Embed Size (px)
Citation preview

JOURNAL OF BACTERIOLOGY, July 1983, p. 113-121 Vol. 155, No. 10021-9193/83/070113-09$02.00/0Copyright 0 1983, American Society for Microbiology
Catabolism of Phenylpropionic Acid and Its 3-HydroxyDerivative by Escherichia coliRICH BURLINGAME AND PETER J. CHAPMAN*
Department of Biochemistry, College ofBiological Sciences, University of Minnesota, St. Paul, MinnesotaSS108
Received 5 January 1983/Accepted 12 April 1983
A number of laboratory strains and clinical isolates of Escherichia coli utilizedseveral aromatic acids as sole sources of carbon for growth. E. coli K-12 usedseparate reactions to convert 3-phenylpropionic and 3-(3-hydroxyphenyl)pro-pionic acids into 3-(2,3-dihydroxyphenyl)propionic acid which, after meta-fissionof the benzene nucleus, gave succinate, pyruvate, and acetaldehyde as products.Enzyme assays and respirometry showed that all enzymes of this branchedpathway were inducible and that syntheses of enzymes required to convert thetwo initial growth substrates into 3-(2,3-dihydroxyphenyl)propionate are underseparate control. E. coli K-12 also grew with 3-hydroxycinnamic acid as solesource of carbon; the ability of cells to oxidize cinnamic and 3-phenylpropionicacids, and hydroxylated derivatives, was investigated. The lactone of 4-hydroxy-2-ketovaleric acid was isolated from enzymatic reaction mixtures and its proper-ties, including optical activity, were recorded.
Most of our present knowledge of bacterialaromatic catabolism stems from investigationswith the genus Pseudomonas. Enteric organismshave also been studied on occasions (1, 19-21,29), but the ability of Escherichia coli to degradecertain aromatics was not realized until Cooperand Skinner (8) grew a strain of this organismwith 3- and 4-hydroxyphenylacetic acids anddelineated the catabolic pathway used. We nowreport that many laboratory strains and clinicalisolates of E. coli can grow with various aromat-ic acids. In particular, E. coli K-12 can growwith 3-phenylpropionic or 3-(3-hydroxyphenyl)-propionic acids as sources of carbon, and wedescribe in detail the reactions used for degrad-ing these compounds. This information shouldfacilitate studies of the genetics of aromaticcatabolism for a system that is well character-ized and amenable to genetic manipulations.
Portions of this work were reported previous-ly (R. Burlingame and P. J. Chapman, Abstr.Annu. Meet. Am. Soc. Microbiol. 1981, K150,p. 162).
MATERIALS AND METHODSBacterial strains and cel extracts. E. coli KK334, a
prototrophic derivative of strain K-12, was a gift fromJames Fuchs, University of Minnesota, St. Paul. Aci-netobacter sp. strain PC/4 (NCIB 9781), isolated in1963 by Dagley et al. (10), was originally described asAchromobacter sp., but has since been identified as aspecies of Acinetobacter in this laboratory by usingthe transformation assay of Juni (26). Clinical isolates
of E. coli were obtained from University of Minnesotahospitals; other E. coli laboratory strains were giftsfrom sources acknowledged. Cells were grown withaeration at 30°C in the basal medium of Hareland et al.(23) with the following additions, per liter: yeastextract, 0.05 g (Difco Laboratories, Detroit, Mich.);Casamino Acids, 0.05 g; and the aromatic acid servingas major source of carbon, 0.5 g. The yield of cells,harvested in midexponential phase, was 2 to 2.5 g (wetweight) per liter of culture; when not used immediatelythey were stored at -20°C. For growth on plates, thesame basal medium was used, yeast extract and Casa-mino Acids were omitted, and agar (1.5%) was added.Crude cell extracts containing 25 to 35 mg of protein
per ml of 0.05 M K+-Na+ phosphate buffer, pH 7.0,were prepared from cells broken in a Hughes bacterialpress as described previously (11, 37). Cell extractswere fractionated by slow addition of saturated(NH4)2SO4, pH 7, at 0°C. For the fraction designatedAS-1, the precipitate obtained at 20%o saturation with(NH4)2SO4 was removed by centrifugation, and thesupernatant solution was then brought to 309% satura-tion. This precipitate was collected and dissolved inthe same volume of 0.05 M phosphate buffer, pH 7, asthat of the original extract. Fractions designated AS-2and AS-3 were precipitated, respectively, between 20and 40% saturation and between 40 and 80%o saturationwith (NH4)2SO4.
Chemicals. 3-Phenylpropionic acid (Fig. 1, I), 3-hydroxycinnamic acid, 3-(4-hydroxyphenyl)propionicacid, and 3-(3,4-dihydroxyphenyl)propionic acid werefrom Aldrich Chemical Co. (Milwaukee, Wis.). Allenzymes and coenzymes were from Sigma ChemicalCo. (St. Louis, Mo.). 3-(2,3-Dihydroxyphenyl)pro-pionic acid (Fig. 1, IV) was synthesized by the proce-dure of Blakley and Simpson (4); samples of this
113

114 BURLINGAME AND CHAPMAN
COOH COOH] COOH COOH
OH LOH
IOH
HINON^NII2 COOHH/'NONENZYMATIC d\N
POOH COOHOH A OOH
V~OHMAJOR MINOR e H a2
COOH
COOH VII~ H3 .L COON H20 COON
CH3' 0 'H J 00 O
x Ix VIII VI
FIG. 1. Reaction sequence proposed for the degra-dation of 3-phenylpropionic acid and 3-(3-hydroxy-phenyl)propionic acid to succinate, acetaldehyde, andpyruvate by E. c6li K-12. The metabolites are asfollows: 3-phenylpropionic acid (I), cis-3-(3-carbox-yethyl)-3,5-cyclohexadiene-1,2-diol (II), 3-(3-hydroxy-phenyl)propionic acid (III), 3-(2,3-dihydroxyphen-yl)propionic acid (IV), 2-hydroxy-6-ketonona-2,4-dienedioic acid (V), 2-keto-4-pentenoic acid (enol)(VI), succinic acid (VII), 4-hydroxy-2-ketovaleric acid(VIII), acetaldehyde (IX), pyruvate (X). Enzymesreferred to in the text are given the symbols a throughg.
compound and of 2,3-dihydroxycinnamic acid weregenerous gifts from E. R. Blakley, National ResearchCouncil of Canada, Saskatoon, Saskatchewan. 3-(3-Hydroxyphenyl)propionic acid (Fig. 1, III) was pre-pared by catalytic hydrogenation of 3-hydroxycinna-mic acid in glacial acetic acid-ethyl acetate (3:1) withPtO2 as catalyst. The following compounds were pre-pared as previously described: 4-methyl-2-ketobutyro-lactone (33), 3-(2-hydroxyphenyl)propionic acid (36),and the ring-fission product, 2-hydroxy-6-ketonona-2,4-dienedioic acid (Fig. 1, V), the latter by using theenzymatic procedure of Dagley et al. (10). CompoundV was recrystallized from acetone-petroleum ether;solutions were also conveniently prepared directlyfrom compound IV by the action of heat-treated cellextract (10) ofAcinetobacter sp. strain PC/4. Solutionsof 2-keto-4-pentenoic acid (Fig. 1, VI, keto form) wereprepared by the procedure of Collinsworth et al. (7),and solutions of 4-hydroxy-2-ketovalexic acid (Fig. 1,VIII) were prepared by mild alkaline hydrolysis of 4-methyl-2-ketobutyrolactoxe (11).Enzyme assays. Hydroxylation of 3-(3-hydroxy-
phenyl)propionate (Fig. 1, enzyme c) was assayed byadding 0.4 ,umol of substrate to 3, ml of a solutioncontaining 150 Fmol of K+-Na+ phosphate buffer (pH7), 0.5 1Lmol of NADH, and 10 to 75 pl of crude cellextract. The cha4ge in absorbance at 340 nm wasmonitored and corrected for o*odation ofNADH in theabsence of substrate. Dioxygenase activity against 3-(2,3-dihydroxyphenyl)propionate (Fig. 1, enzyme d)was measured with an oxygen electrode (23); thereaction was started by adding 10 to 80 p.l of cellextract to a reaction mixture (1.5 ml) containing 0.4p.mol of substrate and 75 p.mol of air-saturated phos-
phate buffer, pH 8, at 30°C. Activity of the ring-fissionproduct hydrolase (Fig. 1, enzyme e) was assayed bymeasuring the decrease in absorbance of the substrate,compound V, at 394 nm (e = 15,600 at pH 8) uponaddition of 0.5 to 2.5 ,ul of cell extract to reactionmixtures (3 ml) containing 150 p.mol of phosphatebuffer, pH 8, and 0.25 p.mol of compound V. 2-Keto-4-pentenoate hydratase (Fig. 1, enzyme J) was assayedessentially as described by Collinsworth et al. (7). 4-Hydroxy-2-ketovalerate aldolase (enzyme g) was de-termined spectrophotometrically at 340 nm from therate of release of acetaldehyde from the substrate.Reaction mixtures (3 ml) contained 150 ,umol of phos-phate buffer, (pH 7.5), 10 p.mol of 4-hydroxy-2-keto-valerate, 0.5 p.mol of NADH, 7.5 U of alcohol dehy-drogenase, and 20 to 80 ,u1 of cell extract. Rates werecorrected for NADH oxidation in the absence ofsubstrate.Chromatography of catabolites. Compounds were
chromatographed on thin-layer silica gel plates (35).For phenolic acids, the solvent systems were as fol-lows:'A, benzene-acetic acid-water (25:5:1); and B,petroleum ether (bp 60 to 70°C)-acetone-acetic acid(75:25:1). Compounds were located by spraying withdiazotized p-nitroaniline (34) or with Gibbs reagent,2,6-dibromoquinone-4-chloroimide (38), followed by5% NaHCO3. Keto acids and aldehydes were chroma-tographed as their 2,4-dinitrophenylhydrazones in thefollowing solvents (6, 11): C, benzene-ethyl formate-propionic acid (18:1:1); D, benzene-ethyl formate-propionic acid (70:30:15.4); E, benzene-tetrahydrofu-ran-acetic acid (15:9:1); F, petroleum ether-diethylether (3:1); and G, cyclohexane-nitrobenzene-petro-leum ether (6:3:2).
Preparation of 4-methyl-2-ketobutyrolactone and suc-cdn!c.dd, uIng cell extract fraction AS-2. Fraction AS-2, which was essentially devoid of enzyme g (Fig. 1),degraded compound IV to compounds VII and VIII;the hydroxy acid was isolated as its lactone after acidtreatment. In the procedure described, compound IVwas exposed to the enzyme solution at near neutralityand was then diluted and shaken in air at pH 8, whenformation of ring-fission product V was evident fromthe appearance of a yellow color. Other proce4ures,such as adding compound IV initially at pH 8 in dilutesolution, were not successful since dark-colored reac-tion side products accumulated. The method adoptedconsisted in adding slowly, with stirring, 5 ml of anaqueous solution of 1.5 mmol of 3-(2,3-dihydroxy-phenyl)propionic acid (IV) to 24 ml of cell extractfraction AS-2. Before addition, the pH of the solutionof IV was brought to pH 5 with NaOH, and duringaddition it was kept at pH 7.0 to 7.5 with 0.1 N NaOH.The resulting solution was then diluted to 375 ml with0.05 M phosphate buffer, pH 8, when it became abright yellow color which subsequently faded. Gentleshaking caused a reappearance of the yellow color,and the solution was intermittently shaken until thereaction was complete as shqwn by no further changesin absorbance at 394 and 266 nm. Concentrated HCI(34 ml) was added, the solution was centrifuged, andthe supernatant was heated to 95C for 5 min toconvert compound VIII into its lactone; the solutionwas then extracted continuously with ether for 36 h.The ethereal solution was decanted from a semicrys-talline crust which was triturated several times withsmall volumes of warm benzene. The combined ether
J. BACTERIOL.

PHENYLPROPIONATE METABOLISM IN E. COLI 115
and benzene solutions were dried over MgSO4, sol-vent was removed under reduced pressure, and theresulting material was again triturated as before withbenzene. This benzene solution was decolorized withactivated charcoal and evaporated to dryness, and theresidue was sublimed at 60°C and 0.2 mmHg (26.6 Pa)to give 56.7 mg of 4-methyl-2-ketobutyrolactone. In asecond experiment designed to optimize succinateyields, ether extraction was continued for 4 days, andthe crust remaining after the first trituration wasdissolved in a minimum volume of hot ethyl acetate,decolorized, and crystallized at -20°C to give 43.6 mgof crystalline material, later identified as succinic acid,from 182 mg of compound IV.
Analytical methods. Nuclear magnetic resonancespectra were obtained with a Bruker 270-MHz spec-trometer. Procedures for mass spectrometry of com-pounds and their trimethylsilyl derivatives were aspreviously described (35); diazomethane from thebase-catalyzed decomposition of N-methyl-N'-nitro-sourea (2) was used to prepare methyl esters orphenolic ethers for mass spectrometry. Circular di-chroism measurements were made with a Jasco J-41Cspectropolarimeter.
RESULTS
Aromatic acids supporting growth ofE. coli. Inaddition to phenylacetic and 3- and 4-hydroxy-phenylacetic acids (8), we found that 3-phenyl-propionic, 3-(3-hydroxyphenyl)propionic, and 3-hydroxycinnamic acids could support growth ofvarious strains of E. coli (Table 1). None of thestrains tested was able to grow with 2-hydroxy-phenylacetic acid, cinnamic acid, or its 2- or 4-hydroxy derivatives, or with the 2- or 4-hydroxyderivatives of 3-phenylpropionic acid. Amongclinical isolates, 48 of 67 (72%) could use at leastone of these compounds for growth and 27 (40%)could use all six. Strains that could grow with 3-hydroxyphenylacetic acid could also grow with
4-hydroxyphenylacetic acid, consistent with thesuggestion of Cooper and Skinner (8) that asingle enzyme hydroxylates either isomer togive the ring-fission substrate 3,4-dihydroxy-phenylacetic acid. Moreover, all strains thatused 3-(3-hydroxyphenyl)propionate also grewwith 3-hydroxycinnamate, suggesting that thesame enzymes are used for catabolizing thesetwo compounds. Of the clinical isolates, all 38which grew with 3-phenylpropionic acid couldalso grow with 3-(3-hydroxyphenyl)-propionateand 3-hydroxycinnamate; 2 additional strainsthat grew with the latter failed to grow with 3-phenylpropionic acid.
Oxidation of aromatic acids by E. coli KK334.After growth (division time, 7.5 h at 30°C) with3-phenylpropionate (Fig. 1, I), washed cells ofE. coli KK334 readily oxidized the growth sub-strate and also 3-(3-hydroxyphenyl)propionate(III), 3-(2,3-dihydroxyphenyl)propionate (IV), 3-hydroxycinnamate, and 2,3-dihydroxycinna-mate (Table 2). When grown with either 3-(3-hydroxyphenyl)propionate (division time, 3.5 hat 30°C) or 3-hydroxycinnamate, cells readilyoxidized the alternative growth substrate, and inaddition the 2,3-dihydroxy derivatives of 3-phenylpropionic and cinnamic acids were alsooxidized; however, unsubstituted 3-phenylpro-pionate (I) was oxidized at a much lower rate:evidently growth with compound III fails toinduce enzymes responsible for initial attackupon compound I.
Identification and isolation of compounds pres-ent in culture filtrates. A 7-liter portion of medi-um, centrifuged free from cells grown with 3-phenylpropionic acid, was saturated with NaCl,acidified to pH 2 with H2SO4, and extractedthree times with one-tenth volume of diethyl
TABLE 1. Growth properties of E. coli strainsAromatic acids utilized for growth
E. coli strain e Ned Phenylacetic 3-Hydroxy- 4-Hydroxy- 3-Phenyl- 3-(3-Hydroxy- 3-Hydroxy-examned Phenacida phenylacetic phenylacetic propionic phenyl)propionic cinnamicacid acid acida acid acid'
B 1 - + + + + +C 1 - + + + + +W 2 + + + + + +K-12 2 + - - + + +NCTC 5928 1 + - - + + +Clinical isolates (n = 67) 19
5 + - - _ _ _2 - - - - + +8 - - - + + +1 - + + _ _ _2 + + + _ _ _3 + - - + + +
27 + + + + + +
Growth can be slow with some strains at 37°C but is more rapid at 30°C.b Growth is slow at 37°C and is accompanied by reddish-brown coloration of the medium.
VOL. 155, 1983

116 BURLINGAME AND CHAPMAN
TABLE 2. Oxidation of aromatic acids by whole cells of E. coli KK334Rate of oxygen consumption (p. of 02 h- mg [dry wtf-') after
growth withb:Substrate oxidizeda 3-Phenyl- 3-(3-Hydroxy- 3-Hydroxy-
propionic phenyl)pro- cinnamicacid pionic acid acid
3-Phenylpropionic acid 240 0 25Cinnamic acid 205 0 253-(3-Hydroxyphenyl)propionic acid 293 267 1653-Hydroxycinnamic acid 205 267 1303-(2,3-Dihydroxyphenyl)propionic acid 227 240 2452,3-Dihydroxycinnamic acid 227 240 245
a Reaction vessels contained, in a total volume of 2.8 ml: 20 ,umol of phosphate buffer (pH 7.0), 140 pg ofchloramphenicol, 20 mg (wet weight) of cells, and 3 ,umol of substrate. The center well contained 0.2 ml of 20%KOH.
b Corrected for endogenous respiration rate.
ether. The ethereal solution was washed with asmall volume of water and dried over MgSO4.Solvent was removed by evaporation under re-duced pressure. The residue was washed withsmall samples of ice-cold water, the aqueoussolution was re-extracted with ether, and theextract was dried over MgSO4. Solvent wasremoved by rotary evaporation, and the result-ing dark oil was applied to preparative thin-layerchromatography plates and developed twice insolvent A. Three major banks (Rf = 0.29, 0.23,0.16) were visible under UV light and when anarrow lane was sprayed with diazotized p-nitroaniline. These bands were scraped from theplate, eluted with ethyl acetate, and evaporatedto dryness, leaving dark oils which could not berecrystallized. When subjected to thin-layerchromatography in solvents A and B, thesecompounds had, respectively, the same Rf val-ues and colors with diazotized p-nitroaniline asauthentic samples of 3-(2-hydroxyphenyl)pro-pionic, 3-(3-hydroxyphenyl)propionic (III), and3-(2,3-dihydroxyphenyl)propionic (IV) acids.The identity of each isolate was confirmed bygas chromatography-mass spectrometry. Themass spectra (at 20 eV) of each methyl ester andtrimethylsilyl derivative, prepared from eachone of the three samples of biological origin,were essentially identical to those of the corre-sponding derivatives prepared from the threerespective authentic aromatic acids. For 3-(2-hydroxyphenyl)propionic acid, either biologicalor reference compound, the methyl ester gavethe following major peaks: mle (relative intensi-ty) = 180 (15), 149 (15), 148 (100), 120 (68), 107(11), and 91 (15). The trimethylsilyl derivativehad major peaks at: mle = 310 (42), 295 (49), 253(25), 192 (82), 179 (20), 177 (21), 147 (100), and 73(27). For 3-(3-hydroxyphenyl)propionic acid(III), the methyl ester gave peaks at mle = 180(72), 149 (17), 120 (100), and 107 (26). Thetrimethylsilyl derivative had major peaks at: mle
= 310 (63), 205 (67), 192 (100), and 177 (14). For3-(2,3-dihydroxyphenyl)propionic acid IV, themethyl ester gave peaks at: mle = 224 (100), 193(9), 167 (19), 164 (48), 151 (35), and 136 (17). Thetrimethylsilyl derivative gave peaks at: mle =398 (100), 383 (62), 179 (87), 147 (37), and 73 (25).The spectra of the derivatives of the three com-pounds were clearly distinguished from those ofthe derivatives of their isomers, namely, 3-(4-hydroxyphenyl)-propionic and 3-(3,4-dihydrox-yphenyl)propionic acids. No evidence was ob-tained, either from chromatography or massspectrometry, for the presence of either of theseisomers in culture fluids.
Activities of ceil extracts. (i) Conversion ofcompounds I and III into compound IV. Dioxy-genase (d) (Fig. 1) was inhibited at concentra-tions of 2,2'-bipyridyl that did not significantlyaffect enzymes (a, b, or c); accordingly, theaccumulation of ring-fission substrate IV fromits precursors could be investigated. Reactionmixtures (7 ml) containing 7 ,umol of substrate,14 ,umol of 2,2'-bipridyl, 35 ,umol of NADH, 23mg of cell extract protein, and 350 ,tmol ofphosphate buffer, pH 7.3, were incubated inErlenmeyer flasks with reciprocal shaking at26°C. Samples (1 ml) were removed at 0, 5, 10,20, 30, and 60 min from the start of the reaction,and each was added to 1 ml of 10%o metaphos-phoric acid. Solutions were cooled to 4°C, cen-trifuged to remove protein, and extracted threetimes with 3-ml portions of diethyl ether; ex-tracts were then dried over MgSO4, filtered, andevaporated under a stream of dry air. The resi-due from each fraction was dissolved in 0.1 ml ofethyl acetate, and approximately 10 ,ul of eachsolution was chromatographed on silica gel,using solvent A. Phenolic acids were locatedwhen sprayed with Gibbs reagent; reaction con-trols, from which aromatic acids were omitted assubstrates for the cell extract, gave no visiblespots.
J. BACTERIOL.
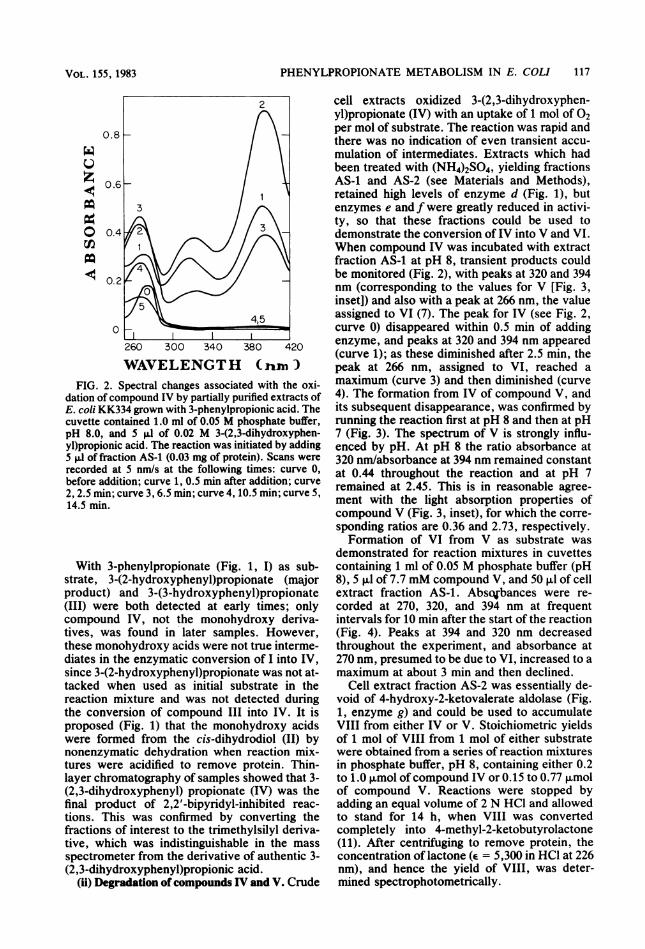
PHENYLPROPIONATE METABOLISM IN E. COLI 117
¢
u
¢
;Q
0(A)
260 300 340 380 420
WAVELENGTH (nim)FIG. 2. Spectral changes associated with the oxi-
dation of compound IV by partially purified extracts ofE. coli KK334 grown with 3-phenylpropionic acid. Thecuvette contained 1.0 ml of 0.05 M phosphate buffer,pH 8.0, and 5 p.l of 0.02 M 3-(2,3-dihydroxyphen-yl)propionic acid. The reaction was initiated by adding5 pJ of fraction AS-1 (0.03 mg of protein). Scans wererecorded at 5 nm/s at the following times: curve 0,before addition; curve 1, 0.5 min after addition; curve2, 2.5 min; curve 3, 6.5 min; curve 4, 10.5 min; curve 5,14.5 min.
With 3-phenylpropionate (Fig. 1, I) as sub-strate, 3-(2-hydroxyphenyl)propionate (majorproduct) and 3-(3-hydroxyphenyl)propionate(III) were both detected at early times; onlycompound IV, not the monohydroxy deriva-tives, was found in later samples. However,these monohydroxy acids were not true interme-diates in the enzymatic conversion of I into IV,since 3-(2-hydroxyphenyl)propionate was not at-tacked when used as initial substrate in thereaction mixture and was not detected duringthe conversion of compound III into IV. It isproposed (Fig. 1) that the monohydroxy acidswere formed from the cis-dihydrodiol (II) bynonenzymatic dehydration when reaction mix-tures were acidified to remove protein. Thin-layer chromatography of samples showed that 3-(2,3-dihydroxyphenyl) propionate (IV) was thefinal product of 2,2'-bipyridyl-inhibited reac-tions. This was confirmed by converting thefractions of interest to the trimethylsilyl deriva-tive, which was indistinguishable in the mass
spectrometer from the derivative of authentic 3-(2,3-dihydroxyphenyl)propionic acid.
(ii) Degradation ofcompounds IV and V. Crude
cell extracts oxidized 3-(2,3-dihydroxyphen-yl)propionate (IV) with an uptake of 1 mol of 02per mol of substrate. The reaction was rapid andthere was no indication of even transient accu-mulation of intermediates. Extracts which hadbeen treated with (NH4)2SO4, yielding fractionsAS-1 and AS-2 (see Materials and Methods),retained high levels of enzyme d (Fig. 1), butenzymes e and f were greatly reduced in activi-ty, so that these fractions could be used todemonstrate the conversion of IV into V and VI.When compound IV was incubated with extractfraction AS-1 at pH 8, transient products couldbe monitored (Fig. 2), with peaks at 320 and 394nm (corresponding to the values for V [Fig. 3,inset]) and also with a peak at 266 nm, the valueassigned to VI (7). The peak for IV (see Fig. 2,curve 0) disappeared within 0.5 min of addingenzyme, and peaks at 320 and 394 nm appeared(curve 1); as these diminished after 2.5 min, thepeak at 266 nm, assigned to VI, reached amaximum (curve 3) and then diminished (curve4). The formation from IV of compound V, andits subsequent disappearance, was confirmed byrunning the reaction first at pH 8 and then at pH7 (Fig. 3). The spectrum of V is strongly influ-enced by pH. At pH 8 the ratio absorbance at320 nm/absorbance at 394 nm remained constantat 0.44 throughout the reaction and at pH 7remained at 2.45. This is in reasonable agree-ment with the light absorption properties ofcompound V (Fig. 3, inset), for which the corre-sponding ratios are 0.36 and 2.73, respectively.Formation of VI from V as substrate was
demonstrated for reaction mixtures in cuvettescontaining 1 ml of 0.05 M phosphate buffer (pH8), 5 ,ul of 7.7 mM compound V, and 50 ,ul of cellextract fraction AS-1. Absc%bances were re-corded at 270, 320, and 394 nm at frequentintervals for 10 min after the start of the reaction(Fig. 4). Peaks at 394 and 320 nm decreasedthroughout the experiment, and absorbance at270 nm, presumed to be due to VI, increased to amaximum at about 3 min and then declined.
Cell extract fraction AS-2 was essentially de-void of 4-hydroxy-2-ketovalerate aldolase (Fig.1, enzyme g) and could be used to accumulateVIII from either IV or V. Stoichiometric yieldsof 1 mol of VIII from 1 mol of either substratewere obtained from a series of reaction mixturesin phosphate buffer, pH 8, containing either 0.2to 1.0 ,umol ofcompound IV or 0.15 to 0.77 ,umolof compound V. Reactions were stopped byadding an equal volume of 2 N HCl and allowedto stand for 14 h, when VIII was convertedcompletely into 4-methyl-2-ketobutyrolactone(11). After centrifuging to remove protein, theconcentration of lactone (e = 5,300 in HCl at 226nm), and hence the yield of VIII, was deter-mined spectrophotometrically.
VOL. 15S, 1983
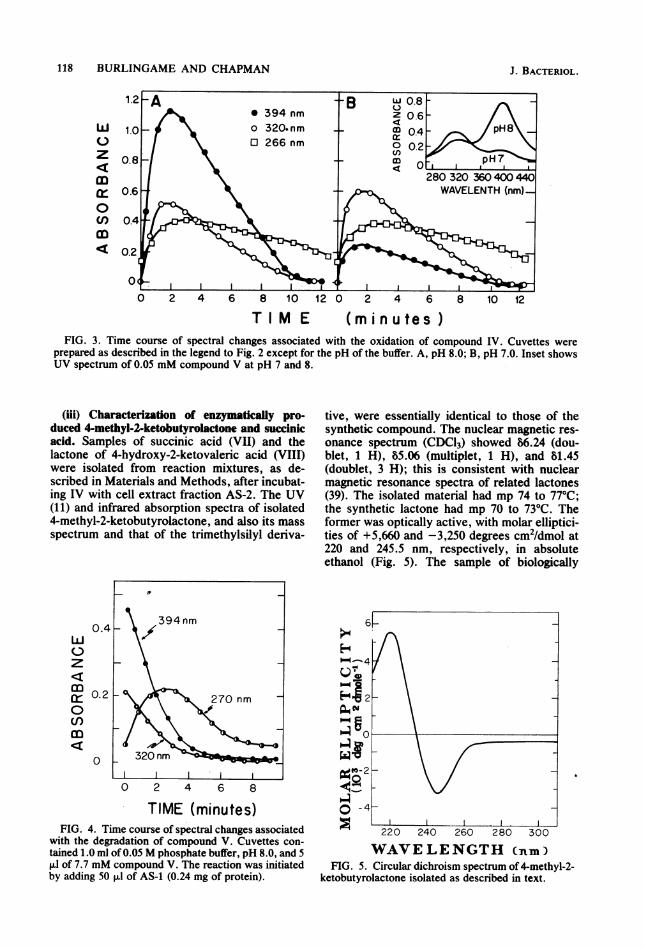
118 BURLINGAME AND CHAPMAN
W 1.0 0 320.nm 04q pH8o ~~~~0266nm c0.2
Z 0.8 Ff pH71220Inmt = <4°0z U)~~~~~~~~280 320 360 400 440
c 0.6 WAVELENTH (nm)-0
C/) 0.4 t
0 2 4 6 8 10 12 0 2 4 6 8 10 12
T I M E (minutes)FIG. 3. Time course of spectral changes associated with the oxidation of compound IV. Cuvettes were
prepared as described in the legend to Fig. 2 except for the pH of the buffer. A, pH 8.0; B, pH 7.0. Inset showsUV spectrum of 0.05 mM compound V at pH 7 and 8.
(iii) Characterization of enznymatically pro-duced 4-methyl-2-ketobutyrolactone and succinicacid. Samples of succinic acid (VII) and thelactone of 4-hydroxy-2-ketovaleric acid (VIII)were isolated from reaction mixtures, as de-scribed in Materials and Methods, after incubat-ing IV with cell extract fraction AS-2. The UV(11) and infrared absorption spectra of isolated4-methyl-2-ketobutyrolactone, and also its massspectrum and that of the trimethylsilyl deriva-
0.4LLu0z
m( 0.2
U)mo
TIME (minutes)FIG. 4. Time course of spectral changes associated
with the degradation of compound V. Cuvettes con-tained 1.0 ml of 0.05 M phosphate buffer, pH 8.0, and 5RI of 7.7 mM compound V. The reaction was initiatedby adding 50 ,ul of AS-1 (0.24 mg of protein).
tive, were essentially identical to those of thesynthetic compound. The nuclear magnetic res-onance spectrum (CDC13) showed 86.24 (dou-blet, 1 H), 85.06 (multiplet, 1 H), and 81.45(doublet, 3 H); this is consistent with nuclearmagnetic resonance spectra of related lactones(39). The isolated material had mp 74 to 77°C;the synthetic lactone had mp 70 to 73°C. Theformer was optically active, with molar elliptici-ties of +5,660 and -3,250 degrees cm2/dmol at220 and 245.5 nm, respectively, in absoluteethanol (Fig. 5). The sample of biologically
220 240 260 280 300
WAVELENGTH (nm)FIG. 5. Circular dichroism spectrum of 4-methyl-2-
ketobutyrolactone isolated as described in text.
J. BACTERIOL.
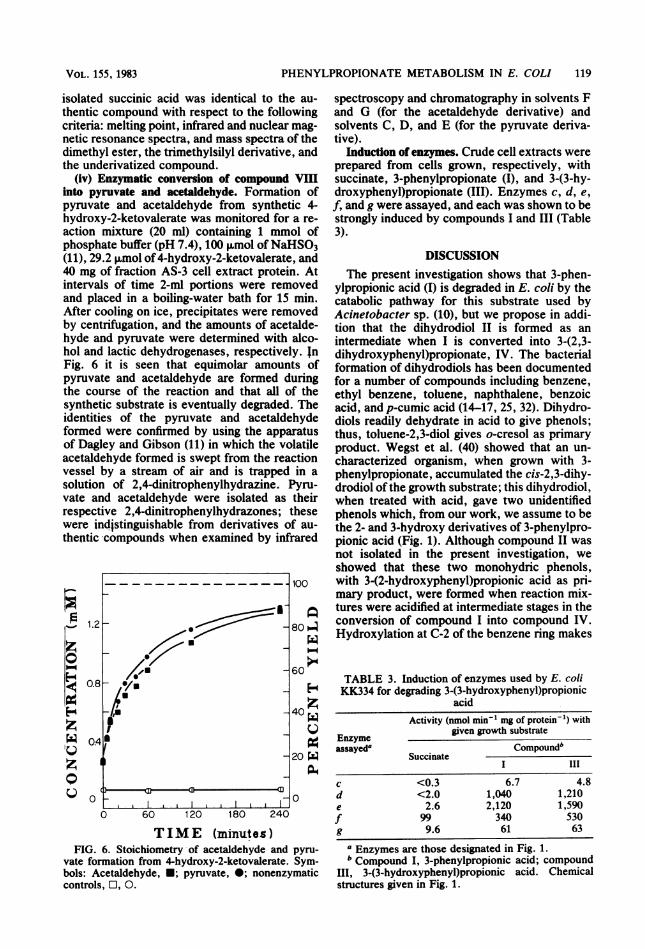
PHENYLPROPIONATE METABOLISM IN E. COLI 119
isolated succinic acid was identical to the au-thentic compound with respect to the followingcriteria: melting point, infrared and nuclear mag-netic resonance spectra, and mass spectra of thedimethyl ester, the trimethylsilyl derivative, andthe underivatized compound.
(iv) Enzymatic conversion of compound VIIIinto pyruvate and acetaldehyde. Formation ofpyruvate and acetaldehyde from synthetic 4-hydroxy-2-ketovalerate was monitored for a re-action mixture (20 ml) containing 1 mmol ofphosphate buffer (pH 7.4), 100 ,umol ofNaHSO3(11), 29.2 giwmol of4-hydroxy-2-ketovalerate, and40 mg of fraction AS-3 cell extract protein. Atintervals of time 2-ml portions were removedand placed in a boiling-water bath for 15 min.After cooling on ice, precipitates were removedby centrifugation, and the amounts of acetalde-hyde and pyruvate were determined with alco-hol and lactic dehydrogenases, respectively. inFig. 6 it is seen that equimolar amounts ofpyruvate and acetaldehyde are formed duringthe course of the reaction and that all of thesynthetic substrate is eventually degraded. Theidentities of the pyruvate and acetaldehydeformed were confirmed by using the apparatusof Dagley and Gibson (11) in which the volatileacetaldehyde formed is swept from the reactionvessel by a stream of air and is trapped in asolution of 2,4-dinitrophenylhydrazine. Pyru-vate and acetaldehyde were isolated as theirrespective 2,4-dinitrophenylhydrazones; thesewere indistinguishable from derivatives of au-thentic compounds when examined by infrared
1.2 F-z
09
E-4zriuz9Q
0I I
100
80 4
0-
60
40 Z
20 Wp.4
0
0 60 120 180 240
TIME (minutes)FIG. 6. Stoichiometry of acetaldehyde and pyru-
vate formation from 4-hydroxy-2-ketovalerate. Sym-bols: Acetaldehyde, *; pyruvate, 0; nonenzymaticcontrols, O, 0.
spectroscopy and chromatography in solvents Fand G (for the acetaldehyde derivative) andsolvents C, D, and E (for the pyruvate deriva-tive).
Induction of enzymes. Crude cell extracts wereprepared from cells grown, respectively, withsuccinate, 3-phenylpropionate (I), and 3-(3-hy-droxyphenyl)propionate (III). Enzymes c, d, e,f, and g were assayed, and each was shown to bestrongly induced by compounds I and III (Table3).
DISCUSSIONThe present investigation shows that 3-phen-
ylpropionic acid (I) is degraded in E. coli by thecatabolic pathway for this substrate used byAcinetobacter sp. (10), but we propose in addi-tion that the dihydrodiol II is formed as anintermediate when I is converted into 3-(2,3-dihydroxyphenyl)propionate, IV. The bacterialformation of dihydrodiols has been documentedfor a number of compounds including benzene,ethyl benzene, toluene, naphthalene, benzoicacid, and p-cumic acid (14-17, 25, 32). Dihydro-diols readily dehydrate in acid to give phenols;thus, toluene-2,3-diol gives o-cresol as primaryproduct. Wegst et al. (40) showed that an un-characterized organism, when grown with 3-phenylpropionate, accumulated the cis-2,3-dihy-drodiol of the growth substrate; this dihydrodiol,when treated with acid, gave two unidentifiedphenols which, from our work, we assume to bethe 2- and 3-hydroxy derivatives of 3-phenylpro-pionic acid (Fig. 1). Although compound II wasnot isolated in the present investigation, weshowed that these two monohydric phenols,with 3-(2-hydroxyphenyl)propionic acid as pri-mary product, were formed when reaction mix-tures were acidified at intermediate stages in theconversion of compound I into compound IV.Hydroxylation at C-2 of the benzene ring makes
TABLE 3. Induction of enzymes used by E. coliKK334 for degrading 3-(3-hydroxyphenyl)propionic
acidActivity (nmol min-1 mg of protein-') with
given growth substrate
assayed' CompoundbSuccinate
I III
c <0.3 6.7 4.8d <2.0 1,040 1,210e 2.6 2,120 1,590f 99 340 530g 9.6 61 63
a Enzymes are those designated in Fig. 1.b Compound I, 3-phenylpropionic acid; compound
III, 3-(3-hydroxyphenyl)propionic acid. Chemicalstructures given in Fig. 1.
f:~~~~aU1
VOL. 155, 1983

120 BURLINGAME AND CHAPMAN
available the reactions of Fig. 1 to 3-(3-hydroxy-phenyl)propionate (III). This hydroxylase hasnot been characterized, but Levy (27) and Stick-land and Massey (36) purified and examinedflavoproteins from Arthrobacter sp. and Pseu-domonas sp., respectivley, that oxidize 3-(2-hydroxyphenyl)propionate to compound IV.The latter is also the ring-fission substrate whentrans-cinnamic acid is metabolized by Pseudo-monas sp. (4), so that in this organism thesubstrate is reduced before hydroxylation andring-fission occur. None of the strains of E. coliwe examined could grow with cinnamate, al-though whole cells ofE. coli KK334 when grownwith 3-phenylpropionate, rapidly oxidized thissubstrate (Table 2) to completion (data notshown), suggesting that the same enzymes areinvolved in the degradation of both compounds.Further, cells grown with 3-(3-hydroxyphen-yl)propionate failed to oxidize either 3-phenyl-propionate or cinnamate. When 3-hydroxycin-namate and 2,3-dihydroxycinnamate were oxi-dized by 3-phenylpropionate-grown cells, atransient yellow compound was formed havingthe special characteristics of a ring-fission prod-uct. Hlowever, these properties differed signifi-cantly from those of compound V, suggestingthat side chain reduction of the cinnamate deriv-atives of Table 2 does not take place before ring-fission.Cooper and Skinner (8) suggested that the
strains of E. coli they used could grow with 4-hydroxyphenylacetic acid because this com-pound, being a product of anaerobic degradationof tyrosine, would be encountered in fecal envi-ronments. 3-Phenylpropionic and 3-(3-hydroxy-phenyl)propionic acids are likewise compoundsthat have been found in the urine and feces ofanimals. They are formed by the action of intes-tinal microflora on plant constituents such asferulic and caffeic acids, the flavonoids catechin,myricetin, and hesperetin, and the amino acidsphenylalanine and tyrosine and certain of theirmetabolites (3, 5, 9, 12, 13, 18, 22, 24, 30, 31). Itthus seems likely that E. coli encounters thecompounds of Table 1 in the animal'gut.
Biologically formed 4-hydroxy-2-ketovalerate(VIII) was not isolated by previous investiga-tors. As expected, the physical properties of ourisolated compound were essentially the same asthose of the synthetic, except for melting point,which was several degrees higher: this is com-monly observed for other compounds with cen-ters of asymmetry.The present work, with that of Cooper and
Skinner (8), shows that E. coli can degrade atleast six aromatic acids. All of the laboratorystrains and 72% of the clinical isolates testedcould grow with at least one of these acids.Reaction sequences for aromatic catabolism are
initiated by oxygenases, and this ability to acti-vate dioxygen- suggests a wider natural distribu-tion for E. coli than the anaerobic environment(28) of the animal gut.
ACKNOWLEDGMENTSThis research was supported by Public Health Service grant
ES AI 00678 from the National Institute of EnvironmentalHealth Sciences. R.B. wag supported by Public Health Ser-vice National Research Service Award T32 GM 07323 fromthe National Institutes of Health.We are grateful to Tom Krick for skilled assistance in the
operation of the mass spectrometry facilities provided andmaintained by the Minnesota Agricultural Experiment Sta-tion. We thank James Puchs, Huber Warner, D. Peter Snus-tad, and Marlys Lund for strains; E. R. Blakley for chemicals;Placida Venegas for technical assistance; and Dan Kunz andMark Donnelly for helpful discussions. We are especiallyindebted to Stanley Dagley for reviewing the manuscript.
LITERATURE CITED
1. Adachl, T., Y. Murooka, and T. Harada. 1973. Derepres-sion of arylsulfatase synthesis in Aerobacter aerogenes bytyramine. J. Bacteriol. 116:19-24.
2. Arndt, F. 1943. Diazomethane, p. 165-167. In A. H. Blatt(ed.), Organic syntheses, collective Vol. 2. John Wiley &Sons, Inc., New York.
3. Bakke, 0. M. 1971. Degradation of DOPA by intestinalmicroorganisms in vitro. Acta Pharmacol. Toxicol.30:115-121.
4. Blakley, E. R., and J. F. Simpson. 1963. The microbialmetabolism of cinnamic acid. Can. J. Microbiol. 10:175-185.
5. Bonrd, O., T. Mldvodt, and L. R. Gjesang. 1973. Pheno-lic metabolism in urine and faeces from rats given radioac-tive '4C-L-DOPA. Acta Pharmacol. Toxicol. 33:308-316.
6. Byrne, G. A. 1965. The separation of 2,4-dinitrophenylhy-drazones by thin layer chromatography. J. Chromatogr.20;528-540.
7. Colllnsworth, W. L., P. J. Chapman, and S. Dagley. 1973.Stereospecific enzymes in the degradation of aromaticcompounds by Pseudomonas putida. J. Bacteriol.113:922-931.
8. Cooper, R. A., and M. A. Skinner. 1980. Catabolism of 3-and 4-hydroxyphenylacetate by the 3,4-dihydroxyphenyl-acetate pathway in Escherichia coli. J. Bacteriol. 143:302-306.
9. Curtius, H. C., M. Mettler, and L. Ettllnger. 1976. Studyof the intestinal tyrosine metabolism using stable isotopesand gas chromatography-mass spectrometry. J. Chroma-togr. 126:569-580.
10. Dagley, S., P. J. Chapman, and D. T. Gibson. 1965. Themetabolism of 3-phenylpropionic acid by an Achromo-bacter. Biochem. J. 97:643-650.
11. Dagley, S., and D. T. Gibson. 1965. The bacterial degrada-tion of catechol. Biochem. J. 95:466-474.
12. Das, N. P. 1971. Studies on flavonoid metabolism: adsorp-tion and metabolism of (+)-catechin in man. Biochem.Pharmacol. 20:3435-3445.
13. Das, N. P., and S. P. Sothy. 1971. Studies of flavonoidmetabolism: biliary and urinary excretion of metabolitesof (+)-[U-m4C]catechin. Biochem. J. 125:417-423.
14. DeFrank, J. J., and D. W. Ribbons. 1977. p-Cymenepathway in Pseudomonas putida: initial reactions. J.Bacteriol. 129:1356-1364.
15. Gibson, D. T., B. Gschwendt, W. K. Yeh, and V. M.Kobal. 1973. Initial reactions in the oxidation of ethylben-zene by Pseudomonas putida. Biochemistry 12:1520-1528.
16. Gibson, D. T., M. Hensley, H. Yoshoka, and T. J. Mabry.1970. Formation of (+)-cis-2,3-dihydroxy-1-methylcyclo-hexa4,6-diene from toluene by Pseudomonas putida.Biochemistry 9:1626-1630.
J. BACTERIOL.

PHENYLPROPIONATE METABOLISM IN E. COLI 121
17. Gibson, D. T., J. R. Koch, and R. E. Kallio. 1968. Oxida-tive degradation of aromatic hydrocarbons by microorga-nisms. I. Enzymatic formation of catechol from benzene.Biochemistry 7:2653-2662.
18. Goldin, B. R., M. A. Peppercorn, and P. Goldman. 1973.Contributions of host and intestinal microflora in themetabolism of L-DOPA by the rat. J. Pharmacol. Exp.Ther. 186:160-166.
19. Grant, D. J. W. 1967. Kinetic aspects of the growth ofKlebsiella aerogenes with some benzenoid carbonsources. J. Gen. Microbiol. 46:213-224.
20. Grant, D. J. W. 1970. The oxidative degradation of ben-zoate and catechol by Klebsiella aerogenes (Aerobacteraerogenes). Antonie van Leeuwenhoek J. Microbiol.Serol. 36:161-177.
21. Grant, D. J. W., and J. C. Patel. 1969. The non-oxidativedecarboxylation of p-hydroxybenzoic acid, gentisic acid,protocatechuic acid, and gallic acid by Klebsiella aero-genes (Aerobacter aerogenes). Antonie van Leeuwen-hoek J. Microbiol. Serol. 35:325-343.
22. GrUliths, L. A., and G. E. Smith. 1972. Metabolism ofmyricetin and related compounds in the rat. Metaboliteformation in vivo and by the intestinal microflora in vitro.Biochem. J. 130:141-151.
23. Hareland, W. A., R. L. Crawford, P. J. Chapman, and S.Dagey. 1975. Metabolic function and properties of 4-hydroxyphenylacetic acid 1-hydroxylase from Pseudomo-nas acidovorans. J. Bacteriol. 121:272-285.
24. lIonohan, T., R. L. Hale, J. P. Brown, and R. E. Wino-grad, Jr. 1976. Synthesis and metabolic fate of hesperitin-3-14(. J. Agric. Food Chem. 24:906-911.
25. Jeffrey, A. M., H. J. C. Yeh, D. M. Jerina, T. R. Patel,J. F. Davey, and D. T. Gibson. 1975. Initial reactions inthe oxidation of naphthalene by Pseudomonas putida.Biochemistry 14:575-584.
26. Juni, E. 1972. Interspecies transformation of Acineto-bacter: genetic evidence for a ubiquitous genus. J. Bacte-riol. 112:917-931.
richia coli and the human gut: some ecological consider-ations. J. Appl. Bacteriol. 51:1-16.
29. Patel, J. C., and D. J. W. Grant. 1969. The formation ofphenol in the degradation of p-hydroxybenzoic acid byKlebsiella aerogenes (Aerobacter aerogenes). Antonievon Leeuwenhoek J. Microbiol. Serol. 35:53-64.
30. P crn, M. A., and P. Goldman. 1971. Caffeic acidmetabolism by bacteria of the human gastrointestinaltract. J. Bacteriol. 108:996-1000.
31. Poilltt, R. J. 1974. Phenylpropionic acid in the urine ofpatients with phenylketonuria and normals. Clin. Chim.Acta 55:317-322.
32. Relner, A. M., and G. D. Hegeman. 1971. Metabolism ofbenzoic acid by bacteria. Accumulation of (-)-3,5-cyclo-hexadiene-1,2-diol-1-carboxylic acid by a mutant strain ofAlcaligenes eutrophus. Biochemistry 10:2530-2536.
.33. Rossi, A., and H. Schinz. 1948. Alcuni a-cheto-y-lattonicon sostituenti alchilici in posizione -y. Helv. Chim. Acta31:473-492.
34. Smith, I. 1960. Phenolic acids, p. 291-307. In I. Smith(ed.), Chromatographic and electrophoretic techniques,vol. 1. William Heinmann, London.
35. Sparnins, V. L., P. J. Chapman, and S. Dagley. 1974.Bacterial degradation of 4-hydroxyphenylacetic acid andhomoprotocatechuic acid. J. Bacteriol. 120:159-167.
36. Stickland, S., and V. Massey. 1973. The purification andproperties of the flavoprotein melilotate hydroxylase. J.Biol. Chem. 248:2944-2952.
37. Tack, B. F., P. J. Chapman, and S. Dagley. 1972. Purifica-tion and properties of 4-hydroxy-4-methyl-2-oxoglutaratealdolase. J. Biol. Chem. 247:6444-6449.
38. Waddi, D. 1965. Spray reagents for thin-layer chromatog-raphy, p. 483-502. In E. Stahl (ed.), Thin-layer chroma-tography. Academic Press, Inc., New York.
- 39. Wasserman, H. H., and S. L. Ives. 1978. Reaction ofsinglet oxygen with eneamino lactones. Conversion oflactones to a-keto lactones. J. Org. Chem. 43:3238-3240.
27. Levy, C. C. 1967. Melilotate hydroxylase. Purification of 40. Wegst, W., U. Tittemann, J. Eberspacher, and F. Lingens.the enzyme and the nature of the prosthetic group. J. Biol. 1981. Bacterial conversion of phenylalanine and aromaticChem. 242:747-753. carboxylic acids into dihydrodiols. Biochem. J. 194:679-
28. Mason, T. G., and G. Richardson. 1981. A review. Esche- 684.
VOL. 155, 1983





























