Embed Size (px)
Citation preview

VALÉRIA FERNANDES DE SOUZA
Administração repetida de baixas doses de reserpina:
um possível modelo para o estudo de déficits cognitivos e motores associados à Doença de
Parkinson
Tese apresentada à Universidade
Federal do Rio Grande do Norte,
para obtenção de título de doutor no
curso de pós-graduação em
Psicobiologia.
NATAL/RN
2011

II
VALÉRIA FERNANDES DE SOUZA
Administração repetida de baixas doses de reserpina: um possível modelo para o estudo de déficits cognitivos e
motores associados à Doença de Parkinson
Tese apresentada à
Universidade Federal do Rio Grande
do Norte, para obtenção de título de
doutor no curso de pós-graduação
em Psicobiologia.
ORIENTADORA: Profa. Dra. Regina Helena da Silva
NATAL/RN


III
Título: Administração repetida de baixas doses de reserpina: um possível modelo para o estudo de déficits cognitivos e motores associados à Doença de Parkinson
Autor: Valéria Fernandes de Souza
Data da defesa: 15/ 09/ 2011
Banca Examinadora:
___________________________________
Prof. Vanessa Costhek Abílio
Universidade Federal de São Paulo, UNIFESP
___________________________________
Prof. Ângela Maria Ribeiro
Universidade Federal de Minas Gerais, UFMG
___________________________________
Prof. Elaine Cristina Gavioli
Universidade Federal do Rio Grande do Norte, UFRN
___________________________________
Prof John Fontenele Araujo
Universidade Federal do Rio Grande do Norte, RN
___________________________________
Prof. Regina Helena da Silva
Universidade Federal do Rio Grande do Norte, RN

IV
“O homem não teceu a teia da vida, ele é dela apena um fio. O que ele fizer estará
fazendo para si mesmo. O que ele fizer para si mesmo estará fazendo para a Teia.”
Chefe Seattle

V
AGRADECIMENTOS
À Universidade Federal do Rio Grande do Norte pela oportunidade concedida.
A minha orientadora, Profa. Regina Helena da Silva, por acreditar e tornar
possível a realização desse sonho e por sua amizade.
Aos meus colegas do Laboratório de Estudo de Memória (LEME), pela
contribuição nos experimentos e amizade. Em especial, ao Ronaldo, Thieza, Anderson,
Alicia e Geison pela colaboração nos experimentos e nas análises dos vídeos.
A colaboração da Profa. Angela Maria Ribeiro (Laboratório de Neurociências e
Comportamento, LaNeC/UFMG) na coleta de meus dados, incentivo nas horas difíceis
e por sua amizade.
A minha família, em especial minha mãe, pelo seu apoio e amor incondicional
que sempre me incentivaram na busca da realização de meus sonhos. Meu pai, que
sempre cultivou na minha criação a busca pela curiosidade e estudos, obrigada. Ao
meu irmão. A Tula que sempre manteve sua fidelidade e amizade. Aos meus parentes
(Tias, Tios, Primos e Primas) pelo reconhecimento do meu esforço, pelo carinho e
apoio. Muito obrigada!!!
Aos meus amigos do LaNeC pela troca constante de conhecimento e alegria em
trabalhar dentro de um laboratório.
Aos meus amigos e amigas Potiguares que tornaram minha estada em Natal
agradável.
Às minhas amigas e amigos da minha terra natal (Belo Horizonte) que estão
comigo na caminhada da vida em vários momentos, sempre demonstrando apoio,
carinho.

VI
Gostaria muito de agradecer aos animais que deram suas vidas em prol do meu
trabalho de doutoramento e para a melhoria da ciência brasileira.
A todos os momentos difíceis que enfrentei para chegar a conclusão deste
trabalho pois eles me ensinaram a buscar soluções, ter força e determinação que
servirão como características essenciais para exercer minha profissão. Além disso, a
todos os momentos maravilhosos que me deram motivação, alegria e a certeza que
tudo passa.
Enfim, a todos que diretamente ou indiretamente me ajudaram a concluir esse
trabalho.

VII
SUMÁRIO
Página
1. Introdução..................................................................................................................
13
1.1. Apresentação........................................................................................................... 14
1.2. Introdução Geral...................................................................................................... 14
1.2.1. Doença de Parkinson.............................................................................................
14
1.2.2. Transtornos motores na Doença de Parkinson...................................................... 17
1.2.3. Déficits cognitivos na Doença de Parkinson.......................................................... 22
1.2.4. Sistemas de neurotransmissão na Doença de Parkinson...................................... 24
1.2.5. Estudo da doença de Parkinson em modelos animais.......................................... 27
1.2.6. Estresse oxidativo e doença de Parkinson............................................................ 35
1.3. Justificativa.............................................................................................................. 39
1.4. Objetivo geral.......................................................................................................... 41
1.4.1. Objetivos específicos.................................................................................. 41
2. Experimentos.............................................................................................................. 42
2.1. Experimento I................................................................................................. 43
2.2. Experimento II................................................................................................ 72
3. Discussão geral e conclusões.................................................................................. 104
4. Referências................................................................................................................. 113
5. Anexo.......................................................................................................................... 135

VIII
Resumo
A doença de Parkinson (DP) é um dos transtornos cerebrais neurodegenerativos
mais comuns e se caracteriza primariamente por uma progressiva degeneração dos
neurônios dopaminérgicos nigroestriatais. Os sintomas principais dessa doença são
aqueles de origem motora (bradicinesia, rigidez, tremor em repouso), porém alterações
na cognição, no humor e no sistema sensorial também podem ser observadas.
Modelos animais que tentam mimetizar características clínicas da DP vêm sendo
utilizados para compreender as alterações comportamentais e mecanismos neuronais
subjacentes ao distúrbio neurofisiológicos dessa doença Contudo, a maioria dos
modelos promove um comprometimento motor intenso e imediato, compatível com
estágios avançados da doença, invalidando estes estudos quanto à avaliação da
natureza progressiva da manifestação sintomatológica (motora ou cognitiva) da DP.
A administração de reserpina (um depletor de monoaminas) em roedores tem
sido considerada um modelo animal para o estudo da DP. Recentemente verificamos
que a reserpina (em doses menores que as usualmente empregadas para produzir os
sintomas motores) promove um déficit de memória em uma tarefa de discriminação
aversiva, sem alterar a atividade motora. A partir desse estudo sugeriu-se que a
administração desse fármaco em doses baixas pode ser útil para o estudo dos déficits
de memória encontrados na DP. Corroborando esse dado, em outro estudo, a
administração aguda subcutânea de reserpina, em doses que não afetam a função
motora, levou a alterações em memória que envolve contexto emocional enquanto as
sem conotação emocional não foram afetadas.
Os objetivos do presente trabalho foram estudar os déficits cognitivos e motores
associados à administração repetida de baixas doses de reserpina e desenvolver um

IX
possível modelo que mimetize uma neurodegeneração progressiva. Para isso, ratos
Wistar machos com idade de 5 meses foram submetidos a um tratamento repetido, em
dias alternados, com veículo ou diferentes doses de reserpina. Parâmetros cognitivos e
motores, bem como possíveis alterações na função neuronal, foram avaliados ao longo
do tratamento. Os principais resultados encontrados foram: a administração repetida de
0,1 mg/Kg de reserpina em ratos é capaz de induzir o aparecimento gradual de sinais
motores compatíveis com as características progressivas encontrados em pacientes
com DP; os sinais motores foram acompanhados por um aumento dos níveis de
estresse oxidativo no estriado; alterações nas concentrações de glutamato no estriato
nos grupos tratados com doses repetidas de 0,1 e 0,2 mg/Kg foram observadas cinco
dias após o final do tratamento; em animais tratados com doses repetidas de 0,1 mg/kg,
déficits cognitivos foram observados apenas após o surgimento dos sinais motores,
mas não em avaliações feitas anteriormente ao surgimento desses sinais; na dose de
0,2 mg/kg a avaliação cognitiva foi comprometida pela presença de déficits motores
intensos. Dessa forma, os dados obtidos indicam que o protocolo de tratamento com a
reserpina utilizado neste trabalho seja uma alternativa viável para os estudos do
processo progressivo de aparecimento de sinais parkinsonianos em ratos,
principalmente no que diz respeito aos sinais motores. Quanto aos sinais cognitivos,
sugere-se que mais estudos são necessários, possivelmente em outros modelos
comportamentais e/ou alterando-se o esquema de tratamento.

X
Abstract
Parkinson's disease (PD) is one of the most common neurodegenerative brain
disorders and is characterized primarily by a progressive degeneration of dopaminergic
neurons nigroestriatais. The main symptoms of this disease are motor alterations
(bradykinesia, rigidity, tremor at rest), which can be highly disabling in advanced stages
of the condition. However, there are symptomatic manifestations other than motor
impairment, such as changes in cognition, mood and sensory systems.
Animal models that attempt to mimic clinical features of PD have been used to
understand the behavioral and neural mechanisms underlying neurophysiological
disturbance of this disease. However, most models promote an intense and immediate
motor impairment, consistent with advanced stages of the disease, invalidating these
studies for the evaluation of its progressive nature.
The administration of reserpine (a monoamine depletor) in rodents has been
considered an animal model for studying PD. Recently we found that reserpine (in doses
lower than those usually employed to produce the motor symptoms) promotes a memory
deficit in an aversive discrimination task, without changing the motor activity. It was
suggested that the administration of this drug in low doses can be useful for the study of
memory deficits found in PD. Corroborating this data, in another study, acute
subcutaneous administration of reserpine, while preserving motor function, led to
changes in emotional context-related (but not neutral) memory tasks.
The goal of this research was to study the cognitive and motor deficits in rats
repeatedly treated with low doses of reserpine, as a possible model that simulates the
progressive nature of the PD. For this purpose, 5-month-old male Wistar rats were
submitted to a repeated treatment with vehicle or different doses of reserpine on

XI
alternate days. Cognitive and motor parameters and possible changes in neuronal
function were evaluated during treatment. The main findings were: repeated
administration of 0.1 mg / kg of reserpine in rats is able to induce the gradual
appearance of motor signs compatible with progressive features found in patients with
PD; an increase in striatal levels of oxidative stress and changes in the concentrations of
glutamate in the striatum were observed five days after the end of treatment; in animals
repeatedly-treated with 0. 1 mg/kg, cognitive deficits were observed only after the onset
of motor symptoms, but not prior to the onset of these symptoms; 0.2 mg / kg reserpine
repeated treatment has jeopardized the cognitive assessment due to the presence of
severe motor deficits. Thus, we suggest that the protocol of treatment with reserpine
used in this work is a viable alternative for studies of the progressive appearance of
parkinsonian signs in rats, especially concerning motor symptoms. As for the cognitive
symptoms, we suggest that more studies are needed, possibly using other behavioral
models, and / or changing the treatment regimen.

XII
Lista de abreviações
ALDH2: adeído desidrogenase mitocondrial
DOPAC: ácido 3-4-dihidroxifenilacetico
DOPAL: 3-4-dihidroxifenilacetaldeido
DP: Doença de Parkinson
DSM-IV: ―Diagnostic and Statistic Manual of Mental Disorders of the American
Psychiatry Association IV‖
4HNE: 4-hidroxi-2-nonenal
GSH: glutationa
O2-: superóxido
OH-: radical hidroxila
6-OHDA: 6-hidroxidopamina
MAO-B: Monoamina oxidase B
MAO: monoamina oxidase
MDA: malondialdeído
NMDA: N-metil-D-aspartato
NE: norepinefrina
NO-: oxido nítrico
MPP+: 1-metil-4-fenil-piridina
MPTP: 1-metil-4-fenil-1,2,3,6-tetrahidropiridina
ROS: “oxygen reactive species”
SN: Sistema nervoso
VMAT2: transportadores vesiculares de monoaminas 2

13
1.Introdução

14
1.1. Apresentação
Esta tese foi organizada em formato de artigos científicos. Dessa forma,
apresentamos uma introdução geral e, em seguida, os experimentos I e II, estes
expostos como manuscritos para submissão. Finalizamos com uma discussão geral
e conclusão, onde os resultados obtidos nos experimentos realizados foram
unificados.
1.2. Introdução Geral
1.2.1. Doença de Parkinson
A Doença de Parkinson (DP) foi descrita pela primeira vez por James
Parkinson em 1817 (2002). Atualmente, é um dos transtornos cerebrais
neurodegenerativos mais comuns, atingindo em torno de 1% da população com 65
anos ou mais (Bennett et al. 1996, Mayeux 2003).
Transtornos motores como tremores, bradicinesia (lentificação dos
movimentos), rigidez e anormalidades na postura ou na marcha são considerados as
características primárias da DP (Grossman 1999, Korczyn 2001, Nieoullon 2002).
Contudo, existem outras manifestações sintomáticas além das motoras, dentre as
quais podemos citar alterações na cognição, no humor e no sistema sensorial
(Higginson et al. 2001, Korczyn 2001, Richard et al. 2004, Zgaljardic et al. 2004,
Perbal et al. 2005, Shohamy et al. 2005, Koerts et al. 2007, Monchi et al. 2007,

15
Huang et al. 2007, Schmitt-Eliassen et al. 2007). Além disso, por ser uma doença de
evolução progressiva, dependendo do estágio da doença pode ser observado
disfunção autonômica, alterações de personalidade, distúrbios do sono, dificuldade
na fala e disfunção sexual (Mayeux 2003, Klochgether 2004).
Classicamente, a maioria dos pesquisadores tem utilizado a idade de 40 anos
para classificar os pacientes quanto às manifestações clínicas da DP (Quinn et al.
1987). Quinn et al. (1987) propuseram que casos da DP que se iniciassem entre a
idade de 21-40 anos deveriam ser denominados de portadores da ―DP de início
precoce‖. Além disso, resultados de estudos com portadores da ―DP de início
precoce‖ indicaram uma relação com fatores genéticos relacionados ao risco de
desenvolvimento da doença, especialmente se houver um histórico familiar positivo
(Quinn et al. 1987, Schrag & Schott 2006). A maioria destes pacientes também
apresenta idiopatia de corpos de Lewys (Schrag & Schott 2006). Os pacientes que
manifestam os sintomas da DP com 70 anos ou mais são classificados como de
inícios tardio (Jankovic et al. 1990).
Estudos têm indicado existir uma heterogeneidade clínica em pacientes com
DP que sugerem diferentes mecanismos bioquímicos e degenetarivos (Jankovic &
Kapadia 2001, Lewis et al. 2005, Reijnders et al. 2009). Relatos da literatura
mostram evidências que a progressão da degeneração não é linear (Jankovic &
Kapadia 2001, Nurmi et al. 2001). No início da doença, há uma progressão mais
rápida e em estágios mais avançados, uma taxa de deterioração desacelerada
(Jankovic & Kapadia 2001). Alguns pesquisadores propõem uma classificação para
estes pacientes sendo estes separados em quatro principais subtipos de evolução
da doença: (1) que se inicia na juventude; (2) de rápida progressão; (3) de tremor
não dominante associado a psicopatologias; (4) de tremor dominante. Os sintomas

16
relacionados a déficits cognitivos, depressão, apatia, alucinações podem ser
inclusos no subtipo tremor não dominante, associado a psicopatologias que também
acompanham distúrbios motores como hipocinesia (movimentos lentos ou
ausentes), rigidez, instabilidade postural e distúrbios da marcha. Essas
características fisiopatológicas subjacentes aos subtipos indicam possíveis
implicações neuropatológicas diferenciadas (Reijnders et al. 2009).
Estudos que enfocam deficits motores e cognitvos na DP apresentam
resultados consideravelmente variados (Aarsland et al. (2004, 2007), Borek et al.
2006, Reijnders et al. 2009). Alterações cognitivas que se manifestam antes das
disfunções motoras em pacientes com DP são descritos na literatura (Fenelon 1997,
Shults 2003). Por outro lado, é comum encontrar a associação de um declínio
funcional e rápido das funções motoras com a presença de prejuízos cognitivo que
caracteriza um quadro demencial em indivíduos com DP (Aarsland et al. (2004,
2007)). Geralmente, o início do desenvolvimento do quadro demencial pode oscilar
do diagnóstico até 10 anos ou mais após detectada a doença. Existem vários fatores
preditores do declínio cognitivo, dentre eles podemos destacar os sintomas motores
graves, a presença de alucinações, a presença de corpos de Lewys e os distúrbios
na fala (Aarsland et al. 2004, Burn et al. 2006). Apesar da heterogeneidade dos
pacientes com DP, é encontratado um consenso na literatura quanto à influência dos
déficits cognitivos na qualidade de vida destes pacientes (Aarsland et al. (2004,
2007), Borek et al. 2006).
Existe uma grande dificuldade em identificar os fatores de risco que podem
causar a DP embora exista um consenso de que seja uma doença multifatorial.
Contudo, alguns estudos destacaram fatores como a predisposição genética,
infecção viral (influenza A), traumas físicos e exposição a substâncias tóxicas tais

17
como 6-hidroxidopamina (6-OHDA) e 1-metil-4-fenil-1,2,3,6-tetrahidropiridina (MPTP)
(Calne 2007, Mayeux 2003). Adicionalmente, estudos sobre as taxas de mortalidade
e prevalência da DP mostram que a incidência é maior nos homens do que nas
mulheres. As razões para o aumento do risco de homens desenvolverem DP não
são conhecidas (Wooten et al. 2004).
1.2.2. Transtornos motores na Doença de Parkinson
Os primeiros sintomas manifestados durante o desenvolvimento da DP
podem ser tremor de repouso unilateral no braço ou na perna. Contudo, sintomas
como a bradicinesia (lentificação e escassez de movimentos), incapacidade de
realizar movimentos (acinesia), membros rígidos, andar e postura inclinados, podem
também estar presentes em fases iniciais da doença (Mayeux 2003). Outra
dificuldade motora frequentemente relatada por pacientes com DP é a incapacidade
de realizar movimentos suaves e coordenados com as mãos, que dificulta o
desempenho na escrita e o desenvolvimento de movimentos precisos (Van Gemmert
et al. 2001).
O sintoma de bradicinesia afeta todos os movimentos voluntários e
involuntários. Os movimentos automáticos e habituais, tais como a movimentação
dos braços durante a caminhada, o piscar dos olhos e a deglutição da saliva, são
fortemente reduzidos. Outras características da bradicinesia são: dificuldade em
iniciar movimentos voluntários, lentidão e passos pequenos ao andar. Os pacientes
com DP também apresentam uma menor mobilidade na expressão facial e uma fala
monotônica, que leva a déficits na habilidade de comunicação, apesar de poder

18
apresentar uma função intelectual preservada (Hallett & Khoshbin 1980, Klockgether
2004). A presença de bradicinesia nas extremidades superiores manifesta-se como
micrografia e, com o processo degenerativo, as pessoas afetadas desenvolvem
dificuldade na execução de movimentos finos, como o de abotoar roupas
(Klockgether 2004).
O tremor parkinsoniano normalmente manifesta-se durante o repouso e afeta
principalmente os membros superiores, podendo também afetar as pernas e, com
menos frequência a cabeça. Nos casos típicos de DP, o tremor de repouso possui
uma frequência de 4-7 Hz. Este sintoma não é necessariamente incapacitante, mas
muitos pacientes sofrem porque o tremor os estigmatiza como portadores da DP
(Klockgether 2004).
A rigidez muscular é definida como um aumento da resistência da
movimentação passiva em consequência da rigidez da articulação, que se manifesta
em toda a amplitude do movimento (Xia et al. 2009). Em pacientes nos quais a
rigidez é acompanhada pelo tremor de repouso, um tipo muito característico de
resistência pode ser observado e tem sido denominado como rigidez de roda
denteada. Relatos subjetivos dos pacientes com rigidez a descrevem como
sensações de rigidez e diminuição da capacidade de relaxar os músculos dos
membros (Klockgether 2004).
Na maioria dos casos de pacientes com DP, os sintomas de tremor, rigidez e
bradicinesia estão presentes. Entretanto, a extensão e a gravidade destes sintomas
apresentam variações (Louis et al. 1999, Bertram et al. 2005) . Um sintoma que
claramente está presente é a incapacidade de realizar movimentos suaves e
coordenados (Bertram et al. 2005).

19
O diagnóstico de DP utilizado na clínica geralmente inclui a presença de
bradicinesia e pelo menos uma das três características primárias que são: (1) rigidez
muscular dos membros; (2) tremor postural ou residual; (3) instabilidade postural ou
transtorno postural. Apesar de por vezes presentes, características como demência
ou disfunções autonômicas não contemplam os sintomas utilizados para o
diagnóstico (Mayeux 2003). Assim sendo, os sintomas motores têm sido ressaltados
como os mais importantes transtornos associados com a DP (Klockgether 2004).
Geralmente, os sintomas motores da DP são atribuídos à perda progressiva
dos neurônios dopaminérgicos da substância negra, que leva ao comprometimento
dos tratos extrapiramidais que controlam movimentos corporais complexos
(Grossman 1999, Korczyn 2001, Nieoullon 2002). Entretanto, os mecanismos
subjacentes da depleção de dopamina relacionados aos distúrbios motores não
estão completamente esclarecidos e continuam sob investigação. Alguns estudos
que correlacionam disfunções motoras, dopamina e DP têm sugerido que estes
fatores envolvem alterações do funcionamento dos circuitos cortico-estriatais
(Antonini et al. 1997, Costa et al. (2004, 2006)).
Os pesquisadores Glendinning & Enka (1994) relatam que os mecanismos
subjacentes a alterações da unidade motora na DP são ainda pouco entendidos.
Entretando, estes pesquisadores apontam uma diminuição na atividade muscular e
alteração da unidade motora devido: (1) a irregularidade e intermitência dos padrões
de descargas nas unidades motoras; (2) ao fato de que os músculos antagonistas
(os quais possuem ação anatômica oposta à dos músculos agonistas e usualmente
no movimento permanencem relaxados permitindo a maior facilidade do movimento)
são coativos. Uma possível hipótese para estas mudanças está em um desequilíbrio
entre os impulsos excitatórios e inibitórios para os neurônios motores.

20
Resultados de estudos post mortem em pacientes com DP suregem que
características parkinsonianas diferentes podem ter alterações diferenciadas nos
circuitos neuronais. Estes estudos mostram que pacientes com parkinsonismo do
tipo acinético-rígido possuem perdas mais significativas de células da porção
ventrolateral da substância negra e do locus coeruleus em comparação à pacientes
com parkinsonismo com tremor predominante (Paulus & Jellinger 1991, Jellinger
1999).
Geralmente, os sintomas motores da DP são atribuídos à perda progressiva
dos neurônios dopaminérgicos da substância negra, que levam ao comprometimento
dos tratos extrapiramidais que controlam movimentos corporais complexos
(Grossman 1999, Korczyn 2001, Nieoullon 2002). Entretanto, os mecanismos
subjacentes da depleção de dopamina relacionados distúrbios motores não estão
completamente esclarecidos e continuam em discussão. Alguns estudos que
correlacionam disfunções motoras, dopamina e DP têm sugerido que estes fatores
envolvem alterações do funcionamento dos circuitos cortico-estriatais (Antonini et al.
1997, Costa et al. (2004, 2006), Cilia et al. 2007). De fato, num estudo desenvolvido
por Cilia et al. 2007, no qual foram avaliadas características clínicas e imagem de
ressonância magnética do cérebro de um paciente com tremor palatal (caracterizado
clinicamente por contrações rítmicas e involuntárias dos músculos do palato mole) e
ataxia progressiva (falta de coordenação dos movimentos, podendo afetar a força
muscular e o equilíbrio de uma pessoa), foi encontrada redução dos transportadores
de dopamina no estriado direito. Este estudo também revelou uma degeneração
hipertrófica dos núcleos olivares e significante hipometabolismo nos núcleos rubros,
sugerindo que os sintomas de tremor palatal e ataxia progressiva podem estar

21
relacionados a danos nas vias dentato-rubro-olivar e a disfunções dopaminérgicas
nigro-estriatais.
Um estudo utilizando tomografia por emissão de pósitrons (TEP) com
marcadores fluoroso F18, fluorodopa (FDOPA) e raclopride (RACLO) utilizados para
estudar o metabolismo da glicose estriatal e de DOPA, e marcação de receptor D2
de dopamina mostraram em seus resultados sintomas motores e atrofia de múltiplos
sistemas dopaminérgicos. Este achado indica que a degeneração de sistemas
dopaminérgicos pré e pós-sinápticos estriatais é responsável pelas alterações
motoras em humanos (Antonini et al. 1997).
Outras evidências do envolvimento do sistema dopaminérgico com as
alterações motoras estão no efeito de medicamentos utilizados no tratamento de tais
sintomas, os quais aumentam a função dopaminérgica. Estudos têm demonstrado
que medicamentos pró-dopaminérgicos (exemplo: L-DOPA, selegelina, entre outros)
melhoram a rigidez de pacientes com DP (Benecke et al. 1987, Xia et al. 2009).
A gravidade do transtorno motor pode também estar associada ao declínio
cognitivo (Aarsland et al. (2004, 2005), Burn et al. 2006). Um estudo indicou uma
relação entre pacientes com instabilidade na marcha e dificuldade postural com o
risco de desenvolver um quadro demencial (Burn et al. 2006). Além disso, Louis et
al. (1999) também encontraram que sintomas como rigidez, bradicinesia, tremor e
instabilidade postural em pacientes com DP são preditores do desenvolvimento de
demência. Nesse sentido, podemos observar que as diversidades de sintomas
motores podem ser consequência de uma neurodegeneração e alterações
neuroquímicas que ainda não estão esclarecidas.

22
1.2.3. Déficits cognitivos na Doença de Parkinson
Como escrito no item anterior evidências surgerem que os sintomas motores
são devido à perda progressiva dos neurônios dopaminérgicos da substância negra
que promovem depleção dos níveis de dopamina estriatal (Johnston et al. 1999,
Lindner et al. 1999, Ridley et al. 2006). Contudo, os distúrbios motores podem estar
acompanhados também por prejuízos intelectuais que afetam significativamente a
qualidade de vida de uma pessoa acometida (Korczyn 2001, Nieoullon 2002,
Scherfler et al. 2004, Zgaljardic et al. 2004). Em alguns casos, esses prejuízos
cognitivos se manifestam antes das alterações motoras, e sugere-se que estejam
envolvidos com circuitos neuronais diferentes (Fenelon 1997, Shults 2003). Além
disso, as alterações cognitivas têm sido correlacionadas a disfunções nas projeções
das vias dopaminérgicas envolvidas em funções de áreas fronto-corticais, tais como
planejamento de ações e a memória operacional (Pillon et al. (1997, 1997a) , Cools
et al. 2002).
Estudos de neuroimagem em pacientes com Parkinson evidenciam uma base
neural específica para os danos cognitivos encontrados nesses casos clínicos
(Owen et al. 1998, Cools et al. 2002, Koerst et al. 2007). Conforme já mencionado,
esses déficits cognitivos na DP têm sido freqüentemente atribuídos a prejuízos em
projeções dopaminérgicas corticais (Cools et al. 2002). Contudo, outras evidências
sugerem que os déficits também podem estar relacionados a danos em regiões
subcorticais (Pillon et al. 1996, Pillon et al. 1997). Além disso, a própria via nigro-
estriatal (onde ocorre a degeneração característica da doença causadora dos
sintomas motores) pode estar relacionada a alguns tipos de funções cognitivas

23
(Perry et al. 2004, Albouy et al. 2008, Ferreira et al. 2008). Finalmente, deve-se
ressaltar que embora as disfunções executivas sejam as mais bem estudadas em
pacientes com DP, o DSM-IV (―Diagnostic and Statistic Manual of Mental Disorders
of the American Psychiatry Association IV‖) coloca déficits de memória como
característica básica da demência associada à DP.
O declínio cognitivo mais acelerado na DP tem sido relacionado a alguns
fatores preditivos como idade avançada, ocorrência de alucinações, presença de
sintomas motores graves (Aarsland et al. 2004).
A prevalência de demência na DP possui resultados variados em estudos
encontrados na literatura científica onde métodos de avaliações distintos são usados
na população estudada. Entretando, estima-se que a demência afete
aproximadamente 40% dos pacientes com DP e a incidência nestes pacientes é de
até seis vezes maior que em pessoas saudáveis. Existe uma forte discussão sobre
alterações em outras vias neuronais envolvidas na demência da DP, pois danos na
via nigroestriatal não são suficientes para explicar o desenvolvimento da demência.
Vários sintomas da DP são constantemente associados a fatores de risco do
desenvolvimento da demência, entre eles estão: a idade avançada; idade avançada
e início de sintomas motores; início precoce de confusões mentais relacionados à
levodopa ou psicose; presença de comprometimento axial e da fala; sintomas
motores graves, em especial bradicinesia; escores em teste de cognição baixos, em
especial na fluência verbal; e depressão (Murat 2003, Aarsland et al. (2004, 2007),
Borek et al. 2006). Assim sendo, a etiologia dos déficits cognitivos associados à DP
ainda não está muito bem esclarecida.

24
1.2.4. Sistemas de neurotransmissão na Doença de Parkinson
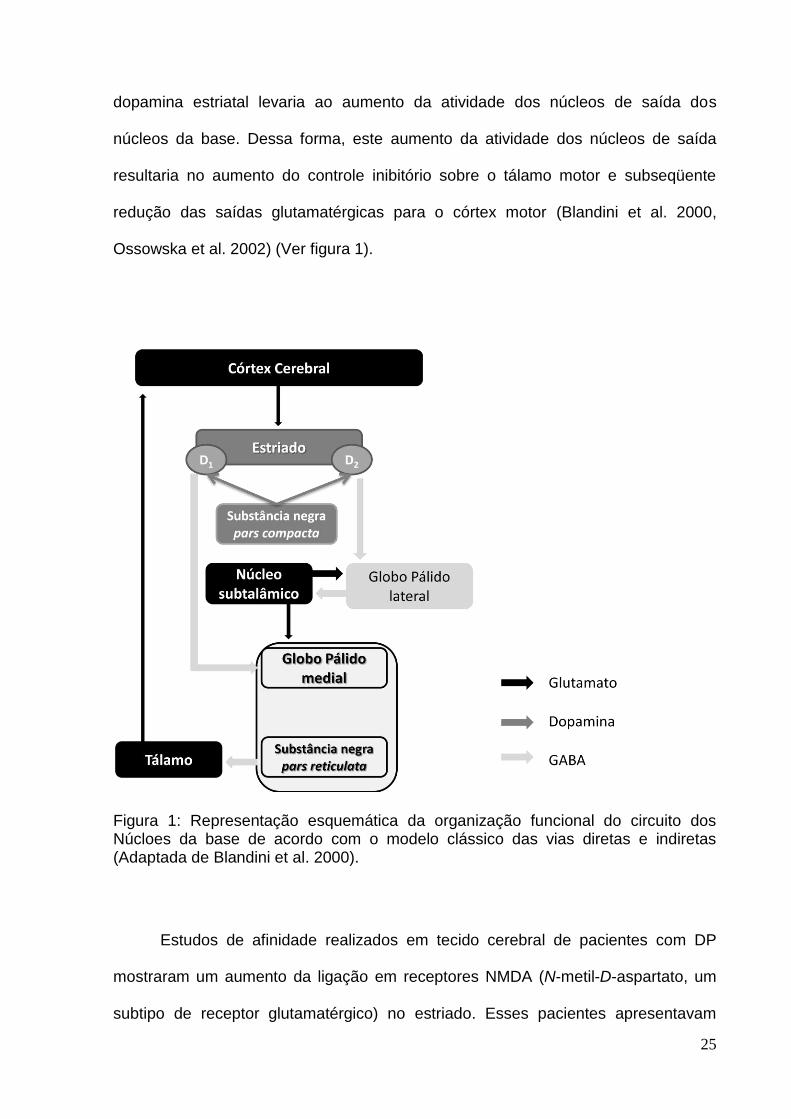
A execução correta dos movimentos depende do circuito dos núcleos da base
que processam sinais que chegam do córtex. Assim sendo, alguns pesquisadores
desenvolveram um modelo de funcionamento dos núcleos da base a fim de
compreender melhor os mecanismos envolvidos na execução dos movimentos na
condição normal e em transtornos como a DP (DeLong & Wichmann 2007, Blandini
et al. 2000). De acordo com o modelo do circuito dos núcleos da base, a entrada do
sinal do circuito seria na substância negra pars compacta que projeta vias
dopaminérgicas para o estriado. Os neurônios estriatais expressam receptores
dopaminérgicos do tipo D1 e D2 , os quais são distintos funcionalmente. O subgrupo
de neurônios estriatais que expressa receptores D1 projeta vias GABAérgica para a
substância negra pars reticulata e para globo pálido medial, denominada via direta.
O subgrupo de neurônios estriatais que expressa receptores D2 projeta vias
Gabaérgicas para o globo pálido lateral, denominada via indireta, que envia
projeções GABAérgicas para o núcleo subtalâmico. O núcleo subtalâmico, por sua
vez, envia eferências glutamatérgicas para o globo pálido medial e globo pálido
lateral. A substância negra pars reticulata e o globo pálido medial formam um núcleo
que envia projeções Gabaérgicas (inibitórias) que atingem o tálamo motor que
projeções glutamatérgicas para o córtex motor, fechando o circuito (Blandini et al.
2000) (Ver figura 1).
O processo de neurodegeneração dopaminérgica na substância negra que
ocorre na DP resulta em uma consequente diminuição da dopamina no estriado,
desencadeando alterações secundárias, as quais contribuem para os complexos
sintomas parkinsonianos subjacentes. Foi postulado que uma perda gradativa de
25
dopamina estriatal levaria ao aumento da atividade dos núcleos de saída dos
núcleos da base. Dessa forma, este aumento da atividade dos núcleos de saída
resultaria no aumento do controle inibitório sobre o tálamo motor e subseqüente
redução das saídas glutamatérgicas para o córtex motor (Blandini et al. 2000,
Ossowska et al. 2002) (Ver figura 1).
Figura 1: Representação esquemática da organização funcional do circuito dos Núcloes da base de acordo com o modelo clássico das vias diretas e indiretas (Adaptada de Blandini et al. 2000).
Estudos de afinidade realizados em tecido cerebral de pacientes com DP
mostraram um aumento da ligação em receptores NMDA (N-metil-D-aspartato, um
subtipo de receptor glutamatérgico) no estriado. Esses pacientes apresentavam

26
características neurológicas e psiquiátricas do quadro clínico da DP (Ulas et al.
1994). Os resultados ressaltam uma possível relação de alterações das vias
glutamatérgicas que fazem parte do circuito dos núcleos da base com os complexos
sintomas parkinsonianos (Lange et al. 1997, Ulas et al. 1994).
Outra possível relação do glutamato com a DP está na hipótese de esse
transmissor desencadear um processo gradual de eliminação de células resultante
da ativação de programas de apoptose. Uma via seria através da excitotoxidade por
estimulação excessiva do receptor NMDA pelo glutamato, que causaria morte
celular. Outra via seria através da capacidade do glutamato em induzir a formação
de espécies reativas de oxigênio (oxygen reactive species, ROS) que resultariam em
danos nas células neuronais (Tan et a. 1998, Blandini et al. 2000).
Entretanto, existem controvérsias na literatura quanto a este modelo de
funcionamento do circuito dos gânglios da base, que tem sido uma proposta para
explicar aspectos dos distúrbios motores associados a alterações anatômicas e
neuroquímicas. Um estudo realizado com pacientes com sintomas parkinsonianos
avaliou a concentração de neurotransmissores (GABA e Glutamato) em 18 regiões
das vias tálamo-cortical do circuito dos gânglios da base e compararou com tecidos
das mesmas regiões de indivíduos que morreram sem históricos de distúrbios
neurológicos ou psiquiátricos. Os resultados desta pesquisa mostram uma
diminuição da concentração de GABA apenas na região centromedial do tálamo nos
pacientes com sintomas parkinsonianos (Gerlach et al. 1996).

27
1.2.5. Estudo da doença de Parkinson em modelos animais
Modelos animais vêm sendo utilizados para estudar as alterações causadas
pela DP. Alguns modelos tentam mimetizar características clínicas da DP em
roedores, a fim de compreender melhor os mecanismos subjacentes ao distúrbio
neurofisiológicos dessa doença. Contudo, há muitas controvérsias quanto aos
modelos que expressariam a natureza progressiva da DP e dos estágios ―pré-clinico‖
e ―clínico‖.
Nas últimas décadas, alguns modelos famacológicos foram criados e os mais
estudados utilizam toxinas tais como 6-hidroxidopamina (6-OHDA) e 1-metil-4-fenil-
1,2,3,6-tetrahidropiridina (MPTP). Estas duas substâncias promovem lesões
específicas de células do sistema nervoso (SN), promovendo um comprometimento
motor intenso e imediato, sem estabelecer um processo gradativo
neurodegenerativo (Meredith et al. 2008).
A neurotoxina, 6-OHDA, tem uma estrutura similar à dopamina e à
norepinefrina (NE), o que proporciona uma alta afinidade pelos transportadores de
catecolaminas das membranas. Dessa forma, essa toxina é transportada para
dentro do neurônio, onde promove reações de oxidação e produção de paraquinona
e peróxido hidrogênio, ambos com alta toxicidade. A 6-OHDA não atravessa a
barreira hematoencefálica, por isso deve ser administrada diretamente no tecido
nervoso, onde causa lesões específicas nos neurônios liberadores de dopamina e
norepinefrina (Meredith et al. 2008).
A 6-OHDA induz a morte de células neuronais dentro das primeiras 12 horas
após a administração. A depleção de dopamina estabelece-se entre o segundo e o
terceiro dia após a administração. Em geral, esta neurotoxina normalmente é

28
administrada unilateralmente, que induz o aparecimento de um comportamento
esteriotipado de rotação contralateral a lesão (Marin et al. 2007, Blandini et al. 2008),
e o hemisfério contralateral é utilizado como controle. As injeções bilaterais da 6-
OHDA são evitadas devido à alta taxa de mortalidade dos animais submetidos a
este procedimento (Ferro et al. 2005, Blandini et al. 2008), pelo menos nas doses
usuais. Outro aspecto interessante é que os efeitos funcionais induzidos pela lesão
por 6-OHDA não dependem apenas do total de doses injetadas, mas também do
local ou sub-região em que a toxina provoca morte celular.
A MPTP é uma toxina que causa sintomas motores, semelhantes à DP, por
destruir especificamente neurônios dopaminérgicos do SN. Nas últimas décadas,
sua administração tem sido usada como um modelo mais eficaz para estudar
mudanças moleculares subjacentes as disfunções mitocondriais da DP. Após a
administração de MPTP, ocorre uma perda rápida de neurônios dopaminérgicos,
apresentando-se assim os transtornos motores característicos da doença de
Parkinson (Meredith et al. 2008a). A MPTP, uma vez no tecido nervoso, é oxidada
para 1-metil-4-fenil-2,3-dihidropiridinium (MPDP+) pela monoamina oxidase B (MAO-
B). A MPDP+ é, então, convertida em MPP+ (1-metil-4-fenil-piridina, uma molécula
altamente tóxica), que pode entrar nas células dopaminérgicas por ser um substrato
com alta afinidade pelos transportadores dopaminérgicos. Dentro das células
dopaminérgicas: (1) pode ser armazenado nas vesículas sendo transportado pelos
transportadores vesiculares de monoaminas 2 (VMAT2); (2) pode ser armazenado
dentro das mitocôndrias, através de um mecanismo dependente do potencial
transmembrana mitocondrial e, dentro da mitocôndria, agir bloqueando o
componente I de transporte de elétrons que induz o aumento de espécies reativas
de oxigênio (não mostrado na Figura 2) e diminuição da síntese de ATP e (3) pode

29
permanecer no citosol celular interagindo com as enzimas (Ver figura 2) (Dauer et al.
2003).
Recentemente, alguns estudos têm proposto modelos crônicos com o MPTP
em roedores, através da infusão crônica intra-cerebral. Contudo, os estudos crônicos
com MPTP têm encontrado algumas limitações, como uma alta mortalidade dos
animais, pelo nível de toxidade da substância, e uma alta variabilidade dos sinais
que caracterizam a DP. Apesar dessas limitações esse modelo tem evidenciado
algumas vantagens, como a presença de alterações mitocondriais e a possibilidade
de avaliar processos neuroprotetores em estágios do desenvolvimento dos sinais da
DP (Sonsalla et al. 2008, Meredith et al. 2008 a).
Figura 2: Representação esquemática das vias intracelulares do MPP+ dentro das células dopaminérgicas (Adaptada de Dauer et al. 2003).
Mitocôndria Vesícula sináptica
Enzimas MPP+ bloqueia a cadeia
de transporte de elétrons.

30
Estudos em animais que expressam 5% do transportados vesicular de
monoaminas 2 (VMAT 2) tem sido proposto como um novo modelo para o estudo da
DP (Taylor et al. 2009). Os animais deficientes de VMAT 2 apresentam aumento do
estresse oxidativo, perda progressiva dos terminais de dopamina assim como
acumulação de -sinucleína (Caudle et al. 2007). Além disso, disfunções
monoaminérgicas também são encontradas, os níveis de dopamina, de
norepinefrina e de serotonina são severamente diminuídos (Caudle et al. 2007,
Taylor et al. 2009). Alterações no sono, gastrointestinais, sintomas de ansiedade e
depressão foram observados em resultados de estudos com camundongos
deficientes de VMAT 2 (Taylor et al. 2009).
Outro modelo utilizado para se estudar a DP é a administração de reserpina
em roedores, baseado nos efeitos de agentes de depleção de monoaminas sobre a
atividade motora (Colpaert 1987, Kim et al. 1999, Alves et al. 2000, Silva et al. 2002,
Skalisy et al. 2002). Tanto o modelo farmacológico da administração de reserpina
dos animais deficientes de VMAT 2 quanto o modelo resultam na disfunção do
transportados vesicular de monoaminas. A reserpina é uma droga que evita o
armazenamento de monoaminas nas vesículas sinápticas, através do bloqueio dos
transportadores da membrana que captam as monoaminas para dentro da vesícula
(Liu et al. 1996, Verheij & Cools 2007). Dessa forma, as vesículas sinápticas
permanecem vazias e consequentemente não há neurotransmissores para serem
liberados na fenda sináptica, quando um potencial de ação atinge o botão sináptico
(Rang et al. 2004). Contudo, é importante ressaltar que o tratamento com reserpina,
como um modelo de DP, apresenta limitações, pois a administração da droga não
provoca depleção de neurotransmissores apenas na via nigroestriatal e nem age
exclusivamente em vias dopaminérgicas. Outro aspecto é o fato da administração

31
única de reserpina não promover uma degeneração neuronal progressiva. Por outro
lado, a reserpina pode promover um aumento no estresse oxidativo celular,
possivelmente pelo aumento da metabolização da dopamina acumulada no
citoplasma pela enzima monoaminoxidase (Abílio et al. 2002). Dessa forma, a
administração repetida de doses reduzidas dessa substância poderia ser um melhor
modelo para estudar uma doença neurodegenerativa progressiva.
A maioria dos modelos farmacológicos desenvolvidos utiliza avaliações
comportamentais que são realizadas para observação de sinais motores e cognitivos
semelhantes às características clínicas da DP. As avaliações dos sinais motores são
estudadas através de: avaliações da passada (Kirik et al.1998, Chang et al. 1999),
parâmetros motores no campo aberto (distância percorrida, frequência em levantar
as patas dianteiras, latência em iniciar o movimento, tempo de imobilidade,
velocidade) (Peixoto et al. 2005, Perry et al. 2005, Reksidler et al. 2008), tempo no
comportamento de catalepsia (Namba et al. 1981, Perry et al. 2005), distância
percorrida e velocidade em labirintos (Carvalho et al. 2006), entre outros. As
avaliações dos sinais cognitivos são realizadas através de: tarefas do labirinto
aquático de Morris (Da Cunha et al. 2002, Bellissimo et al. 2004, Perry et al. 2004),
teste do medo condicionado ao contexto (Fernandes et al. 2008), teste da esquiva
discriminativa em labirinto em cruz elevado (Carvalho et al. 2006), entre outros.
As lesões induzidas pela 6-OHDA em parte do corpo estriado dorso medial
provocam alterações no comportamento motor de uma forma geral. Entretanto,
lesão na via ventrolateral dos núcleos caudado-putamen provocam alteração no
início do movimento e na orientação sensório-motora (Cousins & Salamone 1996,
Kirik et al.1998). Além disso, um estudo demonstrou que uma depleção de dopamina

32
no estriado superior a 80% induz reduções significativas da capacidade dos ratos
em ajustar os passos enquanto que redução dopaminérgica estriatal inferior a 80%
não provoca déficits detectáveis (Chang et al. 1999).
Ratos com lesão na substância negra, induzida pela MPTP, 24 horas após a
administração aguda mostram redução no número de quadrantes percorridos e na
freqüência em levantar as patas dianteiras, parâmetros avaliados no campo aberto.
Contudo, este efeito não persiste ao longo do tempo, indicando um possível
mecanismo de compensação do circuito neuronal (Perry et al. 2005). Outro trabalho
com administração repetida de MPTP demonstrou que as perdas de neurônios
dopaminérgicos na substância negra compacta foram significativas somente após a
primeira aplicação. Porém, as alterações motoras (diminuição distância percorrida e
velocidade no campo aberto e aumento do tempo no comportamento de catalepsia)
permaneceram estáveis durante o tratamento com três aplicações. Uma provável
explicação para a alteração motora não regredir está na consequente diminuição
dopaminérgica nas vias estriatais, reafirmada pela diminuição expressiva da enzima
tirosina hidroxilase, da primeira aplicação ao último dia de análises (Reksidler et al.
2008).
Alguns trabalhos demonstraram que déficits de memória ocorrem ainda na
fase inicial da DP, quando sinais motores são pouco observados (Da Cunha et al.
(2001, 2002), Bellissimo et al. 2004, Perry et al. 2004, Fernandes et al. 2008). Neste
sentido, trabalhos nos quais foram utilizado o tratamento com MPTP como modelo
animal de DP, aplicado agudamente na substância negra pars compacta, os dados
mostraram déficits na aquisição da memória e nos processos de retenção no teste
de esquiva ativa (Da Cunha et al. (2001, 2002)), assim como prejuízos na memória
espacial de ratos na tarefa do labirinto aquático de Morris (Da Cunha et al. 2002,

33
Bellissimo et al. 2004, Perry et al. 2004). Contudo, os estudos de Bellissimo et al.
(2004) e Da Cunha et al. (2002) também demonstraram a possibilidade da
participação do comprometimento motor, a negligência sensorial e/ou prejuízo da
representação espacial contralateral neste prejuízo da tarefa do labirinto aquático.
Os resultados destes estudos ainda revelaram que os ratos com lesão pelo MPTP
apresentaram perda acentuada de células dopaminérgicas da substância negra
(parte compacta), assim como uma significativa depleção da dopamina no estriado
(Da Cunha et al. ( 2001, 2002), Bellissimo et al. 2004).
Um estudo comparativo dos modelos de Parkinson, utilizando MPTP (100 µg)
e 6-OHDA (6 µg), infundidos bilateralmente na região central da substância negra de
ratos adultos, detectou que ambas as neurotoxinas causavam perdas significativas
de células marcadas pela tirosina hidroxilase, assim como levaram a depleção de
dopamina no estriado. Entretanto, este estudo demonstrou que a 6-OHDA causa
perda de células mais intensa e ampla, além de levar o animal a ter uma perda de
peso mais intensa e mortalidade mais acentuada que a do MPTP (Ferro et al. 2005).
A administração de reserpina promove sinais parkinsonianos como acinesia,
rigidez, tremores e déficits cognitivos visuoespaciais (Colpaert 1987, Johnston et al.
1999, Lindner et al. 1999, Skalisz et al. 2002, Delfino et al. 2004, Peixoto et al. 2005,
Carvalho et al. 2006, Aguiar Jr et al. 2009). Além disso, a hipocinesia induzida pela
reserpina parece estar relacionada ao decréscimo de dopamina, e um estudo
demonstrou que a L-DOPA é capaz de reverter estes efeitos de catalepsia (Namba
et al. 1981). Além de induzir uma diminuição na atividade locomotora, a reserpina
também causa concomitantemente anedonia (uma menor resposta a recompensas)
e estes sintomas estão associados à DP (Skalisz et al. 2002). Dessa forma, a
administração de reserpina constitui um modelo farmacológico de DP capaz de

34
mimetizar não somente os aspectos motores, mas também outros sintomas
presentes no desenvolvimento da patologia referida.
Carvalho et al. (2006) demonstraram que os efeitos da reserpina induzem um
prejuízo no desempenho da memória na esquiva discriminativa em labirinto em cruz
elevado, uma tarefa que associa uma estimulação aversiva com um determinado
local do labirinto. É importante ressaltar que nesse estudo (Carvalho et al. 2006),
foram utilizadas doses de reserpina menores que as usuais (0,1 a 0,5 mg/kg) e que
o comprometimento cognitivo foi observado mesmo em doses que não afetaram a
função motora. O fato de doses pequenas de reserpina induzirem déficits cognitivos,
sem alterarem a atividade motora está de acordo com a observação de que déficits
cognitivos podem preceder os sinais motores tanto em pacientes com DP (Cooper et
al. 1991, Owen et al. 1992) quanto em modelos animais (Schneider & Pope-
Coleman 1995, Carvalho et al. 2006). De fato, evidências sugerem que pequenas
perturbações na transmissão dopaminérgica levariam a déficits cognitivos, enquanto
que um alto nível de alteração nessa neurotransmissão levaria a déficits motores, os
quais poderiam até sobrepor prejuízos cognitivos pré-existentes (Schneider & Pope-
Coleman 1995, Pillon et al. 1997, Owen et al. 1998).
Recentemente, em nosso laboratório, com base no estudo de Carvalho et al.
(2006) citado acima, investigamos os efeitos da reserpina (0,1 - 0,5 mg/Kg) no
desempenho de ratos no reconhecimento de objetos, na memória operacional
espacial (alternação espontânea) e na memória emocional (condicionamento
contextual da resposta de medo). Na tarefa de reconhecimento de objetos e de
alternação espontânea os animais não foram afetados pelo tratamento com
reserpina, ao contrário do condicionamento contextual da resposta de medo, que foi
prejudicado. Associados a estudos prévios, esses resultados sugerem que uma

35
depleção moderada de monoaminas pode preferencialmente induzir déficits em
tarefas que envolvem contextos emocionais (Fernandes et al. 2008). Tomados em
conjunto, os estudos até o momento realizados sugerem que o efeito amnésico da
reserpina em ratos pode ser uma abordagem comportamental para o estudo dos
sintomas cognitivos da DP podendo estar correlacionados com disfunções nas
projeções dopaminérgicas envolvidas no controle de funções de áreas fronto-
corticais e nigroestriatais. Tais estudos prévios, entretanto, foram realizados com a
administração sistêmica (subcutânea) aguda de reserpina, de modo que seria
interessante verificar os efeitos da reserpina em um tratamento prolongado, o que
poderia mimetizar com mais fidedignidade as etapas relacionadas com déficits
cognitivos que surgiriam ao longo do processo neurodegenerativo progressivo da
DP.
1.2.6. Estresse oxidativo e doença de Parkinson
O oxigênio (O2) é uma molécula essencial para a vida dos seres vivos,
contudo, é capaz de produzir espécies altamente reativas (radicais livres)
denominado ―espécies reativas de oxigênio‖ (reactive oxygen species – ROS), que
ocorrem durante a fosforilação oxidativa mitocondrial. Exemplos de radicais livres
derivados de reações com o O2 são o superóxido (O2-), o radical hidroxila (OH-) e o
oxido nítrico (NO-). Entretanto, existem defesas naturais do organismo para proteger
contra as ROS. Normalmente, as células mantêm um controle homeostático sobre o
estado oxidativo, equilibrando a produção de ROS e das defesas antioxidantes.

36
Quando o equilíbrio é afetado, favorecendo a produção de ROS, ocorre o estresse
oxidativo, o que resulta no acúmulo de moléculas oxidativas que alteram a atividade
normal da célula (Bains & Shaw 1997, Tsang & Chung 2009). Todos os
componentes celulares são vulneráveis à ação das ROS, mas a membrana é um
dos mais atingidos em decorrência da peroxidação lipídica, que gera alterações
estruturais e na permeabilidade iônica. Além disso, a peroxidação lipídica pode ser
catalizada por íons ferro (Ferreira & Matsubara 1997, Rauhala et al. 1996). Os
neurônios são particularmente susceptíveis ao estresse oxidativo, pois podem
apresentar altas taxas de atividade metabólicas oxidativas e baixos níveis de
enzimas antioxidantes, o que pode resultar na morte celular (Bains & Shaw 1997,
Tsang & Chung 2009).
O desequilíbrio entre eventos oxidativos e as defesas antioxidantes pode
gerar estresse oxidativo que por sua vez pode induzir a morte neuronal. Dessa
forma, este desequilíbrio pode aumentar a produção de ROS e reduzir agentes
antioxidantes, como as moléculas de glutationa (GSH) (Tsang & Chung 2009).
Nesse sentido, um dos fatores propostos como mecanismo de perdas de células
nigroestriatais na DP é o estresse oxidativo neuronal (Beal 2003). Contudo, o
estresse oxidativo também é proposto como causa do processo de envelhecimento
normal (Cadenas & Davies 2000, Beal 2002), assim como em doenças
neurodegenerativas relacionadas ao envelhecimento, como a doença de Alzheimer
entre outras (Beal (2000, 2002)).
Alguns prováveis indicativos da relação entre o estresse oxidativo e a DP têm
sido relatados, como redução dos níveis de glutationa (GSH, agente óxido-redutor)
no mesencéfalo, indicando um aumento dos níveis de radicais livres, aumento de
teor de ferro na substância negra, propiciando reações de peroxidação lipídica e

37
alterações no complexo I da cadeia respiratória mitocondrial (Bains & Shaw 1997,
Tsang & Chung 2009). Além disso, o metabolismo de dopamina é uma fonte de ROS
nos neurônios nigroestriatais (Tsang & Chung 2009). O processo oxidativo da
dopamina é catalizado pela ação da enzima monoamina-oxidase (MAO). A
metabolização da dopamina produz quinonas e semi-quinonas que podem atuar
como oxidantes, sustentando a hipótese da formação da ROS (Tsang & Chung
2009). Alguns pesquisadores acreditam que o aumento da reciclagem (―turnover‖) da
dopamina está associado a eventos oxidativos na célula, através do aumento da
produção de peróxido de hidrogênio. O peróxido de hidrogênio é formado durante a
degradação da dopamina pela enzima monoamina oxidase (MAO) ou pela oxidação
do anel catecol (Ver figura 3). Um produto desta reação é o H2O2 (Ver figura 3) que
pode interagir com metais de transição (por exemplo, o ferro) e formar radicais
hidroxilas que causam danos em proteínas, lipídeos e no DNA celular. Essa reação
pode ser bloqueada por antioxidantes como a glutationa (Rabinovic & Hastings
1998). Além do H2O2 também é produzido o 3,4-dihidroxifenilacetaldeido (DOPAL)
(Ver figura 3) que em seguida passa por uma oxidação mediada pela ALDH2
(adeído desidrogenase mitocondrial) produzindo o ácido 3-4-dihidroxifenilacetico
(DOPAC) (Jinsmaa et al. 2009, Marchitti et al. 2010). Em estudos recentes foi
evidenciada a existência de enzimas que podem compensar a oxidação do DOPAL
podendo inibir o ALDH2 (Marchitti et al. 2010). Portanto, a peroxidação lipídica e os
pordutos desta que são 4HNE (4-hidroxi-2-nonenal) e MDA (malondialdeído) podem
prejudicar o catabolismo de dopamina celular via inibição do ALDH2, produzindo
nível elevados de aldeídos (Jinsmaa et al. 2009).

38
Figura 3: Formação de peróxido de hidrogênio durante a degradação da dopamina por uma reação de oxidação catalisada pela enzima monoamina oxidase. (Adaptado de Spina & Cohen 1989)
O MPTP, depois de ser convertido em MPP+ pela MAO B no cérebro, induz a
formação de radicais livres, como o radical hidroxila (OH-). A elevação do nível do
radical hidroxila leva a peroxidação lipídica que, como já foi explicado, pode levar à
morte celular. Assim sendo, alguns trabalhos têm relacionado o modelo do MPTP
como um método interessante de estudar a hipótese do estresse oxidativo e os
possíveis mecanismos patofisiológicos da DP (Obata 2002).
Alguns autores sugerem que as lesões dopaminérgicas nigroestriatais
induzidas pela 6-OHDA ocorrem pela geração de peróxido de hidrogênio e radicais
hidroxilas (Heikkila & Cohen 1971, Riobó et al. 2002).
A reserpina parece exercer um efeito sobre estes eventos oxidativos citados
acima. Resultados de estudos demonstraram que a reserpina induz uma queda dos
níveis de glutationa do estriado (Abílio et al. 2003, Teixeira et al. 2008), aumento da
peroxidação lipídica e da atividade de catalase do estriado (Abílio et al. 2002, Nade
et al. 2009) e de glutationa oxidada no estriado e no córtex pré-frontal (Spina &
Cohen 1989).
Dessa forma, podemos concluir que existem evidências dos danos do
estresse oxidativo em cérebro de pacientes com DP (Beal 2002), e em modelos
farmacológicos da doença, tais como o MPTP (Obata, 2002), a 6-OHDA (Riobó et al.
2002) e a reserpina (Bilska et al. 2007, Spina & Cohen 1989). Contudo, a relação
Dopamina
O
2
H2
O
H2O
2 NH
3
3,4-dihidroxifenilacetaldeido
(DOPAL)
+ + +
MAO
+

39
entre o estresse oxidativo e fatores relacionados à degeneração progressiva,
alterações motoras e cognitivas não estão esclarecidos.
Em face do exposto nesta introdução, propomos o estudo da administração
repetida de baixas doses de reserpina como um modelo de DP que possa abranger
as características comportamentais e bioquímicas citadas acima, em semelhança
aos sintomas observados em humanos afetados por esta patologia.
1.3. Justificativa
A maioria dos modelos utilizados para estudar os déficits cognitivos e motores
da DP é baseada em efeitos de administrações agudas de substâncias como 6-
OHDA e MPTP. Contudo, esses dois modelos acarretaram em perdas específicas e
imediatas de células do sistema nervoso (SN) além de um acentuado número de
mortes de animais, não apresentando um processo neurodegenerativo progressivo
(Ferro et al. 2005, Meredith et al. 2008).
Uma alternativa proposta nesse trabalho seria a utilização de um modelo
farmacológico crônico, administrando-se repetidamente baixas doses de reserpina,
que possibilitasse o aparecimento progressivo de sinais semelhantes aos sintomas
encontrados na DP. Estudos prévios com administração aguda de reserpina têm
mostrado alterações motoras (Skalisz et al. 2002, Aguiar Jr et al. 2009), mas
também um comprometimento cognitivo que ocorreria independentemente do
declínio motor, como quadros clínicos encontrados em humanos com DP (Carvalho
et al. 2006, Fernandes et al. 2008). Além disso, nesses estudos anteriores com

40
animais, a administração aguda subcutânea de reserpina (em doses que não afetam
a função motora) levou a alterações de memória que envolve contexto emocional
enquanto as sem conotação emocional não foram afetadas. Outro fator importante a
ser considerado é que além da dopamina, há evidências da participação de outros
sistemas de neurotransmissão, como o GABA e o glutamato na gênese dos
sintomas parkinsonianos, tanto em modelos animais quanto em humanos (Bezard et
al, 1997; Bianchi et al, 2003; Bonsi et al, 2007; DeLong e Wichmann, 2007). Existe
ainda o fator da alteração de mecanismos de neuroproteção a eventos oxidativos
induzidos pela reserpina (Bilska et al. 2007, Spina & Cohen 1989), os quais
corroboram os danos do estresse oxidativo em cérebro de pacientes com DP (Beal
2002), e em modelos farmacológicos como o MPTP (Obata 2002) e a 6-OHDA
(Riobó et al. 2002). Contudo, a relação entre o estresse oxidativo, o envolvimento de
sistemas de neurotransmissão não dopaminérgicos, fatores relacionados à
degeneração progressiva e alterações motoras e cognitivas na DP ainda não está
completamente esclarecida.
Tomados em conjunto, dados prévios sugerem que o efeito induzido pela
reserpina sobe a memória e parâmetros motores de ratos pode ser uma abordagem
adequada para o estudo dos sintomas cognitivos e motores da DP. Entretanto, até o
momento, tais estudos foram realizados apenas com a administração aguda de
reserpina. Assim sendo, propomos o estudo da administração repetida de baixas
doses de reserpina como um modelo farmacológico de DP que possa abranger as
características citadas acima, assemelhando aos sintomas observados em humanos
afetados por esta patologia.

41
1.4. Objetivo geral
No presente trabalho, propomos desenvolver um possível modelo
farmacológico em ratos que mimetize uma neurodegeneração progressiva
semelhante às encontradas em pacientes com DP, através da administração
repetida de baixas doses de reserpina.
1.4.1. Objetivos específicos
Avaliamos os efeitos da administração repetida de reserpina (em doses que
causariam pouco ou nenhum comprometimento motor agudamente) sobre:
1. O desempenho de ratos em modelos comportamentais de memória ao longo
do tratamento;
2. A atividade motora ao longo do tratamento;
3. Os níveis de GABA e glutamato em regiões cerebrais possivelmente
relacionadas com o surgimento de déficits cognitivos ou sintomas motores ao
final do tratamento;
4. Os níveis de peroxidação lipídica como indicativo de dano causado por
processos oxidativos decorrentes da administração repetida de reserpina.

42
2.Experimentos

43
2.1. Experimento I: Artigo científico que será submetido ao periódico Psychology &
Neuroscience
BEHAVIORAL AND NEUROCHEMICAL EFFECTS OF REPEATED ADMINISTRATION OF LOW DOSES OF
RESERPINE: A PROGRESSIVE MODEL FOR THE STUDY OF PARKINSON’S DISEASE?
Valéria S. Fernandes1, Anderson H.F.F. Leão1, Angela Maria Ribeiro2, Alessandra M. Ribeiro1, Regina H. Silva1,*
1Memory Studies Laboratory, Physiology Department, Federal University of Rio Grande do Norte, Natal, Brazil 2Departamento de Bioquímica e Imunologia, Laboratório de Neurociência Comportamental e Molecular – LaNeC. Universidade Federal de Minas Gerais, Brazil

44
Abstract
Parkinson's Disease (PD) has been studied in models that attempt to mimic the
neurophysiologic and behavioral changes found in the development of this disease.
However, in general, these protocols induce an immediate severe motor impairment,
similar to advanced stages of PD. The administration of reserpine (a monoamine
depletor) in rodents has been considered a model for studying PD. In this study,
repeated treatment with 0.1 and 0.2 mg/kg (but not 0.05 mg/kg) reserpine have
induced progressive motor alterations in rats when compared with the vehicle-treated
group, as shown by the evaluation of the catalepsy behavior across the treatment.
Additionaly, animals repeatedly treated with 0.1 mg/kg reserpine showed
concomitant memory impairment when tested in the plus-maze discriminative
avoidance task. At the end of the treatment (5 days after the 15th injection) striatal
GABA and gluatamate levels were determined. While no changes were observed for
the GABAergic system, a decrease in glutamate striatal concentration was found in
0.1 and 0.2 mg/kg reserpine-treated animals. Thus, repeated treatment with low
doses of reserpine appears to be promising as a model of PD, since it induces
progressive motor alterations. By the end of the treatment, these motor symptoms
were accompanied by cognitive and neurochemical changes. However, more studies
are needed to verify if the memory deficits and neurochemical alterations would
present a progressive profile as well.
Keywords:
Reserpine, Parkinson‘s Disease, cognition, GABA, glutamate, animal model

45
1. Introduction
Parkinson's disease (PD) is a progressive neurodegenerative disease in which
the ability to perform voluntary movements is gradually lost. The clinical condition of
PD includes rigidity, tremor and bradykinesia (slowness of movement) (Klockgether,
2004). However, cognitive changes can also be observed in patients with PD
(Aarsland et al., 2004; Mahieux et al., 1998; Verbaan et al., 2007).
Animal models have been used to study the changes caused by PD. Some
studies try to mimic the clinical features of PD in rodents in order to better understand
the neurophysiological mechanisms underlying the disorder (Meredith et al. 2008).
However, the the effectiveness of models regarding the progressive nature of the
"preclinical" and "clinical" stages of PD is controversial (Deumens et al., 2002).
Indeed, in recent decades some pharmacological animal models of PD have been
developed, with the most studied toxins being 6-hydroxydopamine (6-OHDA) and 1-
methyl 4-phenyl 1,2,3,6-tetrahydropyridine (MPTP) (Meredith et al., 2008; Schober,
2004). These two models have shown specific loss of cells related to PD in the
central nervous system (CNS), although not presenting a neurodegenerative
process, instating already an advanced stage of the disease upon administration
(Meredith et al., 2008).
The administration of reserpine (an irreversible blocker of monoamine
vesicular carrier) in rodents has been considered an animal model for the study of
PD (Colpaert, 1987; Kim et al., 1999; Alves et al., 2000; Silva et al., 2002; Skalisy et
al., 2002). Reserpine interferes with the storage of monoamines in intracellular
vesicles, causing monoamine depletion in nerve terminals and transient
hypolocomotion and muscular rigidity, depending on the dose (Colpaert, 1987).

46
Recently, lower doses of reserpine have also been found to promote a memory
deficit in an aversive discrimination task without any effects on motor activity,
suggesting that the administration of this drug in low doses can be useful to study
memory deficits found in PD (Carvalho et al., 2006). Similar results were found in a
different aversively motivated behavioral model, the contextual fear conditioning
(Fernandes et al., 2008). In summary, the data from the literature suggest that acute
reserpine is able to induce motor and cognitive alterations similar to those found in
PD patients, although in different dose ranges.
Although hypofunction of the dopaminergic nigrostriatal system is considered
to be the core of the physiopathology of PD, there is evidence that other
neurotransmitter systems are involved in the symptoms of the disease (Bezard et al.,
1997; Bianchi et al., 2003; Bonsi et al., 2007; DeLong & Wichmann, 2007). Studies
have suggested that the depletion of dopamine (DA) in the striatum consequently
leads to inhibition of the GABAergic striato-pallidal projections, as well as changes in
thalamo-nigral glutamatergic projections (Filloux & Townsend, 1993; DeLong &
Wichmann, 2007). Indeed, a study with rats treated with 6-OHDA showed that loss of
dopaminergic neurons in the forebrain induce increase on the GABA levels in
pallidum globe (Bianchi et al., 2003). Furthermore, studies with glutamatergic drugs
(in particular, group II mGluR agonists) show improvement of motor signs in mice
treated with MPTP (Bonsi et al., 2007). However, these studies in animals were
performed with acute pharmacological models, and have shown controversial results.
Further, studies with brain tissue from PD patients show altered levels of GABA in the
medial center thalamus (Gerlach et al., 1996). These studies indicate that the
relationship between dopaminergic, glutamatergic, GABAergic and behavioral
changes is somewhat complex, and still unclear.

47
Considering the importance of an animal model that simulates the progressive
nature of the disease, we evaluated the effects of a repeated treatment with low sub-
effective doses of reserpine on motor and cognitive behaviors. In addition, we also
addressed possible changes in GABAergic and glutamatergic systems as a
consequence of this treatment.
2. Methods
2.1. Subjects
Five-month old male Wistar rats (n= 29) were used. All animals were
maintained in groups of four or five per cage, under a 12 h light 12 h dark cycle and
at a constant temperature of 25 1 C, with food and water available ad libitum. The
rats were handled according to Brazilian law procedures for the use of animals in
scientific research (Law Number 11.794) and all procedures were approved by the
local research ethics committee (final opinion number 149/2008). All efforts were
made to minimize animal pain, suffering or discomfort, and to minimize the number of
rats used.
2.2. Drug treatment, general procedures and experimental design
Reserpine (methyl reserpate 3,4,5-trimetothoxycinnamic acid ester: Sigma
Chemical Co. St. Louis, MO) was dissolved in glacial acetic acid and diluted to the
correct concentration in distilled water. Vehicle consisted of the same amount of

48
acetic acid and water as in the reserpine solution. These solutions were injected
subcutaneously (s.c.).
Rats received 15 s.c. injections of vehicle (VEH; n=8), 0.05 mg/Kg (RES 0.05;
n=7), 0.1 mg/Kg (RES 0.1; n=7) or 0.2 mg/kg (RES 0.2; n=7) of reserpine, at a
volume of 1 ml/kg body weight, on alternate days. During treatment rats went through
the following procedures: (1) assessment of catalepsy behavior 24h after the 3rd, 6th,
9th, 12th and 15th injections; (2) plus-maze discriminative avoidance task 24h and 48 h
after the 10th injection; (3) assessment of orofacial movements 24 h after the 14th
injection; (4) Contextual fear conditioning 48 and 72 h after the 15th injection; (5)
evaluation of GABAergic and glutamatergic parameters in the striatum 5 days after
the 15th injection (Figure 1).
Figure 1: Experimental design
Every rat was submitted to 10 min of gentle handling once a day for five days
before the beginning of the experimental procedures. The analyses of catalepsy

49
behavior and orofacial movements were performed by direct observation (by
researchers blind to the treatment). All other behavioral sessions were recorded by a
camera placed above the apparatus and the behavioral parameters were registered
by an animal video-tracking software (Any maze Stoelting, USA).
All apparatus were washed with a water–alcohol (5%) solution before
behavioral testing to eliminate possible bias due to odors left by previous subjects.
2.3. Apparatus
2.3.1. Catalepsy Test:
The catalepsy was assessed placing the animal‘s front paws on a horizontal
bar positioned at 9 cm above the bench surface. The duration of catalepsy, which
was defined as an immobile posture, keeping the two front paws on the bar, was
measured within a maximum of 180 s.
2.3.2. Plus-maze discriminative avoidance task:
The apparatus employed was a modified elevated plus-maze made of wood
containing two enclosed arms (50 X 15 X 40 cm) opposite to two open arms (50 X 15
cm). A 100-watt lamp was placed over the middle of one of the enclosed arms
(aversive enclosed arm). In the training session, each rat was placed in the centre of
the apparatus and, over a period of 10 min, every time the animal entered the
enclosed arm containing the lamp, an aversive situation was produced until the
animal left the arm. The aversive stimuli were the 100-watt light and an 80 dB noise

50
applied through a speaker placed over the aversive enclosed arm. In the test
session, held 24h later, the rats were again placed in the apparatus for 10 min,
without receiving the aversive stimulation, with the lamp and the speaker still present
over the aversive arm, but turned off. Distance traveled in the apparatus (used for
motor activity evaluation) and time spent in each arm (aversive, non-aversive and
open arms) were registered. Percent time in aversive arm (time spent in aversive
enclosed arm/time spent in both enclosed arms) and percent time spent in open
arms (time spent in open arms/time spent in both open and enclosed arms)
considering the whole duration of behavioral sessions were used to evaluate memory
and anxiety, respectively (Silva et al., 2000). Percent time spent in the aversive
enclosed arm assessed minute by minute across the training and test sessions were
used to evaluate learning and extinction of the task, respectively (Ribeiro et al.,
2010).
2.3.3. Orofacial movements assessment:
Rats were placed individually in wired cages (29 cm × 24 cm × 21 cm) with
mirrors positioned under the floor and behind the back wall of the cage to allow
behavioral quantification when the animal faced away from the observer. The number
of tongue protrusions (projection of the tongue out of the oral cavity), vacuous
chewing movement frequency (mouth openings in the vertical plane not directed
toward physical material), and facial twitching (duration (in seconds) of twitching of
the facial musculature) were measured continuously for 15 min.

51
2.4. Biochemical analysis: Evaluation of GABA and glutamate levels:
After the animals were sacrificed by decapitation, the brains were quickly
removed from the cranial cavity, weighed and dissected according to the stereotactic
coordinates provided by Paxinos & Watson (Paxinos & Watson 1997). The sample of
striatum was then stored at -80 ° C to achieve the biochemical assays.
Samples of striatum were weighed and homogenized in 15 volumes of
methanol: water (85:15 v / v) in automatic homogenizing. Then the homogenate was
centrifuged at 4 ° C for 15 minutes at 7800x g (Sorvall RC-5B). The supernatant
obtained after centrifugation was collected and kept on ice until subjected to
derivatization.
Due to the absence of electroactive or fluorescent characteristics inherent in
the amino acid glutamate and GABA, several works have used the technique of pre-
column derivatization for the chromatographic separation and identification of these
compounds. One of the most widely used derivatising agents is o-phthalaldehyde
(OPA), which reacts with primary amines in the presence of thiol and generates
electroactive and fluorescent derivatives (Freitas et al. 2009). The derivatization
reaction was made by mixing 100 mL of sample, 20 mL of methanolic OPA (5 mg /
mL) prepared daily, 75 mL of borate buffer (pH 9.9) and 5 mL of 3-
mercaptopropiônico acid (MPA). The resulting solution was stirred and injected into
the chromatographic system after 1 minute at room temperature.
The chromatographic system used to determine of GABA and glutamate
consisted of a Shimadzu chromatograph (LC-10AD, Tokyo, Japan) with 200 mL
injector valve (Rheodyne 7725-I, California, USA) and fluorescence detector (FLD-
Shimadzu spectrofluorometric detector RF-551, Tokyo, Japan) coupled to a pump

52
LC-10. The wavelengths of excitation and emission used were 337 nm and 454 nm,
respectively. A reversed phase chromatographic column C18 (150 mm x 4.6 mm ID)
and a guard column (E. Merck RT 250-4, ER Darmsdt, Germany) were used in the
analysis. The isocratic mobile phase consisted of a 0.05 M solution of sodium
acetate, tetrahydrofuran and methanol (50:1:49 v / v), pH 4.0. The mean elution of
GABA and glutamate is 8.0 to 3.0 minutes, respectively, the concentrations in µg / g
of tissue were calculated using peak areas and their standard curves which was
provided by an integrator (R7Ae Shimadzu C-plus) coupled to the chromatographic
system (Freitas et al., 2009).
2.5. Statistics
All data were tested for homogeneity of variances (Levene's test) and
normality (Kolmogorov-Smirnov test) and parametric tests were performed for all
data. Data on the percentage of time spent in the aversive arm (measured every
minute, in training and test sessions) and catalepsy behavior across the treatment
(24h after the 3rd, 6th, 9th, 12th and 15th injections) were analyzed by analysis of
variance (ANOVA) with repeated measures. For catalepsy behavior analysis,
between-subject comparisons were held in each timepoint with one-way ANOVA with
sequential Bonferroni‘s post hoc. Other data were analyzed by one-way ANOVA
followed by Duncan's test for post hoc analysis. The results were expressed as
means ± SE. The level of significance in all tests was p <0.05.

53
3. Results
3.1- Effects of repeated administration of low doses of reserpine on the
catalepsy behavior:
Catelpsy behavior results are shown in figure 2. ANOVA with repeated
measures revealed time (quantity of injections) [F(5, 125)=22.36; p=0.000],
treatment (reserpine (0.05, 0.1 or 0.2 mg/Kg) or vehicle) [F(3, 25)=16.92; p=0.000]
and time X treatment interaction [F(5, 125)=7.54; p=0.000] effects. Analyzing the
data of each observation, we found no significant effects of treatment until the
observation held after the 6th injection, although treatment with 0,2 mg/kg reserpine
induced a marginally significant effect after six injections (p=0.068 compared to
vehicle). Significant effects of treatment were observed after the 9th, 12th and 15th
subcutaneous injections [F (3, 25) = 8.32, p = 0.001; F (3, 25) = 11.22, p = 0.000; F
(3, 25) = 8.28, p = 0.000, respectively]. The Bonferroni‘s post hoc analysis showed
that RES 0.1 and RES 0.2 groups presented increased immobility time in the bar
when compared to the VEH and RES 0.05 groups after the 9th, 12th and 15th
injections.

54
Fig. 2: Effects of repeated administration of low doses of reserpine (RES- 0.05, 0.1 or
0.2 mg/Kg) or vehicle (VEH) on catalepsy behavior before the beginning (basal) and
24h hours after the 3rd, 6th, 9th, 12th and 15th injections. Data are expressed as
mean±S.E.M. (s). ANOVA with repeated measures revealed time, treatment and time
x treatment interaction effects. *p<0.05 compared to VEH and RES 0.05 group
(ANOVA and Bonferroni‘s test).
3.2- Effects of repeated administration of low doses of reserpine on plus-maze
discriminative avoidance task
In the training session, we observed a significant effect of treatment in the
distance traveled in the apparatus (Figure 3 A; ANOVA, F (3, 25) = 15.98, p = 0.000).
The post hoc analysis (Duncan‘s test) revealed that RES 0.1 and 0.2 groups showed
decreased motor activity when compared to groups VEH and RES 0.05 in the
training session (24 h after administration of the 10th injection). In the test session, 48
0
20
40
60
80
100
0 3rd 6th 9th 12th 15th
Ca
tale
ps
y d
ura
tio
n (
s)
VEH RES 0.05 RES 0.1 RES 0.2

55
h after administration of the 10th injection, there was no significant difference between
groups in the distance traveled (Fig. 3 B; ANOVA, F (3, 25) = 0.51, p = 0.681).
Fig. 3: Effects of repeated administration of low doses of reserpine (RES - 0.05, 0.1
or 0.2 mg/Kg) or vehicle (VEH) on the plus-maze discriminative avoidance apparatus
during training (A) and test (B) sessions performed 24 and 48 h after the 10th
injection, respectively. * p <0.05 compared with vehicle and RES 0.05 group (ANOVA
and Duncan‘s test)
In the training session (24h after the 10th injection), we found no significant
differences in the percentage of time in the open arms (%TO) (Figure 4 A; ANOVA, F
(3, 25) = 0.39, p = 0.761). However, in the test (48h after the 10th injection), a
significant effect of treatment was found for %TO. The post hoc analysis (Duncan
test) revealed that RES 0.2 group showed a significant increase in the percentage of
time in open arms in the test session when compared with all other groups (Fig. 4 B,
ANOVA, F (3, 25) = 8.99, p = 0.000).

56
Fig. 4: Effects of repeated administration of low doses of reserpine (RES - 0.05, 0.1
or 0.2 mg/Kg) or vehicle (VEH) on percent time in the open arms (%TO) of the plus-
maze discriminative avoidance apparatus during training (A) and test (B) sessions,
performed 24 and 48 h after the 10th injection, respectively. * p <0.05 compared with
all other groups (ANOVA and Duncan‘s test)
Regarding the percent time in the aversive arm, we found no significant effects
when the whole training session duration was considered for analysis (Figure 5 A,
ANOVA, F (3, 25) = 1.99, p = 0. 142). When the same analysis was applied to the
test session, we found a significant effect of treatment for percentage of time in the
aversive arm. The post hoc analysis (Duncan test) showed that RES 0.1 group
showed increased aversive arm exploration when compared with groups VEH and
RES 0.05 (Figure 5 B, ANOVA, F (3, 25) = 3.25, p = 0. 039).
In the training session, significant effects of time (minutes) [F (9, 225)= 8.41;
p=0.000] and the time X treatment interaction [F (9, 225)= 2.38; p=0.019] were found
when the percentage of time in the aversive enclosed arm (%TAV) was evaluated
across the session. No effect of the treatment (RES (0.05, 0.1 or 0.2 mg/Kg) or VEH)
[F (3, 25)= 2.75; p=0.064] was found (see Fig 5 C).

57
In the test session, a significant effect of the time X treatment interaction [F (9,
225)= 2.04; p=0.029] was found when the percentage of time in the aversive
enclosed arm (%TAV) was evaluated across the session. No effects of time (minutes)
[F (9,225)= 1.38; p=0.246] or treatment (RES (0.05, 0.1 or 0.2 mg/Kg) or VEH) [F
(3,25)= 0.96; p=0.429] were found (see Fig 5 D).

58
Fig. 5: Effects of repeated administration of low doses of reserpine (RES - 0.05, 0.1
or 0.2 mg/Kg) or vehicle (VEH) on the percent time spent in the aversive arm (%TAV)
of the discriminative avoidance apparatus. Data are expressed as the mean±S.E.M.
for the whole training (A) and test (B) sessions and minute by minute during training

59
(C) and test (D) sessions, performed 24 and 48 h after the 10th injection, respectively.
ANOVA with repeated measures revealed time (minutes) effect, in training session,
and time x treatment interaction effects, in training and test sessions (C and D)* p
<0.05 compared with vehicle and RES 0.05 group (ANOVA and Duncan‘s test)
3.3- Effects of repeated administration of low doses of reserpine on orofacial
movements
The analysis of the orofacial movements, held 24 h after the 14th injection,
showed significant differences between groups for the number of vacuous chewing
movements (Figure 6 A, F (3, 25) = 60.46, p = 0.000), the duration of facial twitching
(Figure 6 B, F (3, 25) = 40.99, p = 0.000) and the number of tongue protrusions
(Figure 6 C, F (3, 25) = 13.34, p = 0.000). The post hoc analysis (Duncan‘s test)
showed that, in all orofacial movement measures, RES 0.1 and RES 0.2 groups
showed significantly increased values when compared with the VEH and RES 0.05
groups. In addition, RES 0.2 group presented increased duration of facial twitching
when compared to RES 0.1 animals.

60

61
Figure 6: Effects of repeated administration of low doses of reserpine (RES - 0.05,
0.1 or 0.2 mg/Kg) or vehicle (VEH) on orofacial movements 24 h after the 14th
subcutaneous injection. Data are expressed as the mean±S.E.M. of the number of
vacuous chewing movements (A), the duration of facial twitching (B) and the number
of tongue protrusions (C). * p <0.05 compared with vehicle group and RES 0.05
group, # p <0.05 compared with RES 0.1 group (ANOVA and Duncan‘s test).
3.4- Effects of repeated administration of low doses of reserpine on GABA and
glutamate striatal levels
Significant differences were observed between groups for concentrations (µg/g)
of glutamate in the striatum, 5 days after the administration of 15 subcutaneous
injections (Figure 7 A; ANOVA, F (3, 25) = 4.72, p = 0.010). The post hoc analysis
(Duncan test) showed that reserpine RES 0.1 and 0.2 groups showed decreased
glutamate striatal levels when compared with the VEH and RES 0.05 groups.
However, we found no significant difference between groups of concentrations in
µg/g of GABA was observed (Figure 7 B, ANOVA, F (3, 25) = 0.88, p = 0.466) in the
striatum, after the administration of 15 subcutaneous injections.

62
Figure 7: Effects of repeated administration of low doses of reserpine (RES - 0.05,
0.1 or 0.2 mg/Kg) or vehicle (VEH) on the concentration of glutamate or GABA in
striatum 5 days after the 15th injection. Data are expressed as the mean±S.E.M. of
the concentrations of glutamate (A) and GABA (B) present in striatum samples. *p
<0.05 compared with vehicle and RES 0.05 groups (ANOVA and Duncan‘s test).

63
4. Discussion
This study evaluated the effect of repeated treatment with low doses of
reserpine on motor and cognitive parameters. The results revealed that repeated
treatment with 0.05 mg/kg of reserpine did not change the performance of cognitive
and motor tasks or in the concentrations of glutamate and GABA in the striatum. On
the other hand, the results obtained by repeated treatment with higher doses of
reserpine (RES 0.1 and 0.2 mg / kg) showed changes in the performance of animals
on contextual fear conditioning and plus-maze discriminative avoidance behavior.
More importantly, repeated treatment with 0.1 and 0.2 mg/kg of reserpine showed a
progressive onset of motor alterations, as shown by increased on catalepsy behavior
from the 9th injection onwards (Fig. 2), decreased distance traveled in the
discriminative avoidance apparatus 24 h after the 10th injection (Fig. 3A) and
increase in orofacial movements 24 h after the 14th injection (Fig 6 A, B and C). We
also found a significant decrease of striatal glutamate in levels in both groups RES
0.1 and 0.2 five days after the 15th injection (Fig. 7A).
The motor symptoms are characteristic of the clinical presentation of PD and
are associated with the neurodegenerative process of the basal ganglia (Klockgether,
2004; Johnston et al., 1999; Lindner et al., 1999; Mayeux, 2003, Ridley et al., 2006).
Studies in rodents with drugs that change the dopaminergic system (haloperidol,
MPTP, 6-OHDA, reserpine) induces the appearance of signs of hypokinesia and
rigidity (catalepsy) similar to Parkinsonian symptoms (Shiozaki et al., 1999; Díaz et
al., 2001; Góngora-Alfaro et al., 2009; Corona et al., 2010). Additionaly, the presence
of hypokinesia can be addressed by the evaluation of locomotor activity after these
pharmacological treatments (Carvalho et al., 2006; Díaz et al., 2001; Capitelli et al.,
2008). As mentioned above, the present study revealed that rats exposed to a

64
repeated administration of 0.1 and 0.2 mg/kg reserpine showed both kinds of
alterations (increased catalepsy behavior - Fig. 2; decreased distance traveled in the
plus-maze discriminative avoidance apparatus- Fig. 3A). Previous studies have
demonstrated these effects of reserpine on catalepsy behavior and locomotor
activity. However, in these studies, one or two injections of considerably higher doses
(1.0 or 5.0 mg / kg) were administred (Shiozaki et al., 1999; Dutra et al., 2002) to
produce catalepsy and hipolocomotion. Therefore, the results found in the present
work suggest that the alterations observed during treatment could indicate
progressive features of those signs.
The evaluation of orofacial movements has been used in animal studies of
tardive dyskinesia, using reserpine to induce the appearance of oral dyskinesia
(Neisewander et al., 1991; Neisewander et al., 1994; Abílio et al., 2003; Abílio et al.,
2004). The tardive dyskinesia is characterized by severe motor symptoms where the
face, mouth and tongue are often involved (called orofacial dyskinesia) and can
manifest as a side effect of neuroleptics both in humans and in animals (Hansen et
al., 1997; Andreassen et al., 2000). On the other hand, some researchers apply the
evaluation of orofacial movements as a model of parkinsonism, especially of tremor-
related symptoms (Salamone et al., 1996; Salamone et al., 2008). In this study, we
observed that after administration of the 14th injection, groups RES 0.1 and 0.2 (but
not RES 0.05) showed an increase in orofacial movements (Fig.6 A, B and C).
Studies have shown that changes in the ventrolateral striatal dopamine system (Jicha
et al., 1991) or changes in striatal levels of oxidative stress (Abílio et al., 2003; Abílio
et al., 2004) may be related to the increase of orofacial movements. Both changes in
striatal dopaminergic system as changes in levels of oxidative stress in brain are
factors related to PD (Nieoullon et al., 2002; Beal, 2003). In addition, there is

65
evidence that orofacial movements alterations co-exist with other kinds of motor
changes, including parkinsonism (Harten et al., 1997). On the other hand, a study of
specific dopamine lesions in the ventrolateral striatum in rats showed oral motor
disorders in the absence of locomotor deficits (Jicha et al., 1991). Thus, different
changes in the dopaminergic system can cause various motor signs showing that it is
unclear which is the neurochemical mechanism underlying the simultaneous onset of
orofacial dyskinesia and other motor alterations.
Besides evaluating memory, we used the plus maze discriminative avoidance
task (performed after the 10th injection) to assess learning, memory, anxiety and
motor behavior, since it has been shown that these evaluations can be performed
concomitantly, and by different parameters, in this paradigm (Silva et al., 2002; Silva
et al., 2000). In the training session, we observed that RES 0.1 and 0.2 groups
showed a decrease in distance traveled in the maze (Fig. 3A). The possibility that
this motor impairment could have interfered with the acquisition of the task should be
considered, since the animals have to enter the aversive arm during training to learn
the assossiation with the aversive stimuli. However, the RES 0.1 animals did
explored the aversive enclosed arm in the first three minutes of the session,
gradually avoing this arm across the training, indicating they have learned the task
(as shown by the evaluation of behavior minute by minute throughout the training
session in Fig 5C). On the other hand, the animals from RES 0.2 group almost did
not enter the aversive arm during the training session. Thus, these results suggest
that the RES 0.2 group had an impairment in the acquisition of the task due to the
motor deficit.
The evaluation of the data obtained in the test session has shown two altered
parameters as a consequence of reserpine repeated treatment. First, animals treated

66
with 0.2 mg/kg reserpine presented increased percent time in the open arms (Fig
4B). This result would usually indicate changes in anxiety-like behavior. However, the
severe motor impairment presented by this group at this point of the treatment could
be the reason of this increase. Indeed, the observation of individual data of this
parameter (data not shown) revealed that most animals minimally advanced from the
central platform to one of the open arms and remained there. The other alteration
induced by reserpine treatment was the increase in percent time in the aversive
enclosed arm, specifically at 0.1 mg/kg (Fig. 5C). In this respect, we found that these
animals showed motor deficits during the acquisition of the task, which could
jeopardize an interpretation related to the cognitive aspect of animal behavior in the
test session. However, as mentioned before, the animals in this group did learn the
task, so the increased aversive arm exploration in the test session is probably
reflects a retrieval deficit. Nevertheless, it would be interesting to verify if reserpine
would induce cognitive impairments earlier during treatment, when the motor
impairment would be absent or mild.
One of the possible consequences of the neurodegeneration of the
nigrostriatal pathway in PD would be the modulation in the neural circuitry of the
basal ganglia, through alterations of the GABAergic and glutamatergic systems
(Blandini et al., 2000; Ossowska et al., 2002). Thus, we investigated the levels of
glutamate and GABA in the striatum region and found a significant decrease in the
concentration of glutamate groups RES 0.1 and 0.2. This result corroborates the
work of Day et al (2006) that showed that in a period of 5 days after initiation of
dopamine depletion, there were significant losses striatopallidal glutamatergic
synapses. Moreover, the loss of glutamatergic synapses after depletion of dopamine
has already been described in previous studies in animal models and in patients with

67
Parkinson's disease (McNeill et al., 1988; Ingham et al., 1998; Dunah et al., 2000).
On the other hand, no alterations were found in GABA striatal levels after treatment,
corroborating reports that show absence of alterations in the striatal GABAergic
system in humans with PD (Gerlach et al., 1996).
In summary, we observed motor alterations during repeated treatment with 0.1
and 0.2 mg/kg reserpine consistent with a progressive development of motor
impairment. The RES 0.1 group showed a concomitant cognitive impairment,
although the possible progressive nature of this alteration remains to be investigated.
Finally, motor changes (observed in tests of catalepsy) were accompanied by a
decreased level of glutamate (but not GABA) in the striatum, which is consistent with
neurochemical changes previously shown in animal models and in patients with PD
(see above). Thus, although further investigation is undoubtedly necessary, repeated
treatment with low doses of reserpine appears to be a promising animal model to
study the progressive motor changes of PD.
5. Acknowledgments
The authors would like to thank Ana Raquel Borges Pereira Caixeta, Fabio
Antonio Vigil and Patricia da Silva Oliveira (LaNeC/UFMG) for methodological
assistance and Dra. Vanessa C. Abílio (LiNC/UFSP) for helpful suggestions. This
research was supported by fellowships from Conselho Nacional de Desenvolvimento
Científico e Tecnológico (CNPq) and Coordenação de Aperfeiçoamento de Pessoal
de Nível Superior (CAPES).

68
6. References
Aarsland, D., Andersen K., Larsen, J.P., Perry, R., Wentzel-Larsen, T., Lolk, A. & Kragh-Sorensen, P. (2004). The Rate of Cognitive Decline in Parkinson Disease. Arch Neurol, 61, 1906-1911.
Abílio, V.C., Araujo, C.C.S., Bergamo, M., Calvente, P.R.V. & D‘Almeida, V. de A.R.R. (2003). Vitamin E attenuates reserpine-induced oral dyskinesia and striatal gssg/gsh ratio enhancement in rats. Prog Neuro- Psychopharmacol Biol Psychiat, 27,109–14.
Abílio, V.C., Silva, R.H., Carvalho, R.C., Grassl, C., Calzavara, M.B. & Registro, S. (2004). Important role of striatal catalase in aging- and reserpine-induced oral dyskinesia. Neuropharmacology, 47, 263–72.
Alves, C.S.D, Andreatini, R., da Cunha, C., Tufik, S. & Vital, M.A.B.F. (2000). Phosphatidylserine reserves reserpine-induced amnesia. European Journal of Pharmacology, 404, 161-167.
Andreassen, O.A. & Jorgensen, H.A. (2000). Neurotoxicity associated with neuroleptic-induced oral dyskinesias in rats Implications for tardive dyskinesia? Progress in Neurobiology, 61, 525-541.
Beal, M.F. (2003). Mitochondria, Oxidative damage, and Inflammation in Parkinson's disease. Ann. N. Y. Acad. Sci., 991, 120-131.
Bezard, E., Boraud, T., Bioulac, B. & Gross, C.E. (1997). Compensatory effectsof glutamatergic inputs to the substantia nigra pars compacta in experimental parkinsonism. Neuroscience, 81, 399–404.
Bianchi, L., Galeffi, F., Bolam, J.P. & Corte, D.L. (2003). The effect of 6-hydroxydopamine lesions on the relase of amino acids in the direct and indirect pathways of the basal ganglia: a dual microdialysis probe analysis. European Journal of Neuroscience, 18, 856-868.
Blandini, F., Nappi, G., Tassorelli, C. & Martignoni, E. (2000). Functional changes of the basal ganglia circuitry in Parkinson's disease. Progress in Neurobiology, 62, 63-88.
Bonsi, P., Cuomo, D., Picconi, B., Sciamanna, G., Tscherter, A., Tolu, M., Bernardi, G., Calabresi, P. & Pisani, A. (2007). Striatal metabotropic glutamate receptors as a target for pharmacotherapy in Parkinson‘s disease. Amino Acids, 32, 189–195.
Capitelli, C., Sereniki, A., Lima, M.M.S., Reksidler, A.B., Tufik, S. & Vital, M.A.B.F. (2008). Melatonin attenuates tyrosine hydroxylase loss and hypolocomotion in MPTP-lesioned rats. European Journal of Pharmacology, 594, 101–108.
Carvalho, R.C., Patti, C.C., Takatsu-Coleman, A.L., Kameda, S.R., Souza, C.F., Garcez-do-Carmo, L. & Abílio, V.C. (2006). Effects of reserpina on the pluz-maze discriminative avoidance task: Dissociation between memory and motor impairments. Brain Research, 1122, 176-183.
Colpaert, F.C. (1987). Pharmacological characteristics of tremor, rigidity and hypokinesia induced by reserpine in rat. Neuropharmacology, 26, 1431-1440.
Corona, J.C., Gimenez-Cassina, A., Lim, F. & Díaz-Nido, J. (2010). Hexokinase II Gene Transfer Protects against Neurodegeneration in the Rotenone and MPTP Mouse Models of Parkinson‘s Disease. Journal of Neuroscience Research, 88, 1943–1950.

69
Day, M., Wang, Z., Ding, J., An, X., Ingham, C., Shering, A.A., Wokosin, F.D., Ilijic, E., Sun, Z., Sampson, A.R., Mugnaini, E., Deutch, A., Sesack, Y.S. R., WArbuthnott, G. & Surmeier, D.J. (2006). Selective elimination of glutamatergic synapses on striatopallidal neurons in Parkinson disease models. Nature Neuroscience, 9, 251- 259.
DeLong, M.R. & Wichmann, T. (2007). Circuits and Circuit Disorders of the Basal Ganglia. Arch Neurol., 64, 20-24.
Deumens, R., Blokland, A. & Prickaerts, J. (2002). Modeling Parkinson‘s Disease in Rats: An Evaluation of 6-OHDA Lesions of the Nigrostriatal Pathway. Experimental Neurology, 175, 303–317.
Díaz, M.R., Abdala, P., Barroso-Chinea, P., Obeso, J. & Gonzalez-Hernande, T. (2001). Motor behavioural changes after intracerebroventricular injection of 6-hydroxydopamine in the rat: an animal model of Parkinson‘s disease. Behavioural Brain Research, 122, 79–92.
Dunah, A.W., Wang, Y., Yasuda, R.P., Kameyama, K., Huganir, R.L., Wolfe, B.B. & Standaert, D.G. (2000). Alterations in Subunit Expression, Composition, and Phosphorylation of Striatal N-Methyl-D-Aspartate Glutamate Receptors in a Rat 6-Hydroxydopamine Model of Parkinson‘s Disease. Molecular Pharmacology, 57, 342–352.
Dutra, R.C., Andreazza, A.P., Andreatini, R., Tufik, S. & Vital, M.A.B.F. (2002). Behavioral effects of MK-801 on reserpine-treated mice. Progress in Neuro-Psychopharmacology & Biological Psychiatry, 26, 487– 495.
Fernandes, V.S., Ribeiro, A.M., Melo, T.G., Godinho, M., Barbosa, F.F., Medeiros, D.S., Munguba, H. & Silva, R.H. (2008). Memory impairment induced by low doses of reserpine in rats: Possible relationship with emotional processing deficits in Parkinson disease. Progress in Neuro-Psychopharmacology & Biological Psychiatry, 32, 1479–1483.
Filloux, F. & Townsend, J.J. (1993). Pre-and Postsynaptic Neurotoxic effects of dopamine demonstrated by Intrastriatal injection. Experimental Neurology, 119, 79-88.
Freitas Silva, D.M., Ferraz, V.P. & Ribeiro, A.M. (2009). Improved high-performance liquid chromatographic method for GABA and glutamate determination in regions of the rodent brain. Journal of Neuroscience Methods, 177, 289–293.
Gerlach, M., Gsell, W., Kornhuber, J., Jellinger, K., Krieger, V., Pantucek, F., Vock, R. & Riederer, P. (1996). A post mortem study on neurochemical markers of dopaminergic, GABA-ergic and glutamatergic neurons in basal ganglia-thalamocortical circuits in Parkinson syndrome. Brain Research, 741, 142-152.
Góngora-Alfaro, J.L., Moo-Puc, R.E., Villanueva-Toledo, J.R., Alvarez-Cervera F.J., Bata-García, J.L., Heredia-López, F.J. & Pineda, J.C. (2009). Long-lasting resistance to haloperidol-induced catalepsy in male rats chronically treated with caffeine. Neuroscience Letters, 463, 210–214.
Hansen, E., Casey, D.E. & Hoffma, W.F. (1997). Neuroleptic Intolerance. Schizophrenia Bulletin, 23, 567- 582.
Harten, P.N.van, Hoek, H.W., Matroos, G.E., Koeter, M. & Kahn, R.S. (1997). The inter-relationships of tardive dyskinesia, parkinsonism, akathisia and tardive dystonia: the Curaçao Extrapyramidal Syndromes Study II. Schizophrenia Research, 2, 235-242.
Ingham, C.A., Hood, S.H., Taggart, P. & Arbuthnott, G.W. (1998). Plasticity of Synapses in the Rat Neostriatum after Unilateral Lesion of the Nigrostriatal Dopaminergic Pathway. The Journal of Neuroscience, 18, 4732–4743.

70
Jicha, G.A. & Salamone, J.D. (1991). Vacuous Jaw Movements and Feeding Deficits in Rats with Ventrolateral Striatal Dopamine Depletion: Possible Relation to Parkinsonian Symptoms. The Journal of Neuroscience, 12, 3822-3929.
Johnston, R.E., Schallert, T. & Becker, J.B.B. (1999). Akinesia and postural abnormality after unilateral dopamine depletion. Behavioural Brain Research, 104, 189-196.
Kim, H., Gyu-Seek, R., Oh, S. & Park, W. (1999). NMDA receptor antagonists inhibit apomorphine-induced climbing behavior not only in intact mice but also in reserpine-treated mice. Behevioural Brain Research, 100, 135-142.
Klockgether, T. (2004) Parkinson’s disease: clinical aspects. Cell Tissue Res, 318, 115–120.
Lindner, M.D., Cain, C.K., Plone, M.A., Frydel, B.R., Blaney, T.J., Emerich, D.F. & Hoane, M.R. (1999). Incomplete nigrostriatal dopaminergic cell loss and partial reductions in striatal dopamine produce akinesia, rigidity, tremor and cognitive deficits in middle-aged rats. Behavioural Brain Research, 102, 1-16.
Mahieux, F., Fénelon, G., Flahault, A., Manifacier, M.J., Michelet, D. & Boller, F. (1998). Neuropsychological prediction of dementia in Parkinson‘s disease. J Neurol Neurosurg Psychiatry, 64, 178–183.
Mayeux R. (2003). Epidemiology of neurodegeneration. Annu. Rev. Neurosci., 26, 81–104.
McNeill, T.H., Brown, S.A., Rafols, J.A. & Shoulson, I. (1988). Atrophy of medium spiny I striatal dendrites in advanced Parkinson's disease. Brain Res., 455,148-52.
Meredith, G.E., Totterdell, S., Potashkin, J.A. & Surmeier, D.J. (2008). Modeling PD pathogenesis in mice: Advantages of a chronic MPTP protocol. Parkinsonism and Related Disorders, 14, 112-115.
Neisewander, J.L., Castañeda, E. & Davis, D.A. (1994). Dose-dependent differences in the development of reserpine-induced oral dyskinesia in rats: support for a model of tardive dyskinesia. Psychopharmacology, 116, 79-84.
Neisewander, J.L., Lucki, I. & McGonigle, P. (1991). Neurochemical changes associated with the persistence of spontaneous oral dyskinesia in rats following chronic reserpine treatment. Brain Research, 558, 27-35.
Nieoullon, A. (2002). Dopamine and the regulation of cognition and attention. Progress in Neurobiology, 67, 53–83.
Ossowska, K., Konieczny, J., Wardas, J., Gol ´embiowska, K., Wolfarth, S. & Pilc, A. (2002). The role of striatal metabotropic glutamate receptors in Parkinson‘s disease. Amino Acids, 23, 193–198.
Paxinos, G. & Watson, C. (1997). The rat brain in stereotaxic coordinates. San Diego: Academic Press.
Ribeiro, A.M., Barbosa, F.F., Godinho, M.R., Fernandes, V.S., Munguba, H., Melo, T.G., Barbosa, M.T., Eufrasio, R.A., Cabral, A., Izídio, G.S. & Silva, R.H. (2010). Sex differences in aversive memory in rats: Possible role of extinction and reactive emotional factors. Brain and Cognition, 74, 145–151.
Ridley, R.M., Cummings, R.M., Leow-Dyke, A. & Baker, H.F. (2006). Neglect of memory after dopaminergic lesions in monkeys. Behavioural Brain Research, 166, 253-262.
Salamone, J. & Baskin, P. (1996). Vacuous Jaw Movements Induced by Acute Reserpine and Low-Dose Apomorphine: Possible Model of Parkinsonian Tremor. Pharmacology Biochemistry and Behavior, 53, 179-183.

71
Salamone, J.D., Ishiwari, K., Betz, A.J., Farrar, A.M., Mingote, S.M., Font, L., Hockemeyer, J., Muller, C.E. & Correa, M. (2008). Dopamine/adenosine interactions related to locomotion and tremor in animal models: Possible relevance to parkinsonism. Parkinsonism and Related Disorders, 14, S130-S134.
Schober, A. (2004). Classic toxin-induced animal models of Parkinson’s disease: 6-OHDA and MPTP. Cell Tissue Res, 318, 215–224.
Shiozaki, S., Ichikawa, S., Kitamura ,J.N.S., Yamada K. & Kuwana, Y. (1999). Actions of adenosine A2A receptor antagonist KW-6002 on drug-induced catalepsy and hypokinesia caused by reserpine or MPTP. Psychopharmacology, 147, 90–95.
Silva, R.H., Abílio, V.C., Torres-Leite, D., Bergamo, M., Chinen, C.C., Claro, F.T., Carvalho, R.C. & Frussa-Filho, R. (2002). Concomitant development of oral dyskinesia and memory deficits in reserpine-treated male and female mice. Behavioural Brain Research, 132, 171–177.
Silva, R.H. & Frussa-Filho, R. (2000). The plus-maze discriminative avoidance task: a new model to study memory–anxiety interactions. Effects of chlordiazepoxide and caffeine. J Neurosci Methods, 102, 117–25.
Skalisy, L.L., Beijamini, V., Joca, S.L., Vital, M.A.B.F., da Cunha, C. & Andreatini, R. (2002). Evaluation of the face validity of reserpine administration as an animal modelo of depression-Parkinson‘s disease association. Progress in Neuro-Psychopharmacology & Biological Psychiatry, 26, 879-883.
Verbaan, D., Marinus, J., Visser, M., van Rooden, S.M.A., Stiggelbout, M., Middelkoop, H.A.M.J. & van Hilten, J. (2007). Cognitive impairment in Parkinson‘s disease. J Neurol Neurosurg Psychiatry, 78, 1182–1187.

72
2.2. Experimento II: Artigo científico que será submetido ao periódico Progress in
Neuropsychopharmacology & Biological Psychiatry
Repeated treatment with reserpine as a possible progressive model of
Parkinson’s disease
Valéria S. Fernandes1, José R. Santos1, Thieza G. Melo1, Anderson H.F.F. Leão1,
André M. Medeiros1, Geison S. Izídio1, Alicia Cabral1, Rosana A. Ribeiro2, Vanessa
C. Abílio2,3, Alessandra M. Ribeiro1, Regina H. Silva1,*
1Memory Studies Laboratory, Physiology Department, Federal University of Rio
Grande do Norte, Natal, Brazil
2Department of Pharmacology, Universidade Federal de São Paulo, São Paulo,
Brazil.
3Laboratório Interdisciplinar de Neurociência Clínica (LiNC), Department of
Psychiatry, Universidade Federal de São Paulo, São Paulo, Brazil.

73
ABSTRACT
Animal models are widely used to study alterations caused by the Parkinson‘s
Disease (PD). However, in general, pharmacological models do not express the
progressive nature of the disease, causing immediate severe motor impairment after
acute administration. Reserpine administration in rodents has been suggested as a
pharmacological model of PD based on the effects of this monoamine-depleting
agent on motor activity. We found that repeated administration with a low dose (0.1
mg/Kg) of reserpine in rats induces a gradual appearance of motor signs.
Furthermore, these motor signs were accompanied by increased levels of striatal lipid
peroxidation. However, treatment with reserpine was unable to induce cognitive
abnormalities and alterations in hippocampal lipid peroxidation. Thus, repeated
treatment with low alternated doses of reserpine progressively induced alterations in
motor function, indicating a possible application of this model in the study of the
progressive nature the motor signs in PD.
Keywords: reserpine, Parkinson‘s Disease, cognition, movement disorders,
oxidative stress, animal model

74
1- Introduction
Parkinson‘s disease (PD) is a progressive neurodegenerative disorder,
characterized by bradykinesia, tremor, rigidity and postural abnormalities
(Klockgether, 2004). However, cognitive impairments can also be observed in PD
patients (Mahieux et a., 1998; Aarsland et al., 2004; Verbaan et al., 2007). The
pattern of cognitive disturbances associated with PD includes learning impairments
(Schmitt-Eliassen et al., 2007), deficits of executive functions such as planning or
working memory (Morris et al.,1988; Cools et al., 2002; Cox et al., 2002) and
attentional deficits (Bronnick et al., 2006).
Animal models are extensively used to study alterations caused by the PD
(Beal, 2001). However, in general, pharmacological models do not express the
progressive nature of the disease, causing immediate severe motor impairment with
a single administration (Da Cunha et al., 2002; Bellissimo et al., 2004; Henderson et
al., 2005).
The administration of reserpine to rodents has been suggested as a
pharmacological model of PD based on the effects of this monoamine-depleting
agent on motor activity. Reserpine interferes with the storage of monoamines in
intracellular vesicles, causing monoamine depletion in nerve terminals and transient
hypolocomotion and muscular rigidity, depending on the dose (Colpaert 1987;
Gerlach and Riederer, 1996). The range dose usually used to induce such motor
alterations in rodents is 1 to 5 mg/kg, and the severe motor impairment prevents
other kinds of behavioral evaluations, such as memory tests and other
cognitive/emotional assessments. However, previous results from our group
(Fernandes et al., 2008) have shown that a single administration of reserpine in low
doses (0.1-0.5 mg/kg) can induce deficits in emotional memory without causing motor
alterations. Carvalho et al. (2006) obtained similar results, in a different behavioral
model. These findings corroborate studies with PD patients showing deficits in
emotional processing while motor deficits were absent (Schneider et al., 2003;
Salgado-Pineda et al., 2005; Bowers et al., 2006). Therefore, the previous studies
suggest that depending on the dose, reserpine is able to induce changes in rodents
similar to the symptoms found in humans with PD.
The reserpine is an irreversible inhibitor of the vesicular monoamine
transporter 2 (VMAT2). The blockage of dopamine vesicular uptake results in the

75
accumulation of neurotoxic dopamine oxidation byproducts (Caudle et al., 2008). For
instance, dopamine (DA) reacts with molecular oxygen to form dopamine-quinones
which can deplete glutathione, generating reactive oxygen species (ROS) during this
process (Tsang and Chung, 2009). When the production of ROS exceeds the ability
of the antioxidant system to eliminate them, oxidative damage occurs (Cadenas and
Davies, 2000). These neuronal damages caused by oxidative stress can induce
alterations in both motor (Faria et al., 2005; Teixeira et al., 2009) and cognitive skills
(Chen et al., 2010).
The brain is particularly sensitive to oxidative damage when compared to other
organs or systems, mainly because it contains high levels of membrane lipids,
excitotoxic amino acids, low levels of antioxidant defenses and autoxidizable
neurotransmitters (Cadenas and Davies, 2000).Therefore, it is quite understandable
that many studies have been performed to investigate the role of oxidative injury in
neurodegenerative diseases including PD (Cadenas and Davies, 2000; Beal, 2003).
In fact, evidences of oxidative stress damage are found in both brain tissue from PD
patients (Beal, 2002) and in pharmacological models such as the 1-methyl 4-phenyl
1,2,3,6-tetrahydropyridine (MPTP) (Obata, 2002), 6-hydroxydopamine (6-OHDA)
(Riobó et al., 2002) and reserpine (Spina and Cohen, 1989; Bilska et al., 2007)
models. However, the relationship among oxidative stress, PD‘s progressive
degeneration, motor and cognitive deficits remains unclear.
Considering the importance of PD, the progressive nature of the disease and
the possible relationship with oxidative stress, we evaluate the repeated
administration of reserpine as a possible pharmacological model with progressive
effects similar to those in patients with PD. Therefore we submit Wistar rats to a
repeated treatment with a sub-effective dose of reserpine and evaluate motor
behavior and memory performance. Furthermore, we evaluated oxidative stress in
the striatum and hippocampus by measuring lipid peroxidation.
2- Materials and methods
2.1- Subjects
Five-month old male Wistar rats (n= 74) were used. All animals were
maintained in groups of four or five per cage, under a 12 h light 12 h dark cycle and

76
at a constant temperature of 25 1 C, with food and water available ad libitum. The
rats were handled according to Brazilian law procedures for the use of animals in
scientific research (Law Number 11.794) and all procedures were approved by the
local research ethics committee (final opinion number 149/2008). All efforts were
made to minimize animal pain, suffering or discomfort, and to minimize the number of
rats used.
2.2- Drug treatment, general procedures and experimental design
Reserpine (methyl reserpate 3,4,5-trimetothoxycinnamic acid ester: Sigma
Chemical Co. St. Louis, MO) was dissolved in glacial acetic acid and diluted to the
correct concentration in distilled water. Vehicle consisted of the same amount of
acetic acid and water as in the reserpine solution. These solutions were injected
subcutaneously (s.c.).
Rats received 7-10 subcutaneous injections of vehicle (VEH) or 0.1 mg / kg of
reserpine (RES), at a volume of 1 ml/kg body weight, on alternate days. During
treatment rats went through the following procedures: (1) catalepsy test two days
before the beginning, and every day throughout the treatment, i.e., 24 h and 48 h
after each injection (n=13 (VEH); n=12 (RES)); (2) quantification of open field
behavior 24 h after the 4th injection (n=17 per group); (3) assessment of orofacial
movements before starting the treatment, 24 h after the 5th and 10th injections and 48
h after the 10th injection (n=8 per group); (4) training and test sessions of novel object
recognition task, 24 h and 48 h after the 5th injection, respectively (n= 7 per group);
(5) training and test sessions of plus-maze discriminative avoidance task, 24h and 48
h after the 7th injection, respectively (n=23 per group); (6) body weighing 24 h before
1st, 3rd, 5th and 7th injections, (7) quantification of striatal and hippocampal lipid
peroxidation 48 h after the 7th (n=10 per group) and 10th injections (n = 12 per group)
(Figure 1).

77
Fig. 1. Outline: experimental design
Every rat was submitted to 10 min of gentle handling once a day for five days
before the beginning of the experimental procedures. The behavioral analysis of the
catalepsy test and orofacial movements assessment were performed manually by
direct observation. All other behavioral sessions were recorded by a camera placed
above the apparatus and the behavioral parameters were registered by an animal
video-tracking software (Anymaze, Stoelting, USA).
During behavioral sessions, all apparatuses were washed with a water–
alcohol (5%) solution before behavioral testing to eliminate possible bias due to
odors left by previous subjects.
During treatment rats went through the following procedures:
2.3- Behavioral testing
2.3.1- Catalepsy Test
The catalepsy was assessed placing the animal‘s front paws on a horizontal
bar positioned at 9 cm above the bench surface. The duration of catalepsy, which

78
was defined as an immobile posture, keeping the two front paws on the bar, was
measured with a maximum of 180 s. Three trials were carried out for each animal in
each observation day and the results were analyzed considering the mean value of
the three trials.
2.3.2- Open field
The apparatus, made of wood and painted in black, was a circular open-field
arena (84 cm in diameter) with 32 cm high walls. We quantified the distance traveled
(in meters), the frequency of rearing (partial or total rising onto hind limbs), immobility
duration (time of complete absence of paw movements), the latency to start
movement (time spent to move one or more paws from the initial position) and the
time in center (time spent in the center of the open field).
2.3.3- Orofacial movements
Rats were placed individually in wired cages (40 cm×40.5 cm×20 cm) with
mirrors positioned under the floor and behind the back wall of the cage to allow
behavioral quantification when the animal was faced away from the observer. The
number of tongue protrusions (projection of the tongue out of the oral cavity),
vacuous chewing movement frequency (mouth openings in the vertical plane not
directed toward physical material), and facial twitching (duration, in seconds, of
twitching of the facial musculature) were measured continuously for 15 min.
2.3.4- Novel object recognition task
The task was carried out in a circular open-field arena (84 cm in diameter) with
32 cm high walls, made of wood and painted in black. The objects used were a sugar
bowl and a plastic stem glass, which were alternately familiar or new to avoid the
effect of possible preference. In the training session, rats were exposed to the
experimental chamber and two copies of an object for 10 min. In the test session, 24
h after the training, one object was replaced for a new object and the rats were
allowed to explore them for ten min. The time to explore each object was measured.

79
Exploration behavior included touching with forepaws or nose, sniffing and biting
each object.
2.3.5- Plus-maze discriminative avoidance task
The apparatus employed was a modified elevated plus-maze made of wood
containing two enclosed arms (50 X 15 X 40 cm) opposite to two open arms (50 X 15
cm). A 100-watt lamp was placed over the middle of one of the enclosed arms
(aversive enclosed arm). In the training session, each rat was placed in the centre of
the apparatus and, over a period of 10 min, every time the animal entered the
enclosed arm containing the lamp, an aversive situation was produced until the
animal left the arm. The aversive stimuli were the 100-watt light and an 80 dB noise
applied through a speaker placed over the aversive enclosed arm. In the test
session, held 24h later, the rats were again placed in the apparatus for 10 min,
without receiving the aversive stimulation, with the lamp and the speaker still present
over the aversive arm, but turned off. Distance traveled in the apparatus (used for
motor activity evaluation) and time spent in each arm (aversive, non-aversive and
open arms) were registered. Percent time in aversive arm (time spent in aversive
enclosed arm/time spent in both enclosed arms) and percent time spent in open
arms (time spent in open arms/time spent in both open and enclosed arms)
considering the whole duration of behavioral sessions were used to evaluate memory
and anxiety, respectively (Silva & Frussa-Filho, 2000). Percent time spent in the
aversive enclosed arm assessed minute by minute across the training and test
sessions were used to evaluate learning and extinction of the task (Ribeiro et al.,
2010).
2.4- Tissue preparation and oxidative stress parameters
Rats were decapitated, 48 h after the 7th (n=10 per group) and 10th injections (n
= 12 per group); and their brains were removed and put on ice. The striatum and
hippocampus were dissected. Each tissue sample of the hippocampus and striatum
was homogenized in ice-cold 0.1 M phosphate buffer (1:50, w:v). A duplicate of each
sample was used to determine MDA by measurement of fluorescent product formed
from the reaction of this aldehyde with thiobarbituric acid, as described by Tanizawa

80
et al. (1981). The results are expressed as nmol MDA/g tissue, calculated by plotting
the obtained fluorescence (excitation at 315 nm, emission at 553 nm) against an
MDA concentration standard curve.
2.5. Statistical analysis
Catalepsy behavior across the treatment (before, 24 h and 48 hours after each
injection), the orofacial movements (before starting the treatment, 24 h after the 5th
and 10th injections and 48 h after the 10th injection), the percentage of time in
aversive arm across behavioral sessions of the discriminative avoidance task and the
body weight (24 h before 1st, 3rd, 5th and 7th injections), were compared using ANOVA
with repeated measures. When necessary, pair wise comparisons were held with
multiple t tests with sequential Bonferroni‘s correction. The independent samples t
test was used to analyze differences between groups RES and VEH in all
parameters of open field behavior and also distance traveled, percent time spent in
the aversive or in the open arms in the discriminative avoidance task. In the novel
object recognition task, comparisons within groups for percentage of time to explore
old x new objects were performed with paired-samples t test. Results are expressed
as mean±SE and the significant threshold considered was p< 0.05.
3- Results
3.1- Effects of repeated administration of reserpine on catalepsy behavior
ANOVA with repeated measures revealed time (days of treatment) [F(21,
483)=18.16; p=0.000], treatment (reserpine or vehicle) [F(1, 23)=12.19; p=0.002] and
time X treatment interaction effects [F(21, 483)=9.29; p=0.000]. Rats repeatedly
treated with reserpine showed progressive increases in the catalepsy behavior,
which were significantly different from VEH on days 16 (48 h after the 7th injection) (t=
3.44; p= 0.002), and from day 18 onwards: 48 h after the 8th injection (t=4.24;
p=0.000), 24 h after the 9th injection (t=5.52; p=0.000), 48 h after the 9th injection
(t=4.43; p=0.000), 24 h after the 10th injection (t=7.40; p=0.000) and 48 h after the
10th injection (t= 6.41; p=0.000) (See Fig 2).

81
Fig.2. Effects of repeated administration of reserpine (RES - 0.1 mg/Kg) or vehicle (VEH) on catalepsy behavior. Data are expressed as mean±S.E.M. (s). ANOVA with repeated measures revealed time, treatment and time x treatment interaction effects. *p<0.05 compared to VEH group (independent samples t test with Bonferroni‘s correction).
3.2- Effects of repeated administration of reserpine in an open field
No effects of repeated administration of reserpine were found in open field
behavior. The distance traveled (t= 0.74; p=0.466), the frequency of rearing (t=0.91;
p= 0.368), the immobility duration (t=0.24; p=0.816), the latency to start movement
(t=0.69; p=0.498) and the time spent in the center of the open field (t=0.53; p=0.597)
of the reserpine group were not different from those presented by the vehicle group
(Fig. 3 A, B, C, D and E, respectively).

82
Fig.3. Effects of repeated administration of reserpine (RES - 0.1 mg/Kg) or vehicle (VEH) on open field behavior evaluated 24 h after the 4th injection. Data are expressed as the mean±S.E.M. of distance traveled (A), rearing frequency (B), immobility duration (C), latency to start the movement (D) and time in center (E).
3.3- Effects of repeated administration of reserpine on orofacial movements
ANOVA with repeated measures revealed time (days of treatment) [F(3,
42)=16.35; p=0.000], treatment (reserpine or vehicle) [F(1, 14)=7.66; p=0.015] and
time X treatment interaction effects [F(3, 42)=7.58; p=0.001] for the number of
vacuous chewing movements. Significant increases due to reserpine treatment
compared to VEH were detected on days 21 (24 h after the 10th injection) (t=3.37; p=
0.005) and 22 (48 h after the 10th injection) (t=2.90; p= 0.012) (see Fig. 4 A).
Regarding duration of facial twitching, ANOVA with repeated measures
revealed a treatment (reserpine or vehicle) effect [F (1,14)= 7.52; p=0.016]. No effect
A
B
C
D
E

83
of time (days or treatment) [F(3, 42)=0.83; p=0.473] or time X treatment interaction
[F(3, 42)=1.75; p=0.178] were found. Significant increases due to reserpine treatment
compared to VEH were detected on day 21 (24 h after the 10th injection) (t=3.73; p=
0.002) (see Fig 4 B).
For the number of tongue protrusions, ANOVA with repeated measures
revealed a treatment (reserpine or vehicle) effect [F (1,14)= 6.45; p=0.024]. No effect
of time (days of treatment) [F(3, 42)=2.57; p=0.091] or time X treatment interaction
[F(3, 42)=1.29; p=0.291] were found. Significant increases due to reserpine treatment
compared to VEH were detected on days 21 (24 h after the 10th injection) (t=2.98; p=
0.010) and 22 (48 h after the 10th injection) (t=2.92; p= 0.011) (see Fig 4 C).

84
Fig.4. Effects of repeated administration of reserpine (RES - 0.1 mg/Kg) or vehicle (VEH) on orofacial movements. Data are expressed as the mean±S.E.M. of the

85
number of vacuous chewing movements (A), the duration of facial twitching (B) and the number of tongue protrusions (C). ANOVA with repeated measures revealed treatment effects for all parameters, as well as time and time x treatment interaction effects for vacuous chewing. *p<0.05 compared to VEH group (independent samples t test with Bonferroni‘s correction).
3.4- Effects of repeated administration of reserpine on novel object recognition task
Both groups showed an increased percentage of novel object exploration
compared to the old object (t= 3.73; 3.36 and p= 0.010; 0.015 for VEH and RES,
respectively, Fig 5), indicating adequate performance in the task.
Fig.5. Effects of repeated administration of reserpine (RES - 0.1 mg/Kg) or vehicle (VEH) on novel object recognition task performed 48 h after the 5th injection. Data are expressed as the mean±S.E.M. *p<0.05 compared to percent of old object exploration (paired-samples t-test).
3.5- Effects of repeated administration of reserpine on plus-maze discriminative
avoidance task
Repeated treatment with reserpine decreased the distance traveled in the
maze 24 h after the 7th injection (t= 3.32 and p= 0.002, Fig 5A), in the training
session, and 48 h after the 7th injection (t= 2.39 and p= 0.021, Fig 5C), in the test
session, when compared to the vehicle group. However, as shown in Fig. 6 (B and
* *

86
D), no effects of repeated administration of reserpine were found on the latency to
start the movement, in the training or test sessions.
Fig.6. Effects of repeated administration of reserpine (RES - 0.1 mg/Kg) or vehicle (VEH) on plus-maze discriminative avoidance training (A,B) and test (C,D) sessions performed 24 and 48 h after the 7th injection, respectively. Data are expressed as the mean±S.E.M. of the distance traveled (m) (A, C) and latency to start the movement (s) (B,D). *p<0.05 compared to VEH group (independent samples t test).
Regarding anxiety-like behavior, the percentage of time in the open arms
(%TO) in the training (t= 0.44 and p= 0.665) and in the test (t= 1.08 and p= 0. 286)
presented by the reserpine group were not different from those presented by the
vehicle group (Fig. 7 A, and B, respectively).
Fig.7. Effects of repeated administration of reserpine (RES - 0.1 mg/Kg) or vehicle (VEH) on percent time in the open arms (%TO) of the plus-maze discriminative
A B

87
avoidance apparatus during training (A) and test (B) sessions, performed 24 and 48 h after the 7th injection, respectively. Data are expressed as the mean±S.E.M.
No effects of repeated administration of reserpine were found on the percent
time in the aversive enclosed arm (%TAV), in the training (t= 1.77; p=0.084) and test
(t=0.63; p=0.533) (see Fig 8 A and B) when the whole sessions were considered for
analysis.
In the training session, a significant effect of time (minutes) [F (9,396)= 8.19;
p=0.000] was found when the percentage of time in the aversive enclosed arm
(%TAV) was evaluated across the session. No effect of the treatment (reserpine or
vehicle) [F (1,44)= 0.64; p=0.429] and time X treatment interaction [F (9,396)= 1.34;
p=0.251] were found (see Fig 8 C).
In the test session, significant effects of time (minutes) [F( 9,396)=2.58;
p=0.024] was found when the percentage of time in the aversive enclosed arm
(%TAV) was evaluated across the session. No effect of the treatment (reserpine or
vehicle) [F (1,44)= 1.25; p=0.27] and the time X treatment interaction [F(9,396)=1.03;
p=0.406] were found (see Fig 8 D).

88
Fig.8. Effects of repeated administration of reserpine (RES - 0.1 mg/Kg) or vehicle (VEH) on percent time in the aversive enclosed arm (%TAV) of the plus-maze discriminative avoidance apparatus during the whole sessions (A, B) or minute by minute across the sessions (C,D), for training (A, C) and test (B,D), performed 24 and 48 h after the 7th injection, respectively. Data are expressed as the mean±S.E.M. ANOVA with repeated measures revealed time (minutes) effects in C and D.
C
D
A B

89
3.6- Effects of repeated administration of reserpine in the body weight
Regarding the body weight, ANOVA with repeated measures revealed no effect
of time (number of injections) [F(4, 96)=0.73; p=0.536], treatment (reserpine or
vehicle) [F (1,24)= 0.82; p=0.374] or time X treatment interaction [F(4, 96)=0.66;
p=0.572] (see Fig 9).
Fig.9. Effects of repeated administration of reserpine (RES - 0.1 mg/Kg) or vehicle in the body weight across the treatment. Data are expressed as the mean±S.E.M.
3.7- Effects of repeated administration of reserpine on striatal and hippocampal lipid
peroxidation
Fig. 10 (A and B) shows striatal and hippocampal levels of lipid peroxidation,
respectively, 48 h after 7th and 10th injection of rats repeatedly treated with reserpine.
No effects of repeated administration of reserpine were found after the 7th injection in
striatum (t= 1.35, p = 0.195) or hippocampus (t= 1.15, p = 0.269), and after the 10th
injection in hippocampus (t= 2.09; p=0.055). However, the reserpine-treated rats
showed increased levels of lipid peroxidation in the striatum 48 h after the 10th
injection (t=3.00; p=0.011) (see Fig 10 A).
0
100
200
300
400
500
2 6 10 14 20
Bo
dy w
eig
ht
(g)
Days of treatment
VEHRES

90
Fig.10. Effects of repeated administration of reserpine (RES - 0.1 mg/Kg) or vehicle (VEH) on striatal (A) and hippocampal (B) levels of lipid peroxidation. Data are expressed as the mean±S.E.M. of MDA levels per gram of tissue.*p<0.05 compared to VEH group (independent samples t test).

91
4- Discussion
In this study, we investigated the effects of repeated administration with a low
dose of reserpine on motor and cognitive parameters. We observed that this
repeated treatment with reserpine induced a progressive motor impairment. In fact,
these results can be seen in the evaluation of catalepsy behavior performed before,
24 and 48 h after each injection (Figure 2). In addition, the motor parameters
evaluated in the open field were not altered in the RES group 24 h after 4th injection
(Figure 3) but hypolocomotion was detected when the distance traveled in the
discriminative avoidance maze was measured 24 and 48 h after the 7th injection
(Figures 6 A and C). However, none of the memory tests performed were affected by
the treatment with reserpine (Figures 5 and 8). Furthermore, the present study
demonstrates that repeated reserpine treatment can induce motor abnormalities and
concomitant increases in striatal levels of lipid peroxidation, an indicative of oxidative
stress-induced neuronal damages (Figure 10).
PD is a neurodegenerative disorder of the basal ganglia characterized for a
complex situation of behavioral disorders, including tremor, rigidity and bradkinesia
(Johnston et al., 1999; Lindner et al., 1999; Ridley et al., 2006). These motor
symptoms have been highlighted as those that characterize the clinical status of an
affected person, so they are considered the most important disorders associated with
PD (Mayeux, 2003; Klockgether, 2004). In rodents, dopamine hypofunction lead to
parkinsonian symptoms as the emergence of signs of akinesia and rigidity
(catalepsy). The evaluation of catalepsy has been used as an important parameter
for the detection of motor signs in animal models of PD (Chinen and Frussa-Filho,
1999; Diaz et al., 2001). These signs can be induced not only by drugs that block
dopamine receptors such as haloperidol (Shiozaki et al., 1999; Gongora-Alfaro et al.,
2009) but also by substances that are potential inhibitors of mitochondrial complex I
as MPTP (Shiozaki et al., 1999) and rotenone (Corona et al., 2010), or the neurotoxin
6-OHDA (Diaz et al., 2001). Additionaly, these effects are also present after
monoamine vesicle depletion induced by reserpine (Shiozaki et al., 1999).
Reserpine interferes with the storage of catecholamines by blocking the presynaptic
vesicular carriers, resulting in depletion of monoamines in nerve terminals (Caudle et
al., 2008) and induction of hypolocomotion and muscular rigidity (Colpaert 1987;
Gerlach and Riederer, 1996). This study revealed that rats exposed to repeated

92
administration of reserpine at 0.1 mg / kg showed a gradual increase of cataleptic
immobility time when compared with the control group (Figure 2). Previous studies
have demonstrated that a short treatment with high doses of reserpine (1.0 mg / kg
every other day for 4 days) (Dutra et al., 2002) or an acute injection of an even higher
dose (5 mg / kg) (Shiozaki et al., 1999) produce catalepsy as well hypolocomotion.
However, in the present study, it is unlikely that the increase in catalepsy behavior is
due to the acute effect of the previous administration since it is still present even 48 h
after the last injection from the 7th injection onwards. Thus, a progressive neuronal
effect of the repeated treatment leading to the motor impairment could be
hypothesized.
Data from catalepsy evaluation are corroborated by the fact that the repeated
treatment with a low-dose of reserpine was not able to impair motor parameters
evaluated after the 4th injection in the open field (distance traveled, rearing frequency,
immobility duration and latency to start movement) (Figure 3). Previous research has
demonstrated that acute administration of higher doses of reserpine induced
locomotor alterations (Faria et al., 2005; Peixoto et al., 2005; Tadaiesky et al., 2006).
In this respect, hypokinesia is an important feature of animal models of PD and is
often related to a significant loss of dopamine cells (Da Cunha et al., 2002; Delfino et
al., 2004, Ferro et al., 2005; Capitello et al., 2008).The present results suggest that
there was no acute effect of the dose used on motor behavior, and the continuation
of the repeated treatment was necessary to produce motor abnormalities that were
observed in the catalepsy test only after the 7th injection (Figure 2). In this respect,
another motor parameter used in this study was the distance traveled and the latency
to start the movement in the elevated plus-maze used in the discriminative avoidance
task, which was performed 24 and 48 h after the 7th injection (Figure 6).
Corroborating the data from the catalepsy evaluation, animals treated with reserpine
showed a significant decrease in distance traveled in the maze in both sessions.
However, no significant differences between groups were found in latency to start
movement. Studies with MPTP model evaluated the latency in initiating the first
movement and demonstrated that there was an increase 24 h after administration of
the toxin, however, this change disappeared after 7 days (Capitelli et al., 2008).
Thus, these differences in results found in motor discriminative avoidance test may
reflect a gradual period of motor alteration, as can be found in humans with PD (Burn
et al., 2006).

93
Previous studies have suggested oral dyskinesia induced in rodents by
reserpine as a possible animal model of tardive dyskinesia (Neisewander et al.,
1991; Neisewander et al., 1994; Abilio et al., 2003 and 2004). The tardive dyskinesia
is a side effect of long-term treatment with neuroleptics characterized by severe
motor symptoms where the face, mouth and tongue are frequently involved (orofacial
dyskinesia) (Hansen et al., 1997; Andreassen and Jorgensen, 2000). On the other
hand, some authors advocate the induction of orofacial movements as a model of the
tremor-related symptoms found in patients with PD (Salamone and Baskin, 1996,
Salamone et al., 2008). These authors argue that the difference of tremor seen in
rodents is restricted to the observation of the jaw which does not occur in humans
where this symptom can be observed in the limbs (Salamone and Baskin, 1996).
These movement alterations in rodents can be induced by a series of conditions
related to the neurochemistry and pathophysiology of parkinsonism such as depletion
of dopamine levels caused by reserpine (Salamone and Baskin , 1996, Salamone et
al., 2008), dopamine antagonists such as haloperidol (Andreassen et al., 2003) and
neurotoxins such as 6-OHDA (Jicha and Salamone, 1991). Here we found that the
treatment with repeated administration with a low-dose of reserpine was able to
induce an increase in orofacial movements 24h after the 10th injection (Figure 4). It
should be noted that neither orofacial movements nor catalepsy behavior were
altered 24 h after the 5th injection (Figures 2 and 4). However, 24h after the 10th
injection, concomitant motor alterations were observed in the catalepsy test and the
oral dyskinesia evaluation. Indeed, previous research has shown that acute reserpine
administration (at higher doses) induced decreased locomotion and increased
duration of immobility concomitantly to the oral dyskinesia (Faria et al., 2005; Peixoto
et al., 2005). In addition, there are descriptions of cases of PD patients who have
concomitant usual motor symptoms (bradykinesia, disorders in walking, among
others) and impaired oromotor control (Robertson et al., 2001). On the other hand,
Sussman et al. (1997) showed that reserpine-induced oral dyskinesia persisted
despite repletion of dopamine in the caudate-putamen, suggesting that the persistent
neuropathological change underlying this behavior occurs in a neural pathway other
than the dopaminergic nigrostriatal pathway. Thus, the pathophysiological
characteristics of orofacial movements are still controversial. However, the data
presented here indicate a progressive increase in orofacial movements
simultaneously to catalepsy behavior. Additionaly, this result is corroborated by

94
recent data from our laboratory showing the same pattern of concomitant
appearance of both kinds of symptoms across a repeated treatment with 6-OHDA
(unpublished results).
As mentioned before, besides motor symptoms, PD patients also have other
manifestations such as cognitive, mood and sensory system alterations (Higginson et
al., 2001; Korczyn, 2001; Zgaljardic et al., 2004; Koerts et al., 2007). We have
recently verified that single administrations of reserpine — at doses that do not
modify motor function — impair memory in the discriminative avoidance task (a
rodent model of aversive discrimination – Carvalho et al., 2006), while no effects of
the same acute doses were detect in the novel object recognition task (Fernandes et
al., 2008). Thus, in the present study we investigated the effects of the repeated
treatment with 0.1 mg/kg reserpine on the performance of rats in these two tasks.
Due to evidence that cognitive deficits can precede the appearance of the motor
symptoms in the progress of the disease, we attempt to evaluate cognitive deficits
before an expressive motor impairment was instated.
The novel object recognition task (performed after the 5th injection) is based on
the fact that rats recognize a previously presented object, and therefore would spend
more time exploring the new object presented in the test session (Figure 5). The
preference for exploring new objects was shown by both groups, indicating that the
repeated administration of reserpine did not affect this kind of memory. Similar results
were found with acute administration of reserpine (0.1, 0.25 or 0.5 mg / kg)
(Fernandes et al., 2008). In this respect, the lack of alteration in this task is in
accordance with some clinical studies showing intact recognition memory in PD
patients (Gabrieli, 1996; Postle et al., 1997).
Besides evaluating memory, we used the plus maze discriminative avoidance
task (performed after the 7th injection) to assess learning, anxiety and motor
behavior, since it has been shown that these evaluations can be performed
concomitantly, and by different parameters, in this paradigm (Silva and Frussa-Filho,
2000; Silva et al., 2002a). The results have shown that there were no significant
differences in the percentage of time in the aversive arm between the VEH and RES
groups, both in the training and test sessions, indicating that repeated treatment with
reserpine was not able to promote changes in learning or retrieval of the aversive
task (Figure 8). Moreover, by evaluating the distance traveled in the maze, we
observed that animals treated with reserpine showed motor deficits during the

95
acquisition and retrieval of the task (Figure 6), corroborating the increase in catalepsy
duration also shown at this point of the treatment (Figure 2). This motor activity
decrement, however, did not interfere with the analysis of the data related to the
cognitive aspect of the task, since rats‘ performances were evaluated by time spent
in the aversive enclosed arm. Indeed, previous studies conducted with this paradigm
have shown the viability of separate and reliable analysis and interpretation of the
two parameters (Silva & Frussa-Filho, 2000; Silva et al., 2002a,b; Carvalho et al.,
2006; Kameda et al., 2007;Niigaki et al., 2010). Furthermore, the evaluation of
behavior minute by minute throughout the training session indicated that even in the
presence of motor deficits, animals treated with reserpine learned the task, as shown
by a decrement of aversive arm exploration by the end of the session (Figure 8 C).
Similarly, the analysis of aversive arm exploration throughout the test session
indicated that animals treaded with reserpine or vehicle showed retrieval of the task
(low aversive arm exploration in the first minutes) followed by extinction of the task
(increase in exploration) (Figure 8 D).
In summary, in the present study, the repeated administration with a low-dose
of reserpine did not produce changes in the memory task involving an emotional
context, as opposed to what has been previously observed after single injections in
this same paradigm (Silva et al., 2002a; Carvalho et al., 2006) or in the contextual
fear conditioning task (Fernandes et al., 2008). In this respect, research has shown
that excessive or insufficient levels of dopamine may have a negative effect on
emotional memory (Cools et al., 2002; Halbig et al., 2008). Thus, the previous
studies performed with acute treatments (with doses from 0.1 to 1 mg/kg) could
reflect the effects of acute dopamine depletion on emotional memory. Although the
dose used in this study is within this range (0.1 mg / kg), it was given several times,
and it is possible that the decrease in the levels of dopamine depletion was
outweighed by up regulation of D1 and D2 receptors in the caudate-putamen
(Neisewander et al., 1991) or by other compensatory mechanisms of plasticity
(Castaneda et al., 1990; Bezard and Gross 1998; Berzard et al., 2001; Cropley et al.,
2006). Also, the repeated treatment, which was efficient in inducing progressive
motor impairment, did not induce cognitive impairments in any of the paradigms used
here. Thus, a dissociation between the cognitive deficits induced by reserpine
treatment (observed in previous studies) and a possible degenerative process
induced by the repeated treatment conducted here is suggested. It is important to

96
note, however, that cognitive impairments can be observed in rats injured with MPTP
(Da Cunha et al., 2002) and PD patients (Bowers et al., 2006; Halbig et al., 2008).
Thus, it would be interest to verify the effects of the repeated treatment used here in
other animal models of memory, or even in other aspects of cognitive function.
Another parameter evaluated in the plus-maze discriminative avoidance task
was the time of exploration of the open arms of the maze, indicative of anxiety-like
behavior (Silva & Frussa-Filho, 2000). Results showed that treatment with reserpine
did not induce alterations in anxiety-like behavior (Figure 7), corroborating the
previous studies that investigated the effects of reserpine in this task (Silvia et al.,
2002a; Carvalho et al., 2006).
Increased oxidative stress with cumulative free radical damage is present in
brain aging and neurodegenerative diseases such as PD (Cadenas and Davies,
2000; Beal, 2003). In this respect, treatment with reserpine can result in the
accumulation of neurotoxic dopamine oxidants that can induce the production of
ROS exceeding the ability of the antioxidant system to eliminate them, thus resulting
in oxidative damage (Cadenas and Davies, 2000; Caudle et al., 2008). Recent
studies showed that acute administration of high doses of reserpine increases lipid
peroxidation on striatum and antioxidant agents are able to reverse the behavioral
effects induced by reserpine (Abílio et al., 2003, 2004; Faria et al., 2005). Herein, the
repeated treatment with a low dose of reserpine has shown to induce an increased
striatal level of lipid peroxidation 48 h after the 10th injection (Figure 10A), when an
important motor impairment was also present (Figure 2). On the other hand,
hippocampal levels of lipid peroxidation were not modified by the treatment.
Interestingly, the absence of memory impairment may be related to lack of neuronal
damage caused by oxidative stress in the hippocampus (Figure 10B). These findings
suggest that the treatment used here may induce a progressive neuronal damage
similar to what is found in patients with PD, at least considering motor aspects of the
pathology.
Although quantitative assessment was not conducted, there were no important
peripheral autonomic changes in reserpine-treated animals throughout the treatment.
Additionally, no change was found in body weight of rats during the repeated
treatment of reserpine (Figure 9), and all animals survived to the treatment. In this
respect, Ferro et al. (2005) found a significant change in weight in pharmacological
models of MPTP and 6-OHDA when compared to control groups, with 20% death of

97
treated animals. In light of these findings, we suggest that reserpine may be a more
favorable drug to the development of a pharmacological progressive model, which
requires repeated treatment over time, compared to MPTP or 6-OHDA models. On
the other hand, it is important to mention that reserpine, as a pharmacological model
of PD, is considered to be unspecific, because this drug acts on the depletion of all
monoamines. However, there is evidence in the literature that the physiopathology of
PD itself is not exclusively related to dopamine, since other neurotransmitter systems
have shown to be involved in the PD symptoms such as the serotonergic and the
GABAergic systems, among others (Ossowska et al., 2002; Borah et a., 2007).
Reserpine is an irreversible inhibitor of the vesicular monoamine transporter
(VMAT). As mentioned above the action of reserpine prevents the storage of
monoamines in synaptic vesicles (Caudle et al., 2008). There are studies suggesting
that animals express 5% of VMAT as a promising model for the study of PD. The
VMAT deficient animals have increased oxidative stress, progressive loss of
dopamine terminals and accumulation of -synuclein (Caudle et al. 2007). In addition,
monoaminergic dysfunctions are also found levels of dopamine, norepinephrine and
serotonin are severely diminished (Caudle et al. 2007, Taylor et al. 2009). Sleep
disturbances, gastrointestinal symptoms of anxiety and depression were observed in
results from studies with mice deficient for VMAT (Taylor et al. 2009). As can be seen
the VMAT2-deficient animals exhibit similar alterations in the animals treated with
reserpine. Data from this study show increased oxidative stress and data from other
studies show alteration of dopamine levels in rats treated with reserpine (Sussman et
al., 1997) as well as can be seen in animals deficient for VMAT. In addition, results of
studies using Western blot analysis showed reductions markers for VMAT2
immunoreactivity in putamen, caudate and nucleus accumbens of PD brain
compared to control cases (Miller et al., 1999). These findings show that alterations
in VMAT can be one of the factors related to the development of PD.
In conclusion, we found that repeated administration with a low dose of
reserpine in rats induces a gradual appearance of motor signs compatible with the
progressive nature of PD (Klockgether, 2004). These motor signs are accompanied
by increased levels of oxidative stress in the striatum that support studies showing an
increase in free radicals as a possible factor in PD (Cadenas and Davies, 2000; Beal,
2003). Nevertheless, the treatment protocol applied was not able to induce cognitive
deficits, at least in the behavioral models used, which was corroborated by the

98
absence of oxidative damage in the hippocampus. More studies are required to verify
the progressive possible changes of the dopaminergic and other neurotransmitter
systems in the PD model proposed here. Further investigation with other behavioral
models could also clarify if the cognitive deficits related to PD can be observed in this
new progressive pharmacological model.
Acknowledgements
The authors would like to thank Claudenice Moreira dos Santos and Josué
Cândido Macedo for capable technical assistance. This research was supported by
fellowships from Conselho Nacional de Desenvolvimento Científico e Tecnológico
(CNPq), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES),
Fundação de Apoio à Pesquisa do Estado do Rio Grande do Norte (FAPERN), and
Pró-reitoria de Pesquisa da Universidade Federal do Rio Grande do Norte
(PROPESQ/UFRN).
5- References
Aarsland D, Andersen K, Larsen JP, Perry R, Wentzel-Larsen T, Lolk A, Kragh-Sorensen P. The Rate of Cognitive Decline in Parkinson Disease. Arch Neurol 2004; 61: 1906-1911.
Abílio VC; Araujo CCS; Bergamo M; Calvente PRV; D‘Almeida V de ARR. Vitamin E attenuates reserpine-induced oral dyskinesia and striatal gssg/gsh ratio enhancement in rats. Prog Neuro- Psychopharmacol Biol Psychiat 2003; 27:109–14.
Abílio VC, Silva RH, Carvalho RC, Grassl C, Calzavara MB, Registro S. Important role of striatal catalase in aging- and reserpine-induced oral dyskinesia. Neuropharmacology 2004; 47:263–72.
Andreassen OA, Jorgensen HA. Neurotoxicity associated with neuroleptic-induced oral dyskinesias in rats Implications for tardive dyskinesia? Progress in Neurobiology 2000; 61: 525-541.
Andreassen OA., Ferrante RJ, Aamo TO, Bealf MF, Jorgensen H A. Oral dyskinesia and histopathological alterations in substantia nigra after long-term haloperidol treatment of old rats. Neuroscience 2003; 122: 717–725.
Beal MF. Experimental Models of Parkinson‘s disease. Nature/Neuroscience 2001; 2; 325-332.
Beal MF. Oxidatively modified proteins in aging and disease. Free Radical Biology & Medicine 2002; 32:797–803.
Beal MF. Mitochondria, Oxidative damage, and Inflammation in Parkinson's disease. Ann. N. Y. Acad. Sci. 2003; 991: 120-131.

99
Bellissimo MI, Kouzmine I, Ferro MM, Oliveira BH de, Canteras NS, Da Cunha C. Is the unilateral lesion of the left substantia nigra pars compacta sufficient to induce working memory impairment in rats? Neurobiology of Learning and Memory 2004; 82; 150–158.
Bezard E, Gross CE. Compensatory mechanisms in experimental and human parkinsonism: towards a dynamic approach. Progress in Neurobiology 1998; 55: 93-116.
Bezard E, Dovero S, Prunier C, Ravenscroft P, Chalon S, Guilloteau D, Crossman AR, Bioulac B, Brotchie J M, Gross CE. Relationship between the Appearance of Symptoms and the Level of Nigrostriatal Degeneration in a Progressive 1-Methyl-4-Phenyl- 1,2,3,6-Tetrahydropyridine-Lesioned Macaque Model of Parkinson‘s Disease. The Journal of Neuroscience 2001; 21: 6853–6861.
Bilska A, Dubiel M, Sokolowska-Jezewicz M, Lorenc-Koci E, Wlodek L. Alpha-lipoic acid differently affects the reserpine-induced oxidative stress in the striatum and prefrontal cortex o rat brain. Neuroscience 2007; 146: 1758–1771.
Borah A, Kochupurackal P, Mohanakumar. Long-Term L-DOPA Treatment Causes Indiscriminate Increase in Dopamine Levels at the Cost of Serotonin Synthesis in Discrete Brain Regions of Rats. Cell Mol Neurobiol 2007; 27:985–996.
Bowers D, Miller K, Mikos A, Kirsch-Darrow L, Springer U, Fernandez H, Foote K, Okun M. Startling facts about emotion in Parkinson‘s disease: blunted reactivity to aversive stimuli. Brain 2006; 129; 3356–3365.
Bronnick K, Ehrt U, Emre M, De Deyn PP, Wesnes K, Tekin S, Aarsand D. Attentional deficits affect activities of daily living in dementia-associated with Parkinson‘s disease. J. Neurol Neurosurg. Psychiatry 2006; 77: 1136- 1142.
Burn DJ, Rowan EN, Allan LM, Molloy S, O‘Brien JT, McKeith IG. Motor subtype and cognitive decline in Parkinson‘s disease, Parkinson‘s disease with dementia, and dementia with Lewy Bodies. J Neurol Neurosurg Psychiatry 2006; 77:585–589.
Cadenas E, Davies KJA. Mitochondrial free radical generation, oxidative stress, and aging. Free Radical Biology & Medicine 2000; 29: 222–230.
Capitelli C, Sereniki A, Lima MMS, Reksidler A B, Tufik S, Vital MABF. Melatonin attenuates tyrosine hydroxylase loss and hypolocomotion in MPTP-lesioned rats. European Journal of Pharmacology 2008; 594: 101–108.
Carvalho RC, Patti CC, Takatsu-Coleman AL, Kameda SR, Souza CF, Garcez-do-Carmo L. Effects of reserpina on the pluz-maze discriminative avoidance task: dissociation between memory and motor impairments. Brain Res 2006; 1122: 176–83.
Castaneda E, Whishaw IQ, Robinson TE. Changes in striatal dopamine neurotransmission assessed with microdialysis following recovery from a bilateral 6-OHDA lesion: variation as a function of lesion size. J Neurosci. 1990; 10: 1847-54.
Caudle, W.M., Richardson, J.R., Wang, M.Z., Taylor, T.N., Guillot, T.S., McCormack, A.L., Colebrooke, R.E., Monte, D.A., Emson, P.C. & Miller, G.W. Reduced Vesicular Storage of Dopamine Causes Progressive Nigrostriatal Neurodegeneration. The Journal of Neuroscience, 2007; 27: 8138–8148.
Caudle WM, Colebrooke RE, Emson PC, Miller GW. Altered vesicular dopamine storage in Parkinson‘s disease: a premature Demise. Trends in Neurosciences 2008; 31: 303- 308.

100
Chen Q, Niu Y, Zhang R, Guo H, Gao Y, Li Y, Liu R. The toxic influence of paraquat on hippocampus of mice: Involvement of oxidative stress. Neuro Toxicology 2010; 31: 310–316.
Chinen CC, Frussa-Filho R. Conditioning to Injection Procedures and Repeated Testing Increase SCH 23390-Induced Catalepsy in Mice. N. Europsychopharmacology 1999; 21: 670-678.
Colpaert, FC. Pharmacological characteristics of tremor, rigidity and hypokinesia induced by reserpine in rat. Neuropharmacology 1987; 26: 1431-1440.
Cools R, Stefanova E, Barker RA, Robbins TW, Owen AM. Dopaminergic modulation of high-level cognitition in Parkinson‘s disease: the role of the prefrontal cortex revealed by PET. Brain 2002; 125: 584-594.
Corona JC, Gimenez-Cassina A, Lim F, Díaz-Nido J. Hexokinase II Gene Transfer Protects against Neurodegeneration in the Rotenone and MPTP Mouse Models of Parkinson‘s Disease. Journal of Neuroscience Research 2010; 88:1943–1950.
Cox SML, Stefanova E, Johnsrude IS, Robbins TW, Owen AM. Preference formation and working memory in Parkinson‘s disease and normal ageing. Neuropsychologia 2002; 40: 317-326.
Cropley VL, Fujita M, Innis RB, Nathan PJ. Molecular Imaging of the Dopaminergic System and its Association with Human Cognitive Function. Biol Psychiatry 2006; 59: 898–907.
Da Cunha C, Angelucci MEM, Canteras NS, Wonnacott S, Takahashi RN. The Lesion of the Rat Substantia Nigra pars compacta Dopaminergic Neurons as a Model for Parkinson‘s Disease Memory Disabilities. Cellular and Molecular Neurobiology 2002; 22: 227-237.
Delfino MA, Stefano AV, Ferrario JE, Tavavini IRE, Murer MG, Gershanik OS. Behavioral sensitization to different dopamine agonists in a parkinsonian rodent model of drug-induced dyskinesias. Behavioural Brain Research 2004; 152; 297-306.
Díaz MR, Abdala P, Barroso-Chinea P, Obeso J, Gonzalez-Hernande T. Motor behavioural changes after intracerebroventricular injection of 6-hydroxydopamine in the rat: an animal model of Parkinson‘s disease. Behavioural Brain Research 2001; 122: 79–92.
Dutra RC, Andreazza AP, Andreatini R, Tufik S, Vital MABF. Behavioral effects of MK-801 on reserpine-treated mice. Progress in Neuro-Psychopharmacology & Biological Psychiatry 2002; 26; 487– 495.
Faria RR, Abílio VC, Grassl C, Chinen CC, Negrão LTR, de Castro JPMV, Fukushiro DF, Rodrigues MSD, Gomes Zanier PH, Registro S, de Carvalho RdeC, D‘Almeida V, Silva RH, Ribeiro RA; Frussa-Filho R. Beneficial effects of vitamin C and vitamin E on reserpine-induced oral dyskinesia in rats: Critical role of striatal catalase activity. Neuropharmacology 2005; 48: 993-1001.
Fernandes VS, Ribeiro AM, Melo TG, Godinho M, Barbosa FF, Medeiros DS, Munguba H, Silva RH. Memory impairment induced by low doses of reserpine in rats: Possible relationship with emotional processing deficits in Parkinson disease. Progress in Neuro-Psychopharmacology & Biological Psychiatry 2008; 32: 1479–1483.
Ferro MM, Bellissimo MI, Anselmo-Franci JÁ, Angellucci MEM, Canteras NS, Da Cunha C. Comparison of bilaterally 6-OHDA- and MPTP-lesioned rats as models of the early phase of Parkinson‘s disease: Histological, neurochemical,

101
motor and memory alterations. Journal of Neuroscience Methods 2005; 148: 78–87.
Gabrieli JDE. Memory systems analyses of mnemonic disorders in aging and age-related diseases. Proc Natl Acad Sci 1996; 93: 13534–40.
Gerlach M, Riederer P. Animal models of Parkinson's disease: an empirical comparison with the phenomenology of the disease in man. J Neural Transm, 1996; 103: 987-1041.
Góngora-Alfaro JL, Moo-Puc, RE, Villanueva-Toledo JR, Alvarez-Cervera FJ, Bata-García JL, Heredia-López FJ, Pineda JC. Long-lasting resistance to haloperidol-induced catalepsy in male rats chronically treated with caffeine. Neuroscience Letters 2009; 463: 210–214.
Halbig TD, Kopp UA, Wodarz F, Borod JC, Gracies JM, Ebersbach G, Kupsch A. Dopaminergic modulation of emotional memory in Parkinson‘s disease. J Neural Transm 2008; 115:1159–1163.
Hansen E, Casey DE, Hoffma WF. Neuroleptic Intolerance. Schizophrenia Bulletin 1997; 23: 567- 582.
Higginson CI, Fields JA, Troster AI. Which Symptoms of Anxiety Diminish After Surgical Interventions for Parkinson Disease? Neuropsychiatry, Neuropsychology, and Behavioral Neurology 2001; 14: 117-121.
Henderson JM, Stanic D, Tomas D, Patch J, Horne MK, Bourke D, Finkelstein DI. Postural changes after lesions of the substantia nigra pars reticulata in hemiparkinsonian monkeys. Behavioural Brain Research 2005; 160: 267–276.
Jicha GA, Salamone JD. Vacuous Jaw Movements and Feeding Deficits in Rats with Ventrolateral Striatal Dopamine Depletion: Possible Relation to Parkinsonian Symptoms. The Journal of Neuroscience 1991; 12: 3822-3929.
Johnston RE, Schallert T, Becker JBB. Akinesia and postural abnormality after unilateral dopamine depletion. Behavioural Brain Research 1999; 104: 189-196.
Kameda SR, Frussa-Filho R, Carvalho RC, Takatsu-Coleman AL, Ricardo VP, Patti CL, Calzavara MB, Lopez GB, Araujo NP, Abílio VC, Ribeiro R A, D‘Almeida V, Silva RH. Dissociation of the effects of ethanol on memory, anxiety, and motor behavior in mice tested in the plus-maze discriminative avoidance task. Psychopharmacology 2007; 192:39–48.
Klockgether T. Parkinson’s disease: clinical aspects. Cell Tissue Res. 2004; 318: 115–120.
Koerst J, Leenders KL, Koning M, Portman AT, Beilen NV. Striatal dopaminergic activity (FDOPA-PET) associated with cognitive items of depression scale (MADRS) in Parkinson‘s disease. Europen Journal of Neuroscience 2007; 25: 3132-3136.
Korczyn A D. Dementia in Parkinson‘s disease. J. Neurol. 2001; 248: III/1- III/4. Lindner MD, Cain CK, Plone MA, Frydel BR, Blaney TJ, Emerich DF, Hoane MR.
Incomplete nigrostriatal dopaminergic cell loss and partial reductions in striatal dopamine produce akinesia, rigidity, tremor and cognitive deficits in middle-aged rats. Behavioural Brain Research 1999; 102: 1-16.
Lowry OH, Rosebrough N, Farr AL, Randall RL. Protein measurement with Folin phenol reagent. Journal of Biological Chemestry 1951;193: 265–275.
Mahieux F, Fénelon G, Flahault A, Manifacier MJ, Michelet D, Boller F. Neuropsychological prediction of dementia in Parkinson‘s disease. J Neurol Neurosurg Psychiatry 1998; 64:178–183.
Mayeux R. Epidemiology of neurodegeneration. Annu. Rev. Neurosci. 2003; 26: 81–104.

102
Miller GW, Erickson JD, Perez JT, Penland SN, Mash DC, Rye DB, Levey AI. Immunochemical Analysis of Vesicular Monoamine Transporter (VMAT2) Protein in Parkinson's Disease. Experimental Neurology. 1999; 156: 138-148.
Morris RG, Downes JJ, Sahakian BJ, Evenden JL, Heald A, Robbinsii TW. Planning and spatial working memory in Parkinson's Disease. Journal of Neurology, Neurosurgery, and Psychiatry 1988; 51: 757-766.
Neisewander JL, Lucki I, McGonigle P. Neurochemical changes associated with the persistence of spontaneous oral dyskinesia in rats following chronic reserpine treatment Brain Research 1991; 558: 27-35.
Neisewander JL, Castañeda E, Davis DA. Dose-dependent differences in the development of reserpine-induced oral dyskinesia in rats: support for a model of tardive dyskinesia. Psychopharmacology 1994; 116: 79-84.
Niigaki ST, Silva RH, Patti CL, Cunha JLS, Kameda SR, Correia-Pinto JC, Takatsu-Coleman AL, Levin R, Abílio VC, Frussa-Filho R. Amnestic effect of cocaine after the termination of its stimulant action. Progress in Neuro-Psychopharmacology & Biological Psychiatry 2010; 34: 212–218.
Obata, T. Dopamine efflux by MPTP and hydroxyl radical generation. J Neural Transm 2002; 109: 1159–1180.
Ossowska K, Konieczny J, Wardas J, Gol ´embiowska K, Wolfarth S, Pilc A. The role of striatal metabotropic glutamate receptors in Parkinson‘s disease. Amino Acids 2002; 23: 193–198.
Peixoto MF, Araujo NP, Silva RH, Castro JPMV, Fukushiro DF, Faria RR, Zanier-Gomes PH, Medrano WA, Frussa-Filho R, Abílio VC. Effects of gabaergic drugs on reserpine-induced oral dyskinesia. Behavioural Brain Research 2005; 160: 51–59.
Postle BR, Locascio JJ, Corkin S, Growdon JH. The time course of spatial and object learning in Parkinson's disease. Neuropsychologia 1997; 35:1413–22.
Ribeiro AM, Barbosa FF, Godinho MR, Fernandes VS, Munguba H, Melo TG, Barbosa MT, Eufrasio RA, Cabral A, Izídio GS, Silva RH. Sex differences in aversive memory in rats: Possible role of extinction and reactive emotional factors. Brain and Cognition 2010; 74: 145–151.
Ridley RM, Cummings RM, Leow-Dyke A, Baker HF. Neglect of memory after dopaminergic lesions in monkeys. Behavioural Brain Research 2006; 166: 253-262.
Riobó NA, Schopfer FJ, Boveris AD, Cadenas E, Poderoso JJ. The reaction of nitric oxide with 6-hydroxydopamine: implications for Parknson‘s disease. Free Radical Biology & Medicine 2002; 32: 115–121.
Robertson LT, Horak FB, Anderson VC, Burchiel KJ, Hammerstad JP. Assessments of Axial Motor Control during Deep Brain Stimulation in Parkinsonian Patients. Neurosurgery 2001; 48: 544–552.
Salamone J, Baskin P.Vacuous Jaw Movements Induced by Acute Reserpine and Low-Dose Apomorphine: Possible Model of Parkinsonian Tremor. Pharmacology Biochemistry and Behavior 1996; 53: 179-183.
Salamone JD, Ishiwari K, Betz AJ, Farrar AM, Mingote SM, Font L, Hockemeyer J, Muller CE, Correa M. Dopamine/adenosine interactions related to locomotion and tremor in animal models: Possible relevance to parkinsonism. Parkinsonism and Related Disorders 2008; 14: S130-S134.
Salgado-Pineda P, Delaveau P, Blin O, Nieoullon A. Dopaminergic contribution to the regulation of emotional perception. Clin Neuropharmacol 2005; 28: 228–37.

103
Schneider K, Habel U, Volkmann J, Regel S, Kornischka J, Sturm V, et al. Deep brain stimulation of the subthalamic nucleus enhances emotional processing in Parkinson disease. Arch Gen Psychiatry 2003; 60: 296–302.
Schimitt-Eliassen, J; Ferstl, R; Wiesner, C; Deuschl, G; Witt, K. Feedback-based versus observational classification learning in healthy aging and Parkinson‘s disease. Brain Research, 2007; 1142: 178-188.
Shiozaki S, Ichikawa S, Kitamura JNS, Yamada K, Kuwana Y. Actions of adenosine A2A receptor antagonist KW-6002 on drug-induced catalepsy and hypokinesia caused by reserpine or MPTP. Psychopharmacology 1999; 147: 90–95.
Silva RH, Frussa-Filho R. The plus-maze discriminative avoidance task: a new model to study memory–anxiety interactions. Effects of chlordiazepoxide and caffeine. J Neurosci Methods 2000; 102: 117–25.
Silva RH, Abílio VC, Torres-Leite D, Bergamo M, Chinen CC, Claro FT, Carvalho RC, Frussa-Filho R. Concomitant development of oral dyskinesia and memory deficits in reserpine-treated male and female mice. Behavioural Brain Research 2002a; 132: 171–177.
Silva RH, Kameda SR, Carvalho RC, Rigo GS, Costa KLB, Taricano ID, Frussa-Filho R. Effects of amphetamine on the plus-maze discriminative avoidance task in mice. Psychopharmacology 2002b; 160: 9–18.
Skalisy LL, Beijamini V, Joca SL, Vital MABF, da Cunha C, Andreatini R. Evaluation of the face validity of reserpine administration as an animal modelo of depression-Parkinson‘s disease association. Progress in Neuro-Psychopharmacology & Biological Psychiatry 2002; 26: 879-883.
Spina MB, Cohen G. Dopamine turnover and glutathione oxidation: Implications for Parkinson disease. Proc. Natl. Acad. Sci. USA. 1989; 86: 1398-1400.
Sussman AN, Tran-Nguyen LTL, Neisewander JL. Acute reserpine administration elicits long-term spontaneous oral Dyskinesia. European Journal of Pharmacology 1997; 337: 157–160.
Tadaiesky MT, Andreatini R, Vital MABF. Different effects of 7-nitroindazole in reserpine-induced hypolocomotion in two strains of mice. European Journal of Pharmacology 2006; 535: 199–207.
Tanizawa H, Sazuka Y, Tabino Y. Micro-determination of lipoperoxide inthe mouse myocardium by thiobarbituric acid fluorophotometry. Chemical Pharmaceutical Bulletin 1981; 29: 2910–2914.
Taylor, T.N., Caudle, W. M., Shepherd, K.R., Noorian, A., Jackson, C.R., Iuvone, P. M., Weinshenker, D., Greene, J.G. & Miller, G.W. Nonmotor Symptoms of Parkinson‘s Disease Revealed in an Animal Model with Reduced Monoamine Storage Capacity. The Journal of Neuroscience2009; 29: 8103– 8113.
Teixeira AM, Reckziegel P, Müller L, Pereira RP, Roos DH, Rocha JBT, Bürger ME. Intense exercise potentiates oxidative stress in striatum of reserpine-treated animals. Pharmacology, Biochemistry and Behavior 2009; 92: 231–235.
Tsang AHK, Chung KKK. Oxidative and nitrosative stress in Parkinson's disease. Biochimica et Biophysica Acta 2009; 1792: 643–650.
Verbaan D, Marinus J, Visser M, van Rooden SMA, Stiggelbout M, Middelkoop HAMJ, van Hilten J. Cognitive impairment in Parkinson‘s disease. J Neurol Neurosurg Psychiatry 2007; 78:1182–1187.
Zgaljardic DJ, Foldi NS, Borod JC. Cognitive and behavioral dysfunction in Parkinson's disease: neurochemical and clinicopathogical contributions. J. Neural Transm 2004; 111: 1287–301.

104
3.Discussão geral
e conclusão

105
3. Discussão geral e conclusão
Neste trabalho, investigamos os efeitos da administração repetida com doses
baixas de reserpina em parâmetros motores e cognitivos. Podemos observar nos
resultados tanto do experimento I quanto no experimento II que o tratamento
repetido com reserpina (0,1 e 0,2 mg/Kg) é capaz de induzir alterações motoras. No
experimento I, observamos alteração no comportamento de catalepsia avaliado vinte
e quatro horas após a nona, a décima segunda e a decima quinta injeções para os
grupos RES 0,1 e 0,2. No experimento II, pode ser observado na avaliação do
comportamento de catalepsia, realizada antes, 24 e 48 horas após cada injeção, que
a alteração motora aparenta ser progressiva. Na avaliação de outros parâmetros
motores, como os movimentos orofaciais, também podemos observar uma alteração
vinte e quatro horas após a décima quarta injeção, no experimento I, e vinte e quatro
e quarenta e oito horas após a décima injeção, no experimento II. Nos parâmetros
cognitivos observamos no experimento I, as alterações encontradas foram
acompanhadas por déficits motores nos grupos RES 0,1 e 0,2. Entretanto, no
experimento II, foram encontrados déficits motores sem alterações cognitivas no
grupo RES 0,1 vinte e quatro e quarenta e oito horas após a sétima injeção. Os
fatores bioquímicos analisados revelaram um aumento dos níveis de estresse
oxidativo no estriado dos animais tratados com RES 0,1, quarenta e oito horas após
a décima injeção (experimento II) e, uma diminuição da concetração de glutamato
estrital dos animais dos grupos RES 0,1 e 0,2, cinco dias após a décima quinta
injeção (experimento I).
No experimento I, observamos que os ratos tratados com doses repetidas de
reserpina (0,1 e 0,2 mg/Kg) apresentaram alteração motora durante o periodo de

106
tratamento. Estes dados podem ser observados na avaliação do comportamento de
catalepsia (24 horas após a nona, a décima segunda e a décima quinta injeções), na
distância percorrida no labirinto em cruz da esquiva discriminativa (na sessão de
treino, 24 horas após a décima injeção), nas tentativas de escapar no teste de medo
condicionado ao contexto (48 e 72 horas após a décima quinta injeção). Contudo, os
animais do grupo RES 0,05 não apresentaram alterações motoras durante o
tratamento. Os resultados dos grupos RES 0,1 e 0,2 indicaram que o tratamento
proposto pode apresentar estágios com ausência ou presença de alterações
motoras. Entretanto, somente com estes dados não era possível determinar se as
alterações motoras eram decorrentes de uma modificação progressiva permanente
nas vias motoras ou de um efeito imediato de uma determinada injeção de
reserpina. Dessa forma, no experimento II, escolhemos a dose de 0,1 mg/ Kg de
reserpina pois esta foi capaz de induzir estágios nas alterações motoras (ausência e
presença de alterações motoras) e avaliamos o efeito da reserpina sobre a
motricidade (por meio da avaliação da catalepsia, um parâmetro importante para a
detecção de sinais motores em modelos animais de PD) (Chinen & Frussa-Filho
1999, Diaz et al. 2001) vinte e quatro horas e quarenta e oito horas após as
administrações, durante todo o tratamento. Assim, no experimento II, os resultados
demonstraram que a administração repetida de 0,1 mg/kg de reserpina induziu um
aumento gradual do tempo de imobilidade cataléptica, quando comparado com o
grupo controle. Estes dados indicam uma possível semelhança ao processo
neurodegenerativo encontrados em humanos com DP onde as alterações gradativas
dos circuitos dos núcleos da base provocam um aparecimento progressivo dos
sintomas parkinsonianos (distúrbios comportamentais nos quais a rigidez, o tremor e
a bradicinesia são caracteríticos) (Johnston et al. 1999, Lindner et al. 1999,

107
Deumens et al. 2002, Ridley et al. 2006, Meredith et al. 2008a). Além disso,
evidências sugerem que o comportamento de catalepsia em animais está
diretamente relacionado à alterações no sistema dopaminérgico (Chinen et al. 1999,
Gongora-Alfaro et al. 2009). Um estudo em ratos com administração intraventricular
de 6-OHDA demonstrou um efeito dose dependente no tempo de permanência da
posição caléptica (Díaz et al. 2001). Dessa forma, os dados apresentados no
experimento II parecem corroborar estudos que mostram que a reserpina induz
alterações motoras (Kannari et al. 2000, Farley et al. 2006). Outro dado importante é
que no experimento II encontramos um aumento dos níveis de peroxidação lipídica
no estriado concomitante ao aumento do comportamento cataléptico sugerindo uma
relação entre os fatores.
Outro parâmetro motor avaliado nos experimento I e II foi os movimentos
orofaciais. Esta avaliação foi realizada através de três parâmetros que são: o
número de protrusões de língua; o número de movimentos de mastigação que não
fosse direcionado a nenhum objeto (vacuous chewing movements); e a duração de
tremores de queixo (em segundos). O comportamento de mastigação é um
movimento repetitivo da mandíbula (de freqüência de 03-06 Hz) que pode ser
induzido por alterações dopaminérgicas (Jicha & Salamone 1999, Díaz et al. 2001) e
tem sido proposto por alguns pesquisadores como um modelo de tremor
parkinsoniano (Salamone & Baskin 1996, Dutra et al. 2002). No experimento I,
observamos que os animais dos grupos RES 0,1 e 0,2 apresentaram um aumento
em todos os parâmetros orofacias avaliados, vinte e quatro horas após a décima
quarta injeção. Aparentemente, este resultado foi concomitante com as alterações
motoras encontradas na avaliação do comportameto cataléptico. Entretanto, não foi
possível confirmar esta hipótese já que não havíamos feito a mensuração da

108
catalepsia neste período do tratamento. Dessa forma, no experimento II, avaliamos
os movimentos orofaciais vinte e quatro horas após a quinta e a décima injeção, e
quarenta e oito horas após a décima injeção. Os resultados obtidos mostraram que
os animais do grupo RES 0,1 apresentaram alterações nos movimentos orofaciais
24 e 48 horas após a décima injeção, concomitantemente às alterações motoras das
análises do comportamento de catalepsia. Estudos anteriores com administração
aguda de reserpina corroboram com estes resultados encontrados (Faria et al. 2005,
Peixoto et al. 2005). Além disso, Sussman et al. (1997) demonstraram que a
discinesia oral induzida pela reserpina persistiu por 84 dias e produziu uma
dimunição de 74% da dopamina da via caudado-putâmen três dias após a injeção
mas esta diminuição não foi observada 84 dias após a injeção. Dessa forma, as
mudanças neurobiológicas subjacentes ao comportamento em debate ainda não
estão bem compreendidas.
Um parâmetro cognitivo avaliado no experimento I foi o modelo da esquiva
discriminativa em labirinto em cruz elevado. Este modelo permite a avaliação
concomitantemente, por parâmetros diferentes, da memória, da ansiedade e do
comportamento motor (Silva & Frussa-Filho 2000, Silva et al 2002a). Os resultados
mostraram que o grupo RES 0,05 não apresentou alteração nos parâmentros
avaliados. Entretando, o grupo RES 0,1 apresentou alteração na porcentagem de
tempo de permanência no braço aversivo concomitantemente às alterações
motoras. As alterações motoras encontradas no grupo RES 0,1 não prejudicaram a
aquisição da tarefa, no treino, prejuízo que foi constatado no grupo RES 0,2. Dessa
forma, no experimento II, decidimos utilizar a dose de 0,1mg/Kg de reserpina em um
período do tratamento onde não houvesse (ou fossem mínimas) alterações motoras
na avaliação de catalepsia. Assim sendo, a esquiva discriminativa foi realizada vinte

109
e quatro e quarenta e oito horas após a sétima injeção. A administração repetida de
0,1mg/Kg de reserpina não induziu alteração na memória, mas houve alteração na
distância percorrida no labirinto. Estes resultados são contrários a outros estudos
nos quais os autores utilizaram um tratamento agudo de reserpina no mesmo
modelo comportamental (Silvia et al. 2002a, Carvalho et al. 2006). A este respeito
existem evidências na literatura mostrando que níveis excessivos ou insuficientes de
dopamina podem alterar a memória emocional (Cools et al. 2002, Halbing et al.
2008). Além disso, estudos indicam que as vias dopaminérgicas têm propriedades
intrínsecas capazes de ativar mecanismos compensatórios que são diferenciados
dependendo nos níveis de alteração (Castaneda et al. 1990, Bezard & Gross 1998,
Berzard et al. 2001, Mawlawi et al. 2001, Cropley et al. 2006). Assim sendo, a dose e
o protocolo de tratamento utilizado no experimento II provavelmente pode ter
induzido um decréscimo dos níveis de dopamina que foram contrabalanceados por
um mecanismo compensatório de plasticidade nos sistemas neuronais relacionados
à memória. Portanto, o tratamento repetido com reserpina, foi eficiente na indução
de déficits motores, mas não em alterações cognitivas, pelo menos nos paradigmas
utilizados neste trabalho. Entretanto, deficiências cognitivas foram observadas em
ratos lesados com MPTP nos testes de esquiva ativa e labirinto aquático (Da Cunha
et al. 2002) e em pacientes com DP (Bowers et al. 2006, Halbig et al. 2008). Dessa
forma, seria interessente verificar os efeitos do tratamento repetido de reserpina 0,1
mg/kg em outros modelos de memória ou mesmo em outras funções cognitivas.
No desenvolvimento da DP ocorrem mortes de neurônios dopaminérgicos,
entretanto, existe um período que precede o aparecimento dos primeiros sinais
clínicos onde o sistema mantem certo nível de funcionamento (Bezard & Gross
1998). Tudo indica que as vias dopaminérgicas possuem propriedades intrínsecas

110
capazes de acionar mecanismos compensatórios (Castaneda et al. 1990, Bezard &
Gross 1998, Berzard et al. 2001, Mawlawi et al. 2001, Cropley et al. 2006). Estes
mecanismos compensatórios parecem ser diferenciados dependendo do nível de
perda de dopamina (Bezard & Gross 1998). Estes mecanismos podem ser através
da regulação dos receptores de dopamina assim como super sensibilização deste ou
aumento de liberação de dopamina em terminais remanescentes como demonstrado
em animais lesados com 6-OHDA (Castaneda et al. 1990). Entretanto, existem
contradições quanto à relação entre perdas de células dopaminérgicas e sintomas
clínicos (Bezard & Gross, 1998). Este fato sugere que os sintomas clínicos poderiam
estar vinculados não somente com as alterações dopaminérgicas, mas também com
a atuação de outros sistemas não-dopaminérgicos (Bezard & Gross 1998, Blandini
et al. 2000). Assim sendo, no experimento I, foi observada diminuição na
concentração do nível de glutamato estriatal dos grupos RES 0,1 e 0,2 cinco dias
após a décima quinta injeção. Este resultado corrobora o estudo de Day et al. (2006)
que mostraram que ocorre perda de sinapses glutamatérgicas no estriato-pallidal
após a depleção dopaminérgica. Seria interessante ter realizado análises
bioquímicas dos níveis de dopamina no estriado dos ratos do experimento I.
Contudo, por problemas de ordem técnica, não foi possível realizar estas análises.
Em vista destes fatores, e de evidências que indicam uma relação de estresse
oxidativo cerebral com a fisiopatologia da doença de Parkinson (Cadenas & Davies
2000, Beal 2003), resolvemos avaliar os danos oxidativos no estriado e no
hipocampo causado pela reserpina no experimento II. Assim sendo, no experimento
II, foi observado um aumento nos níveis de estresse oxidativo do estriato, 24 horas
após a décima injeção, o que mostra que possíveis danos celulares podem estar
ocorrendo nesta área. De uma forma geral, uma hipótese que podemos levantar dos

111
dados encontrados nos dois experimentos seria: a redução da atividade
dopaminérgica nigroestriatal induzida por danos oxidativos alteraria a atividade do
globo pallidum e da substância nigra reticutala, resultando na diminuição da
atividade talâmina e cortical, que finalmente, levaria a redução da atividade
glutamatérgica corticoestriatal. Roberts et al. (1982) sugerem que um possível
mecanismo compensatório para a redução da atividade glutamatérgica
corticoestriatal que seria o aumento de receptores NMDA levando a uma
supersensibilidade que compensaria a regulação da liberação de dopamina.
A reserpina é uma droga que evita o armazenamento de monoaminas nas
vesículas sinápticas através da inibição da ação dos transportadores da membrana
que captam as monoaminas para dentro da vesícula (Liu et al. 1996, Verheij & Cools
2007). Dessa forma, as vesículas sinápticas permanecem vazias e
conseqüentemente não há neurotransmissores para serem liberados na fenda
sináptica quando um potencial de ação atinge o botão sináptico (Rang et al. 2004).
Então, a atuação da reserpina não se restringe apenas nas vias dopaminérgicas,
atuando também nas vias noradrenérgicas e serotoninérgicas. Este fator poderia ser
uma limitação da utilização desta droga como um modelo farmacológico de
Parkinson se não houvesse relatos na literatura demonstrando que existem também
alterações em outras vias monoaminérgicas na DP (Devos et al. 2010, Fox et al.
2009).
Podemos concluir através dos resultados obtidos nos experimentos I e II que:
1. A administração repetida de 0,1 mg/Kg de reserpina em ratos é capaz de
induzir o aparecimento gradual de sinais motores compatíveis com as
características progressivas encontrados em pacientes com DP (Klockgether
2004);

112
2. Os sinais motores induzidos através da administração repetida de 0,1 mg/Kg
de reserpina em ratos foram acompanhados por um aumento dos níveis de
estresse oxidativo no estriado que está de acordo com trabalhos que
sustentam a hipótese do aumento de radicais livres estarem relacionados à
DP (Cadenas & Davies 2000, Beal 2003, Abílio et al. 2004, Faria et al. 2005,
Teixeira et al. 2009);
3. Foram encontradas alterações nas concentrações de glutamato no estriato
nos grupos tratados com doses repetidas de 0,1 e 0,2 mg/Kg, cinco dias após
a décima quinta injeção, corroborando os estudos prévios;
4. O protocolo de tratamento aplicado não foi capaz de induzir déficits
cognitivos sem alteração motora, dados corroborados pela ausência de
alteração dos níveis de estresse oxidativo no hipocampo.
Dessa forma, são necessários mais estudos para a compreensão das
mudanças nos sistemas de neurotransmissão durante o processo de
aparecimento dos sinais parkinsonianos no modelo proposto.

113
4.Referências

114
4. Referências
Aarsland ,D., Andersen, K., Larsen, J. P., Perry, R., Wentzel-Larsen ,T., Lolk , A.,
& Kragh-Sorensen ,P. 2004 .The Rate of Cognitive Decline in Parkinson
Disease. Arch Neurol., 61, 1906-1911.
Aarsland, D., Ehrt, K. B. U., Deyn, P. P. De, Emre, S. T., M & Cummings, J. L.
2007. Neuropsychiatric symptoms in patients with Parkinson‘s disease and
dementia: frequency, profile and associated care giver stress. J Neurol
Neurosurg Psychiatry,78, 36–42.
Abílio, V. C., Vera, J. A. Jr., Ferreira, L. S., Duarte, C. R., Carvalho, R. C., Grassl,
C., Martins, C. R., Torres-Leite, D., Bignotto, M., Tufik, S., Ribeiro, R. de A
& Frussa-Filho, R. 2002. Effects of melatonin on orofacial movements in rats.
Psychopharmacology, 161, 340-7.
Abílio, V.C., Araujo, C.C.S., Bergamo, M., Calvente, P.R.V. & D’Almeida, V.de A.
R.R. 2003. Vitamin E attenuates reserpine-induced oral dyskinesia and striatal
gssg/gsh ratio enhancement in rats. Prog Neuro- Psychopharmacol Biol
Psychiat, 27,109–14.
Abílio, V.C., Silva, R.H., Carvalho, R.C.,Grassl, C., Calzavara, M.B. Registro, S.
2004. Important role of striatal catalase in aging- and reserpine-induced oral
dyskinesia. Neuropharmacology, 47,263–72.
Aguiar Jr., Aderbal S., Araújo, Andréa L., da-Cunha, Thaise R., Speck, A. E.,
Ignácio, Z. M., De-Mello, N. & Prediger, R. D. S. 2009. Physical exercise
improves motor and short-term social memory deficits in reserpinized rats. Brain
Research Bulletin, 79, 452–457.

115
Albouy, G., Sterpenich, V., Balteau, E., Vandewalle, G., Desseilles, M., Dang-Vu,
T., Darsaud, A., Ruby, P., Luppi, P.H., Degueldre, C., Peigneux, P., Luxen,
A. & Maquet, P. 2008. Both the hippocampus and striatum are involved in
consolidation of motor sequence memory. Neuron., 58, 261-72.
Alves, C. S. D., Andreatini, R., da Cunha, C., Tufik, S. & Vital, M.A.B.F. 2000.
Phosphatidylserine reserves reserpine-induced amnesia. European Journal of
Pharmacology, 404, 161-167.
Antonini, A., Leenders, K. L., Vontobel, P., Maguire, R. P., Missimer, J., Psylla,
M. & Gunther, I. 1997. Complementary PET studies of striatal neuronal
function in the differential diagnosis between multiple system atrophy and
Parkinson‘s disease. Brain, 120, 2187–2195.
Bains, J. S. & Shaw, C. A. 1997. Neurodegenerative disorders in humans: the role
of glutathione in oxidative stress-mediated neuronal death. Brain Research
Reviews, 25, 335–358.
Beal, M. F. 2000. Oxidative Metabolism. Ann. N. Y. Acad. Sci., 164-169.
Beal, M. F. 2002. Oxidatively modified proteins in aging and disease. Free Radical
Biology & Medicine, 32, 797–803.
Beal, M. F. 2003. Mitochondria, Oxidative damage, and Inflammation in Parkinson's
disease. Ann. N. Y. Acad. Sci., 991, 120-131.
Bellissimo, M. I., Kouzmine, I., Ferro, M. M., Oliveira, B. H. de, Canteras, N. S. &
Da Cunha, C. 2004. Is the unilateral lesion of the left substantia nigra pars
compacta sufficient to induce working memory impairment in rats?
Neurobiology of Learning and Memory, 82, 150–158.
Benecke, R., Rothwell, J. C., Dick, J. P. R., Day, B. L. & Marsden, C. D. 1987.
Simple and complex movements off and on treatment in patients with

116
Parkinson's disease. Journal of Neurology, Neurosurgery, and Psychiatry, 50,
296-303.
Bennett, D. A., Beckett, L. A., Murray, A. M.; Shannon, K. M., Goetz, C. G.;
Pilgrim, D. M. & Evans, D. A. 1996. Prevalence of parkinsonian signs and
associated mortality in a community population of older people. The New
England Journal of Medicine, 334, 71- 76.
Bertram, C. P.; Lemay, M. & Stelmach, G. E. 2005. The effect of Parkinson_s
disease on the control of multi-segmental coordination. Brain and Cognition, 57,
16–20.
Bezard, E. & Gross, C. E. 1998. Compensatory mechanisms in experimental and
human parkinsonism: towards a dynamic approach. Progress in Neurobiology,
55, 93-116.
Bezard, E., Dovero, S, Prunier, C., Ravenscroft, P., Chalon, S., Guilloteau, D.,
Crossman, A. R., Bioulac, B., Brotchie, J. M. & Gross, C. E. 2001.
Relationship between the Appearance of Symptoms and the Level of
Nigrostriatal Degeneration in a Progressive 1-Methyl-4-Phenyl- 1,2,3,6-
Tetrahydropyridine-Lesioned Macaque Model of Parkinson‘s Disease. The
Journal of Neuroscience, 21, 6853–6861.
Bezard, E., Boraud, T., Bioulac, B. & Gross, C. E. 1997. Compensatory effects of
glutamatergic inputs to the substantia nigra pars compacta in experimental
parkinsonism. Neuroscience, 81, 399–404.
Bianchi, L., Galeff, J.P. & Corte, L. D. 2003. The effect of 6-hydroxydopamine
lesions on the release of amino acids in the direct and indirect pathways of the
basal ganglia: a dual microdialysis probe analysis. European Journal of
Neuroscience, 18, 856-868.

117
Bilska, A., Dubiel, M., Sokolowska-Jezewicz, M., Lorenc-Koci, E. & Wlodek, L.
2007. Alpha-lipoic acid differently affects the reserpine-induced oxidative stress
in the striatum and prefrontal cortex of rat brain. Neuroscience, 146, 1758–
1771.
Blandini, F., Armentero, M. & Martignoni, E. 2008. The 6-hydroxydopamine model:
News from the past. Parkinsonism and Related Disorders, 14, 124-129.
Blandini, F., Nappi, G, Tassorelli, C & Martignoni, E. 2000. Functional changes of
the basal ganglia circuitry in Parkinson's disease. Progress in Neurobiology,
62,63-88.
Bonsi, P., Cuomo, D., Picconi, B., Sciamanna G., Tscherter, A., Tolu, M.,
Bernardi, G., Calabresi, P. & Pisani, A. 2007. Striatal metabotropic glutamate
receptors as a target for pharmacotherapy in Parkinson‘s disease. Amino Acids,
32, 189–195.
Borek, L. L., Amick, M. M. & Friedman, J. H. 2006. Non-Motor Aspects of
Parkinson‘s Disease. CNS Spectr., 11, 541-554.
Bowers, D, Miller, K, Mikos, A, Kirsch-Darrow, L, Springer, U, Fernandez, H,
Foote, K & Okun, M. 2006. Starling facts about emotion in Parkinson‘s disease:
blunted reactivity to aversive stimuli. Brain, 129, 3356-3365.
Brayne, I. L. 2006. A systematic review of depression and mental illness preceding
Parkinson‘s disease. Acta Neurol Scand, 113, 211–220.
Burn, D. J., Rowan, E. N., M Allan, L., Molloy, S., O’Brien, J. T. & McKeith, I. G.
2006. Motor subtype and cognitive decline in Parkinson‘s disease, Parkinson‘s
disease with dementia, and dementia with Lewy bodies. J. Neurol. Neurosurg.
Psychiatry,77,585-589.

118
Cadenas, E. & Davies, K. J. A. 2000. Mitochondrial free radical generation,
oxidative stress, and aging. Free Radical Biology & Medicine, 29, 222–230.
Calne, D.B. 2007. Parkinson‘s disease is not one disease. Parkinsonism and
Related Disordes, 7, 3-7.
Carvalho, R. C., Patti, C. C., Takatsu-Coleman, A. L., Kameda, S. R., Souza, C.
F., Garcez-do-Carmo, L. & Abílio, V. C. 2006. Effects of reserpina on the pluz-
maze discriminative avoidance task: Dissociation between memory and motor
impairments. Brain Research, 1122, 176-183.
Castaneda, E., Whishaw, I.Q. & Robinson, T.E. 1990. Changes in striatal
dopamine neurotransmission assessed with microdialysis following recovery
from a bilateral 6-OHDA lesion: variation as a function of lesion size. J
Neurosci., 10, 1847-54.
Chang, J. W., Wachtel, S. R., Young, D. & Kang, U.J. 1999. Biochemical and
anatomical characterization of forepaw adjusting steps in rat models of
Parkinson‘s disease: studies on medial forebrain bundle and striatal lesions.
Neuroscience, 88, 617–628.
Chaudhuri, K. R. & Schapira, A. H. V. 2009. Non-motor symptoms of Parkinson‘s
disease: dopaminergic pathophysiology and treatment. Lancet Neurol, 8, 464–
74.
Caudle, W.M., Richardson, J.R., Wang, M.Z., Taylor, T.N., Guillot, T.S.,
McCormack, A.L., Colebrooke, R.E., Monte, D.A., Emson, P.C. & Miller,
G.W. 2007. Reduced Vesicular Storage of Dopamine Causes Progressive
Nigrostriatal Neurodegeneration. The Journal of Neuroscience, 27, 8138–8148.

119
Chia, L., Cheng, L., Chuo, L. J., Cheng, F. C. & Cu, J. S. 1995. Studies of
dementia, depression, electrophysiology and cerebrospinal fluid monoamine
metabolites in patients with Parkinson‘s disease. Journal of the Neurological
Sciences, 133, 73-78.
Chinen, C. C. & Frussa-Filho, R. 1999. Conditioning to Injection Procedures and
Repeated Testing Increase SCH 23390-Induced Catalepsy in Mice. N.
Europsychopharmacology, 21,670-678.
Cilia, R., Righini, A., Marotta, G., Benti, R., Marconi, R., Isaias, I. U., Pezzoli, G. &
Antonini, A. 2007. Clinical and imaging characterization of a patient with
idiopathic progressive ataxia and palatal tremor. European Journal of
Neurology, 14, 944-946.
Colpaert, F.C. 1987. Pharmacological characteristics of tremor, rigidity and
hypokinesia induced by reserpine in rat. Neuropharmacology, 26, 1431-1440.
Cools, R., Stefanova, E., Barker, R.A., Robbins, T.W. & Owen, A.M. 2002.
Dopaminergic modulation of high-level cognitition in Parkinson‘s disease: the
role of the prefrontal cortex revealed by PET. Brain,125, 584-594.
Cools, R. 2006. Dopaminergic modulation of cognitive function- implications for L-
Dopa treatment in Parkinson‘s disease. Neuroscience and Biobehavioral
Reviews, 30, 1-23.
Cools, R., Stefanova, E., Barker, R.A., Robbins, T.W. & Owen, A. M. 2002.
Dopaminergic modulation of high-level cognitition in Parkinson‘s disearse: the
role of the preforntal cortex revealed by PET. Brain, 125, 584-594.
Cooper, J., Sagar, H.J., Jordan, N., Harvey, N.S & Sullivan, E.V. 1991. Cognitive
impairment in early, untreated Parkinson‘s disease and its relationship to motor
disability. Clin. Neuropsychol, 14, 38-55.

120
Costa, R. M., Cohen, D. & Nicolelis, M. A. L. 2004. Differential Corticostriatal
Plasticity during Fast and Slow Motor Skill Learning in Mice. Current Biology,
14, 1124–1134.
Costa, R. M. , Lin, S.C., Sotnikova,T. D. , Cyr, M., Gainetdinov, R. R., Caron, M
G. & Nicolelis, M. A.L. 2006. Rapid Alterations in Corticostriatal Ensemble
Coordination during Acute Dopamine-Dependent Motor Dysfunction. Neuron,
52, 359–369.
Cousins, M. S. & Salamone, J. D.. 1996. Invovement of ventolateral striatal
dopamine in movement initiation and execution: a microdialysis and behavioral
investigation. Neuroscience, 70, 849-859.
Cropley, V.L., Fujita, M., Innis, R.B. & Nathan, P.J. 2006. Molecular Imaging of the
Dopaminergic System and its Association with Human Cognitive Function. Biol
Psychiatry, 59, 898–907.
Cubo, E., Bernard, B., Leurgans, S. & Raman, R. 2000. Cognitive and Motor
Function in Patients with Parkinson‘s Disease with and without Depression.
Clinical Neuropharmacology, 23, 331–334.
Da Cunha, C., Gevaerd, M.S., Vital, M.A.B.F., Miyoshi, E., Andreatini, R.,
Silveira, R., Takahashi, R.N. & Canteras, N.S. 2001. Memory disruption in
rats with nigral lesions induced by MPTP: a model for early Parkinson‘s disease
amnésia. Behavioural Brain Research, 124, 9–18.
Da Cunha, C., Angelucci, M.E.M., Canteras, N.S., Wonnacott, S. & Takahashi, R.
N. 2002. The Lesion of the Rat Substantia Nigra pars compacta Dopaminergic
Neurons as a Model for Parkinson‘s Disease Memory Disabilities. Cellular and
Molecular Neurobiology, 22, 227-237.

121
Day, M., Wang, Z., Ding, J., An, X., Ingham, C.A., Shering, A.F., Wokosin, D.,
Ilijic, E., Sun, Z., Sampson, A.R., Mugnaini, E., Deutch, A.Y., Sesack, S.R.,
WArbuthnott, G. & Surmeier, D.J. 2006. Selective elimination of glutamatergic
synapses on striatopallidal neurons in Parkinson disease models. NATURE
NEUROSCIENCE, 9, 251-259.
Dauer, W. & Przedborski, S. 2003 .Parkinson‘s Disease: Mechanisms and Models.
Neuron, 39, 889–909.
Delfino, M.A., Stefano, A.V., Ferrario, J.E., Tavavini, I.R.E., Murer, M.G. &
Gershanik, O.S. 2004. Behavioral sensitization to different dopamine agonists
in a parkinsonian rodent model of drug-induced dyskinesias. Behavioural Brain
Research, 152, 297-306.
DeLong, M.R. & Wichmann, T. 2007. Circuits and Circuit Disorders of the Basal
Ganglia. Arch Neurol., 64, 20-24.
Deumens, R., Blokland, A. & Prickaerts, J. 2002. Modeling Parkinson‘s Disease in
Rats: An Evaluation of 6-OHDA Lesions of the Nigrostriatal Pathway.
Experimental Neurology, 175, 303–317.
Devos, D., Defebvrea, L. & Bordet, R. 2010. Dopaminergic and non-dopaminergic
pharmacological hypotheses for gait disorders in Parkinson‘s disease,
Fundamental & Clinical Pharmacology, 24, 407–421.
Díaz, M.R., Abdala, P., Barroso-Chinea, P., Obeso, J. & Gonzalez-Hernande, T.
2001. Motor behavioural changes after intracerebroventricular injection of 6-
hydroxydopamine in the rat: an animal model of Parkinson‘s disease.
Behavioural Brain Research, 122,79–92.

122
Dutra, R.C., Andreazza, A.P., Andreatini, R., Tufik, S. & Vital, M.A.B.F. 2002.
Behavioral effects of MK-801 on reserpine-treated mice. Progress in Neuro-
Psychopharmacology & Biological Psychiatry, 26, 487– 495.
Ulas, J., Weihmuller, F., Brunner, L.C., Joyce, J.N., Marshall, J.F. & Cotman, C.
W. 1994. Selective Increase of NMDA-Sensitive Glutamate Binding in the
Striatum of Parkinson‘s Disease, Alzheimer‘s Disease, and Mixed Parkinson‘s
Disease/Alzheimer‘s Disease Patients: An Autoradiographic Study. The Journal
of Neuroscience, 14, 6317-6324.
Faria, R.R., Abílio, V.C., Grassl, C., Chinen, C.C., Negrão, L.T.R., Castro, J.P.M.
V., Fukushiro, D.F., Rodrigues, M.S.D., Gomes, P.H.Z., Registro, S.,
Carvalho, R.C., D’Almeida, V., Silva, R.H., Ribeiro, R.A. & Frussa-Filho, R.
2005. Beneficial effects of vitamin C and vitamin E on reserpine-induced oral
dyskinesia in rats: Critical role of striatal catalase activity. Neuropharmacology,
48,993-1001.
Farley, C. M., Baella, S., Wacan, A.J.J., Crawford, C.A. & McDougall, S.A. 2006 .
Pre- and postsynaptic actions of a partial D2 receptor agonist in reserpinized
young rats: Longevity of agonistic effects. BRAIN RESEARCH, 1124, 37– 44.
Fénelon, G. 1997. Parkinson disease. Medical treatment. Presse Med, 26, 1357-61.
Fernandes, V.S., Ribeiro, A.M., Melo, T.G., Godinho, M., Barbosa, F.F.,
Medeiros, D.S., Munguba, H. & Silva, R.H. 2008. Memory impairment induced
by low doses of reserpine in rats: possible relationship with emotional
processing deficits in Parkinson disease. Progress in Neuro-
Psychopharmacology & Biological Psychiatry, 32,1479–1483.

123
Ferreira, A.L.A. & Matsubara, L.S. 1997. Radicais livres: conceitos, doenças
relacionadas, sistema de defesa e estresse oxidativo. Rev Ass Med Brasil, 43,
61-8.
Ferreira, T.L., Shammah-Lagnado, S.J., Bueno, O.F., Moreira, K.M., Fornari, R.V
& Oliveira, M.G. 2008. The indirect amygdala-dorsal striatum pathway
mediates conditioned freezing:Insights on emotional memory networks.
Neuroscience, 153,84-94.
Ferro, M.M., Bellissimo, M.I., Anselmo-Franci, J.A., Angellucci, M.E.M.,
Canteras, N. S & Da Cunha, C. 2005. Comparison of bilaterally 6-OHDA- and
MPTP-lesioned rats as models of the early phase of Parkinson‘s disease:
Histological, neurochemical, motor and memory alterations. Journal of
Neuroscience Methods, 148, 78–87.
Fox, S.H., Chuang, R. & Brotchie, J.M. 2009. Serotonin and Parkinson‘s Disease:
On Movement, Mood, and Madness. Movement Disorders, 24, 1255–1266.
Gerlach, M., Gsell, W., Kornhuber, J., Jellinger, K., Krieger, V., Pantucek, F.,
Vock, R. & Riederer, P. 1996. A post mortem study on neurochemical markers
of dopaminergic, GABA-ergic and glutamatergic neurons in basal ganglia-
thalamocortical circuits in Parkinson syndrome. Brain Research, 741, 142-152.
Glendinning, D.S. & Enoka, R.M. 1994. Motor unit behavior in Parkinson's disease.
Phys Ther., 74, 61-70.
Góngora-Alfaro, J.L., Moo-Puc, R.E., Villanueva-Toledo, J.R., Alvarez-Cervera,
F.J., Bata-García, J.L., Heredia-López, F.J. & Pineda, J.C. 2009. Long-lasting
resistance to haloperidol-induced catalepsy in male rats chronically treated with
caffeine. Neuroscience Letters, 463, 210–214.

124
Grossman, M. 1999. Sentence Processing in Parkinson‘s Disease. Brain and
Cognition, 40, 387–413.
Halbig, T.D., Kopp, U.A., Wodarz, F., Borod, J.C., Gracies, J.M., Ebersbach, G. &
Kupsch, A. 2008. Dopaminergic modulation of emotional memory in
Parkinson‘s disease. J Neural Transm, 115, 1159–1163.
Hallett, M. & Khoshbin, S. 1980. A physiological mechanism of bradykinesia. Brain ,
103, 301-314.
Heikkila, R. & Cohen, G. 1971. Further Studies on the Generation of Hydrogen
Peroxide by 6-Hydroxydopamine Potentiation by Ascorbic Acid. Molecular
Pharmacology, 8, 241-248.
Higginson, C.I., Fields, J.A. & Troster, A.I. 2001. Which Symptoms of Anxiety
Diminish After Surgical Interventions for Parkinson Disease? Neuropsychiatry,
Neuropsychology, and Behavioral Neurology, 14, 117-121.
Huang, C., Tang, C., Feigin, A., Lesser, M., Ma, Y., Pourfar, M., Dhawan, V. &
Eidelberg, D. 2007. Changes in network activity with the progression of
Parkinson‘s disease. Brain, 130, 1834-1846.
Jankovic,J. & Kapadia, A.S. 2001. Functional Decline in Parkinson Disease. Arch
Neurol. , 58, 1611-1615.
Jankovic J., McDermott M., Carter J., Gauthier S., Goetz C., Golbe L., Huber S.,
Koller W., Olanow C., Shoulson I. & et al. 1990. Variable expression of
Parkinson's disease: a base-line analysis of the DATATOP cohort. . Neurology,
40,1529-34.
Jellinger K.A. 1999. Post mortem studies in Parkinson's disease--is it possible to
detect brain areas for specific symptoms? J Neural Transm Suppl.,56, 1-29.

125
Jicha, G.A. & Salamone, J.D. 1999. Vacuous Jaw Movements and Feeding Deficits
in Rats with Ventrolateral Striatal Dopamine Depletion: Possible Relation to
Parkinsonian Symptoms. The Journal of Neuroscience, 12, 3822-3929.
Jinsmaa Y., Florang V.R., Rees, J.N., Anderson, D.G., Strack, S. & Doorn J.A.
2009. Products of Oxidative Stress Inhibit Aldehyde Oxidation and Reduction
Pathways in Dopamine Catabolism Yielding Elevated Levels of a Reactive
Intermediate. Chem Res Toxicol., 22, 835–841.
Johnston, R.E., Schallert, T., Becker, J.B.B. 1999. Akinesia and postural
abnormality after unilateral dopamine depletion. Behavioural Brain Research,
104, 189-196.
Kannari, K., Tanaka, H., Maeda, T., Tomiyama, M., Suda, T. & Matsunaga, M.
2000. Reserpine Pretreatment Prevents Increases in Extracellular Striatal
Dopamine Following L-DOPA Administration in Rats with Nigrostriatal
Denervation. J. Neurochem., 74, 263–269.
Kim, H., Gyu-Seek, R., Oh, S. & Park, W. 1999. NMDA receptor antagonists inhibit
apomorphine-induced climbing behavior not only in intact mice but also in
reserpine-treated mice. Behevioural Brain Research, 100, 135-142.
Kirik, D., Rosenblad, C. & Bjorklund, A. 1998. Characterization of Behavioral and
Neurodegenerative Changes Following Partial Lesions of the Nigrostriatal
Dopamine System Induced by Intrastriatal 6-Hydroxydopamine in the Rat.
Experimental Neurology, 152, 259–277.
Klockgether, T. 2004. Parkinson’s disease: clinical aspects. Cell Tissue Res., 318,
115–120.
Koerst, J., Leenders, K.L., Koning, M., Portman, A.T. & Beilen, N.V. 2007. Striatal
dopaminergic activity (FDOPA-PET) associated with cognitive items of

126
depression scale (MADRS) in Parkinson‘s disease. Europen Journal of
Neuroscience, 25, 3132-3136.
Korczyn, A.D. 2001. Dementia in Parkinson‘s disease. J. Neurol., 248 , III/1- III/4.
Lange, K.W., Kornhuber, J. & Riederer, P. 1997. Dopamine/Glutamate in
Parkinson‘s Disease. Neuroscience and Biobehavioral Reviews, 4, 393-400.
Lewis, S.J.G., Foltynie,T., Blackwell, A.D., Robbins, T.W., Owen, A.M. & Barker,
R.A. 2005. Heterogeneity of Parkinson‘s disease in the early clinical stages
using a data driven approach. J Neurol Neurosurg Psychiatry, 76, 343–348.
Lindner, M.D., Cain, C.K., Plone, M.A., Frydel, B.R., Blaney, T.J., Emerich, D.F. &
Hoane, M.R. 1999. Incomplete nigrostriatal dopaminergic cell loss and partial
reductions in striatal dopamine produce akinesia, rigidity, tremor and cognitive
deficits in middle-aged rats. Behavioural Brain Research, 102, 1-16.
Liu, Y., Peter, D., Merickel, A., Krantz, D., Finn, J. P. & Edwards, R.H. 1996.A
molecular analysis of vesicular amine transport. Behavioural Brain Research,
73, 51-58.
Louis, E.D., Tang, M.X., Cote, L., Alfaro, B., Mejia, H. & Marder, K. 1999.
Progression of Parkinsonian Signs in Parkinson Disease. Arch Neurol., 56, 334-
337.
Marchitti, S.A., Orlicky D.J., Brocker C., & Vasiliou V. 2010. Aldehyde
Dehydrogenase 3B1 (ALDH3B1): Immunohistochemical Tissue Distribution and
Cellular-specific Localization in Normal and Cancerous Human Tissues. J
Histochem Cytochem, 58, 765–783.
Marin, C., Aguilar, E., Mengod, G., Corte´s, R. & Obeso, J.A. 2007. Concomitant
short- and long-duration response to levodopa in the 6-OHDA-lesioned rat: a

127
behavioural and molecular study. European Journal of Neuroscience, 25, 259–
269.
Mayeux, R. 2003.Epidemiology neurodegeneration. Annu. Rev. Neurosci, 62, 81-
104.
Mawlawi, O., Martinez, D., Slifstein, M., Broft, A., Chatterjee, R., Hwang, D.R.,
Huang, Y., Simpson, No., Ngo, K., Van Heertum, R., Laruelle M. 2001.
Imaging Human Mesolimbic Dopamine Transmission With Positron Emission
Tomography: I. Accuracy and Precision of D2 Receptor Parameter
Measurements in Ventral Striatum. Journal of Cerebral Blood Flow and
Metabolism, 21, 1034–1057.
Meredith, G.E., Sonsalla, P.K. & Chesselet, M.F. 2008. Animal models of
Parkinson‘s disease progression. Acta Neuropathol, 115, 385–398.
Meredith, G.E., Totterdell, S., Potashkin, J.A. & Surmeier D.J. 2008 a. Modeling
PD pathogenesis in mice: Advantages of a chronic MPTP protocol.
Parkinsonism and Related Disorders, 14, 112-115.
Monchi, O., Petrides, M., Mejia-Constain, B. & Strafella, A.P. 2007. Cortical
activity in Parkinsons‘s disease during executive processing depends on striatal
involvement. Brain, 130, 233-244.
Murat, E. 2003. Dementia associated with Parkinson‘s disease. Lancet Neurology, 2,
229–37.
Nade, V. S., Dwivedi, S., Kawale, L. A., Upasani, C. D. & Yadav, A. V.. 2009.
Effect of Hibiscus rosa sinensis on reserpine-induced neurobehavioral and
biochemical alteration in rat. Indian Journal of Experimental Biology, 47, 559-
563.

128
Namba, M.M., Quock, R.M. & Malone, M.H. 1981. Effects of narcotic antagonists on
L-dopa reversal of reserpine-induced catalepsy and blepharoptosis in mice. Life
Sciences, 28, 1629-1636.
Nieoullon, A. 2002. Dopamine and the regulation of cognition and attention.
Progress in Neurobiology, 67, 53-83.
Nurmi. E., Ruottinen, H.M., Bergman, J., Haaparanta, M., Solin, O., Sonninen, P.
& Rinne, J.O. 2001. Rate of progression in Parkinson's disease: a 6-[18F]
fluoro-L-dopa PET study. Mov Disord., 16, 608-615.
Obata, T. 2002. Dopamine efflux by MPTP and hydroxyl radical generation. J Neural
Transm, 109, 1159–1180.
Ossowska, K., Konieczny, J., Wardas, J., Gol ´embiowska, K., Wolfarth, S. &
Pilc, A. 2002. The role of striatal metabotropic glutamate receptors in
Parkinson‘s disease. Amino Acids, 23, 193–198.
Owen, A.M., James, M., Leigh, P.N., Summers, B.A., Marsden, C.D., Quinn, N.P.,
Lange, K.W. & Robbins, T.W. 1992. Fronto-striatal cognitive deficits at different
stages of Parkinson's disease. Brain, 6, 1727-51.
Owen, A.M., Doyon, J., Dagher, A., Sadikot A. & Evans, A.C. 1998. Abnormal
basal ganglia outflow in Parkinson‘s disease identified with PET: Implications for
higher cortical functions. Brain, 121, 949–965.
Parkinson, J. 2002. An essay on the shaking palsy. J Neuropsychiatry Clin
Neurosci, 14, 223-236.
Paulus, W. & Jellinger, K. 1991. The neuropathologic basis of different clinical
subgroups of Parkinson's disease. J Neuropathol Exp Neurol, 50,743-55.
Peixoto, M.F., Araujo, N.P., Silva, R.H., Castro, J.P.M.V., Fukushiro, D.F., Faria,
R.n R., Zanier-Gomes, P.H., Medrano, W.A., Frussa-Filho, R. & Abílio, V.C.

129
2005. Effects of gabaergic drugs on reserpine-induced oral dyskinesia.
Behavioural Brain Research, 160, 51–59.
Perbal, S., Deweer, B., Pillon, B., Vidailhet, M., Dubois, B. & Pouthas, V. 2005.
Effects of internal clock and memory disorders on duration reproductions and
duration productions in pacients with Parkinson‘s disease. Brain and Cognition,
58, 35-48.
Perry, J.C., Da Cunha, C., Anselmo-Franci, J., Andreatini, R., Miyoshi, E., Tufik,
S. & Vital, M.A. 2004. Behavioural and neurochemical effects of
hosphatidylserine in MPTP lesion of the substantia nigra of rats. Eur J
Pharmacol, 26, 225-33.
Perry, J.C., Hipólide, D.C., Tufik, S., Martins, R.D., Da Cunha, C., Andreatini, R.
& Vital, M.A.B.F. 2005. Intra-nigral MPTP lesion in rats: Behavioral and
autoradiography studies. Experimental Neurology, 195, 322 – 329.
PILLON, B., ERTLE, S., DEWEER, B., SARAZIN, M., AGID, Y. & DUBOIS, B. 1996.
Memory for spatial location is affected in Parkinson's disease.
Neuropsychologia, 34, 77 -85.
Pillon, B., Deweer, B., Vidailhet, M., Bonnet, A.M., Hahn-Barma, V. & Dobois, B.
1997. Is impaired memory for spatial location in Parkinson‘s disease domain
specific or dependent on ‗strategic‘ processes Neuropsychologia, 36:1, 1-9.
Pillon, B., Ertle, S., Deweer, B., Bonnet, A. M., Vidailhet, M. & Dubois, B. 1997a.
Memory for spatial location in ―de novo‖ parkinsonian patients. Neurochologia,
35:3, 221-228.
Quinn, N., Critchley, P. & Marsden, C.D. 1987. Young onset Parkinson's disease.
Mov Disord., 2, 73-91.

130
Rabinovic, A.D. & Hastings, T.O.1998. Role of Endogenous Glutathione in the
Oxidation of Dopamine. J. Neurochem., 71, 2071-2078.
Rang, H.P., Dale, M.M., Ritter, J.M. & Moore, P.K. 2004. Farmacologia. Rio de
Janeiro: Elsevier, 5 ed., 903p.
Rauhala, P., Szirahi, I. & Chiueh, C.C. 1996. Peroxidation of brain lipids in vitro:
nitric oxide versus hydroxyl radicals. Free Radical Biology & Medicine, 21, 391-
394.
Reijnders, J.S.A.M., Ehrt, U., Lousberg, R., Aarsland, D. & Leentjens, A.F.G.
2009. The association between motor subtypes and psychopathology in
Parkinson‘s disease. Parkinsonism and Related Disorders, 15, 379–382.
Reksidler, A.B., Lima, M.M.S., Dombrowski, P., Andersen, M.L., Zanata, S.M.,
Andreatini, R., Tufik, S. & Vital, M.A.B.F. 2008. Repeated intranigral MPTP
administration: A new protocol of prolonged locomotor impairment mimicking
Parkinson‘s disease. Journal of Neuroscience Methods, 167, 268–277.
Richard, I.H., Frank, S., McDermott, M.P., Wang, H., Justus, A.W., LaDonna, K.
A. & Kurlan, R. 2004. The Ups and Downs of Parkinson Disease: A prospective
study of mood and anxiety flutuatuions. Cog. Behav. Neurol, 17, No. 4, 201-206.
Ridley, R.M., Cummings, R.M., Leow-Dyke, A., Baker, H.F. 2006. Neglect of
memory after dopaminergic lesions in monkeys. Behavioural Brain Research,
166, 253-262.
Riobó, N.A., Schopfer, F.J., Boveris, A.D., Cadenas, E. & Poderoso, J.J. 2002.
The reaction of nitric oxide with 6-hydroxydopamine: implications for
Parkinson‘s disease. Free Radical Biology & Medicine, 32, 115–121.

131
Roberts, P.J., McBean, G.J., Sharif, N.A. & Thomas, E.M. 1982. Striatal
glutamatergic function: modifications following specific lesions. Brain Res., 235,
83-91.
Salamone, J. & Baskin, P. 1996. Vacuous Jaw Movements Induced by Acute
Reserpine and Low-Dose Apomorphine: Possible Model of Parkinsonian
Tremor. Pharmacology Biochemistry and Behavior, 53, 179-183.
Scherfler, C., Khan, N.I., Pavese, N., Eunson, L., Graham, E., Lees, A.J., Quinn,
N.P., Wood, N.W., Brooks, D.J. & Piccini, P.P. 2004. Striatal and cortical pré-
and postsynaptic dopaminergic dysfuntion in sporadic parkin-linked
parkinsonism. Brain, 127, 1332-1342.
Schimitt-Eliassen, J., Ferstl, R., Wiesner, C., Deuschl, G. & Witt, K. 2007.
Feedback-based versus observational classification learning in healthy aging
and Parkinson‘s disease. Brain Research, 1142, 178-188.
Schneider, J.S. & Pope-Coleman, A. 1995. Cognitive deficits precede motor deficits
in a slowly progressing model of parkinsonism in the monkey.
Neurodegeneration, 4: 245-255.
Schrag, A. & Schott, J.M. 2006. Epidemiological, clinical, and genetic
characteristics of early-onset parkinsonism. Lancet Neurol, 5, 355–363.
Shohamy, D., Myers, C.E., Grossman, S., Sage, J. & Gluck, M.A. 2005. The role
of dopamine in cognitive sequence learning: evidence from Parkinon‘s disease.
Behavioural Brain Research, 159, 191-199.
Shults, C.W. 2003. Treatments of Parkinson Disease. Arch Neurol, 60: 1680:1684.
Silva, R.H. & Frussa-Filho, R. 2000. The plus-maze discriminative avoidance task: a
new model to study memory–anxiety interactions. Effects of chlordiazepoxide
and caffeine. Journal of Neuroscience Methods, 102, 117–125.

132
Silva, R.H., Abílio, V.C., Torres-Leite, D., Bergamo, M., Chinen, C.C., Claro, F.T.,
Cravalho, R. de C. & Frussa-Filho, R. 2002. Concomitant development of oral
dyskinesia and memory deficits in reserpine-treated male and female mice.
Behavioural Brain Research, 132, 171-177.
Silva, R.H., Abílio, V.C., Torres-Leite, D., Bergamo, M., Chinen, C.C., Claro, F.T.,
Carvalho, R.C. & Frussa-Filho, R. 2002a. Concomitant development of oral
dyskinesia and memory deficits in reserpine-treated male and female mice.
Behavioural Brain Research, 132, 171–177.
Skalisy, L.L., Beijamini, V., Joca, S.L., Vital, M.A.B.F., da Cunha, C. &
Andreatini, R. 2002. Evaluation of the face validity of reserpine administration
as an animal modelo of depression- Parkinson‘s disease association. Progress
in Neuro-Psychopharmacology & Biological Psychiatry, 26, 879-883.
Sonsalla, P.K., Zeevalk, G.D. & German, D.C. 2008. Chronic intraventricular
administration of 1-methyl-4-phenylpyridinium as a progressive model of
Parkinson‘s disease. Parkinsonism and Related Disorders, 14, 116-118.
Spina, M.B. & Cohen, G. 1989. Dopamine turnover and glutathione oxidation:
Implications for Parkinson disease. Proc. Natl. Acad. Sci., 86, 1398-1400.
Tan, S., Wood, M. & Maher P. 1998. Oxidative Stress Induces a Form of
Programmed Cell Death with Characteristics of Both Apoptosis and Necrosis in
Neuronal Cells. J. Neurochem., 71, 95—105.
Taylor, T.N., Caudle, W. M., Shepherd, K.R., Noorian, A., Jackson, C.R., Iuvone,
P. M., Weinshenker, D., Greene, J.G. & Miller, G.W. 2009. Nonmotor
Symptoms of Parkinson‘s Disease Revealed in an Animal Model with Reduced
Monoamine Storage Capacity. The Journal of Neuroscience, 29, 8103– 8113.

133
Tsang, A.H.K. & Chung, K.K.K. 2009. Oxidative and nitrosative stress in
Parkinson's disease. Biochimica et Biophysica Acta, 1792, 643–650.
Teixeira, A.M., Reckziegel, P., Müller, L., Pereira, R.P., Roos, D.H., Rocha, J.
B.T. & Bürger, M.E. 2009. Intense exercise potentiates oxidative stress in
striatum of reserpine-treated animals. Pharmacology, Biochemistry and
Behavior, 92, 231–235.
Teixeira, A.M., Trevizol, F., Colpo, G., Garcia, S.C., Charão, M., Pereira, R.P.,
Fachinetto, R., Rocha, J. B.T. & Bürger, M.E. 2008. Influence of chronic
exercise on reserpine-induced oxidative stress in rats: Behavioral and
antioxidant evaluations. Pharmacology, Biochemistry and Behavior, 88, 465–
472.
Van Gemmert, A.W.A., Teulings, H.L. & Stelmach, G.E. 2001. Parkinsonian
Patients Reduce Their Stroke Size with Increased Processing Demands. Brain
and Cognition, 47, 504–512.
Veheij, M.M.M. & Cools, A.R. 2007.Differential contribution of storage pools to the
extracellular amount of accumbal dopamine in high and low responders to
novelty: effects of reserpine. Journal of Neurochemistry, 100, 810-821.
Xia, R., Sun, J. & Threlkeld, A. 2009. Joseph. Analysis of interactive effect of stretch
reflex and shortening reaction on rigidity in Parkinson‘s disease. Clinical
Neurophysiology, 120, 1400–1407.
Wooten, G.F., Currie,L.J., Bovbjerg V.E., Lee, J.K. & Patrie, J. 2004. Are men at
greater risk for Parkinson‘s disease than women? J Neurol Neurosurg
Psychiatry, 75, 637–639.

134
Zgaljardic, D.J., Foldi, N.S. & Borod, J.C. 2004. Cognitive and behavioral
dysfunction in Parkinson‘s disease: neurochemical and clinicopathogical
contributions. J. Neural Transm., 111, 1287-1301.

135
5. Anexo