Embed Size (px)
DESCRIPTION
Central nervous system malignancies. Commonest malignant solid tumors in childhood 20% of cancers in age < 15 years Annually 20 – 26/ 1 million children below the age of 16 years - PowerPoint PPT Presentation
Citation preview

Central nervous system malignancies

• Commonest malignant solid tumors in childhood• 20% of cancers in age < 15 years• Annually 20 – 26/ 1 million children below the
age of 16 years• Age –stratified incidence is:
<1year - 27/ 1 million 1 – 4 - 31/ 1 million 5 – 9 - 27/ 1 million 10 – 14 - 20/ 1 million
• Slightly higher frequency in boys 1.25:1 (especially for medulloblastoma and germinoma)
Epidemiology

Etiology and pathogenesis
• Association between primary CNS tumors and following conditions/ genetic disorders : 1. Neurofibromatosis (NF) type 1 and 2 2. Tuberous sclerosis 3. Von Hippel- Lindau syndrome 4. Gordlin’s, Cowden’s, Turcot’s syndromes 5. Li-Fraumeni syndrome (mutation of suppressor oncogene p53)
• Deletion of chromosome 17 or 20 (medulloblastoma)• Exposition of the brain to ionizing radiation i.e. after
cranial radiotherapy in leukemia

Pathology• Supratentorial lesions (30 – 40%):
1. Cerebral hemisphere ( astrocytoma, ependymoma, glioblastoma, meningioma) 2. Sella or chiasm ( craniopharyngioma, pituitary adenoma, optic nerve glioma)
• Infratentorial lesions (60 – 70%) 1. Cerebellum (medulloblastoma, astrocytoma, meningioma) 2. Brain stem ( astrocytoma, ependymoma, glioblastoma)

Classification
• Based on histogenesis and predominance of cell type
• Degree of malignancy is defined by grading system e.i. WHO grade based on cellular morphology, mitotic index, anaplasia and necrosis: grades I and II represent benign tumors grades III and IV - malignant tumors

Clinical presentation
• Depends on : -age -anatomical site -tumor type
• -raised intracranial pressure (ICP) -localizing neurological deficits

Signs of increased ICP
• Direct tumor infiltration
• Compression of normal structures
• Secondary to obstruction of the cerebrospinal fluid (CSF)

• Older children: -inilially behavioural changes and declining school performance prior to development of the more classical features of headache, nausea and vomiting , headaches start as generalized and intermittent > increase in both intensity and frequency with time - the child may awake with headache at night, with the pain generally being worse in the morning and improving during the day with an upright posture -School-age children complain of visual disturbances

• Infants and younger children: plasticity of the developing skull and inability to communicate symptoms > -infant may be irritable, with failure to thrive, associated with anorexia and vomiting -regression of developmental milestones -increase head circumference with widened sutures and a tense anterior fontanelle „sun-setting” sign

Symptoms and signs according to anatomical site of CNS tumors
• Supratentorial (30-40%) Cerebral hemisphere- hemiparesis, spasticity, seizures (focal or generalized) - para/suprasellar – endocrinopathy (growth failure, diabetes insipidus, pubertal abnormality) - hypothalamus – diencephalic syndrome (infants), developmental and behavioural abnormalities - optic pathway – visual field acuity, color vision deficits, optic atrophy, nystagmus, head tilt - pineal - Parinaud’s syndrome, sleep abnormalities - thalamus, basal ganglia – pain, sensory loss, memory disturbances
- intraventicular - meningeal

• Infratentorial (60 – 70%) - posterior fossa – ataxia, nystagmus, dysmetria (presents as clumsiness or worse handwriting) - brainstem – multiple cranial nerve palsies, hemiparesis, spasticity, mood changes
• Spinal (2 – 5%) - primary intramedullary – pain (local back and root pain), motor and sensory disturbance - spinal metastases – scoliosis, sphincter (bowel, bladder) disturbances, reflex changes

Diagnostic evaluation
• Magnetic resonance and computed tomography – basic imaging techniques for brain tumors
• Positron emission tomography – help to distinguish tumors or lesions with a volume greater than 1 cm3
• Conventional radiography of the skull: bone structure, separating off sutures ( due to ICP), calcification within the brain
• Special methods (for special indications): - brain scintigraphy - angiography -ultrasonography -myelography

Additional diagnosis
• Cerebral fluid analysis ( to determine spread of the tumor to the spinal fluid)
• Electroencephalography
• Stereotactic biopsy

Therapy
• Neurosurgery for maximum tumor removal and low morbidity depending on the location and extent of the tumor -often preoperative relief of intracranial pressure by ventriculoperitoneal or ventriculoarterial shunt - preoperative reduction of tumor edema by corticosteroids - in patients with seizures - anticonvulsive therapy

• Radiotherapy – extension and volume of irradiation depend on the biology and histology of the tumor, age of the child and combination with chemotherapy and neurosurgery - irradiation in children < 3 years of life only in special cases
• Chemotherapy – depends on tumor type, location and age -efficacy and penetration depend on vascularization of the tumor
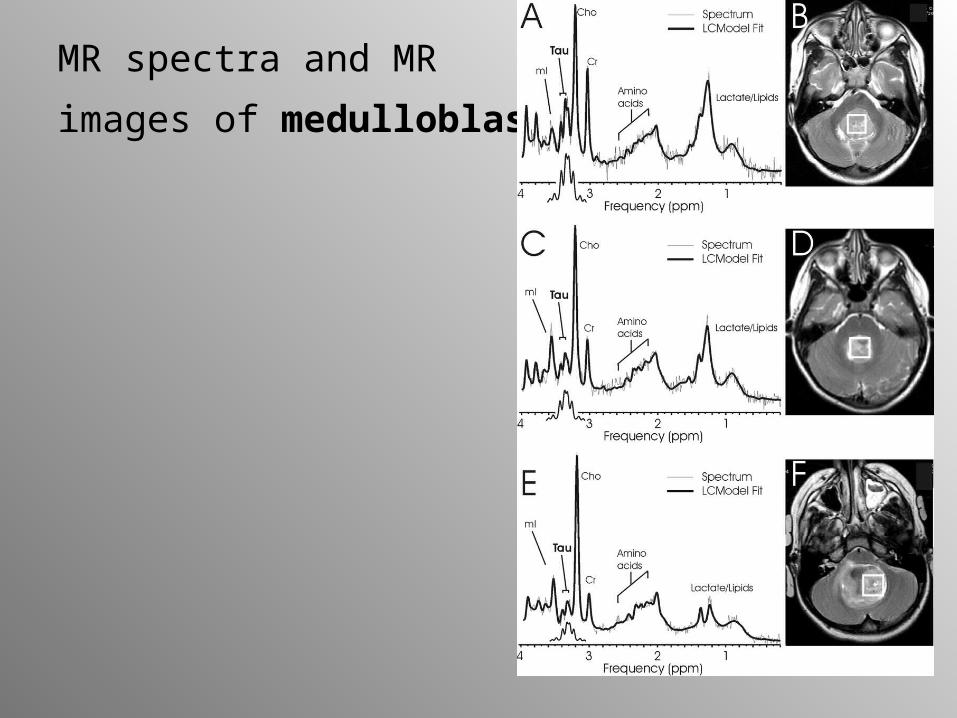
MR spectra and MR
images of medulloblastoma
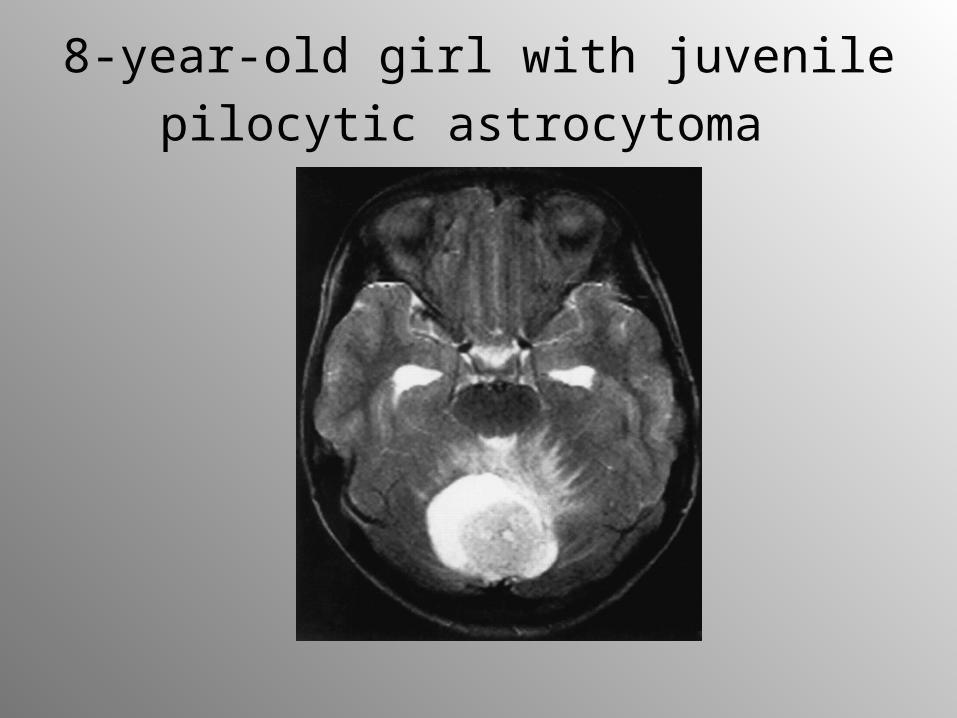
8-year-old girl with juvenile pilocytic
astrocytoma
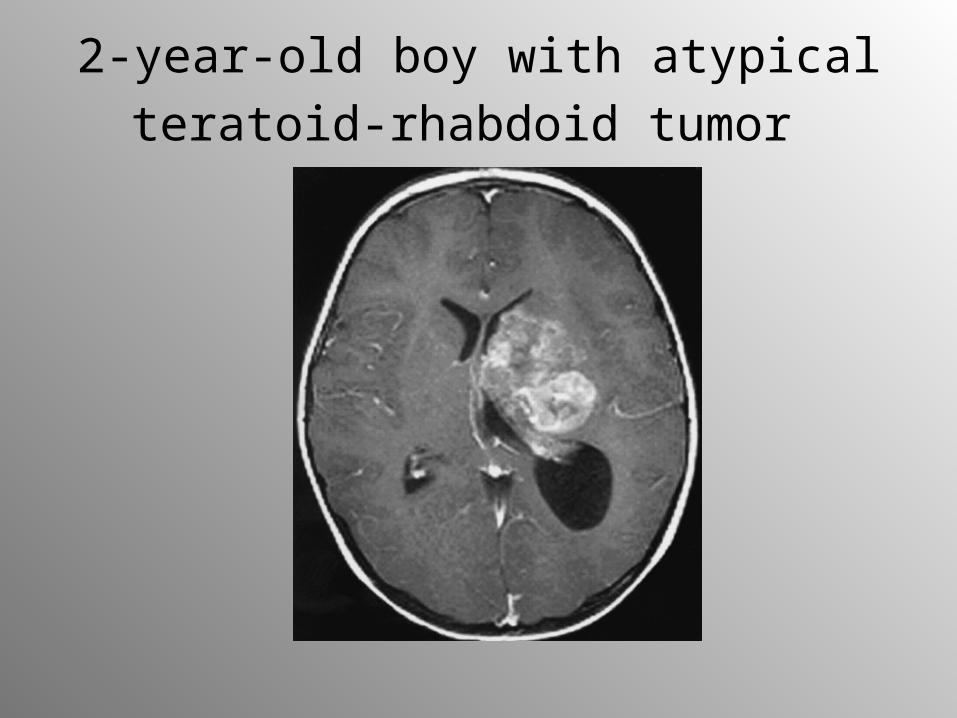
2-year-old boy with atypical teratoid-
rhabdoid tumor

Neuroblastoma

• Malignant, embryonal tumor derived from precursor cells of sympathetic ganglia and adrenal medulla
• Other types of tumors derived from sympathetic nervous system: -ganglioneuroblastoma -ganglioneuroma -pheochromocytoma
• Possibility to spontaneous regression and differentiation to benign tumor in infants less 1 year of age extremely malignant in older children

Epidemiology
• 8% of all neoplasms in children
• Most frequent malignant neoplasm in infants
• Mean age at diagnosis 2 – 5 years

Pathology
Two distinct entities: • Infant:
1. possibility of spontaneous regression (apoptosis or differentiation into ganglioneuroblastoma) 2. chemosensitive, chemocurable
• Older 3. chemoresistant malignancies

Molecular cytogenetics• MYCN oncogene amplification. MYCN is located on chromosome 2p.
Independent prognostic factor: in stage III EFS for patients with a single copy is curable 80%; for those with amplification MYCN – 20%
• DNA ploidy: hyperploidy = good prognosis
• Nerve growth factor receptor: ligands for high –affinity tyrosine kinase receptors TRKA, TRKB, TRKC: -TRKA expression is associated with MYCN single copy, low stage and good prognosis - TRKA (-) + MYCN amplification= very poor survival
• Structural and numerical abnormalities of chromosome 1

Clinical manifestation
• Occurence in any area with sympathetic nervous system Primary location: -abdomen 65% -adrenal medulla or sympathetic ganglia 46% -posterior mediastinum 15% -pelvic 4% -head and neck 3% -others 8%

Common symptoms
• Weight loss• Fever • Abdominal disturbances• Irratability• Pain of bones and joints• Child not stand up, not walk• Pallor• Lassitude

Symptoms associated with catecholamine production
• Paroxysmal attacks of sweating, flushing, pallor
• Headache
• Hypertension
• Palpitation

Paraneoplastic syndromes
• VIP syndrome: untreatable diarrhea,with low level of potassium
• Opsoclonus- myoclonus• Anemia, trombocytopenia, leukopenia
( in bone marrow infiltration or massive hemorrhage)

Local symptoms
• Abdomen: -intra-abdominal tumor
–paravertebral and presacral -neurological dysfunction -abdominal distension• Liver:
-hepatomegaly• Chest, posterior mediastinum, vertebrae:
-compression of trachea > coughing, dyspnea -infiltration in vertebral foramina > dumbbell tumor -compression of nerves >disturbances of gait, muscle weakness, parasthesia, bladder dysfunction, constipation
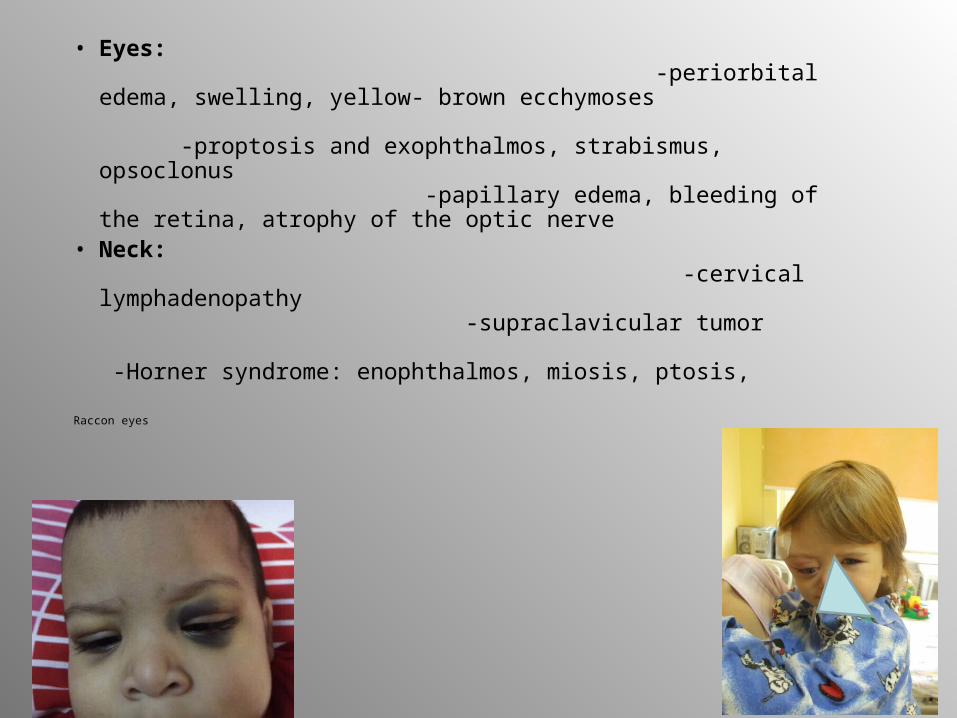
• Eyes: -periorbital edema, swelling, yellow- brown ecchymoses -proptosis and exophthalmos, strabismus, opsoclonus -papillary edema, bleeding of the retina, atrophy of the optic nerve
• Neck: -cervical lymphadenopathy -supraclavicular tumor -Horner syndrome: enophthalmos, miosis, ptosis,
Raccon eyes

• Skin: - subcutaneous nodules of blue color > reddish > white owing to vasoconstriction from release of catecholamines after palpation - nodules are mainly observed in neonates or infants with disseminated NBL
• Bone: -pain involvement mainly in the skull and long bones - in X-rays – lytic defects with irregular margins and periosteal reaction
• Bone marrow: -trombocytopenia, anemia

Metastases
• Lymphatic and/or hematogenous spread
• Often initially present in children (40 – 50% children < 1 year and 70% children > 1 year)
• Metastatic spread mostly in bone marrow, bone, liver, skin
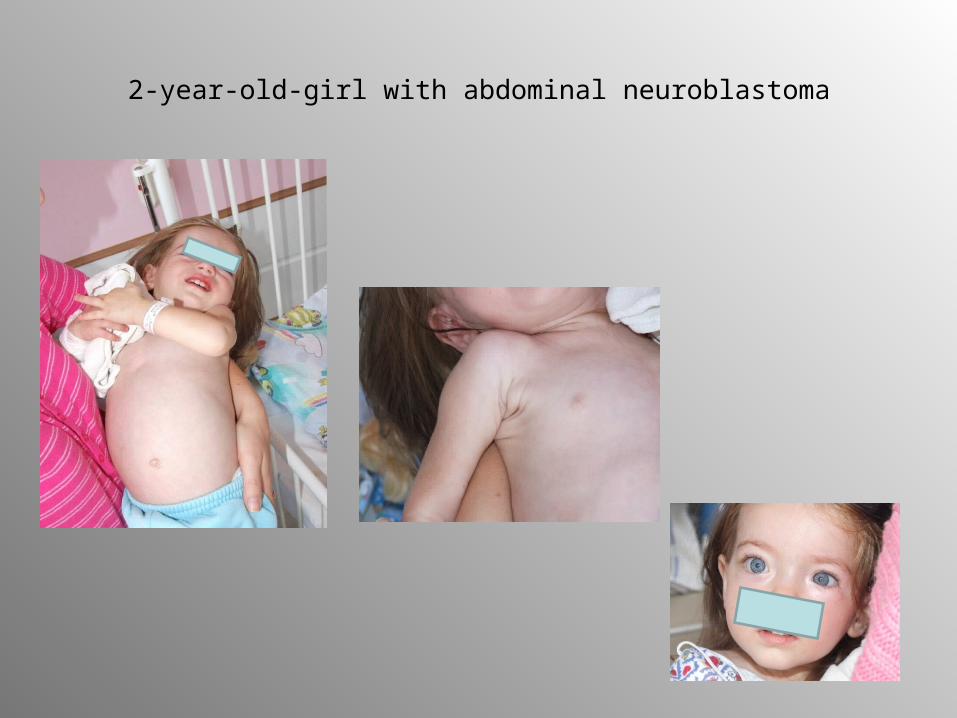
2-year-old-girl with abdominal neuroblastoma
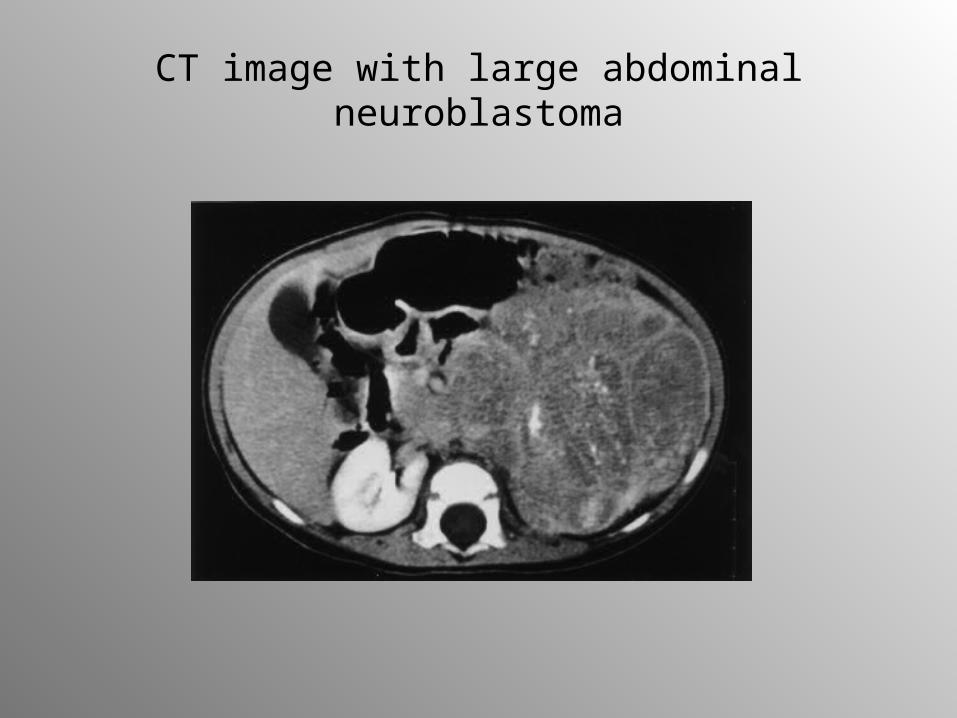
CT image with large abdominal neuroblastoma
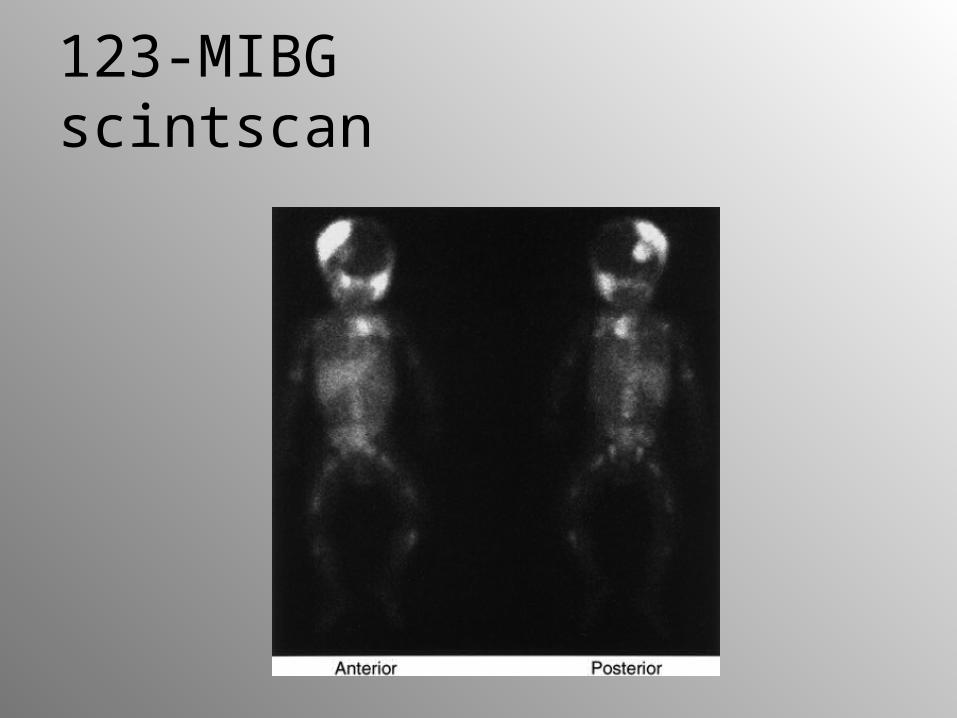
123-MIBG scintscan

International Staging System for NBL(INSS)
• 1 - localized tumor with complete excision,lymph nodes negative
• 2a - localized tumor without incomplete gross excision, representative, ipsilateral nonadherent lymph nodes negative for tumor microscopically
• 2b – ipsilateral nonadherent lymph nodes positive for tumor. Enlarged contralateral lymph nodes negative microscopically
• 3 – unresectable unilateral tumor infiltrating across the midline,with or without regional lymph node involvement or localized unilateral tumor with contralateral regional lymph node involvement or – midline tumor with bilateral extension by infiltration or by lymph node involvement
• 4 – any primary tumor with dissemination to distant lymph nodes, bone, bone marrow, liver, skin or other organs (except as defined for stage 4S
• 4s localized primary tumor (as defined for stages 1, 2a, 2b) with dissemination limited to skin, liver or bone marrow; limited to infants aged less than 1 year)

Laboratory findings
• Tumor markers: -catecholamines: vanillylmandelic acid (VMA), homovanillic acid (HVA) dopamine in urine/ plasma adrenaline, noradrenaline -neuron-specific enolase (glycolitic enzyme of brain and neuroendocrine tissues) -NSE
• Ferritin • Lactate dehydrogenease (LDH) • Bone marrow (aspiration and biopsy)

Locoregional involvement
• Computed tomography scan and/or • Ultrasound and/or• magnetic resonance imaging →
localize the mass, provide measurements, give anatomical information about intra- and extraperitoneal structures, differentiate cystic from solid tumors, define the extent of a primary tumor and its relationship with other structures, detect small calcification
Evaluation of metastases
• Bone marrow metastases – bone marrow aspiration and trephine biopsy
• Skeletal metastases - X-ray, Tc-99 scintigraphy,
• mIBG scintigraphy demonstrates primary, residual tumor masses,diffuse bone marrow infiltration, skeletal, lymph node and soft tissue metastases
• FDG-PET scanning

Therapy
Depends on: age, stage, localization and molecular features at diagnosis
• Surgery• ChemotherapyIV stage• Radiotherapy• Target radiotherapy (I-131-mIBG)• Differentiation therapy (retinoids 13-cis and all-trans)• Immunotherapy: anti-GD2 antibodies• Auto-BMT

Prognosis
• Depends on: -age (favorable if less than 18 months of age at diagnosis), -stage and localization (favorable in primary NBL of thorax, presacral and cervical) -involvement of lymph nodes (poor prognosis)
• Low-risk group - 90% long-term survival• Intermediate and high-risk groups:
-response to initial treatment 60-70% of children with complete or partial remission -after consolidation therapy (high-dose chemotherapy+ autologous stem cell support) –EFS after 3 years is 40-60%

Hepatic tumors

• Malignant: -hepatoblastoma 43% -hepatocellular carcinoma 23% -sarcoma 6%
• Benign: -vascular (haemangioma,haemangioendothelioma) 13% -hamartoma 6% -others 9%

Incidence (malignant)
• -1.2 –5% of all neoplasms in childhood
• Boys: girls 1.4 –2 :1• High incidence in genetically associated syndromes:
-Beckwith-Wiedemann syndrome, familial adematous polyposis, trisomy 18, glycogen storage disease, hereditary tyrosinemia, Li-Fraumeni syndrome
• HBL –mostly in infants, rarely after the age of 3 years• Hepatocellular Ca – in older children, in adolescents,
after hepatitis B
Clinical symptoms
• Abdominal mass and/ or abdominal distention• Anorexia, weight loss• Lethargy• Jaundice and ascites• Abdominal pain• Pallor
• Precocious puberty (hepatoblastoma), virilization (due to gonadotrophin production by the tumor)
• Generalized osteoporosis (HBL)- tumor cells secrete osteoclast-activating factor

Laboratory diagnosis
• Serum AFP elevated in 80 - 90% of children with HBL and in 60-70% with hepatocellular Ca
• B-HCG elevated in both • Increased bilirubin • Increased serum aspartate aminotransferase (AST),
alanine transaminase (ALT) because of associated hepatitis or cirrhosis
• Anemia, thrombocytosis, trombocytopenia (rarely)• biopsy
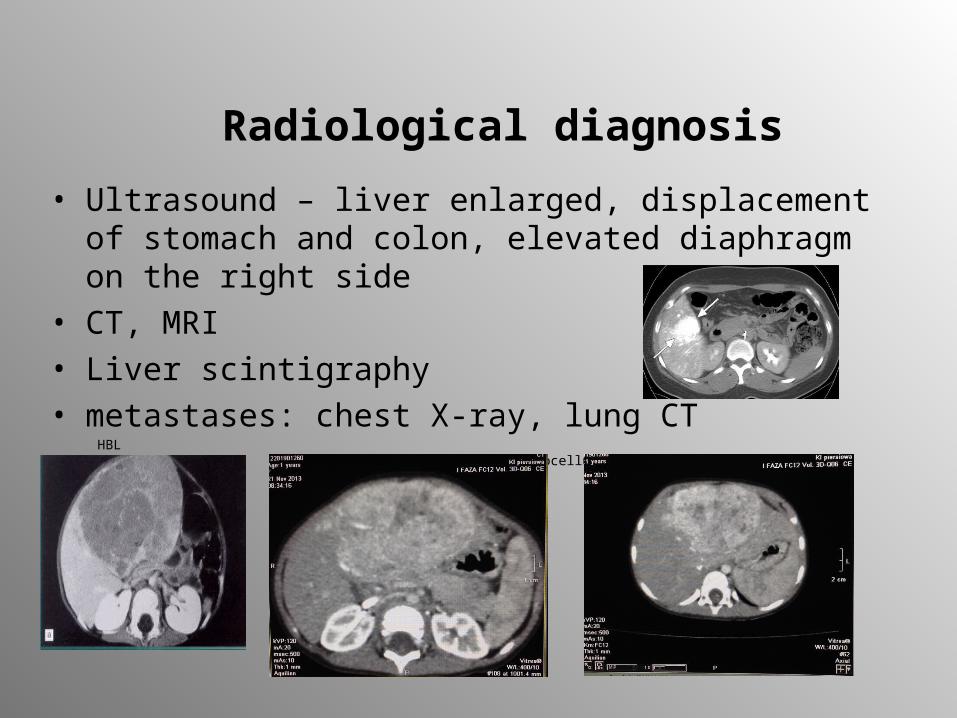
Radiological diagnosis
• Ultrasound – liver enlarged, displacement of stomach and colon, elevated diaphragm on the right side
• CT, MRI• Liver scintigraphy• metastases: chest X-ray, lung CT
HBL hepatocellular carcinoma

Treatment
• Presurgical chemotherapy (makes HBL resectable)• Complete resection (initially > 50% of liver tumors are
not totally resectable)• Liver transplantation
Prognosis: • Depends on stage of the tumor
after complete tumor resection and chemotherapy 65-75 % children with HBL and 40-60% with hepatocellular Ca survive

Germ cell tumors (GCT)

• Develop from embryonal germ cells• Represent ectodermal, endodermal and mesodermal
lineages• Approximately 3% of childhood malignancies• Annually 2.4 – 3.8/ 1 million/per year• 2/3 GCTs in children occur in extragonadal sites• The commonest GCT - sacrococcygeal teratoma• Bimodal age distribution:
one peak in children < 3y ears of age (sacrococcygeal tumors, yolk sac tumors of testis) - the later- the later GCT of the ovary, testis, and intracranial sites

Histological classification of gonadal and extragonadal tumors
• Germ cell and germinoma/ dysgerminoma and embryonal yolk sac tumor (pluripotent cells) a)extraembryonic structures - yolk sac or endodermal sinus tumor - choriocarcinoma b) embryonal ecto-, meso-, endodermal origin tissues represented -teratoma c) embryonal carcinoma
• Gonadal germ cells and stroma tumor (Sertoli and Leydig cells) • Epithelial cells (ovarian origin) and granulosa cell tumor or mixted
form as well as epithelial cell tumors more common in adults

Clinical symptoms
• Testicular GCT :scrotal enlargement,hydrocele• Ovarian tumors: abdominal pain, acute abdomen,
abdominal mass• Extragonadal GCT: main sites of involvement:
-sacrococcygeal: t.1- (47%) predominantly external, t.2 -both external and intrapelvic t.3 -external, pelvix and abdominal t.4 –entitely presacral, -mediastinal (dyspnea, wheezing, thoracic pain, superior vena cava syndrome), -intracranial (visual disturbances, diabetes insipidus, hypopituitarism, anorexia, precocious puberty)
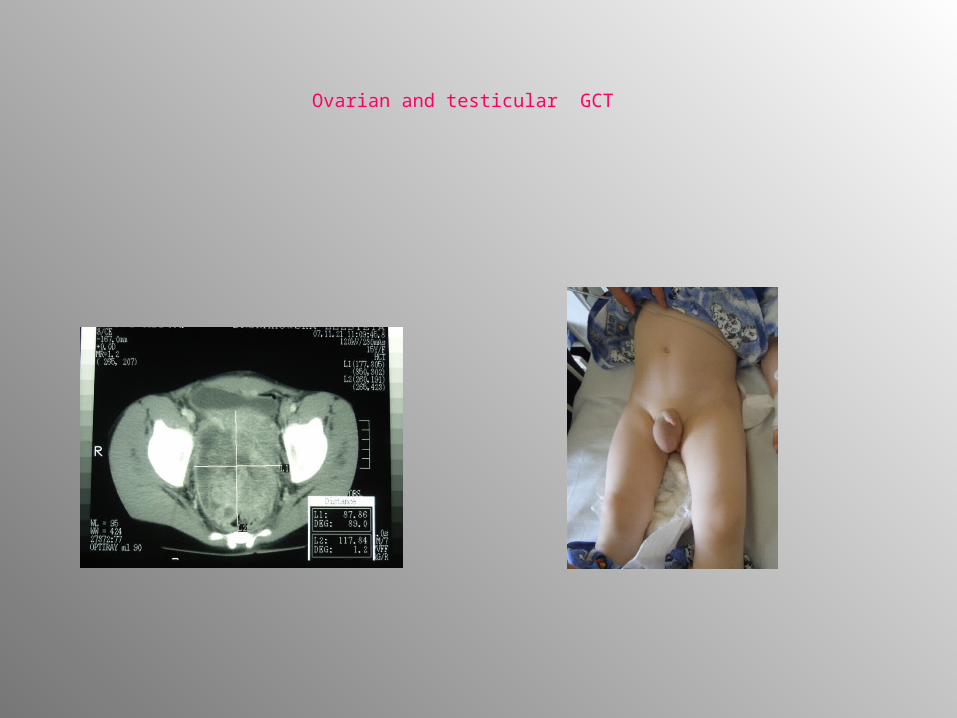
Ovarian and testicular GCT
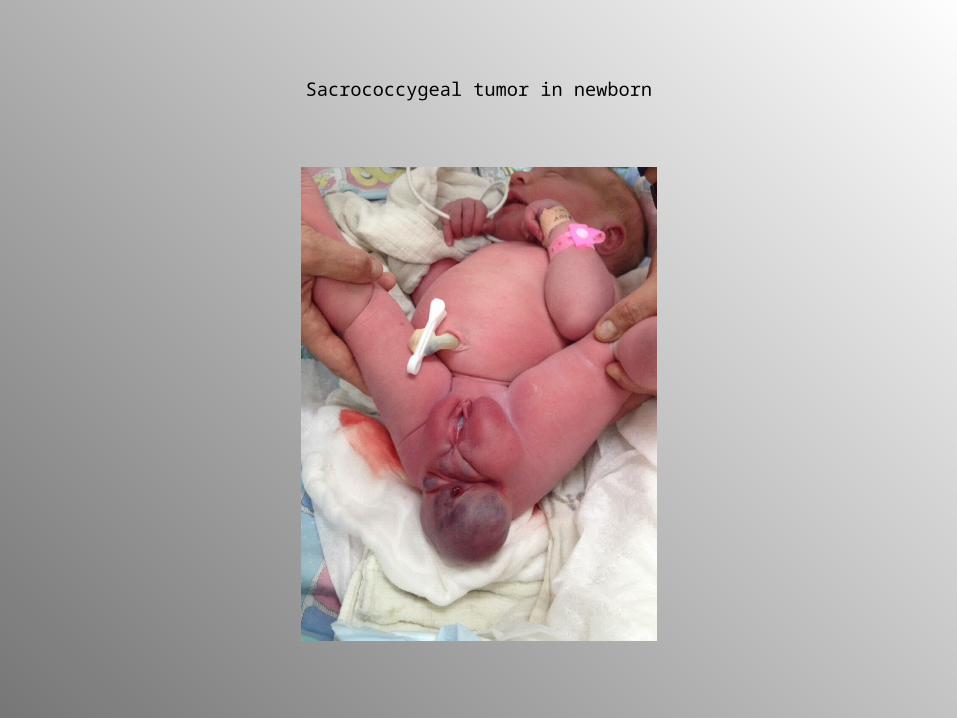
Sacrococcygeal tumor in newborn

Diagnostics
Radiological diagnosis: ultrasound, computed tomography or magnetic resonance imaging
• Tumor markers: alpha-fetoprotein (AFP)- globulin produced in the fetal yolk sac, in embryonal hepatocytes and in gastrointestinal tract. Increase in malignant GCT, hepatoblastoma
• Newborns 48,000+/-34,000IU• Up to 1 month 9,000+/-12,000• Up to 2 months 320 +/-280• Up to 4 months 74 +/-56• Up to 6 months 12+/-10• Up to 8 months 8+/-5
beta -human chorionic gonadotropin (HCG) – increase in germinoma/ dysgerminoma, choriocarcinoma

Therapy
• Depend on:• stage, location, tumor markers level• Surgery in stage I• Advanced stages: chemotherapy, surgery, radiotherapy
Survival
95% - sacrococcygeal teratoma
80% -yolk sac tumor

Wilms’ tumor
Nephroblastoma

• Malignant embryonal tumor of renal tissue consisting of varying proportion of blastema, stroma and epithelium
• Epidemiology: 6% of all neoplasms in children 80 % of children at diagnosis are less than 5 years old peak incidence between 2nd and 3rd year of age - incidence slightly higher in boys

Genetics
Chromosomal association:• Chromosome 11p13 with Wilms tumor suppressor gene WT1
in 10-30% of nephroblastoma • Chromosome 11p15 with Wilms tumor suppressor gene WT2• Chromosome 17q with familial FWT-2
Association with congenital anomalies: -WAGR syndrome: Wilms tumor, aniridia, genital malformation, mental retardation) -Denys+Drash syndrome – pseudohermaphroditism, glomerulopathy, mutation on chromosome 11p -Beckwith Wiedemann syndrome- hemihypertrophy, macroglossia, omphalocele, visceromegaly, associated with WT2

Clinical manifestation
• Abdominal mass- visible and/or palpable - 70%. Palpation must be done with care – risk of tumor rupture or dissemination!
• Fever• Vomiting, anorexia• Hematuria (micro or macro) 20-25%• Hypertension in renin-producing tumors• Pain 44%• Special symptoms in association with congenital
anomalies

Laboratory diagnosis
• Urine: hematuria• Chemistry: high serum calcium in children with rhabdoid
nephroblastoma• Acquired von Willebrand coagulopathy in about 8% of
patients• Differential diagnosis of NBL: 24-h urine catecholamine
analysis• Biopsy – only in children with unclear presentation or
diagnosis
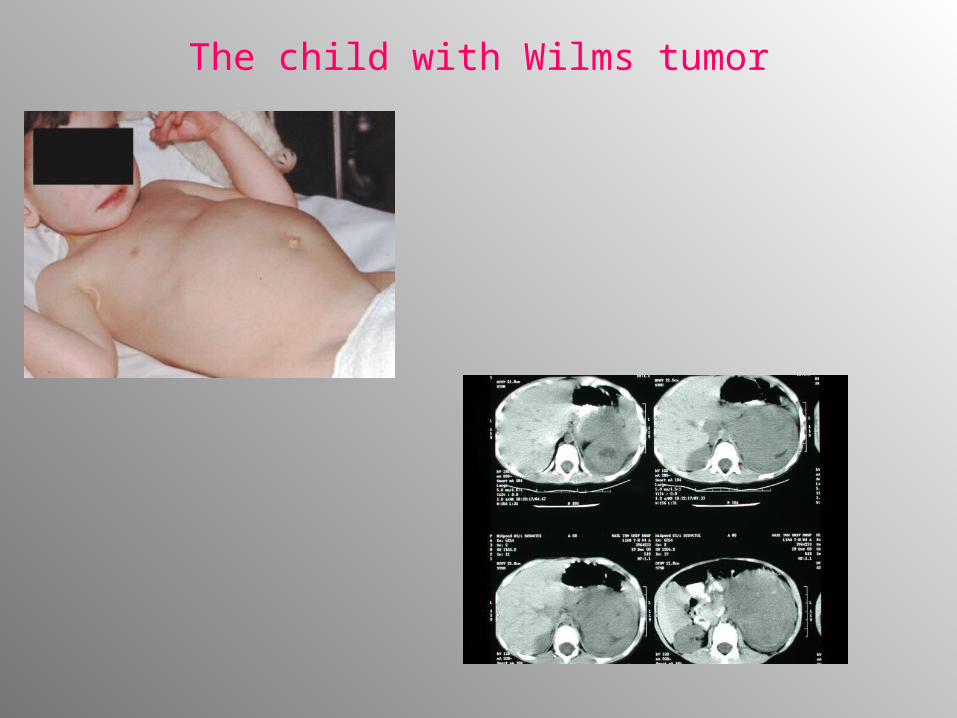
The child with Wilms tumor

Radiological diagnosis
• Ultrasound• Computed tomography• Magnetic resonance imaging
=anatomy of tumor extend of any spread within abdomen Metastases:
• Chest radiographs (posteroanterior and lateral) to exclude pulmonary metastases
• Angiography may be indicated in bilateral nephroblastoma• Radioisotope scans , skeletal survey in patients with suspected
skeletal metastates• CNS MRI in clear-cell sarcoma or rhabdoid kidney sarcoma and in
patients with possible brain metastases
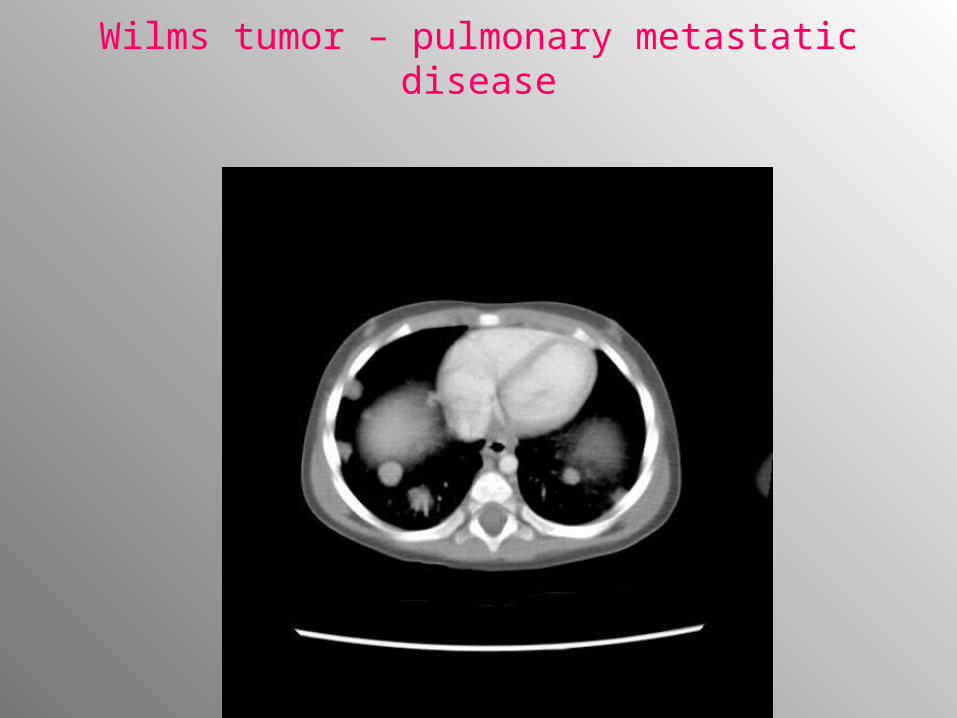
Wilms tumor – pulmonary metastatic disease
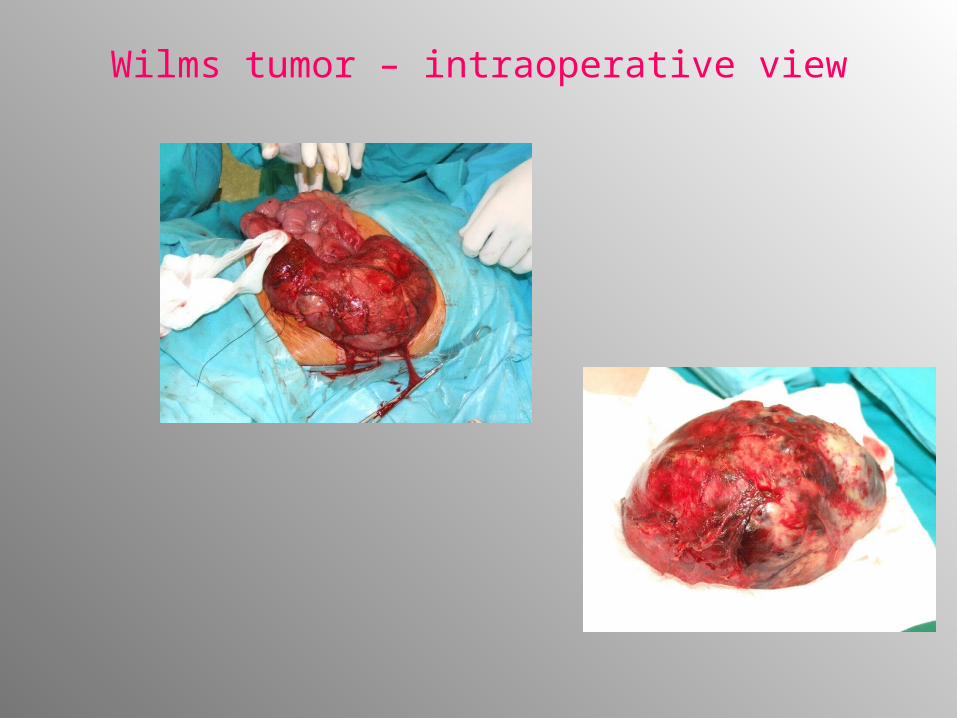
Wilms tumor – intraoperative view

Prognostic factors
• Histological appearance „ favourable, unfavourable”• Histology: rhabdoid tumor• Diffuse anaplasia• Viable malignant cells after preoperative chemotherapy• Infiltration of tumor capsule• Invasion of tumor cells into vessels• Nonradical surgical resection of tumor• Lymph nodes involvement• Tumor rupture• Metastatic spread• Large tumor volume

Stage
National Wilms’ Tumor Study Group staging system I – tumor confined to the kidney and completely resected II- tumor extend beyond the kidney but is completely resected (none at margins, no lymph nodes). At least one of the following has occured: a) penetration of the renal capsule b) invasion of the renal sinus vessels c)biopsy of tumor before removal
III- gross or microscopic residual tumor remains postoperatively including inoperable tumor, tumor at surgical margins, tumor spillage involving peritoneal surfaces, regional lymph node metastases or transected tumor thrombus IV- hematogenous metastases or lymph node metastases outside the abdomen (lung, liver, bone, brain) V – bilateral renal Wilms’tumor at onset
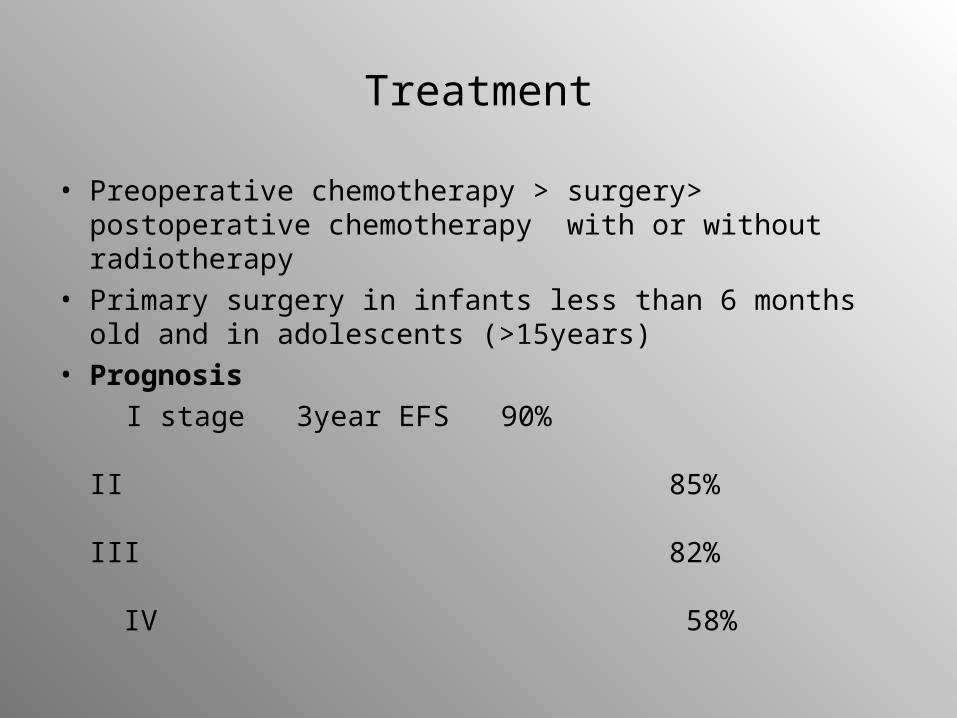
Treatment
• Preoperative chemotherapy > surgery> postoperative chemotherapy with or without radiotherapy
• Primary surgery in infants less than 6 months old and in adolescents (>15years)
• Prognosis
I stage 3year EFS 90% II 85% III 82% IV 58%