Embed Size (px)
Citation preview

CHAPTER 11
Genetic manipulation of animals
Introduction
The genetic manipulation of animals has revolu-tionized our understanding of biology, by making itpossible to test gene expression and function at thewhole-animal level. Gene-transfer techniques canbe used to produce transgenic animals, in which everycell carries new genetic information, as well asdesigner mutants with specific preselected modi-fications to the genome. The whole animal is theultimate assay system in which to investigate genefunction, particularly for complex biological pro-cesses, such as development.
In the case of plants, gene transfer to tissues orcultured cells is often the first step in the productionof a transgenic organism, since whole fertile plantscan regenerate from such cells and explants underthe appropriate culture conditions. Two fundamentaldifferences between plants and animals make thisstrategy impossible in animals. First, animal cellsbecome progressively restricted in terms of develop-mental potency as development proceeds, whichmeans that differentiated animal cells are normallyunable to fully dedifferentiate and recapitulate thedevelopmental programme.* Secondly, in most animals, the somatic cells and germ cells (the cellsthat give rise to gametes) separate at an early devel-opmental stage. Therefore, the only way to achievegerm-line transformation in animals is to introduce
DNA into totipotent cells prior to the developmentalstage at which the germ line forms.
In most cases, this involves introducing DNAdirectly into the developing oocyte, egg or earlyembryo. In mice, it is also possible to use culturedembryonic stem cells (ES cells), which are derivedfrom the preimplantation embryo and can con-tribute to all the tissues of the developing animal(including the germ line) if introduced into a hostembryo at the correct developmental stage. Thesecells are also remarkably amenable to homologousrecombination, which allows them to be used forgene targeting, the accurate replacement of a seg-ment of the endogenous genome with a homologoussegment of exogenous DNA. Gene targeting can beused to replace endogenous genes with a completelynon-functional copy (a null allele) or to make subtlechanges, both allowing the function of the endogen-ous gene to be tested. The same technology can beused for the opposite purpose, i.e. to replace amutant allele with a functional copy.
While rapid progress has been made with a rangeof model organisms, especially the fruit fly Drosophilamelanogaster, the mouse and, more recently, theAfrican clawed frog, Xenopus, great potential existsin the production of transgenic farm animals withimproved or novel traits. The technology for intro-ducing DNA into animals such as chickens, pigs,cattle and sheep is still in its infancy and is much less efficient than that of mice. Furthermore, it hasproved impossible to isolate amenable ES cells fromany species except mice and chickens. The desire togenerate transgenic livestock has driven research in a different direction, that of nuclear transfer.Although differentiated animal cells are develop-mentally restricted, their nuclei still contain all thegenetic information required to recapitulate thewhole of development. Dolly, the first sheep pro-duced following nuclear transfer from a differenti-ated somatic cell to an enucleated egg, has opened
* Recently, adult haemopoietic stem cells have been shown tocontribute to fetal blood-cell development when injected intothe blastocysts of mouse embryos (Geiger et al. 1998). Undersimilar circumstances, bone marrow and neural stem cellshave been shown to contribute to specific developing tissues(Petersen et al. 1999, Clarke et al. 2000). This suggests thatmammalian development may be more plastic than previ-ously assumed, and could open the way for novel routes to animal transgenesis.
POGC11 9/11/2001 11:15 AM Page 202

Genetic manipulation of animals 203
the way for animal cloning and the rapid productionof élite transgenic herds.
Genetic manipulation of mammals
Methods for producing transgenic mice
The ability to introduce DNA into the germ line ofmice is one of the greatest achievements of the twentieth century and has paved the way for the transformation of other mammals. Geneticallymodified mammals have been used not only to studygene function and regulation, but also as bioreactors producing valuable recombinant proteins, e.g. intheir milk. Several methods for germ-line transforma-tion have been developed, all of which require theremoval of fertilized eggs or early embryos fromdonor mothers, brief culture in vitro and then theirreturn to foster-mothers, where development con-tinues to term. These methods are discussed belowand summarized in Fig. 11.1.
Pronuclear microinjection
Direct microinjection of DNA was the first strategyused to generate transgenic mice. Simian virus 40(SV40) DNA was injected into the blastocoele cavities of preimplantation embryos by Jaenisch andMintz (1974). The embryos were then implantedinto the uteri of foster-mothers and allowed todevelop. The DNA was taken up by some of theembryonic cells and occasionally contributed to thegerm line, resulting in transgenic mice containingintegrated SV40 DNA in the following generation.Transgenic mice have also been recovered followingthe injection of viral DNA into the cytoplasm of thefertilized egg (Harbers et al. 1981).
The technique that has become established is theinjection of DNA into one of the pronuclei of the egg(reviewed by Palmiter & Brinster 1986). The tech-nique is shown in Fig. 11.2. Just after fertilizaton,the small egg nucleus (female pronucleus) and thelarge sperm nucleus (male pronucleus) are discrete.
Blastocyst
DNA transfection
Recombinantretroviral infection
Embryonicdevelopment
Early cleavage
Fertilized egg
Cell transfer
Nuclearreplacement
Embryonic stem(ES) cells
Unfertilizedmouse eggAdult cells
Attach DNAto sperm
DNAmicroinjection
Fig. 11.1 Summary of methods for producingtransgenic mammals.
POGC11 9/11/2001 11:15 AM Page 203

204 CHAPTER 11
Since the male pronucleus is larger, this is usuallychosen as the target for injection. About 2 pl of DNAsolution is transferred into the nucleus through afine needle, while the egg is held in position with asuction pipette. The injected embryos are cultured invitro to the morula stage and then transferred topseudopregnant foster-mothers (Gordon & Ruddle,1981). The procedure requires specialized microin-jection equipment and considerable dexterity fromthe handler. The exogenous DNA may integrateimmediately or, less commonly, may remain extra-chromosomal for one or more cell divisions. Thusthe resulting animal may be transgenic or may bechimeric for transgene insertion. The technique is reliable, although the efficiency varies, so that5–40% of mice developing from manipulated eggscontain the transgene (Lacy et al. 1983). However,once the transgene is transmitted through the germline, it tends to be stably inherited over many genera-tions. The exogenous DNA tends to form head-to-tailarrays prior to integration, and the copy numbervaries from a few copies to hundreds. The site of integration appears random and may depend on theoccurrence of natural chromosome breaks. Extensivedeletions and rearrangements of the genomic DNAoften accompany transgene integration (Bishop &Smith 1989).
Recombinant retroviruses
As discussed in Chapter 10, recombinant retro-viruses provide a natural mechanism for stablyintroducing DNA into the genome of animal cells.Retroviruses are able to infect early embryos and EScells (see below), so recombinant retroviral vectorscan be used for germ-line transformation (Robertsonet al. 1986). An advantage over the microinjectiontechnique is that only a single copy of the retroviralprovirus is integrated, and the genomic DNA sur-rounding the transgenic locus generally remainsintact. The infection of preimplantation embryoswith a recombinant retrovirus is technically straight-forward and, once the infected embryos are implantedin the uterus of a foster-mother, can lead to germ-line transmission of the transgene. However, thereare also considerable disadvantages to this method,including the limited amount of foreign DNA thatcan be carried by the virus, the possible interferenceof viral regulatory elements with the expression ofsurrounding genes and the susceptibility of the virusto de novo methylation, resulting in transgene silenc-ing (see Box 13.2). The founder embryos are alwayschimeric with respect to transgene integration(reviewed by Jaenisch 1988). Retroviral transductionis therefore not favoured as a method for generatingfully transgenic animals, but it is useful for generat-ing transgenic sectors of embryos. For example, theanalysis of chicken-limb buds infected with recom-binant retroviruses has allowed many of the genesinvolved in limb development to be functionallycharacterized (see review by Tickle & Eichele 1994).
Transfection of ES cells
ES cells are derived from the inner cell mass of themouse blastocyst and thus have the potential to contribute to all tissues of the developing embryo(Evans & Kaufman 1981, Martin 1981). The abilityof ES cells to contribute to the germ line was firstdemonstrated by Bradley et al. (1984) and requiresculture conditions that maintain the cells in an undif-ferentiated state ( Joyner 1998). Since these cells canbe serially cultured, like any other established cellline, DNA can be introduced by transfection or viral transduction and the transformed cells can be selected using standard markers, as discussed in
Fig. 11.2 Pronuclear microinjection of a fertilized mouse egg. The two pronuclei are visible, and the egg is held using a suction pipette. The DNA is introduced through a fine glass needle. (Photograph courtesy of Roberta Wallace,Roslin Institute.)
POGC11 9/11/2001 11:15 AM Page 204

Genetic manipulation of animals 205
Chapter 10. This is an important advantage, sincethere is no convenient way to select for eggs orembryos that have taken up foreign DNA, so,instead, each potential transgenic mouse must betested by Southern-blot hybridization or the poly-merase chain reaction (PCR) to confirm transgeneintegration. ES cells are also particularly efficient atcarrying out homologous recombination (see below),so, depending on the design of the vector, DNA intro-duced into ES cells may integrate randomly or maytarget and replace a specific locus.
Whichever strategy is chosen, the recombinantES cells are then introduced into the blastocoele of ahost embryo at the blastocyst stage, where they mixwith the inner cell mass. This creates a true chimericembryo (i.e. an embryo comprising cells from differ-ent sources). The contribution of ES cells to the germline can thus be confirmed using visible markers.Most ES cell lines in common use are derived frommouse strain 129, which has the dominant coatcolour agouti. A popular strategy is to use hostembryos from a mouse strain such as C57BL/6J,which has a recessive black coat colour. Colon-ization of the embryo by vigorous ES cells can be substantial, generating chimeras with patchworkcoats of black and agouti cell clones. If the ES cellshave contributed to the germ line, mating chimericmales with black females will generate heterozygoustransgenic offspring with the agouti coat colour,confirming germ-line transmission of the foreignDNA. Most ES cells in use today are derived frommale embryos, resulting in a large sex bias towardsmale chimeras (McMahon & Bradley, 1990). This is desirable because male chimeras sire many moreoffspring than females.
Gene targeting with ES cells
Pronuclear microinjection and retroviral transferare useful for the addition of DNA to the mousegenome. However, in many cases it is more desirableto replace endogenous gene sequences with exogen-ous DNA, since this would allow the introduction of specific mutations into any preselected gene. Inyeast, gene targeting by homologous recombinationoccurs with high efficiency (Chapter 9). In contrast,when the first gene-targeting experiments in animalcells were carried out in the 1980s, only a very low
frequency of targeted recombination was achieved.These experiments involved the correction of muta-tions in selectable markers such as neo, which hadbeen introduced into cell lines as transgenes by standard methods (e.g. Thomas et al. 1986). Smithieset al. (1985) were the first to demonstrate targetingof an endogenous gene. They introduced a modifiedβ-globin gene containing the bacterial marker supFinto a human fibroblast × mouse erythroleukaemiacell line and screened large numbers of potentialrecombinants by reisolating the modified locus, usingsupF as a cloning tag. This experiment demonstratedthat the frequency of homologous recombinationwas up to 1000-fold lower than that of random inte-gration. Targeting occurs with significantly higherefficiency in certain cell lines, including mouse EScells. The combination of pluripotency, amenabilityfor in vitro manipulation and capacity for homo-logous recombination makes ES cells uniquely suitable for the generation of targeted mutant mice,i.e. mice carrying the same mutation in every celland transmitting it through the germ line. Gene targeting in ES cells was first achieved by Thomasand Capecchi (1987), who disrupted the hprt genewith the neo marker and selected recombinant cellsusing either G418 or 6-thioguanine, a toxic guanineanalogue that is only incorporated into DNA if the nucleotide salvage pathway is functional (see p. 178). Doetschman et al. (1987) also successfullytargeted the hprt locus, although they used a mutantrecipient cell line and repaired the locus with homo-logous DNA, subsequently selecting on HAT medium(see p. 177).
Design of targeting vectors
Targeting vectors are specialized plasmid vectors,which promote homologous recombination whenintroduced into ES cells. This is achieved by theinclusion of a homology region, i.e. a region that ishomologous to the target gene, allowing the target-ing vector to synapse with the endogenous DNA.Both the size of the homology region and the level of sequence identity have been shown to play animportant role in the efficiency of gene targeting(Hasty et al. 1991b, Deng & Capecchi 1992, Te Rieleet al. 1992). Recombination is also more efficient ifthe vector is linearized prior to transfection.
POGC11 9/11/2001 11:15 AM Page 205

206 CHAPTER 11
Most gene targeting experiments have been usedto disrupt endogenous loci, resulting in targeted nullalleles (this strategy is often termed ‘gene knockout’).Two types of targeting vector have been developedfor this purpose: insertion vectors and replacement(or transplacement) vectors (Thomas & Capecchi1987; Fig. 11.3). Insertion vectors are linearizedwithin the homology region, resulting in the inser-tion of the entire vector into the target locus. Thistype of vector disrupts the target gene but leads to a duplication of the sequences adjacent to theselectable marker. This is not always a desirableconfiguration, since duplication of the target sequencescan lead to a subsequent homologous recombina-tion event that restores the wild-type genotype(Fiering et al. 1995). Replacement vectors aredesigned so that the homology region is collinearwith the target. The vector is linearized outside thehomology region prior to transfection, resulting incrossover events in which the endogenous DNA isreplaced by the incoming DNA. With this type ofvector, only sequences within the homology region(not the vector backbone) are inserted, so, to achievegene knockout, the homology region itself must be interrupted. Insertion and replacement vectors are thought to be equally efficient, but replace-ment vectors have been used in the majority ofknockout experiments. In both cases, however, it is possible for transcription to occur through the targeted locus, producing low amounts of RNA. This may be spliced in such a way as to remove thetargeted exon, resulting in a residual amount offunctional protein (e.g. Dorin et al. 1994, Dahme et al. 1997).
Selection strategy
The first gene-targeting experiments involved theselectable Hprt locus, which is present on the X chromosome, allowing targeted events to be selectedin male ES cells without the requirement for homo-zygosity. The first non-selectable genes to be targetedwere int-2 (also known as fgf-3) (Mansour et al.1988) and the oncogene c-abl (Schwartzberg et al.1989). In each case, it was necessary to include aselectable marker in the targeting vector to identifytransformed cells. In the case of insertion vectorsthis is placed anywhere on the vector backbone,
while in replacement vectors the marker must interrupt the homology region. The neo marker hasbeen most commonly used, allowing transformed ES cells to be selected using G418. However, otherdominant markers are equally applicable (e.g. seeVon Melchner et al. 1992), and Hprt can be used incombination with hprt− mutant ES cells (Matzuk et al. 1992).
The use of a single marker fails to discriminatebetween targeted cells and those where the con-struct has integrated randomly. This problem can beaddressed by combined positive–negative selectionusing neo and the herpes simplex virus (HSV) Tkgene (Mansour et al. 1988). If the neo marker is usedto interrupt the homology region in a replacementvector, transformed cells can be selected using G418.The HSV Tk gene is placed outside the homologyregion, such that it is inserted by random integrationbut not by homologous recombination. Therefore,cells that have undergone homologous recombina-tion will survive in the presence of the toxic thymi-dine analogues ganciclovir or FIAU,* while in thosecells containing randomly integrated copies of the Tk gene, the analogues will be incorporated into theDNA, resulting in cell death.
A different strategy is to make expression of theneo gene dependent on homologous recombination(e.g. see Schwartzberg et al. 1989, Mansour et al.1993). Another alternative to positive–negativeselection, which is used in many laboratories, is simply to screen large numbers of G418-resistanttransfected cells by PCR to identify genuine re-combinants (Hogan & Lyons 1988, Zimmer & Gruss1989). Screening can be carried out relatively quicklywithout recourse to cloning.
Introducing subtle mutations
While gene targeting has often been used to disruptand hence inactivate specific endogenous genes byintroducing large insertions, more refined approachescan be used to generate subtle mutations. The precise effects of minor deletions or point mutationscannot be assessed using the simple targeting strat-egies discussed above, which necessarily leave the
* 1-(2-deoxy-2-fluoro-β-d-arabinofuranosyl)-5-iodouracil.
POGC11 9/11/2001 11:15 AM Page 206
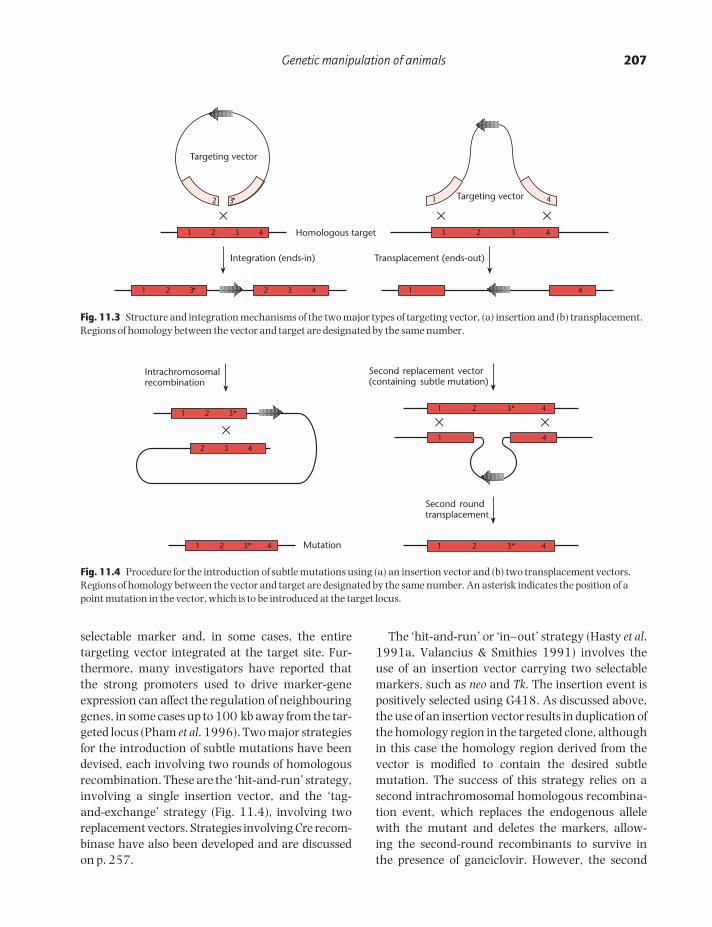
Genetic manipulation of animals 207
selectable marker and, in some cases, the entire targeting vector integrated at the target site. Fur-thermore, many investigators have reported thatthe strong promoters used to drive marker-geneexpression can affect the regulation of neighbouringgenes, in some cases up to 100 kb away from the tar-geted locus (Pham et al. 1996). Two major strategiesfor the introduction of subtle mutations have beendevised, each involving two rounds of homologousrecombination. These are the ‘hit-and-run’ strategy,involving a single insertion vector, and the ‘tag-and-exchange’ strategy (Fig. 11.4), involving tworeplacement vectors. Strategies involving Cre recom-binase have also been developed and are discussedon p. 257.
The ‘hit-and-run’ or ‘in–out’ strategy (Hasty et al.1991a, Valancius & Smithies 1991) involves the use of an insertion vector carrying two selectablemarkers, such as neo and Tk. The insertion event ispositively selected using G418. As discussed above,the use of an insertion vector results in duplication ofthe homology region in the targeted clone, althoughin this case the homology region derived from thevector is modified to contain the desired subtle mutation. The success of this strategy relies on a second intrachromosomal homologous recombina-tion event, which replaces the endogenous allelewith the mutant and deletes the markers, allow-ing the second-round recombinants to survive in the presence of ganciclovir. However, the second
Targeting vector
3*2
×1 2 3 4 Homologous target
Integration (ends-in)
1 2 3* 2 3 4
Targeting vector1
×1 2 3 4
Transplacement (ends-out)
1 4
4
×
Fig. 11.3 Structure and integration mechanisms of the two major types of targeting vector, (a) insertion and (b) transplacement.Regions of homology between the vector and target are designated by the same number.
1 2 3* 4
1 2 3*
×
Intrachromosomalrecombination
2 3 4
Mutation
1 2 3* 4
× ×1 4
Second replacement vector(containing subtle mutation)
1 2 3* 4
Second roundtransplacement
Fig. 11.4 Procedure for the introduction of subtle mutations using (a) an insertion vector and (b) two transplacement vectors.Regions of homology between the vector and target are designated by the same number. An asterisk indicates the position of apoint mutation in the vector, which is to be introduced at the target locus.
POGC11 9/11/2001 11:15 AM Page 207

208 CHAPTER 11
homologous recombination event occurs at a verylow frequency and, in 50% of cases, restores thelocus to its original wild-type configuration.
The ‘tag-and-exchange’ strategy also requires twohomologous recombination events, but in this casetwo replacement-type vectors are used. For exam-ple, Moore et al. (1995) demonstrated the principleusing neo for positive selection and HSV Tk for nega-tive selection. The first ‘tag’ vector was designed tomutate the target gene by inserting a large cassettecontaining the selectable markers. This event waspositively selected with G418. The second ‘exchange’vector introduced the desired mutation and eliminatedthe selectable markers, allowing the second-roundrecombinants to be selected for the absence of Tk.
Applications of genetically modified mice
Applications of transgenic mice
Transgenic mice, i.e. mice containing additionaltransgenes, as opposed to those with targeted muta-tions, have been used to address many aspects ofgene function and regulation. A vast literature hasaccumulated on this subject, and genes concerningevery conceivable biological process have beeninvestigated (e.g. see Houdebine 1997). As well astheir use for basic scientific investigation, transgenicmice can be used for more applied purposes, such as models for human disease and the production of valuable pharmaceuticals. Many mouse models for human diseases have been generated by geneknockout (see below), but gain-of-function modelshave also been generated by adding transgenes. Forexample, much information concerning the patho-logy of prion diseases has arisen from the study oftransgenic mice expressing mutant prion trans-genes (reviewed by Gabizon & Taraboulos 1997).Transgenic mice expressing oncogenes have beenextensively used to study cancer (reviewed byMacleod & Jacks 1999).
For illustrative purposes, we now consider someearly experiments that demonstrated how trans-genic mice can be used for the analysis of gene function and regulation, but also highlighted somelimitations of the transgenic approach. Brinster et al.(1981) constructed plasmids in which the promoterof the mouse metallothionein-1 (MMT ) gene was
fused to the coding region of the HSV Tk gene. The thymidine kinase (TK) enzyme can be assayedreadily and provides a convenient reporter of MMTpromoter function. The endogenous MMT promoteris inducible by glucocorticoid hormones and heavymetals, such as cadmium and zinc, so it was envisagedthat the hybrid transgene, MK (metallothionein-thymidine kinase), would be similarly regulated.The gene was injected into the male pronucleus offertilized eggs, which were then incubated in vitro inthe presence or absence of cadmium ions (Brinster et al. 1982). As expected, TK activity was found to beinduced by the metal. By making a range of deletionsof mouse sequences upstream of the MMT promotersequences, the minimum region necessary for induc-ibility was localized to a stretch of DNA 40–180nucleotides upstream of the transcription-initiationsite. Additional sequences that potentiate both basaland induced activities extended to at least 600 bpupstream of the transcription-initiation site. Themouse egg was therefore being used in the same wayas transfected cell lines, to dissect the activity of a func-tional promoter (Gorman et al. 1982b; see Box 10.l).
The same MK fusion gene was injected intoembryos, which were raised to transgenic adults(Brinster et al. 1981). Most of these mice expressedthe MK gene and in such mice there were from oneto 150 copies of the gene. The reporter activity wasinducible by cadmium ions and showed a tissue distribution very similar to that of metallothioneinitself (Palmiter et al. 1982b). Therefore these experi-ments showed that DNA sequences necessary forheavy-metal induction and tissue-specific expressioncould be functionally dissected in both eggs andtransgenic mice. For unknown reasons, there wasno response to glucocorticoids in either the egg orthe transgenic-mouse experiments.
In a dramatic series of experiments, Palmiter et al.(1982a) fused the MMT promoter to the rat growth-hormone gene. This hybrid gene (MGH ) was con-structed using the same principles as the MK fusion.Of 21 mice that developed from microinjected eggs,seven carried the MGH fusion gene and six of thesegrew significantly larger than their littermates. Themice were fed zinc to induce transcription of the MGHgene, but this did not appear to be absolutely necess-ary, since they showed an accelerated growth ratebefore being placed on the zinc diet. Mice containing
POGC11 9/11/2001 11:15 AM Page 208

Genetic manipulation of animals 209
high copy numbers of the MGH gene (20–40 copiesper cell) had very high concentrations of growthhormone in their serum, some 100–800 times abovenormal. Such mice grew to almost double the weightof littermates at 74 days old (Fig. 11.5).
The similarities between the tissue distribution ofnormal MMT expression and that of the hybridtransgenes encouraged the hope that transgenicmice would provide a general assay for functionallydissecting DNA sequences responsible for tissue-specific or developmental regulation of a variety ofgenes. However, there were also some unexpectedfindings. For example, independently derived trans-genic mice carrying the MK transgene showedsignificant variations in the levels and patterns oftransgene expression. Furthermore, while trans-genic founders transmitted the construct to theirprogeny as expected, when reporter activity wasassayed in these offspring the amount of expressioncould be very different from that in the parent.
Examples of increased, decreased or even totallyextinguished expression were found. In some, butnot all, cases, the changes in expression correlatedwith changes in methylation of the gene sequences(Palmiter et al. 1982b). These results provided thefirst examples of two complex phenomena, positioneffects (Box 11.1) and de novo transgene silencing(Box 13.2), which often affect integrated transgenes.
Yeast artificial chromosome (YAC) transgenic mice
Studies of the MMT promoter and others havedemonstrated the principle that transgenes withminimal flanking sequences tend not to be expressedin the same manner as the corresponding endogen-ous gene. In many cases, it has also been shown that authentic patterns and levels of protein expres-sion occur only when the intact gene is used, andthis can span tens or hundreds of kilobase pairs of DNA (Box 11.1). The transfer of large DNA segments to the mouse genome has been achievedby transformation with yeast artificial chromosome(YAC) vectors. Jakobovits et al. (1993) were the firstto report transformation of ES cells with a YAC vector, via fusion with yeast sphaeroplasts. The vec-tor contained the entire human HPRT locus, nearly700 kb in length. The disadvantage of this method is that the endogenous yeast chromosomes were co-introduced with the vector. Alternative strategiesinvolve isolation of the vector DNA by pulsed-fieldgel electrophoresis (p. 10), followed by introductionof the purified YAC DNA into mouse eggs by pro-nuclear microinjection or transfection into ES cells.The latter technique is more suitable because micro-injection involves shear forces that break the DNAinto fragments. YAC transfer to ES cells has beenachieved by lipofection, as discussed in Chapter 10.YAC transgenics have been used to study gene regulation, particularly by long-range regulatoryelements, such as locus-control regions (reviewedby Lamb & Gerhart 1995). They have also been usedto introduce the entire human immunoglobulin locusinto mice, for the production of fully humanizedantibodies (Mendez et al. 1997). It is also possible tointroduce chromosomes and chromosome fragmentsinto ES cells, using a technique called microcell-mediated fusion. This involves the prolonged mitotic
Fig. 11.5 Transgenic mouse containing the mousemetallothionein promoter fused to the rat growth-hormonegene. The photograph shows two male mice at about 10 weeks old. The mouse on the left contains the MGH geneand weighs 44 g; his sibling without the gene weighs 29 g. In general, mice that express the gene grow two to three timesas fast as controls and reach a size up to twice the normal.(Photograph by courtesy of Dr R.L. Brinster.)
POGC11 9/11/2001 11:15 AM Page 209

210 CHAPTER 11
Independently derived transgenic animals and plantscarrying the same expression construct often showvariable levels and patterns of transgene expression.In many cases, such variation is dependent on thesite of transgene integration, and this phenomenonhas been termed the position effect (reviewed byWilson et al. 1990). Position effects result from the influence of local regulatory elements on thetransgene, as well as the architecture of thesurrounding chromatin. For example, an integratedtransgene may come under the influence of a localenhancer, resulting in the alteration of its expressionprofile to match that of the correspondingendogenous gene. The position dependence of the phenomenon has been demonstrated in mice by isolating the entire transgenic locus from such an anomalous line and microinjecting it into thepronuclei of wild-type eggs, resulting in ‘secondary’transgenic lines with normal transgene expressionprofiles (Al-Shawi et al. 1990). Position effects arealso revealed by enhancer-trap constructs, whichcontain a minimal promoter linked to a reporter gene (O’Kane & Gehring 1987; see Chapter 13).
Unlike the specific influences of nearby regulatoryelements, chromatin-mediated position effects aregenerally non-specific and repressive. They reflect the integration of the transgene into a chromosomalregion containing repressed chromatin(heterochromatin). The molecular features ofheterochromatin, including its characteristicnucleosome structure, deacetylated histones and, in many cases, hypermethylated DNA, spread intothe transgene, causing it to be inactivated (Huber et al. 1996, Pikaart et al. 1998). In some cases,variegated transgene expression has been reporteddue to cell-autonomous variations in the extent ofthis spreading process (reviewed by Heinkoff 1990).Negative chromosomal position effects can betroublesome in terms of achieving desirabletransgene expression levels and patterns; thus a number of different strategies have been used to combat them.
Incorporating dominantly actingtranscriptional control elements
Certain regulatory elements are thought to act asmaster-switches, regulating the expression of genesor gene clusters by helping to establish an openchromatin domain. The locus control region (LCR) of the human b-globin gene cluster is one example(Forrester et al. 1987). Transgenic mice carrying a human b-globin transgene driven by its ownpromoter show a low frequency of expression and, inthose mice that do express the transgene, only a lowlevel of the mRNA is produced (e.g. Magram et al.1985, Townes et al. 1985). However, inclusion of theLCR in the expression construct confers high-leveland position-independent expression (Grosveld et al.1987). There is evidence that LCRs induce chromatinremodelling over large distances. For example, themurine immunoglobulin heavy-chain LCR has beenshown to induce histone deacetylation in a linked c-myc gene (Madisen et al. 1998). This suggests that LCRs could protect against position effects byconverting heterochromatin to open euchromatin atthe site of transgene integration (Festenstein et al.1996, Milot et al. 1996). The interested reader canconsult several comprehensive reviews of LCRresearch (Bonifer 1999, Grosveld 1999, Li et al.1999).
Using boundary elements/matrix attachment regions
Boundary elements (insulators) are sequences thatcan block the activity of enhancers when placedbetween the enhancer and a test transgene driven bya minimal promoter. For example, an ‘A element’ withinsulator activity is found upstream of the chickenlysozyme gene. This inhibits the activity of a reportergene when interposed between the promoter and anupstream enhancer, but not when placed elsewherein the construct (Stief et al. 1989). However, by flanking the entire construct with a pair of
continued
Box 11.1 Position effects
POGC11 9/11/2001 11:15 AM Page 210

Genetic manipulation of animals 211
arrest of cultured human cells, using an inhibitorsuch as colchicine. Eventually, the nucleus breaksup into vesicles containing individual chromosomes,which can be rescued as microcells comprising anuclear vesicle surrounded by a small amount ofcytoplasm and a plasma membrane (Fournier &Ruddle 1977). Transgenic mice have been generatedusing ES cells that were fused to human microcells,and evidence for germ-line transmission and ex-
pression of the human chromosome was obtained(Tomizuka et al. 1997).
Applications of gene targeting
Since the first reports of gene targeting in ES cells, anever-increasing number of targeted mutant micehave been produced. These have been discussed in several comprehensive reviews (Brandon et al.
A elements, the transgene is protected fromchromosomal position effects (Stief et al. 1989). Thisprotective effect works not only in cell lines, but alsoin transgenic animals (McKnight et al. 1992) andplants (Mlynarova et al. 1994). Many boundaryelements are associated with matrix-attachmentregions (MARs), sequences dispersed throughout thegenome that attach to the nuclear matrix, dividingchromosomes into topologically independent loops(reviewed by Spiker & Thompson 1996). It istherefore possible that transgenes flanked by suchelements are maintained in an isolated chromatindomain into which heterochromatin cannot spread.However, not all boundary elements are associatedwith MARs (e.g. see Mirkovitch et al. 1984). Similarly,some MARs do not function as boundary elementsbut as facilitators of gene expression (e.g. Van derGeest & Hall 1997).
Using large genomic transgenes
Conventional transgenes generally comprisecomplementary DNAs (cDNAs) or intronless‘minigenes’ expressed under the control of viralpromoters or cell-type-specific regulatory elements.Such transgenes are highly sensitive to positioneffects. Over the last few years, there has been anincreasing appreciation that the regulation ofeukaryotic gene expression is far more complex and involves much more upstream and downstreamDNA than previously thought (reviewed by Bonifer1999, 2000). The correct, high-level expression of transgenes is favoured by the use of genomicconstructs that include introns and large amounts
of flanking sequence from the source gene (e.g. seeBonifer et al. 1990, Lien et al. 1997, Nielsen et al.1998). Such constructs are likely to include multipleenhancers, dominant regulatory elements, such asLCRs, and boundary elements, which all act togetherto protect the transgene from position effects.
Dominantly acting transgenes (transgene rescue)
Some conventional transgenes, including b-globinand a-fetoprotein (Chada et al. 1986, Kollias et al.1986, Hammer et al. 1987) are very sensitive toposition effects and de novo silencing. Other genesappear to be less sensitive to these phenomena, e.g.immunoglobulin and elastase (Storb et al. 1984,Swift et al. 1984, Davis & MacDonald 1988).Although the reason for this is not clear, the lesssensitive transgenes are assumed to in some wayinduce or define an open chromatin domain. In somecases, such sequences have been used to protectmore susceptible transgenes from negative positioneffects by introducing the two transgenessimultaneously (e.g. see Clark et al. 1992).
Site-specific integration
Site-specific recombination systems (see Chapter 13)can be used to introduce transgenes into a locusknown to lack negative position effects, if a targetsite for the recombinase can be introduced at such a locus. The Cre-loxP system has been used to thiseffect in mammalian cells (see Fukushige & Sauer1992).
Box 11.1 continued
POGC11 9/11/2001 11:15 AM Page 211

212 CHAPTER 11
1995a,b,c, Soriano 1995, Muller 1999) and a num-ber of Internet databases have been established tokeep track of the results (see Sikorski & Peters 1997).The phenotypes of homozygous, null mutant miceprovide important clues to the normal function ofthe gene. Some gene knockouts have resulted in surprisingly little phenotypic effect, much less severethan might have been expected. For example, myoD,whose expression in transfected fibroblasts causesthem to differentiate into muscle cells, and whichwas therefore a good candidate as a key regulator ofmyogenesis, is not necessary for development of aviable animal (Rudnicki et al. 1992). Similarly, theretinoic acid γ receptor is not necessary for viablemouse development in knockout mice (Lohnes et al.1993), even though this receptor is a necessarycomponent of the pathway for signalling by retinoidsand has a pattern of expression quite distinct fromother retinoic acid receptors in embryos. Such ob-servations have prompted speculation that geneticredundancy may be common in development, andmay include compensatory up-regulation of somemembers of a gene family when one member is inactivated. An example of this may be the up-regulation of myf-5 in mice lacking myoD (Rudnickiet al. 1992). Gene knockouts have also been used asmouse models of human diseases, such as cysticfibrosis, β-thalassaemia and fragile X syndrome(reviewed by Bedell et al. 1997; see Chapter 14).
While most gene-targeting experiments in micehave been used to introduce mutations into genes(either disruptive insertional mutations or subtlechanges), the scope of the technique is much wider.The early gene-targeting experiments demonstratedthat this approach could also be used to correctmutated genes, with obvious applications in genetherapy. Homologous recombination has also beenused to exchange the coding region of one gene forthat of another, a strategy described as ‘gene knock-in’. This has been used, for example, to test the ability of the transcription factors Engrailed-1 andEngrailed-2 to compensate for each other’s functions.Hanks et al. (1995) replaced the coding region of theengrailed-1 gene with that of engrailed-2, and showedthat the engrailed-1 mutant phenotype could be rescued. A more applied use of gene knock-in is the replacement of parts of the murine immuno-globulin genes with their human counterparts,
resulting in the production of humanized antibodiesin transgenic mice (Moore et al. 1995). The Cre-loxPsite-specific recombinase system has been used exten-sively in ES cells to generate mice in which condi-tional or inducible gene targeting is possible and toproduce defined chromosome deletions and trans-locations as models for human disease. We shall discuss the many applications of Cre-loxP and othersite-specific recombinase systems in Chapter 13.
Other mammals and birds
Traditional techniques
The three major routes for producing transgenic micehave also been used in other mammals and birds,particularly in farm animals. The efficiency of eachprocedure is much lower than in mice. Pronuclearmicroinjection in mammals such as sheep and cows,for example, typically results in less than 1% of theinjected eggs giving rise to transgenic animals. Addedto this, the recovery of eggs from donor animals andthe reimplantation of transformed eggs into foster-mothers is a less efficient procedure and requires, atgreat expense, a large number of donors and reci-pients. The eggs themselves are also more difficult tomanipulate – they are very delicate and tend to beopaque. It is often necessary to centrifuge the eggs inorder to see the pronuclei. In chickens, it is possibleto remove eggs just after fertilization and microinjectDNA into the cytoplasm of the germinal disc, wherethe male and female pronuclei are to be found.However, it is not possible to return the manipulatedeggs to a surrogate mother, so they must be culturedin vitro. Using this procedure, Love et al. (1994)obtained seven chicks, equivalent to about 5% of the eggs injected, that survived to sexual maturity.One cockerel transmitted the transgene to a smallproportion of his offspring, indicating that he waschimeric for transgene integration.
The use of retroviruses to produce transgenicchickens has been reported by Bosselman et al.(1989). These investigators injected a replication-defective recombinant reticuloendotheliosis viruscarrying the neo gene into laid eggs and found that approximately 8% of male birds carried vectorsequences. The transgene was transmitted throughthe germ line in a proportion of these birds and
POGC11 9/11/2001 11:15 AM Page 212

Genetic manipulation of animals 213
was stably expressed in 20 transgenic lines. It is necessary for the nuclear envelope to break down for most retroviral infections (lentiviruses such ashuman immunodeficiency virus (HIV) are excep-tional) and, since this only occurs during mitosis,most retroviruses are unable to infect non-dividingcells (Chapter 10). The nuclear envelope also breaksdown during meiosis, and this was exploited byChan et al. (1998) to produce transgenic cattle following the injection of replication-defective retro-viral vectors into the perivitelline space of isolatedbovine oocytes. Retroviral integration occurred dur-ing the second meiotic division, resulting in the production of a number of transgenic offspring.Remarkably, the same technique has recently beenused to generate the first ever transgenic primate, arhesus monkey named ANDi (Chan et al. 2001). In primates, the technique was inefficient. Two hundred and twenty-four oocytes were injected toproduce one live transgenic monkey; a number offurther transgenic fetuses failed to develop to term.
After more than a decade of research, it has alsoproved impossible to derive reliable ES cell lines fromany domestic species other than mice and, morerecently, chickens (Pain et al. 1999, Prelle et al.1999)*. However, there have been great advancesin the isolation and transfection of primordial germcells (PGCs), the embryonic cells that give rise togametes. These can be transfected directly, or cul-tured as embryonic germ cells (EG cells), which aremorphologically very similar to ES cells and couldprovide a route for the direct transformation of thegerm line (Resnick et al. 1992). Chicken PGCs havebeen isolated from the germinal crescent, infectedwith a recombinant retrovirus and replaced in theembryo, leading to the development of chimericbirds producing transgenic offspring (Vick et al.1993). Mammalian PGCs have also been trans-formed, although it has been difficult to persuade thecells to contribute to the germ line once introducedinto the host animal (Labosky et al. 1994).
Intracytoplasmic sperm injection
The injection of sperm heads directly into the cyto-plasm of the egg (intracytoplasmic sperm injection
(ICSI)) can overcome infertility in humans. It hasbeen shown that sperm heads bind spontaneously tonaked plasmid DNA in vitro, suggesting that sperminjections could be used to achieve transformation.This was demonstrated by Perry et al. (1999), whomixed mouse sperm with plasmid DNA carrying the gene for green fluorescent protein (GFP). Thesesperm were injected into unfertilized oocytes, and aremarkable 94% of the resulting embryos showedGFP activity. Random transfer of these embryos topseudopregnant females resulted in development to term, and in about 20% of cases the mice weretransgenic. This method could be adaptable to otheranimals. Rhesus monkey oocytes fertilized in thesame manner gave rise to a number of embryos withGFP activity, but this only lasted until the blastulastage, suggesting that there was no stable integra-tion. However, several monkeys developed to term,showing that the procedure was compatible withnormal development (Chan et al. 2000).
Nuclear transfer technology
The failure of traditional transgenesis techniques toyield routine procedures for the genetic modificationof mammals other than mice has driven researchersin search of other methods. Fifty years ago, Briggsand King (1952) established the principle of nucleartransfer in amphibians by transplanting nuclei fromthe blastula of the frog Rana pipens to an enucleatedegg, obtaining a number of normal embryos in theprocess. In Xenopus laevis, nuclei from various typesof cell in the swimming tadpole can be transplantedto an egg that has been UV-irradiated to destroy theperipheral chromosomes, and similar results areobtained (reviewed by Gurdon 1986, 1991). Theimportant principle here is that, while animal cellsbecome irreversibly committed to their fate as development proceeds, the nuclei of most cells stillretain all the genetic information required for theentire developmental programme and can, underappropriate circumstances, be reprogrammed by thecytoplasm of the egg to recapitulate development. Inall species, it appears that the earlier the develop-mental stage at which nuclei are isolated, the greatertheir potential to be reprogrammed. Nuclear trans-plantation can be used to generate clones of animalswith the same genotype by transplanting many* Human ES cells and Human ES cell lines are available.
POGC11 9/11/2001 11:15 AM Page 213

214 CHAPTER 11
somatic nuclei from the same individual into a seriesof enucleated eggs (King & Briggs 1956). This allowsanimals with specific and desirable traits to be propagated. If possible in mammals, this would haveobvious applications in farming.
Nuclear transfer in mammals has been practisedwith success for the last decade, although rabbitsand farm animals, such as sheep, pigs and cows, arefar more amenable to the process than mice. In eachcase, donor nuclei were obtained from the morula orblastocyst-stage embryo and transferred to an egg oroocyte from which the nucleus had been removedwith a pipette (Smith & Wilmut 1989, Willadsen1989, Collas & Robl 1990, McLaughlin et al. 1990).The donor nucleus can be introduced by promotingfusion between the egg and a somatic cell. A briefelectric pulse is often used to achieve this, as it alsoactivates embryonic development by stimulatingthe mobilization of calcium ions.
A major advance was made in 1995, when twolive lambs, Megan and Morag (Fig. 11.6), were pro-duced by nuclear transfer from cultured embryoniccells (Campbell et al. 1996). This demonstrated theprinciple that mammalian nuclear transfer was pos-sible using a cultured cell line. The same group laterreported the birth of Dolly (Fig. 11.7), followingnuclear transfer from an adult mammary epithelialcell line (Wilmut et al. 1997). This was the firstmammal to be produced by nuclear transfer from adifferentiated adult cell, and aroused much debate
among both scientists and the public concerning thepossibility of human cloning (see Johnson 1998). Itwas suggested that a critical factor in the success ofthe experiment was the quiescent state of the cells in culture, allowing synchronization between thedonor and recipient cell cycles. For the production ofDolly, this was achieved by lowering the level ofserum in the culture medium, causing the cells towithdraw from the cell cycle due to lack of growthfactors. However, the success rate was very low:only one of 250 transfer experiments produced aviable lamb. Similar transfer experiments have sincebeen carried out in mice, cows, pigs and goats(Cibelli et al. 1998, Wakayama et al. 1998, Baguisi et al. 1999, Polejaeva et al. 2000).
The success of nuclear transfer in domestic mam-mals provides a new route for the production oftransgenic animals. This involves the introductionof DNA into cultured cells, which are then used as asource of donor nuclei for nuclear transfer. Such acell-based strategy has many advantages over tradi-tional techniques, such as microinjection, includingthe ability to screen transformed cells for high-leveltransgene expression prior to the nuclear-transferstep. The production of a transgenic mammal bynuclear transfer from a transfected cell line was first
Fig. 11.6 Megan and Morag, the first sheep produced bynuclear transfer from cultured cells. Reproduced by kindpermission of the Roslin Institute, Edinburgh.
Fig. 11.7 Dolly and her lamb Bonnie. Dolly was the firstmammal to be generated by nuclear transfer from an adult cell. Reproduced by kind permission of the RoslinInstitute, Edinburgh.
POGC11 9/11/2001 11:15 AM Page 214

Genetic manipulation of animals 215
achieved by Schnieke et al. (1997), who introducedthe gene for human factor IX into fetal sheep fibrob-lasts and transferred the nuclei to enucleated eggs.The resulting sheep, Polly, produces the recombinantprotein in her milk and can therefore be used as abioreactor (Chapter 14). More recently, McCreath et al. (2000) succeeded in producing a transgenicsheep by nuclear transfer from a somatic cell whosegenome had been specifically modified by gene targeting. A foreign gene was introduced into theCOL1A1 locus and was expressed at high levels inthe lamb.
DNA transfer to other vertebrates
Gene transfer to Xenopus
Xenopus oocytes as a heterologous expression system
Since Gurdon et al. (1971) first showed that oocytessynthesized large amounts of globin after they hadbeen microinjected with rabbit globin mRNA, theXenopus oocyte expression system has been a valu-able tool for expressing a very wide range of proteinsfrom plants and animals (Colman 1984). X. laevis isan African clawed frog. Oocytes can be obtained in large numbers by removal of the ovary of an adult female. Each fully grown oocyte is a large cell(0.8–1.2 mm diameter) arrested at first meioticprophase. This large cell has a correspondingly largenucleus (called the germinal vesicle), which is locatedin the darkly pigmented hemisphere of the oocyte.
Due to the large size of the oocytes, mRNA – eithernatural or synthesized by transcription in vitro,using phage-T7 RNA polymerase (Melton 1987) –can be readily introduced into the cytoplasm ornucleus by microinjection. This is achieved using afinely drawn glass capillary as the injection needle,held in a simple micromanipulator. DNA can also beinjected. The oocyte nucleus contains a store of the three eukaryotic RNA polymerases, enough tofurnish the needs of the developing embryo at leastuntil the 60 000-cell stage. The RNA polymerasesare available for the transcription of injected exogen-ous DNA. Using this system, it has therefore beenpossible to express complementary DNAs (cDNAs)linked to a heat-shock promoter or to mammalian
virus promoters (Ballivet et al. 1988, Ymer et al.1989, Swick et al. 1992). In addition, vaccinia virusvectors (Chapter 10) can be used for gene expressionin the cytoplasm (Yang et al. 1991).
An important aspect of the oocyte expression system is that recombinant proteins are usually correctly post-translationally modified and directedto the correct cellular compartment. For example,oocytes translate a wide variety of mRNAs encod-ing secretory proteins, modify them and correctlysecrete them (Lane et al. 1980, Colman et al. 1981).Foreign plasma-membrane proteins are generallytargeted to the plasma membrane of the oocyte,where they can be shown to be functional. The firstplasma-membrane protein to be expressed in thissystem was the acetylcholine receptor from the elec-tric organ of the ray, Torpedo marmorata (Sumikawaet al. 1981). Injected oocytes translated mRNAextracted from the electric organ and assembledfunctional multi-subunit receptor molecules in theplasma membrane (Barnard et al. 1982). Followingthis work, the oocyte has become a standard hetero-logous expression system for plasma-membraneproteins, including ion channels, carriers and recep-tors. The variety of successfully expressed plasma-membrane proteins is very impressive. However,there are examples of foreign channels and receptorsbeing non-functional in oocytes, either due to lack of coupling to second-messenger systems in theoocyte, incorrect post-translational modification, orother reasons (reviewed in Goldin 1991).
Xenopus oocytes for functional expression cloning
Functional expression cloning using oocytes wasfirst developed by Noma et al. (1986), using a strategyoutlined in Fig. 11.8. The following example, thecloning of the substance-K receptor, is illustrative. Ithas been found that oocytes can be made responsiveto the mammalian tachykinin neuropeptide, sub-stance K, by injecting an mRNA preparation frombovine stomach into the oocyte cytoplasm. The pre-paration contains mRNA encoding the substance-Kreceptor protein, which is evidently expressed as afunctional protein and inserted into the oocytemembrane. Masu et al. (1987) exploited this prop-erty to isolate a cDNA clone encoding the receptor.The principle was to make a cDNA library from
POGC11 9/11/2001 11:15 AM Page 215

216 CHAPTER 11
stomach mRNA, using a vector in which the cDNAwas flanked by a promoter for the SP6 or T7 RNApolymerase. This allowed in vitro synthesis of mRNAfrom the mixture of cloned cDNAs in the library.
The receptor clone was identified by testing forreceptor expression following injection of syntheticmRNA into the oocyte cytoplasm. Repeated sub-division of the mixture of cDNAs in the library led to the isolation of a single cloned cDNA. The strat-egy described above can only be applied to cloning single-subunit proteins, not proteins composed ofdifferent subunits or proteins whose function inoocytes requires more than one foreign polypeptide.This limitation was overcome by Lubbert et al.(1987), who used a hybrid depletion procedure toclone a serotonin-receptor cDNA.
A prerequisite for using the oocyte in functionalexpression cloning is a knowledge of the oocyte’sown ion channels, carriers and receptors. Endogen-ous activity may mask or interfere with the sought-after function (for a review, see Goldin 1991).
Transient gene expression in Xenopus embryos
Messenger RNA, synthesized and capped in vitro,can be microinjected into dejellied Xenopus embryosat the one- or two-cell stage. The mRNA is distri-buted among the descendants of the injected cellsand is expressed during early development. This
approach has been exploited very widely for examin-ing the developmental effects resulting from theoverexpression of normal or altered gene products(reviewed by Vize & Melton 1991).
DNA can be introduced into Xenopus embryos in the same manner. However, unlike the situationin mammals, where the injected DNA integratesrapidly into the genome, exogenous DNA in Xenopuspersists episomally and undergoes extensive replica-tion (Endean & Smithies 1989). Bendig and Williams(1983) provide a typical example of this process.They injected a recombinant plasmid carrying Xenopusglobin genes into the egg and showed that theamount of plasmid DNA increased 50- to 100-foldby the gastrula stage. In later development, theamount of DNA per embryo decreased, and most of the persisting DNA co-migrated with high-molecular-weight chromosomal DNA. This differencebetween mammals and amphibians probably re-flects their distinct modes of early development. In mammals, cleavage divisions are slow and asyn-chronous. Gene expression occurs throughout earlydevelopment and supplies the embryo with the proteins it requires at a steady rate. Conversely,there is no transcription in the early Xenopus embryoand yet the cleavage divisions are rapid and syn-chronous. DNA replication relies on stored maternalgene products, so there is a stockpile of chromatinassembly proteins and replication enzymes. Exogen-ous DNA injected into Xenopus eggs is thereforeassembled immediately into chromatin and under-goes replication in tune with the rapid DNA synthesisalready occurring in the nucleus (Leno & Laskey1991). Etkin et al. (1987) have analysed the replica-tion of a variety of DNAs injected into Xenopusembryos. It was found that various plasmidsincrease to different extents. This was not simplyrelated to the size of the plasmid, but also reflectedthe presence of specific sequences that inhibitedreplication. Replication has also been found todepend upon the conformation and number ofmolecules injected (Marini et al. 1989).
Transgenic Xenopus
DNA injected into early Xenopus embryos is ex-pressed in a mosaic fashion during development,regardless of the promoter used, which limits the use
Poly(A)+RNA
cDNA library
Microinjection
Poly(A)+RNA
Sizefractionation
Synthesis
Xenopusoocyte
Microinjection
ExtractionAffinity chromatography
Tissue
In vitro transcriptionand capping
Assay forfunctionalexpression
Fig. 11.8 Strategy for functional expression cloning, usingXenopus oocytes as a heterologous expression system.
POGC11 9/11/2001 11:15 AM Page 216

Genetic manipulation of animals 217
of this system for the analysis of gene expression andfunction. Some of the DNA does become incor-porated into the genome and may be transmittedthrough the germ line (Rusconi and Schaffner1981). However, integration occurs at a very lowfrequency and, given the long generation interval of Xenopus laevis (12–18 months from egg to adult), this is not an efficient way to generate trans-genic frogs.
A simple and efficient process for large-scaletransgenesis in Xenopus has become available onlyin the last few years (Kroll & Amaya 1996). In thistechnique, known as restriction-enzyme-mediatedintegration (REMI), linearized plasmids containingthe transgene of interest are mixed with decon-densed sperm nuclei and treated with limitingamounts of a restriction enzyme to introduce nicksin the DNA. The nuclei are then transplanted intounfertilized Xenopus eggs, where the DNA is repaired,resulting in the integration of plasmid DNA into thegenome. This technique allows the production of upto 700 transgenic embryos per person per day, mostof which survive at least to the swimming-tadpolestage. The decondensed nuclei are extremely fragile,so careful handling and transplantation withinabout 30 min are required for a good yield of normaltransgenic embryos. In some cases, viable trans-genic adults have been derived from the tadpolesand transgenic X. laevis lines have been established(Bronchain et al. 1999, Marsh-Armstrong et al.1999). A disadvantage of X. laevis is that the speciesis tetraploid. Offield et al. (2000) have thereforeestablished transgenic lines of the closely related butdiploid species Xenopus tropicalis, which also has ashorter generation interval than its tetraploid cousin.
Since Xenopus is used worldwide as a develop-mental model organism, transgenic Xenopus techno-logy has been rapidly adopted in many laboratories and is being used to examine (or in many cases re-examine) the roles of developmental genes. Thusfar, the sophisticated tools used in transgenic micehave not been applied to Xenopus, but this is only amatter of time. Recently, an inducible expressionsystem based on the use of a Xenopus heat-shock promoter was described, allowing inducible controlof the GFP gene. This system has been used to invest-igate Wnt signalling in early Xenopus development(Wheeler et al. 2000). As discussed above, one of the
early successes in transgenic mouse methodologywas the expression of rat growth hormone, resultingin transgenic mice up to twice the size of their non-transgenic siblings. The role of growth hormone inamphibian metamorphosis has now been examinedby expressing Xenopus growth hormone in trans-genic frogs. The transgenic tadpoles developed at thesame rate as control tadpoles, but typically grew totwice the normal size (Huang & Brown 2000). Aftermetamorphosis, the transgenic frogs also grew muchmore quickly than controls and showed skeletaldefects.
Gene transfer to fish
Fish transgenesis can be used to study gene functionand regulation, e.g. in model species, such as thezebrafish (Danio rerio) and medaka (Oryzias latipes),and to improve the traits of commercially importantspecies, such as salmon and trout. Gene-transfertechnology in fish has lagged behind that of mam-mals, predominantly due to the lack of suitable regulatory elements to control transgene expression.The first transgenic fish carried transgenes driven by mammalian or viral regulatory elements, andtheir performance varied considerably. For example,attempts to express growth-hormone genes in troutinitially met with little success, and this may havebeen due to the inability of fish cells to correctly pro-cess mammalian introns (Betancourt et al. 1993).However, fish are advantageous assay systems forseveral reasons, including their fecundity, the factthat fertilization and development are external andthe ease with which haploid and uniparental diploidembryos can be produced (Ihssen et al. 1990).
Like frogs, the injection of DNA into fish eggs andearly embryos leads to extensive replication andexpression from unintegrated transgenes, so thatfish, like frogs, can be used for transient expressionassays (Vielkind 1992). Some of the DNA integratesinto the genome, leading to germ-line transmissionand the production of transgenic fish lines (reviewedby Iyengar et al. 1996). There has been recentprogress in the development of transgenic fish with enhanced growth characteristics, particularlythrough the use of expression constructs that arederived from the same species (e.g. Rahman et al.1998; reviewed by Dunham 1999). It is likely that
POGC11 9/11/2001 11:15 AM Page 217

218 CHAPTER 11
transgenic fish will be the first genetically modifiedanimals to enter the food-chain.
DNA transfer to invertebrates
Transgenic flies
Drosophila P elements
P elements are transposable DNA elements that, undercertain circumstances, can be highly mobile in thegerm line of D. melanogaster. The subjugation of thesesequences as specialized vector molecules in Droso-phila was a landmark in Drosophila genetics. Throughthe use of P-element vectors, any DNA sequence canbe introduced into the genome of the fly.
P elements cause a syndrome of related geneticphenomena called P–M hybrid dysgenesis (Binghamet al. 1982, Rubin et al. 1982). Dysgenesis occurswhen males of a P (paternally contributing) strainare mated with females of an M (maternally contrib-uting) strain, but not when the reciprocal cross ismade. The syndrome predominantly affects thegerm line and induces a high rate of mutation andfrequent chromosomal aberrations, resulting inabnormal (dysgenic) hybrid offspring. In extremecases, there is failure to produce any gametes at all.
Hybrid dysgenesis occurs because P strains contain transposable genetic elements, P elements,which are mobilized in the eggs of M-strain females(eggs that are permissive for P-element transposi-tion are described as ‘M-cytotype’). The P elementsdo not cause dysgenesis in crosses within P strains,because they are not mobilized in P-cytotype eggs.This is because the P element encodes a repressor ofits own transposase, which prevents transposition.When a sperm from a P-cytotype male fertilizes theegg of an M-cytotype female, the absence of repres-sor in the egg results in temporary derepression ofthe transposase, such that P-element transpositionoccurs at a high frequency. The high rate of muta-tion characteristic of the dysgenesis syndromereflects the insertion of P elements into multiplegenetic loci.
Several members of the P-element family havebeen cloned and characterized (O’Hare & Rubin1983). The prototype is a 2.9-kb element, whileother members of the family appear to have arisen
by internal deletion events. The elements are char-acterized by perfect 31-bp inverted terminal repeats,which are recognized by the transposase. The proto-type element contains a single gene, comprising fourexons, encoding the transposase (a truncated ver-sion of the transposase may act as the repressor).The transposase primary transcript is differentiallyspliced in germ cells and somatic cells, such thatfunctional transposase is produced only in germcells. Laski et al. (1986) showed this clearly by making a P-element construct in which the differen-tially spliced third intron was precisely removed.This element showed a high level of somatic transposi-tion activity. Naturally occurring short P elementsare generally defective, because they do not encodefunctional transposase. However, they do possessthe inverted terminal repeats and can be activated in trans by transposase supplied by a non-defective P element in the same nucleus.
Spradling and Rubin (1982) devised an approachfor introducing P-element DNA into Drosophila chromosomes. Essentially, a recombinant plasmidcomprising a 2.9 kb P element together with someflanking Drosophila DNA sequences, cloned in thepBR322 vector, was microinjected into the posteriorpole of M-cytotype embryos. The embryos wereinjected at the syncytial blastoderm stage, when thecytoplasm has not yet become partitioned into individual cells (Fig. 11.9). The posterior pole waschosen because this is where the germ line ori-ginates, and P-element DNA in this region wasexpected to be incorporated into the genome in aproportion of the germ cells.
A screen of progeny lines showed that P elementshad indeed integrated at a variety of sites in each ofthe five major chromosomal arms, as revealed by in situ hybridization to polytene chromosomes. P-element integration occurred by transposition, notby random integration. This was proved by probingSouthern blots of restricted DNA and showing thatthe integrated P element was not accompanied bythe flanking Drosophila or pBR322 DNA sequencespresent in the recombinant plasmid (Spradling & Rubin 1982). The injected plasmid DNA musttherefore have been expressed at some level beforeintegration, so as to provide transposase.
These experiments showed that P elements couldtranspose with a high efficiency from injected plasmids
POGC11 9/11/2001 11:15 AM Page 218

Genetic manipulation of animals 219
into diverse sites in the chromosomes of germ cells.At least one of the integrated P elements in eachprogeny line remained functional, as evidenced bythe hypermutability it caused in subsequent crossesto M-cytotype eggs.
Development of P-element vectors for gene transfer
Rubin and Spradling (1982) exploited their findingthat P elements can be artificially introduced in theDrosophila genome. A possible strategy for using the
P element as a vector would be to attempt to identifya suitable site in the 2.9 kb P-element sequencewhere insertion of foreign DNA could be made with-out disrupting genes essential for transposition.However, an alternative strategy was favoured. Arecombinant plasmid was isolated which compriseda short (1.2 kb), internally deleted member of the P-element family together with flanking Drosophilasequences, cloned in pBR322. This naturally defect-ive P element did not encode transposase (O’Hare & Rubin 1983). Target DNA was ligated into thedefective P element. The aim was to integrate thisrecombinant P element into the germ line of injectedembryos by providing transposase function in trans.Two approaches for doing this were initially tested.In one approach a plasmid carrying the recombin-ant P element was injected into embryos derivedfrom a P–M dysgenic cross, in which transposaseactivity was therefore expected to be high. A disadvantage of this approach was that frequentmutations and chromosomal aberrations wouldalso be expected. In the other approach, the plasmidcarrying the recombinant P element was co-injectedwith a plasmid carrying the non-defective 2.9 kbelement.
In the first experiments of this kind, embryos homo-zygous for the rosy mutation were microinjectedwith a P-element vector containing a wild-type rosygene. Both methods for providing complementingtransposase were effective. Rosy+ progeny, recognizedby their wild-type eye colour, were obtained from20–50% of injected embryos. The chromosomes ofthese flies contained one or two copies of the integ-rated rosy transgene. The rosy gene is a particularlyuseful genetic marker. It produces a clearly visiblephenotype: Rosy− flies have brown eyes instead ofthe characteristic red colour of Rosy+ flies. The rosygene encodes the enzyme xanthine dehydrogenase,which is involved in the production of a precursor ofeye pigments. The rosy gene is not cell-autonomous:expression of rosy anywhere in the fly, for example ina genetically mosaic fly developing from an injectedlarva, results in a wild-type eye colour. Selectablemarkers have been used instead of visible markers to identify transformed flies. These include the alcohol dehydrogenase gene adh (Goldberg et al.1983) and neo (Steller & Pirrotta 1985). Other eye-colour markers have been used, including white and
About 12 nuclei migrate tothe posterior pole where theyform germ line precursor cellsor pole cells
Individualblastodermcells formedBlastoderm cell
Pole cells(germ lineprecursors)
Chorion(protectivecoat)
Micropyle(through whichsperm enters)Fertilized egg
Plasma membrane Female pronucleusMale pronucleus
Diploid cleavage nuclei
Anteriorend
Posteriorend
Germ plasm
Syncytialblastoderm
DNA injected
9 sy
nchr
onou
s nu
clea
r di
visi
ons
Fig. 11.9 Early embryogenesis of Drosophila. DNA injected at the posterior end of the embryo just prior to pole-cellformation is incorporated into germ-line cells.
POGC11 9/11/2001 11:15 AM Page 219

220 CHAPTER 11
vermilion (Ashburner 1989, Fridell & Searles 1991),as well as alternative visible markers, such as rough(which restores normal eye morphology) and yellow(which restores normal body pigmentation and isparticularly useful for scoring larvae) (Locket et al.1992; Patton et al. 1992).
A simple P-element vector is shown in Fig. 11.10(Rubin & Spradling 1983). It consists of a P elementcloned in the bacterial vector pUC8. Most of the Pelement has been replaced by the rosy gene, but theterminal repeats essential for transposition havebeen retained. The vector includes a polylinker sitefor inserting foreign sequences. Transposition of therecombinant vector into the genome of injected larvae is brought about by co-injecting a helper Pelement, which provides transposase in trans butwhich cannot transpose itself because of a deletionin one of its terminal inverted repeats. Such an element is referred to as a wings-clipped element
(Karess & Rubin 1984). An alternative strategy is toinject purified transposase protein (Kaufman & Rio1991). The capacity of P-element vectors is large,although increasing the size of the recombinant element appears to reduce the transposition fre-quency. Inserts of over 40 kb have been successfullyintroduced into flies (Haenlin et al. 1985) and thishas allowed the construction of cosmid librariesusing P-element vectors (Speek et al. 1988).
As well as their use for germ-line transformation,P elements have been exploited for insertional muta-genesis, as cloning tags and as entrapment vectorsto detect genes and regulatory elements. Similarapplications have been applied to other transposableelements, such as the Ac–Ds transposons of maize,and to other gene-transfer systems, such as retro-viruses and the T-DNA of Agrobacterium tumefaciens.These diverse uses of gene-transfer vectors are dis-cussed in Chapter 13.
Fig. 11.10 P-element derivatives as avector system. (See text for details.)
Structure of P-element derivative: Carnegie 20Structure of P-element derivative: Carnegie 20
7.2 kb rosy fragmentIR pUC8 vector
Polylinker
Insert DNA as required
31 bp inverted repeat (IR)
Lacks own transposase gene
Structure of helper P-element: pπ 25.7 wings clippedStructure of helper P-element: pπ 25.7 wings clipped
IR
23 bp of IR deletedtherefore cannot be transposed
Encodes transposase
0 1 2 3
POGC11 9/11/2001 11:15 AM Page 220





























