essential fordevelopment of the spinal cord, in which Shh signaling isknown to play a key role in the pattern formation.
To investigate the function of the mouse Ptc moleculeand its C-terminal cytoplasmic domain in detail, we char-acterized the phenotypes of the compound heterozygotes ofmes and ptc knockout alleles, which survive up to birth.They exhibited developmental defects more severe thanthose of mes homozygotes, but much milder than those ofhomozygotes of the ptc knockout allele. Therefore, use ofthe compound heterozygotes has paved a new way forstudies of the biological roles of ptc in later stages ofdevelopment, especially in organogenesis processes. In thisreport, using this system, we demonstrate that theC-terminal cytoplasmic domain of mouse Ptc is not essen-tial for spinal cord development, but plays an indispensablerole in both repressing body weight and anteroposterior axis
formation of the limb buds.Copyright © 2001 by Academic Press. All right
MATERIALS AND METHODS
Mice
C57BL/6J, B6C3Fe-a/a-mes/1 (Sweet et al., 1996) and B6,129-Ptchtm1Mps (Goodrich et al., 1997) were purchased from the Jackson
aboratory (Bar Harbor, ME) and maintained at the Nationalnstitute of Genetics (NIG, Mishima, Japan). C57BL/6J-mes/1 wasenerated by backcrossing of B6C3Fe-a/a-mes/1 to C57BL/6J forour to six generations. B6,129-Ptchtm1Mps was used under a licensedatent of DuPont, Japanese Patent 2,058,915. B6,129-Ptchtm1Mps was
backcrossed one or four generations to C57BL/6J and used in thisstudy. The MSM strain, derived from the Japanese wild mouse,Mus musculus molossinus, was established and maintained atNIG.
PCR Genotyping
Genomic DNA for genotyping was prepared from ear, liver, tail,or amnion of embryos. The oligonucleotide primer pair used todetect the deletion site of ptc in the mes mutation was as follows:mesdF, 59-TCCAAGTGTCGTCCGGTTTG-39 and mesdR, 59-GTGGCTTCCACAATCACTTG-39. For genotyping of the ptc2
heterozygotes, the neomycin resistance gene that is inserted in theptc locus of B6,129-Ptchtm1Mps was used. The oligonucleotide primerpair used to detect the neomycin gene in the ptc2 mutants was asfollows: neoP1, 59-GGCTATTCGGCTATGACTGG-39 and neoP2,59-GAGATGACAGGAGATCCTGC-39.
Linkage Analysis
For linkage analysis of mes, (MSM 3 B6C3Fe-a/a-mes/mes)F1
mice were backcrossed to B6C3Fe-a/a-mes/mes. Since homozygousfemales fail to deliver viable offspring (Sweet et al., 1996), only thehomozygous males were used to generate the F1 and the backcrossprogeny. Genomic DNA was prepared from the liver or the ear, andmicrosatellite loci were genotyped by simple sequence lengthpolymorphism (SSLP). Microsatellite primer pairs were purchasedfrom Research Genetics (Huntsville, AL). The ptc gene was typedby Southern analysis using an 841-bp DNA fragment of the59-coding region of ptc, which was obtained from Dr. M. P. Scott(Goodrich et al., 1996).
Section in Situ Hybridization
Section in situ hybridization was performed essentially follow-ing the method of Birren et al. (1993). Embryos were fixed in 4%PFA–PBS for 6 h at 4°C and replaced in PBS containing 30% sucroseovernight at 4°C. Embryos were embedded in OCT compound(Sakura Finetechnical Co. Ltd., Japan), frozen in liquid nitrogen,and sectioned at 25 mm. The following cDNA clones were used astemplates for synthesizing digoxigenin-labeled riboprobes: Shh(Echelard et al., 1993), Pax1 (Deutsch et al., 1988), Pax3 (Gouldinget al., 1991), and Nkx6.1 (Qiu et al., 1998). An about 2-kbp fragmentof Nkx2.2 and a 0.9-kbp fragment of Isl1 were also used.
Whole-Mount in Situ Hybridization
Whole-mount in situ hybridization using digoxigenin-labeledRNA was performed essentially as described elsewhere (Prince andLumsden, 1994). The probe for Gli was transcribed from a 1.6-kbp
fragment (Platt et al., 1997).s of reproduction in any form reserved.

msft
d
ido
tn
97Analysis of a patched Hypomorph Mutant, mes
FIG. 1. Gene mapping of the mes mutation. (A) Segregation panel of backcross of (MSM 3 B6C3Fe-a/a-mes/mes)F1 3 B6C3Fe-a/a-mes/es. Microsatellite marker loci examined are listed to the left side of the haplotype panel. The number of animals with each haplotype is
hown at the bottom of each column. The open squares represent the mes allele and the solid squares the MSM allele. Polydactyly was usedor phenotyping of the mes mutation. (B) Genetic map around mes in Chromosome 13. Microsatellite marker loci examined are shown at
he right side of the map. In a total of 241 backcross progeny (BCN2), mes was not genetically separated from the patched (ptc) gene.FIG. 2. A 32-bp deletion in the C-terminal cytoplasmic domain of ptc in the mes mutant. (A) PCR products of genomic DNAs fromifferent inbred strains and the mes mutant, which were amplified by the primer pair around one of the SacI sites. (B) Nucleotide sequence
of the deletion region of ptc in mes. ptc cDNA obtained from a mes homozygous embryo was sequenced and a 32-bp deletion was identifiedn the region underlined. The two arrows indicate the primer pair used in the PCR-amplification. The numbers at the ends of the sequenceenote the nucleotide number from the start codon of the ptc gene. The SacI site in question is boxed. (C) Amino acid sequence alignmentf the C-terminal cytoplasmic domain of Ptc. Various Ptc proteins except for mes Ptc were aligned using the clustal program in DDBJ (NIG,
Mishima, Japan). Amino acid residues identical in mouse, human, and chick Ptc are shaded, and the region of putative transmembranedomain 12 (TM12) is indicated by an underline. Aberrant amino acid stretch caused by mes mutation is boxed. (D) Putative topologicalmodel of two forms of Ptc proteins and Ptcmes. (Left) The C-terminal cytoplasmic domain of mouse Ptc1 has a long extension of 273 aminoacids (blue line). The structure of Ptc1 orthologs of higher vertebrates, chick and human, are highly conserved. (Middle) The C-terminalcytoplasmic domain of mouse Ptc2 lacks the C-terminal extension and has a short stretch of 71 amino acids. (Right) The Ptcmes protein losthe most of the C-terminal cytoplasmic domain, retaining the last 53 amino acids after the last transmembrane domain and gaining the
onsense 68 amino acids (red line).Copyright © 2001 by Academic Press. All rights of reproduction in any form reserved.

98 Makino et al.
FIG. 2—Continued
Copyright © 2001 by Academic Press. All rights of reproduction in any form reserved.

o
a
limb buds (H). Limb buds are oriented with the anterior side toward the
TA
only one foot, as described previously (Goodrich et al., 1997).
99Analysis of a patched Hypomorph Mutant, mes
Copyright © 2001 by Academic Press. All right
X-Gal Staining of Embryos
X-Gal staining of embryos was performed essentially followingthe method of Whiting et al. (1991). Embryos were stained for 4 hto overnight, depending on their size. Specimens were fixed in 4%PFA–PBS overnight at 4°C, dehydrated and embedded in paraffin,and sectioned at 10 mm.
Analysis of Cell Proliferation in Vivo
Pregnant females were injected intraperitoneally with a mix-ture of BrdU and FUdr (50 and 10 mg per kg body weight,respectively). Samples were collected after 2 h, and fixed in 4%PFA–PBS overnight at 4°C. Then, dehydrated samples were
e of limb buds in ptc2/ptcmes embryos. (A–C) Dorsal view of thethe ptc2/1 embryos (A) exhibit the wild-type phenotype. Mostdigit I. The ptc2/ptcmes embryos (C) showed complete duplicationbryos. Forelimbs of the ptcmes/ptcmes and the ptc2/ptcmes embryos
re oriented with the anterior side toward the top of the panels. (D,D). In ptc2/ptcmes hindlimb buds (E), Shh was ectopically expressed.0 dpc. (F–I) Ectopic expression of Gli was observed at the anteriorition to the normal posterior expression in ptc2/1 (F). This ectopicarrow), in addition to the posteriorly restricted expression in 1/1
FIG. 3. Ectopic activation of Shh signaling in the anterior mesenchymhindlimbs stained with Alcian blue and Alizarin Red. Almost all ofptcmes/ptcmes embryos (B) showed bifurcation of the distal phalange of thef the digit I, which is much more severe than that of the ptcmes/mes em
showed similar phenotypes to the hindlimbs (data not shown). Limbs aE) Shh was normally expressed in the posterior mesenchyme of ptc2/1 (t the anterior margin of the limb buds very weakly in some cases at 12
mesenchyme in all ptc2/ptcmes embryos to be examined (G, arrow), in addexpression of Gli in ptc2/ptcmes embryos was maintained by 12.0 dpc (I,
ABLE 1llelism Test of mes for ptc
Phenotype
No. of embryos generated from the crossof ptc2/1 3 mes/1
ptc2/1mes/1
ptc2/11/1
1/1mes/1
ptc2/11/1 Total
Polydactyly of allfour feet
37a 0 0 0 37
Normal 0 53b 62 58 173
a All ptc2/1 mes/1 embryos showed preaxial polydactyly.b One of 53 ptc2/1 embryos exhibited preaxial polydactyly in
top of the panels. Scale bar, 1 mm in (A–C) and 0.5 mm in (D–I).
s of reproduction in any form reserved.
(dVuh
m
hstD
cig
f
p
100 Makino et al.
embedded in paraffin, sectioned (4 mm), and stained as describedBellusci et al., 1997). Mouse anti-BrdU antibody (Sigma) wasiluted 1:2000; and immunostaining was carried out with aector stain elute kit (Vector Laboratory, Inc., Burlingame, CA)sing DAB as the substrate. Sections were counterstained withematoxylin.
RESULTS
Fine Localization of mes in Chromosome 13
Based on a total of 241 backcross progeny generated froma cross with an inbred strain MSM, mes was mapped to a1.7-cM interval flanked by two microsatellite markers,D13Mit318 and D13Mit187. mes was separated by onerecombinant from the proximal marker D13Mit318, andseparated by three recombinants from the distal markerD13Mit187 (Figs. 1A and 1B). Using a database (Encyclope-dia, The Jackson Laboratory), we searched known genesthat could be potential candidates for mes. Among thesegenes, we found that ptc is tightly linked to mes. Subse-quent linkage analysis based on restriction fragment lengthpolymorphism (RFLP) of the ptc gene showed no recombi-nant between mes and ptc (Fig. 1B).
Molecular Characterization of the ptc Gene in themes Mutant
For molecular characterization of the ptc gene in the mesutant, we tried to amplify ptc cDNA from mes mRNA by
RT-PCR, and intended to clone the fragment into a plasmidvector. In this experiment, we soon noticed that the ptcgene of mes lacked one of two SacI sites that are expectedfrom the cDNA sequence of the wild-type ptc gene. Basedon this observation, we analyzed the PCR products ampli-fied from genomic DNAs of various mouse strains astemplates with a primer pair around the SacI site inquestion (Fig. 2B). Because mes arose in the CBA/J strainand has been maintained in the hybrid background (C3H 3C57BL/6)F1, the PCR product of the mes mutants should
ave a size identical to that of the CBA, C3H, or C57BL/6Jtrain. In this analysis, the PCR product from mes appearedo be smaller than those of the other three strains (Fig. 2A).
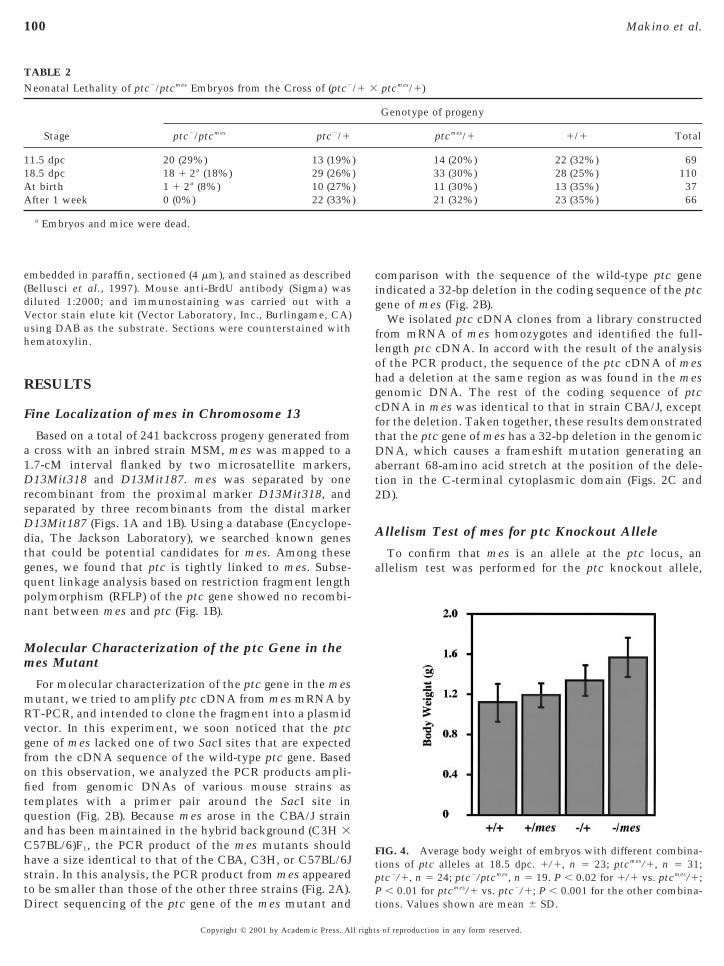
TABLE 2Neonatal Lethality of ptc2/ptcmes Embryos from the Cross of (ptc2
Stage ptc2/ptcmes ptc2/1
11.5 dpc 20 (29%) 13 (19%18.5 dpc 18 1 2a (18%) 29 (26%At birth 1 1 2a (8%) 10 (27%After 1 week 0 (0%) 22 (33%
a Embryos and mice were dead.
irect sequencing of the ptc gene of the mes mutant and
Copyright © 2001 by Academic Press. All right
omparison with the sequence of the wild-type ptc genendicated a 32-bp deletion in the coding sequence of the ptcene of mes (Fig. 2B).We isolated ptc cDNA clones from a library constructed
rom mRNA of mes homozygotes and identified the full-length ptc cDNA. In accord with the result of the analysisof the PCR product, the sequence of the ptc cDNA of meshad a deletion at the same region as was found in the mesgenomic DNA. The rest of the coding sequence of ptccDNA in mes was identical to that in strain CBA/J, exceptfor the deletion. Taken together, these results demonstratedthat the ptc gene of mes has a 32-bp deletion in the genomicDNA, which causes a frameshift mutation generating anaberrant 68-amino acid stretch at the position of the dele-tion in the C-terminal cytoplasmic domain (Figs. 2C and2D).
Allelism Test of mes for ptc Knockout Allele
To confirm that mes is an allele at the ptc locus, anallelism test was performed for the ptc knockout allele,
FIG. 4. Average body weight of embryos with different combina-tions of ptc alleles at 18.5 dpc. 1/1, n 5 23; ptcmes/1, n 5 31;tc2/1, n 5 24; ptc2/ptcmes, n 5 19. P , 0.02 for 1/1 vs. ptcmes/1;
P , 0.01 for ptcmes/1 vs. ptc2/1; P , 0.001 for the other combina-
ptcmes/1)
Genotype of progeny
ptcmes/1 1/1 Total
14 (20%) 22 (32%) 6933 (30%) 28 (25%) 11011 (30%) 13 (35%) 3721 (32%) 23 (35%) 66
/1 3
))))
tions. Values shown are mean 6 SD.
s of reproduction in any form reserved.

(iehcwe2
tp
osgaatwtp
ui
a
bltd
ws
fapa5o
rpsm
Dp
101Analysis of a patched Hypomorph Mutant, mes
hereafter designated as ptc2. In this allele, the 59-part of theptc gene, including the putative start codon, was replacedby lacZ and a neomycin resistance gene (Goodrich et al.,1997). Since both mes and ptc2 are recessive mutations,mes heterozygotes never show any visible phenotype; like-wise, ptc2 heterozygotes rarely show preaxial polydactylyonly about 1% of heterozygotes exhibit polydactyly). If mess allelic for ptc, mice that had both mutant alleles werexpected to exhibit the polydactyly phenotype, like mesomozygotes. In this study, mes heterozygotes wererossed with ptc2 heterozygotes, and the resultant progenyere investigated for the polydactyly phenotype at the
mbryonic stage from 15.5 dpc until birth (Table 1). Out of10 progeny, all 37 embryos that had both mes and ptc2
alleles showed preaxial polydactyly in all four feet, whichwas more severe than the phenotype of mes homozygotes(Figs. 3B and 3C). The rest of the embryos never showedpolydactyly. These results indicated that mes is an allele athe ptc locus. Therefore, we hereafter designate mes astcmes.
Neonatal Lethality of Compound Heterozygotes(ptc2/ptcmes)
In order to examine whether the compound heterozy-gotes, hereafter designated as ptc2/ptcmes, survive until birthr die at embryonic stages, the embryos generated from theame cross as in the allelism test were analyzed for theirenotype at different developmental stages (Table 2). Thisnalysis showed that the ptc2/ptcmes embryos were observedt approximately the Mendelian segregation ratio until athe stage of 18.5 dpc, although the number of the embryosas slightly lower than those with the three other geno-
ypes. At birth, there was only one viable progeny oftc2/ptcmes. Histological analysis revealed that this mouse
had a malformation of the lung (data not shown). The resultsuggested that most of ptc2/ptcmes embryos did not die in
tero, but rather died soon after birth, probably due to annability to breathe.
Increased Body Weight of ptc2/ptcmes Embryos
Increased body weight caused by ptc mutations is acommon characteristic in humans and mice (Goodrich etl., 1997; Gorlin, 1987; Hahn et al., 1998; Milenkovic et al.,
1999). We analyzed the body weight of 18.5-dpc embryosobtained from the same cross as was used in the allelismtest. We found that ptc2/ptcmes, ptc2/1, and ptcmes/1 em-ryos were about 41, 19, and 7% heavier than the 1/1ittermates, respectively (Fig. 4). This result indicated thathe normal ptc gene negatively regulates body weight in aose-dependent manner.Hematoxylin and eosin-stained sections of the ptc2/ptcmes
embryos at 13.5 dpc showed that the mesenchyme of thetrunk overgrew around the neural tube, esophagus, andaorta, especially under the dorsal skin (Figs. 5A and 5B). Wefound that cell proliferation rate in the dorsal mesenchyme
of ptc2/ptcmes embryos at 12.5 dpc was not significantlyCopyright © 2001 by Academic Press. All right
higher than that of ptc2/1 embryos (Figs. 5I and 5J), when itas monitored by BrdU uptake. Gross reduction of apopto-
is was not observed in the dorsal trunk of ptc2/ptcmes
embryos at 12.5 dpc (data not shown). Sclerotome is derivedfrom ventromedial somites and is thought to be directlyinduced by Shh (Fan and Tessier-Lavigne, 1994; Johnson etal., 1994). In later development, sclerotomal cells migrateand differentiate into skeletal elements, including the ver-tebral column and ribs. To examine whether the overgrownmesenchymal cells in the ptc2/ptcmes embryos are derivedrom the sclerotome, we analyzed the expression pattern ofsclerotomal marker, Pax1. Section in situ hybridization oftc2/ptcmes embryos at 11.5 dpc revealed no significantlteration of the expression domain of Pax1 (Figs. 5C andD). This observation indicated that the increased numberf mesenchymal cells in the ptc2/ptcmes trunk does not
result from the migration of overgrown sclerotomal cells.We also examined the expression patterns of the Shh and
ptc genes in the ptc2/ptcmes embryos at 11.5 and 12.5 dpc,espectively. No significant alteration of the expressionattern of either gene was observed, and no ectopic expres-ion was observed in the dorsal neural tube and dorsalesenchyme of the trunk (Figs. 5E–5H).
Dorsal–Ventral Axis Formation in DevelopingSpinal Cord of mes Homozygotesand ptc2/ptcmes Embryos
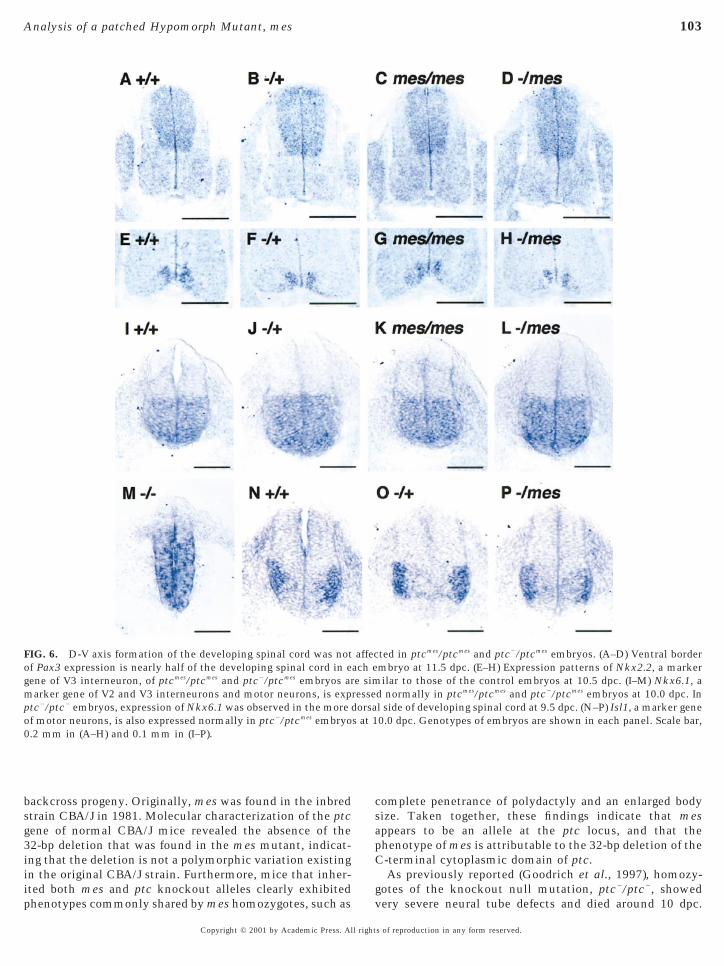
Since it is known that Shh signaling controls the fate ofneural progenitor cells along the dorsal–ventral (D-V) axisof the neural tube (Ericson et al., 1996, 1997; Goodrich etal., 1997), we analyzed the expression of several markergenes, Pax3, Isl1, Nkx2.2, and Nkx6.1, in the mutantembryos. In normal embryos, Pax3 is expressed in thedorsal half of the developing spinal cord (Fig. 6A). Isl1 andNkx2.2 are expressed in the progenitor cells of the ventraldeveloping spinal cord, corresponding to the motor neu-rons and V3 interneurons, respectively (Figs. 6E and 6N).Nkx6.1 is expressed at the three classes of ventral neu-rons, V2 interneurons, motor neurons, and V3 interneu-rons (Fig. 6I). Unexpectedly, no gross alteration of theexpression domain of these four genes was observed inthe developing spinal cord of the ptcmes/ptcmes, ptc2/ptcmes
(Figs. 6C, 6D, 6G, 6H, 6K, 6L, and 6P), as compared withptc2/1 and 1/1 embryos. This result indicated that the
-V axis formation of the developing spinal cord of bothtcmes/ptcmes and ptc2/ptcmes embryos was not affected.
We concluded that the C-terminal cytoplasmic domainhas a dispensable role in normal pattern formation of thedeveloping spinal cord.
Ectopic Activation of Shh Signaling in the AnteriorMesenchyme of the Limb Buds of ptc2/ptcmes
Embryos
Many mouse mutants that exhibit preaxial polydactylyshow ectopic activation of Shh signaling in the anterior
mesenchyme of the limb buds (Masuya et al., 1995).s of reproduction in any form reserved.

(SiT
odPtm
scEiee
102 Makino et al.
Since ptcmes/ptcmes and ptc2/ptcmes embryos exhibited pre-axial polydactyly in both fore- and hindlimbs with perfectpenetrance, we first analyzed the expression of Shh. At11.5 dpc, ectopic expression of Shh was not observeddata not shown). At 12.0 dpc, very weak expression ofhh was observed at the anterior margin of the limb budsn a part of the embryos to be examined (Figs. 3D and 3E).o ascertain whether Shh signaling was activated in the
anterior mesenchyme, we analyzed the expression of Gli,which is known to be a downstream gene of Shh. As aresult, Gli was expressed substantially at the anterior
FIG. 5. Expression of marker genes was not affected in the trections of ptc2/1 (A) and ptc2/ptcmes (B) embryos at 13.5 dpc.ells. (C, D) A sclerotomal marker gene, Pax1, is expressed simctopic expression of Shh is not observed in the dorsal trunk ofn the notochord and the floor plate of the ptc2/ptcmes embryo ixpression pattern of ptc in the trunk of the ptc2/ptcmes embryo (Hxpression of ptc is not observed in the dorsal mesenchyme of t
is not observed between the ptc2/1 (I) and the ptc2/ptcmes (J) em(A–H), and 0.1 mm in (I) and (J).
mesenchyme at 11.5 dpc prior to the ectopic expression
Copyright © 2001 by Academic Press. All right
f Shh, and at 12.0 dpc as well (Figs. 3F–3I). This resultemonstrated that the C-terminal cytoplasmic domain oftc, which is lost in the mes mutation, has a function inhe negative regulation of Shh signaling in the anterioresenchyme.
DISCUSSION
mes Is a ptc AlleleIn this study, no recombinant between the mes and ptc
of ptc2/ptcmes embryos. (A, B) Hematoxylin and eosin-stainedwheads indicate abnormally thickened region of mesenchymaly in ptc2/1 (C) and ptc2/ptcmes embryos (D) at 11.5 dpc. (E, F)dpc ptc2/ptcmes embryos (F), and the expression pattern of Shhilar to that of the ptc2/1 embryo (E) at this stage. (G, H) The
also similar to that of the ptc2/1 embryo (G) at 12.5 dpc. Ectopicc2/ptcmes embryo. (I, J) Gross difference in the proliferation rates in the dorsal mesenchyme at 12.5 dpc. Scale bar, 0.5 mm in
unkArroilarl11.5s sim) is
he ptbryo
genes was found in the linkage analysis based on 241
s of reproduction in any form reserved.
ogm
0
103Analysis of a patched Hypomorph Mutant, mes
backcross progeny. Originally, mes was found in the inbredstrain CBA/J in 1981. Molecular characterization of the ptcgene of normal CBA/J mice revealed the absence of the32-bp deletion that was found in the mes mutant, indicat-ing that the deletion is not a polymorphic variation existingin the original CBA/J strain. Furthermore, mice that inher-ited both mes and ptc knockout alleles clearly exhibited
FIG. 6. D-V axis formation of the developing spinal cord was notf Pax3 expression is nearly half of the developing spinal cord in eene of V3 interneuron, of ptcmes/ptcmes and ptc2/ptcmes embryos ararker gene of V2 and V3 interneurons and motor neurons, is exp
ptc2/ptc2 embryos, expression of Nkx6.1 was observed in the moreof motor neurons, is also expressed normally in ptc2/ptcmes embryo.2 mm in (A–H) and 0.1 mm in (I–P).
phenotypes commonly shared by mes homozygotes, such as
Copyright © 2001 by Academic Press. All right
complete penetrance of polydactyly and an enlarged bodysize. Taken together, these findings indicate that mesappears to be an allele at the ptc locus, and that thephenotype of mes is attributable to the 32-bp deletion of theC-terminal cytoplasmic domain of ptc.
As previously reported (Goodrich et al., 1997), homozy-gotes of the knockout null mutation, ptc2/ptc2, showed
ted in ptcmes/ptcmes and ptc2/ptcmes embryos. (A–D) Ventral bordermbryo at 11.5 dpc. (E–H) Expression patterns of Nkx2.2, a markerilar to those of the control embryos at 10.5 dpc. (I–M) Nkx6.1, a
d normally in ptcmes/ptcmes and ptc2/ptcmes embryos at 10.0 dpc. Inl side of developing spinal cord at 9.5 dpc. (N–P) Isl1, a marker gene0.0 dpc. Genotypes of embryos are shown in each panel. Scale bar,
affecach ee simressedorsas at 1
very severe neural tube defects and died around 10 dpc.
s of reproduction in any form reserved.

rsal
nticmpHc
idstsfmtfm
(aNdXM
t
eotl
Mdcwmc
mdrdtsanb
ttw
104 Makino et al.
Homozygotes of the mes mutation, ptcmes/ptcmes, exhibitather milder phenotypes of preaxial polydactyly andlightly increased body size (Sweet et al., 1996). Thus, mespparently retains ptc activity to some extent and is mostikely a hypomorphic allele of ptc.
The C-Terminal Cytoplasmic Domain Lost in Ptcmes
Is Not Essential for Patterning of the DevelopingSpinal Cord, but Indispensable for AnteroposteriorAxis Formation of Limb Buds
It was reported that the ptc2 homozygous embryos,which were partially rescued by the wild-type ptc transgeneand survived until 14.5 dpc, showed a striking defect of D-Vaxis formation of the neural tube (Milenkovic et al., 1999).The fact that the same embryos showed normal overallmorphology other than in the limbs and neural tubesappears to rule out the possibility that only quantitativereduction of the Ptc activity results in failure of the D-Vaxis formation of the neural tube in the mes mutant. Itsuggests that the C-terminal cytoplasmic domain deleted inPtcmes has a qualitatively distinct function in the axisformation of the limb buds, but not in pattern formation inspinal cord development.
Ubiquitous expression of truncated Ptc that had lostalmost all of the C-terminal cytoplasmic domain of Dro-sophila Ptc could not rescue the cuticle phenotype of ptc
ull mutant embryos (Johnson et al., 2000). Moreover, theruncated Ptc dominantly abolished the ability of Ptc tonhibit downstream genes of Hh signaling, while the trun-ated Ptc still retained the ability to sequester the Hholecule. These results indicate that the C-terminal cyto-
lasmic domain of Drosophila Ptc is indispensable for theh signaling. In contrast, neither mes homozygotes nor
ompound heterozygotes (ptc2/ptcmes) have gross anomaliesin the D-V axis formation of the developing spinal cord, andthe effect of the truncation of Ptcmes is not a dominant trait.How can we interpret the difference in these two experi-ments? The truncated Drosophila Ptc retains only the first14 amino acids after the last transmembrane domain,whereas the Ptcmes retains the first 53 amino acids. Thus, its possible that the first 53 amino acids of the C-terminalomain have an important role of Ptc function in Shhignaling pathway. Alternatively, as the result was based onhe effect of overexpression of the truncated Ptc on Hhignaling in the case of Drosophila, the data might resultrom a nonphysiologically high expression level of the
utant form of Drosophila Ptc. Finally, it is also possiblehat the difference may simply indicate divergence of theunction of the C-terminal cytoplasmic domain between
ouse and Drosophila.Ptc is highly conserved from Drosophila to humans
Concordet et al., 1996; Goodrich et al., 1996; Johnson etl., 1996; Kuwabara et al., 2000; Marigo et al., 1996a;akano et al., 1989; Takabatake et al., 2000). Ptc paralogs,
esignated as Ptc2, have been also identified in zebrafish,enopus, chick, mouse, and human (Carpenter et al., 1998;
otoyama et al., 1998; Takabatake et al., 2000). The SCopyright © 2001 by Academic Press. All right
runcated form of Ptc encoded by the ptcmes allele is almostidentical in length to Ptc2 in mouse and human, XenopusPtc1, and zebrafish Ptc. The C-terminal cytoplasmic do-main lost in Ptcmes has been retained during the vertebratevolution with the emergence of various tetrapod specificrgans, such as lungs and limbs. In this context, it is notablehat the ptc2/ptcmes embryos showed abnormality in theung and limb development.
Increased Body Weight in the ptc2/ptcmes EmbryosPrevious studies (Goodrich et al., 1997; Hahn et al., 1998;ilenkovic et al., 1999) suggested that ptc is a major
eterminant of body weight in utero. The present studyonfirmed that ptc indeed functions to downregulate bodyeight in normal development. Since body weight isainly determined by the number of cells that an animal
ontains (Conlon and Raff, 1999), ptc negatively regulatesthe proliferation of cells in embryos. In this study, we couldnot detect ectopic expression of Shh or a gross hyper-proliferation of sclerotomal cells in the ptc2/ptcmes em-bryos. Hence, the increased proliferation of mesenchymalcells observed in the trunk of the ptc2/ptcmes embryos wasunlikely to have resulted as a consequence of either ectopicexpression or an increase of Shh ligand. Instead, this obser-vation suggests that Ptc protein acts as a negative regulatorof cell proliferation in the dorsal trunk where Shh cannotmigrate.
One of other important determinants of the body weightof developing mouse embryos is insulin-like growth factors,IGFI and IGFII. Homozygotes of knockout mutations ofIGFI, -II, and the type I IGF receptor (IGF1R) exhibit severegrowth deficiency (Baker et al., 1993; DeChiara et al., 1990;Liu et al., 1993; Powell-Braxton et al., 1993). In IGF1R
utants, which show a very severe phenotype, growtheficiency appeared from the stage at 11.5 dpc. It waseported that ptc2/1 mice have a high incidence of rhab-omyosarcomas (RMS), which is the most common soft-issue sarcoma in children. In mice with RMS, overexpres-ion of IGFII was observed (Hahn et al., 1998). Furthermore,n increased level of IGFII expression was observed inormal embryonic tissues of mice which had lost one oroth ptc alleles (Hahn et al., 2000). This fact implies that
the IGF signal has a role in the increased body weight of theptc2/ptcmes embryos. However, the serum IGFI level of meshomozygotes (ptcmes/ptcmes) is not increased compared withhat of the control littermates (Sweet et al., 1996). Itherefore remains an open question whether increased bodyeight of ptc2/ptcmes embryos resulted from deregulation of
the expression level of IGFs.
ACKNOWLEDGMENTS
We thank Dr. C.-c. Hui for reading the manuscript and providinguseful comments, Drs. M. Nakafuku and Y. Hashimoto for helpfulsuggestions and discussions. We are grateful to Dr. U. Deutsch forthe Pax1 and Pax3 probes, Dr. A. P. McMahon for Shh probe, Dr. T.
aito for Isl1 probe, Dr. M. Nakafuku for Nkx2.2 probe, Dr. A.s of reproduction in any form reserved.

arapCSJI
105Analysis of a patched Hypomorph Mutant, mes
Joyner for Gli probe, and Dr. J. Rubenstein for Nkx6.1 probe. We arelso grateful to Drs. T. Saito and M. Tamura for technical adviceegarding section in situ hybridization. We also thank Ms. M. Ariind K. Aida for maintenance of mice. This study was supported inart by grants in aid from the Ministry of Education, Science andulture of Japan, and by Special Coordination Funds for Promotingcience and Technology of the Science and Technology Agency,apan. This paper is contribution no. 2446 from the Nationalnstitute of Genetics, Japan.
REFERENCES
Alcedo, J., Ayzenzon, M., Von Ohlen, T., Noll, M., and Hooper, J. E.(1996). The Drosophila smoothened gene encodes a seven-passmembrane protein, a putative receptor for the hedgehog signal.Cell 86, 221–232.
Baker, J., Liu, J. P., Robertson, E. J., and Efstratiadis, A. (1993). Roleof insulin-like growth factors in embryonic and postnatalgrowth. Cell 75, 73–82.
Bellusci, S., Furuta, Y., Rush, M. G., Henderson, R., Winnier, G.,and Hogan, B. L. (1997). Involvement of Sonic hedgehog (Shh) inmouse embryonic lung growth and morphogenesis. Develop-ment 124, 53–63.
Birren, S. J., Lo, L., and Anderson, D. J. (1993). Sympatheticneuroblasts undergo a developmental switch in trophic depen-dence. Development 119, 597–610.
Bitgood, M. J., and McMahon, A. P. (1995). Hedgehog and Bmpgenes are coexpressed at many diverse sites of cell- cell interac-tion in the mouse embryo. Dev. Biol. 172, 126–138.
Capdevila, J., Pariente, F., Sampedro, J., Alonso, J. L., and Guerrero,I. (1994). Subcellular localization of the segment polarity proteinpatched suggests an interaction with the wingless receptioncomplex in Drosophila embryos. Development 120, 987–998.
Carpenter, D., Stone, D. M., Brush, J., Ryan, A., Armanini, M.,Frantz, G., Rosenthal, A., and de Sauvage, F. J. (1998). Character-ization of two patched receptors for the vertebrate hedgehogprotein family. Proc. Natl. Acad. Sci. USA 95, 13630–13634.
Chiang, C., Litingtung, Y., Lee, E., Young, K. E., Corden, J. L.,Westphal, H., and Beachy, P. A. (1996). Cyclopia and defectiveaxial patterning in mice lacking Sonic hedgehog gene function.Nature 383, 407–413.
Concordet, J. P., Lewis, K. E., Moore, J. W., Goodrich, L. V.,Johnson, R. L., Scott, M. P., and Ingham, P. W. (1996). Spatialregulation of a zebrafish patched homologue reflects the roles ofsonic hedgehog and protein kinase A in neural tube and somitepatterning. Development 122, 2835–2846.
Conlon, I., and Raff, M. (1999). Size control in animal development.Cell 96, 235–244.
DeChiara, T. M., Efstratiadis, A., and Robertson, E. J. (1990). Agrowth-deficiency phenotype in heterozygous mice carrying aninsulin- like growth factor II gene disrupted by targeting. Nature345, 78–80.
Deutsch, U., Dressler, G. R., and Gruss, P. (1988). Pax 1, a memberof a paired box homologous murine gene family, is expressed insegmented structures during development. Cell 53, 617–625.
Echelard, Y., Epstein, D. J., St-Jacques, B., Shen, L., Mohler, J.,McMahon, J. A., and McMahon, A. P. (1993). Sonic hedgehog, amember of a family of putative signaling molecules, is impli-cated in the regulation of CNS polarity. Cell 75, 1417–1430.
Ericson, J., Morton, S., Kawakami, A., Roelink, H., and Jessell,
T. M. (1996). Two critical periods of Sonic Hedgehog signalingCopyright © 2001 by Academic Press. All right
required for the specification of motor neuron identity. Cell 87,661–673.
Ericson, J., Rashbass, P., Schedl, A., Brenner-Morton, S., Kawakami,A., van Heyningen, V., Jessell, T. M., and Briscoe, J. (1997). Pax6controls progenitor cell identity and neuronal fate in response tograded Shh signaling. Cell 90, 169–180.
Fan, C. M., and Tessier-Lavigne, M. (1994). Patterning of mamma-lian somites by surface ectoderm and notochord: Evidence forsclerotome induction by a hedgehog homolog. Cell 79, 1175–1186.
Goodrich, L. V., Johnson, R. L., Milenkovic, L., McMahon, J. A.,and Scott, M. P. (1996). Conservation of the hedgehog/patchedsignaling pathway from flies to mice: Induction of a mousepatched gene by Hedgehog. Genes Dev. 10, 301–312.
Goodrich, L. V., Milenkovic, L., Higgins, K. M., and Scott, M. P.(1997). Altered neural cell fates and medulloblastoma in mousepatched mutants. Science 277, 1109–1113.
Gorlin, R. J. (1987). Nevoid basal-cell carcinoma syndrome. Medi-cine (Baltimore) 66, 98–113.
Goulding, M. D., Chalepakis, G., Deutsch, U., Erselius, J. R., andGruss, P. (1991). Pax-3, a novel murine DNA binding proteinexpressed during early neurogenesis. EMBO J. 10, 1135–1147.
Hahn, H., Wojnowski, L., Specht, K., Kappler, R., Calzada-Wack, J.,Potter, D., Zimmer, A., Muller, U., Samson, E., and Quintanilla-Martinez, L. (2000). Patched target Igf2 is indispensable for theformation of medulloblastoma and rhabdomyosarcoma. J. Biol.Chem. 275, 28341–28344.
Hahn, H., Wojnowski, L., Zimmer, A. M., Hall, J., Miller, G., andZimmer, A. (1998). Rhabdomyosarcomas and radiation hypersen-sitivity in a mouse model of Gorlin syndrome. Nat. Med. 4,619–622.
Hooper, J. E. (1994). Distinct pathways for autocrine and paracrineWingless signalling in Drosophila embryos. Nature 372, 461–464.
Hooper, J. E., and Scott, M. P. (1989). The Drosophila patched geneencodes a putative membrane protein required for segmentalpatterning. Cell 59, 751–765.
Ingham, P. W., Taylor, A. M., and Nakano, Y. (1991). Role of theDrosophila patched gene in positional signalling. Nature 353,184–187.
Johnson, R. L., Laufer, E., Riddle, R. D., and Tabin, C. (1994).Ectopic expression of Sonic hedgehog alters dorsal-ventral pat-terning of somites. Cell 79, 1165–1173.
Johnson, R. L., Milenkovic, L., and Scott, M. P. (2000). In vivofunctions of the patched protein: Requirement of the C terminusfor target gene inactivation but not Hedgehog sequestration. Mol.Cell 6, 467–478.
Johnson, R. L., Rothman, A. L., Xie, J., Goodrich, L. V., Bare, J. W.,Bonifas, J. M., Quinn, A. G., Myers, R. M., Cox, D. R., Epstein,E. H., Jr., and Scott, M. P. (1996). Human homolog of patched, acandidate gene for the basal cell nevus syndrome. Science 272,1668–1671.
Kuwabara, P. E., Lee, M. H., Schedl, T., and Jefferis, G. S. (2000). AC. elegans patched gene, ptc-1, functions in germ-line cytokine-sis. Genes Dev. 14, 1933–1944.
Liu, J. P., Baker, J., Perkins, A. S., Robertson, E. J., and Efstratiadis,A. (1993). Mice carrying null mutations of the genes encodinginsulin-like growth factor I (Igf-1) and type 1 IGF receptor (Igf1r).Cell 75, 59–72.
Marigo, V., Davey, R. A., Zuo, Y., Cunningham, J. M., and Tabin,C. J. (1996a). Biochemical evidence that patched is the Hedgehogreceptor. Nature 384, 176–179.
Marigo, V., Scott, M. P., Johnson, R. L., Goodrich, L. V., and Tabin,
s of reproduction in any form reserved.

M
M
M
N
P
P
106 Makino et al.
C. J. (1996b). Conservation in hedgehog signaling: induction of achicken patched homolog by Sonic hedgehog in the developinglimb. Development 122, 1225–1233.
Masuya, H., Sagai, T., Wakana, S., Moriwaki, K., and Shiroishi, T.(1995). A duplicated zone of polarizing activity in polydactylousmouse mutants. Genes Dev. 9, 1645–1653.ilenkovic, L., Goodrich, L. V., Higgins, K. M., and Scott, M. P.(1999). Mouse patched1 controls body size determination andlimb patterning. Development 126, 4431–4440.otoyama, J., Takabatake, T., Takeshima, K., and Hui, C. (1998).Ptch2, a second mouse Patched gene is co-expressed with Sonichedgehog. Nat. Genet. 18, 104–106.urone, M., Rosenthal, A., and de Sauvage, F. J. (1999). Hedgehogsignal transduction: From flies to vertebrates. Exp. Cell Res. 253,25–33.akano, Y., Guerrero, I., Hidalgo, A., Taylor, A., Whittle, J. R., andIngham, P. W. (1989). A protein with several possible membrane-spanning domains encoded by the Drosophila segment polaritygene patched. Nature 341, 508–513.
latt, K. A., Michaud, J., and Joyner, A. L. (1997). Expression of themouse Gli and Ptc genes is adjacent to embryonic sources ofhedgehog signals suggesting a conservation of pathways betweenflies and mice. Mech. Dev. 62, 121–135.
owell-Braxton, L., Hollingshead, P., Warburton, C., Dowd, M.,Pitts-Meek, S., Dalton, D., Gillett, N., and Stewart, T. A. (1993).IGF-I is required for normal embryonic growth in mice. GenesDev. 7, 2609–2617.
Prince, V., and Lumsden, A. (1994). Hoxa-2 expression in normaland transposed rhombomeres: Independent regulation in the
neural tube and neural crest. Development 120, 911–923.Copyright © 2001 by Academic Press. All right
Qiu, M., Shimamura, K., Sussel, L., Chen, S., and Rubenstein, J. L.(1998). Control of anteroposterior and dorsoventral domains ofNkx-6.1 gene expression relative to other Nkx genes duringvertebrate CNS development. Mech. Dev. 72, 77–88.
Riddle, R. D., Johnson, R. L., Laufer, E., and Tabin, C. (1993). Sonichedgehog mediates the polarizing activity of the ZPA. Cell 75,1401–1416.
Stone, D. M., Hynes, M., Armanini, M., Swanson, T. A., Gu, Q.,Johnson, R. L., Scott, M. P., Pennica, D., Goddard, A., Phillips,H., Noll, M., Hooper, J. E., de Sauvage, F., and Rosenthal, A.(1996). The tumour-suppressor gene patched encodes a candidatereceptor for Sonic hedgehog. Nature 384, 129–134.
Sweet, H. O., Bronson, R. T., Donahue, L. R., and Davisson, M. T.(1996). Mesenchymal dysplasia: A recessive mutation on chro-mosome 13 of the mouse. J. Hered. 87, 87–95.
Takabatake, T., Takahashi, T. C., Takabatake, Y., Yamada, K.,Ogawa, M., and Takeshima, K. (2000). Distinct expression of twotypes of Xenopus Patched genes during early embryogenesis andhindlimb development. Mech. Dev. 98, 99–104.
van den Heuvel, M., and Ingham, P. W. (1996). smoothened encodesa receptor-like serpentine protein required for hedgehog signal-ling. Nature 382, 547–551.
Whiting, J., Marshall, H., Cook, M., Krumlauf, R., Rigby, P. W.,Stott, D., and Allemann, R. K. (1991). Multiple spatially specificenhancers are required to reconstruct the pattern of Hox-2.6 geneexpression. Genes Dev. 5, 2048–2059.
Received for publication April 16, 2001Revised July 29, 2001
Accepted July 31, 2001
Published online September 26, 2001s of reproduction in any form reserved.





























