Embed Size (px)
Citation preview

Atmospheric Environment 33 (1999) 2269—2277
Black carbon concentrations in precipitation and near surfaceair in and near Halifax, Nova Scotia
Petr Chylek*, L. Kou, B. Johnson, F. Boudala, G. Lesins
Atmospheric Science Program, Departments of Physics and Oceanography, Dalhousie University, Halifax, Nova Scotia, Canada B3H 3J5
Received 10 October 1997; accepted 15 April 1998
Abstract
Black carbon (soot) concentrations have been measured in rain water, snow samples and near surface air at severallocations in Nova Scotia, Canada. The average black carbon concentration in near surface air in summer was found to be0.54 kgm~3 compared to 1.74 kgm~3 in the winter season. These values are comparable to black carbon concentrationsfound in other mid-size urban areas. The black carbon concentration in rain water and snow samples varied between anundetectable amount to about 20 kg kg~1 of rain (or melt) water. The relatively low concentrations of black carbon inprecipitation are attributed to extratropical cyclones that often develop off-shore to the east and south of Nova Scotia inrelatively clean conditions of the marine boundary layer. ( 1999 Elsevier Science Ltd. All rights reserved.
Keywords: Aerosols; Black Carbon; Soot; Scavenging
1. Introduction
Black carbon (soot) is an important constituent ofatmospheric aerosols, because it is primarily responsiblefor aerosol absorption of solar radiation. While non-absorbing or only lightly absorbing aerosols contributetowards the cooling of the earth—atmosphere system,highly absorbing black carbon tends to warm the atmo-sphere. The total effect of aerosols at a given location andatmospheric situation may be either to cool or to warmdepending on the optical properties and concentrationsof individual aerosol components. Due to the variety ofaerosol sources, transport processes and relatively shortatmospheric lifetimes (typically of a few days), the con-centration of individual aerosols is spatially and tem-porally inhomogeneous and is not well known.
*Corresponding author. Tel.: 902 494 1456; fax: 902 494 5191;e-mail: [email protected].
There are several measured black carbon concentra-tions in near surface air (Chesselet et al., 1981; Wolff et al.,1981; Heintzenberg, 1982a; Andreae et al., 1984; Dzubayet al., 1984; Hansen et al., 1988; Clarke, 1989; Cachieret al., 1990; Heintzenberg and Bigg, 1990; Jennings andO’Dowd, 1990; Mukai et al., 1990; Hopper et al., 1991;Yaaqub et al., 1991; Jennings et al., 1993; O’Dowd et al.,1993; Heintzenberg and Leck, 1994; Pinnick et al., 1993;Cachier et al., 1994; Parungo et al., 1994; Bodhaine,1995; van Dingenen et al., 1995; Chylek et al., 1996;Kuhlbusch et al., 1996; Jennings et al., 1997). A fewmeasurements were reported for the troposphere (Chyleket al., 1996; Kou, 1996) and the stratosphere (Chuan andWoods, 1984; Pueschel et al., 1992; Blake and Kato,1995). There are also limited data available concerningthe black carbon concentration in cloud water droplets(Twohy et al., 1989; Chylek et al., 1996; Kou, 1996), rainand snow (Ogren and Charleson, 1984; Clarke andNoone, 1985; Chylek et al., 1987; Warren and Clarke,1990; Cachier and Ducret, 1991; Ducret and Cachier,
1352-2310/99/$ - see front matter ( 1999 Elsevier Science Ltd. All rights reserved.PII: S 1 3 5 2 - 2 3 1 0 ( 9 8 ) 0 0 1 5 4 - X

1992), and ice core samples (Chylek et al., 1987; 1992a, b;1995).
Recently, an inventory of black carbon sources (Pen-ner et al., 1993) and 3D models of black carbon transportand concentrations have been developed (Liousse et al.,1996; Cooke and Wilson, 1996). An effort to incorporateblack carbon into general circulation models for climatestudies is in progress. Radiative transfer calculations sug-gest that black carbon can significantly modify the re-gional and global cooling effect of other aerosols (Chylekand Wong, 1995; Haywood and Shine, 1995; Haywoodet al., 1997; Schult et al., 1997) and affect the albedo ofclouds (Chylek et al., 1984; Heintzenberg and Wendish,1996). Due to the role that black carbon plays in theradiation balance of the earth atmosphere system, addi-tional information concerning the concentration of blackcarbon and its removal processes is needed.
To contribute towards the black carbon data base thatcan be used for radiative modeling as well as for valida-tion of black carbon transport and concentration models,we present measurements of black carbon concentrationin rainwater and snow for several locations at or nearHalifax, Nova Scotia. To complement the data of blackcarbon concentrations in precipitation, we have alsomeasured black carbon concentration in near surface airin Halifax during the summer and winter seasons.
2. Black carbon analysis
Black carbon (soot) is a byproduct of fossil fuel andbiomass burning. The carbon present in the form ofaerosol particles in the atmosphere is usually divided intoorganic and elemental carbon. A recent review (Horvath,1993) describes various methods used to determine blackcarbon. Traditionally, it is assumed that organic carbonis nonabsorbing in the range of solar wavelengths andthat the absorption is due to elemental carbon. Recently,it has been shown that this assumption is not quitecorrect (Chow et al., 1993).
Malm et al. (1994) has found that elemental carbonoxidized at temperatures above 700°C shows little or nocorrelation with the black carbon absorption coefficient.On the other hand, organic carbon oxidized at temper-atures between 450 and 550°C exhibits a high correlationwith the absorption coefficient. This is consistent withthe finding that the transmission and the reflectance ofquartz filters, containing samples of atmospheric aero-sols, changes only between the temperatures of about450—700°C during the heating of the filter in the furnace(Chylek et al., 1987; Chow et al., 1993). This suggests thatoptically active carbon, that is, carbon that stronglyabsorbs solar radiation, is composed partially by organicand partially by elemental carbon. Malm et al. (1994)claim that up to 50% of the black carbon absorption isactually attributed to organic carbon. Since we are inter-
ested in climatic effects and radiative forcing of atmo-spheric black carbon (and not so much in individualchemical species), we adopt an operational definition ofblack carbon as being that part of organic and elementalcarbon that absorbs solar radiation. Consequently, weuse the instrumentation that is aimed at determining theamount of this light absorbing carbon in the atmosphereand in the precipitation.
The method we use to measure black carbon ona quartz filter is to slowly heat the filter up to 700°C andto measure light transmittance before and after carbonhas been oxidized to CO
2. Aerosol particles are concen-
trated in a small region around the center of the filter andthrough scanning, transmittance is referenced to theblank filter edge. The change of filter transmission thatresults from being heated between 400 and 700°C iscalibrated using a known amount of several differentsizes of commercially available carbon black with radiusbetween 0.05 and 0.10 km. In this sense the amount ofblack carbon that we report is equal to the amount ofcarbon black that causes the same absorption as blackcarbon on the analyzed filter. More details of the methodare described elsewhere (Chylek et al., 1992a; Kou, 1996).
3. Black carbon concentration in near surface air inurban Halifax
Air samples were collected during the Summer of 1995and Winter of 1996 in the downtown Halifax (BarringtonStreet) field collection station operated by the Environ-mental Department of Nova Scotia. The main samplingtube was placed facing downward at a level of about 15mabove the street. The quartz filters were exposed to a slowflow of air (of the order of 0.5 l min~1) for up to a week.For several samples the total amount of the collectedparticulate mass was also determined by weighing thefilter before and after the collection.
Table 1 show the results of the black carbon measure-ments for the ‘‘Summer’’ season (including the measure-ments taken in the late Spring and early Fall of 1995).The range of black carbon concentration is relativelynarrow between 0.43 and 0.68 kg m~3 with a median of0.55 and a mean value of 0.54 kgm~3, suggesting thatweekly averages of black carbon during the warm seasonare relatively stable, probably dominated by local vehicu-lar traffic.
During the cold season the black carbon concentrationin Halifax is increased due to domestic and commercialheating. The range in the ‘‘Winter’’ season (includingearly Spring) for black carbon concentrations in air isbetween 1.1 and 3.1 kgm~3, with a median and a meanvalue of 1.7 kgm~3 (Table 2). The winter average blackcarbon concentration is by about a factor of three higherthan the summer average. The major contribution to thewinter black carbon concentrations is local heating (oil).
2270 P. Chylek et al. / Atmospheric Environment 33 (1999) 2269—2277
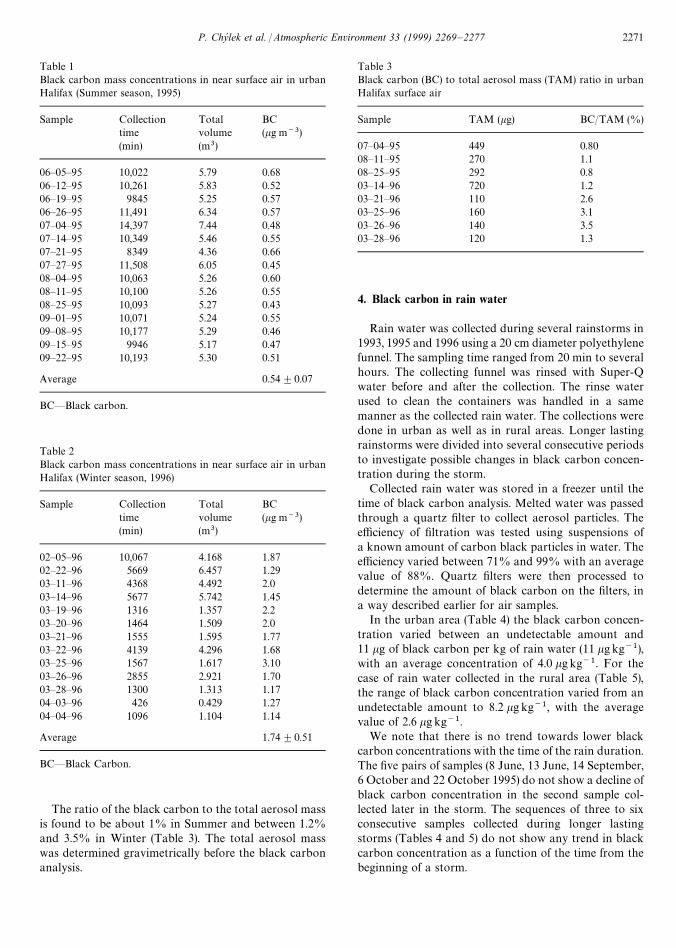
Table 1Black carbon mass concentrations in near surface air in urbanHalifax (Summer season, 1995)
Sample Collection Total BCtime volume (kg m~3)(min) (m3)
06—05—95 10,022 5.79 0.6806—12—95 10,261 5.83 0.5206—19—95 9845 5.25 0.5706—26—95 11,491 6.34 0.5707—04—95 14,397 7.44 0.4807—14—95 10,349 5.46 0.5507—21—95 8349 4.36 0.6607—27—95 11,508 6.05 0.4508—04—95 10,063 5.26 0.6008—11—95 10,100 5.26 0.5508—25—95 10,093 5.27 0.4309—01—95 10,071 5.24 0.5509—08—95 10,177 5.29 0.4609—15—95 9946 5.17 0.4709—22—95 10,193 5.30 0.51
Average 0.54$0.07
BC—Black carbon.
Table 2Black carbon mass concentrations in near surface air in urbanHalifax (Winter season, 1996)
Sample Collection Total BCtime volume (kg m~3)(min) (m3)
02—05—96 10,067 4.168 1.8702—22—96 5669 6.457 1.2903—11—96 4368 4.492 2.003—14—96 5677 5.742 1.4503—19—96 1316 1.357 2.203—20—96 1464 1.509 2.003—21—96 1555 1.595 1.7703—22—96 4139 4.296 1.6803—25—96 1567 1.617 3.1003—26—96 2855 2.921 1.7003—28—96 1300 1.313 1.1704—03—96 426 0.429 1.2704—04—96 1096 1.104 1.14
Average 1.74$0.51
BC—Black Carbon.
The ratio of the black carbon to the total aerosol massis found to be about 1% in Summer and between 1.2%and 3.5% in Winter (Table 3). The total aerosol masswas determined gravimetrically before the black carbonanalysis.
Table 3Black carbon (BC) to total aerosol mass (TAM) ratio in urbanHalifax surface air
Sample TAM (kg) BC/TAM (%)
07—04—95 449 0.8008—11—95 270 1.108—25—95 292 0.803—14—96 720 1.203—21—96 110 2.603—25—96 160 3.103—26—96 140 3.503—28—96 120 1.3
4. Black carbon in rain water
Rain water was collected during several rainstorms in1993, 1995 and 1996 using a 20 cm diameter polyethylenefunnel. The sampling time ranged from 20 min to severalhours. The collecting funnel was rinsed with Super-Qwater before and after the collection. The rinse waterused to clean the containers was handled in a samemanner as the collected rain water. The collections weredone in urban as well as in rural areas. Longer lastingrainstorms were divided into several consecutive periodsto investigate possible changes in black carbon concen-tration during the storm.
Collected rain water was stored in a freezer until thetime of black carbon analysis. Melted water was passedthrough a quartz filter to collect aerosol particles. Theefficiency of filtration was tested using suspensions ofa known amount of carbon black particles in water. Theefficiency varied between 71% and 99% with an averagevalue of 88%. Quartz filters were then processed todetermine the amount of black carbon on the filters, ina way described earlier for air samples.
In the urban area (Table 4) the black carbon concen-tration varied between an undetectable amount and11 kg of black carbon per kg of rain water (11 kg kg~1),with an average concentration of 4.0 kg kg~1. For thecase of rain water collected in the rural area (Table 5),the range of black carbon concentration varied from anundetectable amount to 8.2 kg kg~1, with the averagevalue of 2.6 kg kg~1.
We note that there is no trend towards lower blackcarbon concentrations with the time of the rain duration.The five pairs of samples (8 June, 13 June, 14 September,6 October and 22 October 1995) do not show a decline ofblack carbon concentration in the second sample col-lected later in the storm. The sequences of three to sixconsecutive samples collected during longer lastingstorms (Tables 4 and 5) do not show any trend in blackcarbon concentration as a function of the time from thebeginning of a storm.
P. Chylek et al. / Atmospheric Environment 33 (1999) 2269—2277 2271
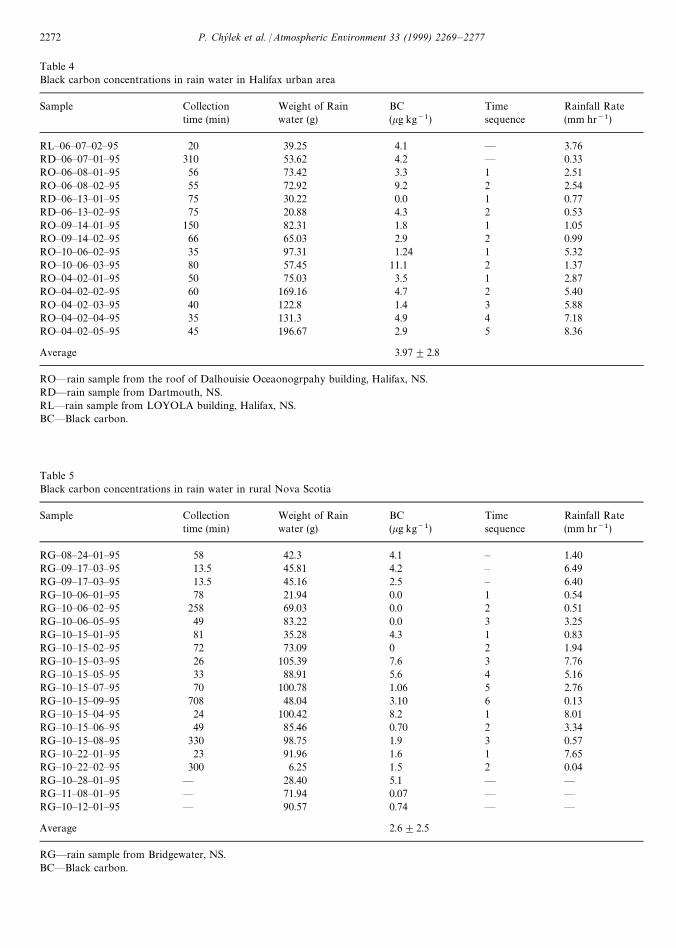
Table 4Black carbon concentrations in rain water in Halifax urban area
Sample Collection Weight of Rain BC Time Rainfall Ratetime (min) water (g) (kg kg~1) sequence (mm hr~1)
RL—06—07—02—95 20 39.25 4.1 — 3.76RD—06—07—01—95 310 53.62 4.2 — 0.33RO—06—08—01—95 56 73.42 3.3 1 2.51RO—06—08—02—95 55 72.92 9.2 2 2.54RD—06—13—01—95 75 30.22 0.0 1 0.77RD—06—13—02—95 75 20.88 4.3 2 0.53RO—09—14—01—95 150 82.31 1.8 1 1.05RO—09—14—02—95 66 65.03 2.9 2 0.99RO—10—06—02—95 35 97.31 1.24 1 5.32RO—10—06—03—95 80 57.45 11.1 2 1.37RO—04—02—01—95 50 75.03 3.5 1 2.87RO—04—02—02—95 60 169.16 4.7 2 5.40RO—04—02—03—95 40 122.8 1.4 3 5.88RO—04—02—04—95 35 131.3 4.9 4 7.18RO—04—02—05—95 45 196.67 2.9 5 8.36
Average 3.97$2.8
RO—rain sample from the roof of Dalhouisie Oceaonogrpahy building, Halifax, NS.RD—rain sample from Dartmouth, NS.RL—rain sample from LOYOLA building, Halifax, NS.BC—Black carbon.
Table 5Black carbon concentrations in rain water in rural Nova Scotia
Sample Collection Weight of Rain BC Time Rainfall Ratetime (min) water (g) (kg kg~1) sequence (mm hr~1)
RG—08—24—01—95 58 42.3 4.1 — 1.40RG—09—17—03—95 13.5 45.81 4.2 — 6.49RG—09—17—03—95 13.5 45.16 2.5 — 6.40RG—10—06—01—95 78 21.94 0.0 1 0.54RG—10—06—02—95 258 69.03 0.0 2 0.51RG—10—06—05—95 49 83.22 0.0 3 3.25RG—10—15—01—95 81 35.28 4.3 1 0.83RG—10—15—02—95 72 73.09 0 2 1.94RG—10—15—03—95 26 105.39 7.6 3 7.76RG—10—15—05—95 33 88.91 5.6 4 5.16RG—10—15—07—95 70 100.78 1.06 5 2.76RG—10—15—09—95 708 48.04 3.10 6 0.13RG—10—15—04—95 24 100.42 8.2 1 8.01RG—10—15—06—95 49 85.46 0.70 2 3.34RG—10—15—08—95 330 98.75 1.9 3 0.57RG—10—22—01—95 23 91.96 1.6 1 7.65RG—10—22—02—95 300 6.25 1.5 2 0.04RG—10—28—01—95 — 28.40 5.1 — —RG—11—08—01—95 — 71.94 0.07 — —RG—10—12—01—95 — 90.57 0.74 — —
Average 2.6$2.5
RG—rain sample from Bridgewater, NS.BC—Black carbon.
2272 P. Chylek et al. / Atmospheric Environment 33 (1999) 2269—2277
The relative difference in black carbon average con-centration in rain water in urban and rural areas (fromTables 4 and 5) 2(4.0!2.6)/(4.0#2.6)"0.42 is not solarge as the corresponding relative differences betweenblack carbon concentrations in air near the ground2(0.54!0.11)/(0.54#0.11)"1.32 (black carbon concen-tration in near surface rural air is from Chylek et al.,1996). The differences in concentrations of black carbonin surface air reflect the strength of local sources and itcharacterizes the amount of black carbon that is avail-able for scavenging. Since the black carbon concentra-tion in rain water is independent of the time from thebeginning of the storm, and since it is not directly propor-tional to the near-surface black carbon concentrations, itsuggests, that the major part of black carbon that isremoved by the rain gets into cloud droplets and/or icecrystals during the nucleation and activation processesrather than by scavenging.
There are three main processes contributing towardsthe scavenging of uncharged aerosols by water drops: (1)Brownian motion, which dominates for aerosol radii lessthan about 0.05 km, (2) inertial collection, which becomessignificant only for aerosols larger than a few microns insize, and (3) phoretic forces which are important forintermediate sizes.
The size distribution of the small black carbon par-ticles has been determined only on a very few occasions(Heintzenberg, 1982b; Pueschel et al., 1992; Parungo etal., 1994; Blake and Kato, 1995). Most of black carbonparticles are of a submicron size range with a typicalmode radius between 0.03 and 0.06 km. We considera black carbon particle of radius 0.05 km and density of1 g cm~3, to account for a composition of graphite andempty space (Horvath, 1993).
The half-life, t, of an interstitial aerosol with respect tothe convective Brownian capture by water drops is givenby (Section 17.4.2 of Pruppacher and Klett, 1997)
t"ln (2)oa2
1.35Dwf(1)
where o is the density of water, a is the mean radius of thewater drop, D is the diffusivity of black carbon in air, w isthe liquid water content. The ventilation coefficient, f, isan empirical enhancement of the particle flux due to theterminal velocity of water drops. Let us consider a case ofan average rain drop radius, a"1.0 mm, w"0.5 gm~3
(this corresponds to a rainfall rate of about 10 mmh~1)and f"9. For a 0.05 km radius carbon particle, D is ofthe order of 10~9 m2 s~1 and this gives a half-life forblack carbon of over 108 s, much too long for any signifi-cant scavenging by Brownian motion.
Phoretic forces will enhance the scavenging by rain-drops falling in sub-saturated air by a factor of about 2 atrelative humidities '75% (see Fig. 2 from Wang et al.,1978). Inertial scavenging by the rain drops at terminal
velocities is also negligible because the collision efficiencyis essentially zero for aerosol particles with radiusr(1 km. These theoretical considerations are in agree-ment with our measurements.
5. Black carbon concentration in snow
Snow samples were collected during several snowstorms during the 1995—1996 winter (Table 6) in theHalifax urban area and in a rural area near Bridgewater,Nova Scotia. Fresh snow was collected on polyethylenecontainers and scooped into glass bottles. Samples werekept frozen until analysis.
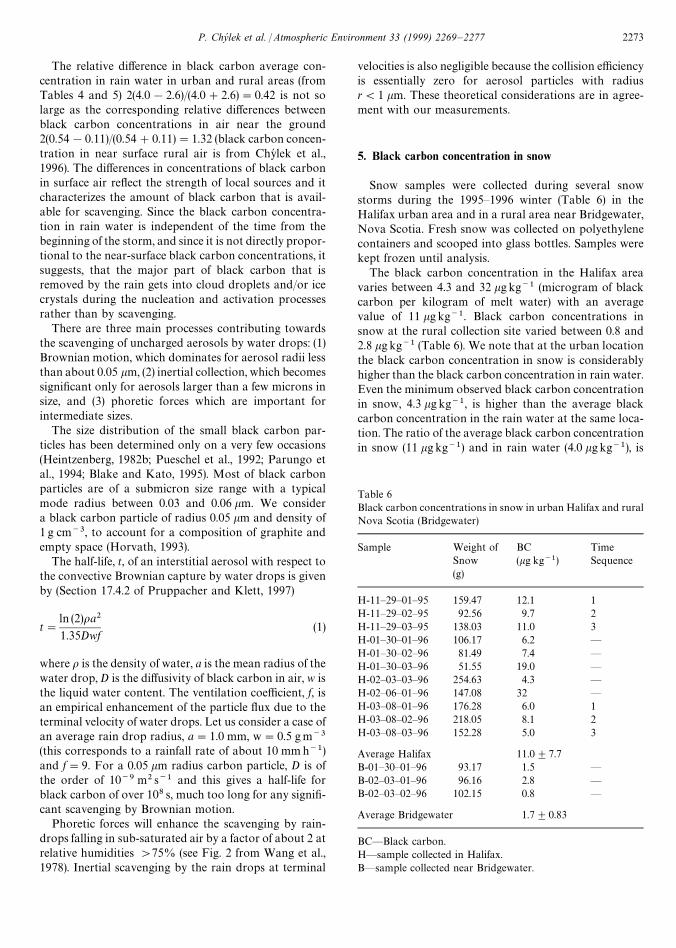
The black carbon concentration in the Halifax areavaries between 4.3 and 32 kg kg~1 (microgram of blackcarbon per kilogram of melt water) with an averagevalue of 11 kg kg~1. Black carbon concentrations insnow at the rural collection site varied between 0.8 and2.8 kg kg~1 (Table 6). We note that at the urban locationthe black carbon concentration in snow is considerablyhigher than the black carbon concentration in rain water.Even the minimum observed black carbon concentrationin snow, 4.3 kg kg~1, is higher than the average blackcarbon concentration in the rain water at the same loca-tion. The ratio of the average black carbon concentrationin snow (11 kg kg~1) and in rain water (4.0 kg kg~1), is
Table 6Black carbon concentrations in snow in urban Halifax and ruralNova Scotia (Bridgewater)
Sample Weight ofSnow
BC(kg kg~1)
TimeSequence
(g)
H-11—29—01—95 159.47 12.1 1H-11—29—02—95 92.56 9.7 2H-11—29—03—95 138.03 11.0 3H-01—30—01—96 106.17 6.2 —H-01—30—02—96 81.49 7.4 —H-01—30—03—96 51.55 19.0 —H-02—03—03—96 254.63 4.3 —H-02—06—01—96 147.08 32 —H-03—08—01—96 176.28 6.0 1H-03—08—02—96 218.05 8.1 2H-03—08—03—96 152.28 5.0 3
Average Halifax 11.0$7.7B-01—30—01—96 93.17 1.5 —B-02—03—01—96 96.16 2.8 —B-02—03—02—96 102.15 0.8 —
Average Bridgewater 1.7$0.83
BC—Black carbon.H—sample collected in Halifax.B—sample collected near Bridgewater.
P. Chylek et al. / Atmospheric Environment 33 (1999) 2269—2277 2273

Table 7Black carbon concentrations in cloud water
Experiment Mean BC Range BC LWC Reference(kg kg~1) (kg kg~1) (g m~3)
NARE (1993) 40 10—61 0.15—0.44 Chylek et al. (1996)RACE (1995) 16 8—41 0.10—0.56 Kou (1996)CALIFORNIA 23—79 0.24—0.31 Twohy et al. (1989)
BC—Black carbon.LWC—Liquid water content.
equal to 2.8. This value is quite close to the ratio betweenthe winter (1.7 kgm~3) and summer (0.54 kgm~3) blackcarbon concentrations in near surface air, which is equalto 3.1. Since the rain samples were collected during non-winter months, the similarity of the ratios can be ex-pected. The two time sequences of the collected samples(29 November 1995 and 8 March 1996) do not againshow any dependence on the time since the beginning ofthe snow storm.
The source for most non-summer precipitation overNova Scotia is nimbostratus with (1) weak uplift overa large horizontal area, (2) low cloud water content and(3) probable complete glaciation above the freezing level.In this type of synoptic system the black carbon aerosolsmay not be scavenged at all because the precipitationparticles are too large to present enough total surfacearea for Brownian capture and because of a lack ofpersistent cloud water. The relatively small amount ofblack carbon found in the precipitation may have beenscavenged near the top of clouds where numerous tiny icecrystals have resided long enough for capture to occur.
6. Black carbon in cloud water
For the purpose of the following discussion we reviewhere briefly the past measurements of black carbon con-centration in cloud water. Cloud water was collected inmarine stratus clouds over Nova Scotia in 1993 as a partof the NARE (North Atlantic Regional Experiment) fieldobservation program (Chylek et al., 1996) and in 1995during the RACE (Radiation, Aerosols, Cloud Experi-ment) measurements (Kou, 1996). The summary of thesemeasurements together with the concentration of blackcarbon in cloud water found in marine stratus off thecoast of southern California (Twohy et al., 1989) is givenin Table 7. The range of black carbon concentrations incloud water found in the three sets of measurements isfrom 8 to about 80 kg kg~1.
To estimate the rate of scavenging for the case of clouddroplets, we assume an average droplet radius around8 km and a liquid water content of 0.5 gm~3. Since the
terminal velocities of cloud droplets are negligible, wetake f"1. Using equation (1), this leads to an aerosolhalf-life of about 18 h. If the air flow carries the blackcarbon into a non-precipitating cloud and if the blackcarbon resides there for several hours, we can expecta portion of aerosols to be captured by cloud droplets.
7. Discussion
The measured summer black carbon concentra-tion in near surface air in urban Halifax (average of0.54 kgm~3) was found to be about factor of fivehigher that black carbon concentration (average of0.11 kgm~3) in rural Nova Scotia (Chylek et al., 1996).The winter time black carbon concentration in Halifaxincreases by an additional factor of three (to an averagevalue of 1.7 kgm~3) due to residential and commercialheating. The black carbon concentrations found in Hali-fax are of the same order of magnitude as black carbonconcentrations in other North American cities. Forexample, the black carbon concentration in air over theWashington DC area was found to be about 1.6 kgm~3
in summer and 2.0 kgm~3 in winter (Malm et al., 1994).Since the winter temperatures in the Washington DCarea are much milder than in Halifax, the winter increasein the black carbon concentration in Washington DC isonly about 25% compared to over 300% increase inHalifax.
The fact that the black carbon concentration in rainand snow is independent of the time from the beginningof the precipitation suggests that the amount of blackcarbon contained in the rain and snow is dominated bythe amount of black carbon present in the atmosphere atnucleation time or that gets incorporated into the clouddroplets by diffusion during the cloud’s lifetime. Theblack carbon measurements in air and precipitation sug-gest that scavenging by precipitation cannot explain theblack carbon concentrations found in the rain and snow.
A simple calculation allows us to estimate the fractionof cloud droplets that have black carbon inside them. Todo that we need some information concerning black
2274 P. Chylek et al. / Atmospheric Environment 33 (1999) 2269—2277

carbon size. Let us assume that on average there is oneblack carbon particle in each cloud droplet. Consideringthe mean droplet radius of marine stratus to be between6 and 8 km, we obtain the average black carbon concen-tration in cloud droplets between 244 and 579 kgkg~1
(if each droplet contains one black carbon particle of0.05 km radius). The range of measured black carbonconcentration in marine stratus (Table 7) is at least bya factor of 10 lower than the values obtained. Thus, themeasurements suggest that no more than 10% of themarine stratus cloud droplets and no more than 10% ofthe cloud condensation nuclei should contain a blackcarbon particle.
Next, we can estimate the fraction of black carbon thatis inside cloud droplets to form an internal mixture ofblack carbon and cloud water. During the NAREmeasurements (Chylek et al., 1996), the average atmo-spheric black carbon concentration in clean and pollutedatmospheres was around 0.03 and 0.23 kgm~3. The aver-age marine stratus liquid water content was about0.25 gm~3. If all the black carbon were internally mixedwithin the cloud drops, the expected black carbon con-centrations in cloud water would be 120 kg kg~1 forclean atmosphere and 920 kg kg~1 for the case of averagepolluted atmosphere. The mean observed black carbonconcentration in marine stratus cloud water duringthe NARE measurements (Table 7) is, however, only40 kgkg~1, suggesting that only between 4% and 33% ofblack carbon can be expected to be in the droplets(internal mixture).
We note that the measured black carbon concentra-tion in marine stratus cloud water (Table 7) is abouta factor of 10 higher than the black carbon concentrationin rain water (Tables 4 and 5). The rain water, however,did not come from marine stratus cloud but from deepstratiform clouds (usually with higher liquid water con-tent). Since a considerable portion of precipitating cloudwill be above the marine boundary layer, the air in theregion of cloud formation can be expected to be cleanercontaining less black carbon (unless a source of blackcarbon, like forest fore or extensive biosphere burning, isnearby).
Source regions for conservative tracers in dry air canbe estimated using back-trajectory analysis of measuredwind fields. Establishing the source regions of black car-bon found in rain and snow is more problematic becausethe mesoscale variability of clouds and precipitating sys-tems are not properly resolved by analyzed wind fieldswhich are primarily determined by the synoptic radio-sonde network. However, it is known that during thesummer months, the large-scale flow over Nova Scotia isoften dominated by weak cold frontal passages from thewest or northwest with only a few off-shore cyclogeneticevents (Merrill and Moody, 1996). The warm sector airthat is advected immediately ahead of the approachingcold fronts allow polluted air from the US northeast to
move over Nova Scotia. This scenario is less frequentduring the stormy winter season.
Most of the non-summer precipitation that falls onNova Scotia is associated with extratropical cyclonesthat typically develop off-shore to the east and south ofthe Atlantic coast. The high frequency of such cyclo-genetic events has been documented by Roebber (1984).The relatively low black carbon concentrations found inNova Scotia precipitation as compared to other NorthAmerican sites may be explained by having the conden-sed water originate from relatively clean conditions of themarine boundary layer in the warm sector to the south-east of a surface low, far to the east of the AmericanAtlantic seaboard.
References
Andreae, M.O., Andreae, T.W., Ferek, R.J., Raemdonck H.,1984. Long range transport of soot carbon in the marineatmosphere. Science of Total Environment 36, 73—80.
Andreae, M.O., Anderson, B.E., Blacke, D.R., Bradshaw, J.D.,Collins, J.E., Gregory, G.L., Sachse G.W., Shipham, M.C.,1994. Influence of plumes from biomass burning on atmo-spheric chemistry over equatorial and tropical south Atlanticduring CITE 3. Journal of Geophysical Research 99,12,793—12,808.
Blake, D., Kato, K., 1995. Latitudinal distribution of blackcarbon soot in the upper troposphere and lower stratosphere.Geophysical Research 100, 7195—7202.
Bodhaine, B.A., 1995. Aerosol absorption measurements at Bar-row, Mauna Loa and the South Pole. Journal of GeophysicalResearch 100, 8967—8975.
Cachier, H., Bermond, M.P., Buat-Menard, P., 1990. Organicand black carbon aerosol over marine regions of the Northernhemisphere. In: Newman, L., Wang, W., Kiang, C.S. (Eds.),Proceedings of International Conference on Global Atmo-spheric Chemistry, pp. 249—261. Brookhaven National Labor-atory Press, Brookhaven, NY.
Cachier, H., Ducret, J., 1991. Influence of biomass burning onequatorial African rain. Nature 352, 228—230.
Cachier, H., Liousse, C., Cachier, A., Ardouin, B., Polian, G.,Kazan, V., Hanson, A.D.A, 1994. Black carbon aerosols at theremote site of Amsterdam Island. Paper presented at TheFifth International Conference on Carbonaceous Particles inthe Atmosphere, U.S. Department of Energy, Berkeley, CA.
Chesselet, R., Fontugne, M., Buart-Menard, P., Ezat U.,Lambert, C.E., 1981. The origin of particulate organic inthe marine atmosphere as indicated by its stable carbonisotopic composition. Geophysical Research Letters 8,345—348.
Chow, C.J., John, G.W., Watson, G.J., Parichett, C.L., Pierson,R.W., Frazier, A.C., Purcell G.R., 1993. The DRI ther-mal/optical reflectance carbon analysis system: description,evaluation and applications in U.S. air quality studies.Atmospheric Environment 27A, 1185—1201.
Chuan, L.R., Woods, C.D., 1984. The appearance of carbonaerosol particles in the lower stratosphere. Geophysical Re-search Letters 11, 553—556.
P. Chylek et al. / Atmospheric Environment 33 (1999) 2269—2277 2275

Chylek, P., Ramaswamy, V., Cheng, R.J., 1984. Effect of graph-itic carbon on the albedo of clouds. Journal of AtmosphericScience 41, 3076—3084.
Chylek, P., Srivastava, V., Cahenzli, L., Pinnick, R.G., Dod, R.L.,Novakov, T., Cook, T.L., Hinds, B.D., 1987. Aerosol andgraphitic carbon content of snow. Journal of GeophysicalResearch 92, 9801—9809.
Chylek, P., Johnson, B., Wu, H., 1992a. Black carbon concentra-tion in Byrd Station ice core: from 13,000 to 700 years beforepresent. Annals of Geophysics 10, 625—629.
Chylek, P., Johnson, B., Wu, H., (1992b) Black carbon concen-tration in Greenland Dye-3 ice core. Geophysical ResearchLetters 19, 1951—1953.
Chylek, P., Johnson, B., Damiano, P., Taylor, K., Clement, P.,1995. Biomass burning record and black carbon in the GISP2Ice Core. Geophysical Research Letters 22, 89—92.
Chylek, P., Wong, J., 1995. Effect of absorbing aerosols onglobal radiation. Geophysical Research Letters 22, 929—931.
Chylek, P., Banic, C.M., Johnson, B., Damiano, P.A., Isaac,G.A., Leaitch, W.R., Liu, P.S.K., Boudala, F.S., Winter, B.,Ngo, D., 1996. Black carbon: atmospheric concentrationsand cloud water content measurements over southernNova Scotia. Journal of Geophysical Research 101,29,105—29,111.
Clarke, A.D., 1989. Aerosol light absorption by soot in remoteenvironments. Aerosol Science and Technology 10, 161—171.
Clarke, A.D., Noone, K.J., 1985. Soot in the arctic snowpack:a cause for perturbations in radiative transfer. AtmosphericEnvironment 19, 2045—2053.
Cooke, W.F., Wilson, J.J.N., 1996. A global black carbonaerosol model. Journal of Geophysical Research 101,19,395—19,409.
Ducret, J., Cachier, H., 1992. Particulate carbon in rain atvarious temperate and tropical locations. Journal of Atmo-spheric Chemistry 15, 55—67.
Dzubay, T.J., Stevens, R.K., Haagenson, 1984. Composition andorigins of aerosol at a forested mountain in Soviet Georgia.Environmental Science and Technology 18, 873—883.
Hansen, A.D.A., Bodhaine, B.A., Dutton, E.G., Schnell, R.C.,1988. Aerosol black carbon measurement at the South pole:initial results, 1986—1987. Geophysical Research Letters 15,1193—1196.
Haywood, M.J., Shine, K.P., 1995. The effect of anthropogenicsulfate and soot aerosol on the clear sky planetary radiationbudget. Geophysical Research Letters 22, 603—606.
Haywood, M.J., Roberts, L.D., Slingo, A., Edwards, M.J., Shine,K.P., 1997. General circulation model calculations of thedirect radiative forcing by anthropogenic sulfate andfossil-fuel soot aerosol. Journal of Climate 10, 1562—1577.
Heintzenberg, J., 1982a. Measurement of light absorption andelemental carbon in atmospheric aerosol samples from remotelocations. In: Particulate Carbon: Atmospheric Life Cycle,Wolff, G.T. and Klimisch, R.K. (eds.) pp. 371—377. PlenumPress, New York.
Heintzenberg, J., 1982b. Size-segregated measurements of par-ticulate elemental carbon and aerosol light absorption atremote Arctic locations. Atmospheric Environment 16,2461—2469.
Heintzenberg, J., Bigg, E.K., 1990. Tropospheric transport oftrace substance in the Southern Hemisphere. Tellus 42B,355—363.
Heintzenberg, J., Leck, C., 1994. Seasonal variation of the atmo-spheric aerosol near the top of the marine boundary layerover Spitsbergen related to the Arctic sulfur cycle. Tellus 46B,52—67.
Heintzenberg, J., Wendisch, M., 1996. On the sensitivity of cloudalbedo to the partitioning of the particulate absorbers incloudy air. Beitrage zur Physik der Atmosphere 69, 491—499.
Hopper, J.E, Barrie, L.A, Trivett, N.B., Worthy, D.J., 1991.Continuous measurements of black carbon and relatedspecies at Alert, Canada. Paper presented to The FourthInternational Conference on Carbonaceous Aerosols,Austrian Federal Ministries of Environment, Science andTechnology. Vienna, 3—5 April.
Horvath, H., 1993. Atmospheric light absorption — a review.Atmospheric Environment 27A, 293—317.
Jennings, G.S., O‘Dowd, C.D., 1990. Volatility of aerosol atMace Head on the West Coast of Ireland. Journal of Geo-physical Research 95, 937—948.
Jennings, G.S., McGovern, F.M., 1993. Carbon mass concentra-tion measurements at Mace Head on the west coast of Ireland.Atmospheric Environment 27A, 1229—1239.
Jennings, G.S., Geever, M., McGovern, F.M., Francis, J.,Spain, G., Donaghy, T., 1997. Micro—physical and physico—chemical characterization of atmospheric marine and conti-nental aerosol at Mace Head. Atmospheric Environment 31,2795—2808.
Kou, L., 1996. Black carbon: atmospheric measurements andradiative effect. Ph.D thesis, Dalhousie University, Halifax,Nova Scotia, Canada.
Kuhlbusch, T.A.J., Andreae, M.O., Cachier, H., Goldammer,J.G., Lacaux, J-P., Shea, R., Crutzen, P.J., 1996. Black carbonformation by savanna fires: measurements and implicationsfor the global carbon cycle. Journal of Geophysical Research101, 23,651—23,666.
Liousse, C., Penner, J.E., Chuang, C., Walton, J.J, Eddleman, H.,Cachier, H., 1996. A global three—dimensional model study ofcarbonaceous aerosols. Journal of Geophysical Research 101,19,411—19,432.
Malm, C.W., Sister, F.J., Huffman, D., Eldred, A.R., Cahill, A.T.,1994. Spatial and seasonal trends in particle concentrationand optical extinction in the United States. Journal of Geo-physical Research 99, 1347—1370.
Merrill, J.T., Moody, J.L., 1996. Synoptic meteorology andtransport during the North Atlantic Regional Experiment(NARE) intensive: overview. Journal of Geophysical Research101, 28,903—28,928.
Mukai, H., Ambe, Y., Shibata, K., Muku T., Takeshita, K.,Fukuma, T., Takahashi, J., Mizota, S., 1990. Long-term vari-ation of chemical composition of atmospheric aerosols on theOki Islands in the Sea of Japan. Atmospheric Environment24, 1379—1390.
O’Dowd, C.D., Smith, M.H., Jennings, S.G., 1993. Submicronparticle, radon and soot carbon characteristics over thenortheast Atlantic. Journal of Geophysical Research 98,1123—1135.
Ogren, J.A., Charlson, R.J., Groblicki, P.J., 1983. Determinationof elemental carbon in rainwater. Analytical Chemistry 55,1569—1572.
Ogren, J.A., Charlson, R.J., 1984. Measurement of the removalrate of elemental carbon from the atmosphere. Science ofTotal Environment 36, 329—338.
2276 P. Chylek et al. / Atmospheric Environment 33 (1999) 2269—2277

Parungo, F., Nagamoto, C., Zhou, M.Y., Hanson, A.D.A.,Harris J., 1994. Aeolian transport of aerosol black carbon fromChina to the ocean. Atmospheric Environment 28, 3251—3260.
Penner, J.E., Eddleman, H., Novakov, T., 1993. Towards thedevelopment of a global inventory for black carbon emissions.Atmospheric Environment 27A, 1277—1295.
Pinnick, R.G., Fernandez, G., Martinez-Andazola, E., Hinds,B.D., Hansen, A.D.A., Fuller, K., 1993. Aerosol in the aridsouthwestern United States: measurements of mass loading,volatility, size distribution, absorption characteristics, blackcarbon content and vertical structure to 7 km above sea level.Journal of Geophysical Research 98, 2651—2666.
Pruppacher, H.R., Klett, J., 1997. Microphysics of Clouds andPrecipitation, 2nd edn. Kluwer Academic Publishers, Dor-drecht.
Pueschel, R.F., Blake, D.F., Snetsinger, K.G., Hansen, A.D.A.,Verma, S., Kato, K., 1992. Black carbon soot aerosol in thelower stratosphere and upper troposphere. Geophysical Re-search Letters 19, 1659—1662.
Roebber, P.J., 1984. Statistical analysis and updated climatologyof explosive cyclones. Monthly Weather Review 112, 1577—1589.
Schult, I., Feicher, J., Cooke, F.W., Wilson, J.J.N., 1997. Theeffect of black carbon and sulfate aerosols on global radiationbudget. Journal of Geophysical Research 102, 30,107—30,118.
Twohy, C.H., Clarke, A.D., Warren, S.G., Radke, L.F., Charlson,R.J., 1989. Light-absorbing material extracted from clouddroplets and its effect on cloud albedo. Journal of Geophysi-cal Research 94, 8623—8631.
van Dingenen, R., Raes, F., Jensen, N.R., 1995. Evidence foranthropogenic impact on number concentration and sulfatecontent of cloud-processed aerosol particles over theNorth Atlantic. Journal of Geophysical Research 100,21,057—21,067.
Wang, P.K., Grover, S.N., Prupacher, H.R., 1978. On the effectof electric charges on the scavenging of aerosol particles byclouds and small raindrops. Journal of Atmospheric Science35, 1735—1743.
Warren, S.G., Clarke, A.D., 1990. Soot in the atmosphere andsnow surface of Antarctica. Journal of Geophysical Research95, 1811—1816.
Wolff, G.T., Countless, R.J., Groblickie, P.J., Ferman, M.A.,Cadle, S., Muhlbaier, 1981. Visibility-reducing species in theDenver ’Brown Cloud’: II sources and temporal patterns.Atmospheric Environment 15, 2485—2502.
Yaaqub, R.R., Davies, T.D., Jickells, T.D., Miller, J.M., 1991.Trace elements in daily collected aerosols at site in southeastEngland. Atmospheric Environment 25, 985—996.
P. Chylek et al. / Atmospheric Environment 33 (1999) 2269—2277 2277





























