Embed Size (px)
Citation preview

Chemical modi®cation of starch based biodegradable polymericblends: e�ects on water uptake, degradation behaviour and
mechanical properties
DoÈ ne DemirgoÈ z a,b, Carlos Elvira a,*, JoaÄ o F. Mano a,Antonio M. Cunha a, Erhan Piskin b, Rui L. Reis a
aDepartment of Polymer Engineering, University of Minho, Campus AzureÂm, 4810-058 GuimaraÄes, PortugalbDepartment of Chemical Engineering, Hacettepe University, Beytepe Campus, 06532 Ankara, Turkey
Received 13 April 2000; accepted 13 June 2000
Abstract
The main disadvantages of biodegradable polymers obtained from renewable sources are their dominant hydrophilic character,
fast degradation rate and, in some cases, unsatisfactory mechanical properties particularly under wet environments. One possiblesolution to this problem is to reduce the water-uptake ability of these materials and to enhance the respective mechanical behaviourby chemical modi®cation. In this work, three based starch blends with: (i) a copolymer of ethylene and vinyl alcohol (SEVA-C), (ii)
cellulose acetate (SCA), and (iii) poly-e-caprolactone (SPCL); were chemically modi®ed by chain crosslinking. This modi®cation isbased on the reaction between the starch hydroxyl groups and tri-sodium tri-meta phosphate. The obtained compounds werecharacterized by FTIR and the respective properties were assessed and compared to the original materials by means of the hydra-tion degree, the degradation behaviour, contact angle measurements and mechanical testing. The results show that the water-uptake
of these blends could be reduced up to 15% and that simultaneously sti�er materials with a less pronounced degradation rate canbe obtained. # 2000 Elsevier Science Ltd. All rights reserved.
1. Introduction
Natural origin and synthetic polymers (and therespective composites) are currently extensively used asbiomaterials and in biodegradable applications, becausethey are available in a wide variety of compositions andtheir properties can be tailored to meet speci®cdemands. Furthermore, the stability of the majority ofthe polymers obtained from the petrochemical industrycan cause disposal problems. Within this scope, it hasbeen noticeable in the last few years an increasinginterest in biodegradable polymers derived fromannually renewable feedstock such as starch and starch-based materials [1±4].Starch-based blends present an enormous potential to
be widely used in the biomedical and the environmental®elds, as they are totally biodegradable, inexpensive(when compared to other biodegradable polymers) and
available in large quantities [1,5]. Furthermore, starch-based polymers can also be converted into complexgeometries using standard equipment developed for theprocessing of synthetic polymers. The properties, appli-cations and the processing procedures of biodegradablestarch-based thermoplastic blends, like starch/poly-caprolactone, starch/cellulose acetate and starch/(ethyl-ene-vinyl alcohol copolymer), as been already reportedin literature [4,6±8,11]. However, new areas are emer-ging including drug delivery systems, hydrogels, bonecements and bone replacement/®xation devices [6±14].All these developments require the possibility of
accurately control the polymer water sensitivity, degra-dation rate and, consequently, the mechanical perfor-mance. In the orthopaedic ®eld, the aim is to developsystems that will be able to sustain their integrity andmechanical performance in the presence of aqueousmedia in the ®rst implantation stages and start todegrade thereafter. This performance has to be achievedkeeping the materials able to degrade under controlledkinetics [15±25].
0141-3910/00/$ - see front matter # 2000 Elsevier Science Ltd. All rights reserved.
PI I : S0141-3910(00 )00102-6
Polymer Degradation and Stability 70 (2000) 161±170
www.elsevier.nl/locate/polydegstab
* Corresponding author.
E-mail address: [email protected] (C. Elvira).

In order to improve this kind of behaviour, chemicalmodi®cations based on well-known organic reactionswere used in this work. Nevertheless, it is worthmentioning that such type of modi®cations have neverbeen tried before on the types of blends studied in thiswork. Special attention was devoted to the surfacemodi®cation as material surface properties are determi-nant in biomedical applications (bioactivity, cell adhe-sion and proliferation, protein interactions, etc.). Infact, it is the surface of the material that interacts withproteins and cells and, consequently, the implant beha-viour will deeply depend on its properties [26].So, the present work is an attempt to ®nd an adequate
method to reduce the water sensitivity and the degra-dation rate of starch-based formulations by crosslinkingsome of the hydroxyl groups present in the blends.These chemical modi®cations are expected to have alsoa direct impact over surface and mechanical propertiesof the modi®ed compounds.
2. Experimental
2.1. Materials
The studied starch-based thermoplastic blends, kindlysupplied by Novamont, Novara, Italy, were: (i) a cornstarch/ethylene-vinyl alcohol (SEVA-C); (ii) a cornstarch/cellulose acetate (SCA); and (iii) a corn starch/e-caprolactone, (SPCL).Powders, ®lms (prepared by compression moulding at
120±130�C) and ASTM dumbell tensile test specimens(prepared by injection moulding using a previouslydescribed procedure 4, with a cross-section of 2�4 mm)were used in this work.
2.2. Crosslinking reaction
The crosslinking reactions were carried out, asdescribed in the literature for native starch 28, using tri-sodium tri-meta phosphate, Na3P3O9 (Aldrich-Ger-many) as a crosslinking reagent in the following con-centrations (relatively to the weight of the blend): 15.7,7.8 and 3.9%. These concentrations were used onpreliminary tests aimed at choosing the ideal Na3P3O9
concentration to use on all subsequent studies. Thisideal concentration was found to be 15.7% (seeResults).First, the Na3P3O9 was dissolved in an aqueous solu-
tion being the pH value adjusted to 10±11 by usingNa2CO3 (CO) or NaOH (NA), both from Aldrich-Ger-many. The starch-based blend was then added (2 g) tothe solution (20 ml) and the reaction was carried out at50�C for 5 h under continuous stirring.
The reaction was initially performed using the blends inthe powder form. However, some studies were alsodeveloped on the obtained ®lms and on the ASTM speci-mens. All the tensile samples were previously immersed ina water solution at room temperature for 3 days in order toremove the processing aids contained in the blends.
2.3. Characterization by Fourier transformed infra-redspectroscopy (FTIR)
FTIR spectra were recorded on a Perkin-Elmer 1600Series spectrometer. Spectra were taken with a resolutionof 2 cmÿ1 and were averaged over 32 scans. Samples werethoroughly grounded with exhaustively dried KBr andpellets were prepared by compression under vacuum.
2.4. Hydration degree and degradation behaviour
The water-uptake and the degradation of the materi-als (in the form of tensile specimens) was studied over aperiod of 90 days. Materials were conditioned to mini-mum weight at 37�C in an oven with desiccant prior tobeing immersed in 100 ml of a simulated physiologicalisotonic solution (0.154 M NaCl aqueous solution at pH7.4). The specimens were removed at regular intervals of3, 7, 14, 30, 60 and 90 days, being taken out of thesolution, blotted on ®lter paper to remove surface solu-tion and further rinsed with distilled water and weighed.After being removed from the solution, the specimenswere dried, in an oven at 60�C, to constant weight inorder to determine the eventual weight loss (an averageof value of two readings was used). Equilibrium hydra-tion degree was considered when no weight change (�0.001 g) was observed, after 2±3 days of immersion. Thehydration degree of the blends was also determined incrosslinked ®lms with thickness between 90 and 220 mm,considering the equilibrium hydration degree obtainedwhen no weight change was observed.
2.5. Mechanical properties
The tensile tests were performed on an Instron 4505machine, ®tted with resistive extensometer (gauge length 10mm) in a controlled environment (23�C, 55% RH). Thecrosshead velocity was 5 mm/min (8.3 � 10ÿ5 m/s) until1% strain (to determine the secant modulus with higherprecision) and then increased to 50mm/min (8.3�10ÿ4m/s)until fracture. The samples were stored under controlledtemperature and moisture conditions until testing, andat least six specimens were used. The fracture surfaceswere examined by scanning electron microscopy (SEM).
2.6. DMA analysis
Dynamic mechanical analysis (DMA) was also usedto characterize the mechanical behaviour of the mod-
162 D. DemirgoÈz et al. / Polymer Degradation and Stability 70 (2000) 161±170

i®ed blends. The tests were carried out using a tensileloading scheme, by means of scanning the temperaturefrom ÿ20 to 160�C, in a Perkin-Elmer 7 series ThermalAnalysis System (specimen size 20�5�3 mm). A fre-quency of 1 Hz and a constant heating rate of 4�C minÿ1
were used. A static stress of 0.12 MPa was applied to thespecimens over a dynamic stress of 0.1 MPa.
2.7. Contact angle measurements
The contact angle between a liquid and the surface ofthe ASTM specimens was measured at room tempera-ture by means of a Contact Angle Measuring SystemG10 by depositing a drop of distilled water and meth-ylene iodide (CH2I2) on the material to study. The aimwas to evaluate the polar and dispersive components ofthe surface energy. The quantity of liquid was measuredwith a syringe monitored by a micrometric screw. Theresults correspond to the average value of at least tenmeasurements.
3. Results and discussion
3.1. Crosslinking reaction
Non-modi®ed SEVA-C, SCA and SPCL blends arecompletely soluble in dimethylsulfoxide (DMSO).However, after the reaction only a partial solubilizationwas observed in all blends, which is a clear indication ofthe high percentages of crosslinking obtained. Con-centrations of Na3P3O9 (7.8 and 3.9%) were only usedin terms of ®nding the most optimal conditions toachieve a relatively high crosslinking density. As theamount of crosslinker was increased, the crosslinkingmass percentages were found to be higher, using thehighest concentration of Na3P3O9 (15.7%) in all theexperiments and performed studies. Consequently, thisconcentration was used for preparing samples for allother tests. Table 1 presents the amount of crosslinkedmaterial for the three blends under study, following thereactions performed with NaOH or Na2CO3, disclosingpercentages higher then 90% for SCA and SPCLblends, and of about 50% for SEVA-C.The high percentage of crosslinked mass obtained
with the described experimental conditions for SCA and
SPCL can be attributed to: (i) the partial hydrolysis inbasic media of the corresponding ester groups (acetateand lactone, respectively); and (ii) the hydroxyl groupswhich can react with Na3P3O9 and also be involved inthe crosslinking reaction (see Fig. 1). On the other hand,the low crosslinked mass percentage in the case ofSEVA-C blend can be attributed to the compatibility ofthe blend in which starch and the ethylene-vinyl alcoholare forming an interpenetrating network, and con-seqently both hydroxyl groups from starch and vinylalcohol can be involved in the crosslinking reaction withmore di�culty. The presence of the non-reactive ethyl-ene units also reduced the crosslinked mass percentage,as less accessible hydroxyl groups are present in theblend. It was also observed in all cases that the cross-linked mass percentage is higher when NaOH is used tocatalyse and to adjust the pH of the reaction [27].
3.2. Characterization by FTIR
The crosslinking reaction (Fig. 1) was charaterized bycomparing the FTIR spectra of both reacted and non-reacted samples as the original material does not con-tain any phosphate group in its structure. The typicalstretching vibrations of P�O and P±O (at about 1200±1100 cmÿ1) were observed in the spectra of all thecrosslinked samples. It was also noticeable that thebands assigned to the starch hydroxyl stretching vibra-tions (at 3400 cmÿ1) were decreased in intensity after thecrosslinking treatment as a consequence of the reactioninvolving two starch hydroxyl groups per one phos-phate. This can be observed in the spectra of non-treatedand crosslinked SPCL exhibited in Fig. 2.The most important signals assigned to the corre-
sponding stretching vibrations (±OH, P�O and P±O) ofSEVA-C, SCA and SPCL samples are summarised inTable 2. No remarkable di�erences were observed in thebands assigned to the carbonyl stretching vibration (atabout 1700 cmÿ1) in SCA (of acetate groups) and SPCL(from poly-caprolactone), with respect to the corre-sponding crosslinked blends.
3.3. Hydration degree and degradation behaviour
Both the hydration degree and the degradation beha-viour are the most important properties of these mate-rials when they are aimed to be used in biomedical orenvironmental applications, as their life time will be
Table 1
Crosslinked mass percentage of the starch based blends after reacting
with 15.7% of Na3P3O9 (w/w) with respect to the blend, at 50�C for 5
h in the presence of NaOH (NA) or Na2CO3 (CO)
Sample SEVA-C-
NA
SEVA-C-
CO
SCA-
NA
SCA-
CO
SPCL-
NA
SPCL-
CO
Crosslinked
mass (%)
62 50 91 63 96 94Fig. 1. Crosslinking reaction of starch with tn-sodium tri-meta phos-
phate through the hydroxyl starch groups in sodium carbonate or
sodium hydroxide aqueous solution.
D. DemirgoÈz et al. / Polymer Degradation and Stability 70 (2000) 161±170 163
governed by these two processes which are intimatelycorrelated.The water-uptake percentage of the blends was deter-
mined by using the following equation [28]:
%Water-uptake �WW ÿWi
Wi� 100%
where Wi is the initial weight of the sample and Ww isthe weight of the wet sample at di�erent times.Table 3 presents the equilibrium hydration degree of
both tensile specimens and ®lms of SEVA-C, SCA andSPCL and of the corresponding crosslinked blendsusing NaOH or Na2CO3 in the reaction. No remarkabledi�erences are noticeable between the hydration degreesof the ASTM specimens and of the ®lms, following thesame tendency in all cases.When NaOH is used in the reaction a reduction in the
equilibrium hydration degree is observed for all theblends, especially in the case of SCA going from 49.5 to33.5% (in tensile specimens) as a result of the highercrosslinked mass when NaOH was used.When sodium carbonate was added to the reaction
media, the equilibrium hydration degrees were in thesame range than those found for the untreated materials.Fig. 3 shows the swelling isotherms of the ®lms of
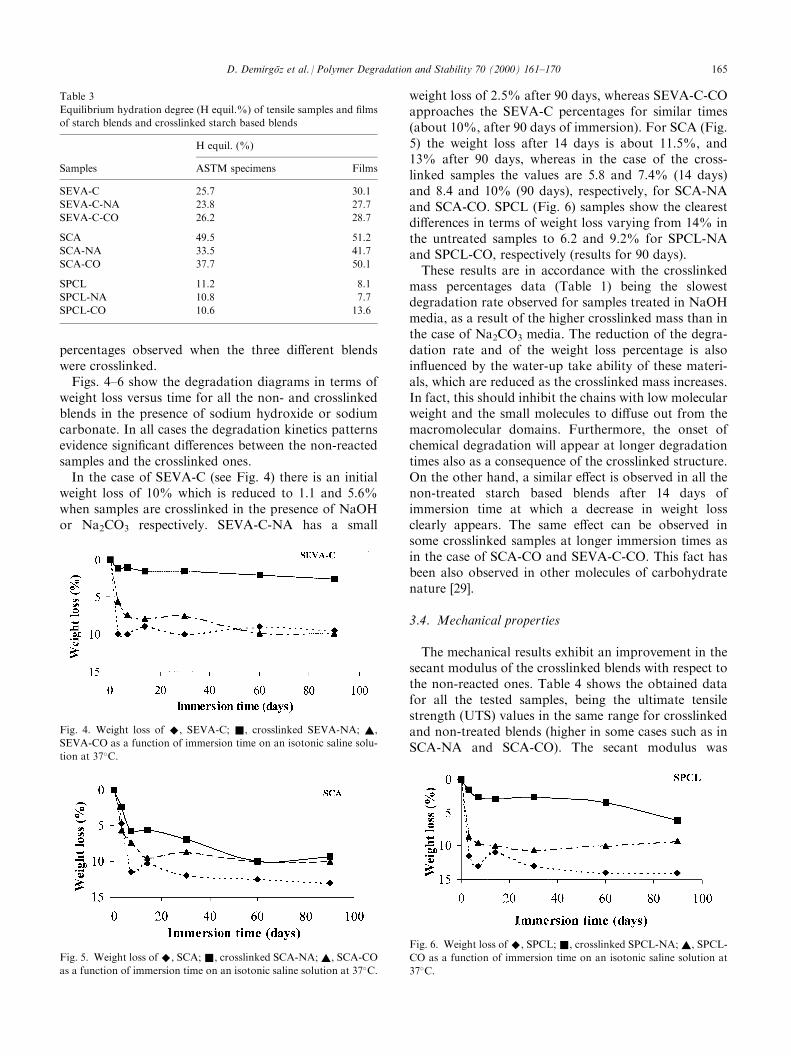
untreated and crosslinked SEVA-C. It is possible to
observe that the respective swelling kinetics are verydi�erent, being much faster in the case of the cross-linked samples. However, the equilibrium hydrationdegree is slightly lower in both cases. This should beassociated to the presence of the ionic phosphate groupson the surface, which leads initially to a fast water-up-take, but is later limited by the crosslinked nature of theblends. The phosphate groups (shown in the ionic formin Fig. 1) have, as the hydroxyl ones, a hydrophiliccharacter, being capable to be associated to watermolecules by hydrogen bonding or ionic solvating. Asthe crosslinked mass percentage is increased the watermolecules di�usion and the consequent accessibility tothe polymeric chains becomes more di�cult and the¯exibility of the polymer chains is reduced.The percentage weight loss of the material was deter-
mined by the equation:
%Weight loss �Wi ÿWf
Wi� 100%
where Wf is the ®nal weight of the dry sample and Wi isthe initial weight of the specimen at di�erent immersiontimes.It has been reported [29] that the degradation of
SEVA-C is composed by: (i) an initial step of leachingof the plasticisers (necessary to enable the material pro-cessing); (ii) the elimination of the low molecular weightchains resulting from the thermo-oxidative degradationoccurred during the processing stages; and (iii) thechemical degradation of the starch phase. So, it isexpected the considerable decrease of the weight loss
Table 2
Most signi®cant FTIR signals of the starch based blends and crosslinked starch blends SEVA-C, SCA and SPCL
Stretching vibration SEVA-C signals (cmÿ1) SCA signals (cmÿ1) SPCL signals (cmÿ1)
Non-treated Crosslinked Non-treated Crosslinked Non-treated Crosslinked
O±H st 3423 3350 3444 3423 3391 3391
Sharp Less intense Sharp Less intense Sharp Less intense
C±H st 2959 2949 2959 2959 2949 2927
P±O st and No signals 1283 No signals 1256 No signals 1046
P�O st 1288 1079
C�O st No signal No signal 1720 1720 1740 1740
Fig. 2. FTIR spectra of SPCL and crosslinked SPCL.
Fig. 3. Swelling isotherms in isotonic saline solutions at 37�C of ®lms
of ^, SEVA; &, SEVA-NA; ~, SEVA-CO.
164 D. DemirgoÈz et al. / Polymer Degradation and Stability 70 (2000) 161±170
percentages observed when the three di�erent blendswere crosslinked.Figs. 4±6 show the degradation diagrams in terms of
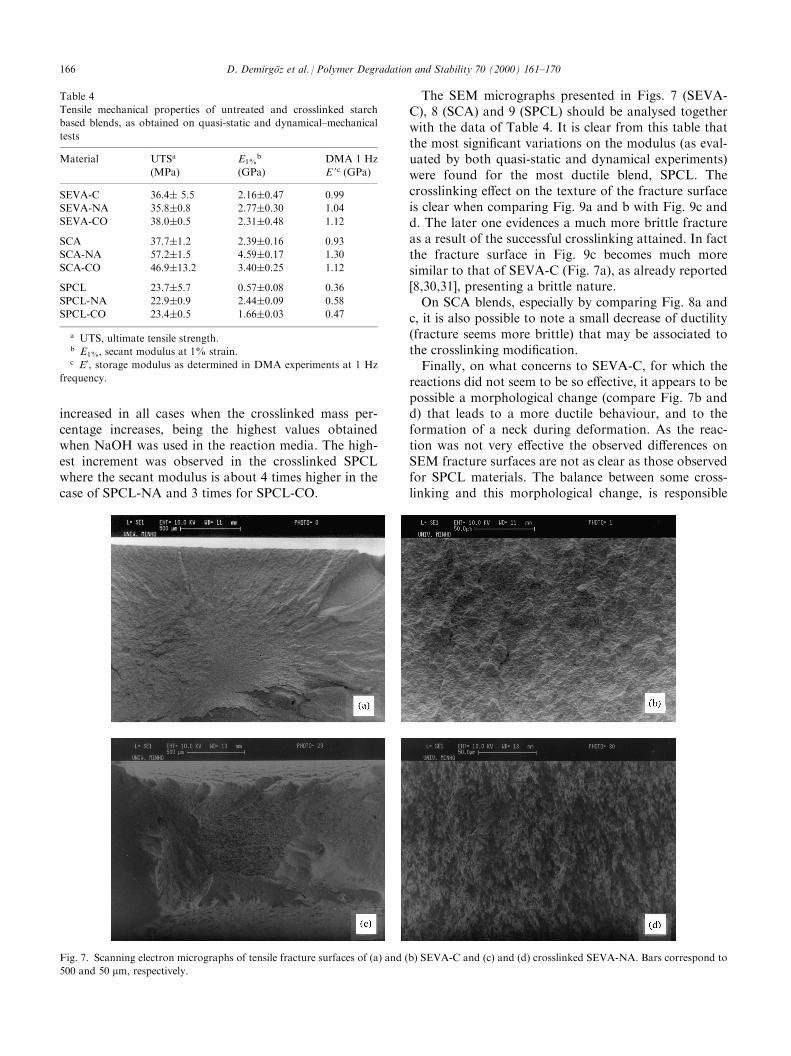
weight loss versus time for all the non- and crosslinkedblends in the presence of sodium hydroxide or sodiumcarbonate. In all cases the degradation kinetics patternsevidence signi®cant di�erences between the non-reactedsamples and the crosslinked ones.In the case of SEVA-C (see Fig. 4) there is an initial
weight loss of 10% which is reduced to 1.1 and 5.6%when samples are crosslinked in the presence of NaOHor Na2CO3 respectively. SEVA-C-NA has a small
weight loss of 2.5% after 90 days, whereas SEVA-C-COapproaches the SEVA-C percentages for similar times(about 10%, after 90 days of immersion). For SCA (Fig.5) the weight loss after 14 days is about 11.5%, and13% after 90 days, whereas in the case of the cross-linked samples the values are 5.8 and 7.4% (14 days)and 8.4 and 10% (90 days), respectively, for SCA-NAand SCA-CO. SPCL (Fig. 6) samples show the clearestdi�erences in terms of weight loss varying from 14% inthe untreated samples to 6.2 and 9.2% for SPCL-NAand SPCL-CO, respectively (results for 90 days).These results are in accordance with the crosslinked
mass percentages data (Table 1) being the slowestdegradation rate observed for samples treated in NaOHmedia, as a result of the higher crosslinked mass than inthe case of Na2CO3 media. The reduction of the degra-dation rate and of the weight loss percentage is alsoin¯uenced by the water-up take ability of these materi-als, which are reduced as the crosslinked mass increases.In fact, this should inhibit the chains with low molecularweight and the small molecules to di�use out from themacromolecular domains. Furthermore, the onset ofchemical degradation will appear at longer degradationtimes also as a consequence of the crosslinked structure.On the other hand, a similar e�ect is observed in all thenon-treated starch based blends after 14 days ofimmersion time at which a decrease in weight lossclearly appears. The same e�ect can be observed insome crosslinked samples at longer immersion times asin the case of SCA-CO and SEVA-C-CO. This fact hasbeen also observed in other molecules of carbohydratenature [29].
3.4. Mechanical properties
The mechanical results exhibit an improvement in thesecant modulus of the crosslinked blends with respect tothe non-reacted ones. Table 4 shows the obtained datafor all the tested samples, being the ultimate tensilestrength (UTS) values in the same range for crosslinkedand non-treated blends (higher in some cases such as inSCA-NA and SCA-CO). The secant modulus was
Table 3
Equilibrium hydration degree (H equil.%) of tensile samples and ®lms
of starch blends and crosslinked starch based blends
H equil. (%)
Samples ASTM specimens Films
SEVA-C 25.7 30.1
SEVA-C-NA 23.8 27.7
SEVA-C-CO 26.2 28.7
SCA 49.5 51.2
SCA-NA 33.5 41.7
SCA-CO 37.7 50.1
SPCL 11.2 8.1
SPCL-NA 10.8 7.7
SPCL-CO 10.6 13.6
Fig. 4. Weight loss of ^, SEVA-C; &, crosslinked SEVA-NA; ~,
SEVA-CO as a function of immersion time on an isotonic saline solu-
tion at 37�C.
Fig. 5. Weight loss of^, SCA; &, crosslinked SCA-NA;~, SCA-CO
as a function of immersion time on an isotonic saline solution at 37�C.
Fig. 6. Weight loss of ^, SPCL;&, crosslinked SPCL-NA;~, SPCL-
CO as a function of immersion time on an isotonic saline solution at
37�C.
D. DemirgoÈz et al. / Polymer Degradation and Stability 70 (2000) 161±170 165
increased in all cases when the crosslinked mass per-centage increases, being the highest values obtainedwhen NaOH was used in the reaction media. The high-est increment was observed in the crosslinked SPCLwhere the secant modulus is about 4 times higher in thecase of SPCL-NA and 3 times for SPCL-CO.
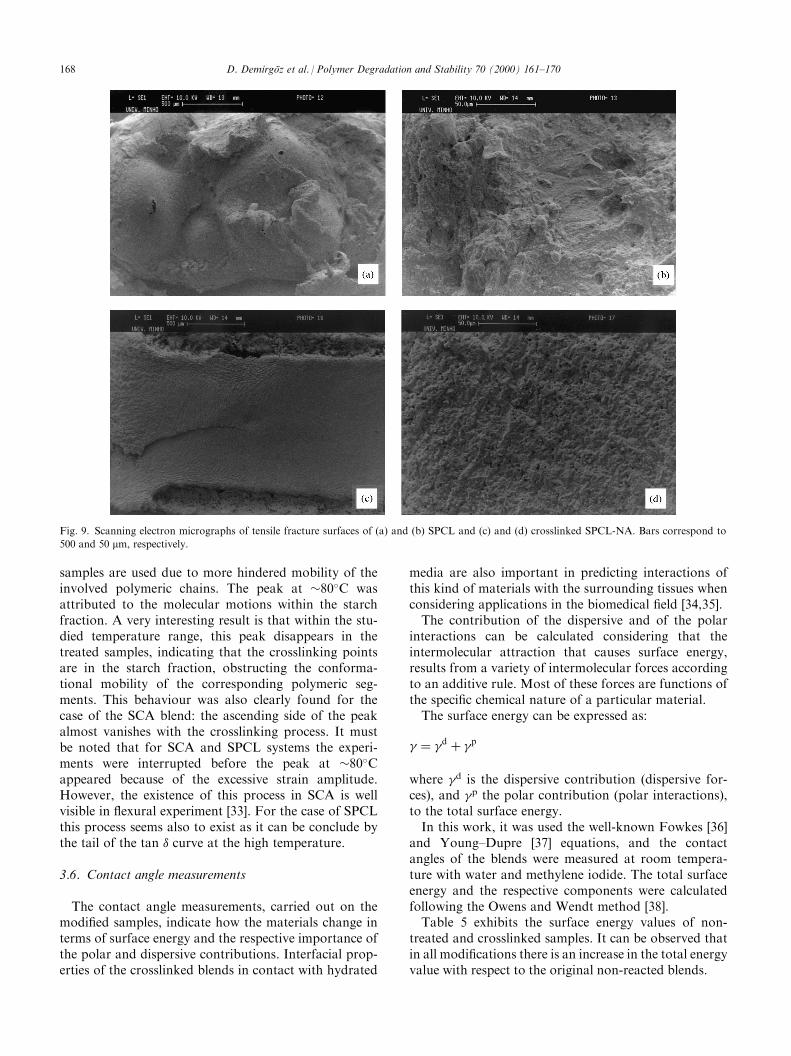
The SEM micrographs presented in Figs. 7 (SEVA-C), 8 (SCA) and 9 (SPCL) should be analysed togetherwith the data of Table 4. It is clear from this table thatthe most signi®cant variations on the modulus (as eval-uated by both quasi-static and dynamical experiments)were found for the most ductile blend, SPCL. Thecrosslinking e�ect on the texture of the fracture surfaceis clear when comparing Fig. 9a and b with Fig. 9c andd. The later one evidences a much more brittle fractureas a result of the successful crosslinking attained. In factthe fracture surface in Fig. 9c becomes much moresimilar to that of SEVA-C (Fig. 7a), as already reported[8,30,31], presenting a brittle nature.On SCA blends, especially by comparing Fig. 8a and
c, it is also possible to note a small decrease of ductility(fracture seems more brittle) that may be associated tothe crosslinking modi®cation.Finally, on what concerns to SEVA-C, for which the
reactions did not seem to be so e�ective, it appears to bepossible a morphological change (compare Fig. 7b andd) that leads to a more ductile behaviour, and to theformation of a neck during deformation. As the reac-tion was not very e�ective the observed di�erences onSEM fracture surfaces are not as clear as those observedfor SPCL materials. The balance between some cross-linking and this morphological change, is responsible
Table 4
Tensile mechanical properties of untreated and crosslinked starch
based blends, as obtained on quasi-static and dynamical±mechanical
tests
Material UTSa
(MPa)
E1%b
(GPa)
DMA 1 Hz
E 0c (GPa)
SEVA-C 36.4� 5.5 2.16�0.47 0.99
SEVA-NA 35.8�0.8 2.77�0.30 1.04
SEVA-CO 38.0�0.5 2.31�0.48 1.12
SCA 37.7�1.2 2.39�0.16 0.93
SCA-NA 57.2�1.5 4.59�0.17 1.30
SCA-CO 46.9�13.2 3.40�0.25 1.12
SPCL 23.7�5.7 0.57�0.08 0.36
SPCL-NA 22.9�0.9 2.44�0.09 0.58
SPCL-CO 23.4�0.5 1.66�0.03 0.47
a UTS, ultimate tensile strength.b E1%, secant modulus at 1% strain.c E0, storage modulus as determined in DMA experiments at 1 Hz
frequency.
Fig. 7. Scanning electron micrographs of tensile fracture surfaces of (a) and (b) SEVA-C and (c) and (d) crosslinked SEVA-NA. Bars correspond to
500 and 50 mm, respectively.
166 D. DemirgoÈz et al. / Polymer Degradation and Stability 70 (2000) 161±170

for the almost invariable values of UTS and the smallincrease of the modulus (E1% and E0). It should bereminded that the modulus is being measured for 1%strain, for which the morphological change is, of course,not yet clear.
3.5. DMA analysis
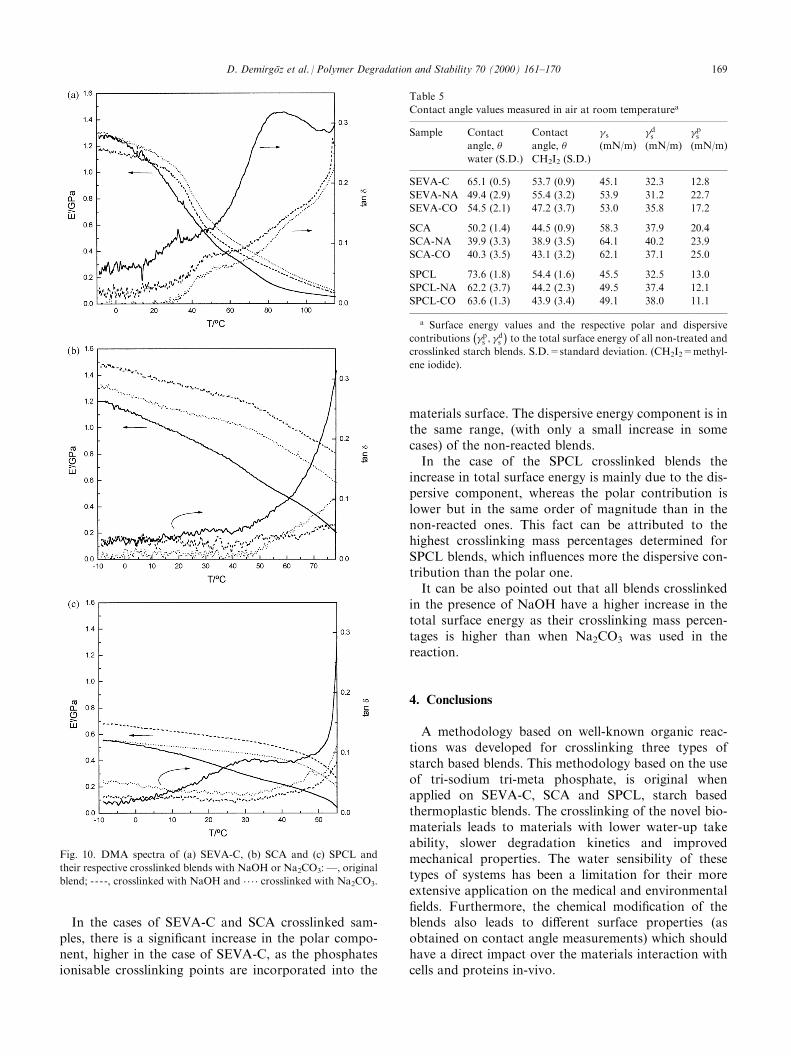
Dynamic mechanical experiments are a valuabletechnique to investigate the mechanical behaviour ofmaterials subjected to cyclic loads and to obtain infor-mation about the relaxation mechanisms that may becorrelated with the dynamics and the microstructure ofthe material.Fig. 10 shows the DMA results for the studied sam-
ples. Storage modulus, E 0, and loss factor, tan �, arepresented for each blend. The ®rst parameter is relatedwith the mechanical energy stored during each loadcycled and per volume unit. The loss factor is sensitiveto the viscoelastic behaviour and is useful to detectmechanical relaxations.For all specimens E 0 decreases with the increasing of
the temperature, re¯ecting a sti�ness loss. It is interest-ing to note that this e�ect (i.e. the variation of the slopeof E 0 curve) is less pronounced for the crosslinked sam-
ples, namely in SPCL specimens (see Fig. 10c) that, ascommented previously, were more e�ectively cross-linked. These results are expected to be associated to thehigher crosslinking density that prevents the liberationof new degrees of translational/rotational freedom dur-ing heating.The values of E 0 at room temperature are also pre-
sented in Table 4. The sti�ness measured by dynamicexperiments is, as expected, very di�erent from thatobtained on quasi-static tensile measurements. How-ever, it may be concluded that the variation of thevalues between di�erent samples is consistent for thetwo techniques used. In fact, as in the quasi-staticresults, it is clear for SEVA-C and SPCL samples anincreasing of sti�ness after the crosslinking reactionsthat is higher when NaOH is used. For the SCA blendthis e�ect is not so evident.The tan � curves may also provide complementary
information. SEVA-C exhibits two relaxation processesin this temperature range, also observed in ¯exuralexperiments [32,33]. The low temperature relaxation, at�30�C is more noticeable in a loss modulus plot (notshown) and was attributed to molecular mobility wherethe copolymer intervenes. As seen in Fig. 10a the pro-cess shifts to higher temperatures when crosslinked
Fig. 8. Scanning electron micrographs of tensile fracture surfaces of (a) and (b) SCA and (c) and (d) crosslinked SCA-NA. Bars correspond to 500
and 50 mm, respectively.
D. DemirgoÈz et al. / Polymer Degradation and Stability 70 (2000) 161±170 167
samples are used due to more hindered mobility of theinvolved polymeric chains. The peak at �80�C wasattributed to the molecular motions within the starchfraction. A very interesting result is that within the stu-died temperature range, this peak disappears in thetreated samples, indicating that the crosslinking pointsare in the starch fraction, obstructing the conforma-tional mobility of the corresponding polymeric seg-ments. This behaviour was also clearly found for thecase of the SCA blend: the ascending side of the peakalmost vanishes with the crosslinking process. It mustbe noted that for SCA and SPCL systems the experi-ments were interrupted before the peak at �80�Cappeared because of the excessive strain amplitude.However, the existence of this process in SCA is wellvisible in ¯exural experiment [33]. For the case of SPCLthis process seems also to exist as it can be conclude bythe tail of the tan � curve at the high temperature.
3.6. Contact angle measurements
The contact angle measurements, carried out on themodi®ed samples, indicate how the materials change interms of surface energy and the respective importance ofthe polar and dispersive contributions. Interfacial prop-erties of the crosslinked blends in contact with hydrated
media are also important in predicting interactions ofthis kind of materials with the surrounding tissues whenconsidering applications in the biomedical ®eld [34,35].The contribution of the dispersive and of the polar
interactions can be calculated considering that theintermolecular attraction that causes surface energy,results from a variety of intermolecular forces accordingto an additive rule. Most of these forces are functions ofthe speci®c chemical nature of a particular material.The surface energy can be expressed as:
� d � p
where d is the dispersive contribution (dispersive for-ces), and p the polar contribution (polar interactions),to the total surface energy.In this work, it was used the well-known Fowkes [36]
and Young±Dupre [37] equations, and the contactangles of the blends were measured at room tempera-ture with water and methylene iodide. The total surfaceenergy and the respective components were calculatedfollowing the Owens and Wendt method [38].Table 5 exhibits the surface energy values of non-
treated and crosslinked samples. It can be observed thatin all modi®cations there is an increase in the total energyvalue with respect to the original non-reacted blends.
Fig. 9. Scanning electron micrographs of tensile fracture surfaces of (a) and (b) SPCL and (c) and (d) crosslinked SPCL-NA. Bars correspond to
500 and 50 mm, respectively.
168 D. DemirgoÈz et al. / Polymer Degradation and Stability 70 (2000) 161±170
In the cases of SEVA-C and SCA crosslinked sam-ples, there is a signi®cant increase in the polar compo-nent, higher in the case of SEVA-C, as the phosphatesionisable crosslinking points are incorporated into the
materials surface. The dispersive energy component is inthe same range, (with only a small increase in somecases) of the non-reacted blends.In the case of the SPCL crosslinked blends the
increase in total surface energy is mainly due to the dis-persive component, whereas the polar contribution islower but in the same order of magnitude than in thenon-reacted ones. This fact can be attributed to thehighest crosslinking mass percentages determined forSPCL blends, which in¯uences more the dispersive con-tribution than the polar one.It can be also pointed out that all blends crosslinked
in the presence of NaOH have a higher increase in thetotal surface energy as their crosslinking mass percen-tages is higher than when Na2CO3 was used in thereaction.
4. Conclusions
A methodology based on well-known organic reac-tions was developed for crosslinking three types ofstarch based blends. This methodology based on the useof tri-sodium tri-meta phosphate, is original whenapplied on SEVA-C, SCA and SPCL, starch basedthermoplastic blends. The crosslinking of the novel bio-materials leads to materials with lower water-up takeability, slower degradation kinetics and improvedmechanical properties. The water sensibility of thesetypes of systems has been a limitation for their moreextensive application on the medical and environmental®elds. Furthermore, the chemical modi®cation of theblends also leads to di�erent surface properties (asobtained on contact angle measurements) which shouldhave a direct impact over the materials interaction withcells and proteins in-vivo.
Fig. 10. DMA spectra of (a) SEVA-C, (b) SCA and (c) SPCL and
their respective crosslinked blends with NaOH or Na2CO3: Ð, original
blend; ----, crosslinked with NaOH and .. . . crosslinked with Na2CO3.
Table 5
Contact angle values measured in air at room temperaturea
Sample Contact
angle, �water (S.D.)
Contact
angle, �CH2I2 (S.D.)
s(mN/m)
ds
(mN/m)
ps
(mN/m)
SEVA-C 65.1 (0.5) 53.7 (0.9) 45.1 32.3 12.8
SEVA-NA 49.4 (2.9) 55.4 (3.2) 53.9 31.2 22.7
SEVA-CO 54.5 (2.1) 47.2 (3.7) 53.0 35.8 17.2
SCA 50.2 (1.4) 44.5 (0.9) 58.3 37.9 20.4
SCA-NA 39.9 (3.3) 38.9 (3.5) 64.1 40.2 23.9
SCA-CO 40.3 (3.5) 43.1 (3.2) 62.1 37.1 25.0
SPCL 73.6 (1.8) 54.4 (1.6) 45.5 32.5 13.0
SPCL-NA 62.2 (3.7) 44.2 (2.3) 49.5 37.4 12.1
SPCL-CO 63.6 (1.3) 43.9 (3.4) 49.1 38.0 11.1
a Surface energy values and the respective polar and dispersive
contributions ps ;
ds
ÿ �to the total surface energy of all non-treated and
crosslinked starch blends. S.D.=standard deviation. (CH2I2=methyl-
ene iodide).
D. DemirgoÈz et al. / Polymer Degradation and Stability 70 (2000) 161±170 169

Acknowledgements
D.D. wishes to acknowledge ICBT, InternationalCentre of Biopolymer Technology, Netherland, ShortTerm Exchange Mission for a research grant supportingher work at the University of Minho, Portugal. We arealso grateful to the technical sta� of the University ofMinho.
References
[1] Glover R. Int Biodeter Biodegrad 1993;31:171.
[2] Ellis JR. Polymer recycling economic realities, ACS Symp. Series
609, 1995. p. 62.
[3] Alexander RJ. Cereal Foods World 1996;1996:41426.
[4] Reis RL, Cunha AM. J Mater Sci: Mater in Medicine 1995;6:78.
[5] Knowles JC, Hastings GW. J Mater Sci: Mater in Medicine
1992;3:352.
[6] Pereira CS, Cunha AM, Reis RL, Vazquez B, San Roman J. J
Mater Sci: Mater in Medicine 1998;9:825.
[7] Reis RL, Cunha AM. Medical Plastics 1995;9:71.
[8] Reis RL, Mendes SC, Cunha AM. Bevis. Polym Int 1997;43:347
[9] Langer RS, Peppas NA. Biomaterials 1981;2:201.
[10] Poutanen K, Forsell P. Trends Polym Sci 1996;4:128.
[11] Bastiolli C, Bellotti V, Del Giudice L, Gilli G. J Environ Polym
Deg 1993;181:1.
[12] Yoshida Y, Uemura T. TRIP 1998;4:143.
[13] Copperauld I. Clin Mater 1989;4:3.
[14] Hayashi T. Prog Polym Sci 1994;19:663.
[15] Roper H, Koch H. Starch/Starke 1990;42:123.
[16] Chinnaswamy R, Hanna MA. Starch/Starke 1991;43:396.
[17] Lai LS, Lakini JL. Biotechol Prog 1991;7:251.
[18] Wiedman W, Strobel E. Starch/Starke 1991;43:138.
[19] Stepto R, Tamka I. Acta Chimia 1987;41:76.
[20] van Soest JJG. Agro-Food Industry, Hi-Tech 1997;17.
[21] Lawton JW. Biodegradable Coatings for Thermoplastic Starch,
Cereals-1997, p. 43.
[22] Jane J. Pure Appl Chem 1995;A32:751.
[23] van Soest JJG, Vliegenthart JFG. TIBTECH 1997;15:208.
[24] Ellis RP, Cochrane MP, Dale MFB, Du�us CM, Lynn A, Mor-
rison IM, et al. J Sci Agric 1998;77:289.
[25] Booma M, Selke SE, Giacin JR. J Elastomers and Plastics
1994;26:104.
[26] Ratner BD, Chilkoti A, Castner DG. Clin Mater 1992;11:25.
[27] Rutenberg MW, Solarek D. In: Whistler RL, Bemiller JN,
Paschall EF, editors. Starch: chemistry and technology. San
Diego: Academic Press, 1984. p. 324±6.
[28] Tighe BJ. Brit Polym J 1986;18:8.
[29] Vaz CM, Reis RL, Cunha AM. 8th Conference on Polymer
Characterization Polychar±8, Denton USA, 2000. p.1.
[30] Reis RL, Cunha AM, Allan PS, Bevis MJ. Adv in Polym Tech
1997;16:163.
[31] Reis RL, Cunha AM, Allan PS, Bevis MJ. Polym Adv Tech
1996;7:784.
[32] Mano JF, Vaz CM, Mendes SC, Reis RL, Cunha AM. J Mater
Sci: Mater in Medicine 1999;10:857.
[33] Mano JF, Reis RL, Cunha AM. J Appl Polym Sci, in press.
[34] Ratner BD, Weathersby PK, Ho�man AS, Kelly MA, Scharpen
LH. J Appl Polym Sci 1978;22:643.
[35] Sun CSP, Hu CB. J Biomed Mater Res 1979;12:996.
[36] Berg JC. Wettability (Surfactant Series) 1993;49:75.
[37] Bikerman JJ. In: Physical surfaces. New York: Academic Press,
1970.
[38] Owens DK, Wendt RC. J Appl Polym Sci 1969;13:1741.
170 D. DemirgoÈz et al. / Polymer Degradation and Stability 70 (2000) 161±170




























![Modified starch - Krishna districtkrishna.nic.in/PDFfiles/MSME/Chemical/MODIFIED STARCH[1].pdf · MODIFIED STARCH CONTENTS ... SECTION XI SWOT ANALYSIS ... Malaysia 19916 Mauritious](https://img.pdfslide.net/doc/110x75/5aa35f827f8b9ada698e223e/modified-starch-krishna-starch1pdfmodified-starch-contents-section-xi-swot.jpg)