Embed Size (px)
Citation preview

Alignment Class III
We continue where we stopped last week: FASTA – BLAST

FASTA-Stages
1. Find k-tups in the two sequences (k=1,2 for proteins, 4-6 for DNA sequences)
2. Score and select top 10 scoring “local diagonals”
a. For proteins, each k-tup found is scored using the PAM250 matrix
b. For DNA, the number of k-tups foundc. Penalize intervening gaps

Finding k-tups
position 1 2 3 4 5 6 7 8 9 10 11protein 1 n c s p t a . . . . . protein 2 . . . . . a c s p r k position in offsetamino acid protein A protein B pos A - posB-----------------------------------------------------a 6 6 0c 2 7 -5k - 11n 1 -p 4 9 -5r - 10s 3 8 -5t 5 ------------------------------------------------------Note the common offset for the 3 amino acids c,s and pA possible alignment is thus quickly found -protein 1 n c s p t a | | | protein 2 a c s p r k
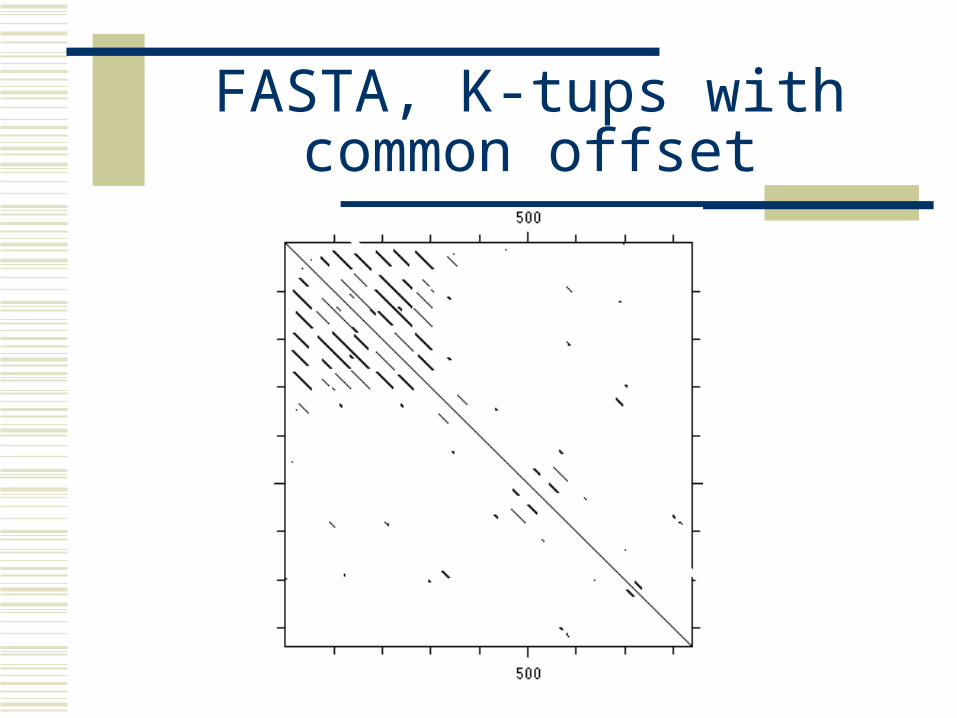
FASTA, K-tups with common offset

FASTA-Stages
3. Rescan top 10 regions, score with PAM250 (proteins) or DNA scoring matrix. Trim off the ends of the regions to achieve highest scores.
4. Try to join regions with gapped alignments. Join if similarity score is one standard deviation above average expected score
5. After finding the best initial region, FASTA performs a global alignment of a 32 residue wide region centered on the best initial region, and uses the score as the optimized score.

BLAST
Basic Local Alignment Search Tool Altschul et al. 1990,1994,1997
Heuristic method for local alignment Designed specifically for database searches Idea: Good alignments contain short lengths
of exact matches

Blast Application
Blast is a family of programs: BlastN, BlastP, BlastX, tBlastN, tBlastX
BlastN - nt versus nt database BlastP - protein versus protein database BlastX - translated nt versus protein database tBlastN - protein versus translated nt database tBlastX - translated nt versus translated nt database
Query: DNA Protein
Database: DNA Protein

Mathematical Basis of BLAST
Model matches as a sequence of coin tosses Let p be the probability of a “head”
For a “fair” coin, p = 0.5 (Erdös-Rényi) If there are n throws, then the expected length
R of the longest run of heads is
R = log1/p (n). Example: Suppose n = 20 for a “fair” coin
R=log2(20)=4.32 Trick is how to model DNA (or amino acid) sequence
alignments as coin tosses.

Mathematical Basis of BLAST
To model random sequence alignments, replace a match with a “head” and mismatch with a “tail”.
For DNA, the probability of a “head” is 1/4 Same logic applies to amino acids
AATCAT
ATTCAGHTHHHT

Mathematical Basis of BLAST
So, for one particular alignment, the Erdös-Rényi property can be applied
What about for all possible alignments? Consider that sequences are being shifted back and forth,
dot matrix plot The expected length of the longest match is
R=log1/p(mn)where m and n are the lengths of the two sequences.

Steps of BLAST
1. Filter out low-complexity regions
where L is length, N is alphabet size, ni is the number of letter i appearing in sequence. Example: AAAT
K=1/4 log4(24/(3!*1!*0!*0!))=0.25
iiN nLLK !/!log/1

Steps of BLAST
2. Query words of length 3 (for proteins) or 11 (for DNA) are created from query sequence using a sliding window
MEFPGLGSLGTSEPLPQFVDPALVSSMEF EFP FPG PGL GLG

Steps of BLAST
3. Using BLOSUM62 (for proteins) or scores of +5/-4 (DNA, PAM40), score all possible words of length 3 or 11 respectively against a query word.
4. Select a neighborhood word score threshold (T) so that only most significant sequences are kept. Approximately 50 hits per query word.
5. Repeat 3 and 4 for each query word in step 2. Total number of high scoring words is approximately 50 * sequence length.

Steps of BLAST
6. Organize the high-scoring words into a search tree
7. Scan each database sequence for match to high-scoring words. Each match is a seed for an ungapped alignment.
M
E
F
E
GP

Steps of BLAST
8. (Original BLAST) extend matching words to the left and right using ungapped alignments. Extension continues as long as score increases or stays same. This is a HSP (high scoring pair).
(BLAST2) Matches along the same diagonal within a distance A of each other are joined and then the longer sequence extended as before.

Steps of BLAST
9. Using a cutoff score S, keep only the extended matches that have a score at least S.
10. Determine statistical significance of each remaining match (from last time).
11. Try to extend the HSPs if possible.
12. Show Smith-Waterman local alignments.

Information theory
Shanon Entropy and information

Entropy
X: discrete Random Variable (RV), p(X) Entropy (or self-information)
Entropy measures the amount of information in a RV
p(x)p(x)logH(X)H(p)Xx
2

Entropy (cont)
p(x)1
log E
p(x)1
p(x)log
p(x)p(x)logH(X)
2
Xx2
Xx2
1p(X)0H(X)
0H(X)
i.e when the value of X
is determinate, hence providing no new information

Joint Entropy
The joint entropy of 2 RV X,Y is the amount of the information needed on average to specify both their values
Xx y
Y)p(X,y)logp(x,Y)H(X,Y

Conditional Entropy
The conditional entropy of a RV Y given another X, expresses how much extra information one still needs to supply on average to communicate Y given that the other party knows X
X)|p(YlogE x)|p(yy)logp(x,
x)|p(yx)log|p(yp(x)
x)X|p(x)H(YX)|H(Y
Xx Yy
Xx Yy
Xx

Chain Rule
X)|H(YH(X) Y)H(X,
),...XX|H(X....)X|H(X)H(X)X...,H(X 1n1n121n1,

Mutual Information
I(X,Y) is the mutual information between X and Y. It is the reduction of uncertainty of one RV due to knowing about the other, or the amount of information one RV contains about the other
Y)I(X, X)|H(Y -H(Y) Y)|H(X-H(X)
Y)|H(XH(Y) X)|H(YH(X) Y)H(X,

Mutual Information (cont)
I is 0 only when X,Y are independent: H(X|Y)=H(X)
H(X)=H(X)-H(X|X)=I(X,X) Entropy is the self-information
X)|H(Y -H(Y) Y)|H(X-H(X) Y)I(X,

Kullback-Leibler Divergence
Relative entropy or KL (Kullback-Leibler) divergence
q(X)p(X)
logE
q(x)p(x)
p(x)log q) ||D(p
p
Xx

Scoring matrices
Identity
PAM
BLOSUM

Scoring Matrices Types
• Identity matrix – exact matches receive one score and non-exat matches a different score (say 1 and 0, or 6 and –1 for local alignment.).
• Mutation data matrix – a scoring matrix compiled based on observation of protein point mutation (PAM, BLOSUM).
• Physical properties matrix – amino acids with with similar properties (e.G. hydrophobicity ) receive high score.
• Genetic code matrix – amino acids are scored based on similarities in the coding triple (codons).

Substitution Matrix
Amino acids substitute easily for another due to similar physicochemical properties
Isoleucine for Valine (both small, hydrophobic) Serine for Threonine (both polar) Such changes – “conservative”
Thus, need a way to increase sensitivity of the alignment algorithm
Solution – substitution matrix Therefore, we need a range of values that depend on the nature of
sequences being compared Identical amino acids > Conservative substitutions >
Nonconservative substitutions

Choice of scoring matrix is dictated by the alignment goals
• Two proteins are homologous if (and only if) they are evolutionarily related (have a common ancestor)
• Homologous proteins are likely to have related functions (and have the same fold)
• Scoring matrices must in some way model our understanding of protein evolution.
• Based on the result of the search we have to be able to decide if the discovered sequence similarity could happen by chance or is a signature of likely homology.





























