Embed Size (px)
Citation preview

Building innovative
drug discovery alliances
Algorithms, Evolution and Network-Based Approaches in Molecular Discovery
ChemAxon UGM, Budapest, May2015
PAGE
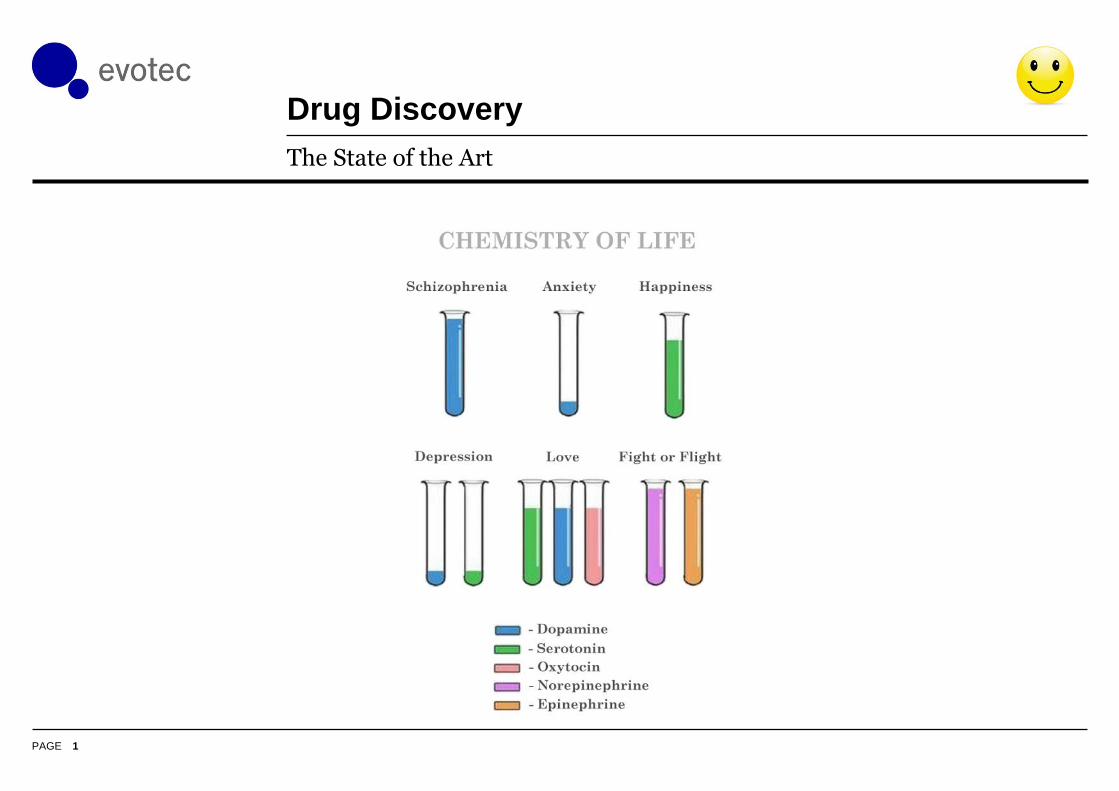
Drug Discovery
1
The State of the Art
PAGE
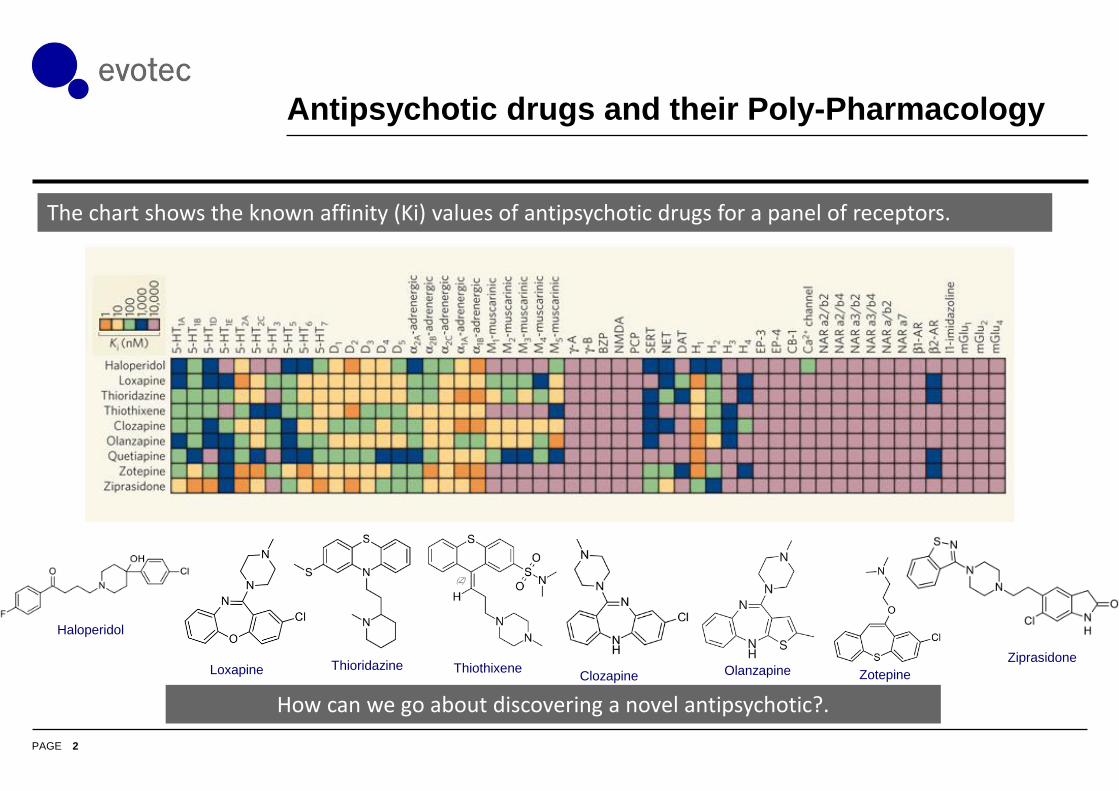
Antipsychotic drugs and their Poly-Pharmacology
2
The chart shows the known affinity (Ki) values of antipsychotic drugs for a panel of receptors.
Haloperidol
Loxapine Clozapine OlanzapineZiprasidone
Thioridazine ThiothixeneZotepine
How can we go about discovering a novel antipsychotic?.
PAGE
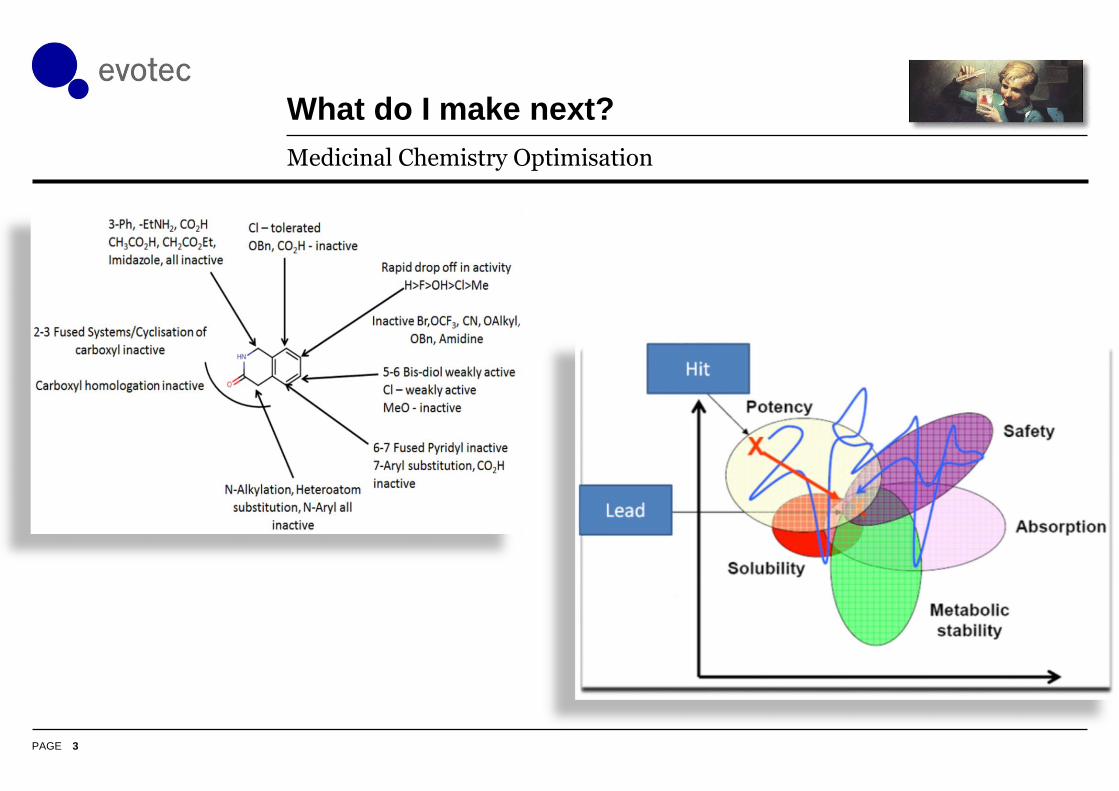
What do I make next?
3
Medicinal Chemistry Optimisation
PAGE
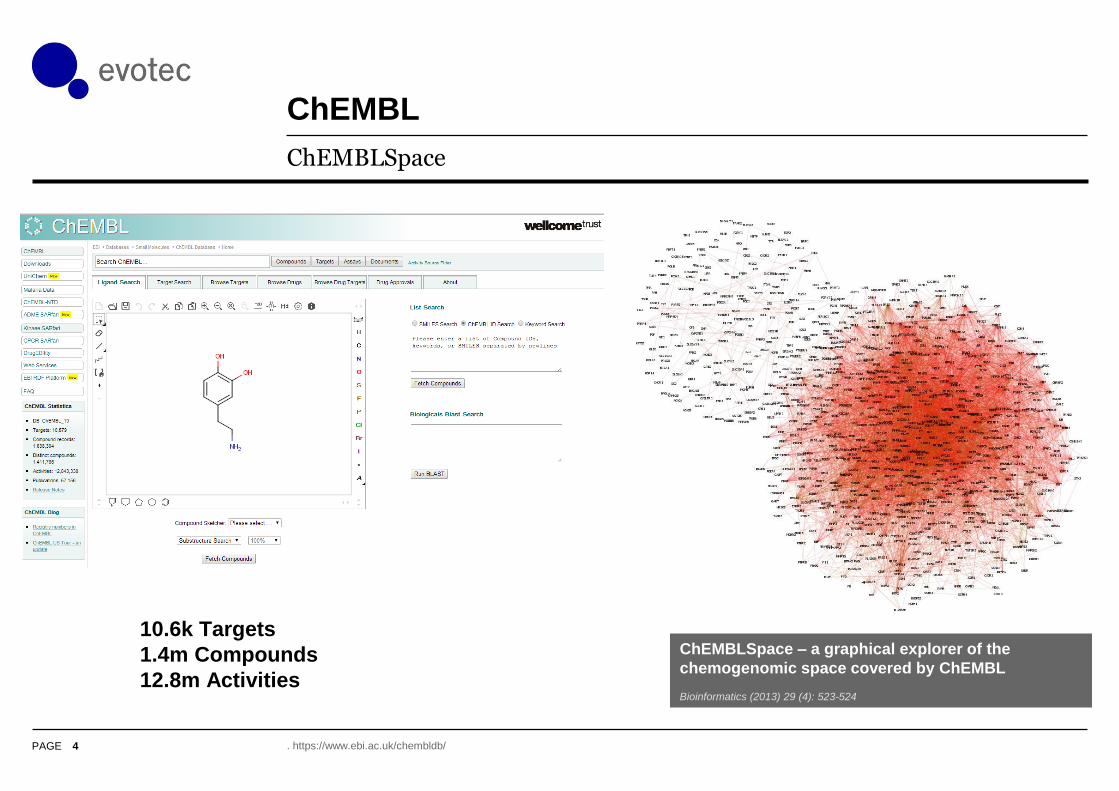
ChEMBL
4
ChEMBLSpace
. https://www.ebi.ac.uk/chembldb/
ChEMBLSpace – a graphical explorer of the
chemogenomic space covered by ChEMBL
Bioinformatics (2013) 29 (4): 523-524
10.6k Targets
1.4m Compounds
12.8m Activities

PAGE
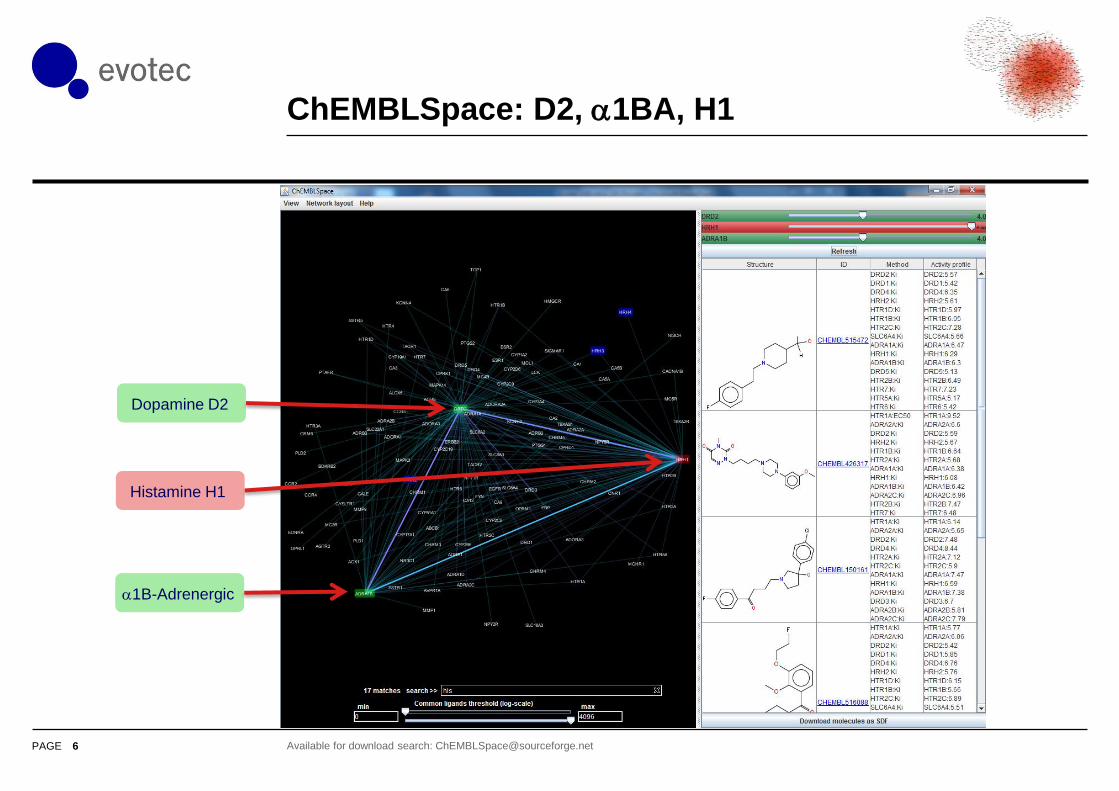
ChEMBLSpace search: D2 & a1BA
5 Available for download search: [email protected]
Dopamine D2
a1B-Adrenergic
PAGE
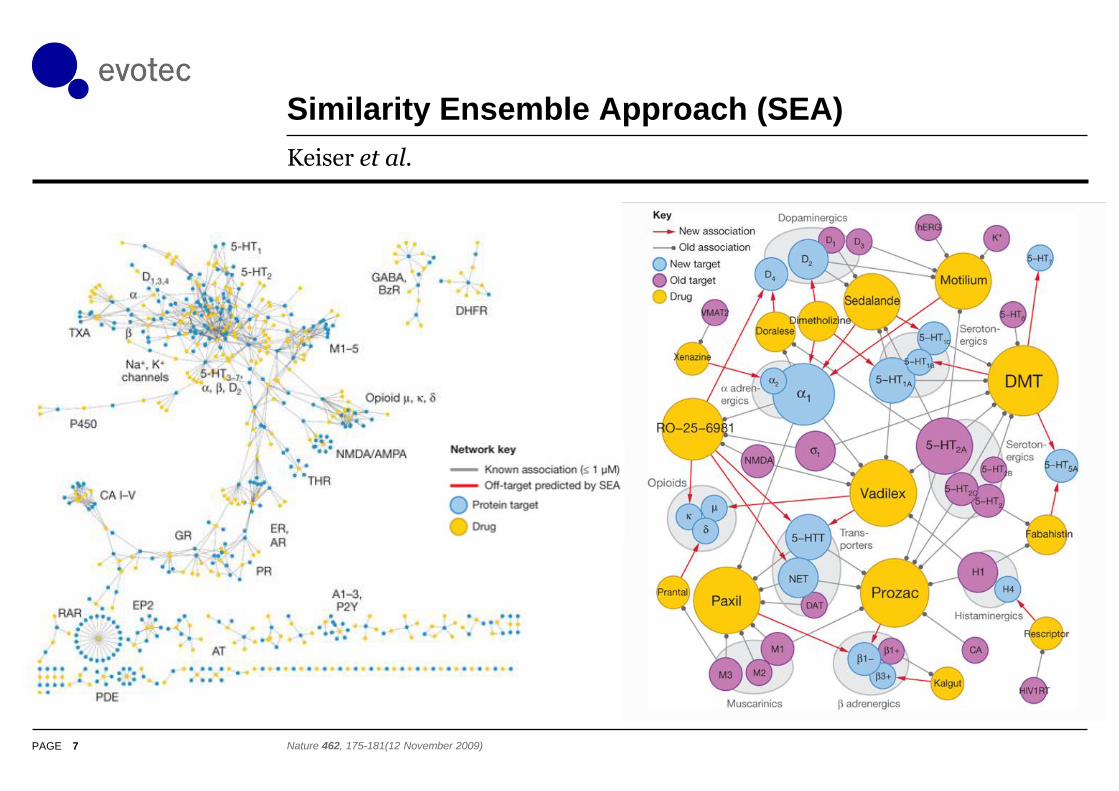
ChEMBLSpace: D2, a1BA, H1
6 Available for download search: [email protected]
Histamine H1
Dopamine D2
a1B-Adrenergic
PAGE
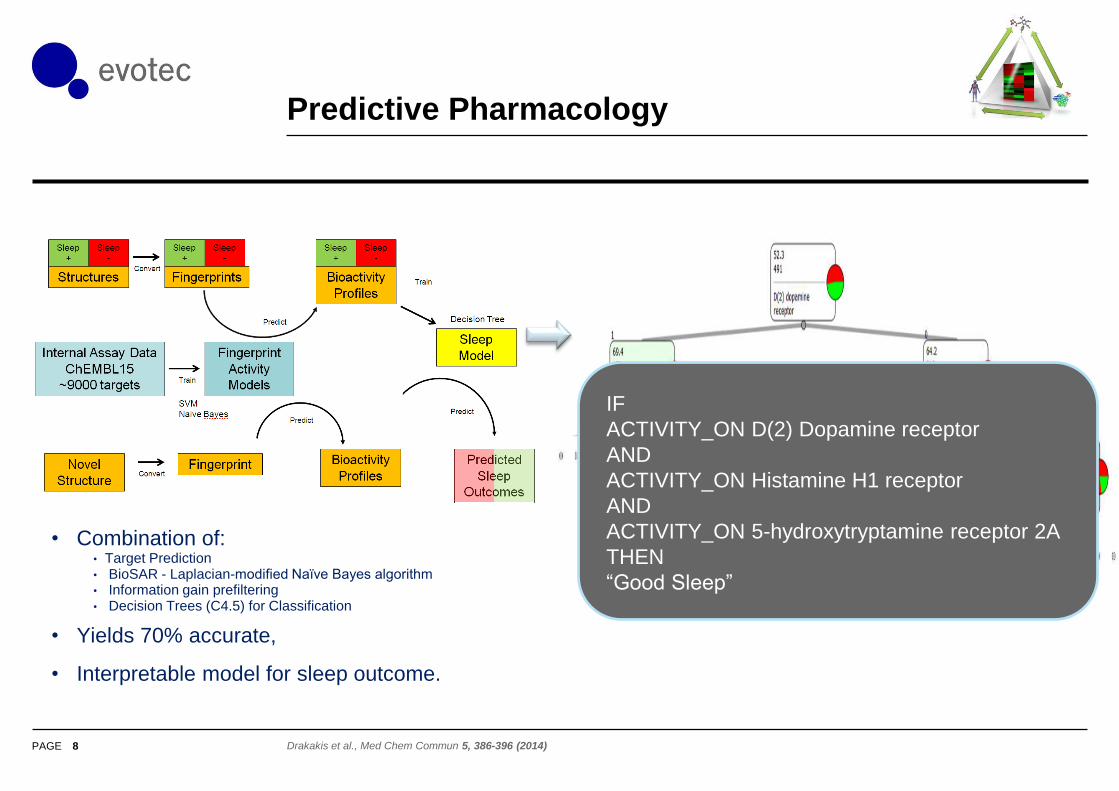
Similarity Ensemble Approach (SEA)
7
Keiser et al.
Nature 462, 175-181(12 November 2009)
PAGE
Predictive Pharmacology
8 Drakakis et al., Med Chem Commun 5, 386-396 (2014)
• Combination of: • Target Prediction • BioSAR - Laplacian-modified Naïve Bayes algorithm • Information gain prefiltering• Decision Trees (C4.5) for Classification
• Yields 70% accurate,
• Interpretable model for sleep outcome.
BIOPRINT
IF
ACTIVITY_ON D(2) Dopamine receptor
AND
ACTIVITY_ON Histamine H1 receptor
AND
ACTIVITY_ON 5-hydroxytryptamine receptor 2A
THEN
“Good Sleep”
PAGE
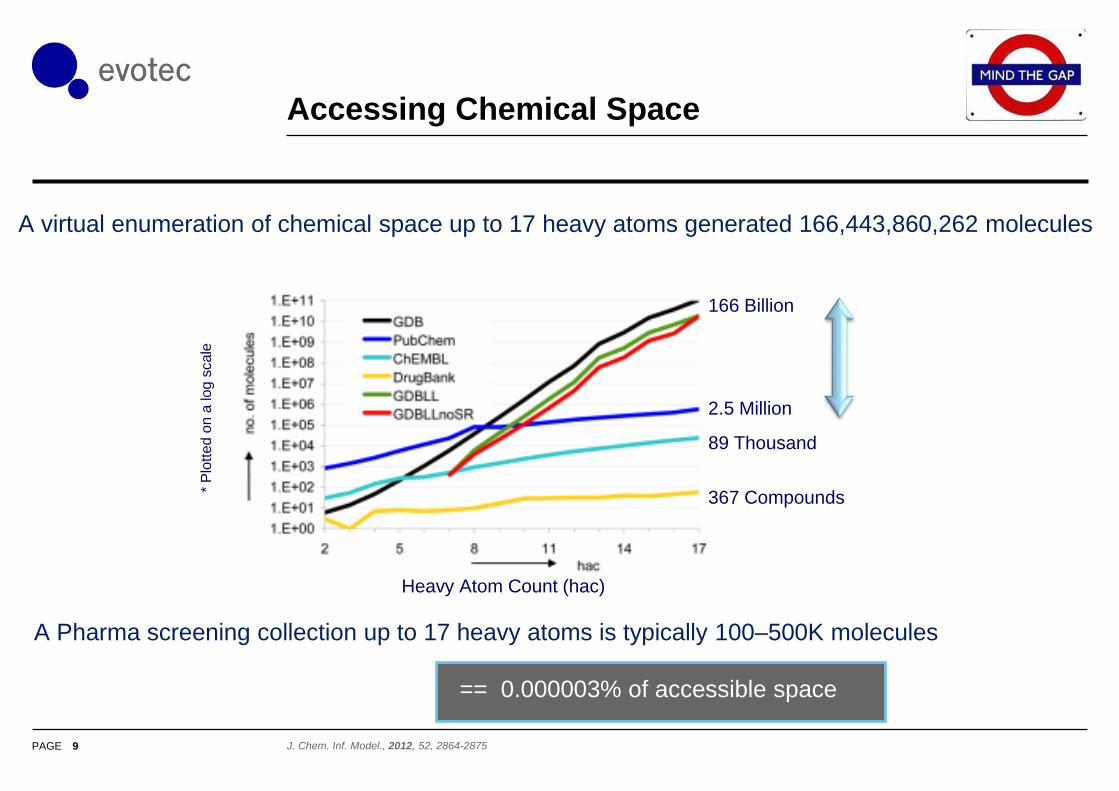
Accessing Chemical Space
9 J. Chem. Inf. Model., 2012, 52, 2864-2875
Heavy Atom Count (hac)
166 Billion
2.5 Million
89 Thousand
367 Compounds
A virtual enumeration of chemical space up to 17 heavy atoms generated 166,443,860,262 molecules *
Plo
tte
d o
n a
lo
g s
ca
le
A Pharma screening collection up to 17 heavy atoms is typically 100–500K molecules
== 0.000003% of accessible space
PAGE
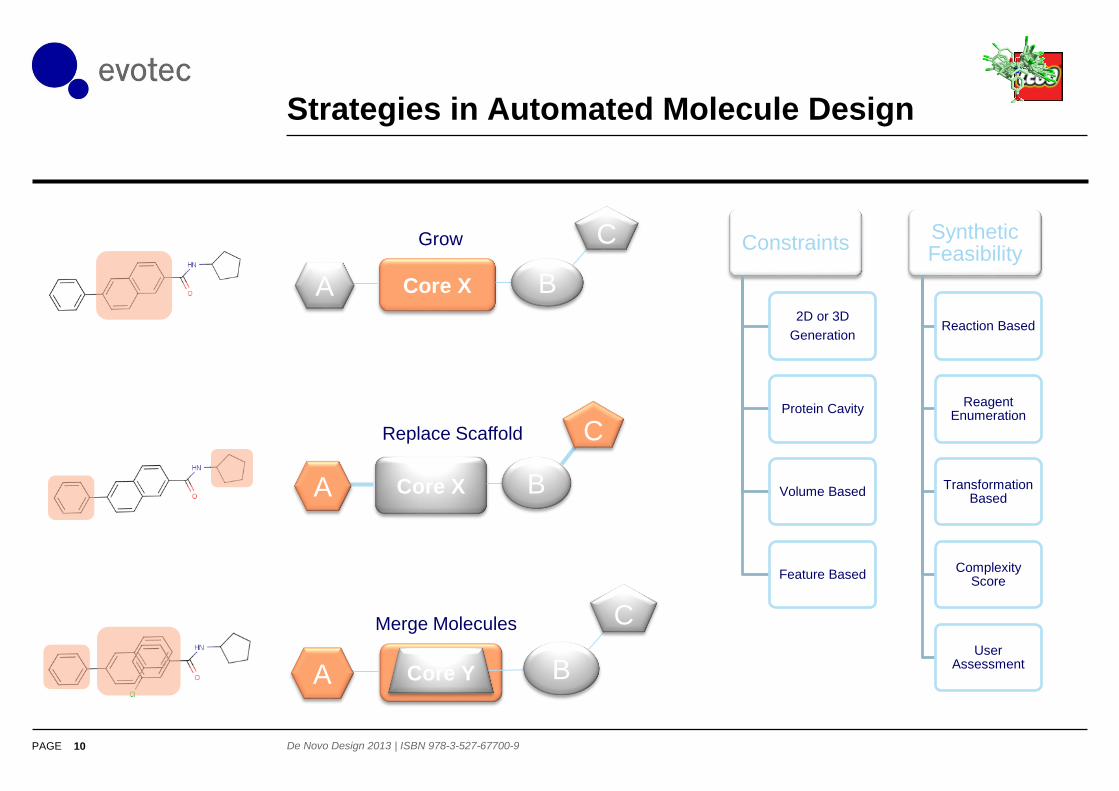
Strategies in Automated Molecule Design
10 De Novo Design 2013 | ISBN 978-3-527-67700-9
Core X BA
C
Core X BA
CGrow
Replace Scaffold
Constraints
2D or 3D
Generation
Protein Cavity
Volume Based
Feature Based
SyntheticFeasibility
Reaction Based
Reagent Enumeration
Transformation Based
Complexity Score
User Assessment
Core X B
C
Core Y
Merge Molecules
A
PAGE
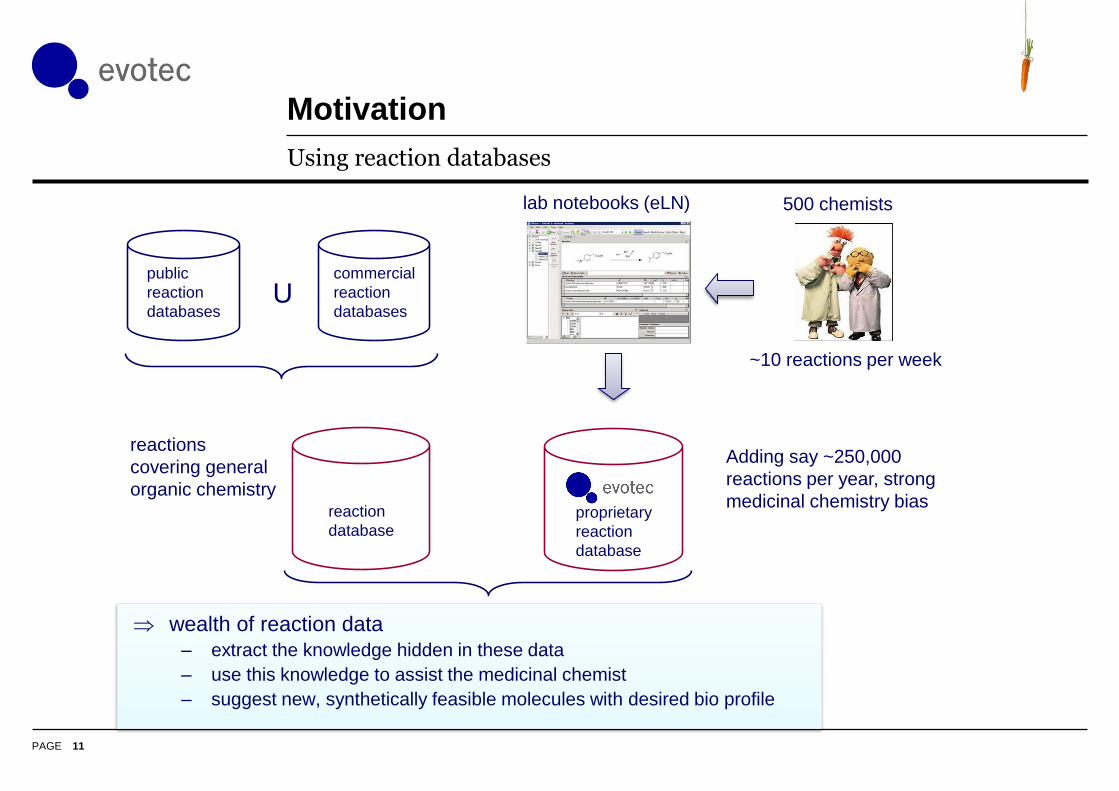
Motivation
11
Using reaction databases
public
reaction
databases
reactions
covering general
organic chemistryreaction
database
commercial
reaction
databasesU
Adding say ~250,000
reactions per year, strong
medicinal chemistry bias
wealth of reaction data
– extract the knowledge hidden in these data
– use this knowledge to assist the medicinal chemist
– suggest new, synthetically feasible molecules with desired bio profile
500 chemists
~10 reactions per week
lab notebooks (eLN)
proprietary
reaction
database
PAGE
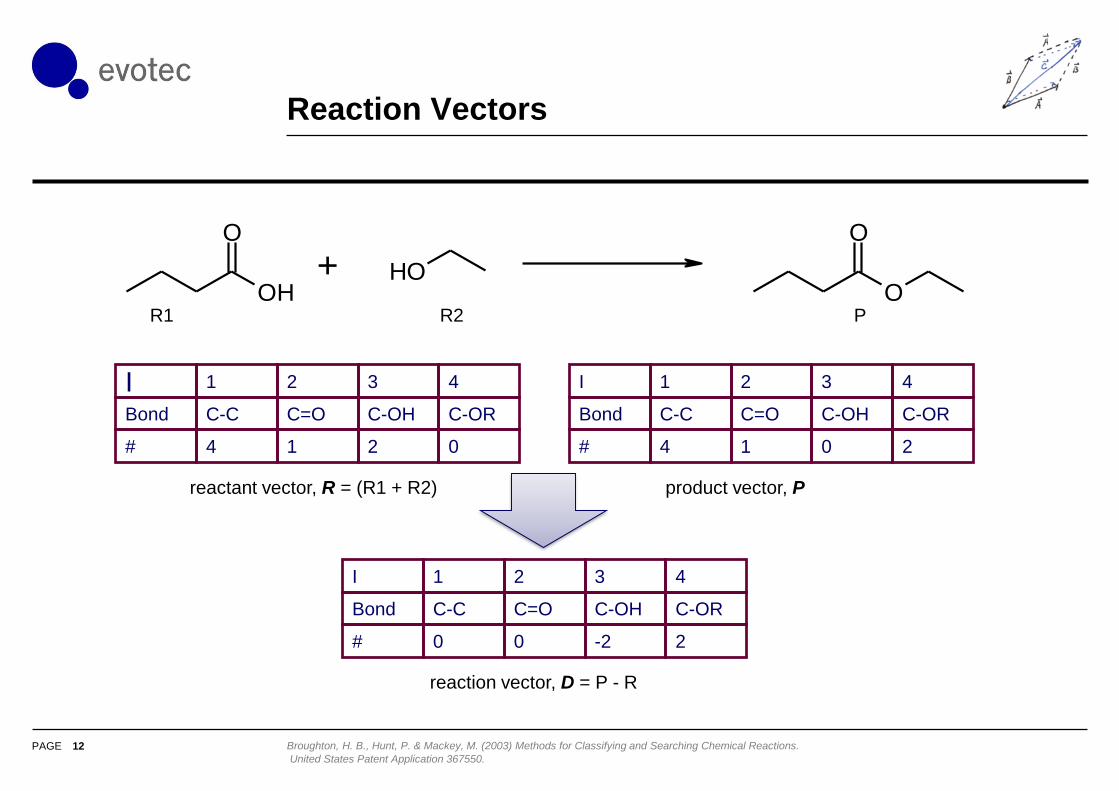
Reaction Vectors
12 Broughton, H. B., Hunt, P. & Mackey, M. (2003) Methods for Classifying and Searching Chemical Reactions.
United States Patent Application 367550.
I 1 2 3 4
Bond C-C C=O C-OH C-OR
# 0 0 -2 2
reactant vector, R = (R1 + R2) product vector, P
reaction vector, D = P - R
OH
O
OHO
O
+
I 1 2 3 4
Bond C-C C=O C-OH C-OR
# 4 1 2 0
I 1 2 3 4
Bond C-C C=O C-OH C-OR
# 4 1 0 2
R1 R2 P
PAGE
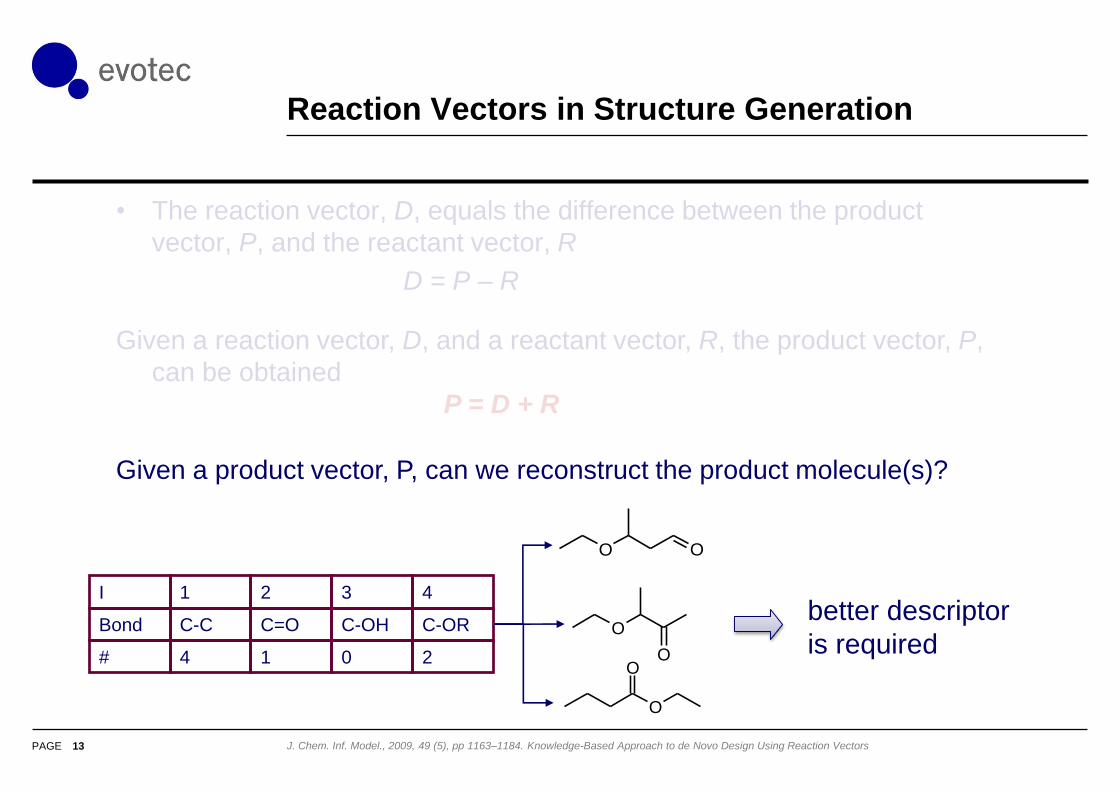
Reaction Vectors in Structure Generation
13 J. Chem. Inf. Model., 2009, 49 (5), pp 1163–1184. Knowledge-Based Approach to de Novo Design Using Reaction Vectors
• The reaction vector, D, equals the difference between the product
vector, P, and the reactant vector, R
D = P – R
I 1 2 3 4
Bond C-C C=O C-OH C-OR
# 4 1 0 2
O O
O
O
O
O
better descriptor
is required
Given a reaction vector, D, and a reactant vector, R, the product vector, P,
can be obtained
P = D + R
Given a product vector, P, can we reconstruct the product molecule(s)?

PAGE
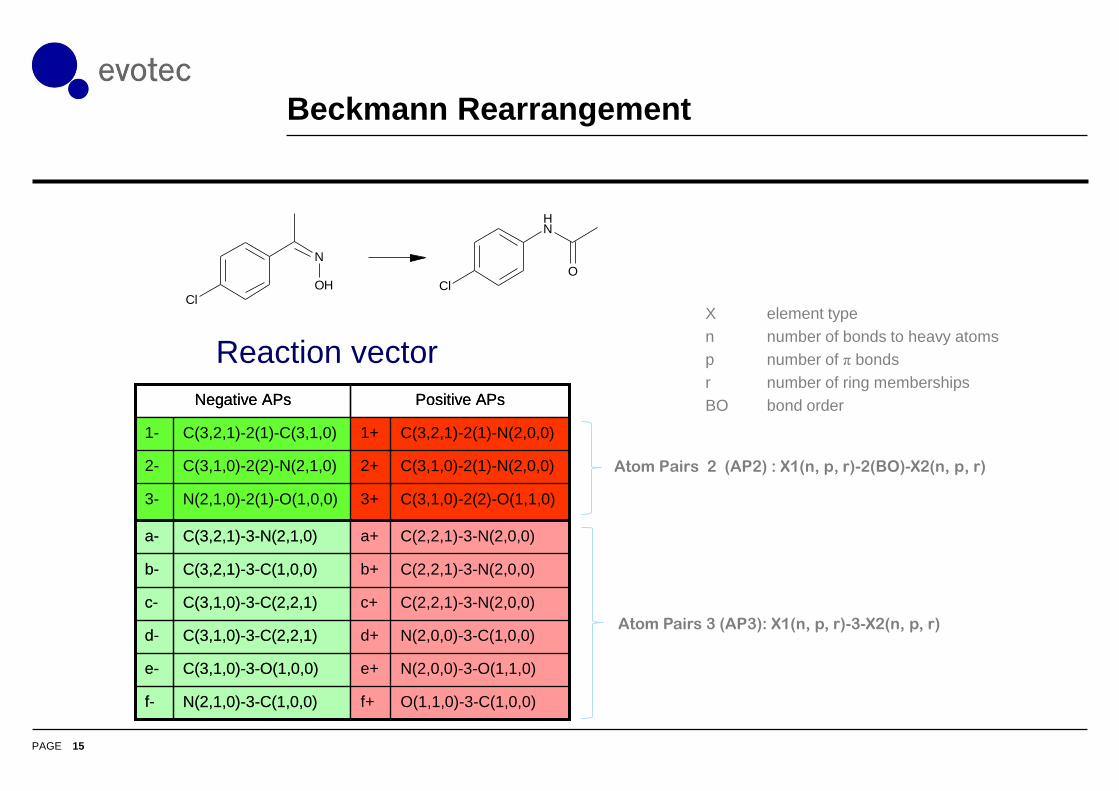
Modified Atom Pairs
14
Atom Pairs 2 (AP2) : X1(n, p, r)-2(BO)-X2(n, p, r)
X: element type
n: number of bonds to heavy atoms
p: number of π bonds
r: number of ring memberships
BO: bond order
Atom Pairs 3 (AP3): X1(n, p, r)-3-X2(n, p, r)
• Extending the bond distance in atom pairs encodes more of
the environment of the reaction centre
PAGE
Beckmann Rearrangement
15
Cl
N
OH Cl
NH
O
Reaction vector
O(1,1,0)-3-C(1,0,0)f+N(2,1,0)-3-C(1,0,0)f-
N(2,0,0)-3-O(1,1,0)e+C(3,1,0)-3-O(1,0,0)e-
N(2,0,0)-3-C(1,0,0)d+C(3,1,0)-3-C(2,2,1)d-
C(2,2,1)-3-N(2,0,0)c+C(3,1,0)-3-C(2,2,1)c-
C(2,2,1)-3-N(2,0,0)b+C(3,2,1)-3-C(1,0,0)b-
C(2,2,1)-3-N(2,0,0)a+C(3,2,1)-3-N(2,1,0)a-
C(3,1,0)-2(2)-O(1,1,0)3+N(2,1,0)-2(1)-O(1,0,0)3-
C(3,1,0)-2(1)-N(2,0,0)2+C(3,1,0)-2(2)-N(2,1,0)2-
C(3,2,1)-2(1)-N(2,0,0)1+C(3,2,1)-2(1)-C(3,1,0)1-
Positive APsNegative APs
O(1,1,0)-3-C(1,0,0)f+N(2,1,0)-3-C(1,0,0)f-
N(2,0,0)-3-O(1,1,0)e+C(3,1,0)-3-O(1,0,0)e-
N(2,0,0)-3-C(1,0,0)d+C(3,1,0)-3-C(2,2,1)d-
C(2,2,1)-3-N(2,0,0)c+C(3,1,0)-3-C(2,2,1)c-
C(2,2,1)-3-N(2,0,0)b+C(3,2,1)-3-C(1,0,0)b-
C(2,2,1)-3-N(2,0,0)a+C(3,2,1)-3-N(2,1,0)a-
C(3,1,0)-2(2)-O(1,1,0)3+N(2,1,0)-2(1)-O(1,0,0)3-
C(3,1,0)-2(1)-N(2,0,0)2+C(3,1,0)-2(2)-N(2,1,0)2-
C(3,2,1)-2(1)-N(2,0,0)1+C(3,2,1)-2(1)-C(3,1,0)1-
Positive APsNegative APs
Atom Pairs 2 (AP2) : X1(n, p, r)-2(BO)-X2(n, p, r)
Atom Pairs 3 (AP3): X1(n, p, r)-3-X2(n, p, r)
X element type
n number of bonds to heavy atoms
p number of π bonds
r number of ring memberships
BO bond order
PAGE
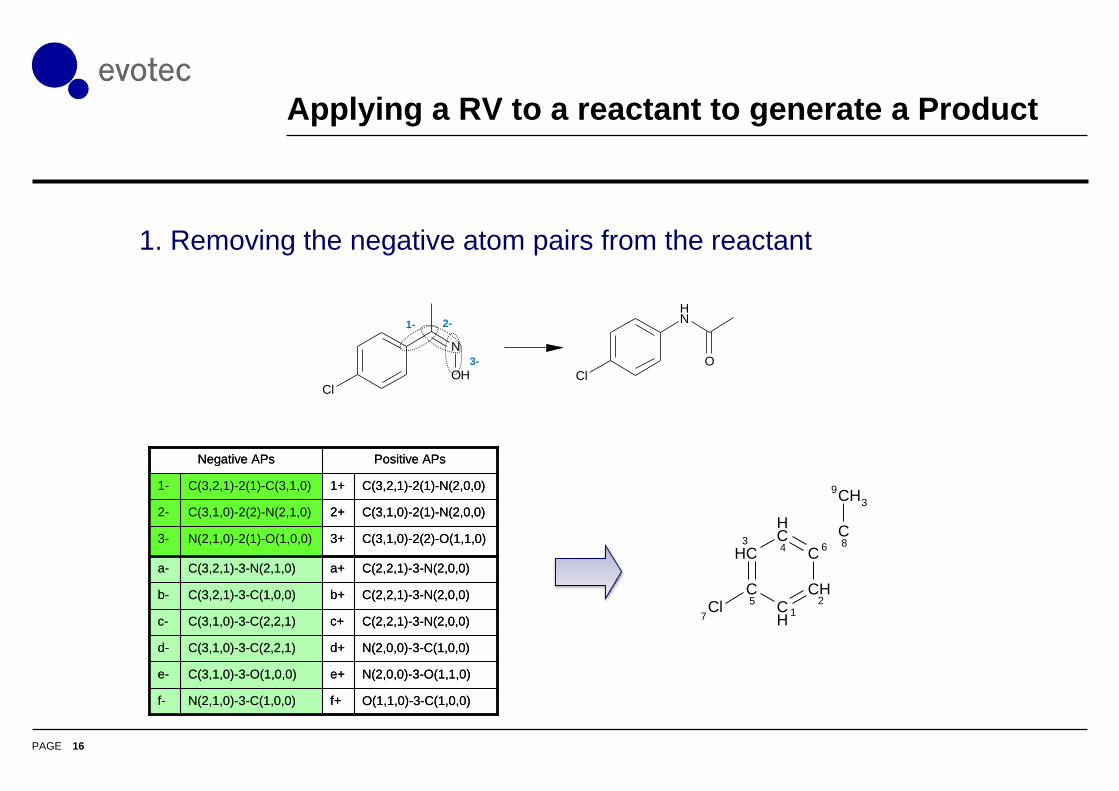
Applying a RV to a reactant to generate a Product
16
CH3
C5
CH
1
CH2
C6
CH
4
Cl7
C8
CH3
9
O(1,1,0)-3-C(1,0,0)f+N(2,1,0)-3-C(1,0,0)f-
N(2,0,0)-3-O(1,1,0)e+C(3,1,0)-3-O(1,0,0)e-
N(2,0,0)-3-C(1,0,0)d+C(3,1,0)-3-C(2,2,1)d-
C(2,2,1)-3-N(2,0,0)c+C(3,1,0)-3-C(2,2,1)c-
C(2,2,1)-3-N(2,0,0)b+C(3,2,1)-3-C(1,0,0)b-
C(2,2,1)-3-N(2,0,0)a+C(3,2,1)-3-N(2,1,0)a-
C(3,1,0)-2(2)-O(1,1,0)3+N(2,1,0)-2(1)-O(1,0,0)3-
C(3,1,0)-2(1)-N(2,0,0)2+C(3,1,0)-2(2)-N(2,1,0)2-
C(3,2,1)-2(1)-N(2,0,0)1+C(3,2,1)-2(1)-C(3,1,0)1-
Positive APsNegative APs
O(1,1,0)-3-C(1,0,0)f+N(2,1,0)-3-C(1,0,0)f-
N(2,0,0)-3-O(1,1,0)e+C(3,1,0)-3-O(1,0,0)e-
N(2,0,0)-3-C(1,0,0)d+C(3,1,0)-3-C(2,2,1)d-
C(2,2,1)-3-N(2,0,0)c+C(3,1,0)-3-C(2,2,1)c-
C(2,2,1)-3-N(2,0,0)b+C(3,2,1)-3-C(1,0,0)b-
C(2,2,1)-3-N(2,0,0)a+C(3,2,1)-3-N(2,1,0)a-
C(3,1,0)-2(2)-O(1,1,0)3+N(2,1,0)-2(1)-O(1,0,0)3-
C(3,1,0)-2(1)-N(2,0,0)2+C(3,1,0)-2(2)-N(2,1,0)2-
C(3,2,1)-2(1)-N(2,0,0)1+C(3,2,1)-2(1)-C(3,1,0)1-
Positive APsNegative APs
Cl
N
OH Cl
NH
O
1. Removing the negative atom pairs from the reactant
1- 2-
3-
PAGE
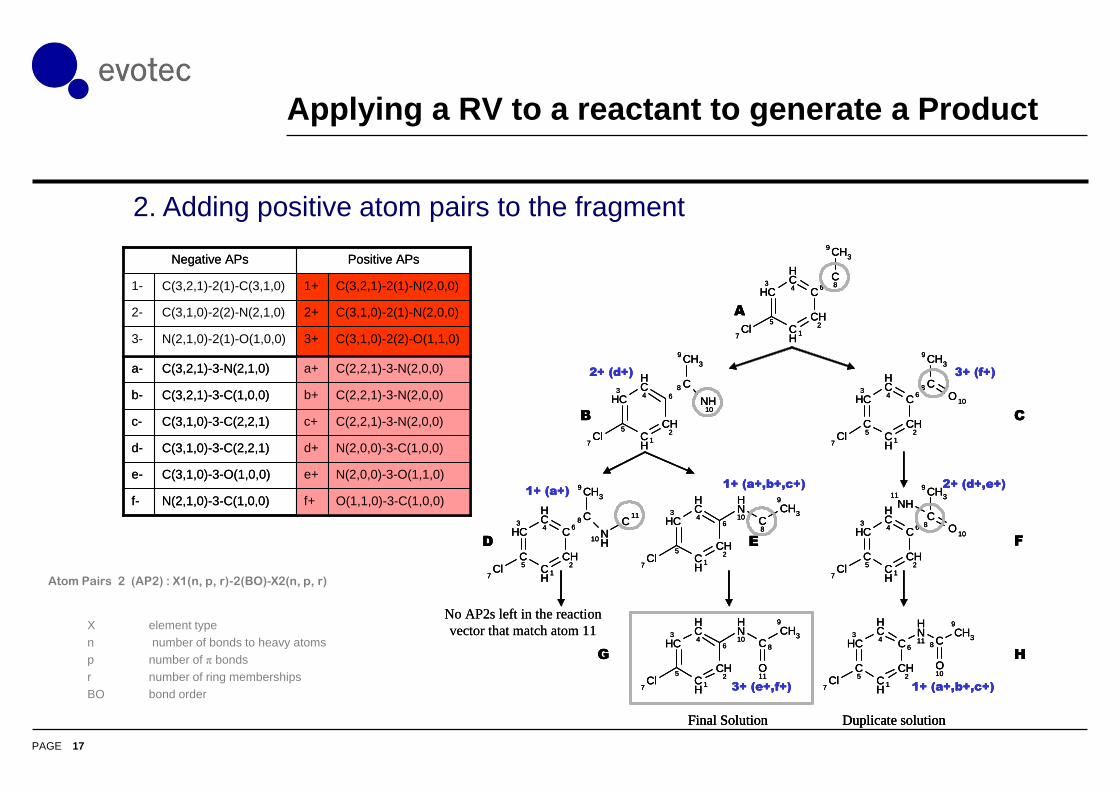
Applying a RV to a reactant to generate a Product
17
No AP2s left in the reaction
vector that match atom 11
CH3
5
CH
1
CH2
C6
CH
4
Cl7
C8
CH3
9
CH3
5
CH
1
CH2
6CH
4
Cl7
C8
CH3
9
NH10
CH3
C5
CH
1
CH2
C6
CH
4
Cl7
C8
CH3
9
O10
CH3
C5
CH
1
CH2
C6
CH
4
Cl7
C8
CH3
9
NH
10
C11
CH3
5
CH
1
CH2
6
CH
4
Cl7
NH
10C8
CH3
9
CH3
C5
CH
1
CH2
C6
CH
4
Cl7
C8
CH3
9
O10
NH11
CH3
5
CH
1
CH2
6
CH
4
Cl7
NH
10C 8
CH3
9
O11
CH3
C5
CH
1
CH2
C 6
CH
4
Cl7
C8
CH3
9
O10
NH
11
Duplicate solutionFinal Solution
2+ (d+) 3+ (f+)
1+ (a+)1+ (a+,b+,c+) 2+ (d+,e+)
3+ (e+,f+) 1+ (a+,b+,c+)
A
B C
D E F
G H
No AP2s left in the reaction
vector that match atom 11
CH3
5
CH
1
CH2
C6
CH
4
Cl7
C8
CH3
9
CH3
5
CH
1
CH2
6CH
4
Cl7
C8
CH3
9
NH10
CH3
C5
CH
1
CH2
C6
CH
4
Cl7
C8
CH3
9
O10
CH3
C5
CH
1
CH2
C6
CH
4
Cl7
C8
CH3
9
NH
10
C11
CH3
5
CH
1
CH2
6
CH
4
Cl7
NH
10C8
CH3
9
CH3
C5
CH
1
CH2
C6
CH
4
Cl7
C8
CH3
9
O10
NH11
CH3
5
CH
1
CH2
6
CH
4
Cl7
NH
10C 8
CH3
9
O11
CH3
C5
CH
1
CH2
C 6
CH
4
Cl7
C8
CH3
9
O10
NH
11
Duplicate solutionFinal Solution
2+ (d+) 3+ (f+)
1+ (a+)1+ (a+,b+,c+) 2+ (d+,e+)
3+ (e+,f+) 1+ (a+,b+,c+)
A
B C
D E F
G H
CH3
5
CH
1
CH2
C6
CH
4
Cl7
C8
CH3
9
CH3
5
CH
1
CH2
6CH
4
Cl7
C8
CH3
9
NH10
CH3
C5
CH
1
CH2
C6
CH
4
Cl7
C8
CH3
9
O10
CH3
C5
CH
1
CH2
C6
CH
4
Cl7
C8
CH3
9
NH
10
C11
CH3
5
CH
1
CH2
6
CH
4
Cl7
NH
10C8
CH3
9
CH3
C5
CH
1
CH2
C6
CH
4
Cl7
C8
CH3
9
O10
NH11
CH3
5
CH
1
CH2
6
CH
4
Cl7
NH
10C 8
CH3
9
O11
CH3
C5
CH
1
CH2
C 6
CH
4
Cl7
C8
CH3
9
O10
NH
11
Duplicate solutionFinal Solution
2+ (d+) 3+ (f+)
1+ (a+)1+ (a+,b+,c+) 2+ (d+,e+)
3+ (e+,f+) 1+ (a+,b+,c+)
A
B C
D E F
G H
O(1,1,0)-3-C(1,0,0)f+N(2,1,0)-3-C(1,0,0)f-
N(2,0,0)-3-O(1,1,0)e+C(3,1,0)-3-O(1,0,0)e-
N(2,0,0)-3-C(1,0,0)d+C(3,1,0)-3-C(2,2,1)d-
C(2,2,1)-3-N(2,0,0)c+C(3,1,0)-3-C(2,2,1)c-
C(2,2,1)-3-N(2,0,0)b+C(3,2,1)-3-C(1,0,0)b-
C(2,2,1)-3-N(2,0,0)a+C(3,2,1)-3-N(2,1,0)a-
C(3,1,0)-2(2)-O(1,1,0)3+N(2,1,0)-2(1)-O(1,0,0)3-
C(3,1,0)-2(1)-N(2,0,0)2+C(3,1,0)-2(2)-N(2,1,0)2-
C(3,2,1)-2(1)-N(2,0,0)1+C(3,2,1)-2(1)-C(3,1,0)1-
Positive APsNegative APs
O(1,1,0)-3-C(1,0,0)f+N(2,1,0)-3-C(1,0,0)f-
N(2,0,0)-3-O(1,1,0)e+C(3,1,0)-3-O(1,0,0)e-
N(2,0,0)-3-C(1,0,0)d+C(3,1,0)-3-C(2,2,1)d-
C(2,2,1)-3-N(2,0,0)c+C(3,1,0)-3-C(2,2,1)c-
C(2,2,1)-3-N(2,0,0)b+C(3,2,1)-3-C(1,0,0)b-
C(2,2,1)-3-N(2,0,0)a+C(3,2,1)-3-N(2,1,0)a-
C(3,1,0)-2(2)-O(1,1,0)3+N(2,1,0)-2(1)-O(1,0,0)3-
C(3,1,0)-2(1)-N(2,0,0)2+C(3,1,0)-2(2)-N(2,1,0)2-
C(3,2,1)-2(1)-N(2,0,0)1+C(3,2,1)-2(1)-C(3,1,0)1-
Positive APsNegative APs
2. Adding positive atom pairs to the fragment
X element type
n number of bonds to heavy atoms
p number of π bonds
r number of ring memberships
BO bond order
Atom Pairs 2 (AP2) : X1(n, p, r)-2(BO)-X2(n, p, r)
PAGE
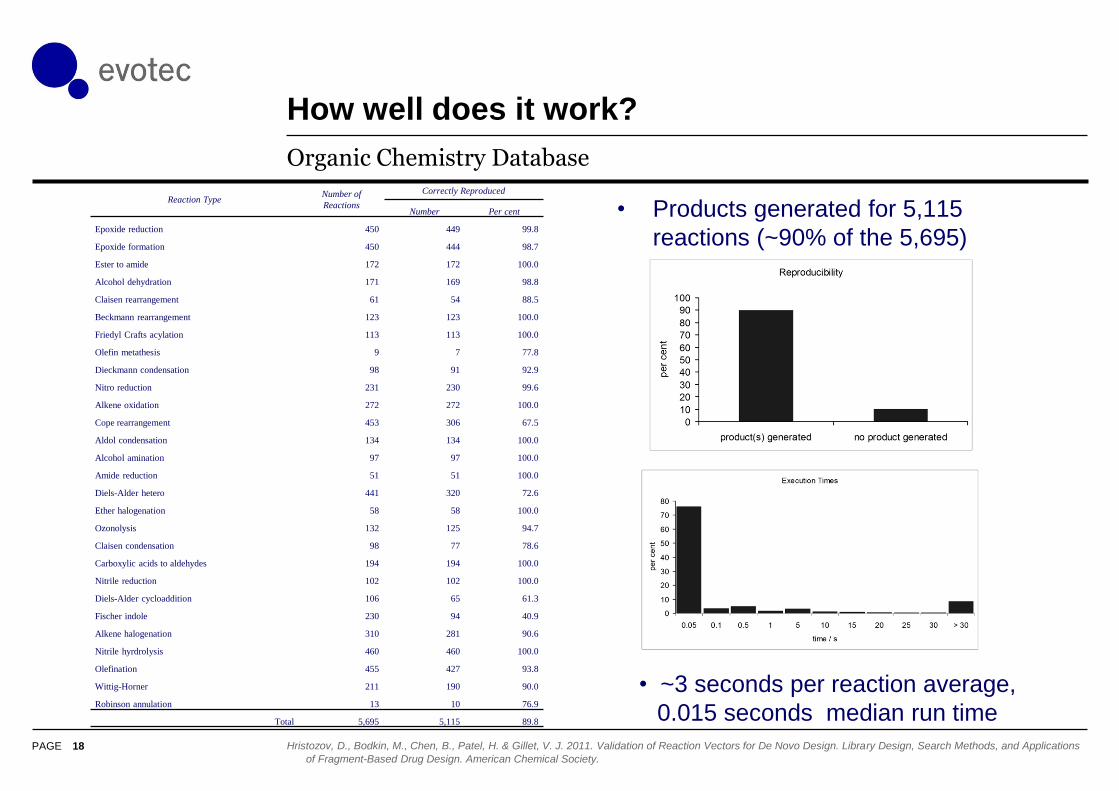
How well does it work?
18
Organic Chemistry Database
Hristozov, D., Bodkin, M., Chen, B., Patel, H. & Gillet, V. J. 2011. Validation of Reaction Vectors for De Novo Design. Library Design, Search Methods, and Applications
of Fragment-Based Drug Design. American Chemical Society.
Reaction TypeNumber of
Reactions
Correctly Reproduced
Number Per cent
Epoxide reduction 450 449 99.8
Epoxide formation 450 444 98.7
Ester to amide 172 172 100.0
Alcohol dehydration 171 169 98.8
Claisen rearrangement 61 54 88.5
Beckmann rearrangement 123 123 100.0
Friedyl Crafts acylation 113 113 100.0
Olefin metathesis 9 7 77.8
Dieckmann condensation 98 91 92.9
Nitro reduction 231 230 99.6
Alkene oxidation 272 272 100.0
Cope rearrangement 453 306 67.5
Aldol condensation 134 134 100.0
Alcohol amination 97 97 100.0
Amide reduction 51 51 100.0
Diels-Alder hetero 441 320 72.6
Ether halogenation 58 58 100.0
Ozonolysis 132 125 94.7
Claisen condensation 98 77 78.6
Carboxylic acids to aldehydes 194 194 100.0
Nitrile reduction 102 102 100.0
Diels-Alder cycloaddition 106 65 61.3
Fischer indole 230 94 40.9
Alkene halogenation 310 281 90.6
Nitrile hyrdrolysis 460 460 100.0
Olefination 455 427 93.8
Wittig-Horner 211 190 90.0
Robinson annulation 13 10 76.9
Total 5,695 5,115 89.8
• ~3 seconds per reaction average,
0.015 seconds median run time
• Products generated for 5,115
reactions (~90% of the 5,695)
PAGE
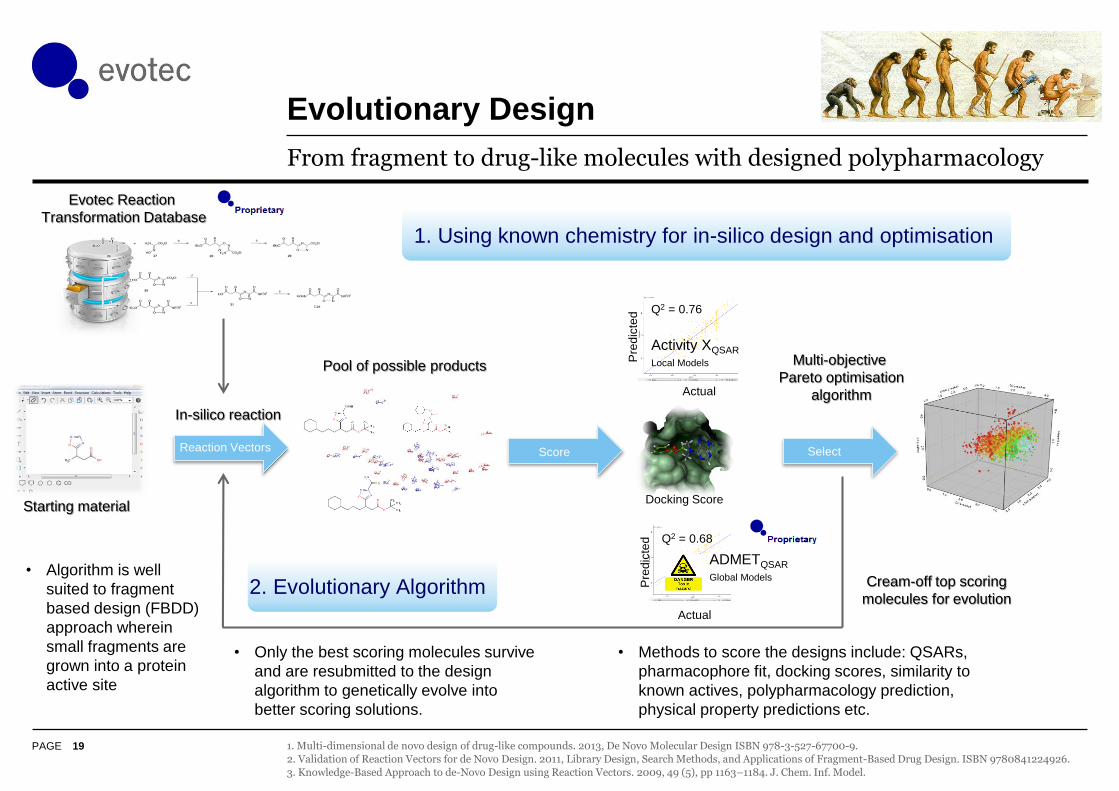
Evolutionary Design
19
From fragment to drug-like molecules with designed polypharmacology
1. Multi-dimensional de novo design of drug-like compounds. 2013, De Novo Molecular Design ISBN 978-3-527-67700-9.
2. Validation of Reaction Vectors for de Novo Design. 2011, Library Design, Search Methods, and Applications of Fragment-Based Drug Design. ISBN 9780841224926.
3. Knowledge-Based Approach to de-Novo Design using Reaction Vectors. 2009, 49 (5), pp 1163–1184. J. Chem. Inf. Model.
Evotec Reaction
Transformation Database
Pool of possible products
In-silico reaction
Q2 = 0.76
Pre
dic
ted
Actual
Activity XQSAR
Local Models
Docking Score
Actual
Pre
dic
ted Q2 = 0.68
ADMETQSAR
Global Models
Multi-objective
Pareto optimisation
algorithm
Reaction Vectors Score Select
1. Using known chemistry for in-silico design and optimisation
Starting material
2. Evolutionary Algorithm Cream-off top scoring
molecules for evolution
• Only the best scoring molecules survive
and are resubmitted to the design
algorithm to genetically evolve into
better scoring solutions.
• Algorithm is well
suited to fragment
based design (FBDD)
approach wherein
small fragments are
grown into a protein
active site
• Methods to score the designs include: QSARs,
pharmacophore fit, docking scores, similarity to
known actives, polypharmacology prediction,
physical property predictions etc.
PAGE
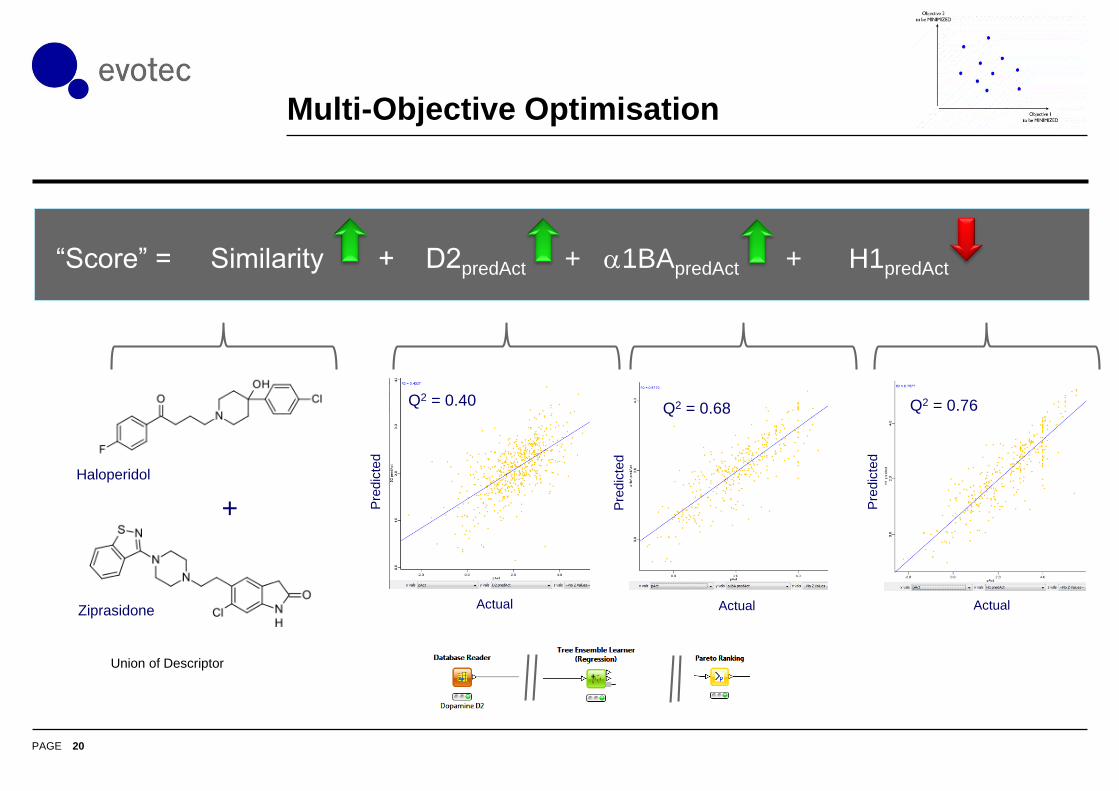
Multi-Objective Optimisation
20
Haloperidol
Ziprasidone
“Score” = Similarity + D2predAct + a1BApredAct + H1predAct
Q2 = 0.76
Pre
dic
ted
ActualActualP
redic
ted
Q2 = 0.68
Actual
Q2 = 0.40
Pre
dic
ted
Union of Descriptor
+
PAGE
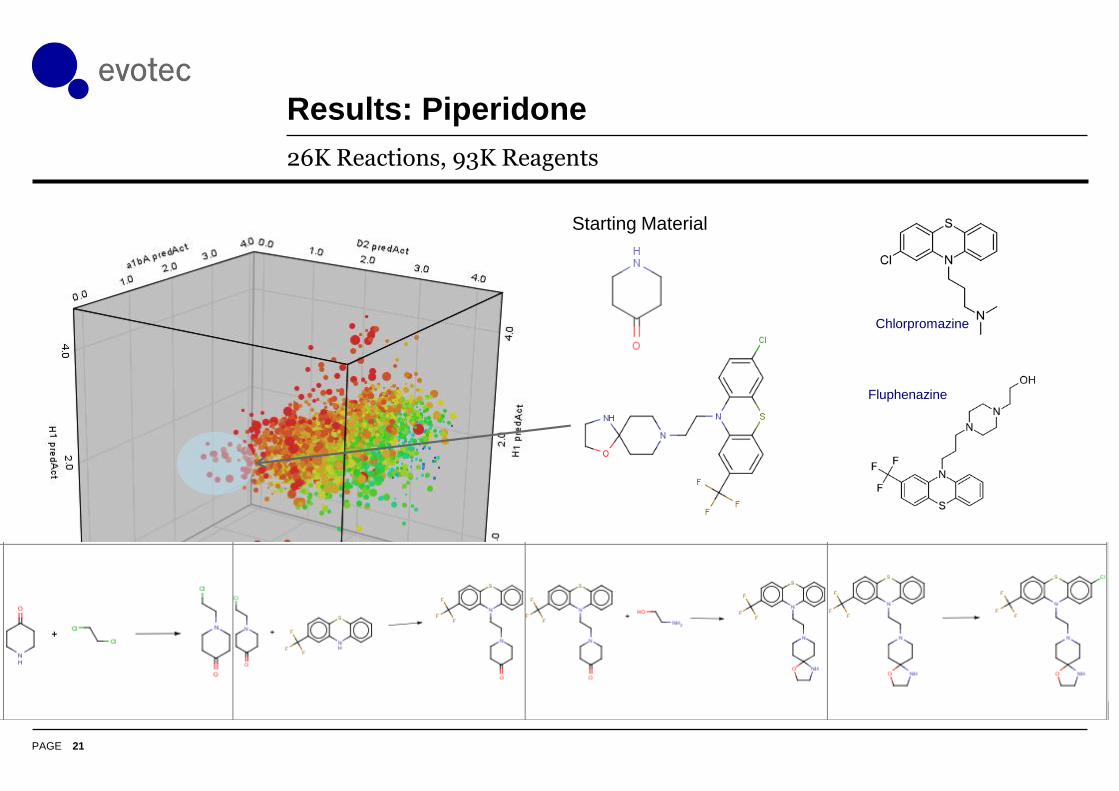
Results: Piperidone
21
26K Reactions, 93K Reagents
Starting Material
Fluphenazine
Chlorpromazine
PAGE
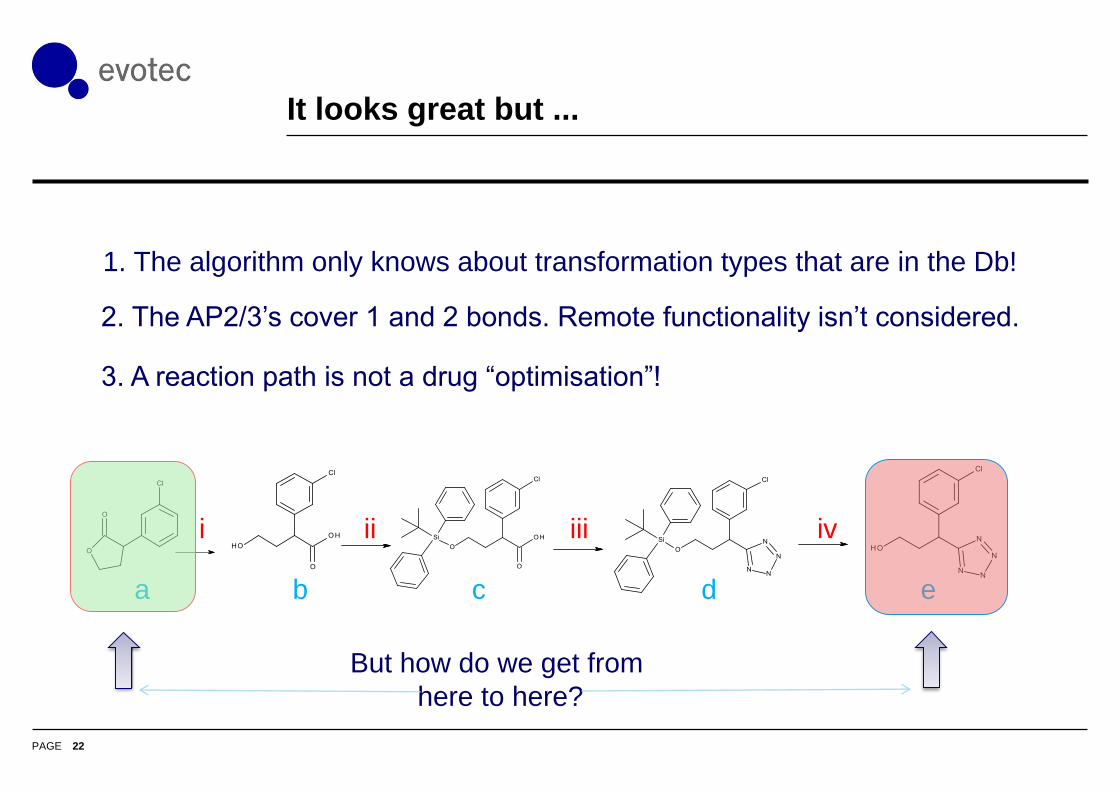
It looks great but ...
22
1. The algorithm only knows about transformation types that are in the Db!
2. The AP2/3’s cover 1 and 2 bonds. Remote functionality isn’t considered.
3. A reaction path is not a drug “optimisation”!
a b c d e
i ii iii iv
But how do we get from
here to here?
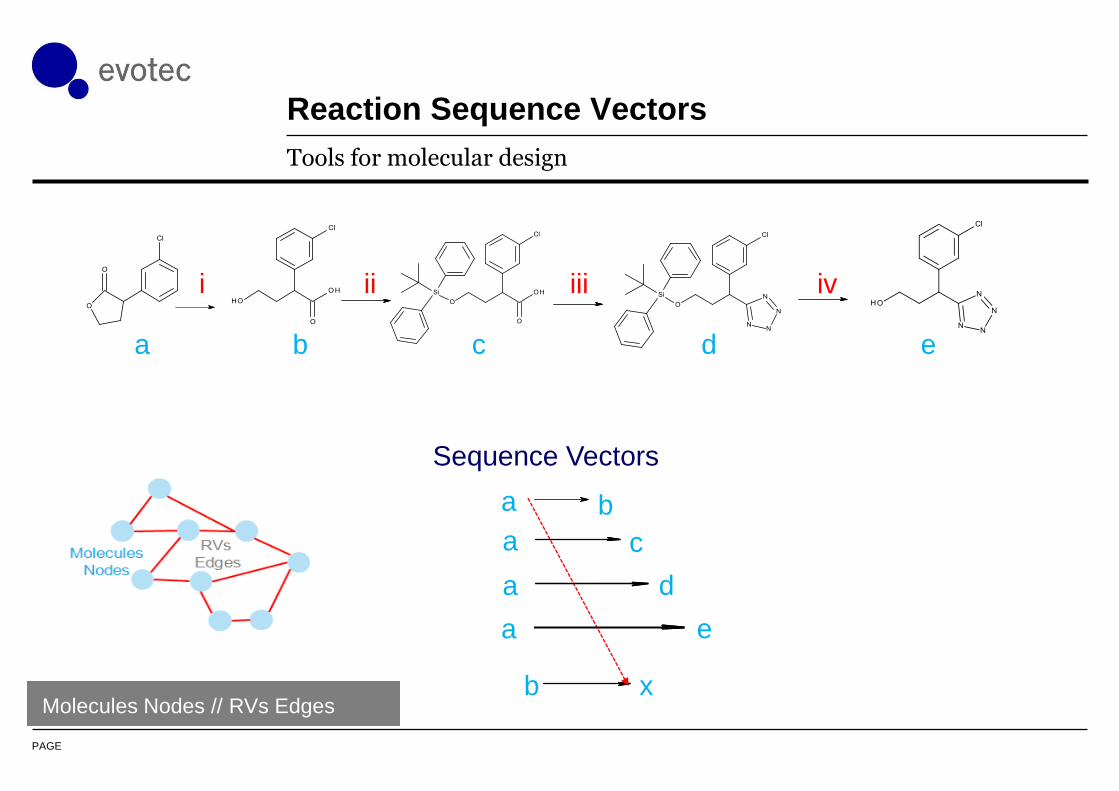
PAGE
Reaction Sequence Vectors
Tools for molecular design
xb
a b c d e
i ii iii iv
ea
a b
ca
da
Sequence Vectors
Molecules Nodes // RVs Edges
PAGE
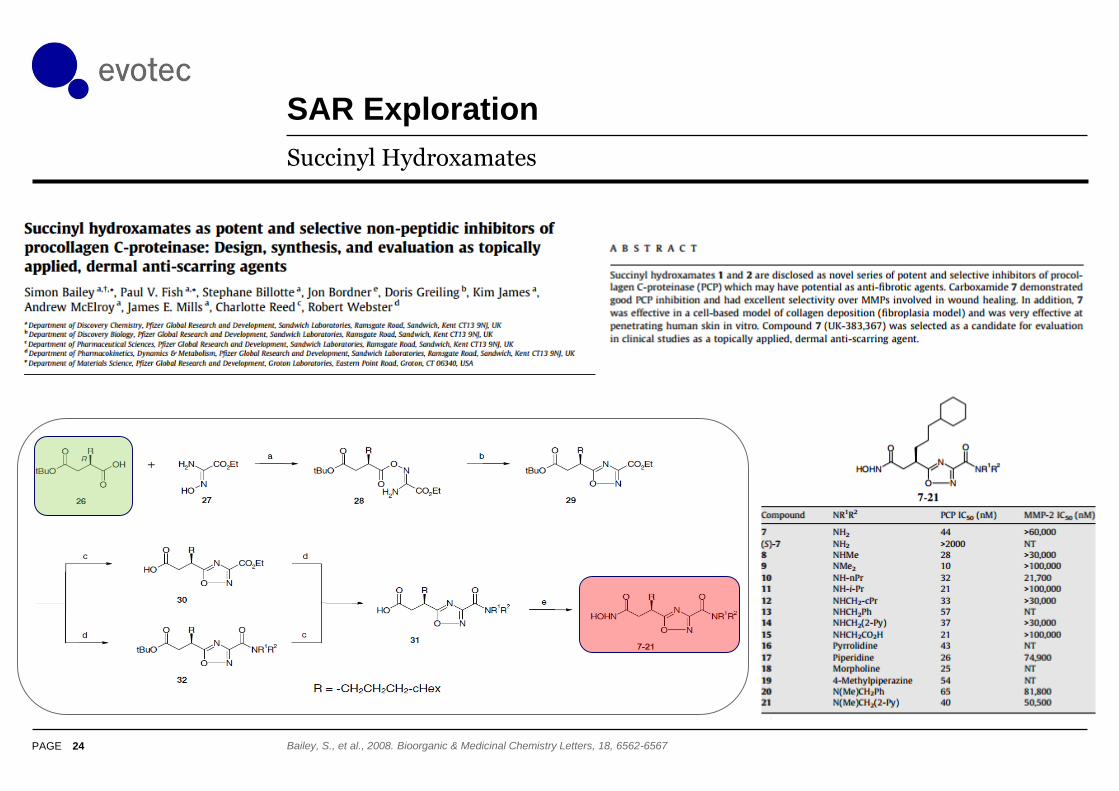
SAR Exploration
24
Succinyl Hydroxamates
Bailey, S., et al., 2008. Bioorganic & Medicinal Chemistry Letters, 18, 6562-6567
PAGE
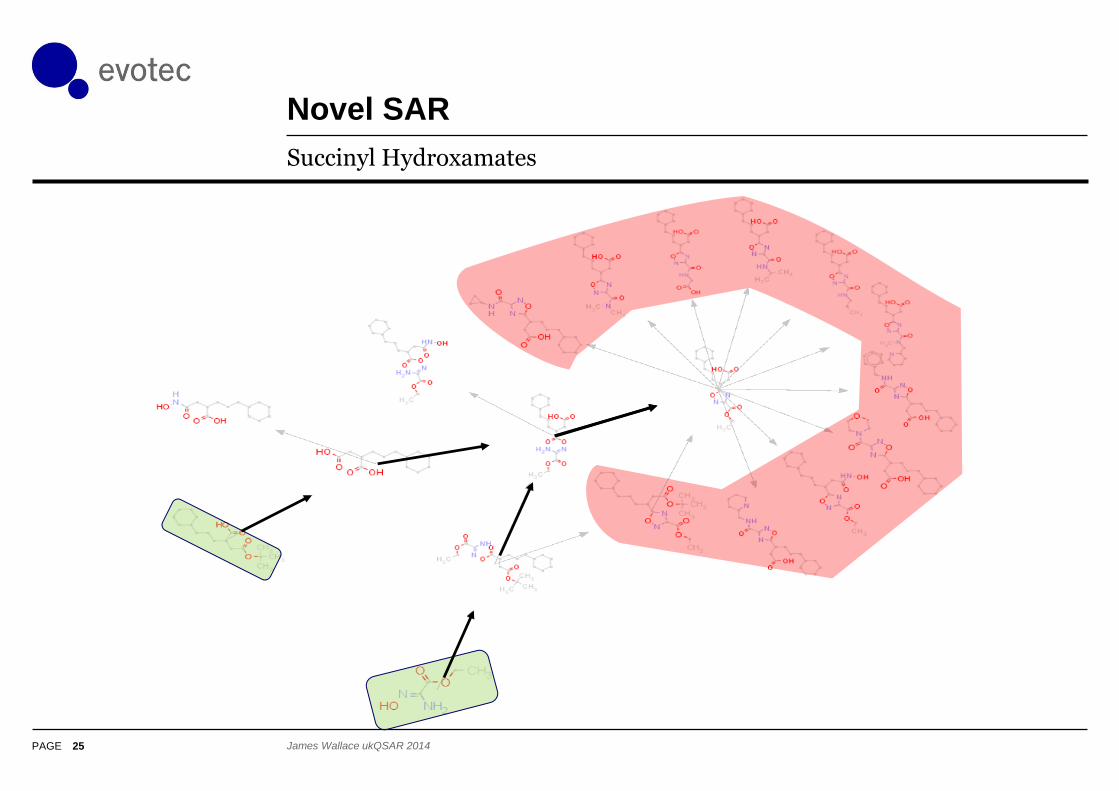
Novel SAR
25
Succinyl Hydroxamates
James Wallace ukQSAR 2014
PAGE
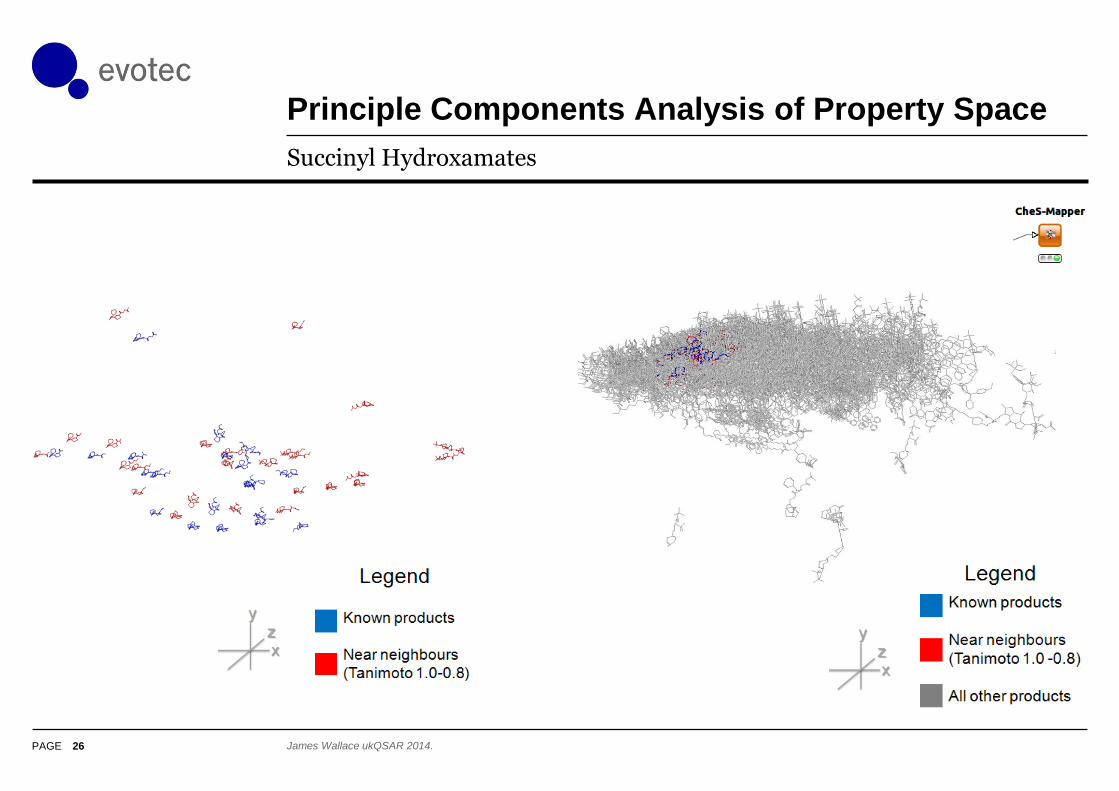
Principle Components Analysis of Property Space
26
Succinyl Hydroxamates
James Wallace ukQSAR 2014.
PAGE
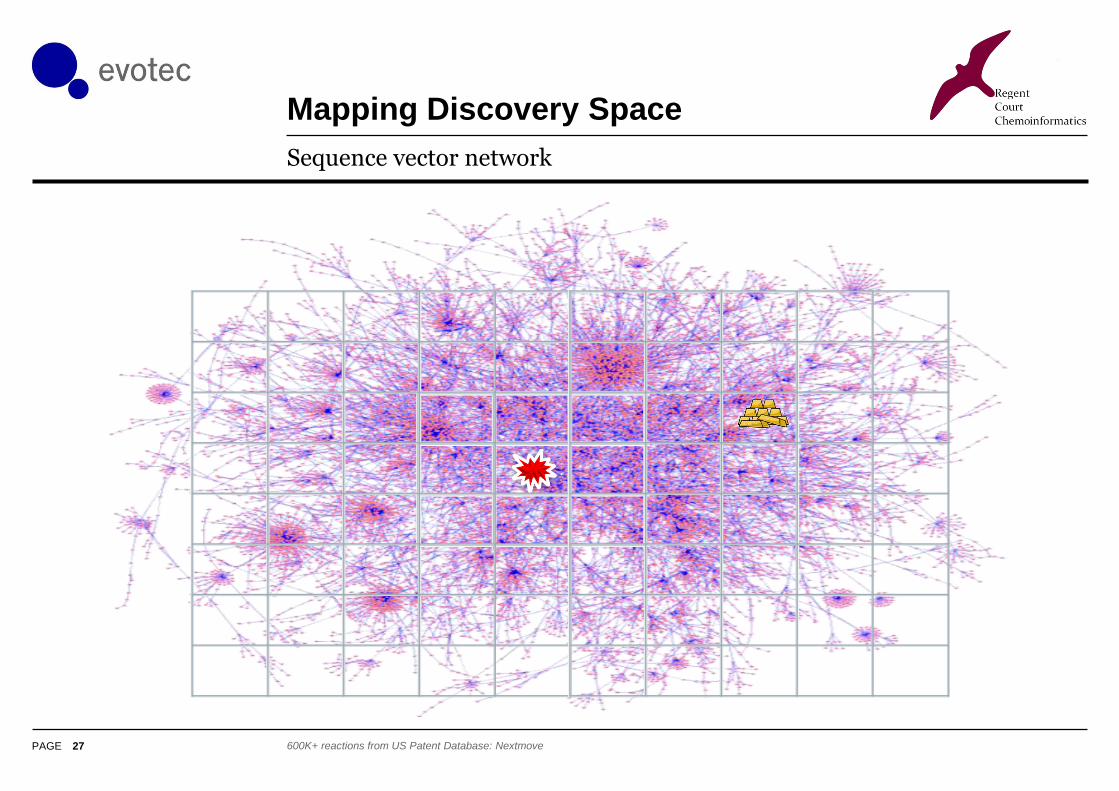
Mapping Discovery Space
27
Sequence vector network
600K+ reactions from US Patent Database: Nextmove
PAGE
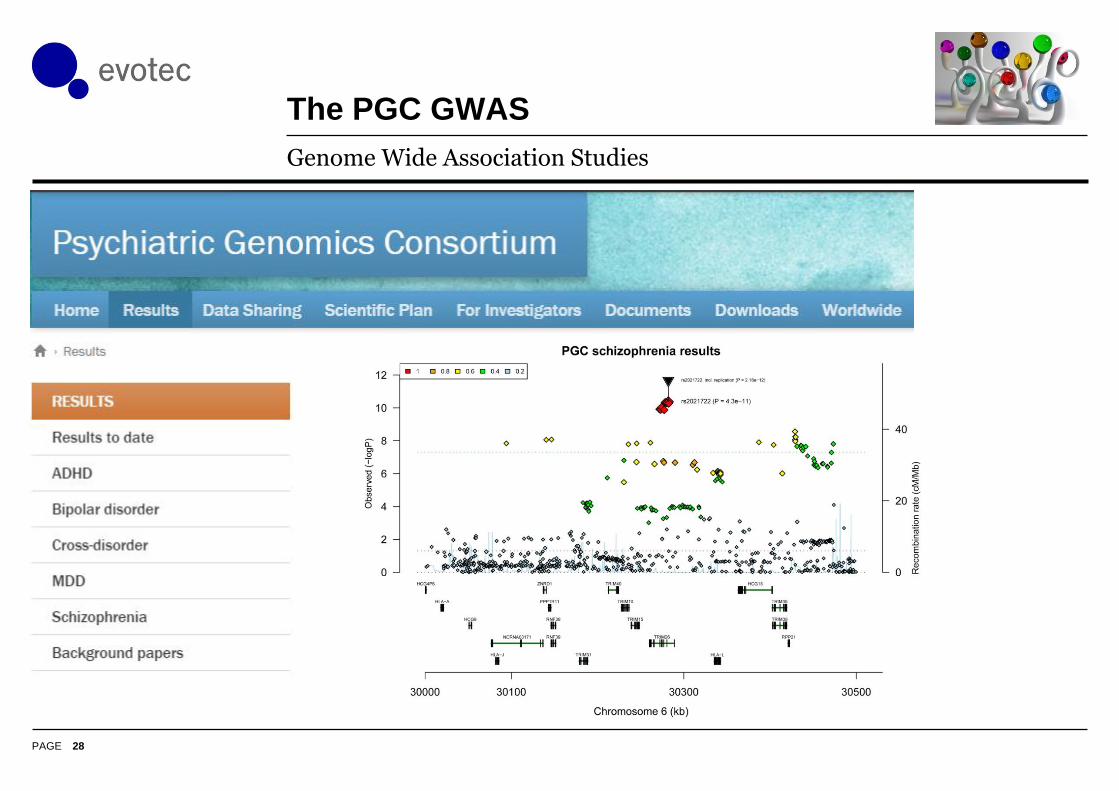
The PGC GWAS
28
Genome Wide Association Studies

PAGE
GPCRs associated with Schizophrenia GWAS genes
29
2 step shortest paths network
Suzanne Brewerton – UKQSAR Lilly 2014
Histamine receptor network
Adrenergic receptor network
Dopamine receptor network
Schizophrenia GWAS genes
GPCR receptor subtypes
Eg, From: Dopamine receptor subtypesTo: Proteins defined by PGC2 schizophrenia GWAS genes
Hubs involved in GPCR
signalling in schizophrenia
Remove proteins degree <10
PAGE
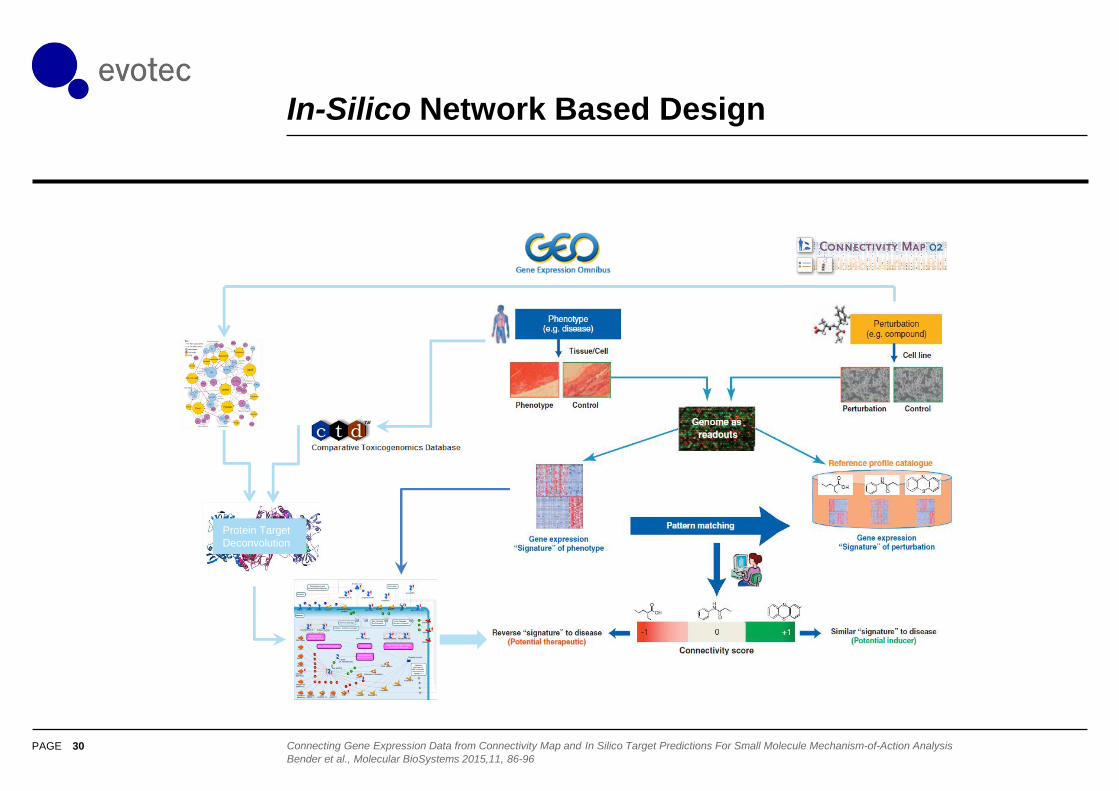
In-Silico Network Based Design
30
Protein Target
Deconvolution
Connecting Gene Expression Data from Connectivity Map and In Silico Target Predictions For Small Molecule Mechanism-of-Action Analysis
Bender et al., Molecular BioSystems 2015,11, 86-96
PAGE
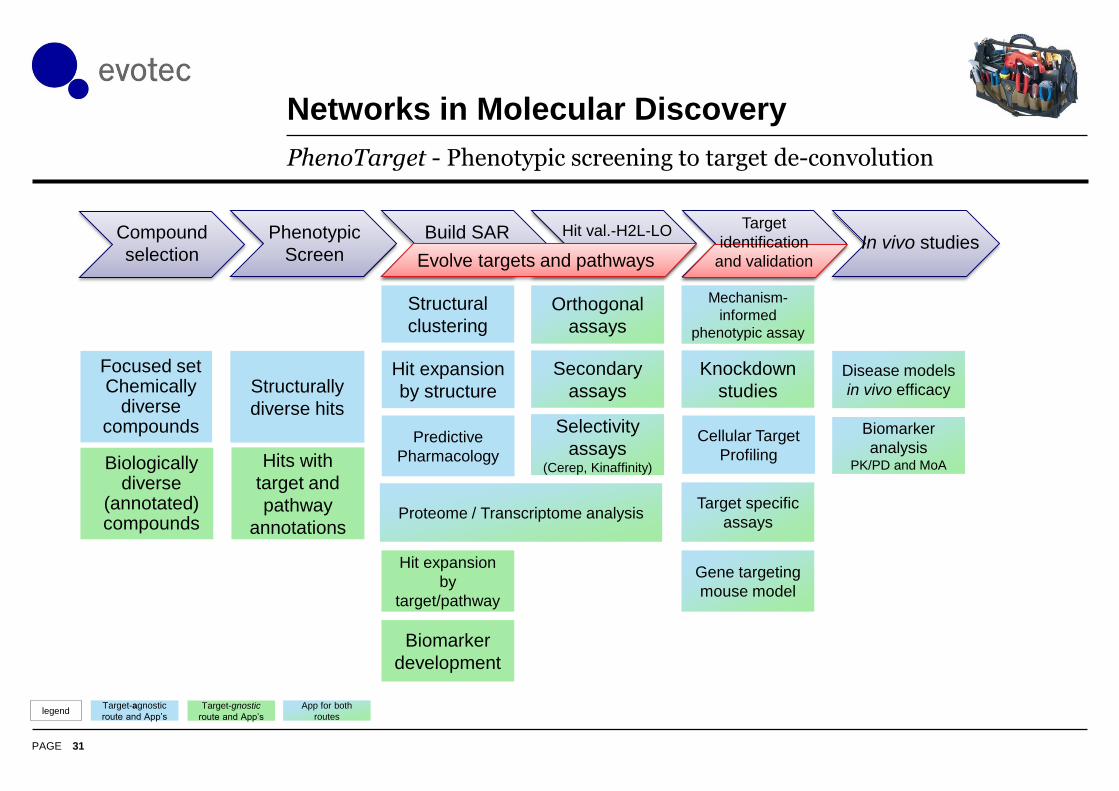
Networks in Molecular Discovery
31
PhenoTarget - Phenotypic screening to target de-convolution
Focused set Chemically
diverse compounds
Biologically diverse
(annotated) compounds
Structurally
diverse hits
Hits with
target and
pathway
annotations
Selectivity
assays(Cerep, Kinaffinity)
Secondary
assays
Orthogonal
assays
Predictive
Pharmacology
Hit expansion
by structure
Structural
clustering
Hit expansion
by
target/pathway
Biomarker
development
Cellular Target
Profiling
Mechanism-
informed
phenotypic assay
Knockdown
studies
Target specific
assays
Gene targeting
mouse model
Compound
selection
Phenotypic
Screen
Build SAR Hit val.-H2L-LO In vivo studies
Biomarker
analysis PK/PD and MoA
Disease models
in vivo efficacy
Proteome / Transcriptome analysis
Evolve targets and pathways
Target
identification
and validation
App for both
routes
Target-gnostic
route and App’s
Target-agnostic
route and App’slegend
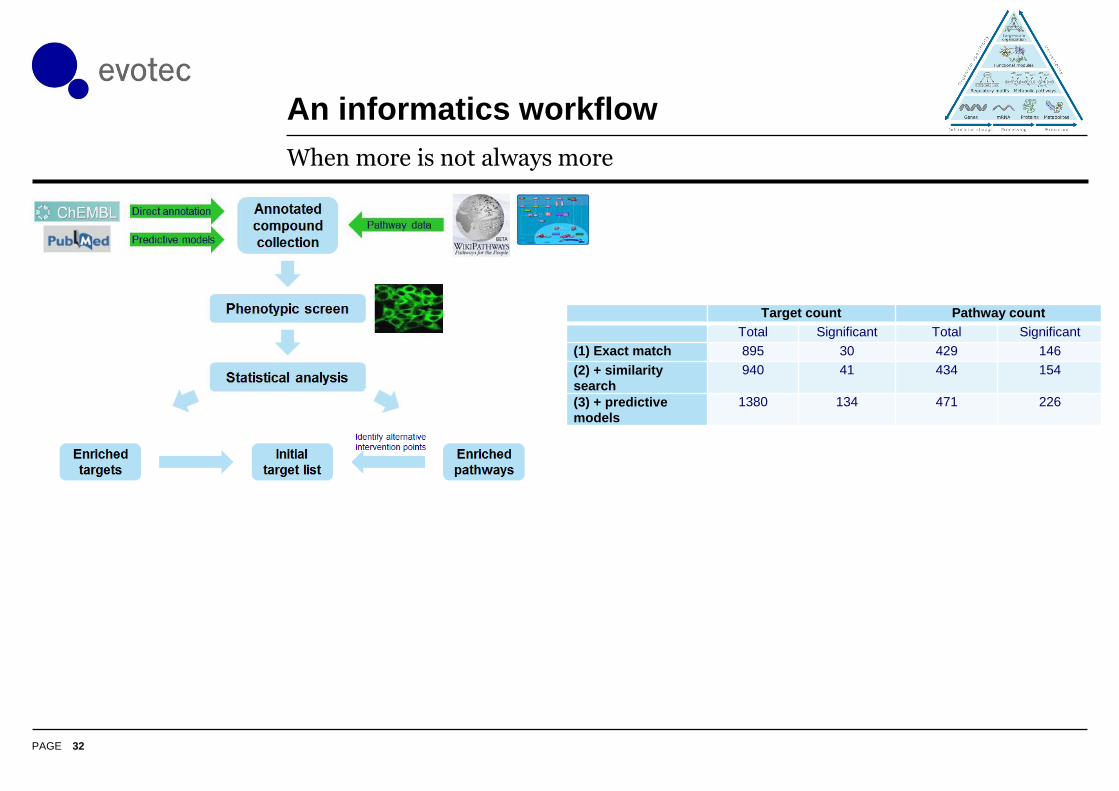
PAGE
An informatics workflow
32
When more is not always more
Target count Pathway count
Total Significant Total Significant
(1) Exact match 895 30 429 146
(2) + similarity
search
940 41 434 154
(3) + predictive
models
1380 134 471 226
PAGE
The Drug Discovery Challenge
33
Summary
Given a disease signature how do we best sample appropriate chemistry space?

PAGE
Happiness
34
What makes you happy?
A man doesn’t know what happiness is until he’s married,
by then it’s too late! (Frank Skinner)

PAGE
Acknowledgments
35
Dimitar Hristozov
David Thorner
David Evans
Nik Fechner
George Papadatos
Suzanne Brewerton
Lewis Vidler
Hina Patel
Ben Allen
James Wallace
John Liebeschuetz
Jason Cole
Colin Groom
Val Gillet
Beining Chen Andreas Bender
Georgios Drakakis
Tim James
Inaki Morai
Jon Hollick
York Rudhard
Alex Böcker
Your contact:
Building innovative
drug discovery alliances
Mike BodkinVP Research Informatics114 Innovation Drive,Milton Park, AbingdonOxfordshire OX14 4RZ, UKT: +44 (0)1235 44 1207
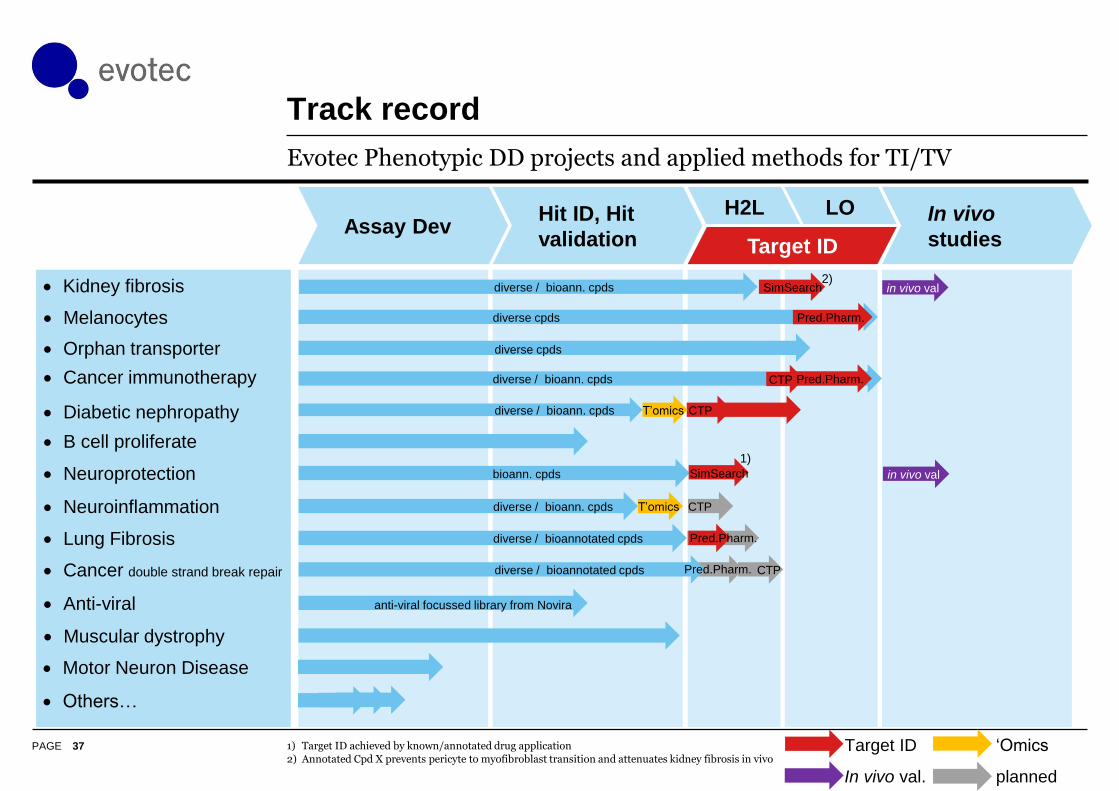
PAGE
Track record
37
Evotec Phenotypic DD projects and applied methods for TI/TV
1) Target ID achieved by known/annotated drug application
2) Annotated Cpd X prevents pericyte to myofibroblast transition and attenuates kidney fibrosis in vivo
Assay DevHit ID, Hit
validation
In vivo
studiesTarget ID
H2L LO
planned
B cell proliferate
Lung Fibrosis Pred.Pharm.diverse / bioannotated cpds
CTPT’omicsdiverse / bioann. cpds Neuroinflammation
Orphan transporter diverse cpds
Melanocytes Pred.Pharm.diverse cpds
Cancer immunotherapy CTPdiverse / bioann. cpds Pred.Pharm.
Diabetic nephropathy CTPT’omicsdiverse / bioann. cpds
Cancer double strand break repair CTPdiverse / bioannotated cpds Pred.Pharm.
diverse / bioann. cpds in vivo val Kidney fibrosis SimSearch2)
in vivo val Neuroprotection 1)
bioann. cpds SimSearch
Anti-viral anti-viral focussed library from Novira
Muscular dystrophy
Motor Neuron Disease
Others…
Target ID
In vivo val.
‘Omics
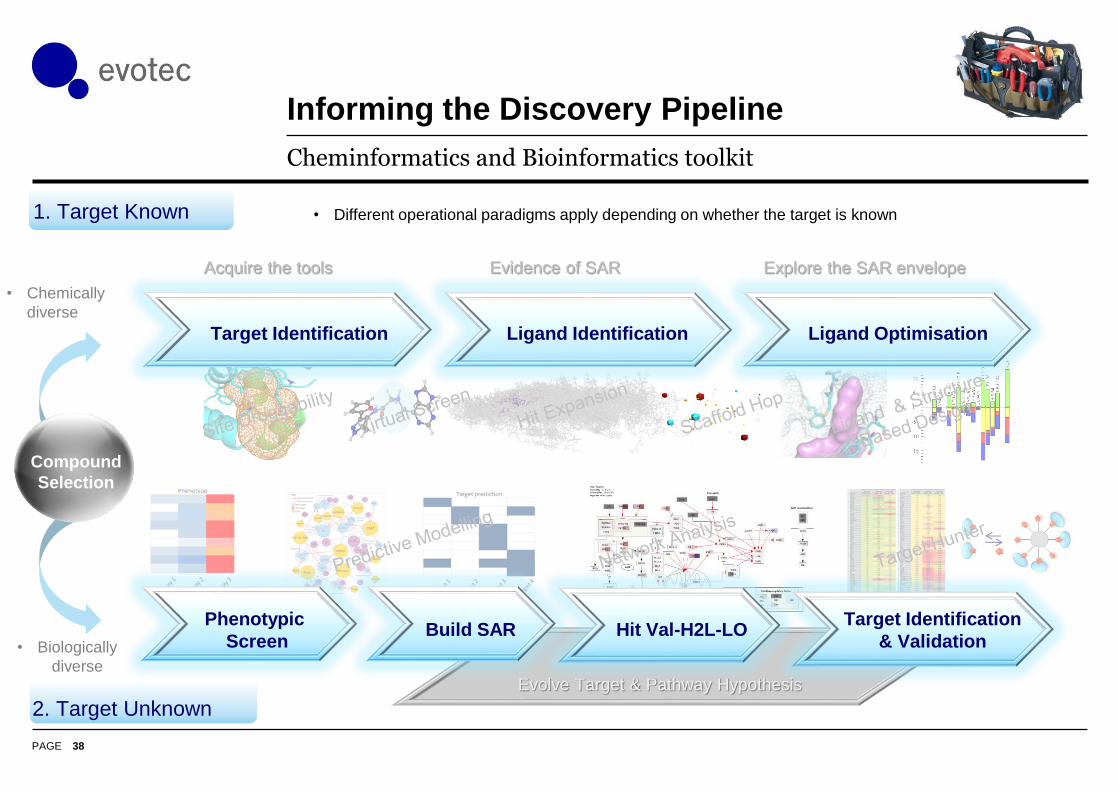
PAGE
Evolve Target & Pathway Hypothesis
Informing the Discovery Pipeline
38
Cheminformatics and Bioinformatics toolkit
Target Identification Ligand Identification Ligand Optimisation
Acquire the tools Evidence of SAR Explore the SAR envelope
Phenotypic
ScreenBuild SAR Hit Val-H2L-LO
Target Identification
& Validation
Compound
Selection
2. Target Unknown
1. Target Known
• Chemically
diverse
• Biologically
diverse
• Different operational paradigms apply depending on whether the target is known





























