Embed Size (px)
Citation preview

Two siblings and a first degree cousin of a consanguinous marriage were afflicted with recessive generalized myotonia (RGMy). All had muscle weakness which was particularly prominent after rest, thinning of the forearms, weakness of anterior compartment muscles, and mus- cular contractures. The first degree cousin was the most severely afflicted with congenital myotonia. Muscle biopsy and electromyog- raphy were consistent with a myopathy. Exercise after rest demon- strated a marked reduction in muscle membrane excitability in all patients.
MUSCLE 81 NERVE 6~143-148 1983
AUTOSOMAL RECESSIVE GENERALIZED MYOTONIA
SALLIE F. SUN, MD, and ERlCH W. STREIB, MD
Only a few families with recessive generalized myotonia (RGMy) have been reported in the United States."" lS2' The frequent presence of conspicuous muscle weakness and wasting, dystro- phic changes on muscle biopsy, and myopathic features electromyographically in RGMy are not generally appreciated and may result in the erro- neous diagnosis of myotonic muscular dystrophy (MyD). This prompted the report of the follow- ing family (Fig. l), including some unusual clini- cal features.
CASE 1
El , a 33-year-old woman, presented with life-long muscle stiffness. This stiffness was most notable in the legs although there was marked difficulty with handgrip release. After her first, and particularly after her second, pregnancy she complained of a progressive feeling of weakness as if her legs would give way. The weakness was most pronounced after rest and often caused staggering after pro- longed sitting. She also experienced intermittent tightness of the eyes, tongue, and throat. She
From the Department of Neurology Universlty of Nebraska Medical Cen- ter 42nd and Dewey Avenue Omaha NE
Address reprint requests to Dr Sun at the Department of Neurology University of Nebraska Medical Center 42nd and Dewey Ave Omaha NE 68105
Acknowledgments We thank Dr Andrew Engel for providing the muscle biopsy slides for patient E, and Kathy Smith for preparation of the manuscript
Received for publication April 13 1982, revised manuscript accepted for publication October 5 1982
1983 John Wiley & Sons Inc 01 48-639X1060210143 $01 2510
denied episodes of localized or generalized paraly- sis. Her stiff-legged gait had been noted by her mother since age 18 months. Neonatal feeding or respiratory difficulties or delayed eye opening were denied.
General physical examination was unremark- able. Neither weakness of cranial nerve innervated muscles, nor lid lag or eyelid myotonia was pres- ent. The muscle bulk of the calves and thighs was prominent. The anterior compartment muscles of the legs were judged as 4/5 on formal testing; she had great difficulty walking on her heels, which could not be altered by warniup period. Toe walk- ing was normal. On arising from a chair her gait was stiff-legged for approximately 10- 15 seconds. In contrast to the increased muscle bulk in the legs, muscle bulk in her upper extremities was not increased, and there was mild thinning of the forearm extensors. Forearm extensor strength and handgrip were slightly reduced. Mild weakness of the triceps and deltoid muscles was also present. There was marked handgrip myotonia. Percussion myotonia was easily elicited from the thenar, tongue, and large muscles of the legs. Tendon reflexes were brisk and symmetrical; sensation was normal. Elbow flexion contractures of approxi- mately 30" were present.
Laboratory findings were as follows. Normal values are given in parentheses: creatine kinase (CK) 155 IU (s70); lactic dehydrogenase (LDH) 78 (c110); SGOT 17 (s25); IgG 1184 mg/100 ml (700-1800); IgA 220 mg/dl (60-450); IgM 126 mg/dl (60-250). Complete blood count, thyroid function studies, calcium and phosphorus levels, BUN, creatinine was normal.
EKG and slit-lamp examination findings were
Recessive Generalized Myotonia MUSCLE & NERVE February 1983 143
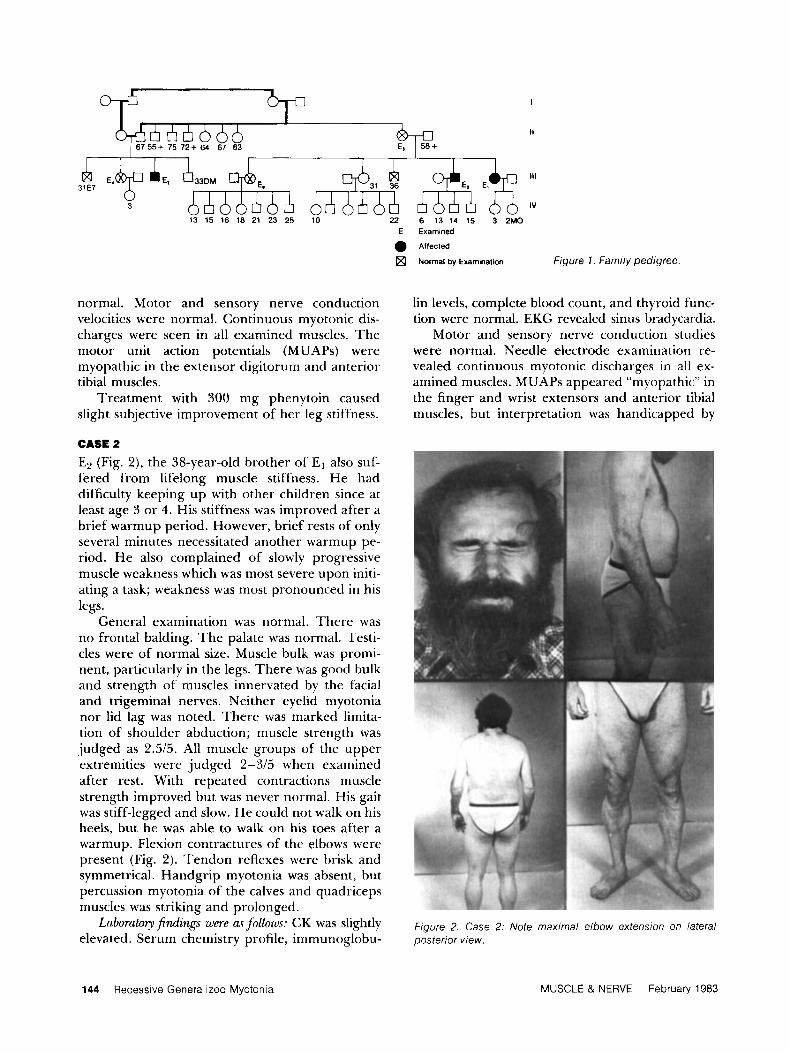
I 55+ 75 ?2+ 64 67 63
Ill
IV
13 15 16 18 21 23 25 10 22 6 13 14 15 3 2MO E Examined
Affected
a Normal by Examination Figure 7 . Family pedigree
normal. Motor and sensory nerve conduction velocities were normal. Continuous myotonic dis- charges were seen in all examined muscles. The motor unit action potentials (MUAPs) were myopathic in the extensor digitorum and anterior tibial muscles.
Treatment with 300 mg phenytoin caused slight subjective improvement of her leg stiffness.
lin levels, complete blood count, and thyroid func- tion were normal. EKG revealed sinus bradycardia.
Motor and sensory nerve conduction studies were normal. Needle electrode examination re- vealed continuous myotonic discharges in all ex- amined muscles. MUAPs appeared “myopathic” in the finger and wrist extensors and anterior tibial muscles, but interpretation was handicapped by
CASE 2
E2 (Fig. 2), the 38-year-old brother of El also suf- fered from lifelong muscle stiffness. He had difficulty keeping up with other children since at least age 3 or 4. His stiffness was improved after a brief warmup period. However, brief rests of only several minutes necessitated another warmup pe- riod. He also complained of slowly progressive muscle weakness which was most severe upon initi- ating a task; weakness was most pronounced in his legs.
General examination was normal. There was no frontal balding. The palate was normal. Testi- cles were of normal size. Muscle bulk was promi- nent, particularly in the legs. There was good bulk and strength of muscles innervated by the facial and trigeminal nerves. Neither eyelid myotonia nor lid lag was noted. There was marked limita- tion of shoulder abduction; muscle strength was judged as 2.5/5. All muscle groups of the upper extremities were judged 2-315 when examined after rest. With repeated contractions muscle strength improved but was never normal. His gait was stiff-legged and slow. He could not walk on his heels, but he was able to walk on his toes after a warmup. Flexion contractures of the elbows were present (Fig. 2). Tendon reflexes were brisk and symmetrical. Handgrip myotonia was absent, but percussion myotonia of the calves and quadriceps muscles was striking and prolonged.
were follo~us: CK was slightly elevated. Serum chemistry profile, immunoglobu-
Laborato?y Figure 2. Case 2: Note maximal elbow extension on lateral posterior view.
144 Recessive Generalized Myotonia MUSCLE & NERVE February 1983

Figure 3. Patient E?. Biopsy of extensor carpiradialis muscle reveals variable muscle fiber size with occasional fiber split- ting. lnternal nuclei are moderately increased in number. Hematoxylin and eosin stain. Bar = 100 Km.
continuous m yotonic activity with even minimal muscle contractions.
The extensor carpi radialis muscle was biop- sied. Muscle fiber size ranged from 16-150 pm with occasional fiber splitting (Fig. 3). Atrophic fibers were scattered randomly among the large ones. Occasionally, degenerating fibers were pres- ent, some of which were invaded by macrophages. Internal nuclei were moderately increased in num- ber. Endomysial and perimysial connective tissue were slightly increased. Type 1 fibers were more abundant than Type 2, and most of the atrophic fibers were type 2 (Fig. 4).
CASE 3 ES (Fig. 5) was the 39-year-old first degree cousin of El and EP. His parents were first cousins (Fig. l),
Figure 5. Case 3: Arm abduction and elbow extension limited due to contractures.
Figure 4. Patient E,: Type 1 fibers are more abundant than type 2. Most of the atrophic fibers are type 2. ATPase stain at pH 9.3. Bar = 700 km.
and his mother was 39 when he was born. A pro- longed delivery occurred at home. He was unable to suck for an indeterminate period and had to be fed with an eyedropper. A dislocated hip and bilat- eral talipes necessitated a plaster cast and braces during the first year of life. Although he partici- pated in most sports, he never ran well. He under- went corrective leg surgery in high school. Muscle strength has slightly worsened since age 14 but he cares for his own home and works as a woodcarver at a state school. He handles hammers, chisels, and chain saws daily without difficulty. He finished high school and the first year of college. He is unmarried and has no children.
He has always been lean and thin as are his father and younger brothers. Insulin dependent diabetes mellitus was first diagnosed at the age of 31.
The patient was an extremely thin man and without any appreciable subcutaneous fat. A mid- systolic murmur was heard over the left sternal border.
Language and intellectual function were nor- mal. His face was gaunt but the masseter and tem- poralis muscles were prominent. He whistled well. Anterior neck flexors were atrophic and moder- ately weak. All skeletal muscles were thin but definite atrophy was noted only in the muscles below the knees. Individual muscle strength was judged as follows: external rotators of the shoul- der and deltoid 515; triceps 415; wrist extensors and flexors 415; finger extensors and small hand muscles 415; hip flexion 3.515; knee extension 4.51 5 ; hamstrings 415; toe and ankle extensors and flexors 1-215. He was areflexic. Percussion myotonia was present in large and small muscles including calves, thighs, thenar, and tongue.
Recessive Generalized Myotonia MUSCLE & NERVE February 1983 145
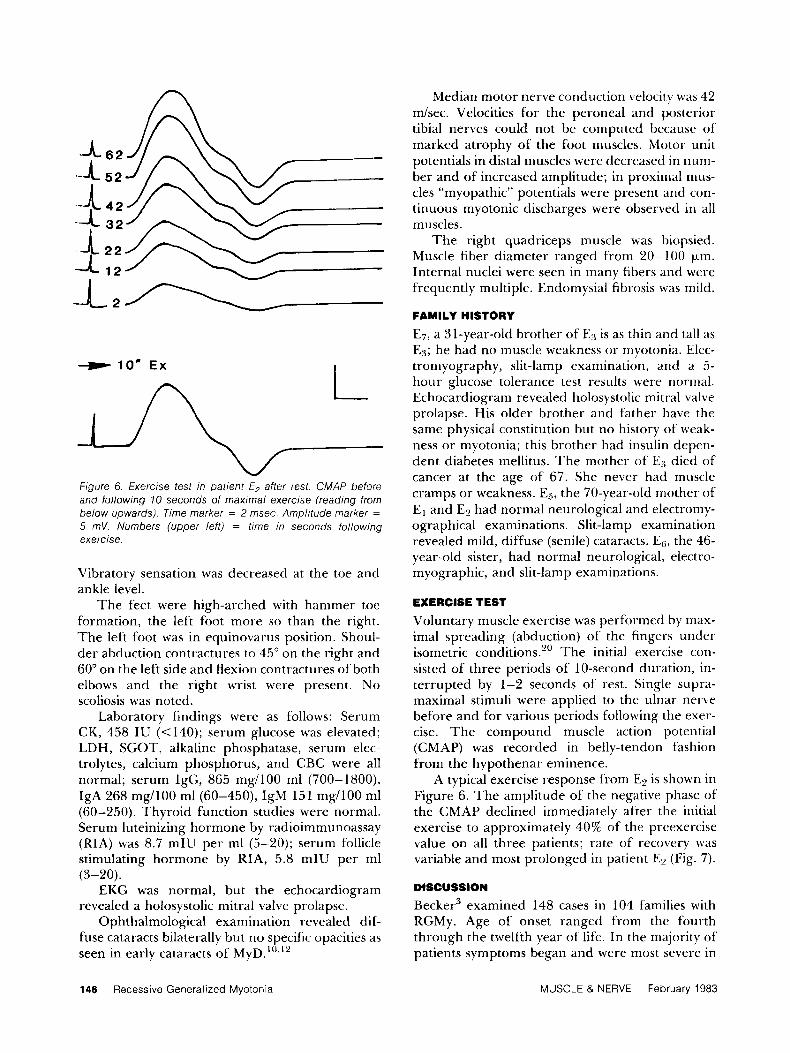
A -A
n 6 2
5 2
42 32
22 12
I - l o r n Ex
Figure 6. Exercise test in patient E2 after rest. CMAP before and following 10 seconds of maximal exercise (reading from below upwards). Time marker = 2 msec. Amplitude marker =
5 mV. Numbers (upper leff) = time in seconds following exercise.
Vibratory sensation was decreased at the toe and ankle level.
The feet were high-arched with hammer toe formation, the left foot more so than the right. The left foot was in equinovarus position. Shoul- der abduction contractures to 45” on the right and 60” on the left side and flexion contractures of both elbows and the right wrist were present. No scoliosis was noted.
Laboratory findings were as follows: Serum CK, 458 IU (<140); serum glucose was elevated; LDH, SGOT, alkaline phosphatase, serum elec- trolytes, calcium phosphorus, and CBC were all normal; serum IgG, 865 mg/100 ml (700-1800), IgA 268 mg/100 ml(60-450), IgM 151 mg/100 ml (60-250). Thyroid function studies were normal. Serum luteinizing hormone by radioimmunoassay (RIA) was 8.7 mIU per ml (5-20); serum follicle stimulating hormone by RIA, 5.8 mIU per ml
EKG was normal, but the echocardiogram revealed a holosystolic mitral valve prolapse.
Ophthalmological examination revealed dif- fuse cataracts bilaterally but no specific opacities as seen in early cataracts of M ~ D . ~ ~ ’ , ~ ~
(3- 2 0).
Median motor nerve conduction velocity was 42 m/sec. Velocities for the peroneal and posterior tibia1 nerves could not be computed because of marked atrophy of the foot muscles. Motor unit potentials in distal muscles were decreased in nurn- ber and of increased amplitude; in proximal mus- cles “myopathic” potentials were present and con- tinuous myotonic discharges were observed in all muscles.
The right quadriceps muscle was biopsied. Muscle fiber diameter ranged from 20-100 Fm. Internal nuclei were seen in many fibers and were frequently multiple. Endomysial fibrosis was mild.
FAMILY HISTORY
E7, a 3 I-year-old brother of E:4 is as thin and tall a s E:l; he had no muscle weakness or myotonia. Elec- tromyography, slit-lamp examination, and a 5- hour glucose tolerance test results were normal. Echocardiogram revealed holosystolic mitral valve prolapse. His older brother and father have the same physical constitution but no history of weak- ness or myotonia; this brother had insulin depen- dent diabetes mellitus. The mother of Eg died of cancer at the age of 67. She never had muscle cramps or weakness. E5, the 70-year-old mother of El and E2 had normal neurological and electromy- ographical examinations. Slit-lamp examination revealed mild, diffuse (senile) cataracts. EG, the 46- year-old sister, had normal neurological, electro- myographic, and slit-lamp examinations.
EXERCISE TEST
Voluntary muscle exercise was performed by max- imal spreading (abduction) of the fingers under isometric conditions.“ The initial exercise con- sisted of three periods of 10-second duration, in- terrupted by 1-2 seconds of rest. Single supra- maximal stimuli were applied to the ulnar nerve before and for various periods following the exer- cise. The compound muscle action potential (CMAP) was recorded in belly-tendon fashion from the hypothenar eminence.
A typical exercise response from E2 is shown in Figure 6. The amplitude of the negative phase of the CMAP declined immediately after the initial exercise to approximately 40% of the preexercise value on all three patients; rate of recovery was variable and most prolonged in patient E2 (Fig. 7).
DISCUSSION
Becker3 examined 148 cases in 104 families with RGMy. Age of onset ranged from the fourth through the twelfth year of life. In the majority of patients symptoms began and were most severe in
146 Recessive Generalized Myotonia MUSCLE & NERVE February 1983
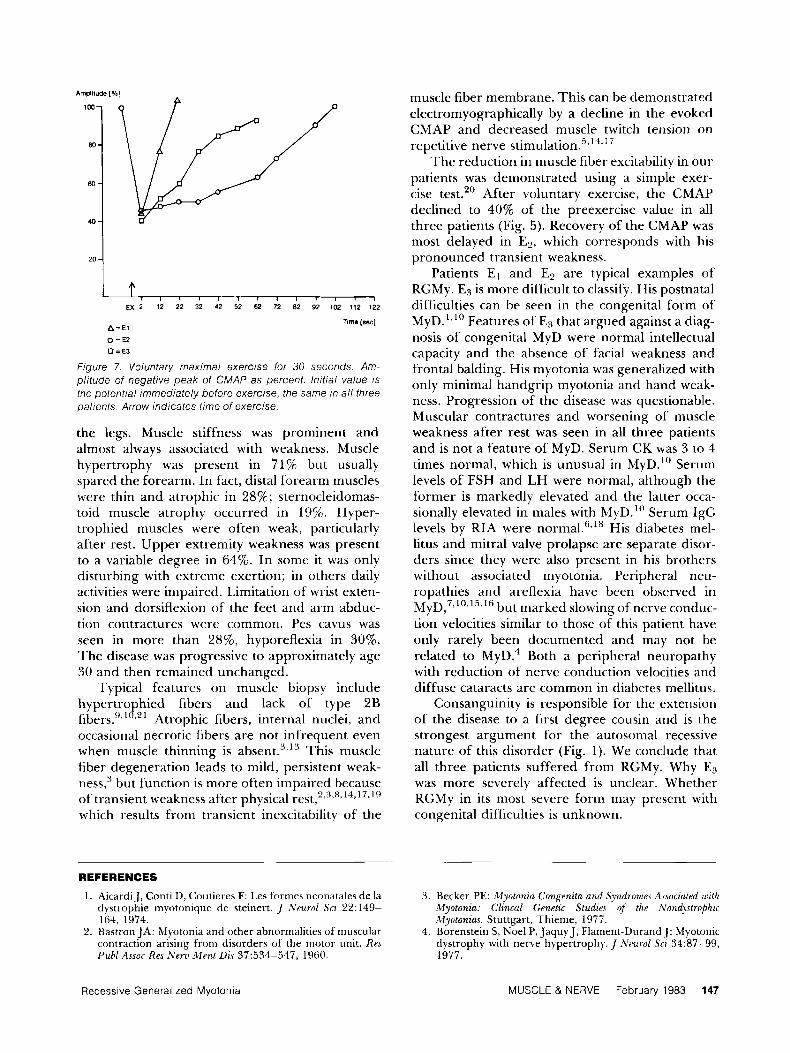
Amplitude [a] 1W-
Bo-
60-
40 -
2 0 .
EX 2 12 22 32 42 52 62 72 82 92 102 112 122
Time [sac] ATE1
0 =E2 0 = E 3
Figure 7. Voluntary maximal exercise for 30 seconds. Am- plitude of negative peak of CMAP as percent. lnitial value is the potential immediately before exercise, the same in a / / three oatients. Arrow indicates time of exercise.
the legs. Muscle stiffness was prominent and almost always associated with weakness. Muscle hypertrophy was present in 71% but usually spared the forearm. In fact, distal forearm muscles were thin and atrophic in 28%; sternocleidomas- toid muscle atrophy occurred in 19%. Hyper- trophied muscles were often weak, particularly after rest. Upper extremity weakness was present to a variable degree in 64%. In some it was only disturbing with extreme exertion; in others daily activities were impaired. Limitation of wrist exten- sion and dorsiflexion of the feet and arm abduc- tion contractures were common. Pes caws was seen in more than 2876, hyporeflexia in 30%. The disease was progressive to approximately age 30 and then remained unchanged.
Typical features on muscle biopsy include hypertrophied fibers and lack of type 2B fibers."'0.21 Atrophic fibers, internal nuclei, and occasional necrotic fibers are not infrequent even when muscle thinning is absent."13 This muscle fiber degeneration leads to mild, persistent weak- n e q 3 but function is more often impaired because of transient weakness after physical re~t , ' , " ,~ , '~ . '~ . ' " which results from transient inexcitability of the
muscle fiber membrane. This can be demonstrated electrornyographically by a decline in the evoked CMAP and decreased muscle twitch tension on repetitive nerve stimulation."'4.'
The reduction in muscle fiber excitability in our patients was demonstrated using a simple exer- cise test.20 After voluntary exercise, the CMAP declined to 40% of the preexercise value in all three patients (Fig. 5). Recovery of the CMAP was most delayed in E P , which corresponds with his pronounced transient weakness.
Patients El and E2 are typical examples of RGMy. E3 is more difficult to classify. His postnatal difficulties can be seen in the congenital form of MYD.'~'' Features of E3 that argued against a diag- nosis of congenital MyD were normal intellectual capacity and the absence of facial weakness and frontal balding. His myotonia was generalized with only minimal handgrip myotonia and hand weak- ness. Progression of the disease was questionable. Muscular contractures and worsening of muscle weakness after rest was seen in all three patients and is not a feature of MyD. Serum CK was 3 to 4 times normal, which is unusual in MyD." Serum levels of FSH and LH were normal, although the former is markedly elevated and the latter occa- sionally elevated in males with MyD." Serum IgG levels by RIA were His diabetes mel- litus and mitral valve prolapse are separate disor- ders since they were also present in his brothers without associated myotonia. Peripheral neu- ropathies and areflexia have been observed in MyD,7,10,1"16 b ut marked slowing of nerve conduc- tion velocities similar to those of this patient have only rarely been documented and may not be related to MyD.' Both a peripheral neuropathy with reduction of nerve conduction velocities and diffuse cataracts are common in diabetes mellitus.
Consanguinity is responsible for the extension of the disease to a first degree cousin and is the strongest argument for the autosomal recessive nature of this disorder (Fig. 1). We conclude that all three patients suffered from RGMy. Why E3 was more severely affected is unclear. Whether RGMy in its most severe form may present with congenital difficulties is unknown.
REFERENCES
1 . Aicardi.], Conti I), Coutieres F: Les formes neonatales de la 3 . Becker PE: Myotonia Congenita and Syndromes A.noczuted with dystrophie myotonique de steinert. ,I "Veurol Scz 22: 149- Myotonia: Clincal Genetic Studie5 qf the Nondystrophic 164, 1974. Myotonzas. Stuttgart, Thienie, 1977.
2. Bastron JA: Myotonia and other abnormalities of muscular 4. Borenstein S, Noel P, Jaquy J , Flament-DurandJ: Myotonic contraction arising from disorders of the motor unit. Res dystrophy with nerve hypertrophy. J Neurol Sci 34:87-99, Pub1 Assoc Res N e m Merit Dic 37:534-547, 1960. 1977.
Recessive Generalized Myotonia MUSCLE & NERVE February 1983 147

5. Brown JC: Muscle weakness after rest in myotonic disor- ders: An electrophysiological study. J Neurol Neurosurg PSJ- chzatry 37:1336-1342, 1974.
6. Bundey S, Carter CO, Soothill JF: Early recognition of heterozygotes for the gene of dystrophia myotonica. J Neurol Neurosurg Psyhiotry 37:1336-1342, 1974.
7. Caccia MR, Negri S, Parivis VP: Myotonic dystrophy with neural involvement. J Neurol Sci 16:253-269, 1974.
8. Castaigne P, Laplane D, Augustin P, Dordain G, Penders C: Myotonia congenitale, faiblesse musculaire corrigke par l’exercise et hypertrophie musculaire. Rev ,Veurol (Paris) 129:52-57, 1973.
9. Crews J, Kaiser KK, Brooke MH: Muscle pathology of my- otonia congenita. J Neurol Sci 283449-457, 1972.
10. Harper PS: Myotontr Dy.rtrophy. Philadelphia, WI3 Saunders, 1979.
1 1. Harper PS, Johnston DM: Kecessively inherited myotonia congenita. J Med Genet 9:2 13-2 15, 1972.
12. Junge J: Ocular changes in dystrophia myotonica, paramy- otonia and myotonia congenita. Doc Ophthalmol 2 1: 1-1 15, 1966.
13. Kuhn E, Fiehn W, Seiler D, Schroder J-M: The autosomal recessive (Becker) form of niyotonia congenita. M u s c l e Nerve 2: 109-1 17, 1979.
14. Lambert EH, Millikan Ch, Eaton LM: Stage of neuromus- cular paralysis in myotonia. Am J Phyzol I il:74 1, I052 (Abstract).
1.5. Panayiotopoulos CP, Scarpalezos S: Dystrophia myotoiiica. Peripheral nerve involvement arid pathogenetir implica- tions../ Neurol Scz 27:l-16. 1976.
16. Pilz I i , Prill A, Volles E: Kombination \’on tnyotonischer Dystrophie mit “idiopathischer” Neuropathie. Z h’eurol 206:253-265, 1974.
17. Kicker K, Meinck H-M, Stunipf H: Neurophysiologische Untersuchungen uber das Stadium passagerer LLhmung bei Myatonia congenita und Dystrophia myotonica.j N e u d 204:135-148, 1973.
18. Roberts DF, Bradley WG: Immunoglubulin levels in dys- trophia myotonica. J Med Genet 14: 16-19, 1977.
19. Sabouraud 0, Bourel M, Chatel M, LebarsJ: Faiblesse mus- culaire corrigee par exercise accompagnant une hyper- trophie musculaire avec myotonie. lieu Neurol (Paris) 121:546-549, 1965.
20. Streib EW, Sun SF: Isometric exercise test in myotonic dis- orders. EEG Clin Neuroflhysiol 52: 120, 1981 (Abstract).
21. Zellweger H, Pavone L, Biondi A, Cimino V, Gullotta F, Hart M, Ionescu V, Mollica F, Schieken R: Autosomdl reces- sive generalized myotonia. Muscle Neme 3: 176-180, 1980.
148 Recessive Generalized Myotonia MUSCLE & NERVE February 1983




![Autosomal recessive ichthyosis with limb reduction defect ... · including autosomal dominant, autosomal recessive and X-linked inheritance [1,2]. Associated cutaneous and extracutaneous](https://img.pdfslide.net/doc/110x75/5ec8c9b91adfdf12ab3e663c/autosomal-recessive-ichthyosis-with-limb-reduction-defect-including-autosomal.jpg)
























