Embed Size (px)
Citation preview

www.elsevier.com/locate/yviro
Virology 329 (20
Activation of HTLV-I gene transcription by protein tyrosine
phosphatase inhibitors
Melanie Langloisa,b, Brigitte Audeta,b, Eric Legaulta,b, Marie-Eve Parea,b, Michel Ouelleta,b,
Jocelyn Roya,b, Nancy Dumaisc, Jean-Michel Mesnardd, David M. Rothsteine,
Susan J. Marriottf, Michel J. Tremblaya,b, Benoit Barbeaua,b,*
aCentre de Recherche en Infectiologie, Centre Hospitalier Universitaire de Quebec, Pavillon CHUL, Ste-Foy (Quebec), Canada G1V 4G2bDepartement de Biologie Medicale, Faculte de Medecine, Universite Laval, Ste-Foy (Quebec), Canada G1V 4G2
cDepartement de Biologie, Faculte des Sciences, Universite de Sherbrooke, Sherbrooke (Quebec), CanadadLaboratoires Infections Retrovirales et Signalisation Cellulaire, CRBM-CNRS UPR 1086, Montpellier 34060, France
eDepartment of Medicine, Yale Medical School, New Haven, CT 06510, USAfDepartment of Molecular Virology and Microbiology, Baylor College of Medicine, Houston, TX 77030, USA
Received 30 March 2004; returned to author for revision 10 May 2004; accepted 8 September 2004
Available online 1 October 2004
Abstract
Human T-cell leukemia virus type I (HTLV-I) transcription generally depends on the ability of the viral Tax protein to bind the CREB
transcription factor and form an active complex by recruiting CBP/p300 coactivators to the long terminal repeat (LTR). Studies have
demonstrated that T-cell activating agents that stimulate CREB are potent inducers of HTLV-I transcription. Herein, we demonstrate that
bpV[pic], a protein tyrosine phosphatase (PTP) inhibitor activates the HTLV-I LTR in the presence and absence of Tax expression. Optimal
activation occurred at 8 h and was synergistic with forskolin or PGE2. Infected cell lines and cells transfected with HTLV-I proviral DNAwere
equally responsive to the synergistic effect of bpV and forskolin on HTLV-I gene expression. Activation of the LTR by bpV[pic] was T-cell
receptor-independent, but required ZAP70, calcineurin activity and functional calcium entry. Inhibition of the SHP-1 PTP was suggested to be
important. Transfection experiments with a CREB dominant-negative mutant and with isolated TRE1- or CREB-responsive reporter constructs
and treatment with the MDL-12,330A adenylate cyclase inhibitor all supported the involvement of a CREB/ATF family member in this bpV-
dependent activation of the HTLV-I LTR, although CREB itself did not seem to be involved. Analysis of HTLV-I reporter constructs containing
mutated CREB-binding sites also implied the involvement of another element in this activation. These results demonstrate for the first time a
powerful effect of PTP inhibitors on HTLV-I LTR activity and suggest participation of both CREB-dependent and -independent pathways in
this activation.
D 2004 Elsevier Inc. All rights reserved.
Keywords: HTLV-I; LTR; CREB; Protein tyrosine phosphatase
Introduction
Human T-cell leukemia virus type I (HTLV-I) is the
etiological agent of adult T-cell leukemia/lymphoma
0042-6822/$ - see front matter D 2004 Elsevier Inc. All rights reserved.
doi:10.1016/j.virol.2004.09.003
* Corresponding author. Centre de Recherche en Infectiologie, RC709,
Centre Hospitalier Universitaire de Quebec, Pavillon CHUL 2705
Boulevard Laurier, Ste-Foy (Quebec), Canada G1V 4G2. Fax: +1 418
654 2715.
E-mail address: [email protected] (B. Barbeau).
(ATLL) (Hinuma et al., 1981; Poiesz et al., 1980; Yoshida
et al., 1982) and the neurological disease HTLV-I-associated
myelopathy (also termed tropical spastic paraparesis)
(HAM/TSP) (Gessain et al., 1985; Osame et al., 1986;
Rodgers-Johnson et al., 1985). A number of other diseases
have been tentatively linked to HTLV-I infection in humans
although their association remains to be more clearly
ascertained (Uchiyama, 1997). HTLV-I is endemic in
specific regions of the world including Japan and the
Caribbean Islands. Between 10 and 20 million individuals
04) 395–411

M. Langlois et al. / Virology 329 (2004) 395–411396
are presently infected with HTLV-I; however, only 5% of
these infected individuals will eventually develop HTLV-I-
related diseases, while the rest will remain asymptomatic for
life (Uchiyama, 1997).
Infected cells are generally considered to be poor
producers of viral particles and only a very low proportion
of virions are infectious (Sommerfelt, 1999). It is thus
believed that HTLV-I replication in infected patients occurs
mainly through mitotic cell division, resulting in an
oligoclonal expansion of infected cells. The viral Tax
protein is an important T-cell proliferation factor thereby
contributing to the expansion of infected cells (for review,
see Smith and Greene, 1991; Yoshida et al., 1995).
However, when uninfected individuals become infected, a
transient burst of viral gene expression from the infected
cells promotes cell-to-cell transmission of HTLV-I, and
might be part of the two-step infection process previously
described in the squirrel monkey model (Kazanji et al.,
2000). Following the hypothesized cell contact-mediated
increase in viral gene expression, HTLV-I transcription is
believed to be mostly silent in the infected individual, which
might be important for allowing efficient immune escape.
Like all retroviruses, HTLV-I is bordered by sequences
known as long terminal repeats (LTRs). The 5VLTR contains
the major viral promoter and thus controls the expression of
HTLV-I genes. Several regions of the promoter have been
identified as important regulators of HTLV-I gene expres-
sion. Perhaps, the best-characterized transcriptional control
elements within the HTLV-I promoter are the Tax-responsive
element 1 (TRE1) sequences, which mediate the powerful
transactivation potential of Tax (Fujisawa et al., 1986;
Paskalis et al., 1986; Shimotohno et al., 1986). The three
imperfect TRE1 repeats span the U3 region of the LTR and
contain suboptimal cyclic AMP response element binding
protein (CREB) binding sites. Tax is thought to stabilize the
association of both CREB and CREB-binding protein (CBP)/
p300 with the LTR, thereby mediating its strong activation of
HTLV-I gene expression (Giebler et al., 1997; Harrod et al.,
1998; Kashanchi et al., 1998; Kwok et al., 1996; Yan et al.,
1998). Additional studies have also identified a second Tax-
responsive element within the LTR, TRE2 (Brady et al.,
1987), which contains sequences specific for the binding of
transcription factors such as NF-nB and Ets-1 (Bosselut et al.,
1990; Gitlin et al., 1991; Numata et al., 1991).
Little is known about regulation of the HTLV-I LTR in the
absence of Tax. This regulation is important in vivo since
Tax expression is generally low in HTLV-I-positive cells
isolated from infected individuals (Brauweiler et al., 1995).
A previous study has suggested that some of the TRE1
repeats are important for basal transcription (Barnhart et al.,
1997). These same sequences are also known to be important
determinants for HTLV-I LTR activation by cAMP-modulat-
ing agents such as forskolin (Nakamura et al., 1989; Poteat et
al., 1989). In contrast, agents that induce T-cell receptor
(TCR) aggregation have been shown to be poor activators of
the HTLV-I promoter (Copeland et al., 1994, 1995). A
phorbol 12-myristate 13-acetate (PMA)-responsive element
has been identified within the HTLV-I promoter and its
modulatory activity is mediated through Sp1 and p53
(Torgeman et al., 1999, 2001b). However, activation of the
LTR in response to PMA was shown to depend on the
apoptotic-inducing effects of PMA as well as other
apoptosis-inducing agents rather than transcriptional effects
(Torgeman et al., 2001a). A study by Lin et al. (1998)
demonstrated that phytohemagglutinin (PHA)/PMA in com-
bination with Tax resulted in high levels of HTLV-I
replication and that this induction was sensitive to inhibitors
of tyrosine phosphorylation.
Tyrosine phosphorylation is a very important event in
intracellular signaling cascades. In fact, early events
following T cell activation involve a sustained increase in
intracellular phosphotyrosine levels. A critical balance
between the activity of protein tyrosine kinases (PTK) and
protein tyrosine phosphatases (PTP) maintains a crucial
intracellular balance of tyrosine phosphorylation (Yakura,
1998). Some of the most important PTKs for T-cell
activation are p56lck, p59fyn, and ZAP70, while important
PTPs in T-cell activation include SHP-1, a downregulator of
T cell activation, and CD45, an abundant molecule having
both negative and positive effects on T-cell receptor-
mediated signaling (Yakura, 1998). Specific PTP inhibitors
have been used to examine the role of PTPs in T cell
activation. Evidence clearly suggests that these inhibitors
are potent T-cell activators, and strongly induce IL-2
secretion and the activity of transcription factors such as
AP-1 and NF-nB (Barbeau et al., 1997; Imbert et al., 1994,
1996a, 1996b; Ouellet et al., 1999, 2003a, 2003b; Secrist et
al., 1993). Recently, we demonstrated that PTP inhibitors
are also powerful activators of NFAT (Fortin et al., 2001).
Based on the results of Lin et al. (1998), we wanted to
determine whether PTP could modulate transcriptional
activity of the HTLV-I LTR. To directly test this hypothesis,
we used powerful PTP inhibitors known as bis-peroxovana-
dium (bpV) compounds (Posner et al., 1994). Incubation of
T cells with these compounds strongly stimulated activity of
the HTLV-I LTR, which could be further induced upon the
addition of forskolin. This activation was partly CREB-
dependent and required most components of the TCR
pathway. In addition, we identified the PTP, SHP-1, as an
upstream regulator of this induction. We conclude that PTPs
play an important role in the control of HTLV-I gene
expression and that inhibition of this activity leads to higher
levels of HTLV-I LTR activity.
Results
BpV[pic] is a powerful activator of HTLV-I LTR
transcription
We have previously used strong PTP inhibitors to study
the impact of tyrosine phosphorylation on gene regulation in

M. Langlois et al. / Virology 329 (2004) 395–411 397
the context of T cells (Barbeau et al., 1997; Fortin et al.,
2001; Ouellet et al., 1999). These compounds are termed
bis-peroxovanadium (bpV) (Posner et al., 1994) and were
demonstrated in our previous studies to strongly inhibit PTP
activity in Jurkat T cells. On the basis of a previous study
establishing that tyrosine phosphorylation might be required
for Tax-dependent HTLV-I LTR regulation (Lin et al.,
1998), we tested our bpV compounds in Jurkat cells to gain
a better understanding of the role played by tyrosine
phosphorylation in HTLV-I LTR gene expression.
Jurkat cells were transfected with an HTLV-I LTR
reporter vector (pHTLV-LUC) and a wild-type Tax expres-
sion vector or the empty vector. Different combinations of
activators were subsequently added to the transfected cells.
As depicted in Fig. 1A, stimulation of T cells by the bpV[pic]
PTP inhibitor led to a significant increase in Tax-dependent
LTR activation to levels higher than those measured in
untreated cells (36.7-fold). The addition of PMA with or
without ionomycin led to a significant but weaker induction
of Tax-dependent LTR activation. However, unexpectedly,
we found that bpV[pic] also induced a potent increase in
LTR gene expression in the absence of Tax expression. In
fact, LTR-dependent gene expression was induced 20-fold
following PTP inhibition, which was significantly greater
than induction measured in cells treated with PMA alone or
in combination with ionomycin (2.4 and 4.2-fold, respec-
tively). This increase in HTLV-I LTR activity could not be
generalized to all promoters as no similar induction in
reporter gene expression was noted when Jurkat cells
transfected with a h-actin promoter-driven h-gal vector
was treated with our PTP inhibitors (data not shown).
We next conducted a time course analysis to determine
the optimal time for HTLV-I LTR activation by bpV[pic].
Fig. 1B demonstrated that optimal HTLV-I LTR induction
occurred at 8 h following bpV[pic] treatment, reached a
plateau at 12 h, and then returned to basal level after 24 h of
stimulation. Such a pattern of induction has also been
observed in previous studies testing several activating
agents in T cells (including PTP inhibitors) (Fortin et al.,
Fig. 1. Activation of the HTLV-I LTR by bpV[pic] occurs in the presence and absen
10 Ag of the wild-type Tax expression vector or pHhPr.1-neo. In panel B, cells w
were either left untreated or stimulated with the following agents: PMA (20 ng/m
lysed and luciferase activity was measured after 8 h and at other time points p
triplicates F SD. Fold inductions in panel B were calculated as the ratio of the
results are representative of three independent experiments.
2001; Ouellet et al., 1999). In comparison, activation by
either PMA or the PMA/ionomycin combination continued
to increase after 24 h of incubation, a result which is
consistent with a previous study (Mor-Vaknin et al., 1997).
Activation of the HTLV-I LTR by bpV[pic] was also
detected in other T-cell lines such as SupT1 and HPB.ALL
(data not shown). These results demonstrate that PTP
inhibitors can independently induce activation of the
HTLV-I LTR in different T-cell lines, and that this LTR
activation is greater than other previously described
stimulation.
Activation of the HTLV-I LTR by bpV[pic] is synergistic
with forskolin
We next investigated possible synergistic activation of
the LTR by bpV[pic] and other known activators of HTLV-I
transcription. A series of different activators was tested
individually or in combination. These included PHA, PMA,
ionomycin, anti-CD3 (OKT3) and anti-CD28 (9.3) anti-
bodies, bpV[pic] and forskolin, a compound that upregu-
lates HTLV-I gene expression by activating the CREB
transcription factor (Nakamura et al., 1989; Poteat et al.,
1989). Jurkat cells were transfected with the pHTLV-LUC
vector and subsequently stimulated as indicated (Fig. 2A).
As demonstrated earlier, bpV[pic] potently induced HTLV-I
LTR-regulated gene expression while other activators
resulted in poor activation. When the PTP inhibitor was
combined with other activators, bpV-dependent induction
was either unaltered (PMA, ionomycin, anti-CD3/anti-
CD28), reduced (PMA/ionomycin) or was weakly enhanced
(PHA, PHA/PMA). The greatest synergistic activation of
the LTR was observed in the presence of forskolin and
bpV[pic]. A similar synergistic activation of the LTR by
bpV[pic] and forskolin was also observed in isolated
peripheral blood mononuclear cells (PBMCs) transfected
with the pHTLV-LUC vector (data not shown).
Synergistic activation of the LTR by forskolin and
bpV[pic] or PMA was compared at different concentrations
ce of Tax. (A) Jurkat cells were co-transfected with pHTLV-LUC (5 Ag) andere transfected with 15 Ag of pHTLV-LUC. At 24 h post-transfection, cells
l), PMA (20 ng/ml)/ionomycin (1 AM) and bpV[pic] (10 AM). Cells were
ost-induction for panel B. Luciferase values are reported as the mean of
mean value of treated samples over the mean of untreated samples. These

Fig. 2. Synergistic activation of the HTLV-I LTR by bpV[pic] and CREB-activating agent. Jurkat cells were transfected with 15 Ag of pHTLV-LUC. At 24 h
post-transfection, cells were either left untreated or stimulated with the following agents: (A) PHA (3 Ag/ml), PMA (20 ng/ml), ionomycin (1 AM), PHA (3
Ag/ml)/PMA (20 ng/ml), PMA (20 ng/ml)/ionomycin (1 AM), OKT3 (anti-CD3) (3 Ag/ml)/9.3 (anti-CD28) (1 Ag/ml), forskolin (100 AM) alone or in
combination with bpV[pic] (10 AM), (B) PMA (20 ng/ml) and bpV[pic] (10 AM) added in combination with increasing concentrations of forskolin (0.1, 1, 10,
100 AM), (C) combinations of increasing concentrations of forskolin (1, 10, 100 AM), bpV[pic] (0.1, 1, 10 AM) and Na3VO4 (0.25, 2.5, 25 mM), (D) bpV[pic]
(10 AM), PGE2 (100 nM), bpV[pic] (10 AM)/PGE2 (100 nM). After 8 h of stimulation (6h for panel A), cells were lysed and luciferase activity was measured.
Luciferase values were reported as the mean of triplicates F SD. Fold inductions were calculated as the ratio of the mean value of treated samples over the
mean value of untreated samples. These results are representative of three independent experiments.
M. Langlois et al. / Virology 329 (2004) 395–411398
in Jurkat cells transfected with pHTLV-LUC (Fig. 2B).
Although a significant increase was apparent for all tested
activators at higher concentrations of forskolin, the greatest
synergistic induction was observed in the presence of the
bpV[pic]/forskolin combination. Also, maximal synergistic
activation with bpV[pic] occurred at a lower concentration
of forskolin (1 AM) than needed to induce synergistic
activation with PMA.
Sodium orthovanadate is another PTP inhibitor, which
has been suggested to be less potent than bpV[pic] (Posner
et al., 1994). LTR activity in response to different
concentrations of bpV[pic] or sodium orthovanadate in the
presence or absence of forskolin was analyzed in Jurkat cells
(Fig. 2C). Sodium orthovanadate did not induce HTLV-I
LTR-mediated gene expression either alone or in the
presence of forskolin, indicating that bpV[pic] was likely
more inhibitory for PTPs and consequently a better activator
of HTLV-I LTR transcription. These results also indicated
that the action of bpV[pic] occurred over a very narrow
concentration range and was not measurable at 1 AM either
with or without forskolin.
Since the synergy of bpV[pic] and forskolin might be
driven by the ability of forskolin to activate CREB, we
tested PGE2, another CREB activator, which we have
recently shown to activate the HTLV-I LTR upon concom-
itant TCR activation (Dumais et al., 2003). Jurkat cells
transfected with pHTLV-LUC were treated with bpV[pic]
alone or in combination with PGE2. As demonstrated in Fig.
2D, the combination of PGE2 and bpV[pic] resulted in
synergistic activation of the HTLV-I LTR supporting a role
for CREB in bpV[pic]-mediated activation of the LTR. As
we have previously demonstrated (Dumais et al., 2003),
PGE2 alone was a poor activator of the HTLV-I LTR.
To determine whether this bpV-dependent activation of
the HTLV-I LTR was reporter-dependent, the GFP reporter
gene was positioned downstream of the HTLV-I LTR.
Typically, less than 1% of Jurkat cells transfected with the
pHTLV-GFP vector showed a positive GFP signal. In Jurkat

M. Langlois et al. / Virology 329 (2004) 395–411 399
cells co-transfected with pHTLV-GFP and the Tax expres-
sion vector, GFP signals could be detected in up to 6% of
the analyzed cells (data not shown). Therefore, Jurkat cells
were transfected with pHTLV-GFP and subsequently
stimulated with different activating agents for 8 h. A small
increase in the number of cells positive for GFP expression
was observed following PMA/ionomycin treatment (1.93%)
(Table 1). Interestingly, a greater increase in positive cells
was measured in bpV[pic]-treated cells (3.12%). The
highest level of GFP-positive cells was observed when
bpV[pic] was used in combination with forskolin (6.62%).
These results demonstrate potent activation of the HTLV-I
LTR in response to a PTP inhibitor as compared to other
known T cell activators. Our results further indicate the
synergistic action of bpV[pic] with CREB-activating agents.
Activation of HTLV-I gene expression from full-length
proviral DNA and integrated proviral DNA by bpV[pic] in
combination with forskolin
To evaluate bpV[pic] activation of HTLV-I LTR tran-
scription in the context of full-length HTLV-I proviral DNA,
a luciferase reporter gene was inserted in frame with the
envelope ATG initiation codon from the K30 proviral DNA
clone without altering the splice donor site for multiply
spliced mRNA. This proviral DNA was directly transfected
into Jurkat cells and the transfected cells were subsequently
treated with activators. As depicted in Fig. 3A, activation by
bpV[pic] and all other tested activators led to a weak
induction, which was less than 2-fold. However, the
combination of bpV[pic] and forskolin induced luciferase
expression by 9.1-fold 8 h after stimulation, and this activity
was greatly enhanced at 24 h post-stimulation (129.7-fold)
and sustained up to at least 72 h post-treatment.
Previous experiments have indicated differences in the
regulation of HTLV-I gene transcription when integrated
into cellular DNAversus an episomal form (Lemasson et al.,
2002; Okada and Jeang, 2002). To confirm our results with
integrated proviral DNA, we performed experiments with
HTLV-I-infected MJ cells, which are CD3- and CD4-
positive T-cells. At different time points after stimulation,
supernatants from the MJ cells were quantitated for the
presence of the HTLV-I p19 antigen. As illustrated in Fig.
3B, at 24 h post-stimulation, only the bpV[pic]/forskolin
combination led to a significant increase in supernatant-
Table 1
Induction of HTLV-I-driven GFP gene expression by bpV[pic]
Treatment Percentage of positive
cells (%)a
NT 1.23
PMA/ionomycin 1.93
bpV[pic] 3.12
bpV[pic]/forskolin 6.62
a Percentage of positive cells for the GFP signal was determined at 8 h after
stimulation by FACS analysis.
associated p19 antigen. While bpV[pic] alone had little
effect on the production of HTLV-I p19 in the supernatant of
MJ cells throughout the time course, bpV[pic]/forskolin led
to a sustained increase in p19 production, to higher levels
than observed following treatment with the combination of
PMA/ionomycin. Forskolin-like PMA had no detectable
effect on the level of extracellular p19 antigen (data not
shown). These results support the notion that bpV com-
pounds can activate the HTLV-I LTR in the context of
episomal DNA as well as the integrated proviral DNA form,
although forskolin was required to observe this induction in
MJ cells.
Effect of early TCR-like cell signaling mediated by bpV
compounds on HTLV-I LTR activation
Our results confirmed that TCR-mediated T cell activa-
tion results in weak activation of the HTLV-I LTR. Since
PTP inhibitors can activate multiple transcription factors
through TCR-like events (Ouellet et al., 2003a,b), we next
examined the effect of bpV[pic] on the activation of HTLV-I
LTR transcription in response to TCR signaling pathways.
To determine whether the T-cell receptor is required for
activation of HTLV-I LTR transcription by bpV[pic], both
Jurkat and J.RT3 (a TCR-negative derivative) cells were
transfected with the pHTLV-LUC reporter construct and
transfected cells were then exposed to various agents.
bpV[pic]-mediated LTR activation (with or without added
forskolin) was decreased in J.RT3 cells as compared to
Jurkat cells, although a significant induction of the HTLV-I
promoter was still observed in J.RT3 cells (Fig. 4A). Other
activating agents showed similar abilities to activate the
LTR in both Jurkat cell lines. These results suggest that the
TCR is not absolutely necessary for bpV[pic] induction of
HTLV-I LTR-mediated gene expression and agree with our
observation of HTLV-I LTR activation in the TCR-negative
SupT1 cell line (data not shown).
ZAP70 is another important player in T cell signaling.
P116 cells are Jurkat cell derivatives that are negative for
ZAP70 (Williams et al., 1998). A stable transfectant (P116
clone 39) has been generated from this clone and
expresses normal levels of ZAP70. P116, P116 clone 39,
and Jurkat cells were transfected with pHTLV-LUC and
then stimulated with the different activators (Fig. 4B).
Comparison of the different cell lines showed that P116
cells had the weakest HTLV-I LTR response to bpV[pic].
P116 clone 39 showed an increased HTLV-I LTR response
to bpV stimulation and reached levels of activation close
to that observed in similarly stimulated and transfected
Jurkat cells. ZAP70 expression was also required for
activation of the HTLV-I LTR by the bpV[pic]/forskolin
combination. In general, activation by PMA, PMA/
ionomycin or forskolin was not statistically different in
the presence or absence of ZAP70. This was expected due
to the fact that these agents activate the signaling pathway
downstream of ZAP70.

Fig. 3. Activation of HTLV-I proviral gene expression by bpV[pic]. (A) Jurkat cells were transfected with 30 Ag of 5VK30-LUC/3VK30-ligated DNA mixture.
At 24 h post-transfection, cells were either left untreated or stimulated with the following conditions: PMA (20 ng/ml), PMA (20 ng/ml)/ionomycin (1 AM),
forskolin (100 AM), bpV[pic] (10 AM) and bpV[pic] (10 AM)/forskolin (100 AM). After 8, 24, 48, and 72 h of stimulation, cells were lysed and luciferase
activity was measured. (B) MJ cells were left untreated or stimulated for 24, 48, and 72 h with the following conditions: PMA (20 ng/ml)/ionomycin (1 AM),
bpV[pic] (10 AM) and bpV[pic] (10 AM)/forskolin (100 AM). Supernatants were then harvested and p19 antigen was quantitated. Luciferase values and p19
antigen levels were reported as the mean of triplicates F SD. In panel A, fold inductions were calculated as the ratio of the mean value of treated samples over
the mean value of untreated samples. These results are representative of three independent experiments.
M. Langlois et al. / Virology 329 (2004) 395–411400
Since the calcineurin serine/threonine phosphatase plays
an important role in activating several transcription factors
in T cells, we assessed the effect of this serine/threonine
phosphatase on bpV[pic]-mediated HTLV-I LTR activation.
Jurkat cells transfected with the pHTLV-LUC vector were
treated with FK506 or CsA, two specific calcineurin
inhibitors, prior to stimulation (Fig. 4C). Both of these
inhibitors reduced the capacity of bpV[pic] and bpV[pic]/
forskolin activators to induce HTLV-I LTR activity. Since
calcium is a crucial signaling mediator in the activity of
calcineurin, we assessed the role of intracellular calcium
mobilization in bpV[pic]-dependent activation of the HTLV-
I LTR. PMA/ionomycin activation of the HTLV-I LTR was
reduced in the transfected CJ1.1 cell line, deficient for
capacitative calcium entry, as compared to the wild type CJ
parental clone (Fig. 4D). Importantly, a similarly reduced
LTR response was observed in this calcium-deficient cell
line when cells were stimulated with the PTP inhibitor (with
or without forskolin), suggesting that calcium plays an
important role in activation of the HTLV-I LTR by PTP
inhibition. These results demonstrate that certain cellular
components involved in TCR-mediated signaling play a role
in the bpV[pic]-mediated signaling cascade that leads to
HTLV-I LTR activation. However, the TCR was not
specifically required in this pathway.
BpV[pic] activation of HTLV-I LTR activity is counteracted
by SHP-1 expression
Activation of the HTLV-I LTR by the PTP inhibitor
suggested the involvement of one or more PTPs in this
activation pathway. We looked at the possible role of two
important PTPs, CD45 and SHP-1, initially focusing on
CD45 as a target of bpV[pic]. JKF cells and the CD45-
deficient K6D cell line derived from JKF cells were
transfected with pHTLV-LUC and subsequently treated
with the activating agents. No significant differences in
LTR response to any of the tested activators including
bpV[pic] were detected (data not shown). The Jurkat-
derived CD45-negative J45.01 cell line was also compared
to Jurkat cells, and again no significant difference in
response was observed when cells transfected with
pHTLV-LUC were treated with our PTP inhibitor (data
not shown). We then turned our attention to SHP-1. This
PTP is required to terminate the TCR-initiated cascade and
has been observed to dephosphorylate several crucial
players in this cascade. To determine whether SHP-1 was
targeted by bpV[pic], a vector expressing SHP-1 or the
empty vector were co-transfected with pHTLV-LUC into
Jurkat cells and cells were then stimulated with the different
activators (Fig. 5). We predicted that overexpression of a
PTP targeted by bpV[pic] should block activation of the
HTLV-I LTR. In fact, although SHP-1 expression did not
affect the response of the HTLV-I LTR following PMA or
PMA/ionomycin induction, its expression significantly
reduced bpV[pic]- and bpV[pic]/forskolin-dependent
HTLV-I LTR activation. These results thus suggest that
SHP-1 inhibition might be responsible for activation of the
HTLV-I LTR following bpV[pic] treatment.
Role of the LTR region containing TRE1 sequences in
bpV[pic]-mediated activation of the HTLV-I transcription
TRE1 sequences in the HTLV-I LTR contain CREB-
binding sites that are important for positive regulation of
HTLV-I transcription. To determine the potential role of
these sequences in bpV-mediated LTR activation, LTR
deletion mutants were generated with or without the TRE1
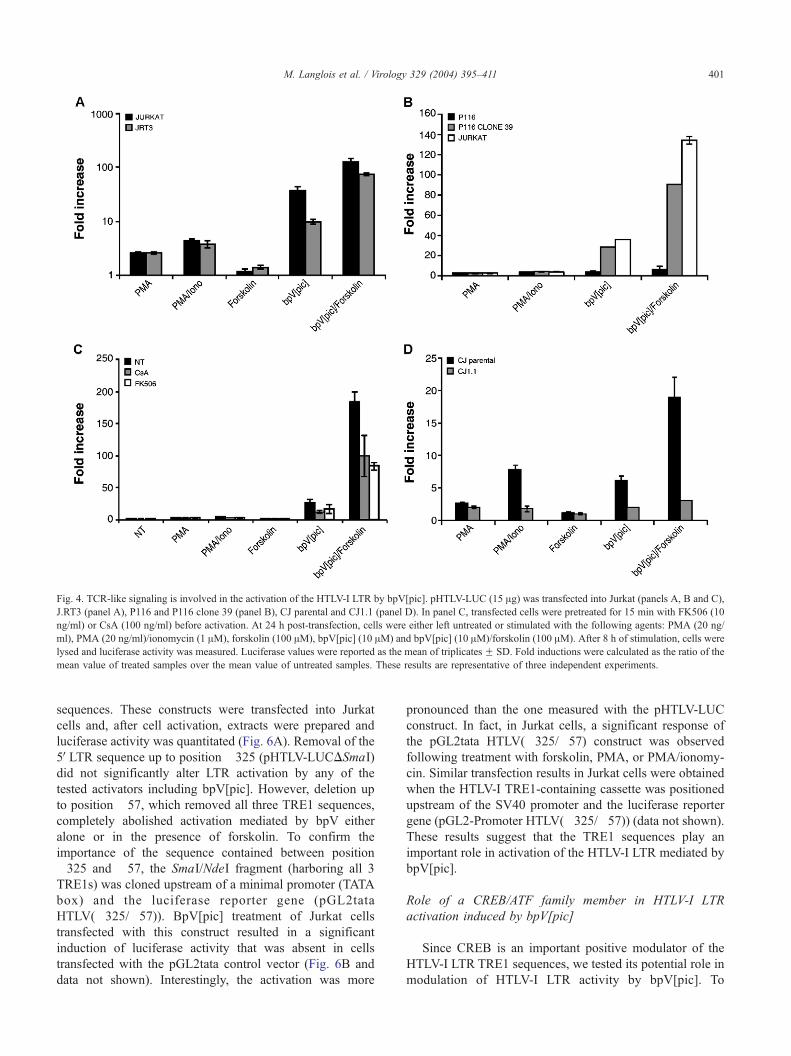
Fig. 4. TCR-like signaling is involved in the activation of the HTLV-I LTR by bpV[pic]. pHTLV-LUC (15 Ag) was transfected into Jurkat (panels A, B and C),
J.RT3 (panel A), P116 and P116 clone 39 (panel B), CJ parental and CJ1.1 (panel D). In panel C, transfected cells were pretreated for 15 min with FK506 (10
ng/ml) or CsA (100 ng/ml) before activation. At 24 h post-transfection, cells were either left untreated or stimulated with the following agents: PMA (20 ng/
ml), PMA (20 ng/ml)/ionomycin (1 AM), forskolin (100 AM), bpV[pic] (10 AM) and bpV[pic] (10 AM)/forskolin (100 AM). After 8 h of stimulation, cells were
lysed and luciferase activity was measured. Luciferase values were reported as the mean of triplicates F SD. Fold inductions were calculated as the ratio of the
mean value of treated samples over the mean value of untreated samples. These results are representative of three independent experiments.
M. Langlois et al. / Virology 329 (2004) 395–411 401
sequences. These constructs were transfected into Jurkat
cells and, after cell activation, extracts were prepared and
luciferase activity was quantitated (Fig. 6A). Removal of the
5VLTR sequence up to position �325 (pHTLV-LUCDSmaI)
did not significantly alter LTR activation by any of the
tested activators including bpV[pic]. However, deletion up
to position �57, which removed all three TRE1 sequences,
completely abolished activation mediated by bpV either
alone or in the presence of forskolin. To confirm the
importance of the sequence contained between position
�325 and �57, the SmaI/NdeI fragment (harboring all 3
TRE1s) was cloned upstream of a minimal promoter (TATA
box) and the luciferase reporter gene (pGL2tata
HTLV(�325/�57)). BpV[pic] treatment of Jurkat cells
transfected with this construct resulted in a significant
induction of luciferase activity that was absent in cells
transfected with the pGL2tata control vector (Fig. 6B and
data not shown). Interestingly, the activation was more
pronounced than the one measured with the pHTLV-LUC
construct. In fact, in Jurkat cells, a significant response of
the pGL2tata HTLV(�325/�57) construct was observed
following treatment with forskolin, PMA, or PMA/ionomy-
cin. Similar transfection results in Jurkat cells were obtained
when the HTLV-I TRE1-containing cassette was positioned
upstream of the SV40 promoter and the luciferase reporter
gene (pGL2-Promoter HTLV(�325/�57)) (data not shown).
These results suggest that the TRE1 sequences play an
important role in activation of the HTLV-I LTR mediated by
bpV[pic].
Role of a CREB/ATF family member in HTLV-I LTR
activation induced by bpV[pic]
Since CREB is an important positive modulator of the
HTLV-I LTR TRE1 sequences, we tested its potential role in
modulation of HTLV-I LTR activity by bpV[pic]. To
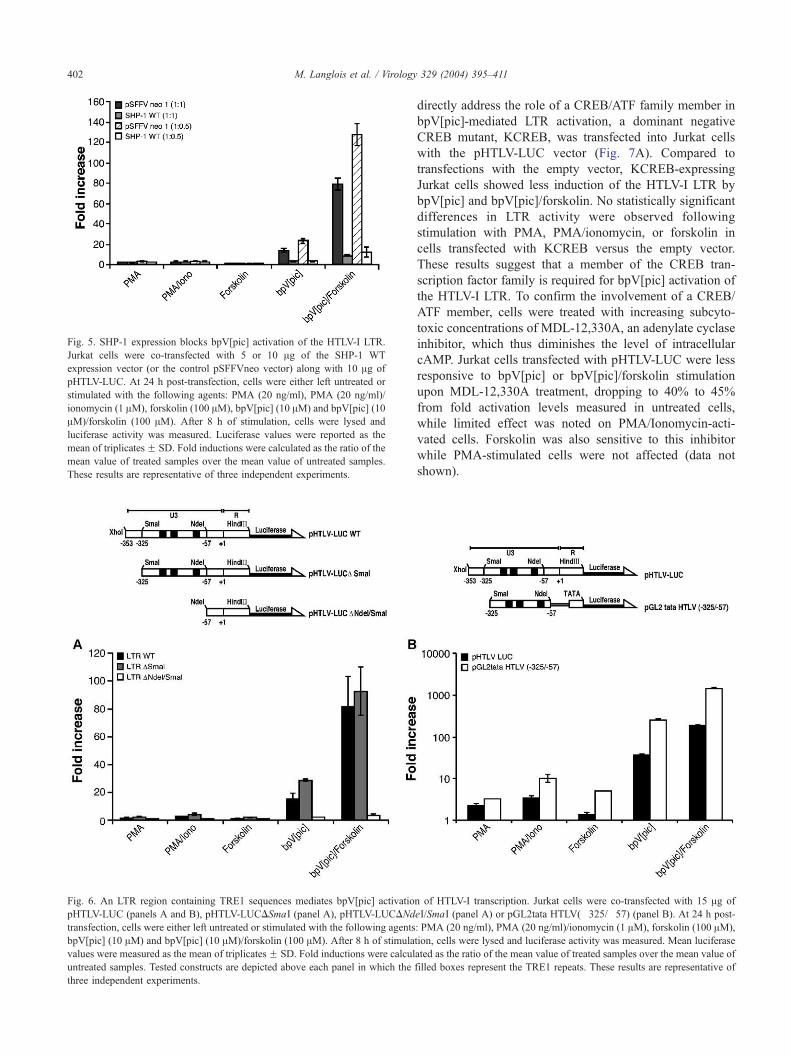
Fig. 6. An LTR region containing TRE1 sequences mediates bpV[pic] activatio
pHTLV-LUC (panels A and B), pHTLV-LUCDSmaI (panel A), pHTLV-LUCDNd
transfection, cells were either left untreated or stimulated with the following agents
bpV[pic] (10 AM) and bpV[pic] (10 AM)/forskolin (100 AM). After 8 h of stimula
values were measured as the mean of triplicatesF SD. Fold inductions were calcul
untreated samples. Tested constructs are depicted above each panel in which the
three independent experiments.
Fig. 5. SHP-1 expression blocks bpV[pic] activation of the HTLV-I LTR.
Jurkat cells were co-transfected with 5 or 10 Ag of the SHP-1 WT
expression vector (or the control pSFFVneo vector) along with 10 Ag of
pHTLV-LUC. At 24 h post-transfection, cells were either left untreated or
stimulated with the following agents: PMA (20 ng/ml), PMA (20 ng/ml)/
ionomycin (1 AM), forskolin (100 AM), bpV[pic] (10 AM) and bpV[pic] (10
AM)/forskolin (100 AM). After 8 h of stimulation, cells were lysed and
luciferase activity was measured. Luciferase values were reported as the
mean of triplicatesF SD. Fold inductions were calculated as the ratio of the
mean value of treated samples over the mean value of untreated samples.
These results are representative of three independent experiments.
M. Langlois et al. / Virology 329 (2004) 395–411402
directly address the role of a CREB/ATF family member in
bpV[pic]-mediated LTR activation, a dominant negative
CREB mutant, KCREB, was transfected into Jurkat cells
with the pHTLV-LUC vector (Fig. 7A). Compared to
transfections with the empty vector, KCREB-expressing
Jurkat cells showed less induction of the HTLV-I LTR by
bpV[pic] and bpV[pic]/forskolin. No statistically significant
differences in LTR activity were observed following
stimulation with PMA, PMA/ionomycin, or forskolin in
cells transfected with KCREB versus the empty vector.
These results suggest that a member of the CREB tran-
scription factor family is required for bpV[pic] activation of
the HTLV-I LTR. To confirm the involvement of a CREB/
ATF member, cells were treated with increasing subcyto-
toxic concentrations of MDL-12,330A, an adenylate cyclase
inhibitor, which thus diminishes the level of intracellular
cAMP. Jurkat cells transfected with pHTLV-LUC were less
responsive to bpV[pic] or bpV[pic]/forskolin stimulation
upon MDL-12,330A treatment, dropping to 40% to 45%
from fold activation levels measured in untreated cells,
while limited effect was noted on PMA/Ionomycin-acti-
vated cells. Forskolin was also sensitive to this inhibitor
while PMA-stimulated cells were not affected (data not
shown).
n of HTLV-I transcription. Jurkat cells were co-transfected with 15 Ag of
eI/SmaI (panel A) or pGL2tata HTLV(�325/�57) (panel B). At 24 h post-
: PMA (20 ng/ml), PMA (20 ng/ml)/ionomycin (1 AM), forskolin (100 AM),
tion, cells were lysed and luciferase activity was measured. Mean luciferase
ated as the ratio of the mean value of treated samples over the mean value of
filled boxes represent the TRE1 repeats. These results are representative of

Fig. 7. Implication of a CREB/ATF factor in bpV[pic]-dependent activation of HTLV-I LTR. Jurkat cells were co-transfected with 5 or 10 Ag of expression
vectors of KCREB (or the pRC/RSV control vector) with 10 Ag of pHTLV-LUC (panel A). In panel B, Jurkat cells were transfected with 15 Ag of pHTLV-LUCand pretreated or not with MDL-12,330A (100 AM and 250 AM) for 1 h prior to activation. In panel C, Jurkat cells were transfected with 15 Ag of pCRE-LUC.
In panels D and E, Jurkat cells were co-transfected with 10 Ag of expression vectors for CREB WT or CREB F/Yor the pcDNA.CF empty vector and 10 Ag of
pGL2tata HTLV(�325/�57) (panel D) or pNF-nB-LUC (panel E). At 24 h post-transfection, cells were either left untreated or stimulated with the following
agents: PMA (20 ng/ml) (except for panels B, D and E), PMA (20 ng/ml)/ionomycin (1 AM), forskolin (100 AM) (except for panels B, D and E), bpV[pic] (10
AM), and bpV[pic] (10 AM)/forskolin (100 AM). After 8 h of stimulation (in panel C, after 2, 4, 6, 8, and 24 h), cells were lysed and luciferase activity was
measured. Luciferase values were reported as the mean value of triplicates F SD. Fold inductions were calculated as the ratio of the mean value of treated
samples over the mean value of untreated samples. In panel B, values are presented as percentage of fold activation with 100% representing folds obtained in
cells not treated with the inhibitor. These results are representative of three independent experiments.
M. Langlois et al. / Virology 329 (2004) 395–411 403

M. Langlois et al. / Virology 329 (2004) 395–411404
To further demonstrate that bpV[pic] activated a member
of the CREB/ATF family, Jurkat cells were transfected with
pCRE-LUC, which contains 4 tandem repeats of the CRE
element (Fig. 7C). Upon activation of the transfected Jurkat
cells, reporter gene expression was responsive to several
stimulating agents. While forskolin and bpV[pic] both
activated this reporter weakly, forskolin alone was a more
potent activator of CREB-dependent transcription than
bpV[pic] alone. However, the combination of bpV[pic]/
forskolin again led to a significant increase in CREB-
dependent luciferase expression. As expected, PMA and
PMA/ionomycin were poor inducers of luciferase gene
expression in these transfected cells.
We directly tested the possible role of CREB family
members in activation of the HTLV-I LTR by bpV[pic] by
overexpressing the ATF/CREB family member in Jurkat
cells. Expression vectors for wild type CREB or a
constitutively active version of CREB were transfected into
Jurkat cells along with pGL2tata HTLV(�325/�57) and
then stimulated with different activators (Fig. 7D). To our
surprise, expression of wild type or constitutively active
CREB resulted in a significant reduction in the activation of
pGL2tata HTLV(�325/�57) by bpV[pic] or bpV[pic]/
forskolin, while no comparable effects were observed in
PMA/ionomycin-stimulated cells. As a control, Jurkat cells
were similarly co-transfected with the CREB expression
vectors and pNF-nB-LUC (Fig. 7E). In contrast to the
results presented above, CREB expression had no effect on
NF-nB-dependent luciferase expression regardless of the
tested activator. This result is likely due to competitive
binding of the overexpressed CREB protein to the LTR,
thereby displacing a functional CREB family member. It
appears unlikely that the reduced HTLV-I LTR activity
observed upon CREB expression and bpV[pic] treatment is
simply an effect of squelching the CBP/p300 co-activators
since a similar effect was not observed on a NF-nBdependent promoter. Together, these results suggest that a
CREB family member, but not CREB itself, is involved in
bpV[pic]-mediated activation of the HTLV-I LTR.
A second factor is required for bpV-induced HTLV-I LTR
activity
To more directly address the role of TRE1 CREB-
binding sites in activation of the HTLV-I LTR following
PTP inhibition, different HTLV-I LTR constructs containing
mutations in 1, 2, or all 3 TRE1 repeats were used (Fig. 8A).
Reduced bpV[pic] activation was reproducibly observed
when these constructs were transfected into Jurkat cells and
was most evident when all three TRE1 sequences were
mutated (Fig. 8B). However, even in this triple TRE1-
mutated construct (pGL3-21PMD/11T), a significant level
of bpV-dependent activation was still apparent and
accounted for more than 50% of the activity observed with
the wild-type LTR. A similar trend was detected with the
bpV[pic]/forskolin combination. PMA and PMA/ionomycin
induced luciferase gene expression to similar levels for all 8
tested LTR constructs. In contrast to these results, co-
transfection of a Tax expression vector with pGL3-21PMD/
11T showed a greater than 90% reduction in luciferase
expression from that observed on pGL3-U3R at both 24 and
48 h post-transfection (Fig. 8C). These data are consistent
with the idea that activation of the HTLV-I LTR by bpV[pic]
is not exclusively dependent on the CREB-binding elements
located in the TRE1 repeats.
Discussion
In this study, we were interested in assessing the
involvement of PTP in the transcriptional regulation of
HTLV-I. A previous study had suggested that tyrosine
phosphorylation plays an important role in HTLV-I tran-
scription by the Tax protein combined with a PHA
stimulation (Lin et al., 1998). Indeed, in this study, we
demonstrated that the use of a PTP inhibitor termed
bpV[pic], which is known to increase intracellular phos-
photyrosine levels, resulted in a significant increase in
HTLV-I LTR activity.
Our first experiments were conducted to assess the role
of bpV[pic] in Tax-mediated LTR activation. These results
indicated that bpV[pic] could significantly induce HTLV-I
LTR activity in both Tax-expressing and non-Tax-express-
ing cells. Time course experiments permitted us to conclude
that this induction was likely mediated by direct activation
of a signaling pathway since the optimal effect was at 8 h,
consistent with several studies from our group using
different activators that act directly on specific transcription
factors such as NFAT and NF-nB (Barbeau et al., 1997;
Fortin et al., 2001; Ouellet et al., 1999). In addition, the
kinetic response of the HTLV-I LTR induced by bpV[pic]
was drastically different (at least in Jurkat cells) from the
activation pattern of PMA, recently linked to apoptosis
(Torgeman et al., 2001a). These results with bpV[pic]
demonstrate the potential of TCR-like signaling cascades
to positively modulate HTLV-I gene expression.
A series of experiments also indicated that the combina-
tion of forskolin and bpV[pic] resulted in increased
activation of the HTLV-I LTR, while other agents in
combination with bpV[pic] generally did not affect the
bpV[pic] response. In contrast, sodium orthovanadate had
no such effect, which is consistent with the fact that bpV
compounds are more efficient PTP inhibitors than sodium
orthovanadate. It is also possible that bpV compounds might
target different PTPs than those inhibited by sodium
orthovanadate. In the presence of bpV[pic], forskolin
efficiently induced the HTLV-I LTR to greater levels than
PMA. Similar to bpV[pic] (discussed below), PHA and
PMA are likely to be weak activators of CREB and might
also activate other HTLV-I-regulatory factors that synergize
with CREB. This is consistent with our finding that
bpV[pic] strongly synergizes with PGE2, another CREB
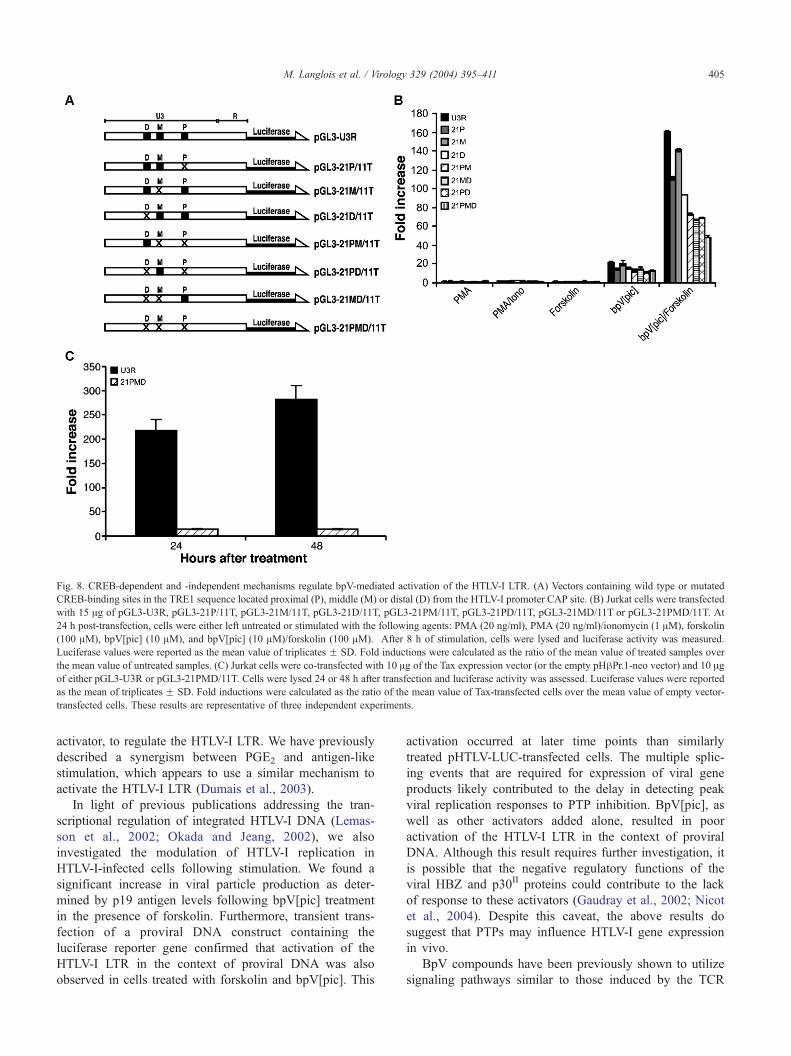
Fig. 8. CREB-dependent and -independent mechanisms regulate bpV-mediated activation of the HTLV-I LTR. (A) Vectors containing wild type or mutated
CREB-binding sites in the TRE1 sequence located proximal (P), middle (M) or distal (D) from the HTLV-I promoter CAP site. (B) Jurkat cells were transfected
with 15 Ag of pGL3-U3R, pGL3-21P/11T, pGL3-21M/11T, pGL3-21D/11T, pGL3-21PM/11T, pGL3-21PD/11T, pGL3-21MD/11T or pGL3-21PMD/11T. At
24 h post-transfection, cells were either left untreated or stimulated with the following agents: PMA (20 ng/ml), PMA (20 ng/ml)/ionomycin (1 AM), forskolin
(100 AM), bpV[pic] (10 AM), and bpV[pic] (10 AM)/forskolin (100 AM). After 8 h of stimulation, cells were lysed and luciferase activity was measured.
Luciferase values were reported as the mean value of triplicates F SD. Fold inductions were calculated as the ratio of the mean value of treated samples over
the mean value of untreated samples. (C) Jurkat cells were co-transfected with 10 Ag of the Tax expression vector (or the empty pHhPr.1-neo vector) and 10 Agof either pGL3-U3R or pGL3-21PMD/11T. Cells were lysed 24 or 48 h after transfection and luciferase activity was assessed. Luciferase values were reported
as the mean of triplicates F SD. Fold inductions were calculated as the ratio of the mean value of Tax-transfected cells over the mean value of empty vector-
transfected cells. These results are representative of three independent experiments.
M. Langlois et al. / Virology 329 (2004) 395–411 405
activator, to regulate the HTLV-I LTR. We have previously
described a synergism between PGE2 and antigen-like
stimulation, which appears to use a similar mechanism to
activate the HTLV-I LTR (Dumais et al., 2003).
In light of previous publications addressing the tran-
scriptional regulation of integrated HTLV-I DNA (Lemas-
son et al., 2002; Okada and Jeang, 2002), we also
investigated the modulation of HTLV-I replication in
HTLV-I-infected cells following stimulation. We found a
significant increase in viral particle production as deter-
mined by p19 antigen levels following bpV[pic] treatment
in the presence of forskolin. Furthermore, transient trans-
fection of a proviral DNA construct containing the
luciferase reporter gene confirmed that activation of the
HTLV-I LTR in the context of proviral DNA was also
observed in cells treated with forskolin and bpV[pic]. This
activation occurred at later time points than similarly
treated pHTLV-LUC-transfected cells. The multiple splic-
ing events that are required for expression of viral gene
products likely contributed to the delay in detecting peak
viral replication responses to PTP inhibition. BpV[pic], as
well as other activators added alone, resulted in poor
activation of the HTLV-I LTR in the context of proviral
DNA. Although this result requires further investigation, it
is possible that the negative regulatory functions of the
viral HBZ and p30II proteins could contribute to the lack
of response to these activators (Gaudray et al., 2002; Nicot
et al., 2004). Despite this caveat, the above results do
suggest that PTPs may influence HTLV-I gene expression
in vivo.
BpV compounds have been previously shown to utilize
signaling pathways similar to those induced by the TCR

M. Langlois et al. / Virology 329 (2004) 395–411406
(Fortin et al., 2001; Ouellet et al., 1999, 2003a,b).
Activation of the HTLV-I LTR by these inhibitors was not
surprisingly found to depend on ZAP70, calcineurin activity
and calcium mobilization, but did not require the expression
of cell surface TCR. These signaling molecules are also
involved in bpV-mediated NF-nB and NFAT activation
(Fortin et al., 2001; Ouellet et al., 1999). These results
support the idea that TCR signaling can activate the HTLV-I
LTR. This is especially relevant since our data suggest that
the PTP targeted by bpV[pic], permitting activation of the
HTLV-I LTR, is likely to be SHP-1, a crucial downregulator
of TCR signaling.
Our results strongly suggest that the TRE1 repeats are
important for PTP-dependent activation of the HTLV-I LTR.
Indeed, isolated TRE1 sequences were sufficient to confer
bpV[pic] responsiveness to a minimal promoter. We also
suggested that CREB activation and binding to the CREB-
binding site within the TRE1 repeats is important for
activation of HTLV-I transcription. Furthermore, point
mutation of the CREB-binding site in all three TRE1
repeats reduced the ability of the PTP inhibitor to induce
transcription. Parallel experiments conducted with the
pCRE-LUC plasmid also showed that it was activated by
bpV[pic] in synergy with forskolin. The ability of our PTP
inhibitors to activate CREB/ATF family members is not
surprising in light of a previous study indicating that PTP
activity inhibits CREB activation (Champion-Arnaud et al.,
1991). It is interesting to note the difference between
activation of the CREB consensus sequence (pCRE-LUC)
and the CREB-binding site in the TRE1 sequence by the
combination of bpV[pic] and forskolin. These dissimilar
responses might reflect a minimal involvement of CREB in
activation of the HTLV-I LTR following PTP inhibition (see
below). We are presently making efforts to identify the
CREB/ATF family member involved in HTLV-I LTR
activation by bpV[pic], and our results suggest that CREB
itself is not a likely candidate. Indeed, Tax-mediated
modulation of HTLV-I gene expression was affected by
CREB expression in a manner similar to that observed with
bpV[pic]-dependent stimulation of the HTLV-I LTR
(Gachon et al., 1998). It is tempting to speculate that
CREB2 might be the relevant factor, although experiments
conducted with a CREB2 expression vector indicated no
increasing effects by this family member on our observed
bpV-mediated induction of HTLV-I LTR activity in Jurkat
cells (data not shown). Other family members are known to
bind to the TRE1 regions in vivo (Lemasson et al., 2002)
and might thus be involved.
Our results strongly argue for the involvement of a
transcription factor that binds outside of the CRE-like
consensus sequence. Based on experiments using the TRE1-
mutated HTLV-I LTRs, this putative factor appears to
contribute more than 50% of the induction mediated by
bpV[pic]. In the presence of forskolin, bpV[pic] induction
was more sensitive to the mutated TRE1 repeats, which
might be expected given the known activation of CREB by
forskolin. In fact, the synergy between bpV[pic] and
forskolin is consistent with the weak CREB activating
potential of bpV[pic] alone and strong enhancement of the
CREB activating potential by forskolin. There are several
possible candidates for the identity of this other factor. First,
the TRE2 sequence harbors a binding site for the GLI2
protein, which was initially suggested to be involved in Tax-
mediated activation of the HTLV-I LTR (Dan et al., 1999;
Tanimura et al., 1998). When the LTR was mutated to knock
out the GLI2-binding site in TRE2, there was no apparent
difference in response of the mutant and wild-type LTRs to
bpV[pic] stimulation (B.A. and B.B., unpublished results).
This suggests that GLI2 is not involved in the activation of
the LTR by bpV[pic]. Previous studies have shown that
PMA is a good activator of HTLV-I LTR transcription and
that this activation depended on a region containing Sp1-
and p53-binding sites (Torgeman et al., 2001b). PTP
inhibitors might activate these factors; however, the effect
of PMA is believed to increase with time and to be
enhanced by PKC inhibition. This type of kinetic activation
was not observed for our PTP inhibitor, although these
results do not completely rule out the possible involvement
of these factors in bpV[pic]-mediated activation of HTLV-I
transcription. Since other transcription factor-binding sites
(including the recently described serum response element
(Wycuff et al., 2004)) have been identified within the
HTLV-I LTR, further studies will be needed to identify the
transcription factor that mediates bpV[pic] activation of the
HTLV-I LTR.
The data presented in this study argues that constitutive
expression of various cellular PTPs provide an important
mechanism to suppress HTLV-I transcription in infected
cells thereby contributing to their immune escape. It remains
possible that an increase in oxidative state might also be
induced through the peroxo group of the bpV[pic] com-
pound as described previously (Baier-Bitterlich et al., 1996;
Sasada et al., 2002), and could partially contribute to
activation of the HTLV-I LTR. However, our analysis of the
signaling pathway activated by bpV[pic] suggests that the
main mechanism behind the modulation of HTLV-I gene
expression by bpV[pic] involves the inhibitory potential of
PTP activity. PTP inhibition (possibly by SHP-1) provides a
possible mechanism for activation of latent HTLV-I proviral
DNA. In fact, it has been shown that HTLV-I-infected cells
are frequently negative for SHP-1 expression (Cheng et al.,
2004; Migone et al., 1998), although the absence of SHP-1
expression is also observed in other leukemic cell lines and
is thus not exclusive to HTLV-I-infected transformed cells.
Since it has been suggested that activation of HTLV-I gene
expression might be sudden and dependent on cell-to-cell
contact, PTP inhibition could potentially turn on a signaling
cascade, resulting in rapid turn on of viral gene expression,
viral particle production and transmission to uninfected
cells.
Our results indicate a strong transcriptional upregulation
of the HTLV-I LTR by the bpV[pic] PTP inhibitor in T cells.

M. Langlois et al. / Virology 329 (2004) 395–411 407
The implication of PTP inhibition in positive regulation of
the HTLV-I gene transcription needs to be further addressed
with respect to key events in HTLV-I replication such as
HTLV-I transmission to uninfected cells. We are presently
addressing these issues to better identify the transcription
factors involved in bpV-dependent modulation of HTLV-I
transcription.
Materials and methods
Cell lines and PBMCs
Jurkat (clone E6-1) (Weiss et al., 1984), and Jurkat-
derived J45.01 (Koretsky et al., 1991), J.RT3 (Morley et al.,
1988) and P116 (Williams et al., 1998) cell lines were
included in this study. J45.01 (generously provided by Dr.
Arthur Weiss, Howard Hughes Medical Center San Fran-
cisco, CA), J.RT3 and P116 (kindly given by Dr. Robert T.
Abraham, The Mayo Clinic Rochester, MN) are deficient in
CD45, TCR and ZAP70/Syk expression, respectively. P116
clone 39 was derived from P116 and has been recomple-
mented for ZAP70 expression. Jurkat derivatives deficient
in a mode of calcium entry dependent on extracellular
calcium known as capacitative calcium entry (CJ.1) and the
parental CJ clone (Fanger et al., 1995) were kindly provided
by Dr. Richard S. Lewis (Stanford University School of
Medicine, Stanford, CA). The CD45-negative K6D cell line
has been previously described and is a clone derived from
the JKF cell line stably expressing antisense CD45 mRNA
(McKenney et al., 1995). The HPB.ALL cell line (Koretsky
et al., 1990) is a TCR-positive T cell line with a more
immature phenotype than Jurkat cells and was generously
provided by Dr. Arthur Weiss. The SupT1 T-cell line (Smith
et al., 1984) is negative for the TCR and was obtained from
the NIH AIDS Repository Reagent Program (Rockville,
MD). The HTLV-I-infected MJ cell line (Popovic et al.,
1983) produces infectious particles and was purchased from
the American Type Culture Collection (Manassas, VA). Cell
lines were maintained in RPMI-1640 medium supplemented
with 10% FBS (Hyclone Laboratories, Logan, UT),
glutamine (2 mM), penicillin G (100 U/ml), and streptomy-
cin (100 Ag/ml). G418 (Invitrogen Life Technologies,
Burlington, Canada) and Hygromycin B (Calbiochem, San
Diego, CA) were added to the appropriate concentrations as
indicated. PBMCs from healthy donors were isolated by
Ficoll-Hypaque density gradient centrifugation as previ-
ously described (Fortin et al., 1999).
Plasmid constructs
The pHTLV-LUC vector (Geleziunas et al., 1998) was
kindly provided by Dr. Warner C. Greene (The J. Gladstone
Institutes, San Francisco, CA) and contains the HTLV-I LTR
cloned upstream of the luciferase reporter gene in the pGL2-
Basic vector (Promega, Madison, WI). The pHTLV-GFP
construct was generated by excising the luciferase gene/
poly(A) segment from pHTLV-LUC and inserting a green
fluorescent protein (GFP)/SV40 poly(A) fragment isolated
from the HindIII/BamHI-digested phGFP-S65T vector (BD
Biosciences Clontech, Palo Alto, CA). Deletion mutants of
pHTLV-LUC were generated by self-ligation after digestion
with SmaI (pHTLV-LUCDSmaI) or SmaI and NdeI
(pHTLV-LUCDNdeI/SmaI). The pGL2tata-LUC vector
was constructed by subcloning a synthetic double-stranded
oligonucleotide containing a TATA box and extremities
compatible with NheI/HindIII (produced from hybridized 5V-CTAGCGGGTATATAATGGATCCA-3V and 5V-AGCTTG-GATCCATTATATACCCG-3V oligonucleotides) into the
digested pGL2-Basic vector (Promega). The pGL2tata
HTLV(�325/�57) construct was then generated by cloning
the SmaI/NdeI blunted fragment from the HTLV-I LTR into
the SmaI site of the pGL2tata-LUC vector. A similar cloning
procedure was used to insert this HTLV-I LTR fragment into
the SmaI-digested pGL2-Promoter vector (Promega) gen-
erating pGL2-Promoter HTLV(�325/�57). Constructs were
also created with previously described HTLV-I LTRs
(Barnhart et al., 1997) being either wild type or mutated
for one, two, or all three CREB consensus sequences located
in the TRE1 regions. These LTRs were excised from their
vector by XhoI/HindIII and cloned into the pGL3-Basic
vector (Promega) using the same restriction site. These
vectors are termed pGL3-U3R, pGL3-21P/11T, pGL3-21M/
11T, pGL3-21D/11T, pGL3-21PM/11T, pGL3-21PD/11T,
pGL3-21MD/112T and pGL3-21PMD/11T, and are wild
type or mutated in the proximal (P), middle (M) and/or distal
(D) TRE1 sequences. Expression vectors for the wild type
Tax protein and the empty vector (pHhPr.1-neo) (Matsu-
moto et al., 1997) were kindly provided by Dr. Masataka
Nakamura (Tokyo Medical and Dental University, Tokyo,
Japan). The pNF-nB-LUC and pCRE-LUC vectors were
obtained from Stratagene (La Jolla, CA) and contain
multiple NF-nB- and CREB-binding sites, respectively,
positioned upstream of a minimal promoter and the
luciferase reporter gene. Vectors encoding wild-type SHP-
1 as well as the empty vector pSFFVneo (Plas et al., 1996)
were provided by Dr. Matthew L. Thomas (Washington
University School of Medicine, Saint Louis, MI). The
CREB dominant negative mutant KCREB and the control
empty vector (Walton et al., 1992) were provided by Dr.
R.H. Goodman (Vollum Institute for Advanced Biochemical
Research, Portland, OR). Vectors expressing wild type
CREB or the constitutively active CREB F/Y mutant (Du
et al., 2000) were obtained from Dr. Marc Montminy (The
Salk Institute for Biological Studies, La Jolla, CA). We
constructed the empty vector termed pcDNA.CF by self-
ligation after excision of the insert.
Generation of luciferase-containing HTLV-I proviral DNA
The 5VK30-Luc vector was derived from the pre-
viously described K30 plasmid harboring a full-length

M. Langlois et al. / Virology 329 (2004) 395–411408
and infectious HTLV-I proviral DNA clone (Zhao et al.,
1995) obtained from the NIH AIDS Repository Reagent
Program. A BamHI fragment was isolated from this
construct (position 5102 to 6107) and cloned into
pBlueScript SK (Stratagene). This fragment contained
the ATG initiation codon for the env, tax, and rex
genes. A two-step PCR mutagenesis protocol directed at
position 5550 to 5552 (48 nucleotides downstream of
the ATG codon) mutated the ATC sequence to GAG
thereby generating a SacI site. Briefly, the 5V-CAGTTCTGCCCCCTCGAGCTCGGTGATTACAGCCC-
CAG-3V/Reverse primer and 5V-CTGGGGCTGTAAT-
CACCGAGCTCGAGGGGGCAGAACTG-3V/Universal
primer combinations were used for amplification of 5V and 3Vends of the BamHI fragment. Both amplified products
were mixed and amplified as a complete fragment with
pBlueScript Reverse and Universal primers. The luciferase
gene was amplified from pGL3basic with oligonucleotides
(5V-TGGAGCTCGTTGGTAAAGCCACCATGGAA-
GACGCCAAAAAC-3V and 5V-ACGAGCTCATCAATG-TATCTTATCATGTCTGCTCGAAGC-3V) to generate SacI
sites at each end, and then cloned in frame with the HTLV-
I ATG codon into the SacI-generated site of the K30
BamHI fragment. Sequencing of each PCR-amplified
product was carried out to ascertain proper sequence.
The BamHI fragment was subsequently cloned into the
K30 vector, after BamHI digestion, to generate 5V K30-LUC. Since the 3V end of the K30 proviral DNA had been
deleted in this construct, complete full-length proviral
DNA was recreated by ligating this 5V K30 end (EcoRI/
SalI digestion) with the 3VK30 fragment (SalI/EcoRI). The
ligation mixture was then directly transfected into T cell
lines.
Antibodies and reagents
BpV[pic] was synthesized as previously described
(Posner et al., 1994) and supplied by Dr. R. Faure (CHUL
Research Center, Ste-Foy, Canada). Briefly, V2O5 was
dissolved in an aqueous KOH solution and then mixed
with 30% H2O2 and the picolinic acid ancillary ligand in
addition to ethanol for optimal precipitation. Character-
ization of the bpV molecule was carried out by infrared 1H
NMR and vanadium-51 (51V) NMR spectroscopy. Stock
solutions of bpV[pic] molecules (1 mM in phosphate-
buffered saline, pH 7.4) were kept at �20 8C until used.
Sodium orthovanadate (Sigma, St Louis. MO) was freshly
dissolved in 10 mM HEPES, pH 7.4. FK506 and cyclo-
sporin A (CsA) were purchased from Fujisawa (Osaka,
Japan) and Sigma, respectively. Antibodies from the anti-
CD3 OKT3 hybridoma were purified with mAbTrap protein
G affinity columns (Pharmacia LKB Biotechnology AB,
Uppsala, Sweden). Purified anti-CD28 antibodies (clone
9.3) were provided by Dr. Jeffrey A. Ledbetter (Bristol-
Myers Squibb Pharmaceutical Research Institute, Princeton,
NJ).
Transfections and reporter gene assays
Transient transfections of T-cell lines using the DEAE-
Dextran method were performed as previously described
(Barbeau et al., 1997). Electroporation of PBMCs was
conducted according to Hughes and Pober’s protocol
(Hughes and Pober, 1996). Transiently transfected cells
were seeded at a density of 105 cells/well in 96-well plates
and left unstimulated or treated for 8 h (unless specified)
with bpV[pic] (10 AM), PHA-P (3 Ag/ml), PMA (20 ng/ml)
(Sigma), ionomycin (1 AM) (Iono; Calbiochem), anti-CD3
antibody (clone OKT3) (3 Ag/ml), anti-CD28 antibody
(clone 9.3) (1 Ag/ml), forskolin (100 AM) (BioMol,
Plymouth Meeting, PA) and prostaglandin E2 (100 nM)
(PGE2; Sigma) in a final volume of 200 Al. For some
experiments, prior to activation, cells were treated with
FK506 (10 ng/ml) or CsA (100 ng/ml) for 15 min or with
MDL-12,330A (100–250 AM) (Calbiochem) for 1 h at 37
8C. Cells were then lysed in a 1� lysis buffer (25 mM Tris
phosphate, pH 7.8, 2 mM DTT, 1% Triton X-100, 10%
glycerol). Luciferase activity was determined as follows.
After a freeze/thaw cycle, 20 Al of cellular extract was
transferred to a 96-well luminometer plate, and luciferase
activity was quantitated on a Dynex MLX microplate
luminometer (MLX; Dynex Technologies, Chantilly, VA)
following the addition of 100 Al of luciferase buffer (20
mM tricine, 1.07 mM (MgCO3)4dMg(OH)2d 5 H2O, 2.67
mM MgSO4, 0.1 mM EDTA, 220 AM Coenzyme A, 4.7
AM d-Luciferin potassium salt, 530 AM ATP, 33.3 mM
DTT). Values from the luminometer are expressed as
Relative Light Units (RLU). Fold induction was calculated
as the ratio between the mean values F standard deviation
(SD) of the luciferase activity of treated over untreated
samples. Cells transfected with the pHTLV-GFP vector
were fixed in a solution of 1� phosphate-buffered saline
(PBS) containing methanol-free 1% paraformaldehyde and
subsequently analyzed by flow cytometry for GFP detection
(EPICS XL, Coulter Corp., Miami, FL).
Detection of supernatant HTLV-I p19 antigen
HTLV-I-producing MJ cells (1 � 106 cells/ml) were
treated with PMA (20 ng/ml)/ionomycin (1 AM), bpV[pic]
(10 AM) and bpV[pic] (10 AM)/forskolin (100 AM).
Supernatants were subsequently harvested at 24, 48, and
72 h post-activation. Quantification of HTLV-I p19 antigen
in the collected supernatant was performed using an ELISA
HTLV-I p19 antigen kit (Zeptometrix, Buffalo, NY) as
described by the manufacturer.
Acknowledgments
We thank Dr. Maurice Dufour for FACS analyses. This
work was supported by a grant from the Fonds de
Recherche en Sante du Quebec (FRSQ) (#003318). Dr.

M. Langlois et al. / Virology 329 (2004) 395–411 409
Michel J. Tremblay holds a Tier 1 Canada Research Chair in
Human Retrovirology. Dr. Benoit Barbeau is the recipient of
a Chercheur-Boursier Junior 2 award from the FRSQ.
References
Baier-Bitterlich, G., Baier, G., Fuchs, D., Bock, G., Hausen, A., Utermann,
G., Pavelka, M., Wachter, H., 1996. Role of 7,8-dihydroneopterin in T-
cell apoptosis and HTLV-1 transcription in vitro. Oncogene 13 (10),
2281–2285.
Barbeau, B., Bernier, R., Dumais, N., Briand, G., Olivier, M., Faure, R.,
Posner, B.I., Tremblay, M.J., 1997. Activation of HIV-1 long
terminal repeat transcription and virus replication via NF-kB-
dependent and -independent pathways by potent phosphotyrosine
phosphatase inhibitors, the peroxovanadium compounds. J. Biol.
Chem. 272, 12968–12977.
Barnhart, M.K., Connor, L.M., Marriott, S.J., 1997. Function of the human
T-cell leukemia virus type 1 21-base-pair repeats in basal transcription.
J. Virol. 71, 337–344.
Bosselut, R., Duvall, J.F., Gegonne, A., Bailly, M., Hemar, A., Brady, J.,
Ghysdael, J., 1990. The product of the c-ets-1 proto-oncogene and the
related Ets2 protein act as transcriptional activators of the long
terminal repeat of human T cell leukemia virus HTLV-1. EMBO J. 9
(10), 3137–3144.
Brady, J., Jeang, K.T., Duvall, J., Khoury, G., 1987. Identification of p40x-
responsive regulatory sequences within the human T-cell leukemia virus
type I long terminal repeat. J. Virol. 61 (7), 2175–2181.
Brauweiler, A., Garl, P., Franklin, A.A., Giebler, H.A., Nyborg, J.K., 1995.
A molecular mechanism for human T-cell leukemia virus latency and
Tax transactivation. J. Biol. Chem. 270, 12814–12822.
Champion-Arnaud, P., Gesnel, M.C., Foulkes, N., Ronsin, C., Sassone-
Corsi, P., Breathnach, R., 1991. Activation of transcription via AP-1 or
CREB regulatory sites is blocked by protein tyrosine phosphatases.
Oncogene 6 (7), 1203–1209.
Cheng, J., Zhang, D., Zhou, C., Marasco, W.A., 2004. Down-regulation of
SHP1 and up-regulation of negative regulators of JAK/STAT signaling
in HTLV-1 transformed cell lines and freshly transformed human
peripheral blood CD4+ T-cells. Leuk. Res. 28 (1), 71–82.
Copeland, K.F.T., Haaksma, A.G.M., Goudsmit, J., Heeney, J.L., 1994.
Calcium-mediated inhibition of phorbol ester and tax trans-activation of
the human T-cell leukaemia virus type 1. J. Gen. Virol. 75, 1523–1631.
Copeland, K.F.T., Hendrikx, P.J., Haaksma, A.G.M., Fiering, S., Van Lier,
R., Goudsmit, J., Heeney, J.L., 1995. Comparison of the response to T-
cell activation by integrated HIV-1 and HTLV-1 LTR-lacZ vectors.
Virology 209, 633–636.
Dan, S., Tanimura, A., Yoshida, M., 1999. Interaction of Gli2 with CREB
protein on DNA elements in the long terminal repeat of human T-cell
leukemia virus type I is responsible for transcriptional activation by Tax
protein. J. Virol. 73, 3258–3263.
Du, K., Asahara, H., Jhala, U.S., Wagner, B.L., Montminy, M., 2000.
Characterization of a CREB gain-of-function mutant with constitutive
transcriptional activity in vivo. Mol. Cell. Biol. 20, 4320–4327.
Dumais, N., Pare, M.E., Mercier, S., Bounou, S., Marriot, S.J.,
Barbeau, B., Tremblay, M.J., 2003. T-cell receptor/CD28 engage-
ment when combined with prostaglandin E2 treatment leads to
potent activation of human T-cell leukemia virus type 1. J. Virol. 77
(20), 11170–11179.
Fanger, C.M., Hoth, M., Crabtree, G.R., Lewis, R.S., 1995. Characte-
rization of T cell mutants with defects in capacitative calcium entry:
genetic evidence for the physiological roles of CRAC channels. J. Cell
Biol. 131 (3), 655–667.
Fortin, J.-F., Barbeau, B., Lundgren, E., Hedman, H., Tremblay, M.J., 1999.
Role of the leukocyte function antigen-1 conformational state in the
process of human immunodeficiency virus type 1-mediated syncytium
formation and virus infection. Virology 257, 228–238.
Fortin, J.F., Barbeau, B., Robichaud, G.A., Pare, M.-E., Lemieux, A.M.,
Tremblay, M.J., 2001. Regulation of nuclear factor of activated T cells
by phosphotyrosyl-specific phosphatase activity: a positive effect on
HIV-1 long terminal repeat-driven transcription and a possible
implication of SHP-1. Blood 97 (8), 2390–2400.
Fujisawa, J., Seiki, M., Sato, M., Yoshida, M., 1986. A transcriptional
enhancer sequence of HTLV-I is responsible for trans-activation
mediated by p40 chi HTLV-I. Embo J. 5 (4), 713–718.
Gachon, F., Peleraux, A., Thebault, S., Dick, J., Lemasson, I., Devaux, C.,
Mesnard, J.M., 1998. CREB-2, a cellular CRE-dependent transcription
repressor, functions in association with Tax as an activator of the human
T-cell leukemia virus type 1 promoter. J. Virol. 72 (10), 8332–8337.
Gaudray, G., Gachon, F., Basbous, J., Biard-Piechaczyk, M., Devaux, C.,
Mesnard, J.M., 2002. The complementary strand of the human T-cell
leukemia virus type 1 RNA genome encodes a bZIP transcription factor
that down-regulates viral transcription. J. Virol. 76 (24), 12813–12822.
Geleziunas, R., Ferrell, S., Lin, X., Mu, Y., Cunningham Jr., E.T., Grant,
M., Connelly, M.A., Hambor, J.E., Marcu, K.B., Greene, W.C., 1998.
Human T-cell leukemia virus type 1 Tax induction of NF-kappaB
involves activation of the IkappaB kinase alpha (IKKalpha) and
IKKbeta cellular kinases. Mol. Cell. Biol. 18 (9), 5157–5165.
Gessain, A., Barin, F., Vernant, J.C., Gout, O., Maurs, L., Calender, A., de
The, G., 1985. Antibodies to human T-lymphotropic virus type-I in
patients with tropical spastic paraparesis. Lancet 2 (8452), 407–410.
Giebler, H.A., Loring, J.E., van Orden, K., Colgin, M.A., Garrus, J.E.,
Escudero, K.W., Brauweiler, A., Nyborg, J.K., 1997. Anchoring of
CREB binding protein to the human T-cell leukemia virus type 1
promoter: a molecular mechanism of Tax transactivation. Mol. Cell.
Biol. 17 (9), 5156–5164.
Gitlin, S.D., Bosselut, R., Gegonne, A., Ghysdael, J., Brady, J.N., 1991.
Sequence-specific interaction of the Ets1 protein with the long terminal
repeat of the human T-lymphotropic virus type I. J. Virol. 65 (10),
5513–5523.
Harrod, R., Tang, Y., Nicot, C., Lu, H.S., Vassilev, A., Nakatani, Y., Giam,
C.Z., 1998. An exposed KID-like domain in human T-cell lymphotropic
virus type 1 Tax is responsible for the recruitment of coactivators CBP/
p300. Mol. Cell. Biol. 18 (9), 5052–5061.
Hinuma, Y., Nagata, K., Hanaoka, M., Nakai, M., Matsumoto, T.,
Kinoshita, K.I., Shirakawa, S., Miyoshi, I., 1981. Adult T-cell leukemia:
antigen in an ATL cell line and detection of antibodies to the antigen in
human sera. Proc. Natl. Acad. Sci. U.S.A. 78 (10), 6476–6480.
Hughes, C.C.W., Pober, J.S., 1996. Transcriptional regulation of the
interleukin-2 gene in normal human peripheral blood T cells. J. Biol.
Chem. 271, 5369–5377.
Imbert, V., Peyron, J.F., Farahi, F.D., Mari, B., Auberger, P., Rossi, B.,
1994. Induction of tyrosine phosphorylation and T-cell activation by
vanadate peroxide, an inhibitor of protein tyrosine phosphatases.
Biochem. J. 297, 163–173.
Imbert, V., Farahifar, D., Auberger, P., Mary, D., Rossi, B., Peyron, J.F.,
1996a. Stimulation of the T-cell antigen receptor-CD3 complex signaling
pathway by the tyrosine phosphatase inhibitor pervanadate is mediated
by inhibition of CD45: evidence for two interconnected Lck/Fyn- or zap-
70-dependent signaling pathways. J. Inflammation 46 (2), 65–77.
Imbert, V., Rupec, R.A., Livolsi, A., Pahl, H.L., Traenckner, E.B., Mueller-
Dieckmann, C., Farahifar, D., Rossi, B., Auberger, P., Baeurle, P.A.,
Peyron, J.F., 1996b. Tyrosine phosphorylation of InBa activates NF-nBwithout proteolytic degradation of InBa. Cell 86 (5), 787–798.
Kashanchi, F., Duvall, J.F., Kwok, R.P., Lundblad, J.R., Goodman, R.H.,
Brady, J.N., 1998. The coactivator CBP stimulates human T-cell
lymphotrophic virus type I Tax transactivation in vitro. J. Biol. Chem.
273 (51), 34646–34652.
Kazanji, M., Ureta-Vidal, A., Ozden, S., Tangy, F., De Thoisy, B., Fiette, L.,
Talarmin, A., Gessain, A., De The, G., 2000. Lymphoid organs as a major
reservoir for human T-cell leukemia virus type 1 in experimentally
infected squirrel monkeys (Saimiri sciureus): proviral expression,
persistence, and humoral and cellular immune responses. J. Virol. 74,
4860–4867.

M. Langlois et al. / Virology 329 (2004) 395–411410
Koretsky, G.A., Picus, J., Thomas, M.L., Weiss, A., 1990. Tyrosine
phosphatase CD45 is essential for coupling T-cell antigen receptor to
the phosphatidyl inositol pathway. Nature 346, 66–68.
Koretsky, G.A., Picus, J., Schultz, T., Weiss, A., 1991. The tyrosine
phosphatase CD45 is required for T cell antigen receptor and CD2-
mediated activation of a protein tyrosine kinase and interleukin 2
production. Proc. Natl. Acad. Sci. U.S.A. 88, 2307–2341.
Kwok, R.P., Laurance, M.E., Lundblad, J.R., Goldman, P.S., Shih, H.,
Connor, L.M., Marriott, S.J., Goodman, R.H., 1996. Control of cAMP-
regulated enhancers by the viral transactivator Tax through CREB and
the co-activator CBP. Nature 380 (6575), 642–646.
Lemasson, I., Polakowski, N.J., Laybourn, P.J., Nyborg, J.K., 2002.
Transcription factor binding and histone modifications on the
integrated proviral promoter in human T-cell leukemia virus-I-infected
T-cells. J. Biol. Chem. 277 (51), 49459–49465.
Lin, H.-C., Dezzutti, C.S., Lal, R.B., Rabson, A.B., 1998. Activation of
human T-cell leukemia virus type 1 tax gene expression in chronically
infected T cells. J. Virol. 72, 6264–6270.
Matsumoto, K., Shibata, H., Fujisawa, J.-I., Inoue, H., Hakura, A.,
Tsukahara, T., Fujii, M., 1997. Human T-cell leukemia virus type 1
Tax protein transforms rat fibroblasts via two distinct pathways. J. Virol.
71, 4445–4451.
McKenney, D.W., Onodera, H., Gorman, L., Mimura, T., Rothstein, D.M.,
1995. Distinct isoforms of the CD45 protein-tyrosine phosphatase
differentially regulate interleukin 2 secretion and activation signal
pathways involving Vav in T cells. J. Biol. Chem. 270, 24249–24254.
Migone, T.S., Cacalano, N.A., Taylor, N., Yi, T., Waldmann, T.A.,
Johnston, J.A., 1998. Recruitment of SH2-containing protein tyrosine
phosphatase SHP-1 to the interleukin 2 receptor; loss of SHP-1
expression in human T-lymphotropic virus type I-transformed T cells.
Proc. Natl. Acad. Sci. U.S.A. 95 (7), 3845–3850.
Morley, B.J., Chin, K.N., Newton, M.E., Weiss, A., 1988. The lysine
residue in the membrane-spanning domain of the beta chain is necessary
for cell surface expression of the T cell antigen receptor. J. Exp. Med.
168 (6), 1971–1978.
Mor-Vaknin, N., Torgeman, A., Galron, D., Lfchelt, M., Flqgel, R.M.,
Aboud, M., 1997. The long terminal repeats of human immunodefi-
ciency virus type-1 and human T-cell leukemia virus type 1 are
activated by 12-O-tetradecanoylphorbol-13-acetate through different
pathways. Virology 232, 337–344.
Nakamura, M., Niki, M., Ohtani, K., Sugamura, K., 1989. Differential
activation of the 21-base-pair enhancer element of human T-cell
leukemia virus type I by its own trans-activator and cyclic AMP.
Nucleic Acids Res. 17 (13), 5207–5221.
Nicot, C., Dundr, M., Johnson, J.M., Fullen, J.R., Alonzo, N., Fukumoto,
R., Princler, G.L., Derse, D., Misteli, T., Franchini, G., 2004. HTLV-1-
encoded p30(II) is a post-transcriptional negative regulator of viral
replication. Nat. Med. 10 (2), 197–201.
Numata, N., Ohtani, K., Niki, M., Nakamura, M., Sugamura, K., 1991.
Synergism between two distinct elements of the HTLV-I enhancer
during activation by the trans-activator of HTLV-I. New Biol. 3 (9),
896–906.
Okada, M., Jeang, K.T., 2002. Differential requirements for activation of
integrated and transiently transfected human T-cell leukemia virus type
1 long terminal repeat. J. Virol. 76 (24), 12564–12573.
Osame, M., Usuku, K., Ijichi, N., Amitani, H., Igata, A., Matsumoto, M.,
Tara, H., 1986. HTLV-I associated myelopathy, a new clinical entity.
Lancet i, 1031–1032.
Ouellet, M., Barbeau, B., Tremblay, M.J., 1999. p56(lck), ZAP-70, SLP-76,
and calcium-regulated effectors are involved in NF-kappaB activation
by bisperoxovanadium phosphotyrosyl phosphatase inhibitors in human
T cells. J. Biol. Chem. 274 (49), 35029–35036.
Ouellet, M., Barbeau, B., Tremblay, M.J., 2003a. Protein tyrosyl
phosphatases in T cell activation: implication for human immunodefi-
ciency virus transcriptional activity. Prog. Nucleic Acid Res. Mol. Biol.
73, 69–105.
Ouellet, M., Roy, J., Barbeau, B., Geleziunas, R., Tremblay, M.J., 2003b.
NF-kappaB induction by bisperoxovanadium compounds requires
CD45, p36(LAT), PKC, and IKK activity and exhibits kinetics of
activation comparable to those of TCR/CD28 coengagement. Bio-
chemistry 42 (27), 8260–8271.
Paskalis, H., Felber, B.K., Pavlakis, G.N., 1986. Cis-acting sequences
responsible for the transcriptional activation of human T-cell leukemia
virus type I constitute a conditional enhancer. Proc. Natl. Acad. Sci.
U.S.A. 83 (17), 6558–6562.
Plas, D.R., Johnson, R., Pingel, J.T., Matthews, R.J., Dalton, M., Roy, G.,
Chan, A.C., Thomas, M.L., 1996. Direct regulation of ZAP-70 by SHP-1
in T cell antigen receptor signaling. Science 272 (5265), 1173–1176.
Poiesz, B., Ruscetti, F.W., Gazdar, A.F., Bunn, P.A., Minna, J.D., Gallo,
R.C., 1980. Detection and isolation of type C retrovirus particles from
fresh and cultured lymphocytes of a patient with cutaneous T-cell
lymphoma. Proc. Natl. Acad. Sci. U.S.A. 77, 7415–7419.
Popovic, M., Sarin, P.S., Robert-Gurroff, M., Kalyanaraman, V.S., Mann,
D., Minowada, J., Gallo, R.C., 1983. Isolation and transmission of
human retrovirus (human t-cell leukemia virus). Science 219 (4586),
856–859.
Posner, B.I., Faure, R., Burgess, J.W., Bevan, A.P., Lachance, D., Zhang-
Sun, G., Fantus, I.G., Ng, J.B., Hall, D.A., Lum, B.S., et al., 1994.
Peroxovanadium compounds. A new class of potent phosphotyrosine
phosphatase inhibitors which are insulin mimetics. J. Biol. Chem. 269
(6), 4596–4604.
Poteat, H., Kadison, P., McGuire, K., Park, L., Park, R.E., Sodroski, J.G.,
Haseltine, W.A., 1989. Response of the human T-cell leukemia virus
type 1 long terminal repeat to cyclic AMP. J. Virol. 63, 1604–1611.
Rodgers-Johnson, P., Gajdusek, D.C., Morgan, O.S., Zaninovic, V., Sarin,
P.S., Graham, D.S., 1985. HTLV-I and HTLV-III antibodies and tropical
spastic paraparesis. Lancet 2 (8466), 1247–1248.
Sasada, T., Nakamura, H., Masutani, H., Ueda, S., Sono, H., Takabayashi,
A., Yodoi, J., 2002. Thioredoxin-mediated redox control of human T
cell lymphotropic virus type I (HTLV-I) gene expression. Mol.
Immunol. 38 (2001), 723–732.
Secrist, J.P., Burns, L.A., Karnitz, L., Koretzky, G.A., Abraham, R.T.,
1993. Stimulatory effects of the protein tyrosine phosphatase
inhibitor, pervanadate, on T-cell activation events. J. Biol. Chem.
268, 5886–5893.
Shimotohno, K., Takano, M., Teruuchi, T., Miwa, M., 1986. Requirement
of multiple copies of a 21-nucleotide sequence in the U3 regions of
human T-cell leukemia virus type I and type II long terminal repeats for
trans-acting activation of transcription. Proc. Natl. Acad. Sci. U.S.A. 83
(21), 8112–8116.
Smith, M.R., Greene, W.C., 1991. Molecular biology of the type 1 human
T-cell leukemia virus (HTLV-1) and adult T-cell leukemia. J. Clin.
Invest. 87, 761–766.
Smith, S.D., Shatsky, M., Cohen, P.S., Warnke, R., Link, M.P., Glader, B.E.,
1984. Monoclonal antibody and enzymatic profiles of human malignant
T lymphoid cells and derived cell lines. Cancer Res. 44, 5657–5660.
Sommerfelt, M.A., 1999. Retrovirus receptors. J. Gen. Virol. 80 (Pt. 12),
3049–3064.
Tanimura, A., Dan, S., Yoshida, M., 1998. Cloning of novel isoforms of
the human gli2 oncogene and their activities to enhance tax-
dependent transcription of the human T-cell leukemia virus type 1
genome. J. Virol. 72, 3958–3964.
Torgeman, A., Mor-Vaknin, N., Aboud, M., 1999. Sp1 is involved in a
protein kinase C-independent activation of human T cell leukemia virus
type I long terminal repeat by 12-O-tetradecanoylphorbol-13-acetate.
Virology 254 (2), 279–287.
Torgeman, A., Ben-Aroya, Z., Grunspan, A., Zelin, E., Butovsky, E.,
Hallak, M., Lochelt, M., Flugel, R.M., Livneh, E., Wolfson, M., Kedar,
I., Aboud, M., 2001a. Activation of HTLV-I long terminal repeat by
stress-inducing agents and protection of HTLV-I-infected T-cells from
apoptosis by the viral tax protein. Exp. Cell Res. 271 (1), 169–179.
Torgeman, A., Mor-Vaknin, N., Zelin, E., Ben-Aroya, Z., Lochelt, M.,
Flugel, R.M., Aboud, M., 2001b. Sp1-p53 heterocomplex mediates
activation of HTLV-I long terminal repeat by 12-O-tetradecanoylphor-

M. Langlois et al. / Virology 329 (2004) 395–411 411
bol-13-acetate that is antagonized by protein kinase C. Virology 281
(1), 10–20.
Uchiyama, T., 1997. Human T cell leukemia virus type I (HTLV-I) and
human diseases. Annu. Rev. Immunol. 15, 15–37.
Walton, K.M., Rehfuss, R.P., Chrivia, J.C., Lochner, J.E., Goodman, R.H.,
1992. A dominant repressor of cyclic adenosine 3V,5V-monophosphate
(cAMP)-regulated enhancer-binding protein activity inhibits the cAMP-
mediated induction of the somatostatin promoter in vivo. Mol.
Endocrinol. 6, 647–655.
Weiss, A., Imboden, J., Shoback, D., Strobo, J., 1984. Role of T3 surface
molecules in human T-cell activation: T3-dependent activation results in
an increase in cytoplasmic free calcium. Proc. Natl. Acad. Sci. U.S.A.
81, 4169–4173.
Williams, B.L., Schreiber, K.L., Zhang, W., Wange, R.L., Samelson, L.E.,
Leibson, P.J., Abraham, R.T., 1998. Genetic evidence for differential
coupling of Syk family kinases to the T-cell receptor: reconstitution
studies in a ZAP-70-deficient Jurkat T-cell line. Mol. Cell. Biol. 18 (3),
1388–1399.
Wycuff, D.R., Yanites, H.L., Marriott, S.J., 2004. Identification of a
functional serum response element in the HTLV-I LTR. Virology 324
(2), 540–553.
Yakura, H., 1998. Phosphatases and kinases in lymphocyte signaling.
Immunol. Today 19, 198–201.
Yan, J.P., Garrus, J.E., Giebler, H.A., Stargell, L.A., Nyborg, J.K., 1998.
Molecular interactions between the coactivator CBP and the human T-
cell leukemia virus Tax protein. J. Mol. Biol. 281 (3), 395–400.
Yoshida, M., Miyoshi, I., Hinuma, Y., 1982. Isolation and characte-
rization of retrovirus from cell lines of human adult T-cell leukemia
and its implication in the disease. Proc. Natl. Acad. Sci. U.S.A. 79,
2031–2035.
Yoshida, M., Suzuki, T., Fujisawa, J., Hirai, H., 1995. HTLV-1 oncoprotein
Tax and cellular transcription factors. Curr. Top. Microbiol. Immunol.
193, 79–89.
Zhao, T.M., Robinson, M.A., Bowers, F.S., Kindt, T.J., 1995. Characte-
rization of an infectious molecular clone of human T-cell leukemia virus
type I. J. Virol. 69, 2024–2030.

![Enhanced Kat3A/Catenin transcription: a common mechanism of … · 2019. 9. 28. · mediated transcription) or non-canonical (planar cell polarity, Ca2+/PKC activation)[42,43]. Canonical](https://img.pdfslide.net/doc/110x75/60c2e324f6a8a620c25ac30f/enhanced-kat3acatenin-transcription-a-common-mechanism-of-2019-9-28-mediated.jpg)



























