Embed Size (px)
Citation preview

Prader-Willi Syndrome: An overview
Iqbal Muhamad, Hamish Cameron, Toby Struthers

PC
• 1/12 old term baby girl present with poor feeding, poor weight gain, low muscle tone and sleepiness since birth.

HPC
• Feeding problem since day 1• Sleepiness interfere with feeding• Poor suck requiring naso-gastric (NG) feeds while
in QM• Poor suck may be related to being generally low
tone• A decrease of about 8.6% birth weight at
discharge• Able to fully bottle feed q3-4 hourly at time of
discharge

HPC
• Feeding problem continues post discharge
• Food: EBM and BF only
• Sleepiness – need to be woken up for feeds for most of the time. Q3h during the day and q4h during the night.
• Sleeping while feeding – only able to suck about 30-40mls each time before falling asleep again

HPC
• Not been particularly spilly
• No choking on feeds
• Normal bowel motions
• Usually has 5 to 6 wet nappies per day
• She had no breathing problems/cyanotic spells/apnoeas/sweatiness/fevers

Pregnancy history
• Born to a G2P2 mother
• Born at term 37+5/40 by LSCS for drop off in EFW 50th to 5th centile – birth weight 2740g
• Apgar 8 at 1 minute, 10 at 5 minutes
• Vitamin K at birth
• No maternal post-natal issues

Growth and Development Hx
• Growth:– Birth weight – 2740g
– 1 week old – 2505g (-235g)
– 2 week old – 2610g (+105g)
– 1 month old – 2755g (+145g)
• Development:– Generally low tone
– Poor eye contact with parents – sleepiness
– Parents think baby recognises their voices
– Weak cry – minimal vocalization

Other Medical Issues/Medication Hx
• Fitted with Pawlick hip harness @ <7/7 of age as of DDH of R hip – to stay on for 6/52
• Not on regular medications/NKDA
• Not immunised yet as too young

Social Hx
• Lives with mother and father and older sister 2 and ½ year old in Dunedin
• Good social support
• Smoke free environment
• Own private transport and phone
Family History
• Samoan father, European mother
• No family history genetic disorders
• Healthy older sister

Examination:
• Afebrile, HR 136bpm, RR 36bpm Sats 100% at 21% FiO2• Wt 2755g (<3rd centile), L 52cm (25th centile), Head
Circumference 36cm (25th centile)• Mostly asleep during examination, mildly reactive and not
crying – opened eyes briefly only during length examination• Eyes were not fixing or following stimulus• Slightly almond shaped eyes• Small chin but not obviously disproportional to size of face• Generally low tone/hypotonia with marked head lag• Normal patellar reflexes bilaterally• Pawlick hip harness• All other examinations are unremarkable

Impression
• Poor feeding, poor weight gain, sleepiness, hypotonia with possible mild dysmorphic features
• ? Genetic disorder– Prader-Willi– Spinal muscular atrophy– (look up diff hypotonia and poor feeding)– Congenital myotonic dystrophy– Bardet-Biedl– Cohen syndrome– Albright hereditary osteodystrophy
• ? Metabolic disorder

Investigations
• Guthrie tests (PKU, MSUD, galactosemia, biotinidase deficiency, congenital hypothyrodism, CF, CAH, fatty acid and amino-acid disorders) – were all normal
• Blood tests - unremarkable
• Genotypic cytogenetic karyotyping – blood sample

Chromosome studies
• Microarray analysis showed a 5.7Mb deletion involving the long arm of chromosome 15q11.2-13.1
• Further testing was performed indicating that the paternal allele had been deleted
• Diagnosis consistent with Prader WilliSyndrome

Epidemiology of Prader WilliSyndrome
• Internationally the prevalence is anywhere between 1/10,000-1/30,000
• Affects males and females equally

History
• In 1887, John Langdon Down described the first girl with probable Prader-Willi Syndrome (PWS) during adolescence manifest by – mental impairment– short stature– Hypogonadism– Obesity and he termed the
condition polysarcia
John Langdon Down 1828-1896

History
• 70 years later
• 1956: PWS was properly described by Endocrinologists Prader, Labhart and Willi. Published findings included:– Diminished foetal activity.– Hypotonia and feeding problems in infancy.– Hypogonadism.– Short stature and retarded bone age.– Small hands and feet, delayed developmental
milestones, characteristic faces, cognitive impairment.
– Gross obesity in early childhood due to insatiable hunger, and a tendency to develop diabetes in adolescence and adulthood.
www.pwsausa.org

Since then
• In 1960s, further studies and case reports were published, adding more to the understanding of the syndrome. No genetic basis identified
• In 1981, Ledbetter et al, identified microdeletions within chromosome 15 as the site for PWS
Deletions of chromosome 15 as a cause of the Prader-Willi syndrome. Ledbetter DH, Riccardi VM, Airhart SD, Strobel RJ, Keenan BS, Crawford JD. N Engl J Med. 1981;304(6):325.
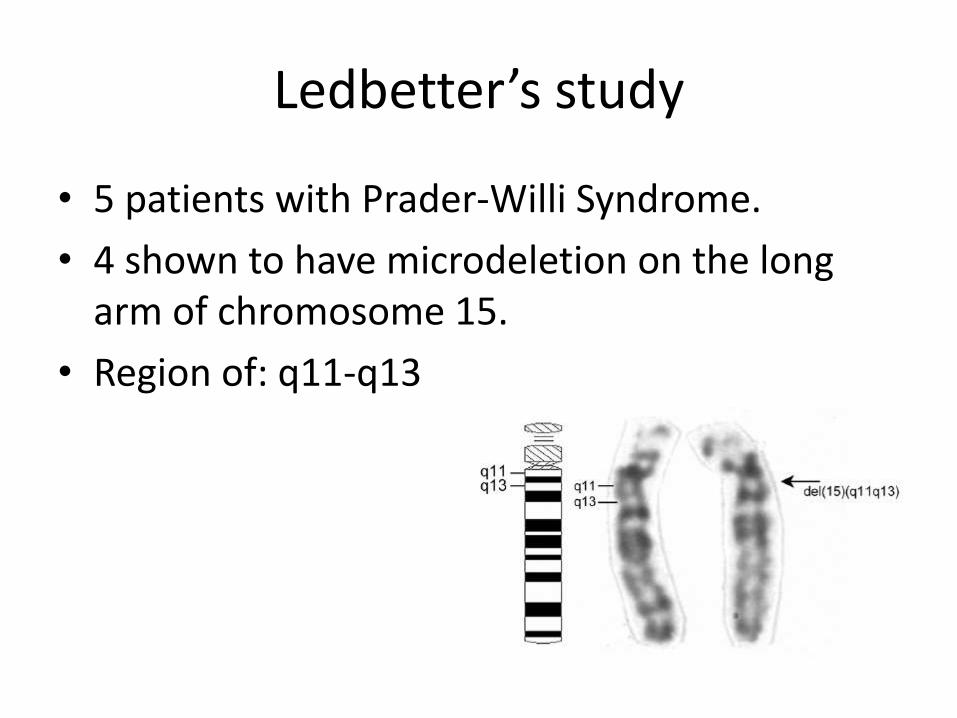
Ledbetter’s study
• 5 patients with Prader-Willi Syndrome.
• 4 shown to have microdeletion on the long arm of chromosome 15.
• Region of: q11-q13

Current genetic understanding
• Due to abnormality affecting 15q11.2-13
• First ever genetic disorder explained by genomic imprinting
– Paternal chromosome 15 must be affected

Current Genetic Understanding
• Majority of cases arise sporadically
• PWS only develops if the paternal chromosome is affected
• If the same region on the maternal chromosome is affected then Angelmansyndrome results.

Mutations
• Vast majority of cases are sporadic
• Approximately 70% of cases are due to deletion of 15q11.2-13
• Almost 30% are due to maternal uniparentaldisomy. Chromosomal non-disjunction
• Approximately 1% are due to mutations in imprinting centre, translocations or other small mutations

Which Genes are affected?
• SNRPN gene
• P gene
• UBE3A gene
• necdin gene
• Ghrelin has been implicated by several studies
Correlation with phenotype unclear

What is the role of genetic testing
1. Secure a diagnosis
2. Establish risk for future pregnancies
1. < 1% if a deletion or maternal disomy
2. Around 50% if mutation affects the imprinting control centre
3. 25% if chromosomal translocation is present.
This makes up small minority of cases

Genetic testing
1. Methylation analysis detects about 99% of cases. But doesnt identify cause.
2. Then need to determine if cause is
1. Deletion: FISH
2. Uniparental disomy: analysis of microsattelitemarkers. Both child and parents
3. If these both negative, then further testing for more obscure mutations.

Prader – (Labhart-) Willi Syndrome, PWS
• First described in 1956
• Genetic disorder associated
– Developmental delay
– Obesity
– Obsessive behaviour related to food consumption
• Clinically is dysmorphic syndrome affecting the CNS, particularly the hypothalamus

Clinical features of PWS
• Prenatal
– Reduced foetal movements
– Breech presentations
– Polyhydramnios
• Neonatal
– Hypotonia especially neck
– Abnormal weak/absent cry
– Poor feeding – poor suck tube feeding

Clinical features of PWS
• Infancy – Motor activity gradually
– Ongoing feeding problems failure to thrive
– Developmental delay ( obvious at 1 year)• Gross motor
• Speech – high pitched nasal
– Facial • Narrow biparietal diameter, thin upper lip,
almond palpebral fissures, narrow nose
– Depigmentation of the skin

Clinical features of PWS
• Childhood – Late acquisition of major motor milestones– Hyperphagia– Obesity – lean BM and fat mass– Growth impairment
• Impaired linear growth • Short stature
– Cognitive impairment • Most mild to moderate disability • School performance: skill at jigsaw excellent,
reading reasonable, arithmetic poor
– Behavioural problems• Temper tantrums• Stubborness
Clinical features of PWS
• Late childhood / adolescence – Delayed sexual development
• Delayed menarche, delayed testicular dissent
– Failure of pubertal growth spurt – Behavioural and learning difficulties become prominent– Behavioural
• Obsessional – trivial changes in routine lead to tantrums• Difficultly handling money , narcolepsy
– Complications of obesity • OSA, DM , Artherosclerosis
– Scoliosis • Corrective surgery
– Hypogonadism • Sterility• Osteoporosis

Clinical features of PWS
• Adulthood
– Historically death in 5th decade
– Complications of Obesity
• CVD, DM, orthopaedic complications
– Dependence on others
– Possible psychiatric disorders
• Severe affective disorder – Depression or Bipolar
• Florid psychosis

Hyperphagia (excessive appetite)
• PWS patients consume up to 3x normal caloric intake • Mechanism unknown
– Possibly ghrelin (hunger stimulating peptide)
• 30% are > 200% ideal body weight • Food seeking behaviours
– Eating rubbish, eating frozen food, stealing for food
• Binge eating and decreased ability to vomit • Choking episodes associated with voracious eating • Cases of gastric rupture
– Abdominal discomfort and vomiting – May present late due to increased pain threshold

Diagnosis of PWS
• Characteristic clinical features
• Clinical diagnostic criteria, ( Holm 1993)– PWS highly likely child < 3 years if 5 points are scored
– Major (one point)• neonatal and infantile hypotonia, feeding problems in infancy,
excessive weight gain after infancy, characteristic facial features
– Minor (half point)• Decreased fetal movement, characteristic behavioural problems,
sleep disturbance or sleep apnoea, short stature, small hands or feet
• Genetic testing – Karyotype and methylation studies, FISH

Management of PWS
• Feeding and diet control
• Endocrine
– Hypothalamic and pituitary dysfunction
– GH deficiency
– Hypogonadism
• Osteoporosis
• Obesity related problems
• Social supports

Management of PWS
• Neonates and infant feeding– Oromotor assessment, swallow study, thickened feeds– Aim for moderate weight gain– Nutrition for brain myelination and cognitive development– Intensive physical therapy and occupational therapy for muscle
strength– Speech therapy
• Swallowing, communication and enunciation
• Older children and adults– Limit food intake
• Low caloric diets • Vitamin and mineral supplementation• Physical barriers to food , close supervision• Coordination of home and school
– Anorectic ( appetite suppressants) and surgery not beneficial

PWS in childhood

Management of PWS
• Hypothalamic and pituitary dysfunction– Short stature , central obesity, hypogonadism,
osteoporosis
• GH treatment (Endocrinologist)– For short stature– Benefits –linear growth, bony density, improved
fat free mass – Early treatment may improve outcomes – Contraindicated
• >225% ideal body mass, DM, severe sleep apnoea, respiratory compromise

Management of PWS
• Hypogonadism – Low LH, high FSH
– Most sterile
– 2/3 cryptorchidism – surgery indicated
– Endocrinologist for sex hormone replacement
• Osteoporosis– Serial DEXA scan at aged 5 then 2-3 yearly
– Low bone density • Consider sex hormone/ GH replacement

Management of PWS
• Obesity related problems
– DM, dyslipidemia, cardiovascular disease, GORD, non alcohol fatty liver disease , OSA
• Social supports – peers, family
• Additional education / special needs
• Multidisciplinary management
– Healthcare and education workers

Prognosis
• Recurrent risk <1 % unless parental translocation (rare)
• Lifespan shortened only by complication of obesity
• Management extremely difficult





























