Embed Size (px)
Citation preview

Biological and Biomechanical Changes in Muscle Contractures Richard L. Lieber
Chief Scientific Officer and Senior Vice President Rehabilitation Institute of Chicago
Chicago, IL 60611
Relevant References (references shown in bold attached to handout)
1. Normal muscle has a stereotypical sarcomere length operating range. Lieber, R.L. G.J. Loren and J. Fridén. (1994). In vivo measurement of human wrist extensor muscle sarcomere length changes. J. Neurophysiol. 71:874-881.
Fridén, J. and R.L. Lieber. (1994). Physiological consequences of surgical lengthening of extensor carpi radialis brevis MTJ for tennis elbow. J. Hand Surg. 19A:269-274.
Loren, G.J., S.D. Shoemaker, T.J. Burkholder, M.D. Jacobson, J. Fridén, and R. L. Lieber. (1996). Human wrist motors: Biomechanical design and application to tendon transfers. J. Biomech. 29:331-342.
Lieber, R.L, Pontén, E and J. Fridén. (1996). Sarcomere length changes after flexor carpi ulnaris-to-extensor digitorum communis tendon transfer. J. Hand Surg. 21A:612-618.
Lieber, R.L. , Ljung, B.-O., and J. Fridén. (1997). Intraoperative sarcomere measurements reveal differential design of human wrist extensor muscles. J. Exp. Biol. 200:19-25.
Lieber, R.L. and J. Fridén. (1997). Intraoperative measurement and biomechanical modeling of the flexor carpi ulnaris-to-extensor carpi radialis longus tendon transfer. J. Biomech. Eng. 119:386-391.
Ward, S.R., C.W. Kim, C.M. Eng, L.J. Gottschalk, A. Tomiya, S.R. Garfin, and R.L. Lieber. (2009). Architectural analysis and intraoperative measurements demonstrate the unique design of the multifidus for lumbar spine stability. J. Bone Joint Surg. [Am.] 91:176-185.
Mittal, R.K., G. Sheean, G. Padda, R. Lieber and R. Rajasekaran. (2011). The external anal sphincter operates at short sarcomere length in humans. Neurogast. and Motil. 23:643-648.
2. Contracture have shortened muscles with highly stretched sarcomeres: Lieber, R.L. and J. Fridén. (2002). Spasticity causes a fundamental rearrangement of muscle-joint interaction. Muscle & Nerve 25:265-270.
Pontén, E. and R.L. Lieber. (2007). Intraoperative sarcomere length measurements provide insight into the development of wrist flexion contractures. Muscle & Nerve 36:47-54.
Smith, L.R., K.S. Lee, S.R. Ward, H.G. Chambers, R.L. Lieber. (2011) Hamstring contractures in children with spastic cerebral palsy result from a stiffer ECM and increased in vivo sarcomere length. J. Physiol. (Lond.) 589:2625-2639.
3. Contractures contain muscles with altered extracellular matrix and deranged transcriptional regulation:
Fridén, J. and R.L. Lieber. (2003). Spastic muscle cells are shorter and stiffer than normal cells.

Muscle & Nerve 26:157-164.
Lieber, R.L., E. Runesson, F. Einarsson and J. Fridén. (2003). Inferior mechanical properties of spastic muscle bundles due to hypertrophic but compromised extracellular matrix material. Muscle & Nerve 28:464-471.
Smith, L., E. Pontén, Y. Hedström, S.R. Ward, S. Subramaniam and R.L. Lieber. (2009). Novel transcriptional profile in wrist muscles from cerebral palsy patients. BMC Medical Genomics.14:44-54.
Smith, L.R., H.G. Chambers, S. Subramaniam, and R.L. Lieber. (2012) Transcriptional abnormalities of hamstring muscle contractures in children with cerebral palsy. PLoS One Biology 7:e40686.
Smith, L.R., H.G. Chambers, and R.L. Lieber. (2013) Reduced satellite cell population may lead to contractures in children with cerebral palsy. Dev. Med. Child Neurol. 55:264-270.







BioMed CentralBMC Medical Genomics
ss
Open AcceResearch articleNovel transcriptional profile in wrist muscles from cerebral palsy patientsLucas R Smith1, Eva Pontén2, Yvette Hedström3, Samuel R Ward4, Henry G Chambers5, Shankar Subramaniam1 and Richard L Lieber*1,6Address: 1Department of Bioengineering, University of California San Diego, La Jolla, California, USA, 2Dept of Woman and Child Health, Pediatric Orthopedic Surgery, Karolinska Institute, Stockholm, Sweden, 3Department of Clinical Neurophysiology, Uppsala University, Uppsala, Sweden, 4Department of Radiology, University of California San Diego, La Jolla, California, USA, 5Rady Children's Hospital San Diego, San Diego, California, USA and 6Department of Orthopaedic Surgery, University of California San Diego, La Jolla, California, USA
Email: Lucas R Smith - [email protected]; Eva Pontén - [email protected]; Yvette Hedström - [email protected]; Samuel R Ward - [email protected]; Henry G Chambers - [email protected]; Shankar Subramaniam - [email protected]; Richard L Lieber* - [email protected]
* Corresponding author
AbstractBackground: Cerebral palsy (CP) is an upper motor neuron disease that results in a progressive movement disorder.Secondary to the neurological insult, muscles from CP patients often become spastic. Spastic muscle is characterized byan increased resistance to stretch, but often develops the further complication of contracture which represents aprominent disability in children with CP. This study's purpose is to characterize alterations of spastic muscle on thetranscriptional level. Increased knowledge of spastic muscle may lead to novel therapies to improve the quality of life forchildren with CP.
Method: The transcriptional profile of spastic muscles were defined in children with cerebral palsy and compared tocontrol patients using Affymetrix U133A chips. Expression data were verified using quantitative-PCR (QPCR) andvalidated with SDS-PAGE for select genes. Significant genes were determined using a 2 × 2 ANOVA and results requiredcongruence between 3 preprocessing algorithms.
Results: CP patients clustered independently and 205 genes were significantly altered, covering a range of cellularprocesses. Placing gene expression in the context of physiological pathways, the results demonstrated that spastic musclein CP adapts transcriptionally by altering extracellular matrix, fiber type, and myogenic potential. Extracellular matrixadaptations occur primarily in the basal lamina although there is increase in fibrillar collagen components. Fiber type ispredominately fast compared to normal muscle as evidenced by contractile gene isoforms and decrease in oxidativemetabolic gene transcription, despite a paradoxical increased transcription of slow fiber pathway genes. We also foundcompeting pathways of fiber hypertrophy with an increase in the anabolic IGF1 gene in parallel with a paradoxical increasein myostatin, a gene responsible for stopping muscle growth. We found evidence that excitation-contraction couplinggenes are altered in muscles from patients with CP and may be a significant component of disease.
Conclusion: This is the first transcriptional profile performed on spastic muscle of CP patients and these adaptationswere not characteristic of those observed in other disease states such as Duchenne muscular dystrophy andimmobilization-induced muscle atrophy. Further research is required to understand the mechanism of muscle adaptationto this upper motor neuron lesion that could lead to the development of innovative therapies.
Published: 14 July 2009
BMC Medical Genomics 2009, 2:44 doi:10.1186/1755-8794-2-44
Received: 28 January 2009Accepted: 14 July 2009
This article is available from: http://www.biomedcentral.com/1755-8794/2/44
© 2009 Smith et al; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Page 1 of 16(page number not for citation purposes)

BMC Medical Genomics 2009, 2:44 http://www.biomedcentral.com/1755-8794/2/44
BackgroundCerebral palsy (CP) is a disorder in which children expe-rience a non-progressive brain lesion that results in per-manent and progressive secondary postural andmovement disorders [1]. CP has an incidence of 2.0–2.5occurrences per 1000 live births in developed nations,making it the most common cause of physical disability inchildren [2]. There is a spectrum of disease states in CPthat affect upper and lower limbs to varying degrees. Sincethe primary lesion in CP is in the central nervous system,most CP research has been focused on the neurologicaldisorder [3-5]. However, since the secondary effects of CPdisrupt posture and movement, most conservative andsurgical treatments address the musculoskeletal system[6].
It is clear that skeletal muscles from CP patients are alteredsecondary to the neurological lesion. There are many neu-rological symptoms secondary to the brain lesion includ-ing dystonia, ataxia, athetosis and particularly spasticity[7,8]. Loss of upper motor neuron (UMN) inhibition onthe lower motor neurons (LMN) results in spasticity,altered muscle tone and increased/impaired motor unitfiring. Loss of UMN excitation of LMNs leads to negativefeatures of UMN syndrome that include weakness, fatiga-bility, poor balance, and occasionally, sensory deficits.Although the mechanism is unknown, spastic muscleoften shortens to create muscle contractures, which is aprimary disability of CP that leads to further complica-tions [6]. There are many clinical approaches to managingspasticity to increase function, potentially decrease mus-cle contractures and most importantly improve quality oflife. Oral medications, physical therapy techniques, chem-ical neurectomies with phenol or alcohol, chemodenerva-tion using neurotoxins (BTX), and surgical neurectomieshave all been utilized to decrease spasticity in childrenwith CP [9]. Unfortunately, while there has been somesuccess in this management, many children ultimatelyrequire orthopedic surgery to lengthen the tendons ofcontracted muscle so that arm or leg function can beincreased. If the adaptation of the muscle tissue weremore completely understood, it might lead to novel med-ical treatments of contractures.
Skeletal muscle from children with CP has been character-ized at a variety of levels, with most studies reporting mus-cle tissue and muscle fiber atrophy, decreased musclecross-sectional area, muscle shortening, and decreasedspecific tension [10,11]. All of these changes implicatephysiological mechanisms of growth being involved inthe pathology of muscle from CP patients. Interestingly,recent intraoperative studies of human muscles revealedabnormally long muscle sarcomere lengths in vivo [12]that were associated with muscle tissue of altered proper-ties. Specifically, muscle fiber sarcomere length under no
load (i.e., slack sarcomere length) was significantlydecreased while the muscle tissue itself contained a hyper-trophic extracellular matrix of poor material quality[11,13]. These changes implicate the mechanical forcegenerating system of the muscle cell as well its extracellu-lar matrix tissue. Muscle has been shown to adapt itsmechanical function to neurological input [14], howeverthe mechanism by which UMN lesion could lead to alter-ations in muscle myogenesis, force generation, forcetransmission and extracellular matrix properties isunknown. While there is evidence that neurotrophic fac-tors dramatically affect muscle properties [15,16], there isneither mechanistic understanding as to how such factorsmight alter tissue properties, nor information as to whichspecific biosynthetic pathways might lead to thesechanges.
To develop an understanding of the physiological proc-esses altered in spastic muscle secondary to CP, weexploited the fact that muscle tissue from a previous study,in which the clinical severity of the spasticity was clearlyestablished, was available for transcriptional profiling[17]. We used GeneChip technology to contrast the tran-scriptome from CP patients with age-matched controlpatients whose muscles were completely normal. We per-formed a variety of analyses to identify a robust set ofgenes that were significantly altered in CP and interpretedthese genes in their biological context to explain previ-ously defined muscle changes. We also compared ourtranscriptional data to two other disease states to deter-mine whether spasticity secondary to CP results in aunique muscle disorder at the gene expression level.
MethodsMuscle SamplingChildren were recruited for this study because they werereceiving tendon transfers of the flexor carpi ulnaris(FCU) muscle into the extensor carpi radialis brevis(ECRB), the extensor carpi radialis longus muscle, or theextensor digitorum communis muscles [18]. All patientshad CP and developed a contracture indicating surgery,despite receiving conservative treatment that includedsplinting and occupational therapy. Parental consent andchild's assent was obtained in accordance with our insti-tutional review boards. From the original sample size of23 [18], a subset (n = 6 children, average age 12.8 ± 1.5years) was selected to cover a range of clinical severitiesdetermined from the House [19], Ashworth [20], andZancolli [21] classification systems as well as characteris-tics of sarcomere length and range of motion. Control tis-sue was obtained incidentally (n = 2 children, average age8.5 ± 2.1 years) from both the FCU and ECRB muscles inchildren with no previous history of any neural injurywho were undergoing surgery for forearm fracture repair.None of the surgeries injured the control muscles in any
Page 2 of 16(page number not for citation purposes)

BMC Medical Genomics 2009, 2:44 http://www.biomedcentral.com/1755-8794/2/44
way. We suggest that these samples represent true musclecontrols for the following reasons: 1) the surgeon verifiedthat the muscles from which biopsies were taken were inpristine condition and showed no signs of damage, 2) sur-gery was emergent in these children, and therefore, con-trol samples were obtained within 24 hours of fracture, 3)controls showed no significant effect for many of the tran-scripts associated with trauma or immobilization andwere, in fact, often altered in the opposite direction (datanot shown) (21, 60). Just prior to harvesting of the spasticmuscle biopsies, sarcomere length of the FCU was meas-ured by laser diffraction in vivo. While the wrist was heldin neutral, a small fiber bundle was transilluminated witha HeNe light. The sarcomere length could be calculatedfrom the diffraction pattern obtained [18]. CP biopsieswere snap frozen in isopentane chilled by liquid nitrogen(-159°C), and stored at -80°C until analyzed (Table 1).No patient had undergone serial casting prior to surgery,two patients (AN and BF) had BTX injections into the FCUseveral months prior to surgery, and one patient (AQ) hada prior BTX injection in the biceps.
RNA preparation and gene expression profilingRNA was extracted using a combination of standard Trizol(Invitrogen, Carlsbad, CA) and RNeasy (Qiagen, Valencia,CA) protocols. Briefly, 30 mg of frozen muscle washomogenized in a rotor-stator homogenizer on ice in 0.5ml of Trizol; 0.1 ml of chloroform was added to the solu-tion, which was then vigorously vortexed for 15 s fol-lowed by centrifugation at 4°C for 15 min. The upperaqueous layer was removed and mixed with an equal vol-ume of 70% ethanol before being added to the RNeasyspin column. After the column was washed, it was incu-bated with RNAse-free DNAse (Qiagen) for 15 min andthen washed again three more times before being elutedas described in the manufacturer's protocol. RNA concen-tration was determined by the absorbance at 260 nm, and
the 260 nm-to-280 nm absorbance ratio was calculated todefine RNA purity.
Microarray data analysisAffymetrix microarrays ("GeneChip" HG-U133A; Affyme-trix, Santa Clara, CA) were used for each muscle biopsy (n= 16 chips; 2 muscles × 8 patients) and the data are avail-able [GEO: GSE11686]. RNA processing for the Gene-Chip, including stringent quality control measures, wasperformed by the Gene Chip Core at the Department ofVeterans Affairs San Diego Health Care System, (SanDiego, CA). GeneSpring software (version 7.3; Silcon-Genetics, Redwood City, CA) was used to identify thosegenes that were significantly altered in CP. Initially, a12.5% (2/16 chips) present call on MAS5 (Affymetrix)was used to filter out poorly performing probe sets in theanalyses. Three independent probe set algorithms wereused for signal generation and normalization: MAS5,RMA, and GCRMA. Recent reports support requiring con-cordance among different probe set algorithms as anapproach to reduce false positives in data sets [22-24].Each feature was normalized per chip (to the median ofall features on each chip) and per gene (to the median ofthat feature on all chips). Normalized gene values weresubjected to a 2 × 2 Welch ANOVA of muscle type (FCUvs. ECRB) and disease state (CP vs. CTRL) with a requiredstatistical significance (P < 0.05) with a Benjamini andHochberg False Discovery Rate (FDR) multiple testingcorrection for present features. Thus 5% of the genesdeemed significant for an individual preprocessing algo-rithm are suspected to be false positives. Features thatpassed in all three preprocessing algorithms were deemedsignificantly altered in CP.
The condition tree was created using a Pearson Correla-tion similarity score and average linkage clustering algo-rithm for all samples on present features. For severityanalysis, a Welch ANOVA for each severity parameter was
Table 1: Primers used for quantitative PCR
Gene Transcript Base pairs Sense Primer (5'-3') Antisense Primer (5'-3')
COL1A2 NM_000089 225 TCCAAAGGAGAGAGCGGTAA GCCACTTGCACCACGACTACOL4A2 NM_001846 269 CTGGGTGGCGGAGTTTGTG GCTGATGTGTGTGCGGATGAGCOL4A3 NM_000091 157 CACCAGCTCTGATGCCAATG AGAGAAATCCAGCCGTGAGGDMD NM_004019 151 GACCAGCACAACCTCAAGCAA TCAGCAGCCAGTTCAGACACAFBXO32 NM_058229 376 GTCCAAAGAGTCGGCAAGT TTGGGTAACATCGGACAAGTGAPDH NM_002046 172 TCTGACTTCAACAGCGACAC TGGTCCAGGGGTCTTACTCIGF1 NM_000618 355 AGCAGTCTTCCAACCCAATTA CACGGACAGAGCGAGCTGIGFBP5 NM_000599 339 CCAAACACACCCGCATCT CAGCTTCATCCCGTACTTGTCGDF8 NM_005259 167 TATCACGCTACAACGGAAAC GGAGTCTCGACGGGTCTCMYH1 NM_005963 132 AAGAGCAGGGAGGTTCACAC TTATCTCCAAAAGTCATAAGTACANEB NM_004543 131 CCGTGCCATGTATGACTATAT CGGTCCTGCCAGTCCTCTGPVALB NM_002854 329 GATGACAGACTTGCTGAACGC CTTAGCTTTCAGCCACCAGAGTRIM63 NM_032588 393 GAGGATTCCCGTCGAGTGAC AATGGCTCTCAGGGCGTCT
Table of primers used for QPCR analysis. Genbank accession number and PCR product length given in parentheses after each transcript name
Page 3 of 16(page number not for citation purposes)
BMC Medical Genomics 2009, 2:44 http://www.biomedcentral.com/1755-8794/2/44
run on the MAS5 data of the features deemed significantlyaltered in CP without the control patients and a requiredstatistical significance of (P < 0.05) also with an FDR mul-tiple testing correction.
Promoter sequence analysis was conducted using Gene-Spring on the list of genes altered in CP. The upstreamsequence from -10 to -1000 base pairs was analyzed for anucleotide sequence of from 6 to 10 nucleotides long andcontaining at most 2 N values in the middle. Significancewas determined based on the number of times the givensequence appeared in the upstream sequence of all othergenes and was corrected for multiple testing. The analysiswas performed on the whole list of genes altered in CPand the sub lists of up- or down-regulated.
Quantitative real-time PCRQPCR was performed to validate expression levels ofselected genes to the GeneChip data and to providemRNA expression levels for genes not contained on theHG-U133A chip. After RNA was extracted from the muscleas described previously and diluted 1:5 with DNase/RNase free water (Invitrogen), 1 μl of each sample wasreverse transcribed using standard protocols (SuperscriptIII; Invitrogen). cDNA was amplified with the CepheidSmartCycler (Sunnyvale, CA) with primers specific to thegenes of interest (Table 2). All primers were tested forcross-reactivity with other transcripts using nBLAST andOligo (version 6.6; Molecular Biology Insights, Cascade,CO). All samples were run at least in triplicate, along witha standard curve. The PCR reaction vessel (25 μl) con-tained 1× PCR buffer, 2 mM MgCl2 (Invitrogen), 0.2 mM
sense and antisense primers, 0.2 mM dNTP, 0.2×SYBRgreen, and 1 U of platinum Taq polymerase (Invitro-gen). Amplification conditions were as follows: An initialhold at 95°C for 2 min was followed by 40 cycles of dena-turing at 95°C for 15 s, followed by annealing/extensionat 68°C for 40 s. The success of each reaction was deducedbased on the observation of a single reaction product onan agarose gel and a single peak on the DNA melting tem-perature curve determined at the end of the reaction. Toexpress QPCR results, we used the standard curve methodwith the "cycles to threshold" value representing thenumber of PCR cycles at which the SYBRgreen signal wasincreased above the threshold. Each sample's value wasmeasured in triplicate, normalized to the housekeepinggene GAPDH, and then averaged. QPCR data were nor-malized to the median value of the gene to permit com-parison to the GeneChip data.
Myosin protein content biochemistryMyosin heavy chain protein content was measured (Table2) for comparison to the GeneChip data as previouslydescribed [25]. Three bands were identified correspond-ing to MyHC I, MyHC IIa/fetal and MyHC IIx/embryonic.(Using this methodology, embryonic MyHC cannot beseparated from MyHC IIx and fetal MyHC cannot be sep-arated from MyHC IIa.) The gels were scanned in a softlaser densitometer (Molecular Dynamics Sunnyvale, CA,USA). The relative proportion of each MyHC isoform wasdetermined by using a densitometric system (ImageQuantTL Software v 2003.01, Amersham Biosciences, Uppsala,Sweden).
Table 2: Subject information and clinical evaluations
Sub Sex Age Muscle SL (μm) House Sev Ash Zan PEFF (°) PESF (°) AEFF (°) MyHC gel
AI M 13 FCU 6.36 1 SS 4 3 -5 -90 -70 xECRB x
AN F 12 FCU 4.56 3 SS 3 2B 50 45 -90ECRB
AO F 11 FCU 4.01 5 SM 2 2B 65 60 -50 xECRB x
AQ M 15 FCU 4.01 7 M 2 2B 80 80 -50ECRB
AT M 14 FCU 3.57 0 SS 3 3 45 40ECRB
BF F 12 FCU 5.31 6 SM 3 2A 85 85 55 xECRB x
BP M 10 FCU - - C 1 0 90 90 90 xECRB
AZ M 7 FCU - - C 1 0 90 90 90 xECRB x
Table of the eight subjects used in this study with 2 muscles (FCU and ECRB) per patient with sex and age recorded. Measures of severity were taken to include: intraoperative measurement of sarcomere length (SL) on the FCU, House clinical assessment of activity, Severity (Sev) grouped by House (Severe Severe (SS), Severe Moderate (SM), Mild (M), and Control (C)), Ashworth (Ash) clinical assessment, Zancolli (Zan) classification based on finger extension, passive extension with flexed fingers (PEFF) passive extension with straight fingers (PESF) and active extension with flexed fingers (AEFF). Samples that were available for MyHC SDS-PAGE gels are noted in the final column
Page 4 of 16(page number not for citation purposes)

BMC Medical Genomics 2009, 2:44 http://www.biomedcentral.com/1755-8794/2/44
Gene Ontology analysesGene ontology analysis provides a means of converting alist of differentially expressed genes into a hierarchical listof gene ontologies that are significantly altered. We usedthe web-based software GOTree http://bioinfo.vanderbilt.edu/gotm/; [26]) to compare the list of featuresaltered in CP to the list of features present on the HG-U133A chip. In this analysis, a P-value is generated foreach ontology based on hyperbolic comparison of thenumber of genes present in that list to the number ofgenes expected to be present based on the size of the list.The analysis was performed on the entire list of genesaltered in CP, with a required P < 0.01.
Biological pathway analysesTo gain understanding into the biological context of tran-script changes, we investigated the way in which geneswere involved in various muscle pathways. We analyzedpathways from databases including: Ingenuity PathwayAnalysis (IPA; http://www.ingenuity.com), Kyoto Ency-clopedia of Genes and Genomes (KEGG; http://www.genome.ad.jp/kegg/pathway.html), and Gene Mapand Pathway Profiler (GenMAPP; http://www.genmapp.org/). These pathways permit establishment ofpathways specific to muscle involving critical musclefunctions such as: neuromuscular junction function, exci-tation-contraction coupling, muscle contraction, extracel-lular matrix formation, muscle hypertrophy/atrophy,myogenesis, and fiber type switching. Based on the path-way databases and relying heavily our own literaturereview, we created pathways specific to muscle with par-ticular emphasis on genes altered in CP. To quantify geneexpression for pathway analysis, MAS5 data were normal-ized to the averaged control data within each muscle typeand across CP samples. This value is termed the expres-sion ratio.
Finally, to compare the CP transcriptome to other condi-tions we examined the pathways specific to muscle againsttranscriptome deposited for three other disease states,Duchenne Muscular Dystrophy (DMD; GSE465; [27]),immobilization (IMB; GSE8872; [28]), and hereditaryspastic paraplegia (HSP; GSE1300; [29]). The DMD exper-iment used muscle from patients age 6–9 years and fur-ther details are described in the reference [27], but wecompared data only from those U95A chipset. The IMBexperiment used medial gastrocniemus muscle from adultpatients and further details are described in the reference[28], but we compared only data from voluntary controlsand ankle facture patients immobilized for 4–9 days. TheHSP experiment used vastus lateralis muscle from adultpatients and further details are described in the reference[29], but we used the U133A chipset and controls (1–10)from GSE3307. As these data sets are from subjects of dif-ferent ages and muscles, and are acute (in the case of 4–9days of immobilization), direct comparison to our CP
dataset is somewhat problematic; however, we are able toinvestigate whether similar transcriptional trends arepresent for these muscle conditions. The expression ratiofor each feature was taken as the MAS5 ratio of the averagedisease state:average control state of the particular studyso disease values are normalized to their own controls.The genes expression ratio of a pathway for each disease(CP, DMD, IMB, HSP) was log averaged across the path-way with inverse expression values used for inhibitors. Wesimilarly investigated a list of genes involved in satellitecell states of quiescence and activation [30-32].
ResultsOf the 22,283 probe sets on the HG-U133A GeneChip,11,312 met the criteria of being "present" on 2/16 Gene-Chips and were therefore considered for further analysis.The number of genes that were significant for CP (P <0.05) on the 2 × 2 Welch ANOVA of disease state andmuscle (CP vs. CTRL; flexor carpi ulnaris (FCU) vs. exten-sor carpi radialis brevis (ECRB)) with FDR among thethree preprocessing algorithms were: 495 for MicroarraySuite Version 5.0 (MAS5), 1,141 for Robust MultiarrayAnalysis (RMA), and 1,207 for GCRMA. The overlap ofthese 3 preprocessing algorithms produced a final list of205 genes (319 features) that were considered signifi-cantly altered secondary to CP (Sup. Table 1). Of these,more were up-regulated (143 genes, 220 features) thandown-regulated (62 genes, 99 features). Table 3 reportsthe 72 genes subset of these 205 genes that were consid-ered relevant to specific muscle functions. Genes in theTables (Table 1; Sup. Table 1) are reported with the P-value for each preprocessing algorithm as well as theexpression ratio. The 2 × 2 ANOVA yielded no genes sig-nificant (P < 0.05) for muscle type and only one gene witha significant interaction, MYH1. This was due to controlECRB tissue having a very low MYH1 mRNA content. Thisimportant result supports our previous contention that,even though these children present with wrist flexion con-tracture, the FCU and ECRB are equally affected and thewrist flexion simply results from the large size of the FCU[18].
Promoter sequence analysis performed on each genealtered in CP did not reveal any 6–10 base pair sequencesthat were overrepresented 10–1000 base pairs upstreamof the gene. Of course, regulation can occur fartherupstream than 1000 base pairs and regulation sequencescan be outside of the 6–10 base pair range. Thus, furthersequence analysis may reveal significant promoter orenhancer sequences, but none were identified using thesecriteria.
Condition tree correlates with clinical severity scores and treatmentThe condition tree resulted in the control patients beingclustered together separate from CP patients (Figure 1).
Page 5 of 16(page number not for citation purposes)

BMC Medical Genomics 2009, 2:44 http://www.biomedcentral.com/1755-8794/2/44
Figure 1 illustrates the condition tree based on all presentgenes. The tree shows that patients are grouped together inmost cases rather than by muscle type suggesting morebetween-patient than between-muscle variability.
We had hoped that clinical severity [1,19-21] would allowus to define transcriptionally, the severity of CP or differ-ences between flexor and extensor muscles. In this way,clinical parameters would be seen as representative of the
Table 3: Significantly altered genes in functional categories
P-Values P-ValuesGENE Ratio MAS5 RMA GCRMA GENE Ratio MAS5 RMA GCRMA
Neuromuscular Junction Muscle Contraction and Structure
KCNN3 12.98 0.013 0.011 0.018 MYH1 8.57 0.001 0.001 0.000COL4A3 2.89 0.043 0.014 0.019 MYH4 4.33 0.001 0.002 0.015LAMB2 1.74 0.042 0.018 0.015 NEB 2.54 0.015 0.008 0.013
Excitation Contraction Coupling MYBPC2 2.16 0.027 0.004 0.004
PVALB 62.60 0.001 0.004 0.001 DMD 2.11 0.023 0.020 0.027ATP2B2 2.60 0.033 0.023 0.021 LDB3 1.93 0.030 0.015 0.015
TRDN 2.38 0.028 0.019 0.017 Metabolism/Mitochondria Related
ATP2C1 2.30 0.046 0.047 0.041 WARS 0.64 0.044 0.040 0.019PDE4DIP 2.10 0.005 0.003 0.004 CAV1 0.64 0.037 0.013 0.017CALM1 1.70 0.005 0.004 0.008 CERK 0.63 0.033 0.006 0.014CACNB1 1.51 0.045 0.014 0.017 A2M 0.62 0.015 0.016 0.010FKBP1A 0.56 0.039 0.016 0.011 MDH1 0.60 0.050 0.017 0.011
Myogenesis/Fiber type pathways PECI 0.58 0.029 0.024 0.012
GDF8 3.65 0.025 0.032 0.007 MRPL35 0.57 0.040 0.043 0.037IGF1 2.63 0.013 0.010 0.008 MRPS18B 0.56 0.009 0.002 0.001IGFBP5 2.48 0.002 0.000 0.000 SLC25A20 0.55 0.044 0.049 0.022PLCB1 2.34 0.028 0.014 0.007 CPT2 0.54 0.044 0.022 0.013RASA4 2.26 0.037 0.027 0.029 MRPS12 0.53 0.044 0.008 0.010PPP3CA 1.95 0.012 0.003 0.005 UCP2 0.51 0.030 0.023 0.015PBX1 1.86 0.028 0.010 0.007 MLYCD 0.48 0.017 0.004 0.001CALM1 1.70 0.005 0.004 0.008 PPIF 0.48 0.014 0.011 0.010MBNL1 1.55 0.033 0.015 0.024 ADM 0.47 0.019 0.012 0.018MEF2A 1.53 0.028 0.006 0.007 UCP3 0.47 0.014 0.010 0.007NEO1 1.44 0.015 0.005 0.001 ALDH6A1 0.46 0.023 0.013 0.007HMGB1 1.34 0.015 0.013 0.015 ACSL1 0.43 0.014 0.008 0.004
Extracellular Matrix GOT1 0.42 0.039 0.027 0.021
MFAP5 3.64 0.012 0.004 0.014 TST 0.41 0.015 0.002 0.001COL4A3 2.89 0.043 0.014 0.019 MT1G 0.37 0.015 0.009 0.011COL21A1 2.86 0.021 0.022 0.015 LPL 0.35 0.013 0.010 0.007KAL1 2.57 0.002 0.003 0.003 RETSAT 0.35 0.002 0.006 0.005MATN2 2.24 0.013 0.011 0.007 MT1M 0.34 0.015 0.006 0.004CILP 2.09 0.035 0.043 0.034 MT1X 0.32 0.014 0.012 0.010SMC3 1.88 0.021 0.026 0.019 GLUL 0.31 0.018 0.017 0.013ECM2 1.86 0.037 0.035 0.021 MT2A 0.29 0.015 0.013 0.011LAMB2 1.74 0.042 0.018 0.015 MT1H 0.29 0.015 0.015 0.012COL4A1 0.51 0.048 0.032 0.015 MT1F 0.27 0.019 0.012 0.009BSG 0.48 0.033 0.026 0.030 MT1E 0.26 0.012 0.006 0.005COL4A2 0.43 0.012 0.010 0.006 LIPE 0.22 0.028 0.001 0.002
Table of genes within function groups related to skeletal muscle. Groups defined by ratio is the expression ratio of CP:CTRL. P-values are listed for the three separate preprocessing algorithms used (MAS5, RMA, GCRMA).
Page 6 of 16(page number not for citation purposes)

BMC Medical Genomics 2009, 2:44 http://www.biomedcentral.com/1755-8794/2/44
Page 7 of 16(page number not for citation purposes)
Top: Condition tree created using Pearson Correlation for the similarity score and an average linkage clustering algorithmFigure 1Top: Condition tree created using Pearson Correlation for the similarity score and an average linkage cluster-ing algorithm. The tree was created based on all present features. MAS5 data were used with expression values normalized to each features median. Features are ordered from highest expression ratio to lowest. Bottom: Clinical conditions color-coded with values for each sample.

BMC Medical Genomics 2009, 2:44 http://www.biomedcentral.com/1755-8794/2/44
state of the muscle tissue. These parameters were appliedto an analysis of all of the 319 features altered in CP(Additional File 1 Table S1) but only sarcomere lengthand active wrist extension with fingers flexed had featuresthat were significant, containing the same two genes,RBM9 and RHOBTB1. Heat plot of these data reveals thateven these genes undergo a much larger change in expres-sion from the control sarcomere length (3.37 μm) to CPsarcomere lengths than they do in CP progression (datanot shown). Thus it appears that our study is underpow-ered to reveal transcriptional correlation with clinicalseverity scores.
Treatment with BTX was investigated by comparinginjected muscles vs. non-injected CP muscles, musclefrom patients receiving injection of any muscle vs. non-injected patient muscle, and injected FCU muscles vs.non-injected FCU muscles. None of these analyses yieldedany genes that met our requirement for statistical signifi-cance, and thus we show no significant transcriptionaleffect of BTX injection.
RT-PCR results compared to chip resultsAs a quality control measure, correlation of data betweenthe GeneChip and QPCR was highly significant (P <0.001) indicating internal consistency. To validate theGeneChip data, 10 genes covering a variety of cellularprocesses and expression levels were compared directly totranscript levels determined by QPCR on the same cDNAsamples (Figure 2). For 9/10 genes studied, the directionof the transcript change (i.e., up- or down-regulation) wasconfirmed, and there was a good correlation betweenmethodologies in terms of the magnitude of the effect. Fortwo genes, (PVALB, GDF8) expression levels were evalu-ated relative to GAPDH transcript levels. These genes wereselected based on their significant differences on the chipand their relevance to the disease state. Significant posi-tive correlations were observed for both genes (PVALB, r2
= 0.924, P < 0.001; GDF8, r2 = 0.864, P < 0.001). TheQPCR data were also subjected to 2 × 2 ANOVA (CP vs.CTRL; FCU vs. ECRB) and both PVALB and GDF8 wereconfirmed as significantly up-regulated in CP (Figures 3Cand 3D).
Two genes of particular interest that are related to muscleatrophy, MAFbx (FBXO32) and MURF1 (TRIM63) werenot represented on the chip [28,33]. Their expression wasdetermined in the same manner as the genes describedabove (Figures 3E and 3F). Both of these genes weredown-regulated in CP, but neither reached statistical sig-nificance.
Myosin heavy chain protein-mRNA comparisonThe GeneChip and QPCR provide only transcriptionaldata and we wished to determine whether the transcrip-
tional changes resulted in translational changes in thecase of myosin heavy chain (MHC) for these samples[25,34]. All of the muscles were of a mixed fiber type,however the control ECRB tissue showed no evidence oftype 2X MHC. The spastic muscles had a higher propor-tion of fast fibers than the controls of the correspondingmuscle, with most of the increase in type 2X MHC. Com-parison between protein and mRNA was confounded bythe fact that MHC expression was normalized as percentof total myosin while mRNA was normalized to the
QPCR results compared to GeneChip results for several individual genes shown from CP (open bars) and CTRL (filled bars) patientsFigure 2QPCR results compared to GeneChip results for sev-eral individual genes shown from CP (open bars) and CTRL (filled bars) patients. Error bars represent SEM. QPCR data are from dilute (1:100) samples to test multiple genes and represent transcript level relative to total RNA (fg/μg). GeneChip data are normalized to the median value for each gene and averaged across CP or CTRL samples.
Page 8 of 16(page number not for citation purposes)

BMC Medical Genomics 2009, 2:44 http://www.biomedcentral.com/1755-8794/2/44
median of that individual transcript across subjects. Inspite of this difference, we still expected to see the sametrend across samples, which was the case for type 1 MHC(gene MYH8) and type 2X MHC (gene MYH1) where pro-tein and mRNA levels were significantly correlated (Fig-ures 4A and 4C; P < 0.05) while type 2A MHC (geneMYH4) did not quite reach significance (Figure 4B; P =0.065). Taken as a whole, these results suggest that, in thecase of the MHC, protein levels reflected transcript levels.
Gene ontology analysisThirty-eight different ontologies were overrepresentedbased on the 143 up-regulated genes (Additional File 2Table S2; Additional File 3 Figure S1). The biological proc-esses that stood out as most relevant to the disease stateincluded striated muscle contraction, muscle develop-ment, cytoskeletal anchoring, negative regulation ofmetabolism, protein ubiquitination, and RNA processing.The cellular components of these genes were generallygrouped into muscle components and ECM components,
(A/B) Sample-to-sample correlation between the QPCR and GeneChip resultsFigure 3(A/B) Sample-to-sample correlation between the QPCR and GeneChip results. (A) PVALB, parvalbumin; (B) GDF8, myostatin. The solid line is a best fit regression line. (C-F) QPCR results showing the difference between CP and CTRL patients for specific genes, (C) PVALB, (D) GDF8, (E) FBXO32/MaFBX/Atrogin, (F) TRIM63/MuRF. Values are expressed determined relative to GAPDH and normalized to the median value for each individual gene. (*) represents sig-nificant difference (P < 0.05). Error bars represent SEM.
Comparison of the GeneChip mRNA data to protein SDS-PAGE gel data for three myosin heavy chains commonly expressed in human skeletal muscleFigure 4Comparison of the GeneChip mRNA data to protein SDS-PAGE gel data for three myosin heavy chains commonly expressed in human skeletal muscle. A: Type 1 MHC (MYH6), B: Type 2A MHC (MYH2), C: Type 2X MHC (MYH1). mRNA data are normalized to the median value for each gene on the chip using MAS5 preprocessing and protein data are normalized to total MyHC content.
Page 9 of 16(page number not for citation purposes)

BMC Medical Genomics 2009, 2:44 http://www.biomedcentral.com/1755-8794/2/44
particularly the basement membrane. Twenty-eight differ-ent ontogenies were overrepresented based on the 99down-regulated genes (Additional File 4 Table S3; Addi-tional File 5 Figure S2). The two major functions of thedown-regulated biological processes were fatty acidmetabolism and transport. This corresponded with themolecular function ontologies involved in fatty acid/acylCoA binding and also contained cadmium and copperion binding. Cellular component categories were almostexclusively related to the mitochondria, however it wasinteresting that sheet forming collagen type IV of the base-ment membrane also was over represented using this ana-lytical approach.
Gene pathways related to muscle functionTo understand muscle tissue adaptation to CP from aphysiological perspective, we analyzed gene expressionratio patterns within muscle-specific pathways of geneproducts that interact in a given muscle function.
Because CP is a neurological disorder, a pathway describ-ing the neuromuscular junction (NMJ) was created (NMJ;Figure 5A). No postsynaptic genes were significantlyaltered in CP including subunits of the nACHR receptor.
Collagen type IV subunits (COL4A3; 2.89 and COL4A4;3.26) and laminin (LAMB2; 1.74) of the synaptic basallamina were significantly up-regulated. A Ca2+-activatedK+ channel (KCNN3; 12.98), was dramatically up-regu-lated in CP.
The process of converting the action potential into musclecontraction is referred to as excitation-contraction cou-pling (ECC; Figure 5B). The β1 regulatory subunit of theL-type voltage gated Ca2+ channel was significantly up-reg-ulated in CP (CACNB1; 1.59). Although the ryanodinereceptor responsible for releasing Ca2+ from the sarcoplas-mic reticulum (SR) was not altered, the genes FKBP1A(0.56) and PDE4D (0.68) that prevent channel leaking,were significantly down-regulated [35]. Myomegalin(PDE4DIP; 2.10) was altered significantly in CP and isthought to anchor PDE4D near the SR [36]. Calmodulin(CALM1; 1.70) was significantly up-regulated. The mostdrastic change with CP on gene expression was in the up-regulation of muscle-relaxing protein, parvalbumin(PVALB; 62.6). The process of pumping Ca2+ back into theSR is assisted by triadin (TRDN; 2.39), which was signifi-cantly up-regulated.
Pathways specific to muscle analysis of transcription in CP muscleFigure 5Pathways specific to muscle analysis of transcription in CP muscle. Pathways A-G involved in muscle function. Color is determined by the expression ratio. Up-regulated genes are red and down-regulated genes are green. Green connec-tors represent activation and red connectors represent inhibition in the direction of the arrow. Bolded genes represent those that are significantly altered in all three preprocessing algorithms. Italic genes (RAS, MAPK) are sets of genes involved in muscle MAPK pathway in muscle, but are not colored by expression because many individual genes are applicable and not altered in CP. Pathways represented are A: NMJ, B: ECC, C: MC, D: ECM, E: MYG, F: FT, and G: IGF1.
Page 10 of 16(page number not for citation purposes)

BMC Medical Genomics 2009, 2:44 http://www.biomedcentral.com/1755-8794/2/44
Muscle contraction obviously requires the myosin heavychain motor and a cytoskeletal framework (MC; Figure5C). MYH1 (type 2X MHC; 8.57) was significantly up-reg-ulated in CP along with MYH4 (type 2B MHC; 4.33) agene not normally expressed in humans [37]. The devel-opmental MHCs, MYH3 (embryonic MHC; 15.74) andMYH8 (perinatal MHC; 7.74) showed large up-regulation.MYH7 (type 1 MHC; 0.72) was unchanged. The MYBPC2(2.16) is a fast isoform of myosin binding protein and wasup-regulated in CP. Several sarcomeric structural compo-nents were also up-regulated as well: dystrophin (DMD;2.11), nebulin (NEB; 2.54), and muscle LIM domainbinding protein 3 (LDB3; 1.93).
We suspected that ECM transcription would be alteredbased on previous biomechanical results ([11,13,38];ECM; Figure 5D). Fibrillar collagens all increased mod-estly. Interestingly, basal laminar collagen IV was alteredwith COL4A1 (0.51) and COL4A2 (0.43) decreasing sig-nificantly while COL4A3 (2.89) and COL4A4 (3.26)increased significantly. Basigin, (BSG; 0.48) was signifi-cantly down-regulated. Various other ECM componentswere also up-regulated in CP: ECM2 (1.86), KAL1 (2.57),MATN2 (2.24), MFAP5 (3.64), CILP (2.09) and SMC3(1.88).
Myogenesis describes the pathway that produces musclegrowth (MYG; Figure 5E). IGF1 (2.63) was up-regulatedalong with IGFBP5 (2.48) (IGF1; Figure 5F). Myostatin(GDF8; 3.65), an inhibitor of myogenesis, was also signif-icantly up-regulated. Other significantly up-regulatedgenes implicated in myogenesis are NEO1 (1.44, [39]),PLCB1 (2.34, [40]), PBX1 (1.86, [41]), and HMGB1 (1.65,[42]), MBNL1 (1.74, [43]), and MAPK6 (1.77, [44]).However, the muscle regulatory factors (MYOD1 1.22;MYF6 0.87; MYF5 1.06; MYOG 1.75) did not show a sig-nificant transcriptional change. Mitogen activated proteinkinases (MAPK) have been proposed as a major pathwayin muscle hypertrophy [45], however our study showedminimal transcriptional affects on this signaling family.Another segment of myogenesis relates to satellite cellactivation, proliferation and incorporation into adultmuscle fibers. None of the markers for quiescent or acti-vated satellite cells (quiescence: PAX7, FOXK1, MET,CDH15, NCAM1, VCAM1, SDC3, SDC4; activation:MYF5, MYOD1, MYOG, MYF6, PCNA, CDKN1A, MYH3,MYH8) were significantly altered in CP suggesting mini-mal involvement of satellite cells in the disease. Addi-tional cell cycle transcripts were investigated, but did notshow a significant change.
Although slow fiber creation is related to myogenesis,there is also a specific pathway for slow oxidative musclefiber type determination (FT; Figure 5F). Sensing and sig-naling factors, CALM1 (1.70) and calcineurin (PPP3CA;
1.95) respectively, had significantly increased transcrip-tion along with transcription factor MEF2A (1.53), butNFATs and other MEF2 expressions levels wereunchanged.
Cerebral palsy compared to other muscle pathologiesTo determine whether the CP transcriptome was uniqueor simply a secondary adaptation of decreased activity inthese children (as might be observed with immobilization(IMB)), or whether the response was a generic musclepathology (Duchenne Muscular Dystrophy (DMD) beingthe most-commonly studied), or was similar to spasticmuscle in an alternative more developed muscle (Heredi-tary Spastic Paraplegia (HSP) being a spastic conditionwith adult subjects) we compared our GeneChip data tothese three muscle pathologies for which GeneChip datawere available [27-29]. To make these comparisons, theexpression ratio values for the pathways were comparedamongst the three conditions (Table 4). While averagingover an entire pathway may be misleading (similar scoresmay result from different gene expression patterns), differ-ent scores do emphasize pathways that are unique amongdisease states. This analysis revealed significant satellitecell activation, as expected, in DMD [27] as well asincreased NMJ components (primarily nicotinic acetyl-choline receptor subunits) and loss of contractile materialas expected in IMB [28]. HSP represents muscle adapta-tion to altered neuronal input, although there was a neg-ative correlation in most pathways, ECC seemed to behandled in a similar manner. CP was unique relative tothe other two pathologies based on the IGF1 pathwayincrease, slow fiber activation, and increased expression ofECC activators and inhibitors. Thus, the correlation datasupport the assertion that CP is unique relative to otherdisease states.
Table 4: Changes in pathways correlated with other muscle disease states
CP DMD IMB HSPMEAN MEAN CORR MEAN CORR MEAN CORR
NMJ 2.39 0.98 -0.24 1.19 -0.05 0.97 -0.02ECC 0.66 0.90 0.12 1.26 0.14 1.39 0.62MC 2.91 2.18 0.91 0.65 -0.74 1.07 -0.48ECM 1.63 1.73 -0.23 0.69 0.44 0.66 0.02FT 1.35 0.70 -0.21 0.92 0.37 0.91 -0.79IGF1 1.19 1.58 N/A 1.04 N/A 0.76 N/AMYG 1.14 1.11 -0.16 1.01 -0.24 1.01 -0.09SCQ 1.19 1.18 -0.67 1.11 0.66 1.23 0.07SCA 2.17 3.73 0.91 0.86 -0.81 0.94 -0.54
Quantification of gene pathways in various disease states (CP, DMD, IMB, HSP). Quantification represents MEAN (geometric mean of expression ratios in specific muscle pathways defined in Figure 5 using inverse values for pathway inhibitors) and CORR (correlation of CP with the other disease states, reported with an R value). Satellite cells markers are separated into genes expressed in the quiescent (SCQ) and activated states (SCA).
Page 11 of 16(page number not for citation purposes)

BMC Medical Genomics 2009, 2:44 http://www.biomedcentral.com/1755-8794/2/44
DiscussionThe purpose of this study was to define the muscle tran-scriptional adaptations in children with cerebral palsy(CP) to gain insights into the cellular mechanisms thatmight explain muscular adaptation in this neurologicalcondition. We show that the transcriptional profile of CPmuscle is fundamentally different compared to normalcontrols (Figure 1). Previous CP muscle studies of intra-operative sarcomere length [12], in vitro tissue biome-chanics [38,46], and immunohistochemical andbiochemical assays suggested adaptation of extracellularmatrix regulation [11,13], myogenenic pathways [10,38],and fiber type determination pathways [25] in this condi-tion. Our transcriptional analyses provide potential expla-nations of the cellular bases for these adaptations. Basedon a general understanding of muscle physiology andbiology, we placed the gene expression patterns into thecontext of six major muscle physiological systems – theneuromuscular junction (Figure 5A), excitation-contrac-tion coupling (Figure 5B), muscle contraction (Figure5C), extracellular matrix regulation (Figure 5D), myogen-esis (Figure 5E) and fiber type determination (Figure 5F).As will be seen, one feature of CP is that conflicting ten-dencies occur within and between these various systems.
The initial insult in CP is located in the central nervoussystem, but this primary insult leads to a secondary effecton the skeletal muscle system. Thus the NMJ, as the nerve-muscle interface, may play a role in CP. Studies haveshown disrupted NMJ in that acetylcholine receptorsappear outside the NMJ area more often in CP, althoughthey were unable to find any change in transcriptional reg-ulation [47,48]. However, KCNN3 was the 2nd most up-regulated gene on the entire chip and this gene plays a rolein causing after-hyperpolarizations which may be a cellu-lar attempt to limit the excessive motor unit firing that hasbeen reported in spastic muscle [49]. Interestingly,KCNN3 is usually expressed in immature muscle andinhibited after innervation, which may indicate a sort of"immature state" of this muscle [50]. Although their local-ization in these samples is unknown the standard colla-genous component of the synaptic basal lamina(COL4A3; COL4A4) was transcriptionally increased, theopposite activity of primary muscle basal lamina colla-gens (COL4A1; COL4A2) [51]. If these synaptic collagenIV subunits occurred outside the NMJ it would suggest afurther degree of NMJ disorganization, alternatively theycould be another indicator of muscle in an "immaturestate."
We also uncovered significant evidence of altered calciumhandling secondary to CP. Our data appear to reflectchronically increased intracellular calcium since the L-type voltage gated Ca2+ channel (CACNB1) was up-regu-lated (leading to activation of the ryanodine receptor) and
leakage through the ryanodine receptor would beincreased by down-regulation of two genes that preventleakage (FKBP1A; PDE4D). Another "attempt" by themuscle to re-regulate [Ca]i can be inferred by the up-regu-lation of TRDN, which reclaims Ca2+ to the SR by localiz-ing calsequestrin within the SR [52]. Chronically alteredcalcium levels and subsequent activation of the intramus-cular calcium-activated proteases (Calpains) would causedramatic muscle lesions, although they are not transcrip-tionally regulated in CP. Indeed, a relatively new class ofCalpain-mediated myopathies has recently beendescribed [53,54]. Perhaps in response to this chronicchange in [Ca]i a huge 63-fold increase in PVALB, a Ca2+
binding protein was induced in order to force musclerelaxation [55]. This dramatic adaptation could have sig-nificant effects on the [Ca]I and may even lower it belowcontrol levels and alter muscle contractile properties.
Of the proteins involved in calcium induced force genera-tion, MHC isoforms are the most responsive to CP. Theyare primarily responsible for determining muscle fiberstype [56] and undergo a transformation in the direction ofa slow-to-fast phenotype. This shift included immaturemyosins, which saw large increases, although they wereonly significant in 2/3 algorithms, and lends further evi-dence to muscle in an "immature state." The many oxida-tive metabolic genes that are down-regulated in CP (Table3) support this slow-to-fast transition. The ontology anal-ysis revealed the loss of metabolic and mitochondrialrelated transcripts represented the majority of down regu-lated ontologies (Additional File 4 Table S3). Althoughprevious research is mixed on whether spastic musclesbecome more fast or slow, our data is in concordance withrecent research that fast fibers dominate spastic muscle inCP [25,34]. Paradoxically, this transformation occursdespite an overall increase in gene transcription related tothe determination of the slow fiber phenotype, particu-larly calmodulin (CALM1) and calcineurin (PPP3CA)[57]. A potential explanation may be that the dramaticPVALB expression actually leads to a decrease in intracel-lular calcium, thus turning off the initiation of the slowgene program. The validity of the calcineurin/NFAT path-way for transcription of a slow muscle fiber program hasalso come under question [58,59].
The slow fiber program represents only one segment ofmyogenesis that is controlled by many other genes. Whilethe majority of the pathway elements (receptors, secondmessengers, signaling molecules) involved in myogenesiswere not changed, two of the most important initial fac-tors were both up-regulated – insulin-like growth factor(IGF1) and myostatin (GDF8). Interestingly they produceopposing effects on myogenesis with IGF1 leading tohypertrophy and myostatin opposing growth [60,61].What this means for the net level of myogenesis is unclear.
Page 12 of 16(page number not for citation purposes)

BMC Medical Genomics 2009, 2:44 http://www.biomedcentral.com/1755-8794/2/44
Satellite cells are an important contributor to musclegrowth, but their role in CP is difficult for us to discern asneither quiescent nor activated satellite cell markers werealtered transcriptionally.
Muscle development was indicated in the ontology analy-sis (Additional File 2 Table S2) and some genes related tomyogenesis were up-regulated (Table 3). While the mus-cle regulatory factors were not significantly altered, appar-ently fewer "growth" proteins must be activated sincemuscle growth in children with CP is decreased [10]. Thereduction of parallel growth would lead to decreased mus-cle strength in CP patients. Reduced longitudinal growthwould limit range of motion, and this has been suggestedas the cause for extraordinarily long in vivo sarcomerelengths in children with wrist flexion contractures [12].The increase in GDF8 could be responsible for this lack ofgrowth in spastic CP muscle and thus represent a potentialtherapeutic target. Other evidence pointing toward mus-cle degradation is in the expression ontology of proteinubiquitination being increased, based on the up-regula-tion of 4 related genes (FBXO3, PCNP, RBBP6, andUBE2V2) and supported by an up-regulation of CACYBP,a gene involved in calcium dependent ubiquitination. Theopposing actions of IGF1 to increase muscle mass are alsocontrolled by a number of IGF binding proteins and werevealed IGFBP5 was significantly up-regulated in CP,however the effects of IGFBP5 in muscle have been ques-tioned [62,63]. These results make the activation of theIGF1 pathway difficult to decipher at the transcriptionallevel. Furthermore, the hypertrophic effect of IGF1 is pri-marily from an increase in translation efficiency, whichcould have broad effects but would be unobservable inour study.
One of IGF1's broad anabolic effects could be a contribu-tion to the increased ECM in muscle from CP patients[64,65]. While the ECM is altered transcriptionally, it isunclear which components are most affected. The fibrillarcomponents of collagen in muscle are primarily collagentypes I and III and each alpha chain of these collagen typeswere slightly up-regulated. The most dramatic changeswere in the collagens of the basal lamina discussed in ref-erence to the NMJ. Overall the basal lamina has beendemonstrated as an area of excessive growth, and thusmay be important in understanding muscle pathology[13]. Gene ontology analysis revealed a set of genes asso-ciated with the ECM that were all significantly up-regu-lated (Additional File 2 Table S2). This supports thehypothesis of a prolific ECM in spastic muscle of CPpatients. The decreased transcription of basigin (BSG;0.48) could also lead to extensive ECM through thereduced activation of MMPs [66]. Basigin may also impli-cate a disorganized ECM lacking full functionality as MMPactivity is usually increased along with increases in ECMproduction. However TIMP's are the primary MMP inhib-
itors and did not show a corresponding transcriptionalincrease [67].
It is important to note the distinct pathology of CP, asspastic muscle does not fit neatly into any of the other"altered use" muscle models [68]. The transcriptional con-trol of muscle in CP was qualitatively different comparedto DMD, IMB, or HSP (Table 4). DMD actually showedthe most similarity to CP, particularly among contractilegenes and satellite cell markers. DMD is known to haveactivated satellite cells and regenerating muscle and thiscorrelation implicates the same in CP, although no satel-lite cell activation markers were significantly up-regulatedin CP. IMB and HSP had an opposing effect on satellitecells, which shows this is not consistent with all diseasestates. IMB has been shown to result in muscle atrophy,fibrosis, and a shift from slow to fast muscle fibers.Although we were not able to compare our results to ahuman overactivity microarray study we clearly did notshow the increase in slow fibers and mitochondrial tran-scripts expected. In fact HSP, which may be expected tothe most similar to CP, resulted in little correlation (R <0.1) with CP in all pathways except ECC. This suggeststhat ECC alterations may be a defining characteristic ofspastic muscle. It is also interesting to note that, in theother cases, IGF1 and GDF8 acted alternatively – IGF1increased while GDF8 decreased in DMD and converselyfor IMB and HSP. This highlights the unique adaptation ofCP, where myogenesis is turned on and off simultane-ously.
While we are able to demonstrate the transcriptionaleffects of CP we also investigated this effect on two sepa-rate muscles and at different levels of clinical severity. Ten-don transfer surgery is relatively common procedure forCP patients and is implicated when there is a muscularimbalance around a joint. It involves transferring the dis-tal tendon of a muscle on the side of a joint considered tohave a contracture or relative over activity to a tendon onthe opposing side of the joint. Transfer of FCU to ECRB tocorrect wrist position is one of the common tendon trans-fer surgeries. Thus FCU is considered the more pathologicmuscle and we might have expected a different transcrip-tional profile. However, we were unable to show any tran-scriptional differences between the muscles, indicatingthat both wrist flexors and extensors have a similar adap-tation to CP. While the FCU is known to exhibit contrac-tures in CP, we conclude that the contracture is developeddue to its architecture, not due to a fundamental differ-ence is secondary adaptation to the altered neuronal inputof CP. The FCU is a larger muscle than the ECRB and thelarger wrist flexor muscles may simply dominate the dis-ease state based on their size. We were also unable to showsignificant transcriptional differences among various clin-ical severity scores in CP patients. This may be because CPtranscriptional profiles are either on or off. More likely
Page 13 of 16(page number not for citation purposes)

BMC Medical Genomics 2009, 2:44 http://www.biomedcentral.com/1755-8794/2/44
our study was unable to resolve a severity effect as thestudy is biased towards the most severe cases (patientsrecruited based on corrective surgery) or the study is sim-ply underpowered. We would likely need more patientsacross the range of clinical severity scores to define thegenes most closely correlated with severity. However thelow power of the severity analysis is increased in our com-parisons of CP vs. control muscle. Further, a discussion ofstatistical power does not apply to significant differencesdetected in CP vs. control muscle. We do acknowledge,however, that we are clearly not detecting all transcriptsthat are altered in CP.
Our study has some inherent limitations, one of which isthe small sample size noted above, especially in the caseof control patients. As with any human study there is ahigh degree of heterogeneity among the samples. Thesepatients have been treated in a variety of ways, and it isimportant to note that our transcriptional profile is notsolely based on CP, but includes conservative treatment.We must also point out that this muscle is in a chronic dis-ease state, making it difficult to discern the primary effectsof CP from compensatory mechanisms that have takenplace. As with any GeneChip study, we discuss only tran-scriptional control and any observation is subject to post-transcriptional modification.
Despite these inherent limitations we have been able tohighlight areas where future work on spastic CP musclemay lead to innovative therapies. Our altered calciumhandling data points to chronically elevated calcium lev-els which are highly dangerous since they may activateendogenous proteases. Fortunately a variety of calciumchannel blockers have been developed and tested whichcould be of use in treating CP. Another potential applica-tion of current techniques could come from antifibrotictherapy to combat the increase in ECM componentswhich is suggested by the transcriptomes. Of the mostpromising may be myostatin inhibiters, currently underinvestigation, since growth is inhibited in muscle from CPpatients and myostatin, a major inhibiter of musclegrowth is significantly up-regulated. This transcriptionalstudy helps point the way to these and other areas of pro-tein modifications, cell signaling, and biomechanicswhere future investigations should be focused.
ConclusionDramatic transcriptional alterations occur in muscle sec-ondary to CP. These transcriptional changes ultimatelylead to derangement of the ECM components of spasticmuscle along with alteration of transcripts involved inmyogenesis. A number of genes alter their expression inorder to create a slow-to-fast transition of MHC isoformsand metabolic profile. GeneChip analysis has alsoallowed us to demonstrate the many changes in Ca2+ han-dling occur in CP that was not suggested previously.
Together we are able to postulate the mechanisms knownto affect muscle function in CP and predict new ones. Thiswill aid future research into CP muscle and therapies totreat CP patients.
Abbreviations(CP): Cerebral palsy; (ECC): excitation contraction cou-pling pathway; (ECM): extra-cellular matrix pathway;(ECRB): extensor carpi radialis brevis; (DMD): Duchennemuscular dystrophy; (FCU): flexor carpi ulnaris; (FT):fiber type pathway; (GCRMA): GC robust multichip anal-ysis; (GEO): Gene Expression Omnibus; (HSP): heredi-tary spastic paraplegia; (IGF1): IGF1 pathway; (IMB):immobilization; (LMN): lower motor neuron; (MAS5):microarray suite version 5.0; (MC): muscle contractionpathway; (MYG): myogenesis pathway; (NMJ): neu-romuscular junction pathway; (QPCR): quantitativepolymerase chain reaction; (RMA): robust multichip anal-ysis; (SCA): satellite cell activation markers; (SCQ): satel-lite cell quiescence markers; (SR): sarcoplasmic reticulum;(UMN): upper motor neuron.
Competing interestsThe authors declare that they have no competing interests.
Authors' contributionsLRS carried out the RNA isolation, qPCR experiments,genechip analysis, and drafted the manuscript. EP pro-vided the biopsies and assisted in review of the manu-script. YH carried out the myosin heavy chain contentexperiments. SRW participated in critical review of themanuscript. HC provided expertise on CP and criticalreview of the manuscript. SS provided expertise on gene-chip analysis and critical review of the manuscript. RLLconceived of the study, and participated in its design andcoordination and supervised the writing of the manu-script. All authors have read and approve of this manu-script.
Additional material
Additional file 1Significantly altered genes in CP. List of all features with a significant p-value (< 0.05) using each preprocessing algorithm (MAS5, RMA, GCRMA). Ratio of CP/CTRL is determined using MAS5.Click here for file[http://www.biomedcentral.com/content/supplementary/1755-8794-2-44-S1.xls]
Additional file 2Significantly up-regulated Gene Ontologies in CP. List of all Gene Ontologies significantly up-regulated genes in CP. O: observed genes, E: expected genes, R: ratio of observed/expected, P: p-value.Click here for file[http://www.biomedcentral.com/content/supplementary/1755-8794-2-44-S2.xls]
Page 14 of 16(page number not for citation purposes)

BMC Medical Genomics 2009, 2:44 http://www.biomedcentral.com/1755-8794/2/44
AcknowledgementsWe thank Drs. Ju Chen, Randy Gastwirt, Allen Ryan, and Nick Webster for advice representing and interpreting our data: We also thank Shannon Bremner for technical support. This work was supported by NIH grants AR40539, HD44822, and the United States Department of Veterans Affairs. National Institutes of Health (AR40539, HD44822); United States Department of Veterans Affairs.
References1. Rosenbaum P, Paneth N, Leviton A, Goldstein M, Bax M, Damiano D,
Dan B, Jacobsson B: A report: the definition and classificationof cerebral palsy April 2006. Dev Med Child Neurol Suppl 2007,109:8-14.
2. Johnson A: Cerebral palsies: epidemiology and causal path-ways. Archives of Disease in Childhood 2000, 83(3):279A.
3. Hoon AH Jr: Neuroimaging in cerebral palsy: Patterns of braindysgenesis and injury. Journal of child neurology 2005,20(12):936-939.
4. Korzeniewski SJ, Birbeck G, DeLano MC, Potchen MJ, Paneth N: Asystematic review of neuroimaging for cerebral palsy. Journalof child neurology 2008, 23(2):216-227.
5. Wood E: The child with cerebral palsy: diagnosis and beyond.Seminars in pediatric neurology 2006, 13(4):286-296.
6. Kerr Graham H, Selber P: Musculoskeletal aspects of cerebralpalsy. J Bone Joint Surg Br 2003, 85(2):157-166.
7. Sanger TD: Toward a definition of childhood dystonia. Currentopinion in pediatrics 2004, 16(6):623-627.
8. Sanger TD, Delgado MR, Gaebler-Spira D, Hallett M, Mink JW: Clas-sification and definition of disorders causing hypertonia inchildhood. Pediatrics 2003, 111(1):e89-97.
9. Koman LA, Smith BP, Shilt JS: Cerebral palsy. Lancet 2004,363(9421):1619-1631.
10. Elder GC, Kirk J, Stewart G, Cook K, Weir D, Marshall A, Leahey L:Contributing factors to muscle weakness in children withcerebral palsy. Dev Med Child Neurol 2003, 45(8):542-550.
11. Lieber RL, Runesson E, Einarsson F, Friden J: Inferior mechanicalproperties of spastic muscle bundles due to hypertrophic butcompromised extracellular matrix material. Muscle & nerve2003, 28(4):464-471.
12. Lieber RL, Friden J: Spasticity causes a fundamental rearrange-ment of muscle-joint interaction. Muscle Nerve 2002,25(2):265-270.
13. Booth CM, Cortina-Borja MJ, Theologis TN: Collagen accumula-tion in muscles of children with cerebral palsy and correla-tion with severity of spasticity. Dev Med Child Neurol 2001,43(5):314-320.
14. Buller AJ, Pope R: Plasticity in mammalian skeletal muscle. Phil-osophical transactions of the Royal Society of London 1977,278(961):295-305.
15. Hyatt JP, Roy RR, Baldwin KM, Edgerton VR: Nerve activity-inde-pendent regulation of skeletal muscle atrophy: role of MyoDand myogenin in satellite cells and myonuclei. American journalof physiology 2003, 285(5):C1161-1173.
16. Hyatt JP, Roy RR, Baldwin KM, Wernig A, Edgerton VR: Activity-unrelated neural control of myogenic factors in a slow mus-cle. Muscle Nerve 2006, 33(1):49-60.
17. Ponten E, Friden J, Thornell LE, Lieber RL: Spastic wrist flexors aremore severely affected than wrist extensors in children withcerebral palsy. Dev Med Child Neurol 2005, 47(6):384-389.
18. Ponten E, Gantelius S, Lieber RL: Intraoperative muscle meas-urements reveal a relationship between contracture forma-tion and muscle remodeling. Muscle Nerve 2007, 36(1):47-54.
19. House JH, Gwathmey FW, Fidler MO: A dynamic approach to thethumb-in palm deformity in cerebral palsy. J Bone Joint Surg Am1981, 63(2):216-225.
20. Ashworth B: Preliminary Trial of Carisoprodol in MultipleSclerosis. The Practitioner 1964, 192:540-542.
21. Zancolli EA, Goldner LJ, Swanson AB: Surgery of the spastic handin cerebral palsy: report of the Committee on Spastic HandEvaluation (International Federation of Societies for Surgeryof the Hand). The Journal of hand surgery 1983, 8(5 Pt 2):766-772.
22. Park JJ, Berggren JR, Hulver MW, Houmard JA, Hoffman EP: GRB14,GPD1, and GDF8 as potential network collaborators inweight loss-induced improvements in insulin action inhuman skeletal muscle. Physiological genomics 2006,27(2):114-121.
23. Seo J, Gordish-Dressman H, Hoffman EP: An interactive poweranalysis tool for microarray hypothesis testing and genera-tion. Bioinformatics (Oxford, England) 2006, 22(7):808-814.
24. Seo J, Hoffman EP: Probe set algorithms: is there a rational bestbet? BMC bioinformatics 2006, 7:395.
25. Ponten EM, Stal PS: Decreased capillarization and a shift to fastmyosin heavy chain IIx in the biceps brachii muscle fromyoung adults with spastic paresis. Journal of the neurological sci-ences 2007, 253(1–2):25-33.
26. Zhang B, Schmoyer D, Kirov S, Snoddy J: GOTree Machine(GOTM): a web-based platform for interpreting sets of inter-esting genes using Gene Ontology hierarchies. BMC bioinfor-matics 2004, 5:16.
27. Chen YW, Zhao P, Borup R, Hoffman EP: Expression profiling inthe muscular dystrophies: identification of novel aspects ofmolecular pathophysiology. J Cell Biol 2000, 151(6):1321-1336.
28. Chen YW, Gregory CM, Scarborough MT, Shi R, Walter GA,Vandenborne K: Transcriptional pathways associated withskeletal muscle disuse atrophy in humans. Physiological genomics2007, 31(3):510-520.
29. Molon A, Di Giovanni S, Chen YW, Clarkson PM, Angelini C,Pegoraro E, Hoffman EP: Large-scale disruption of microtubulepathways in morphologically normal human spastin muscle.Neurology 2004, 62(7):1097-1104.
30. Barjot C, Laplace-Marieze V, Gannoun-Zaki L, McKoy G, Briand M,Vigneron P, Bacou F: Expression of lactate dehydrogenase,myosin heavy chain and myogenic regulatory factor genes inrabbit embryonic muscle cell cultures. J Muscle Res Cell Motil1998, 19(4):343-351.
31. Hawke TJ, Garry DJ: Myogenic satellite cells: physiology tomolecular biology. J Appl Physiol 2001, 91(2):534-551.
32. Ishido M, Kami K, Masuhara M: In vivo expression patterns ofMyoD, p21, and Rb proteins in myonuclei and satellite cellsof denervated rat skeletal muscle. American journal of physiology2004, 287(2):C484-493.
33. de Palma L, Marinelli M, Pavan M, Orazi A: Ubiquitin ligasesMuRF1 and MAFbx in human skeletal muscle atrophy. Joint,bone, spine: revue du rhumatisme 2008, 75(1):53-57.
Additional file 3Significantly up-regulated Gene Ontologies tree in CP. Hierarchical list of Gene Ontologies in CP with red lettering representing significantly up-regulated Gene Ontologies.Click here for file[http://www.biomedcentral.com/content/supplementary/1755-8794-2-44-S3.gif]
Additional file 4Significantly down-regulated Gene Ontologies in CP. List of all Gene Ontologies significantly down-regulated genes in CP. O: observed genes, E: expected genes, R: ratio of observed/expected, P: p-value.Click here for file[http://www.biomedcentral.com/content/supplementary/1755-8794-2-44-S4.xls]
Additional file 5Significantly down-regulated Gene Ontologies in CP. Hierarchical list of Gene Ontologies in CP with red lettering representing significantly down-regulated Gene Ontologies.Click here for file[http://www.biomedcentral.com/content/supplementary/1755-8794-2-44-S5.gif]
Page 15 of 16(page number not for citation purposes)

BMC Medical Genomics 2009, 2:44 http://www.biomedcentral.com/1755-8794/2/44
34. Ponten E, Lindstrom M, Kadi F: Higher amount of MyHC IIX in awrist flexor in tetraplegic compared to hemiplegic cerebralpalsy. Journal of the neurological sciences 2008, 266(1–2):51-56.
35. Bellinger AM, Reiken S, Dura M, Murphy PW, Deng SX, Landry DW,Nieman D, Lehnart SE, Samaru M, LaCampagne A, et al.: Remode-ling of ryanodine receptor complex causes "leaky" channels:a molecular mechanism for decreased exercise capacity. Pro-ceedings of the National Academy of Sciences of the United States of Amer-ica 2008, 105(6):2198-2202.
36. Verde I, Pahlke G, Salanova M, Zhang G, Wang S, Coletti D, OnufferJ, Jin SL, Conti M: Myomegalin is a novel protein of the golgi/centrosome that interacts with a cyclic nucleotide phos-phodiesterase. The Journal of biological chemistry 2001,276(14):11189-11198.
37. Smerdu V, Karsch-Mizrachi I, Campione M, Leinwand L, Schiaffino S:Type IIx myosin heavy chain transcripts are expressed intype IIb fibers of human skeletal muscle. The American journalof physiology 1994, 267(6 Pt 1):C1723-1728.
38. Friden J, Lieber RL: Spastic muscle cells are shorter and stifferthan normal cells. Muscle & nerve 2003, 27(2):157-164.
39. Kang JS, Yi MJ, Zhang W, Feinleib JL, Cole F, Krauss RS: Netrins andneogenin promote myotube formation. The Journal of cell biol-ogy 2004, 167(3):493-504.
40. Faenza I, Ramazzotti G, Bavelloni A, Fiume R, Gaboardi GC, Follo MY,Gilmour RS, Martelli AM, Ravid K, Cocco L: Inositide-dependentphospholipase C signaling mimics insulin in skeletal muscledifferentiation by affecting specific regions of the cyclin D3promoter. Endocrinology 2007, 148(3):1108-1117.
41. de la Serna IL, Ohkawa Y, Berkes CA, Bergstrom DA, Dacwag CS,Tapscott SJ, Imbalzano AN: MyoD targets chromatin remode-ling complexes to the myogenin locus prior to forming a sta-ble DNA-bound complex. Mol Cell Biol 2005, 25(10):3997-4009.
42. Sorci G, Riuzzi F, Arcuri C, Giambanco I, Donato R: Amphoterinstimulates myogenesis and counteracts the antimyogenicfactors basic fibroblast growth factor and S100B via RAGEbinding. Mol Cell Biol 2004, 24(11):4880-4894.
43. Lin X, Miller JW, Mankodi A, Kanadia RN, Yuan Y, Moxley RT, Swan-son MS, Thornton CA: Failure of MBNL1-dependent post-natalsplicing transitions in myotonic dystrophy. Human moleculargenetics 2006, 15(13):2087-2097.
44. Janiesch PC, Kim J, Mouysset J, Barikbin R, Lochmuller H, Cassata G,Krause S, Hoppe T: The ubiquitin-selective chaperone CDC-48/p97 links myosin assembly to human myopathy. Nat CellBiol 2007, 9(4):379-390.
45. Bennett AM, Tonks NK: Regulation of distinct stages of skeletalmuscle differentiation by mitogen-activated protein kinases.Science 1997, 278(5341):1288-1291.
46. Lieber RL, Steinman S, Barash IA, Chambers H: Structural andfunctional changes in spastic skeletal muscle. Muscle & nerve2004, 29(5):615-627.
47. Theroux MC, Akins RE, Barone C, Boyce B, Miller F, Dabney KW:Neuromuscular junctions in cerebral palsy: presence ofextrajunctional acetylcholine receptors. Anesthesiology 2002,96(2):330-335.
48. Theroux MC, Oberman KG, Lahaye J, Boyce BA, Duhadaway D,Miller F, Akins RE: Dysmorphic neuromuscular junctions asso-ciated with motor ability in cerebral palsy. Muscle Nerve 2005,32(5):626-632.
49. Suresh NL, Ellis MD, Moore J, Heckman H, Rymer WZ: Excitatorysynaptic potentials in spastic human motoneurons have ashort rise-time. Muscle Nerve 2005, 32(1):99-103.
50. Kimura T, Takahashi MP, Fujimura H, Sakoda S: Expression and dis-tribution of a small-conductance calcium-activated potas-sium channel (SK3) protein in skeletal muscles frommyotonic muscular dystrophy patients and congenital myo-tonic mice. Neuroscience letters 2003, 347(3):191-195.
51. Hughes BW, Kusner LL, Kaminski HJ: Molecular architecture ofthe neuromuscular junction. Muscle & nerve 2006,33(4):445-461.
52. Guo W, Campbell KP: Association of triadin with the ryanodinereceptor and calsequestrin in the lumen of the sarcoplasmicreticulum. The Journal of biological chemistry 1995,270(16):9027-9030.
53. Kramerova I, Kudryashova E, Venkatraman G, Spencer MJ: Calpain3 participates in sarcomere remodeling by acting upstream
of the ubiquitin-proteasome pathway. Hum Mol Genet 2005,14(15):2125-2134.
54. Kramerova I, Kudryashova E, Wu B, Spencer MJ: Regulation of theM-cadherin-beta-catenin complex by calpain 3 during termi-nal stages of myogenic differentiation. Mol Cell Biol 2006,26(22):8437-8447.
55. Wilwert JL, Madhoun NM, Coughlin DJ: Parvalbumin correlateswith relaxation rate in the swimming muscle of sheepsheadand kingfish. The Journal of experimental biology 2006, 209(Pt2):227-237.
56. Miller JB, Crow MT, Stockdale FE: Slow and fast myosin heavychain content defines three types of myotubes in early mus-cle cell cultures. The Journal of cell biology 1985, 101(5 Pt1):1643-1650.
57. Schiaffino S, Sandri M, Murgia M: Activity-dependent signalingpathways controlling muscle diversity and plasticity. Physiol-ogy (Bethesda, Md) 2007, 22:269-278.
58. Giger JM, Haddad F, Qin AX, Baldwin KM: Effect of cyclosporin Atreatment on the in vivo regulation of type I MHC geneexpression. J Appl Physiol 2004, 97(2):475-483.
59. Parsons SA, Wilkins BJ, Bueno OF, Molkentin JD: Altered skeletalmuscle phenotypes in calcineurin Aalpha and Abeta gene-targeted mice. Mol Cell Biol 2003, 23(12):4331-4343.
60. Kollias HD, McDermott JC: Transforming growth factor-{beta}and myostatin signaling in skeletal muscle. Journal of appliedphysiology (Bethesda, Md: 1985) 2008, 104(3):579-587.
61. Dasarathy S, Dodig M, Muc SM, Kalhan SC, McCullough AJ: Skeletalmuscle atrophy is associated with an increased expression ofmyostatin and impaired satellite cell function in the porta-caval anastamosis rat. Am J Physiol Gastrointest Liver Physiol 2004,287(6):G1124-G1130.
62. Cobb LJ, Salih DA, Gonzalez I, Tripathi G, Carter EJ, Lovett F, HoldingC, Pell JM: Partitioning of IGFBP-5 actions in myogenesis: IGF-independent anti-apoptotic function. Journal of cell science 2004,117(Pt 9):1737-1746.
63. Ewton DZ, Coolican SA, Mohan S, Chernausek SD, Florini JR: Mod-ulation of insulin-like growth factor actions in L6A1 myob-lasts by insulin-like growth factor binding protein (IGFBP)-4and IGFBP-5: a dual role for IGFBP-5. J Cell Physiol 1998,177(1):47-57.
64. Rolfe KJ, Cambrey AD, Richardson J, Irvine LM, Grobbelaar AO,Linge C: Dermal fibroblasts derived from fetal and postnatalhumans exhibit distinct responses to insulin like growth fac-tors. BMC developmental biology 2007, 7:124.
65. Tarcin O, Gedik N, Karakoyun B, Tahan V, Sood G, Celikel C, TozunN: Serum Prolidase and IGF-1 as Non-invasive Markers ofHepatic Fibrosis During Four Different Periods After Bile-duct Ligation in Rats. Dig Dis Sci 2008, 53(7):1938-1945.
66. Schmidt R, Bultmann A, Ungerer M, Joghetaei N, Bulbul O, Thieme S,Chavakis T, Toole BP, Gawaz M, Schomig A, et al.: Extracellularmatrix metalloproteinase inducer regulates matrix metallo-proteinase activity in cardiovascular cells: implications inacute myocardial infarction. Circulation 2006, 113(6):834-841.
67. Nagase H, Visse R, Murphy G: Structure and function of matrixmetalloproteinases and TIMPs. Cardiovascular research 2006,69(3):562-573.
68. Foran JR, Steinman S, Barash I, Chambers HG, Lieber RL: Structuraland mechanical alterations in spastic skeletal muscle. DevMed Child Neurol 2005, 47(10):713-717.
Pre-publication historyThe pre-publication history for this paper can be accessedhere:
http://www.biomedcentral.com/1755-8794/2/44/prepub
Page 16 of 16(page number not for citation purposes)


J Physiol 589.10 (2011) pp 2625–2639 2625
The
Jour
nalo
fPh
ysio
logy
Hamstring contractures in children with spastic cerebralpalsy result from a stiffer extracellular matrixand increased in vivo sarcomere length
Lucas R. Smith1, Ki S. Lee2, Samuel R. Ward3, Henry G. Chambers4 and Richard L. Lieber1,5,6
1Departments of Bioengineering, 3Radiology and 5Orthopaedics, University of California, San Diego, CA, USA2Department of Orthopaedic Surgery, Severance Children’s Hospital, Yonsei University College of Medicine, Seoul, South Korea4Department of Orthopaedics, Rady Children’s Hospital, San Diego, CA, USA6 Department of Veterans Affairs Medical Center, San Diego, CA, USA
Non-technical summary Muscle spasticity, due to an upper motoneuron lesion, often leads tomuscle contractures that limit range of motion and cause increased muscle stiffness. However,the elements responsible for this muscle adaption are unknown. Here we show that muscle tissueis stiffer in contracture compared to age-matched children, implicating the extracellular matrix(ECM). However, titin, the major load-bearing protein within muscle fibres, is not altered incontracture, and individual fibre stiffness is unaltered. Increased ECM stiffness is even morefunctionally significant given our finding of long in vivo sarcomeres which leads to much larger invivo forces in muscle contracture. These results may lead to novel therapeutics for treating spasticmuscle contracture.
Abstract Cerebral palsy (CP) results from an upper motoneuron (UMN) lesion in the developingbrain. Secondary to the UMN lesion, which causes spasticity, is a pathological response by muscle –namely, contracture. However, the elements within muscle that increase passive mechanicalstiffness, and therefore result in contracture, are unknown. Using hamstring muscle biopsiesfrom pediatric patients with CP (n = 33) and control (n = 19) patients we investigated passivemechanical properties at the protein, cellular, tissue and architectural levels to identify the elementsresponsible for contracture. Titin isoform, the major load-bearing protein within muscle cells,was unaltered in CP. Correspondingly, the passive mechanics of individual muscle fibres werenot altered. However, CP muscle bundles, which include fibres in their constituent ECM, werestiffer than control bundles. This corresponded to an increase in collagen content of CP musclesmeasured by hydroxyproline assay and observed using immunohistochemistry. In vivo sarcomerelength of CP muscle measured during surgery was significantly longer than that predicted forcontrol muscle. The combination of increased tissue stiffness and increased sarcomere lengthinteract to increase stiffness greatly of the contracture tissue in vivo. These findings provideevidence that contracture formation is not the result of stiffening at the cellular level, but stiffeningof the ECM with increased collagen and an increase of in vivo sarcomere length leading to higherpassive stresses.
(Received 2 December 2010; accepted after revision 21 March 2011; first published online 21 March 2011)Corresponding author R. L. Lieber: Department of Orthopaedic Surgery (0863), UC San Diego and VA Medical Centre,9500 Gilman Drive, Mail Code 0863, La Jolla, CA 92093-0863, USA. Email: [email protected]
Abbreviations ACL, anterior cruciate ligament; CP, cerebral palsy; ECM, extracellular matrix; UMN, uppermotoneuron.
C! 2011 The Authors. Journal compilation C! 2011 The Physiological Society DOI: 10.1113/jphysiol.2010.203364

2626 L. R. Smith and others J Physiol 589.10
Introduction
Cerebral palsy (CP) describes a spectrum of movementdisorders caused by upper motoneuron (UMN) lesionsthat occur in the developing brain (Rosenbaum et al.2007). CP is the most common childhood movementdisorder with a prevalence of 3.6 cases per 1000 in theUS (Yeargin-Allsopp et al. 2008). Although the primaryUMN insult is not progressive, the resulting musclepathology does progress (Kerr Graham & Selber, 2003).Pathological muscle in CP is described as spastic, which isa velocity-dependent resistance to stretch due to reducedinhibition of the stretch reflexes (Crenna, 1998). Despitebest clinical practices, children with CP often developcontractures that limit their range of motion, decreasetheir mobility and may be painful. While muscle spasticityand hyper-activity are commonly seen in cerebral palsy,contracture represents a unique muscle adaptation inwhich the muscle increases passive stiffness such thatrange of motion around a joint is limited without activeforce production of the muscle. Thus, muscle contracturesrepresent a major disability to those affected by CP inparticular and those with UMN lesions in general (Bacheet al. 2003).
The skeletal muscle mechanism by which spasticityresults in contracture is not known. Transcriptional datasuggest many physiological pathways are altered in contra-cture (Smith et al. 2009). One consistent finding is thatspastic muscles from children with CP are weaker thanthose of typically developing control children due toa combination of decreased neuronal drive, decreasedmuscle size and decreased specific tension (Elder et al.2003; Rose & McGill, 2005; Stackhouse et al. 2005).Previous studies also demonstrated that increasedresistance to stretch in spastic muscle has both an activeand passive component (Sinkjaer & Magnussen, 1994;Mirbagheri et al. 2001; Lorentzen et al. 2010). However,the passive elements responsible for this increased stiffnesshave not been identified and these presumably representthe therapeutic targets of physical therapy (Wiart et al.2008), surgery (Beals, 2001) and neurotoxin injection(Lukban et al. 2009). To date, these treatments do notprevent contracture formation (Tilton, 2006).
It should be noted that the term ‘contracture’ is typicallyreferred to in the muscle physiology literature as anincrease in tension of isolated muscles or fibres in responseto external activation by caffeine or potassium (Savage& Atanga, 1988). Caffeine induces calcium release fromthe sarcoplasmic reticulum and potassium depolarizesthe muscle as methods to activate the crossbridge cyclethat produces muscle active tension (Conway & Sakai,1960; Hodgkin & Horowicz, 1960). However, the commonclinical use of the term ‘contracture’ does not refer tosuch activation. Rather, a clinical ‘contracture’ representsa condition where a muscle becomes extremely stiff,
limiting range of motion, perhaps causing pain, anddeforming joints. These contractures often result fromupper motoneuron lesions such as those that occurafter stroke, head injury or cerebral palsy and representtremendous challenges to treat (O’Dwyer et al. 1996;Farmer & James, 2001). Often, clinical contractures resultfrom chronic activation of a muscle, referred to as‘spasticity’ and the net result is a stiff muscle that limitsthe range of motion around a joint in the absence of anyactive component of crossbridge cycling (Fergusson et al.2007).
As muscle architecture is the most importantdeterminant of muscle force-generating capacity andexcursion, previous studies have sought to describe themacroscopic structural adaptation of muscle in CP. It hasbeen suggested that contracture results from shortenedmuscles and thus multiple studies have used ultrasoundtechnology to measure fascicle length in contracturedmuscle and, while these experiments confirm reducedCP muscle volume, evidence for shortened fascicles isinconclusive (Shortland et al. 2002; Malaiya et al. 2007;Mohagheghi et al. 2007, 2008). A major drawback ofultrasound studies is that there is no normalization offascicle length to sarcomere length so it is conceivablethat a CP muscle and control muscle could have exactlythe same fascicle lengths, yet have different numbersof sarcomeres in series and correspondingly differentfunctional mechanical properties. This would be invisibleto the ultrasound method. Direct measurement ofintraoperative sarcomere length revealed that sarcomerelengths are indeed longer in CP muscle, suggestingincreased passive stiffness (Lieber & Friden, 2002; Pontenet al. 2007).
Another proposed mechanism for increased passivestiffness in contractured muscle involves alteration of thetissue itself. Previous studies demonstrated that individualfibres from contractured muscles are stiffer than controls,indicating an alteration within the muscle cell (Friden& Lieber, 2003). This increased stiffness from within thefibre was hypothesized to arise from titin, considered themajor passive load-bearing protein within the musclefibre (Prado et al. 2005). Further studies confoundedthis result showing that bundles of fibres, which includeextracellular matrix (ECM), from contractured muscleswere more compliant compared to controls, and thusunable to explain the increased stiffness on the wholemuscle scale (Lieber et al. 2003). A drawback of ourprevious mechanical studies is that they studied a varietyof human muscles, and we have since shown thathealthy human muscles have different passive mechanicalproperties (Ward et al. 2009b) as was shown for rabbitmuscle (Prado et al. 2005).
To avoid complications that arise when makingcomparisons across different muscles, we have takenadvantage of the fact that children who are undergoing
C! 2011 The Authors. Journal compilation C! 2011 The Physiological Society

J Physiol 589.10 Passive mechanical properties of muscle contracture 2627
Table 1. Patient parameters
Group N Age Sex GMFCS Popliteal angle Pass Mech N SL OH-Pro MyHC Titin
Control 19 15.8 ± 1.8 8 M11 F
N/A N/A 14 N/A 12 6 6
CP 33 9.6 ± 4.2 23 M10 F
I(2),II(13),III(2),IV(6),V(10)
114 ± 15 17 11 12 6 6
Patient parameters for the control and CP groups; control patients do not have Gross Motor Function Classification System (GMFCS;Palisano et al. 1997) or popliteal angle measurements. The right columns are the number of subjects whose biopsies were used inthe various analysis; many biopsies were used for multiple analysis: passive mechanics sample size (Pass Mech N), in vivo sarcomerelength (SL), hydroxyproline (OH-pro), myosin heavy chain biopsy analysis (MyHC) and titin biopsy analysis (Titin).
anterior cruciate ligament (ACL) reconstruction witha hamstring autograft have muscle trimmed from thetendon graft that can be harvested and directly comparedto the same hamstring muscles from children with CPundergoing surgery. We hypothesized that the passivemechanical properties of spastic muscle are altered in CPand that this could arise across the levels of: protein (titin),cellular (fibre), tissue (fibre bundle including ECM),and/or architecture (sarcomere length). This work willprovide further insight into the debilitating mechanismof muscle contracture and drive research on targetedtherapies to treat contractures.
Methods
Muscle biopsy collection
Ethical approval for this study conformed to the standardsof the Declaration of Helsinki and was approved by theInstitutional Review Board of the University of California,San Diego Human Research Protection Program. Afterobtaining consent from parents and age-appropriateassent from children, subjects with CP (n = 33) wererecruited for this study because they were undergoinghamstring lengthening surgery that involved gracilis andsemitendinosus muscles. Control children (n = 19) withno history of neurological disorder were recruited becausethey were undergoing ACL reconstructive surgery witha hamstring autograft using gracilis and semitendinosustendons that were excised along with a portion of musclethat could be obtained as it was trimmed from the tendon.All patients with CP had developed a contracture requiringsurgery, despite receiving conservative treatment. Patientswere classified based on clinical measures of the GrossMotor Function Classification System (Palisano et al.1997), popliteal angle, limbs affected and treatmentmeasures of previous surgical lengthening or botulinumtoxin injection (Table 1). Muscle biopsies were obtainedand either snap frozen in isopentane chilled by liquidnitrogen ("159#C), and stored at "80#C, or placedin glycerinated muscle relaxing solution and stored at"20#C.
In vivo sarcomere lengths
Custom muscle biopsy clamps, modified for pediatricuse with 0.5 cm jaw spacing were used to determine invivo sarcomere length (Fig. 1). We previously validatedthis method against intraoperative laser diffraction (Wardet al. 2009a). After skin incision and prior to lengthening,gracilis and semitendinosus were identified. A smallsegment of each muscle was atraumatically isolated byblunt dissection. The custom clamp was then slippedover the bundle, with care to prevent undue tension onthe muscle. The child’s leg was positioned with 90 degof hip flexion and 90 deg of knee flexion, and neutralhip abduction–adduction, the clamp was engaged, andthe section of muscle within the jaws of the clamp wasre-sected and immediately placed in Formalin to fix thebiopsy specimen in its in vivo configuration. After 2 days offixation, muscle bundles were isolated on glass slides andsarcomere length was measured by laser diffraction (seebelow). For control patients receiving ACL reconstruction,hamstring muscles are not accessible in their in vivoposition, which precludes the use of the biopsy clampsto obtain control values. Thus, for estimation of controlsarcomere lengths, we extracted these values from ourprevious musculoskeletal model (Arnold et al. 2010).
Muscle mechanical testing
Biopsies for mechanics were stored in a glycerinatedrelaxing solution overnight, composed of (mM):potassium propionate (170.0), K3EGTA (5.0), MgCl2
(5.3), imidazole (10.0), Na2ATP (21.2), NaN3 (1.0),glutathione (2.5), 50 µM leupeptin, and 50% (v/v)glycerol. For dissection of fibre or fibre bundle samples,muscles were removed from storage solution and trans-ferred to a relaxing solution at pCa 8.0 and pH 7.1consisting of (mM): imidazole (59.4), KCH4O3S (86.0),Ca(KCH4O3S)2 (0.13), Mg(KCH4O3S)2 (10.8), K3EGTA(5.5), KH2PO4 (1.0), Na2ATP (5.1), and 50.0 µM
leupeptin. Single fibre segments (1.5–3 mm in length)were carefully dissected and mounted in a chamber ina custom apparatus at room temperature (20#C). Fibreswere secured using 10-0 monofilament nylon suture on
C! 2011 The Authors. Journal compilation C! 2011 The Physiological Society

2628 L. R. Smith and others J Physiol 589.10
one end to a force transducer (Model 405A, sensitivity10 V g"1, Aurora Scientific, Ontario, Canada) and onthe other end to a titanium wire rigidly attached to arotational bearing (Newport MT-RS; Irvine, CA, USA;Supplementary Fig. S1). Segments displaying obviousabnormalities or discoloration were not used. The samplewas transillumated by a 7 mW He–Ne laser to permitsarcomere length measurement by laser diffraction (Lieberet al. 1984). Resolution of this method is approximately5 nm (Baskin et al. 1979). The system was calibratedwith a 2.50 µm plastic blazed diffraction grating priorto experimentation (Diffraction Gratings, Inc., Nashville,TN, USA).
The fibre was brought to slack length, defined whenpassive tension was just measurable above the noiselevel of the force transducer. Sample dimensions weremeasured optically with a cross-hair reticule mountedon a dissecting microscope and micromanipulators onan x–y mobile stage. The fibre was then loaded withstrains of approximately 0.25 µm sarcomere"1 at 100 fibrelengths s"1. Each stretch was held for 2 or 3 minduring which stress relaxation was measured, beforea sequential stretch was made. Fibres were stretchedin total to approximately 100% strain and were savedfor titin analysis after mechanical testing. Force datawere converted to stress by dividing force by the base-line cross-sectional area value determined assuming a
cylindrical sample with an average diameter determinedfrom three separate points along the fibre. Samples werediscarded if they did not produce a clear diffractionpattern, if any irregularities appeared along their lengthduring testing, or if they were severed or slipped at eithersuture attachment point during the test. Muscle bundleswere mechanically tested in the same manner as fibres andconsisted of approximately 20 fibres and their constitutiveECM.
Mechanical data analysis
All analysis was performed using Matlab (Mathworks Inc.,Natick, MA, USA). Relaxed stress after 2 or 3 min was usedto fit a relaxed stress vs. sarcomere length curve. This curvewas fitted with a line for fibres, but with a quadratic forbundles, as there was notable non-linearity in bundle data.For sample fits, sarcomere lengths below slack length areassigned a stress of 0. This produces a ‘toe region’ due toaveraging of the fits across the range of slack sarcomerelengths tested in fibres, generally below 2.5 µm sarcomerelength. Only the data beyond the toe region in which mostfibres are generating tension are depicted (Fig. 2A andC). Tangent modulus was calculated at given sarcomerelength by taking the derivative of the relaxed stress vs.sarcomere length fit at that length. Comparisons of tangent
Figure 1. Images of biopsy collectionmethod using clamps for in vivo sarcomerelength determinationA, hamstring muscle is exposed. B, clamp issecured around gracilis muscle with jointposition at 90 deg of hip and knee flexion.C, close-up view of muscle clamps around thebiopsy tissue. D, biopsy is dissected frommuscle while clamped and fixed in Formalin forsubsequent sarcomere length measurement.
C! 2011 The Authors. Journal compilation C! 2011 The Physiological Society

J Physiol 589.10 Passive mechanical properties of muscle contracture 2629
stiffness were conducted with a three-way ANOVA forpathology (CP vs. control), muscle (gracilis vs. semi-tendinosus; repeated measure), and scale (fibre vs. bundle;repeated measure) with results considered significant atP < 0.05. All data are presented in the text as mean ± SEMunless otherwise noted.
Protein gels
Titin isoform and MyHC content were analysed on gelsfrom both single fibres after mechanical experiments
and from sections of biopsies. Single fibres were storedat "80#C until analysed and boiled for 2 min in 10 µlsodium dodecyl sulfate-vertical agarose gel electrophoresis(SDS-VAGE) sample buffer (comprised of 8 M urea, 2 M
thiourea, 3% SDS w/v, 75 mM dithiothreitol (DTT), 0.03%bromophenol blue and 0.05 M Tris-Cl, pH 6.8; Warren et al.2003). For biopsies, a myofibril-rich fraction ($10 mg wetweight) of individual biopsies (n = 24 biopsies from 12patients for titin and MyHC) was homogenized in samplebuffer using the Bullet Blender (Next Advance, Inc., AverillPark, NY, USA).
Figure 2. Passive tension as a function of sarcomere length for fibres and bundles, after stress relaxationPlots represent the average of the fits from each individual sample ± SEM. The stress vs. sarcomere length fit waslinear for fibres with a R2 value of 0.962 ± 0.003 (A and B) and quadratic for bundles with a R2 value of0.985 ± 0.002 (C and D). A, gracilis fibres show no difference between CP and control. B, semitendinosus fibresshow no difference between CP and control. C, CP gracilis bundles show a significant increase in stress at highsarcomere lengths compared to control. D, CP semitendinosus bundles show a significant increase in stress at highsarcomere lengths compared to control.% inside symbol designates the approximate sarcomere length at 90 degof hip and knee flexion.
C! 2011 The Authors. Journal compilation C! 2011 The Physiological Society

2630 L. R. Smith and others J Physiol 589.10
To quantify titin isoforms, the molecular mass of titinin muscle samples was determined using SDS-VAGE. Anacrylamide plug was placed at the bottom of the gel to holdthe agarose in place. The final composition of this plug was12.8% acrylamide, 10% v/v glycerol, 0.5 M Tris-Cl, 2.34%N ,N &-diallyltartardiamide, 0.028% ammonium persulfateand 0.152% tetramethylethylenediamine (TEMED). Thecomposition of the agarose gel was 1% w/v SeaKem Goldagarose (Lonza, Basel, Switzerland), 30% v/v glycerol,50 mM Tris-base, 0.384 M glycine and 0.1% w/v sodiumdodecyl sulfate (SDS). Titin standards were obtained fromhuman cadaver soleus (3700 kDa) and rat cardiac muscle(2992 kDa). The standard titin molecular masses are basedon sequence analysis of the 300 kb titin gene with acoding sequence contained within 363 exons (Labeit &Kolmerer, 1995; Freiburg et al. 2000). These tissues werealso homogenized and stored at "80#C until analysis.Before loading onto the gel, a titin standard ‘cocktail’was created with the following ratio: 1 unit of humansoleus standard:3 units rat cardiac standard:6 units samplebuffer. Sample wells were then loaded with both biopsy andrat cardiac homogenate. Human soleus and rat cardiactitin homogenates were loaded into standard lanes. Thisenabled titin quantification on each gel as previouslydescribed (Warren et al. 2003). Gels were run at 4#C for5 h at 15 mA constant current.
To quantify MyHC isoform distribution, homogenizedprotein solution was resuspended to 0.125 µg µl"1 protein(BCA protein assay, Pierce, Rockford, IL, USA) in asample buffer consisting of DTT (100 mmol l"1), SDS(2%), Tris-base (80 mmol l"1) pH 6.8, glycerol (10%)and bromophenol blue (0.01% w/v). Samples were boiled(2 min) and stored at "80#C. Before loading onto thegel, protein was further diluted 1:15 (0.008 µg µl"1) inthe same sample buffer to account for the approximately50-fold greater sensitivity of the silver stain. Ten micro-litres of each sample were loaded in each lane. Totalacrylamide concentration was 4% and 8% in the stackingand resolving gels, respectively (bisacrylamide, 1:50). Gels(16 cm ' 22 cm, 0.75 mm thick) were run at a constantcurrent of 10 mA for 1 h, and thereafter at constantvoltage of 275 V for 22 h at 4–6#C. Gels were silverstained (BioRad, Hercules, CA, USA). MyHC bands wereidentified and quantified with densitometry (GS-800,BioRad). The progression of the band was compared andidentified based on its relative molecular weight to that ofa human protein standard prepared (as described above)from a normal semitendinosus biopsy that showed all threehuman MHC bands (IIa, IIx and I).
Hydroxyproline content
Collagen percentage was determined using a colourimetricanalysis of hydroxyproline content. Briefly, muscle
samples were hydrolysed in 6 N HCl for 18 h, neutralized,and samples were treated with a chloramine T solutionfor 20 min at room temperature followed by a solutionof p-diaminobenzaldehyde for 30 min at 60#C. Sampleabsorbance was read at 550 nm in triplicate andcompared to a standard curve to determine the hydro-xyproline content. Hydroxyproline content was convertedto collagen using a constant (7.46) that defines thenumber of hydroxyproline residues in a molecule ofcollagen.
Immunohistochemistry
Biopsies previously snap-frozen in isopentane were usedfor immunohistochemistry. Cross-sections (10 µm thick)taken from the midportion of the tissue block were cut on acryostat at "25#C (Microm HM500, Walldorf, Germany).Serial sections were stained with haematoxylin–eosin toobserve general tissue morphology. To investigate ECMcomponents sections were labelled with primary anti-bodies to fibrillar collagen type I (rabbit polyclonal,Rockland, Gilbertsville, PA, USA) and laminin (rabbitpolyclonal, Sigma, St Louis, MI, USA). The secondary anti-body used for visualization was an Alexa Fluor 594 goatanti-rabbit immunoglobulin G (Invitrogen, Carlsbad, CA,USA).
Fibre cross-sectional areas were measured fromlaminin-stained slides using a custom-written macro inImageJ (NIH, Bethesda, MD, USA). Filtering criteria wereapplied to insure measurement of actual muscle fibres.These criteria rejected regions with areas below 50 µm2
or above 5600 µm2 to eliminate neurovascular structuresand ‘optically fused’ fibres, respectively. Fibres touchingthe edge of the field were excluded as they were assumedto be incomplete. Regions with circularity below 0.30 orabove 1.0 were excluded to prevent inclusion of fibres thatwere obliquely sectioned.
Results
Passive mechanics
Passive mechanical properties were determined for threefibres per muscle and two muscles per subject, for bothcontrol children (n = 14) and children with CP (n = 17).Fibre diameter was smaller for CP (66.3 ± 2.6 µm) thanfor control (80.2 ± 2.6 µm; P < 0.001) as previouslydescribed (Friden & Lieber, 2003). Slack sarcomere lengthfor control (2.31 ± 0.04 µm) fibres was not significantlydifferent from CP (2.31 ± 0.04 µm (SD); P > 0.9) fibres.The stiffness of CP fibres was not significantly differentfrom control for gracilis (control 21.5 ± 1.8 kPa µm"1; CP21.5 ± 2.4 kPa µm"1; Figs 2A and 3A) or semitendinosus
C! 2011 The Authors. Journal compilation C! 2011 The Physiological Society

J Physiol 589.10 Passive mechanical properties of muscle contracture 2631
(control 22.4 ± 1.8 kPa µm"1; CP 23.6 ± 1.7 kPa µm"1;Figs 2B and 3A), nor was it significantly different betweenmuscles.
Passive mechanical properties were determined forthree bundles on the same biopsy as for fibres. Fibre bundlediameters were not significantly different between control(366.1 ± 15.0 µm) and CP (354.9 ± 15.2 µm; P > 0.4)bundles, nor were slack sarcomere lengths (control:2.27 ± 0.03 µm; CP 2.29 ± 0.03 µm; P > 0.5). CPbundles had higher stresses at longer sarcomere lengthsfor both gracilis and semitendinosus muscle. Whencomparing the tangent stiffness at 4.0 µm the stiffnessof CP bundles was significantly greater than control(P < 0.05) for both gracilis (control 36.1 ± 3.9 kPa µm"1;CP 60.4 ± 11.8 kPa µm"1; Figs 2C and 3B) andsemitendinosus (control 25.2 ± 2.9 kPa µm"1; CP40.7 ± 4.9 kPa µm"1; Figs 2D and 3B).
The three-way ANOVA with muscle (semitendinosus/gracilis; repeated measure), scale (fibre/bundle; repeatedmeasure) and condition (CP/control) on tangent stiffnessrevealed a main effect of all three independent measures(P < 0.05 for condition and muscle; P < 0.001 scale).The results also showed a significant interaction betweenmuscle and scale (P < 0.05), with post hoc tests revealinggracilis bundles are stiffer than semitendinosus, and asignificant interaction of condition and scale, with post hoctests revealing an effect of CP only at the bundle level forsemitendinosus (P < 0.05).
To determine whether the mechanical changes wererelated to the clinical observations made on the patients,stiffness was correlated with clinical severity score. Therewas no significant correlation between stiffness and eitherGross Motor Function Classification System or poplitealangle at either the fibre or bundle level (SupplementalFig. 2A). To determine consistency within patients, acorrelation was run between gracilis and semitendinosusstiffness within the same patient, or fibre stiffness tobundle stiffness within the same biopsy, but again therewere no significant correlations (Supplemental Fig. 2B andC). There was also a concern that there might be anage effect since the control subjects were slightly olderthan CP subjects (Table 1) but no significant correlationwas found, validating the comparison of control toCP subjects with different ages (Supplemental Fig. 2D).Further, the mechanics were compared from patients whounderwent a previous hamstring lengthening surgery orbotulinum toxin injection prior to biopsy as these mayaffect stiffness. No significant difference was observed forprior botulinum toxin injection for bundles (P > 0.3)or fibres (P > 0.8). Only one patient with mechanicsmeasured had undergone a previous lengthening surgery,which was not an outlier among any mechanicalmeasure.
Figure 3. Tangent stiffness of fibres and bundlesSamples are represented with either a linear fit for fibres or aquadratic fit for bundles. A, tangent stiffness values at 4.0 µm forsingle fibres are not changed with CP for either gracilis orsemitendinosus muscles. B, tangent stiffness values at 4.0 µm forfibre bundles are significantly greater in CP compared to controlbundles in both gracilis and semitendinosus (P < 0.05). C, tangentstiffness values at measured average in vivo sarcomere length for CPbundles or the predicted in vivo sarcomere length for controlbundles show highly elevated values in CP for a joint configurationof 90 deg hip and knee flexion.
C! 2011 The Authors. Journal compilation C! 2011 The Physiological Society

2632 L. R. Smith and others J Physiol 589.10
In vivo sarcomere lengths
In vivo sarcomere length from cerebral palsy patientsof contractured hamstring muscles (n = 22) was3.54 ± .14 µm for gracilis and 3.62 ± .13 µm for semi-tendinosus at 90 deg of hip and knee flexion. For controlcomparison, model results were used as described inMethods (Arnold et al. 2010). Both gracilis and semi-tendinosus had significantly longer sarcomere lengths at90 deg of hip and knee flexion than predicted by the modelby about 0.5 µm (Fig. 4A, P < 0.05 for semitendinosusand gracilis). Combining the sarcomere length valueswith passive mechanical properties demonstrates that CPmuscle tissue at these joint angles bears a higher passiveload compared to control muscle (Fig. 3C).
To determine whether the sarcomere length wasassociated with functional changes, clinical measures werecorrelated to sarcomere length. There was a significantcorrelation between in vivo sarcomere length and bothGross Motor Function Classification System (P < 0.05)and sarcomere length (P < 0.05), indicating that moreseverely involved patients had longer in vivo sarcomerelengths (Fig. 4B). There was also a significant negativecorrelation between popliteal angle and in vivo sarcomerelength indicating that longer sarcomere lengths werepresent in joints with more severe contractures (Fig. 4C).Together these correlations provide further evidence thatin vivo sarcomere lengths are elevated in CP.
To compare predicted in vivo stiffness of CP musclescompared to control we also evaluated the tangent stiffnessat the average in vivo sarcomere length of 90 deg of hip andknee flexion for each muscle and condition from the dataabove. Combining the in vivo sarcomere lengths with the
mechanical data shows that in vivo stiffness is predictedto be much larger for CP muscle (Fig. 3C). As fibres hadlinear stress–strain relationships, the tangent stiffness doesnot vary with sarcomere length and is thus the same resultas the tangent stiffness at 4.0 µm.
Titin isoforms
To determine if titin size was related to overall musclestiffness we measured titin molecular mass from a biopsy.The results of a two-way ANOVA showed no significantdifference (P > 0.05) for CP with mean values of gracilis(control 3588 ± 18 kDa; CP 3667 ± 22 kDa) and semi-tendinosus (control 3625 ± 19 kDa; CP 3658 ± 26 kDa;Fig. 5) among the samples measured (n = 24, 6 per musclecondition). The mass of CP titin was actually larger thanthat of control suggesting, if anything, a more compliantisoform and unable to account for any increased passivestiffness of the muscle as a whole.
The effect of titin isoform was also investigated on singlefibres that had previously undergone passive mechanicaltesting. A two-way ANOVA showed no significantdifference (P > 0.1) between titin isoform sizes for gracilis(control 3758 ± 24 kDa; CP 3772 ± 36 kDa) or semi-tendinosus (control 3729 ± 40 kDa; CP 3797 ± 40 kDa)among the single fibres measured (n = 55). CP fibreshaving equivalent titin isoform size to control fibres isconsistent with the fact that CP and control fibres haveequivalent stiffness. The effect of titin isoform size onthe variability in mechanical stiffness of fibres was alsoinvestigated, but there was not a significant correlationbetween titin size and fibre stiffness within single fibres
Figure 4. In vivo sarcomere length of gracilis and semitendinosusA, measured in vivo sarcomere length with 90 deg of hip and knee flexion ± SEM for CP subjects in gracilisand semitindenosus (P < 0.05). Solid white line represents predicted sarcomere length for control children.B, correlation between in vivo sarcomere length measured for CP subjects and their Gross Motor FunctionClassification System (GMFCS) shows a positive significant correlation (P < 0.05), meaning subjects with longer invivo sarcomeres are more severely affected patients. C, correlation between in vivo sarcomere length and poplitealangle is negative and significant (P < 0.05), meaning subjects with less knee extension have longer sarcomerelengths.
C! 2011 The Authors. Journal compilation C! 2011 The Physiological Society
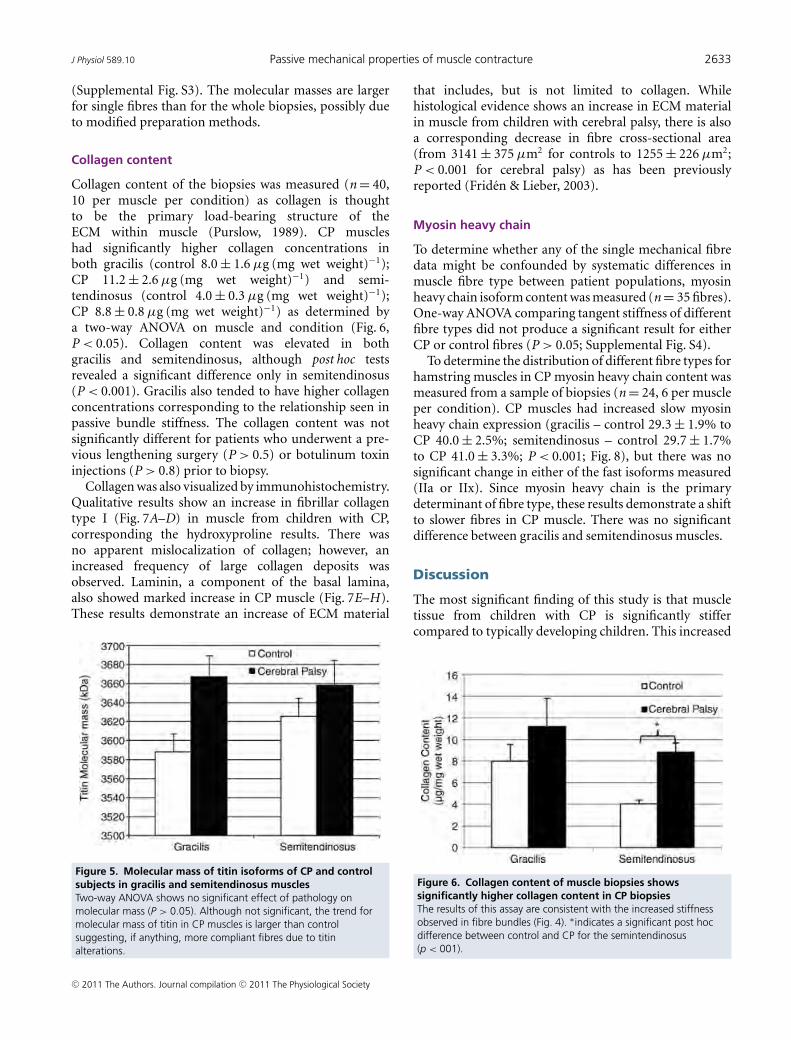
J Physiol 589.10 Passive mechanical properties of muscle contracture 2633
(Supplemental Fig. S3). The molecular masses are largerfor single fibres than for the whole biopsies, possibly dueto modified preparation methods.
Collagen content
Collagen content of the biopsies was measured (n = 40,10 per muscle per condition) as collagen is thoughtto be the primary load-bearing structure of theECM within muscle (Purslow, 1989). CP muscleshad significantly higher collagen concentrations inboth gracilis (control 8.0 ± 1.6 µg (mg wet weight)"1);CP 11.2 ± 2.6 µg (mg wet weight)"1) and semi-tendinosus (control 4.0 ± 0.3 µg (mg wet weight)"1);CP 8.8 ± 0.8 µg (mg wet weight)"1) as determined bya two-way ANOVA on muscle and condition (Fig. 6,P < 0.05). Collagen content was elevated in bothgracilis and semitendinosus, although post hoc testsrevealed a significant difference only in semitendinosus(P < 0.001). Gracilis also tended to have higher collagenconcentrations corresponding to the relationship seen inpassive bundle stiffness. The collagen content was notsignificantly different for patients who underwent a pre-vious lengthening surgery (P > 0.5) or botulinum toxininjections (P > 0.8) prior to biopsy.
Collagen was also visualized by immunohistochemistry.Qualitative results show an increase in fibrillar collagentype I (Fig. 7A–D) in muscle from children with CP,corresponding the hydroxyproline results. There wasno apparent mislocalization of collagen; however, anincreased frequency of large collagen deposits wasobserved. Laminin, a component of the basal lamina,also showed marked increase in CP muscle (Fig. 7E–H).These results demonstrate an increase of ECM material
Figure 5. Molecular mass of titin isoforms of CP and controlsubjects in gracilis and semitendinosus musclesTwo-way ANOVA shows no significant effect of pathology onmolecular mass (P > 0.05). Although not significant, the trend formolecular mass of titin in CP muscles is larger than controlsuggesting, if anything, more compliant fibres due to titinalterations.
that includes, but is not limited to collagen. Whilehistological evidence shows an increase in ECM materialin muscle from children with cerebral palsy, there is alsoa corresponding decrease in fibre cross-sectional area(from 3141 ± 375 µm2 for controls to 1255 ± 226 µm2;P < 0.001 for cerebral palsy) as has been previouslyreported (Friden & Lieber, 2003).
Myosin heavy chain
To determine whether any of the single mechanical fibredata might be confounded by systematic differences inmuscle fibre type between patient populations, myosinheavy chain isoform content was measured (n = 35 fibres).One-way ANOVA comparing tangent stiffness of differentfibre types did not produce a significant result for eitherCP or control fibres (P > 0.05; Supplemental Fig. S4).
To determine the distribution of different fibre types forhamstring muscles in CP myosin heavy chain content wasmeasured from a sample of biopsies (n = 24, 6 per muscleper condition). CP muscles had increased slow myosinheavy chain expression (gracilis – control 29.3 ± 1.9% toCP 40.0 ± 2.5%; semitendinosus – control 29.7 ± 1.7%to CP 41.0 ± 3.3%; P < 0.001; Fig. 8), but there was nosignificant change in either of the fast isoforms measured(IIa or IIx). Since myosin heavy chain is the primarydeterminant of fibre type, these results demonstrate a shiftto slower fibres in CP muscle. There was no significantdifference between gracilis and semitendinosus muscles.
Discussion
The most significant finding of this study is that muscletissue from children with CP is significantly stiffercompared to typically developing children. This increased
Figure 6. Collagen content of muscle biopsies showssignificantly higher collagen content in CP biopsiesThe results of this assay are consistent with the increased stiffnessobserved in fibre bundles (Fig. 4). %indicates a significant post hocdifference between control and CP for the semintendinosus(p < 001).
C! 2011 The Authors. Journal compilation C! 2011 The Physiological Society
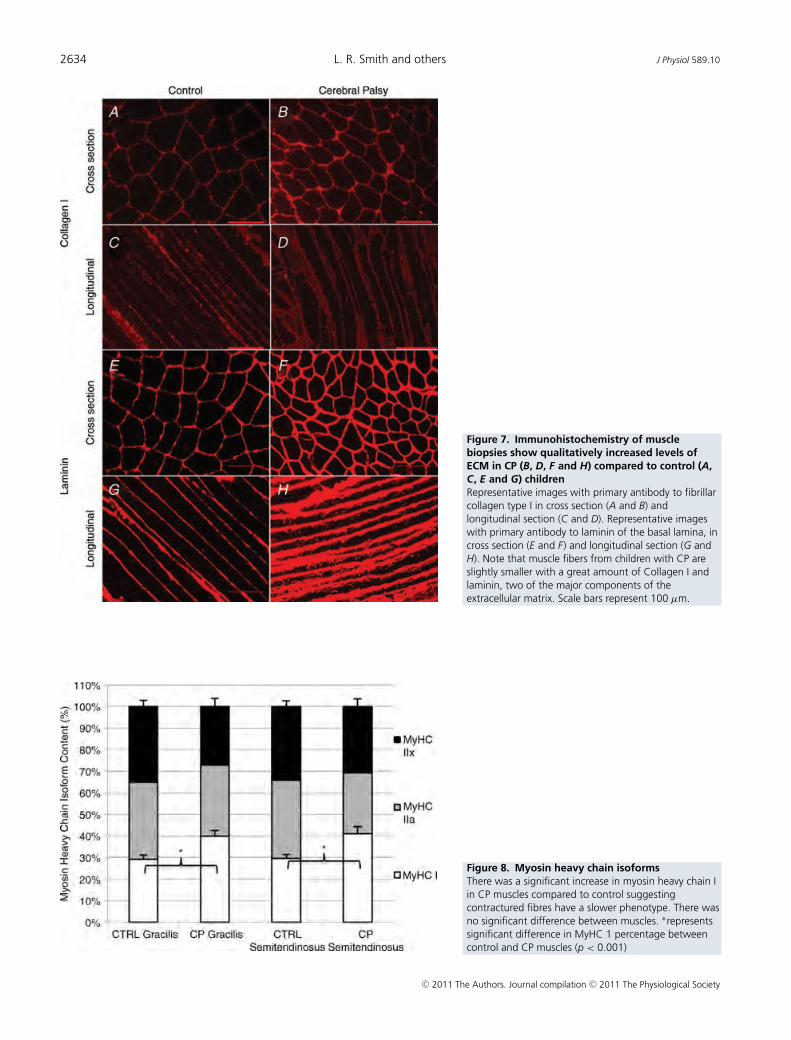
2634 L. R. Smith and others J Physiol 589.10
Figure 7. Immunohistochemistry of musclebiopsies show qualitatively increased levels ofECM in CP (B, D, F and H) compared to control (A,C, E and G) childrenRepresentative images with primary antibody to fibrillarcollagen type I in cross section (A and B) andlongitudinal section (C and D). Representative imageswith primary antibody to laminin of the basal lamina, incross section (E and F) and longitudinal section (G andH). Note that muscle fibers from children with CP areslightly smaller with a great amount of Collagen I andlaminin, two of the major components of theextracellular matrix. Scale bars represent 100 µm.
Figure 8. Myosin heavy chain isoformsThere was a significant increase in myosin heavy chain Iin CP muscles compared to control suggestingcontractured fibres have a slower phenotype. There wasno significant difference between muscles. %representssignificant difference in MyHC 1 percentage betweencontrol and CP muscles (p < 0.001)
C! 2011 The Authors. Journal compilation C! 2011 The Physiological Society

J Physiol 589.10 Passive mechanical properties of muscle contracture 2635
passive stiffness is accompanied by an increase in collagencontent and is made even more functionally significant inthat in vivo sarcomere length of CP hamstring muscles issignificantly longer compared to predictions for controlchildren. Taken together, these data provide a mechanisticexplanation for the increased joint and muscle stiffnessobserved in these contracture patients. While fibre bundleswere different between CP and control muscle, we foundno significant difference in mechanical properties at thesingle fibre level of muscle and no change in titin isoformsize. Thus, we conclude that, for human hamstringmuscles, increased passive tension in contracture is due toa change in ECM stiffness and increased in vivo functionalsarcomere length rather than any intracellular alteration.
Bundle mechanics
While fibres contribute to passive tension of muscle,muscle ECM plays an important role in passive mechanics,especially at longer sarcomere lengths. The results show asignificant increase in the tangent stiffness of fibre bundlesfrom CP patients. While fibres were fitted well with alinear stress–sarcomere length relationship, fibre bundlesrequired a non-linear quadratic fit. This non-linearityresults in similar tissue stiffness at small strains, butsignificantly increased stiffness at long sarcomere lengthsof the CP muscle tissue. Muscle contractures often limitjoint range of motion suggesting that there are largein vivo strains on the muscle. We thus believe that,in vivo, the ECM bears a large portion of the passivemuscle load.
Collagen is considered the primary load-bearingstructure within muscle ECM (Purslow, 1989). We hypo-thesized that an increase in collagen content of CPmuscle could lead to the increased passive stiffness seenin bundles. Using a hydroxyproline assay to test thehypothesis, the results demonstrate a significant increasein collagen within CP muscle. Although these data arepresented in micrograms of collagen per milligram ofmuscle wet weight, they are similar to previous datapresenting collagen as a percent of dry weight (Bendall,1967), using the assumption that muscle is approximately80% water (Ward & Lieber, 2005). Increased collagenwas also observed by immunohistochemistry along withanother ECM component, laminin, a critical componentfor cellular attachment to the basal lamina. These resultsare in agreement with a previous study showing increasedcollagen content within CP muscle (Booth et al. 2001).Many additional factors may be playing a role in theincreased ECM stiffness. The organization of collagen,the distribution of collagen types, or the proteoglycancontent all could be altered in CP to create a stiffer ECMand represent areas of further investigation. Immuno-histochemistry of other muscle proteins (!-actinin,
desmin, dystrophin) revealed no obvious differencesbetween patient groups.
One previous study investigated the mechanics offibre bundles from contractured muscle tissue (Lieberet al. 2003). Despite finding stiffer fibres we found morecompliant bundles in CP muscle. This result is difficultto reconcile with the increased passive stiffness of thewhole muscle that has been reported. The previous studywas conducted on biopsies taken from various musclesthat were not matched between populations, which couldaccount for some of these differences and all muscles werefrom upper extremities, which may respond differentlyto spasticity. In addition, the ECM from upper extremitymuscles was highly deranged in the contractured muscles(see Fig. 2 of Lieber et al. 2003) making area fractionmeasurements from these specimens difficult. It is possiblethat the area fraction of ECM was overestimated, resultingin artificially low values for bundle modulus. Finally, thenon-linear behaviour of upper extremity muscles wasquantified by only fitting data to the linear portion ofthe sarcomere length–stress curve. The current methodrepresents a more accurate method for handling analysisof the non-linear relationships.
Sarcomere organization
Sarcomere length operating ranges of semitendinosusand gracilis muscles are unknown, although musclesare typically believed to operate on the plateau of thelength–tension curve (2.5–2.7 µm for human skeletalmuscle; Walker & Schrodt, 1974). A previous studydemonstrated that spastic muscle operates at longersarcomere lengths than control (Lieber & Friden, 2002),which would lead to a larger observed passive stiffnesssuch as that seen in contracture. We measured in vivosarcomere lengths of patients with CP at a defined jointangle and compared these values to sarcomere lengthsof control subjects that were calculated based on in vivosarcomere lengths, moment arms and muscle–tendonlengths (Arnold et al. 2010). The results showed that theCP sarcomere lengths are significantly longer than thosepredicted from the model. The lengths measured werealso much longer than optimal sarcomere length, lendingfurther evidence to the idea that they are overly stretched incontracture. With CP subjects operating at longer lengthsof the passive length–tension relationship, this means thatthe muscle is experiencing higher stresses not only due tomaterial property changes, but also due to this shift alongthe passive length–tension curve. This difference becomesmore pronounced as the knee extends and the hip flexes,and may limit range of motion for children with contrac-tures.
It is often stated that muscle adds or subtracts serialsarcomeres to optimal sarcomere length in vivo (Williams
C! 2011 The Authors. Journal compilation C! 2011 The Physiological Society

2636 L. R. Smith and others J Physiol 589.10
& Goldspink, 1973). Long in vivo sarcomeres suggestan inability of the muscle to add sarcomeres in series,which would be exacerbated during growth spurts, whichhave been associated with the onset of muscle contra-ctures (Janice & Alwyn, 2005). The very long sarcomerelengths observed in vivo clearly imply that muscles fromchildren with CP are under high stress. The source ofthe force that creates or opposes this stress is not known.However, we have speculated, based on analysis of thetranscriptome, that muscles from children with CP areunable to grow serially in response to the stretch imposedby osteogenesis (Smith et al. 2009). It is also possible thatmuscles would decrease their serial sarcomere number,which would provide a resistive force since the changescould be slow and accompanied by reinforcement ofthe muscle fibre by the ECM. Muscle contracture isoften described as a ‘shortened’ muscle; our finding ofincreased in vivo sarcomere length corresponds with thenotion that muscle shortening is derived from fewer seriessarcomeres, not shortened sarcomeres. Longer in vivosarcomere lengths are an important factor for both passiveand active force production of skeletal muscle. Previousresearch has demonstrated that muscles from childrenwith CP are smaller than those of control children, yetmuscle force production is reduced to an even greaterextent indicating a dysfunction of active muscle forceproduction in CP (Elder et al. 2003; Moreau et al. 2010).A consequence of having longer in vivo sarcomere lengthsfor children with CP is the muscle will be working atdifferent portions along its active length–tension curve(Gordon et al. 1966) compared to control subjects. Basedon measured human filament lengths (Walker & Schrodt,1974) and the increase in $0.5 µm sarcomere length, thedecrease in force from a typically developing child on theplateau of the length–tension curve to a child with CPon the descending limb would be 33%. It is interestingto note that this is on the same scale as the reduction inforce that is not accounted for by decreased muscle size inthese patients (Elder et al. 2003; Moreau et al. 2010). Thus,perhaps altered in vivo sarcomere length operating rangerepresents a significant functional alteration in musclesfrom children CP and demonstrates that these muscles arenot simply changing sarcomere number to ‘re-optimize’the muscle after injury.
Potential mechanisms of contracture formation
It is possible that the changes in ECM and in vivo sarcomerelength take place simultaneously and independently, orthat one precedes and directly affects the other. If thesetwo alterations of CP muscle are not causal, they could bea consequence of the same factors within spastic muscle.Previous research showed that myostatin, a negativeregulator of muscle growth, also stimulates proliferation
of muscle fibroblasts and the release of ECM proteins (Liet al. 2008). Myostatin mRNA has also been shown assignificantly increased in CP muscle of the upper extremity(Smith et al. 2009). Alternatively, transforming growthfactor-"1 has been shown to induce a shift in satellitecells from a myogenic lineage to fibrobasts (Li et al. 2004).This process also has the potential to limit growth throughsatellite cell depletion and increase the ECM secreting cellpopulation.
Longer in vivo sarcomeres of CP muscles demonstratethat there is increased sarcomere strain, which has beenshown to directly induce skeletal muscle injury (Patelet al. 2004). Repeated strain-induced injuries have beenshown to drastically increase collagen content and fibrosisin skeletal muscle (Stauber, 2004). The effects of chronicstrain injuries persist for months or even years andcould be responsible for the effects of muscle in contra-cture. Repeated strain-induced injury also results in lowerforce-producing capacity of muscle, which may provideanother explanation for the reduced specific tension ofCP muscle (Proske & Morgan, 2001). The increasedfibrosis and stiffness of muscle contracture could also be acompensatory mechanism to limit further strain-inducedinjury.
There is also potential for a fibrosis induced fromspasticity to lead directly to a limitation of longitudinalgrowth. Satellite cells responsible for muscle growth relyon migration across the basement membrane duringactivation with the release of matrix metalloproteases(Chen & Li, 2009). Skeletal muscle fibrosis could impedemuscle regeneration by forming a mechanical barrier tothis process (Chen & Li, 2009). Stem cell differentiation isalso sensitive to the elasticity of the matrix in which it isembedded (Engler et al. 2006). Our study demonstratedan altered stiffness of the ECM in contracture tissue thatcould lead to an inhibition of satellite cell activation orproliferation (Boonen et al. 2009; Gilbert et al. 2010) andperhaps even predispose muscle stem cells to differentiatetoward the fibroblast lineage. It is also possible that fibrosisand lack of growth create a vicious cycle that leads tomuscle contracture.
Titin isoforms
Since titin isoform size is related to muscle passive tension(Prado et al. 2005), we hypothesized that shorter titinisoforms would be present in muscle contracture leadingto increased stiffness. However, our results show nodifference in titin size between CP and control muscles.Thus, we conclude that titin is not altered in CP to causecontracture, at least not in a manner that alters size. Titinisoform changes have been reported in cardiac disease(Neagoe et al. 2003), but literature on titin isoform changesin skeletal muscle is sparse. A previous study investigating
C! 2011 The Authors. Journal compilation C! 2011 The Physiological Society

J Physiol 589.10 Passive mechanical properties of muscle contracture 2637
titin isoform in spastic muscles of stroke patients alsofound no change in isoform size (Olsson et al. 2006).
Fibre mechanics
While titin isoform size contributes to single fibremechanics, other proteins or organization of fibre materialcould be responsible for an increased passive tension atthe cellular level. Two previous studies did demonstratestiffer muscle fibres for spastic patients. These studies eachhad important differences, however. In one the musclefibres tested were from a range of muscles which wasnot the same in the spastic and control groups (Friden& Lieber, 2003). This is a confounding issue because itis known that different muscles have different passivemechanical properties (Prado et al. 2005). Another showedincreased stiffness only in fast fibres with an increase inthe proportion of fast fibres (Olsson et al. 2006). Wedid not see a corresponding shift to fast fibres of ourmuscles, and in fact showed a significant increase in typeI myosin heavy chain of CP muscles indicating a shift toslower fibres (Fig. 8). Overall, previous studies have showndisagreement on whether spastic muscles gain a faster orslower phenotype, which could be muscle specific (Brooke& Engel, 1969; Scelsi et al. 1984; Jakobsson et al. 1991).However, the most straightforward interpretation of ourdata is that over-activity resulting from spasticity drivesa shift to slower fibres. It is also important to note thatthe previous study was performed with vastus lateralismuscle biopsies (Olsson et al. 2006), which show muchless spasticity and contracture development comparedto the medial hamstrings studied here (Damiano et al.2002; Pierce et al. 2008). Future studies across a widerrange of muscles are required to ultimately resolve theseambiguities.
Study limitations
One important limitation of this study is the subjectheterogeneity. CP is a spectrum disorder and here wehave primarily examined only the commonly sharedparameters of this range of subjects with spastic CP.A more detailed analysis could be attempted usingpatient stratification by clinical parameters, Gross MotorFunction Classification System, popliteal angle, limbsaffected, age and treatment regimens. However, due tolimitations of the sample size and the high variabilityof parameters in working with human subjects, this wasnot possible. We were able to demonstrate a significantcorrelation between severity measures and sarcomerelength, which helped to mitigate the fact that we are relyingon model data for our comparison with in vivo sarcomerelengths control children.
The source of controls for this study is not idealsince the patients had sustained an ACL tear. However,these patients were several months removed from theinjury and had normal mobility at the time of surgery.Our approach represents the best available source ofnormal hamstring muscle from a pediatric population.These subject groups were not perfectly age-matched,although they all came from a pediatric population, asACL surgeries do not occur prior to the teenage yearswhereas hamstring-lengthening surgeries often occurmuch earlier. However, our passive mechanical data didnot correlate with age, suggesting that these small agediscrepancies did not affect our outcomes. The subjectsalso underwent varied previous treatments, of whichprevious hamstring-lengthening surgery and botulinumtoxin injections into the hamstrings was tracked. Thesevariables were analysed in relation to mechanical measuresand collagen content, but no relationship was found.These treatment effects are further complicated by highlyvariable times since treatment.
Summary
It is known that muscle contractures result from the UMNlesion in CP. Here, using a larger and more controlledstudy then previous ones, we showed increased passivestiffness of fibre bundles and increased sarcomere length invivo. Together, these properties create a muscle in CP thatexperiences much higher stresses with increasing musclelength and clearly contributes to the development ofmuscle and joint contractures. Future studies are requiredto understand the mechanistic basis for the sarcomerelength change and increased ECM content in CP as theseclearly represent targets for therapy.
References
Arnold EM, Ward SR, Lieber RL & Delp SL (2010). A model ofthe lower limb for analysis of human movement. AnnBiomed Eng 38, 269–279.
Bache CE, Selber P & Graham HK (2003). (ii) The managementof spastic diplegia. Current Orthopaedics 17, 88–104.
Baskin RJ, Roos KP & Yeh Y (1979). Light diffraction study ofsingle skeletal muscle fibres. Biophys J 28, 45–64.
Beals RK (2001). Treatment of knee contracture in cerebralpalsy by hamstring lengthening, posterior capsulotomy, andquadriceps mechanism shortening. Dev Med Child Neurol43, 802–805.
Bendall JR (1967). The elastin content of various muscles ofbeef animals. J Sci Food Agriculture 18, 553–558.
Boonen KJ, Rosaria-Chak KY, Baaijens FP, Van Der Schaft DW& Post MJ (2009). Essential environmental cues from thesatellite cell niche: optimizing proliferation anddifferentiation. Am J Physiol Cell Physiol 296, C1338–C1345.
C! 2011 The Authors. Journal compilation C! 2011 The Physiological Society

2638 L. R. Smith and others J Physiol 589.10
Booth CM, Cortina-Borja MJ & Theologis TN (2001). Collagenaccumulation in muscles of children with cerebral palsy andcorrelation with severity of spasticity. Dev Med Child Neurol43, 314–320.
Brooke MH & Engel WK (1969). The histographic analysis ofhuman muscle biopsies with regard to fiber types. 2. Diseasesof the upper and lower motor neuron. Neurology 19,378–393.
Chen X & Li Y (2009). Role of matrix metalloproteinases inskeletal muscle: migration, differentiation, regeneration andfibrosis. Cell Adh Migr 3, 337–341.
Conway D & Sakai T (1960). Caffeine contracture. Proc NatlAcad Sci U S A 46, 897–903.
Crenna P (1998). Spasticity and ‘spastic’ gait in children withcerebral palsy. Neurosci Biobehav Rev 22, 571–578.
Damiano DL, Quinlivan JM, Owen BF, Payne P, Nelson KC &Abel MF (2002). What does the Ashworth scale reallymeasure and are instrumented measures more valid andprecise? Dev Med Child Neurol 44, 112–118.
Elder GC, Kirk J, Stewart G, Cook K, Weir D, Marshall A &Leahey L (2003). Contributing factors to muscle weakness inchildren with cerebral palsy. Dev Med Child Neurol 45,542–550.
Engler AJ, Sen S, Sweeney HL & Discher DE (2006). Matrixelasticity directs stem cell lineage specification. Cell 126,677–689.
Farmer SE & James M (2001). Contractures in orthopaedic andneurological conditions: a review of causes and treatment.Disabil Rehabil 23, 549–558.
Fergusson D, Hutton B & Drodge A (2007). The epidemiologyof major joint contractures: a systematic review of theliterature. Clin Orthop Relat Res 456, 22–29.
Freiburg A, Trombitas K, Hell W, Cazorla O, Fougerousse F,Centner T, Kolmerer B, Witt C, Beckmann JS, Gregorio CC,Granzier H & Labeit S (2000). Series of exon-skipping eventsin the elastic spring region of titin as the structural basis formyofibrillar elastic diversity. Circ Res 86, 1114–1121.
Friden J & Lieber RL (2003). Spastic muscle cells are shorterand stiffer than normal cells. Muscle Nerve 27,157–164.
Gilbert PM, Havenstrite KL, Magnusson KE, Sacco A, LeonardiNA, Kraft P, Nguyen NK, Thrun S, Lutolf MP & Blau HM(2010). Substrate elasticity regulates skeletal muscle stem cellself-renewal in culture. Science 329, 1078–1081.
Gordon AM, Huxley AF & Julian FJ (1966). The variation inisometric tension with sarcomere length in vertebrate musclefibres. J Physiol 184, 170–192.
Hodgkin AL & Horowicz P (1960). Potassium contractures insingle muscle fibres. J Physiol 153, 386–403.
Jakobsson F, Edstrom L, Grimby L & Thornell LE (1991).Disuse of anterior tibial muscle during locomotion andincreased proportion of type II fibres in hemiplegia. J NeurolSci 105, 49–56.
Janice MQ & Alwyn A (2005). Musculoskeletal problems incerebral palsy. Current Paediatrics 15, 9–14.
Kerr Graham H & Selber P (2003). Musculoskeletal aspects ofcerebral palsy. J Bone Joint Surg Br 85, 157–166.
Labeit S & Kolmerer B (1995). Titins: giant proteins in chargeof muscle ultrastructure and elasticity. Science 270, 293–296.
Li Y, Foster W, Deasy BM, Chan Y, Prisk V, Tang Y, Cummins J& Huard J (2004). Transforming growth factor-beta1 inducesthe differentiation of myogenic cells into fibrotic cells ininjured skeletal muscle: a key event in muscle fibrogenesis.Am J Pathol 164, 1007–1019.
Li ZB, Kollias HD & Wagner KR (2008). Myostatin directlyregulates skeletal muscle fibrosis. J Biol Chem 283,19371–19378.
Lieber RL, Yeh Y & Baskin RJ (1984). Sarcomere lengthdetermination using laser diffraction. Effect of beam andfiber diameter. Biophys J 45, 1007–1016.
Lieber RL & Friden J (2002). Spasticity causes a fundamentalrearrangement of muscle-joint interaction. Muscle Nerve 25,265–270.
Lieber RL, Runesson E, Einarsson F & Friden J (2003). Inferiormechanical properties of spastic muscle bundles due tohypertrophic but compromised extracellular matrixmaterial. Muscle Nerve 28, 464–471.
Lorentzen J, Grey MJ, Crone C, Mazevet D, Biering-Sorensen F& Nielsen JB (2010). Distinguishing active from passivecomponents of ankle plantar flexor stiffness in stroke, spinalcord injury and multiple sclerosis. Clin Neurophysiol 121,1939–1951.
Lukban MB, Rosales RL & Dressler D (2009). Effectiveness ofbotulinum toxin A for upper and lower limb spasticity inchildren with cerebral palsy: a summary of evidence. J NeuralTransm 116, 319–331.
Malaiya R, McNee AE, Fry NR, Eve LC, Gough M & ShortlandAP (2007). The morphology of the medial gastrocnemius intypically developing children and children with spastichemiplegic cerebral palsy. J Electromyogr Kinesiol 17,657–663.
Mirbagheri MM, Barbeau H, Ladouceur M & Kearney RE(2001). Intrinsic and reflex stiffness in normal and spastic,spinal cord injured subjects. Exp Brain Res 141,446–459.
Mohagheghi AA, Khan T, Meadows TH, Giannikas K,Baltzopoulos V & Maganaris CN (2007). Differences ingastrocnemius muscle architecture between the pareticand non-paretic legs in children with hemiplegiccerebral palsy. Clin Biomech (Bristol, Avon) 22,718–724.
Mohagheghi AA, Khan T, Meadows TH, Giannikas K,Baltzopoulos V & Maganaris CN (2008). In vivogastrocnemius muscle fascicle length in children with andwithout diplegic cerebral palsy. Dev Med Child Neurol 50,44–50.
Moreau NG, Simpson KN, Teefey SA & Damiano DL (2010).Muscle architecture predicts maximum strength and isrelated to activity levels in cerebral palsy. Phys Ther 90,1619–1630.
Neagoe C, Opitz CA, Makarenko I & Linke WA (2003).Gigantic variety: expression patterns of titin isoforms instriated muscles and consequences for myofibrillarpassive stiffness. J Muscle Res Cell Motil 24,175–189.
O’Dwyer NJ, Ada L & Neilson PD (1996). Spasticity andmuscle contracture following stroke. Brain 119,1737–1749.
C! 2011 The Authors. Journal compilation C! 2011 The Physiological Society

J Physiol 589.10 Passive mechanical properties of muscle contracture 2639
Olsson MC, Kruger M, Meyer LH, Ahnlund L, Gransberg L,Linke WA & Larsson L (2006). Fibre type-specific increase inpassive muscle tension in spinal cord-injured subjects withspasticity. J Physiol 577, 339–352.
Palisano R, Rosenbaum P, Walter S, Russell D, Wood E &Galuppi B (1997). Development and reliability of a system toclassify gross motor function in children with cerebral palsy.Dev Med Child Neurol 39, 214–223.
Patel TJ, Das R, Friden J, Lutz GJ & Lieber RL (2004).Sarcomere strain and heterogeneity correlate with injury tofrog skeletal muscle fiber bundles. J Appl Physiol 97,1803–1813.
Pierce SR, Barbe MF, Barr AE, Shewokis PA & Lauer RT (2008).Roles of reflex activity and co-contraction duringassessments of spasticity of the knee flexor and knee extensormuscles in children with cerebral palsy and differentfunctional levels. Phys Ther 88, 1124–1134.
Ponten E, Gantelius S & Lieber RL (2007). Intraoperativemuscle measurements reveal a relationship betweencontracture formation and muscle remodeling. Muscle Nerve36, 47–54.
Prado LG, Makarenko I, Andresen C, Kruger M, Opitz CA &Linke WA (2005). Isoform diversity of giant proteins inrelation to passive and active contractile properties of rabbitskeletal muscles. J Gen Physiol 126, 461–480.
Proske U & Morgan DL (2001). Muscle damage from eccentricexercise: mechanism, mechanical signs, adaptation andclinical applications. J Physiol 537, 333–345.
Purslow PP (1989). Strain-induced reorientation of anintramuscular connective tissue network: implications forpassive muscle elasticity. J Biomech 22, 21–31.
Rose J & McGill KC (2005). Neuromuscular activation andmotor-unit firing characteristics in cerebral palsy. Dev MedChild Neurol 47, 329–336.
Rosenbaum P, Paneth N, Leviton A, Goldstein M, Bax M,Damiano D, Dan B & Jacobsson B (2007). A report: thedefinition and classification of cerebral palsy April 2006. DevMed Child Neurol Suppl 109, 8–14.
Savage AO & Atanga KG (1988). Caffeine- andpotassium-induced contractures of mouse isolated soleusmuscle: effects of verapamil, manganese, EGTA and calciumwithdrawal. Clin Exp Pharmacol Physiol 15, 901–911.
Scelsi R, Lotta S, Lommi G, Poggi P & Marchetti C (1984).Hemiplegic atrophy. Morphological findings in the anteriortibial muscle of patients with cerebral vascular accidents.Acta Neuropathol 62, 324–331.
Shortland AP, Harris CA, Gough M & Robinson RO (2002).Architecture of the medial gastrocnemius in children withspastic diplegia. Dev Med Child Neurol 44, 158–163.
Sinkjaer T & Magnussen I (1994). Passive, intrinsic andreflex-mediated stiffness in the ankle extensors ofhemiparetic patients. Brain 117, 355–363.
Smith LR, Ponten E, Hedstrom Y, Ward SR, Chambers HG,Subramaniam S & Lieber RL (2009). Novel transcriptionalprofile in wrist muscles from cerebral palsy patients. BMCMed Genomics 2, 44.
Stackhouse SK, Binder-Macleod SA & Lee SC (2005).Voluntary muscle activation, contractile properties, andfatigability in children with and without cerebral palsy.Muscle Nerve 31, 594–601.
Stauber WT (2004). Factors involved in strain-induced injuryin skeletal muscles and outcomes of prolonged exposures. JElectromyogr Kinesiol 14, 61–70.
Tilton AH (2006). Therapeutic interventions for toneabnormalities in cerebral palsy. NeuroRx 3, 217–224.
Walker SM & Schrodt GR (1974). I segment lengths and thinfilament periods in skeletal muscle fibers of the Rhesusmonkey and the human. Anat Rec 178, 63–81.
Ward SR & Lieber RL (2005). Density and hydration of freshand fixed human skeletal muscle. J Biomech 38, 2317–2320.
Ward SR, Takahashi M, Winters TM, Kwan A & Lieber RL(2009a). A novel muscle biopsy clamp yields accurate in vivosarcomere length values. J Biomech 42, 193–196.
Ward SR, Tomiya A, Regev GJ, Thacker BE, Benzl RC, Kim CW& Lieber RL (2009b). Passive mechanical properties of thelumbar multifidus muscle support its role as a stabilizer.J Biomech 42, 1384–1389.
Warren CM, Krzesinski PR & Greaser ML (2003). Verticalagarose gel electrophoresis and electroblotting ofhigh-molecular-weight proteins. Electrophoresis 24,1695–1702.
Wiart L, Darrah J & Kembhavi G (2008). Stretching withchildren with cerebral palsy: what do we know and where arewe going? Pediatr Phys Ther 20, 173–178.
Williams PE & Goldspink G (1973). The effect ofimmobilization on the longitudinal growth of striatedmuscle fibres. J Anat 116, 45–55.
Yeargin-Allsopp M, Van Naarden Braun K, Doernberg NS,Benedict RE, Kirby RS & Durkin MS (2008). Prevalence ofcerebral palsy in 8-year-old children in three areas of theUnited States in 2002: a multisite collaboration. Pediatrics121, 547–554.
Author contributions
Study conception and design: L.R.S., S.R.W., H.G.C., R.L.L.Sample collection: L.R.S., K.S.L., H.G.C. Data analysis: L.R.S.Manuscript preparation: L.R.S., R.L.L. Manuscript editing:L.R.S, K.S.L., S.R.W., H.G.C, R.L.L. All authors approvedthe final version for publication. This work was completedat University of California, San Diego and Rady Children’sHospital, San Diego, CA, USA.
Acknowledgements
This work was supported by grants from the National Instituteof Health (AR057393), the Department of Veterans Affairs andthe Department of Defense (DoD) through the National DefenseScience & Engineering Graduate Fellowship (NDSEG) Program.We also acknowledge Dr Eric Edmonds for assistance collectingbiopsies, Gretchen Meyer for assistance in analysis of mechanicaldata, Austin Carr for assistance with titin and MyHC gels, RandyGastwirt for assistance with the hydroxyproline assay, VeraDebelynska for assistance with histology, and Shannon Bremnerfor technical assistance. We thank Dr. Scott Delp and EdithArnold for making control sarcomere length calculations. Theauthors have no conflicts of interest regarding the publication ofthis work.
C! 2011 The Authors. Journal compilation C! 2011 The Physiological Society





























