Embed Size (px)
Citation preview

International Journal of Biological Sciences and Applications
2015; 2(5): 48-66
Published online October 20, 2015 (http://www.aascit.org/journal/ijbsa)
ISSN: 2375-3811
Keywords Natural products,
Phosphonic Esters,
Phosphinic Acids,
2-AEP, Bialaphos,
Fosfomycin,
Phosphinothricin,
FR900098,
Phosphatidylethanolamine
Received: September 28, 2015
Revised: October 15, 2015
Accepted: October 17, 2015
Biosynthesis of Natural Products Containing Phosphonic Esters and Acids: Review
Azza A. Kamel
Chemical Industries Division, National Research Centre (NRC), Elbohouth Street, Dokki, Cairo,
Egypt
Email address [email protected]
Citation Azza A. Kamel. Biosynthesis of Natural Products Containing Phosphonic Esters and Acids:
Review. International Journal of Biological Sciences and Applications.
Vol. 2, No. 5, 2015, pp. 48-66.
Abstract Natural products containing carbon-phosphorus bond (phosphonic esters and phosphinic
acids) have found widespread use in medicine and agriculture. As such, natural products
are the active components not only of most traditional medicines but also many modern
medicines. Furthermore, because the structural diversity of natural products exceeds that
readily achievable by chemical synthesis, and synthetic analogs can be prepared with
improved potency and safety, natural products are often used as starting points for drug
discovery. In fact, natural products are the inspiration for approximately one half of U.S.
Food and Drug Administration (FDA)-approved drugs. Recent years have seen a renewed
interest in the biochemistry and biology of these compounds with the cloning of the
biosynthetic gene clusters for several family members. This article aims to assay the
classification and biosynthesis of some natural products containing C-P bond. The current
knowledge regarding the metabolic pathways and enzymes involved in the production of a
number of natural products containing C-P bond (6-representative examples) was briefly
scrutinized. These compounds including: 2-AEP, bialaphos, fosfomycin, phosphinothricin,
FR900098, and phosphatidylethanolamine. The emphasis is on the recent information
added to this topic. Although the author has attempted the review to be encyclopedic with
respect to the topic, the article is not exhaustive.
1. Introduction
Since very ancient ages, man used plants and then crude extracts in the treatment of
diseases, as poisons, toxins, hormones, and stimulants. Recently, scientists have been
interested in both structure elucidation and synthesis of natural products, isolated from
plants, animals, insects, or lower organisms. The ability to extend nature's chemistry
through combinatorial biosynthesis—altering functional groups, regiochemistry and
scaffold backbones through the manipulation of biosynthetic enzymes—offers unique
opportunities to create natural product analogs [1].
A natural product is a chemical compound or substance produced by a living organism
that is found in nature. In the broadest sense, natural products include any substance
produced by life. Natural products can also be prepared by chemical synthesis (both
semi-synthesis and total synthesis) and have played a central role in the development of
the field of organic chemistry by providing challenging synthetic targets. The term natural
product has also been extended for commercial purposes to refer to cosmetics, dietary
supplements, and foods produced from natural sources without added artificial ingredients
[2-4].

49 Azza A. Kamel: Biosynthesis of Natural Products Containing Phosphonic Esters and Acids: Review
Chemistry of natural products is the branch of organic
chemistry that deals with the study of organic compounds
isolated from natural sources, such as plants and animals. This
science comprises extraction, isolation, structure elucidation,
synthesis and biogenesis. The work in this field is divided into
two major branches: a) the isolation and structure elucidation
of natural products; b) the synthesis of natural products [2].
Natural products containing carbon-phosphorus bond
(phosphonic and phosphinic acids) have found widespread use
in medicine and agriculture as phosphorus plays a
fundamental role in the microbial cell from the physiological
and biochemical points of view [2,3]. It is suggested [4] that
phosphonates, as well as phosphites, emerged at an early stage
of the Earth's evolution and could be prebiotic phosphorus
carriers. They are usually small molecules characterized by
the presence of a stable covalent C–P bond and have been
isolated from a wide variety of external life forms.
Furthermore, a significant percentage of biogenic phosphorus
is believed to occur in the C–P linkage. However, more and
more data show the ability of bacteria to utilize the more
reduced organophosphorus compounds as phosphorus sources,
in particular, phosphonates characterized by a single
carbon-to-phosphorus (C-P) bond. In contrast to the more
labile O-P, N-P, and S-P linkages, the C-P bond is extremely
resistant to chemical hydrolysis, thermal decomposition, and
photolysis. Only prokaryotic microorganisms are able to
cleave this bond [5].
To date, only a few number of the complete phosphonate
biosynthetic pathways has been unrevealed. In this article six
representative examples of natural products containing C-P
bond are shed light including 2-AEP, bialaphos, fosfomycin,
phosphinothricin, FR900098, and phosphatidylethanolamine.
2. Classification of Natural Products
Fig. 1. Biosynthesis of primary and secondary metabolites (From Wikipedia, the free encyclopedia).
International Journal of Biological Sciences and Applications 2015; 2(5): 48-66 50
Following Albrecht Kossel's original proposal in 1891 [6],
natural products are often divided into two major classes, the
primary and secondary metabolites [7,8] .
In contrast, secondary metabolites have an extrinsic
function that mainly affects other organisms.
Secondary metabolites are not essential to survival but do
increase the competitiveness of the organism within its
environment. Because of their ability to modulate biochemical
and signal transduction pathways, some secondary
metabolites have useful medicinal properties.
Natural products especially within the field of organic
chemistry are often defined as primary and secondary
metabolites (Fig. 1). A more restrictive definition limiting
natural products to secondary metabolites is commonly used
within the fields of medicinal chemistry and pharmacognosy
[8].
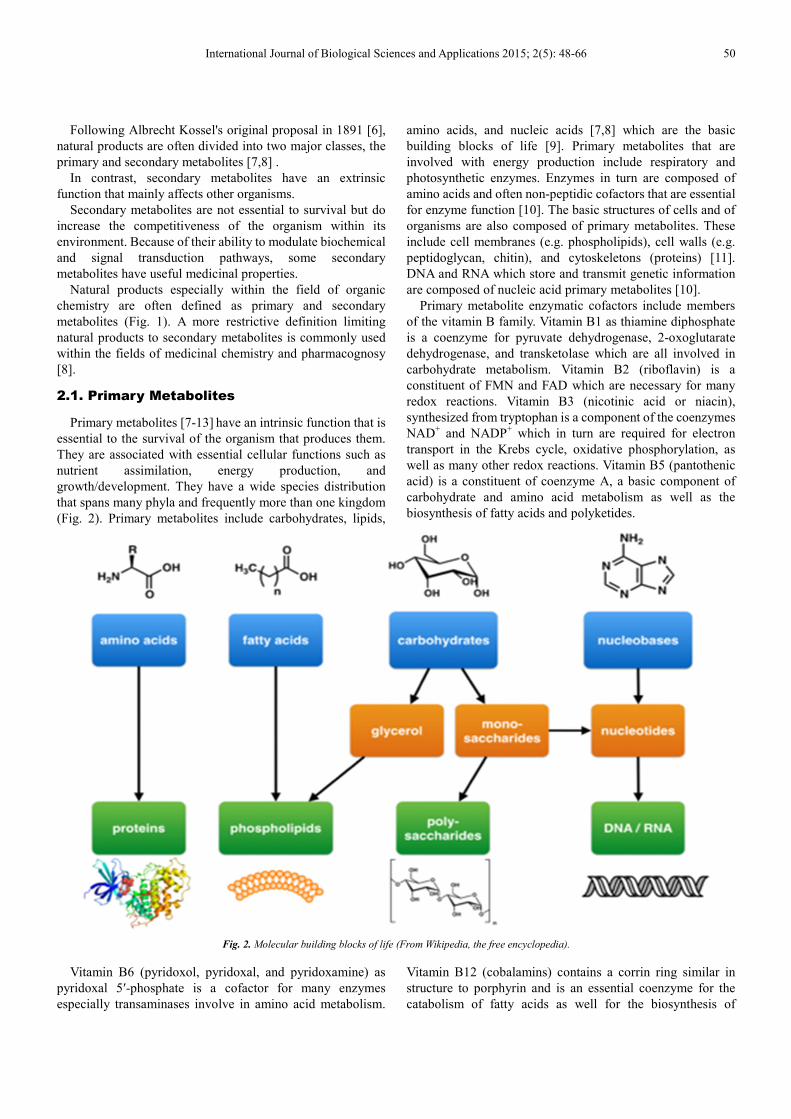
2.1. Primary Metabolites
Primary metabolites [7-13] have an intrinsic function that is
essential to the survival of the organism that produces them.
They are associated with essential cellular functions such as
nutrient assimilation, energy production, and
growth/development. They have a wide species distribution
that spans many phyla and frequently more than one kingdom
(Fig. 2). Primary metabolites include carbohydrates, lipids,
amino acids, and nucleic acids [7,8] which are the basic
building blocks of life [9]. Primary metabolites that are
involved with energy production include respiratory and
photosynthetic enzymes. Enzymes in turn are composed of
amino acids and often non-peptidic cofactors that are essential
for enzyme function [10]. The basic structures of cells and of
organisms are also composed of primary metabolites. These
include cell membranes (e.g. phospholipids), cell walls (e.g.
peptidoglycan, chitin), and cytoskeletons (proteins) [11].
DNA and RNA which store and transmit genetic information
are composed of nucleic acid primary metabolites [10].
Primary metabolite enzymatic cofactors include members
of the vitamin B family. Vitamin B1 as thiamine diphosphate
is a coenzyme for pyruvate dehydrogenase, 2-oxoglutarate
dehydrogenase, and transketolase which are all involved in
carbohydrate metabolism. Vitamin B2 (riboflavin) is a
constituent of FMN and FAD which are necessary for many
redox reactions. Vitamin B3 (nicotinic acid or niacin),
synthesized from tryptophan is a component of the coenzymes
NAD+ and NADP
+ which in turn are required for electron
transport in the Krebs cycle, oxidative phosphorylation, as
well as many other redox reactions. Vitamin B5 (pantothenic
acid) is a constituent of coenzyme A, a basic component of
carbohydrate and amino acid metabolism as well as the
biosynthesis of fatty acids and polyketides.
Fig. 2. Molecular building blocks of life (From Wikipedia, the free encyclopedia).
Vitamin B6 (pyridoxol, pyridoxal, and pyridoxamine) as
pyridoxal 5′-phosphate is a cofactor for many enzymes
especially transaminases involve in amino acid metabolism.
Vitamin B12 (cobalamins) contains a corrin ring similar in
structure to porphyrin and is an essential coenzyme for the
catabolism of fatty acids as well for the biosynthesis of

51 Azza A. Kamel: Biosynthesis of Natural Products Containing Phosphonic Esters and Acids: Review
methionine [12].
First messengers are signaling molecules that control
metabolism or cellular differentiation. These signaling
molecules include hormones and growth factors in turn are
composed of peptides, biogenic amines, steroid hormones,
auxins, gibberellins, etc. These first messengers interact with
cellular receptors which are composed of proteins. Cellular
receptors in turn activate second messengers are used to relay
the extracellular message to intracellular targets. These
signaling molecules include the primary metabolites cyclic
nucleotides, diacyl glyceroletc [13].
2.2. Secondary Metabolites
In contrast to primary metabolites, secondary metabolites
[14,15]
are dispensable and not absolutely required for
survival. Furthermore, they typically have a narrow species
distribution (Fig. 3). Some secondary metabolites have a
broad range of functions. These include pheromones that act
as social signaling molecules with other individuals of the
same species, communication molecules that attract and
activate symbiotic organisms, agents that solubilize and
transport nutrients (siderophores etc.), and competitive
weapons (repellants, venoms, toxins etc.) that are used against
competitors, prey, and predators [14]. For many other
secondary metabolites, the function is unknown. One
hypothesis is that they confer a competitive advantage to the
organism that produces them. An alternative view is that, in
analogy to the immune system, these secondary metabolites
have no specific function, but having the machinery in place to
produce these diverse chemical structures is important and a
few secondary metabolites are therefore produced and
selected for [15].
Fig. 3. Representative examples of each of the major classes of secondary metabolites (From Wikipedia, the free encyclopedia).
3. Classification of Natural Products
Containing C-P Bond
3.1. Phospholipids
Phospholipids [16] are fat derivatives in which one fatty
acid has been replaced by either a phosphonate group or a
phosphate group and one of several nitrogen-containing
molecules. 2-Aminoethylphosphonic acid (2- AEP) is an
example for phospholipids containing a phosphonate group.
Whereas, examples for phospholipids containing a phosphate
group include phosphatidyl ethanolamine (also known as
Cephalin, represented in Fig. 4); phosphatidylcholine;
Lecithin and Phosphocholine.
Fig. 4. Structure of phosphatidyl ethanolamine (From Wikipedia, the free
encyclopedia).
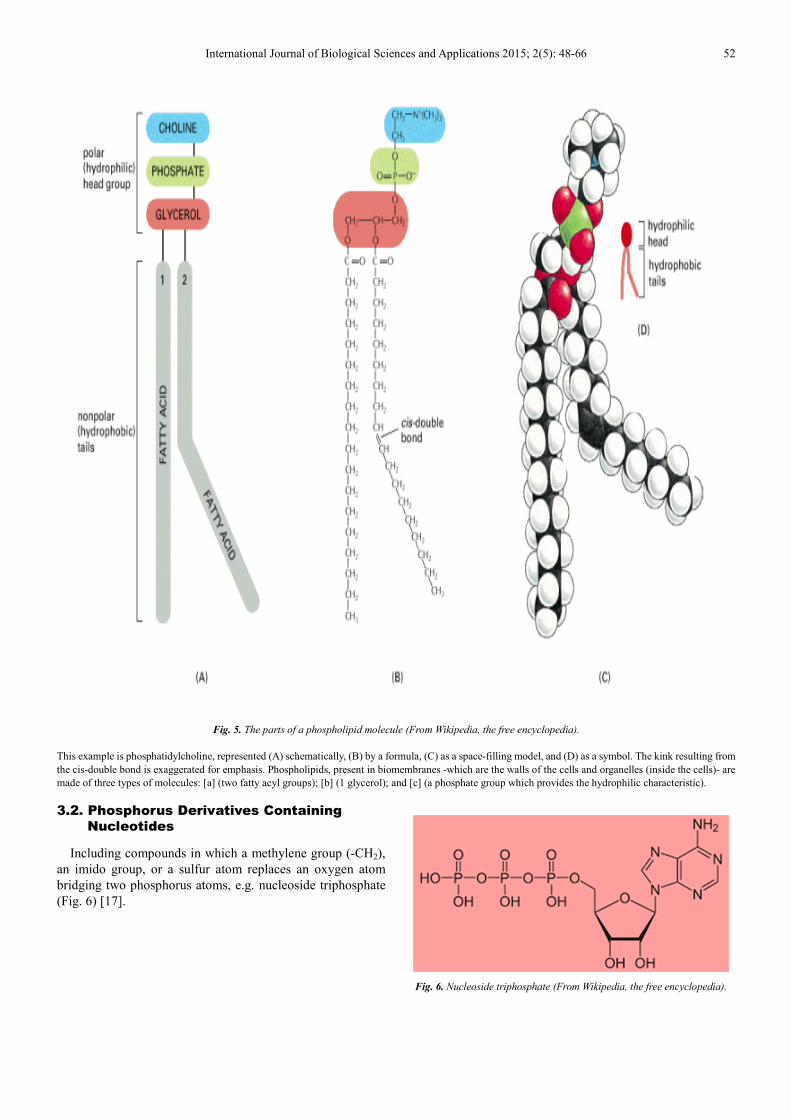
A phospholipid, (as shown in Fig. 5) looks like a bubble
with two long chains attached. There are three major kinds of
lipids in living organisms, one of them are phospholipids as
demonstrated in Fig. 5.
International Journal of Biological Sciences and Applications 2015; 2(5): 48-66 52
Fig. 5. The parts of a phospholipid molecule (From Wikipedia, the free encyclopedia).
This example is phosphatidylcholine, represented (A) schematically, (B) by a formula, (C) as a space-filling model, and (D) as a symbol. The kink resulting from
the cis-double bond is exaggerated for emphasis. Phospholipids, present in biomembranes -which are the walls of the cells and organelles (inside the cells)- are
made of three types of molecules: [a] (two fatty acyl groups); [b] (1 glycerol); and [c] (a phosphate group which provides the hydrophilic characteristic).
3.2. Phosphorus Derivatives Containing
Nucleotides
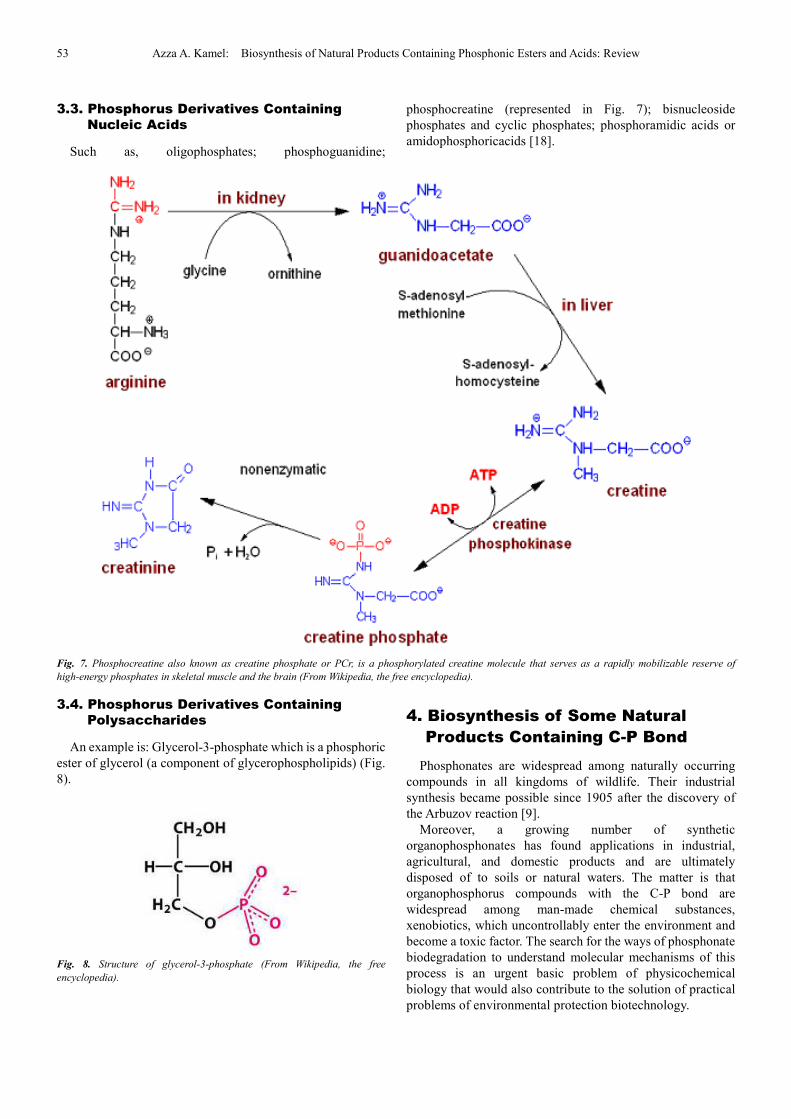
Including compounds in which a methylene group (-CH2),
an imido group, or a sulfur atom replaces an oxygen atom
bridging two phosphorus atoms, e.g. nucleoside triphosphate
(Fig. 6) [17].
Fig. 6. Nucleoside triphosphate (From Wikipedia, the free encyclopedia).
53 Azza A. Kamel: Biosynthesis of Natural Products Containing Phosphonic Esters and Acids: Review
3.3. Phosphorus Derivatives Containing
Nucleic Acids
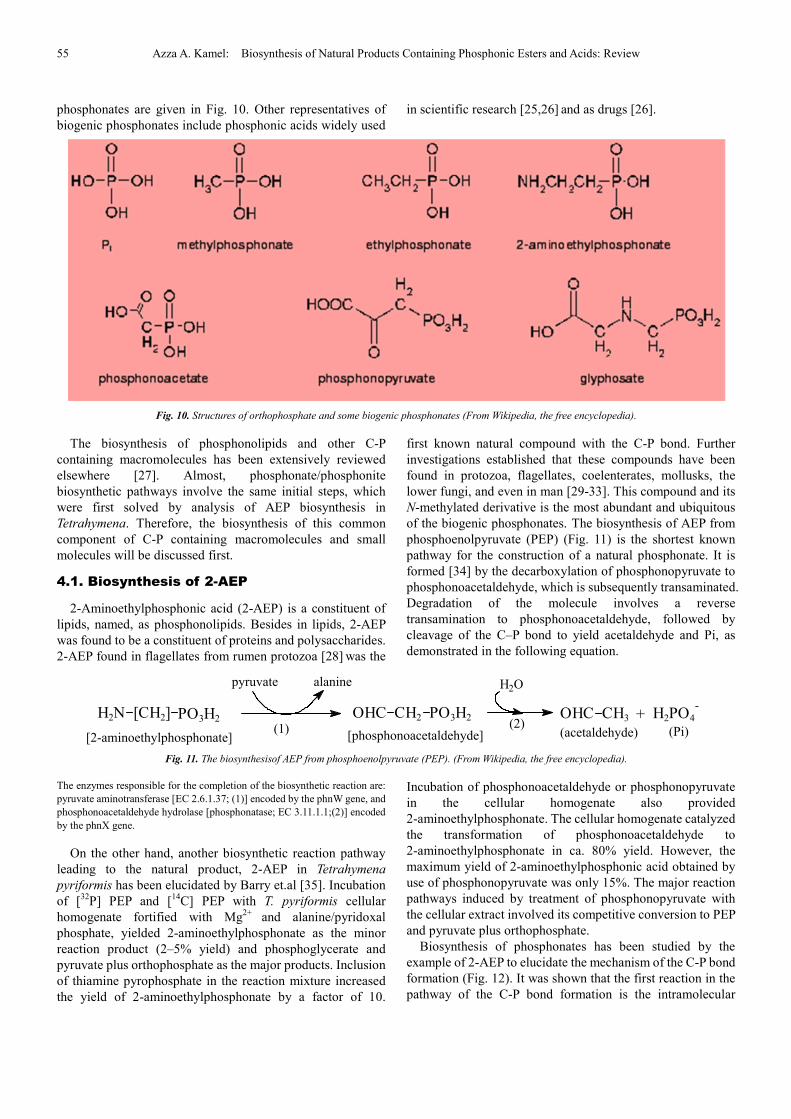
Such as, oligophosphates; phosphoguanidine;
phosphocreatine (represented in Fig. 7); bisnucleoside
phosphates and cyclic phosphates; phosphoramidic acids or
amidophosphoricacids [18].
Fig. 7. Phosphocreatine also known as creatine phosphate or PCr, is a phosphorylated creatine molecule that serves as a rapidly mobilizable reserve of
high-energy phosphates in skeletal muscle and the brain (From Wikipedia, the free encyclopedia).
3.4. Phosphorus Derivatives Containing
Polysaccharides
An example is: Glycerol-3-phosphate which is a phosphoric
ester of glycerol (a component of glycerophospholipids) (Fig.
8).
Fig. 8. Structure of glycerol-3-phosphate (From Wikipedia, the free
encyclopedia).
4. Biosynthesis of Some Natural
Products Containing C-P Bond
Phosphonates are widespread among naturally occurring
compounds in all kingdoms of wildlife. Their industrial
synthesis became possible since 1905 after the discovery of
the Arbuzov reaction [9].
Moreover, a growing number of synthetic
organophosphonates has found applications in industrial,
agricultural, and domestic products and are ultimately
disposed of to soils or natural waters. The matter is that
organophosphorus compounds with the C-P bond are
widespread among man-made chemical substances,
xenobiotics, which uncontrollably enter the environment and
become a toxic factor. The search for the ways of phosphonate
biodegradation to understand molecular mechanisms of this
process is an urgent basic problem of physicochemical
biology that would also contribute to the solution of practical
problems of environmental protection biotechnology.

International Journal of Biological Sciences and Applications 2015; 2(5): 48-66 54
The ability to degrade phosphonates is relatively
widespread, occurring in Gram-positive and Gram-negative
bacteria as well as in fungi. Three classes of enzyme capable
of breaking the C-P bond of phosphonates are known:
PA-hydrolase, an enzyme specific for PA breakdown;
phosphonatase, which specifically degrades 2-AEP; and
C-P-lyase, which cleaves the C-P bond in a broad spectrum of
phosphonates. The C-P-lyase activity can be detected in the
whole organisms; nevertheless, it has never been convincingly
assayed in cell extracts, and this has limited attempts to
understand the mechanism of the enzyme, which has been
suggested to involve a redox-dependent free radical
mechanism. The C–P-lyase complex has a broad substrate
specificity [19] and can act upon unsubstituted alkyl and aryl
organophosphonates (general formula R–PO3H2). The two C–
P hydrolases that act on phosphonoacetaldehyde (OHC–CH2–
PO3H2) and phosphonoacetate (HOOC–CH2–PO3H2) are
essentially specific to their respective substrates (Fig. 9) [20].
Fig. 9. The hydrolytic cleavage of the C–P bond of phosphonopyruvate (McGrath, 2013 [20]).
A fourth C–P bond-metabolizing enzyme,
phosphoenolpyruvate phosphomutase (PEP mutase),
catalyzes the intramolecular rearrangement and
interconversion of 3-phosphonopyruvate (COOH–CO–CH2–
PO3H2) and phosphoenolpyruvate (COOH–C(=CH2)–O–
PO3H2). Phosphonopyruvate formation by this route is a key
step in the biosynthesis of all known natural products that
contain the C–P bond [21].
One of the most widely distributed of such biogenic C–P
compounds is 2-amino-3-phosphonopropionic acid,
commonly called phosphonoalanine (HOOC–CH-(NH2)–
CH2–PO3H2); it is formed through the transamination of
phosphonopyruvate by many lower organisms, such as the sea
anemone Zoanthussociatus and the protozoan Tetrahymena
pyriformis. The earlier studies on its biodegradation showed
that 47 of 100 randomly chosen environmental bacterial
isolates had the ability to utilize phosphonoalanine as a sole
phosphorus source for growth [22]. Subsequently, Ternan et al.
[23] reported the isolation of an environmental
Burkholderiacepacia strain capable of growth on
phosphonoalanine as sole source of carbon, nitrogen, and
phosphorus. Cell extracts of the isolate contained a previously
unknown Pi-independent activity designated
phosphonopyruvate hydrolase (PPH) that catalyzed the
hydrolytic cleavage of the C–P bond of phosphonopyruvate to
yield pyruvate and Pi (Fig. 9).
Other representatives of biogenic phosphonates are
antibiotics synthesized by Streptomyces. They include
phosphonomycin (1,2-cis-epoxyprolylphosphonic acid)
(known also as fosfomycin), an inhibitor of biosynthesis of
UDP-N-acetylmuramic acid essential for microbial cell wall
formation and bialaphos (L-alanyl-L-alanyl-phosphinothricin),
an inhibitor of glutamine synthetase in Escherichia coli and
plants. Biogenic phosphonates also include
phosphonopyruvateand phosphonoacetate (PA) [24].
The structures of some of the simplest biogenic
55 Azza A. Kamel: Biosynthesis of Natural Products Containing Phosphonic Esters and Acids: Review
phosphonates are given in Fig. 10. Other representatives of
biogenic phosphonates include phosphonic acids widely used
in scientific research [25,26] and as drugs [26].
Fig. 10. Structures of orthophosphate and some biogenic phosphonates (From Wikipedia, the free encyclopedia).
The biosynthesis of phosphonolipids and other C-P
containing macromolecules has been extensively reviewed
elsewhere [27]. Almost, phosphonate/phosphonite
biosynthetic pathways involve the same initial steps, which
were first solved by analysis of AEP biosynthesis in
Tetrahymena. Therefore, the biosynthesis of this common
component of C-P containing macromolecules and small
molecules will be discussed first.
4.1. Biosynthesis of 2-AEP
2-Aminoethylphosphonic acid (2-AEP) is a constituent of
lipids, named, as phosphonolipids. Besides in lipids, 2-AEP
was found to be a constituent of proteins and polysaccharides.
2-AEP found in flagellates from rumen protozoa [28] was the
first known natural compound with the C-P bond. Further
investigations established that these compounds have been
found in protozoa, flagellates, coelenterates, mollusks, the
lower fungi, and even in man [29-33]. This compound and its
N-methylated derivative is the most abundant and ubiquitous
of the biogenic phosphonates. The biosynthesis of AEP from
phosphoenolpyruvate (PEP) (Fig. 11) is the shortest known
pathway for the construction of a natural phosphonate. It is
formed [34] by the decarboxylation of phosphonopyruvate to
phosphonoacetaldehyde, which is subsequently transaminated.
Degradation of the molecule involves a reverse
transamination to phosphonoacetaldehyde, followed by
cleavage of the C–P bond to yield acetaldehyde and Pi, as
demonstrated in the following equation.
Fig. 11. The biosynthesisof AEP from phosphoenolpyruvate (PEP). (From Wikipedia, the free encyclopedia).
The enzymes responsible for the completion of the biosynthetic reaction are:
pyruvate aminotransferase [EC 2.6.1.37; (1)] encoded by the phnW gene, and
phosphonoacetaldehyde hydrolase [phosphonatase; EC 3.11.1.1;(2)] encoded
by the phnX gene.
On the other hand, another biosynthetic reaction pathway
leading to the natural product, 2-AEP in Tetrahymena
pyriformis has been elucidated by Barry et.al [35]. Incubation
of [32
P] PEP and [14
C] PEP with T. pyriformis cellular
homogenate fortified with Mg2+
and alanine/pyridoxal
phosphate, yielded 2-aminoethylphosphonate as the minor
reaction product (2–5% yield) and phosphoglycerate and
pyruvate plus orthophosphate as the major products. Inclusion
of thiamine pyrophosphate in the reaction mixture increased
the yield of 2-aminoethylphosphonate by a factor of 10.
Incubation of phosphonoacetaldehyde or phosphonopyruvate
in the cellular homogenate also provided
2-aminoethylphosphonate. The cellular homogenate catalyzed
the transformation of phosphonoacetaldehyde to
2-aminoethylphosphonate in ca. 80% yield. However, the
maximum yield of 2-aminoethylphosphonic acid obtained by
use of phosphonopyruvate was only 15%. The major reaction
pathways induced by treatment of phosphonopyruvate with
the cellular extract involved its competitive conversion to PEP
and pyruvate plus orthophosphate.
Biosynthesis of phosphonates has been studied by the
example of 2-AEP to elucidate the mechanism of the C-P bond
formation (Fig. 12). It was shown that the first reaction in the
pathway of the C-P bond formation is the intramolecular
[2-aminoethylphosphonate] [phosphonoacetaldehyde]
H2O
(2)H2N [CH2] PO3H2 OHC CH2 PO3H2
pyruvate alanine
(1)+
(acetaldehyde) (Pi)
OHC CH3 H2PO4
-

International Journal of Biological Sciences and Applications 2015; 2(5): 48-66 56
restructuring of phosphoenolpyruvate to phosphonopyruvate
(Fig. 12, III A), catalyzed by
phosphoenolpyruvate-phosphomutase. Phosphonopyruvate
under the action of enzyme
phosphonopyruvate-decarboxylase is then converted to
phosphonoacetaldehyde which is the key intermediate in the
biosynthesis formation of many other phosphonates.
Phosphonoacetaldehyde can be converted either to
hydroxymethylphosphonic acid during the bialophos
biosynthesis (described in Section 4.2.) or to
2-hydroxypropylphosphonic acid during the phosphonomycin
biosynthesis (described in Section 4.3.). The finding of
enzymes responsible for the C-P bond biosynthesis suggested
that under appropriate conditions in a cell they could also
realize back reactions, i.e., to participate in decomposition of
the C-P bond. Indeed, as it was shown later,
phosphoenolpyruvate-phosphomutase is involved in
biodegradation of phosphonoalanine. It is suggested that the
ability to synthesize phosphonates could give
microorganisms- due to the presence of the chemically stable
and phosphatase-resistant C-P bond in these compounds-
certain biological advantages for survival in a potentially
phosphate-limited marine environment [5].
Fig. 12. Enzymes cleaving the C-P bond (From Wikipedia, the free encyclopedia).
H2N
PO3H2
H CH3
+ P1
O
H
PO3H2
O2AEP-pyruvateaminotransferase Phosphonatase
Phosphonatase( i )
HO
PO3H2
O
HO CH3P1
O
+
( ii ) Phosphonoacetate hydrolase
2-aminoethylphosphonate 2-phophonoacetaldehyde acetaldehyde
phosphonacetate acetate
HOOC C
PO3H2
H2
CH2
HOOC C
PO3H2
H2
O
phsphoenolpyruvate phosphonpyruvate
( iii) Phosphonopyruvate hydrolase
Aphsphoenolpyruvate
mutase
B
HOOC C
PO3H2
H2
O
H3C COOH
O
+ P1
phosphonpyruvate
phosphonpyruvatehydrolase
pyruvate
R P
PO3H2
OH
OH
O
H3C R P1+
P1+C-P lyase
( IV ) C-P lyase
R = H, CH3, H2N CH2-
C-P lyase

57 Azza A. Kamel: Biosynthesis of Natural Products Containing Phosphonic Esters and Acids: Review
4.2. Biosynthesis of Bialaphos
Bialaphos (Fig.13):
(2S)-2-amino-4-[hydroxy(methyl)phosphinoyl]butyryl-L-ala
nyl-L-alanine; or (L-alanyl-L-alanyl-phosphinothricin), is a
natural herbicide produced by the bacteria Streptomyces
hygroscopicus [36] and Streptomyces viridochromogenes.
Bialaphos is a protoxin, non-toxic inhibitor of glutamine
synthetase in Escherichia coli and plants. When it is
metabolized by the plant, the glutamic acid analog
L-phosphinothricin is released which inhibits glutamine
synthetase. This results in the accumulation of ammonium and
disruption of primary metabolism [37]. Bialaphos is made up
of two alanine residues and phosphinothricin and is commonly
used as a gene sector in plants.
Fig. 13. Structure of bialaphos (From Wikipedia, the free encyclopedia).
The genes involved in the alanylation step in the
biosynthesis of a herbicide, bialaphos have been isolated and
studied by Nakashita et al [24]. It is produced by Streptomyces
hygroscopicus. Three bialaphos-non-producing mutants,
NP60, NP61 and NP62, isolated from S. hygroscopicus by
treatment with N-methyl-N'-nitro-N-nitrosoguanidine were
defective for the alanylation step and were not restored to
productivity by any locus of the gene cluster previously
cloned. Three plasmids were isolated using NP60, NP61 and
NP62 as recipients. The genes which restored productivity to
NP61 and NP62 hybridized to the contiguous region of the
bialaphos biosynthetic gene cluster. The gene cluster involved
in the bialaphos production was about 35 Kb long. The gene
which restored productivity to NP60 did not hybridize to the
bialaphos biosynthetic gene cluster. VM3 and VM4, putative
alanylation blocked mutants, were derived from a bialaphos
producer by gene replacement of an unidentified region of the
biosynthetic gene cluster with an in vitro altered DNA
sequence. The genes which restored productivity to VM3 and
VM4 were located between the genes which code for
phosphinomethylmalic acid synthase and
demethylphosphinothricin acetyltransferase in the cluster.
These results suggest that multiple genes are involved in the
alanylation step [38].
4.3. Biosynthesis of Fosfomycin
The research activities of the last years concentrated on
fosfomycin “referred also as phosphonomycin” (Fig. 14).
(1R,2S)-Epoxypropylphosphonic acid known as fosfomycin,
is a member of a class of compounds called phosphonic acids,
which is clinically utilized, highly effective antibiotic.
Fosfomycin is a low molecular weight natural product that
was originally isolated by Merck researchers from
Streptomyces fradiae, S. wedmorensis, and S.
viridochromogenes and was later shown to also be produced
by Pseudomonas syringae, and Pseudomonas viridiflava. In
the United States, fosfomycintromethamine
(tris(hydroxymethyl)aminomethane) is used under the name
Monurol® and is an FDA-approved drug that has become the
first choice for treatment of certain types of infections [39-44].
Attractively, fosfomycin has been proven effective for the
treatment of cephalosporin- and penicillin-resistant
Streptococcus pneumonia and is effective against
methicillin-resistant [41] and vancomycin-resistant [42]
strains of Staphylococcus aureus. In other countries like Japan,
Germany, Spain and France the compound is also used for
gastrointestinal infections [43].
Fig. 14. Structure of fosfomycin (From Wikipedia, the free encyclopedia).
Fosfomycin inactivates
UDP-N-acetyl-glucosamine-3-O-enolpyruvyltransferase
(MurA), an essential enzyme that catalyzes the first
committed step in cell wall biosynthesis [45-47], by covalent
alkylation of an active site cysteine [48,49].
Pioneering studies from the Seto laboratory on the
biosynthesis of fosfomycin, using genetic techniques [50]
complemented with in vivo experiments using isotopically
labeled precursors provided evidence for the biosynthetic
pathway shown in Fig. 15.
Antibiotics containing C-P bond as a class are virtually
uncharacterized on the genetic and biochemical level.
Although the biosynthetic pathway for fosfomycin production
in S. wedmorensis has been proposed, heterologous
production of fosfomycin has never been achieved, suggesting
the possibility that additional required genes remain to be
identified [51]. The enzymes encoded by fom 1-4 are
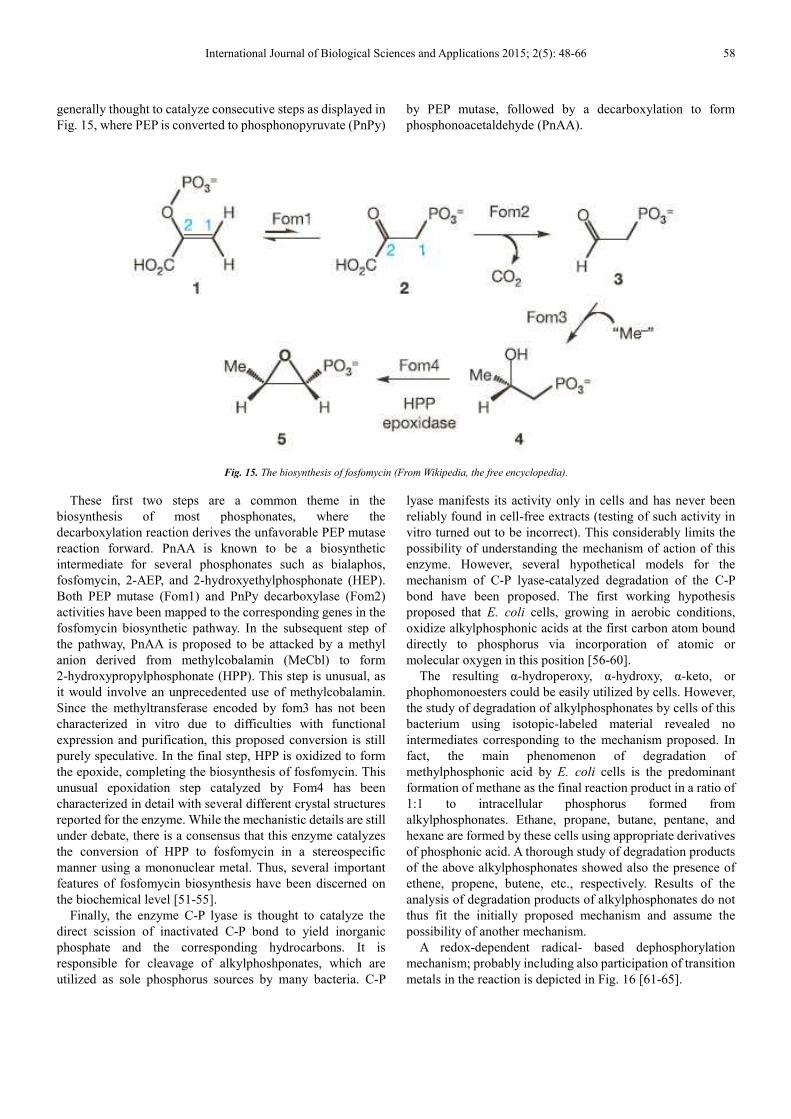
International Journal of Biological Sciences and Applications 2015; 2(5): 48-66 58
generally thought to catalyze consecutive steps as displayed in
Fig. 15, where PEP is converted to phosphonopyruvate (PnPy)
by PEP mutase, followed by a decarboxylation to form
phosphonoacetaldehyde (PnAA).
Fig. 15. The biosynthesis of fosfomycin (From Wikipedia, the free encyclopedia).
These first two steps are a common theme in the
biosynthesis of most phosphonates, where the
decarboxylation reaction derives the unfavorable PEP mutase
reaction forward. PnAA is known to be a biosynthetic
intermediate for several phosphonates such as bialaphos,
fosfomycin, 2-AEP, and 2-hydroxyethylphosphonate (HEP).
Both PEP mutase (Fom1) and PnPy decarboxylase (Fom2)
activities have been mapped to the corresponding genes in the
fosfomycin biosynthetic pathway. In the subsequent step of
the pathway, PnAA is proposed to be attacked by a methyl
anion derived from methylcobalamin (MeCbl) to form
2-hydroxypropylphosphonate (HPP). This step is unusual, as
it would involve an unprecedented use of methylcobalamin.
Since the methyltransferase encoded by fom3 has not been
characterized in vitro due to difficulties with functional
expression and purification, this proposed conversion is still
purely speculative. In the final step, HPP is oxidized to form
the epoxide, completing the biosynthesis of fosfomycin. This
unusual epoxidation step catalyzed by Fom4 has been
characterized in detail with several different crystal structures
reported for the enzyme. While the mechanistic details are still
under debate, there is a consensus that this enzyme catalyzes
the conversion of HPP to fosfomycin in a stereospecific
manner using a mononuclear metal. Thus, several important
features of fosfomycin biosynthesis have been discerned on
the biochemical level [51-55].
Finally, the enzyme C-P lyase is thought to catalyze the
direct scission of inactivated C-P bond to yield inorganic
phosphate and the corresponding hydrocarbons. It is
responsible for cleavage of alkylphoshponates, which are
utilized as sole phosphorus sources by many bacteria. C-P
lyase manifests its activity only in cells and has never been
reliably found in cell-free extracts (testing of such activity in
vitro turned out to be incorrect). This considerably limits the
possibility of understanding the mechanism of action of this
enzyme. However, several hypothetical models for the
mechanism of C-P lyase-catalyzed degradation of the C-P
bond have been proposed. The first working hypothesis
proposed that E. coli cells, growing in aerobic conditions,
oxidize alkylphosphonic acids at the first carbon atom bound
directly to phosphorus via incorporation of atomic or
molecular oxygen in this position [56-60].
The resulting α-hydroperoxy, α-hydroxy, α-keto, or
phophomonoesters could be easily utilized by cells. However,
the study of degradation of alkylphosphonates by cells of this
bacterium using isotopic-labeled material revealed no
intermediates corresponding to the mechanism proposed. In
fact, the main phenomenon of degradation of
methylphosphonic acid by E. coli cells is the predominant
formation of methane as the final reaction product in a ratio of
1:1 to intracellular phosphorus formed from
alkylphosphonates. Ethane, propane, butane, pentane, and
hexane are formed by these cells using appropriate derivatives
of phosphonic acid. A thorough study of degradation products
of the above alkylphosphonates showed also the presence of
ethene, propene, butene, etc., respectively. Results of the
analysis of degradation products of alkylphosphonates do not
thus fit the initially proposed mechanism and assume the
possibility of another mechanism.
A redox-dependent radical- based dephosphorylation
mechanism; probably including also participation of transition
metals in the reaction is depicted in Fig. 16 [61-65].
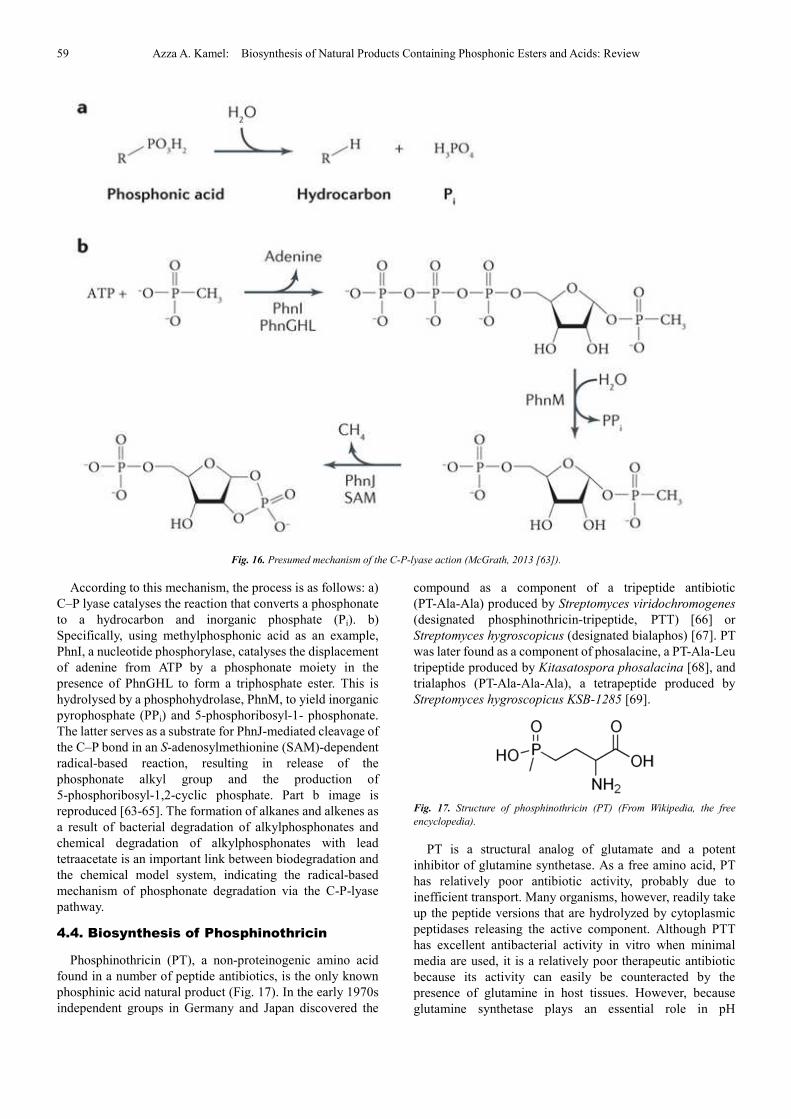
59 Azza A. Kamel: Biosynthesis of Natural Products Containing Phosphonic Esters and Acids: Review
Fig. 16. Presumed mechanism of the C-P-lyase action (McGrath, 2013 [63]).
According to this mechanism, the process is as follows: a)
C–P lyase catalyses the reaction that converts a phosphonate
to a hydrocarbon and inorganic phosphate (Pi). b)
Specifically, using methylphosphonic acid as an example,
PhnI, a nucleotide phosphorylase, catalyses the displacement
of adenine from ATP by a phosphonate moiety in the
presence of PhnGHL to form a triphosphate ester. This is
hydrolysed by a phosphohydrolase, PhnM, to yield inorganic
pyrophosphate (PPi) and 5-phosphoribosyl-1- phosphonate.
The latter serves as a substrate for PhnJ-mediated cleavage of
the C–P bond in an S-adenosylmethionine (SAM)-dependent
radical-based reaction, resulting in release of the
phosphonate alkyl group and the production of
5-phosphoribosyl-1,2-cyclic phosphate. Part b image is
reproduced [63-65]. The formation of alkanes and alkenes as
a result of bacterial degradation of alkylphosphonates and
chemical degradation of alkylphosphonates with lead
tetraacetate is an important link between biodegradation and
the chemical model system, indicating the radical-based
mechanism of phosphonate degradation via the C-P-lyase
pathway.
4.4. Biosynthesis of Phosphinothricin
Phosphinothricin (PT), a non-proteinogenic amino acid
found in a number of peptide antibiotics, is the only known
phosphinic acid natural product (Fig. 17). In the early 1970s
independent groups in Germany and Japan discovered the
compound as a component of a tripeptide antibiotic
(PT-Ala-Ala) produced by Streptomyces viridochromogenes
(designated phosphinothricin-tripeptide, PTT)
[66]
or
Streptomyces hygroscopicus (designated bialaphos) [67]. PT
was later found as a component of phosalacine, a PT-Ala-Leu
tripeptide produced by Kitasatospora phosalacina [68], and
trialaphos (PT-Ala-Ala-Ala), a tetrapeptide produced by
Streptomyces hygroscopicus KSB-1285 [69].
Fig. 17. Structure of phosphinothricin (PT) (From Wikipedia, the free
encyclopedia).
PT is a structural analog of glutamate and a potent
inhibitor of glutamine synthetase. As a free amino acid, PT
has relatively poor antibiotic activity, probably due to
inefficient transport. Many organisms, however, readily take
up the peptide versions that are hydrolyzed by cytoplasmic
peptidases releasing the active component. Although PTT
has excellent antibacterial activity in vitro when minimal
media are used, it is a relatively poor therapeutic antibiotic
because its activity can easily be counteracted by the
presence of glutamine in host tissues. However, because
glutamine synthetase plays an essential role in pH
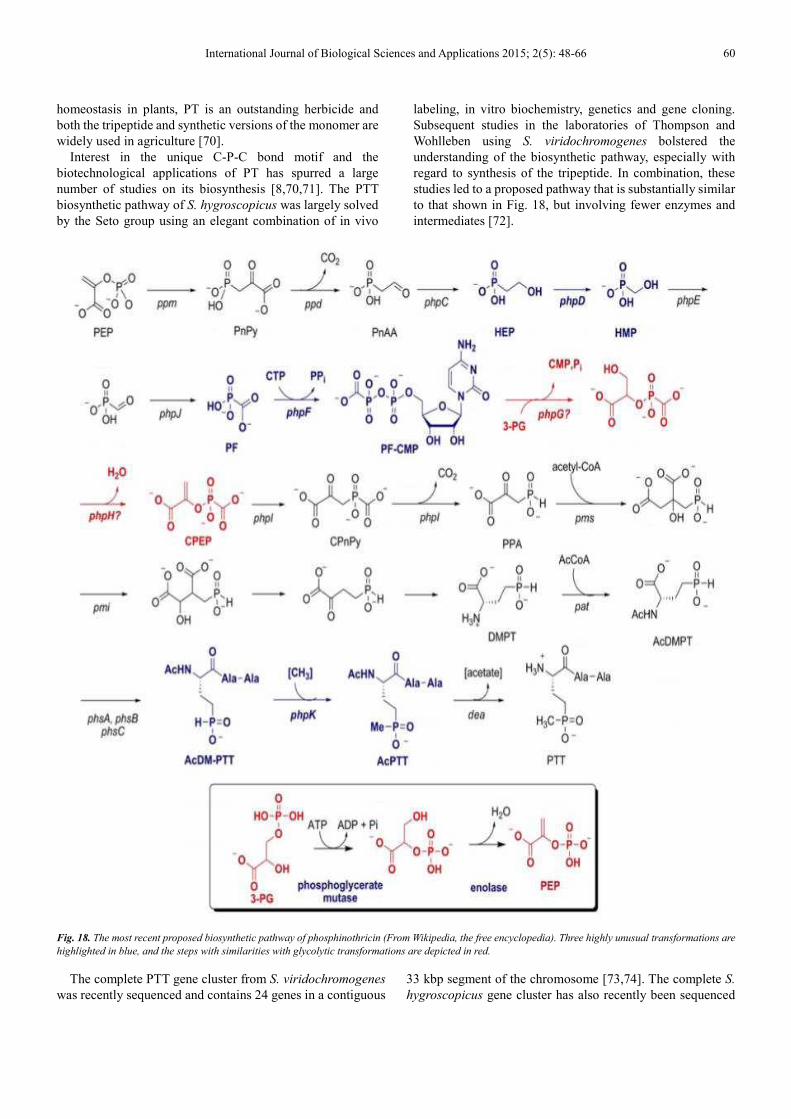
International Journal of Biological Sciences and Applications 2015; 2(5): 48-66 60
homeostasis in plants, PT is an outstanding herbicide and
both the tripeptide and synthetic versions of the monomer are
widely used in agriculture [70].
Interest in the unique C-P-C bond motif and the
biotechnological applications of PT has spurred a large
number of studies on its biosynthesis [8,70,71]. The PTT
biosynthetic pathway of S. hygroscopicus was largely solved
by the Seto group using an elegant combination of in vivo
labeling, in vitro biochemistry, genetics and gene cloning.
Subsequent studies in the laboratories of Thompson and
Wohlleben using S. viridochromogenes bolstered the
understanding of the biosynthetic pathway, especially with
regard to synthesis of the tripeptide. In combination, these
studies led to a proposed pathway that is substantially similar
to that shown in Fig. 18, but involving fewer enzymes and
intermediates [72].
Fig. 18. The most recent proposed biosynthetic pathway of phosphinothricin (From Wikipedia, the free encyclopedia). Three highly unusual transformations are
highlighted in blue, and the steps with similarities with glycolytic transformations are depicted in red.
The complete PTT gene cluster from S. viridochromogenes
was recently sequenced and contains 24 genes in a contiguous
33 kbp segment of the chromosome [73,74]. The complete S.
hygroscopicus gene cluster has also recently been sequenced
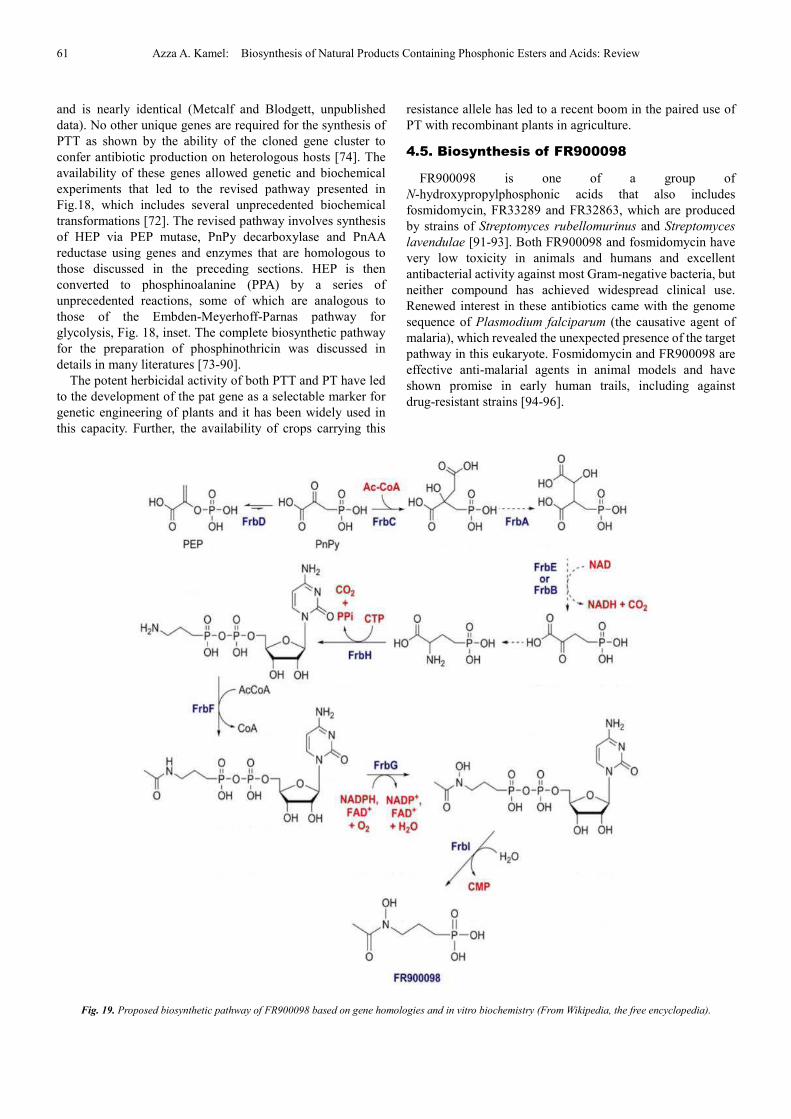
61 Azza A. Kamel: Biosynthesis of Natural Products Containing Phosphonic Esters and Acids: Review
and is nearly identical (Metcalf and Blodgett, unpublished
data). No other unique genes are required for the synthesis of
PTT as shown by the ability of the cloned gene cluster to
confer antibiotic production on heterologous hosts [74]. The
availability of these genes allowed genetic and biochemical
experiments that led to the revised pathway presented in
Fig.18, which includes several unprecedented biochemical
transformations [72]. The revised pathway involves synthesis
of HEP via PEP mutase, PnPy decarboxylase and PnAA
reductase using genes and enzymes that are homologous to
those discussed in the preceding sections. HEP is then
converted to phosphinoalanine (PPA) by a series of
unprecedented reactions, some of which are analogous to
those of the Embden-Meyerhoff-Parnas pathway for
glycolysis, Fig. 18, inset. The complete biosynthetic pathway
for the preparation of phosphinothricin was discussed in
details in many literatures [73-90].
The potent herbicidal activity of both PTT and PT have led
to the development of the pat gene as a selectable marker for
genetic engineering of plants and it has been widely used in
this capacity. Further, the availability of crops carrying this
resistance allele has led to a recent boom in the paired use of
PT with recombinant plants in agriculture.
4.5. Biosynthesis of FR900098
FR900098 is one of a group of
N-hydroxypropylphosphonic acids that also includes
fosmidomycin, FR33289 and FR32863, which are produced
by strains of Streptomyces rubellomurinus and Streptomyces
lavendulae [91-93]. Both FR900098 and fosmidomycin have
very low toxicity in animals and humans and excellent
antibacterial activity against most Gram-negative bacteria, but
neither compound has achieved widespread clinical use.
Renewed interest in these antibiotics came with the genome
sequence of Plasmodium falciparum (the causative agent of
malaria), which revealed the unexpected presence of the target
pathway in this eukaryote. Fosmidomycin and FR900098 are
effective anti-malarial agents in animal models and have
shown promise in early human trails, including against
drug-resistant strains [94-96].
Fig. 19. Proposed biosynthetic pathway of FR900098 based on gene homologies and in vitro biochemistry (From Wikipedia, the free encyclopedia).

International Journal of Biological Sciences and Applications 2015; 2(5): 48-66 62
The FR900098 biosynthetic gene cluster was identified by
screening a large insert fosmid library from S. rubellomurinus
with degenerate PCR primers designed to amplify PEP
mutase-encoding (ppm) genes [97]. Further screening of
ppm-positive clones led to the identification of a clone that
conferred production of FR900098 to the heterologous host
Streptomyces lividans and, therefore, contains all of the
unique genes needed for synthesis of the antibiotic. Sequence
analysis of the clone revealed the presence of genes encoding
a putative PEP mutase and a number of genes related to those
encoding enzymes of the TCA cycle. The predicted functions
of these genes, in combination with in vitro biochemistry and
mass spectrometric identification of intermediates provide
strong support for the proposed biosynthetic pathway shown
in Fig. 19.
Unlike other phosphonate biosynthetic pathways, PnPy
decarboxylase does not provide the thermodynamic driving
force needed to pull the unfavorable PEP mutase reaction.
Instead, a homolog of homocitrate synthase, FrbC, catalyzes
the exergonic condensation of acetyl-CoA and
phosphonopyruvate to form 2-phosphonomethylmalate. This
activity has been verified in vivo using synthetic PnPy as
substrate, and also in a coupled reaction with the S.
rubellomurinus PEP mutase (FrbD) using PEP as the starting
material [97]. Subsequent steps are predicted to convert this
intermediate into 2-oxo-4-phosphonobutyrate, an analog of
2-oxo-glutarate, although this has yet to be demonstrated. An
unknown host encoded transaminase is thought to convert this
intermediate to 2-amino-4-phosphonobutyrate. FrbH is a
two-domain protein containing discrete
nucleotidyl-transferase and PLP-dependent
decarboxylase/aminotranserase domains that was predicted to
convert 2-amino-4-phosphonobutyrate into
3-aminopropylphosphonate. In vitro decarboxylation activity
of FrbH requires CTP and produces
5′-CMP-3-aminopropylphosphonate. The biological rationale
for this nucleotide modification is unclear; however, the
CMP-modified product is competent as a substrate for in vitro
acetylation with FrbF and N-hydroxylation by FrbG, whereas
free 3-aminopropylphosphonate is not. Removal of the CMP
is catalyzed by FrbI in vitro, although this gene is not required
in vivo, suggesting that other cellular phosphodiesterases can
fulfill this function as well.
Interestingly, the FR900098 gene cluster also includes a
gene (dxrB) that encodes a homolog of DXR, the target of the
antibiotic. The dxrB gene was proposed to encode an
FR900098-insensitive allele of DXR, and thus to be involved
in self-resistance to the antibiotic [97]. This idea has yet to be
tested experimentally, but if true, the structure of this protein
may lend insight into the nature of the interaction of the
antibiotic and the enzyme allowing design of more effective
derivatives.
4.6. Biosynthesis of
Phosphatidylethanolamine
Phosphatidylethanolamines (sometimes abbreviated PE)
are a class of phospholipids found in biological membranes. It
can mainly be found in the inner (cytoplasmic) leaflet of the
lipid bilayer [98].
Phosphatidylethanolamines are found in all living cells,
composing 25% of all phospholipids. In human physiology,
they are found particularly in nervous tissue such as the white
matter of brain, nerves, neural tissue, and in spinal cord, where
they make up 45% of all phospholipids [99]. In humans [100],
metabolism of PE is thought to be important in the heart.
When blood flow to the heart is restricted, the asymmetrical
distribution of PE between membrane leaflets is disrupted,
and as a result the membrane is disrupted. Additionally, PE
plays a role in the secretion of lipoproteins in the liver. This is
because vesicles for secretion of VLDLs coming off of the
Golgi have a significantly higher PE concentration when
compared to other vesicles containing VLDLs [101]. PE has
also shown to be able to propagate infectious prions without
the assistance of any proteins or nucleic acids, which is a
unique characteristic of it [102]. PE is also thought to play a
role in blood clotting, as it works with phosphatidylserine to
increase the rate of thrombin formation by promoting binding
to Factor V and Factor X, two proteins which catalyze the
formation of thrombin from prothrombin. PEs play a role in
membrane fusion and in disassembly of the contractile ring
during cytokinesis in cell division. Additionally, it is thought
that PE regulates membrane curvature. PE acts as an important
precursor, substrate, or donor in several biological pathways
[99].
As a polar head group, PE creates a more viscous lipid
membrane compared to phosphatidylcholine (PC). For
example, the melting temperature of di-oleoyl-PE is -16 °C
while the melting temperature of di-oleoyl-PC is -20 °C. If the
lipids had two palmitoyl chains, PE would melt at 63°C while
PC would melt already at 41 °C [100]. Lower melting
temperatures correspond, in a simplistic view, to more fluid
membranes.
Where phosphatidylcholine is the principal phospholipid in
animals, PE is the principal one in bacteria. One of the primary
roles for PE in bacterial membranes is to spread out the
negative charge caused by anionic membrane phospholipids.
In the bacterium E. coli, PE plays a role in supporting lactose
permease's active transport of lactose into the cell, and may
play a role in other transport systems as well. PE plays a role
in the assembly of lactose permease and other membrane
proteins. It acts as a 'chaperone' to help the membrane proteins
correctly folds their tertiary structures so that they can
function properly. When PE is not present, the transport
proteins have incorrect tertiary structures and do not function
correctly [103]. PE also enables bacterial multidrug
transporters to function properly. PE allows the formation of
intermediates that are needed for the transporters to properly
63 Azza A. Kamel: Biosynthesis of Natural Products Containing Phosphonic Esters and Acids: Review
open and close.
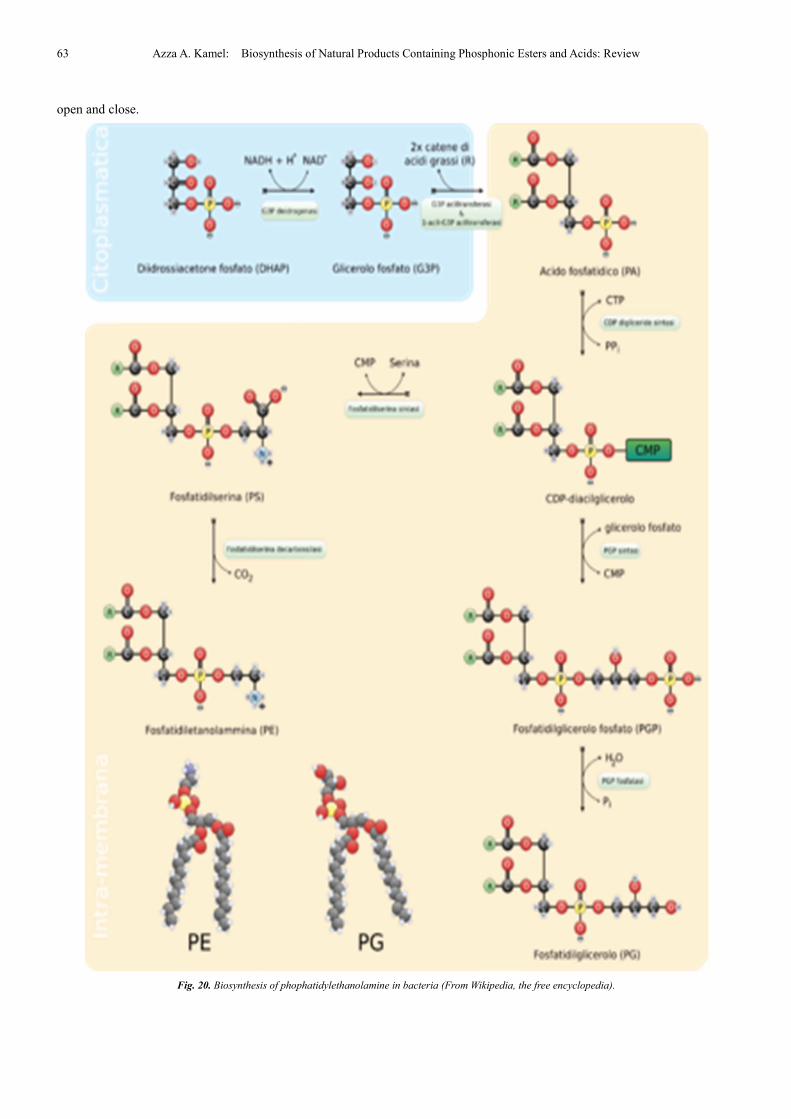
Fig. 20. Biosynthesis of phophatidylethanolamine in bacteria (From Wikipedia, the free encyclopedia).

International Journal of Biological Sciences and Applications 2015; 2(5): 48-66 64
Phosphatidylethanolamines in food break down to form
PE-linked Amadori products as a part of the Maillard reaction
[104,105]. These products accelerate membrane lipid
per-oxidation, causing oxidative stress to cells that come in
contact with them [106]. Oxidative stress is known to cause
food deterioration and several diseases. Significant levels of
Amadori-PE products have been found in a wide variety of
foods such as chocolate, soybean milk, infant formula, and
other processed foods. The levels of Amadori-PE products are
higher in foods with high lipid and sugar concentrations that
have high temperatures in processing [105]. Additional
studies have found that Amadori-PE may play a role in
vascular disease [106,107], act as the mechanism by which
diabetes can increase the incidence of cancer, and potentially
play a role in other diseases as well. Amadori-PE has a higher
plasma concentration in diabetes patients than healthy people,
indicating it may play a role in the development of the disease
or be a product of the disease [108].
They are synthesized by the addition of Cytidine
diphosphate (CDP)-ethanolamine to diglycerides, releasing
Cytidine monophosphate (CMP). S-Adenosyl methionine can
subsequently methylate the amine of
phosphatidylethanolamines to yield phosphatidylcholines.
The phosphatidylserine decarboxylation pathway and the
CDP-ethanolamine pathways are used to synthesize PE.
Phosphatidylserine decarboxylase (PSD) is the enzyme that is
used to decarboxylate phosphatidylserine in the first pathway.
The phosphatidylserine decarboxylation pathway is the main
source of synthesis for PE in the membranes of the
mitochondria. PE produced in the mitochondrial membrane is
also transported throughout the cell to other membranes for
use. In a process that mirrors phosphatidylcholine synthesis,
PE is also made via the CDP-ethanolamine pathway, using
ethanolamine as the substrate. Through several steps taking
place is both the cytosol and endoplasmic reticulum, the
synthesis pathway yields the end product of PE [104]. PE is
also found abundantly in soya or egg lecithin and is produced
commercially using chromatographic separation (Fig. 20).
5. Conclusion and Prospective
Natural products continue to play a pivotal role in
drug-discovery efforts and in the understanding of human
health.
Phosphonate containing natural products constitute very
important part of biomolecules such as: phospholipids, nucleic
acids, proteins and polysaccharides, as well as nucleotide
cofactors that involved in energy transport and catalysis of
many cell processes.
The wide occurrence of phosphonates among biogenic and
abiogenic (natural and man-made) organophosphorus
compounds makes the question about the catabolism of these
compounds topical and at the same time potentially resolvable.
The diversity of phosphonate structures, occurrence among
these natural compounds with activated C-P bond
(aminophosphonates, acetylphosphonates); and other
compounds with non-activated, more stable C-P bond
(alkylphosphonates) determine the great number of pathways
of their catabolism.
A wide range of microorganisms, mainly bacteria, was
shown able to degrade different phosphonates. The
fundamental knowledge of the mechanism of biodegradation
of organophosphorus compounds with the C-P bond will serve,
in turn, as a basis of biotechnology for environmental
protection and biodegradation of toxic organophosphorus
compounds with stable C-P bond, which uncontrollably enter
the environment as pesticides, herbicides, and other products
of economic activity. However, only the pathways of activated
C-P bond degradation have been characterized rather
completely, and enzymes catalyzing this degradation have
been identified and characterized.
The most problematic aspect of phosphonate
biodegradation -from both fundamental and biotechnological
points of view- is the degradation of the most stable C-P bond
of alkylphosphonates. Neither the mechanism of degradation
of this bond nor the appropriate enzyme (or polyenzyme
complex) C-P-lyase has been finally determined and
characterized so far.
In summary, this article provides a brief survey on the
classification and biosynthesis of some natural products
containing C-P bond.
References
[1] Kim E, Moore BS, Yoon YJ. Nature Chemical Biology, 11, 649 (2015).
[2] Williams DA, Lemke TL. Foye's Principles of Medicinal Chemistry (5th ed.), Chapter 1, Natural Products, Philadelphia, Lippincott Williams Wilkins, p. 25, ISBN 0-683-30737-1 (2002).
[3] Kulakova AN, Wisdom GB, Kulakov LA, Quinn, JP. J. Biol. Chem., 278, 23426 (2003).
[4] Hanson JR. Natural products: the secondary metabolite. Cambridge, Royal Society of Chemistry, Hanson JR. (ed), ISBN 0-85404-490-6 (2003).
[5] Kononova SV, Nesmeyanova MA. Biochemistry (Moscow), 67, 184 (2002).
[6] Kossel A. Archiv für Physiologie (in German), 181 (1891).
[7] Kliebenstein DJ. Plant, Cell and Environment, 27, 675 (2004).
[8] Karlovsky P. Soil Biology, 14, 1 (2008).
[9] Rogers K. The components of life: from nucleic acids to carbohydrates (1st ed.), New York, NY, Britannica Educational Publishing in association with Rosen Educational Services, ISBN 978-1-61530-324-3 (2011).
[10] Cox DL, Nelson MM. Lehninger Principles of biochemistry (6th ed.). New York, N.Y., Freeman WH. (ed), ISBN 978-1-4641-0962-1 (2013).
[11] Boal D. Mechanics of the cell (4th ed.), Cambridge, UK, Cambridge University Press, ISBN 978-052179681-1 (2006).

65 Azza A. Kamel: Biosynthesis of Natural Products Containing Phosphonic Esters and Acids: Review
[12] Dewick PM. Medicinal natural products: a biosynthetic approach (3rd ed.), Chichester, Wiley, ISBN 978-0-470-74167-2 (2009).
[13] Sitaramayya A. Introduction to cellular signal transduction, Boston, Birkhäuser, ISBN 978-0-8176-3982-2 (1999).
[14] Demain AL, Fang A. Adv. Biochem. Eng. Biotechnol. 69, 1 (2000).
[15] Firn RD, Jones CG. Mol. Microbiol., 37, 989 (2000).
[16] Alberts B, Johnson A, Lewis, J, Raff, M. Molecular Biology of the Cell 4th edition, Taylor &France Group (2007).
[17] Get information, facts, and pictures about Genetic Code at Encyclopedia.com.MeyerRR. "Genetic Code." Genetics. 2003. Retrieved September 13(2015) from Encyclopedia.com, by The Gale Group, Inc, http://www.encyclopedia.com/doc/1G2-3406500118.
[18] Schlattner U, Tokarska-Schlattner M, Wallimann T. Biochemica et BiophysicaActa, 27 (2005).
[19] Wanner BL. Biodegradation, 5, 175 (1994).
[20] McGrath JW, Wisdom GB, McMullan G, Larkin, MJ, Quinn JP. Eur. J. Biochem. 234, 225 (1995).
[21] Ternan NG, McGrath JW, McMullan G, Quinn, JP. World J. Microbiol. Biotechnol., 14, 635 (1998).
[22] McGrath JW, Ternan NG, Quinn JP. Lett. Appl. Microbiol. 24, 69 (1997).
[23] Ternan NG, Hamilton JTG, Quinn, JP. Arch. Microbiol., 173, 35 (2000).
[24] Nakashita H, Watanabe K, Hara O, Hidaka T, Seto H. J. Antibiot. (Tokyo), 50, 212 (1997).
[25] Viktorova, LS, Arzumanov, AA, Shirokova, EA, Yasko, MV, Aleksandrova, LA, Shipitsyn, AV, Skoblov AY, Krayevsky, AA. Mol. Biol. (Moscow), 32, 162 (1998).
[26] Fleisch, W. Drugs, 42, 919 (1991).
[27] Kelly K.Phospholipid Biosynthesis,The AOCS Lipid Library, Retrieved September 3 (2012).
[28] Sviridov, AV, Zelenkova, NF, Vinokurova, NG, Ermakova, IT, Leontievsky, AA. Biochemistry (Moscow), 76, 720 (2011).
[29] Bhat SV, Nagasampagi BA, Sivakumar M. Chemistry of Natural Products. Berlin, New York, Springer, ISBN 81-7319-481-5 (2005).
[30] McGrath JW, Chin JP, Quinn JP. Nature Reviews Microbiology, 11, 412 (2013).
[31] McGrath JW, Ternan NG, Quinn JP. Letters in Applied Microbiology, 24 (1997).
[32] Ternan NG, McGrath JW, Quinn JP.Appl. Environ. Microbiol., 64, 2291, (1998).
[33] Sarkar M, Hamilton CJ, Fairlamb AH. The Journal of Biological Chemistry, 278, 22703 (2003).
[34] Tan SA, Tan LG., Clin. Physiol. Biochem., 7, 303 (1989).
[35] Barry RJ, Bowman E, McQueney M, Dunaway-Mariano D., Biochem. Biophys. Res. Commun. 153, 177 (1988).
[36] Murakami T, Anzai H, Imai S, Satoh A, Nagaoka K, Thompson CJ., MGG Molecular & General Genetics, 205, 42 (1986).
[37] Duke O, Dayan E. Toxins (Basel), 3, 1038 (2011).
[38] Kamigiri K, Hidaka T, Imai S, Murakami T, Seto H. J. Antibiot.,45, 781 (1992).
[39] Stein GE. Ann. Pharmacother., 32, 215 (1998).
[40] Reeves DS. Infection, 204, S313 (1992).
[41] Nakazawa H, Kikuchi Y, Honda T, Isago T, Nozaki M. J. Infect. Chemother.9, 304 (2003).
[42] Cassone M, Campanile F, Pantosti A, Venditti M, Stefani S. Microb. Drug Resist., 10, 43 (2004).
[43] Allerberger F, Klare I. J. Antimicrob. Chemother., 43, 211(1999).
[44] Falagas ME, Giannopoulou KP, Kokolakis GN, Rafailidis PI. Clin Infect Dis., 46, 1069 (2008).
[45] Skarzynski T, Mistry A, Wonacott A, Hutchinson SE, Kelly VA, Duncan K. Structure, 4, 1465 (1996).
[46] Schonbrunn E, Svergun DI, Amrhein N, Koch MH. Eur. J. Biochem., 253, 406 (1998).
[47] De Smet KA, Kempsell KE, Gallagher A, Duncan K, Young DB. Microbiology, 145, 3177 (1999).
[48] Marquardt JL, Brown ED, Lane WS, Haley TM, Ichikawa Y. Biochemistry, 33, 10646 (1994).
[49] Kim DH, Lees WJ, Kempsell KE, Lane WS, Duncan K, Walsh CT. Biochemistry, 35, 4923 (1996).
[50] Hidaka T, Goda M, Kuzuyama T, Takei N, Kidaka M, Seto H. Mol. Gen. Genet., 249, 274 (1995).
[51] Kuzuyama T, Hidaka T, Kamigiri K, Imai S, Seto H. J. Antibiot., 45, 1812 (1992).
[52] Hammerschmidt F. Angew. Chem. Int. Ed. Engl. 33, 341 (1994).
[53] Hammerschmidt F, Kählig H. J. Org. Chem. 56, 2364 (1991).
[54] Woodyer RD, Shao Z, Metcalf WM, Thomas PM, Kelleher NL. Chem. Biol.13, 1171 (2006).
[55] Woodyer RD, Li G, Zhao H, van der Donk WA. Chem Commun., 359 (2007).
[56] Shao Z, Blodgett JA, Circello BT, Eliot AC, Woodyer R. J. Biol. Chem., 283, 23161 (2008).
[57] Montella C, Bellsolell L, Perez-Luque R, Badia J, Baldoma L. J. Bacteriol. 187, 4957 (2005).
[58] Sofia HJ, Chen G, Hetzler BG, Reyes-Spindola JF, Miller NE. Nucleic Acids Res., 29, 1097 (2001).
[59] van der Donk WA. J Org Chem.71, 9561 (2006).
[60] Layer G, Heinz DW, Jahn D, Schubert WD. Curr. Opin. Chem. Biol.8, 468 (2004).
[61] Frey PA. Annu. Rev. Biochem., 70, 121 (2001).
[62] Frey PA, Magnusson OT. Chem. Rev., 103, 2129 (2003).

International Journal of Biological Sciences and Applications 2015; 2(5): 48-66 66
[63] McGrath JW, Chin JP, Quinn JP. Nature Reviews Microbiology, 11, 412 (2013).
[64] Mosimann H, Kräutler B. Angew. Chem. Int. Ed. 39, 393 (2000).
[65] Glasenapp-Breiling M, Montforts FP. Angew. Chem. Int. Ed. Engl. 39, 721 (2000).
[66] Schwartz D, Berger S, Heinzelmann E, Muschko K, Welzel K, Wohlleben W. Appl Environ Microbiol., 70, 7093 (2004).
[67] Peck SC, van der Donk WA. Curr Opin Chem Biol., 17, 580 (2013).
[68] Peck SC, Gao J, van der Donk WA. Methods Enzymol., 516, 101 (2012).
[69] Kato H, Nagayama K, Abe H, Kobayashi R, Ishihara E. AgricBiol Chem., 55, 1133 (1991).
[70] Thompson CJ, Seto H., Biotechnology, 28, 197 (1995).
[71] Thompson CJ, Seto H. Bialaphos. In: Vining LC, Stuttard C. (eds). Genetics and Biochemistry of Antibiotic Production, Newton MA, Butterworth-Heinemann, pp 197 (1995).
[72] Blodgett JA, Thomas PM, Li G, Velasquez JE, van der Donk WA. Nat. Chem. Biol., 3, 480 (2007).
[73] Schwartz D, Berger S, Heinzelmann E, Muschko K, Welzel K, Wohlleben W. Appl Environ Microbiol., 70, 7093 (2004).
[74] Blodgett JA, Zhang JK, Metcalf WW. Antimicrob Agents Chemother., 49, 230, (2005).
[75] Dunwell JM, Purvis A, Khuri S. Phytochemistry, 65, 7 (2004).
[76] Costas M, Mehn MP, Jensen MP, Que L., Chem Rev., 104, 939 (2004).
[77] Grogan G. Biochem J., 388, 721 (2005).
[78] Xing G, Diao Y, Hoffart LM, Barr EW, Prabhu KS. Proc Natl Acad Sci USA. 103, 6130 (2006).
[79] Burzlaff NI, Rutledge PJ, Clifton IJ, Hensgens CM, Pickford M. Nature, 401, 721 (1999).
[80] Hidaka T, Hidaka M, Uozumi T, Seto H. Mol Gen Genet., 233, 476 (1992).
[81] Babbitt PC, Hasson MS, Wedekind JE, Palmer DR, Barrett WC. Biochemistry, 35, 16489 (1996).
[82] Potters MB, Solow BT, Bischoff KM, Graham DE, Lower BH. J. Bacteriol., 185, 2112 (2003).
[83] Hidaka T, Imai S, Hara O, Anzai H, Murakami T. J. Bacteriol. , 172, 3066 (1990).
[84] Freeman S, Pollack SJ, Knowles JR. J. Am. Chem. Soc.,114, 377 (1992).
[85] Pollack SJ, Freeman S, Pompliano DL, Knowles JR. Eur. J. Biochem.,209, 735 (1992).
[86] Schwartz D, Alijah R, Nussbaumer B, Pelzer S, Wohlleben W. Appl Environ Microbiol., 62, 570 (1996).
[87] Grammel N, Schwartz D, Wohlleben W, Keller U.
Biochemistry., 37 (1998).
[88] Alijah R, Dorendorf J, Talay S, Puhler A, Wohlleben W. Appl. Microbiol. Biotechnol., 34, 749 (1991).
[89] Schwartz D, Grammel N, Heinzelmann E, Keller U, Wohlleben W. Antimicrob. Agents Chemother., 49, 4598 (2005).
[90] Eys S, Schwartz D, Wohlleben W, Schinko E. Antimicrob. Agents Chemother., 52, 1686 (2008).
[91] Hartley FR. Introduction. In: The Chemistry of Organophosphorus Compounds, Hartley FR (ed), John Wiley & Sons, Ltd. (1990).
[92] Dahl O. The Preparation and Properties of Tervalent Phosphorus Acid Derivatives. In: Hartley FR. (ed), The Chemistry of Organophosphorus Compounds. New York: John Wiley; pp. 1–45 (1996).
[93] Edmunson RS. The Synthesis of Phosphonic and Phosphinic Acids and Their Derivatives. In: Hartley FR, editor. The Chemistry of Organophosphorus Compounds. New York: John Wiley; NY., USA, 48–144 (1996).
[94] Missinou MA, Borrmann S, Schindler A, Issifou S, Adegnika AA. Lancet., 360, 1941 (2002).
[95] Wiesner J, Borrmann S, Jomaa H. Parasitol Res.,902, S71 (2003).
[96] Jomaa H, Wiesner J, Sanderbrand S, Altincicek B, Weidemeyer C. Science,285, 1573 (1999).
[97] Eliot AC, Griffin BM, Thomas PM, Johannes TW, Kelleher NL. Chem Biol., 15, 765 (2008).
[98] Vance JE, Tasseva G. Biochimica et Biophysica Acta, 1831, 543 (2012).
[99] Emoto K, Kobayashi T, Yamaji A, Aizawa H, Yahara I, Inoue K, Umeda M. Proceedings of the National Academy of Sciences, 93, 12867 (1996).
[100] Vance JE. The Journal of Lipid Research, 49, 1377 (2008).
[101] Deleault NR, Piro JR, Walsh DJ, Wang F, Ma J, Geoghegan JC, Supattapone S. Proceedings of the National Academy of Sciences, 109, 8546 (2012).
[102] Majumder R, Liang X, Quinn-Allen MA, Kane WH, Lentz BR. Journal of Biological Chemistry , 286, 35535 (2011).
[103] Gbaguidi B, Hakizimana P, Vandenbussche G, Ruysschaert JM. Cellular and Molecular Life Sciences, 64, 1571 (2007).
[104] Oak JH, Nakagawa K, Miyazawa T. The Journal of Lipid Research, 43, 523 (2002).
[105] Oak J-H., Nakagawa K., Miyazawa T. FEBS Letters, 481, 26 (2000).
[106] Oak J-H., Nakagawa K, Oikawa S, Miyazawa T. FEBS Letters, 555, 419 (2003).
[107] Eitsuka T, Nakagawa K, Ono Y, Tatewaki N, Nishida H, Kurata T, Shoji N, Miyazawa T. FEBS Letters, 586, 2542 (2012).
[108] Ariizumi K, Koike T, Ohara S, Inomata Y, Abe Y, Iijima K, Imatani A, Oka T, Shimosegawa T. World Journal of Gastroenterology, 14, 3212 (2008).





























