Embed Size (px)
Citation preview

INTRODUCTION
Traditionally, protein kinases have been divided into two non-overlapping families: the protein tyrosine kinases and theserine/threonine kinases (Hanks and Quinn, 1991; Hanks et al.,1988). The protein tyrosine kinase family largely consists ofthe receptor protein kinases and cytoplasmic tyrosine kinaseslinked to intracellular signaling mechanisms (Ullrich andSchlessinger, 1990). The remainder of protein kinases belongto the serine/threonine family of protein kinases and areinvolved in the regulation of a wide variety of cellular functions(Hanks and Hunter, 1995). These two distinct families wereoriginally identified based on conserved amino acid motifswithin their catalytic domains (Hanks et al., 1988). Due to theabsolute specificity for their respective substrate residues, itwas believed that protein kinases exclusively phosphorylatedeither tyrosine residues or serine/threonine residues. The recentdiscovery of protein kinases capable of phosphorylating allthree hydroxyl amino acids, termed the dual specificity kinases,has forced a re-evaluation of protein kinase specificity(Lindberg et al., 1992). Surprisingly, the catalytic domain of
the dual specificity kinases is indistinguishable from that of theserine/threonine kinases.
One of the first dual specificity kinases to be discovered wasCLK1 (cdc2-like kinase (Ben-David et al., 1991; Howell et al.,1991; Johnson and Smith, 1991). CLK1, also termed STY, wasinitially isolated in a screen designed to identify tyrosinekinases (Ben-David et al., 1991; Howell et al., 1991). CLK1has been shown to autophosphorylate on serine, threonine andtyrosine residues and phosphorylate exogenous substrates onserine and threonine residues (Ben-David et al., 1991; Duncanet al., 1995; Howell et al., 1991). Tyrosine phosphorylation ofCLK1 has been proposed to be an important determinant ofenzyme activity, as the majority of CLK1 activity can beimmunoprecipitated with antibodies to phosphotyrosine(Howell et al., 1991). CLK1 has recently been shown to belongto a small subfamily of protein kinases which contains at leastfour highly conserved isoforms CLK1, CLK2, CLK3, andCLK4 (Hanes et al., 1994; Nayler et al., 1997). CLKhomologues have been isolated from a number of organismsincluding: Arabidopsis, Drosophila, mouse and human (Ben-David et al., 1991; Duncan et al., 1995; Hanes et al., 1994;
3241Journal of Cell Science 113, 3241-3253 (2000)Printed in Great Britain © The Company of Biologists Limited 2000JCS1491
CLK1 was one of the first identified dual specificity kinasesand is the founding member of the ‘LAMMER’ family ofkinases. We have established the substrate site specificity ofCLK1. We report here that truncation of the N terminusof CLK1 resulted in a dramatic increase in CLK1enzymatic activity, indicating that the N terminus acts as anegative regulatory domain. The N-terminal truncationresulted in a 45-fold increase in Vmax, suggesting that thisdomain does not contain a pseudo-substrate motif, but mayact to conformationally constrain the catalytic activity ofCLK1. Tyrosine phosphorylation has been proposed to becritical for CLK1 activity, however, CLK1 activity wasunaffected by exposure to tyrosine phosphatases.Treatment of CLK1 with the serine/threonine specificphosphatase PP2A, resulted in a 2- to 6-fold increase inenzymatic activity. Incubation of CLK1 with tyrosinephosphatases in combination with PP2A abolished CLK1activity. These data suggest that CLK1 is regulated by threedistinct mechanisms that serve to both positively and
negatively regulate CLK1 activity. CLK1 activity ispositively regulated by phosphorylation on either tyrosineresidues or serine/threonine residues, and is negativelyregulated by steric constraints mediated by the N-terminaldomain, as well as, by phosphorylation on a subset ofserine/threonine residues within the catalytic domain.CLK1 mRNA is expressed at low levels in all tissues andcell lines examined. The full-length and truncated spliceforms are expressed at roughly equivalent levels in mosttissues. The ratio of the two splice variants of CLK1 can bealtered by treatment with cycloheximide. CLK1 proteinexpression is limited to a small subset of highly localizedneuronal populations in the rat brain. Contrary to previousstudies using overexpression systems, we show that CLK1protein is primarily found in the cytoplasm of these cells,with only a small fraction localized to the nucleus.
Key words: Phosphatase, Cytoplasmic, Neuronal
SUMMARY
Biochemical characterization and localization of the dual specificity kinase
CLK1
Harry J. Menegay, Michael P. Myers, Fred M. Moeslein and Gary E. Landreth*
Alzheimer Research Laboratory, Department of Neurosciences, Case Western Reserve University, School of Medicine, 10900Euclid Ave., Cleveland, Ohio 44106, USA*Author for correspondence (e-mail: [email protected])
Accepted 1 July 2000; published on WWW 22 August

3242
Howell et al., 1991; Johnson and Smith, 1991). All fourisoforms have a highly conserved domain structure. Theconserved kinase domain is located at the C terminus of themolecule and contains the signature amino acid motif‘EHLAMMERILG’ in subdomain X, which has led thesekinases to be dubbed ‘LAMMER’ kinases (Hanes et al., 1994;Yun et al., 1994). The 160 amino acid N-terminal domain hasbeen proposed to comprise a putative regulatory domain andincludes a bipartite nuclear localization signal (Duncan et al.,1995). With the exception of the nuclear localization signals,the N-termini of the CLK isoforms are only distantly related(Hanes et al., 1994). Surprisingly, mRNAs for all four CLKisoforms are alternatively spliced to produce proteins in whichthe kinase domain is missing, resulting in the expression ofproteins which comprise only the putative N-terminalregulatory sequences (Duncan et al., 1995; Hanes et al., 1994).
Mutations in the DrosophilaCLK homologue, darkener ofapricot (DOA) suggest that CLK family members play animportant role during development. Mutations in DOA areembryonic lethal and lead to defects in differentiation,including abnormalities in segmentation, eye formation, andneuronal development (Yun et al., 1994). The importance ofCLK family members during neuronal development issupported by the finding that expression of CLK1 in PC12 cellscauses the cells to undergo neuronal differentiation (Myers etal., 1994). The ability of CLK1 to differentiate PC12 cells wasassociated with the CLK1-dependent activation of members ofthe MAP kinase cascade, suggesting that CLK1 normallyfunctions in signal transduction cascades (Myers et al., 1994).Indeed, the Arabadopsis CLK1 homologue, AFC1,complements mutations in the yeast MAP kinases KSS1 andFUS3 (Bender and Fink, 1994). Both CLK1 and CLK2 haverecently been shown to phosphorylate and activate the tyrosinephosphatase PTP1B (Moeslein et al., 1999). CLK1 may alsoplay a role in regulating RNA splicing, as CLK1 has beenfound to form complexes with and phosphorylate members ofthe SR family of splicing factors (Colwill et al., 1996a,b) andhas been shown to affect the splicing of mRNA (Duncan et al.,1997). It has recently been demonstrated that this affect is theresult of the phosphorylation of SR proteins, but not otheressential splicing factors (Prasad et al., 1999).
A better understanding of the biochemical characteristics ofCLK1 will provide additional insights into the function andregulation of this potentially important family of kinases.Previous studies have investigated the expression of CLK1mRNA in some tissues, but have not examined expression ofthe endogenous protein. This study describes the identificationof the consensus phosphorylation site of CLK1 and examinesthe role that phosphorylation and the N-terminal domain playin regulating CLK1 activity. We also describe the expressionpattern and localization of CLK1 in the rat tissues and in celllines.
MATERIALS AND METHODS
Expression of CLK1 and trCLK1 in bacteriaCLK1 was expressed either as a hexa-histidine or a GST fusionprotein. Briefly, the full length CLK1 was amplified by PCR andcloned into the BamHI sites of pQE10 (Qiagen Corp.) and pGEX-2T(Pharmacia) and transformed into the JM109 E. colistrain. CLK1 was
amplified using a plasmid containing the full length CLK1 cDNA asa template and the high fidelity Vent polymerase (New EnglandBiolabs). Expression of CLK1 was induced by stimulating 500 ml ofmid-log phase bacteria with 1 mM IPTG for 4 hours at 30°C. Thecells were harvested by centrifugation, resuspended in 3 ml ofsonication buffer (50 mM NaH2PO4, 10 mM Tris, 100 mM NaCl, pH8.0) containing 20 µg/ml polymyxin B (Sigma) and incubated on icefor 10 minutes. The resuspended cells were sonicated 3× 1 minute andthen centrifuged for 10 minutes to clear the lysate. Cleared lysatesfrom hexahistidine-CLK1 cells were bound in batch to 1 ml of TalonAffinity Resin (Clontech), while GST-CLK1 lysates were bound to 1ml of glutathione Sepharose (Pharmacia). The resins were washed 3times with 10 ml of sonication buffer and then 3 more times withsonication buffer at pH 7.4. Hexahistidine-CLK1 was eluted with animidizole elution buffer (250 mM imidizole, 10 mM Tris, 30%ethylene glycol, pH 8.0) while GST-CLK1 was eluted with aglutathione elution buffer (10 mM glutathione, 10 mM Tris, 30%ethylene glycol, pH 8.0). A CLK1 N-terminal truncation (trCLK1)was created by PCR using the following PCR primers(5′CGGGATCCGGGGAAGAGTCACCGAAGG3′, 5′CCCGGAT-TCCTCACGTATGCTTTTTAAGTGG3′) which truncate the 128 N-terminal amino acids leaving the entire CLK1 kinase domain intact.The PCR was performed using a plasmid containing the full lengthCLK1 and Vent polymerase. The PCR product was cloned into pGEX-2T and the GST-trCLK1 fusion protein purified as described.
Protein kinase assaysThe protein kinase assays employed in these studies utilized 0.5 µgof recombinant CLK1 in the presence of 1 µg of myelin basic protein(MBP), 10 mM MgCl2, 2 mM MnCl2, 10 µM [γ-32P]ATP (44dpm/fmol) in kinase buffer (10 mM Tris, 1 mM EGTA, 100 µMsodium ortho-vanadate, 20 mM p-nitrophenyl phosphate (PNP), pH7.4) in a reaction volume of 50 µl. The reactions were incubated atroom temperature for 20 minutes and stopped with the addition of 3×Laemmli sample buffer and boiled. The reaction products wereseparated by SDS-PAGE and visualized by autoradiography of thedried gels using a phosphoimager (Molecular Dynamics).Incorporation of radioactivity into MBP was quantitated byCherenkov counting of MBP-containing gel fragments. MBP peptides(LC Laboratories), S6 Peptide (UBI), PKCζ substrate peptide (fromDr M. Wooten), Tau peptide (from Dr S. Feinstein), Myosin peptide(from Dr T. Egelhof), and Crebtide were used at a concentration of50 µM under standard reaction conditions. Peptide kinase reactionswere stopped by the addition of BSA to 0.2% and TCA to 7% andincubated on ice for 15 minutes. Following a 10 minute centrifugationat 10,000 g, aliquots of the supernatants were applied to WhatmanP81 paper which were then washed 3× 5 minutes in 75 mM H3PO4as previously described (Glass et al., 1978). The incorporatedradioactivity was determined by Cherenkov counting of the washedpapers.
Sequencing of MBP peptidesMyelin basic protein (50 µM) was phosphorylated in a kinase reactioncontaining 5 µg of trCLK1 in kinase buffer containing 10 µM ATP(44 dpm/fmol), 10 mM MgCl2, and 2 mM MnCl2 for 2 hours. Thereaction products were separated by SDS-PAGE and then transferredto PVDF as previously described (Myers et al., 1994). Thephosphorylated MBP was excised from the membrane and digestedwith endoproteinase AspN. The digested peptides were separated byreverse phase HPLC and the phosphorylated peptides were sequencedby Dr C. Beach (University of Kentucky).
Phosphatase sensitivityFull length and trCLK1 were treated for 20 minutes at roomtemperature with 2 units of PP2A, YPTP1, or PTP-1B in phosphatasebuffer (10 mM Tris, 1 mM EGTA, 1 mM EDTA, 1 mM DTT, 20 mMMgCl2, pH 7.4). Phosphatase inhibitors were added to yield final
H. J. Menegay and others

3243Characterization and localization of CLK1
concentrations of 5 µM okadaic acid, 100 mM ortho-vanadate, and 20mM p-nitrophenylphosphate (PNP). The resulting mixture wassupplemented with [32P]ATP (44 dpm/fmol), MnCl2 to 2 mM and 1µg of MBP and assayed as above.
Phosphoamino acid analysisMBP, histone VIIIS (arginine rich), and casein were phosphorylatedby trCLK1 under standard reaction conditions and incubated at 20°Cfor 40 minutes. The reaction products were separated by SDS-PAGEand transferred to PVDF membrane. The membrane was stained withCoomassie blue and then destained in 10% glacial acetic acid, 15%methanol and autoradiographs were obtained. The stained radioactiveproteins were excised from the gel, subjected to Cherenkov counting,submerged in 100 µl of 6 N HCl and incubated for 45 minutes at110°C and phosphoamino acid analysis was performed essentially aspreviously described (Boyle et al., 1991). Standards were visualizedby spraying the dried plates with 0.1% ninhydrin. Radioactive aminoacids were visualized by autoradiography.
Phosphopeptide analysis/two-dimensionalphosphopeptide mappingTrCLK1 was autophosphorylated under standard reaction conditionsand incubated at 20°C for 40 minutes. The reaction products wereseparated by SDS-PAGE and transferred to nitrocellulose membrane.Autoradiographs were obtained from the membrane and a fragmentof the membrane, which contained the radioactive protein, was cutout. The 32P-labeled proteins were proteolytically digested asdescribed by Luo et al. (1991). Briefly, the immobilized proteins weredigested with either trypsin or chymotrypsin or double digested withboth enzymes. The digestion was performed by addition of 10 µg ofenzyme/200 µl for 1.5 hours at 37°C, at which point 10 µg ofenzyme/200 µl were added, and a second 1.5 hour digest at 37°C wasperformed. The supernatants were transferred to another tube anddried in a Speed-vac. Radioactivity incorporated into the pellets ofdigested peptides was determined by Cherenkov counting, and thepellets were resuspended at 1000 cpm/µl.
The 32P-labeled phosphopeptides were resolved in the firstdimension by electrophoresis at pH 1.9 on TLC plates as described(Boyle et al., 1991). The plates were dried, and then subjected toascending chromatography in the second dimension using a buffercomposed of isobutanol, pyridine, acetic acid, and water(75:15:50:60). The plates were dried and the phosphopeptides werevisualized by autoradiography.
RNA extraction and RT-PCRRNA was isolated from cell lines and mouse tissues using the methodof Chomczynski and Sacchi (1987). Total RNA (1 µg) was used in areverse transcription reaction using SuperScript II reversetranscriptase (Gibco) primed by oligo(dT). A 50-µl volume PCRreaction was carried out using a fraction (1/100) of the reversetranscription reaction using Taq DNA polymerase (BoehringerMannheim) including 50 µCi [α-32P]dCTP. The CLK1 PCR wascarried out for 30 cycles, while the GAPDH PCR was carried out for25 cycles. Additional PCR reactions were done at higher and lowercycle numbers to assure the amplification analyzed was in the linearrange. Primers used were: upstream primer 5′-ATG AGA CAT TCAAAG AGA ACT TAC TGT CCT-3′, and downstream primer 5′-CCGAAT TCC TGC TAC ACG TCT ACC TCC CAC-3′. Products wereseparated on polyacrylamide gels, dried, and imaged usingphosphorimager plates (Molecular Dynamics). The predicted size ofthe full-length kinase containing PCR product is 560 bp and thetruncated kinase-less PCR product is 453 bp. Parallel PCR reactionswere performed using primers to GAPDH as RNA loading controls.
Analysis of CLK1 isoform mRNAPC12 cells were treated with 500 mM NaCl or 10 µg/mlcycloheximide for 60 minutes, or were exposed to 300 J/m2 of UV
radiation (290-320 nm) with a Stratalinker (Stratagene) and thenincubated for an additional 60 minutes. RNA was then isolated andCLK1 and GAPDH message were amplified and analyzed as above.
Antibody productionMembers of the CLK family, while highly conserved in their kinasedomain, are very divergent their N-terminal domain. A syntheticpeptide (KRTYCPDWDERD) was synthesized which corresponds toa part of this N-terminal region that is not conserved among otherCLK family members and has no significant sequence homology withother genes in the GenBank database. The peptide was conjugated toKLH using the Imject Supercarrier System for Peptides (Pierce). Thisconjugated peptide was injected into rabbits with Freund’s CompleteAdjuvant to generate a polyclonal antiserum in rabbits. Affinitypurified CLK1 antibody was prepared from the serum by passing theserum over two columns. The first column consisted of BSAconjugated to CNBr-activated Sepharose 4B (Pharmacia Biotech) toremove non-specific interacting antibodies (Porath and Axen, 1976).The second column was prepared by first conjugatingCLK1-synthetic-peptide to BSA using 1-ethyl-3(3-dimethylaminopropyl)carbodiimide-HCl (Pharmacia Biotech). Theproduct of this reaction was then conjugated to CNBr-activatedSepharose 4B. CLK1 specific antibodies were eluted from the secondcolumn with 100 mM glycine (pH 2.5), or 100 mM triethanolamine(pH 11.5) and were then neutralized in 100 mM Tris (pH 7.4). Theeluted antibodies were concentrated with a Centricon filter to workingconcentration (0.1 µg/µl). The affinity purified CLK1 antisera wasimmunoreactive by western analysis with both the synthetic CLK1-peptide as well as recombinant full-length CLK1 protein expressed inbacteria, however it was unable to immunoprecipitate the protein.
Protein expression in rat tissuesTissue lysates were made by freezing dissected adult rat tissues inliquid nitrogen and pulverizing the tissue with a pestle and mortar.The powdered tissues were then resuspended in lysis buffer (1% NP-40, 2 mM NaF, 500 µM sodium vanadate, 200 µg/mlphenylmethylsulfonyl fluoride, 2 µg/ml aprotinin, 5 µg/ml leupeptin,150 mM NaCl, 50 mM Tris, pH 8.0) and sonicated for 3 times for 30seconds. Cell lysates were prepared by resuspending pelleted cells inlysis buffer followed by sonication for 30 seconds. Proteins from eachtissue or cell line (50 µg) were separated on SDS-polyacrylamide gels,and transferred to PVDF membrane by electroblotting. The blots wereprobed with the purified CLK1 antisera (0.5 µg), or CLK1 antisera(0.5 µg) that had been preincubated with the immunizing CLK1peptide (10 ng). CLK1 was detected with a horseradish peroxidase-conjugated goat anti-rabbit antibody and visualized by ECL reagents(Amersham).
Immunostaining of tissuesAdult rats were perfused with Bouin’s fixative and dissected tissueswere then incubated for 24 hours in the same fixative (Humason,1962). Following fixation the tissues were submersed for 72 hours in18% sucrose/PBS. The tissues were frozen in OCT (Miles Inc.) ondry ice, and 10 µM sections were prepared on a cryostat. The tissuesections were then probed with the purified CLK1 antibody, orpurified CLK1 antibody which had been preincubated with the peptidethe antibody was generated against. Visualization was by either aTexas Red-conjugated or fluorescein-conjugated secondary antibody(Jackson ImmunoResearch Laboratories Inc.) using a Leitzmicroscope or Zeiss confocal microscope.
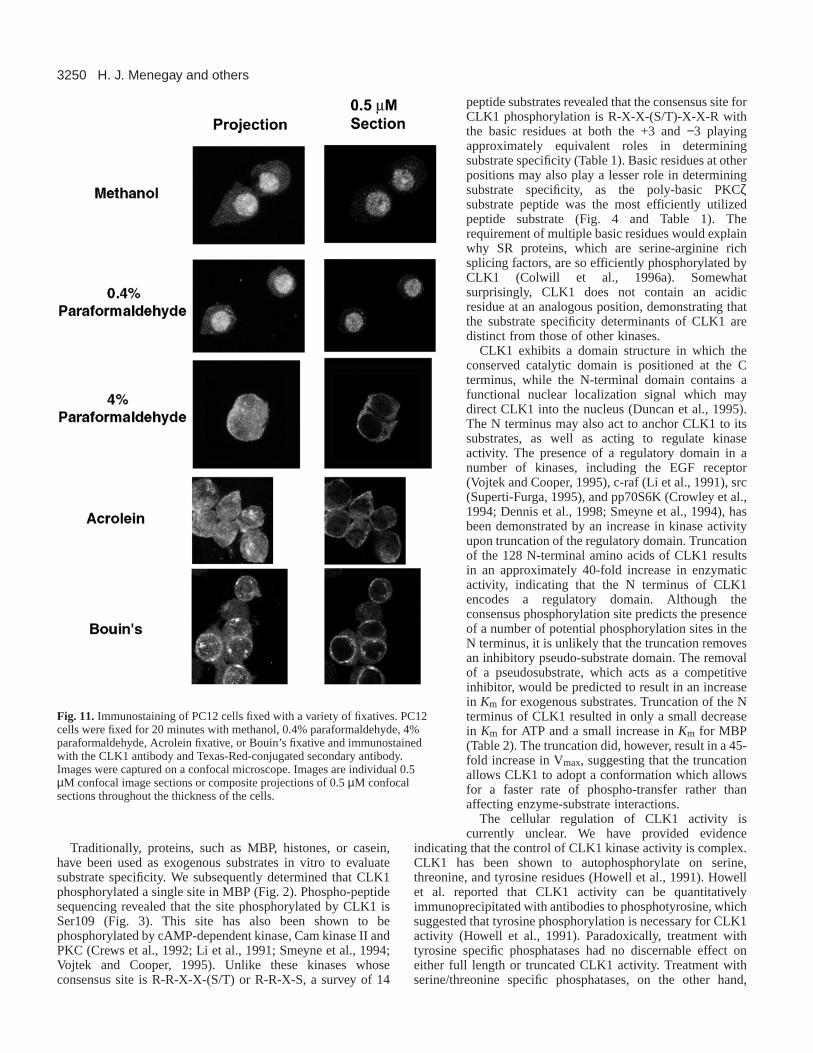
Expression of CLK1 in PC12 cellsPC12 cells grown on coverslips were fixed in methanol at –20°C, orin 0.4% paraformaldehyde, 4% paraformaldehyde, Acrolein fixative,or Bouin’s fixative on ice for 20 minutes. After extensive washing thecells were probed with the CLK1 antibody and visualized with Texas-Red-conjugated secondary antibody on a confocal microscope.

3244
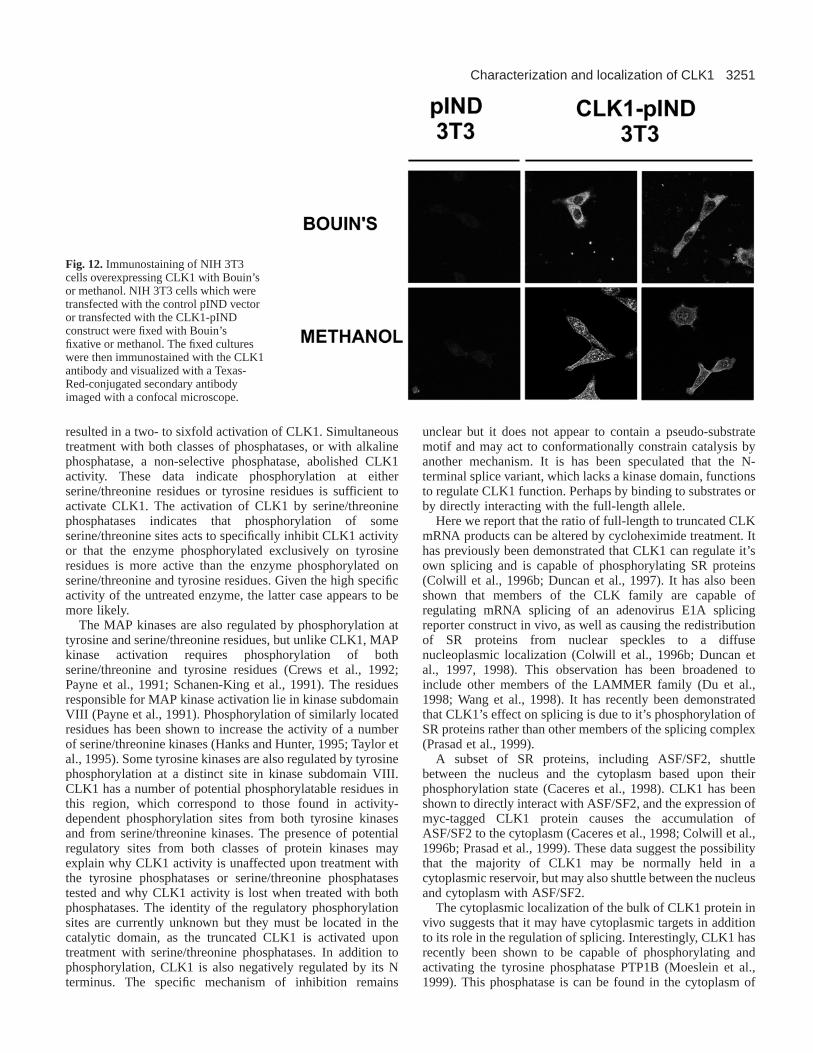
Overexpression of CLK1 in 3T3 cellspIND plasmid (Invitrogen) containing the CLK1 gene and pVgRXRwere cotransfected into NIH-3T3 cells with calcium phosphate. ThepVgRXR plasmid constitutively expresses the ecdysone receptor,while pIND contains an ecdysone-responsive promotor. It was foundthat steroid induction with ecdysone was not necessary for CLK1protein production in this system. Transfected cells were selected forthree weeks in media containing Geneticin (G418) and zeocin toobtain stably expressing cells. The cells were then grown oncoverslips. Localization of the CLK1 protein was observed by eitherfixation in Bouin’s fixative for 20 minutes on ice, or in methanol at–20°C for 20 minutes, followed by extensive washing and probingwith the CLK1 antibody. CLK1 was visualized using a Texas-Redconjugated secondary antibody on a confocal microscope.
RESULTS
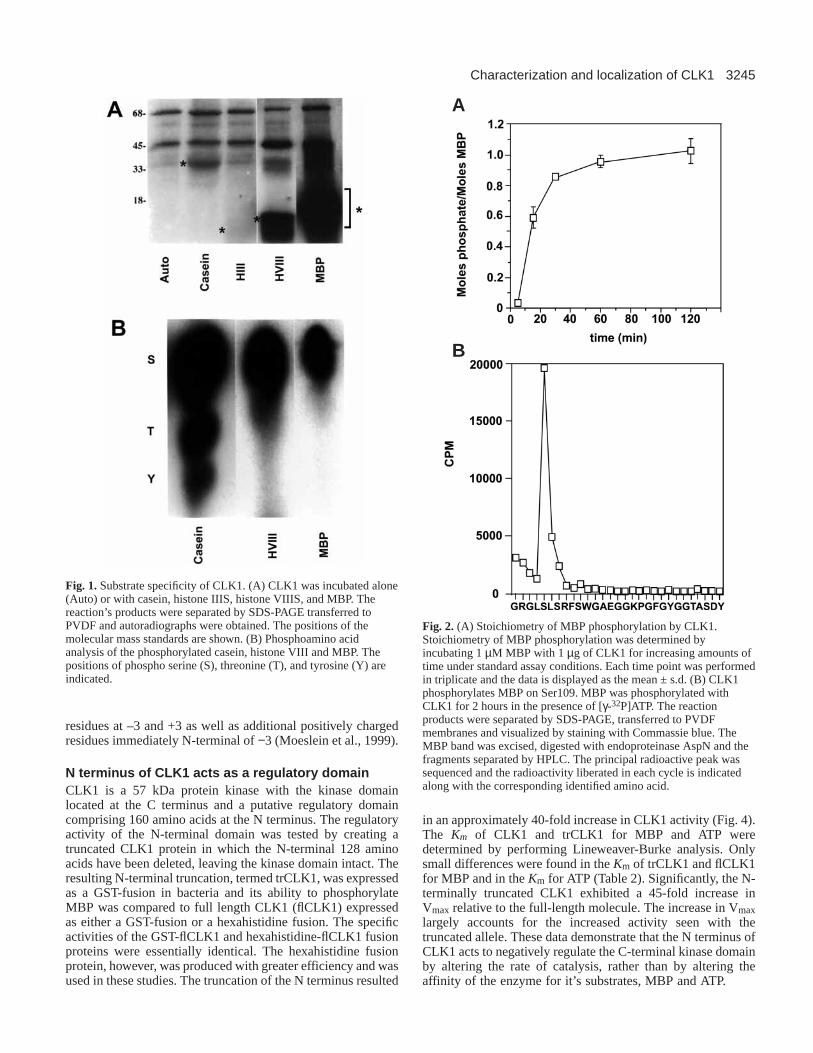
CLK1 substrate specificityA battery of substrates were tested for their ability to bephosphorylated by CLK1. CLK1 phosphorylated casein,histone VIIIS (arginine rich histones), and MBP, with thehistones and MBP being the best substrates (Fig. 1A).Phosphoamino acid analysis of these substrates revealed thatCLK1 phosphorylated MBP and Histone VIIIS predominantlyon serine residues. Casein was phosphorylated on serine, andat lower levels on threonine and tyrosine residues (Fig. 1B).CLK1 did not phosphorylate histone IIIS (lysine rich histones),lysozyme, or enolase (data not shown).
Determination of the CLK1 phosphorylation site inMBPFurther biochemical characterization was performed usingMBP as a CLK1 substrate. The stoichiometry ofphosphorylation was determined by incubating 1 µM MBPwith CLK1 for increasing periods of time. The phosphorylationof MBP reached maximum levels within 1 hour and plateauedat 1 mole 32P/mole MBP (Fig. 2A), indicating that CLK1phosphorylated MBP at a single site. The site phosphorylatedby CLK1 was determined by radiosequencing of thephosphorylated MBP. Following phosphorylation anddigestion with endoproteinase LysC, the major radioactivepeptide was sequenced (Fig. 2B). The majority of theradioactivity eluted in cycle 5, corresponding to Serine 109 inMBP. Very little radioactivity eluted in cycle 7 (Ser 111), cycle10 (Ser 114), or cycle 27 (Ser 131), which identified Ser 109as the site of phosphorylation in MBP. A synthetic peptide from
this region of MBP (MBP104-118) was efficientlyphosphorylated by CLK1 (Fig. 4), verifying that Ser 109 wasphosphorylated by CLK1. This major site of phosphorylationon MBP by CLK1 is different from that of another LAMMERfamily member DOA, whose major site of phosphorylation onMBP is serine 164 (SGSPMAR) (Lee et al., 1996).
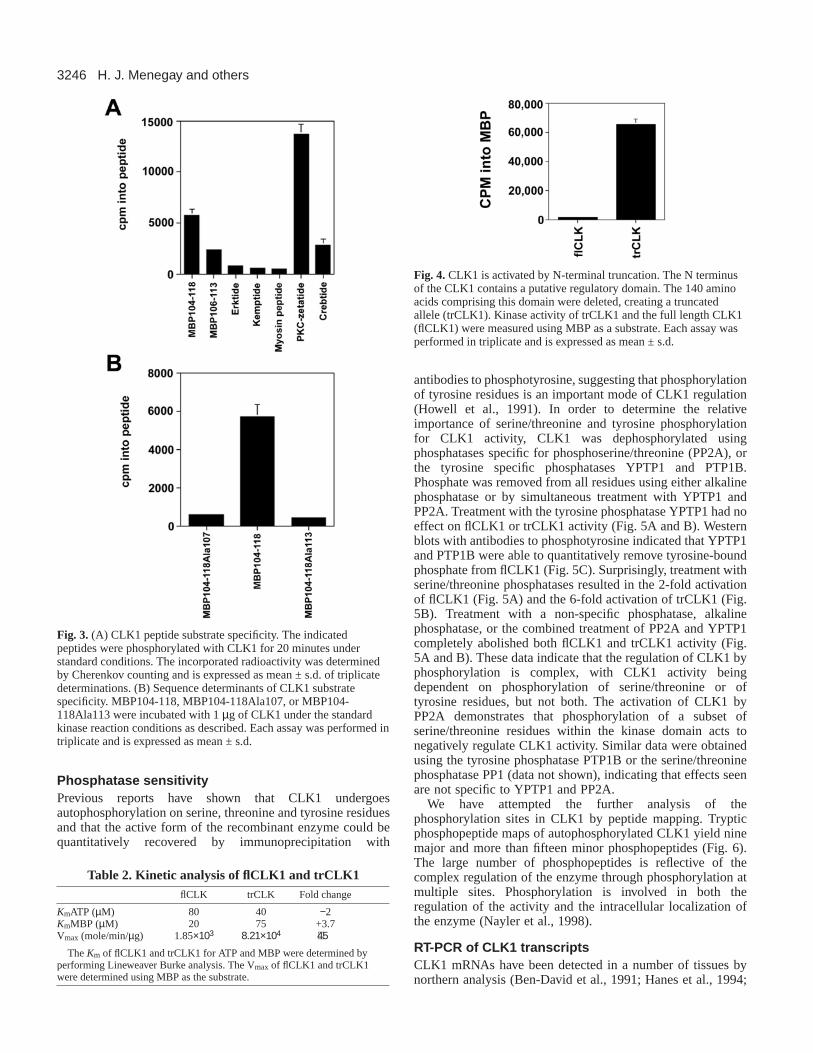
Determination of CLK1 phosphorylation consensussiteIn order to determine the consensus site of phosphorylation,the ability of CLK1 to phosphorylate other peptides was tested.CLK1 efficiently phosphorylated MBP104-118 (GKGRGLS-LSRFSWGA), PKCζ substrate (ERMRPRKRKRQGSVRRR),Crebtide (REILSRRPSYRK), but did not efficientlyphosphorylate kemptide (KLRRASLG), or a peptide derivedfrom myosin (KKRAARATSNVFA) (Fig. 3A). CLK1 was alsounable to phosphorylate a casein kinase substrate peptide(RREEETEEE), MBPtide (APRTPGGRR), a peptide derivedfrom c-fos (RKGSSSNEPSSD) or a battery of tyrosine kinasepeptides, including a src substrate peptide(KVEKIGEGTYGVVYK), Raytide-ELtm (OncogeneSciences), or polyGluTyr (data not shown). These dataindicated that a basic residue is necessary at positions +3 and−3 relative to the phosphorylated residue (Table 1) and that thetyrosine kinase activity of CLK1 is likely to be highlyrestricted. This conclusion was confirmed by using mutantMBP104-118 peptides in which either arginine 107 or arginine113 were changed to Alanine. As predicted by the peptidespecificity data, these peptides were not efficientlyphosphorylated by CLK1 (Fig. 3B). Mutation at arginine 107resulted in a 90% decrease in phosphorylation and increasedthe Km 7.8 fold (Table 1). Similarly, mutation of arginine 113resulted in a 92.5% decrease in phosphorylation and increasedthe Km 11-fold (Table 1). The apparent decrease in activity wasnot due to a decrease in the peptides affinity forphosphocellulose, as similar results were obtained when thephosphorylated peptides were analyzed by SDS-PAGE (datanot shown). These data indicate that basic residues at both −3and +3 are critical determinants of CLK1 substrate specificity.The polybasic PKCζ substrate peptide was the best peptidesubstrate tested (Km=452 nM) indicating that basic residues atother positions flanking the phosphorylation site enhanceCLK1 substrate recognition and phosphorylation. However, thebasic residues at −3 and +3 play the major role in forming aCLK1 phosphorylation site. This consensus site for CLK1 issimilar to that of CLK2 which prefers positively charged
H. J. Menegay and others
Table 1. CLK1 substrate specificityPeptide Sequence Relative activity Km (µM)
Consensus R-X-X- S*-X-X- RMBP104-118 G-K-G- R-G-L- S*-L-S- R-F-S-W-G-A 100 61MBP104-118Ala107 G-K-G- A-G-L- S*-L-S- R-F-S-W-G-A 10 480MBP104-118Ala113 G-K-G- R-G-L- S*-L-S- R-F-S-W-G-A 7.5 674PKC-zetatide E-R-M-P-R-K- R-Q-G- S*-V-R- R-R 239 0.45Crebteide R-E-I-L-S- R-R-P- S*-Y-R- K 49 80Erktide A-P-R- T*-P-G- G-R-R 13 n.d.Kemptide K-L- R-R-A- S*-L-G 9.7 n.d.Myosin peptide K-K-R-A- A-R-A- T*-S-N- V-F-A 7.5 n.d.
The sequence of the tested peptides are listed, along with their Km’s for CLK1. The activity toward the individual peptides is normalized to that obtained usingMBP104-118 as a peptide substrate. Amino acids in bold indicate the positions identified as important in forming the consensus phosphorylation site and thephosphorylated residue is indicated by an asterisk.
3245Characterization and localization of CLK1
residues at –3 and +3 as well as additional positively chargedresidues immediately N-terminal of −3 (Moeslein et al., 1999).
N terminus of CLK1 acts as a regulatory domainCLK1 is a 57 kDa protein kinase with the kinase domainlocated at the C terminus and a putative regulatory domaincomprising 160 amino acids at the N terminus. The regulatoryactivity of the N-terminal domain was tested by creating atruncated CLK1 protein in which the N-terminal 128 aminoacids have been deleted, leaving the kinase domain intact. Theresulting N-terminal truncation, termed trCLK1, was expressedas a GST-fusion in bacteria and its ability to phosphorylateMBP was compared to full length CLK1 (flCLK1) expressedas either a GST-fusion or a hexahistidine fusion. The specificactivities of the GST-flCLK1 and hexahistidine-flCLK1 fusionproteins were essentially identical. The hexahistidine fusionprotein, however, was produced with greater efficiency and wasused in these studies. The truncation of the N terminus resulted
in an approximately 40-fold increase in CLK1 activity (Fig. 4).The Km of CLK1 and trCLK1 for MBP and ATP weredetermined by performing Lineweaver-Burke analysis. Onlysmall differences were found in the Km of trCLK1 and flCLK1for MBP and in the Km for ATP (Table 2). Significantly, the N-terminally truncated CLK1 exhibited a 45-fold increase inVmax relative to the full-length molecule. The increase in Vmaxlargely accounts for the increased activity seen with thetruncated allele. These data demonstrate that the N terminus ofCLK1 acts to negatively regulate the C-terminal kinase domainby altering the rate of catalysis, rather than by altering theaffinity of the enzyme for it’s substrates, MBP and ATP.
Fig. 1.Substrate specificity of CLK1. (A) CLK1 was incubated alone(Auto) or with casein, histone IIIS, histone VIIIS, and MBP. Thereaction’s products were separated by SDS-PAGE transferred toPVDF and autoradiographs were obtained. The positions of themolecular mass standards are shown. (B) Phosphoamino acidanalysis of the phosphorylated casein, histone VIII and MBP. Thepositions of phospho serine (S), threonine (T), and tyrosine (Y) areindicated.
Fig. 2. (A) Stoichiometry of MBP phosphorylation by CLK1.Stoichiometry of MBP phosphorylation was determined byincubating 1 µM MBP with 1 µg of CLK1 for increasing amounts oftime under standard assay conditions. Each time point was performedin triplicate and the data is displayed as the mean ± s.d. (B) CLK1phosphorylates MBP on Ser109. MBP was phosphorylated withCLK1 for 2 hours in the presence of [γ-32P]ATP. The reactionproducts were separated by SDS-PAGE, transferred to PVDFmembranes and visualized by staining with Commassie blue. TheMBP band was excised, digested with endoproteinase AspN and thefragments separated by HPLC. The principal radioactive peak wassequenced and the radioactivity liberated in each cycle is indicatedalong with the corresponding identified amino acid.
A
B
3246
Phosphatase sensitivityPrevious reports have shown that CLK1 undergoesautophosphorylation on serine, threonine and tyrosine residuesand that the active form of the recombinant enzyme could bequantitatively recovered by immunoprecipitation with
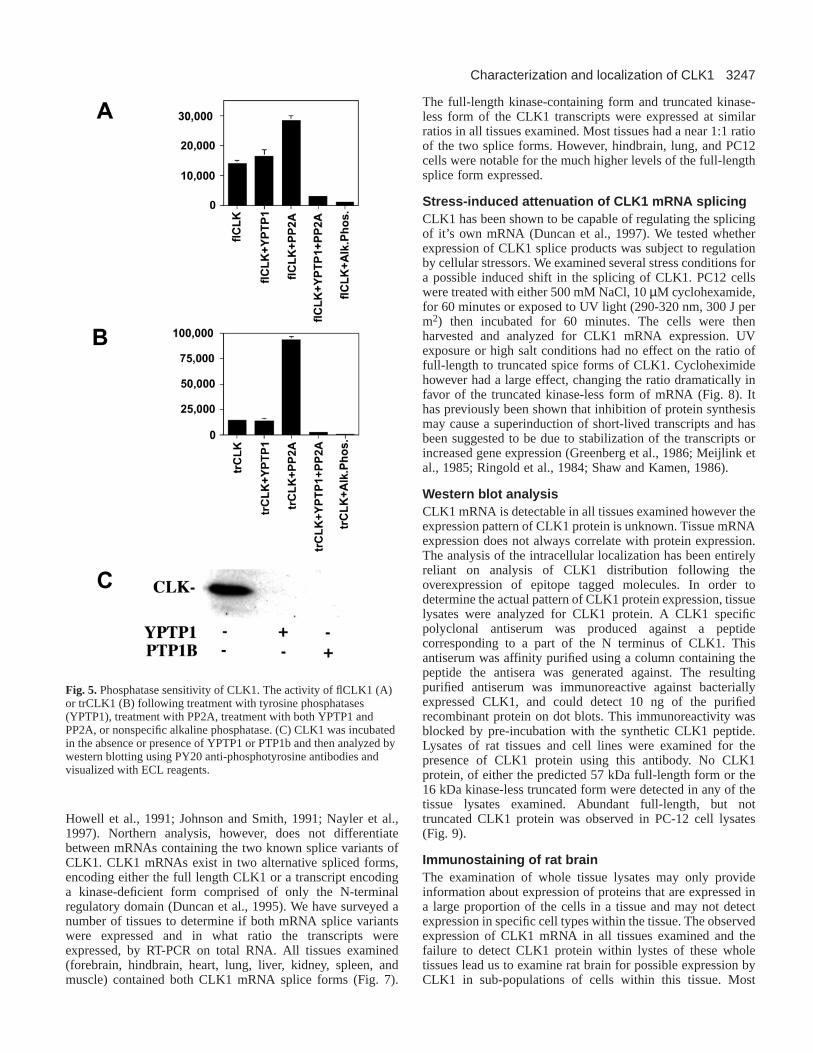
antibodies to phosphotyrosine, suggesting that phosphorylationof tyrosine residues is an important mode of CLK1 regulation(Howell et al., 1991). In order to determine the relativeimportance of serine/threonine and tyrosine phosphorylationfor CLK1 activity, CLK1 was dephosphorylated usingphosphatases specific for phosphoserine/threonine (PP2A), orthe tyrosine specific phosphatases YPTP1 and PTP1B.Phosphate was removed from all residues using either alkalinephosphatase or by simultaneous treatment with YPTP1 andPP2A. Treatment with the tyrosine phosphatase YPTP1 had noeffect on flCLK1 or trCLK1 activity (Fig. 5A and B). Westernblots with antibodies to phosphotyrosine indicated that YPTP1and PTP1B were able to quantitatively remove tyrosine-boundphosphate from flCLK1 (Fig. 5C). Surprisingly, treatment withserine/threonine phosphatases resulted in the 2-fold activationof flCLK1 (Fig. 5A) and the 6-fold activation of trCLK1 (Fig.5B). Treatment with a non-specific phosphatase, alkalinephosphatase, or the combined treatment of PP2A and YPTP1completely abolished both flCLK1 and trCLK1 activity (Fig.5A and B). These data indicate that the regulation of CLK1 byphosphorylation is complex, with CLK1 activity beingdependent on phosphorylation of serine/threonine or oftyrosine residues, but not both. The activation of CLK1 byPP2A demonstrates that phosphorylation of a subset ofserine/threonine residues within the kinase domain acts tonegatively regulate CLK1 activity. Similar data were obtainedusing the tyrosine phosphatase PTP1B or the serine/threoninephosphatase PP1 (data not shown), indicating that effects seenare not specific to YPTP1 and PP2A.
We have attempted the further analysis of thephosphorylation sites in CLK1 by peptide mapping. Trypticphosphopeptide maps of autophosphorylated CLK1 yield ninemajor and more than fifteen minor phosphopeptides (Fig. 6).The large number of phosphopeptides is reflective of thecomplex regulation of the enzyme through phosphorylation atmultiple sites. Phosphorylation is involved in both theregulation of the activity and the intracellular localization ofthe enzyme (Nayler et al., 1998).
RT-PCR of CLK1 transcriptsCLK1 mRNAs have been detected in a number of tissues bynorthern analysis (Ben-David et al., 1991; Hanes et al., 1994;
H. J. Menegay and others
Fig. 3. (A) CLK1 peptide substrate specificity. The indicatedpeptides were phosphorylated with CLK1 for 20 minutes understandard conditions. The incorporated radioactivity was determinedby Cherenkov counting and is expressed as mean ± s.d. of triplicatedeterminations. (B) Sequence determinants of CLK1 substratespecificity. MBP104-118, MBP104-118Ala107, or MBP104-118Ala113 were incubated with 1 µg of CLK1 under the standardkinase reaction conditions as described. Each assay was performed intriplicate and is expressed as mean ± s.d.
Fig. 4.CLK1 is activated by N-terminal truncation. The N terminusof the CLK1 contains a putative regulatory domain. The 140 aminoacids comprising this domain were deleted, creating a truncatedallele (trCLK1). Kinase activity of trCLK1 and the full length CLK1(flCLK1) were measured using MBP as a substrate. Each assay wasperformed in triplicate and is expressed as mean ± s.d.
Table 2. Kinetic analysis of flCLK1 and trCLK1flCLK trCLK Fold change
KmATP (µM) 80 40 −2KmMBP (µM) 20 75 +3.7Vmax (mole/min/µg) 1.85×103 8.21×104 +45
The Km of flCLK1 and trCLK1 for ATP and MBP were determined byperforming Lineweaver Burke analysis. The Vmax of flCLK1 and trCLK1were determined using MBP as the substrate.
3247Characterization and localization of CLK1
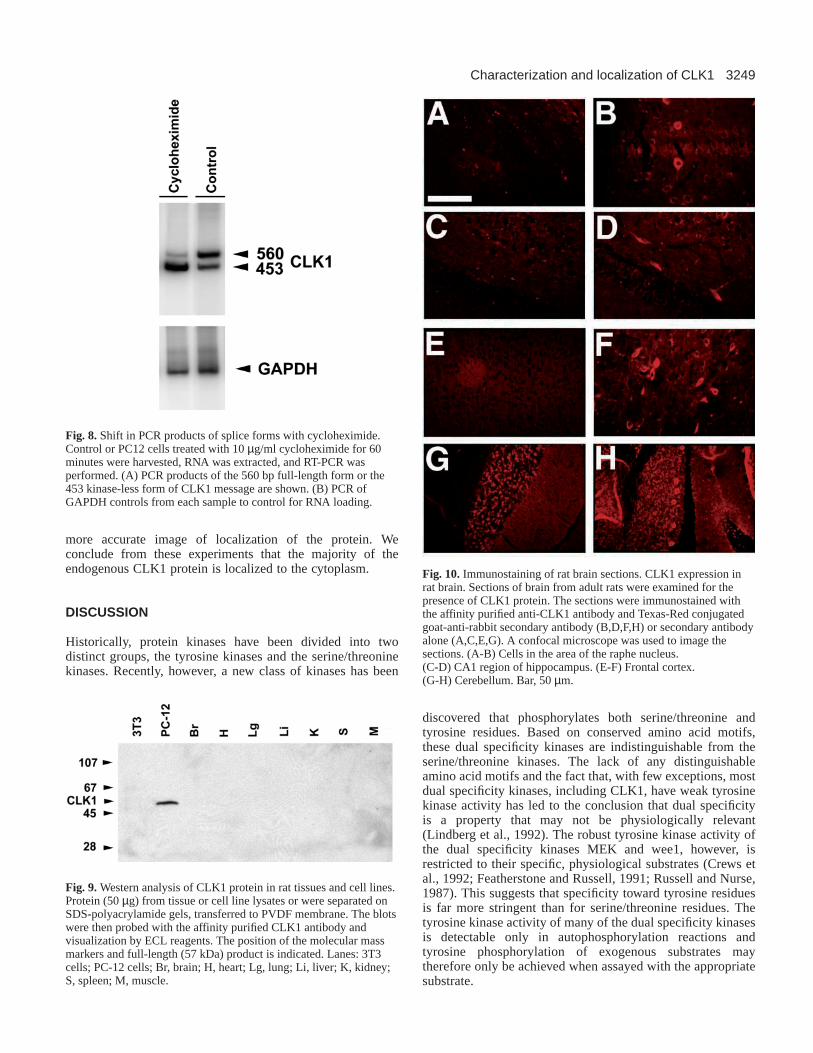
Howell et al., 1991; Johnson and Smith, 1991; Nayler et al.,1997). Northern analysis, however, does not differentiatebetween mRNAs containing the two known splice variants ofCLK1. CLK1 mRNAs exist in two alternative spliced forms,encoding either the full length CLK1 or a transcript encodinga kinase-deficient form comprised of only the N-terminalregulatory domain (Duncan et al., 1995). We have surveyed anumber of tissues to determine if both mRNA splice variantswere expressed and in what ratio the transcripts wereexpressed, by RT-PCR on total RNA. All tissues examined(forebrain, hindbrain, heart, lung, liver, kidney, spleen, andmuscle) contained both CLK1 mRNA splice forms (Fig. 7).
The full-length kinase-containing form and truncated kinase-less form of the CLK1 transcripts were expressed at similarratios in all tissues examined. Most tissues had a near 1:1 ratioof the two splice forms. However, hindbrain, lung, and PC12cells were notable for the much higher levels of the full-lengthsplice form expressed.
Stress-induced attenuation of CLK1 mRNA splicingCLK1 has been shown to be capable of regulating the splicingof it’s own mRNA (Duncan et al., 1997). We tested whetherexpression of CLK1 splice products was subject to regulationby cellular stressors. We examined several stress conditions fora possible induced shift in the splicing of CLK1. PC12 cellswere treated with either 500 mM NaCl, 10 µM cyclohexamide,for 60 minutes or exposed to UV light (290-320 nm, 300 J perm2) then incubated for 60 minutes. The cells were thenharvested and analyzed for CLK1 mRNA expression. UVexposure or high salt conditions had no effect on the ratio offull-length to truncated spice forms of CLK1. Cycloheximidehowever had a large effect, changing the ratio dramatically infavor of the truncated kinase-less form of mRNA (Fig. 8). Ithas previously been shown that inhibition of protein synthesismay cause a superinduction of short-lived transcripts and hasbeen suggested to be due to stabilization of the transcripts orincreased gene expression (Greenberg et al., 1986; Meijlink etal., 1985; Ringold et al., 1984; Shaw and Kamen, 1986).
Western blot analysisCLK1 mRNA is detectable in all tissues examined however theexpression pattern of CLK1 protein is unknown. Tissue mRNAexpression does not always correlate with protein expression.The analysis of the intracellular localization has been entirelyreliant on analysis of CLK1 distribution following theoverexpression of epitope tagged molecules. In order todetermine the actual pattern of CLK1 protein expression, tissuelysates were analyzed for CLK1 protein. A CLK1 specificpolyclonal antiserum was produced against a peptidecorresponding to a part of the N terminus of CLK1. Thisantiserum was affinity purified using a column containing thepeptide the antisera was generated against. The resultingpurified antiserum was immunoreactive against bacteriallyexpressed CLK1, and could detect 10 ng of the purifiedrecombinant protein on dot blots. This immunoreactivity wasblocked by pre-incubation with the synthetic CLK1 peptide.Lysates of rat tissues and cell lines were examined for thepresence of CLK1 protein using this antibody. No CLK1protein, of either the predicted 57 kDa full-length form or the16 kDa kinase-less truncated form were detected in any of thetissue lysates examined. Abundant full-length, but nottruncated CLK1 protein was observed in PC-12 cell lysates(Fig. 9).
Immunostaining of rat brainThe examination of whole tissue lysates may only provideinformation about expression of proteins that are expressed ina large proportion of the cells in a tissue and may not detectexpression in specific cell types within the tissue. The observedexpression of CLK1 mRNA in all tissues examined and thefailure to detect CLK1 protein within lystes of these wholetissues lead us to examine rat brain for possible expression byCLK1 in sub-populations of cells within this tissue. Most
Fig. 5.Phosphatase sensitivity of CLK1. The activity of flCLK1 (A)or trCLK1 (B) following treatment with tyrosine phosphatases(YPTP1), treatment with PP2A, treatment with both YPTP1 andPP2A, or nonspecific alkaline phosphatase. (C) CLK1 was incubatedin the absence or presence of YPTP1 or PTP1b and then analyzed bywestern blotting using PY20 anti-phosphotyrosine antibodies andvisualized with ECL reagents.

3248
tissues examined (heart, lung, liver, kidneys, spleen, muscle,skin, bone, blood vessles) had no detectable staining (data notshown), however there was robust staining in a subpopulationof cells in the adult rat brain. Cells exhibiting a neuronalphenotype were stained throughout the brain (Fig. 10). NotablyPurkinje cells in the cerebellum, cells of the inferior olive,raphe nucleus, hippocampus, and frontal cortex a showedexpression of CLK1 protein. Significantly, CLK1 was foundprincipally within the cytoplasm of the neurons. This stainingcould be blocked with the preincubation of the antisera withthe CLK1 peptide the antibody was generated against.
Immunostaining of PC12 cellsThe immunohistochemical detection of CLK1 within thecytoplasm of neurons with little or no nuclear staining was insharp contrast to previous studies examining the localization ofCLK family members within cells. These previous studies haveshown that the protein is principally localized to ‘speckles’ inthe nucleus, presumptive sites of storage for splicing factors(Huang and Spector, 1996; Jimenez-Garcia and Spector, 1993;Mattaj, 1994). However, these studies have only been done inCLK overexpression systems that could misrepresentlocalization of the native protein by expressing the protein atmuch higher levels than the endogenous protein. CLK1 proteincontains nuclear localization motifs within its N terminus andexpression in excess of any cytoplasmic binding protein mayresult in artifactual nuclear localization of the protein. We haverecently shown that another CLK family member, CLK3 islocalized to the cytoplasm, rather than the nucleus of cells(Menegay et al., 1999). Although the localization of CLK3 tothe nucleus may lead to inferences about the localization of afamily member, their regulation may be very different. Whilethe kinase domains of CLK1 and CLK3 are fairly wellconserved with 62% identity, their N-terminal domains, whichcontain their putative nuclear localization are very poorlyconserved, with only 33% identity (Hanes et al., 1994). In orderto determine the localization of native CLK1 protein weexamined the distribution of CLK1 in PC12 cells. PC12 cellswere fixed in a variety of fixatives and examined with theCLK1 antibody. Fixation in methanol resulted in detection ofnuclear staining of the overexpressed and epitope tagged CLK1protein (Fig. 11). Fixation in 0.4% paraformaldehyde alsoresulted in a similar pattern of staining. Fixation in faster actingfixatives however resulted in cytoplasmic localization of
CLK1. In cells fixed in 4% paraformaldehyde or Acroleinfixative, the majority of the CLK1 staining was cytoplasmic,and the nuclei exhibited low levels of immunoreactivitycompared to the cytoplasm. Fixation in Bouin’s fixativeresulted in cytoplasmic staining, however much of the CLK1epitope appeared to be localized to perinuclear spots in thecytoplasm.
Immunostaining of CLK1 transfected 3T3 cellsTo confirm the results from the PC-12 cells, and to verify thatthe protein the antibody was reacting with was indeed CLK1,NIH 3T3 cells were transfected with a CLK1 expressionconstruct. Upon fixation in Bouin’s fixative staining in CLK1-expressing NIH-3T3 cells was limited to the cytoplasm.Fixation with methanol resulted in staining mainly in thenucleus, although there was staining in the cytoplasm of thesecells (Fig. 12). The nuclear staining was mainly localized to‘speckles’ as have been previously reported in otheroverexpresssion studies. Fixation in methanol precipitates butdoes not cross-link proteins and may allow for their movementduring the fixation process. Bouin’s fixative, however, acts toboth precipitate and cross-links proteins and may provide a
H. J. Menegay and others
Fig. 6.Phosphopeptide analysis. HA-TaggedCLK1 protein expressed in bacteria wasautophosphorylated and digested with cyanogenbromide, trypsin, chymotrypsin, or both trypsinand chymotrypsin. The digestion products wereseparated on a 2-D gel.
Fig. 7.CLK1 mRNA expression in adult rat tissues and cell linesdetected by RT-PCR. (A) Total RNA was isolated from cell lines andrat tissues. The RNA (1 µg) was reverse transcribed, then amplifiedby PCR. The PCR of full-length kinase domain containingtranscripts results in a 560 bp product, while the truncated, kinase-defidient transcripts yields a 453 bp product. Lanes: F, forebrain; Hb, Hindbrain, H, Heart; Lg, Lung; Li, Liver; K, Kidney; S, Spleen;M, Muscle; 3T3 cells; PC-12 cells. (B) PCR of GAPDH controlsfrom each sample to control for RNA loading.
3249Characterization and localization of CLK1
more accurate image of localization of the protein. Weconclude from these experiments that the majority of theendogenous CLK1 protein is localized to the cytoplasm.
DISCUSSION
Historically, protein kinases have been divided into twodistinct groups, the tyrosine kinases and the serine/threoninekinases. Recently, however, a new class of kinases has been
discovered that phosphorylates both serine/threonine andtyrosine residues. Based on conserved amino acid motifs,these dual specificity kinases are indistinguishable from theserine/threonine kinases. The lack of any distinguishableamino acid motifs and the fact that, with few exceptions, mostdual specificity kinases, including CLK1, have weak tyrosinekinase activity has led to the conclusion that dual specificityis a property that may not be physiologically relevant(Lindberg et al., 1992). The robust tyrosine kinase activity ofthe dual specificity kinases MEK and wee1, however, isrestricted to their specific, physiological substrates (Crews etal., 1992; Featherstone and Russell, 1991; Russell and Nurse,1987). This suggests that specificity toward tyrosine residuesis far more stringent than for serine/threonine residues. Thetyrosine kinase activity of many of the dual specificity kinasesis detectable only in autophosphorylation reactions andtyrosine phosphorylation of exogenous substrates maytherefore only be achieved when assayed with the appropriatesubstrate.
Fig. 8.Shift in PCR products of splice forms with cycloheximide.Control or PC12 cells treated with 10 µg/ml cycloheximide for 60minutes were harvested, RNA was extracted, and RT-PCR wasperformed. (A) PCR products of the 560 bp full-length form or the453 kinase-less form of CLK1 message are shown. (B) PCR ofGAPDH controls from each sample to control for RNA loading.
Fig. 9.Western analysis of CLK1 protein in rat tissues and cell lines.Protein (50 µg) from tissue or cell line lysates or were separated onSDS-polyacrylamide gels, transferred to PVDF membrane. The blotswere then probed with the affinity purified CLK1 antibody andvisualization by ECL reagents. The position of the molecular massmarkers and full-length (57 kDa) product is indicated. Lanes: 3T3cells; PC-12 cells; Br, brain; H, heart; Lg, lung; Li, liver; K, kidney;S, spleen; M, muscle.
Fig. 10.Immunostaining of rat brain sections. CLK1 expression inrat brain. Sections of brain from adult rats were examined for thepresence of CLK1 protein. The sections were immunostained withthe affinity purified anti-CLK1 antibody and Texas-Red conjugatedgoat-anti-rabbit secondary antibody (B,D,F,H) or secondary antibodyalone (A,C,E,G). A confocal microscope was used to image thesections. (A-B) Cells in the area of the raphe nucleus. (C-D) CA1 region of hippocampus. (E-F) Frontal cortex. (G-H) Cerebellum. Bar, 50 µm.
3250
Traditionally, proteins, such as MBP, histones, or casein,have been used as exogenous substrates in vitro to evaluatesubstrate specificity. We subsequently determined that CLK1phosphorylated a single site in MBP (Fig. 2). Phospho-peptidesequencing revealed that the site phosphorylated by CLK1 isSer109 (Fig. 3). This site has also been shown to bephosphorylated by cAMP-dependent kinase, Cam kinase II andPKC (Crews et al., 1992; Li et al., 1991; Smeyne et al., 1994;Vojtek and Cooper, 1995). Unlike these kinases whoseconsensus site is R-R-X-X-(S/T) or R-R-X-S, a survey of 14
peptide substrates revealed that the consensus site forCLK1 phosphorylation is R-X-X-(S/T)-X-X-R withthe basic residues at both the +3 and −3 playingapproximately equivalent roles in determiningsubstrate specificity (Table 1). Basic residues at otherpositions may also play a lesser role in determiningsubstrate specificity, as the poly-basic PKCζsubstrate peptide was the most efficiently utilizedpeptide substrate (Fig. 4 and Table 1). Therequirement of multiple basic residues would explainwhy SR proteins, which are serine-arginine richsplicing factors, are so efficiently phosphorylated byCLK1 (Colwill et al., 1996a). Somewhatsurprisingly, CLK1 does not contain an acidicresidue at an analogous position, demonstrating thatthe substrate specificity determinants of CLK1 aredistinct from those of other kinases.
CLK1 exhibits a domain structure in which theconserved catalytic domain is positioned at the Cterminus, while the N-terminal domain contains afunctional nuclear localization signal which maydirect CLK1 into the nucleus (Duncan et al., 1995).The N terminus may also act to anchor CLK1 to itssubstrates, as well as acting to regulate kinaseactivity. The presence of a regulatory domain in anumber of kinases, including the EGF receptor(Vojtek and Cooper, 1995), c-raf (Li et al., 1991), src(Superti-Furga, 1995), and pp70S6K (Crowley et al.,1994; Dennis et al., 1998; Smeyne et al., 1994), hasbeen demonstrated by an increase in kinase activityupon truncation of the regulatory domain. Truncationof the 128 N-terminal amino acids of CLK1 resultsin an approximately 40-fold increase in enzymaticactivity, indicating that the N terminus of CLK1encodes a regulatory domain. Although theconsensus phosphorylation site predicts the presenceof a number of potential phosphorylation sites in theN terminus, it is unlikely that the truncation removesan inhibitory pseudo-substrate domain. The removalof a pseudosubstrate, which acts as a competitiveinhibitor, would be predicted to result in an increasein Km for exogenous substrates. Truncation of the Nterminus of CLK1 resulted in only a small decreasein Km for ATP and a small increase in Km for MBP(Table 2). The truncation did, however, result in a 45-fold increase in Vmax, suggesting that the truncationallows CLK1 to adopt a conformation which allowsfor a faster rate of phospho-transfer rather thanaffecting enzyme-substrate interactions.
The cellular regulation of CLK1 activity iscurrently unclear. We have provided evidence
indicating that the control of CLK1 kinase activity is complex.CLK1 has been shown to autophosphorylate on serine,threonine, and tyrosine residues (Howell et al., 1991). Howellet al. reported that CLK1 activity can be quantitativelyimmunoprecipitated with antibodies to phosphotyrosine, whichsuggested that tyrosine phosphorylation is necessary for CLK1activity (Howell et al., 1991). Paradoxically, treatment withtyrosine specific phosphatases had no discernable effect oneither full length or truncated CLK1 activity. Treatment withserine/threonine specific phosphatases, on the other hand,
H. J. Menegay and others
Fig. 11.Immunostaining of PC12 cells fixed with a variety of fixatives. PC12cells were fixed for 20 minutes with methanol, 0.4% paraformaldehyde, 4%paraformaldehyde, Acrolein fixative, or Bouin’s fixative and immunostainedwith the CLK1 antibody and Texas-Red-conjugated secondary antibody.Images were captured on a confocal microscope. Images are individual 0.5µM confocal image sections or composite projections of 0.5 µM confocalsections throughout the thickness of the cells.
3251Characterization and localization of CLK1
resulted in a two- to sixfold activation of CLK1. Simultaneoustreatment with both classes of phosphatases, or with alkalinephosphatase, a non-selective phosphatase, abolished CLK1activity. These data indicate phosphorylation at eitherserine/threonine residues or tyrosine residues is sufficient toactivate CLK1. The activation of CLK1 by serine/threoninephosphatases indicates that phosphorylation of someserine/threonine sites acts to specifically inhibit CLK1 activityor that the enzyme phosphorylated exclusively on tyrosineresidues is more active than the enzyme phosphorylated onserine/threonine and tyrosine residues. Given the high specificactivity of the untreated enzyme, the latter case appears to bemore likely.
The MAP kinases are also regulated by phosphorylation attyrosine and serine/threonine residues, but unlike CLK1, MAPkinase activation requires phosphorylation of bothserine/threonine and tyrosine residues (Crews et al., 1992;Payne et al., 1991; Schanen-King et al., 1991). The residuesresponsible for MAP kinase activation lie in kinase subdomainVIII (Payne et al., 1991). Phosphorylation of similarly locatedresidues has been shown to increase the activity of a numberof serine/threonine kinases (Hanks and Hunter, 1995; Taylor etal., 1995). Some tyrosine kinases are also regulated by tyrosinephosphorylation at a distinct site in kinase subdomain VIII.CLK1 has a number of potential phosphorylatable residues inthis region, which correspond to those found in activity-dependent phosphorylation sites from both tyrosine kinasesand from serine/threonine kinases. The presence of potentialregulatory sites from both classes of protein kinases mayexplain why CLK1 activity is unaffected upon treatment withthe tyrosine phosphatases or serine/threonine phosphatasestested and why CLK1 activity is lost when treated with bothphosphatases. The identity of the regulatory phosphorylationsites are currently unknown but they must be located in thecatalytic domain, as the truncated CLK1 is activated upontreatment with serine/threonine phosphatases. In addition tophosphorylation, CLK1 is also negatively regulated by its Nterminus. The specific mechanism of inhibition remains
unclear but it does not appear to contain a pseudo-substratemotif and may act to conformationally constrain catalysis byanother mechanism. It is has been speculated that the N-terminal splice variant, which lacks a kinase domain, functionsto regulate CLK1 function. Perhaps by binding to substrates orby directly interacting with the full-length allele.
Here we report that the ratio of full-length to truncated CLKmRNA products can be altered by cycloheximide treatment. Ithas previously been demonstrated that CLK1 can regulate it’sown splicing and is capable of phosphorylating SR proteins(Colwill et al., 1996b; Duncan et al., 1997). It has also beenshown that members of the CLK family are capable ofregulating mRNA splicing of an adenovirus E1A splicingreporter construct in vivo, as well as causing the redistributionof SR proteins from nuclear speckles to a diffusenucleoplasmic localization (Colwill et al., 1996b; Duncan etal., 1997, 1998). This observation has been broadened toinclude other members of the LAMMER family (Du et al.,1998; Wang et al., 1998). It has recently been demonstratedthat CLK1’s effect on splicing is due to it’s phosphorylation ofSR proteins rather than other members of the splicing complex(Prasad et al., 1999).
A subset of SR proteins, including ASF/SF2, shuttlebetween the nucleus and the cytoplasm based upon theirphosphorylation state (Caceres et al., 1998). CLK1 has beenshown to directly interact with ASF/SF2, and the expression ofmyc-tagged CLK1 protein causes the accumulation ofASF/SF2 to the cytoplasm (Caceres et al., 1998; Colwill et al.,1996b; Prasad et al., 1999). These data suggest the possibilitythat the majority of CLK1 may be normally held in acytoplasmic reservoir, but may also shuttle between the nucleusand cytoplasm with ASF/SF2.
The cytoplasmic localization of the bulk of CLK1 protein invivo suggests that it may have cytoplasmic targets in additionto its role in the regulation of splicing. Interestingly, CLK1 hasrecently been shown to be capable of phosphorylating andactivating the tyrosine phosphatase PTP1B (Moeslein et al.,1999). This phosphatase is can be found in the cytoplasm of
Fig. 12.Immunostaining of NIH 3T3cells overexpressing CLK1 with Bouin’sor methanol. NIH 3T3 cells which weretransfected with the control pIND vectoror transfected with the CLK1-pINDconstruct were fixed with Bouin’sfixative or methanol. The fixed cultureswere then immunostained with the CLK1antibody and visualized with a Texas-Red-conjugated secondary antibodyimaged with a confocal microscope.

3252
cells, and may be a cytoplasmic target of CLK1. AlthoughCLK1 and another CLK family member, CLK3, have beenshown to be localized to the nucleus in overexpression systems,we recently demonstrated that in vivo the CLK3 protein islocalized to the cytoplasm of developing spermatozoa and tothe acrosome of mature sperm (Menegay et al., 1999). The N-terminal domains of the CLK family, containing their nuclearlocalization signals, are not conserved (Nayler et al., 1997).Their variability in this region suggests that their localizationmay be regulated very differently, however we believe that bothof these CLK family members are generally cytoplasmicproteins.
We demonstrate that the apparent subcellular distribution ofCLK1 protein varies dramatically based upon the fixationagent. These differences are likely due to the rate of fixationand cross-linking between these fixing agents. Methanolprecipitates proteins, while Bouin’s fixative both precipitatesand cross-links. A light fixative such as 0.4%paraformaldehyde gives a similar pattern of staining asmethanol, while a faster fixation in 4% paraformaldehydepreserves a pattern similar to that of Bouin’s. This suggests tous that the pattern observed with Bouin’s fixative may be morerepresentative of the pattern of CLK1 localization in vivo.
This work was supported by grants from the National Institutes ofHealth (NS31987) and M.P.M. was supported by a training grant fromthe NIH (HD 07204-25). We are indebted to Dr T. Egelhof, Dr M.Wooten, Dr R. Erikson, and Dr M. Greenberg for kindly providingreagents. We are also indebted to Dr C. Beach for providing excellenttechnical guidance and for performing the peptide sequencing.
REFERENCES
Ben-David, Y., Letwin, K., Tannock, L., Bernstein, A. and Pawson, T.(1991).A mammalian protein kinase with potential for serine/threonine and tyrosinephosphorylation is related to cell cycle regulators. EMBO J. 10, 317-325.
Bender, J. and Fink, G. R.(1994). AFC1, a LAMMER kinase from Arabidopsisthaliana, activates STE12-dependent processes in yeast. Proc. Nat. Acad. Sci.USA91, 12105-12109.
Boyle, W. J., van der Geer, P. and Hunter, T.(1991). Phosphopeptide mappingand phosphoamino acid analysis by two-dimensional separation on thin-layercellulose plates. Meth. Enzymol.201, 110-149.
Caceres, J. F., Screaton, G. R. and Krainer, A. R.(1998). A specific subsetof SR proteins shuttles continuously between the nucleus and the cytoplasm.Genes Dev.12, 55-66.
Chomczynski, P. and Sacchi, N.(1987). Single-step method of RNA isolationby acid guanidinium thiocyanate-phenol-chloroform extraction. Anal.Biochem.162, 156-159.
Colwill, K., Feng, L. L., Yeakley, J. M., Gish, G. D., Caceres, J. F., Pawson,T. and Fu, X. D. (1996a). SRPK1 and Clk/Sty protein kinases show distinctsubstrate specificities for serine/arginine-rich splicing factors. J. Biol. Chem.271, 24569-24575.
Colwill, K., Pawson, T., Andrews, B., Prasad, J., Manley, J. L., Bell, J. C. andDuncan, P. I.(1996b). The Clk/Sty protein kinase phosphorylates SR splicingfactors and regulates their intranuclear distribution. EMBO J.15, 265-275.
Crews, C. M., Alessandrini, A. and Erikson, R. L. (1992). The primarystructure of MEK, a protein kinase that phosphorylates the ERK gene product.Science258, 478-480.
Crowley, C., Spencer, S. D., Nishimura, M. C., Chen, K. S., Pitts-Meek, S.,Armanini, M. P., Ling, L. H., MacMahon, S. B., Shelton, D. L., Levinson,A. D. et al. (1994). Mice lacking nerve growth factor display perinatal loss ofsensory and sympathetic neurons yet develop basal forebrain cholinergicneurons. Cell 76, 1001-1011.
Dennis, P. B., Pullen, N., Pearson, R. B., Kozma, S. C. and Thomas, G.(1998). Phosphorylation sites in the autoinhibitory domain participate inp70(s6k) activation loop phosphorylation. J. Biol. Chem. 273, 14845-14852.
Du, C., McGuffin, M. E., Dauwalder, B., Rabinow, L. and Mattox, W.(1998).
Protein phosphorylation plays an essential role in the regulation of alternativesplicing and sex determination in Drosophila. Mol. Cell 2, 741-750.
Duncan, P. I., Howell, B. W., Marius, R. M., Drmanic, S., Douville, E. M.and Bell, J. C.(1995). Alternative splicing of STY, a nuclear dual specificitykinase. J. Biol. Chem. 270, 21524-21531.
Duncan, P. I., Stojdl, D. F., Marius, R. M. and Bell, J. C.(1997). In vivoregulation of alternative pre-mRNA splicing by the Clk1 protein kinase. Mol.Cell Biol. 17, 5996-6001.
Duncan, P. I., Stojdl, D. F., Marius, R. M., Scheit, K. H. and Bell, J. C.(1998).The Clk2 and Clk3 dual-specificity protein kinases regulate the intranucleardistribution of SR proteins and influence pre-mRNA splicing. Exp. Cell Res.241, 300-308.
Featherstone, C. and Russell, P.(1991). Fission yeast p107wee1 mitoticinhibitor is a tyrosine/serine kinase. Nature349, 808-811.
Glass, D. B., Masaracchia, J. R. and Kemp, B. E.(1978). Anal. Biochem.87,566-575.
Greenberg, M. E., Hermanowski, A. L. and Ziff, E. B. (1986). Effect ofprotein synthesis inhibitors on growth factor activation of c-fos, c-myc, andactin gene transcription. Mol. Cell Biol.6, 1050-1057.
Hanes, J., von der Kammer, H., Klaudiny, J. and Scheit, K. H.(1994).Characterization by cDNA cloning of two new human protein kinases.Evidence by sequence comparison of a new family of mammalian proteinkinases. J. Mol. Biol.244, 665-672.
Hanks, S. K. and Hunter, T.(1995). Protein kinases 6. The eukaryotic proteinkinase superfamily: kinase (catalytic) domain structure and classification.FASEB J.9, 576-596.
Hanks, S. K. and Quinn, A. M. (1991). Protein kinase catalytic domainsequence database: identification of conserved features of primary structureand classification of family members. Meth. Enzymol. 200, 38-62.
Hanks, S. K., Quinn, A. M. and Hunter, T.(1988). The protein kinase family:conserved features and deduced phylogeny of the catalytic domains. Science241, 42-52.
Howell, B. W., Afar, D. E., Lew, J., Douville, E. M., Icely, P. L., Gray, D. A.and Bell, J. C. (1991). STY, a tyrosine-phosphorylating enzyme withsequence homology to serine/threonine kinases. Mol. Cell Biol.11, 568-572.
Huang, S. and Spector, D. L.(1996). Dynamic organization of pre-mRNAsplicing factors. J. Cell. Biochem.62, 191-197.
Humason, G. L. (1962). Animal Tissue Techniques. W. H. Freeman andCompany.
Jimenez-Garcia, L. F. and Spector, D. L.(1993). In vivo evidence thattranscription and splicing are coordinated by a recruiting mechanism. Cell 73,47-59.
Johnson, K. W. and Smith, K. A.(1991). Molecular cloning of a novel humancdc2/CDC28-like protein kinase. J. Biol. Chem. 266, 3402-3407.
Lee, K., Du, C., Horn, M. and Rabinow, L. (1996). Activity andautophosphorylation of LAMMER protein kinases. J. Biol. Chem. 271, 27299-27303.
Li, P., Wood, K., Mamon, H., Haser, W. and Roberts, T.(1991). Raf-1: akinase currently without a cause but not lacking in effects. Cell 64, 479-482.
Lindberg, R. A., Quinn, A. M. and Hunter, T. (1992). Dual-specificity proteinkinases: will any hydroxyl do? Trends Biochem. Sci.17, 114-119.
Luo, K. X., Hurley, T. R. and Sefton, B. M. (1991). Cyanogen bromidecleavage and proteolytic peptide mapping of proteins immobilized tomembranes. Meth. Enzymol.201, 149-252.
Mattaj, I. W. (1994). RNA processing. Splicing in space [news; comment].Nature372, 727-728.
Meijlink, F., Curran, T., Miller, A. D. and Verma, I. M. (1985). Removal ofa 67-base-pair sequence in the noncoding region of protooncogene fosconverts it to a transforming gene. Proc. Nat. Acad. Sci. USA82, 4987-4991.
Menegay, H., Moeslein, F. and Landreth, G.(1999). The dual specificityprotein kinase CLK3 is abundantly expressed in mature mouse spermatozoa.Exp. Cell Res.253, 463-473.
Moeslein, F. M., Myers, M. P. and Landreth, G. E.(1999). The CLK familykinases, CLK1 and CLK2, phosphorylate and activate the tyrosinephosphatase, PTP-1B. J. Biol. Chem. 274, 26697-26704.
Myers, M. P., Murphy, M. B. and Landreth, G. (1994). The dual-specificityCLK kinase induces neuronal differentiation of PC12 cells. Mol. Cell Biol.14, 6954-6961.
Nayler, O., Stamm, S. and Ullrich, A.(1997). Characterization and comparisonof four serine- and arginine-rich (SR) protein kinases. Biochem. J.326, 693-700.
Nayler, O., Schnorrer, F., Stamm, S. and Ullrich, A.(1998). The cellularlocalization of the murine serine/arginine-rich protein kinase CLK2 isregulated by serine 141 autophosphorylation. J. Biol. Chem. 273, 34341-34348.
Payne, D. M., Rossomando, A. J., Martino, P., Erickson, A. K., Her, J. H.,
H. J. Menegay and others

3253Characterization and localization of CLK1
Shabanowitz, J., Hunt, D. F., Weber, M. J. and Sturgill, T. W.(1991).Identification of the regulatory phosphorylation sites in pp42/mitogen-activated protein kinase (MAP kinase). EMBO J.10, 885-892.
Porath, J. and Axen, R.(1976). Immobilization of enzymes to agar, agarose,and Sephadex supports. Meth. Enzymol. 44, 19-45.
Prasad, J., Colwill, K., Pawson, T. and Manley, J. L.(1999). The proteinkinase Clk/Sty directly modulates SR protein activity: both hyper- andhypophosphorylation inhibit splicing. Mol. Cell Biol.19, 6991-7000.
Ringold, G. M., Dieckmann, B., Vannice, J. L., Trahey, M. and McCormick,F. (1984). Inhibition of protein synthesis stimulates the transcription of humanbeta-interferon genes in Chinese hamster ovary cells. Proc. Nat. Acad. Sci.USA81, 3964-3968.
Russell, P. and Nurse, P.(1987). Negative regulation of mitosis by wee1+, agene encoding a protein kinase homolog. Cell 49, 559-567.
Schanen-King, C., Nel, A., Williams, L. K. and Landreth, G.(1991). Nervegrowth factor stimulates the tyrosine phosphorylation of MAP2 kinase inPC12 cells. Neuron6, 915-922.
Shaw, G. and Kamen, R.(1986). A conserved AU sequence from the 3′untranslated region of GM-CSF mRNA mediates selective mRNAdegradation. Cell 46, 659-667.
Smeyne, R. J., Klein, R., Schnapp, A., Long, L. K., Bryant, S., Lewin, A.,
Lira, S. A. and Barbacid, M. (1994). Severe sensory and sympatheticneuropathies in mice carrying a disrupted Trk/NGF receptor gene [seecomments]. Nature368, 246-249.
Superti-Furga, G. (1995). Regulation of the Src protein tyrosine kinase. FEBSLett. 369, 62-66.
Taylor, S. S., Radzio-Andzelm, E. and Hunter, T.(1995). How do proteinkinases discriminate between serine/threonine and tyrosine? Structuralinsights from the insulin receptor protein-tyrosine kinase. FASEB J.9, 1255-1266.
Ullrich, A. and Schlessinger, J.(1990). Signal transduction by receptors withtyrosine kinase activity. Cell 61, 203-212.
Vojtek, A. B. and Cooper, J. A. (1995). Rho family members: activators ofMAP kinase cascades. Cell 82, 527-529.
Wang, H. Y., Lin, W., Dyck, J. A., Yeakley, J. M., Songyang, Z., Cantley, L.C. and Fu, X. D. (1998). SRPK2: a differentially expressed SR protein-specific kinase involved in mediating the interaction and localization of pre-mRNA splicing factors in mammalian cells. J. Cell Biol.140, 737-750.
Yun, B., Farkas, R., Lee, K. and Rabinow, L.(1994). The Doa locus encodesa member of a new protein kinase family and is essential for eye andembryonic development in Drosophila melanogaster. Genes Dev.8, 1160-1173.





![Channel Characterization for RF Localization Inside Human Body · Performance evaluation needs channel models [1] M. A. Assad, A Real-Time Laboratory Testbed for Evaluating Localization](https://img.pdfslide.net/doc/110x75/5f192b23b7a8605b25199059/channel-characterization-for-rf-localization-inside-human-performance-evaluation.jpg)






![Genetic Localization and Molecular Characterization of …jb.asm.org/content/181/7/2199.full.pdf · Genetic Localization and Molecular ... [grams per liter] glucose, 15; soluble starch,](https://img.pdfslide.net/doc/110x75/5aeb76e07f8b9a585f8da78e/genetic-localization-and-molecular-characterization-of-jbasmorgcontent18172199fullpdfgenetic.jpg)
















