Embed Size (px)
Citation preview

Caracterització de les alteracions moleculars de FLT3 en pacients pediàtrics afectes
de leucèmia aguda
Correlació amb l’activitat i expressió dels transportadors de fàrmacs derivats de nucleòsids
Albert Català i Temprano
Aquesta tesi doctoral està subjecta a la llicència Reconeixement- NoComercial – SenseObraDerivada 3.0. Espanya de Creative Commons. Esta tesis doctoral está sujeta a la licencia Reconocimiento - NoComercial – SinObraDerivada 3.0. España de Creative Commons. This doctoral thesis is licensed under the Creative Commons Attribution-NonCommercial-NoDerivs 3.0. Spain License.

1
CARACTERITZACIÓ DE LES ALTERACIONS MOLECULARS DE FLT3
EN PACIENTS PEDIÀTRICS AFECTES DE LEUCÈMIA AGUDA.
CORRELACIÓ AMB L’ACTIVITAT I EXPRESSIÓ DELS
TRANSPORTADORS DE FÀRMACS DERIVATS DE NUCLEÒSIDS
Tesi presentada per:
Albert Català i Temprano
Per aspirar al grau de Doctor en Medicina
Tesi dirigida per:
Dra. Mireia Camós i Guijosa i Dr. Marçal Pastor-Anglada
Facultat de Medicina
Universitat de Barcelona
Març del 2017

2

3
Dedicatòria
Als meus pares
A la Beti, en Lucas i en Pol

4

INDEX
5
ÍNDEX
I. AGRAÏMENTS 7
II. ABREUJAMENTS O ACRÒNIMS 13
III. GLOSARI DE GENS 19
IV. AJUDES 27
V. INTRODUCCIÓ: LEUCÈMIA AGUDA EN EL NEN I L’ADOLESCENT 31
1. Leucèmia aguda en el nen i l’adolescent
1.1. Hematopoesi fisiològica 33
1.2. Leucèmia aguda: definició i incidència 34
1.3. Etiologia 35
1.4. Fisiopatologia 41
1.5. Formes de presentació clínica 49
1.6. Diagnòstic 52
1.7. Biologia 55
Classificació 55
Subtipus genètics i moleculars 59
1.8. Factors pronòstics 66
1.9. Estratègies terapèutiques 74
Bases generals del tractament en LLA i LMA 74
Nous tractaments 79
1.10. Grups de pacients amb abordatge diferencial 81
Lactants 81
Pacients amb la Síndrome de Down 82
Adolescents i adults joves 83
2. FLT3 en leucèmia aguda pediàtrica 86
Implicacions pronòstiques de les alteracions de FLT3 en LA 88
Fàrmacs inhibidors de FLT3 89
3. Transportadors de nucleòsids en leucèmia aguda pediàtrica 92

INDEX
6
VI. HIPÒTESI I OBJETIUS 97
Hipòtesi 99
Objectius 100
VII. MATERIAL I MÈTODE 101
VIII. RESULTATS 109
Caracterització de les alteracions molecular s del receptor FLT3 en
pacients pediàtrics afectes de leucèmia aguda: incidència, correlació
amb variables clíniques i analítiques i impacte pronòstic
Correlació de FLT3 amb el transport intracel ·lular de citarabina per
hENT1. Impacte de diferent s inhibidors de FLT3 sobre el transport i
efectivitat de la citarabina
IX. ANNEX DE RESULTATS 135
Article: “FLT3 is implicated in cytarabine transport by human equilibrative
nucleoside transporter 1 in pediatric acute leukemia” 137
X. DISCUSSIÓ 155
XI. CONCLUSIONS 169
XII. BIBLIOGRAFIA 173

AGRAÏMENTS
7
I. AGRAÏMENTS

AGRAÏMENTS
8

AGRAÏMENTS
9
AGRAÏMENTS
Diferents motivacions m’han portat a redactar aquest treball de tesi:
Primer de tot perquè és un treball que ha comportat un temps molt important de la
meva vida professional i personal. Hi ha un esforç immens darrere d’aquest projecte,
tant personal, com de molta gent que m’hi ha ajudat en algun moment.
Per compromís amb la meva família, en Marçal sempre m’havia insistit en la
importància d’aquest projecte. És una cosa que he “mamat” des de petit..
Per últim, però potser més important, per la meva manera d’entendre la vida; el poder
comprendre el perquè de les coses, la interrogació constant.. Haver contribuït amb
aquesta tesi a comprendre una mica millor la biologia de les leucèmies, és un plaer
indescriptible. Espero que aquest neguit m’acompanyi durant tota la meva vida
professional i no professional.
Persones a qui vull agrair aquest treball:
Primer de tot, als meus directors de tesi, la Mireia i en Marçal:
Mire, la meva “pepito grillo”, està clar que sense tu això no seria possible. Amb tu es
va acabar la meva llarga “travesía en el desierto”. He conegut poca gent amb tanta
empenta, intel·ligència i dedicació. Espero continuar baixant al lab per compartir
projectes científics, i algun aperitiu..
Marçal, que et puc dir que no sàpigues, probablement una de les persones que més
m’ha impressionat i que més influència ha tingut en la meva vida científica (i no
científica). Després de tants sopars sentint parlar de transportadors.., alguna cosa se’m
va quedar, no ?

AGRAÏMENTS
10
Als meus companys de la clínica diària: Rubén y Anna, gracias por toda vuestro apoyo
y amistad, por cubrirme en la consulta y hacer que el Lunes no sea un drama..
Susana, no tinc paraules, com a professora i companya has estat i seràs excel·lent.
Treballar amb tots vosaltres és un luxe. Us trobaré molt a faltar.
Teresa, de tu he après a nivell professional i com a persona. Gràcies per sempre estar
allà.
Jesús, siempre serás “el jefe”. Tú me diste la oportunidad de trabajar en esto y nunca
dejaré de agradecértelo, ya lo sabes.
Montse, la meva companya “mieloide” i de penes de tesi. Està clar que la senzillesa no
està renyida amb l’excel·lència, a tots nivells..
Gràcies a tots els màsters i fellows que m’heu ajudat aquests anys: Ana, Montse, Mia,
Isabela, Georgina, Raquel, Nuria, María, Ignacio, Vicky, Geno, Antonio.. El vostre treball
és imprescindible perquè tot plegat funcioni. Mis compañeros de despacho: Ali,
Vicente y Moira, gracias por compartir muchos desayunos y muchas risas (y alguna
lagrima).
Gràcies a la Isabel Badell i a totes les companyes de TPH: Júlia, Cata, Maria, Luisa.
Gràcies a la gent del lab: Camino, gracias por ayudarme con las PCRs y otros
menesteres, contigo la parte experimental se aceleró significativamente. Gràcies a la
Rosa, la Celi, en Justo, a l’Emili, a la Lourdes, Susanna..
També vull recordar-me de l’Esperanza Tuset i de la Carmen de Torres. Con vosotras
empecé esta andadura..
Gràcies a les meves compis de tesi: Nerea & Roberta.
No vull descuidar-me de totes les infermeres, auxiliars, administratives.., que han
compartit durant aquest anys moltes alegries i alguna pena: Pepi, Anna, Mari, Cris
vàries, Mercé, Ade, Ave, Laura, Yoli, Viky, Isa, Jose, Montse, Marisa, Carmen, Tina,
Sveta, Natàlia, Rocío, Saray, Mariones,..

AGRAÏMENTS
11
La meva família és també còmplice de tot plegat:
A la meva mare, sempre vas estar quant et necessitava. Em vas ensenyar a ser
perseverant i a no rendir-me davant les adversitat i sobretot a ser honest i just.
Irene, la meva germaneta, tu també m´has acompanyat en aquest camí, gràcies per tot
el teu carinyo i estimació.
Jan, gràcies també per resoldre’m els dubtes informàtics i altres..
Pare, gràcies pel teu recolzament i per inculcar-me la recerca del “com” i del “per-
què”.
Beti, el motor de mi vida, tu impronta ha calado muy hondo.. Contigo todo es más fácil
y está claro que no hubiera habido tesis sin ti; por tu soporte con los niños, con la casa,
con las mudanzas, con las obras !.. y, sobre todo, por soportarme a mí. Espero que lo
continúes haciendo el resto de nuestras vidas.
Per en Lucas i en Pol, de moment l’únic que enteneu és que el papa es passa moltes
(massa) hores a la feina.. espero que algun dia sigueu capaços d’entendre tot el que
faig i pugueu gaudir de les vostres feines tant com jo ho faig de la meva.
Tot i que tot plegat soni a acomiadament o a final.. res més lluny, ara comença el més
interessant !

AGRAÏMENTS
12

ABREUJAMENTS I ACRÒNIMS
13
II. ABREUJAMENTS I ACRÒNIMS

ABREUJAMENTS I ACRÒNIMS
14

ABREUJAMENTS I ACRÒNIMS
15
ABREUJAMENTS I ACRÒNIMS
AD: autosòmic dominant
ADNc: ADN complementari
AN: anàlegs de nucleòsids
AR: autosòmic recessiu
Ara-G: anàleg de l’arabinòsid de guanina
Ara-GTP: arabinòsid de guanosin trifosfat
AVC: accident vascular cerebral
BFM: Berlin Frankfurt Münster
CARTs: chimeric antigen receptor-modified T cells
CBF: Core binding factor
CD: cluster of differentiation
CID: coagulació intravascular disseminada
CNA: copy number alterations
CNT: concentrative nucleoside transporter
COG: Children Oncology Group
DCFI: Dana Farber Cancer Institute
dCK: deoxycytidine kinase
DIC: drug interaction coefficient, coeficient d'interacció de fàrmacs
DNA: deoxyribonucleic acid o ADN (àcid desoxiribonucleic)
DNE: donant no emparentat
EGIL: European Group of Immunological classification of Leukemias
EM: enzim metabolitzador (d’anàlegs de nucleòsids)
ENT: equilibrative nucleoside transporter
EsPhALL: European Intergroup Stu dy on Post Induction Treatment of Philadelphia
Positive Acute Lymphoblastic Leukaemia with Imatinib .
ETP: Early-T cell Precursor o precursor immadur de les cèl·lules T
FAB: classificació de leucèmies French American British
FISH: fluorescence in situ hybridization o hibridació in situ fluorescent
FLT3wt: FLT3 wild type (no mutat)
FRALLE: French Acute Lymphoblastic Leukaemia Study Group

ABREUJAMENTS I ACRÒNIMS
16
GMALL: German Multicenter Acute Lymphoblastic Leukemia
HeH: Alta hiperdiploïdia (51-67 cromosomes)
HLA: Human Leukocyte Antigen
HSC: Hematopoetic Stem Cell
I-BFM: grup internacional BFM
ITD: internal tandem duplication
LA: leucèmia aguda
LAIP: leukemia-associated immunophenotype
LCR: líquid cefaloraquidi
LDH: lactat deshidrogenasa
LLA: leucèmia limfoblàstica aguda
LLA-B: leucèmia limfoblàstica aguda de fenotip B
LLA Ph+: leucèmia limfoblàstica aguda amb cromosoma Philadelphia
LLA-T: leucèmia limfoblàstica aguda de fenotip T
LMA: leucèmia mieloblàstica aguda
LMC: leucèmia mieloide crònica
LPA: leucèmia promielocítica aguda
LSCs: leukemic stem cells o cèl·lules mare leucèmiques
MECR: malaltia de l’empelt contra el receptor
MRM: malaltia residual mínima
MO: moll de l’os
MPAL: mixed phenotype acute leukemia o leucèmies de fenotip mixt
MTHFR: enzim metilen-tetrahidrofolat reductasa
NCI: National Cancer Institute
NOPHO: Nordic Society for Pediatric Hematology and Oncology
PCR: polymerase chain reaction o reacció en cadena de la polimerasa
RQ-PCR: real time quantitative PCR o PCR quantitativa a temps real
PETHEMA: Protocolo para el Estudio y Tratamiento de las Hemopatías Malignas
PKA: proteïna cinasa A
PKC: proteïna cinasa C
PL: punció lumbar
PNP: purine nucleoside phosphorylase

ABREUJAMENTS I ACRÒNIMS
17
RC: remissió completa
RCT: receptor de cèl·lula T
RNA: ribonucleic acid o ARN (àcid ribonucleic)
S: (herència) esporàdica
SD: síndrome de Down
SEER: US National Cancer Institute’s Surveillance, Epidemiology and End Results (SEER)
program
SG: supervivència global
SHOP / SEHOP: Sociedad Española de Hematología y Oncología Pediátrica
SJCRH: Saint Jude Children’s Research Hospital
SLE: supervivència lliure d’esdeveniment
SLT: síndrome de lisi tumoral
SMT: síndrome mieloproliferativa transitòria
SNC: sistema nerviós central
SOC: standard of care
TCR: T-cell receptor o RCT, receptor de cèl·lules T
TKD: tyrosin kinase domain
TKi: tyrosin kinase inhibitor o inhibidor de tirosín-cinases
TN: transportador de nucleòsid
TPH: trasplantament de progenitors hemopoètics
TPMT: enzim tiopurin-metiltransferasa
UKALL: United Kingdom Acute Lymphoblastic Leukemia Group
WGA: whole genome amplification
WHO: World Health Organization (Organització Mundial de la Salut, OMS)

ABREUJAMENTS I ACRÒNIMS
18

GLOSARI DE GENS
19
III. GLOSARI DE GENS

GLOSARI DE GENS
20

GLOSARI DE GENS
21
GLOSARI DE GENS
Símbol
aprovat*
Nom aprovat Localització Altres símbols
ABL1 v-abl Abelson murine leukemia viral
oncogene homolog1
9q34.1 ABL, JTK7, C-
ABL, p150, v-abl
AKT1 v-akt murine thymoma viral oncogene
homolog 1
14q32.32-
q32.33
AKT
ARID5B AT rich interactive domain 5B (MRF1-like) 10q11.22
BCL11B B-cell CLL/lymphoma 11B (zinc finger
protein)
14q32
BCL2 B-cell CLL/lymphoma 2 18q21.3
BCL9 B-cell CLL/lymphoma 9 1q21
BCR Break cluster region 22q11
BMI1 BMI1 polycomb ring finger oncogene 10p13
CBS cystathionine-beta-synthase 21q22.3 HIP4
CDKN1B cyclin-dependent kinase inhibitor 1B (p27,
Kip1)
12p13.1-p12
CDKN2A cyclin-dependent kinase inhibitor 2A 9p21 CDK4 inhibitor
p16INK4, ARF,
p14ARF
CDKN2B cyclin-dependent kinase inhibitor 2B (p15,
inhibits CDK4)
9p21 CDK4 inhibitor
p15INK4b, p15INK4
CEBPE CCAAT /enhancer binding protein (C/EBP),
epsilon
14.q11.2
CEBPA CCAAT/enhancer binding protein (C/EBP),
alpha
19q13.1
CEBPB CCAAT/enhancer binding protein (C/EBP),
beta
20q13.1
CEBPD CCAAT/enhancer binding protein (C/EBP),
delta
8p11.2-p11.1
CEBPG CCAAT/enhancer binding protein (C/EBP),
gamma
19q13.2
CREBBP CREB binding protein 16p13.3

GLOSARI DE GENS
22
Símbol
aprovat*
Nom aprovat Localització Altres símbols
CRLF2 cytokine receptor-like factor 2 Xp22.3 i
Yp11.3
CTGF connective tissue growth factor 6q23.2 CCN2
EBF1 connective tissue growth factor 5q34 EBF
EED embryonic ectoderm development 11q14.2-
q22.3
EML1 echinoderm microtubule associated
protein like 1
14q32
EPOR erythropoietin receptor 19p13.3-
p13.2
EPS8 epidermal growth factor receptor
pathway substrate 8
12p12.3
EPS15 epidermal growth factor receptor
pathway substrate 15
1p32
ERG v-ets erythroblastosis virus E26 oncogene
homolog (avian)
21q22.3
ETV6 ets variant 6 12p13 TEL
EZH2 enhancer of zeste homolog 2 (Drosophila) 7q35-q36
FBXW7 F-box and WD repeat domain containing
7, E3 ubiquitin protein ligase
4q31.23
FLT3 fms-related tyrosine kinase 3 13q12
GATA1 GATA binding protein 1 Xp11.23
GATA2 GATA binding protein 2 3q21.3
GATA3 GATA binding protein 3 10p15
HLF hepatic leukemia factor 17q22
HOXA@ homeobox A cluster 7p15.2 HOXA
ID4 inhibitor of DNA binding 4, dominant
negative helix-loop-helix protein
6p22.3
IGF2BP1 insulin-like growth factor 2 mRNA binding
protein 1
17q21.32
IGH immunoglobulin heavy locus 14q32.33
IKZF1 IKAROS family zinc finger 1 (Ikaros) 7pter-7qter

GLOSARI DE GENS
23
Símbol
aprovat*
Nom aprovat Localització Altres símbols
IKZF2 IKAROS family zinc finger 2 (Helios) 2q13-1
IKZF3 IKAROS family zinc finger 3 (Aiolos) 17q11.2
IL3 interleukin 3 (colony-stimulating factor,
multiple)
5q23-q31
IL7R interleukin 7 receptor 5p13
IRX2 iroquois homeobox 2 5p15.33
JAK1 Janus kinase 1 1p32.3-p31.3
JAK2 Janus kinase 2 9p24
JAK3 Janus kinase 3 19p13-p12
LEF1 lymphoid enhancer-binding factor 1 4q23-q25
LHX3 LIM homeobox 3 9q34.3
LMO1 LIM domain only 1 (rhombotin 1) 11p15
LMO2 LIM domain only 1 (rhombotin like-2) 11p13
LMO3 LIM domain only 1 (rhombotin like-1) 12p13
LYL1 lymphoblastic leukemia derived sequence
1
19p13.2
MLL myeloid/lymphoid or mixed-lineage
leukemia (trithorax homolog, Drosophila)
11q23 KMT2A, ALL-1,
TRX1, HTRX1,
CXX7, MLL1A,
MLLT1 myeloid/lymphoid or mixed-lineage
leukemia (trithorax homolog, Drosophila);
translocated to, 1
19p13.3 ENL, LTG19,
YEATS1
MLLT3 myeloid/lymphoid or mixed-lineage
leukemia (trithorax homolog, Drosophila);
translocated to, 3
9p21 AF9, YEAST3
MLLT4 myeloid/lymphoid or mixed-lineage
leukemia (trithorax homolog, Drosophila);
translocated to, 4
6q27 AFF1, AF4,
PBM1
MLLT10 myeloid/lymphoid or mixed-lineage
leukemia (trithorax homolog, Drosophila);
translocated to, 10
10p12 AF10

GLOSARI DE GENS
24
Símbol
aprovat*
Nom aprovat Localització Altres símbols
MYCv-myb myeloblastosis viral oncogene homolog (avian)
8q24 C-MYC
NEGR1 neuronal growth regulator 1 1p31.1
NF1 neurofibromin 1 17q11.2
NKX2.1 NK2 homeobox 1 14q13.3
NKX2.2 NK2 homeobox 2 20p11.22
NOTCH1 NOTCH1 9q34.3 TAN1, hN1
NRAS neuroblastoma RAS viral (v-ras) oncogene
homolog
1p13.2 N-ras, ALPS4
NUP214 nucleoporin 214kDa 9q34
OLIG2 oligodendrocyte transcription factor 2 21q22.11 BHLHB1
P2RY8 purinergic receptor P2Y, G-protein
coupled, 8
Xp22.33 and
Yp11.3
PAX5 paired box 5 9p13.2
PBX1 pre-B-cell leukemia homeobox 1 1q23.3
PDE4B Phosphodiesterase 4B 1p31.3
PDGFRB platelet-derived growth factor receptor,
beta polypeptide
5q33.1 PDGFR1
PHF6 PHD finger protein 6 Xq26.3
PIK3CA phosphatidylinositol-4,5-bisphosphate 3-
kinase, catalytic subunit alpha
3q26.3 PI3K
PICALM phosphatidylinositol binding clathrin
assembly protein
11q14 CALM

GLOSARI DE GENS
25
Símbol
aprovat*
Nom aprovat Localització Altres símbols
PIP4K2A phosphatidylinositol-5-phosphate 4-
kinase, type II, alpha
10p12.2 PIPK
PNP purine nucleoside phosphorylase 14q13.1 PUNP
PTEN phosphatase and tensin homolog 10q23 MMAC1
RB1 retinoblastoma 1 13q14.2
RUNX1 runt-related transcription factor 1 21q22.3 AML1, CBFA2
SET SET nuclear oncogene 9q34
SLC29A1 solute carrier family 29 member 1 6p21.2-p21.1 hENT1
SLC29A2 solute carrier family 29 member 2 11q13.2 hENT2
STAT@ signal-transducer and activator of
transcription protein familyVariable
STIL SCL/TAL1 interrupting locus 1p32
SUZ12 SUZ12 polycomb repressive complex 2
subunit
17q21
TACC2 transforming, acidic coiled-coil containing
protein 2
10q26
TAL1 T-cell acute lymphocytic leukemia 1 1p32 SCL, TCL5, bHLHa17
TAL2 T-cell acute lymphocytic leukemia 2 9q32
TCF3 transcription factor 3 19p13.3 E2A
TCRA T cell receptor alpha locus 14q11.2
TCRB T cell receptor beta locus 7q34
TCRD T cell receptor delta locus 14q11.2
TCRG T cell receptor gamma locus 7p14
TLX1 T-cell leukemia homeobox 1 10q24.32 HOX11, TCL3
TLX3 T-cell leukemia homeobox 3 5q35.1 HOX11L2
TP53 tumor protein p53 17p13.1 P53
TPD52 tumor protein D52 8q21
TPMT thiopurine S-methyltransferase 6p22.3
TYK2 tyrosine kinase 2 19p13.2
WT1 Wilms tumor 1 11p13
*Segons nomenclatura HUGO (http://www.gene.ucl.ac.uk/nomenclature)

AJUDES
26

AJUDES
27
IV. AJUDES

AJUDES
28

AJUDES
29
AJUDES
Ajudes institucionals
Projecte PI12/2417 Plan Nacional de I+D+I, cofinançat per Instituto de Salud Carlos III
(ISCIII) – Subdirección General de Evaluación y Fomento de la Investigación Sanitaria –
, Fondo Europeo de Desarrollo Regional (FEDER), SAF2011-23660 i SAF2014-52067-R.
Asociación Española Contra el Cáncer AECC, convocatòria Càncer Infantil 2012.
Fundación Sandra Ibarra de Solidaridad Frente al Cáncer, convocatòria Beca
Karactermanía 2013.
Fundación Cris contra el Cáncer.
Fundación Pelayo.
Mútua de Granollers.
Ajudes no institucionals
Projecte “Força Miquel”, Associació “Mua” i Associació “Candela, polsera solidària” i
aportacions de famílies de pacients, particulars i empreses, a través de l’Obra Social
de l’Hospital Sant Joan de Déu.

INTRODUCCIÓ
30

INTRODUCCIÓ
31
V. INTRODUCCIÓ: LEUCÈMIA AGUDA EN EL NEN I L’ADOLESCENT

INTRODUCCIÓ
32

INTRODUCCIÓ
33
1. LEUCÈMIA AGUDA EN EL NEN I L’ADOLESCENT
1.1 Hematopoesi fisiològica
L’hemopoesi es produeix en diferents òrgans i/o teixits al llarg dels p rimers mesos de
vida embrionària. S’inicia al sac de Yolk durant el període més prematur i
progressivament es va desplaçant principalment al fetge i marginalment a la melsa
durant el segon trimestre. El moll de l’os no constitueix el principal òrgan involu crat
fins al tercer trimestre. Aquest esdevindrà el principal òrgan responsable de
l’hemopoesi durant tota la vida de l’individu. En els nens l’hemopoesi predomina en el
moll de l’os dels ossos llargs, mentre que en l’adult predomina en els ossos plans com
el crani, la pelvis o les vèrtebres.
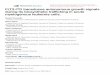
Totes les cèl·lules hemopoètiques s’originen a partir d’una cèl·lula mare pluripotencial
(figura 1). A partir d’aquesta cèl·lula se’n produiran d’altres que s’aniran diferenciant a
les diferents cèl·lules que conformen el sistema hemopoètic i amb les seves particulars
responsabilitats fisiològiques.
La vida mitjana de les cèl·lules que conformen el sistema hematopoètic és molt
variable, oscil·lant entre els 120 dies de vida que pot tenir un hematie a les 8 hores
d’un granulòcit. Per tant, per un normal funcionament, és imprescindible que el
sistema hemopoètic sigui un procés continu de proliferació i diferenciació.

INTRODUCCIÓ
34
Figura 1. Hematopoesi fisiològica. Adaptat de [1].
1.2 Leucèmia aguda: definició i incidència
Les leucèmies agudes són un grup heterogeni de malalties neoplàsiques
caracteritzades per la transformació maligna i expansió clonal de progenitor s
hematopoètics immadurs de línia limfoide (leucèmia limfoblàstica aguda, LLA) o
mieloide (leucèmia mieloblàstica aguda, LMA). Dins les LLA podem distingir les
leucèmies de línia B (LLA-B) i les de línia T (LLA-T).
La incidència anual en l’edat pediàtrica (per milió d’habitants) de la LLA i la LMA és de
39,9 i 8,1, respectivament ( US Cancer Institute’s Surveillance, Epidemiology and End
Results (SEER) program [2][3]). En les LLA existeix un pic d’incidència entre els 2-5 anys,
mentre que la LMA és més freqüent per sota de ls dos anys i després es manté estable
en l’edat pediàtrica i augmenta progressivament amb l’edat (figura 2) [4][5]. Les
leucèmies agudes són les neoplàsies més freqüents en l’edat pediàtrica, representen
un 32% dels càncers durant aquest p eríode (menors de 15 anys) [6]. La forma limfoide
és la més freqüent i la LLA constitueix el 83% de les leucèmies pediàtriques. Existeixen
diferències geogràfiques i racials (figura 3), sent menys freqüents en població de raça
negra, especialment les L LA, i més freqüents en raça hispànica, on són especialment

INTRODUCCIÓ
35
freqüents alguns subtipus d e LLA d’alt risc o la leucèmia promielocítica aguda (LPA)
[4][7].
Figura 2 . Incidència de la LLA i la LMA segons grups d’edat ( SEER program ,
http://seer.cancer.gov).
Figura 3. Incidència de la LLA pediàtrica segons ètnia. Reproduït de Xu H., et al. [8].
1.3 Etiologia
Les leucèmies es desenvolupen perquè hi ha un aug ment de la capacitat proliferativa i
una aturada en la diferenciació durant el procés de maduració del progenitor
hemopoètic. Depenent del precursor hematopoètic que pateix la transformació
leucèmica, el seu estadi maduratiu i la seva capacitat de diferenc iació cap a un
determinat llinatge, diferenciarem leucèmies de línia limfoide o mieloide.

INTRODUCCIÓ
36
Donada la gran diversitat biològica que presenten les leucèmies agudes és molt
improbable que tots els casos tinguin una sola etiologia; resulta més versemblant que
les leucèmies es deguin a la convergència de múltiples factors genètics i ambientals,
que podrien variar segons el subtipu s de leucèmia (figura 4). Així, s’hipotetitza que la
susceptibilitat g enètica modulada per factors ambientals seria la responsable d’ un
estat pre -leucèmic; l ’adquisició de nous e sdeveniments genètics promouria la
progressió a una LA.
Figura 4. Diagrama de causalitat de LA pediàtrica. L’e sdeveniment genèt ic primari
modulat per la convergència d’exposicions a factors externs (p.e . infeccions,
genotòxics...) i la susce ptibilitat gen ètica hereditària, promourien una sèrie
d’esdeveniments genètics secundaris que donarien lloc a una LA. Adaptat de [9].
Tot i que les causes específiques de la majoria de casos de leucèmia no es coneixen,
varis factors ambientals i demogràfics s’han associat a un augment de risc de patir
aquestes malalties. Aquests factors va n des de determinades substàncies químiques
com el benzè a les radiacions ionitzants [10]. En el cas de la leucèmia del lactant,
sembla que l'exposició transplacentària a inhibidors de la topoisomerasa -II pot tenir un
paper important en l'origen de la leucèmia. S'ha observat, tant en leucèmies del

INTRODUCCIÓ
37
lactant com en les leucèmi es secundàries a inhibidors de topoisomerasa -II amb
reordenament del gen MLL, que presenten més freqüentment el punt de trencament
en l’intró 11 [11]. Aquest intró conté múltiples dominis d'unió a una topoisomerasa -II i
això li conferiria una gran vulnerabilitat a determinades substàncies genotòxiques.
Diferents components naturals i sintètics tenen activitat enfront de la topoisomerasa-II
(quinolones, flavonoides, etc., presents en determinats aliments i begudes com el te).
Tot i que l’activitat d’aquests és molt més feble que la dels fàrmacs quimioteràpics, la
capacitat detoxificadora podria ser inferior en el fetus en algunes gestants que
tinguessin polimorfismes genètics associats a una menor activitat enzimàtica [12].
En la majoria de casos no hi ha factors genètics hereditaris coneguts i només en un
percentatge molt petit de casos (al voltant d’5% de casos) les LA en l’edat pediàtrica es
desenvolupen en pacients amb una malaltia genètica subjacent amb predisposició a la
leucèmia (taula 1) [13].
Taula 1. Síndromes predisponents a LLA o LMA [14]
Síndrome predisponent
Gen Locus Herència Tipus de leucèmia associada
Síndrome de Down No gen específic
S SMT, LMA <3 anys, LLA-B >3
anysSíndromes en relació a gens supressors
Síndrome de Li-Fraumeni
TP53, CHEK2
17p13.1,22q12.1 AD LLA-B hipodiploides
Síndromes d’inestabilitat cromosòmica
Mismatch repaircancer syndrome
MLH1, MSH2,MSH6, PMS2
3p22.2, 2p21,2p16, 7p22.2
AD LLA-T
Anèmia de Fanconi FANC A-E,BRCA
16q24.3, 9q22 AR >LMA
Atàxia telangiectàsia ATM 11q22.3 AR LLA-T

INTRODUCCIÓ
38
Síndrome de Nijmegen
NBS1 8q21.3 AR LLA-T
Síndrome de Bloom BLM 15q26.1 AR ?
Mutacions via RAS, inclòs NF1NF1/PTPN11/NRAS/KR
AS
17q11.2/12q24.1/12p12/1p22-p32
AD LLA/LMMJ/LLA ?
Síndromes congènites d’insuficiència medul·lar Anèmia de Blackfan-Diamond
RPS19, RPL5,RPL11
19q13.2,1p22.1, 1p35
AD, AR, S >LMA
Disqueratosi congènita
DKC1, TERC, TERT,
RTEL1, TINF2,
NOP10,WRAP53
3q26,5p15.33,20q13.3,14q.12,15q14.q
15,17p13.1
AD, AR, S, LX
>LMA
Síndrome TAR RBM8A 1q21.1 AR, S ?
Síndrome de Shwachman- Diamond
SBDS 7q11.21 AR >LMA
Neutropènia congènita
ELANE/HAX1
19q13.31q21.3 AR,AD LMA
Immunodeficiències
Síndrome de Wiskott-Aldrich
WASP Xp11.23 LX LLA
Agammaglobulinèmia de Brutton
BTK Xq22.1 LX LLA
LLA familiar
Síndrome PAX5 PAX5 9p13.1 AD LLA-B
Síndrome ETV6 ETV6 12p13 AD LLA-B
Síndromes LMA/SMD familiars
Síndrome GATA2 GATA2 3q21.3 AD,S LMA, SMD amb monosomia 7
CEBPA CEBPA 19q13.11 AD LMARUNX1 RUNX1 21q22.12 AD LMAAD: autosòmic dominant; AR: autosòmic recessiu; LX: lligat al cromosoma X; S: esporàdic

INTRODUCCIÓ
39
Destaquem alguna particularitat etiològica segons si es tracta de LLA o LMA.
Factors etiològics en LLA
Recentment s'ha descrit l'associació entre certs polimorfismes genètics en la població i
risc augmentat de desenvolupar LLA pediàtrica. Així, determinats polimorfismes en els
gens ARID5B, IKZF1, CEBPE, BMI1 -PIP4K2A i CDKN2A, s'associen a una major
predisposició a patir una LLA [8]. Els polimorfismes en el gen ARID5B associats a un
major risc de desenvolupar LLA són menys freqüents en la raça negra i més freqüents
en els hispano-americans, el que podria explicar les diferències en la incidència de LL A
entre races [15]. A més, determinats polimorfismes genètics s'associen específicament
a subgrups biològics de LLA; així, determinades variants polimòrfiques en els gens
ARID5B i IKZF1 s'associen a la LLA amb hiperdiploïdia [16]. Les alteracions germinals en
factors de transcripció limfoides com PAX5 també s’han relacionat amb un estat pre -
leucèmic que pot donar lloc a una LLA en un 30% dels portadors [17].
Les infeccions han estat descrites en l’últim segle com un dels principals factors
causants de leucèmia en l’edat pediàtrica. D ues hipòte sis han tractat d’explicar
aquesta idea: la hipòtesi de la “barreja de poblacions” de Kinlen [18][19] i la de les
“infeccions endarrerides” de Greaves [20][21]. De forma resumida, aquests estudis
suggereixen que la falta d’exposició a infeccions en el període postnatal precoç pot
donar lloc a una resposta aberrant del sistema immunològic quan hi ha una exposició a
patògens comuns en un perío de més tardà. Molt recentment s’ha descrit que
l’exposició a determinats patògens donaria lloc a una leucèmia aguda en portadors de
mutacions germinals en PAX5 [17].
Factors etiològics en LMA
Des d’un punt de vista etiològi c, les LMA es poden classificar segons si són de novo, o
secundàries a malalties predisponents; en aquest sentit, els nens amb la síndrome de
Down tenen un risc 700 vegades més elevat que la població general de desenvolupar
una leucèmia megacarioblàstica. Altres patologies com síndromes congènites
d’insuficiència medul·lar (ex. Anèmia de Fanconi, disqueratosi congènita , síndrome de

INTRODUCCIÓ
40
Shwachman–Diamond) o la síndrome de Bloom [22]–[26][27], també s’han associat a
un risc incrementat de LMA. L’exposició prèvia a radioteràpia o a agents
quimioteràpics com agents alquilants o epipodofilotoxines pot donar lloc a la
denominada therapy-related LMA [28][29], i l’exposició a determinats factors
ambientals genotòxics també han estat esmentats com a possibles factors precipitants
[30]. En els últims anys s’han descrit mutacions germinals en factors de transcripció
hemopoètics com CEBPA, GATA2, o RUNX1 [31][32][33], responsables del que es
coneixen com LMA familiars. Tal és la rellevància d’aquest fenomen que la OMS ha
incorporat en la última classificació, com a noves entitats, les neoplàsies mieloides
amb predisposició germinal [6].

INTRODUCCIÓ
41
1.4 Fisiopatologia
1.4.1 Mecanismes de leucemogènesi
Si bé el nostre coneixement sobre l'etiologia de la le ucèmia és encara molt limitat, el s
coneixements sobre la cèl·lula leucèmica i els mecanismes de leucemogènesi han
experimentat un gran avenç en les últimes dècades. Aquest fet es deu en gran mesura
a la facilitat en l'obtenció de cèl·lules leucèmiques a pa rtir de mostres de sang
perifèrica o de medul·la òssia i al gran desenvolupament de millors i més eficients
tècniques de seqüenciació genòmica. L'estudi d’aquestes cèl·lules ha permès
identificar alteracions citogenètiques i moleculars recurrents, ha possi bilitat conèixer
millor els mecanismes de leucemogènesi i així mateix, definir diferents subtipus
biològics de LA amb importància pronòstica i terapèutica.
La transformació leucèmica de la cèl·lula hemopoètica requereix una pèrdua de
control de la prolife ració normal, un bloqueig en la diferenciació, resistència a les
senyals apoptòtiques i un augment en la capacitat d’auto-renovació (self-renewal).
Tot i la gran varietat de lesions genètiques implicades en la patogènesi de la LLA i LMA,
les alteracions f onamentals són les translocacions cromosòmiques i les aneuploïdies
(hiperdiploïdia o hipodiploïdi a) [34][35], que representen els denominats
esdeveniments primaris, insuficients normalment per si mateixos per donar lloc a la
leucèmia, i que necessiten de l’addició d’esdeveniments secundaris cooperants, com
mutacions o delecions (figura 5). Els esdeveniments primaris defineixen els principals
subtipus de leucèmies i presenten característiques biològiques pròpies (veure apartat
Classificació), amb patrons genòmics distintius demostrats per estudis d’alta capacitat
com els microarrays o les noves tècniques de seqüenciació de nova generació (next
generation sequencing, NGS) [36][37][38]. Les translocacions cromosòmiques donen
lloc a recombinacions il·legítimes de gens normalment separats, podent afectar
l’expressió d’un oncogen, com per exemple, fusionant un oncogen amb el gen de la
cadena pesada de les immunoglobulines ( IGH) o el receptor de cèl·lules T ( TCR), que
actuarien com a potenciadors. De totes maneres, són més habituals la formació de
gens híbrids o quimèrics que donen lloc a la producció de proteïnes amb propietats
aberrants, fre qüentment amb una activitat cinasa augmentada ( BCR-ABL1) o que

INTRODUCCIÓ
42
actuen com a factors nous de transcripció (per exemple, ETV6-RUNX1 o RUNX1 -
RUNX1T1). Les translocacions en la leucèmia aguda solen ser balancejades o
recíproques i normalment no concorren amb un gran número d’alteracions genètiques
addicionals, a diferència d’altres tipus de neoplàsies [39].
Diferents autors han intentat explicar on i quan es produeixen aquestes alteracions
genètiques i si per elles soles explicarien tot el fenotip leucèmic.
S’ha assumit des de fa molt temps que les cèl·lules mare són les principals dianes on es
produeixen el e sdeveniments genètics clau, ja sigui en leucèmies o en altres tipus de
càncer. De totes maneres, és difícil preci sar on i en quin moment es produeix aquesta
lesió dins la jerarquia de les cèl·lules mare, principalment perquè l’impacte funcional
amb la conseqüent expansió clonal pot tenir lloc en progenitors més diferenciats del
que s’ha produït la lesió inicial. Per exemple, la fusió BCR-ABL1 en la leucèmia mieloide
crònica (LMC) té un origen en la cèl·lula mare multipotencial, però indueix un fenotip
exclusivament granulocític. Aquests fets estarien relacionats amb el context cel·lular
que es dona en cada moment del desenvolupament de la cèl·lula hemopoètica [40].
Amb la finalitat de comprendre millor aquest procés, el grup de Mel G reaves i altres
grups d'investigadors han realitzat estudis per identificar l'origen de la cèl·lula
leucèmica [41]. Per fer -ho, varen estudiar la seqüència temporal en el procés de la
leucemogènesi, varen comparar cèl·lules pre -leucèmiques (portador es només de
l'esdeveniment primari) amb les cèl·lules leucèmiques (amb les alteracions genètiques
afegides o secundàries). Van realitzar, per un costat, estudis en bessons univitel·lins en
que només un d’ells desenvolupava la leucèmia i, per un altre, estu dis en pacients amb
LA dels que es disposava de mostra de sang en el moment de néixer (obtinguda de les
cartes de Guthrie o de cèl·lules criopreservades de sang de cordó umbilical). Així, van
poder demostrar no només l'origen en l'úter d'una proporció de c asos de LA pediàtrica
on té lloc la primera alteració genètica, sinó també com algunes cèl·lules adquireixen,
posteriorment al part, lesions genètiques secundàries que desencadenen la leucèmia.
L'estudi amb bessons amb LLA i reordenament ETV6-RUNX1 (TEL-AML1) ha permès
inferir la història natural d’aquest subtipus de LLA. Tots dos bessons compartirien el
clon pre-leucèmic (amb el reordenament ETV6-RUNX1) per pas transplacentari a l'úter.
En un bessó es desenvoluparia la leucèmia mitjançant l'adquisició d'an omalies

INTRODUCCIÓ
43
genètiques secundàries i en l’altre (bessó sa), el clon pre -leucèmic podria no adquirir
noves lesions i quedar aturat en la seva trajectòria evolutiva [42] [41] [43]. En el cas de
les LLA amb reordenament del gen MLL el grau de concordança en el
desenvolupament en els dos bessons és superior, el que suggereix que la capacitat
oncogènica de MLL és major.
En resum, en un progenitor hematopoètic es produeixen determinades alteracions
genètiques, denominades esdeveniments primaris, a les quals se sumen més t ard
diferents lesions secundàries, que cooperen fins a donar lloc a la leucèmia (figura 5).
Per tant, de manera similar al model dels dos hits de Knudson [41], algunes d’aquestes
lesions es considerarien iniciadores de la leucèmia o primàries ( founding), com la
translocació t(9;22)(q34;q11.2)/ BCR-ABL1 o els reordenaments del gen MLL, i podrien
tenir lloc en una etapa molt precoç del desenvolupament, inclús in utero. Algunes
d’elles per si soles poden tenir una alta capacitat leucemogènica, com és el cas dels
reordenaments MLL, mentre que d’altres, com ETV6-RUNX1, requeririen més
esdeveniments genètics secundaris per donar lloc a un fenotip leucèmic complet.
Aquest fet podria explicar, en part, l’epidemiologia de les leucèmies en la infància. Així,
les LA amb MLL reordenat, amb més capacitat leucemogènica, tindrien un temps de
latència més curt i, per tant, es presenten a edats més precoces.

INTRODUCCIÓ
44
Figura 5. Als denominats esdeveniments primaris se sumen més tard diferents lesions
secundàries, que cooperen fins a donar lloc a la leucèmia. Adaptat de [9].
Quant a les LMA, tradicionalment s’havia postulat, segons el s treballs de Kelly i
Gilliland [44], que aquestes leucèmies s’originarien a partir d’almenys dos
esdeveniment genètics somàtics; un d’ells conferiria al progenitor hemopoètic un
avantatge proliferatiu (mutacions tipus I), mentre que l’altre donaria lloc a una
desregulació de la diferenciació (mutacions tipus II). Les tipus I impliquen mutacions
activadores a nivell de vies de senyalització on intervenen FLT3, C-KIT, N-RAS, K-RAS i
PTPN11 (figura 6) [45]–[50], mentre que les mutacions tipus II estarien constituïdes
principalment per translocacions cromosòmiques ( RUNX1-RUNX1T1, CBFB -MYH11,
PML-RARalfa, etc.) o mutacions a RUNX1, CEBPA i NPM1.

INTRODUCCIÓ
45
Figura 6. Hipòtesi dels dos hits de les LMA. Segons aquesta hipòtesi farien falta dos
tipus de mutacions complementàries per induir i promoure una LMA. Adaptat de [51].
Tot i això, amb el desenvolupament de la seqüenciació genòmica de la LMA s’ha vist
que aquest paisatge és molt més complex i fins un 40% de les LMA no seguirien aquest
patró [52]. Així, en els últims anys s’han descrit mutacions recurrents en fins a 9 grups
funcionals o vies gèniques ( The Cancer Genome Atlas Research Network, 2013 [53]). A
diferència d’altres tumors, s’ha demostrat en models murins que únicament dues
mutacions en alguns d’aquest gens serien suficients per generar un a LMA [54]. S’ha
descrit que exi steixen patrons d’expressió gènica característics per diferents grups de
LMA pediàtrica. A més, l’expressió alterada d’alguns gens podria tenir importància
pronòstica, com BRE, EVI1, IGSF4, BAALC i ERG [55]–[57] [58]. Tanmateix, sembla que
determinats esdeveniments epigenètics també podrien estar implicats en la
fisiopatologia d’aquesta malaltia. Aquests esdeveniments actuen a nivell de la
metilació de l’A DN, modificació d’histones, remodelació del nucleosoma o actuant
sobre els microARN (miARNs). En relació amb la metilació de l’ADN s’han descrit
mutacions recurrents a nivell de TET2, IDH1/IDH2 i DNMT3A [59], [60] . Els miARNs són
fragments curts de ARN no codificant que inhibeixen l’expressió gènica, i que poden
inhibir l’expressió tant de proto -oncogens com de gens supressors de tumors [61]. La
desregulació dels miARNs pot contribuir a la leucemogènesi a través de la interacció de
gens supressors de tumors que participen en la proliferació i diferenciació. En aquest

INTRODUCCIÓ
46
sentit, seria freqüent la desregulació de miARNs en LMA en nens. S’han descrit patrons
d’expressió de miARNs específics segons el subtipus genètic [62] [63].
1.4.2. Clonalitat en la leucèmia aguda. Evolució clonal
La teoria que actualment preval és que una única cèl·lula mutada (tipus stem cell ) o
cèl·lula iniciadora de leucèmia amb capacitat d’auto -renovació, donaria lloc a una
prole de cèl·lules malignes poc diferenciades i que conformarien el gruix ( bulky)
leucèmic, adquirint una organització jeràrquica similar a la que presenta la h emopoesi
fisiològica. La transformació maligna pot donar -se en una cèl ·lula mare primitiva o en
cèl·lules més diferenciades, que adquireixen característiques pròpies d’una cèl·lula
mare. Vàries línies de recerca recolzen la hipòtesi clonal de l’origen leuc èmic, com per
exemple la demostració de clons genètics de TCR o reordenaments
d’immunoglobulines en LLA ó el mateix anàlisi del cariotip leucèmic. Recentment,
però, s'ha revisat el concepte de la leucemogènesi i la linealitat de la seva evolució
[64]. La teoria clàssica d'un únic clon inicial amb una desenvolupament lineal en la que
van apareixent subclons amb noves mutacions, es va veient substituïda per una
explicació darwiniana, on l’evolució clonal tindria una arquitectura ramificada.
D’aquesta manera, al diagnòstic de la leucèmia ja existirien div ersos subclons de
cèl·lules mare leucèmiques ( Leukemic Stem Cells, LSC) amb diferents alteracions
genotípiques [65]. Les LSC són les responsables del manteniment de la leucèmia i són
menys accessibles al tractaments quimioteràpics convencionals (figura 7) [66].

INTRODUCCIÓ
47
Figura 7. Les LSC són les responsables del manteniment de la leucèmia i són menys
accessibles al tractaments quimioteràpics convencionals. Les LSC es poden originar a
partir de cèl·lules mare hemopoètiques ( Hematopoetic Stem Cell , HSC ) o en
progenitors immadurs. Adaptat de [66].
El tractament de la leucèmia té una gran importància sobre la selecció i evolució de
determinats subclons que adquireixen noves mutacions que, entre d’altres, poden
conferir resistència al tractament, en són un exemple l’adquisi ció de mutacions a
CREBBP o NT5C2 i la resistència a glucocorticoid es o anàlegs de nucleòsids [67]. A les
recaigudes es poden trobar clons derivats d’un clon ancestral diferent al clon
majoritari que va originar la leucèmia [68]. Aquesta teoria té importants implicacions
en el seguiment i en el tractament dirigit enfront de dianes terapèutiques (figura 8).

INTRODUCCIÓ
48
Figura 8. Relació clonal entre el diagnòstic i la recaiguda en mostres de pacients
recaiguts amb LLA. L es recaigudes es poden relacionar amb el clon majoritari al
diagnòstic o amb evolucions clonals d’aquest, amb subclons presents al di agnòstic que
s’han seleccionat o amb clons ancestrals presents abans del diagnòstic. Adaptat de
[68].

INTRODUCCIÓ
49
1.5 Formes de presentació clínica
La presentació clínica de la LA és variable i, en general, els símptomes i signes al
diagnòstic són deguts a l a infiltració de les cèl·lules leucèmiques a la medul·la òssia i
altres òrgans. Tot i que pot presentar -se de forma insidiosa, la LA infantil sol fer -ho de
forma aguda, amb una història de menys de tres mesos des de l'inici de la clínica fins al
diagnòstic [69]. Una de les troballes més freqüents és la febre. Si bé la febre pot ser de
causa infecciosa, sovint es degut a la pròpia leucèmi a i es resol en 24 -72 hores des de
l'inici del tractament. En ocasions, el nen presenta malestar i astènia general. També
sol haver-hi símptomes relacionats amb una síndrome anèmica i/o hemorràgica per la
plaquetopènia. En el cas de les LLA, en una tercera part dels casos el símptoma
principal és el dolor ossi o articular, que pot ser migratori o localitzat en una
extremitat. En nens petits, aquest dolor pot manifestar -se únicament en forma de
coixesa. La LLA -T sol donar-se en nens més grans i freqüentment es presenta amb una
càrrega tumoral elevada, massa mediastínica, hepato -esplenomegàlia i
hiperleucocitosi. A causa de la massa mediastínica, alguns nens tenen dispnea i
ortopnea.
L’afectació extramedul·lar pot ser evident en ambdós tipus de leucèmia en for ma de
hepato i/o esplenomegàlia o nefromegàlia (més en el cas de les LLA). De forma
ocasional en les LMA, la manifestació predominant és per efecte d’un tumor
extramedul·lar; a aquestes lesions se les ha denominat cloromes, sarcomes
granulocítics o mielobl astomes i solen originar -se en ossos amb una activitat
hemopoètica intensa i periosti prim, com són els del sol de les òrbites o les vèrtebres.
La hipertròfia gingival pot veure’s en un 10% de pacients i, com l’aparició de nòduls
subcutanis, és més caracte rístic de les LA amb component monocític. Les lesions
cutànies es poden presentar de forma única o múltiple i es solen presentar en lactants.
En l'exploració física les troballes més freqüents són la pal·lidesa, les equimosis i/o
petèquies, les adenopatie s i/o hepato -esplenomegàlia. En els homes s'han d’explorar
els testicles, ja que poden estar infiltrats. En menys del 5% dels casos es troba
infiltració del sistema nerviós central (SNC) al diagnòstic. Aquesta és més freqüent en
les LLA -T, en els nens meno rs de 2 anys, en les leucèmies amb reordenament BCR-
ABL1 i, en el cas de les LMA, en aquelles amb component monocític. Pot manifestar -se

INTRODUCCIÓ
50
amb clínica d'hipertensió endocranial o amb paràlisi d’un parell cranial. Més rarament,
es pot observar infiltració ocu lar, en forma d'hemorràgia o infiltració leucocitària a la
retina o al nervi òptic. S'han descrit altres manifestacions clíniques secundàries a l a
infiltració extramedul·lar com el priapisme o la compressió de la medul·la espinal per
masses epidurals, més freqüents en les LMA que en les LLA. Els lactants amb
reordenament del gen MLL solen presentar-se amb hiperleucocitosi, lesions cutànies,
organomegàlies i infiltració del SNC.
L’hemograma sol ser diagnòstic per la presència de blasts en sang perifèrica.
Generalment s'observa anèmia, plaquetopènia i leucopènia o leucocitosi. En un 10%
dels casos l'hemograma al diagnòstic és normal o presenta alteracions mínimes. Amb
menor freqüència que la LMA, la LLA al diagnòstic pot ocasionar situacions clíniques
d'emergència amb compromís vital que requereixin un tractament urgent. Així, la
clínica de debut pot ser una insuficiència respiratòria per compromís de la via aèria
causat per una massa mediastínica, característic de les LLA -T. La hiperleucocitosi, més
habitual en les LLA, pot donar fenòmens de leucostasi, tot i que això és més freqüent
en LMA. La leucostasi és una complicació greu que es produeix per ocupació de la llum
vascular per part dels blasts. El mecanisme patogènic es produiria per lesió endotelial
probablement mediat per cito cines [70]. Les principals manifestacions clíniques
d’aquest quadre es donen a nivell de SNC i van des de la cefalea al coma, passant a
accident vascular cere bral (AVC) isquèmic o hemorràgic, insuficiència respiratòria en
forma d’hipoxèmia o destret respiratori. Les alteracions de la coagulació poden
esdevenir autèntiques emergències, sent un exemple la coagulopatia que es produeix
en el cas de les LPA i en alg uns casos de LMA monocítica [71]. En el cas de la LPA la
causa de la coagulopatia és complexa i v e donada per l ’alliberament de cito cines
provinents dels blasts, una hiperfibrinolisi i un e stat de coagulació intravascular
disseminada (CID) [72]. El tractament amb àcid trans -retinoic (ATRA), introduït de
forma precoç, ha aconseguit disminuir de forma significativa l’elevada mortalitat
produïda per aquest fenomen [73]. En el cas de les LLA, la coagulopatia és
característica dels casos que presenten la translocació t(17;19)( TCF3-HLF) [74].
Finalment, la síndrome de lisi tumoral (SLT), és més típica de les LLA
hiperleucocitòsiques, especialment la LLA -T i LLA del lactant. Aquesta es produeix per
l’alliberació massiva de metabòlits intracel·lulars a l’espai vascular com a conseqüència

INTRODUCCIÓ
51
del ràpid recanvi cel·lular i apoptosi produïda de forma espontània o després de
l’exposició a fàrmacs citostàtics. Analíticament es caracteritza per hiperuricèmia,
hiperpotassèmia, hiperfosfatèmia i hipocalcèmia. En fases més avançades pot donar
lloc a una insuficiència renal, arítmies o símptomes neuromusculars.

INTRODUCCIÓ
52
1.6 Diagnòstic
El diagnòstic precís de les leucèmies és essencial pe r al tractament i el seguiment de la
malaltia, és a dir, de com respon al tractament. Per tant, aquest procés determinarà el
pronòstic de cada pacient.
Actualment les eines diagnòstiques van més enllà de la caracterització morfològica de
la cèl·lula leucèmica i es basen en estudis immunofenotípics mitjançant el citòmetre de
flux, estudis citogenètics i la detecció i quantificació d’alteracions genètiques
mitjançant estudis de biologia molecular.
Estudi del moll de l’os:
l’estudi del moll de l’os (MO) re sulta essencial, ja que en un 10% dels pacients amb
leucèmia aguda no es detecten blasts en la sang perifèrica al diagnòstic [75]. A més, la
morfologia a sang perifèrica pot ser diferent a la que es troba en el MO. Aquesta
mostra s’obté habitualment per punció -aspiració del MO. Malgrat això, en algun cas
pot ser necessari fer una biòpsia (per exemple, en cas d’empaquetament del MO,
fibrosi en d eterminats subtipus de leucèmia, moll de l’os molt hipocel·lular...).
L’obtenció d’aquestes mostres ens permetrà realitzar els estudis morfològics i
biològics.
Estudi morfològic:
L’estudi inicial de les leucèmies s’inicia amb l’observació mitjançant el mi croscopi òptic
de les mostres procedents del moll de l’os i de la sang perifèrica. És necessari fer una
tinció d’aquestes mostres; habitualment s’utilitza la tinció de May -Grünwald-Giemsa.
L’estudi morfològic ens permetrà fer el diagnòstic de la leucèmia a guda (>25%
infiltració blàstica en el cas de LLA, >30% en la LMA pediàtrica) i fer una primera
classificació segons la línia cel·lular i el grau de diferenciació.
L’estudi citoquímic pot ajudar a fer un diagnòstic morfològic més acurat i reproduïble
segons els criteris diagnòstics de les classificacions de l’OMS i la FAB, però el seu ús
s’ha vist substituït progressivament per la tècnica de citometria de flux [76][77].

INTRODUCCIÓ
53
Estudi immunofenotipic del blasts:
Mitjançant la citometria de flux podem determinar el llinatge dels blasts i l’estadi
maduratiu. Això permet fer una classificació immunofenotípica (veure en apartat
classificació) i quantificar l’índex d’ADN per valorar la ploïdia cel·lular; a més, en e ls
últims anys ha esdevingut una eina clau en el seguiment de la Malaltia Residual
Mínima (MRM). Per tant, ens proporciona informació d’importància pronòstica i inclús
la identificació de potencials dianes terapèutiques.
Estudis citogenètics/moleculars:
Els estudis de cariotip convencional permeten la detecció d’alteracions numèriques i/o
estructurals en les cèl·lules leucèmiques. Aquesta tècnica permet identificar
alteracions genètiques d’importància pronòstica i, a més, permet identificar la
localització de lesions moleculars que potencialment estan implicades en la
transformació i proliferació cel·lular.
La tècnica de FISH ( Fluorescent in Situ Hybridization ) ajuda a la detecció d'anomalies
genètiques no detectades per citogenètica convencional, per manc a d'obtenció de
metafases o per tractar -se d'alteracions d’una mesura inferior a la resolució de la
citogenètica convencional (alteracions críptiques). Existeixen altres tècniques de
citogenètica molecular que permeten augmentar la resolució de les alterac ions
genètiques. Entre elles, cal destacar la tècnica SKY ( Spectral Karyotyping), que és una
variant de la FISH que permet tenyir els 23 cromosomes simultàniament amb diferents
sondes, la CGH (Comparative Genomic Hybridization ) i els SNParrays (Single Nucleotide
Polymorphisms arrays).
Sovint, les lesions genètiques només poden ser detectades per tècniques moleculars,
com la tècnica de la reacció en cadena de la polimerasa (PCR). Així, podem detectar el
productes del reordenament de diferents gens amb implic ació pronòstica, ja sigui en
LLA o LMA (veure apartat 1.8 Factors pronòstics ). També ens permet fer un seguiment
molt precís de la MRM.

INTRODUCCIÓ
54

INTRODUCCIÓ
55
1.7 Biologia
1.7.1 Classificació
La classificació moderna de les leucèmies requereix la integració de l a morfologia,
l’immunofenotip (citometria de flux) i de les alteracions genètiques detectades per les
anàlisi citogenètiques o moleculars. L’observació al microscopi de la cèl·lula leucèmica
continua sent la prova gold standard per establir el diagnòstic. Aquest fet propicia que
la classificació establerta pel grup cooperatiu FAB (French -American-British) [76], que
integra criteris morfològics, citoquímics i immunofenotípics, continuï sent aplicada en
alguns casos de LMA.
El descobriment d’alteracions genètiques i moleculars ha permès identificar subtipus
biològics específics de leucèmies agudes. Això s’ha traduït en una classificació més
causal de les leucèmies agudes i ha permès un refinament dels grups de risc i la
identificació de potencials dianes terapèutiques. Fins en un 20% dels casos no es
poden detectar marcadors citogenètics o moleculars per les tècniques habituals;
nogensmenys, les noves tècniques de NGS han permès classificar moltes de les
leucèmies des del punt de vista de les alteracions genètiques que presenten [78].
Classificació de les LLA
La classificació morfològica i citoquímica o classificació de la FAB [76][79] no s'utilitza
actualment en l'estratificació dels pacients amb LLA. Les classificacions que major
utilitat tenen en l'actualitat són la classificació immunològica segons el grup EGIL
(European Group of immunological classification of leukemias ) [80] i la classificació de
l’OMS (Organització Mundial de la Salut, o WHO, World Health Organization ) [77]. En
la classificació EGIL (taula 2) es sistematitza la LLA segons el grau de maduresa de la
cèl·lula leucèmica. El fenotip més freqüent en la LLA pediàtrica és el B comú, definit
per la positivitat de CD10. La classificació de l’OMS (taula 3) integra aspectes clínics,
morfològics, immunofenotípics i genètics i reconeix com a entitats diferent s subgrups
de LLA [77].

INTRODUCCIÓ
56
Taula 2. Classificació immunològica (EGIL) de les LLA [80].
Classificació immunològica de la LLA segons grup EGIL
LLA de línia B: CD22+ i/o CD79a+ i/o CD19+
Pro-B (B-I): TdT+, CD10-, Ig citoplasma-, Ig membrana-, CD38+.
Comú (B-II): TdT+, CD10+, Ig citoplasma-, Ig membrana-, CD38+
Pre-B (B-III): TdT+, CD10+/-, Ig citoplasma +, Ig membrana-, CD38+/-
B madura (B-IV): CD20+, TdT-, CD10 -, Ig citoplasma -, cadenes lleugeres de superfície o citoplasmàtiques +, CD38-
LLA de línia T: CD3 de citoplasma +
Pro-T (T-I): CD7+, CD2-, CD5-, CD8-, CD1a-
Pre-T (T-II): CD2+ i/o CD5+ i/o CD8+, CD1a-, CD71+
T cortical: CD1a+, CD3 de superfície + o -, CD71-
T madura: CD3 de superfície+, CD1a-, CD2+, CD5+, CD4/8+
Taula 3. Classificació de la OMS 2016 de les neoplàsies precursores limfoides [6].
Classificació de l’OMS de les neoplàsies de precursors limfoides
Leucèmia/limfoma limfoblàstic B
Leucèmia/limfoma limfoblàstic B, no especificat
Leucèmia/limfoma limfoblàstic B amb alteracions genètiques recurrents
Leucèmia limfoblàstica B amb t(9;22)(q34;q11.2); BCR-ABL1
Leucèmia limfoblàstica B amb t(v;11q23); reordenament del gen MLL (KMT2A)
Leucèmia limfoblàstica B amb t(12;21)(p13;q22); ETV6-RUNX1 (TEL-AML1)
Leucèmia limfoblàstica B amb hiperdiploïdia
Leucèmia limfoblàstica B amb hipodiploïdia
Leucèmia limfoblàstica B amb t(5;14)(q31;q32); IL3-IGH
Leucèmia limfoblàstica B amb t(1;19)(q23;p13.3); TCF3-PBX1
Entitats provisionals:
Leucèmia/limfoma limfoblàstic B, BCR-ABL1 like
Leucèmia/limfoma limfoblàstic B amb iAMP21
Leucèmia/limfoma linfoblàstic T
Entitats provisionals:
Leucèmia limfoblàstica Early T-cell precursorLeucèmia/limfoma limfoblàstic Natural Killer (NK)

INTRODUCCIÓ
57
Classificació de les LMA
La classificació clàssica FAB creada als anys 70 [76], basada en criteris morfològics
(llinatge i grau de maduració) i citoquímics està sent substituïda per la classificació de
l’OMS [77]. S’ha intentat fer una classificació més útil, incorpor ant també aspectes
genètics, immunofenotípics i clínics.
Així, l’última classificació defineix les entitats considerant alteracions citogenètiques
recurrents, la presència de displàsia multilinial i els antecedents de tractaments
quimioteràpics. Aquesta c lassificació manté les categories FAB per aquells casos que
no poden ser classificats segons aquest criteris.
La nova classificació de l’OMS, però, no discrimina entre nens i adults; tot i que permet
categoritzar un 60 -70% de la població pediàtrica, exist eixen entitats recentment
descobertes molt característiques d’aquest grup d’edat: algunes d’elles han estat
incorporades [77]), com la translocació t(1;22)(p13;q13)/ RBM15-MKL1, característica
de pacients amb la síndrome de Down, però n’hi ha d’altres que encara no han estat
recollides a la nova classificació, com per exemple la translocació
t(7;12)(q36;p13)/MNX1-ETV6 en els lactants, la t(5;11)(q35;p15.5) /NUP98-NSD1
[81][82], present fins en un 10% de casos de leucèmia megacarioblàstica, o la inversió
críptica inv16(p13.3q24.3)/CBA2T3-GLIS2. També cal esmentar que per primer cop s’ha
incorporat en la nova classificació de la OMS el grup de neoplàsies mieloides amb
predisposició germinal (veure taula 1).
Una altra particularitat de les LMA pediàtriques és que en nens s’ha proposat mantenir
el llindar de 30% de blasts per tal de fer el diagnòstic de leucèmia aguda (llindar
disminuït al 20% en pacients adults en la classificació de l’OMS) [82]. Com a excepció,
si existeixen alteracions citogenètiques específiques (t(8;21) , inv(16)/t(16;16), t(15;17))
o en casos de pacients amb la síndrome de Down, es classifica com una LMA,
independentment del percentatge de blasts.

INTRODUCCIÓ
58
Taula 4. Classificació de les LMA segons la classificació FAB i associació a alteracions
citogenètiques recurrents.
FAB Nom % en pediatria
Associació a alteracions citogenètiques
M0M1M2M3M4 M4EoM5M6M7
LMA amb poca diferenciacióLMA sense maduracióLMA amb maduracióLeucèmia promielocítica agudaLeucèmia mielomonocítica agudaM4 + eosinofília al moll de l’osLeucèmia monoblàstica/monocíticaLeucèmia eritroideLeucèmia aguda megacarioblàstica
2-510-1525-305-1015-251015-251-35-10
t(8;21)(q22;q22)t(15;17)q22;q12)
inv(16)(p13;q22)/t(16;16)(p13;q22)
rMLL
t(1;22)(p13;q13)
Taula 5. Taula adaptada de la classificació de l’OMS [77].
Classificació WHO de LMA i neoplàsies relacionades
LMA amb alteracions genètiques recurrents
LMA amb t(8;21)(q22;q22.1); RUNX1-RUNX1T1LMA amb inv(16)(p13.1q22) o t(16;16)(p13.1;q22); CBF-MYH11LMA amb t(15;17)(q24;q21); PML-RARALMA amb t(9;11)(p21.3;q23.3); MLLT3-KMT2ALMA amb t(6;9)(p23;q34.1); DEK-NUP214LMA amb inv(3)(q21.3q26.2) o t(3;3)(q21.3;q26.2);GATA2, MECOMLMA (megacarioblàstica) amb t(1;22)(p13.3;q13.3); RBM15-MKL1LMA amb mutació a NPM1LMA amb mutació bial·lèlica a CEBPA
Entitats provisionals:LMA amb BCR-ABL1LMA amb mutació a RUNX1
LMA amb canvis en relació a mielodisplàsiaLMA en relació a tractamentLMA no específica (NOS)
LMA amb poca diferenciacióLMA sense maduracióLMA amb maduracióLeucèmia mielomonocítica agudaLeucèmia monoblàstica/monocíticaLeucèmia eritroideLeucèmia aguda megacarioblàsticaLeucèmia aguda basofílicaPanmielosi amb mielofibrosi aguda

INTRODUCCIÓ
59
Sarcoma mieloideProliferació mieloide en relació a la Síndrome de Down
Mielopoesi anòmala transitòriaLeucèmia mieloide associada a la Síndrome de Down
Leucèmies agudes de llinatge ambiguLeucèmia aguda indiferenciadaLeucèmia aguda de fenotip mixt (MPAL) amb t(9;22)(q34.1;q11.2); BCR-ABL1MPAL amb t(v;11q23.3); reordenament de MLL/KMT2AMPAL, B/mieloide, NOSMPAL, T/mieloide, NOS
1.7.2 Subtipus genètics i moleculars
Subtipus en LLA
Les alteracions cromosòmiques, numèriques o estructurals, constitueixen un dels trets
distintius de les LLA. Aquestes alteracions representen e sdeveniments genètics
primaris que determinen subtipus amb característiques clínico -biològiques
diferenciades que recullen les principals classificacions de les leucèmies. L’alta
hiperdiploïdia (HeH, entre 51 -67 cromosomes), amb guany de forma no aleatòria
d’almenys 5 cromo somes (inclouen els cromosomes X, 4, 6, 10, 14, 17, 18, 21),
succeeix en un 25 -30% dels pacients pediàtrics amb LLA -B precursora i s’associa a un
pronòstic favorable. La hipodiploïdia (<44 cromosomes) és poc freqüent, 2 -3% dels
casos, i sol associar un mal pronòstic. Recentment s’ha observat que en el 90% de
casos de baixa hipodiploïdia (32 -39 cromosomes), es troben mutacions de p53; dins
aquests casos, la meitat presenta la mutació en forma germinal, el que tradueix que la
leucèmia en aquests pacients és u na forma de presentació de la síndrome de Li
Fraumeni [83].
Els reordenaments cromosòmics, habitualment translocacions, donen lloc a gens de
fusió on estan implicats factors de transcripció hematopoètics, modificadors
epigenètics, receptors de citocines o receptors tipus tirosin -cinasa. Els reord enaments
més habituals en LLA -B són la translocació t(12;21)(p13;q22), que dona lloc al gen de
fusió ETV6-RUNX1 (TEL-AML1); t(1;19)(q23;p13)/ TCF3-PBX1 (E2A-PBX1); i la
t(9;22)(q34;q11.2), que dona lloc al cromosoma Philadelphia, “Ph” i codifica el gen de
fusió BCR-ABL1. El gen KMT2A (MLL), localitzat al cromosoma 11q23, es pot reordenar
amb més de 70 gens diferents i pot presentar -se com a LLA -B, LLA -T, LMA o com a

INTRODUCCIÓ
60
leucèmia de fenotip mixt ( Mixed Phenotype Acute Leukemia, MPAL ). La translocació
t(17;19)(q22;p13)/TCF3-HLF és molt infreqüent, però és molt important detectar -la, ja
que comporta un pronòstic molt advers i requereix tractament d’alt risc. Els últims
anys es va descriure l’amplificació intracromos òmica del cromosoma 21 (iAMP21), que
representa el 2% de les LLA -B i és més freqüent en adolescents. Es defineix per la
troballa de ≥5 senyals del gen RUNX1 en els estudis de FISH, i en metafases s’ha
comprovat que aquestes senyals corresponen a amplificacions d’una zona
determinada d’un sol cromosoma 21. És important la seva identificació, ja que s’ha
demostrat que aquests pacients presenten una supervivència significativament
superior si es tracten amb més intensitat [84].
En un 20% de les LLA -B dels nens no es troba cap d’aquestes alteracions; aquests
casos, denominats LLA -B B-other, tenen altres an omalies genètiques, moltes de les
quals han estat identificades gràcies a les noves tècniques d’alta capacitat com els
microarrays d’expressió o les tècniques de NGS. Així, s’han identificat uns casos amb
firma gènica similar al reordenament BCR-ABL1, però sense ser portadors de la
translocació; aquests casos s’han denominat LLA -B BCR-ABL like i en ells es troben una
multitud d’alteracions genètiques que activen la via de cinases [85], [86].
D’altra banda, molt recentment s’han identif icat mitjançant RNA -Seq nous gens de
fusió, com els que imp liquen als gens DUX4, que comporten una desregulació del
factor de transcripció ERG, els gens de fusió de MEF2D, o els casos amb fusió de
ZNF384 [87]–[90] (figura 9).

INTRODUCCIÓ
61
Figura 9. A. Representació mitjançant Circos Plot dels diferents subtipus genètics de
LLA-B. Cada cinta uneix el partners de les fusions gèniques; el seu gruix és proporcional
a la freqüència de la fusió . Els gens estan endreçats segons la seva posició genòmica
(del cromosoma 1 -22, seguits dels cromosomes X i Y). B. Mateixa representació pel
subgrup de LLA-B B other. Reproduït de [89].
Com s’ha comentat prèviament, a aquestes alteracions denominades primàries,
formades prin cipalment per grans anomalies cromosòmiques, s’afegeixen alteracions
secundàries, que cooperen per donar lloc al desenvolupament de la leucèmia.
Aquestes inclouen delecions o amplificacions intragèniques o mutacions i afecten
principalment a factors de tra nscripció importants en la diferenciació limfoide normal,
a oncogens o gens relacionats amb proliferació. Tot i que no són plenament
específiques, algunes alteracions es troben preferentment en alguns subtipus, com les
delecions del gen IKZF1 (IKAROS), presents en el 80% de les LLA -B Ph+ i en el 30% de
les LLA -B BCR-ABL1 like , o les mutacions de RAS¸ que són més freqüents en els
reordenaments del gen MLL o en les hiperdiploïdies (figura 10).

INTRODUCCIÓ
62
Figura 10. Interacció entre subtipus principals i alteracions s ecundàries afegides en
LLA-B. Reproduït de [89].
Les LLA-T són molt heterogènies biològicament. S’observen mutacions activadores de
NOTCH1 i FBWX7 en més del 50% de casos. També es poden identificar reordenaments
que impliquen al TCR i diferents gens de fusió que involucren factors de transcripció
com TLX1 (HOX11), TLX3 (HOX11L2), LYL1, TAL1 i a MLL [91][92][9]. Els últims anys es
va identificar mitjançant estudis d’expressió gènica un subtipus deno minat LLA -T
“early T cell precursor ” (ETP), que s’originaria en una cèl·lula hematopoètica molt
immadura, amb la capacitat de diferenciar -se a llinatge limfoide T i mieloide. Així
doncs, aquests casos es caracteritzen per un perfil fenotípic i mutacional típ ic, amb
marcadors d’immaduresa, de línia T i mieloide. S’ha descrit un pronòstic advers per
aquests pacients, tot i que recentment s’ha vist que això podria modular -se per la
resposta al tractament i per l’expressió o absència de gens HOXA [93], [94].

INTRODUCCIÓ
63
Subtipus de LMA
Les alteracions genètiques i la resposta al tractament són un dels principals factors
pronòstics en LMA i són utilitzats àmpliament en diferents protocols de tractament
actuals per estratificar els pacients en grups de risc. La taula 7 resumeix les alteracions
genètiques en LMA pediàtrica; algunes d’elles amb rellevància clínica establerta i
d’altres amb rellevància incerta. Les tècniques de seqüenciació massiva del genoma
han permès determinar un espectre complert de les lesions leucemogèniques en l a
LMA i han confirmat que, de forma similar a les LLA, les LMA tenen menor número
d’alteracions genètiques que altres neoplàsies [95]. Nogensmenys, s’han identificat
noves lesions que configuren un paisatge genètic molt h eterogeni i complex [96][97]
(figura 11). Tot i que els pacients pediàtrics amb LMA comparteixen les principals
alteracions genètiques amb els pacients de major edat, és cert que les freqüències
varien i que existeixen una sèrie d’alteracions genètiques específiques de la infància.
D’aquesta manera, en lactants predominen les alteracions del gen MLL i trobem de
forma específica algunes translocac ions, com la t(1;22)(p13;q13)/ OTT-MAL, la
t(11;15)(p15;q35)/NUP98-JARID1A o la inv(16)(p13.3 -q24.3)/CBFA2T3-GLIS2, totes
elles associades a leucèmia megacarioblàstica, o les translocacions críptiques
t(7;12)(q36;p13)/ETV6-HLXB9 o la t(5;11)(q35;p15)/ NUP98-NSD1. En nens, les
alteracions desfavorables són més infreqüents que en adults; per contra, les
translocacions de bon pronòstic que afecten al core binding factor (CBF), com la
t(8;21)(q22;q22)/RUNX1-RUNX1T1 o la inv(16)(p13.1;q22)/t(16;16)(p13.1;q22)/ CBFB-
MYH11, són més freqüents en la població pediàtrica. Els reordenaments del gen MLL
estan presents en el 50% de lactants i fins en un 25% dels casos de LMA pediàtrica.

INTRODUCCIÓ
64
Figura 11. Freqüències estimades de les alteracions genètiques recurrents en LMA
pediàtrica. Estimat a partir de les publicacions: [98][99][100][101][102][103]

INTRODUCCIÓ
65
Taula 7. Alteracions genètiques en LMA pediàtrica.
Cariotip Gen afectat % Significat clínic
t(8;21)(q22;q22) RUNX1-RUNX1T1 15 Pronòstic favorableinv(16)(p13.1;q22)t(16;16)(p13.1;q22)
CBFB-MYH11 10 Pronòstic favorableNo candidats per TPH en RC1
t(15;17)(q22;q21) PML-RARA 6-10 Pronòstic favorablet(9;11)(p22;q23) MLLT3-MLL 7 Pronòstic intermig-
favorablet(10;11)(p12;q23) MLLT10-MLL 3 Majoritàriament
lactants. Pronòsticdesfavorable
t(6;9)(p23;q34) DEK-NUP214 <2 Pronòsticdesfavorable
inv(3)(q21q26.2) ot(3;3)(q21;q26.2)
RPN1-EVI1 <1 Pronòsticdesfavorable
-7 Desconegut 1 Mal pronòstic11q23 Reordenaments
MLL (KMT2A)20 Segons el gen amb
que es reordena
t(1;22)(p13;q13) RBM15-MKL1 Leucèmia megacarioblàstica, lactants. Pronòstic intermig
AML amb mutació a NPM1 NPM1 5-10 (15-20 en CN)
Pronòstic favorable
AML amb mutació bial·lèlica a CEBPA
CEBPA 5 (14 en CN) Pronòstic favorable
FLT3-ITD FLT3 10 (18 en CN) Depenent del context mutacional
t(7;12)(q36;p13)/t(7;12)(q32;p13) MXX1/ETV6 Lactants Pronòsticdesfavorable
t(5;11)(q35;p15.5) NUP98/NSD1 Pronòsticdesfavorable. Majoritàriament en CN
inv(16)(p13.3q24.3) CBFA2T3-GLIS2 Majoritàriament CN. LA megacarioblàsticaPronòstic desfavorable
t(11;12)(p15;p13) NUP98/KDM5A Leucèmia megacarioblàsticaPronòstic desfavorable

INTRODUCCIÓ
66
1.8 Factors pronòstics:
Actualment el pronòstic de la majoria de les LLA i, en menor mesura, de les LMA en
pediatria, ha millorat de forma significativa en les últimes dècades. Aquesta millora ve
donada fonamentalment per una millor estratificació en grups de risc, en funció del
risc de recaiguda de cada pacient. Aquesta estratificació perm etrà aplicar estratègies
terapèutiques adaptades al risc, de manera que s'intensif icarà el tractament en els
pacients que tenen uns factors pronòstics d' Alt Risc i es reduirà en aquells de Baix Risc
de recaiguda.
Els factors pronòstics venen determinats per les característiques del pacient, de la
malaltia i del tractament per se . Diferents factors de risc, biològics o clínics, no es
reprodueixen de forma igual en tots els grups cooperatius i alguns d’ells han deixat de
ser factors de mal pronòstic aplicant règims quimioteràpics més actuals, indicant la
importància del tractament per si mateix. Per tant, esdevé rellevant el tipus de
tractament rebut; els pacients adolescents amb LLA tractats amb protocols pediàtrics
solen tenir millor resposta que els tractats amb protocols d’adults [104], [105].
D’altra banda, la resposta al tractament ve determinada per la farmacocinètica dels
fàrmacs citostàtics, és a dir, com s’absorbeixen, distribueixe n, metab olitzen i
s’excreten, i la sensibilitat cel·lular a determinats fàrmacs. La farmacodinàmia és la
relació entre la farmacocinètica i l’efecte farmacològic. Existeix una gran variabilitat
interpersonal farmacocinètica i farmacodinàmica en molts fàrma cs antileucèmics en
pediatria. La farmacogenètica o farmacogenòmica és el conjunt de factors constitutius
del individu que expliquen les variacions farmacocinètiques i farmacodinàmiques de
les medicacions en cada individu. En són exemple els polimorfismes d’enzims que
intervenen en el metabolisme dels citost àtics [106], [107] . Els pacients amb s índrome
de Down presenten una gran sensibilitat a la citarabina (Ara -C), en part perquè tenen
uns polimorfismes en el gen CBS que es relacionen amb nivells elevats de cistationina
beta sintetasa. A més, els blasts dels pacients amb síndrome de Down presenten
nivells més elevats de citidina desaminasa i això fa que acumulin més ARA -CTP i, per
tant, siguin més sensibles a aquest fàrmac [108]. De la mateixa manera, polimorfismes
en determinats gens també poden contribuir a augmentar la toxicitat de determinats
fàrmacs i augmentar les taxes de mort prematura durant el tractament [109].

INTRODUCCIÓ
67
Respecte als facto rs relacionats amb la biologia de la malaltia i la sensibilitat
farmacològica, s’ha demostrat que la sensibilitat a determinats fàrmacs es pot associar
al subtipus genètic. En aquest sentit, els pacients amb LA i reordenament del gen MLL
(LA-MLL+) presente n una major sensibilitat a anàlegs de deoxinucleòsids com
citarabina i cladribina. Aquest fenomen s’ha comprovat tant en LLA com LMA,
suggerint un mecanisme de sensibilitat similar per aquest fàrmac en aquests tipus de
leucèmies [110].
A més, la manera en que es combinen fàrmacs existents amb nous fàrmacs també pot
ser rellevant per a la resposta al tractament [111], tal i com també es demostra en
aquest treball de tesi.
Les classificacions clàssiques de les LA, ja sigui en LLA o LMA, no tenen importància
pronòstica per se i l’interès pronòstic que se’ls havia atribuït clàssicament estaria
relacionat amb l’associació morfològica a determinats subtipus biològics. La nova
classificació de la OMS i ntenta suplir aquestes mancances incorporant esdeveniments
genètics i clínics. En els últims anys, s’han identificat diferents alteracions genètiques
que defineixen subtipus biològics amb importància pronòstica. Aquestes alteracions
han estat incorporades als sistemes d’estratificació en grups de risc en la majoria de
protocols terapèutics actuals. A més, algunes d’aquestes alteracions podrien constituir
noves dianes terapèutiques.
Factors pronòstics en LLA:
Per a l’estratificació pronòstica dels pacients amb LLA s’utilitzen factors clínics i
biològics presents al diagnòstic i factors evolutius que mesuren la resposta al
tractament.
L’edat (lactant o ≥ 10 anys d'edat), el recompte de leucòcits (≥20 o 50 x 10⁹/L segons el
protocol), la raça (hispans i negres), el sexe masculí i el llinatge T són factors de mal
pronòstic, tot i que el seu efecte es pot veure matisat pels nous esquemes
quimioteràpics i un millor tractament de suport.
Les diferències racials en el pronòstic no han estat només vinculades als factors
socioeconòmics, sinó també a les diferències en variacions genètiques: els
polimorfismes en els gens PDE4B i ARID5B estan associats amb l’ascendència de natius

INTRODUCCIÓ
68
americans [112], i els reordenaments de CRLF2 tenen una major represent ació en
nord-americans d’origen hispànic i s’han associat a un pitjor pronòstic.
L’edat és el principal factor pronòstic clínic. Així, els pacients entre 1 i 10 anys
constitueixen un grup de millor pronòstic, mentre que el risc és més elevat en lactants
menors d’un any i en adolescents i adults. Els lactants amb reordenament del gen MLL,
especialment els menors de 6 mesos d'edat amb hiperleucocitosi superior a 300 x
10⁹/L al moment del diagnòstic, tenen un pronòstic ombrívol. L’edat comporta una
menor presència de comorbiditats en el moment del diagnòstic i una millor tolerància
als tractaments, però la diferència en la supervivència entre pacients adults i pediàtrics
també pot explicar -se per les característiques genètiques de la leucèmia. Així, els
pacients adolescents i els adults tenen una major prevalença de alteracions
biològiques d'alt risc (per exemple, BCR-ABL1, LLA -T), una menor incidència de
subtipus favorable s (com l’alta hiperdiplo ïdia o la t(12;21)/ ETV6-RUNX1), una pitjor
adherència i tolerància al tractament (veure l’apartat 1.10 adolescents i adults joves )
[113].
Donada la gran evolució dels estudis moleculars i genètics en aquestes malalties, s’han
pogut establir subgrups genètics amb importà ncia pronòstica. Algunes d’aquestes
s’associen a bon pronòstic i permeten disminuir la intensitat del tractament; en canvi,
d’altres determinen l’estratificació dels pacients dins grups d’alt risc, com per exemple
la presencia del reordenament BCR-ABL1 (LLA Ph+), la hipodiploïdia (<44 cromosomes)
o el reordenament del gen MLL. En els últims anys s’han descrit noves alteracions
genètiques amb pronòstic definit (taula 9), algunes de les quals ja s’han incorporat als
protocols terapèutics actuals per estratif icar els pacients, com l’amplificació
intracromosòmica del cromosoma 21 (iAMP21) o la translocació
t(17;19)(q22;p13)/TCF3-HLF.

INTRODUCCIÓ
69
Taula 9. Alteracions citogenètiques amb valor pronòstic en la LLA pediàtrica. Gentilesa
de la Dra. Ortega i la Dra. Camós.
PRONÒSTIC ALTERACIÓ CROMOSÒMICA Especificacions Freqüència Ref.
FAVORABLE
t(12;21)(p13;q22)/ETV6-RUNX1
2/3 presenten pèrdua de l’ al·lel normal del gen ETV6
20-25% trisomia 2115-20% duplicació der(21)
25 %114, 115
Alta HIPERDIPLOÏDIA (HeH)51 a 65/67 cromosomes
Trisomies X, 4, 6, 10, 17, 18/ Tetrasomies 14 y 21
Índex de DNA 1,11-1,4825-30%
116, 117
INTERMIG
t(1;19)(q23;p13)/TCF3-PBX1 (E2A-PBX1)
50% desequilibrada der(19)t(1;19)
2-6%118, 119
t11q23 / MLL reordenatExcloent la t(4;11), que és de
mal pronòstic
t(11;19)(q23;p13.3) / MLL-ENL
Altres partners: 6q27 (MLLT4/AF6), 9p21 (MLLT3/AF9),10p12
(MLLT10/AF10),1p32(EPS15)
9%120-122
iAMP21Amplificació de 21q22.11-
21q22.122-5%
123, 124
Cariotip normalMínim 20 metafases
analitzades15% 125
AltresAlteracions estructurals, 45
cromosomes, no creixement, etc.
125
DESFAVORABLE
HIPODIPLOÏDIA <45
cromosomesÍndex de DNA
<0,8
Alta HIPODIPLOÏDIA
40-44 cromosomes
<1% 126
Baixa HIPODIPLOÏDIA
30-39 cr. / quasi triploïdia
60-78 cr.
Monosomies cr. 3,7,15,16,17 / Disomies cr.
1,6,11 y 18 solen doblar la dotació
cromosòmica fins gairebé triploïdia
3-5% 126
Casi HAPLOÏDIA
<30 cromosomes
< 30 cromosomesEs retenen els cr X/Y, 10, 14, 18, 21 solen doblar-se fins 54 cr
1% 126
t(4;11)(q21;q23) / MLL-AFF1 (AF4)
2-3% 123
t(17;19)(q22;p13)/ TCF3-HLF (E2A-HLF)
<1% 74
Segons les publicacions: [114]–[122][74], [123]–[126].

INTRODUCCIÓ
70
La resposta al tractament ha esdevingut un dels principals factors pronòstics en LLA
[127]. Reflecteix els factors inherents a la malaltia (biologia de la leucèmia), els
dependents de l’hoste (genètica del paci ent) i el tractament rebut. La resposta al
tractament pot ser avaluada mitjançant el microscopi òptic, i així, la presència de
blasts en sang perifèrica després d’una setmana de prednisona o de 15 dies de
tractament quimioteràpic en el moll de l’os constit ueixen factors de risc independents.
Per detectar la malaltia residual mínima (MRM) submicroscòpica, existeixen mètodes
més sensibles i específics, com la citometria de flux o tècniques moleculars. Així, la
citometria de flux segueix la MRM mitjançant la i dentificació de l’immunofenotip
associat a la leucèmia ( Leukemic Associated Immuno -Phenotype, LAIP ), assolint una
elevada sensibilitat (1x10 -4). D’altra banda, per tècniques de PCR quantitativa es pot
seguir la MRM estudiant el reordenament específic de ca da pacient de la cadena
pesada de les immunoglobulines o del receptor de cèl·lules T o bé els transcrits dels
gens de fusió si estan presents, amb una sensibilitat que pot ser d’1x10 -5-1x10-6.
L’estudi de la MRM en moments precoços del tractament (final de la inducció o
consolidació) i en punts més tardans ha demostrat ser el factor pronòstic més
important [128] [129]. A més, en la majoria de protocols, ha permès readaptar la
intensitat del tractament en els diferents punts d’avaluació [127]. La detecció de la
MMR per PCR permet una quantificació més sensible en comparació a la citometria
(0,001% vs 0,01%). De totes manere s, la citometria és un mètode més ràpid,
normalment més econòmic, aplicable a la majoria de pacients i permet adoptar
decisions terapèutiques de forma més immediata [130]. En l’actualitat, les nove s
tecnologies d’alta complexitat com la NGS s’estan començant a implantar en l’estudi
de la MRM en diferents grups cooperatius.
Factors pronòstics en LMA
La citogenètica convencional, les alteracions moleculars i la resposta al tractament són
importants pr edictors de la supervivència i s’utilitzen àmpliament com a factors
classificadors de grups de risc [131]. La taula 10 resumeix les alteracions
genètiques/moleculars amb rellevància pronòstica.
Els pacients amb les translocacion s t(8;21)(q22;q22)/ RUNX1-RUNX1T1,

INTRODUCCIÓ
71
inv(16)(p13.1;q22)/CBFB-MYH11 o t(16;16)(p13.1;q22)/ CBFB-MYH11 (denominades
Core Binding Factor Leukemias ) es classifiquen com de baix risc [132]. L’impacte
favorable d’aquestes alteracions citogenètiques ha estat confirmat en pacients adults i
pediàtrics per diferents grups cooperatius [133]. Les alteracions genètiques amb alt
risc de recaiguda en nens inclouen la monosomia del cromosoma 7 [134] i les
mutacions internes en tàndem ( Internal Tandem Duplication, ITD) del gen FLT3, FLT3-
ITD [135], sobretot en aquells casos amb rati de mutació elevad a [136]. Sembla que
aquests pacients es podrien beneficiar d’un transplantament de moll d’os al·logènic
(al·lo-TPH) en primera remissió completa [137].
Existeix un grup molt infreqüent d’alteracions citogenètiques que probablement
s’associen a mal pronòstic. Donada la raresa d’aquestes alteracions és difícil establir -ne
el veritable impacte pronòstic, entre elles la translocació t(6;9)(p23;q34)/ DEK-NUP214,
la t(8;16)(p11;p13)/ KAT6A-CREBBP, o la t(16;21)(q24;q22)/ RUNX1-CBFA2T3 [138]–
[140].
Les LMA amb reordenaments del gen MLL constitueixen el grup genètic més
heterogeni. Balgobind et al, després d’analitzar 756 casos pediàtrics amb aquesta
alteració, varen concloure que el pronòstic ve determinat en gran mesura pel partner
amb que es tran sloca aquest gen [141]. Així, les translocacions t(6;11)(q27;q23),
t(10;11)(p12;q23) i t(10;11)(p11.2;q23) s’associaven a mal pronòstic, mentre que la
t(1;11)(q21;q23) s’associava a un pronòstic molt favorable.
Les mutacions als gens WT1, IDH1 i IDH2 sembla que no tindrien una importància
pronòstica de forma independent en pacients pediàt rics [142], [97]. Les mutacions a
RUNX1, TET2, DNMT3A i els casos amb sobreexpressió de BAALC i ERG són molt poc
freqüents en nens i no s’ha pogut establir la seva importància pronòstica [143].
Taula 10. grups de risc en LMA pediàtrica segons les alteracions citogenètiques [81],
[101], [49], [144].

INTRODUCCIÓ
72
Pronòstic Alteració genètica Freqüència/característiques
Favorable t(8;21)(q22;q22)/RUNX1-RUNX1T1* 12-14%
Inv(16)(p13.1;q22)/t(16;16)(p13.1;q22)/CBFB-
MYH11*
8%
t(15;17)(q22;q21)/PML-RARA* 6-10%
Cariotip normal (CN)
amb
Mutacions de NPM1* 5-10% (14-22% en CN)
Favorable si no està present FLT3-
ITD
Mutacions bial·lèliques
de CEBPA*
5% (14% en CN)
t(1;11)(q21;q23)/MLL-MLLT1 (AF1Q) <1% de la LMA pediàtrica(3% dels
reordenaments de MLL†)
GATA1s En pacients amb la síndrome de
Down i LMA-M7
Intermig Cariotip normal 20-30%
t(9;11)(p22;q23)/MLL-MLLT3 (AF9)* 7%
t(1;22)(p13;q13)/RBM15-MKL1* Lactants amb LMA-M7
Anomalies genètiques no classificades com
favorable o desfavorable
Desfavorable -7‡ / -5, del(5q) 3-5%
Inv(3)(q21q26.2)/t(3;3)(q21;q26.2)/RPN1-MECOM
(EVI1-MDS-EAP)*
<1%
t(6;9)(p23;q34)/DEK-NUP214* <2%
t(8;16)(p13;p16)/MYST3-CREBBP <1%
t(7;12)(q36;p13)/ETV6-HLXB9 (TEL-MNX1)§ 30% de la LMA del lactant
t(4;11)(q21;q23)/MLL-MLLT2 (AF4) <1%
(2% dels reordenaments de MLL†)
t(6;11)(q27;q23)/MLL-MLLT4 (AF6) 1%
(5% dels reordenaments de MLL†)
t(10;11)(p12;q23)/MLL-MLLT10 (AF10)ǁ 3% (principalment en lactants)
(13% dels reordenaments de
MLL†)
t(5;11)(q35;p15.5)/NUP98-NSD1¶ 16%
t(9;22)(q34;q11.2)/BCR-ABL1 Rara
Cariotip complex# 6%
FLT3-ITD** 10% (18% en CN)

INTRODUCCIÓ
73
*Subtipus reconeguts en la classificació de la OMS-2008.† Reordenaments del gen MLL en el 25% de la LMA pediàtrica i en el 50% de lactants amb LMA.ΐ��džĐůŽĞŶƚ�ůĞƐ�ĂůƚĞƌĂĐŝŽŶƐ�ŐĞŶğƟƋƵĞƐ�ƌĞĐƵƌƌĞŶƚƐ�ĚĞĮŶŝĚĞƐ�ĞŶ�ůĂ�ĐůĂƐƐŝĮĐĂĐŝſ�ĚĞ�ůĂ�KD^�ϮϬϬϴ͘§ Translocació críptica en la majoria de casos, o associada a del(12p).ǁ S’han descrit resultats heterogenis com la t(10;11)(p12;q23) i pot considerar-se de risc intermig o desfavorable.¶ Translocació críptica.# Definit como ≥3 anomalies cromosòmiques en absència de translocacions/inversions recurrents definides en la classificació de la OMS-2008.** FLT3-ITD amb un rati de mutació al·lèlica baix podria considerar-se com risc intermig, tot i que aquest aspecte és controvertit.
Probablement el factor pronòstic més important en els pacients amb LMA és la
resposta al tractament [145]. Tradicionalment la resposta al tractament s’ha avaluat
mitjançant criteris morfològics. Aquests mètodes, però, vigents en molts protocols
actuals, poden resultar poc precisos. Diferents grups cooperatius han demostrat que
la MRM al final d’un o dos cicles d’inducció és el factor predictor més important del
risc de recaiguda en LMA [146]–[148], el que fa que cada cop es tingui més present
l’avaluació de la MRM i que aquesta s’utilitzi com a factor estratificador terapèutic, ja
sigui mitjançant la citometria de flux (factible en la majoria de pacients amb LMA) o
l’anàlisi de gens de fusió per PCR.

INTRODUCCIÓ
74
1.9 Estratègies terapèutiques
Tractament de les LLA
Les LLA pediàtriques són una de les neoplàsies on més ha augmen tat la supervivència
en els últims anys mercès a , sobretot, una millor estratificació en grups de r isc dels
pacients i l’adaptació de la intensitat del tractament quimioteràpic per a cada cas
segons el seu risc de recidiva (figura 12). Actualment, més del 80% dels nens de països
desenvolupats amb LLA es pot curar, en gran part per les noves combinacions de
fàrmacs quimioteràpics, molts d’ells coneguts des de fa dècades, administrats d'acord
amb els protocols internacionals [149]. La creació de grans grups cooperatius
internacionals per a l’estudi i tractament d’aquestes pat ologies ha permès avançar
gràcies a l’aplicació de protocols en el context d’assaigs clínics. A Espanya, l’aplicació
de protocols terapèutics consecutius dins el context de la Sociedad Española de
Hematología y Oncología Pediátrica (SEHOP), s’ha traduït ai xí mateix en un increment
progressiu de la supervivència global (SG) (figura 13).
Figura 12. Supervivència global de pacients pediàtrics amb LLA segons l’any de
tractament en la institució SJCRH. Reproduït de Pui y Evans [150].

INTRODUCCIÓ
75
Figura 13. Supervivència global de pacients pediàtrics amb LLA segons l’any de
tractament obtinguda amb els protocols successius de la SEHOP. Gentilesa de la Dra.
Badell i Dra. Rives, en representació del grup de LLA de la SEHOP.
Fases del tractament en la LLA
El tractament inclou la inducció a la remissió, seguit de tractament de consolidació o
intensificació per eliminar residus de la leucèmia, la prevenció de la l eucèmia a nivell
del SNC i el tractament de manteniment per assegurar la continuació de la remissió. En
la majoria dels estudis, els pacients s'estratifiquen en grups de risc, basats en
característiques biològiques de la leucèmia, característiques clínique s i analítiques i la
resposta precoç al tractament d’inducció.
L'objectiu del tractament d’inducció és induir una remissió completa (RC) mitjançant
l’eradicació de més del 99% de la població inicial de cèl·lules leucèmiques i la
restauració de la hematopoe si normal. Amb els protocols més actuals de tractament
s’assoleixen taxes de RC post-inducció (és a dir, ≤ 5% de blasts a la medul·la òssia) en el
97-99% dels nens. Els pacients que no aconsegueixen arribar a la RC al final de la
inducció tenen un pronòsti c molt desfavorable. Aquests pacients, així com els que
tenen una MRM alta (>1%) al finalitzar la inducció o persistent després de la
consolidació són candidats a tractaments més intensius, que inclouen el al·lo -TPH
[151],[152].
Amb la restauració de l'hemopoesi normal, els pacients en RC seguiran tractaments de
consolidació i intensificació. Per raons no del tot aclarides, els nens amb LLA
requereixen tractament de continuació o manteniment a llarg termini. Els intents de

INTRODUCCIÓ
76
escurçar la durada del tractament s'han traduït en un alt risc de recaiguda després del
cessament de la teràpia [153][149], amb l’excepció dels pacients amb LLA -B madura
(tipus Burkitt) que segueixen protocols específics més curts.
La resposta al tractament en la recaiguda és molt inferior a la dels acabats de
diagnosticar, constituint la principal causa de mort en aquests pacients. El temps en RC
fins la recaiguda, la localització de la recaiguda, l’immunofenotip i la presència de
determinades alteracions genètiques d’alt risc s’han establert com a factors pronòstics
en aquest grup de pacients. En la majoria dels nens amb LLA recaiguda es pot assolir
una segona remissió completa (RC 2). No obstant això, en molts pacients, la
quimioteràpia no és suficient per mantenir aquesta remissió. Per tant, el TPH ha estat
utilitzat com a tractament intensiu posterior.
Fases del tractament en la LMA
Tot i que es troba encara lluny dels resultats aconseguits en LLA, la SG dels nens amb
LMA ha millorat notablement en els últims anys, amb SG>60% (figura 14)
[154],[155],[147],[156],[157],[158]. Aquesta millora en la supervivència ha estat
possible gràcies a l’increment en la intensitat del tractament quimioteràpic, una millor
estratificació dels pacients en grups de risc, la implementació de mesures de suport
més eficaces, així com una notable millora en la selecció de donants per la realització
del TPH. Tot i així, fins a un 30 -40% dels pacients recauen, i la recaiguda continua sent
la principal causa de mort en aquest pacients [159].

INTRODUCCIÓ
77
Figura 14. Supervivència global de pacients amb LMA <20 anys diagnosticats en els
períodes indicats. Dades obtingudes de www.seer.cancer.gov/popdata.
La majoria de grups cooperatius estratifiquen els pacients en dos o tres grups de risc,
segons característiques genètiques i la resposta precoç al tractament, especialment
per citomorfologia. A diferencia de les LLA, el seguiment de la MRM en aquest grup de
pacients és complex i no està estandarditzat. És excepció el cas de la leucèmia
promielocítica aguda (LPA), on la detecció del transcrit PML-RARalfa s’utilitza de forma
generalitzada pel seguiment i estratificació d’aquests pacients [160].
El tractament de les LMA inclou, en la majoria de protocols, un o dos cicles d’inducció
seguits de dos a tres cicles de consolidació o post -remissió. A diferència de la LLA, la
majoria de grups han abandonat el tractament de mante niment, exceptuant el
subgrup de les LPA [161].
Ja des del 1980 l’esquema “3+7” (daunorubicina, 45 mg/m 2 al dia durant 3 dies;
citarabina, 100 mg/m2 al dia durant 7 dies) va demostrar induir la remissió en un 60 -70
% dels pac ients, esdevenint el tractament de primera línia o standard of care (SOC) ,
com a tractament d’inducció [162]. Les taxes de remissió varen augmentar fins >80%
en els anys 90. No està clar si aquesta millora va ser deguda a l’adició de nous fàrmacs
com la tioguanina o l’etopòsid [163], o a la incorporació de millors mesures de suport o
una barreja d’ambdues.
En els últims anys s’ha intentat augmentar la supervivència amb la introducció de

INTRODUCCIÓ
78
cladribina [164], fludarabina [165] o gemtuzumab ozogamicin [166]; també s’ha
intentat mitjançant la sub stitució de la daunorubicina per idarubicina o mitoxantron a,
o la intensificació de la dosi de citarabina [147], o intentant una reducció dels temps de
cada cicle de tractament. Les dosis d’antraciclínics també han estat objecte d’estudi en
pacients adults, sense aportar una evidència clara de millors resultats amb altes dosis
[167],[168]. Cap d’aquestes mesures ha demostrat una millora significativa. En l’estudi
del grup I -BFM 2004 es va n randomitzar altes dosis de daunorubicina liposomal (80
mg/m2) versus dosis estàndard d’ida rubicina (12 mg/m 2), en combinació amb
citarabina i etopòsid durant la inducció [159]. Es va observar una tendència a una
millor supervivència i menor toxicitat en el grup que va rebre daunoribicina liposomal.
Tot i la manca d’evidència de que intervencions individuals hagin augmentat les taxes
de remissió significativament, la RC en els protocols actuals es situa al voltant del 90%.
Tractament de post-remissió
Els assaigs conduïts per diferents grups cooperatius com el CCG [169], el grup I -BFM
[170], el grup POG [171] i el MRC [172] varen demostrar el benefici de l’administració
d’altes dosis de citarabina com a tractament de post -remissió en nens amb LMA,
mentre que les baixes dosis de tractament de manteniment empitjoraven les taxes de
supervivència [173].
En els últims anys s’han desenvolupat diversos assaigs amb la finalitat de determinar la
duració òptima d’aquesta etapa, el benefici que podien aportar nous fàrmacs, la
importància de la monitorització de la MRM o el benefici del TPH. La majoria d’aquests
estudis només han aportat beneficis modestos en la supervivència d’aquests pacients.
Només unes mesures de suport molt curoses i intenses, l’adaptació del tractament al
grau de resposta de cada pacient, la quimioteràpia intensiva i/o el TPH en alguns casos,
han contribuït als bons resultats reportats per diversos grups cooperatius com el
Japanese Childhood AML Cooperative Group [155]), St. Jude [147]), COG [165], MRC
[174], I-BFM [175], o NOPHO [156].
El TPH és un tema en constant revisió; les indicacions d’al·lo -TPH en pacients en
primera RC en LMA són controvertides [176]. En general, els grups nord -americans
recomanen aquesta indicació en un n ombre superior de pacients que els grups
europeus [177],[176]. Les recomanacions dels grups europeus es basen en estudis que

INTRODUCCIÓ
79
conclouen que l’eventual benefici de reduir el risc de recaiguda en els pacient s que
reben un TPH en RC1 és inferior al potencial risc de mortalitat relacionat amb el
tractament ( Treatment Related Mortality, TRM), greus complicacions a llarg termini
(malaltia de l’empelt contra el receptor –MECR- crònica, infertilitat, alteracions de l
creixement, etc.) i unes reduïdes taxes de rescat en cas de recaiguda post TPH. De
totes maneres, les millores en el camp del TPH estan disminuint notablement aquests
inconvenients [178],[179],[180][137]. La majoria de pacients en RC2 són candidats a
TPH [181].
Nous tractaments
En els últims anys s'han desenvolupat nous fàrmacs; alguns són fórmules millorades de
fàrmacs ja existents, però mereix destacar l’aparició de fàrmacs dirigits contra d ianes
moleculars específiques i, sobretot en el camp de les LLA, el desenvolupament
d’estratègies terapèutiques basades en la immunoteràpia. Entre els nous fàrmacs
quimioteràpics trobem nous anàlegs dels nucleòsids com la clofarabina i nelarabina i
noves f ormulacions de citostàtics com la vincristina, citarabina i dauno rubicina
liposòmiques. També s'estan realitzant assaigs clínics amb anticossos monoclonals
conjugats amb immunotoxines o quimioteràpics (com l’inotuzumab), anticossos
biespecífics (blinatumom ab) i altres [182]. Per altra banda, s'estan desenvolupant
noves estratègies d'immunoteràpia que d irigeixen cèl·lules T autòlogues enfront de la
cèl·lula tumoral i indueixen la seva destrucció. Amb aquesta finalitat, es modifiquen els
limfòcits T del pacient mitjançant la transfecció d’un receptor antigènic quimèric, amb
especificitat anti -CD19, produint els denominats CART19 (Chimeric Antigen Receptor -
modified T cells) [183],[184]. Aquests assaigs han mostrat resultats molt favorables en
pacients amb malalties molt avançades [185].
Respecte als nous tractament de la LMA, diferents fàrmacs es troben en fases més o
menys avançades d’implementació, entre ells cal dest acar: noves formulacions de
fàrmacs existents com els compostos liposomals de daunorubicina o citarabina,
d’especial interès la daunorubicina liposomal per la menor toxicitat cardíaca;
modificadors de la regulació epigenètica com els inhibidors de DOT1L, que han mostrat
eficàcia en leucèmies amb reordenament del gen MLL; inhibidors de la via ubiquitin -
proteosoma com bortezomib, lenalinomida o la talidomida; inhibidors de vies de

INTRODUCCIÓ
80
senyalització com els inhibidors de FLT3 (veure l’apartat FLT3 en leucèmia agud a
pediàtrica) o cKIT [186]; inhibidors de cicle cel·lular com flavopiridol, tipifarnib [187];
modificadors del metabolisme cel·lular com la rapamicina , el sirolimus o els inhibidors
de MEK; moduladors de resistència de fàrmacs com inhibidors de MCL1 i ABCB1 [188] i
els immunoconjugats com el gemtuzumab ozogamicina [189].
Tot i la intensitat dels tractaments actuals, al límit de la tolerabilitat farmacològica, és
improbable que puguem millorar les taxes de cur ació de les leucèmies agudes
pediàtriques a expenses d’una intensificació dels tractaments existents. Així, tot i
incorporar a milers de pacients en assaigs clínics, no hi ha hagut avenços terapèutics
rellevants en molts anys. Un dels principals inconvenients ha estat la heterogeneïtat de
les leucèmies agudes i la raresa d’alguns subtipus pediàtrics, sobretot en el grup de les
LMA, fent que els estudis per demostrar beneficis de fàrmacs específics siguin molt
més costosos. Un dels pocs exemples d’avenç impo rtant ha estat la LPA, on s’ha
utilitzat l’estratègia de tractar-la com a una entitat única i diferent als demés subtipus i
on la incorporació de l’ATRA en diferents fases del tractament s’ha traduït en un
augment significatiu de les taxes de curació.
A més, és poc probable que un sol d’aquest s nous principis tingui una gran activitat en
front a qualsevol tipus de leucèmia de forma global i, per tant, els esquemes de
quimioteràpia convencional continuaran tenint un paper fonamental en el tractament
de les leucèmies agudes. En els propers anys, caldrà dissenyar nous esquemes
terapèutics que incorporin els nous fàrmacs als esquemes quimioteràpics actuals,
millorant la seva eficàcia. La present tesi posa de manifest com es pot millorar
l’eficàcia d’un esquema de tractament amb un citostàtic convencional, la citarabina, i
nous fàrmacs, com són els inhibidors de FLT3.

INTRODUCCIÓ
81
1.10 Grups de pacients amb abordatge diferencial
Leucèmia aguda en el lactant
Tot i que la leucèmia aguda és el càncer més freqüent en la infà ncia i representa el
33% dels casos diagnosticats, la seva incidència és baixa, i és encara més infreqüent en
nens menors d'un any d'edat (0,5 -1/100.000 hab itants/any). En els lactants, a
diferència de nens més grans, la freqüència de la LMA és força simil ar a la de la LLA, i
dins aquesta gairebé totes són de llinatge B. La leucèmia del lactant es presenta
freqüentment de forma explosiva i amb una gran càrrega tumoral: hiperleucocitosi,
grans megàlies i extensió extra -hematològica, especialment en el SNC i la pell.
Independentment del llinatge, la cèl·lula leucèmica és sovint molt immadura
citològicament i immunològicament [190], i presenta unes característiques genètiques
distintives de la leucèmia del nen més gran. Així, un 80% dels lactants amb LLA i un
50% dels casos de LMA presenta com esdeveniment genètic el reordenaments del gen
MLL. A nivell genòmic també presenten una signatura particular; Armstrong i
col·laboradors van demostrar que la sobreexpressió d el gen FLT3 és la característica
més distintiva de LA amb el gen MLL reordenat [37]. Els aspectes farmacogenètics i
farmacodinàmics de la quimioteràpia que s’administra a aquest grup d’edat són molt
més desconeguts que en nens més grans i poden influir de forma significativa en
l’eficàcia o la toxicitat aguda o crònica en aquests pacients [191]. Des del punt de vista
clínic són leucèmies que presenten una sensibilitat exquisida als anàlegs de nucleòsids
com la citarabina i una resistència in vitro als cortic oesteroids i l’asparraginasa. La
citarabina es transporta principalment, però no exclusivament, pel transportador de
nucleòsids hENT1 (Human Equilibrative Nucleoside Transporter), sent aquest
transportador de membrana plasmàtica un important contribuent en la
biodisponibilitat d’Ara -C i, probablement, en la seva acció final. De fet, ha estat
reportat en pacients amb LLA-MLL+ una expressió elevada d’ hENT1. Aquest fet pot
explicar, almenys en part, la seva elevada sensibilitat a l’Ara -C [192]. El mateix grup va
demostrar que el reordenament de MLL no estava implicat en aquest fenomen,
assenyalant que altr es mecanismes estan implicats en la sensibilitat a l’Ara -C en
aquests pacients [193]. Totes aquestes dades s'han tingut en compte per dissenyar els

INTRODUCCIÓ
82
esquemes de quimioteràpia actuals en el protocol de tractament internacional per a
lactants amb LLA, Interfant-99 i 2006 [194].
La immaduresa immunològica pròpia del lactant, sumada als efectes
immunosupressors del tractament, fa que aquest grup de pacient s presenti una
elevada morbiditat infecciosa, el que impacta de forma important en la supervivència
global. Per tant, el poder disposar de tractaments més dirigits en front a dianes
moleculars com FLT3 pot disminuir la toxicitat i augmentar l’eficàcia dels tractaments
convencionals i, per tant, augmentar-ne la supervivència.
Leucèmia aguda en el nen amb síndrome de Down
Els nens amb la síndrome de Down (SD) tenen un risc de patir una LLA 14 vegades
superior al d’altres nens [195]. La LLA en aquests nens té unes característiques
biològiques diferents; són menys freqüents tant les alteracions genètiques de bon
pronòstic (hiperdiploïdia i reordenament ETV6-RUNX1), com les de mal pronòstic com
la LLA Ph +, el reordenament de MLL o la LLA-T. No obstant això, fins al 50% de casos
presenten sobreexpressió de CRLF2 i mutacions de JAK2, que podrien conferir pitjor
pronòstic [196]. La supervivència d’aquests pacients és una mica inferior, tant per una
major taxa de recaigudes com per una major mortalitat tòxica, principalment deguda a
infeccions i toxicitat cardíaca.
En el cas de les LMA en nens amb la SD, per sota dels 5 anys d’edat presenten de
forma característica el subtipus de leucèmia aguda megacarioblàstica (M7, segons
classificació FAB) [197]. Els nens amb SD presenten un risc 500 vegades superior
respecte als nens no SD de desenvolupar una leucèmia megacarioblàstica. També
presenten un risc més elevat de presentar leucèmia eritroblàstica aguda. Les
alteracions citogenètiques en les LMA en pacients amb la SD son diferents. Així, en la
SD són més freqüents les trisomies dels cromosomes 8 i 11; la duplicació dup(1p) o la
dup(7q); delecions del(6q), del(7p) i del(16q) [198]. Per contra, altres alteracions més
freqüents en leucèmies megacarioblàstiques en pacients sense SD, com la t(1;22), són
rares en nens amb SD.
De forma molt interessant, fins un 10% dels nens amb la SD presenten una proliferació
transitòria de cèl·lules blàstiques durant els primers mesos de vida. Aquests blasts són

INTRODUCCIÓ
83
morfològicament indistingibles d’una LMA. Aquest fenomen es coneix com a síndrome
mieloproliferativa transitòria (SMT) o mielopoesi anòmala transitòria, i també es pot
veure en nens fenotípicament normals però amb adquisició congènita o adquirida d’un
cromosoma 21 o i(21q) en mosaic. Tot i que habitualment presenten un curs benigne,
alguns pacients poden requerir tractaments amb dosis baixes de citostàtics. És
important el seguiment po sterior, ja que un 30% d’aquests nens desenvoluparan una
LMA durant el tres primers anys de vida. De forma característica, tant els pacients amb
SD i SMT o LMA presenten mutacions somàtiques en el factor de transcripció GATA1
[199]. En diferents estudis s’ha demostrat l’origen prenatal d’aquestes mutacions
[200]. El mecanisme pel qual hi ha una progressió de SMT a LMA es desconeix.
Diferents grups cooperatius han demostr at una elevada taxa de curació en pacients
amb SD i LMA, especialment els que presenten leucèmies megacarioblàstiques i els
que són menors de 4 anys. Aquest últim fet estaria relacionat amb que a partir del 4
anys són molt menys freqüents les mutacions en GATA1 en aquest grup de pacients
[199], [201].
De forma similar als nens amb la SD i LLA, l’elevada toxicitat secundària als tractaments
és un factor important que ha limitat la curació d’aquests pacients. És per això que
diferents grups han aconseguit augmentar la supervivència amb una més acurada
definició de les dosis de quimioteràpics, especialment d’antraciclínics [202]. També ha
resultat fonamental la intensificació de les mesures de suport.
Leucèmia aguda en l’adolescent i l’adult jove
Les LLA en aquest grup d’edat presenten unes particularitats q ue mereix comentar
com a una entitat particular. Des del punt de vista biològic es caracteritzen per
presentar amb menys freqüència lesions genètiques de bon pronòstic, com són la
hiperdiploïdia o la t(12;21), més típica de nens petits. Així, la hiperdiplo ïdia es troba
present en un 10-15% dels adolescents i la t(12;21) és rara. En canvi, lesions de
pronòstic desfavorable, com són la t(9;22), la hipodiploïdia o les LLA -T, augmenten la
seva freqüència amb l’edat del pacient [203]. Les amplificacions intracromosòmiques
del cromosoma 21 (iAMP21) i les translocacions que impliquen el locus de les cadenes
pesades de les immunoglobulines (IGH) són també més freqüents [204]. Els blasts

INTRODUCCIÓ
84
d’aquesta població d’edat mostren amb major freqüència alteracions del receptor
CRLF2, associades a pitjor pronòstic [205]. Aquest grup d’edat és el que presenta mé s
pacients sense lesions genètiques conegudes.
L’immunofenotip T amb massa mediastínica és més habitual en aquest grup. En un
70% dels casos es troben anomalies cromosòmiques i en un nombre important afecten
als locus dels receptors de cèl·lules T (TCR). A quest fet comporta reordenaments que
afecten a oncogens com HOX1, TAL1, LY1, LMO1 o LMO2, donant lloc a l’activació de
vies específiques amb potencials implicacions terapèutiques.
A més del factors biològics diferencials, experiments in vitro demostren una menor
sensibilitat a la prednisona, vincristina i L -asparaginasa en blasts d’adults joves. A més
d’això, en alguns estudis, pacients entre 15 -30 anys tenen una major càrrega de MMR,
comparat amb nens [206].
En aquest grup d’edat la toxicitat també seria major per una sèrie de factors, cada cop
més coneguts, dependents de l’hoste: diferències en el metabolisme de fàrmacs
quimioteràpics, menor reserva medul·lar, major toxicitat extramedul·lar. Tots aquests
factors impacten de forma directa en la supervivència global d’aquests pacients.
Un fet rellevant i que mereix un comentari, és que els pacients d’aquests grups d’edats
tractats amb protocols pediàtrics tenen millor pronòstic que si són tractats amb
protocols d’adu lts [207]. Diferents factors podrien explicar aquest fenomen. Entre
d’altres, cal destacar el disseny i la intensitat dels protocols pediàtrics; en aquests, es
dona una importància clau a fàrmacs no mielosupressors com la L -asparaginasa, la
vincristina o els glucocorticoid es. La major intensitat i duració de profilaxi a nivell de
SNC també podria ser rellevant [208]. La cura amb que s’administren aquests protocols
en centres pediàtrics i l’experiència en el maneig d’aquest tipus de pacients també
podria ser important.

INTRODUCCIÓ
85
Taula 8. Fenòmens biològics i clínics en adolescents i adults joves amb LLA. Adaptat de
[209][210].
Citogenètica Baixa incidència: hiperdiploïdia i t(12;21)
Alta incidència: hipodiploïdia, t(9;22), cariotips complexes,
translocacions IgH@
Genètica molecular Més freqüentment: CRLF2, JAK, IKZF1, CDKN2A/B
Alta incidència de: iAMP21, B other, BCR-ABL1-like
Immunofenotip Alta incidència d’ early T precursor ALL
Resposta al tractament Més MRM després del tractament d’inducció
Toxicitat Més elevada en alguns estudis

INTRODUCCIÓ
86
2. FLT3 en leucèmia aguda pediàtrica
El gen FLT3 (Fms tirosina cinasa 3) o FLK2 (fetal liver Kinase -2), situat al cromosoma
13q12, codifica una proteïna receptor tirosina-quinasa tipus III, com PDGFR, c-FMS o c-
Kit. Dos grups, de forma independent, varen clonar FLT3 en teixit murí al 1991 [211],
[212]. Al 1993 el grup de Rosnet ho va fer en teixit humà. Aquest receptor es
caracteritza per tenir 5 dominis extracel·lulars immunoglobulin-like, un domini
transmembrana i dos dominis citoplasmàtics amb activitat quinasa [213]–[216]. En
condicions normals FLT3 s'expressa en precursors hematopoètics immadurs tant
limfoides com mieloides i progenitors dendrítics. També s’express a en cervell,
placenta, melsa, timus, gònades i fetge, tot i que es desconeix quina és la seva funció
en aquests teixits [212], [217].
En condicions fisiològiques, l’activació del receptor i de la seva via de senyalització és
clau en el desenvolupament de les cèl·lules mare hemopoètiques, progenitors de línia
B, progenitors de cèl·lules dendrítiques i cèl·lules natural Killer (NK) [218].
El seu lligand natural (FLT3 -FL) és una proteïna transmembrana que pot ser alliberada
en forma de proteïna soluble homodimèrica i està produïda per cèl·lules del
microambient del moll de l’os com fibroblasts o altres cèl·lules hemopoètiques [219].
En la seva forma inactiva, FLT3 es troba formant u n monòmer en la membrana
plasmàtica amb una conformació del loop d’activació “tancat”, bloquejant l’accés del
llocs de fosforilació i d’unió a ATP. El seu lligand, FLT3 -FL, és una cito cina que actua
sinèrgicament amb altres cito cines promovent l’expansió d e precursors hemopoètics.
En condicions basals, la concentració soluble de FL és molt baixa, però augmenta de
forma important en les situacions d’aplàsia post quimioteràpia [220]. La unió de FLT3
al seu lligand indueix la dimerització del receptor i l'activació de les v ies STAT5,
RAS/MAP quinasa i PI3k/AKT, que juguen un paper important en la diferenciació,
supervivència i proliferació de les cèl·lules hemopoètiques [214]–[216], [221]).
S'ha demostrat que FLT3 està involucrat en el procé s de leucemogènesi a través de
diferents mecanismes. Diversos estudis han objectivat una expressió de FLT3 superior
a l'observada en precursors hemopoètics normals en el 70 -100% de les LMA i en el
90% de les LLA -B [222]; [223]. En particular, alguns subtipus de leucèmia, com són les
LA amb reordenament del gen MLL o les LLA hiperdiploides (HeH) presenten

INTRODUCCIÓ
87
característicament una major sobreexpressió de FLT3 [224]. Diversos articles han
demostrat que les LA amb sobreexpressió de FLT3 poden mostrar una gran sensibilitat
a inhibidors de FLT3, tot suggerint que la supervivència d’aquestes cèl·lules
leucèmiques depèn en part de l'activació d'aquesta via [225]. Hi ha diferents
mecanismes que condueixen a l’activació constitutiva de FLT3 en les cèl·lules
leucèmiques: mutacions del gen, que provoquen una activació independent de FLT3-FL
[223], i la sobreexpressió del gen no associada a mutacions del receptor, amb
coexpressió de FLT3 -FL per mecanismes autocrins/paracrins o independentment del
lligand (figura 1 5). Les mutacions del receptor FLT3 p oden ser eminentment de dos
tipus: les mutacions anomenades Internal Tandem Duplications (FLT3-ITD), que afecten
el domini juxtamembrana (JM) en forma de duplicació en tàndem o les mutacions
puntuals, que afecten el domini quinasa ( Activation Loop Domain, FLT3-ALM o Tirosin
kinase domain, FLT3-TKD) [226]. Les mutacions FLT3-ITD són resultat d’una inserció de
nucleòtids variable, entre 3 a 400 parells de bases, en el domini JM, que produeix una
inhibició del domini autoinhibit ori d'aquest receptor, causant una activació
constitutiva del mateix. El mecanisme pel qual es produeixen aquestes insercions no és
del tot conegut, però s’ha especulat que podria ser per la formació de seqüències
d’ADN palindròmiques que induirien errors en la replicació de l’ADN i per tant
provocarien la inserció de nucleòtids en tàndem [227]. Les mutacions tipus ITD també
s’han trobat en dominis no juxtamembrana [228].
El trasplantament a ratolins de cèl·lules de moll de l’os, a les que s’hi ha t ransduït FLT3-
ITD, dona lloc a un fenotip mieloproliferatiu, però no leucèmic complet. Per tant, se’n
dedueix que és necessària la cooperació amb altres alteracions per desenvolupar la
leucèmia aguda [229].
Les FLT3 -ALM/TKD corresponen a mutacions puntuals o delecions a nivell del loop
d'activació del domini amb activitat quinasa que alteren la configuració d'aquest
domini de manera similar a com ho fa el substrat FLT3 -FL [213], promovent així
l'activació de FLT3. Les més freqüents són les que involucren als aminoàcids D835 i
I836 del loop d’activació TKD2 [219]; [230]. Rarament, també es poden trobar
mutacions puntuals en el domini JM i TKD1 [231], [232] . Tot i que les mutacions al
domini JM també produeixen una activació constitutiva del receptor, similar a les FLT3 -

INTRODUCCIÓ
88
ITD , aquestes produirien una activació de vies diferent, donant lloc a un menor
potencial transformador leucèmic [233].
Les mutacions FLT3-ITD són presents en 15 -35% de la LMA d'adults, mentre que en els
nens aquest percentatge es redueix a un 10 -15% [216]. El percentatge de FLT3 -ITD és
només del 2 -3% en la LLA -B precursora i pràcticament inexistent en les LLA -T,
exceptuant les que mostren un fenotip tipus Early-T (ETP) [234]; [235]. Les mutacions
FLT3-ALM/TKD representen el 8% en LMA, tant en pacients adults com en pediàtrics
[223]. No obstant això, dins els subgrups de les LA -MLL+ i les LLA -HeH, les mutacions
FLT3-ALM/TKD es troben presents fins en un 20% de pacients [216][236].
Figura 15. Esquema del receptor FLT3 i localització dels diferents tipus de mutacions.
2.1 Implicacions pronòstiques de les alteracions de FLT3 en LA
En diversos estudis s’ha demostrat que les mutacions FLT3 -ITD són un factor de mal
pronòstic en la LMA d'adults [237][219] i en nens. Zwaan i Manara [238]; [239], en les
sèries més grans pediàtriques, amb 234 i 494 pacients respectivament, varen
demostrar que els nens portadors de mutacions FLT3 -ITD tenen pitjor pronòstic, amb
pitjors resposta, pitjor SLE i menor SG. Les ratis elevades d’al·lel mutat (FLT3 -ITD)/no
mutat (FLT3wild type ) al debut de la malaltia s’han associat a un pitjor pronòstic. En

INTRODUCCIÓ
89
casos extrems es pot produir una pèrdua d’expressió de l’ al·lel germinal no mutat
[219]; [240], [241] . El grup pediàtric AIEOP també va demostrar que els pacients amb
ratis al·lèlics elevats tenen un pitjor pronòstic ( SLE, 19.2% vs 63.5%) [239]. Quan es
produeix una recaiguda, aquesta sol ser oligoclonal, i es sol presentar amb ratis més
elevats que al diagnòstic i se mbla que els blasts, almenys in vitro , serien més
dependents de l’activació de la via de FLT3 [242]. En el cas de les leucèmia
promielocítica aguda, la presència de FLT3 -ITD sembla associar-se amb la presència del
transcrit BCR3 del gen PML-RARalfa i a amb hiperleucocitosi, però per se no sembla
tenir importància pronòstica [243] [244].
D'altra banda, l'anàlis i de les mutacions FLT3 -ALM/TKD no ha demostrat valor
pronòstic, encara que això és contradictori. Armstrong et al. [236] en una sèrie de 71
casos pediàtrics de LLA , troba mutacions en 10 pacients, sobretot en el grup de LLA
HeH. Tot i la pe tita mida de la sèrie, és interessant el fet que un 20% dels pacients
recaiguts eren portadors de mutacions a FLT3. Tot i aquests resultats, serien necessaris
més estudis per establir un valor pronòstic de les FLT3-ALM/TKD en les LLA.
Ozeki i col·laboradors [222], varen analitzar la importància clínica de la sobre-expressió
de FLT3 en LMA d'adults mitjançant RT -PCR quantitativa i varen trobar una associació
entre la sobreexpressió de FLT3 i mal pronòstic de la malaltia e n termes de SG i RC.
Recentment, un altre grup publicava l'impacte de la sobreexpressió de FLT3 en lactants
amb LA -MLL+ [245]. La SLE als 24 mesos en el grup que sobre -expressava FLT3 era
significativament inferior al grup que no el sobre-expressava (36% vs 71%). No s’ha
analitzat en profunditat si la sobre -expressió de FLT3 en altres subgrups de LA
pediàtrica pot tenir un impacte pronòstic. Així, les LLA HeH, encara que tenen una
major supervivència que les LA -MLL+, fins un 20 % presenten recaigudes en algun
moment de la seva evolució [246]. No s’ha determinat quin impac te podria tenir FLT3
en aquest subgrup.
2.2 Fàrmacs inhibidors de FLT3
No és d’estranyar, per tant, que en els últims anys s’hagin dissenyat varies molècules
inhibidores de FLT3 (al voltant de 12) que han demostrat la seva eficàcia in vitro en
diferents estudis i algunes d'elles com midostaurin, lestaurtinib, sorafenib, quizartinib,
gilteritinib i crenolanib, es troben en fases avançades d’assaigs clínics (fases II/III)

INTRODUCCIÓ
90
[222]; [247]; [248]; [249]–[252]. Quizartinib i gilteritinib són els que han demostrat
major eficàcia quan són utilitzats en monoteràpia, almenys en estudis amb pacients
recaiguts o refractaris. E ls resultats de l’assaig en fase III (CALGB 10603/RATIFY)
demostrà un augment de supervivència significatiu en els pacients als quals se’ls va
donar midostaurin conjuntament amb el seu tractament convencional de
quimioteràpia [253]. De forma similar, sorafenib en combinació amb qui mioteràpia
d’inducció va millorar la SLE [254].
L’addició d’inhibidors de FLT3 pod ria tenir alguna utilitat en tractaments de
manteniment post -trasplantament, però això no s’ha demostrat amb estudis
randomitzats [255], [256].
En un estudi in vitro , es va trobar que la inhibició de FLT3 mitjançant un d’aquests
fàrmacs (AG1296) mostrava igual eficàcia en línies cel·lulars de LMA amb
sobreexpressió de FLT3 i que no presentaven mutacions d'aquest receptor [222]. En
aquest sentit, també en la LLA pediàtrica s’ha demostrat que les cèl·lules leucèmiques
que sobreexpressen FLT3, independentment del seu estat mutac ional, són sensibles a
PKC412 ( midostaurin) i CEP -701 ( lestaurtinib) [223], [225] . Diferents estudis en línies
cel·lulars mostren l'efecte sinèrgic de l'i nhibidor de FLT3 sunitinib (SU11248) amb Ara -
C i daunorubicina en línies cel·lulars de LMA portadores de mutacions FLT3 -ITD [257].
Aquesta sinèrgia ha quedat també evidenciada en assaigs clínics [111], [258]. De totes
maneres, existeix poca informació sobre quina pot ser la millor combinació de fàrmacs
citostàtics amb inhibidors i s i el disseny d’aquest esquemes terapèutics dona marge
per una major eficàcia i que, per tant, acabin redundant en la supervivència d’aquests
pacients.
Hi ha un interès en relació als assaigs clínics que s’estan duent a terme amb inhibidors
de FLT3 més p otents i específics com quizartinib, crenolanib i gilteritinib (ASP-2215).
Quizartinib (AC220) és un inhibidor de segona generació altament selectiu enfront
FLT3. Diferents assaigs en fase II han demostrat la seva eficàcia en pacients refractaris
o recaig uts [259]–[261]. Desafortunadament, un 50% dels pacients presentava una
recaiguda en els següents 3 mesos. Diferents estudis suggereixen que la resistència a
quizartinib vindria determinada per l ’adquisició de mutacions al domini tiros ín-quinasa
de FLT3 [262], [263] . Un dels nous inhibidors que podria ser resistent a aquest
mecanisme és crenolanib [264].

INTRODUCCIÓ
91
Crenolanib ha mostrat activitat en front de la mutació D835 en el loop d’activació, una
de les responsables de la resistència que es desenvolupa a quizartinib. Els assajos en
pacients recaiguts o refractaris no han mostrat una eficàcia significativament superior
respecte a la resta d’inhibidors [264]. Actualment es troba en fase d’assaig en
combinació amb quimioteràpia d’inducció de primera línia (NCT02283177).
Gilteritinib (ASP-2215) és un potent inhibidor tant de FLT3 -ITD com de FLT3 -ALM/TKD.
L’assaig fase I/II en pacients recaiguts/ refractaris va mostrar bons resultats en els
pacients mutats [265].
Per ta nt, els nous inhibidors de FLT3 s’han mostrat notablement actius enfront a
pacients amb leucèmies recaigudes/refractàries. La relativa curta durada de la
resposta obtinguda amb fàrmacs com quizartinib o gilteritinib fa que puguin ser
potencialment utilitzats com a pont a un TPH. És necessari continuar explorant noves
opcions que permetin millorar la RC o la SG, ja sigui mitjançant la combinació amb
nous fàrmacs, agents hipometilants o quimioteràpia convencional [186].

INTRODUCCIÓ
92
3.Transportadors de nucleòsids en leucèmia pediàtrica
3.1 Transportadors de nucleòsids
Els fàrmacs derivats de nucleòsids són fàrmacs àmpliament utilitzats en el tractament
de les leucèmies agudes. Aquests han d e ser transportats a l’interior de la cèl·lula per
exercir la seva acció farmacològica.
La internalització dels nucleòsids naturals i dels seus anàlegs farmacològicament actius
només es pot dur a terme mitjançant transportadors de membrana plasmàtica, ja que
la naturalesa físico -química d’aquestes molècules fa que siguin altament hidrofíliques.
Els transportadors de nucleòsids (TNs) pertanyen a dues famílies gèniques de la
superfamília anomenada SLC (per Solute Carrier), SLC28 i SLC29. La família SLC28 inclou
tres gens, SLC28A1, SLC28A2 i SLC28A3, que codifiquen les proteïnes transportadores
hCNT1, hCNT2 i hCNT3. L’acrònim CNT prové de Concentrative Nucleoside Transporter ,
ja que tots tres són transportadors concentratius que utilitzen el gradient
transmembrana de sodi per tal de poder concentrar els nucleòsids o els fàrmacs en
contra de possibles gradients de concentració. hCNT1 és un transportador de
nucleòsids de pirimidina i d’anàlegs com la gemcitabina; hCNT2 és un transportador de
nucleòsids de purina i d’anàlegs com la ribavirina, mentre que hCNT3 és un
transportador d’àmplia selectivitat de substrat. La família SLC29 inclou 4 gens,
SLC29A1, SLC29A2, SLC29A3 i SLC29A4. Només els dos primers codifiquen per
transportadors de nucleòsids de membrana plasmà tica, hENT1 i hENT2, si bé cal
remarcar que SLC29A3, el gen que codifica per al transportador lisosomal hENT3, és
l’únic d’ambdues famílies que s’ha associat fins ara amb una malaltia rara en edat
pediàtrica, la síndrome H. L’acrònim hENT s’escau pel fet q ue aquestes proteïnes
faciliten el transport equilibratiu (no concentratiu) de nucleòsids i dels seus anàlegs a
favor de gradient de concentració. Tant hENT1 com hENT2 presenten una àmplia
selectivitat de substrat, mentre que hENT2 facilita millor que hENT 1 el transport de
nucleobases. De manera genèrica podríem dir que les proteïnes hENT són d’expressió
pràcticament ubiqua, mentre que els hCNT s’expressen molt en cèl·lules altament
diferenciades, especialment de barreres epitelials com la intestinal, renal , hepàtica i
placentària. En cèl·lules del sistema immune, curiosament, es va poder detectar també

INTRODUCCIÓ
93
l’expressió dels prototípics transportadors epitelials de la família SLC28, essent hCNT2 i
hCNT3, però no hCNT1, els que s’expressen a limfòcits B [266]; [267]; [268]. Les
característiques d’aquestes dues famílies de proteïnes han estat àmpliament revisades
a la literatura [269]; [270]; [271]; [272]; [273]. Tot i els importants avenços en el
coneixement de la distribució tissular i la farmacologia de les proteïnes hTN, encara hi
ha una comprensió limitada dels mecanismes que regulen la seva expressió i l'activi tat
[274].
3.2 Mecanismes d’acció i metabolisme de fàrmacs derivats de nucleòsids
La pràctica totalitat dels fàrm acs derivats de nucleòsids poden ser considerats com a
genotòxics, en tant que interfereixen en el metabolisme de nucleòtids i en la capacitat
de replicació del DNA i, eventualment, també en la síntesi de RNA [275], [276], [277]).
Precisament per això, molts d’ells induiran una aturada del cicle cel·lular en la transició
G1/S i promouran l’apoptosi. En la majoria dels casos, però, el que e l malalt rep són
anàlegs de nucleòsids que hauran de ser internalitzats i fosforilats per tal de generar
les formes actives dels fàrmacs, algunes de les quals poden ja en la seva forma
monofosforilada interferir en les vies de recuperació de nucleòsids, pe r exemple
inhibint enzims com la ribonucleòtid reductasa, mentre que les formes trifosfat seran
les que s’incorporaran als àcids nucleics exercint l’acció genotòxica [275]; [277].
El fet que en algunes condicions aquests transportadors de fàrmacs puguin lim itar la
seva biodisponibilitat i, conseqüentment, la seva acció citotòxica, fa que puguin ser
eventualment considerats com a biomarcadors de resposta tumoral. Les evidències
clíniques a aquest respecte en l’àmbit de tumors sòlids han estat recentment revis ades
[274], essent hENT1 el cas més ben estudiat fins ara, en el context de la resposta a la
gemcitabina en el tractament del càncer de pàncrees.
Tanmateix, a l’igual que per a tumors sòlids, semblen ser els transportadors de la
família SLC29 els que més poden condicionar la resposta terapèutica a fàrmacs
derivats de nucleòsids. Així, hENT2 sembla condicionar la resp osta a la fludarabina en
cèl·lules de leucèmia limfàtica crònica (LLC) [278]; [279], mentre que hENT1, al igual
que en tumor sòlids, semblava ser un important condicionant de l’acció citotòxica de la
gemcitabina en limfoma de cèl·lules del mantel l [280]. La pos sibilitat que els
transportadors de la família SLC28 puguin condicionar la resposta a fàrmacs en

INTRODUCCIÓ
94
malalties limfoproliferatives només s’ha posat de manifest recentment per al cas de
hCNT3, en una subpoblació de pacients de LLC caracteritzats per ser wild type per a
p53, ser resistents a fludarabina i presentar nivells funcionals de hCNT3 baixos, tot
limitant presumiblement l’acció del fàrmac [281]. De fet, la possibilitat de regular
farmacològicament aquest transportador s’ha suggerit com una possible alternativa
terapèutica per millorar quadres de resistència a la fludarabina associats a baixa funció
de hCNT3 [282]; [281].
L’Ara-C és un anàleg de nucleòsid que requereix, per ser internalitzat en les cèl·lules
diana, de les proteïnes de transpo rt de membrana de la família hNT, codificades pels
gens SLC28 i SLC29. L’Ara-C és transportat a través de la membrana de la cèl·lula,
principalment pel transportador de nucleòsids equilibratiu hENT1, codificat per
SLC29A1 [283]. Un cop dins de la cèl·lula l’Ara -C ha d’experimentar una activació
metabòlica mitjançant la seva fosforilació per diferents enzims del me tabolisme
intracel·lular, i així poder exercir la seva acció citotòxica. El pas limitant en aquest
procés és la seva fosforilació per la quinasa deoxicitidina quinasa (dCK) (figura 16) .
Figura 16. Model de com realitzen la seva acció els anàlegs de nucle òsids (AN) des de
que són transportats a l’interior de la cèl·lula mitjançant els TN. Adaptat de [284].

INTRODUCCIÓ
95
L’Ara-C, fàrmac àmpliament utilitzat en el tractament de la leucèmia aguda pediàtrica,
és un bon substrat de hCNT3 (tot i que sembla que també pot interaccionar amb
hCNT1), però també ho és per un dels transportadors equilibratius com hENT1 i, potser
en menor mesura, hENT2 [285]. hENT1 és un bon candidat a modular l’acció citotòxica
de la citarabina en leucèmia aguda pediàtrica, tal i com s’ha anticipat en estudis
preliminars [286]. hENT1 és també un transportador potencialment modulable [267];
[281] i, de fet, molt recentment, s’ha vist que pot estar sota control per fosforilació,
tant via PKA com PKC [287]; [288].
Enllà dels fenòmens de transport, la capacitat d’activar mitjançant fo sforilació o bé
d’inactivar la forma fosforilada del fàrmac mitjançant enzims de tipus nucleotidasa
sembla també jugar un paper important en l’acció citotòxica dels derivats de
nucleòsids [281], [285] ; [277]. Derivats de pirimidines com la gemcitabina i la pròpia
citarabina s eran bons substrats de l’enzim dCK. Les formes nucleotídiques
monofosforilades d’aquests fàrmacs generades per la dCK poden ser inactivades per
acció de la 5’-Nucleotidasa citosòlica (cNII). Des d’aquest punt de vista ambdós enzims
i el seu balanç semblen poder ser un element determinant del mecanisme d’acció dels
fàrmacs derivats de nucleòsids de tipus pirimidínic. La resistència a la gemcitabina pot
estar altament condicionada pels nivells de dCK [289], de la mateixa manera que la
cNII en si mateixa és una diana farmacològica interessant en tant que la seva inhibició
podria contribuir a mantenir nivells alts de les formes actives d’aquests fàrmacs [290].
De fet, la inhibició de la cNII també s’ha postulat com un meca nisme més d’acció per a
la fludarabina [291]. Des del punt de vista d’utilitzar aquests enzims com a
biomarcadors, s’ha comprovat que l’expressió de la dCK, al igual que la de hENT1 tal i
com indicàvem a dalt, també sembla poder ser un biomarcador útil en el cas de malalts
de càncer de pàncrees en monoteràpia amb gemcitabina, tot i que la millor
estratificació de supervivència s’obté quan ambdós biomarcadors es combinen, essent
major la supervivència, tal i com calia preveure, quan l’expressió de tots dos gens
(hENT1 i dCK) és alta [292]. És evident que hi ha altres enzims del metabolisme de
nucleòsids i nucleòtids que poden també condicionar la resposta a fàrmacs, tals com la
ribonucleòtid reductasa o la citidina desaminasa, però als efectes del treball que aquí

INTRODUCCIÓ
96
es presenta, els transportadors i les primeres etapes del metabolisme de fàrmacs són
els elements que s’han estudiat.
Taula 11. Transportadors de nucleòsids i tipus d’anàleg que transporten . Adaptada de [276].
Ara-C Fludarabina Clofarabina Cladribina
ENT1 X X X X
ENT2 X X X
CNT1 X NT NT NT
CNT2 NT NT X NT
CNT3 X X X
NT: No transporta
Així, la captació en les cèl·lules és un pas clau en la biodisponibilitat i l'acció
farmacològica dels anàlegs de nucleòsids [283] i, per tant, diversos estudis han
establert una correlació entre els nivells d'expressió d’ hENT1, la sensibilitat als fàrmacs
i la supervivència [14]. En aquest sentit, l'expressió d’ hENT1 elevada s'ha correlacionat
amb l'alta sensibilitat a l’ Ara-C dels lactants amb LA -MLL+, així com en pacients adults
amb LMA [192]; [293]; [294]. Per contra, els nivells d'expressió baixos d’hENT1 s'han
relacionat amb la resistència a l’Ara -C en LMA infantil. En general, aquestes dades
estan en línia amb l'evidència que la sensibilitat in vitro a l'Ara -C a la infància i en la
LMA en adults depèn d’hENT1 [294].
En aquest sentit, atès que els casos d'LA -MLL+ presenten alts nivells de FLT3 i alta
sensibilitat a l’Ara -C, FLT3 podria ser un candidat adequat per modular l'expressió i
l'activitat dels hTN, contribuint d'aquesta manera a la quimiosensibilitat de les
cèl·lules leucèmiques. A més, el fet que els transportadors de nucleòsids i dels seus
fàrmacs derivats no siguin necessàriament proteïnes d’expressió constitutiva, ans el
contrari, puguin estar fortament regulades, obre la possibilitat de poder utilitzar la
seva modulació com un element potenciador de l’acció terapèutica , com seria el cas
de hCNT3 en la LLC. Tanmateix, també obre la possibilitat que en teràpies combinades
on les dianes són receptors proteïna -quinasa com FLT3, el fàrmac co -administrat (p.e.
inhibidors de FLT3) pugui modular la funció dels transportadors de manera no
desitjable. Aquestes qüestions han estat objecte d’estudi en el present treball de tesi.

HIPÒTESI I OBJECTIUS
97
VI. HIPÒTESI I OBJECTIUS

HIPÒTESI I OBJECTIUS
98

HIPÒTESI I OBJECTIUS
99
HIPÒTESI
La leucèmia aguda pediàtrica és una malaltia heterogènia des del punt de vista clínic i
biològic. Diferents estudis han demostrat el paper rellevant del gen FLT3 en el procés
de leucemogènesi. Les mutacions que afecten aquest receptor han estat reconegudes
com a factors de mal pronòstic, però el paper de la sobreexpressió de FLT3 en sèries
grans de pacients pediàtrics no ha estat suficientment estudiat. Actualment con eixem
que un dels trets distintius de les LA -MLL+ és la sobreexpressió de FLT3. Aquest tipus
de leucèmies es caracteritzen per presentar una elevada sensibilitat a la citarabina i
aquest fet, en part, sembla relacionar -se amb una expressió elevada del seu principal
transportador de membrana, hENT1. Tanmateix, existeix molt poca informació sobre el
perfil d’expressió de transportadors de nucleòsids en els diferents subtipus de
leucèmia aguda pediàtrica.
Així, a l’hora de dissenyar els estudis que composen e l present treball de Tesi
Doctoral, es varen plantejar les següents hipòtesis:
1. La sobreexpressió de FLT3 en els diferents subgrups de LA pediàtrica podria tenir un
impacte pronòstic i contribuir a una millor estratificació dels grups de risc.
2. FLT3 intervé en el transport de citarabina mitjançant la modulació de l’expressió de
les proteïnes transportadores i els enzims implicats en metabolisme intracel·lular
d’anàlegs de nucleòsids en leucèmia aguda pediàtrica. D’acomplir -se aquesta hipòtesi,
podríem dissenyar millors esquemes amb combinacions de fàrmacs inhibidors de FLT3
i anàlegs de nucleòsids com la citarabina.

HIPÒTESI I OBJECTIUS
100
OBJECTIUS
Principal:
• Caracteritzar les alteracions moleculars de FLT3 en pacients pediàtrics afectes de
leucèmia aguda i estudi ar la seva possible correlació amb l’activitat i expressió dels
transportadors de fàrmacs derivats de nucleòsids.
Específics:
• Caracteritzar les diferents alteracions de FLT3 en LLA i LMA en una sèrie de pacients
pediàtrics: incidència, impacte pronòstic i correlació amb altres variables clínico -
biològiques.
• Analitzar la relació entre l’expressió de FLT3 i els TN i enzims de metabolisme
intracel·lular (EMs) dels anàlegs de nucleòsids en diferents subtipus de leucèmia aguda
pediàtrica.
• Estudiar la relació entre FLT3 i l’activitat funcional dels transportadors.
• Analitzar l’impacte de diferents inhibidors de FLT3 sobre el transport i efectivitat de la
citarabina.

MATERIAL I MÈTODE
101
VII. MATERIAL I MÈTODE

MATERIAL I MÈTODE
102

MATERIAL I MÈTODE
103
MATERIAL I MÈTODE
Pacients i línies cel·lulars
S’han analitzat mostres de moll de l’os de pacients pediàtrics amb diferents subtipus
de leucèmia aguda diagnosticats a l’Hospital Sant Joan de Déu entre els anys 2003 i el
2013. Les principals característiques dels pacients an alitzats es resumeixen en les
taules 13 i 15. Tots el pacients havien estat uniformement tractats segons els
protocols de la Sociedad Española de Hematología y Oncología Pediátricas (SEHOP),
en el cas de les LLA: SHOP-LAL-99 & 05 i en el cas de les LMA SHOP-LAM-00 & 07.
També han estat emprades tres línies cel ·lulars: SEM ( línia cel·lular procedent d’un
nen amb LLA -B amb t(4;11) i reordenament MLL. Aquesta línia sobreexpressa FLT3
sense tenir mutacions; DSMZ ACC 546 ); MV4 -11 (línia cel·lular procedent d’un nen
amb LMA amb t(4;11) i reordenament MLL. Aquesta línia presenta una mutació tipus
FLT3-ITD; DSMZ ACC 102) i K562 (línia procedent d’una crisi blàstica d’una LMC , amb
expressió de FLT3 baixa i sense mutacions; DSMZ ACC 10).
Extracció de ADN i ARN i transcripció reversa
Per a l’anàlisi del present treball es van utilitzar mostres del diagnòstic de medul·la
òssia o sang perifèrica amb >80% de blasts. La separació de les cèl·lules mononuclears
es va realitzar amb gradient de densitat amb Ficoll -Hypaque (Sigma, St Louis MO, EUA)
i l'ADN es va extreure amb el kit d'extracció de DNA Qiaquick o Gentra Puregene
(Qiagen, Hilden, Alemanya). L'ARN total es va extreure amb TriPure (Roche
Diagnostics, Indianapolis, IN) i la síntesi d'ADN complementari (ADNc) es va re alitzar a
partir 1 µg de RNA total, utilitzant el kit Qiagen cDNA Quantitect (Qiagen, Hilden,
Alemanya).
Determinació de l’estat mutacional de FLT3
L’estudi de les mutacions tipus FLT3 -ITD es va realitzar mitjançant l'amplificació del
domini juxtamembrana als exons 14 i 15, utilitzant encebadors marcats amb
fluorescència, i la posterior anàlisi en un seqüenciador 3130XL Genetic Analyzer
(Applied Biosystems, Life Technologies, Foster City, CA, EUA) [219];[237]. Per detectar
mutacions en els codons 835 i 836, es va amplificar l'exó 20 de FLT3 i es va digerir el
producte amb l'enzim EcoRV, segons un procediment ja descrit [237]. Tots els casos

MATERIAL I MÈTODE
104
positius van ser seqüenciats directament per mètode Sanger per confi rmar la
presència de mutacions, utilitzant el BigDye Terminator v3.1 Cycle Sequencing Kit
(Applied Biosystems, Life Technologies).
Quantificació de mRNA de FLT3 i expressió de proteïnes
La quantificació del mRNA relacionat amb el gen FLT3 es va realitzar mitjançant PCR en
temps real quantitativa (RQ -PCR) utilitzant la sonda comercial TaqMan®
Hs00174690_m1 (Applied Biosystems, Life Technologies) en un Light Cycler -480 II
(Roche Diagnostics, Indianapolis, IN), d'acord amb les instruccions del fabricant. La
quantificació relativa es va calcular amb el mètode 2 -ΔΔCt, utilitzant β -glucuronidasa
(GUS) (NM_000181.2, Ref. 4310888E) com a gen end ogen i cèl·lules CD34+ normals
com a calibrador.
Els nivells de proteïna FLT3 abans i després del tractament amb l'inhibidor de FLT3
PKC412 es van quantificar per Western blo t. Per a aquest propòsit, les cèl·lules es van
tractar amb PKC412 (0.045μM) durant 2, 4 i 24 hores i van ser llisades mitjançant un
tampó de lisi que conté 20 nM Tris -HCl, 150 mM NaCl, 1 mM EDTA, 1% de Triton,
inhibidor de la proteasa X -100 suplementat amb ortovanadat de sodi 1 mM (Mini
completes; Roche, Basilea, Suïssa) i còctel inhibidor de la fosfatasa (PhosSTOP; Roche).
Cinquanta grams de proteïnes es van separar mitjançant SDS -PAGE en gel estàndard al
10% i es va transferir a membranes Immobilon -P (Mil lipore, Berford, MA, EUA). Les
membranes es van incubar amb anticossos anti -FLT3 (8F2) i anti -phospo-FLT3 (Tyr591)
(Senyalització Cel·lular, MA, EUA). Després de rentar amb TBS -Tween, les membranes
es van incubar amb peroxidasa (HRP) conjugat amb anticosso s secundaris. Les bandes
immunoreactives es van detectar mitjançant quimioluminiscència (ECL, Amersham
Pharmacia Biotech, NJ, EUA).
Quantificació dels transportadors de nucleòsids (TNs) i Enzims de Metabolisme
intracel·lular (EMs)
La quantificació de l’ex pressió dels TNs i els EMs es va realitzar mitjançant RQ -PCR de
hCNTs i hENTs. Es van realitzar amb encebadors i sondes d'Applied Biosystems,
utilitzant la Màster Mix TaqMan universal, 700 nmol/L de sonda i 150 nmol/L de cada
encebador en el sistema ABI Pr ism 7700 de detecció de seqüències (Applied
Biosystems). El nivell de mRNA relatiu de cada gen es va calcular amb el mètode 2 -ΔΔCt,

MATERIAL I MÈTODE
105
es va normalitzar al nivell d’expressió de les cèl·lules β -glucuronidasa (GUS) i es varen
utilitzar cèl·lules CD34+ normals de medul·la òssia com a calibrador. Els resultats
absoluts de la RQ -PCR es van obtenir a partir de la interpolació dels ∆CTs de cada
mostra en la línia de regressió estandarditzada per al nombre de còpies d'ADNc de
cada gen. Els nivells de proteïna hENT1 v an ser semi -quantificats per Western Blot,
utilitzant un anticòs comercial policlonal anti -hENT1 (STJ96396) a partir de Saint John’s
Laboratory Ltd. (Londres, Regne Unit).
Mesura d’activitat de transport del hTN
El transport de nucleòsids va ser mesurat en les línies cel·lulars MV4 -11, SEM i K562
utilitzant la tècnica prèviament descrita [282]. Les cèl·lules van ser rentades dos cops i
resuspeses en una solució tampó NaCl 137mM o colin a 137mM. Els experiments de
transport es van iniciar barrejant una suspensi ó de les cèl·lules amb un 10% del volum
final del mateix tampó suplementat amb radionucleòsids - [3H]Ara-C o [ 3H]citidina – a
una concentració final de 1μM (activitat específica d’1μCi/nmol), a una activitat
específica de 4000 dpm/pmol. La incubació es va parar en 1 minut (condicions de
velocitat lineal inicial) rentant les cèl·lules tres cops en 1 mL de tampó fred, compost
per NaCl 137mM i Tris (hidroximetil) aminometan -HEPES (pH 7.4) 10mM. Les cèl·lules
es van dissoldre en 1ml de Triton -X-100 i es van fer alíquotes per a la determinació de
proteïnes, segons el protocol Bradford (Bio -Rad, Hercules, CA) i mesures de
radioactivitat.
Amb la presència de Na +, els hENTs i hCNTs són funcionals, però només els hCNTs
requereixen aquest ió per la translocació de substracte. Per tant, el transport depenent
de sodi de hCNT va ser mesurat mitjançant la substracció dels índex de transport
mesurats en el medi de cloridi de colina (exclusivament relacionat a l’activitat d’hENT1
i hENT2), dels mesurats en el tampó que cont enia sodi (en el qual tant hENT com
hCNT són actius).
Els experiments d’inhibició creuada van ser realitzats com es descriu prèviament, però
es va afegir al medi d’incubació un segon radionucleòsid a concentracions saturants
(100µM), el qual competeix pel transportador i bloqueja el transport del substracte
marcat. Afegint guanosina extra al medi de transport de [ 3H]citidina, l’activitat hCNT3
es bloqueja, però no la de hCNT1. L’activitat de transport d’hCNT1 va ser determinada

MATERIAL I MÈTODE
106
sostraient l’activitat en le s condicions [ 3H]citidina + guanosina, de la de la [ 3H]citidina
sola i l’activitat d’hCNT3, de la sostracció de la de [ 3H]citidina + guanosina d’una
obtinguda quan s’utilitzava [ 3H] citidina sola. El transport equilibratiu (Na +
independent) inhibit per NBT I (1μM) correspon a l’activitat d’hENT1, mentre que el
transport resistent a NBTI correspon a l’activitat residual d’hENT2 més el component
de difusió i unió que, en general, són negligibles.
Estudis de citotoxicitat i detecció d’apoptosi mitjançant assaigs MTT
Per als assajos de citotoxicitat de rutina, 2x104 cèl·lules es van cultivar amb la presència
o absència dels inhibidors de FLT3. Per conèixer el paper que els transportadors de
nucleòsids poden jugar en citotoxicitat induïda per citarabina, les cèl ·lules es van
incubar durant 15 minuts, ja sigui en presència o en absència d'NBTI (1μM per a la
inhibició d’hENT1) o floridzina 250 micres (per a la inhibició de hCNT1). Posteriorment
es va afegir Ara -C (3μM i 1μM per MV4 -11 i SEM, respectivament) i es va calcular la
citotoxicitat després de 48 hores utilitzant 3 -(4,5-dimetiltiazol-2-il)-2,5-
difeniltetrazoliumbromida (MTT; Sigma -Aldrich). La densitat òptica (OD) es va mesurar
a 550 nm. Per a l'anàlisi de la citotoxicitat amb la combinació de fàrmacs, les c èl·lules
van ser cultivades amb PKC412 (0.045μM) o AC220 (0,5 nM) i Ara-C (10μM), seguint el
disseny experimental descrit en la secció Resultats, dirigit a avaluar el paper de l'ordre
de l'administració del fàrmacs en l'eficàcia quimioterapèutica. El Coeficient d'Interacció
de Fàrmacs ( Drug Interaction Coefficient, DIC ) es va calcular segons la tècnica
prèviament descrita [295].
Anàlisi estadística
Per a la c orrelació de les variables clíniques i biològiques, es va utilitzar la prova χ2 i la
prova exacta de Fisher per a la comparació de variables categòriques i la prova t de
Student o la prova de Mann-Whitney U-test per proves no paramètriques. A més, es va
utilitzar una prova de Kruskal -Wallis per comparar els nivells d'expressió gènica de
FLT3 i TNs entre els diferents subgrups genètics. La probabilitat de la supervivència
lliure d’esdeveniment (SLE) i la supervivència global (SG) es va calcular d'acord amb e l
mètode de Kaplan -Meier, i la prova de log -rank es va utilitzar per avaluar diferències
entre les distribucions de supervivència. La SG es va calcular a partir de la data del
diagnòstic fins a la data de la mort o l'últim seguiment, i la SLE es va calcula r a partir de

MATERIAL I MÈTODE
107
la data del diagnòstic fins a l’aparició d’un esdeveniment (recidiva, mort, altres
esdeveniments o última visita). En el cas dels pacients que van rebre un TPH, la SLE es
va censurar a la data del transplantament. El model de regressió de Cox es va utilitzar
per avaluar el valor predictiu de múltiples variables al moment del diagnòstic en
relació amb SG i SLE en l’anàlisi multivariant. El valor de la p es va considerar
significatiu quan <0,05. Totes les anàlisis estadístiques es van realitzar amb el paquet
estadístic SPSS 24.0 (SPSS Inc., Chicago, IL, EUA).
Principis ètics
La investigació ha estat portada a terme de conformitat amb les normes ètiques i
d'acord amb la Declaració d'Hèlsinki i amb les directrius nacionals i internacionals.
L’estudi ha estat aprovat pel comitè ètic de la nostra institució. Tots els pacients i/o
representants legals han firmat un consentiment informat.

RESULTATS
108

RESULTATS
109
VIII.RESULTATS

RESULTATS
110

RESULTATS
111
Treball 1: Caracterització de les diferents alteracions de FLT3 en LLA i LMA en una
sèrie de pacients pediàtrics: incidència, impacte pronòstic i correlació amb altres
variables clínico-biològiques
En aquest treball s’analitza l’estat mutacional i nivell d’ex pressió en mostres de 204
pacients pediàtrics afectes de diferents subtipus de leucèmia aguda (LLA -B: 144; LLA-T:
23; LMA: 36 i una MPAL), uniformement tractats en el nostre centre segons protocols
de la Sociedad Española de Hematología y Oncología Pediátr icas (SEHOP). A més de la
caracterització de l’estat de FLT3 també vam analitzar la correlació amb altres variables
clínico-biològiques i l’impacte pronòstic de la sobreexpressió de FLT3.
En la nostra sèrie de pacients el número de mutacions de FLT3 va se r escàs. Vàrem
observar que l’expressió de FLT3 es correlacionava amb el llinatge de la leucèmia i el
subtipus genètic; així, els pacients amb LLA-MLL+ són els que mostraren una expressió
més elevada. En el cas de les LLA -B la sobreexpressió s’associava si gnificativament a
l’edat (lactants), leucocitosi i reordenament MLL. Cal destacar que la sobreexpressió de
FLT3 no va mostrar una importància pronòstica (SG ni SLE) en els pacients amb LLA -B,
LLA-T ni LMA. Nogensmenys, sí que vàrem demostrar un impacte pro nòstic d’altres
factors ja descrits clàssicament com l’edat, la hiperleucocitosi, l’afectació del SNC, la
categorització en subgrups de risc genètic, la presència del reordenament de MLL i la
presència de MRM al final d’inducció.

RESULTATS
112
Els resultats principals del treball són:
Característiques clínico-biològiques de la sèrie de pacients pediàtrics amb leucèmia
aguda analitzats
Per a l’anàlisi vam incloure 204 pacients diagnosticats de leucèmia aguda a l’Hospital
Sant Joan de Déu des de 2003 fins al 2013. Es va ren analitzar 122 nens i 82 nenes, amb
una mitjana d’edat de 5,4 anys ( extrems 0-17,4 anys). Vam incloure 15 lactants (7,4%).
Els pacients van rebre tractament segons protocols de la Sociedad Española de
Hematología y Oncología Pediátricas (SEHOP), en el cas de les LLA: SHOP-LAL 99, SHOP
05 i SEHOP-PETHEMA 2013 i en el cas de les LMA SHOP -LAM-00 & 07. Els lactants amb
LLA varen rebre tractament segons el protocol internacional INTERFANT 06. Les
principals característiques clíniques i biològiques al diagnòst ic i evolutives es troben
descrites a la taula 12.
Taula 12. Característiques clíniques i analítiques dels pacients analitzats.

RESULTATS
113
N=204
Edat, anys, mediana (extrems) 5,4 (0-17,4)
Sexe, n (%)- Masculí- Femení
122 (60)82 (40)
SNC, n (%)- SNC1- SNC2a
- SNC3- NR
164 (80)27 (13,5)11 (5,5)
2 (1)
Leucòcits, x109/L, mediana (extrems) 10,15 (0,3-741)
Hemoglobina, g/L, mediana (extrems) 8,1 (2,8-19)
Plaquetes, x109/L, mediana (extrems) 64,5 (2-588)
Blasts, %, mediana (extrems)- Moll de l’os- Sang perifèrica
90 (13-100)45 (0-99)
Immunofenotip, n (%)- LLA-B precursora- LLA-T- LMA- MPALb
144 (71)23 (11)
36 (17,5)1 (0,5)
Genètica, n (%)- LLA, 168
o HeHo altresc
o ETV6-RUNX1o MLL+o TCF3-PBX1o BCR-ABL1o LLA-T
- LMA, 36o t(8;21)(q22;q22)o inv16/t(16;16)( p13;q22)o t(15;17)(q24;q21)o t(9;11)(p21;q23)o altres MLLo t(6;9)(p22;q34)o LMA amb canvis relacionats
amb SMDo normalo altreso no metafases
47 (23)41 (20)
27 (13,5)10 (5)9 (4,5)10 (5)
23 (11)
1 (0,5)3 (1,5)2 (1)2 (1)8 (4)
1 (0,5)3 (1,5)
5 (2,5)8 (4)
3 (1,5)
Expressiód FLT3, n, mediana (extrems) 6,85 (0,015-694,50)

RESULTATS
114
Mutacions tipus FLT3-ITD, n: 203 (%) 4 (2)
Mutacions tipus FLT3 ALK/TKD, n:187 (%) 4 (2,2)
Protocol rebut- LLA
o SHOP LAL 99o SHOP LAL 05o SEHOP PETHEMA 2013o INTERFANT 06
- LMAo SHOP LAM 00o SHOP LAM 07o LPA PETHEMA 05
2690457
16162
Remissió completa, n: (%)Refractarietat, n: (%)Mort en inducció, n: (%)
200 (98)1 (0,5)3 (1,5)
LLA, n (%)MRM dia +15 ≥10% (n: 168)MRM post inducció ≥0,01% (n: 166)
17 (10)26 (16)
TPH: AutòlegAl·logènic
13 (6,4)37 (18,2)
Seguiment, anys, mediana (extrems) 5,8 (0,56-15,51)
Recidiva, n (%) 32 (16)
Mort, n (%) 32 (16)
SG als 5 anys, % ± 95% IC 84 ± 2,7
SLE als 5 anys, % ± 95% IC 74 ± 3,5
a: SNC2 + SNC2 traumàtiques; b: pacient amb leucèmia aguda indi ferenciada (MPAL) amb reordenament MLL; c: LLA-B B-other; d: unitats arbitràries, veure apartat material i mètodeSNC: sistema nerviós central; NR: no realitzat l’estudi; MPAL: mixed phenotype acute leukemia; MRM: malaltia residual mínima; SG: supervivència global; SLE: supervivència lliure d’esdeveniment
Baix número de mutacions de FLT3 en la sèrie de pacients analitzats
Segons es descriu al capítol de material i mètodes “Determinació de l’estat mutacional
de FLT3” es va analitzar la presència de mutacion s tipus ITD en 203 pacients de la sèrie
i de mutacions puntuals en el codó D835 o delecions dins el codó i836 en 18 7 pacients
de la mateixa sèrie. Vàrem trobar 4 mutacions tipus FLT3 -ITD en la nostra sèrie, 2 en
LLA B (un pacient amb LLA -B HeH i una nena a mb una iAMP21); una nena amb una

RESULTATS
115
LLA-T tipus ETP i, en el cas de les LMA, l’única mutació que es va trobar va ser en una
adolescent amb una translocació t(6;9)(p23;q34).
Respecte a les mutacions puntuals, es van detectar 2 mutacions en l’estudi mitjançant
PCR i digestió amb l’enzim Eco-RV, corresponents a 2 casos de LLA-B HeH.
Correlació entre l’expressió de FLT3 amb paràmetres clínico -biològics: e ls nivells
d’expressió de FLT3 varien segons el llinatge i la genètica de la leucèmia
Es va valorar la possibl e correlació entre l’expressió de FLT3 amb les principals
variables clínico-biològiques, com el sexe, l’edat, el nombre de leucòcits o la infiltració
del SNC. L’expressió mediana de FLT3 en la sèrie global va ser de 6,85 (0,015 -694,5,
unitats arbitràries d’expressió respecte el calibrador, veure material i mètode). No vam
trobar una correlació significativa amb cap de les variables clíniques esmentades dels
pacients analitzats, però vam observar una expressió mediana significativament
diferent segons el lli natge, de manera que els pacients amb LLA -B precursora eren els
que més FLT3 expressaven, seguits pels pacients amb LMA; els pacients amb LLA -T
presentaren els nivells més baixos de FLT3 (LLA-B: 9,64; LLA -T: 1,46; LMA: 4,74;
p<0,0001) (figura 1 7). També v am observar una expressió diferent segons la genètica
(p<0.0001). Cal destacar que els pacients amb LLA -MLL+ són els que mostraren una
expressió més elevada (mediana 41,27; extrems 3,16 -228,45), seguits dels casos amb
LLA-B HeH (mediana 20,36; extrems 0,79 -312,53), B-other (mediana 9,3; extrems 0,71-
276,54) i les LMA -MLL+ (mediana 8,15; extrems 1,12 -98,65). Els nivells més baixos de
FLT3 els vàrem trobar en els pacients amb LLA -T (mediana d’expressió 1,46; extrems
0,21-64,23).

RESULTATS
116
Figura 1 7. Nivells d’exp ressió de FLT3 (mRNA) segons el subgrup genètic en els
pacients de la sèrie. La quantificació es va fer mitjançant RQ -PCR segons s’especifica en
l’apartat de material i mètodes. Els límits de les caixes corresponen als valors entre els
percentils p25 i p75 i la línia del mig representa el valor de la mediana. A sota: mediana
d’expressió de cada subgrup.
Mediana: 41,28 20,36 2,37 2,89 4,81 9,30 1,46 8,16 3,79
Donat que en alguns treballs s’ha definit la sobreexpressió de FLT3 com aquells valors
per sobre del percentil 75, vam analitzar la nostra sèrie considerant aquest punt de
tall. De nou, l’única variable estadísticament significativa correlacionada amb la
sobreexpressió de FLT3 en la sèrie global va ser el llinatge (taula 13).

RESULTATS
117
Taula 13. Correlació de les principals variables clínico biològiques al debut amb els
nivells d’expressió de FLT3 (FLT3 alt (≥p75) vs FLT3 baix (<p75)).
Variables clínico-biològiques N total: 204 pFLT3 alt (≥p75)
FLT3baix(<p75)
Sexefemení masculí
1635
6687
0,13
Edat (anys)<11-10≥10
53511
1010637
0,71
Leucòcits (x109/L)<50≥50
3912
12231
0,46
Infiltració SNCSNC1SNC2SNC3
4281
1221910
0,4
LlinatgeLLA-BLLA-TLMA
4434
1002032
0,04
Correlació entre l’expressió de FLT3 amb paràmetres clínico -biològics en els diferents
subgrups de pacients
En el subgrup de pacients amb LLA-B precursora (n=144), la sobreexpressió de FLT3
(≥p75) es va associar de forma significativa a l’edat (lactants vs nens més gr ans,
p=0,024), a la leucocitosi ≥20x10 9/L (p=0,026), al fenotip pro -B (p<0,0001) i al
reordenament del gen MLL (p=0,002) i la categoria molecular (p=0,001). No vam trobar
correlació significativa amb la resta de variables al diagnòstic ni amb els nivells d e
MRM en el seguiment.
No vam trobar cap correlació significativa entre l’expressió de FLT3 i paràmetres
clínico-biològics al debut o evolutius en els pacients amb LLA-T (n=23).
Vàrem estratificar els nostres pacients amb LMA (n=36) en grups de risc genè tic (veure
taula apartat factors de risc en LMA ). Així, a la nostra sèrie vàrem identificar pacients
de baix risc: LMA CBFs i LPA (n=7); pacients d’alt risc, amb monosomia del cromosoma
7 i t(6;9)(p22;q34) (n=5) i pacients de risc intermig (n=24). Tot i qu e el nombre de

RESULTATS
118
pacients en cada grup és reduït, vàrem observar que en el grup d’alt risc la mediana de
d’expressió de FLT3 era més elevada (p=0,049, figura 18).
Figura 18. Nivells d’expressió de FLT3 (mRNA) segons el subgrup genètic de risc en
LMA. La qua ntificació es va fer mitjançant RQ -PCR segons s’especifica en l’apartat de
material i mètodes. Les caixes corresponent als valors entre el p25 i p75 i la línia del
mig representa el valor de la mediana. A sota: mediana d’expressió de cada subgrup.
Mediana: 8.33 3.01 29.1
La sobreexpressió de FLT3 no va ser un factor pronòstic significatiu en la nostra sèrie
de pacients pediàtrics amb leucèmia aguda
Els pacients amb LLA -B de la nostra sèrie van presentar una SG als 5 anys del 91,3%
±2,5%, els pacients amb LLA -T del 78,3% ±8,6% i els pacients amb LMA 56 ±9%
(p<0,0001, figura 1 9). La SLE als 5 anys va ser del 80% ±4%, 77% ±9% i 50% ±9%, per
LLA-B, LLA-T i LMA, respectivament (p<0,0001).
L’anàlisi de la sobreexpressió de FLT3 (≥p75) en la sèrie global de 204 pacients no va
mostrar un impacte pronòstic significatiu sobre la SG ni la SLE. Les anàlisis del subgrup
de pacients afectes de LLA (144 pacients amb LLA -B precursora i 23 pacients amb LLA -
T) i LMA (36 pacients) es mostren de forma separada a continuació.

RESULTATS
119
Figura 19. Supervivència global dels pacients pediàtrics amb leucèmia aguda analitzats
a la nostra sèrie.
Pacients afectes de LLA-B:
Després d’una mediana de seguiment de 5,8 anys (extrems 0,75 -15,23 anys) els
pacients amb LLA -B de la nostra sèrie varen presentar una SG als 5 anys del 91,3% ±
2,5% i una SLE als 5 anys del 80% ± 4%.
a) Quant a l’estudi dels factors predictius de la SG en l’anàlisi univariat, les variables
que van resultar significatives van ser l’edat, el subgrup de risc genètic i la presència de
MRM al final d’inducció (p=0,02; p=0,006; p=0,019; respectivament, taula 14, figura
20). Respecte l’edat, els lactants (<1 any) i els pacients majors de 10 anys presentaren
un pitjor pronòstic respecte el grup de pacients entre 1 i 9 anys. Quant a la genètica,
vam estratificar els pacients en grups de diferent risc segons els criteris utilitzats en els
nostres protocols de tractament (baix risc genètic: LLA -B HeH i ETV6-RUNX1; alt risc
genètic: reordenaments de MLL, BCR-ABL1 i hipodiploïdia <44 cromosomes; risc
genètic intermig: la resta de pacients). En aquests subgrups, vàrem observar una pitjor
SG en els pacients de risc intermig. Els pacients del grup genètic d’alt risc van presentar
una supervivència molt favorable, similar a la SG del grup de baix risc. Tots els pacients
16,014,012,010,08,06,04,02,00,0
Anys
1,0
0,8
0,6
0,4
0,2
0,0
Pro
bab
ilita
t
Supervivència global
LLA-B precursora
LLA-T
LMA
MPAL
p<0,0001

RESULTATS
120
amb reordenament BCR-ABL1, inclosos al grup d’alt risc, van rebre imatinib de forma
concomitant amb la quimioteràpia; els 6 pacients amb LLA -Ph+ inclosos al protocol
SHOP-LAL-05 van rebre un TPH al·logènic en primera remissió completa. En el protocol
posterior, SEHOP-PETHEMA 2013, el criteri de TPH al·logènic es va restringir a aquells
pacients amb persistència de MRM. Dels 4 pacients amb LLA -Ph+ inclosos en aquest
protocol, només una pacient va anar a un TPH al·logènic per persistència de la MRM en
punts tardans del seguiment. En la nostra sèrie, la SG de tots els pacients amb LLA -Ph+
va ser del 100%.
De forma similar, l’anàlisi de la SG estudiant cadascuna de les alteracions genètiques
per s eparat també va mostrar diferències estadísticament significatives (p=0,04). De
nou, els pacients amb reordenaments ETV6-RUNX1, BCR-ABL1 i LLA-B HeH presentaren
SG als 5 anys per sobre del 95%, superiors als pacients amb reordenament del gen MLL
(90% ± 10% ), els pacients amb LLA -B B other (83± 6,5%) i els pacients amb
reordenaments TCF3-PBX1 (78± 14%).
Finalment, la detecció de MRM per citometria de flux (>0 ,01%) al final de la inducció
també va tenir un impacte significativament advers sobre la SG en la n ostra sèrie de
pacients. Els pacients amb SNC1 van mostrar una millor SG respecte les altres
categories (SNC2 i SNC3) (p=0,023).
L’anàlisi de la sobreexpressió de FLT3 (≥p75) en la nostra sèrie de pacients amb LLA -B
precursora va mostrar una tendència a u na millor SG (p=0,085), probablement per la
seva associació a les LLA -B HeH. La resta de variables analitzades: leucocitosi al
diagnòstic (leucòcits ≥20, 50, 100 o 200x10 9/L), la detecció de ≥10% de blasts al MO al
dia +15 per citometria de flux o la presè ncia de reordenament de MLL no varen
mostrar un impacte significatiu sobre la SG.
En l’anàlisi multivariat no es va veure que cap dels factors descrits presentés una
importància significativa a nivell de la SG.
b) Anàlisi univariat per la SLE: vàrem anali tzar també l’impacte sobre la SLE de cada
una d’aquestes variables (taula 14). Així, vàrem veure que l’edat (p<0,0001), la
hiperleucocitosi extrema al diagnòstic (leucòcits ≥200x10 9/L, p=0,006), l’afectació del
SNC (p<0,0001), la categorització en subgrups de risc genètic (p<0,0001), la presència

RESULTATS
121
de reordenament de MLL (p=0,002) i la presència de MRM al final d’inducció (≥0,01%,
p<0,0001), van impactar negativament sobre la SLE (taula 14, figura 20).
En l’anàlisi multivariat, les diferents variables que va n assolir la significació estadística
van ser l’afectació del SNC (HR 2,83 (1,44 -5,83 IC95%), p=0,005); el subtipus de risc
genètic (HR 3,4 (1,56 -7,41 IC95%), p=0,002) i la presència de MRM al final de la
inducció (HR 5,33 (2,13-13,3 IC95%), p<0,0001).
Taula 14. Factors pronòstics rellevants en la nostra sèrie de pacients amb LLA-B.
Variable NSG als 5 anys (%)
Univariat SG
Multivariat SG
SLE als 5 anys
(%)
Univariat SLE
Multivariat SLE
Edat<11-10≥10
810630
75 ±1596 ±280 ±8
0,02pns
66 ±2090 ±4
62 ±11
<0,0001 pns
Leucòcits*<200x109/L≥200x109/L
1395
92 ±380 ±18
0,30 pns 84 ±460 ±21
<0,0001 pns
SNCSNC1SNC2SNC3
123165
94 ±270 ±1580 ±18
0,08pns
85 ±476 ±1530 ±24
<0,0001HR 2,83
(1,44-5,83)p=0,005
Subgrup genètic**
Baix riscRisc intermigAlt risc
745020
97 ±281 ±695 ±5
0,006pns
93 ±364 ±8
44 ±22
<0,0001HR 3,4
(1,56-7,41)p=0,002
Reordenament MLL
MLL+MLL-
10134
90 ±9,592 ±3
0,926pns 37 ±20
82 ±4
0,002 pns
MRM dia +15<10%≥10%
10115
94 ±386 ±10
0,35 pns 81 ±567 ±16
0,482 pns
MRM final inducció
<0.01%≥0,01%
11521
94 ±275 ±12
0,019pns 87 ±4
66 ±15
<0,0001HR 5,33
(2,13-13,3)p<0,0001
FLT3Alt (≥p75)Baix (<p75)
36108
97 ±389 ±3
0,168pns 84
±4,580 ±8
0,41 pns
*Leucòcits 20, 50 i 100x109/L, pns. ** Baix risc: HeH, ETV6-RUNX1; alt risc: BCR-ABL1, MLL+, hipodiploïdia; risc intermig: la resta d’alteracions genètiques o cariotip normal. Pns: “p” no significativa.

RESULTATS
122
Figura 20. Supervivència global i lliure d’esdeveniment (SG i SLE) dels pacients amb
LLA-B precursora de la nostra sèrie. Les variables que van tenir un impacte sobre la SG
(20A, 20C, 20E) i sobre la SLE (20B, 20D, 20F) van ser l’edat, el grup de risc genètic i la
presència de MRM positiva (≥0,01%) al final de la inducció, respectivament. La
hiperleucocitosi ≥200x10 9/L, la infiltració del SNC i el reordenament del gen MLL
també van impactar significativament sobre la SLE (veure taula 14).

RESULTATS
123
Pacients afectes de LLA-T:
Després d’una mediana de seguiment de 4,4 anys ( extrems 1,88-12,2 anys) els pacients
de la nostra sèrie amb LLA -T varen presentar una SG als 5 anys del 78,3% ± 8,6% i una
SLE als 5 anys del 77% ± 9%.
El factor que va resultar predictiu de la SG de forma significativa va ser la presència de
MRM (≥0,01%) al final d’inducció (p=0,004, taula 15). El pacients amb fenotip cortical
van presentar una tendència a una millor SG (p=0,056).
La resta de variables analitzades, com l’edat, la hiperleucocitosi al diagnòsti c (leucòcits
≥200x109/L) o la infiltració del SNC, no varen mostrar un impacte significatiu sobre la
SG en els pacients amb LLA-T. Tampoc vàrem veure un impacte pronòstic en quant a la
sobreexpressió de FLT3.
En l’anàlisi multivariat, cap dels factors es mentats va assolir una significació estadística
per la SG.
Vàrem analitzar també l’impacte sobre la SLE de cadascuna d’aquestes variables. Així,
vàrem veure que la hiperleucocitosi al diagnòstic (leucòcits ≥200x10 9/L) i la presència
de MRM al final d’induc ció presentaven un impacte negatiu sobre la SLE (p=0,019 i
p<0,0001; respectivament, taula 15).
En l’anàlisi multivariat no es va confirmar el paper predictiu de cap dels factors descrits
per la SG o la SLE.

RESULTATS
124
Taula 15. Factors pronòstics rellevants en la nostra sèrie de pacients amb LLA-T.
Pacients afectes de LMA:
Després d’una mediana de seguiment de 8,4 anys ( extrems 0,56-15,51 anys), els
pacients de la nostra sèrie amb LMA varen pre sentar una SG als 5 anys del 60% ± 8,3%
i una SLE als 5 anys del 51% ± 8,5%.
Degut al baix nombre de pacients analitzats (n=36) i a l’heterogeneïtat dels subgrups
genètics analitzats, cal prendre els resultats amb precaució. Els factors que van resultar
predictius per la SG i la SLE van ser l’edat (p=0,018 i p=0,039, respectivament) i el s
leucòcits ≥100x10 9/L (p=0,039 i p=0,022, respectivament). No vàrem trobar correlació
entre la sobreexpressió de FLT3 i la supervivència dels nostres pacients.
Variable N SG als 5 anys (%)
Univariat Multivariat SLE als 5 anys (%)
Univariat SLE
Multivariat SLE
Edat<11-10≥10
0149
-79 ±1178 ±14
0.9 pns-69 ±1289 ±10
0,28 pns
Leucòcits <200x109/L≥200x109/L
176
82 ±967 ±19
0.32 pns 88 ±840 ±22 0,019
pns
SNCSNC1SNC2SNC3
1821
72 ±10100100
0,45 pns70 ±11100100
0,42 pns
FenotipCorticalAltres
176
88 ±850 ±20
0,056 pns 81 ±1067 ±19 0,472
pns
MRM dia +15<10%≥10%
211
81 ±8,60
0,06 pns 76 ±9100
0,65 pns
MRM final inducció<0,01%≥0,01%
185
89 ±7,540 ±22
0.004 pns 94 ±6
20 ±18
<0,0001 pns
FLT3Alt (≥p75)Baix (<p75)
203
80 ±967 ±27 0,66 pns
73±10100
0,36 pns
Pns: p no significativa

RESULTATS
125

RESULTATS
126
Treball 2: Correlació de FLT3 amb el transport intracel·lular de citarabina per hENT1.
Impacte de diferents inhibidors de FLT3 sobre el transport i efectivitat de la
citarabina
Es tracta d’un treball on s’analitza la relació funcional entre el receptor FLT3 i els
principals transportadors d’anàlegs de nucleòsids en mostres de 50 pacients
pediàtrics amb diferents subtipus de leucèmia aguda (LLA -B: 44; LLA -T: 2; LMA: 4),
uniformement tractats segons protocols de la Sociedad Española de Hematología y
Oncología Pediátricas (SEHOP), en el cas de les LLA: SHOP -LAL-99, 05, 13 i en el cas de
les LMA SHOP-LAM-00 & 07.
Descrivim la correlació positiva entre FLT3 i hENT1 a nivell genòmic, proteic i
funcional, és a dir, com FLT3 modula el transport de citarabina a l’interior de la
cèl·lula. A més, demostrem com la inhibició de FLT3 mitjançant fàrmacs inhib idors
(PKC402, AC220) disminueix significativament el transport de citarabina, posant de
manifest que l’ordre com s’administren aquests fàrmacs és important i pot tenir
efectes sobre la seva eficàcia.
Aquest treball ha estat publicat al Juliol de 2016 a la revista Oncotarget [296].

RESULTATS
127
Els resultats principals del treball són:
Característiques clíniques i biològiques dels pacients i línies cel·lulars estudiats
Per a l’anàlisi vam incloure 55 pacients diagnosticats de leucèmia aguda a l’Hospital
Sant Joan de Déu des de 2003 fins a 2013. Vàrem analitzar 26 nens i 24 nenes, amb
una mitjana d’edat de 4,3 anys ( extrems 0-16 anys). Vam incloure 3 lactants. Els
pacients van rebre tractament segon s protocols de la Sociedad Española de
Hematología y Oncología Pediátricas (SEHOP), en el cas de les LLA: SHOP -LAL-99, 05,
13 i en el cas de les LMA SHOP -LAM-00 & 07. Els lactants amb LLA varen rebre
tractament segons el protocol internacional INTERFANT 06.

RESULTATS
128
Taula 15. Característiques clíniques i analítiques dels pacients analitzats.
N=50
Edat, anys, mediana (extrems) 4,3 (0-16)
Sexe, n (%)- Masculí- Femení
26 (52)24 (48)
SNC, n (%)- SNC1- SNC2ta
- SNC3
46 (92)3 (6)1 (2)
Leucòcits, x109/L, mediana (extrems) 17,6 (1.1-331.2)
Hemoglobina, g/L, mediana (extrems) 7,7 (2.9-11.7)
Plaquetes, x109/L, mediana (extrems) 52 (2-520)
Blasts, mediana (extrems)- Moll de l’os- Sang perifèrica
93 (58-100)55 (0-99)
Immunofenotip, n (%)- LLA-B precursora- LLA-Tb
- LMA
44 (88)2 (4)4 (8)
Genètica, n (%)- LLA:
o HeHo Altresc
o ETV6-RUNX1o MLL+o TCF3-PBX1o BCR-ABL1
- LMA:o MLL+o Altresd
19 (38)16 (32)
4 (8)3 (6)2 (4)2 (4)
3 (6)1 (2)
a: SNC2t: punció lumbar traumàtica.b: Dos pacients amb Early T-cell Precursor T-ALL, un d’ells era portador d’una mutació FLT3-ITD.c: Altres pacients amb LLA -B precursora (n=14), on s’inclouen casos amb cariotip normal (n=5), pacients amb <20 metafases analitzables (n=6) i pacients amb alteració del 9p (n=3).Els dos pacients amb LLA-T tenien un cariotip normal.d: Altres casos de LMA: es va incloure un cas amb 47,XX,t(6;9)(p23;q34),+13[6]/46,XX,t(6;9)(p23;q34)[6,] portador d’una mutació FLT3-ITD.
Correlació significativa entre els nivells de mRNA de hENT1 i FLT3
Es van analitzar els nivells de mRNA dels principals TN i EM en les mostres dels 50
pacients analitzats i es va trobar una correlació significativa amb els nivells de FLT3 i

RESULTATS
129
hENT1 (r=0,34, p=0,017 , Figura 21). També es va trobar una correlació significativa entre
l’expressió de FLT3 i DCK (r=0,66, p<0,0001, Figura 22).
No es va trobar relació entre l’expressió dels transportadors i dels EM i les dades
clíniques i analítiques dels pacients de la sèrie.
En l’anàlisi pels diferents subtipus de leucèmia es varen observar nive lls
significativament elevats de hENT1 en pacients amb LMA-MLL+ i nivells baixos de DCK i
Cn-II en els pacients amb LMA i LLA-T.
Figura 21. Correlació entre hENT1 i nivells d’expressió de FLT3 (mRNA) en mostres de
pacients pediàtrics (N=50) afectes de LA . En la figura es mostra el coeficient de
correlació “r” .

RESULTATS
130
Figura 22: Correlació entre DCK i nivells d’expressió de FLT3 (mRNA) en mostres de
pacients pediàtrics (N=50) afectes de LA. En la figura es mostra el coeficient de
correlació “r” .
Caracterització dels nivells d’expressió dels principals TN i perfils de transportabilitat
en les línies cel·lulars
Donat que el perfil d’expressió dels TNs no ha estat molt estudiat en cèl·lules
leucèmiques, vam determinar el perfil d’expressió d’aquests e n les línies cel·lulars
(SEM, MV4 -11 i K562) i vam veure que hi havia expressió d’ hENT1, hENT2 i hCNT1.
Donat que en les tres línies cel·lulars s’expressaven el TNs es van analitzar els perfils de
transportabilitat. Vàrem observar que les tres línies prese ntaven un transport de
citidina depenent bàsicament d’hENT1 (Figura 23A). Tot i que de menys importància,
també es va detectar activitat transportadora associada a hENT2 i hCNT1. MV4 -11
també presentava certa activitat transportadora dependent de hCNT3. Po steriorment
també es va comprovar que el transport de citarabina en la línia cel·lular MV4 -11 era
dependent d’aquests transportadors (Figura 23B)

RESULTATS
131
Figura 23. Caracterització de l’activitat dels principals TN implicats en el transport de la
citarabina. (A) activitat de transport de [3H]-citidina en les tres línies cel·lulars (MV4 -
11, K562 i SEM) ( B) activitat de transport de [3H] -Ara-C en MV4 -11. Les dades
s’expressen com la mitja ± error estàndard de 3 experiments independents.
Efecte de FLT3 sobre l’expressió i l’activitat d’ hENT1
Per tal de determinar l’efecte modulador que podria tenir FLT3 sobre hENT1 vàrem
determinar l’efecte de la inhibició de FLT3 mitjançant dos molècules inhibidores de
FLT3: PKC -412 i AC220. Es va comprovar que ambdues molèc ules produïen una
disminució significativa de l’expressió, a nivell de mRNA i proteïna, i de l’activitat de
transport de citarabina d’hENT1 en les línies cel·lulars MV4 -11 i SEM. Pràcticament no
es va observar efecte sobre els altres transportadors hENT2 i hCNT1 (Figura 24)

RESULTATS
132
Figura 24. Efecte de la inhibició de FLT3 sobre el transport d’Ara -C en la línia cel·lular
MV4-11. Es va determinar el transport d’Ara -C després de 16 hores d’incubació amb
PKC-412. En el cas d’hENT1 es va trobar una diferència signi ficativa ( ***p < 0.001 )
respecte a les cèl·lules no incubades amb l’inhibidor. Les dades s’expressen com a
percentatge ± error estàndard de tres experiments independents, cada un realitzat en
quadruplicat.
Efecte de la inhibició de FLT3 en la citotoxicitat mediada per Ara-C
Donat que vàrem demostrar que FLT3 podria regular per algun mecanisme el transport
de nucleòsids modulant l’expressió d’hENT1, vàrem voler determinar com es podria
alterar la cito toxicitat mediada per citarabina quan es combina aque sta amb un
inhibidor de FLT3. La incubació de MV4 -11 amb un inhibidor de FLT3 (PKC -412 o
AC220) i posterior exposició a citarabina no produïa una citotoxicitat superior a quan
s’exposaven les cèl·lules a l’inhibidor únicament. Canviant les condicions, quan
s’exposaven inicialment les cèl·lules a la citarabina i posteriorment a l’inhibidor hi
havia una citotoxicitat significativament superior, demostrant per tant un efecte
additiu quan s’utilitzava aquesta combinació (Figura 25).

RESULTATS
133
Figura 2 5. Efecte de l a inhibició de FLT3 en la citotoxicitat induïda per Ara -C. La
viabilitat cel·lular es va determinar mitjançant assaig MTT. En (A) les cèl·lules MV4 -11
van ser tractades primer amb l’inhibidor PKC -412 (16 hores), seguit de 6 hores
d’exposició a Ara-C. En (B) es va invertir l’ordre dels fàrmacs, primer exposició a Ara -C i
posteriorment a PKC-412. Significació estadística en relació a les cèl·lules control (*p <
0.1; **p < 0.01; ***p < 0.001).
A B

RESULTATS
134

ANNEX ALS RESULTATS
135
IX. ANNEX ALS RESULTATS

ANNEX ALS RESULTATS
136

DISCUSSIÓ
155
X. DISCUSSIÓ

DISCUSSIÓ
156

DISCUSSIÓ
157
DISCUSSIÓ
Els treballs plantejats en aquesta tesi tenien com objectiu principal caracteritzar les
alteracions moleculars del receptor FLT3 en pacients pediàtrics afectes de leucèmia
aguda i la correlació amb l’activitat i expressió dels transportadors de fàrmacs derivats
de nucleòsids, atès que la citarabina és un dels tractaments fonamentals en aquest
malalts.
Els objectius específics pretenien determinar:
- la incidència de les diferents alteracions de FLT3 (mutacions i sobreexpressió) en
diferents subtipus de leucèmia pediàtrica en una sèrie rellevant de pacients
tractats en el nostre centre i quin impacte podrien tenir en la supervivència en
pacients tractats de forma homogènia;
- la relació entre l’expressió de FLT3 i dels TN i enzims de metabolisme intracel·lular
(EMs) dels anàlegs de nucleòsids en diferents subtipus de leucèmia aguda
pediàtrica;
- la relació entre FLT3 i l’activitat dels transportadors;
- l’impacte de diferents inhibidor s de FLT3 sobre el transport i efectivitat de la
citarabina.

DISCUSSIÓ
158
Caracterització de les alteracions moleculars del receptor FLT3 en pacients pediàtrics
afectes de leucèmia aguda: incidència, correlació amb variables clíniques i
analítiques i impacte pronòstic
Tot i que les mutacions de FLT3 en LA ha estat objecte de múltiples treballs i revisions,
l’expressió d’aquest i la seva correlació amb variables clíniques, biològiques i
pronòstiques és més desconeguda. En el nostre treball hem analitzat l’expressió i
l’estat mutacional de 204 pacients pediàtrics afectes de LA (145 LLA -B, 23 LLA -T i 36
LMA), tots ells tractats de forma homogènia i en un únic centre.
En l’anàlisi de correlació de la sobreexpressió de FLT3 amb variables clinico-biològiques
com l’edat, sexe o número de leucòcits en la sèrie global, les úniques correlacions
estadísticament significatives van ser amb el llinatge (LLA -B vs LLA-T vs LMA) i amb les
categories moleculars. Quan vàrem fer l’anàlisi per subgrups, vam observar dins el
grup de pac ients amb LLA -B precursora, que la sobreexpressió (>p75) de FLT3 es
correlacionava de forma significativa amb l’edat (lactants), leucocitosi >20x10 9/L, el
fenotip pro-B i el reordenament MLL. Per tant, la sobreexpressió de FLT3 reflexa, tal i
com s’ha reportat prèviament, el subgrup de pacients d’alt risc amb LLA -B precursora i
reordenament de MLL de presentació típica en lactants. D’acord amb altres treballs
publicats [297]; [298]; [299]; [225], en la nostra sèrie vam observar una corre lació
estadísticament significativa de la sobreexpressió de FLT3 segons el subgrup genètic en
LLA-B: els pacients amb LLA -MLL+ i LLA HeH van present ar nivells de FLT3
significativament més elevats respecte als altres subgrups.
En els pacients amb LLA -T no vàrem trobar cap correlació significativa amb l’expressió
de FLT3. En el subgrup de pacients amb LMA vam observar únicament una major
expressió de FLT3 en els pacients estratificats en el grup de risc genètic alt.
Aquests resultats van en concordança amb altres estudis, tot i que tant Ozeki com
Kuchenbauer [222]; [300] observen una tendència a que els pacients amb LMA i nivells
més elevats de FLT3 presenten leucocitosi en el debut. Aquest fet també el descriu
Brown en LLA pediàtrica [223].
Cal destacar la baixa freqüència de mutacions identificades en la nostra sèrie de
pacients, tant FLT3-ITD com FLT3 -ALK/TKD, que ha limitat l’estudi estadístic de
correlacions amb altres variables. Quant a les LLA -B, només vam trobar mutacions

DISCUSSIÓ
159
tipus FLT3-ITD (1,4%) en 2 pacients, un pacient amb LLA-HeH i un pacient amb iAMP21.
Aquestes dades serien co nsistents amb altres sèries pediàtriques publicades [301].
Vam identificar 2 pacients amb LLA-B amb mutacions tipus FLT3-ALM/TKD, en casos de
subtipus LLA -B HeH. Aquest tipus de mutacions s’han associat més freqüentment en
les LLA -HeH i LLA -MLL+ [299]. En el grup de les LLA -T vàrem confirmar una mutació
tipus FLT3 -ITD en una pacient amb una LLA amb immunofenotip tipus ETP. Les
mutacions de FLT3 són rares en LLA-T, exceptuant el subgrup de les ETP, en el qual són
més habituals les mutacions més típiques de la línia mieloide [235], [302].
En el grup de les LMA vam observar una mutació entre tots els pacients analitzats
(1/36, 2,7%). En altres series publicades les mutacions tipus FLT3 -ITD en les LMA
arriben fins a un 10 -15%, solen ser més freqüents en nens més grans i en LMA t(15 ;17)
[301]. El grup italià AIEOP va analitzar 494 pacients pediàtrics i fins un 10% presentà
mutacions. La majoria d’aquests pacients tenien més de 10 anys [239]. En la nostra
sèrie de pacients amb LMA, l’únic pacient que presentava una mutació tipus FLT3 -ITD
era una noia adolescent amb una LMA amb translocació t(6;9)(p22;q34). S’ha descrit
que aquesta entitat associa habitualment mutacions FLT3 -ITD [303], [304] . En els
nostres pacients amb LMA no vam identificar mutacions FLT3 -ALM/TKD. Entre els
possibles motius de la baixa incidència de mutacions tipus FLT3 -ITD en la nostra sèrie
cal esmentar el reduït nombre de pacients analitzats amb LMA, que incloïa només un
cas de LPA i que la majo ria dels nostres pacients eren nens molt petits, amb una edat
mitjana de menys de 3 anys i només un 10% de pacients majors de 10 anys inclosos a
la sèrie.
En quant a l’anàlisi de l’ impacte pronòstic de les diferents variables analitzades a la
nostra sèrie , no vam observar que la sobreexpressió de FLT3 tingués importància
pronòstica en els diferents grups de pacients (LLA-B, LLA-T o LMA).
Donada l’heterogeneïtat genètica dels diferents subgrups de LLA-B vam realitzar
l’anàlisi segons els diferents subgrup s citogenètics. En cap dels subgrups de LLA -B vam
trobar que la sobreexpressió de FLT3 alterés el pronòstic dels pacients, tot i que els
pacients amb LLA -HeH i els subgrup de pacients amb LLA -B B-other amb major
expressió de FLT3 varen presentar una millor supervivència, però no de forma
significativa. Caldria ampliar el nombre de casos de cada subgrup per confirmar
aquestes dades. En el cas de les LLA -MLL+, l’heterogeneïtat del reordenaments MLL va

DISCUSSIÓ
160
fer impossible analitzar les t(4;11) per separat (n=3 paci ents: 2 lactants, un
adolescent). Després d’una mediana de seguiment de 5,4 anys, els 10 pacients amb
LLA-MLL+ van presentar una SG del 90 ±10% i una SLE del 60 ±15,5%. Set pacients van
rebre un transplantament al·logènic, sis d’ells en primera RC. Quatre pacients van
recaure, dos d’ells després del TPH al·logènic; una d’aquestes pacients es troba
actualment en tractament amb immunoteràpia tipus CAR-T.
Cal destacar que en el grup de les LLA-T vam incloure una pacient amb una LLA -T ETP
amb mutació a FLT3, fenomen descrit amb anterioritat [235], [302].
En el cas de les LMA, vam demostrar una associació entre la sobreexpressió de FLT3 i
la classificació pronòstica citogenètica. Així, vam veure que els pacients classificats com
d’alt risc són els que presentaven nivells més alts de FLT3. Un dels pacients d’aquest
grup correspon a una pacient adolescent amb una LMA t(6;9) i mutació en FLT3.
Les mutacions en FLT3 han demostrat ser un factor pronòstic rellevant en pacients
afectes de LMA; les mutacions tipus FLT3 -ITD han estat reconegudes com a factor
advers en pacients pediàtrics i adults [305]; [299]; [306]; [222]; [135]. Així, els pacients
portadors de mutacions FLT3 -ITD són considerats pacients d’alt risc que es poden
beneficiar d’un TPH al·logènic en primera RC [305];[135]; [307].
La sobreexpressió de FLT3 dona lloc a una fosforilació i activa ció de FLT3 de forma
similar a quan es troba mutat [299]; [306];[222];[223]. La sobreexpressió de FLT3,
independentment de l’estat mutacional, tot i que molt menys analitzada, també s’ha
correlacionat amb pitjor pronòstic en pacients amb LLA i LMA . Així, Ozeki, en la seva
sèrie de 181 pacients adults amb LMA, va demostrar que la sobreexpressió de FLT3 era
el principal factor pronòstic, independentment de l’estat mutacional [222].
Curiosament, en aquesta sèrie de pacients publicada, el subgrup citogenètic no tenia
impacte pronòstic. El grup de Menéndez va demostrar en una sèrie de pacients adults i
pediàtrics que FLT3 tenia importància pronòstica significativa només en els pacients
amb LLA amb translocació t(4;11) i reorde nament MLL-AF4 [297]. El grup COG, en la
seva sèrie de 97 lactants, va identificar la sobreexpressió de FLT3 com a factor de mal
pronòstic en els pacients sense reordenament de MLL [306]. De la mateixa manera,
Stam i col·laboradors també van observar en una sèrie de 32 lactants que la
sobreexpressió de FLT3 condicionava un pitjor pronòstic [224]. El baix nombre de
lactants i la baixa freqüència del nombre d’esdeveniments en aquest grup de pacients

DISCUSSIÓ
161
de la nostra sèrie tampoc ens ha permès extreure correlacions significatives, tal i com
descriuen els grups esmentats.
Més enllà d’aquestes observacions clíniques, és poc conegut el mecanisme biològic
que determina l’alt risc en aquest grup de pacients. El grup d’Amstrong va demostrar
que la sobreexpressió de FLT3 és el principal fenomen genètic distintiu dels pacients
amb LA-MLL+ [37]. A més, varen observar una sobreexpressió significativa de FLT3 en
els lactants amb LLA -MLL. És rellevant el fet que els lactants amb LLA presenten una
elevada sensibilitat a la citarabina, fenomen que es té en compte en els protocols
específics de tractament (INTERFANT 99 & 06) [194].
En resum , en la nostra sèrie de pacients, la sobreexpressió de FLT3 es va associar
significativament al llinatge de la leucèmia (LLA -B>LMA>LLA-T) i al subgrup molecular,
sent les LLA -MLL+ i les LLA HeH, les que presentaren valors significativament més
elevats de FLT3. Tot i no representar per si mateix un factor pronòstic significatiu, la
sobreexpressió de FLT3 es va associar a grups de pacients d’alt risc, com els lactants
amb reordenament MLL i pacients amb LMA d’alt risc citogenètic.
Les mutacions FLT3-ITD són infreqüents, tot i que això podria ser degut al baix nombre
de pacients inclosos a la sèrie i a la seva het erogeneïtat, especialment en els pacients
amb LMA. Quant a les mutacions FLT3 -ALM/TKD, serien més freqüents en els pacients
amb LLA -B HeH, d’acord amb el prèviament descrit a la literatura. En la nostra sèrie,
els dos pacients amb mutacions FLT3 -ALM/TKD pertanyien al subgrup de LLA -B HeH.
En els nostres pacients, la sobreexpressió de FLT3 no va mostrar un impacte pronòstic
significatiu sobre la SG ni la SLE.
En l’anàlisi de l’impacte pronòstic de les diferents variables analitzades a la nostra
sèrie, no vam observar que la sobreexpressió de FLT3 tingués importància pronòstica
en els diferents grups de pacients.

DISCUSSIÓ
162
Correlació de FLT3 amb el transport intracel·lular de citarabina per hENT1. Impacte
de diferents inhibidors de FLT3 sobre el transport i efectivitat de la citarabina
El transport intracel·lular de la citarabina es realitza fonamentalment, però no de
forma exclusiva, pel transportador de membrana hENT1. El paper d’aquest
transportador resulta essencial en la biodisponibilitat i per tant, l’acció cit otòxica
d’aquest fàrmac. De fet, s’han demostrat alts nivells de ARNm en pacients LLA -MLL, i
això pot explicar, en part, l’alta sensibilitat a la citarabina que presenten aquest grup
de pacients, reportada per Stam i el seu grup [192]. Aquest mateix grup
d’investigadors va demostrar que els reordenaments del gen MLL no estaven
directament implicats en l a sensibilitat a la citarabina en aquests pacients [308], fet
que posa en evidència que altres mecanismes són els responsables.
En el nostre treball hem demostrat, en una cohort de 50 pa cients pediàtrics amb
leucèmia aguda, una correlació positiva entre l’expressió (nivells de ARNm) de FLT3 i
hENT1. Aquest resultats serien coherents amb observacions prèvies que mostraven
que els subgrups de pacients amb alta sensibilitat a la citarabina f reqüentment
sobreexpressaven FLT3 [299]; [234]; [236]. Per tant, els resultats del nostre treball
demostrant la correlació entre l’expressió d’ambdós paràmetres permet suggerir que
FLT3 pot tenir un rol important en la sensibilitat a la citarabina mitjançant la modulació
del seu transport per part d’hENT1.
En aquest treball també vàrem analitzar en la mateixa cohort de pacients els nivells de
ARNm d’altres transportadors putatius d’Ara-C (hENT2, hCNT1, hCNT3) i enzims del seu
metabolisme intracel·lular com DCK i cN-II. En aquesta sèrie, el baix nombre de
pacients va limitar els estudis per trobar una correlació entre els TN i EM amb factors
clínics ni biològics. En altres estudis s’ha observat una correlació entre el perfil
d’expressió de TN i el pronòstic en determinats tumors sòlids i neoplàsies
hematològiques. [283]; [294]; [285]. El nombre de pacients analitzats, però, no ens va
permetre establir correlacions pronòstiques.
També vàrem trobar una correlació p ositiva entre FLT3 i DCK, la quinasa responsable
de la primera fosforilació dels ANs un cop entren a la cèl·lula via els TNs. Els nivells
baixos de DCK es correlacionen amb resistència a Ara -C en LLA i LMA [293]; [193];

DISCUSSIÓ
163
[309]. Aquesta observació suggeriria que DCK no actuaria com a pas limitant en la
fosforilació de Ara -C (Ara -CTP) en pacients amb sobreexpressió de FLT3 i, per tant,
afavorint l’activació d’Ara -C. Aquesta troballa també és coherent amb els resultats
d’altres estudis, on es demostra la correlació in vitro entre l’expressió d’hENT1 i DCK i
la sensibilitat a AN en pacients pediàtrics i adults amb LMA [293]. A més, recentment,
en un assaig del grup cooperatiu GIMEMA -EORTC, on s’estudiava l’efecte d’altes dosis
de citarabina, es va demostra r una millor resposta i supervivència amb aquest
tractament en pacients amb LMA de molt alt risc, on s’inclouen pacients FLT3 -ITD
[310].
Els AN són un dels principals tractaments en LMA i també s’utilitzen àmpliament en
casos de LLA recaiguda o refractaria mostrant resultats diversos [311]; [312]; [313];
[314]. Donades les nostres troballes, on demostrem una correlació significativa entre
FLT3 i el transportador d’Ara -C hENT1, l’expressió de FLT3 podria ser utilitzat com
marcador de sensibilitat a aquests AN.
S’han desenvolupat diferents inhibidors de FLT3 en els últi ms anys, alguns d’ells en
fases clíniques molt avançades. Cal destacar l’èxit de l’assaig CALGB 10603/RATIFY, on
es demostrà per primera vegada en un estudi prospectiu i randomitzat que l’addició de
midostaurin com a tractament de primera línia en combinac ió amb el tractament
quimioteràpic estàndard té un efecte beneficiós.
Els inhibidors de FLT3 sembla que inhibirien la proliferació de la cèl·lula leucèmica al
reprimir l’efecte de la cinasa, donant lloc a una aturada del cicle cel·lular [315] [316]. A
més, un dels inhibidors més selectius i potents de FLT3, quizartinib, promou una
diferenciació del blaste a nivell del moll de l’os [317].
L’efecte de quizartinib en monoteràpia no ha mostrat resultats gai re esperançadors i
els resultats obtinguts han estat, en el millor dels casos, transitoris. Aquest efecte
transitori es deu principalment a l’aparició de resistències per diferents mecanismes.
Sembla que els inhibidors de FLT3 en monoteràpia tindrien indic ació en un grup
seleccionat de pacients, com per exemple, com a pont a un segon trasplantament en
un grup de pacients difícilment tractables [318]; [319]; [248]; [320]. Tot i això, es
desconeix quins són els millors esquemes en combinació i si l’addició d’altres f àrmacs
com els hipometilants podrien ser beneficiosos. Per aquest fet, també hem volgut

DISCUSSIÓ
164
analitzar com la inhibició de FLT3 podria alterar el transport, i per tant, la citotoxicitat
per aquest fàrmac d’us tan comú en leucèmia aguda. Primer vàrem demostrar q ue
l’Ara-C es transportava per hENT1, però també utilitzava hENT2 i hCNT1.
Posteriorment vàrem observar com la inhibició de FLT3 per PKC412 produïa una
repressió significativa de hENT1 en les línies cel·lulars SEM i MV4 -11, donant lloc a una
reducció del t ransport d’aquest fàrmac. El mateix efecte s’observava amb l’inhibidor
de segona generació AC220 (quizartinib), molt més selectiu i potent que l’anterior.
Aquest efecte sembla específic sobre hENT1, ja que l’adició de PKC -412 no modifica la
funció dels al tres transportadors (hENT2 i hCNT1). Aquests resultats indiquen que
l’expressió i l’activitat del transportador d’Ara-C hENT1 es troba regulada per FLT3.
Jin i col·laboradors, utilitzant cèl·lules HF6 transduïdes amb la mutació FLT3 -ITD, varen
observar que hi havia una repressió d’hENT1 [286]). Nosaltres, de forma oposada, vam
demostrar que en línies cel·lulars en les quals de forma endògena hi ha una activació
de FLT3 (FLT3 -ITD o sobreexpressen FLT3, FLT3wt), FLT3 regula positivament
l’expressió i activitat d’hENT1. Aquest res ultats són coherents amb altres treballs i
dades clíniques, com s’ha comentat anteriorment.
Dos treballs previs han demostrat in vitro que els tractaments combinats utilitzant
inhibidors de FLT3 i quimioteràpia tenen una eficàcia diferent en funció de l’o rdre en
que s’administren [258] [111]. En aquests treballs observaven efectes antagònics quan
les línies cel·lulars i mostres de pacients amb mutacions FLT3 -ITD eren incubades
inicialment amb un inhibidor de FLT3 i posterior ment eren exposades a citarabina. Els
mateixos resultats s’observaven en lactants amb alts nivells de FLT3 sense mutacions.
Segons aquests treballs, aquest efecte seria el resultat de l’aturada del cicle cel·lular en
fase G1-S produït per l’inhibidor de FL T3, el que ocasionaria una pertorbació en l’acció
del fàrmac genotòxic, com l’Ara-C. La possibilitat de que la modulació per part de FLT3
sobre hENT1 sigui el resultat d’una alteració sobre el cicle cel·lular no pot ser
descartada, però sembla improbable a l nostre parer. S’ha demostrat que hENT1
proporciona nucleòsids extracel·lulars per la síntesi de ADN en macròfags del moll de
l’os d’origen murí [321], però hCNT1 és el transportador depenent de cicle, que mostra
una regulació positiva en fase S [322]. A més, en el s nostres experiments, la inhibició
de FLT3 indueix una repressió d’hENT1, però no altera en absolut ni la expressió ni

DISCUSSIÓ
165
l’activitat de hCNT1. Per tant, la modulació d’hENT1 per part de FLT3 no seria un efecte
indirecte de la pertorbació del cicle cel·lular per part dels inhibidors de FLT3.
Per altra banda, els nostres resultats són rellevants perquè recolzen la hipòtesi que la
seqüència d’administració en esquemes de tractaments combinats és molt important.
Així, sota condicions de repressió d’hENT1, l’efe cte citotòxic desencadenat pels
inhibidors PKC -412 o AC220 no es veu potenciat pel tractament amb Ara -C, situació
contrària a quan les cèl·lules són primerament exposades a l’acció d’Ara -C, quan
l’activitat d’hENT1 no estat encara inhibida per l’inhibidor de FLT3. Sota aquestes
condicions, l’acció del tractament combinat és significativament més eficaç. Aquesta
troballa posa de manifest la importància d’un millor coneixement de la regulació dels
transportadors de fàrmacs quan es combinen amb inhibidors tirosín-cinasa.
En resum , en aquest treball demostrem que el transport intracel·lular d’Ara -C en el
blast es realitza mitjançant els TN hENT1, hENT2 i hCNT1. A més, demostrem que FLT3
regula l’expressió i activitat del principal transportador d’Ara -C, hENT1 , i per tant,
també la quimiosensibilitat a aquest fàrmac. També hem provat que la seqüencia amb
que s’administren els inhibidors de FLT3 amb Ara -C és important. Així, l’eficàcia es
potencia quan s’administra l’Ara -C abans que l’inhibidor. Tot plegat, les nostres dades
contribueixen a entendre com FLT3 pot influir sobre la quimiosensibilitat i poder
millorar els esquemes terapèutics que combinen aquests fàrmacs.
Com a resum global del projecte de tesi presentat, vàrem trobar una baixa freqüència
dels dife rents tipus de mutacions de FLT3, d’acord amb el prèviament descrit. Vam
observar que la sobreexpressió de FLT3 s’associava a pacients amb LLA-B, LLA HeH i LA
MLL, però no va tenir un impacte pronòstic en la nostra sèrie de pacients. Cal destacar
que en el cas de LMA la sobreexpressió de FLT3 es va associar al subgrup d’alt risc
citogenètic. D’altra banda, vàrem confirmar que FLT3 està implicat en el transport
d’Ara-C modulant l’expressió del seu principal transportador hENT1. Els inhibidors de
FLT3 poden d isminuir el transport de citarabina i, per tant, la seva eficàcia. En aquest
resultat hem mostrat la importància de l’ordre de l’administració d’aquests fàrmacs en
el disseny dels nous protocols terapèutics.

DISCUSSIÓ
166

DISCUSSIÓ
167
Limitacions del treball
La leucèmia aguda, to t i ser la neoplàsia més freqüent en l’edat pediàtrica, és una
malaltia de baixa incidència en nens. A més, és una malaltia heterogènia a nivell clínic i
biològic. En aquest treball hem pogut reunir una sèrie important de pacients tractats
de forma homogèn ia amb protocols consecutius en un sol centre, però tot i així, no
disposem d’un nombre suficient de casos per analitzar per separat diferents subgrups
de baixa freqüència en la nostra sèrie.
Beneficis de la investigació
Aquest treball de tesi ha permès a l doctorand establir una línia de recerca en
col·laboració amb el grup del Professor Pastor -Anglada. Aquesta té per objectius
fonamentals aprofundir en el coneixement farmacogenòmic i farmacodinàmic de
fàrmacs emprats en el tractament de la leucèmia aguda en pacients en edat pediàtrica
en relació amb els diferents transportadors de nucleòsids i EMs. El coneixement
farmacodinàmic sobre molts fàrmacs emprats en pediatria és molt escàs, especialment
en el grup dels lactants i, per tant, són imprescindibles les línies de recerca en aquest
sentit. A més, caldrà establir cóm es pot veure alterada la eficàcia dels citostàtics
convencionals amb la introducció de nous fàrmacs dirigits contra dianes moleculars
específiques.
Ens encaminem a una medicina cada cop més “ personalitzada” o adaptada a les
particularitats biològiques de la malaltia, però també a les característiques pròpies del
pacient; per tant, la farmacogenètica tindrà un paper fonamental a l’hora de dissenyar
els tractaments pels nostres pacients.
El pres ent treball de tesi i els futurs estudis que se’n puguin derivar, enforteixen el
grup de recerca sobre hemopaties malignes de l’HSJD, encapçalat per la Dra. Camós. El
nostre grup, conjuntament amb el del Dr. Ramírez de l’Hospital Niño Jesús a Madrid,
està liderant la coordinació a Espanya dels grups de recerca clínics, translacionals i
bàsics en leucèmia limfoblàstica aguda pediàtrica i del recentment creat de LMA
pediàtrica. Aquests esforços a nivell clínic , amb la coordinació del Grup de Leucèmies
de la S EHOP inicialment per la Prof. Badell i actualment per part de la Dra. Rives, del
nostre centre i del Dr. Dapena, de l’Hospital Vall d’Hebron, i també a nivell biològic,
han permès la incorporació d’Espanya com a país en els principals Grups Cooperatius

DISCUSSIÓ
168
Internacionals ( I-BFM Study Group i NOPHO AML Study Group) , fet imprescindible per
avançar en el coneixement i maneig de les leucèmies pediàtriques.
Més enllà d’aquests beneficis a mitjà termini cal esmentar que, de forma més
específica, els nostres resultat s poden tenir una aplicabilitat directa i immediata en els
protocols de tractament que combinen inhibidors de FLT3 amb quimioteràpia
convencional, molts d’ells en fases molt avançades de desenvolupament clínic.
Perspectives de futur
Amb aquest treball s’obren possibles noves vies de treball:
- Establir quins pacients pediàtrics afectes de LA es poden beneficiar d’inhibidors de
FLT3. A més dels pacients amb mutacions caldrà determinar l’eficàcia en pacients que
sobreexpressen FLT3, ja que la seva eficàcia n o està plenament establerta en aquests
casos. Podria tenir interès amb grups d’especial mal pronòstic i amb sobreexpressió de
FLT3 com els lactants amb leucèmia MLL.
- Caracterització del perfil d’expressió dels transportadors d’anàlegs de nucleòsids en
leucèmia aguda. Al igual que en altres neoplàsies, podria tenir un interès pronòstic i
terapèutic. Tot i que nosaltres ho hem analitzat en la nostra sèrie de pacients, el
nombre de pacients no permet extreure dades concloents i, per tant, faria falta
ampliar aquest estudi a un grup més ampli i amb diferents subtipus de leucèmia.
- Identificar el mecanisme pel qual FLT3 regula al transportador de nucleòsids hENT1 i
a dCK pot tenir interès per tal d’establir vies de sensibilització a anàlegs de nucleòsids
en leucèmia aguda.
- En els assaigs que combinen inhibidors de FLT3 amb anàlegs de nucleòsids com la
citarabina, s’haurà de tenir en compte en el seu disseny els resultats dels nostres
estudis. És a dir, l’inhibidor pot reduir l’eficàcia de la citarabina i p er tant l’odre en que
s’administra cada fàrmac és important.
- Existeixen diferents esquemes de terapèutics on es combinen altres inhibidors
tirosin-cinasa amb fàrmacs anàlegs de nucleòsids. Per tant, resultarà d’interès
identificar si altres inhibidors t irosín-cinasa modulen l’expressió i/o activitat del
transport de nucleòsids per tal d’optimitzar l’eficàcia d’aquests esquemes.

CONCLUSIONS
169
XI. CONCLUSIONS

CONCLUSIONS
170

CONCLUSIONS
171
CONCLUSIONS
• En la nostra sèrie, la sobreexpressió de FLT3 s’associà al llinatge de la leucèmia i
al subgrup genètic: les LLA-MLL+ i les LLA HeH són les que presentaven nivells
més elevats de FLT3.
• La freqüència de mutacions de FLT3 en la nostra sèrie de pacients amb leucèmia
aguda pediàtrica va ser baixa.
• La sobreexpressió de FLT3 no va tenir un impacte pronòstic independent en els
nostres pacients, però es va correlacionar amb subgrups d’alt risc: lactants
amb LLA-MLL+ i pacients amb LMA d’alt risc citogenètic.
• FLT3 està implicat en el transport d’Ara-C, modulant l’expressió del seu principal
transportador hENT1.
• Els inhibidors de FLT3 poden disminuir el transport de citarabina i, per tant, la
seva eficàcia. Aquest fet demostra la importància de l’ordre de l’administració
d’aquests fàrmacs en el disseny dels nous protocols terapèutics.

BIBLIOGRAFIA
172

BIBLIOGRAFIA
173
XII.BIBLIOGRAFIA

BIBLIOGRAFIA
174

BIBLIOGRAFIA
175
BIBLIOGRAFIA
[1] E. M. Pietras, D. Reynaud, Y.-A. Kang, D. Carlin, F. J. Calero-Nieto, A. D. Leavitt, J. M. Stuart, B.
Göttgens, and E. Passegué, “Functionally Distinct Subsets of Lineage-Biased Multipotent
Progenitors Control Blood Production in Normal and Regenerative Conditions,” Cell Stem Cell,
vol. 17, no. 1, pp. 35–46, Jul. 2015.
[2] M. A. Smith, L. A. G. Ries, J. G. Gurney, and J. A. Ross, “Leukemia - SEER Pediatric Monograph,”
pp. 17–34, 1990.
[3] E. Ward, C. Desantis, A. Robbins, B. Kohler, and A. Jemal, “Childhood and Adolescent Cancer
Statistics , 2014,” vol. 0, no. 0, pp. 1–21, 2014.
[4] J. G. Gurney, R. K. Severson, S. Davis, and L. L. Robison, “Incidence of cancer in children in the
United States. Sex-, race-, and 1-year age-specific rates by histologic type,” Cancer, vol. 75, no. 8,
pp. 2186–2195, Apr. 1995.
[5] M. S. Linet and S. S. Devesa, “Descriptive epidemiology of childhood leukaemia.,” Br. J. Cancer,
vol. 63, no. 3, pp. 424–9, Mar. 1991.
[6] D. A. Arber, A. Orazi, R. Hasserjian, J. Thiele, M. J. Borowitz, M. M. Le Beau, C. D. Bloomfield, M.
Cazzola, J. W. Vardiman, E. Jabbour, H. Kantarjian, S. O’Brien, J. Radich, C. Abboud, M. Baccarani,
M. Deininger, G. Rosti, R. Hehlmann, M. Deininger, T. Barbui, J. Thiele, A. Vannucchi, A. Tefferi,
A. Tefferi, P. Guglielmelli, D. Larson, A. Tefferi, J. Thiele, A. Vannucchi, T. Barbui, J. Maxson, J.
Gotlib, D. Pollyea, T. Barbui, J. Thiele, H. Gisslinger, T. Barbui, J. Thiele, A. Vannucchi, A. Tefferi, J.
Thiele, H. Kvasnicka, L. Müllauer, V. Buxhofer-Ausch, B. Gisslinger, H. Gisslinger, T. Barbui, J.
Thiele, F. Passamonti, H. Gisslinger, G. Jeryczynski, B. Gisslinger, G. Barosi, V. Rosti, E. Bonetti, A.
Madelung, H. Bondo, I. Stamp, A. S. Group, U. Gianelli, A. Bossi, I. Cortinovis, P. Valent, P. Valent,
H. Horny, L. Escribano, V. Patterer, S. Schnittger, W. Kern, T. Haferlach, C. Haferlach, B. Bain, S.
Ahmad, E. Rumi, J. Milosevic, D. Selleslag, A. Orazi, U. Germing, F. Locatelli, C. Niemeyer, T.
Mughal, N. Cross, E. Padron, R. Itzykson, O. Kosmider, A. Renneville, M. Meggendorfer, U.
Bacher, T. Alpermann, M. Patnaik, A. Tefferi, S. Jaiswal, P. Fontanillas, J. Flannick, G. Genovese,
A. Kähler, R. Handsaker, C. Ricci, E. Fermo, S. Corti, E. Schuler, M. Schroeder, J. Neukirchen, N.
Cervera, R. Itzykson, E. Coppin, A. Storniolo, W. Moloney, D. Rosenthal, C. Cox, J. Bennett, L.
Boiocchi, R. Espinal-Witter, J. Geyer, L. Boiocchi, U. Gianelli, A. Iurlo, S. Wang, R. Hasserjian, P.
Fox, R. Piazza, S. Valletta, N. Winkelmann, C. Gambacorti-Passerini, C. Donadoni, A. Parmiani, C.
M. D. W. G. of the I. C. G. C. and of the A. I. per la R. sul C. G. I. M. Mieloproliferative, C. M. D. W.
G. of the I. C. G. Consortium, R. Bejar, K. Stevenson, B. Caughey, A. I. per la R. sul C. G. I. M.
Mieloproliferative, S. Passmore, I. Hann, C. Stiller, E. W. G. on M. S. in C. (EWOG-MDS), E.
Verburgh, R. Achten, V. Louw, U. Germing, C. Strupp, A. Giagounidis, A. Maassen, C. Strupp, A.
Giagounidis, L. Senent, L. Arenillas, E. Luño, J. Ruiz, G. Sanz, L. Florensa, P. Font, J. Loscertales, C.
Benavente, S. Parmentier, J. Schetelig, K. Lorenz, R. E. L. (REL) C. Network, P. Greenberg, H.
Tuechler, J. Schanz, J. Vardiman, J. Thiele, D. Arber, U. Germing, M. Lauseker, B. Hildebrandt, M.

BIBLIOGRAFIA
176
Mallo, J. Cervera, J. Schanz, J. Schanz, H. Tüchler, F. Solé, C. M. D. W. G. of the I. C. G.
Consortium, T. Haferlach, Y. Nagata, V. Grossmann, D. Steensma, R. Bejar, S. Jaiswal, B. Kwok, J.
Hall, J. Witte, C. Cargo, N. Rowbotham, P. Evans, R. Bejar, K. Stevenson, O. Abdel-Wahab, R.
Bejar, R. Bejar, K. Stevenson, B. Caughey, M. Mallo, M. Del Rey, M. Ibáñez, L. Saft, M. Karimi, M.
Ghaderi, M. Jädersten, L. Saft, A. Smith, M. del Rey, R. Benito, C. Fontanillo, M. Gerstung, A.
Pellagatti, L. Malcovati, L. Malcovati, M. Karimi, E. Papaemmanuil, M. Patnaik, C. Hanson, N.
Sulai, A. West, L. Godley, J. Churpek, N. C. R. I. A. L. W. Group, S. Gröschel, M. Sanders, R.
Hoogenboezem, H. Yamazaki, M. Suzuki, A. Otsuki, C. Soupir, J. Vergilio, P. D. Cin, S. Konoplev, C.
Yin, S. Kornblau, E. Nacheva, C. Grace, D. Brazma, C. G. A. R. Network, J. Patel, M. Gönen, M.
Figueroa, H. Döhner, D. Weisdorf, C. Bloomfield, B. Wouters, B. Löwenberg, C. Erpelinck-
Verschueren, W. van Putten, P. Valk, R. Delwel, T. Pabst, M. Eyholzer, J. Fos, B. Mueller, H. Hou,
L. Lin, C. Chen, H. Tien, C. Green, K. Koo, R. Hills, A. Burnett, D. Linch, R. Gale, I. Hollink, M. van
den Heuvel-Eibrink, S. Arentsen-Peters, B. Falini, K. Macijewski, T. Weiss, M. Díaz-Beyá, M.
Rozman, M. Pratcorona, U. Bacher, S. Schnittger, K. Macijewski, S. Schnittger, F. Dicker, W. Kern,
J. Tang, H. Hou, C. Chen, J. Mendler, K. Maharry, M. Radmacher, V. Gaidzik, L. Bullinger, R.
Schlenk, G. C. de C. H. and S. C. G. (Grupo C. P. el E. y T. de las L. A. Mieloblásticas), O. Weinberg,
M. Seetharam, L. Ren, C. Haferlach, C. Mecucci, S. Schnittger, R. Schlenk, E. Taskesen, Y. van
Norden, J. Churpek, R. Marquez, B. Neistadt, R. Walter, M. Othus, A. Burnett, Z. Zuo, L.
Medeiros, Z. Chen, V. Grossmann, U. Bacher, C. Haferlach, A. Porwit, J. Vardiman, R. Hasserjian,
Z. Zuo, C. Garcia, S. Wang, R. Hasserjian, A. Yilmaz, G. Saydam, F. Sahin, Y. Baran, A. Roy, I.
Roberts, P. Vyas, B. Lange, N. Kobrinsky, D. Barnard, K. Yoshida, T. Toki, Y. Okuno, E. Matutes, W.
Pickl, M. V. Veer, W. van den Ancker, M. Terwijn, T. Westers, C. Kawajiri, H. Tanaka, S.
Hashimoto, H. Shimizu, A. Yokohama, N. Hatsumi, L. Holmfeldt, L. Wei, E. Diaz-Flores, V.
Mühlbacher, M. Zenger, S. Schnittger, P. di L. I. W. in C. A. L. Leukemia, N. Heerema, A. Carroll,
M. Devidas, M. Den Boer, M. vanSlegtenhorst, R. De Menezes, C. O. Group, J. Boer, J.
Marchante, W. Evans, K. Roberts, R. Morin, J. Zhang, R. Harvey, C. Mullighan, I. Chen, K. Roberts,
Y. Li, D. Payne-Turner, B. Weston, M. Hayden, K. Roberts, J. Meijerink, R. Ohgami, D. Arber, J.
Zehnder, Y. Natkunam, R. Warnke, E. Coustan-Smith, C. Mullighan, M. Onciu, M. Neumann, S.
Heesch, C. Schlee, M. Neumann, E. Coskun, L. Fransecky, J. Zhang, L. Ding, L. Holmfeldt, T. Inukai,
N. Kiyokawa, D. Campana, K. Patrick, R. Wade, N. Goulden, B. Wood, S. Winter, and K.
Dunsmore, “The 2016 revision to the World Health Organization classification of myeloid
neoplasms and acute leukemia.,” Blood, vol. 127, no. 20, pp. 2391–405, May 2016.
[7] E. R. Glazer, C. I. Perkins, J. L. Young, R. D. Schlag, S. L. Campleman, and W. E. Wright, “Cancer
among Hispanic children in California, 1988-1994: comparison with non-Hispanic white
children.,” Cancer, vol. 86, no. 6, pp. 1070–9, Sep. 1999.
[8] H. Xu, C. Cheng, M. Devidas, D. Pei, Y. Fan, W. Yang, G. Neale, P. Scheet, E. G. Burchard, D. G.
Torgerson, C. Eng, M. Dean, F. Antillon, N. J. Winick, P. L. Martin, C. L. Willman, B. M. Camitta, G.
H. Reaman, W. L. Carroll, M. Loh, W. E. Evans, C.-H. Pui, S. P. Hunger, M. V Relling, and J. J. Yang,

BIBLIOGRAFIA
177
“ARID5B genetic polymorphisms contribute to racial disparities in the incidence and treatment
outcome of childhood acute lymphoblastic leukemia.,” J. Clin. Oncol., vol. 30, no. 7, pp. 751–7,
Mar. 2012.
[9] H. Inaba, M. Greaves, and C. G. Mullighan, “Acute lymphoblastic leukaemia,” Lancet, vol. 381,
no. 9881, pp. 1943–1955, 2013.
[10] R. Snyder, “Leukemia and benzene.,” Int. J. Environ. Res. Public Health, vol. 9, no. 8, pp. 2875–93,
Aug. 2012.
[11] C. Meyer, J. Hofmann, T. Burmeister, D. Gröger, T. S. Park, M. Emerenciano, M. Pombo de
Oliveira, A. Renneville, P. Villarese, E. Macintyre, H. Cavé, E. Clappier, K. Mass-Malo, J. Zuna, J.
Trka, E. De Braekeleer, M. De Braekeleer, S. H. Oh, G. Tsaur, L. Fechina, V. H. J. van der Velden, J.
J. M. van Dongen, E. Delabesse, R. Binato, M. L. M. Silva,A. Kustanovich, O. Aleinikova, M. H.
Harris, T. Lund-Aho, V. Juvonen, O. Heidenreich, J. Vormoor, W. W. L. Choi, M. Jarosova, A.
Kolenova, C. Bueno, P. Menendez, S. Wehner, C. Eckert, P. Talmant, S. Tondeur, E. Lippert, E.
Launay, C. Henry, P. Ballerini, H. Lapillone, M. B. Callanan, J. M. Cayuela, C. Herbaux, G.
Cazzaniga, P. M. Kakadiya, S. Bohlander, M. Ahlmann, J. R. Choi, P. Gameiro, D. S. Lee, J. Krauter,
P. Cornillet-Lefebvre, G. Te Kronnie, B. W. Schäfer, S. Kubetzko, C. N. Alonso, U. zur Stadt, R.
Sutton, N. C. Venn, S. Izraeli, L. Trakhtenbrot, H.O. Madsen, P. Archer, J. Hancock, N. Cerveira,
M. R. Teixeira, L. Lo Nigro, A. Möricke, M. Stanulla, M. Schrappe, L. Sedék, T. Szczepański, C. M.
Zwaan, E. A. Coenen, M. M. van den Heuvel-Eibrink, S. Strehl, M. Dworzak, R. Panzer-Grümayer,
T. Dingermann, T. Klingebiel, and R. Marschalek, “The MLL recombinome of acute leukemias in
2013,” Leukemia, vol. 27, no. 11, pp. 2165–2176, Nov. 2013.
[12] A. Biondi, G. Cimino, R. Pieters, and C.-H. Pui, “Biological and therapeutic aspects of infant
leukemia,” Blood, vol. 96, no. 1, 2000.
[13] J. E. Churpek, K. Pyrtel, K.-L. Kanchi, J. Shao, D. Koboldt, C. A. Miller, D. Shen, R. Fulton, M.
O’Laughlin, C. Fronick, I. Pusic, G. L. Uy, E. M. Braunstein, M. Levis, J. Ross, K. Elliott, S. Heath, A.
Jiang, P. Westervelt, J. F. DiPersio, D. C. Link, M. J. Walter, J. Welch, R. Wilson, T. J. Ley, L. A.
Godley, and T. A. Graubert, “Genomic analysis of germ line and somatic variants in familial
myelodysplasia/acute myeloid leukemia,” Blood, vol. 126, no. 22, 2015.
[14] K. Schmiegelow, “Treatment-related toxicities in children with acute lymphoblastic leukaemia
predisposition syndromes.,” Eur. J. Med. Genet., vol. 59, no. 12, pp. 654–660, Dec. 2016.
[15] W. Yang, L. R. Treviño, J. J. Yang, P. Scheet, C.-H. Pui, W. E. Evans, and M. V Relling, “ARID5B SNP
rs10821936 is associated with risk of childhood acute lymphoblastic leukemia in blacks and
contributes to racial differences in leukemia incidence,” Leukemia, vol. 24, no. 4, pp. 894–896,
Apr. 2010.
[16] E. Papaemmanuil, F. J. Hosking, J. Vijayakrishnan, A. Price, B. Olver, E. Sheridan, S. E. Kinsey, T.
Lightfoot, E. Roman, J. A. E. Irving, J. M. Allan, I. P. Tomlinson, M. Taylor, M. Greaves, and R. S.
Houlston, “Loci on 7p12.2, 10q21.2 and 14q11.2 are associated with risk of childhood acute
lymphoblastic leukemia,” Nat. Genet., vol. 41, no. 9, pp. 1006–1010, Sep. 2009.

BIBLIOGRAFIA
178
[17] A. Martín-Lorenzo, J. Hauer, C. Vicente-Dueñas, F. Auer, I. González-Herrero, I. García-Ramírez,
S. Ginzel, R. Thiele, S. N. Constantinescu, C. Bartenhagen, M. Dugas, M. Gombert, D. Schäfer, O.
Blanco, A. Mayado, A. Orfao, D. Alonso-López, J. D. Las Rivas, C. Cobaleda, M. B. García-Cenador,
F. J. García-Criado, I. Sánchez-García, and A. Borkhardt, “Infection Exposure Is a Causal Factor in
B-cell Precursor Acute Lymphoblastic Leukemia as a Result of Pax5-Inherited Susceptibility,”
Cancer Discov., vol. 5, no. 12, 2015.
[18] L. J. Kinlen, “INFECTIVE CAUSE OF CHILDHOOD LEUKAEMIA,” The Lancet, vol. 333, no. 8634.
Elsevier, pp. 378–379, 1989.
[19] L. Kinlen, “EVIDENCE FOR AN INFECTIVE CAUSE OF CHILDHOOD LEUKAEMIA: COMPARISON OF A
SCOTTISH NEW TOWN WITH NUCLEAR REPROCESSING SITES IN BRITAIN,” Lancet, vol. 332, no.
8624, pp. 1323–1327, 1988.
[20] M. F. Greaves, and F. E. Alexander, “An infectious etiology for common acute lymphoblastic
leukemia in childhood?,” Leukemia, vol. 7, no. 3, pp. 349–60, Mar. 1993.
[21] L. Kinlen and A. Balkwill, “Infective cause of childhood leukaemia and wartime population mixing
in Orkney and Shetland, UK,” Lancet, vol. 357, no. 9259, 2001.
[22] G.-L. Moldovan, A. D. D’Andrea, and A. D. D’Andrea, “How the fanconi anemia pathway guards
the genome.,” Annu. Rev. Genet., vol. 43, pp. 223–49, 2009.
[23] M. Payne, I. D. Hickson, and I. D. Hickson, “Genomic instability and cancer: lessons from analysis
of Bloom’s syndrome.,” Biochem. Soc. Trans., vol. 37, no. Pt 3, pp. 553–9, Jun. 2009.
[24] L. Walker, D. Thompson, D. Easton, B. Ponder, M. Ponder, I. Frayling, D. Baralle, and D. Baralle,
“A prospective study of neurofibromatosis type 1 cancer incidence in the UK.,” Br. J. Cancer, vol.
95, no. 2, pp. 233–8, Jul. 2006.
[25] H. Hasle, “Malignant diseases in Noonan syndrome and related disorders.,” Horm. Res., vol. 72
Suppl 2, pp. 8–14, Dec. 2009.
[26] P. S. Rosenberg, C. Zeidler, A. A. Bolyard, B. P. Alter, M. A. Bonilla, L. A. Boxer, Y. Dror, S. Kinsey,
D. C. Link, P. E. Newburger, A. Shimamura, K. Welte, and D. C. Dale, “Stable long-term risk of
leukaemia in patients with severe congenital neutropenia maintained on G-CSF therapy.,” Br. J.
Haematol., vol. 150, no. 2, pp. 196–9, Jul. 2010.
[27] A. C. Xavier and J. W. Taub, “Acute leukemia in children with Down syndrome,” Haematologica,
vol. 95, no. 7, 2010.
[28] E. S. Sandler, D. J. Friedman, M. M. Mustafa, N. J. Winick, W. P. Bowman,and G. R. Buchanan,
“Treatment of children with epipodophyllotoxin-induced secondary acute myeloid leukemia.,”
Cancer, vol. 79, no. 5, pp. 1049–54, Mar. 1997.
[29] B. Weiss, A. Vora, J. Huberty, R. A. Hawkins, and K. K. Matthay, “Secondary myelodysplastic
syndrome and leukemia following 131I-metaiodobenzylguanidine therapy for relapsed
neuroblastoma.,” J. Pediatr. Hematol. Oncol., vol. 25, no. 7, pp. 543–7, Jul. 2003.
[30] C. M. McHale, L. Zhang, and M. T. Smith, “Current understanding of the mechanism of benzene-
induced leukemia in humans: implications for risk assessment.,” Carcinogenesis, vol. 33, no. 2,

BIBLIOGRAFIA
179
pp. 240–52, Feb. 2012.
[31] M. L. Smith, J. D. Cavenagh, T. A. Lister, and J. Fitzgibbon, “Mutation of CEBPA in Familial Acute
Myeloid Leukemia,” N. Engl. J. Med., vol. 351, no. 23, pp. 2403–2407, Dec. 2004.
[32] C. N. Hahn, C.-E. Chong, C. L. Carmichael, E. J. Wilkins, P. J. Brautigan, X.-C. Li, M. Babic, M. Lin, A.
Carmagnac, Y. K. Lee, C. H. Kok, L. Gagliardi, K. L. Friend, P. G. Ekert, C. M. Butcher, A. L. Brown, I.
D. Lewis, L. B. To, A. E. Timms, J. Storek, S. Moore, M. Altree, R. Escher, P. G. Bardy, G. K. Suthers,
R. J. D’Andrea, M. S. Horwitz, and H. S. Scott, “Heritable GATA2 mutations associated with
familial myelodysplastic syndrome and acute myeloid leukemia,” Nat. Genet., vol. 43, no. 10, pp.
1012–1017, Sep. 2011.
[33] W. J. Song, M. G. Sullivan, R. D. Legare, S. Hutchings, X. Tan, D. Kufrin, J. Ratajczak, I. C. Resende,
C. Haworth, R. Hock, M. Loh, C. Felix, D. C. Roy, L. Busque, D. Kurnit, C. Willman, A. M. Gewirtz,
N. A. Speck, J. H. Bushweller, F. P. Li, K. Gardiner, M. Poncz, J. M. Maris,and D. G. Gilliland,
“Haploinsufficiency of CBFA2 causes familial thrombocytopenia with propensity to develop
acute myelogenous leukaemia.,” Nat. Genet., vol. 23, no. 2, pp. 166–75, Oct. 1999.
[34] M. F. Greaves and J. Wiemels, “Origins of chromosome translocations in childhood leukaemia,”
Nat. Rev. Cancer, vol. 3, no. 9, pp. 639–649, Sep. 2003.
[35] A. T. Look, “Oncogenic Transcription Factors in the Human Acute Leukemias,” Science (80-. ).,
vol. 278, no. 5340, 1997.
[36] E.-J. Yeoh, M. E. Ross, S. A. Shurtleff, W. K. Williams, D. Patel, R. Mahfouz, F. G. Behm, S. C.
Raimondi, M. V Relling, A. Patel, C. Cheng, D. Campana, D. Wilkins, X. Zhou, J. Li, H. Liu, C.-H. Pui,
W. E. Evans, C. Naeve, L. Wong, and J. R. Downing, “Classification, subtype discovery, and
prediction of outcome in pediatric acute lymphoblastic leukemia by gene expression profiling,”
Cancer Cell, vol. 1, no. 2, pp. 133–143, 2002.
[37] S. A. Armstrong, J. E. Staunton, L. B. Silverman, R. Pieters, M. L. den Boer, M. D. Minden, S. E.
Sallan, E. S. Lander, T. R. Golub, and S. J. Korsmeyer, “MLL translocations specify a distinct gene
expression profile that distinguishes a unique leukemia,” Nat. Genet., vol. 30, no. 1, pp. 41–47,
Jan. 2002.
[38] C. Schoch, A. Kohlmann, S. Schnittger, B. Brors, M. Dugas, S. Mergenthaler, W. Kern, W.
Hiddemann, R. Eils, and T. Haferlach, “Acute myeloid leukemias with reciprocal rearrangements
can be distinguished by specific gene expression profiles.,” Proc. Natl. Acad. Sci. U. S. A., vol. 99,
no. 15, pp. 10008–13, Jul. 2002.
[39] C. S. Grove and G. S. Vassiliou, “Acute myeloid leukaemia: a paradigm for the clonal evolution of
cancer?,” Dis. Model. Mech., vol. 7, no. 8, pp. 941–951, Aug. 2014.
[40] F. G. Barr, “Translocations, cancer and the puzzle of specificity.,” Nat. Genet., vol. 19, no. 2, pp.
121–4, Jun. 1998.
[41] M. Greaves, “Pre-natal origins of childhood leukemia.,” Rev. Clin. Exp. Hematol., vol. 7, no. 3, pp.
233–45, Sep. 2003.
[42] J. Wiemels, G. Cazzaniga, M. Daniotti, O. Eden, G. Addison, G. Masera, V. Saha, A. Biondi, and M.

BIBLIOGRAFIA
180
Greaves, “Prenatal origin of acute lymphoblastic leukaemia in children,” Lancet, vol. 354, no.
9189, pp. 1499–1503, 1999.
[43] J. Wiemels, M. Kang, and M. Greaves, “Backtracking of leukemic clones to birth.,” Methods Mol.
Biol., vol. 538, pp. 7–27, 2009.
[44] L. M. Kelly, and D. G. Gilliland, “Genetics of myeloid leukemias.,” Annu. Rev. Genomics Hum.
Genet., vol. 3, pp. 179–98, 2002.
[45] N. Boissel, H. Leroy, B. Brethon, N. Philippe, S. de Botton, A. Auvrignon, E. Raffoux, T. Leblanc, X.
Thomas, O. Hermine, B. Quesnel, A. Baruchel, G. Leverger, H. Dombret, C. Preudhomme, C.
Acute Leukemia French Association (ALFA), and Leucémies Aiguës Myéloblastiques de l’Enfant
(LAME) Cooperative Groups, “Incidence and prognostic impact of c-Kit, FLT3, and Ras gene
mutations in core binding factor acute myeloid leukemia (CBF-AML).,” Leukemia, vol. 20, no. 6,
pp. 965–70, Jun. 2006.
[46] I. H. I. M. Hollink, M. M. van den Heuvel-Eibrink, S. T. C. J. M. Arentsen-Peters, M. Zimmermann,
J. K. Peeters, P. J. M. Valk, B. V Balgobind, E. Sonneveld, G. J. L. Kaspers, E. S. J. M. de Bont, J.
Trka, A. Baruchel, U. Creutzig, R. Pieters, D. Reinhardt, and C. M. Zwaan, “Characterization of
CEBPA mutations and promoter hypermethylation in pediatric acute myeloid leukemia.,”
Haematologica, vol. 96, no. 3, pp. 384–92, Mar. 2011.
[47] Z. Li, F. J. Godinho, J.-H. Klusmann, M. Garriga-Canut, C. Yu, and S. H. Orkin, “Developmental
stage-selective effect of somatically mutated leukemogenic transcription factor GATA1.,” Nat.
Genet., vol. 37, no. 6, pp. 613–9, Jun. 2005.
[48] I. H. I. M. Hollink, C. M. Zwaan, M. Zimmermann, T. C. J. M. Arentsen-Peters, R. Pieters, J. Cloos,
G. J. L. Kaspers, S. S. N. de Graaf, J. Harbott, U. Creutzig, D. Reinhardt, M. M. van den Heuvel-
Eibrink, and C. Thiede, “Favorable prognostic impact of NPM1 gene mutations in childhood
acute myeloid leukemia, with emphasis on cytogenetically normal AML.,” Leukemia, vol. 23, no.
2, pp. 262–70, Feb. 2009.
[49] B. V Balgobind, I. H. I. M. Hollink, S. T. C. J. M. Arentsen-Peters, M. Zimmermann, J. Harbott, H. B.
Beverloo, A. R. M. von Bergh, J. Cloos, G. J. L. Kaspers, V. de Haas, Z. Zemanova, J. Stary, J.-M.
Cayuela, A. Baruchel, U. Creutzig, D. Reinhardt, R. Pieters, C. M. Zwaan, and M. M. van den
Heuvel-Eibrink, “Integrative analysis of type-I and type-II aberrations underscores the genetic
heterogeneity of pediatric acute myeloid leukemia.,” Haematologica, vol. 96, no. 10, pp. 1478–
87, Oct. 2011.
[50] I. H. I. M. Hollink, M. M. van den Heuvel-Eibrink, M. Zimmermann, B. V Balgobind, S. T. C. J. M.
Arentsen-Peters, M. Alders, A. Willasch, G.-J. J. L. Kaspers, J. Trka, A. Baruchel, U. Creutzig, R.
Pieters, D. Reinhardt, and C. M. Zwaan, “No prognostic impact of the WT1 gene single nucleotide
polymorphism rs16754 in pediatric acute myeloid leukemia.,” J. Clin. Oncol., vol. 28, no. 28, pp.
e523-6-e528, Oct. 2010.
[51] L. M. Kelly and D. G. Gilliland, “GENETICS OF MYELOID LEUKEMIAS,” 2002.
[52] D. Grimwade, A. Ivey, and B. J. P. Huntly, “Molecular landscape of acute myeloid leukemia in

BIBLIOGRAFIA
181
younger adults and its clinical relevance,” Blood, vol. 127, no. 1, 2016.
[53] “Home - The Cancer Genome Atlas - Cancer Genome - TCGA.” [Online]. Available:
https://cancergenome.nih.gov/.
[54] C. S. Grove and G. S. Vassiliou, “Acute myeloid leukaemia: a paradigm for the clonal evolution of
cancer?,” Dis. Model. Mech., vol. 7, no. 8, 2014.
[55] M. C. H. Hermkens, M. M. van den Heuvel-Eibrink, S. T. C. J. M. Arentsen-Peters, A. Baruchel, J.
Stary, D. Reinhardt, M. Zimmerman, V. de Haas, R. Pieters, and C. M. Zwaan, “The clinical
relevance of BAALC and ERG expression levels in pediatric AML.,” Leukemia, vol. 27, no. 3, pp.
735–7, Mar. 2013.
[56] B. V Balgobind, S. Lugthart, I. H. Hollink, S. T. J. C. M. Arentsen-Peters, E. R. van Wering, S. S. N.
de Graaf, D. Reinhardt, U. Creutzig, G. J. L. Kaspers, E. S. J. M. de Bont, J. Stary, J. Trka, M.
Zimmermann, H. B. Beverloo, R. Pieters, R. Delwel, C. M. Zwaan, and M. M. van den Heuvel-
Eibrink, “EVI1 overexpression in distinct subtypes of pediatric acute myeloid leukemia.,”
Leukemia, vol. 24, no. 5, pp. 942–9, May 2010.
[57] J. E. Kuipers, E. A. Coenen, B. V Balgobind, J. Stary, A. Baruchel, V. de Haas, E. S. J. M. de Bont, D.
Reinhardt, G. J. L. Kaspers, J. Cloos, A. A. Danen-van Oorschot, M. L. den Boer, R. Marschalek, C.
Meyer, R. Pieters, C. M. Zwaan, and M. M. van den Heuvel-Eibrink, “High IGSF4 expression in
pediatric M5 acute myeloid leukemia with t(9;11)(p22;q23).,” Blood, vol. 117, no. 3, pp. 928–35,
Jan. 2011.
[58] B. V Balgobind, C. M. Zwaan, D. Reinhardt, T. J. C. M. Arentsen-Peters, I. H. I. M. Hollink, V. de
Haas, G. J. L. Kaspers, E. S. J. M. de Bont, A. Baruchel, J. Stary, C. Meyer, R. Marschalek, U.
Creutzig, M. L. den Boer, R. Pieters, and M. M. van den Heuvel-Eibrink, “High BRE expression in
pediatric MLL-rearranged AML is associated with favorable outcome.,” Leukemia, vol. 24, no. 12,
pp. 2048–55, Dec. 2010.
[59] A. H. Shih, O. Abdel-Wahab, J. P. Patel, and R. L. Levine, “The role of mutations in epigenetic
regulators in myeloid malignancies.,” Nat. Rev. Cancer, vol. 12, no. 9, pp. 599–612, Sep. 2012.
[60] T. Schoofs, W. E. Berdel, and C. Müller Tidow, “Origins of aberrant DNA methylation in acute
myeloid leukemia.,” Leukemia, vol. 28, no. 1, pp. 1–14, Jan. 2014.
[61] R. C. Lee, R. L. Feinbaum, and V. Ambros, “The C. elegans heterochronic gene lin-4 encodes small
RNAs with antisense complementarity to lin-14.,” Cell, vol. 75, no. 5, pp. 843–54, Dec. 1993.
[62] A. A. Danen-van Oorschot, J. E. Kuipers, S. Arentsen-Peters, D. Schotte, V. de Haas, J. Trka, A.
Baruchel, D. Reinhardt, R. Pieters, C. M. Zwaan, and M. M. van den Heuvel-Eibrink,
“Differentially expressed miRNAs in cytogenetic and molecular subtypes of pediatric acute
myeloid leukemia.,” Pediatr. Blood Cancer, vol. 58, no. 5, pp. 715–21, May 2012.
[63] M. D. Beyá, A. Navarro, T. Díaz, B. Gel, M. Camós, M. Pratcorona, M. Torrebadell, M. Rozman, M.
Maffioli, M. Monzó, and J. Esteve, “A Distinctive MicroRNA Signature Characterizes Acute
Myeloid Leukemia with Translocation (8;16)(p11;p13) and MYST3-CREBBP Rearrangement,”
Blood, vol. 116, no. 21, 2015.

BIBLIOGRAFIA
182
[64] M. Greaves, “Cancer stem cells: Back to Darwin?,” Semin. Cancer Biol., vol. 20, no. 2, pp. 65–70,
2010.
[65] F. Notta, C. G. Mullighan, J. C. Y. Wang, A. Poeppl, S. Doulatov, L. A. Phillips, J. Ma, M. D. Minden,
J. R. Downing, and J. E. Dick, “Evolution of human BCR–ABL1 lymphoblastic leukaemia-initiating
cells,” Nature, vol. 469, no. 7330, pp. 362–367, Jan. 2011.
[66] E. Passegué, C. H. M. Jamieson, L. E. Ailles, and I. L. Weissman, “Normal and leukemic
hematopoiesis: are leukemias a stem cell disorder or a reacquisition of stem cell
characteristics?,” Proc. Natl. Acad. Sci. U. S. A., pp. 11842–9, Sep. 2003.
[67] C. G. Mullighan, J. Zhang, L. H. Kasper, S. Lerach, D. Payne-Turner, L. A. Phillips, S. L. Heatley, L.
Holmfeldt, J. R. Collins-Underwood, J. Ma, K. H. Buetow, C.-H. Pui, S. D. Baker, P. K. Brindle, and
J. R. Downing, “CREBBP mutations in relapsed acute lymphoblastic leukaemia.,” Nature, vol. 471,
no. 7337, pp. 235–9, Mar. 2011.
[68] C. G. Mullighan, L. A. Phillips, X. Su, J. Ma, C. B. Miller, S. A. Shurtleff, and J. R. Downing,
“Genomic Analysis of the Clonal Origins of Relapsed Acute Lymphoblastic Leukemia,” Science
(80-. )., vol. 322, no. 5906, 2008.
[69] C.-H. Pui, “Childhood leukemias, third edition,” 2010.
[70] P. Porcu, L. D. Cripe, E. W. Ng, S. Bhatia, C. M. Danielson, A. Orazi, and L. J. McCarthy,
“Hyperleukocytic Leukemias and Leukostasis: A Review of Pathophysiology, Clinical Presentation
and Management,” Leuk. Lymphoma, vol. 39, no. 1–2, pp. 1–18, Jan. 2000.
[71] M. S. Tallman, P. Lefèbvre, R. M. Baine, M. Shoji, I. Cohen, D. Green, H. C. Kwaan, E. Paietta, and
F. R. Rickles, “Effects of all-trans retinoic acid or chemotherapy on the molecular regulation of
systemic blood coagulation and fibrinolysis in patients with acute promyelocytic leukemia.,” J.
Thromb. Haemost., vol. 2, no. 8, pp. 1341–50, Aug. 2004.
[72] C. Arbuthnot, and J. T. Wilde, “Haemostatic problems in acute promyelocytic leukaemia.,” Blood
Rev., vol. 20, no. 6, pp. 289–97, Nov. 2006.
[73] X. Zhang, H. Zhou, J. Wang, L. Yang, Y. Hu, G. Shen, P. Guo, Z. Qiao, and S. Song, “Arsenic
trioxide, retinoic acid and Ara-c regulated the expression of annexin II on the surface of APL
cells, a novel co-receptor for plasminogen/tissue plasminogen activator.,” Thromb. Res., vol.
106, no. 1, pp. 63–70, Apr. 2002.
[74] K. A. Minson, P. Prasad, S. Vear, S. Borinstein, R. Ho, J. Domm, and H. Frangoul, “t(17;19) in
Children with Acute Lymphocytic Leukemia: A Report of 3 Cases and a Review of the Literature.,”
Case Rep. Hematol., vol. 2013, p. 563291, 2013.
[75] A. Gajjar, R. Ribeiro, M. L. Hancock, G. K. Rivera, H. Mahmoud, J. T. Sandlund, W. M. Crist, and C.
H. Pui, “Persistence of circulating blasts after 1 week of multiagent chemotherapy confers a poor
prognosis in childhood acute lymphoblastic leukemia.,” Blood, vol. 86, no. 4, pp. 1292–5, Aug.
1995.
[76] J. M. Bennett, D. Catovsky, M.-T. Daniel, G. Flandrin, D. A. G. Galton, H. R. Gralnick, and C.
Sultan, “Proposals for the Classification of the Acute Leukaemias French-American-British (FAB)

BIBLIOGRAFIA
183
Co-operative Group,” Br. J. Haematol., vol. 33, no. 4, pp. 451–458, Aug. 1976.
[77] D. A. Arber, A. Orazi, R. Hasserjian, J. Thiele, M. J. Borowitz, M. M. Le Beau, C. D. Bloomfield, M.
Cazzola, and J. W. Vardiman, “The 2016 revision to the World Health Organization classification
of myeloid neoplasms and acute leukemia.,” Blood, vol. 127, no. 20, pp. 2391–405, May 2016.
[78] E. Papaemmanuil, M. Gerstung, L. Bullinger, V. I. Gaidzik, P. Paschka, N. D. Roberts, N. E. Potter,
M. Heuser, F. Thol, N. Bolli, G. Gundem, P. Van Loo, I. Martincorena, P. Ganly, L. Mudie, S.
McLaren, S. O’Meara, K. Raine, D. R. Jones, J. W. Teague, A. P. Butler, M. F. Greaves, A. Ganser,
K. Döhner, R. F. Schlenk, H. Döhner, and P. J. Campbell, “Genomic Classification and Prognosis in
Acute Myeloid Leukemia.,” N. Engl. J. Med., vol. 374, no. 23, pp. 2209–21, Jun. 2016.
[79] J. M. Bennett, D. Catovsky, M. T. Daniel, G. Flandrin, D. A. G. Galton, H. R. Gralnick, and C. Sultan,
“The Morphological Classification of Acute Lymphoblastic Leukaemia: Concordance among
Observers and Clinical Correlations,” Br. J. Haematol., vol. 47, no. 4, pp. 553–561, Apr. 1981.
[80] M. C. Bene, G. Castoldi, W. Knapp, W. D. Ludwig, E. Matutes, A. Orfao, and M. B. van’t Veer,
“Proposals for the immunological classification of acute leukemias. European Group for the
Immunological Characterization of Leukemias (EGIL).,” Leukemia, vol. 9, no. 10, pp. 1783–6, Oct.
1995.
[81] U. Creutzig, M. M. van den Heuvel-Eibrink, B. Gibson, M. N. Dworzak, S. Adachi, E. de Bont, J.
Harbott, H. Hasle, D. Johnston, A. Kinoshita, T. Lehrnbecher, G. Leverger, E. Mejstrikova, S.
Meshinchi, A. Pession, S. C. Raimondi, L. Sung, J. Stary, C. M. Zwaan, G. J. L. Kaspers, D.
Reinhardt, B. AML Committee of the International BFM Study Group, P. Cassileth, D. Head, B.
Cheson, J. Bennett, K. Kopecky, S. Swerdlow, E. Campo, N. Harris, H. Döhner, E. Estey, S.
Amadori, J. Vardiman, J. Thiele, D. Arber, M. Bene, G. Castoldi, W. Knapp, J. Bennett, D.
Catovsky, M. Daniel, H. Hasle, C. Niemeyer, J. Chessells, M. Bene, T. Nebe, P. Bettelheim, J.
Rubnitz, H. Inaba, G. Dahl, B. Wood, M. Arroz, D. Barnett, D. Betts, R. Ammann, A. Hirt, D.
Grimwade, C. Harrison, R. Hills, A. Moorman, C. von Neuhoff, D. Reinhardt, A. Sander, T.
Mercher, M. B.-L. Coniat, K. Nguyen, L. Torres, S. Lisboa, J. Vieira, J. Park, M. Kim, J. Lim, H.
Simmons, L. Oseth, P. Nguyen, M. O’Leary, K. Conklin, B. Hirsch, R. Slater, E. von Drunen, W.
Kroes, A. von Bergh, E. van Drunen, E. van Wering, I. Hollink, M. Van den Heuvel-Eibrink, S.
Arentsen-Peters, H. Hasle, T. Alonzo, A. Auvrignon, D. Breems, W. Van Putten, G. De Greef, U.
Creutzig, T. Buchner, M. Sauerland, D. Gilliland, B. Balgobind, I. Hollink, S. Arentsen-Peters, P.
Ho, T. Alonzo, R. Gerbing, I. Hollink, C. Zwaan, M. Zimmermann, S. Meshinchi, T. Alonzo, D.
Stirewalt, C. Pui, W. Carroll, S. Meshinchi, R. Arceci, P. Brown, E. McIntyre, R. Rau, C.Thiede, E.
Creutzig, D. Reinhardt, G. Ehninger, U. Creutzig, G. Cazzaniga, M. Dell’Oro, C. Mecucci, B.
Balgobind, M. Van den Heuvel-Eibrink, R. De Menezes, P. Ho, R. Zeng, T. Alonzo, I. Hollink, M.
Van den Heuvel-Eibrink, M. Zimmermann, S. Meshinchi, F. Appelbaum, J. Berman, R. Gerbing, T.
Alonzo, N. Boissel, H. Leroy, B. Brethon, L. Shih, D. Liang, C. Huang, M. Loh, M. Reynolds, S.
Vattikuti, B. Goemans, C. Zwaan, M. Miller, J. Pollard, T. Alonzo, R. Gerbing, B. Balgobind, I.
Hollink, D. Reinhardt, B. Balgobind, S. Lugthart, I. Hollink, A. Staffas, M. Kanduri, R. Hovland, F.

BIBLIOGRAFIA
184
Damm, F. Thol, I. Hollink, P. Ho, M. Kutny, T. Alonzo, I. Hollink, Q. Feng, A. D. Oorschot, E.
Mardis, L. Ding, D. Dooling, F. Thol, F. Damm, A. Ludeking, M. Ross, R. Mahfouz, M. Onciu, U.
Creutzig, M. Zimmermann, J.-P. Bourquin, U. Creutzig, M. Zimmermann, J. Ritter, B. Balgobind, S.
Raimondi, J. Harbott, E. Coenen, C. Zwaan, C. Meyer, S. Noronha, J. Farrar, T. Alonzo, J.
Abrahamsson, E. Forestier, J. Heldrup, U. Creutzig, M. Zimmermann, J. Ritter, K. Wheatley, A.
Burnett, A. Goldstone, C. Langebrake, U. Creutzig, M. Dworzak, V. Van der Velden, A. Sluijs-
Geling, B. Gibson, S. Viehmann, A. Teigler-Schlegel, J. Bruch, C. Langebrake, D. Reinhardt, J.
Harbott, H. Ommen, S. Schnittger, J. Jovanovic, K. Alford, K. Reinhardt, C. Garnett, R. Wells, W.
Woods, B. Lampkin, Y. Perel, A. Auvrignon, T. Leblanc, E. Estey, H. Dohner, B. Lowenberg, J.
Griffin, M. Tallman, W. Woods, N. Kobrinsky, J. Buckley, G. Kaspers, H. Fernandez, Z. Sun, X. Yao,
B. Lowenberg, G. Ossenkoppele, W. van Putten, E. van Dalen, H. van der Pal, H. Caron, L. Kremer,
E. van Dalen, E. Michiels, H. Caron, L. Kremer, L. Kremer, H. van der Pal, M. Offringa, E. van
Dalen, P. Voute, M. Grenier, S. Lipshultz, S. Bielack, R. Erttmann, B. Kempf-Bielack, K. Winkler, S.
Lipshultz, A. Giantris, S. Lipsitz, L. Smith, V. Cornelius, C. Plummer, U. Creutzig, J. Ritter, M.
Zimmermann, B. Gibson, D. Webb, A. Howman, S. De Graaf, C. Harrison, K. Wheatley, S.
Lipshultz, J. Alvarez, R. Scully, U. S. F. and D. Administration, A. Burnett, R. Hills, D. Milligan, U.
Creutzig, M. Zimmermann, J. Bourquin, Z. Arlin, D. Case, J. Moore, G. Michel, A. Baruchel, M.
Tabone, R. Krance, C. Hurwitz, D. Head, R. Mayer, R. Davis, C. Schiffer, C. Bloomfield, D.
Lawrence, J. Byrd, S. Lie, J. Abrahamsson, N. Clausen, B. Gibson, K. Wheatley, I. Hann, R. Stevens,
I. Hann, K. Wheatley, R. Gray, U. Creutzig, M. Zimmermann, M. Dworzak, I. Tsukimoto, A. Tawa,
K. Horibe, T. Alonzo, R. Wells, W. Woods, D. Oliansky, J. Rizzo, P. Aplan, Y. Ravindranath, A.
Yeager, M. Chang, W. Woods, S. Neudorf, S. Gold, M. Sanz, D. Grimwade, M. Tallman, J. Horan,
T. Alonzo, G. Lyman, T. Klingebiel, D. Reinhardt, P. Bader, D. Niewerth, U. Creutzig, M. Bierings,
G. Kaspers, Y. Perel, A. Auvrignon, T.Leblanc, S. Bhatia, D. Bresters, I. van Gils, W. Kollen, T.
Gupta, S. Kannan, V. Dantkale, S. Laskar, F. Appelbaum, G. Socie, R. Clift, D. Blaise, F. Appelbaum,
R. Walter, T. Gooley, B. Wood, S. Bonanomi, P. Connor, D. Webb, U. Creutzig, M. Zimmermann,
J. Bourquin, G. Dahl, J. Simone, H. Hustu, C. Mason, U. Creutzig, J. Ritter, M. Zimmermann, G.
Schellong, C. Pui, S. Howard, D. Johnston, T. Alonzo, R. Gerbing, B. Lange, W. Woods, A. Pession,
B. Abbott, J. Rubnitz, X. Tong, J. Mayadev, J. Douglas, B. Storer, F. Appelbaum, R. Storb, D.
Johnston, T. Alonzo, R. Gerbing, B. Lange, W. Woods, S. Ehlers, C. Herbst, M. Zimmermann, Y.
Lee, J. Moon, J. Kim, B. Lowenberg, W. van Putten, M. Theobald, A. Periclou, V. Avramis, H. van
den Berg, A. Hopman, K. Kraakman, D. de Jong, C. Campidelli, C. Agostinelli, R. Stitson, S. Pileri,
N. Harris, E. Jaffe, J. Diebold, D. Reinhardt, U. Creutzig, M. Felice, P. Zubizarreta, E. Alfaro, E.
Matutes, W. Pickl, M. V. Veer, H. Gerr, M. Zimmermann, M. Schrappe, E. Mejstrikova, J.
Volejnikova, E. Fronkova, D. Miller, L. Miller, A. Gamis, T. Alonzo, R. Gerbing, J. Hitzler, J. Cheung,
Y. Li, S. Scherer, A. Zipursky, J. Wechsler, M. Greene, M. McDevitt, Z. Li, F. Godinho, J. Klusmann,
M. Garriga-Canut, C. Yu, S. Orkin, J. Klusmann, U. Creutzig, M. Zimmermann, U. Creutzig, D.
Reinhardt, S. Diekamp, M. Dworzak, J. Stary, M. Zimmermann, K. Kudo, A. Hama, S. Kojima, K.

BIBLIOGRAFIA
185
Kudo, S. Kojima, K. Tabuchi, C. Zwaan, G. Kaspers, R. Pieters, J. Taub, Y. Ge, F. Li, J. Bader, M.
Belson, B. Kingsley, A. Holmes, P. Mehta, T. Ileri, R. Harris, M. Stelljes, A. Corbacioglu, R. Schlenk,
J. Gregory, J. Feusner, G. Mann, D. Reinhardt, J. Ritter, S. de Botton, V. Coiteux, S. Chevret, J.
Ortega, L. Madero, G. Martin, A. Testi, A. Biondi, C. Lo, N. Asou, Y. Kishimoto, H.Kiyoi, M. Sanz,
G. Martin, C. Rayon, U. Creutzig, M. Zimmermann, M. Dworzak, F. Lo-Coco, E. Ammatuna, D.
Grimwade, J. Jovanovic, R. Hills, J. Zhou, Y. Zhang, J. Li, E. Estey, G. Garcia-Manero, A. Ferrajoli, R.
Larson, C. Pui, S. Raimondi, D. Head, D. Aguilera, C. Vaklavas, A. Tsimberidou, S. Wen, L.
Medeiros, S. Corey, G. Kaspers, U. Creutzig, A. Sander, M. Zimmermann, M. Dworzak, K. Stahnke,
J. Boos, C. Bender-Gotze, J. Ritter, M. Zimmermann, U. Creutzig, G. Kaspers, M. Zimmermann, D.
Reinhardt, J. Abrahamsson, N. Clausen, G. Gustafsson, D. Webb, K. Wheatley, G. Harrison, R.
Stevens, I. Hann, V. Gandhi, E. Estey, M. Keating, W. Plunkett, B. Goemans, R. Tamminga, C.
Corbijn, K. Hahlen, G. Kaspers, A. Burnett, R. Hills, D. Milligan, C. Zwaan, D. Reinhardt, M.
Zimmerman, S. Jeha, B. Razzouk, M. Rytting, N. Hijiya, P. Gaynon, E. Barry, E. Sievers, R. Larson,
E. Stadtmauer, C. Zwaan, D. Reinhardt, S. Corbacioglu, J. Delaunay, C. Recher, A. Pigneux, B.
Brethon, K. Yakouben, C. Oudot, D. Reinhardt, S. Diekamp, G. Fleischhack, A. Burnett, R. Hills, A.
Hunter, S. Castaigne, C. Pautas, C. Terre, S. Castaigne, C. Pautas, C. Terre, T. Cooper, J. Franklin,
R. Gerbing, A. Fathi, M. Levis, H. Inaba, J. Rubnitz, E. Coustan-Smith, U. Creutzig, J. Ritter, M.
Budde, A. Sutor, G. Schellong, H. Inaba, Y. Fan, S. Pounds, G. Malaguarnera, M. Giordano, M.
Malaguarnera, O. Cornely, J. Maertens, D. Winston, T. Lehrnbecher, A. Groll, W. Hughes, S.
Kuhn, S. Chaudhary, W. Hughes, G. Rivera, M. Schell, D. Thornton, L. Lott, L. Sung, P. Nathan, S.
Alibhai, G. Tomlinson, J. Beyene, A. Gamis, W. Howells, J. DeSwarte-Wallace, J. Feusner, J.
Buckley, W. Woods, B. Kurt, P. Flynn, J. Shenep, A. Gafter-Gvili, A. Fraser, M. Paul, A. Freifeld, E.
Bow, K. Sepkowitz, M. van de Wetering, M. de Witte, L. Kremer, M. Offringa, R. Scholten, H.
Caron, A. Gafter-Gvili, A. Fraser, M. Paul, L. Leibovici, T. Lehrnbecher, M. Ethier, T. Zaoutis, T.
Lehrnbecher, M. Zimmermann, D. Reinhardt, M. Dworzak, J. Stary, U. Creutzig, G. Schaison, O.
Eden, G. Henze, G. Dores, S. Devesa, R. Curtis, M. Linet, L. Morton, B. Strahm, P. Nollke, M.
Zecca, H. Wandt, U. Schakel, F. Kroschinsky, D. Grimwade, H. Walker, F. Oliver, M. Smith, R. Hills,
D. Grimwade, K. Dohner, H. Dohner, T. Haferlach, U. Bacher, C. Haferlach, W. Kern, S. Schnittger,
R. Schlenk, A. Ganser, K. Dohner, H. Dohner, V. Gaidzik, H. de Jonge, P. Valk, E. de Bont, J. Tyner,
D. Walters, S. Willis, A. Shimada, T. Taki, C. Kubota, F. Delhommeau, S. Dupont, V. Della Valle, T.
Ley, L. Ding, M. Walter, T. Haferlach, C. Schoch, H. Loffler, O. Weinberg, M. Seetharam, L. Ren,
M. Miesner, C. Haferlach, U. Bacher, J. Klusmann, F. Godinho, K. Heitmann, D. Grimwade, H.
Walker, F. Oliver, U. Creutzig, and G. Kaspers, “Diagnosis and management of acute myeloid
leukemia in children and adolescents: recommendations from an international expert panel.,”
Blood, vol. 120, no. 16, pp. 3187–205, Oct. 2012.
[82] H. Hasle, C. M. Niemeyer, J. M. Chessells, I. Baumann, J. M. Bennett, G. Kerndrup, D. R. Head,
and D. R. Head, “A pediatric approach to the WHO classification of myelodysplastic and
myeloproliferative diseases.,” Leukemia, vol. 17, no. 2, pp. 277–82, Feb. 2003.

BIBLIOGRAFIA
186
[83] L. Holmfeldt, L. Wei, E. Diaz-Flores, M. Walsh, J. Zhang, L. Ding, D. Payne-Turner, M. Churchman,
A. Andersson, S.-C. Chen, K. McCastlain, J. Becksfort, J. Ma, G. Wu, S. N. Patel, S. L. Heatley, L. a
Phillips, G. Song, J. Easton, M. Parker, X. Chen, M. Rusch, K. Boggs, B. Vadodaria, E. Hedlund, C.
Drenberg, S. Baker, D. Pei, C. Cheng, R. Huether, C. Lu, R. S. Fulton, L. L. Fulton, Y. Tabib, D. J.
Dooling, K. Ochoa, M. Minden, I. D. Lewis, L. B. To, P. Marlton, A. W. Roberts, G. Raca, W. Stock,
G. Neale, H. G. Drexler, R. a Dickins, D. W. Ellison, S. a Shurtleff, C.-H. Pui, R. C. Ribeiro, M.
Devidas, A. J. Carroll, N. a Heerema, B. Wood, M. J. Borowitz, J. M. Gastier-Foster, S. C.
Raimondi, E. R. Mardis, R. K. Wilson, J. R. Downing, S. P. Hunger, M. L. Loh, and C. G. Mullighan,
“The genomic landscape of hypodiploid acute lymphoblastic leukemia.,” Nat. Genet., vol. 45, no.
3, 2013.
[84] A. V. Moorman, H. Robinson, C. Schwab, S. M. Richards, J. Hancock, C. D. Mitchell, N. Goulden,
A. Vora, and C. J. Harrison, “Risk-Directed Treatment Intensification Significantly Reduces the
Risk of Relapse Among Children and Adolescents With Acute Lymphoblastic Leukemia and
Intrachromosomal Amplification of Chromosome 21: A Comparison of the MRC ALL97/99 and
UKALL2003 Trials,” J. Clin. Oncol., vol. 31, no. 27, pp. 3389–3396, Sep. 2013.
[85] M. L. Den Boer, M. van Slegtenhorst, R. X. De Menezes,M. H. Cheok, J. G. Buijs-Gladdines, S. T.
Peters, L. J. Van Zutven, H. B. Beverloo, P. J. Van der Spek, G. Escherich, M. A. Horstmann, G. E.
Janka-Schaub, W. A. Kamps, W. E. Evans, and R. Pieters, “A subtype of childhood acute
lymphoblastic leukaemia with poor treatment outcome: a genome-wide classification study,”
Lancet Oncol., vol. 10, no. 2, pp. 125–134, Feb. 2009.
[86] C. G. Mullighan, X. Su, J. Zhang, I. Radtke, L. A. A. Phillips, C. B. Miller, J. Ma, W. Liu, C. Cheng, B.
A. Schulman, R. C. Harvey, I.-M. Chen, R. J. Clifford, W. L. Carroll, G. Reaman, W. P. Bowman, M.
Devidas, D. S. Gerhard, W. Yang, M. V. Relling, S. A. Shurtleff, D. Campana, M. J. Borowitz, C.-H.
Pui, M. Smith, S. P. Hunger, C. L. Willman, and J. R. Downing, “Deletion of IKZF1 and Prognosis in
Acute Lymphoblastic Leukemia,” N. Engl. J. Med., vol. 360, no. 5, pp. 470–480, Jan. 2009.
[87] Z. Gu, M. Churchman, K. Roberts, Y. Li, Y. Liu, R. C. Harvey, K. McCastlain, S. C. Reshmi, D. Payne-
Turner, I. Iacobucci, Y. Shao, I.-M. Chen, M. Valentine, D. Pei, K. L. Mungall, A. J. Mungall, Y. Ma,
R. Moore, M. Marra, E. Stonerock, J. M. Gastier-Foster, M. Devidas, Y. Dai, B. Wood, M.
Borowitz, E. E. Larsen, K. Maloney, L. A. Mattano Jr, A. Angiolillo, W. L. Salzer, M. J. Burke, F.
Gianni, O. Spinelli, J. P. Radich, M. D. Minden, A. V. Moorman, B. Patel, A. K. Fielding, J. M. Rowe,
S. M. Luger, R. Bhatia, I. Aldoss, S. J. Forman, J. Kohlschmidt, K. Mrózek, G. Marcucci, C. D.
Bloomfield, W. Stock, S. Kornblau, H. M. Kantarjian, M. Konopleva, E. Paietta, C. L. Willman, M. L.
Loh, S. P. Hunger, and C. G. Mullighan, “Genomic analyses identify recurrent MEF2D fusions in
acute lymphoblastic leukaemia,” Nat. Commun., vol. 7, p. 13331, Nov. 2016.
[88] S. Hirabayashi, K. Ohki, K. Nakabayashi, H. Ichikawa, Y. Momozawa, K. Okamura, A. Yaguchi, K.
Terada, Y. Saito, A. Yoshimi, H. Ogata-Kawata, H. Sakamoto, M. Kato, J. Fujimura, M. Hino, A.
Kinoshita, H. Kakuda, H. Kurosawa, K. Kato, R. Kajiwara, K. Moriwaki, T. Morimoto, K. Nakamura,
Y. Noguchi, T. Osumi, K. Sakashita, J. Takita, Y. Yuza, K. Matsuda, T. Yoshida, K. Matsumoto, K.

BIBLIOGRAFIA
187
Hata, M. Kubo, Y. Matsubara, T. Fukushima, K. Koh, A. Manabe, A. Ohara, and N. Kiyokawa,
“ZNF384 -related fusion genes define a subgroup of childhood B-cell precursor acute
lymphoblastic leukemia with a characteristic immunotype,” Haematologica, vol. 102, no. 1, pp.
118–129, Jan. 2017.
[89] H. Lilljebjörn, R. Henningsson, A. Hyrenius-Wittsten, L. Olsson, C. Orsmark-Pietras, S. von Palffy,
M. Askmyr, M. Rissler, M. Schrappe, G. Cario, A. Castor, C. J. H. Pronk, M. Behrendtz, F.
Mitelman, B. Johansson, K. Paulsson, A. K. Andersson, M. Fontes, and T. Fioretos, “Identification
of ETV6-RUNX1-like and DUX4-rearranged subtypes in paediatric B-cell precursor acute
lymphoblastic leukaemia,” Nat. Commun., vol. 7, p. 11790, Jun. 2016.
[90] J. Zhang, K. McCastlain, H. Yoshihara, B. Xu, Y. Chang, M. L. Churchman, G. Wu, Y. Li, L. Wei, I.
Iacobucci, Y. Liu, C. Qu, J. Wen, M. Edmonson, D. Payne-Turner, K. B. Kaufmann, S. Takayanagi, E.
Wienholds, E. Waanders, P. Ntziachristos, S. Bakogianni, J. Wang, I. Aifantis, K. G. Roberts, J. Ma,
G. Song, J. Easton, H. L. Mulder, X. Chen, S. Newman, X. Ma, M. Rusch, P. Gupta, K. Boggs, B.
Vadodaria, J. Dalton, Y. Liu, M. L. Valentine, L. Ding, C. Lu, R. S. Fulton, L. Fulton, Y. Tabib, K.
Ochoa, M. Devidas, D. Pei, C. Cheng, J. Yang, W. E. Evans, M. V Relling, C.-H. Pui, S. Jeha, R. C.
Harvey, I.-M. L. Chen, C. L. Willman, G. Marcucci, C. D. Bloomfield, J. Kohlschmidt, K. Mrózek, E.
Paietta, M. S. Tallman, W. Stock, M. C. Foster, J. Racevskis, J. M. Rowe, S. Luger, S. M. Kornblau,
S. A. Shurtleff, S. C. Raimondi, E. R. Mardis, R. K. Wilson, J. E. Dick, S. P. Hunger, M. L. Loh, J. R.
Downing, and C. G. Mullighan, “Deregulation of DUX4 and ERG in acute lymphoblastic
leukemia,” Nat. Genet., vol. 48, no. 12, pp. 1481–1489, Oct. 2016.
[91] S. P. Hunger, C. G. Mullighan, H. Inaba, M. Greaves, C. Mullighan, C. O. Group, R. Kuiper, E.
Schoenmakers, S. van Reijmersdal, C. Mullighan, S. Goorha, I. Radtke, C. Mullighan, C. Miller, I.
Radtke, C. Mullighan, J. Collins-Underwood, L. Phillips, C. O. Group, C. Mullighan, J. Zhang, R.
Harvey, V. Tosello, M. Mansour, K. Barnes, P. Van Vlierberghe, T. Palomero, H. Khiabanian, C.
Mullighan, J. Zhang, L. Kasper, P. Van Vlierberghe, A. Ambesi-Impiombato, A. Perez-Garcia, J.
Zhang, L. Ding, L. Holmfeldt, K. Roberts, R. Morin, J. Zhang, L. Holmfeldt, L. Wei, E. Diaz-Flores, A.
Perez-Garcia, A. Ambesi-Impiombato, M. Hadler, G. Tzoneva, A. Perez-Garcia, Z. Carpenter, K. De
Keersmaecker, Z. Atak, N. Li, Z. Atak, V. Gianfelici, G. Hulselmans, E. Papaemmanuil, I. Rapado, Y.
Li, Y. Li, C. Schwab, S. Ryan, K. Roberts, Y. Li, D. Payne-Turner, T. Trimarchi, E. Bilal, P.
Ntziachristos, D. Herranz, A. Ambesi-Impiombato, T. Palomero, M. Mansour, B. Abraham, L.
Anders, C. Harrison, C. and A. L. W. Parties, J. Nachman, N. Heerema, H. Sather, L. Russell, M.
Capasso, I. Vater, M. Den Boer, M. van Slegtenhorst, R. De Menezes, G. Cazzaniga, F. van Delft, L.
Lo Nigro, S. Safavi, E. Forestier, I. Golovleva, K. Anderson, C. Lutz, F. van Delft, C. Mullighan, L.
Phillips, X. Su, X. Ma, M. Edmonson, D. Yergeau, S. J. C. R. H. U. P. C. G. Project, C. Virely, S.
Moulin, C. Cobaleda, I. Joshi, T. Yoshida, N. Jena, J. Zhang, C. Mullighan, R. Harvey, A. van der
Veer, M. Zaliova, F. Mottadelli, R. Harvey, C. Mullighan, I. Chen, A. Yoda, Y. Yoda, S. Chiaretti, D.
Bercovich, I. Ganmore, L. Scott, G. Cario, M. Zimmermann, R. Romey, S. Maude, S. Tasian, T.
Vincent, M. Waibel, V. Solomon, D. Knight, W. Tong, J. Zhang, H. Lodish, C. Shochat, N. Tal, O.

BIBLIOGRAFIA
188
Bandapalli, P. Zenatti, D. Ribeiro, W. Li, L. Treanor, S. Zhou, L. Janke, A. van der Veer, E.
Waanders, R. Pieters, K. Roberts, D. Pei, D. Campana, D. Schinnerl, K. Fortschegger, M. Kauer, B.
Weston, M. Hayden, K. Roberts, E. Yeoh, M. Ross, S. Shurtleff, C. Mullighan, C. Miller, X. Su, R.
Harvey, C. Mullighan, X. Wang, S. Tomlins, D. Rhodes, S. Perner, E. Clappier, M. Auclerc, J.
Rapion, A. Moorman, H. Robinson, C. Schwab, A. Gutierrez, A. Kentsis, T. Sanda, A. Weng, A.
Ferrando, W. Lee, J. O’Neil, J. Grim, P. Strack, I. Lahortiga, K. De Keersmaecker, P. Van
Vlierberghe, A. Gutierrez, T. Sanda, R. Grebliunaite, M. Neumann, S. Vosberg, C. Schlee, E.
Coustan-Smith, C. Mullighan, M. Onciu, T. Inukai, N. Kiyokawa, D. Campana, K. Patrick, R. Wade,
N. Goulden, B. Wood, S. Winter, K. Dunsmore, P. Ntziachristos, A. Tsirigos, P. Van Vlierberghe, G.
Della Gatta, T. Palomero, A. Perez-Garcia, L. Yan, N. Ping, M. Zhu, S. Maude, S. Dolai, C. Delgado-
Martin, C. Graux, J. Cools, C. Melotte, T. Palomero, W. Lim, D. Odom, B. Mar, L. Bullinger, K.
McLean, H. Geng, S. Brennan, T. Milne, M. Figueroa, S. Chen, A. Andersson, J. Jaffe, Y. Wang, H.
Chan, J. Roderick, J. Tesell, L. Shultz, P. Ntziachristos, A. Tsirigos, G. Welstead, J. Hof, S. Krentz, C.
van Schewick, J. Meyer, J. Wang, L. Hogan, M. Faham, J. Zheng, M. Moorhead, E. Papaemmanuil,
F. Hosking, J. Vijayakrishnan, L. Treviño, W. Yang, D. French, R. Prasad, F. Hosking, J.
Vijayakrishnan, H. Xu, W. Yang, V. Perez-Andreu, G. Migliorini, B. Fiege, F. Hosking, J. Yang, C.
Cheng, W. Yang, W. Yang, L. Treviño, J. Yang, J. Yang, C. Cheng, M. Devidas, V. Perez-Andreu, K.
Roberts, R. Harvey, J. Yang, C. Cheng, M. Devidas, T. Akasaka, T. Balasas, L. Russell, S. Shah, K.
Schrader, E. Waanders, M. Zhang, J. Churpek, S. Keel, L. Noetzli, R. Lo, A. Lee-Sherick, C. Pui, W.
Carroll, S. Meshinchi, R. Arceci, C. Pui, D. Campana, D. Pei, E. Waanders, V. van der Velden, C.
van der Schoot, D. Bhojwani, C. Pui, K. Schultz, W. Bowman, A. Aledo, A. Biondi, M. Schrappe, P.
De Lorenzo, N. Venn, V. van der Velden, and M. de Bie, “Redefining ALL classification: toward
detecting high-risk ALL and implementing precision medicine.,” Blood, vol. 125, no. 26, pp.
3977–87, Jun. 2015.
[92] C. G. Mullighan, “The genomic landscape of acute lymphoblastic leukemia in children and young
adults,” Hematology, vol. 2014, no. 1, pp. 174–180, Dec. 2014.
[93] J. Bond, T. Marchand, A. Touzart, A. Cieslak, A. Trinquand, L. Sutton, I. Radford-Weiss, L.
Lhermitte, S. Spicuglia, H. Dombret, E. Macintyre, N. Ifrah, J.-F. Hamel, and V. Asnafi, “An early
thymic precursor phenotype predicts outcome exclusively in HOXA-overexpressing adult T-cell
acute lymphoblastic leukemia: a Group for Research in Adult Acute Lymphoblastic Leukemia
study,” Haematologica, vol. 101, no. 6, pp. 732–740, Jun. 2016.
[94] V. Conter, M. G. Valsecchi, B. Buldini, R. Parasole, F. Locatelli, A. Colombini, C. Rizzari, M. C. Putti,
E. Barisone, L. Lo Nigro, N. Santoro, O. Ziino, A. Pession, A. M. Testi, C. Micalizzi, F. Casale, P.
Pierani, S. Cesaro, M. Cellini, D. Silvestri, G. Cazzaniga, A. Biondi, and G. Basso, “Early T-cell
precursor acute lymphoblastic leukaemia in children treated in AIEOP centres with AIEOP-BFM
protocols: a retrospective analysis,” Lancet Haematol., vol. 3, no. 2, pp. e80–e86, Feb. 2016.
[95] I. Radtke, C. G. Mullighan, M. Ishii, X. Su, J. Cheng, J. Ma, R. Ganti, Z. Cai, S. Goorha, S. B. Pounds,
X. Cao, C. Obert, J. Armstrong, J. Zhang, G. Song, R. C. Ribeiro, J. E. Rubnitz, S. C. Raimondi, S. A.

BIBLIOGRAFIA
189
Shurtleff, and J. R. Downing, “Genomic analysis reveals few genetic alterations in pediatric acute
myeloid leukemia.,” Proc. Natl. Acad. Sci. U. S. A., vol. 106, no. 31, pp. 12944–9, Aug. 2009.
[96] E. R. Mardis, L. Ding, D. J. Dooling, D. E. Larson, M. D. McLellan, K. Chen, D. C. Koboldt, R. S.
Fulton, K. D. Delehaunty, S. D. McGrath, L. A. Fulton, D. P. Locke, V. J. Magrini, R. M. Abbott, T. L.
Vickery, J. S. Reed, J. S. Robinson, T. Wylie, S. M. Smith, L. Carmichael, J. M. Eldred, C. C. Harris, J.
Walker, J. B. Peck, F. Du, A. F. Dukes, G. E. Sanderson, A. M. Brummett, E. Clark, J. F. McMichael,
R. J. Meyer, J. K. Schindler, C. S. Pohl, J. W. Wallis, X. Shi, L. Lin, H. Schmidt, Y. Tang, C. Haipek, M.
E. Wiechert, J. V. Ivy, J. Kalicki, G. Elliott, R. E. Ries, J. E. Payton, P. Westervelt, M. H. Tomasson,
M. A. Watson, J. Baty, S. Heath, W. D. Shannon, R. Nagarajan, D. C. Link, M. J. Walter, T. A.
Graubert, J. F. DiPersio, R. K. Wilson, and T. J. Ley, “Recurring Mutations Found by Sequencing an
Acute Myeloid Leukemia Genome,” N. Engl. J. Med., vol. 361, no. 11, pp. 1058–1066, Sep. 2009.
[97] A. K. Andersson, D. W. Miller, J. A. Lynch, A. S. Lemoff, Z. Cai, S. B. Pounds, I. Radtke, B. Yan, J. D.
Schuetz, J. E. Rubnitz, R. C. Ribeiro, S. C. Raimondi, J. Zhang, C. G. Mullighan, S. A. Shurtleff, B. A.
Schulman, and J. R. Downing, “IDH1 and IDH2 mutations in pediatric acute leukemia,” Leukemia,
vol. 25, no. 10, pp. 1570–1577, Oct. 2011.
[98] B. V Balgobind, S. C. Raimondi, J. Harbott, M. Zimmermann, T. A. Alonzo, A. Auvrignon, H. B.
Beverloo, M. Chang, U. Creutzig, M. N. Dworzak, E. Forestier, B. Gibson, H. Hasle, C. J. Harrison,
N. A. Heerema, G. J. L. Kaspers, A. Leszl, N. Litvinko, L. Lo Nigro, A. Morimoto, C. Perot, R. Pieters,
D. Reinhardt, J. E. Rubnitz, F. O. Smith, J. Stary, I. Stasevich, S. Strehl, T. Taga, D. Tomizawa, D.
Webb, Z. Zemanova, C. M. Zwaan, M. M. van den Heuvel-Eibrink, G. Kaspers, C. Zwaan, D.
Grimwade, H. Walker, F. Oliver, S. Raimondi, M. Chang, Y. Ravindranath, S. Lie, J. Abrahamsson,
N. Clausen, A. von Bergh, B. Emanuel, S. van Zelderen-Bhola, C. Meyer, E. Kowarz, J. Hofmann, G.
Swansbury, R. Slater, B. Bain, A. Moorman, L. Secker-Walker, J. Palle, B. Frost, E. Forestier, J.
Rubnitz, S. Raimondi, X. Tong, C. Zwaan, G. Kaspers, R. Pieters, C. Pui, P. Gaynon, J. Boyett, R.
Pieters, M. Schrappe, P. De Lorenzo, A. Pession, R. Rondelli, G. Basso, U. Creutzig, M.
Zimmermann, T. Lehrnbecher, U. Creutzig, M. Zimmermann, J. Ritter, B. Lange, F. Smith, J.
Feusner, F. Smith, T. Alonzo, R. Gerbing, W. Woods, R. Arceci, Y. Perel, A.Auvrignon, T. Leblanc,
Y. Ravindranath, M. Chang, C. Steuber, R. Ribeiro, B. Razzouk, S. Pounds, N. Hijiya, C. Pui, J.
Rubnitz, B. Gibson, K. Wheatley, I. Hann, N. Katano, M. Tsurusawa, T. Hirota, G. Kaspers, U.
Creutzig, H. Hasle, T. Alonzo, A. Auvrignon, L. Shaffer, N. Tommerup, U. Creutzig, G. Kaspers, N.
Co, W. Tsang, T. Wong, W. Tse, S. Meshinchi, T. Alonzo, W. Blum, K. Mrozek, A. Ruppert, H.
Beverloo, M. Le Coniat, J. Wijsman, K. Mrozek, K. Heinonen, D. Lawrence, R. Aplenc, T. Alonzo, R.
Gerbing, J. Rubnitz, S. Lensing, B. Razzouk, S. Pounds, C. Pui, and R. Ribeiro, “Novel prognostic
subgroups in childhood 11q23/MLL-rearranged acute myeloid leukemia: results of an
international retrospective study.,” Blood, vol. 114, no. 12, pp. 2489–96, Sep. 2009.
[99] P. A. Ho, T. A. Alonzo, R. B. Gerbing, J. Pollard, D. L. Stirewalt, C. Hurwitz, N. A. Heerema, B.
Hirsch, S. C. Raimondi, B. Lange, J. L. Franklin, J. P. Radich, and S. Meshinchi, “Prevalence and
prognostic implications of CEBPA mutations in pediatric acute myeloid leukemia (AML): a report

BIBLIOGRAFIA
190
from the Children’s Oncology Group,” Blood, vol. 113, no. 26, 2009.
[100] I. H. I. M. Hollink, M. M. van den Heuvel-Eibrink, M. Zimmermann, B. V. Balgobind, S. T. C. J. M.
Arentsen-Peters, M. Alders, A. Willasch, G. J. L. Kaspers, J. Trka, A. Baruchel, S. S. N. de Graaf, U.
Creutzig, R. Pieters, D. Reinhardt, and C. M. Zwaan, “Clinical relevance of Wilms tumor 1 gene
mutations in childhood acute myeloid leukemia,” Blood, vol. 113, no. 23, 2009.
[101] C. J. Harrison, R. K. Hills, A. V Moorman, D. J. Grimwade, I. Hann, D. K. H. Webb, K. Wheatley, S.
S. N. de Graaf, E. van den Berg, A. K. Burnett, and B. E. S. Gibson, “Cytogenetics of childhood
acute myeloid leukemia: United Kingdom Medical Research Council Treatment trialsAML 10 and
12.,” J. Clin. Oncol., vol. 28, no. 16, pp. 2674–81, Jun. 2010.
[102] A. K. Andersson, D. W. Miller, J. A. Lynch, A. S. Lemoff, Z. Cai, S. B. Pounds, I. Radtke, B. Yan, J. D.
Schuetz, J. E. Rubnitz, R. C. Ribeiro, S. C. Raimondi, J. Zhang, C. G. Mullighan, S. A. Shurtleff, B. A.
Schulman, J. R. Downing, and J. R. Downing, “IDH1 and IDH2 mutations in pediatric acute
leukemia.,” Leukemia, vol. 25, no. 10, pp. 1570–7, Oct. 2011.
[103] A. Staffas, M. Kanduri, R. Hovland, R. Rosenquist, H. B. Ommen, J. Abrahamsson, E. Forestier, K.
Jahnukainen, Ó. G. Jónsson, B. Zeller, J. Palle, G. Lönnerholm, H. Hasle, L. Palmqvist, H.
Ehrencrona, and H. Nordic Society of Pediatric Hematology and Oncology (NOPHO), “Presence of
FLT3-ITD and high BAALC expression are independent prognostic markers in childhood acute
myeloid leukemia.,” Blood, vol. 118, no. 22, pp. 5905–13, Nov. 2011.
[104] J.-M. Ribera, A. Oriol, M.-A. Sanz, M. Tormo, P. Fernández-Abellán, E. del Potro, E. Abella, J.
Bueno, R. Parody, P. Bastida, C. Grande, I. Heras, C. Bethencourt, E. Feliu, J.-J. Ortega, and J.-J.
Ortega, “Comparison of the results of the treatment of adolescents and young adults with
standard-risk acute lymphoblastic leukemia with the Programa Español de Tratamiento en
Hematología pediatric-based protocol ALL-96.,” J. Clin. Oncol., vol. 26, no. 11, pp. 1843–9, Apr.
2008.
[105] N. Boissel, M.-F. Auclerc, V. Lhéritier, Y. Perel, X. Thomas, T. Leblanc, P. Rousselot, J.-M. Cayuela,
J. Gabert, N. Fegueux, C. Piguet, F. Huguet-Rigal, C. Berthou, J.-M. Boiron, C. Pautas, G. Michel,
D. Fière, G. Leverger, H. Dombret, and A. Baruchel, “Should adolescents with acute
lymphoblastic leukemia be treated as old children or young adults? Comparison of the French
FRALLE-93 and LALA-94 trials.,” J. Clin. Oncol., vol. 21, no. 5, pp. 774–80, Mar. 2003.
[106] J. C. Panetta, A. Gajjar, N. Hijiya, L. J. Hak, C. Cheng, W. Liu, C. H. Pui, and M. V Relling,
“Comparison of Native E. coli and PEG Asparaginase Pharmacokinetics and Pharmacodynamics
in Pediatric Acute Lymphoblastic Leukemia,” Clin. Pharmacol. Ther., vol. 86, no. 6, pp. 651–658,
Dec. 2009.
[107] L. R. Treviño, N. Shimasaki, W. Yang, J. C. Panetta, C. Cheng, D. Pei, D. Chan, A. Sparreboom, K.
M. Giacomini, C.-H. Pui, W. E. Evans, M. V Relling, and M. V Relling, “Germline genetic variation
in an organic anion transporter polypeptide associated with methotrexate pharmacokinetics and
clinical effects.,” J. Clin. Oncol., vol. 27, no. 35, pp. 5972–8, Dec. 2009.
[108] Y. Ge, M. L. Stout, D. A. Tatman, T. L. Jensen, S. Buck, R. L. Thomas, Y. Ravindranath, L. H.

BIBLIOGRAFIA
191
Matherly, and J. W. Taub, “GATA1, cytidine deaminase, and the high cure rate of Down
syndrome children with acute megakaryocytic leukemia.,” J. Natl. Cancer Inst., vol. 97, no. 3, pp.
226–31, Feb. 2005.
[109] D. Bhatla, R. B. Gerbing, T. A. Alonzo, H. Conner, J. A. Ross, S. Meshinchi, X. Zhai, T. Zamzow, P. A.
Mehta, H. Geiger, J. Perentesis, and S. M. Davies, “Cytidine deaminase genotype and toxicity of
cytosine arabinoside therapy in children with acute myeloid leukemia,” Br. J. Haematol., vol.
144, no. 3, pp. 388–394, Feb. 2009.
[110] R. Pieters, M. L. den Boer, M. Durian, G. Janka, K. Schmiegelow, G. J. Kaspers, E. R. van Wering,
and A. J. Veerman, “Relation between age, immunophenotype and in vitro drug resistance in
395 children with acute lymphoblastic leukemia--implications for treatment of infants.,”
Leukemia, vol. 12, no. 9, pp. 1344–8, Sep. 1998.
[111] P. Brown, M. Levis, E. McIntyre, M. Griesemer, and D. Small, “Combinations of the FLT3 inhibitor
CEP-701 and chemotherapy synergistically kill infant and childhood MLL-rearranged ALL cells in a
sequence-dependent manner.,” Leukemia , vol. 20, no. 8, pp. 1368–76, Aug. 2006.
[112] H. Xu, C. Cheng, M. Devidas, D. Pei, Y. Fan, W. Yang, G. Neale, P. Scheet, E. G. Burchard, D. G.
Torgerson, C. Eng, M. Dean, F. Antillon, N. J. Winick, P. L. Martin, C. L. Willman, B. M. Camitta, G.
H. Reaman, W. L. Carroll, M. Loh, W. E. Evans, C.-H. Pui, S. P. Hunger, M. V Relling, and J. J. Yang,
“ARID5B genetic polymorphisms contribute to racial disparities in the incidence and treatment
outcome of childhood acute lymphoblastic leukemia.,” J. Clin. Oncol., vol. 30, no. 7, pp. 751–7,
Mar. 2012.
[113] E. S. Schafer, and S. P. Hunger, “Optimal therapy for acute lymphoblastic leukemia in
adolescents and young adults.,” Nat. Rev. Clin. Oncol., vol. 8, no. 7, pp. 417–24, May 2011.
[114] M. L. Loh, M. A. Goldwasser, L. B. Silverman, W.-M. Poon, S. Vattikuti, A. Cardoso, D. S. Neuberg,
K. M. Shannon, S. E. Sallan, and D. G. Gilliland, “Prospective analysis of TEL/AML1-positive
patients treated on Dana-Farber Cancer Institute Consortium Protocol 95-01.,” Blood, vol. 107,
no. 11, pp. 4508–13, Jun. 2006.
[115] J. E. Rubnitz, D. Wichlan, M. Devidas, J. Shuster, S. B. Linda, J. Kurtzberg,B. Bell, S. P. Hunger, A.
Chauvenet, C.-H. Pui, B. Camitta, J. Pullen, and J. Children’s Oncology Group, “Prospective
analysis of TEL gene rearrangements in childhood acute lymphoblastic leukemia: a Children’s
Oncology Group study.,” J. Clin. Oncol., vol. 26, no. 13, pp. 2186–91, May 2008.
[116] K. Paulsson, E. Forestier, M. K. Andersen, K. Autio, G. Barbany, G. Borgström, L. Cavelier, I.
Golovleva, S. Heim, K. Heinonen, R. Hovland, J. H. Johannsson, E. Kjeldsen, A. Nordgren, L.
Palmqvist, and B. Johansson, “High modal number and triple trisomies are highly correlated
favorable factors in childhood B-cell precursor high hyperdiploid acute lymphoblastic leukemia
treated according to the NOPHO ALL 1992/2000 protocols,” Haematologica, vol. 98, no. 9, 2013.
[117] N. Dastugue, S. Suciu, G. Plat, F. Speleman, H. Cavé, S. Girard, M. Bakkus, M. P. Pagès, K.
Yakouben, B. Nelken, A. Uyttebroeck, C. Gervais, P. Lutz, M. R. Teixeira, P. Heimann, A. Ferster,
P. Rohrlich, M. A. Collonge, M. Munzer, I. Luquet, P. Boutard, N. Sirvent, M. Karrasch, Y.

BIBLIOGRAFIA
192
Bertrand, Y. Benoit, and Y. Benoit, “Hyperdiploidy with 58-66 chromosomes in childhood B-acute
lymphoblastic leukemia is highly curable: 58951 CLG-EORTC results.,” Blood, vol. 121, no. 13, pp.
2415–23, Mar. 2013.
[118] M. K. Andersen, K. Autio, G. Barbany, G. Borgström, L. Cavelier, I. Golovleva, S. Heim, K.
Heinonen, R. Hovland, J. H. Johannsson, B. Johansson, E. Kjeldsen, A. Nordgren, L. Palmqvist, E.
and E. Forestier, “Paediatric B-cell precursor acute lymphoblastic leukaemia with
t(1;19)(q23;p13): clinical and cytogenetic characteristics of 47 cases from the Nordic countries
treated according to NOPHO protocols.,” Br. J. Haematol., vol. 155, no. 2, pp. 235–43, Oct. 2011.
[119] L. Kager, T. Lion, A. Attarbaschi, M. Koenig, S. Strehl, O. A. Haas, M. N. Dworzak, M. Schrappe, H.
Gadner, and G. Mann, “Incidence and outcome of TCF3-PBX1-positive acute lymphoblastic
leukemia in Austrian children,” Haematologica, vol. 92, no. 11, 2007.
[120] C.-H. Pui, D. Campana, D. Pei, W. P. Bowman, J. T. Sandlund, S. C. Kaste, R. C. Ribeiro, J. E.
Rubnitz, S. C. Raimondi, M. Onciu, E. Coustan-Smith, L. E. Kun, S. Jeha, C. Cheng, S. C. Howard, V.
Simmons, A. Bayles, M. L. Metzger, J. M. Boyett, W. Leung, R. Handgretinger, J. R. Downing, W.
E. Evans, and M. V Relling, “Treating childhood acute lymphoblastic leukemia without cranial
irradiation.,” N. Engl. J. Med., vol. 360, no. 26, pp. 2730–41, Jun. 2009.
[121] C.-H. Pui, P. S. Gaynon, J. M. Boyett, J. M. Chessells, A. Baruchel, W. Kamps, L. B. Silverman, A.
Biondi, D. O. Harms, E. Vilmer, M. Schrappe,and B. Camitta, “Outcome of treatment in childhood
acute lymphoblastic leukaemia with rearrangements of the 11q23 chromosomal region.,” Lancet
(London, England), vol. 359, no. 9321, pp. 1909–15, Jun. 2002.
[122] C.-H. Pui, J. M. Chessells, B. Camitta, A. Baruchel, A. Biondi, J. M. Boyett, A. Carroll, O. B. Eden,
W. E. Evans, H. Gadner, J. Harbott, D. O. Harms, C. J. Harrison, P. L. Harrison, N. Heerema, G.
Janka-Schaub, W. Kamps, G. Masera, J. Pullen, S. C. Raimondi, S. Richards, H. Riehm, S. Sallan, H.
Sather, J. Shuster, L. B. Silverman, M. G. Valsecchi, E. Vilmer, Y. Zhou, P. S. Gaynon, and M.
Schrappe, “Clinical heterogeneity in childhood acute lymphoblastic leukemia with 11q23
rearrangements.,” Leukemia, vol. 17, no. 4, pp. 700–6, Apr. 2003.
[123] A. V Moorman, H. Robinson, C. Schwab, S. M. Richards, J. Hancock, C. D. Mitchell, N. Goulden, A.
Vora, and C. J. Harrison, “Risk-directed treatment intensification significantly reduces the risk of
relapse among children and adolescents with acute lymphoblastic leukemia and
intrachromosomal amplification of chromosome 21: a comparison of the MRC ALL97/99 and
UKALL2003 trials.,” J. Clin. Oncol., vol. 31, no. 27, pp. 3389–96, Sep. 2013.
[124] N. A. Heerema, A. J. Carroll, M. Devidas, M. L. Loh, M. J. Borowitz, J. M. Gastier-Foster, E. C.
Larsen, L. A. Mattano, K. W. Maloney, C. L. Willman, B. L. Wood, N. J. Winick, W. L. Carroll, S. P.
Hunger, and E. A. Raetz, “Intrachromosomal amplification of chromosome 21 is associated with
inferior outcomes in children with acute lymphoblastic leukemia treated in contemporary
standard-risk children’s oncology group studies: a report from the children’s oncology group.,” J.
Clin. Oncol., vol. 31, no. 27, pp. 3397–402, Sep. 2013.
[125] A. V Moorman, H. M. Ensor, S. M. Richards, L. Chilton, C. Schwab, S. E. Kinsey, A. Vora, C. D.

BIBLIOGRAFIA
193
Mitchell, C. J. Harrison, and C. J. Harrison, “Prognostic effect of chromosomal abnormalities in
childhood B-cell precursor acute lymphoblastic leukaemia: results from the UK Medical Research
Council ALL97/99 randomised trial.,” Lancet. Oncol., vol. 11, no. 5, pp. 429–38, May 2010.
[126] A. V Moorman, “The clinical relevance of chromosomal and genomic abnormalities in B-cell
precursor acute lymphoblastic leukaemia.,” Blood Rev., vol. 26, no. 3, pp. 123–35, May 2012.
[127] D. Campana, “Minimal residual disease monitoring in childhood acute lymphoblastic leukemia.,”
Curr. Opin. Hematol., vol. 19, no. 4, pp. 313–8, Jul. 2012.
[128] V. Conter, C. R. Bartram, M. G. Valsecchi, A. Schrauder, R. Panzer-Grümayer, A. Möricke, M.
Aricò, M. Zimmermann, G. Mann, G. De Rossi, M. Stanulla, F. Locatelli, G. Basso, F. Niggli, E.
Barisone, G. Henze, W.-D. Ludwig, O. A. Haas, G. Cazzaniga, R. Koehler, D. Silvestri, J. Bradtke, R.
Parasole, R. Beier, J. J. M. van Dongen, A. Biondi, , and M. Schrappe, “Molecular response to
treatment redefines all prognostic factors in children and adolescents with B-cell precursor
acute lymphoblastic leukemia: results in 3184 patients of the AIEOP-BFM ALL 2000 study.,”
Blood, vol. 115, no. 16, pp. 3206–14, Apr. 2010.
[129] M. Schrappe, M. G. Valsecchi, C. R. Bartram, A. Schrauder, R. Panzer-Grümayer, A. Möricke, R.
Parasole, M. Zimmermann, M. Dworzak, B. Buldini, A. Reiter, G. Basso, T. Klingebiel,C. Messina,
R. Ratei, G. Cazzaniga, R. Koehler, F. Locatelli, B. W. Schäfer, M. Aricò, K. Welte, J. J. M. van
Dongen, H. Gadner, A. Biondi, and V. Conter, “Late MRD response determines relapse risk
overall and in subsets of childhood T-cell ALL: results of the AIEOP-BFM-ALL 2000 study.,” Blood,
vol. 118, no. 8, pp. 2077–84, Aug. 2011.
[130] G. Basso, M. Veltroni, M. G. Valsecchi, M. N. Dworzak, R. Ratei, D. Silvestri, A. Benetello, B.
Buldini, O. Maglia, G. Masera, V. Conter, M. Arico, A. Biondi, and G. Gaipa, “Risk of relapse of
childhood acute lymphoblastic leukemia is predicted by flow cytometric measurement of
residual disease on day 15 bone marrow.,” J. Clin. Oncol., vol. 27, no. 31, pp. 5168–74, Nov.
2009.
[131] D. Grimwade and R. K. Hills, “Independent prognostic factors for AML outcome.,” Hematol. Am.
Soc. Hematol. Educ. Progr., vol. 2009, no. 1, pp. 385–95, 2009.
[132] D. Grimwade, H. Walker, F. Oliver, K. Wheatley, C. Harrison, G. Harrison, J. Rees, I. Hann, R.
Stevens, A. Burnett, and A. Goldstone, “The Importance of Diagnostic Cytogenetics on Outcome
in AML: Analysis of 1,612 Patients Entered Into the MRC AML 10 Trial,” Blood, vol. 92, no. 7,
1998.
[133] C. von Neuhoff, D. Reinhardt, A. Sander, M. Zimmermann, J. Bradtke, D. R. Betts, Z. Zemanova, J.
Stary, J.-P. Bourquin, O. A. Haas, M. N. Dworzak, and U. Creutzig, “Prognostic impact of specific
chromosomal aberrations in a large group of pediatric patients with acute myeloid leukemia
treated uniformly according to trial AML-BFM 98.,” J. Clin. Oncol., vol. 28, no. 16, pp. 2682–9,
Jun. 2010.
[134] H. Hasle, T. A. Alonzo, A. Auvrignon, C. Behar, M. Chang, U. Creutzig, A. Fischer, E. Forestier, A.
Fynn, O. A. Haas, J. Harbott, C. J. Harrison, N. A. Heerema, M. M. van den Heuvel-Eibrink, G. J. L.

BIBLIOGRAFIA
194
Kaspers, F. Locatelli, P. Noellke, S. Polychronopoulou, Y. Ravindranath, B. Razzouk, D. Reinhardt,
N. N. Savva, B. Stark, S. Suciu, I. Tsukimoto, D. K. Webb, D. Wojcik, W. G. Woods, M.
Zimmermann, C. M. Niemeyer, and S. C. Raimondi, “Monosomy 7 and deletion 7q in children
and adolescents with acute myeloid leukemia: an international retrospective study.,” Blood, vol.
109, no. 11, pp. 4641–7, Jun. 2007.
[135] S. Meshinchi, T. A. Alonzo, D. L. Stirewalt, M. Zwaan, M. Zimmerman, D. Reinhardt, G. J. L.
Kaspers, N. A. Heerema, R. Gerbing, B. J. Lange, and J. P. Radich, “Clinical implications of FLT3
mutations in pediatric AML,” Blood, vol. 108, no. 12, 2006.
[136] M. Pratcorona, S. Brunet, J. Nomded, J. M. Ribera, M. Tormo, R. Duarte, L. Escoda, and
R. Guàrdia R, Queipo de Llano MP, Salamero O, Bargay J, Pedro C, Martí JM, Torrebadell
M, Díaz-Beyá M, Camós M, Colomer D, Hoyos M, Sierra J, Esteve J; Grupo Cooperativo Para el
Estudio y Tratamiento de las Leucemias Agudas Mieloblásticas. “Favorable outcome of patients
with acute myeloid leukemia harboring a low-allelic burden FLT3 -ITD mutation and concomitant
NPM1 mutation : relevance to post-remission therapy,” Blood, vol. 121, no. 14, pp. 2734–2739,
2013.
[137] H. Hasle, “A critical review of which children with acute myeloid leukaemia need stem cell
procedures,” Br. J. Haematol., vol. 166, no. 1, pp. 23–33, Jul. 2014.
[138] T. Haferlach, A. Kohlmann, H.-U. Klein, C. Ruckert, M. Dugas, P. M. Williams, W. Kern, S.
Schnittger, U. Bacher, H. Löffler, and C. Haferlach, “AML with translocation t(8;16)(p11;p13)
demonstrates unique cytomorphological, cytogenetic, molecular and prognostic features.,”
Leukemia, vol. 23, no. 5, pp. 934–43, May 2009.
[139] I. J. Park, J. E. Park, H. J. Kim, H. J. Jung, W. G. Lee, and S. R. Cho, “Acute myeloid leukemia with
t(16;21)(q24;q22) and eosinophilia: case report and review of the literature.,” Cancer Genet.
Cytogenet., vol. 196, no. 1, pp. 105–8, Jan. 2010.
[140] M. L. Slovak, H. Gundacker, C. D. Bloomfield, G. Dewald, F. R. Appelbaum, R. A. Larson, M. S.
Tallman, J. M. Bennett, D. L. Stirewalt, S. Meshinchi, C. L. Willman, Y. Ravindranath, T. A. Alonzo,
A. J. Carroll, S. C. Raimondi, and N. A. Heerema, “A retrospective study of 69 patients with
t(6;9)(p23;q34) AML emphasizes the need for a prospective, multicenter initiative for rare ‘poor
prognosis’ myeloid malignancies.,” Leukemia, vol. 20, no. 7, pp. 1295–7, Jul. 2006.
[141] B. V. Balgobind, S. C. Raimondi, J. Harbott, M. Zimmermann, T. A. Alonzo, A. Auvrignon, H. B.
Beverloo, M. Chang, U. Creutzig, M. N. Dworzak, E. Forestier, B. Gibson, H. Hasle, C. J. Harrison,
N. A. Heerema, G. J. L. Kaspers, A. Leszl, N. Litvinko, L. Lo Nigro, A. Morimoto, C. Perot, R. Pieters,
D. Reinhardt, J. E. Rubnitz, F. O. Smith, J. Stary, I. Stasevich, S. Strehl, T.Taga, D. Tomizawa, D.
Webb, Z. Zemanova, C. M. Zwaan, and M. M. van den Heuvel-Eibrink, “Novel prognostic
subgroups in childhood 11q23/MLL-rearranged acute myeloid leukemia: results of an
international retrospective study,” Blood, vol. 114, no. 12, 2009.
[142] P. A. Ho, R. Zeng, T. A. Alonzo, R. B. Gerbing, K. L. Miller, J. A. Pollard, D. L. Stirewalt, N. A.
Heerema, S. C. Raimondi, B. Hirsch, J. L. Franklin, B. Lange, S. Meshinchi, and S. Meshinchi,

BIBLIOGRAFIA
195
“Prevalence and prognostic implications of WT1 mutations in pediatric acute myeloid leukemia
(AML): a report from the Children’s Oncology Group.,” Blood, vol. 116, no. 5, pp. 702–10, Aug.
2010.
[143] G. Marcucci, T. Haferlach, and H. Döhner, “Molecular genetics of adult acute myeloid leukemia:
prognostic and therapeutic implications.,” J. Clin. Oncol., vol. 29, no. 5, pp. 475–86, Feb. 2011.
[144] I. H. I. M. Hollink, M. M. van den Heuvel-Eibrink, S. T. C. J. M. Arentsen-Peters, M. Pratcorona, S.
Abbas, J. E. Kuipers, J. F. van Galen, H. B. Beverloo, E. Sonneveld, G.-J. J. L. Kaspers, J. Trka, A.
Baruchel, M. Zimmermann, U. Creutzig, D. Reinhardt, R. Pieters, P. J. M. Valk, and C. M. Zwaan,
“NUP98/NSD1 characterizes a novel poor prognostic group in acute myeloid leukemia with a
distinct HOX gene expression pattern.,” Blood, vol. 118, no. 13, pp. 3645–56, Sep. 2011.
[145] F. Buccisano, L. Maurillo, M. I. Del Principe, G. Del Poeta, G. Sconocchia, F. Lo-Coco, W. Arcese, S.
Amadori, and A. Venditti, “Prognostic and therapeutic implications of minimal residual disease
detection in acute myeloid leukemia.,” Blood, vol. 119, no. 2, pp. 332–41, Jan. 2012.
[146] V. H. J. van der Velden, A. van der Sluijs-Geling, B. E. S. Gibson, J. G. te Marvelde, P. G.
Hoogeveen, W. C. J. Hop, K. Wheatley, M. B. Bierings, G. J. Schuurhuis,S. S. N. de Graaf, E. R. van
Wering, and J. J. M. van Dongen, “Clinical significance of flowcytometric minimal residual disease
detection in pediatric acute myeloid leukemia patients treated according to the DCOG
ANLL97/MRC AML12 protocol.,” Leukemia, vol. 24, no. 9, pp. 1599–606, Sep. 2010.
[147] J. E. Rubnitz, H. Inaba, G. Dahl, R. C. Ribeiro, W. P. Bowman, J. Taub, S. Pounds, B. I. Razzouk, N.
J. Lacayo, X. Cao, S. Meshinchi, B. Degar, G. Airewele, S. C. Raimondi, M. Onciu, E. Coustan-
Smith, J. R. Downing, W. Leung, C.-H. Pui, and D. Campana, “Minimal residual disease-directed
therapy for childhood acute myeloid leukaemia: results of the AML02 multicentre trial,” Lancet
Oncol., vol. 11, no. 6, pp. 543–552, 2010.
[148] M. R. Loken, T. A. Alonzo, L. Pardo, R. B. Gerbing, S. C. Raimondi, B. A. Hirsch, P. A. Ho, J.
Franklin, T. M. Cooper, A. S. Gamis, and S. Meshinchi, “Residual disease detected by
multidimensional flow cytometry signifies high relapse risk in patients with de novo acute
myeloid leukemia: a report from Children’s Oncology Group.,” Blood, vol. 120, no. 8, pp. 1581–8,
Aug. 2012.
[149] C.-H. Pui, J. J. Yang, S. P. Hunger, R. Pieters, M. Schrappe, A. Biondi, A. Vora, A. Baruchel, L. B.
Silverman, K. Schmiegelow, G. Escherich, K. Horibe, Y. C. M. Benoit, S. Izraeli, A. E. J. Yeoh, D.-C.
Liang, J. R. Downing, W. E. Evans, M. V Relling, and C. G. Mullighan, “Childhood Acute
Lymphoblastic Leukemia: Progress Through Collaboration.,” J. Clin. Oncol., vol. 33, no. 27, pp.
2938–48, Sep. 2015.
[150] C. Pui and W. E. Evans, “Treatment of Acute Lymphoblastic Leukemia,” 2006.
[151] C.-H. Pui, Childhood leukemias, 2nd ed. Cambridge;New York: Cambridge University Press, 2006.
[152] M. Schrappe, S. P. Hunger, C.-H. Pui, V. Saha, P. S. Gaynon, A. Baruchel, V. Conter, J. Otten, A.
Ohara, A. B. Versluys, G. Escherich, M. Heyman, L. B. Silverman, K. Horibe, G. Mann, B. M.
Camitta, J. Harbott, H. Riehm, S. Richards, M. Devidas, and M. Zimmermann, “Outcomes after

BIBLIOGRAFIA
196
Induction Failure in Childhood Acute Lymphoblastic Leukemia,” N. Engl. J. Med., vol. 366, no. 15,
pp. 1371–1381, Apr. 2012.
[153] Childhood ALL Collaborative Group, “Duration and intensity of maintenance chemotherapy in
acute lymphoblastic leukaemia: overview of 42 trials involving 12 000 randomised children.,”
Lancet (London, England), vol. 347, no. 9018, pp. 1783–8, Jun. 1996.
[154] U. Creutzig, M. Zimmermann, T. Lehrnbecher, N. Graf, J. Hermann, C. M. Niemeyer, A. Reiter, J.
Ritter, M. Dworzak, J. Stary, and D. Reinhardt, “Less toxicity by optimizing chemotherapy, but
not by addition of granulocyte colony-stimulating factor in children and adolescents with acute
myeloid leukemia: results of AML-BFM 98.,” J. Clin. Oncol., vol. 24, no. 27, pp. 4499–506, Sep.
2006.
[155] I. Tsukimoto, A. Tawa, K. Horibe, K. Tabuchi, H. Kigasawa, M. Tsuchida, H. Yabe, H. Nakayama, K.
Kudo, R. Kobayashi, K. Hamamoto, M. Imaizumi, A. Morimoto, S. Tsuchiya, and R. Hanada, “Risk-
stratified therapy and the intensive use of cytarabine improves the outcome in childhood acute
myeloid leukemia: the AML99 trial from the Japanese Childhood AML Cooperative Study
Group.,” J. Clin. Oncol., vol. 27, no. 24, pp. 4007–13, Aug. 2009.
[156] J. Abrahamsson, E. Forestier, J. Heldrup, K. Jahnukainen, O. G. Jónsson, B. Lausen, J. Palle, B.
Zeller, and H. Hasle, “Response-guided induction therapy in pediatric acute myeloid leukemia
with excellent remission rate.,” J. Clin. Oncol., vol. 29, no. 3, pp. 310–5, Jan. 2011.
[157] J. E. Rubnitz, H. Inaba, G. Dahl, R. C. Ribeiro, W. P. Bowman, J. Taub, S. Pounds, B. I. Razzouk, N.
J. Lacayo, X. Cao, S. Meshinchi, B. Degar, G. Airewele, S. C. Raimondi, M. Onciu, E. Coustan-
Smith, J. R. Downing, W. Leung, C.-H. Pui, and D. Campana, “Minimal residual disease-directed
therapy for childhood acute myeloid leukaemia: resultsof the AML02 multicentre trial.,” Lancet.
Oncol., vol. 11, no. 6, pp. 543–52, Jun. 2010.
[158] C. M. Zwaan, E. A. Kolb, D. Reinhardt, J. Abrahamsson, S. Adachi, R. Aplenc, E. S. J. M. De Bont, B.
De Moerloose, M. Dworzak, B. E. S. Gibson, H. Hasle, G. Leverger, F. Locatelli, C. Ragu, R. C.
Ribeiro, C. Rizzari, J. E. Rubnitz, O. P. Smith, L. Sung, D. Tomizawa, M. M. Van Den Heuvel-eibrink,
U. Creutzig, and G. J. L. Kaspers, “Collaborative Efforts Driving Progress in Pediatric Acute
Myeloid Leukemia,” Journal of Clinical Oncology, vol. 33, no. 27, 2015.
[159] G. J. L. Kaspers, M. Zimmermann, D. Reinhardt, B. E. S. Gibson, R. Y. J. Tamminga, O. Aleinikova,
H. Armendariz, M. Dworzak, S.-Y. Ha, H. Hasle, L. Hovi, A. Maschan, Y. Bertrand, G. G. Leverger,
B. I. Razzouk, C. Rizzari, P. Smisek, O. Smith, B. Stark, and U. Creutzig, “Improved outcome in
pediatric relapsed acute myeloid leukemia: results of a randomized trial on liposomal
daunorubicin by the International BFM Study Group.,” J. Clin. Oncol., vol. 31, no. 5, pp. 599–607,
Feb. 2013.
[160] J. Esteve, L. Escoda, G. Martín, V. Rubio, J. Díaz-Mediavilla, M. González, C. Rivas, C. Álvarez, J. D.
González San Miguel, S. Brunet, J. F. Tomás, M. Tormo, M. J. Sayas, P. Sánchez Godoy, D.
Colomer, P. Bolufer, and M. A. Sanz, “Outcome of patients with acute promyelocytic leukemia
failing to front-line treatment with all-trans retinoic acid and anthracycline-based chemotherapy

BIBLIOGRAFIA
197
(PETHEMA protocols LPA96 and LPA99): benefit of an early intervention,” Leukemia, vol. 21, no.
3, pp. 446–452, Mar. 2007.
[161] Y. Perel, Auvrignon A, Leblanc T, Vannier JP, Michel G, Nelken B, Gandemer V, Schmitt
C, Lamagnere JP, De Lumley L, Bader-Meunier B, Couillaud G, Schaison G, Landman-Parker
J, Thuret I, Dalle JH, Baruchel A, Leverger G; Group LAME of the French Society of Pediatric
Hematology and Immunology “Impact of Addition of Maintenance Therapy to Intensive
Induction and Consolidation Chemotherapy for Childhood Acute Myeloblastic Leukemia: Results
of a Prospective Randomized Trial, LAME 89/91,” J. Clin. Oncol., vol. 20, no. 12, pp. 2774–2782,
Jun. 2002.
[162] J. Yates, O. Glidewell, P. Wiernik, M. Cooper, D. Steinberg, H. Dosik, R. Levy, C. Hoagland, P.
Henry, A. Gottlieb, C. Cornell, J. Berenberg, J. Hutchison, P. Raich, N. Nissen, R. Ellison, R. Frelick,
G. James, G. Falkson, R. Silver, F. Haurani, M. Green, E. Henderson, L. Leone, and J. Holland,
“Cytosine arabinoside with daunorubicin or adriamycin for therapy of acute myelocytic
leukemia: a CALGB study,” Blood, vol. 60, no. 2, 1982.
[163] I. M. Hann, R. F. Stevens, A. H. Goldstone, J. K. H. Rees, K. Wheatley, R. G. Gray, and A. K.
Burnett, “Randomized Comparison of DAT Versus ADE as Induction Chemotherapy in Children
and Younger Adults With Acute Myeloid Leukemia. Results of the Medical Research Council’s
10th AML Trial (MRC AML10),” Blood, vol. 89, no. 7, 1997.
[164] J. E. Rubnitz, K. R. Crews, S. Pounds, S. Yang, D. Campana, V. V Gandhi, S. C. Raimondi, J. R.
Downing, B. I. Razzouk, C.-H. Pui, and R. C. Ribeiro, “Combination of cladribine and cytarabine is
effective for childhood acute myeloid leukemia: results of the St Jude AML97 trial,” Leukemia,
vol. 23, no. 8, pp. 1410–1416, Aug. 2009.
[165] B. J. Lange, F. O. Smith, J. Feusner, D. R. Barnard, P. Dinndorf, S. Feig, N. A. Heerema, C. Arndt, R.
J. Arceci, N. Seibel, M. Weiman, K. Dusenbery, K. Shannon, S. Luna-Fineman, R. B. Gerbing, and
T. A. Alonzo, “Outcomes in CCG-2961, a Children’s Oncology Group Phase 3 Trial for untreated
pediatric acute myeloid leukemia: a report from the Children’s Oncology Group,” Blood, vol. 111,
no. 3, 2008.
[166] T. M. Cooper, J. Franklin, R. B. Gerbing, T. A. Alonzo, C. Hurwitz, S. C. Raimondi, B. Hirsch, F. O.
Smith, P. Mathew, R. J. Arceci, J. Feusner, R. Iannone, R. S. Lavey, S. Meshinchi, A. Gamis, and A.
Gamis, “AAML03P1, a pilot study of the safety of gemtuzumab ozogamicin in combination with
chemotherapy for newly diagnosed childhood acute myeloid leukemia: a reportfrom the
Children’s Oncology Group.,” Cancer, vol. 118, no. 3, pp. 761–9, Feb. 2012.
[167] B. Löwenberg, G. J. Ossenkoppele, W. van Putten, H. C. Schouten, C. Graux, A. Ferrant, P.
Sonneveld, J. Maertens, M. Jongen-Lavrencic, M. von Lilienfeld-Toal, B. J. Biemond, E. Vellenga,
M. van M. Kooy, L. F. Verdonck, J. Beck, H. Döhner, A. Gratwohl, T. Pabst, and G. Verhoef, “High-
Dose Daunorubicin in Older Patients with Acute Myeloid Leukemia,” N. Engl. J. Med., vol. 361,
no. 13, pp. 1235–1248, Sep. 2009.
[168] S. Ohtake, S. Miyawaki, H. Fujita, H. Kiyoi, K. Shinagawa, N. Usui, H. Okumura, K. Miyamura, C.

BIBLIOGRAFIA
198
Nakaseko, Y. Miyazaki, A. Fujieda, T. Nagai, T. Yamane, M. Taniwaki, M. Takahashi, F. Yagasaki, Y.
Kimura, N. Asou, H. Sakamaki, H. Handa, S. Honda, K. Ohnishi, T. Naoe, and R. Ohno,
“Randomized study of induction therapy comparing standard-dose idarubicin with high-dose
daunorubicin in adult patients with previously untreated acute myeloid leukemia: the JALSG
AML201 Study,” Blood, vol. 117, no. 8, 2011.
[169] R. J. Wells, W. G. Woods, B. C. Lampkin, M. E. Nesbit, J. W. Lee, J. D. Buckley, C. Versteeg, and G.
D. Hammond, “Impact of high-dose cytarabine and asparaginase intensification on childhood
acute myeloid leukemia: a report from the Childrens Cancer Group.,” J. Clin. Oncol., vol. 11, no.
3, pp. 538–45, Mar. 1993.
[170] U. Creutzig, J. Ritter, M. Zimmermann, D. Reinhardt, J. Hermann, F. Berthold, G. Henze, H.
Jürgens, H. Kabisch, W. Havers, A. Reiter, U. Kluba, F. Niggli, and H. Gadner, “Improved
treatment results in high-risk pediatric acute myeloid leukemia patients after intensification with
high-dose cytarabine and mitoxantrone: results of Study Acute Myeloid Leukemia-Berlin-
Frankfurt-Münster 93.,” J. Clin. Oncol., vol. 19, no. 10, pp. 2705–13, May 2001.
[171] Y. Ravindranath, C. P. Steuber, J. Krischer, C. I. Civin, J. Ducore, R. Vega, P. Pitel, S. Inoue, E.
Bleher, and C. Sexauer, “High-dose cytarabine for intensification of early therapy of childhood
acute myeloid leukemia: a Pediatric Oncology Group study.,” J. Clin. Oncol., vol. 9, no. 4, pp.
572–80, Apr. 1991.
[172] R. F. Stevens, I. M. Hann, K. Wheatley, and R. G. Gray, “Marked improvements in outcome with
chemotherapy alone in paediatric acute myeloid leukaemia: results of the United Kingdom
Medical Research Council’s 10th AML trial,” Br. J. Haematol., vol. 101, no. 1, pp. 130–140, Apr.
1998.
[173] Y. Perel, A. Auvrignon, T. Leblanc, J.-P. Vannier, G. Michel, B. Nelken, V. Gandemer, C. Schmitt, J.-
P. Lamagnere, L. De Lumley, B. Bader-Meunier, G. Couillaud, G. Schaison, J. Landman-Parker, I.
Thuret, J.-H. Dalle, A. Baruchel, G. Leverger, and G. Group LAME of the French Society of
Pediatric Hematology and Immunology, “Impact of addition of maintenance therapy to intensive
induction and consolidation chemotherapy for childhood acute myeloblastic leukemia: results of
a prospective randomized trial, LAME 89/91. Leucámie Aiqüe Myéloïde Enfant.,” J. Clin. Oncol.,
vol. 20, no. 12, pp. 2774–82, Jun. 2002.
[174] B. E. S. Gibson, D. K. H. Webb, A. J. Howman, S. S. N.De Graaf, C. J. Harrison, and K. Wheatley,
“Results of a randomized trial in children with Acute Myeloid Leukaemia: Medical Research
Council AML12 trial,” Br. J. Haematol., vol. 155, no. 3, pp. 366–376, Nov. 2011.
[175] T. Lehrnbecher, M. Zimmermann, D. Reinhardt, M. Dworzak, J. Stary, and U. Creutzig,
“Prophylactic human granulocyte colony-stimulating factor after induction therapy in pediatric
acute myeloid leukemia,” Blood, vol. 109, no. 3, 2007.
[176] D. Niewerth, U. Creutzig, M. B. Bierings, and G. J.L. Kaspers, “A review on allogeneic stem cell
transplantation for newly diagnosed pediatric acute myeloid leukemia,” Blood, vol. 116, no. 13,
2010.

BIBLIOGRAFIA
199
[177] J. T. Horan, T. A. Alonzo, G. H. Lyman, R. B. Gerbing, B. J. Lange, Y. Ravindranath, D. Becton, F. O.
Smith, W. G. Woods, and Children’s Oncology Group, “Impact of disease risk on efficacy of
matched related bone marrow transplantation for pediatric acute myeloid leukemia: the
Children’s Oncology Group.,” J. Clin. Oncol., vol. 26, no. 35, pp. 5797–801, Dec. 2008.
[178] T. A. Gooley, J. W. Chien, S. A. Pergam, S. Hingorani, M. L. Sorror, M. Boeckh, P. J. Martin, B. M.
Sandmaier, K. A. Marr, F. R. Appelbaum, R. Storb, and G. B. McDonald, “Reduced Mortality after
Allogeneic Hematopoietic-Cell Transplantation,” N. Engl. J. Med., vol. 363, no. 22, pp. 2091–
2101, Nov. 2010.
[179] J. T. Horan, B. R. Logan, M.-A. Agovi-Johnson, H. M. Lazarus, A. A. Bacigalupo, K. K. Ballen, C. N.
Bredeson, M. H. Carabasi, V. Gupta, G. A. Hale, H. J. Khoury, M. B. Juckett, M. R. Litzow, R.
Martino, P. L. McCarthy, F. O. Smith, J. D. Rizzo, and M. C. Pasquini, “Reducing the risk for
transplantation-related mortality after allogeneic hematopoietic cell transplantation: how much
progress has been made?,” J. Clin. Oncol., vol. 29, no. 7, pp. 805–13, Mar. 2011.
[180] U. Creutzig and D. Reinhardt, “current controversies: which patients with acute myeloid
leukaemia should receive a bone marrow transplantation? - A European view,” Br. J. Haematol.,
vol. 118, no. 2, pp. 365–377, Aug. 2002.
[181] C. M. Zwaan, D. Reinhardt, M. Zimmerman, H. Hasle, J. Stary, B. Stark, M. Dworzak, U. Creutzig,
and G. J. L. Kaspers, “Salvage treatment for children with refractory first or second relapse of
acute myeloid leukaemia with gemtuzumab ozogamicin: results of a phase II study,” Br. J.
Haematol., vol. 148, no. 5, pp. 768–776, Mar. 2010.
[182] J. D. E. de Rooij, R. Masetti, M. M. van den Heuvel-Eibrink, J.-M. Cayuela, J. Trka, D. Reinhardt,
M. Rasche, E. Sonneveld, T. A. Alonzo, M. Fornerod, M. Zimmermann, M. Pigazzi, R. Pieters, S.
Meshinchi, C. M. Zwaan, F. Locatelli, A. C. of the I. B. S. Group, C. Pui, W. Carroll, S. Meshinchi, R.
Arceci, A. Xavier, Y. Ge, J. Taub, U. Athale, B. Razzouk, S. Raimondi, J. de Rooij, I. Hollink, S.
Arentsen-Peters, J. Schweitzer, M. Zimmermann, M. Rasche, C. Thiollier, C. Lopez, B. Gerby, A.
Hama, H. Yagasaki, Y. Takahashi, H. Hasle, E. S. for B. and M. T. EBMT, H. Inaba, Y. Zhou, O. Abla,
A. Baruchel, M. Daniel, G. Schaison, R. Berger, A. Carroll, C. Civin, N. Schneider, T. Mercher, M.
B.-L. Coniat, F. N. Khac, M. O’Brien, X. Cao, S. Pounds, T. Gruber, A. L. Gedman, J. Zhang, R.
Masetti, M. Pigazzi, M. Togni, B. Balgobind, C. Zwaan, R. Pieters, M. Van den Heuvel-Eibrink, M.
Pigazzi, R. Masetti, S. Bresolin, T. Mercher, G. Courtois, R. Berger, O. Bernard, J. Hitzler, J.
Cheung, Y. Li, S. Scherer, A. Zipursky, I. Magalhães, A. Splendore, M. Emerenciano, A. Figueiredo,
I. Ferrari, M. Pombo-de-Oliveira, K. Reinhardt, C. Zwaan, M. Dworzak, H. Hasle, T. Alonzo, A.
Auvrignon, C. Harrison, R. Hills, A. Moorman, L. Chilton, R. Hills, C. Harrison, A. Burnett, D.
Grimwade, A. Moorman, J. Sandahl, E. Kjeldsen, J. Abrahamsson, C. von Neuhoff, D. Reinhardt,
A. Sander, F. de C. Hematologique, F. Stölzel, B. Mohr, M. Kramer, M. Bastos-Oreiro, A. Perez-
Corral, C. Martínez-Laperche, T. Taga, A. Saito, K. Kudo, J. Crispino, J. Wechsler, M. Greene, M.
McDevitt, C. Zwaan, G. Kaspers, R. Pieters, K. Yoshida, T. Toki, Y. Okuno, T. Gruber, J. Downing, R.
Ono, D. Hasegawa, S. Hirabayashi, D. Reinhardt, K. Reinhardt, and C. Neuhoff, “Recurrent

BIBLIOGRAFIA
200
abnormalities can be used for risk group stratification in pediatric AMKL: a retrospective
intergroup study.,” Blood, vol. 127, no. 26, pp. 3424–30, Jun. 2016.
[183] R. J. Brentjens, M. L. Davila, I. Riviere, J. Park, X. Wang, L. G. Cowell, S. Bartido, J. Stefanski, C.
Taylor, M. Olszewska, O. Borquez-Ojeda, J. Qu, T. Wasielewska, Q. He, Y. Bernal, I. V Rijo, C.
Hedvat, R. Kobos, K. Curran, P. Steinherz, J. Jurcic, T. Rosenblat, P. Maslak, M. Frattini, M.
Sadelain, and M. Sadelain, “CD19-targeted T cells rapidly induce molecular remissions in adults
with chemotherapy-refractory acute lymphoblastic leukemia.,” Sci. Transl. Med., vol. 5, no. 177,
p. 177ra38, Mar. 2013.
[184] S. A. Grupp, M. Kalos, D. Barrett, R. Aplenc, D.L. Porter, S. R. Rheingold, D. T. Teachey, A. Chew,
B. Hauck, J. F. Wright, M. C. Milone, B. L. Levine, and C. H. June, “Chimeric antigen receptor-
modified T cells for acute lymphoid leukemia.,” N. Engl. J. Med., vol. 368, no. 16, pp. 1509–18,
Apr. 2013.
[185] S. L. Maude, N. Frey, P. A. Shaw, R. Aplenc, D. M. Barrett, N. J. Bunin, A. Chew, V. E. Gonzalez, Z.
Zheng, S. F. Lacey, Y. D. Mahnke, J. J. Melenhorst, S. R. Rheingold, A. Shen, D. T. Teachey, B. L.
Levine, C. H. June, D. L. Porter, S. A. Grupp, andS. A. Grupp, “Chimeric antigen receptor T cells
for sustained remissions in leukemia.,” N. Engl. J. Med., vol. 371, no. 16, pp. 1507–17, Oct. 2014.
[186] E. M. Stein, M. S. Tallman, H. P.-L. G. P. Consortium, A. Burnett, M. Wetzler, B. Löwenberg, C.
Bloomfield, D. Lawrence, J. Byrd, J. Rowe, F. Appelbaum, K. Kopecky, M. Tallman, H. Kantarjian,
X. Thomas, A. Dmoszynska, H. Dombret, J. Seymour, A. Butrym, E. Estey, R. Levine, B.
Löwenberg, H. Fernandez, Z. Sun, X. Yao, J. Holowiecki, S. Grosicki, S. Giebel, L. Mayer, T.
Harasym, P. Tardi, J. Lancet, J. Cortes, D. Hogge, J. Cortes, S. Goldberg, E. Feldman, D. Breems,
W. Van Putten, P. Huijgens, D.-B. C. T. G. for H.-O. (HOVON), F. Ravandi, E. Ritchie, H. Sayar, M.
Dennis, N. Russell, R. Hills, R. Stuart, L. Cripe, M. Maris, J. E. K. P, K. Yee, T. Rosenblat, M.
McDevitt, D. Mulford, M. Sekeres, J. Lancet, B. Wood, E. Feldman, J. Brandwein, R. Stone, S.
Petersdorf, K. Kopecky, M. Slovak, A. Burnett, N. Russell, R. Hills, A. L. F. Association, E. Stein, A.
Stein, R. Walter, H. Döhner, M. Lübbert, W. Fiedler, P. Kottaridis, R. Gale, M. Frew, A. S. G. U. A.
myeloid leukemia, R. Stone, T. Fischer, R. Paquette, D. H. S. RM, M. Ehninger, P. Zarrinkar, R.
Gunawardane, M. Cramer, J. Cortes, H. Kantarjian, J. Foran, M. Levis, A. Perl, H. Dombret, J.
Cortes, A. Perl, H. Dombret, G. Schiller, M. Tallman, S. Goldberg, Y. Alvarado, H. Kantarjian, R.
Luthra, C. Albers, H. Leischner, M. Verbeek, J. Randhawa, H. Kantarjian, G. Borthakur, M. Levis,
A. Perl, J. Altman, C. Green, C. Evans, L. Zhao, P. Paschka, R. Schlenk, V. Gaidzik, G. Marcucci, K.
Maharry, Y. Wu, A. Andersson, D. Miller, J. Lynch, P. Ward, J. Patel, D. Wise, M. Figueroa, O.
Abdel-Wahab, C. Lu, C. Lu, P. Ward, G. Kapoor, C. DiNardo, E. Stein, J. Altman, C. Schoch, S.
Schnittger, M. Klaus, W. Kern, W. Hiddemann, T. Haferlach, K. Bernt, N. Zhu, A. Sinha, A.
Deshpande, L. Chen, M. Fazio, L. Chen, A. Deshpande, D. Banka, C. Chen, R. Koche, A. Sinha, M.
Kühn, M. Hadler, S. Daigle, S. Daigle, E. Olhava, C. Therkelsen, S. Daigle, E. Olhava, C. Therkelsen,
E. Stein, G. Garcia-Manero, D. Rizzieri, M. Konopleva, D. Pollyea, J. Potluri, S. Chan, D. Thomas,
M. Corces-Zimmerman, J. Zuber, J. Shi, E. Wang, G. Blobel, A. Kalota, P. Sanchez, M. Carroll, M.

BIBLIOGRAFIA
201
Dawson, R. Prinjha, A. Dittmann, H. Dombret, C. Preudhomme, C. Berthon, M. Sekeres, and D.
Steensma, “Emerging therapeutic drugs for AML.,” Blood, vol. 127, no. 1, pp. 71–8, Jan. 2016.
[187] F. Rizzolio, T. Tuccinardi, I. Caligiuri, C. Lucchetti, and A. Giordano, “CDK inhibitors: from the
bench to clinical trials.,” Curr. Drug Targets, vol. 11, no. 3, pp. 279–90, Mar. 2010.
[188] M. Ji, J. Li, H. Yu, D. Ma, J. Ye, X. Sun, and C. Ji, “Simultaneous targeting of MCL1 and ABCB1 as a
novel strategy to overcome drug resistance in human leukaemia.,” Br. J. Haematol., vol. 145, no.
5, pp. 648–56, Jun. 2009.
[189] A. K. Burnett, R. K. Hills, D. Milligan, L. Kjeldsen, J. Kell, N. H. Russell, J. A. L. Yin, A. Hunter, A. H.
Goldstone, and K. Wheatley, “Identification of patients with acute myeloblastic leukemia who
benefit from the addition of gemtuzumab ozogamicin: results of the MRC AML15 trial.,” J. Clin.
Oncol., vol. 29, no. 4, pp. 369–77, Feb. 2011.
[190] B. Brethon, H. Cavé, M. Fahd, and A. Baruchel, “[Infant acute leukemia].,” Bull. Cancer, vol. 103,
no. 3, pp. 299–311, Mar. 2016.
[191] G. L. Kearns, S. M. Abdel-Rahman, S. W. Alander, D. L. Blowey, J. S. Leeder, and R. E. Kauffman,
“Developmental pharmacology--drug disposition, action, and therapy in infants and children.,”
N. Engl. J. Med., vol. 349, no. 12, pp. 1157–67, Sep. 2003.
[192] R. W. Stam, M. L. den Boer, J. P. P. Meijerink, M. E. G. Ebus, G. J. Peters, P. Noordhuis, G. E.
Janka-Schaub, S. A. Armstrong, S. J. Korsmeyer, and R. Pieters, “Differential mRNA expression of
Ara-C–metabolizing enzymes explains Ara-C sensitivity in MLL gene–rearranged infant acute
lymphoblastic leukemia,” Blood, vol. 101, no. 4, 2003.
[193] C. M. Galmarini, X. Thomas, K. Graham, A. El Jafaari, E. Cros, L. Jordheim, J. R. Mackey, and C.
Dumontet, “Deoxycytidine kinase and cN-II nucleotidase expression in blast cells predict survival
in acute myeloid leukaemia patients treated with cytarabine,” Br. J. Haematol., vol. 122, no. 1,
pp. 53–60, Jul. 2003.
[194] R. Pieters, M. Schrappe, P. De Lorenzo, I. Hann, G. De Rossi, M. Felice, L. Hovi, T. LeBlanc, T.
Szczepanski, A. Ferster, G. Janka, J. Rubnitz, L. Silverman, J. Stary, M. Campbell, C.-K. Li, G. Mann,
R. Suppiah, A. Biondi, A. Vora, and M. G. Valsecchi, “A treatment protocol for infants younger
than 1 year with acute lymphoblastic leukaemia (Interfant-99): an observational study and a
multicentre randomised trial.,” Lancet (London, England), vol. 370, no. 9583, pp. 240–50, Jul.
2007.
[195] F. Meyr, G. Escherich, G. Mann, T. Klingebiel, A. Kulozik, C. Rossig, M. Schrappe, G. Henze, A. von
Stackelberg, and J. Hitzler, “Outcomes of treatment for relapsed acute lymphoblastic leukaemia
in children with Down syndrome,” Br. J. Haematol., vol. 162, no. 1, pp. 98–106, Jul. 2013.
[196] C. G. Mullighan, J. R. Collins-Underwood, L. A. A. Phillips, M. G. Loudin, W. Liu, J. Zhang, J. Ma, E.
Coustan-Smith, R. C. Harvey, C. L. Willman, F. M. Mikhail, J. Meyer, A. J. Carroll, R. T. Williams, J.
Cheng, N. A. Heerema, G. Basso, A. Pession, C.-H. Pui, S. C. Raimondi, S. P. Hunger, J. R. Downing,
W. L. Carroll, and K. R. Rabin, “Rearrangement of CRLF2 in B-progenitor– and Down syndrome–
associated acute lymphoblastic leukemia,” Nat. Genet., vol. 41, no. 11, pp. 1243–1246, Nov.

BIBLIOGRAFIA
202
2009.
[197] B. Zeller, G. Gustafsson, E. Forestier, J. Abrahamsson, N. Clausen, J. Heldrup, L. Hovi, G.
Jonmundsson, S. O. Lie, A. Glomstein, and H. Hasle, “Acute leukaemia in children with Down
syndrome: a population-based Nordic study,” Br. J. Haematol., vol. 128, no. 6, pp. 797–804, Mar.
2005.
[198] E. Forestier, S. Izraeli, B. Beverloo, O. Haas, A. Pession, K. Michalová, B. Stark, C. J. Harrison, A.
Teigler-Schlegel, and B. Johansson, “Cytogenetic features of acute lymphoblastic and myeloid
leukemias in pediatric patients with Down syndrome: an iBFM-SG study.,” Blood, vol. 111, no. 3,
pp. 1575–83, Feb. 2008.
[199] J. Wechsler, M. Greene, M. A. McDevitt, J. Anastasi, J. E. Karp, M. M. Le Beau, and J. D. Crispino,
“Acquired mutations in GATA1 in the megakaryoblastic leukemia of Down syndrome.,” Nat.
Genet., vol. 32, no. 1, pp. 148–52, Sep. 2002.
[200] A. Shimada, G. Xu, T. Toki, H. Kimura, Y. Hayashi, and E. Ito, “Fetal origin of the GATA1 mutation
in identical twins with transient myeloproliferative disorder and acute megakaryoblastic
leukemia accompanying Down syndrome.,” Blood, vol. 103, no. 1, p. 366, Jan. 2004.
[201] H. Hasle, J. Abrahamsson, M. Arola, A. Karow, A. O’Marcaigh, D. Reinhardt, D. K. H. Webb, E. van
Wering, B. Zeller, C. M. Zwaan, and P. Vyas, “Myeloid leukemia in children 4 years or older with
Down syndrome often lacks GATA1 mutation and cytogenetics and risk of relapse are more akin
to sporadic AML.,” Leukemia, vol. 22, no. 7, pp. 1428–30, Jul. 2008.
[202] J. P. Krischer, S. Epstein, D. D. Cuthbertson, A. M. Goorin, M. L. Epstein, and S. E. Lipshultz,
“Clinical cardiotoxicity following anthracycline treatment for childhood cancer: the Pediatric
Oncology Group experience.,” J. Clin. Oncol., vol. 15, no. 4, pp. 1544–52, Apr. 1997.
[203] A. S. Advani, “Biology and Treatment of Acute Lymphocytic Leukemia in Adolescents and Young
Adults,” Am. Soc. Clin. Oncol. Educ. B., vol. 33, pp. 285–289, 2013.
[204] C. J. Harrison, “Cytogenetics of paediatric and adolescent acute lymphoblastic leukaemia.,” Br. J.
Haematol., vol. 144, no. 2, pp. 147–56, Jan. 2009.
[205] R. C. Harvey, C. G. Mullighan, I.-M. Chen, W. Wharton, F. M. Mikhail, A. J. Carroll, H. Kang, W. Liu,
K. K. Dobbin, M. A. Smith, W. L. Carroll, M. Devidas, W. P. Bowman, B. M. Camitta, G. H. Reaman,
S. P. Hunger, J. R. Downing, and C. L. Willman, “Rearrangement of CRLF2 is associated with
mutation of JAK kinases, alteration of IKZF1, Hispanic/Latino ethnicity, and a poor outcome in
pediatric B-progenitor acute lymphoblastic leukemia.,” Blood, vol. 115, no. 26, pp. 5312–21, Jul.
2010.
[206] E. A. Raetz, M. Devidas, A. J. Carroll, N. A. Heerema, M. J. Borowitz, B. L. Wood, J. M. Gastier-
Foster, C. L. Willman, M. L. Loh, E. C. Larsen, and COG ALL Committee, “Cytogenetic and early-
response characteristics of adolescents and young adults with acute lymphoblastic leukemia
(ALL): A Children’s Oncology Group (COG) study.,” ASCO Meet. Abstr., vol. 28, no. 15_suppl, p.
9509, 2010.
[207] N. Boissel, M.-F. Auclerc, V. Lhéritier, Y. Perel, X. Thomas, T. Leblanc, P. Rousselot, J.-M. Cayuela,

BIBLIOGRAFIA
203
J. Gabert, N. Fegueux, C. Piguet, F. Huguet-Rigal, C. Berthou, J.-M. Boiron, C. Pautas, G. Michel,
D. Fière, G. Leverger, H. Dombret, and A. Baruchel, “Should adolescents with acute
lymphoblastic leukemia be treated as old children or young adults? Comparison of the French
FRALLE-93 and LALA-94 trials.,” J. Clin. Oncol., vol. 21, no. 5, pp. 774–80, Mar. 2003.
[208] G. Gatta, S. Rossi, R. Foschi, A. Trama, R. Marcos-Gragera, G. Pastore, R. Peris-Bonet, C. Stiller,
and R. Capocaccia, “Survival and cure trends for European children, adolescents and young
adults diagnosed with acute lymphoblastic leukemia from 1982 to 2002,” Haematologica, vol.
98, no. 5, 2013.
[209] J.-M. Ribera, A. Oriol, M.-A. Sanz, M. Tormo, P. Fernández-Abellán, E. del Potro, E. Abella, J.
Bueno, R. Parody, P. Bastida, C. Grande, I. Heras, C. Bethencourt, E. Feliu, and J.-J. Ortega,
“Comparison of the results of the treatment of adolescents and young adults with standard-risk
acute lymphoblastic leukemia with the Programa Español de Tratamiento en Hematología
pediatric-based protocol ALL-96.,” J. Clin. Oncol., vol. 26, no. 11, pp. 1843–9, Apr. 2008.
[210] J.-M. Ribera, J. Ribera, and E. Genescà, “Treatment of adolescent and young adults with acute
lymphoblastic leukemia.,” Mediterr. J. Hematol. Infect. Dis., vol. 6, no. 1, p. e2014052, 2014.
[211] W. Matthews, C. T. Jordan, G. W. Wiegand, D. Pardoll, I. R. Lemischka, and I. R. Lemischka, “A
receptor tyrosine kinase specific to hematopoietic stem and progenitor cell-enriched
populations.,” Cell, vol. 65, no. 7, pp. 1143–52, Jun. 1991.
[212] O. Rosnet, and D. Birnbaum, “Hematopoietic receptors of class III receptor-type tyrosine
kinases.,” Crit. Rev. Oncog., vol. 4, no. 6, pp. 595–613, 1993.
[213] D. G. Gilliland, and J. D. Griffin, “The roles of FLT3 in hematopoiesis and leukemia.,” Blood, vol.
100, no. 5, pp. 1532–42, Sep. 2002.
[214] C. E. Carow, M. Levenstein, S. H. Kaufmann, J. Chen, S. Amin, P. Rockwell, L. Witte, M. J.
Borowitz, C. I. Civin, and D. Small, “Expression of the hematopoietic growth factor receptor FLT3
(STK-1/Flk2) in human leukemias.,” Blood, vol. 87, no. 3, pp. 1089–96, Feb. 1996.
[215] F. Birg, M. Courcoul, O. Rosnet, F. Bardin, M. J. Pébusque, S. Marchetto, A. Tabilio, P. Mannoni,
and D. Birnbaum, “Expression of the FMS/KIT-like gene FLT3 in human acute leukemias of the
myeloid and lymphoid lineages.,” Blood, vol. 80, no. 10, pp. 2584–93, Nov. 1992.
[216] M. Levis, and D. Small, “FLT3: ITDoes matter in leukemia.,” Leukemia, vol. 17, no. 9, pp. 1738–52,
Sep. 2003.
[217] O. Rosnet, H. J. Bühring, O. deLapeyrière, N. Beslu, C. Lavagna, S. Marchetto, I. Rappold, H. G.
Drexler, F. Birg, R. Rottapel, C. Hannum, P. Dubreuil, and D. Birnbaum, “Expression and signal
transduction of the FLT3 tyrosine kinase receptor.,” Acta Haematol., vol. 95, no. 3–4, pp. 218–
23, 1996.
[218] C. E. Annesley, and P. Brown, “The Biology and Targeting of FLT3 in Pediatric Leukemia.,” Front.
Oncol., vol. 4, p. 263, 2014.
[219] C. Thiede, C. Steudel, B. Mohr, M. Schaich, U. Schäkel, U. Platzbecker, M. Wermke, M.
Bornhäuser, M. Ritter, A. Neubauer, G. Ehninger, and T. Illmer, “Analysis of FLT3-activating

BIBLIOGRAFIA
204
mutations in 979 patients with acute myelogenous leukemia: association with FAB subtypes and
identification of subgroups with poor prognosis,” Blood, vol. 99, no. 12, 2002.
[220] A. Wodnar-Filipowicz, S. D. Lyman, A. Gratwohl, A. Tichelli, B. Speck,and C. Nissen, “Flt3 ligand
level reflects hematopoietic progenitor cell function in aplastic anemia and chemotherapy-
induced bone marrow aplasia.,” Blood, vol. 88, no. 12, pp. 4493–9, Dec. 1996.
[221] H. J. McKenna, K. L. Stocking, R. E. Miller, K. Brasel, T. De Smedt, E. Maraskovsky, C. R.
Maliszewski, D. H. Lynch, J. Smith, B. Pulendran, E. R. Roux, M. Teepe, S. D. Lyman, and J. J.
Peschon, “Mice lacking flt3 ligand have deficient hematopoiesisaffecting hematopoietic
progenitor cells, dendritic cells, and natural killer cells,” Blood, vol. 95, no. 11, 2000.
[222] K. Ozeki, H. Kiyoi, Y. Hirose, M. Iwai, M. Ninomiya, Y. Kodera, and S. Miyawaki, “Biologic and
clinical significance of the FLT3 transcript level in acute myeloid leukemia Biologic and clinical
significance of the FLT3 transcript level in acute myeloid leukemia,” 2015, 2008. .
[223] P. Brown, M. Levis, S. Shurtleff, D. Campana, J. Downing, and D. Small, “FLT3 inhibition
selectively kills childhood acute lymphoblastic leukemia cells with high levels of FLT3
expression,” Blood, vol. 105, no. 2, 2005.
[224] R. W. Stam, P. Schneider, P. de Lorenzo, M. G. Valsecchi, M. L. den Boer, and R. Pieters,
“Prognostic significance of high-level FLT3 expression in MLL-rearranged infant acute
lymphoblastic leukemia,” Blood, vol. 110, no. 7, 2007.
[225] R. W. Stam, M. L. den Boer, P. Schneider, P. Nollau, M. Horstmann, H. B. Beverloo, E. van der
Voort, M. G. Valsecchi, P. de Lorenzo, S. E. Sallan, S. A. Armstrong, and R. Pieters, “Targeting
FLT3 in primary MLL-gene–rearranged infant acute lymphoblastic leukemia,” Blood, vol. 106, no.
7, 2005.
[226] M. Nakao, S. Yokota, T. Iwai, H. Kaneko, S. Horiike, K. Kashima, Y. Sonoda, T. Fujimoto, S. and S.
Misawa, “Internal tandem duplication of the flt3 gene found in acute myeloid leukemia.,”
Leukemia, vol. 10, no. 12, pp. 1911–8, Dec. 1996.
[227] H. Kiyoi, M. Towatari, S. Yokota, M. Hamaguchi, R. Ohno, H. Saito, and T. Naoe, “Internal tandem
duplication of the FLT3 gene is a novel modality of elongation mutation which causes
constitutive activation of the product.,” Leukemia, vol. 12, no. 9, pp. 1333–7, Sep. 1998.
[228] F. Breitenbuecher, S. Schnittger, R. Grundler, B. Markova, B. Carius, A. Brecht, J. Duyster, T.
Haferlach, C. Huber, and T. Fischer, “Identification of a novel type of ITD mutations located in
nonjuxtamembrane domains of the FLT3 tyrosine kinase receptor,” Blood, vol. 113, no. 17, 2009.
[229] Y. Ishikawa, H. Kiyoi, A. Tsujimura, S. Miyawaki, Y. Miyazaki, K. Kuriyama, T. Naoe, and T. Naoe,
“Comprehensive analysis of cooperative gene mutations between class I and class II in de novo
acute myeloid leukemia.,” Eur. J. Haematol., vol. 83, no. 2, pp. 90–8, Aug. 2009.
[230] F. M. Abu-Duhier, A. C. Goodeve, G. A. Wilson, R. S. Care, I. R. Peake, and J. T. Reilly,
“Identification of novel FLT-3 Asp835 mutations in adult acute myeloid leukaemia.,” Br. J.
Haematol., vol. 113, no. 4, pp. 983–8, Jun. 2001.
[231] K. Spiekermann, K. Bagrintseva, C. Schoch, T. Haferlach, W. Hiddemann, and S. Schnittger, “A

BIBLIOGRAFIA
205
new and recurrent activating length mutation in exon 20 of the FLT3 gene in acute myeloid
leukemia.,” Blood, vol. 100, no. 9, pp. 3423–5, Nov. 2002.
[232] M. M. Schittenhelm, K. W. H. Yee, J. W. Tyner, L. McGreevey, A.D. Haley, A. Town, D. J. Griffith,
T. Bainbridge, R. M. Braziel, A.-M. O’Farrell, J. M. Cherrington, and M. C. Heinrich, “FLT3 K663Q
is a novel AML-associated oncogenic kinase: Determination of biochemical properties and
sensitivity to Sunitinib (SU11248).,” Leukemia, vol. 20, no. 11, pp. 2008–14, Nov. 2006.
[233] C. Reindl, K. Bagrintseva, S. Vempati, S. Schnittger, J. W. Ellwart, K. Wenig, K.-P. Hopfner, W.
Hiddemann, and K. Spiekermann, “Point mutations in the juxtamembrane domain of FLT3 define
a new class of activating mutations in AML,” Blood, vol. 107, no. 9, 2006.
[234] A. Andersson, K. Paulsson, H. Lilljebjörn, C. Lassen, B. Strömbeck, J. Heldrup, M. Behrendtz, B.
Johansson, and T. Fioretos, “FLT3 mutations in a 10 year consecutive series of 177 childhood
acute leukemias and their impact on global gene expression patterns,” Genes, Chromosom.
Cancer, vol. 47, no. 1, pp. 64–70, Jan. 2008.
[235] M. Neumann, E. Coskun, L. Fransecky, L. H. Mochmann, I. Bartram, N. F. Sartangi, S. Heesch, N.
Gökbuget, S. Schwartz, C. Brandts, C. Schlee, R. Haas, U. Dührsen, M. Griesshammer, H. Döhner,
G. Ehninger, T. Burmeister, O. Blau, E. Thiel, D. Hoelzer, W.-K. Hofmann, and C. D. Baldus, “FLT3
mutations in early T-cell precursor ALL characterize a stem cell like leukemia and imply the
clinical use of tyrosine kinase inhibitors.,” PLoS One, vol. 8, no. 1, p. e53190, 2013.
[236] S. A. Armstrong, M. E. Mabon, L. B. Silverman, A. Li, J. G. Gribben, E. A. Fox, S. E. Sallan, and S. J.
Korsmeyer, “FLT3 mutations in childhood acute lymphoblastic leukemia,” Blood, vol. 103, no. 9,
2004.
[237] P. D. Kottaridis, R. E. Gale, S. E. Langabeer, M. E. Frew, D. T. Bowen, and D. C. Linch, “Studies of
FLT3 mutations in paired presentation and relapse samples from patients with acute myeloid
leukemia: implications for the role of FLT3 mutations in leukemogenesis, minimal residual
disease detection, and possible therapy with FLT3 inhibitors,” Blood, vol. 100, no. 7, 2002.
[238] C. M. Zwaan, S. Meshinchi, J. P. Radich, A. J. P. Veerman, D. R. Huismans, L. Munske, M.
Podleschny, K. Hählen, R. Pieters, M. Zimmermann, D. Reinhardt, J. Harbott, U. Creutzig, G. J. L.
Kaspers, and F. Griesinger, “FLT3 internal tandem duplication in 234 children with acute myeloid
leukemia: prognostic significance and relation to cellular drug resistance.,” Blood, vol. 102, no. 7,
pp. 2387–94, Oct. 2003.
[239] E. Manara, G. Basso, M. Zampini, B. Buldini, C. Tregnago, R. Rondelli, R. Masetti, V. Bisio, M.
Frison, K. Polato, G. Cazzaniga, G. Menna, F. Fagioli, P. Merli, A. Biondi, A. Pession, F. Locatelli,
and M. Pigazzi, “Characterization of children with FLT3-ITD acute myeloid leukemia: a report
from the AIEOP AML-2002 study group.,” Leukemia, Jul. 2016.
[240] S. P. Whitman, K. J. Archer, L. Feng, C. Baldus, B. Becknell, B. D. Carlson, A. J. Carroll, K. Mrózek,
J. W. Vardiman, S. L. George, J. E. Kolitz, R. A. Larson, C. D. Bloomfield, and M. A. Caligiuri,
“Absence of the Wild-Type Allele Predicts Poor Prognosis in Adult de Novo Acute Myeloid
Leukemia with Normal Cytogenetics and the Internal Tandem Duplication of FLT3,” Cancer Res.,

BIBLIOGRAFIA
206
vol. 61, no. 19, 2001.
[241] P. D. Kottaridis, R. E. Gale, M. E. Frew, G. Harrison, S. E. Langabeer, A. A. Belton, H. Walker, K.
Wheatley, D. T. Bowen, A. K. Burnett, A. H. Goldstone, and D. C. Linch, “The presence of a FLT3
internal tandem duplication in patients with acute myeloid leukemia (AML) adds important
prognostic information to cytogenetic risk group and response to the first cycle of
chemotherapy: analysis of 854 patients from the United Kin…,” Blood, vol. 98, no. 6, 2001.
[242] K. W. Pratz, T. Sato, K. M. Murphy, A. Stine, T. Rajkhowa, and M. Levis, “FLT3-mutant allelic
burden and clinical status are predictive of response to FLT3 inhibitors in AML.,” Blood, vol. 115,
no. 7, pp. 1425–32, Feb. 2010.
[243] C. Callens, S. Chevret, J.-M. Cayuela, B. Cassinat, E. Raffoux, S. de Botton, X. Thomas, A. Guerci,
N. Fegueux, A. Pigneux, A.-M. Stoppa, T. Lamy, F. Rigal-Huguet, A. Vekhoff, S. Meyer-Monard, A.
Ferrand, M. Sanz, C. Chomienne, P. Fenaux, H. Dombret. European APL Group, “Prognostic
implication of FLT3 and Ras gene mutations in patients with acute promyelocytic leukemia (APL):
a retrospective study from the European APL Group.,” Leukemia, vol. 19, no. 7, pp. 1153–60, Jul.
2005.
[244] E. Barragan, P. Montesinos, M. Camos, M. Gonzalez, M. J. Calasanz, J. Roman-Gomez, M. T.
Gomez-Casares, R. Ayala, J. Lopez, O. Fuster, D. Colomer, C. Chillon, M. J. Larrayoz, P. Sanchez-
Godoy, J. Gonzalez-Campos, F. Manso, M. L. Amador, E. Vellenga, B. Lowenberg, and M. A. Sanz,
“Prognostic value of FLT3 mutations in patients with acute promyelocytic leukemia treated with
all-trans retinoic acid and anthracycline monochemotherapy,” Haematologica, vol. 96, no. 10,
pp. 1470–1477, Oct. 2011.
[245] R. W. Stam, P. Schneider, P. de Lorenzo, M. G. Valsecchi, M. L. den Boer, and R. Pieters,
“Prognostic significance of high-level FLT3 expression in MLL-rearranged infant acute
lymphoblastic leukemia.,” Blood, vol. 110, no. 7, pp. 2774–5, Oct. 2007.
[246] A. V. Moorman, S. M. Richards, M. Martineau, K. L. Cheung, H. M. Robinson, G. R. Jalali, Z. J.
Broadfield, R. L. Harris, K. E. Taylor, B. E. S. Gibson, I. M. Hann, F. G. H. Hill, S. E. Kinsey, T. O. B.
Eden, C. D. Mitchell, and C. J. Harrison, “Outcome heterogeneity in childhood high-hyperdiploid
acute lymphoblastic leukemia,” Blood, vol. 102, no. 8, 2003.
[247] C. L. Sawyers, “Finding the next Gleevec: FLT3 targeted kinase inhibitor therapy for acute
myeloid leukemia,” Cancer Cell, vol. 1, no. 5, pp. 413–415, 2002.
[248] R. M. Stone, D. J. DeAngelo, V. Klimek, I. Galinsky, E. Estey, S. D. Nimer, W. Grandin, D. Lebwohl,
Y. Wang, P. Cohen, E. A. Fox, D. Neuberg, J. Clark, D. G. Gilliland, and J. D. Griffin, “Patients with
acute myeloid leukemia and an activating mutation in FLT3 respond to a small-molecule FLT3
tyrosine kinase inhibitor, PKC412,” Blood, vol. 105, no. 1, 2004.
[249] A.-M. O’Farrell, J. M. Foran, W. Fiedler, H. Serve, R. L. Paquette, M. A. Cooper, H. A. Yuen, S. G.
Louie, H. Kim, S. Nicholas, M. C. Heinrich, W. E. Berdel, C. Bello, M. Jacobs, P. Scigalla, W. C.
Manning, S. Kelsey, and J. M. Cherrington, “An innovative phase I clinical study demonstrates
inhibition of FLT3 phosphorylation by SU11248 in acute myeloid leukemia patients.,” Clin. Cancer

BIBLIOGRAFIA
207
Res., vol. 9, no. 15, pp. 5465–76, Nov. 2003.
[250] B. D. Smith, M. Levis, M. Beran, F. Giles, H. Kantarjian, K. Berg, K. M. Murphy, T. Dauses, J.
Allebach, and D. Small, “Single-agent CEP-701, a novel FLT3 inhibitor, shows biologic and clinical
activity in patients with relapsed or refractory acute myeloid leukemia.,” Blood, vol. 103, no. 10,
pp. 3669–76, May 2004.
[251] K. W. Pratz, J. Cortes, G. J. Roboz, N. Rao, O. Arowojolu, A. Stine, Y. Shiotsu, A. Shudo, S. Akinaga,
D. Small, J. E. Karp, and M. Levis, “A pharmacodynamic study of the FLT3 inhibitor KW-2449
yields insight into the basis for clinical response.,” Blood, vol. 113, no. 17, pp. 3938–46, Apr.
2009.
[252] K. W. Pratz, E. Cho, M. J. Levis, J. E. Karp, S. D. Gore, M. McDevitt, A. Stine, M. Zhao, S. D. Baker,
M. A. Carducci, J. J. Wright, M. A. Rudek, and B. D. Smith, “A pharmacodynamic study of
sorafenib in patients with relapsed and refractory acute leukemias.,” Leukemia, vol. 24, no. 8,
pp. 1437–44, Aug. 2010.
[253] Stone RM, Mandrekar S, Sanford BL, “The multi-kinase inhibitor midostaurin (M) prolongs
survival compared with placebo (P) in combination with daunorubicin (D)/cytarabine (C)
induction (ind), high- dose C consolidation (consol), and as maintenance (maint) therapy in
newly diagnosed acute myeloid leukemia,”. Abtract ASH 2015
[254] C. Röllig, H. Serve, A. Hüttmann, R. Noppeney, C. Müller-Tidow, U. Krug, C. D. Baldus, C. H.
Brandts, V. Kunzmann, H. Einsele, A. Krämer, K. Schäfer-Eckart, A. Neubauer, A. Burchert, A.
Giagounidis, S. W. Krause, A. Mackensen, W. Aulitzky, R. Herbst, M. Hänel, A. Kiani, N.
Frickhofen, J. Kullmer, U. Kaiser, H. Link, T. Geer, A. Reichle, C. Junghanß, R. Repp, F. Heits, H.
Dürk, J. Hase, I.-M. Klut, T. Illmer, M. Bornhäuser, M. Schaich, S. Parmentier, M. Görner, C.
Thiede, M. von Bonin, J. Schetelig, M. Kramer, W. E. Berdel, G. Ehninger Study Alliance
Leukaemia, “Addition of sorafenib versus placebo to standard therapy in patients aged 60 years
or younger with newly diagnosed acute myeloid leukaemia (SORAML): a multicentre,phase 2,
randomised controlled trial.,” Lancet. Oncol., vol. 16, no. 16, pp. 1691–9, Dec. 2015.
[255] Y.-B. Chen, S. Li, A. A. Lane, C. Connolly, C. Del Rio, B. Valles, M. Curtis, K. Ballen, C. Cutler, B. R.
Dey, A. El-Jawahri, A. T. Fathi, V. T. Ho, A. Joyce, S. McAfee, M. Rudek, T. Rajkhowa, S. Verselis, J.
H. Antin, T. R. Spitzer, M. Levis, and R. Soiffer, “Phase I Trial of Maintenance Sorafenib after
Allogeneic Hematopoietic Stem Cell Transplantation for Fms-like Tyrosine Kinase 3 Internal
Tandem Duplication Acute Myeloid Leukemia,” Biol. Blood Marrow Transplant., vol. 20, no. 12,
pp. 2042–2048, 2014.
[256] K. Tarlock, B. Chang, T. Cooper, T. Gross, S. Gupta, S. Neudorf, K. Adlard, P. A. Ho, S. McGoldrick,
T. Watt, T. Templeman, I. Sisler, A. Garee, B. Thomson, A. Woolfrey, E. Estey, S. Meshinchi, J. A.
and J. A. Pollard, “Sorafenib treatment following hematopoietic stem cell transplant in pediatric
FLT3/ITD acute myeloid leukemia.,” Pediatr. Blood Cancer, vol. 62, no. 6, pp. 1048–54, Jun. 2015.
[257] K. W. H. Yee, M. Schittenhelm, A.-M. O’Farrell, A. R. Town, L. McGreevey, T. Bainbridge, J. M.
Cherrington, and M. C. Heinrich, “Synergistic effect of SU11248 with cytarabine or daunorubicin

BIBLIOGRAFIA
208
on FLT3 ITD-positive leukemic cells.,” Blood, vol. 104, no. 13, pp. 4202–9, Dec. 2004.
[258] M. Levis, R. Pham, B. D. Smith, and D. Small, “In vitro studies of a FLT3 inhibitor combined with
chemotherapy: sequence of administration is important to achieve synergistic cytotoxic effects,”
Blood, vol. 104, no. 4, 2004.
[259] Levis MJ, Perl AE, Dombret H, “Final Results of a Phase 2 Open-Label, Monotherapy Efficacy and
Safety Study of Quizartinib (AC220) in Patients with FLT3-ITD Positive or Negative
Relapsed/Refractory Acute Myeloid Leukemia After Second-Line Chemotherapy or
Hematopoietic Stem Cell Transpl,” Abstract ASH 2012.
[260] Cortes JE, Perl AE, Dombret H, “Final Results of a Phase 2 Open-Label, Monotherapy Efficacy and
Safety Study of Quizartinib (AC220) in Patients ≥ 60 Years of Age with FLT3 ITD Positive or
Negative Relapsed/Refractory Acute Myeloid Leukemia [abstract]. Blood. 2012;120(21). Abstract
48,”
[261] Schiller GJ, Tallman MS, Goldberg SL, “Final results of a randomized phase 2 study showing the
clinical benefit of quizartinib (AC220) in patients with FLT3-ITD positive relapsed or refractory
acute myeloid leukemia [abstract]. J Clin Oncol. 2014;32(5s). Abstract 7100
[262] Y. Alvarado, H. M. Kantarjian, R. Luthra, F. Ravandi, G. Borthakur, G. Garcia-Manero, M.
Konopleva, Z. Estrov, M. Andreeff, and J. E. Cortes, “Treatment with FLT3 inhibitor in patients
with FLT3-mutated acute myeloid leukemia is associated with development of secondary FLT3-
tyrosine kinase domain mutations.,” Cancer, vol. 120, no. 14, pp. 2142–9, Jul. 2014.
[263] C. Albers, H. Leischner, M. Verbeek, C. Yu, A. L. Illert, C. Peschel, N. von Bubnoff, and J. Duyster,
“The secondary FLT3-ITD F691L mutation induces resistance to AC220 in FLT3-ITD+ AML but
retains in vitro sensitivity to PKC412 and Sunitinib.,” Leukemia, vol. 27, no. 6, pp. 1416–8, Jun.
2013.
[264] Randhawa JK, Kantarjian HM, Borthakur G, “Results of a Phase II Study of Crenolanib in
Relapsed/Refractory Acute Myeloid Leukemia Patients (Pts) with Activating FLT3 Mutations
[abstract]. Blood. 2014;124(21). Abstract 389,” Blood, 2014.
[265] Perl AE, Altman JK, “phase I/II trial of ASP2215, a selective, potent inhibitor of FLT3/Axl in
patients with relapsed or refractory (R/R) acute myeloid leukemia (AML) [abstract]. J Clin Oncol
2015. 33. Abstract 7003,” J. Clin. Oncol., 2015.
[266] M. Molina-Arcas, B. Bellosillo, F. J. Casado, E. Montserrat, J. Gil, D. Colomer, and M. Pastor-
Anglada, “Fludarabine uptake mechanisms in B-cell chronic lymphocytic leukemia.,” Blood, vol.
101, no. 6, pp. 2328–34, Mar. 2003.
[267] M. Pastor-Anglada, M. Molina-Arcas, F. J. Casado, B. Bellosillo, D. Colomer, and J. Gil,
“Nucleoside transporters in chronic lymphocytic leukaemia.,” Leukemia, vol. 18, no. 3, pp. 385–
93, Mar. 2004.
[268] G. Minuesa, S. Purcet, I. Erkizia, M. Molina-Arcas, M. Bofill, N. Izquierdo-Useros, F. J. Casado, B.
Clotet, M. Pastor-Anglada, and J. Martinez-Picado, “Expression and functionality of anti-human
immunodeficiency virus and anticancer drug uptake transporters in immune cells.,” J.

BIBLIOGRAFIA
209
Pharmacol. Exp. Ther., vol. 324, no. 2, pp. 558–67, Feb. 2008.
[269] P. Cano-Soldado and M. Pastor-Anglada, “Transporters that translocate nucleosides and
structural similar drugs: structural requirements for substrate recognition.,” Med. Res. Rev., vol.
32, no. 2, pp. 428–57, Mar. 2012.
[270] G. Minuesa, I. Huber-Ruano, M. Pastor-Anglada, H. Koepsell, B. Clotet, and J. Martinez-Picado,
“Drug uptake transporters in antiretroviral therapy.,” Pharmacol. Ther., vol. 132, no. 3, pp. 268–
79, Dec. 2011.
[271] M. Molina-Arcas, F. J. Casado, and M. Pastor-Anglada, “Nucleoside transporter proteins.,” Curr.
Vasc. Pharmacol., vol. 7, no. 4, pp. 426–34, Oct. 2009.
[272] M. Pastor-Anglada, P. Cano-Soldado, E. Errasti-Murugarren, and F. J. Casado, “SLC28 genes and
concentrative nucleoside transporter (CNT) proteins.,” Xenobiotica., vol. 38, no. 7–8, pp. 972–94,
Jul. 2008.
[273] M. Pastor-Anglada, P. Cano-Soldado, M. Molina-Arcas, M. P. Lostao, I. Larráyoz, J. Martínez-
Picado, and F. J. Casado, “Cell entry and export of nucleoside analogues.,” Virus Res., vol. 107,
no. 2, pp. 151–64, Feb. 2005.
[274] M. Pastor-Anglada and S. Pérez-Torras, “Nucleoside transporter proteins as biomarkers of drug
responsiveness and drug targets,” Front. Pharmacol., vol. 6, p. 13, Feb. 2015.
[275] M. Pastor-Anglada, A. Felipe, and F. J. Casado, “Transport and mode of action of nucleoside
derivatives used in chemical and antiviral therapies.,” Trends Pharmacol. Sci., vol. 19, no. 10, pp.
424–30, Oct. 1998.
[276] I. Huber-Ruano and M. Pastor-Anglada, “Transport of nucleoside analogs across the plasma
membrane: a clue to understanding drug-induced cytotoxicity.,” Curr. Drug Metab., vol. 10, no.
4, pp. 347–58, May 2009.
[277] L. P. Jordheim, D. Durantel, F. Zoulim, and C. Dumontet, “Advances in the development of
nucleoside and nucleotide analogues for cancerand viral diseases.,” Nat. Rev. Drug Discov., vol.
12, no. 6, pp. 447–64, Jun. 2013.
[278] M. Molina-Arcas, S. Marcé, N. Villamor, I. Huber-Ruano, F. J. Casado, B. Bellosillo, E. Montserrat,
J. Gil, D. Colomer, and M. Pastor-Anglada, “Equilibrative nucleoside transporter-2 (hENT2)
protein expression correlates with ex vivo sensitivity to fludarabine in chronic lymphocytic
leukemia (CLL) cells.,” Leukemia, vol. 19, no. 1, pp. 64–8, Jan. 2005.
[279] M. López-Guerra, L. Trigueros-Motos, M. Molina-Arcas, N. Villamor, F. J. Casado, E. Montserrat,
E. Campo, D. Colomer, and M. Pastor-Anglada, “Identification of TIGAR in the equilibrative
nucleoside transporter 2-mediated response to fludarabine in chronic lymphocytic leukemia
cells.,” Haematologica, vol. 93, no. 12, pp. 1843–51, Dec. 2008.
[280] S. Marcé, M. Molina-Arcas, N. Villamor, F. J. Casado, E. Campo, M. Pastor-Anglada, and D.
Colomer, “Expression of human equilibrative nucleoside transporter 1 (hENT1) and its
correlation with gemcitabine uptake and cytotoxicity in mantle cell lymphoma.,” Haematologica,
vol. 91, no. 7, pp. 895–902, Jul. 2006.

BIBLIOGRAFIA
210
[281] P. X. Fernández-Calotti, M. Lopez-Guerra, D. Colomer, and M. Pastor-Anglada, “Enhancement of
fludarabine sensitivity by all-trans-retinoic acid in chronic lymphocytic leukemia cells.,”
Haematologica, vol. 97, no. 6, pp. 943–51, Jun. 2012.
[282] P. Fernández-Calotti and M. Pastor-Anglada, “All-trans-retinoic acid promotes trafficking of
human concentrative nucleoside transporter-3 (hCNT3) to the plasma membrane by a TGF-
beta1-mediated mechanism.,” J. Biol. Chem., vol. 285, no. 18, pp. 13589–98, Apr. 2010.
[283] J. C. White, J. P. Rathmell, and R. L. Capizzi, “Membrane transport influences the rate of
accumulation of cytosine arabinoside in human leukemia cells.,” J. Clin. Invest., vol. 79, no. 2, pp.
380–7, Feb. 1987.
[284] L. P. Jordheim, D. Durantel, F. Zoulim, and C. Dumontet, “Advances in the development of
nucleoside and nucleotide analogues for cancer and viral diseases,” Nat. Rev. Drug Discov., vol.
12, no. 6, pp. 447–464, May 2013.
[285] P. X. Fernández-Calotti, D. Colomer, and M. Pastor Anglada, “Translocation of nucleoside analogs
across the plasma membrane in hematologic malignancies.,” Nucleosides. Nucleotides Nucleic
Acids, vol. 30, no. 12, pp. 1324–40, Dec. 2011.
[286] G. Jin, H. Matsushita, S. Asai, H. Tsukamoto, R. Ono, T. Nosaka, T. Yahata, S. Takahashi, and H.
Miyachi, “FLT3-ITD induces ara-C resistance in myeloid leukemic cells through the repression of
the ENT1 expression,” Biochem Biophys Res Commun, 18;390(3):1001-6. Dec 2009.
[287] S. J. Hughes, X. Cravetchi, G. Vilas, and J. R. Hammond, “Adenosine A1 receptor activation
modulates human equilibrative nucleoside transporter 1 (hENT1) activity via PKC-mediated
phosphorylation of serine-281.,” Cell. Signal., vol. 27, no. 5, pp. 1008–18, May 2015.
[288] G. Reyes, N. M. I. Nivillac, M. Z. Karim, L. Desouza, K. W. M. Siu, and I. R. Coe, “The Equilibrative
Nucleoside Transporter (ENT1) can be phosphorylated at multiple sites by PKC and PKA.,” Mol.
Membr. Biol., vol. 28, no. 6, pp. 412–26, Sep. 2011.
[289] L. P. Jordheim, C. M. Galmarini, and C. Dumontet, “Gemcitabine resistance due to deoxycytidine
kinase deficiency can be reverted by fruitfly deoxynucleoside kinase, DmdNK, in human uterine
sarcoma cells.,” Cancer Chemother. Pharmacol., vol. 58, no. 4, pp. 547–54, Oct. 2006.
[290] L. P. Jordheim and L. Chaloin, “Therapeutic perspectives for cN-II in cancer.,” Curr. Med. Chem.,
vol. 20, no. 34, pp. 4292–303, 2013.
[291] F. Cividini, R. Pesi, L. Chaloin, S. Allegrini, M. Camici, E. Cros-Perrial, C. Dumontet, L. P. Jordheim,
and M. G. Tozzi, “The purine analog fludarabine acts as a cytosolic 5’-nucleotidase II inhibitor.,”
Biochem. Pharmacol., vol. 94, no. 2, pp. 63–8, Mar. 2015.
[292] R. Maréchal, J.-B. Bachet, J. R. Mackey, C. Dalban, P. Demetter, K. Graham, A. Couvelard, M.
Svrcek, A. Bardier-Dupas, P. Hammel, A. Sauvanet, C. Louvet, F. Paye, P. Rougier, C. Penna, T.
André, C. Dumontet, C. E. Cass, L. P. Jordheim, E.-L. Matera, J. Closset, I. Salmon, J. Devière, J.-F.
Emile, J.-L. Van Laethem, and J.-L. Van Laethem, “Levels of gemcitabine transport and
metabolism proteins predict survival times of patients treated with gemcitabine for pancreatic
adenocarcinoma.,” Gastroenterology, vol. 143, no. 3, pp. 664-74–6, Sep. 2012.

BIBLIOGRAFIA
211
[293] A. Abraham, S. Varatharajan, S. Karathedath, C. Philip, K. M. Lakshmi, A. K. Jayavelu, E.
Mohanan, N. B. Janet, V. M. Srivastava, R. V Shaji, W. Zhang, A. Abraham, A. Viswabandya, B.
George, M. Chandy, A. Srivastava, V. Mathews, and P. Balasubramanian, “RNA expression of
genes involved in cytarabine metabolism and transport predicts cytarabine response in acute
myeloid leukemia,” Pharmacogenomics, vol. 16, no. 8, pp. 877–890, Jun. 2015.
[294] I. Hubeek, R. W. Stam, G. J. Peters, R. Broekhuizen, J. P. P. Meijerink, E. R. van Wering, B. E. S.
Gibson, U. Creutzig, C. M. Zwaan, J. Cloos, D. J. Kuik, R. Pieters, and G. J. L. Kaspers, “The human
equilibrative nucleoside transporter 1 mediates in vitro cytarabine sensitivity in childhood acute
myeloid leukaemia,” Br. J. Cancer, vol. 93, no. 12, pp. 1388–1394, Dec. 2005.
[295] N. Urtasun, A. Vidal-Pla, S. Pérez-Torras, A. Mazo, and A. Mazo, “Human pancreatic cancer stem
cells are sensitive to dual inhibition of IGF-IR and ErbB receptors.,” BMC Cancer, vol. 15, p. 223,
Apr. 2015.
[296] A. Català, M. Pastor-Anglada, L. Caviedes-Cárdenas, R. Malatesta, S. Rives, N. Vega-García, M.
Camós, P. Fernández- Calotti, A. Català, M. Pastor-Anglada, L. Caviedes-Cárdenas, R. Malatesta,
S. Rives, N. Vega-García, M. Camós, and P. Fernández- Calotti, “FLT3 is implicated in cytarabine
transport by human equilibrative nucleoside transporter 1 in pediatric acute leukemia,”
Oncotarget, vol. 7, no. 31, pp. 49786–49799, Nov. 2014.
[297] M. C. Chillón, M. T. Gómez-Casares, C. E. López-Jorge, C. Rodriguez-Medina, A. Molines, M. E.
Sarasquete, M. Alcoceba, J. D. G.-S. Miguel, C. Bueno, R. Montes, F. Ramos, J. N. Rodríguez, P.
Giraldo, M. Ramírez, R. García-Delgado, J. L. Fuster, M. González-Díaz, and P. Menendez,
“Prognostic significance of FLT3 mutational status and expression levels in MLL-AF4+ and MLL-
germline acute lymphoblastic leukemia,” Leukemia, vol. 26, no. May, pp. 2360–2366, Nov. 2012.
[298] S. A. Armstrong, A. L. Kung, M. E. Mabon, L. B. Silverman, R. W. Stam, M. L. Den Boer, R. Pieters,
J. H. Kersey, S. E. Sallan, J. A. Fletcher, T. R. Golub, J. D. Griffin, and S. J. Korsmeyer, “Inhibition of
FLT3 in MLL. Validation of a therapeutic target identified by gene expression based
classification.,” Cancer Cell, vol. 3, no. 2, pp. 173–83, Feb. 2003.
[299] R. W. Stam, M. L. den Boer, P. Schneider, M. Meier, H. B. Beverloo, and R. Pieters, “D-HPLC
analysis of the entire FLT3 gene in MLL rearranged and hyperdiploid acute lymphoblastic
leukemia,” Haematologica, vol. 92, no. 11, 2007.
[300] F. Kuchenbauer, W. Kern, C. Schoch, A. Kohlmann, W. Hiddemann, T. Haferlach, and S.
Schnittger, “Detailed analysis of FLT3 expression levels in acute myeloid leukemia,”
Haematologica, vol. 90, no. 12, 2005.
[301] S. Leow, S. K.-Y. Kham, H. Ariffin, T. C. Quah, and A. E.-J. Yeoh, “FLT3 mutation and expression
did not adversely affect clinical outcome of childhood acute leukaemia-a study of 531 Southeast
Asian children by the Ma-Spore study group,” Hematol. Oncol., vol. 29, no. 4, pp. 211–219, Dec.
2011.
[302] M. Neumann, S. Heesch, N. Gökbuget, S. Schwartz, C. Schlee, O. Benlasfer, N. Farhadi-Sartangi, J.
Thibaut, T. Burmeister, D. Hoelzer, W.-K. Hofmann, E. Thiel, and C. D. Baldus, “Clinical and

BIBLIOGRAFIA
212
molecular characterization of early T-cell precursor leukemia: a high-risk subgroup in adult T-ALL
with a high frequency of FLT3 mutations.,” Blood Cancer J., vol. 2, no. 1, p. e55, Jan. 2012.
[303] J. D. Sandahl, E. A. Coenen, E. Forestier, J. Harbott, B. Johansson, G. Kerndrup, S. Adachi, A.
Auvrignon, H. B. Beverloo, J.-M. Cayuela, L. Chilton, M. Fornerod, V. de Haas, C. J. Harrison, H.
Inaba, G. J. L. Kaspers, D.-C. Liang, F. Locatelli, R. Masetti, C. Perot, S. C. Raimondi, K. Reinhardt,
D. Tomizawa, N. von Neuhoff, M. Zecca, C. M. Zwaan, M. M. van den Heuvel-Eibrink, and H.
Hasle, “t(6;9)(p22;q34)/DEK-NUP214-rearranged pediatric myeloid leukemia: an international
study of 62 patients,” Haematologica, vol. 99, no. 5, 2014.
[304] M. Gupta, J. Ashok Kumar, U. Sitaram, S. Neeraj, A. Nancy, P. Balasubramanian, A. Abraham, V.
Mathews, A. Viswabandya, B. George, M. Chandy, A. Srivastava, and V. M. Srivastava, “The
t(6;9)(p22;q34) in myeloid neoplasms: a retrospective study of 16 cases,” 2010.
[305] H. Hasle, “A critical review of which children with acute myeloid leukaemia need stem cell
procedures,” Br. J. Haematol., vol. 166, no. 1, pp. 23–33, Jul. 2014.
[306] H. Kang, C. S. Wilson, R. C. Harvey, I.-M. Chen, M. H. Murphy, S. R. Atlas, E. J. Bedrick, M.
Devidas, A. J. Carroll, B. W. Robinson, R. W. Stam, M. G. Valsecchi, R. Pieters, N. A. Heerema, J.
M. Hilden, C. A. Felix, G. H. Reaman, B. Camitta, N. Winick, W. L. Carroll, Z. E. Dreyer, S. P.
Hunger, and C. L. Willman, “Gene expression profiles predictive of outcome and age in infant
acute lymphoblastic leukemia: a Children’s Oncology Group study,” Blood, vol. 119, no. 8, 2012.
[307] S. Brunet, M. Labopin, J. Esteve, J. Cornelissen, G. Socié, A. P. Iori, L. F. Verdonck, L. Volin, A.
Gratwohl, J. Sierra, M. Mohty, and V. Rocha, “Impact of FLT3 internal tandem duplication on the
outcome of related and unrelated hematopoietic transplantation for adult acute myeloid
leukemia in first remission: a retrospective analysis.,” J. Clin. Oncol., vol. 30, no. 7, pp. 735–41,
Mar. 2012.
[308] R. W. Stam, I. Hubeek, M. L. den Boer, J. G. C. A. M. Buijs-Gladdines, U. Creutzig, G. J. L. Kaspers,
and R. Pieters, “MLL gene rearrangements have no direct impact on Ara-C sensitivity in infant
acute lymphoblastic leukemia and childhood M4/M5 acute myeloid leukemia,” Leukemia, vol.
20, no. 1, pp. 179–182, Jan. 2006.
[309] T. Kakihar, T. Fukuda, A. Tanaka, I. Emura, K. Kishi, K. Asami, and M. Uchiyama, “Expression of
Deoxycytidine Kinase (dCK) Gene in Leukemic Cells in Childhood: Decreased Expression of dCK
Gene in Relapsed Leukemia,” Leuk. Lymphoma, vol. 31, no. 3–4, pp. 405–409, Jan. 1998.
[310] R. Willemze, S. Suciu, G. Meloni, B. Labar, J.-P. Marie, C. J. M. Halkes, P. Muus, M. Mistrik, S.
Amadori, G. Specchia, F. Fabbiano, F. Nobile, M. Sborgia, A. Camera, D. L. D. Selleslag, F. Lefrère,
D. Magro, S. Sica, N. Cantore, M. Beksac, Z. Berneman, X. Thomas, L. Melillo, J. E. Guimaraes, P.
Leoni, M. Luppi, M. E. Mitra, D. Bron, G. Fillet, E. W. A. Marijt, A. Venditti, A. Hagemeijer, M.
Mancini, J. Jansen, D. Cilloni, L. Meert, P. Fazi, M. Vignetti,S. M. Trisolini, F. Mandelli, and T. de
Witte, “High-dose cytarabine in induction treatment improves the outcome of adult patients
younger than age 46 years with acute myeloid leukemia: results of the EORTC-GIMEMA AML-12
trial.,” J. Clin. Oncol., vol. 32, no. 3, pp. 219–28, Jan. 2014.

BIBLIOGRAFIA
213
[311] F. Locatelli, F. Moretta, S. Rutella, and S. Rutella, “Management of relapsed acute lymphoblastic
leukemia in childhood with conventional and innovative approaches.,” Curr. Opin. Oncol., vol.
25, no. 6, pp. 707–15, Nov. 2013.
[312] J. L. Byrne, E. Dasgupta, M. Pallis, J. Turzanski, K. Forman, D. Mitchell, A. P. Haynes, and N. H.
Russell, “Early allogeneic transplantation for refractory or relapsed acute leukaemia following
remission induction with FLAG.,” Leukemia, vol. 13, no. 5, pp. 786–91, May 1999.
[313] N. Sarper, N. Yalman, and N. Yalman, “FLAG (fludarabine, high-dose cytarabine and G-CSF) for
refractory and high-risk relapsed acute leukemia in children.,” Med. Pediatr. Oncol., vol. 34, no.
2, p. 163, Feb. 2000.
[314] N. Hijiya, E. Barry, and R. J. Arceci, “Clofarabine in pediatric acute leukemia: Current findings and
issues,” Pediatr. Blood Cancer, vol. 59, no. 3, pp. 417–422, Sep. 2012.
[315] M. Levis, J. Allebach, K.-F. Tse, R. Zheng, B. R. Baldwin, B. D. Smith, S. Jones-Bolin, B. Ruggeri, C.
Dionne, and D. Small, “A FLT3-targeted tyrosine kinase inhibitor is cytotoxic to leukemia cells in
vitro and in vivo,” Blood, vol. 99, no. 11, 2002.
[316] M. Stubbs and S. Armstrong, “FLT3 as a Therapeutic Target in Childhood Acute Leukemia,” Curr.
Drug Targets, vol. 8, no. 6, pp. 703–714, Jun. 2007.
[317] A. Sexauer, A. Perl, X. Yang, M. Borowitz, C. Gocke, T. Rajkhowa, C. Thiede, M. Frattini, G. E.
Nybakken, K. Pratz, J. Karp, B. D. Smith, and M. Levis, “Terminal myeloid differentiation in vivo is
induced by FLT3 inhibition in FLT3/ITD AML,” Blood, vol. 120, no. 20, 2012.
[318] M. R. Grunwald and M. J. Levis, “FLT3 Tyrosine Kinase Inhibition as a Paradigm for Targeted Drug
Development in Acute Myeloid Leukemia.,” Semin. Hematol., vol. 52, no. 3, pp. 193–9, Jul. 2015.
[319] M. R. Grunwald and M. J. Levis, “FLT3 inhibitors for acute myeloid leukemia: a review of their
efficacy and mechanisms of resistance,” Int. J. Hematol., vol. 97, no. 6, pp. 683–694, Jun. 2013.
[320] B. D. Smith, M. Levis, M. Beran, F. Giles, H. Kantarjian, K. Berg, K. M. Murphy, T. Dauses, J.
Allebach, and D. Small, “Single-agent CEP-701, a novel FLT3 inhibitor, shows biologic and clinical
activity in patients with relapsed or refractory acute myeloid leukemia,” Blood, vol. 103, no. 10,
2004.
[321] C. Soler, J. García-Manteiga, R. Valdés, J. Xaus, M. Comalada, F. J. Casado, M. Pastor-Anglada, A.
Celada, and A. Felipe, “Macrophages require different nucleoside transport systems for
proliferation and activation.,” FASEB J., vol. 15, no. 11, pp. 1979–88, Sep. 2001.
[322] R. Valdés, F. J. Casado, and M. Pastor Anglada, “Cell-cycle-dependent regulation of CNT1, a
concentrative nucleoside transporter involved in the uptake of cell-cycle-dependent nucleoside-
derived anticancer drugs.,” Biochem. Biophys. Res. Commun., vol. 296, no. 3, pp. 575–9, Aug.
2002.