Embed Size (px)
Citation preview

Ab Initio Molecular Dynamics on Energetic Materials and Detonation Mechanisms
Fan Zhang Defence R&D Canada – Suffield Tom K. Woo University of Western Ontario
Technical Report
DRDC Suffield TR 2003-156
December 2003
Defence Research and Recherche et développement Development Canada pour la défense Canada


Ab Initio Molecular Dynamics on Energetic Materials and Detonation Mechanisms
Fan Zhang Defence R&D Canada – Suffield P.O. Box 4000, Stn Main, Medicine Hat, Alberta, Canada T1A 8K6 Tom K. Woo Department of Chemistry, University of Western Ontario, London, Ontario, Canada N6A 5B7
Defence R&D Canada – Suffield Technical Report DRDC Suffield TR 2003-156 December 2003

© Her Majesty the Queen as represented by the Minister of National Defence, 2003
© Sa majesté la reine, représentée par le ministre de la Défense nationale, 2003

DRDC Suffield TR 2003-156 i
Abstract
The development of ab initio or “first principles” molecular dynamics, along with the rapid development of computer power, has inspired us to apply it to studies of energetic materials and detonation processes. Using Car-Parrinello ab initio molecular dynamics, a multimolecular collision model and a controlled temperature bulk liquid compression approach have been developed to study atomic-level decomposition mechanisms of homogeneous liquid energetic materials under shock and highly static compressed conditions. Liquid nitromethane was chosen as a prototypical molecular explosive. In multimolecular collision simulations, neighbouring molecules act as traps to confine the recoiling fragments produced during the initial collision, thereby allowing them to recombine. This results in higher threshold collision velocities than previously found with binary collision simulations. These threshold velocities (8-10 km/s) are higher than average atomic velocities expected at the detonation shock front, suggesting that molecular dissociation may occur with thermalization after the shock front passes. These threshold velocities also suggest that the excitation of kinetic motion degrees of freedom within the shock front can be a mechanism only for detonation initiation of a new generation of molecular explosives whose detonation velocities would be several times that of current molecular explosives. In bulk liquid compression simulations at controlled temperature (150-300 K), the activation barrier and the electronic energy gap decrease with increasing compression ratios. This provides a necessary condition for the super-compressed detonation of insensitive molecular explosives. Pressure decomposition occurs at three-fold compression accompanied with interatomic proton transfer reactions.

ii DRDC Suffield TR 2003-156
Résumé
Le développement ab initio ou des “principes fondamentaux” de la dynamique moléculaire ainsi que le développement rapide de la puissance des ordinateurs nous a inspirés de les appliquer à la recherche sur les matériaux énergétiques et le processus de la détonation. À l’aide de la dynamique moléculaire ab initio de Car-Parrinello, un modèle de collision multimoléculaire et une méthode de compression de liquide en vrac à température contrôlée ont été mis au point pour examiner des mécanismes de décomposition, au niveau atomique, des matériaux énergétiques de liquide homogène, dans des conditions de choc et de compression hautement statique. Le nitrométhane liquide a été choisi comme explosif moléculaire prototypique. Dans les simulations de collisions multimoléculaires, les molécules avoisinantes agissent comme des trappes, confinant les fragments reculant qui sont produits durant la collision initiale, leur permettant ainsi de se recombiner. Ceci résulte en des vitesses limites d’entraînement plus hautes que celles trouvées précédemment dans les simulations de collisions binaires. Ces vitesses limites d’entraînement (de 8 à 10 km/s) sont plus hautes que les vélocités atomiques moyennes auxquelles on s’attend au front de l’onde de choc de la détonation, suggérant que la dissociation moléculaire peut se passer avec la thermalisation, après le passage du front de l’onde de choc. Ces vitesses limites suggèrent aussi que l’excitation des variances de la motion cinétique, à l’intérieur du front de l’onde de choc, peut être un mécanisme pour l’amorçage seulement de la détonation d’une nouvelle génération d’explosifs moléculaires dont les vitesses limites seraient plusieurs fois celles des explosifs moléculaires actuels. Durant les simulations de compression de liquide en vrac à des températures contrôlées (de 150 à 300 K), la barrière d’activation et l’intervalle d’énergie électronique diminuent avec l’augmentation des rapports de compression. Ceci fournit la condition nécessaire à la détonation par compression maximale d’explosifs moléculaires à risques atténués. La décomposition causée par la pression survient à un niveau de pression qui cause une réduction d’un facteur de trois du volume; cette décompositoin est accompagnée de réactions de transfert de protons interatomiques.

DRDC Suffield TR 2003-156 iii
Executive summary
In the field of molecular condensed matter detonation, we are currently confronted with several difficult problems. Is the initiation of detonation in common molecular explosives controlled by the thermal decomposition induced from shock temperature or by the excitation of non-equilibrium translational degrees of freedom within the shock front? Could the excitation of kinetic motion degrees of freedom and structural bond energy release become main mechanisms for the initiation and propagation of detonation in a new generation of molecular explosives with a detonation velocity several times that of the current explosives? Would pressure dissociation or momentum-induced decomposition at low temperatures be another possible mechanism for detonation resulting in hyper-velocity particles products? Experimental support to answer these questions must be derived under extreme conditions of high pressure from observations inside the shock front, thereby requiring measurements on the time scales of 10-2-1 ps.
The development of ab initio or quantum mechanical molecular dynamics, along with the rapid development of computer power, has offered an alternative to gain insights into these challenging problems and guidance to experimental research on energetic materials and detonation processes. High-energy-density materials must feature metastability and a large energy content that mostly originate in transformations of the molecular, atomic and electronic structures. Successful synthesis of these materials and control of the energy release from the structural bonds strongly rely on the understanding of the chemical processes and physics of structural transformations at the atomic level. Ab initio molecular dynamics can profoundly affect how energy storage and release via structural transformations and bond formation/breaking are understood and applied to defence technology.
For these reasons, under the auspices of Technology Investment Funding Project “Super-compressed Detonation”, we have used Car-Parrinello ab initio molecular dynamics and developed a multimolecular collision model and a controlled temperature bulk liquid compression approach to study decomposition mechanisms of homogeneous liquid energetic materials at the atomic level under shock and highly static compressed conditions. Liquid nitromethane was chosen as a prototypical molecular explosive. The main results of the simulations are summarized below.
1. In multimolecular collision simulations, neighbouring molecules act as traps to confine the recoiling fragments produced during the initial collision, thereby allowing them to recombine. This results in higher threshold collision velocities than previously found with binary collision simulations.
2. These threshold velocities (8-10 km/s) are higher than average atomic velocities expected at the detonation shock front, suggesting that molecular dissociation may occur with thermalization after the shock front passes.
3. These threshold velocities also suggest that the excitation of kinetic motion degrees of freedom within the shock front can be a mechanism for detonation initiation of a new generation of molecular explosives whose detonation velocities would be several fold that of current molecular explosives.

iv DRDC Suffield TR 2003-156
4. In bulk liquid compression simulations at controlled temperatures (300 K), the activation barrier and the electronic energy gap decrease with increasing compression ratios, theoretically suggesting a necessary condition for the super-compressed detonation of insensitive molecular explosives.
5. Pressure decomposition occurs at three-fold compression accompanied with intermolecular proton transfer reactions.
While these models and the simulation approaches on detonation require further development, the results demonstrated the capability and potential of the ab initio or quantum mechanical molecular dynamics for future advances in simulations of high-pressure synthesis studies of new energetic materials and their detonation mechanisms.
Zhang, F. and Woo, T. K. 2003. Ab Initio Molecular Dynamics on Energetic Materials and Detonation Mechanisms. DRDC Suffield TR 2003-156. Defence R&D Canada – Suffield.

DRDC Suffield TR 2003-156 v
Sommaire
Dans le domaine de la détonation de matière moléculaire condensée, nous nous trouvons actuellement confrontés à plusieurs problèmes difficiles. L’amorçage de la détonation des explosifs moléculaires courants est-il contrôlé par la décomposition thermale induite par la température du front de l’onde de choc ou par l’excitation hors équilibre de degrés de liberté de translation à l’intérieur du front de l’onde de choc? Se peut-il que l’excitation des variances de la motion cinétique et la libération d’énergie de liaison structurelle deviennent les mécanismes principaux de l’amorçage et de la propagation de la détonation chez la nouvelle génération d’explosifs moléculaires, ayant une vitesse de détonation plusieurs fois supérieure à celle des explosifs actuels ? Est-ce que la dissociation de pression ou la décomposition induite par l’inertie à des températures faibles, peut être un autre mécanisme possible de détonation, résultant en des produits de particules de haute vitesse ? Le soutien expérimental procurant la réponse à ces questions doit être obtenu sous des conditions extrêmes de haute pression provenant d’observation à l’intérieur du front de l’onde de choc exigeant ainsi des mesures à l’échelle de temps de 10-2-1 ps.
Le développement de la dynamique moléculaire ab initio ou de mécanique quantique ainsi que le développement rapide de la puissance des ordinateurs, offre un autre moyen d’acquérir une meilleure connaissance des problèmes et guide la recherche expérimentale vers les matériaux énergétiques et les processus de détonation. Les matériaux de haute densité et haute énergie doivent être caractérisés par la métastabilité et le contenu important en énergie, le plus souvent créé durant les transformations des structures moléculaires, atomiques et électroniques. Les synthèses réussies de ces matériaux et le contrôle de la libération d’énergie à partir des liens structurels reposent fortement sur la compréhension des processus chimiques et de la physique des transformations structurelles au niveau atomique. La dynamique moléculaire Ab initio peut profondément affecter la manière dont l’accumulation d’énergie et sa libération au moyen de transformations structurelles et de la formation / rupture des liens sont comprises et appliquées à la technologie de la défense.
Pour ces raisons et sous l’égide du projet FIT (Fonds d’investissement en technologie) “Détonation super-compressée”, nous avons utilisé la dynamique moléculaire ab initio de Car-Parrinello et nous avons mis au point un modèle de collision multimoléculaire et une méthode de compression de liquide en vrac, à température contrôlée, pour examiner les mécanismes de décomposition des matériaux énergétiques de liquides homogènes, au niveau atomique, dans des conditions de choc et de compression hautement statique. Le nitrométhane a été choisi comme explosif moléculaire prototypique. Les résultats principaux de ces simulations sont résumés ci-dessous.
1. Dans les simulations de collisions multimoléculaires, les molécules avoisinantes agissent comme des trappes confinant les fragments reculant qui sont produits durant la collision initiale, leur permettant ainsi de se recombiner. Ceci résulte en des vitesses limites d’entraînement plus hautes que celles trouvées précédemment avec les simulations de collisions binaires.

vi DRDC Suffield TR 2003-156
2. Ces vitesses limites d’entraînement (de 8 à 10 km/s) sont plus hautes que les vélocités atomiques moyennes auxquelles on s’attend au front de l’onde de choc de la détonation, suggérant que la dissociation moléculaire peut se passer avec la thermalisation, après le passage du front de l’onde de choc.
3. Ces vitesses limites suggèrent aussi que l’excitation des variances de la motion cinétique, à l’intérieur du front de l’onde de choc, peut être un mécanisme pour l’amorçage seulement de la détonation d’une nouvelle génération d’explosifs moléculaires dont les vitesses limites seraient plusieurs fois celles des explosifs moléculaires actuels.
4. Durant les simulations de compression de liquide en vrac à des températures contrôlées (300 K), la barrière d’activation et l’intervalle d’énergie électronique diminuent avec l’augmentation des rapports de compression, suggérant théoriquement, une condition nécessaire à la détonation par compression maximale d’explosifs moléculaires à risques atténués.
5. La décomposition causée par la pression survient à un niveau de pression qui cause une réduction d’un facteur de trois du volume; cette décompositoin est accompagnée de réactions de transfert de protons interatomiques.
Ces modèles et ces méthodes de simulation de détonation exigent la continuation de la mise au point mais les résultats indiquent déjà la capacité et le potentiel de la dynamique moléculaire ab initio ou de la mécanique quantique, en ce qui concerne les progrès des simulations futures de la recherche, au sujet des synthèses de haute pression des nouveaux matériaux énergétiques et des mécanismes de leur détonation.
Zhang, F. and Woo, T. K. 2003. Ab Initio Molecular Dynamics on Energetic Materials and Detonation Mechanisms. DRDC Suffield TR 2003-156. R & D pour la défense Canada – Suffield.

DRDC Suffield TR 2003-156 vii
Table of contents
Abstract........................................................................................................................................ i
Executive summary ................................................................................................................... iii
Sommaire.................................................................................................................................... v
Table of contents ...................................................................................................................... vii
List of figures...........................................................................................................................viii
1. Introduction ................................................................................................................... 1
2. Theory of Ab Initio Molecular Dynamics...................................................................... 3 2.1 Ab Initio Molecular Dynamics ......................................................................... 3 2.2 Density Functional Theory ............................................................................... 4 2.3 Car-Parrinello Method...................................................................................... 5 2.4 Plane Wave Technique ..................................................................................... 6
3. Computational Methods ................................................................................................ 7
4. Simulations of Shock Initiation ..................................................................................... 8 4.1 Review on Bimolecular Collisions ................................................................... 8 4.2 Multimolecular Collisions ................................................................................ 9
4.2.1 Impact of a Single Molecule on Multiple Molecules .......................... 9 4.2.2 Impact of Multiple Molecules on Multiple Molecules ...................... 12
5. Simulations of Pressure Dissociation .......................................................................... 18 5.1 Structural Properties of Compressed Liquid NM ........................................... 18 5.2 Molecular Decomposition in Compressed Liquid NM .................................. 24
6. Conclusions ................................................................................................................. 29
7. References ................................................................................................................... 31

viii DRDC Suffield TR 2003-156
List of figures
Figure 1. Bimolecular collision orientations with threshold velocities .................................. 8
Figure 2. Schematic of the single molecule on multiple molecule collision simulations....... 9
Figure 3. Snapshots from a 12.0 km/s head-to-tail orientation multimolecular collision simulation. Other neighbouring molecules have been removed for clarity. ......... 11
Figure 4. Schematic of the multiple molecule on multiple molecule collision simulations. 12
Figure 5. Average C-N bond length as a function of time at 2-10 km/s impact velocity..... 13
Figure 6. Average N-O bond length as a function of time at 2-10 km/s impact velocity..... 14
Figure 7. Average C-H bond length as a function of time at 2-10 km/s impact velocity..... 14
Figure 8. Average C-N bond length in static and dynamic simulations at 10 km/s impact velocity.................................................................................................................. 15
Figure 9. Average N-O bond length in static and dynamic simulations at 10 km/s impact velocity.................................................................................................................. 16
Figure 10. Average C-H bond length in static and dynamic simulations at 10 km/s impact velocity.................................................................................................................. 16
Figure 11. High pressure compression of bulk liquid nitromethane ...................................... 18
Figure 12. Pair correlation functions for the (a) C-C and C-N, (b) N-N and N-O, and (c) O-O and O-H pairs of NM at ambient pressure and 300 K. ............................ 20
Figure 13. O-O and O-H pair correlation functions calculated from Car-Parrinello MD simulations of liquid NM compressed by factors of 1.0, 1.5, 2.0, 2.5, and 3.0..... 21
Figure 14. Vibrational spectrum of compressed liquid NM computed with the Fourier Transform of the velocity auto-correlation function: (a) ambient liquid NM, liquid NM compressed by factors of: (b) 1.5, (c) 2.0, (d) 2.5, (e) 3.0................... 22
Figure 15. Time evolution of the average intramolecular bond distances of the 32 NM molecules in the simulation cell during the Car-Parrinello MD simulation.......... 23
Figure 16. Snapshots of the intermolecular H atom transfer events observed in the CPMD simulations of liquid nitromethane compressed by a factor of 3.0. ...................... 25
Figure 17. Schematic of the proton transfer reaction intermolecular H atom transfer events observed in the CPMD simulations of liquid nitromethane compressed by a factor of 3.0. .................................................................................................................... 25

DRDC Suffield TR 2003-156 ix
Figure 18. Reaction energy profiles for the pressure induced proton transfer reaction calculated from linear transit calculations using the incipient O-H bond as the reaction coordinate................................................................................................ 26
Figure 19. Molecular orbitals of nitromethane involved in the proton transfer reaction........ 27
Figure 20. Effect of decreasing C-N bond length on the NM MOs involved in the proton transfer reaction; (a) change in HOMO-LUMO energy gap and (b) change in LUMO................................................................................................................... 28
List of tables
Table 1. Threshold collision velocities from bimolecular and multimolecular collisions........ 10

x DRDC Suffield TR 2003-156
This page intentionally left blank.

DRDC Suffield TR 2003-156 1
1. Introduction
Recent computer technology has changed profoundly the way in which modern scientific research is conducted. At a rather trivial but practically highly relevant level, computer power has sped up considerably the pace of theoretical schemes and approaches in bulk material research at the atomic level over the past decade. The results of the simulations are of great help in guidance of experiments and provide invaluable insight into system behaviour, particularly under extreme conditions of temperature and pressure. In this respect, a major role has been played by molecular dynamics (MD), which since the pioneering efforts of the 1960s [1-2] has developed into a mature and active discipline that has been used as a means of simulating and understanding the properties of real systems. Major progress has been achieved in the development of ab initio or “first principles” MD, in which the potential energy and interatomic forces are derived from accurate electronic calculations of first-principle quantum chemistry that are performed as the simulation proceeds [3-7]. This has greatly improved the predictive power of the simulation and opens the way for the reliable simulation of processes in which chemical bonds are formed and broken. This kind of process is simulated with great difficulty in classical MD based on classical mechanics and empirically derived potentials [8-11].
The development of ab initio MD, along with the rapid development of computer power, has inspired us to apply it to studies of energetic materials and detonation processes. There are several difficult problems currently encountered in the field of molecular condensed matter detonation. Is the initiation of detonation in existing molecular explosives controlled by the thermal decomposition via shock temperature or by the excitation of kinetic motion degrees of freedom within the shock front as speculated by Dremin et al. [8, 12]? Could the excitation of kinetic motion degrees of freedom and structural bond energy release become main mechanisms for the initiation and propagation of detonation in a new generation of molecular explosives, whose detonation velocity is several times that of current explosives? Would pressure dissociation or momentum-induced decomposition at low temperatures be another possible mechanism for detonation to generate high-speed particles products? Experimental support to answer these questions must be derived under extreme conditions of pressure from observations inside the shock front, thereby requiring measurements on the time scales of 10-2-1 ps.
The ab initio or quantum mechanical MD has offered an alternative to gain insights into these challenging problems and guidance to experimental research with energetic materials and detonation processes. High-energy-density materials must feature metastability and a large energy content that mostly originate in transformations of the molecular, atomic and electronic structures. Successful synthesis of these materials and control of the energy release from the structural bonds strongly rely on the understanding of the chemical processes and physics of structural transformations at the atomic level. Ab initio molecular dynamics can profoundly affect how energy storage and release via structural transformations and bond formation/breaking are understood and applied to defence technology.

2 DRDC Suffield TR 2003-156
For these reasons, we have used Car-Parrinello ab initio molecular dynamics and developed a multimolecular collision model and a controlled temperature bulk liquid compression approach to study the decomposition mechanism of homogeneous energetic materials at the atomic level under shock and highly static compressed conditions. Liquid nitromethane (NM) was chosen as a prototypical molecular explosive. This report provides a summary of the methods and simulation results. The models and methods can be extended for future advances in simulations of high-pressure synthesis of new energetic materials and their detonation mechanisms.

DRDC Suffield TR 2003-156 3
2. Theory of Ab Initio Molecular Dynamics
2.1 Ab Initio Molecular Dynamics
The main idea of ab initio or “first principles” MD is to combine the quantum mechanics with molecular dynamics to describe the processes of making and breaking bonds as the simulation proceeds. The theoretical description must take into account the motion of the electrons and the nuclei that is essentially governed by the Schrödinger wave equation for the many-body system inside the simulation cell. The Born-Oppenheimer approximation can be introduced if the total energy mainly originates in the motion of the electrons, the vibrational, rotational and translational motion of the nuclei and the electrostatic interactions between the electrons and nuclei. This approximation follows from the fact that the mass of an electron is much lighter (10-3) than the mass of a nucleus, and therefore electrons are able to adjust their positions instantaneously in the timeframe of the motion of nuclei. In such a case, the nuclei can be treated as classical particles of masses of MI at positions RI moving according to Newton’s equation
I
II R
RR∂
∂−= )(M Iφ&& (1)
with forces which are determined by the potential energy defined by
00 (H( ψψφ )) II RR = (2)
Here, H(RI) is the many-body electronic Hamiltonian calculated with the nuclei at fixed position RI, and ψ0 is the corresponding ground state eigenfunction. The statistical ensemble average of an operator P is obtained from a temporal average
∫∞→=T
0T dt)t(P
T1limP (3)
Thus, the Born-Oppenheimer approximation equation (2) provides a scheme for calculation of potential energy φ(RI). For any fixed position of the nuclei, RI, one solves the Schrödinger equation for the electrons to get ψ0, then the forces on the nuclei can be evaluated by the Hellman-Feynman theorem:
00)(H)( ψψφ
I
I
I
I
RR
RR
∂∂=
∂∂
(4)
These forces can be used to update the nuclei positions, and the solution procedure repeats for a sufficiently long MD trajectory required. A major hurdle in this apparently straightforward procedure is that the Schrödinger equation cannot be solved exactly except in a very few of the simplest cases. Thus approximation schemes have to be found.


DRDC Suffield TR 2003-156 5
where the effective potential is given by
)(-
)()()(rr'r
r'r'rrδρ
ρδρ ][EdVV xceff ++= ∫ (10)
2.3 Car-Parrinello Method
The DFT based Born-Oppenheimer ab initio MD method is subjected to the constraint that the KS orbitals ψi(r) are orthogonal and require that the wave function or electronic structure converges after each MD step. For large systems, this procedure quickly becomes too expensive. An alternative to decoupling the motion of the electrons from that of the nuclei is to solve for the motion of both in terms of a set of coupled equations. Car and Parrinello [3] used the DFT augmented by a Langrangian to describe the coupled dynamics of the nuclei and KS orbitals. The Car-Parrinello Langrangian includes the velocities of the nuclei and KS orbitals:
)(
)([EML
ijji
M
1i
M
1jij
N
1iiiii2
1I
M
1I21
δψψΛ
ψψψµ
−+
−+=
∑∑
∑∑
= =
==
)()(
,()()(2I
rr
RrrrR
&&
&&&I)]
(11)
where N is the number of electrons and M the number of nuclei. The first term is the kinetic energy of nuclei with masses MI, the second term is the fictitious kinetic energy of orbitals due to the electronic degrees of freedom with inertial parameters (masses) µi, the third term is the electronic energy, and the last term is the constraints to maintain orthogonality of orbitals. Upon substitution of (11) into Lagrange’s equation of motion one obtains the Car-Parrinello equation of motion for both the nuclei and the KS orbitals:
I
II R
RR∂
∂−= ),(EM iI
ψ&& (12)
sintconstraityorthogonol),(E
i
iii +
∂∂−=
ψψψµ IR
&& (13)
In the limit that µ approaches zero, the Car-Parrinello equation converges to the KS equation so that E is minimized and the Born-Oppenheimer limit is recovered. In the solution procedure, initially the electrons start out in the ground state, given by the KS equations, for fixed nuclear positions. The nuclei are then allowed to move according to equation (12) at the pre-assigned temperature. The electronic orbitals should follow the motion of the nuclei adiabatically according to equation (13), remaining in the instantaneous ground state. This implies that the fictitious mass parameter µ must be small to ensure the electronic contribution to the error of the forces is negligible. The electronic energy is then updated from equations (5-8) and used in equations (12-13) at the next MD time step. Thus, unlike in the DFT based Born-Oppenheimer ab initio MD where the convergence of wave functions obtained from KS

6 DRDC Suffield TR 2003-156
equations (9-10) must be conducted in each MD time step, the wave functions or the electronic degrees of freedom here are treated as classical degrees of freedom of the nuclei motion that are propagated according to equation (13). This also gives rise to a high degree of energy conservation. While the MD time steps, typically 10-1 fs, are smaller than that used in Born-Oppenheimer ab initio MD (in the order of 1 fs), the Car-Parrinello method is much faster without the converging process at each MD time step. For the system with a finite electronic energy gap, experience has shown that it is possible to choose µ small enough that the adiabaticity is preserved and at the same time reasonably large integration time steps are possible. For zero energy gap systems like metals this adiabaticity preservation becomes impossible. In such a case, the exchange of energy between electrons and ions has to be controlled, for instance, by introducing appropriate thermostats, or the more conventional approach in which E is explicitly minimized at every MD time step.
2.4 Plane Wave Technique
The Car-Parrinello ab initio MD has its roots in solid state physics where a plane wave pseudopotential approach is a natural choice [15]. The KS orbitals are expanded in a plane wave set:
))c ) i rGGrG
•=∑ iexp(((iψ (14)
where G is the reciprocal lattice vector. The plane wave basis is intrinsically periodic and the sum is over the G compatible with the imposed periodic boundary conditions and is bounded by the requirement cut2
1 E≤2G (15)
The cut-off energy Ecut determines the size of the expansion and is used to control the convergence of the calculation.
The plane waves as basis set functions have many advantages. They are unbiased, that is, all points in space are treated equally. They do not depend on the atomic positions and therefore the spurious Puley forces that arise from the position dependence of the basis set are absent [29]. Convergence is easily controlled with a single parameter Ecut. The algorithmic complexity of the calculation is greatly reduced by using Fast Fourier Transformation that can conveniently link the real space representation and G-space representation. Finally, the computational effort follows the simple scaling rule that the number of plane waves is proportional to the volume of the simulation cell.
However, the plane wave basis set has difficulty in describing the rapid oscillations of the wave function in the proximity of the nuclei and therefore it is necessary to use pseudopotentials that integrate out the degrees of freedom associated with the electrons in the inner electronic shells [16]. Thus, the plane wave technique becomes rather inefficient for first row elements and transition metals, where the pseudopotentials cannot be easily constructed smoothly and large cut-off energies (i.e., a large number of plane waves) are required to obtain converged results. This is because the same resolution that is needed close to the nuclei has to be used everywhere due to the unbiased nature of the plane wave set.

DRDC Suffield TR 2003-156 7
3. Computational Methods
The ab initio MD simulations reported in the present work employed the CPMD package based on the Car-Parrinello method within the Kohn-Sham density functional theory framework [17]. The BP86 functional was used and comprised of Becke’s [18] gradient corrected exchange functional in combination with Perdew’s [19] gradient corrected correlation functional. Only the valence electrons were treated explicitly in the simulations, with the valence-core interactions described by the non-local norm-conserving pseudo-potentials of Trouileer and Martins [20] and the valence orbitals expanded in terms of a plane wave basis set with a kinetic energy cutoff of 60 Ry. These parameters provided good energy conservation within 3.5x10-4 Eh/ps. To allow for proper dissociation of the NM molecules, the spin unrestricted DFT formalism was used in all of the simulations. A fictitious electron mass of 800 a.u. and a time step of 0.145 fs were employed. Calculations of the plane-wave set DFT method used here were compared with that of conventional all-electron localized basis set methods and experimental data for a gas-phase single NM molecule. The results showed good agreement in geometric parameters, vibrational frequencies and the zero-point energy corrected dissociation energy [21-22].
A cubic simulation cell containing 32 randomly oriented NM molecules was employed for the Car-Parrinello ab initio MD simulations of compressed liquid NM. Comparison of the classical MD simulations with 32, 108, and 256 NM molecules in the simulation cell indicates that the results obtained with the 32 simulation cell are in good agreement with those obtained with larger simulation cells for moderate pressures where the empirical potential employed is valid. The length of the simulation cell was determined by compressing the volume of the simulation cell of liquid NM at ambient pressure and density by factors of: 1.5, 2.0, 2.5, and 3.0. The initial geometric configurations of the 32 NM molecules in the simulation cell were obtained from well-equilibrated classical simulations for liquid NM compressed by factors of 1.5, 2.0, and 2.5. However, classical MD simulations of NM with the volume of the simulation cell compressed three-fold resulted in unnatural distortions of the NM molecules. In this case the initial geometric configurations of the 32 NM molecules were obtained by incrementally decreasing the volume of the simulation cell from the equilibrated MD simulation of liquid NM compressed by a factor of 2.5. In all of the current MD simulations the temperature was slowly increased from 50 K to 300 K. Initially a short 0.725 ps simulation (corresponding to 5000 time steps) was performed at 50 K, then the temperature was increased to 150 K and another 0.725 ps simulation was carried out. The temperature was then raised to 300 K and the system was equilibrated for a total of 3.6 ps (corresponding to 25000 time steps). Following this equilibration stage a production run was performed at 300 K for another 3.6 ps. Nose-Hoover chain thermostats of length 4 were employed during the equilibration phases of the simulations to ensure proper equipartitioning of the energy. Thermostats were turned off during the production phase of the simulations.

8 DRDC Suffield TR 2003-156
4. Simulations of Shock Initiation
4.1 Review on Bimolecular Collisions Haskins and Cook [23] examined the bimolecular collision of two NM molecules at the DFT level employing Becke’s three parameter hybrid exchange functional with the Perdew-Wang correlation functional. For a head-to-tail orientation, they found a threshold velocity (i.e., the lowest collision velocity which leads to permanent bond cleavage) to be 8.5 km/s, where C-N bond scission occurs.
Our previous effort was made to examine the dependence of the threshold velocity on the initial orientation of the molecules using an orthorhombic simulation cell of 16x12x12 Angstroms [21]. At time zero, one molecule is set to a velocity of between 6.5 and 12.0 km/s toward the second, stationary NM molecule. Shown in Figure 1 are the initial orientations that have been examined. The lowest threshold collision velocity was found to be 7.0 km/s for the offset anti-parallel orientation, while that for the anti-parallel orientation was found to be the highest at 10.5 km/s. The threshold collision velocities for the perpendicular and head-to-tail orientations were found to be 8.0 and 8.5 km/s, which is in agreement with that obtained by Haskins and Cook. In all of the orientations investigated at the threshold velocity, nitromethane decomposition mechanism is dominated by the C-N bond scission of the incoming molecule. A different decomposition mechanism was observed only in one high velocity collision simulation of 11.0 km/s in the offset anti-parallel orientation, in which initial N-O bond cleavage of the stationary NM molecule was observed followed by a series of intramolecular rearrangements leading to the products of NM, NO and OCH3.
NO
OH H
H
NOO
HH
H
NOO
HH
H
NOO
HH
H
NO
OH H
H
NO
O HH
H
perpendicular(8.0 km/s)
head-to-tail(8.5 km/s)
anti-parallel(10.5 km/s)
NO
OH H
H
NO
O HH
H
offset anti-parallel(7.0 km/s)
Figure 1. Bimolecular collision orientations with threshold velocities

DRDC Suffield TR 2003-156 9
4.2 Multimolecular Collisions
4.2.1 Impact of a Single Molecule on Multiple Molecules
The previous bimolecular collision simulations are limited to collision-induced reactions without neighbouring confinement and the collision sequences beyond the initial impact are meaningless due to only the two isolated molecules involved. Multimolecular collision simulations offer a substantial improvement since they allow for possible collision induced reactions involving the neighbouring molecules and beyond the initial impact. Let us first study the multimolecular collisions by the impact of a single molecule on multiple molecules. The simulations are conducted under the same four collision orientations of the colliding NM molecules as the bimolecular collisions previously reported. A total of 13 NM molecules are placed in an orthorhombic simulation cell of 19.0×14.2×14.2 Angstroms such that one molecule is placed at one end of the simulation cell and the remaining twelve molecules are placed at the opposite end of the simulation cell. The simulations are initiated by propelling the separated molecule into a stationary molecule, embedded in a cluster of NM molecules as illustrated in Figure 2. The cluster of 12 molecules is randomly oriented and possesses a density of 1.14 g/cc, corresponding to the density of NM at ambient pressure and temperature. The molecular orientations of the two colliding molecules are varied along with the incident collision velocities ranging from 6 to 12 km/s. Each multimolecular collision simulation is run for 3500 time steps or 0.5 ps.
Figure 2. Schematic of the single molecule on multiple molecule collision simulations
10 DRDC Suffield TR 2003-156
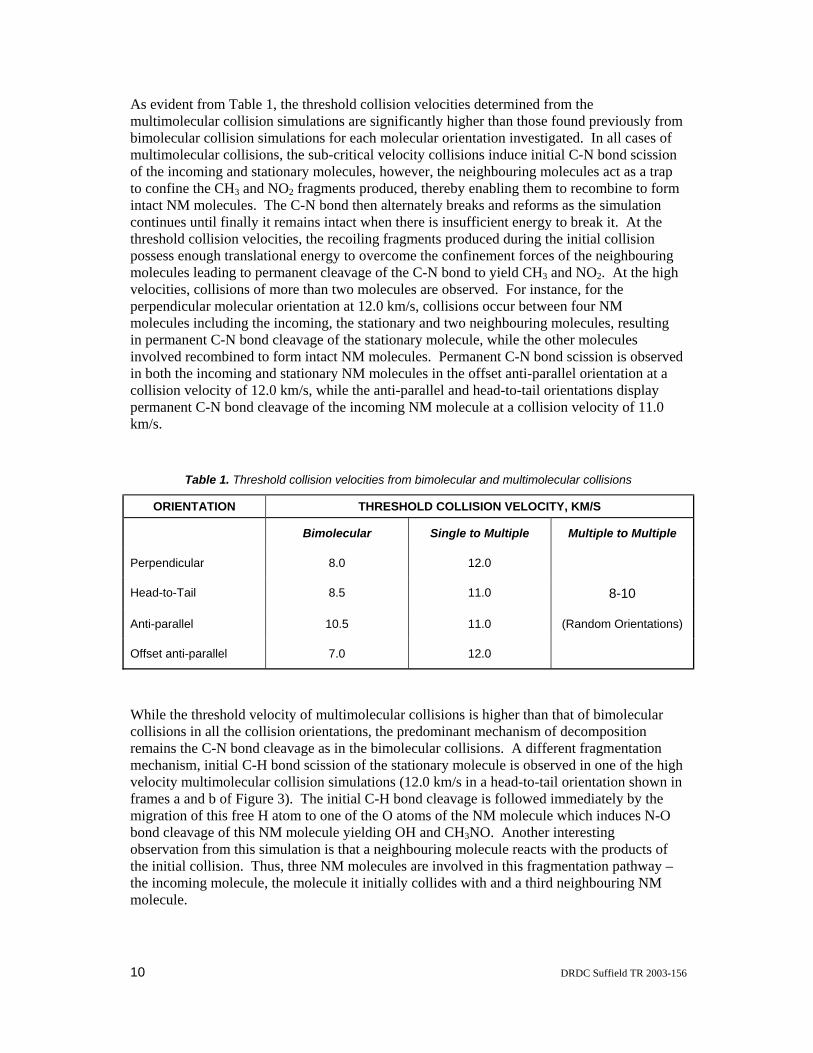
As evident from Table 1, the threshold collision velocities determined from the multimolecular collision simulations are significantly higher than those found previously from bimolecular collision simulations for each molecular orientation investigated. In all cases of multimolecular collisions, the sub-critical velocity collisions induce initial C-N bond scission of the incoming and stationary molecules, however, the neighbouring molecules act as a trap to confine the CH3 and NO2 fragments produced, thereby enabling them to recombine to form intact NM molecules. The C-N bond then alternately breaks and reforms as the simulation continues until finally it remains intact when there is insufficient energy to break it. At the threshold collision velocities, the recoiling fragments produced during the initial collision possess enough translational energy to overcome the confinement forces of the neighbouring molecules leading to permanent cleavage of the C-N bond to yield CH3 and NO2. At the high velocities, collisions of more than two molecules are observed. For instance, for the perpendicular molecular orientation at 12.0 km/s, collisions occur between four NM molecules including the incoming, the stationary and two neighbouring molecules, resulting in permanent C-N bond cleavage of the stationary molecule, while the other molecules involved recombined to form intact NM molecules. Permanent C-N bond scission is observed in both the incoming and stationary NM molecules in the offset anti-parallel orientation at a collision velocity of 12.0 km/s, while the anti-parallel and head-to-tail orientations display permanent C-N bond cleavage of the incoming NM molecule at a collision velocity of 11.0 km/s.
Table 1. Threshold collision velocities from bimolecular and multimolecular collisions
ORIENTATION THRESHOLD COLLISION VELOCITY, KM/S
Bimolecular Single to Multiple Multiple to Multiple
Perpendicular 8.0 12.0
Head-to-Tail 8.5 11.0 8-10
Anti-parallel 10.5 11.0 (Random Orientations)
Offset anti-parallel 7.0 12.0
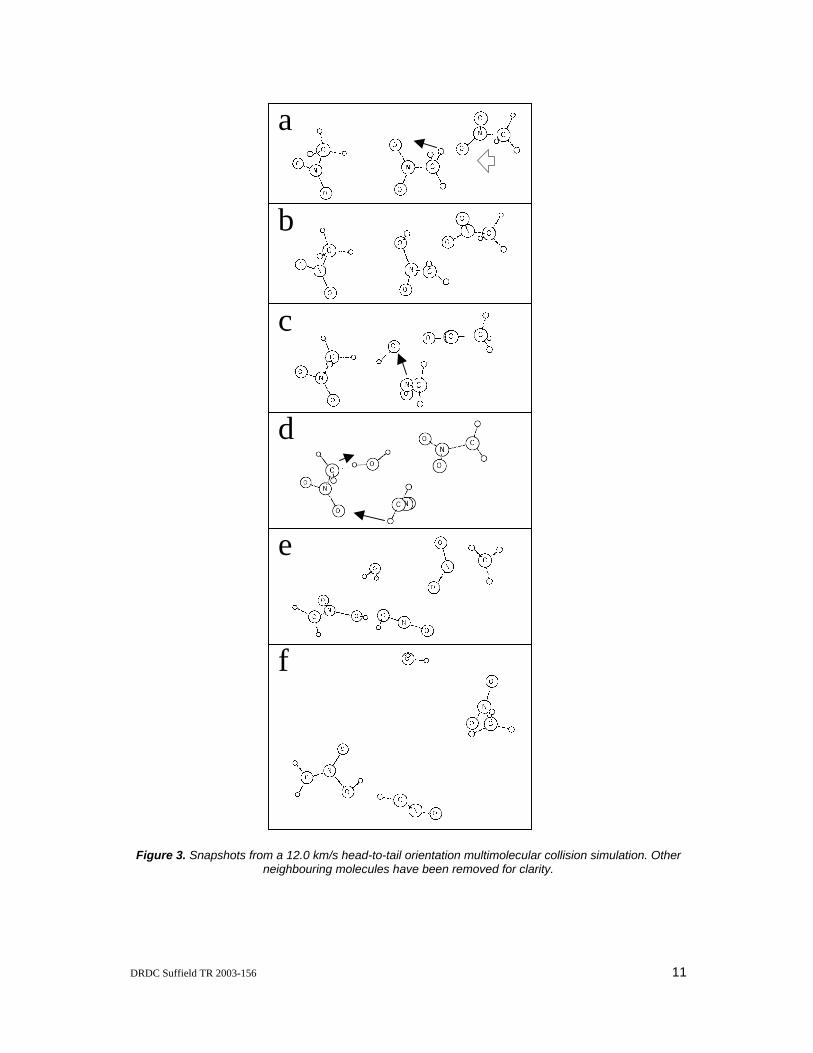
While the threshold velocity of multimolecular collisions is higher than that of bimolecular collisions in all the collision orientations, the predominant mechanism of decomposition remains the C-N bond cleavage as in the bimolecular collisions. A different fragmentation mechanism, initial C-H bond scission of the stationary molecule is observed in one of the high velocity multimolecular collision simulations (12.0 km/s in a head-to-tail orientation shown in frames a and b of Figure 3). The initial C-H bond cleavage is followed immediately by the migration of this free H atom to one of the O atoms of the NM molecule which induces N-O bond cleavage of this NM molecule yielding OH and CH3NO. Another interesting observation from this simulation is that a neighbouring molecule reacts with the products of the initial collision. Thus, three NM molecules are involved in this fragmentation pathway – the incoming molecule, the molecule it initially collides with and a third neighbouring NM molecule.
DRDC Suffield TR 2003-156 11
Figure 3. Snapshots from a 12.0 km/s head-to-tail orientation multimolecular collision simulation. Other neighbouring molecules have been removed for clarity.
a
b
c
d
e
f

12 DRDC Suffield TR 2003-156
4.2.2 Impact of Multiple Molecules on Multiple Molecules
The multimolecular collision simulations are further extended to the collisions between two groups of molecules. The chemical system of 32 NM molecules is contained in a cubic simulation cell of 14.2x14.2x14.2 Angstroms. The mass to volume ratio of 32 molecules in the cube reflects a density of 1.14g/cc for liquid NM. Periodic boundary conditions are used to better represent the bulk material properties and effects of neighbouring molecules. Thus, a molecule will be experiencing short-range and long-range intermolecular forces from its immediate and surrounding neighbours respectively. Periodic boundary conditions also ease the cluster boundary effects since there are no distinct boundaries at the face of each cell under periodic conditions. In contrast, in an identical setup using no periodic boundary conditions, molecules leaving the cell will be effectively in a vacuum and eventually disperse as a result of Brownian motion. The initial orientations and velocities of each of the NM molecules in the simulation cell are derived from a sample that was equilibrated for approximately 4.36ps at 298 K with a Nose-Hoover thermostat. Eight NM molecules, occupying the left most quarter of the simulation cell, are translated linearly forward to impact into the other 24 NM molecules occupying the rest of the cell as shown in Figure 4. In each simulation, all eight NM molecules are assigned uniform initial velocity ranging from 2 to 10 km/s. The simulations are further divided into static and dynamic simulations. The static simulations are similar with the previous simulations of bimolecular collisions and single-to-multiple molecular collisions, in which the kinetic energy and temperature of the molecules are initially set to zero. The eight molecules occupying the left quarter of the cube are then translated forward with their assigned velocity. In the dynamic simulations, all 32 molecules initially possess equilibrium velocity and the total kinetic energy corresponds to the kinetic energy of the equilibrated sample of liquid NM at 298 K and 1 atm. The eight molecules occupying the left quarter of the simulation cube are translated with an assigned velocity in addition to their equilibrium velocity. The dynamic simulations are considered more realistic than the static simulations. All simulations were performed for 5000 time steps or 0.725ps.
Figure 4. Schematic of the multiple molecule on multiple molecule collision simulations
DRDC Suffield TR 2003-156 13
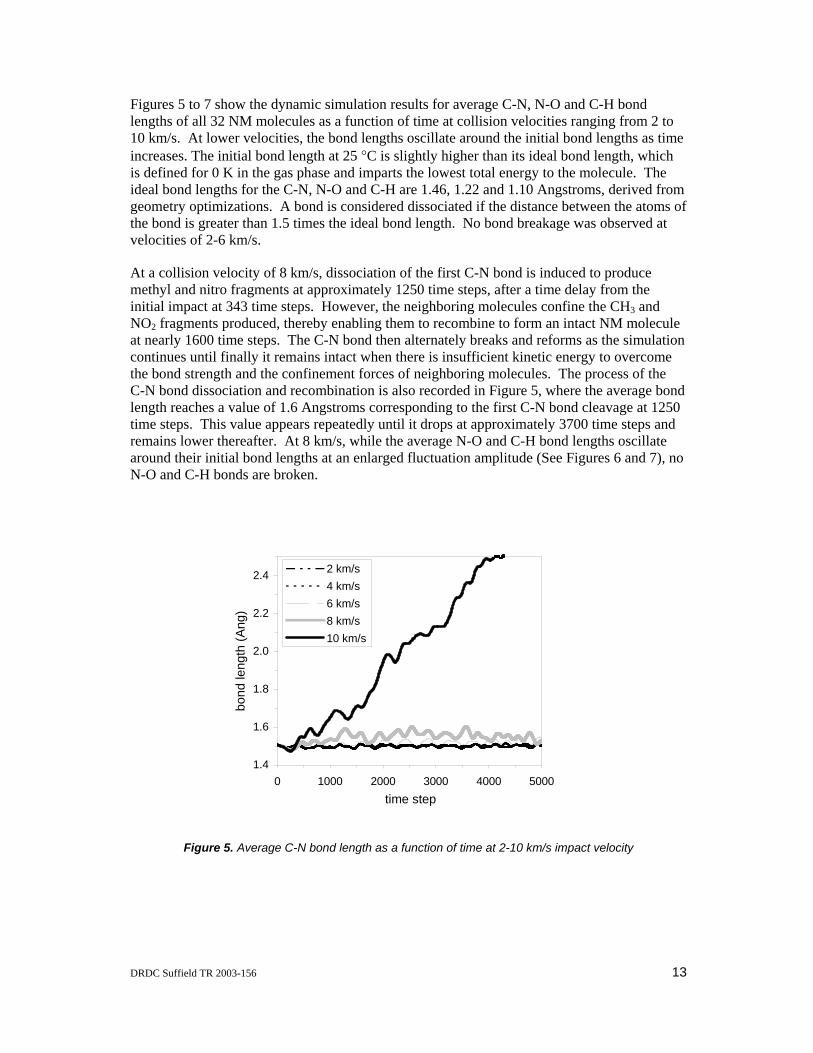
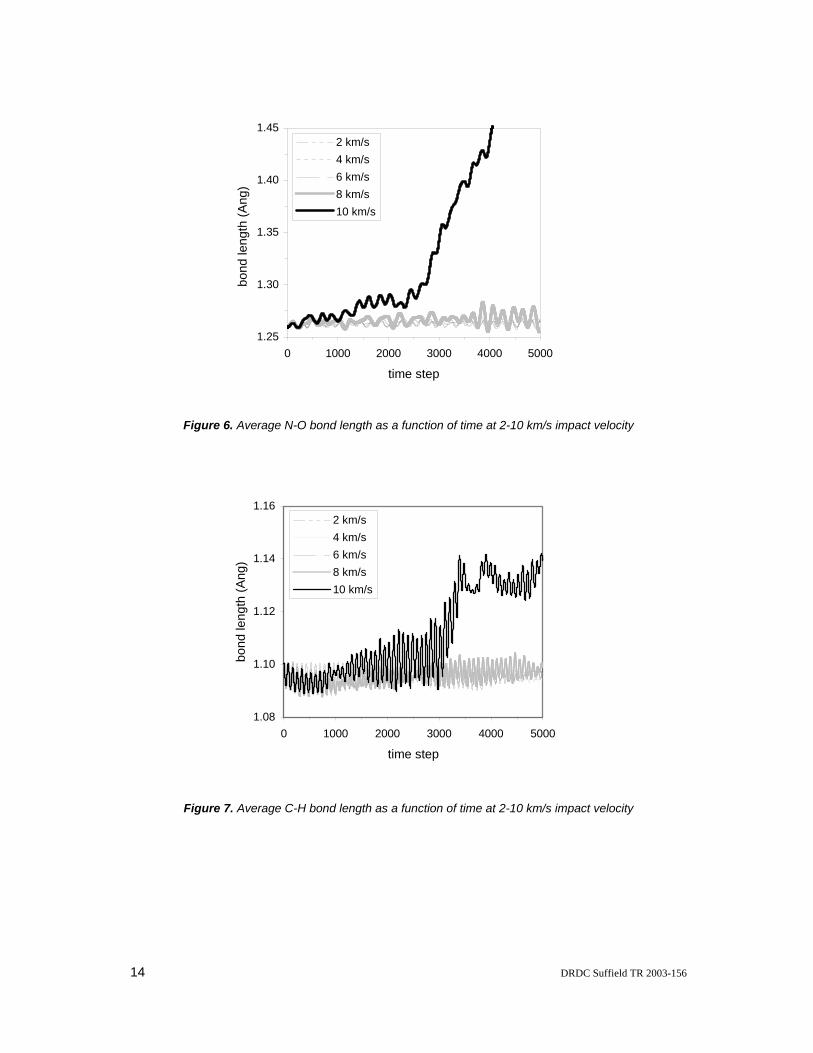
Figures 5 to 7 show the dynamic simulation results for average C-N, N-O and C-H bond lengths of all 32 NM molecules as a function of time at collision velocities ranging from 2 to 10 km/s. At lower velocities, the bond lengths oscillate around the initial bond lengths as time increases. The initial bond length at 25 °C is slightly higher than its ideal bond length, which is defined for 0 K in the gas phase and imparts the lowest total energy to the molecule. The ideal bond lengths for the C-N, N-O and C-H are 1.46, 1.22 and 1.10 Angstroms, derived from geometry optimizations. A bond is considered dissociated if the distance between the atoms of the bond is greater than 1.5 times the ideal bond length. No bond breakage was observed at velocities of 2-6 km/s.
At a collision velocity of 8 km/s, dissociation of the first C-N bond is induced to produce methyl and nitro fragments at approximately 1250 time steps, after a time delay from the initial impact at 343 time steps. However, the neighboring molecules confine the CH3 and NO2 fragments produced, thereby enabling them to recombine to form an intact NM molecule at nearly 1600 time steps. The C-N bond then alternately breaks and reforms as the simulation continues until finally it remains intact when there is insufficient kinetic energy to overcome the bond strength and the confinement forces of neighboring molecules. The process of the C-N bond dissociation and recombination is also recorded in Figure 5, where the average bond length reaches a value of 1.6 Angstroms corresponding to the first C-N bond cleavage at 1250 time steps. This value appears repeatedly until it drops at approximately 3700 time steps and remains lower thereafter. At 8 km/s, while the average N-O and C-H bond lengths oscillate around their initial bond lengths at an enlarged fluctuation amplitude (See Figures 6 and 7), no N-O and C-H bonds are broken.
1.4
1.6
1.8
2.0
2.2
2.4
0 1000 2000 3000 4000 5000time step
bond
leng
th (A
ng)
2 km/s4 km/s6 km/s8 km/s10 km/s
Figure 5. Average C-N bond length as a function of time at 2-10 km/s impact velocity
14 DRDC Suffield TR 2003-156
1.25
1.30
1.35
1.40
1.45
0 1000 2000 3000 4000 5000
time step
bond
leng
th (A
ng)
2 km/s4 km/s6 km/s8 km/s10 km/s
Figure 6. Average N-O bond length as a function of time at 2-10 km/s impact velocity
1.08
1.10
1.12
1.14
1.16
0 1000 2000 3000 4000 5000
time step
bond
leng
th (A
ng)
2 km/s4 km/s6 km/s8 km/s10 km/s
Figure 7. Average C-H bond length as a function of time at 2-10 km/s impact velocity
DRDC Suffield TR 2003-156 15
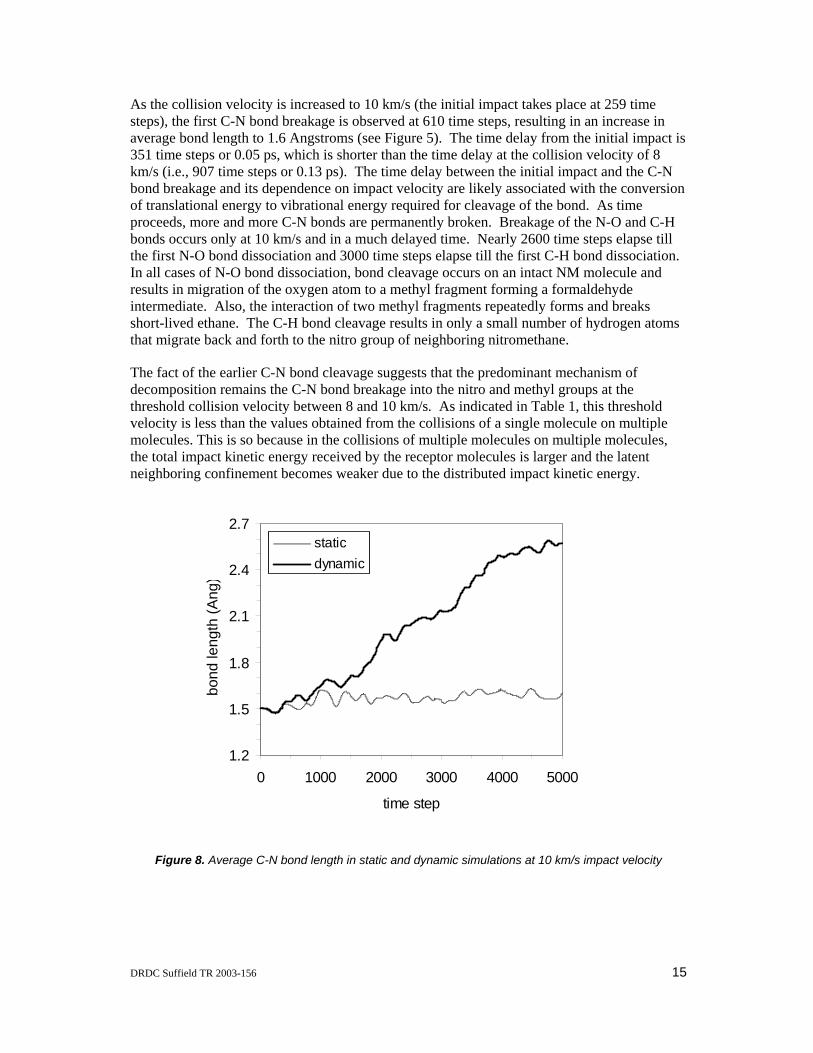
As the collision velocity is increased to 10 km/s (the initial impact takes place at 259 time steps), the first C-N bond breakage is observed at 610 time steps, resulting in an increase in average bond length to 1.6 Angstroms (see Figure 5). The time delay from the initial impact is 351 time steps or 0.05 ps, which is shorter than the time delay at the collision velocity of 8 km/s (i.e., 907 time steps or 0.13 ps). The time delay between the initial impact and the C-N bond breakage and its dependence on impact velocity are likely associated with the conversion of translational energy to vibrational energy required for cleavage of the bond. As time proceeds, more and more C-N bonds are permanently broken. Breakage of the N-O and C-H bonds occurs only at 10 km/s and in a much delayed time. Nearly 2600 time steps elapse till the first N-O bond dissociation and 3000 time steps elapse till the first C-H bond dissociation. In all cases of N-O bond dissociation, bond cleavage occurs on an intact NM molecule and results in migration of the oxygen atom to a methyl fragment forming a formaldehyde intermediate. Also, the interaction of two methyl fragments repeatedly forms and breaks short-lived ethane. The C-H bond cleavage results in only a small number of hydrogen atoms that migrate back and forth to the nitro group of neighboring nitromethane.
The fact of the earlier C-N bond cleavage suggests that the predominant mechanism of decomposition remains the C-N bond breakage into the nitro and methyl groups at the threshold collision velocity between 8 and 10 km/s. As indicated in Table 1, this threshold velocity is less than the values obtained from the collisions of a single molecule on multiple molecules. This is so because in the collisions of multiple molecules on multiple molecules, the total impact kinetic energy received by the receptor molecules is larger and the latent neighboring confinement becomes weaker due to the distributed impact kinetic energy.
1.2
1.5
1.8
2.1
2.4
2.7
0 1000 2000 3000 4000 5000
time step
bond
leng
th (A
ng)
staticdynamic
Figure 8. Average C-N bond length in static and dynamic simulations at 10 km/s impact velocity
16 DRDC Suffield TR 2003-156
0 1000 2000 3000 4000 50001.2
1.25
1.3
1.35
1.4
1.45
Nitrogen - Oxygen Bond Length as a Function of Time: Comparison of Dynamic and Static Data at 10km/s
LEGEND dynamic static
Bond Length (Angstroms)
Time Step0 1000 2000 3000 4000 5000
1.2
1.25
1.3
1.35
1.4
1.45
Bond Length (Ang)
Dynamic
Static
Time Step
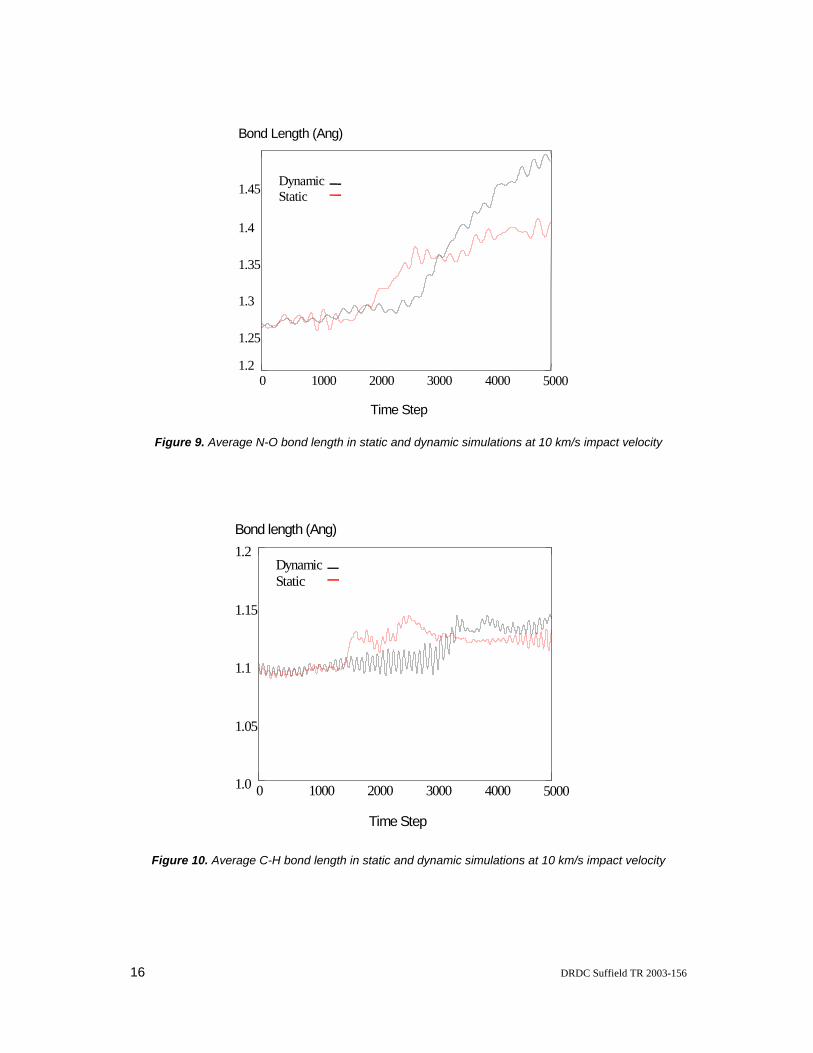
Figure 9. Average N-O bond length in static and dynamic simulations at 10 km/s impact velocity
0 1000 2000 3000 4000 50001
1.05
1.1
1.15
1.2 Carbon - Hydrogen Bond Length as a Function of Time: Comparison of Dynamic and Static Data at 10km/s
Bond Length (Angstroms)
Time Step
LEGEND dynamic static
0 1000 2000 3000 4000 5000 1.0
1.05
1.1
1.15
1.2 Bond length (Ang)
Time Step
Dynamic Static
Figure 10. Average C-H bond length in static and dynamic simulations at 10 km/s impact velocity

DRDC Suffield TR 2003-156 17
The static simulations provide the identical decomposition mechanism and threshold collision velocity based on the C-N bond cleavage. The difference between the static and dynamic simulations lies in the time delay of the decomposition and the rate of bond length increase that becomes apparent at the impact velocity of 10 km/s as shown in Figures 8-10. The difference observed can be explained by the additional kinetic energy at room temperature and pressure available in the dynamic simulations. This additional energy manifests itself vibrationally and promotes decomposition once initial bond breakage begins.

18 DRDC Suffield TR 2003-156
5. Simulations of Pressure Dissociation
5.1 Structural Properties of Compressed Liquid NM
For solid NM, Reed et al. [24] utilized both static DFT calculations and ab initio MD simulations to examine the effects of static compression on the nature of electronic excitations. They concluded that the band gap of solid NM is not lowered enough to produce a significant population of the excited states in the crystal. More recently, Margetis et al. [25] examined how pressure and molecular vacancies affect the atomic structure and electronic properties of solid NM via static self-consistent charge density functional tight-binding calculations. They also found that static compression did not reduce the band gap significantly for electronic excitation to be a dominant process. However, one of the most intriguing findings from their study was that under uniaxial compression of about 25-40 GPa along the b axis, as well as isotropic compression above 100 GPa, the C-H bond became highly stretched leading to a proton dissociation process.
We carried out simulations for ambient liquid NM and liquid NM compressed to factors of 1.5 (V/Vo=0.67), 2.0 (V/Vo=0.50), 2.5 (V/Vo=0.40), and 3.0 (V/Vo=0.33). Figure 11 illustrates a simulation cell.
Figure 11. High pressure compression of bulk liquid nitromethane

DRDC Suffield TR 2003-156 19
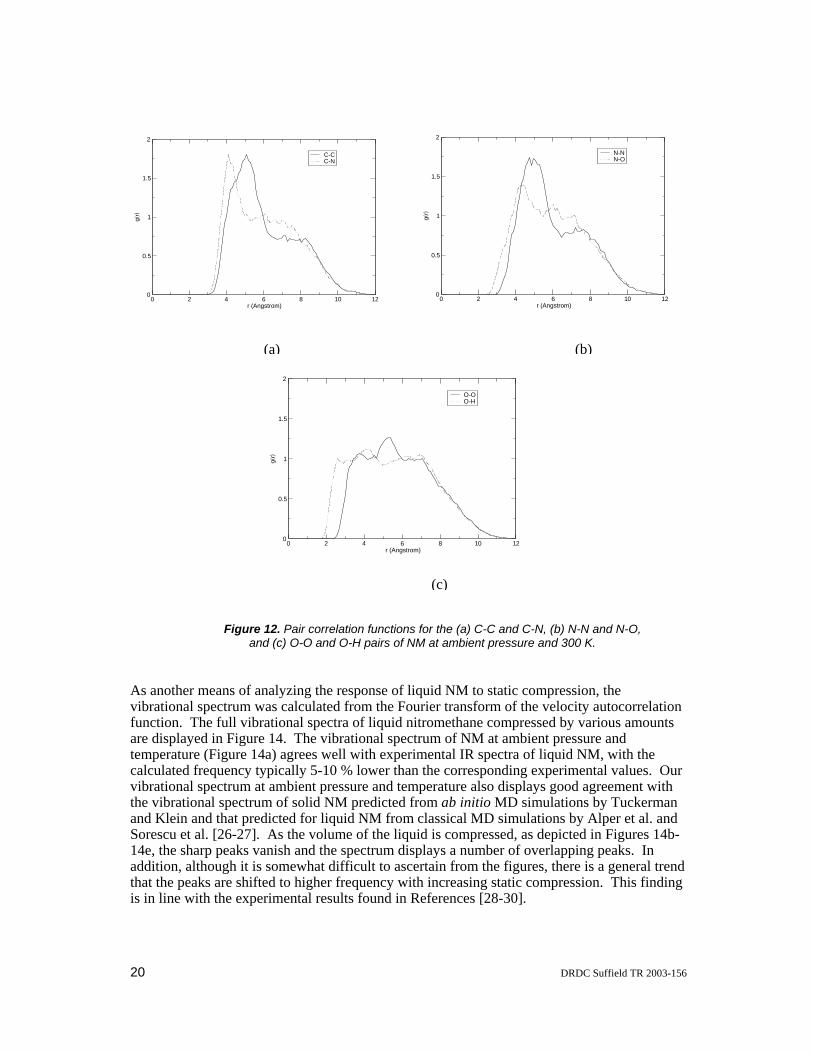
The structural properties of liquid NM in our simulations have been investigated in terms of the various site-site pair correlation functions (radial distribution functions). The pair correlation functions calculated from our MD simulation of liquid NM at ambient pressure and 300 K are displayed in Figure 12. The C-C and N-N pair correlation functions display double peaks indicating that the first solvation shell for a given NM molecule occurs between 4-6 Å and the second occurs around 7-9 Å. This is in good agreement with the C-C and N-N pair correlation functions from the previous classical MD simulations of Thompson, Politzer and co-workers [26-27]. A closer inspection of the C-C pair correlation function indicates that the first peak has a shoulder positioned at 4.3 Å with the maximum of the peak positioned at 5.1 Å. The C-C pair correlation function calculated here agrees very nicely with that obtained from the previous classical MD simulations, both in terms of the general shape as well as the position of the first peak maximum, which was located at 5.2 Å. The C-N pair correlation function also displays excellent agreement with that obtained from the previous classical MD simulations, with the first peak located at 4.1 Å compared to 4.2 Å. The N-N pair correlation function calculated from our MD simulations exhibits a double peak structure with the first peak maximum located at 4.7 Å and the second at 7.6 Å. The positions of these peaks are shifted to slightly shorter distances than observed in the N-N pair correlation functions obtained previously by Thompson and Politzer. Our N-O pair correlation function also displayed two maxima (at 4.3 and 6.0 Å), however, they were less resolved (i.e., much closer together and of approximately the same intensity) than in the N-N pair correlation function, in line with the N-O pair correlation functions obtained by Thompson and co-workers. The structure in the O-O and O-H pair correlation functions is less defined than in the other pair correlation functions. Our O-O pair correlation function contains two closely spaced peaks, centred at 3.7 and 5.3 Å, with the second peak being more intense than the first, in line with that obtained from the previous classical MD simulations. The structure in our O-H pair correlation function is much less defined in line with the results of Politzer et al. who also obtained a plot with very little, if any, structure. However, the O-H pair correlation function obtained by Thompson and co-workers displayed a fairly well defined double peak structure at 2.8 and 4.3 Å, which agrees fairly well with our results that have poorly defined maxima at around 2.6 and 4.2 Å, upon closer examination. Overall there is quite good agreement between the pair correlation functions from the current Car-Parrinello MD simulations and those obtained from the previous classical MD simulations at ambient pressure and 300 K.
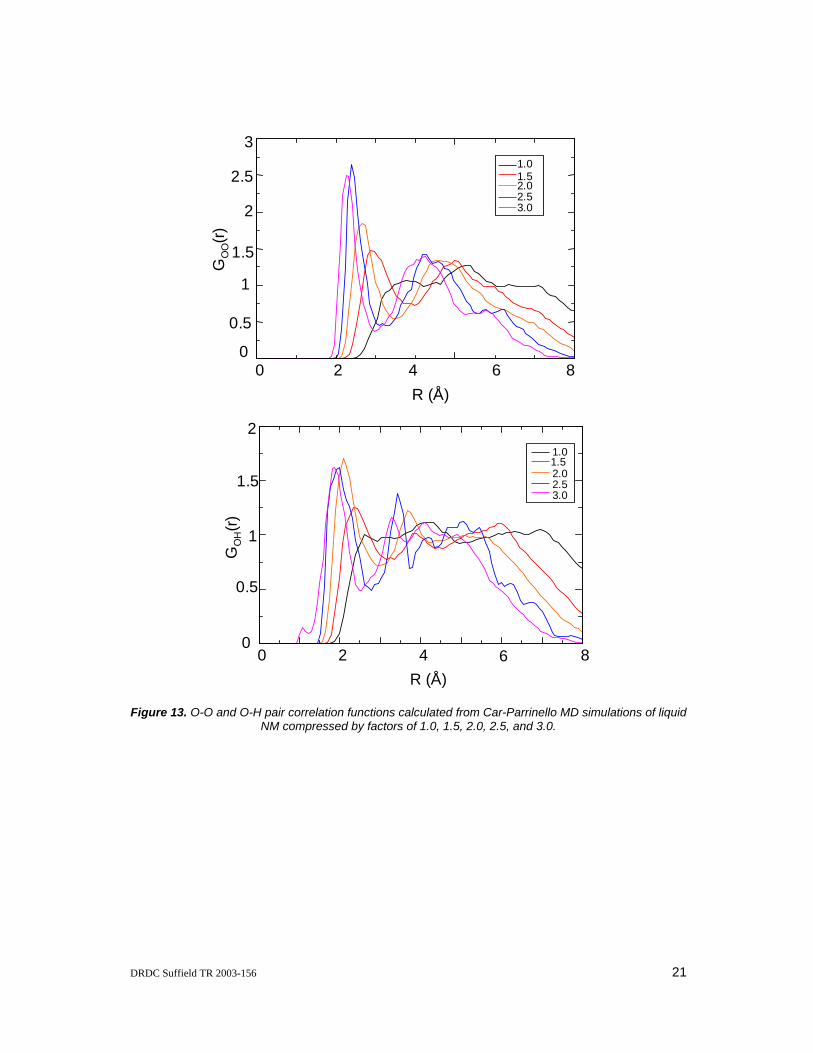
The pair correlation functions calculated from the ab initio MD simulations of NM with the simulation cell compressed by various factors are utilized to gain some insight into how the structure of liquid NM changes under high pressure compression conditions. The same general trends are observed for all of the site-site pair correlation functions and as such only the O-O and O-H pair correlation functions are given in Figure 13. These will be the focus of the following discussion, however, the discussion applies to all of the pair correlation functions. As discussed above, at ambient pressure the O-O and O-H pair correlation functions are rather flat and featureless, indicating little local structure in the liquid. As shown in Figure 13, compression of liquid NM by a factor of 1.5 results in the appearance of two maxima in the O-O and O-H pair correlation functions and these peaks become sharper as the liquid is further compressed, thus indicating that the local structure of the liquid increases upon compression. Furthermore, increasing the compression of the liquid induces a shift of the two peak maxima to shorter distances. Therefore, the intermolecular distances between the individual NM molecules is decreased due to the reduced volume of the simulation cell.
20 DRDC Suffield TR 2003-156
0 2 4 6 8 10 12r (Angstrom)
0
0.5
1
1.5
2
C-CC-N
0 2 4 6 8 10 12r (Angstrom)
0
0.5
1
1.5
2
N-NN-O
0 2 4 6 8 10 12r (Angstrom)
0
0.5
1
1.5
2
O-OO-H
Figure 12. Pair correlation functions for the (a) C-C and C-N, (b) N-N and N-O, and (c) O-O and O-H pairs of NM at ambient pressure and 300 K.
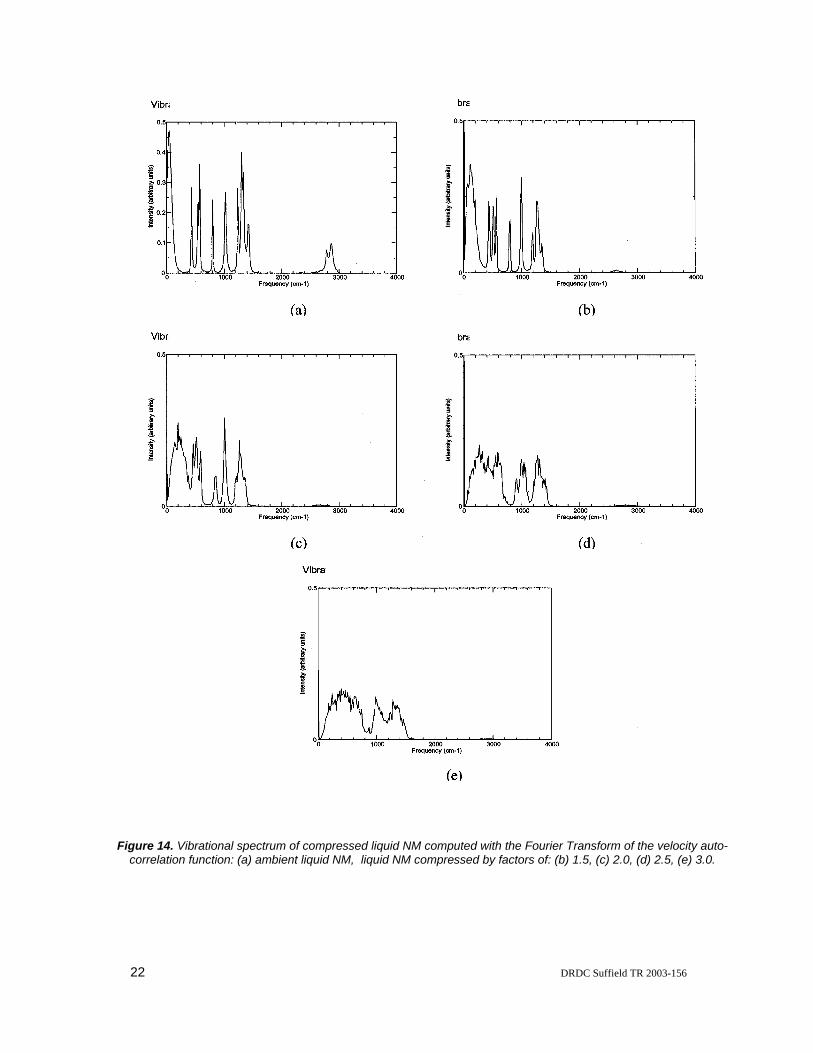
As another means of analyzing the response of liquid NM to static compression, the vibrational spectrum was calculated from the Fourier transform of the velocity autocorrelation function. The full vibrational spectra of liquid nitromethane compressed by various amounts are displayed in Figure 14. The vibrational spectrum of NM at ambient pressure and temperature (Figure 14a) agrees well with experimental IR spectra of liquid NM, with the calculated frequency typically 5-10 % lower than the corresponding experimental values. Our vibrational spectrum at ambient pressure and temperature also displays good agreement with the vibrational spectrum of solid NM predicted from ab initio MD simulations by Tuckerman and Klein and that predicted for liquid NM from classical MD simulations by Alper et al. and Sorescu et al. [26-27]. As the volume of the liquid is compressed, as depicted in Figures 14b-14e, the sharp peaks vanish and the spectrum displays a number of overlapping peaks. In addition, although it is somewhat difficult to ascertain from the figures, there is a general trend that the peaks are shifted to higher frequency with increasing static compression. This finding is in line with the experimental results found in References [28-30].
(a) (b)
(c)
DRDC Suffield TR 2003-156 21
Figure 13. O-O and O-H pair correlation functions calculated from Car-Parrinello MD simulations of liquid NM compressed by factors of 1.0, 1.5, 2.0, 2.5, and 3.0.
R (Å)
0
0.5
1
1.5
2
2.5
3
GO
O(r)
1.0
2.0
3.0
1.5
2.5
0 42 6 8
R (Å)
GO
H(r)
0 2 4 6 80
0.5
1
1.5
21.0
2.0
3.0
1.5
2.5
22 DRDC Suffield TR 2003-156
Figure 14. Vibrational spectrum of compressed liquid NM computed with the Fourier Transform of the velocity auto-correlation function: (a) ambient liquid NM, liquid NM compressed by factors of: (b) 1.5, (c) 2.0, (d) 2.5, (e) 3.0.
DRDC Suffield TR 2003-156 23
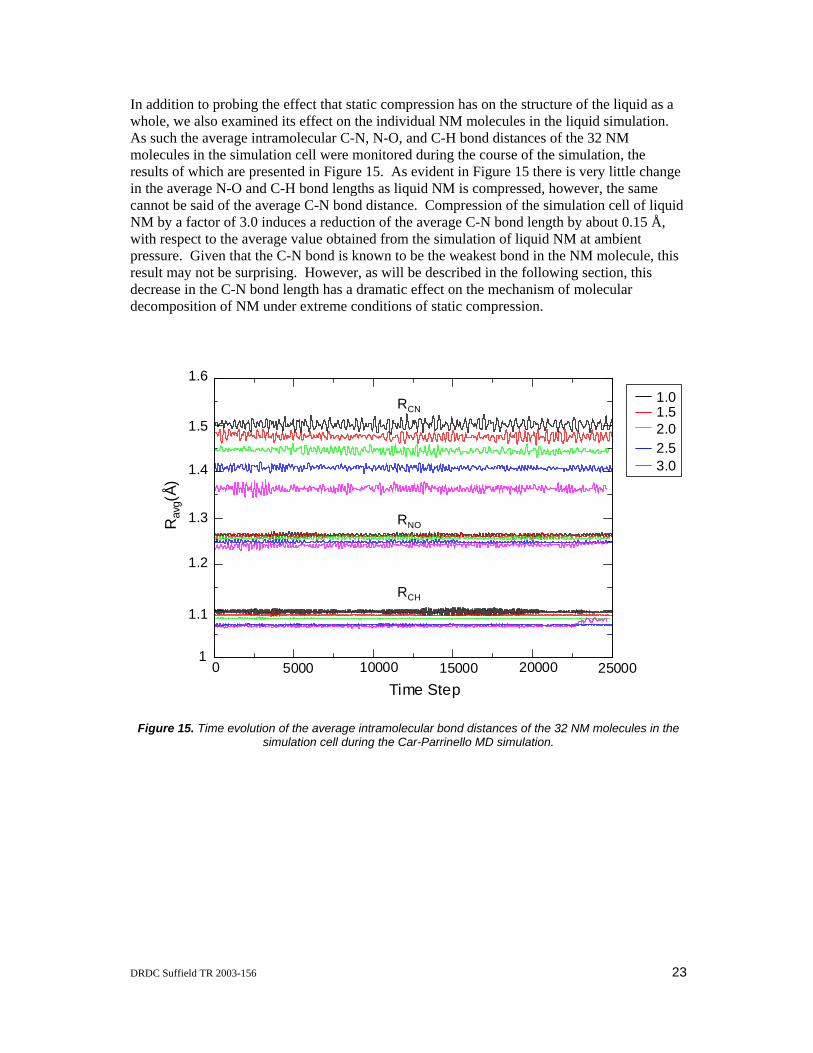
In addition to probing the effect that static compression has on the structure of the liquid as a whole, we also examined its effect on the individual NM molecules in the liquid simulation. As such the average intramolecular C-N, N-O, and C-H bond distances of the 32 NM molecules in the simulation cell were monitored during the course of the simulation, the results of which are presented in Figure 15. As evident in Figure 15 there is very little change in the average N-O and C-H bond lengths as liquid NM is compressed, however, the same cannot be said of the average C-N bond distance. Compression of the simulation cell of liquid NM by a factor of 3.0 induces a reduction of the average C-N bond length by about 0.15 Å, with respect to the average value obtained from the simulation of liquid NM at ambient pressure. Given that the C-N bond is known to be the weakest bond in the NM molecule, this result may not be surprising. However, as will be described in the following section, this decrease in the C-N bond length has a dramatic effect on the mechanism of molecular decomposition of NM under extreme conditions of static compression.
Figure 15. Time evolution of the average intramolecular bond distances of the 32 NM molecules in the simulation cell during the Car-Parrinello MD simulation.
0 5000 10000 15000 20000 25000Time Step
1
1.1
1.2
1.3
1.4
1.5
1.61.01.52.02.53.0
Rav
g(Å)
RCN
RNO
RCH

24 DRDC Suffield TR 2003-156
5.2 Molecular Decomposition in Compressed Liquid NM
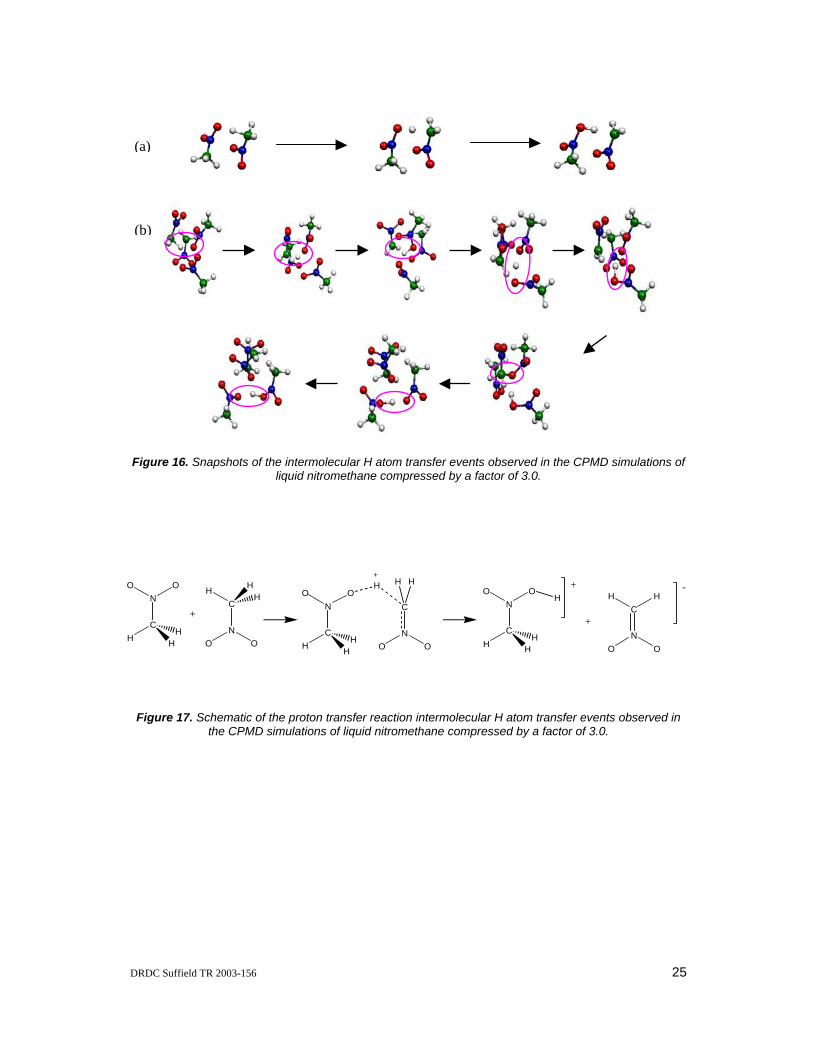
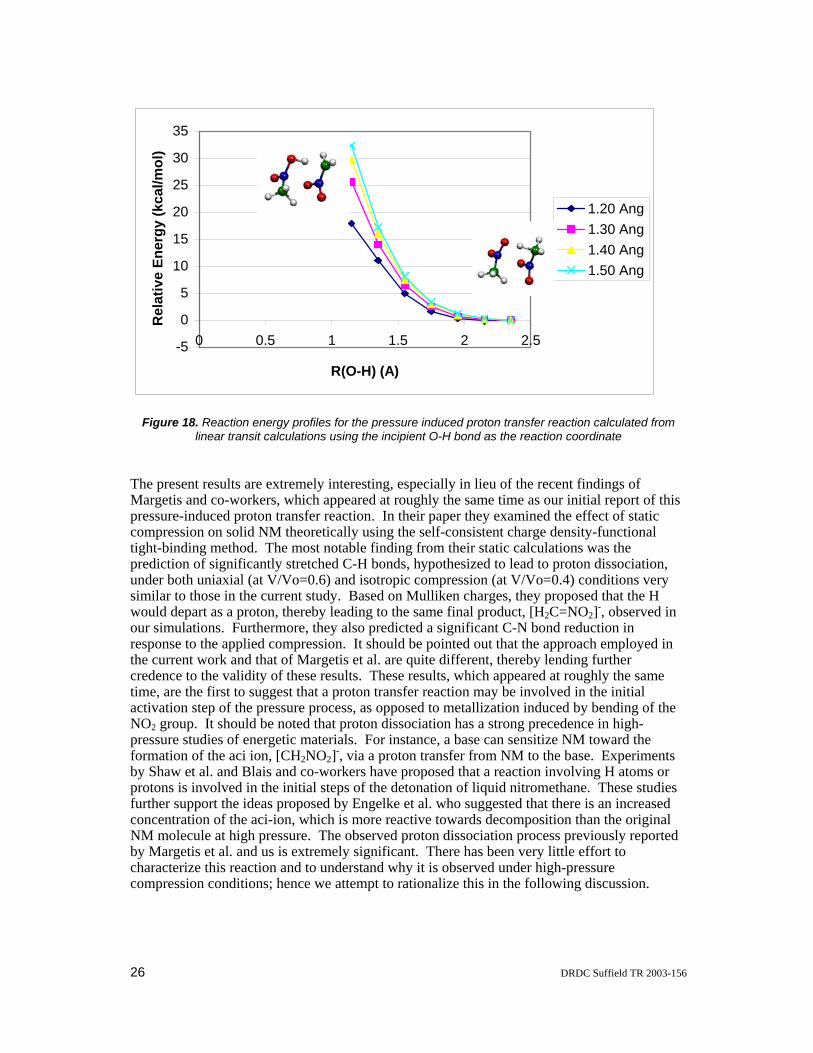
The current CPMD simulations of compressed liquid NM showed no molecular decomposition at 300 K when the simulation cell volume has been compressed by factors of 1.0, 1.5, 2.0, and 2.5. However, when the simulation cell volume has been compressed three-fold, interesting pressure-induced NM molecular decompositions were observed at 150 K (after equilibration for about 3300 time steps) and at 300 K (after equilibration for about 24000 time steps). Both of these processes involve an intermolecular H atom transfer between two closely spaced NM molecules aligned in an anti-parallel alignment. Snapshots of these events taken from the MD trajectories are shown in Figure 16. In the first H atom transfer reaction (illustrated in Figure 16a) the CH3 group of one of the NM molecule re-orients itself such that two of its H atoms lie in the same plane as the NO2 fragment and the third H atom is oriented nearly perpendicular to this plane directed at the O atom of an adjacent NM molecule. This H atom then transfers from the C atom of the first NM molecule to the O atom of the second NM molecule to yield the final products CH2NO2 and CH3NO(OH). The reaction occurs on a time scale of about 15 fs. In the second intermolecular H atom transfer reaction (illustrated in Figure 16b), the initial transfer is followed by the subsequent transfer of the H atom from the O atom of the acceptor NM molecule to the O atoms of two more adjacent NM molecules. Hence the H transfer reaction involves a total of four different NM molecules. In an effort to understand this pressure induced hydrogen transfer process further, the coordinates of the two NM molecules involved in the first H transfer event were extracted and a crude reaction pathway was constructed from BP86/DZVP single point energy calculations. These single point energy calculations employed the ADF quantum chemistry program suite [31]. The changes in bond lengths and Mulliken atomic charges are consistent with a proton transfer reaction as depicted in Figure 17.
DRDC Suffield TR 2003-156 25
Figure 16. Snapshots of the intermolecular H atom transfer events observed in the CPMD simulations of liquid nitromethane compressed by a factor of 3.0.
Figure 17. Schematic of the proton transfer reaction intermolecular H atom transfer events observed in the CPMD simulations of liquid nitromethane compressed by a factor of 3.0.
N
CH H
H
OO
N
CH H
H
OO
N
CH H
H
OO
N
C
OO
+H HH
N
CH H
H
OO
N
CH
OO
H H
+
+ -
+
(b)
(a)
26 DRDC Suffield TR 2003-156
-5
0
5
10
15
20
25
30
35
0 0.5 1 1.5 2 2.5
R(O-H) (A)
Rel
ativ
e En
ergy
(kca
l/mol
)
1.20 Ang1.30 Ang1.40 Ang1.50 Ang
Figure 18. Reaction energy profiles for the pressure induced proton transfer reaction calculated from linear transit calculations using the incipient O-H bond as the reaction coordinate
The present results are extremely interesting, especially in lieu of the recent findings of Margetis and co-workers, which appeared at roughly the same time as our initial report of this pressure-induced proton transfer reaction. In their paper they examined the effect of static compression on solid NM theoretically using the self-consistent charge density-functional tight-binding method. The most notable finding from their static calculations was the prediction of significantly stretched C-H bonds, hypothesized to lead to proton dissociation, under both uniaxial (at V/Vo=0.6) and isotropic compression (at V/Vo=0.4) conditions very similar to those in the current study. Based on Mulliken charges, they proposed that the H would depart as a proton, thereby leading to the same final product, [H2C=NO2]-, observed in our simulations. Furthermore, they also predicted a significant C-N bond reduction in response to the applied compression. It should be pointed out that the approach employed in the current work and that of Margetis et al. are quite different, thereby lending further credence to the validity of these results. These results, which appeared at roughly the same time, are the first to suggest that a proton transfer reaction may be involved in the initial activation step of the pressure process, as opposed to metallization induced by bending of the NO2 group. It should be noted that proton dissociation has a strong precedence in high-pressure studies of energetic materials. For instance, a base can sensitize NM toward the formation of the aci ion, [CH2NO2]-, via a proton transfer from NM to the base. Experiments by Shaw et al. and Blais and co-workers have proposed that a reaction involving H atoms or protons is involved in the initial steps of the detonation of liquid nitromethane. These studies further support the ideas proposed by Engelke et al. who suggested that there is an increased concentration of the aci-ion, which is more reactive towards decomposition than the original NM molecule at high pressure. The observed proton dissociation process previously reported by Margetis et al. and us is extremely significant. There has been very little effort to characterize this reaction and to understand why it is observed under high-pressure compression conditions; hence we attempt to rationalize this in the following discussion.

DRDC Suffield TR 2003-156 27
Figure 19. Molecular orbitals of nitromethane involved in the proton transfer reaction.
To obtain more detailed information regarding the energetics of this proton transfer, a linear transit calculation was conducted along the reaction coordinate corresponding to the newly formed O-H bond. The incipient O-H bond was incrementally decreased from its distance in the original two NM reactant molecules to its distance in the final CH3NO(OH) product (refer to Figure 16a). Our calculations indicate that the proton transfer reaction is highly endothermic, by more than 30 kcal/mol for a pair of gas phase NM molecules, similar to a pair of NM molecules under ambient conditions, having a C-N bond distance of about 1.5 Å. As illustrated in Section 5.1, the C-N bond is significantly shortened under the static high-pressure compression conditions where the proton transfer reaction was observed. Hence, the reaction energetic profile for the proton transfer reaction was recalculated via linear transits involving NM molecules with shorter C-N bond lengths of 1.40 Å, 1.30 Å, and 1.20 Å. The results, displayed in Figure 18, show that compression of the C-N bond from 1.5 Å to 1.2 Å causes a reduction in the endothermicity of the proton transfer reaction from 30 kcal/mol to 18 kcal/mol.
HOMO LUMO

28 DRDC Suffield TR 2003-156
Figure 20. Effect of decreasing C-N bond length on the NM MOs involved in the proton transfer reaction; (a) change in HOMO-LUMO energy gap and (b) change in LUMO
To probe how the C-N bond decrease activates the proton transfer reaction, the molecular orbitals (MO) of the NM molecule involved in the proton transfer reaction were investigated. The proton transfer reaction requires the donation of electron density from a high lying MO of correct symmetry at the O atom to a low lying MO of correct symmetry at the incoming H atom, in this case from the highest occupied MO (HOMO) to the lowest occupied MO (LUMO), as depicted in Figure 19. As illustrated in Figure 20a, as the C-N bond is shortened the HOMO-LUMO energy gap is decreased, thereby making the donation of electron density from the HOMO to the LUMO easier. If one examines the LUMO in more detail, which has contributions from the N-O π* and C-H σ* fragment orbitals, one can see an increased contribution of the σ* C-H fragment orbital in the LUMO as the C-N bond is shortened, as illustrated graphically in Figure 20b. Hence, as the C-N bond is decreased these two effects will combine to activate the C-H bond, thereby facilitating the subsequent proton transfer reaction.
-0.241 au
-0.107 au -0.108 au
-0.235 au
-0.228 au
-0.111 au
0.134 au 0.127 au 0.117 au
RCN=1.5 RCN=1.35 RCN=1.2(a)
(b)

DRDC Suffield TR 2003-156 29
6. Conclusions
The development of ab initio molecular dynamics, along with the rapid development of computer power, has inspired us to apply MD to studies of energetic materials and detonation processes. Multimolecular collision models were developed to study shock-induced dissociation in homogeneous liquid nitromethane using first principles Car-Parrinello molecular dynamics simulations within the Kohn-Sham density functional theory scheme. Simulations were carried out for a variety of collision orientations and collision velocities. The critical velocity for successful dissociation was found to be 11-12 km/s under the impact of a single molecule on multiple molecules and between 8 and 10 km/s under the impact of multiple molecules on multiple molecules. The neighbouring molecules present in the current multimolecular collision simulations act as a trap to confine the recoiling fragments produced during the initial impact, thereby enabling them to recombine and form intact nitromethane molecules. This leads to higher threshold collision velocities than found previously in bimolecular collision simulations (i.e., 7.0-10.5 km/s for collision orientations investigated). Although our rationale for extending the bimolecular collision simulations to multimolecular collision simulations was to account for potential collision induced reactions involving multiple nitromethane molecules, the present simulations indicate that this only occurs at very high incident collision velocities. In accord with previous bimolecular collision simulations, C-N bond cleavage is the primary mechanism of nitromethane dissociation in the multimolecular collision simulations at the threshold velocities. An alternative C-H bond scission fragmentation mechanism was observed only in high collision velocity simulations. The threshold collision velocities determined from the bimolecular and multimolecular collision simulations are much higher than the average atomic velocities expected at the detonation shock front of nitromethane, suggesting that molecular dissociation is likely controlled by thermalization after the shock front passes. The 8-10 km/s threshold collision velocity for successful dissociation also indicates that the detonation initiation via the excitation of kinetic motion degrees of freedom in the shock front would likely correspond to a detonation velocity beyond 15-20 km/s, which is highly challenging for a new generation of explosives. To draw a firm conclusion, more complex multimolecular collision scenarios should be investigated in an effort to more accurately model the stochastic nature of shock front propagation through bulk molecular explosives.
Car-Parrinello ab initio MD simulations under static compression conditions at low temperatures were also employed to investigate the possibility of pressure-induced molecular decomposition of liquid explosives under minimal thermal influence. The simulations of liquid nitromethane were conducted at 150-300 K with the simulation cell volume compressed by factors up to 3.0 (i.e., V/Vo = 0.33) corresponding to a pressure estimated near the megabar range. Analysis of the site-site pair correlation functions calculated from the simulations indicates that the local structure of liquid nitromethane increases. The average intramolecular C-N bond length of the molecules in the simulation cell is significantly reduced, by approximately 0.15 Å, when the simulation cell volume is compressed three-fold. On the other hand, the average N-O and C-H bond distance changes are not significant. No molecular decomposition was observed until the simulation cell volume was compressed three-fold. Under conditions of three-fold compression at 150K or 300K, intramolecular proton transfer with H atom transfer took place, instead of the thermal dissociation where the C-N bond

30 DRDC Suffield TR 2003-156
always breaks first. Analysis of the molecular orbitals involved in the proton transfer reaction demonstrated that decreasing the C-N bond activates the C-H bond toward the loss of a proton such that the HOMO-LUMO energy gap is decreased. Static DFT calculations predicted this proton transfer event to be highly endothermic and to have an activation barrier of about 25 kcal/mol. Linear transit calculations of the proton transfer for nitromethane molecules having different C-N bond lengths revealed that the reaction becomes less endothermic as the C-N bond is shortened with increasing static compression. This also indicates that the activation barrier or dissociation energy decreases with an increase in compression ratios. This theoretically proves a necessary condition for super-compressed detonation in insensitive molecular explosives.
While these models and the simulation approaches require further development, the results demonstrated the capability and potential of the ab initio or quantum mechanical molecular dynamics for future advances in simulations of high-pressure synthesis studies of new energetic materials and their detonation mechanisms.

DRDC Suffield TR 2003-156 31
7. References
1. Rahman, A. (1964) Phys. Rev. A 136: 405.
2. Verlet, L. (1967) Phys. Rev. 159: 98.
3. Car, R. and Parrinello, M. (1985) A unified Approach for Molecular Dynamics and Density Functional Theory. Phys. Rev. Lett. 55: 2471.
4. Car, R. and Parrinello, M. (1989) In: Polian, A. and Loubeyre, P. (eds.) Simple Molecular Systems and Very High Density, Plenum, New York, pp. 455.
5. Galli, G. and Pasquarello, A. (1993) In: Allen, M. P. and Tildesley, D. J.) Computer Simulation in Chemical Physics, Kluwer Acad., pp. 261.
6. Barnett, R. N. and Landman, U. (1993) Phys. Rev. B 48: 2081.
7. Parrinello, M. (1997) From Silicon to RNA: the Coming of Age of Ab Initio Molecular Dynamics. Solid State Communications 102: 107-120.
8. Dremin A. N., Klimenko V.-Yu (1981) On the effect of shock wave front on the reaction origin. In: Bowen JR et al. (eds.) Progress in Astronautics and Aeronautics Vol 75. AIAA Inc., pp 253-268.
9. Brenner, D. W., Robertson, D.H., Elert, M. L. and White, C.T. (1993) Phys. Rev. Lett. 70: 2174.
10. Barrett, J. J. C., Brenner, D. W., Robertson, D. H. and White, C. T. (1996) Shock Compression of Condensed Matter 370.
11. Haskins, P. J., Cook, M. D., Fellows, J. and Wood, A. (2000) Proceedings of the 6th Symposium (International) on Detonation, Snowmass, Colorado, pp. 897.
12. Dremin, A. N. (1992) Shock discontinuity zone effect: the main factor in the explosive decomposition detonation process. Phil. Trans. R. Soc. Lond. A 339:355-364.
13. Hohenberg, P. and Kohn, W. (1964) Phys. Rev. B 136: 864.
14. Kohn W., Sham L. J. (1965) Phys. Rev. A 140: 1133.
15. Yin, M. T. and Cohen, M. L. (1982) Phys. Rev. B 26: 5668.
16. Pickett, W. E. (1989) Comput. Phys. Rep. 9: 115.
17. Hutter, J., Alavi, A., Deutsch, T., Bernasconi, M., Goedecker, S., Marx, D., Tuckerman, M. and Parrinello, M. (1995-1999) CPMD. MPI fur Festkorperforschung and IBM Zurich Research Laboratories.

32 DRDC Suffield TR 2003-156
18. Becke, A. (1988) Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 38:3098.
19. Perdew, J. P. (1986) Density functional approximation for the correlation energy of inhomogeneous electron gas. Phys. Rev. B 33:8822.
20. Trouiller, N., Martins, J. L. (1991) Phys. Rev. B 43:1993.
21. Wei, D., Zhang, F., Woo, T. K. (2002) Ab initio molecular dynamics simulations of molecular collisions of nitromethane. In Am. Inst. Phys. Conference Proceedings 620 (Shock Compression of Condensed Matter, Pt. 1), New York, pp 407-410.
22. Decker, S. A., Woo, T. K., Wei, D. and Zhang, F. (2002) Ab initio molecular dynamics simulations of multimolecular collisions of nitromethane and compressed liquid nitromethane. In: 12th International Symposium on Detonation.
23. Haskins, P. J., Cook, M. D. (1997) Shock-induced reaction in energetic materials. In: Schmidt, Dandekar, Forbes (eds) Shock Compression of Condensed Matter. AIP, New York, pp 305.
24. Reed, E. J., Joannopoulos, J. D. and Fried, L. E. (2000) Phys. Rev. B 62: 16500.
25. Manaa, M. R., Fried, L. E. (1998) DFT and ab Initio Study of the Unimolecular Decomposition of the Lowest Singlet and Triplet States of Nitromethane J. Phys. Chem. A 102:9884.
26. Sorescu, D. C., Rice, B. M. and Thompson, D. L. (2001) J. Phys. Chem. A 105: 9336.
27. Alper, H. E., Abu-Awwad, F. and Politzer, P. J. (1999) Phys. Chem. B 103: 9738.
28. Piermarini, G. J., Block, S. and Miller, P. J. J. (1989) Phys. Chem. 93: 457.
29. Pangilinan, G. I. And Gupta, Y. M. (1994) L. Phys. Chem. 98: 4522.
30. Winey, J. M. and Gupta, Y. M. J. (1997) Phys. Chem. B 101: 10733.
31. ADF2002.01. SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands, 2002.
UNCLASSIFIED SECURITY CLASSIFICATION OF FORM (highest classification of Title, Abstract, Keywords)
DOCUMENT CONTROL DATA (Security classification of title, body of abstract and indexing annotation must be entered when the overall document is classified)
1. ORIGINATOR (the name and address of the organization preparing the document. Organizations for who the document was prepared, e.g. Establishment sponsoring a contractor's report, or tasking agency, are entered in Section 8.)
Defence R&D Canada – Suffield
2. SECURITY CLASSIFICATION (overall security classification of the document, including special
warning terms if applicable) Unclassified
3. TITLE (the complete document title as indicated on the title page. Its classification should be indicated by the appropriate abbreviation (S, C or U) in parentheses after the title).
Ab Initio Molecular Dynamics on Energetic Materials and Detonation Mechanisms (U)
4. AUTHORS (Last name, first name, middle initial. If military, show rank, e.g. Doe, Maj. John E.) Zhang, Fan and Woo, Tom K.
5. DATE OF PUBLICATION (month and year of publication of document)
December 2003
6a. NO. OF PAGES (total containing information, include Annexes, Appendices, etc) 44
6b. NO. OF REFS (total cited in document)
31
7. DESCRIPTIVE NOTES (the category of the document, e.g. technical report, technical note or memorandum. If appropriate, enter the type of report, e.g. interim, progress, summary, annual or final. Give the inclusive dates when a specific reporting period is covered.)
Technical Report
8. SPONSORING ACTIVITY (the name of the department project office or laboratory sponsoring the research and development. Include the address.)
DSTL, Defence R&D Canada
9a. PROJECT OR GRANT NO. (If appropriate, the applicable research and development project or grant number under which the document was written. Please specify whether project or grant.)
R&D Thrust 2nn 21 - TIF
9b. CONTRACT NO. (If appropriate, the applicable number under which the document was written.)
10a. ORIGINATOR'S DOCUMENT NUMBER (the official document number by which the document is identified by the originating activity. This number must be unique to this document.)
DRDC Suffield TR 2003-156
10b. OTHER DOCUMENT NOs. (Any other numbers which may be assigned this document either by the originator or by the sponsor.)
11. DOCUMENT AVAILABILITY (any limitations on further dissemination of the document, other than those imposed by security classification)
( x ) Unlimited distribution ( ) Distribution limited to defence departments and defence contractors; further distribution only as approved ( ) Distribution limited to defence departments and Canadian defence contractors; further distribution only as approved ( ) Distribution limited to government departments and agencies; further distribution only as approved ( ) Distribution limited to defence departments; further distribution only as approved ( ) Other (please specify):
12. DOCUMENT ANNOUNCEMENT (any limitation to the bibliographic announcement of this document. This will normally corresponded to the Document Availability (11). However, where further distribution (beyond the audience specified in 11) is possible, a wider announcement audience may be selected). Unlimited
UNCLASSIFIED SECURITY CLASSIFICATION OF FORM

UNCLASSIFIED SECURITY CLASSIFICATION OF FORM
13. ABSTRACT (a brief and factual summary of the document. It may also appear elsewhere in the body of the document itself. It is highly desirable that the abstract of classified documents be unclassified. Each paragraph of the abstract shall begin with an indication of the security classification of the information in the paragraph (unless the document itself is unclassified) represented as (S), (C) or (U). It is not necessary to include here abstracts in both official languages unless the text is bilingual).
The development of ab initio or “first principles” molecular dynamics, along with the rapid development of computer power, has inspired us to apply it to studies of energetic materials and detonation processes. Using Car-Parrinello ab initio molecular dynamics, a multimolecular collision model and a controlled temperature bulk liquid compression approach have been developed to study atomic-level decomposition mechanisms of homogeneous liquid energetic materials under shock and highly static compressed conditions. Liquid nitromethane was chosen as a prototypical molecular explosive. In multimolecular collision simulations, neighbouring molecules act as traps to confine the recoiling fragments produced during the initial collision, thereby allowing them to recombine. This results in higher threshold collision velocities than previously found with binary collision simulations. These threshold velocities (8-10 km/s) are higher than average atomic velocities expected at the detonation shock front, suggesting that molecular dissociation may occur with thermalization after the shock front passes. These threshold velocities also suggest that the excitation of kinetic motion degrees of freedom within the shock front can be a mechanism only for detonation initiation of a new generation of molecular explosives whose detonation velocities would be several times that of current molecular explosives. In bulk liquid compression simulations at controlled temperature (150-300 K), the activation barrier and the electronic energy gap decrease with increasing compression ratios. This provides a necessary condition for the super-compressed detonation of insensitive molecular explosives. Pressure decomposition occurs at three-fold compression accompanied with interatomic proton transfer reactions.
14. KEYWORDS, DESCRIPTORS or IDENTIFIERS (technically meaningful terms or short phrases that characterize a document and could be helpful in cataloguing the document. They should be selected so that no security classification is required. Identifies, such as equipment model designation, trade name, military project code name, geographic location may also be included. If possible keywords should be selected from a published thesaurus, e.g. Thesaurus of Engineering and Scientific Terms (TEST) and that thesaurus-identified. If it is not possible to select indexing terms which are Unclassified, the classification of each should be indicated as with the title.) Molecular Dynamics Simulation Detonation Energetic Material
UNCLASSIFIED SECURITY CLASSIFICATION OF FORM









![Mixed-effects Maximum Likelihood Difference Scaling · 1860[3][4]. Il formula la loi de Weber-Fechner selon laquelle ”la sensation varie comme le logarithme de l’excitation”](https://img.pdfslide.net/doc/110x75/600441a844a7ff0ba239cf20/mixed-effects-maximum-likelihood-difference-scaling-186034-il-formula-la-loi.jpg)



















