Embed Size (px)
Citation preview

THE JOURNAL OF BIOLOGICAL CHEMISTRY 0 1993 by The American Society for Biochemistry and Molecular Biology, Inc.
Vol. 268, No. 13, Issue of May 5, pp. 9437-9441,1993 Printed in U. S. A.
A Variant of Exotoxin A That Forms Potent and Specific Chemically Conjugated Immunotoxins*
(Received for publication, December 7, 1992)
G. Jilani ChaudryS, R. Jerrold Fultons, and Rockford K. DraperSll From the $Molecular and Cell Biology Program, The University of Texas at Dallas, Richardson, Texas 75083 and the $Inland Laboratories, Inc., Dallas, Texas 75207
To introduce a free sulfhydryl into Pseudomonas aeruginosa exotoxin A (ETA), methionine 161 in do- main I of the toxin was changed to cysteine by site- directed mutagenesis. The free sulfhydryl provides a convenient site for covalent attachment of ETA to other proteins in the production of chimeric toxins. The mutation was then introduced into a variant of ETA that is impaired in receptor binding, termed ETA- 60EF61, that has the dipeptide Glu-Phe inserted be- tween residues 60 and 61. The resulting double mu- tant, ETA-60EF61Cys161, was conjugated to three different monoclonal antibodies via a thioether link- age, and the immunotoxins were tested for cytotoxicity with cells in culture. Each immunotoxin was extremely potent against cells that expressed surface determi- nants for the monoclonal antibodies but had little cy- totoxicity for cells that did not bind the antibodies. For comparison, we also conjugated ricin A chain to each of the three monoclonal antibodies and found that the resulting immunotoxins were at least two-orders of magnitude less potent than the corresponding immu- notoxins made with ETA-60EF61Cys161. This study demonstrates that ETA-60EF61Cys161 makes potent and specific immunotoxins and may potentially be use- ful in selectively eliminating subpopulations of cells in vitro and in vivo.
Pseudomonas aeruginosa exotoxin A (ETA)’ (Mr = 66,583) kills mammalian cells by a process involving at least three steps. First, the toxin binds to a cell surface receptor and is endocytosed. There is recent evidence that the receptor is the a2-macroglobulin receptor/low density lipoprotein receptor- related protein (Kounnas et al., 1992). Second, a carboxyl- terminal fragment of the toxin escapes from an intracellular compartment to enter the cytosol. The identity of the com- partment from which the toxin escapes is not clear, but it
* This work was supported by grants from the National Institutes of Health. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “aduertisernent” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
1 To whom correspondence should be addressed Molecular and Cell Biology Program, University of Texas at Dallas, P. 0. Box 830688, Richardson, TX 75083. Tel.: 214-690-2512; Fax 214-690-2409.
* The abbreviations used are: ETA, exotoxin A; dgRA, deglycosy- lated ricin A chain; DMEM, Dulbecco’s modified Eagle’s medium; ETA-60EF61, exotoxin A with the dipeptide Glu-Phe inserted be- tween the residues 60 and 61; ETA-Cysl61, exotoxin A with a cysteine substituted for methionine 161; ETA-60EF61Cys161, exotoxin A with Glu-Phe inserted between the residues 60 and 61 and a cysteine substituted for methionine 161; PBS, phosphate-buffered saline; SMCC, succinimidyl 4-[N-maleimidomethyl]cyclohexane-l-carbox- ylate; SPDP, N-succinimidyl-3-(2-pyridyldithio)propionate; bp, base pair(s).
may be the endoplasmic reticulum (Pastan et al., 1992; Pel- ham et al., 1992). Third, the toxin then catalyzes the covalent attachment of the adenosine diphosphate ribose (ADP-ribose) moiety of NAD+ to elongation factor 2 in the cytoplasm, thereby arresting protein synthesis (Iglewski and Kabat, 1975). Crystallographic studies of Allured et al. (1986) iden- tified three domains in the ETA protein that correlate with toxin functions (for reviews, see Wick et al., 1990; Pastan and FitzGerald, 1991; Pastan et al., 1992). Domain Ia (residues 1- 252) is the receptor-binding domain. There is also domain Ib (residues 365-399) whose role is unknown. Domain I1 (resi- dues 253-364) functions in membrane penetration and con- tains a loop that is proteolytically cleaved between residues 279 and 280, liberating a 37-kDa carboxyl-terminal fragment that is believed to eventually reach the cytosol (Ogata et al., 1990; Theuer et al., 1992). Domain I11 (residues 400-613) contains the catalytic center for ADP-ribosyl transferase ac- tivity. Adjacent to the carboxyl-terminal lysine, domain I11 also contains the tetrapeptide sequence REDL (residues 609- 612) that is essential for activity (Chaudhary et al., 1990). KDEL can substitute for the REDL sequence, and it has been speculated that after endocytosis the toxin binds to the KDEL receptor and is transported to the endoplasmic reticulum before penetrating to the cytosol (Pastan et al., 1992; Pelham et al., 1992).
There has been much work on combining protein toxins such as ETA with monoclonal antibodies to produce immu- notoxins that selectively attack target cells bearing determi- nants for the antibody. It is important that an immunotoxin attack only the desired target cells while simultaneously re- taining high potency. To be specific, the inherent receptor binding ability of the toxin molecule itself needs to be elimi- nated so that an immunotoxin does not interact with two receptors, the original toxin receptor and the determinant for the monoclonal antibody. Toward this end, we described a variant of ETA containing a dipeptide insert between residues 60 and 61 (termed ETA-60EF61) that strongly impaired the ability of ETA to bind receptors (Chaudry et al., 1989). However, when ETA-60EF61 was chemically derivatized to introduce reactive thiols at primary amines and coupled to transferrin, the resulting toxin was not very potent.2 One explanation for the loss in potency was that the toxin molecule had been damaged by chemical derivatization. To avoid chem- ically derivatizing the toxin, we have introduced a free cys- teine in domain Ia of ETA-60EF61 by site-directed mutagen- esis. The new cysteine residue provides a convenient moiety for conjugating the toxin to other proteins at a defined site in domain Ia. We report here that several immunotoxins made with this new derivative of ETA-60EF61 are highly specific and extremely potent.
G. J. Chaudry, R. J. Fulton, and R. K. Draper, unpublished data.
9437

9438 Exotoxin A-containing Immunotoxins
MATERIALS AND METHODS
Media and Biologicals-LB and LB agar were prepared as described in Maniatis et al. (1982). Tryptic soy broth dialysate was prepared as described by Iglewski and Sadoff (1979) with the following modifica- tions: the medium was dialyzed for 20 h, glycerol was then added (1.5%, w/v), and the medium was deferrated with Chelex-100 (Bio- Rad) for 20 h. The medium was filter sterilized (0.22-pm pore size) and supplemented with 50-75 mM monosodium glutamate and 1 mM MgSOd. Tetracycline was added at 10-20 pg/ml for Escherichia coli and 200 pg/ml for P. aeruginosa. Tryptic soy broth dialysate that was used to produce the toxins contained 300 pg/ml carbenecillin. DNA ligase and restriction enzymes were from Promega, Bethesda Re- search Laboratories, or New England Biolabs. SMCC and SPDP were from Pierce Chemical Co. Anti-ETA rabbit polyclonal antibody was described by Mozola et al. (1984).
Plasmids and Bacterial Strains-Table I describes plasmids used in this work. E. coli DH5a (F”P80dlacZAM15A(lacZYA-argF)U169 recAl endAl hsdR17(r~-,mk+)supE44 1- thi-1 gyrA relA1) was used as the host for site-directed mutagenesis, as well as other recombinant work. The nontoxigenic P. aeruginosa host PAlOBAtoxAl, carrying a deletion in the ETA gene (Chaudry, 1991), was used to produce the plasmid-encoded toxin variants.
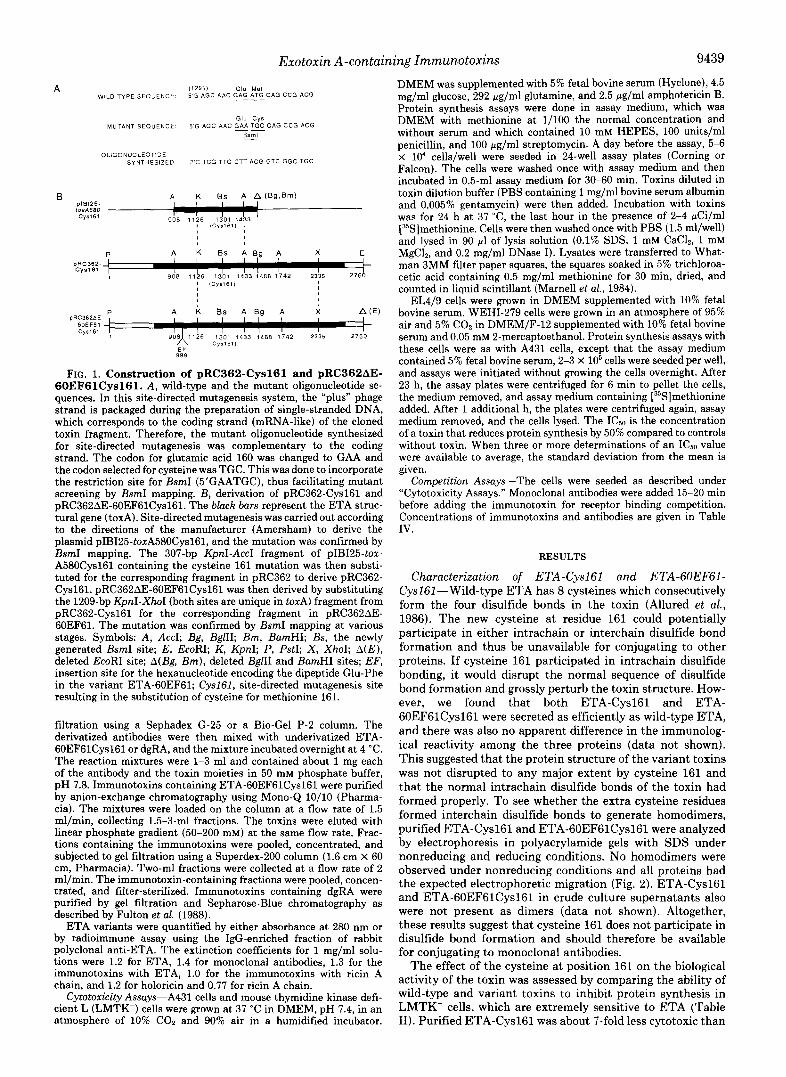
Construetion of pRC362-Cysl61 and pRC362AE-6OEFSlCyslSl- The strategy for site-directed mutagenesis and construction of pRC362-Cys161 and pRC362AE-60EF61Cys161 are summarized in Fig. 1. To substitute cysteine for methionine 161 (codon beginning at nucleotide 1301 of the 2.76-kilobase pair PstI-EcoRI fragment con- taining toxA) in domain I of ETA, the 580-bp Sari-BglII fragment (nucleotides 908-1488) of the toxin gene was first subcloned into the unique Sal1 and BamHI sites of pIBI25 (IBI). This subcloning de- stroyed the BamHI site of the vector and the BglII site of the tonA fragment, resulting in the plasmid pIBI25-toxA580. A 22-mer oligo- nucleotide, 3’CTCGTTGCTTACGGTCGGCTGC (Fig. lA), was syn- thesized and site-directed mutagenesis carried out using the Amer- sham Corp. Kit. The mutant sequence was designed such that it also contained a new restriction site for BsmI (5‘GAATGC), unique in pIB125-toxA580Cys161 (Fig. 1B). E. coli DH5a was transformed with the mutagenized plasmid, selecting for ampicillin resistance, and the plasmid DNA of several Amp’ clones was isolated. The desired mutants were identified by restriction enzyme mapping with BsmI.
pRC362-Cys161 and pRC362AE-60EF61Cysl61 were derived by substituting restriction enzyme fragments as shown in Fig. 1B. All the relevant fragments were separated using ultrapure, low melting point agarose (BRL), and the ligations were also in the same agarose. pRC362-Cys161 was derived by substituting the 307-bp KpnI-AccI fragment (nucleotides 1126-1433) of pIBI25-~0xA580Cys161, which contains the cysteine 161 mutation, for the corresponding fragment in pRC362. pRC362AE-60EF6lCys161 was then derived by substi- tuting the 1209-bp KpnI-XhoI fragment from pRC362-Cys161 for the corresponding fragment in pRC362AE-60EF61, a plasmid that en- codes the toxin variant ETA-60EF61 (Chaudry et d. , 1989). The KpnI and XhoI sites are unique in these plasmids. The constructs were confirmed by BsmI mapping. The plasmids were then introduced into the nontoxigenic P. aeruginosa host PA103AtoxAl to produce the plasmid-encoded toxin variants ETA-Cysl61 and ETA-6OEF- 61Cys161.
Purification of ETA Variants-ETA-Cysl61 and ETA-6OEF- 61Cys161 were purified from culture supernatants as described by Chaudry et al. (1989), except that the nontoxigenic P. aeruginosa host was PA103AtoxA1, a derivative of PA103 in which the tonA was deleted (Chaudry, 1991). The variants were further purified by chro- matography using a Pharmacia LKB Biotechnology Inc. fast protein liquid chromatography instrument. ETA-Cysl61 and ETA- 60EF61Cys161 preparations (in 20 mM Tris, pH 8.2, 1 mM EDTA, 1 mM 2-mercaptoethanol) were diluted with an equal volume of 50 mM sodium phosphate buffer, pH 7.8, and applied to a Q-Sepharose column (Pharmacia) a t a flow rate 1.5 ml/min. The toxins were eluted with a linear 50-200 mM sodium phosphate gradient, and the toxin- containing fractions were pooled. These preparations were then di- luted 3-fold and applied to a Mono-Q 10/10 column (Pharmacia). The column was washed with sodium phosphate buffer (50 mM, pH 7.8) until the absorbance at 280 nm was zero. The toxins were then eluted with a linear gradient of 50-300 mM sodium phosphate, pH 7.6, in a total gradient volume of 50 ml, and the fractions were monitored by checking absorbance at 280 nm. Each toxin preparation resolved into three peaks at sodium phosphate concentrations of 100 mM (peak l ) , 150 mM (peak 2), and 200 mM (peak 3). The fractions were also analyzed by electrophoresis in polyacrylamide gels with SDS, which showed that peak 1 was a 25-kDa protein contaminant, peak 2 was ETA, and peak 3 contained a nonprotein material, presumably pigment. The fractions containing ETA were pooled and concentrated by ultrafiltration (Amicon, 15,000 M. cutoff membrane). The preparations were then extensively dialyzed against 20 mM Tris, pH 8.2, 1 mM EDTA, 2 mM 2-mercaptoethanol, and the toxins were quantitated by absorbance at 280 nm or by a radioimmune assay (Tsaur and Clowes, 1989). The final preparations were analyzed by SDS-polyacrylamide gel (10%) electrophoresis under reducing and nonreducing conditions (Laemmli, 1970) and used for all assays.
Preparation of Ricin and dgRA-Ricin was purified as described by Nicolson et al. (1974) and deglycosylated according to Thorpe et al. (1985). Ricin A chain was prepared as described by Fulton et al. (1986).
Purification of Monoclonal Antibodies-Monoclonal antibodies were produced in tissue culture by growing the hybridoma cell lines a t 37 “C in 5% CO, in a 50:50 mixture of DMEM and Ham’s F-12 media containing 1% Nutridoma (Boehringer Mannheirn) and 0.1% fetal bovine serum. The monoclonal antibodies were purified from media by precipitation with 45% saturated ammonium sulfate, fol- lowed by ion-exchange chromatography essentially as described by Parham et al. (1982), except that Mono-Q (Pharmacia) was used instead of DEAE-cellulose. Monoclonal antibody 5E9, which reacts with the human transferrin receptor, was produced from American Type Culture Collection hybridoma HB 21. Hybridoma cells produc- ing monoclonal antibody 14C3, which reacts with Thy-1 antigen, were kindly provided by Dr. Paul Gottlieb, University of Texas at Austin. Hybridoma cells secreting monoclonal antibody 33-24.12, which reacts with surface IgM, were originally described by Leptin et al. ( 1984).
Construction and Purification of Zmmunotoxins-To couple mono- clonal antibodies to ETA-60EF61Cys161 via a thioether bond, the antibodies were derivatized with SMCC. To couple monoclonal anti- bodies to dgRA via a reducible disulfide bond, the antibodies were derivatized with SPDP. The unreacted linkers were removed by gel
TABLE I Plasmids used in this work
Plasmid Description
pRC360 A pUC9 derivative that contains the 2.76-kilobase pair PstI-EcoRI toxA fragment from P. aeruginosa strain PA103. The fragment, numbered from the PstI site (nucleotide 1) to the EcoRI site (nucleotide 2760), contains the exotoxin structural gene (toxA, nucleotides 746-2660), as well as upstream transcriptional control sequences (Chen et al., 1987).
that allows replication in P. aeruginosa is present. Also the PstI site distal to toxA is deleted (Chen et al., 1987).
pRC362 Similar to pRC360 except that a 1.85-kilobase pair PstI fragment from pR01614 (Olsen et al., 1982)
pRC362AE pRC362AE-60EF61
pRC362 with the EcoRI site at 2760 removed (Tsaur and Clowes, 1989). A derivative of pRC362AE that has the hexanucleotide 5’-CGAATT, encoding the dipeptide Glu-
Phe, inserted into the TaqI site at position 999 in tonA. The plasmid encodes the toxin variant ETA-60EF61 (Chaudry et al., 1989).
pIBI25-toxA580 Derived by subcloning the 580-bp SalI-BglII fragment of toxA in pIBI25. This work. pIBI25-t0xA580Cys161 Derived from pIBI25-toxA580 by substituting the codon for methionine 161 (nucleotide 1301) with a
pRC362-Cy~161 pRC362 with the codon for methionine 161 substituted with a codon for cysteine. This work. codon for cysteine. This work.
pRC362AE-60EF61Cysl61 Derived by combining the lesions in pRC362AE-60EF61 and pRC362-Cys161. This work.
Exotoxin A-containing Immunotoxins 9439
A WILD-TYPE SEOUENCE
MUTANT SEQUENCE
OLIGONUCLEOTIDE SYNTHESIZED
R A
(1291) G l u Me1 5’G AGC AAC GEA4GCAG CCG ACG
5’G AGC AAC E E C A G CCG ACG G l u C y s
E l
3’C TCG TTG CTT ACG GTC GGC TGC
- pd,”d:,”n. 2 Cy0161 908 1 1 2 6 1301 1433
I IC1.1611 I
P A K Bs A B g A X E
8 I C y n l 6 1 ) I
A K 6 s A B 9 A pRC362AE-
i 6OEF61 c y s r s 1
999 EF
FIG. 1. Construction of pRC362-Cys161 and pRC362AE- 60EF61Cys161. A, wild-type and the mutant oligonucleotide se- quences. In this site-directed mutagenesis system, the “plus” phage strand is packaged during the preparation of single-stranded DNA, which corresponds to the coding strand (mRNA-like) of the cloned toxin fragment. Therefore, the mutant oligonucleotide synthesized for site-directed mutagenesis was complementary to the coding strand. The codon for glutamic acid 160 was changed to GAA and the codon selected for cysteine was TGC. This was done to incorporate the restriction site for BsmI (5’GAATGC), thus facilitating mutant screening by BsmI mapping. B, derivation of pRC362-Cys161 and pRC362AE-60EF61Cys161. The black bars represent the ETA struc- tural gene (toxA). Site-directed mutagenesis was carried out according to the directions of the manufacturer (Amersham) to derive the plasmid pIBI25-toxA580Cys161, and the mutation was confirmed by BsmI mapping. The 307-bp KpnI-AccI fragment of pIBI25-toz- A580Cys161 containing the cysteine 161 mutation was then substi- tuted for the corresponding fragment in pRC362 to derive pRC362- Cysl61. pRC362AE-60EF61Cys161 was then derived by substituting the 1209-bp KpnI-XhoI (both sites are unique in torA) fragment from pRC362-Cys161 for the corresponding fragment in pRC362AE- 60EF61. The mutation was confirmed by BsmI mapping at various stages. Symbols: A, AccI; Bg, BglII; Bm, BamHI; Bs, the newly generated BsmI site; E, EcoRI; K, KpnI; P, PstI; X , XhoI; A(E), deleted EcoRI site; A(Bg, Bm), deleted BglII and BamHI sites; EF, insertion site for the hexanucleotide encoding the dipeptide Glu-Phe in the variant ETA-60EF61; Cysl61, site-directed mutagenesis site resulting in the substitution of cysteine for methionine 161.
filtration using a Sephadex G-25 or a Bio-Gel P-2 column. The derivatized antibodies were then mixed with underivatized ETA- 60EF61Cys161 or dgRA, and the mixture incubated overnight at 4 “C. The reaction mixtures were 1-3 ml and contained about 1 mg each of the antibody and the toxin moieties in 50 mM phosphate buffer, pH 7.8. Immunotoxins containing ETA-60EF61Cys161 were purified by anion-exchange chromatography using Mono-Q 10/10 (Pharma- cia). The mixtures were loaded on the column at a flow rate of 1.5 ml/min, collecting 1.5-3-ml fractions. The toxins were eluted with linear phosphate gradient (50-200 mM) at the same flow rate. Frac- tions containing the immunotoxins were pooled, concentrated, and subjected to gel filtration using a Superdex-200 column (1.6 cm x 60 cm, Pharmacia). Two-ml fractions were collected at a flow rate of 2 ml/min. The immunotoxin-containing fractions were pooled, concen- trated, and filter-sterilized. Immunotoxins containing dgRA were purified by gel filtration and Sepharose-Blue chromatography as described by Fulton et al. (1988).
ETA variants were quantified by either absorbance at 280 nm or by radioimmune assay using the IgG-enriched fraction of rabbit polyclonal anti-ETA. The extinction coefficients for 1 mg/ml solu- tions were 1.2 for ETA, 1.4 for monoclonal antibodies, 1.3 for the immunotoxins with ETA, 1.0 for the immunotoxins with ricin A chain, and 1.2 for holoricin and 0.77 for ricin A chain.
Cytotoxicity Assays”A431 cells and mouse thymidine kinase defi- cient L (LMTK-) cells were grown at 37 “C in DMEM, pH 7.4, in an atmosphere of 10% COZ and 90% air in a humidified incubator.
DMEM was supplemented with 5% fetal bovine serum (Hyclone), 4.5 mg/ml glucose, 292 pg/ml glutamine, and 2.5 pg/ml amphotericin B. Protein synthesis assays were done in assay medium, which was DMEM with methionine at 1/100 the normal concentration and without serum and which contained 10 mM HEPES, 100 units/ml penicillin, and 100 pg/ml streptomycin. A day before the assay, 5-6 x lo4 cells/well were seeded in 24-well assay plates (Corning or Falcon). The cells were washed once with assay medium and then incubated in 0.5-ml assay medium for 30-60 min. Toxins diluted in toxin dilution buffer (PBS containing 1 mg/ml bovine serum albumin and 0.005% gentamycin) were then added. Incubation with toxins was for 24 h a t 37 “C, the last hour in the presence of 2-4 pCi/ml [35S]methionine. Cells were then washed once with PBS (1.5 ml/well) and lysed in 90 pl of lysis solution (0.1% SDS, 1 mM CaCl2, 1 mM MgC12, and 0.2 mg/ml DNase I). Lysates were transferred to What- man 3MM filter paper squares, the squares soaked in 5% trichloroa- cetic acid containing 0.5 mg/ml methionine for 30 min, dried, and counted in liquid scintillant (Marnell et al., 1984).
EL4/9 cells were grown in DMEM supplemented with 10% fetal bovine serum. WEHI-279 cells were grown in an atmosphere of 95% air and 5% COz in DMEM/F-12 supplemented with 10% fetal bovine serum and 0.05 mM 2-mercaptoethanol. Protein synthesis assays with these cells were as with A431 cells, except that the assay medium contained 5% fetal bovine serum, 2-3 x lo5 cells were seeded per well, and assays were initiated without growing the cells overnight. After 23 h, the assay plates were centrifuged for 6 min to pellet the cells, the medium removed, and assay medium containing [35S]methionine added. After 1 additional h, the plates were centrifuged again, assay medium removed, and the cells lysed. The ICso is the concentration of a toxin that reduces protein synthesis by 50% compared to controls without toxin. When three or more determinations of an value were available to average, the standard deviation from the mean is given.
Competition Assays-The cells were seeded as described under “Cytotoxicity Assays.” Monoclonal antibodies were added 15-20 min before adding the immunotoxin for receptor binding competition. Concentrations of immunotoxins and antibodies are given in Table IV.
RESULTS
Characterization of ETA-Cysl61 and ETA-60EF61- Cys161-Wild-type ETA has 8 cysteines which consecutively form the four disulfide bonds in the toxin (Allured et al., 1986). The new cysteine at residue 161 could potentially participate in either intrachain or interchain disulfide bond formation and thus be unavailable for conjugating to other proteins. If cysteine 161 participated in intrachain disulfide bonding, it would disrupt the normal sequence of disulfide bond formation and grossly perturb the toxin structure. How- ever, we found that both ETA-Cysl61 and ETA- 60EF61Cys161 were secreted as efficiently as wild-type ETA, and there was also no apparent difference in the immunolog- ical reactivity among the three proteins (data not shown). This suggested that the protein structure of the variant toxins was not disrupted to any major extent by cysteine 161 and that the normal intrachain disulfide bonds of the toxin had formed properly. To see whether the extra cysteine residues formed interchain disulfide bonds to generate homodimers, purified ETA-Cysl61 and ETA-60EF61Cys161 were analyzed by electrophoresis in polyacrylamide gels with SDS under nonreducing and reducing conditions. No homodimers were observed under nonreducing conditions and all proteins had the expected electrophoretic migration (Fig. 2). ETA-Cysl61 and ETA-60EF61Cys161 in crude culture supernatants also were not present as dimers (data not shown). Altogether, these results suggest that cysteine 161 does not participate in disulfide bond formation and should therefore be available for conjugating to monoclonal antibodies.
The effect of the cysteine at position 161 on the biological activity of the toxin was assessed by comparing the ability of wild-type and variant toxins to inhibit protein synthesis in LMTK- cells, which are extremely sensitive to ETA (Table 11). Purified ETA-Cysl61 was about 7-fold less cytotoxic than

9440 Exotoxin A-containing Immunotoxirzs
1-4, -OME 5-9, +OME
1 2 3 4 5 6 7 8 9
FIG. 2. Analysis of wild-type and variant ETA proteins by SDS-polyacrylamide gel electrophoresis under reducing and nonreducing conditions. 9-10 pg of each toxin was applied to each lane of a 10% polyacrylamide gel, and the protein bands were visu- alized by Coomassie Blue staining. Lanes I and 6, ETA-Cysl61; lanes 2 and 7, ETA-60EF61Cys161; lanes 3 and 8, ETA-60EF61; lanes 4 and 9, wild-type ETA; lane 5, molecular weight standards. Lanes 1-4 are under nonreducing conditions and lanes 5-9 under reducing conditions. The molecular mass markers are, from top to bottom, 6- galactosidase (116 kDa), phosphorylase b (97 kDa), bovine serum albumin (66 kDa), ovalbumin (45 kDa), and carbonic anhydrase (29 kDa).
TABLE I1 Cytotoxicity of wild-type ETA and ETA variants on mouse
LMTK- cells
ETA (wild-type) 0.9 ETA-Cysl61 6.9 ETA-60EF61 450 ETA-60EF61C~161 1200
the wild-type toxin, and ETA-60EF61 was approximately 450- fold less cytotoxic. The presence of both mutations in ETA- 60EF61Cys161 reduced cytotoxicity about 1300-fold. I t is not clear why cysteine a t residue 161 reduces cytotoxicity, but considering that the mutation is in domain Ia it probably affects binding to the toxin receptor.
Preparation and Characterization of Zmmunotoxins-The enzymatically active moiety of exotoxin A that enters the cell cytosol is apparently part of a 37-kDa fragment generated by proteolytic cleavage between arginine 279 and glycine 280 in domain I1 (Ogata et al., 1990; Theuer et al., 1992). Considering that cysteine 161 is in domain I, we conjugated ETA- 60EF61Cys161 to monoclonal antibodies via a thioether bond, rather than a reducible disulfide bond, because domain I should be separated from the 37-kDa fragment after proteol- ysis. Conjugates made with thioether bonds should be more stable than those made with reducible disulfide bonds (Gre- gory, 1955; Marsh et al., 1988). To make the immunotoxins, SMCC-derivatized monoclonal antibodies were reacted with underivatized ETA-60EF61Cys161, and heterodimers were purified as described under "Materials and Methods." Elec- trophoresis of conjugates in polyacrylamide gels with SDS indicated that the immunotoxins were highly purified, and
the stoichiometry of ETA-60EF61Cys161 to antibody was one to one (data not shown).
Immunotoxins were made with three different monoclonal antibodies: 5E9, which reacts with the human transferrin receptor; 14C3, which reacts with the murine T-cell marker Thy-1; and 33-24.12 which reacts with surface IgM. Each of the immunotoxins was tested with cells lines that did or did not express determinants for the monoclonal antibodies, as listed in Table 111. Immunotoxins made with ETA- 60EF61Cys161 and each of the three monoclonal antibodies were extremely potent against appropriate target cells, with ICso values of about 1 PM or less. ETA-60EF61Cys161 alone had little cytotoxic activity against any of the cells. dgRA coupled with each of the three monoclonal antibodies was about 100-400-fold less cytotoxic toward the appropriate tar- get cells than immunotoxins containing ETA-60EF61Cys161 while dgRA alone was not cytotoxic. There was little cytotoxic activity of immunotoxins against non-target cell types, one indication that the immunotoxins were highly specific. TO further test specificity, cytotoxicity assays were done in the presence of free competing monoclonal antibodies (Table IV). Cytotoxicity was abolished when antibodies 5E9,14C3, or 33- 24.12 were present with their matching immunotoxins, but not when cross-tested with unmatching immunotoxins.
DISCUSSION
Several lines of evidence suggest that preparing immuno- toxins by chemically derivatizing ETA with SPDP or 2- iminothiolane, which modify lysine residues, would adversely affect the activity of the resulting immunotoxins. 1) The carboxyl-terminal residue of ETA, a lysine a t position 613, is adjacent to the REDL sequence that is necessary for cytotox- icity (Chaudhary et al. 1990), and derivatization of this lysine is likely to impair function of the REDL sequence. 2) In addition to lysine 613, domain I11 of ETA contains 2 other lysines at positions 590 and 606. Derivatization of these could impair passage through a membrane or the enzymatic activity of domain 111, or both. 3) Batra et al. (1989) compared the activity of conjugates made with PE40, a derivative of ETA lacking domain I, with the activity of conjugates made with LysPE40, which contains an extra lysine at the NH2 terminus. Conjugates made with LysPE40 were more toxic than those made with PE40, and the authors suggested that the enhanced potency resulted because the NH2-terminal lysine provided an extra site for conjugation, reducing conjugation at the other lysines in domain 111. 4) Recombinant chimeric toxins containing PE40 and variable regions of the monoclonal an- tibodies or growth factors are markedly more potent than the corresponding chemical conjugates with PE40 (Pastan et al., 1992). 5) Our own experience indicated that conjugating chemically derivatized ETA-60EF61 to transferrin did not make very cytotoxic conjugates.2
To avoid chemically derivatizing ETA with SPDP or 2- iminothiolane, we introduced a free sulfhydryl by substituting methionine 161 with cysteine. The mutation was made a t methionine 161 in domain I for the following reasons. In the crystal structure of wild-type ETA (Allured et al., 1986), methionine 161 is a surface residue, its side chain projecting away from the surface of the toxin molecule. Thus, it is likely that the side chain of cysteine would also project away from the surface, available to react with monoclonal antibodies. Position 161 is distant from the nearest disulfide bonds, one between cysteines 11 and 15 and the other between cysteines 197 and 214, and likely would not disrupt the sequence of existing disulfide bond formation. Position 161 is also on the opposite side of the protein from domains I1 and 111, and thus the ligands conjugated through cysteine 161 should not steri-

Antibody Toxin or immunotoxin EL419 (Thy-l)b
5E9 (anti-TfR) 5E9-ETA-60EF61Cys161 5E9 (anti-TfR) 5E9-dgRA 14C3 (anti-Thy-1) 14C3-ETA-60EF61Cy~161 14C3 (anti-Thy-1) 14C3-dgRA 33-24.12 (anti-IgM) 33-24.12-ETA-60EF61Cy~161 33-24.12 (anti-IgM) 33-24.12-dgRA
ETA-60EF61Cys161 dgRA ETA
1.2 k 0.23 480
1,300 Not tested >25,000
Not tested 17,000 80,000
97 & 6
>7,000 40,000
1.2 f 0.35 200 f 33 >28,000 >22,000
71,000 68,000
938
7,800 6,200 2,700
>44,000 0.3 f 0.04 30 5 10
45,000 98,000
130
a Human transferrin receptor (TfR) is expressed on the surface of A431 cells. * Thy-1 antigen is expressed on the surface of EL4/9 cells.
IgM is expressed on the surface of WEHI-279 cells.
TABLE IV Cytotoxicity of immunotoxins in the presence of matching or
unmatching antibodies Protein synthesis (% of control)
None 1 2 31 14 5E9 101 30 12 14C3 13 91 7 33-24.12 13 41 81
cally hinder the functions of domains I1 and 111. Finally, monoclonal antibodies attached to the new cysteine in domain I should not interfere with translocation of the 37-kDa car- boxyl fragment to the cytosol because domain I is believed to be proteolytically removed prior to translocation. Thus, it should be possible to conjugate antibodies to cysteine 161 by a stable, non-reducible thioether bond. The fact that the immunotoxins made with ETA-60EF61Cys161 via thioether linkages were so potent is consistent with evidence that the COOH-terminal portion of ETA is liberated prior to translo- cation (Ogata et al., 1990; Theuer et al., 1992).
I t was not anticipated that the cysteine substitution would reduce the cytotoxicity of ETA 7-fold, and the reason for the reduction is unclear. However, it seems likely that the new cysteine impairs binding to normal toxin receptors because it is in domain Ia. The presence of cysteine 161 further reduced the cytotoxicity of ETA-60EF61 an additional 3-fold, an advantage because this should decrease the nonspecific cyto- toxicity of immunotoxins.
Three monoclonal antibodies recognizing different recep- tors were used and all three immunotoxins containing ETA- 60EF61Cys161 were extremely potent toward appropriate tar- get cells. As a yardstick to measure the effectiveness of the constructs containing ETA-60EF61Cys161, each monoclonal antibody was also conjugated to dgRA. The corresponding immunotoxins with dgRA were a t least two-orders of magni- tude less potent than those made with ETA-60EF61Cys161. Several lines of evidence indicated that the immunotoxins containing ETA-60EF61Cys161 were highly specific. 1) The immunotoxins were more than four-orders of magnitude more potent than ETA-60EF61Cys161 alone. 2) There was little cytotoxicity for cells that failed to express determinants for
the antibodies. 3) Each immunotoxin was inhibited by the relevant monoclonal antibody.
In summary, the results presented here demonstrate that ETA-60EF61Cys161 forms immunotoxins that are highly po- tent and specific when tested in uitro. Future work will include studies with animal models to see if the potency and specificity of the immunotoxins is retained in uiuo.
Acknowledgments-We thank E. Jordan for constructing strain PAlOBAtoxAl. We are grateful to P. Colbaugh and C. Mikoryak for critically reading the manuscript.
REFERENCES Allured, V. S., Collier, R. J., Carroll, S. F., and Mckay, D. B. (1986) Proc. Natl.
Batra, J. K., Jinno, Y., Chaudhary, V. K., Kondo, T., Willingham, M. C., Acad. Sci. U. 5'. A. 83, 1320-1324
FitzGerald, D. J., and Pastan, I. (1989) Proc. Natl. Acad. Sci. U. S. A. 86,
Chaudry, G. J. (1991) Ph. D. dissertation, Universit of Texas at Dallas Chaudry, G. J., Wilson, R. B., Draper, R. K., and dowes, R. C. (1989) J. Bid.
Chaudhary, V. K., Jinno, Y., FitzGerald, D., and Pastan, L(1990) Proc. Natl.
Chen, S.-T., Jordan, E.,M., Wilson, R. B., Draper, R. K., and Clowes, R. C.
Fulton, R. J., Blakey, D. C., Knowles, P. P., Uhr, J. W., Thorpe, P. E., and
Fulton, R. J., Tucker, T. F., Vitetta, E. S., and Uhr, J. W. (1988) Cancer Res.
Gregory, J. D. (1955) J. Am. Chem. SOC. 77,392-393 Iglewski, B. H., and Kabat, D. (1975) Proc. Natl. Acad. Sci. U. S. A. 72, 2284-
8545-8549
Chem. 264, 15151-15156
Acad. Sci. U. S. A. 87,308-312
(1987) J. Gen. Microbzol. 133, 3081-3091
Vitetta, E. S. (1986) J. Biol. Chem. 261,5314-5319
48,2618-2625
92RR glewski, B. H., and Sadoff, J. C. (1979) Methods Enzymol. 6 8 , 780-793
ounnas, M. Z., Morris, R. E., Thompson, M. R., FitzGerald, D. J., Strickland, D. K., and Saelinger, C. B. (1992) J. Biol. Chem. 267,12420-12423
Laemmli, U. K. (1970) Nature 227,680-685 Le tin, M., Potash M. J. Grutzmann, R., Heusser, C., Shulman, M., Kohler,
Maniatis, T., Fritsch, E. F., and Sambrook, J. (1982) Molecular Cloning: a E. , and Melcheri, F. (1684) Eur. J . Immunol. 14, 534-542
kF%?Spr in Hargor, NY Manual, p 113-114 and 135-139, Cold Spring Harbor Labora-
Marnell, M. H., &a, S.-P., Stookey, M., and Draper, R. K. (1984) Infect. Immun. 44,145-150
Marsh, J. W., Srinivasachar, K., and Neville, D. M., Jr. (1988) in Immunotorim (Frankel, A. E., ed) pp. 213-237, Kluwer Academic Publishers Boston
Mozola, M. A,, Wilson, R. B., Jordan, E. M., Draper, R. K., andclowes, R. C. (1984) J. Bucterlol. 159,683-687
Nicolson, G. L., Blaustein, J., and Etzler, M. E. (1974) Biochemistry 13, 196-
""
2n4 O&i& M., Chaudhary, V. K., Pastan, I., and FitzGerald, D. J. (1990) J. Biol.
Olsen, R., DeBrusscher, H. G., and McCombie, W. R. (1982) J. Bacteriol. 150, Chem. 265,20678-20685
A n - m Parham, P., Androlewicz, M. J., Brodsky, F. M., Holmes, N. J., and Ways, J.
Pastan. I.. and FltzGerald. D. J. (19911 Sclence 254. 1173-1177
" "I
P. (1982) J. Immunol. Methods 6 3 , 133,173
Pastan; I.; Chaudhary, V.; and FitzGerald, D. J. (1992) Annu. Reu. Biochem.
Pelham, H. R. B., Roberts, L. M., and Lord, J. M. (1992) Trends Cell Biol. 2 ,
Theuer, C. P, FitzGerald, D. J., and Pastan, I. (1992) J. Biol. Chem. 267,
Thorpe, P. E., Detre,, S., Foxwell, B., Brown, A., Skilleter, D., Wilson, G.,
Tsaur M.-L. dnd Clowes R. C. (1989) J. Bacteriol. 171,2599-2604 Wick,". J., Hamood, A. N. , and Iglewski, B. H. (1990) Mol. Mierobiol. 4, 527-
61,331-354
183-185
16872-16877
Forrester, J. and Stlrpe F. (1985) Eur. J. Blochem. 147, 197-206
535


![Pentobra: A Potent Antibiotic with Multiple Layers of ...wonglab.seas.ucla.edu/pdf/2015 JID [Schmidt, Wong] Pentobra A Potent... · Pentobra: A Potent Antibiotic with Multiple Layers](https://img.pdfslide.net/doc/110x75/5e79535b6eb666031e579d24/pentobra-a-potent-antibiotic-with-multiple-layers-of-jid-schmidt-wong-pentobra.jpg)


























