Embed Size (px)
Citation preview

APPLIED AND ENVIRONMENTAL MICROBIOLOGY, May 1988, p. 1230-1236 Vol. 54, No. 50099-2240/88/051230-07$02.00/0Copyright © 1988, American Society for Microbiology
Cloning and Characterization of the Tetracycline ResistanceDeterminant of and Several Promoters from within the
Conjugative Transposon Tn919COLIN HILL,2GERARD VENEMA,2 CHARLES DALY,' AND GERALD F. FITZGERALD`*
Department of Food Microbiology, University College, Cork, Ireland,' and Department of Genetics, Centre ofBiological Sciences, NL-9751 NN Haren (Gn), The Netherlands2
Received 2 November 1987/Accepted 2 February 1988
Tn919 is a 15- to 16-kilobase (kb) tetracycline resistance conjugative transposon that was originally isolatedfrom Streptococcus sanguis FC1. The tetracycline resistance determinant (tet) was found on a 4.2-kb HindIIfragment by in vitro deletion analysis. This fragment was subcloned to a pWV01 origin capable of directingreplication in Escherichia coli, Bacillus subtilis, and Streptococcus lactis, and expression was observed in allthree genera. In all cases, expression was weaker when only the 4.2-kb cloned fragment rather than the fulltransposon was present. The resistance gene is of the streptococcal tetM class and codes for a protein ofapproximately 70 kilodaltons. The restriction map resembles that of the tetM gene of Tnl545 (P. Martin, P.Trieu-Cuot, and P. Courvalin, Nucleic Acids Res. 14:7047-7058, 1986), which codes for a protein of 72.5kilodaltons. A number of transposon-derived promoter-bearing fragments were also cloned and sequenced.These closely resemble the consensus sequence of E. coli and B. subtilis promoters. Fusion experiments with atruncated lacZ gene indicate the possibility of an open reading frame for one of the promoters.
Tn919 is one of a number of conjugative transposonsoriginally isolated from members of the genus Streptococcus(4, 8, 24; see reference 9 for a recent review). Thesetransposons, including Tn919, encode tetracycline resis-tance (Tcr), the most widely disseminated drug resistancedeterminant in the streptococci (8, 15, 29). Burdett et al. (2,3) have divided streptococcal Tcr genes into three classes onthe basis of homology studies: tetL is most often associatedwith small nonconjugative plasmids, tetN is associated withlarge conjugative plasmids, and tetM is most usually notplasmid associated. The tetM gene product is believed tofunction inside the cell rather than preventing entry of thedrug as occurs in Escherichia coli-type Tcr. Burdett (2)postulated that the tetM product may be a tetracycline-binding protein or alternatively may function by an alterationof the ribosome. tetM has been widely linked with conjuga-tive transposons including Tn1545 (24) and Tn916 (2) (and,by inference Tn919, since these elements show almost totalhomology [11]). Furthermore, in a number of Tcr strains inwhich transposons have not been identified, homology hasbeen found, by using a tetM gene probe, with a HindlIchromosomal fragment of approximately 5 kilobases (kb),similar to the internal 4.8-kb tetM-bearing fragment of Tn916(3). Unlike Tn916, Tn919 contains an internal 4.2-kb HindlIfragment (11) and Tn1545 has an analogous 4.0-kb fragment(24). A number of tetM determinants have been cloned in E.coli, and these show a degree of similarity in that all possessan internal HindIll site (3, 24, 31, 34). The tetM gene ofTnl545 has been completely sequenced and codes for anopen reading frame (ORF) of 1,917 base pairs, correspondingto a protein with a predicted molecular mass of 72.5 kilodal-tons (kDa) (24). However, the work of Tobian et al. (33)suggests that one of two smaller proteins (33.5 or 35 kDa) isinvolved in tetM resistance.Although considerable effort in our laboratory has concen-
trated on the behaviour of Tn9I9 in lactic streptococci (16,
* Corresponding author.
17), little is known regarding the structure and function ofthis element. In this study, we report the localization andcloning of a 4.2-kb Hindll fragment containing the tetM geneof Tn919. The gene is expressed in Streptococcus lactis andBacillus subtilis in addition to E. coli and codes for a proteinof approximately 70 kDa. The tetM gene of Tn919 has aslightly altered restriction map compared with that ofTn1545. We also report the cloning of promoters from Tn919in B. subtilis, making use of a vector designed for thispurpose by van der Vossen et al. (35). The promoters werecloned in M13 and sequenced and closely resemble theconsensus sequence elucidated for E. coli, B. subtilis, and,most recently, Streptococcus cremoris (36). Fusion experi-ments with lacZ are described which suggest a possible ORFfor the strongest of the Tn919 promoters.
MATERIALS AND METHODS
Bacteria, media, and chemical reagents. Bacterial strainsand their plasmid content, where relevant, are described inTable 1. S. lactis strains were grown in M17 medium (32)supplemented with 0.5% glucose (GM17). B. subtilis and E.coli were routinely grown in LB medium (10). Solid mediacontained 1.5% agar (no. 3; Oxoid Ltd., London, England).Antibiotics present in selective media were added at thefollowing concentrations: for streptococci, tetracycline, 10ptg/ml; for bacilli, tetracycline, 10 pLg/ml; erythromycin, 5jig/ml; chloramphenicol, 5 jig/ml; for E. coli, ampicillin, 100,ug/ml; tetracycline, 5 pLg/ml; kanamycin, 50 jig/ml. Restric-tion enzymes and the Klenow fragment of DNA polymeraseI were obtained from Boehringer Corp., Dublin, Ireland, andused as specified by the manufacturer.
Molecular cloning and transformation. Plasmids were iso-lated as previously described (19). General procedures forDNA manipulations and cloning were essentially as de-scribed by Maniatis et al. (23). DNA fragments were recov-ered from agarose gels by the method of Tautz and Renz(30). Transformation of competent E. coli was by the methodof Mandel and Higa (22). Protoplasts of B. subtilis PSL-1
1230
on June 5, 2020 by guesthttp://aem
.asm.org/
Dow
nloaded from
tet FROM Tn919 1231
TABLE 1. Bacterial strains and genotypes
Relevant Source orgenotype reference
pGKV210pCI139pCI140pCI142pCI144pCI172
PSL-919 None
kan alpha-lacZkan tet
3837This study
38
ampamp tetamp tettetkan tetkan tetamp kan
ampampamp
eryery catery catery catery cattet kan
tet
tet kan
tet
pCI172 tet kan
tet
142111This studyThis studyThis studyThis study18
527M. van der GuchteJ. van der Vossen
2535This studyThis studyThis studyThis studyThis study
This study
12This study
16
7
This study
This study
were transformed as described by Chang and Cohen (6).Transformation of S. lactis MG1363 and IL1403 protoplastswas achieved by following the protocol of van der Vossen etal. (36).
Determining MICs. Cultures were inoculated (0.1%) intoan appropriate medium containing increasing concentrationsof tetracycline (from 0 to 100 ,uglml) and grown for 16 h atoptimum growth temperatures (30°C for S. lactis and 37°Cfor B. subtilis and E. coli), after which the optical density at550 nm (OD550) was read. The tetracycline concentration atwhich the OD550 equalled half the OD550 in the absence oftetracycline was taken as the MIC for each strain. MICs ofchloramphenicol were essentially as described above, ex-
cept that the OD550 was read after 6 h to rule out secondarygrowth after chloramphenicol breakdown.
In vitro transcription and translation. The Amershamprocaryotic DNA-directed cell-free coupled transcription-translation system derived from E. coli was used as specifiedin the protocol supplied by the manufacturer (Amersham
International, Little Chalfont, Buckinghamshire, England).CsCl-purified DNA (0.5 to 1.0 ,ug) was used in the reaction inwhich proteins were labeled with L-[35S]methionine. One-third of the reaction mixture was analyzed on 15% polyacryl-amide gels (20). After electrophoresis, the gels were proc-essed by fluorography as described by Skinner and Griswold(28) and exposed for at least 16 h to RXNIF 100 films (Fuji;J. J. Silber, Dublin, Ireland).DNA sequence analysis. The promoter-bearing fragments
of Tn919 were cloned in M13mpl8 and M13mpl9 (38) andsequenced by the dideoxynucleotide method with [a-35S]dATP (26) essentially as described in the AmershamCloning and Sequencing Handbook (Amersham Interna-tional). M13 and its derivatives were propagated in E. coliJM109 (38).
RESULTSCloning of the tetracycline resistance determinant of Tn919.
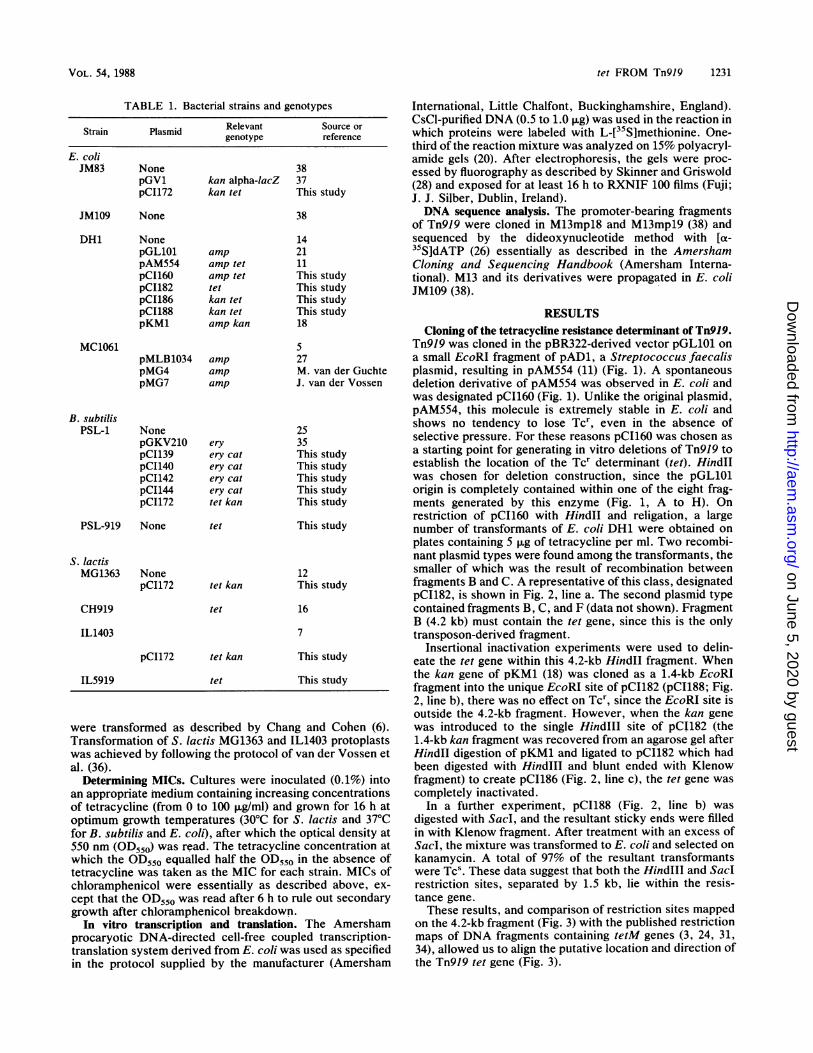
Tn919 was cloned in the pBR322-derived vector pGL101 ona small EcoRI fragment of pAD1, a Streptococcus faecalisplasmid, resulting in pAM554 (11) (Fig. 1). A spontaneousdeletion derivative of pAM554 was observed in E. coli andwas designated pCI160 (Fig. 1). Unlike the original plasmid,pAM554, this molecule is extremely stable in E. coli andshows no tendency to lose Tcr, even in the absence ofselective pressure. For these reasons pCI160 was chosen asa starting point for generating in vitro deletions of Tn919 toestablish the location of the Tcr determinant (tet). HindIIwas chosen for deletion construction, since the pGL101origin is completely contained within one of the eight frag-ments generated by this enzyme (Fig. 1, A to H). Onrestriction of pCI160 with Hindll and religation, a largenumber of transformants of E. coli DH1 were obtained onplates containing 5 ,ug of tetracycline per ml. Two recombi-nant plasmid types were found among the transformants, thesmaller of which was the result of recombination betweenfragments B and C. A representative of this class, designatedpCI182, is shown in Fig. 2, line a. The second plasmid typecontained fragments B, C, and F (data not shown). FragmentB (4.2 kb) must contain the tet gene, since this is the onlytransposon-derived fragment.
Insertional inactivation experiments were used to delin-eate the tet gene within this 4.2-kb Hindll fragment. Whenthe kan gene of pKM1 (18) was cloned as a 1.4-kb EcoRIfragment into the unique EcoRI site of pCI182 (pCI188; Fig.2, line b), there was no effect on Tcr, since the EcoRI site isoutside the 4.2-kb fragment. However, when the kan genewas introduced to the single Hindlll site of pCI182 (the1.4-kb kan fragment was recovered from an agarose gel afterHindII digestion of pKM1 and ligated to pCI182 which hadbeen digested with HindlIl and blunt ended with Klenowfragment) to create pCI186 (Fig. 2, line c), the tet gene wascompletely inactivated.
In a further experiment, pCI188 (Fig. 2, line b) wasdigested with Sacd, and the resultant sticky ends were filledin with Klenow fragment. After treatment with an excess ofSacd, the mixture was transformed to E. coli and selected onkanamycin. A total of 97% of the resultant transformantswere Tcs. These data suggest that both the HindlIl and Sacdrestriction sites, separated by 1.5 kb, lie within the resis-tance gene.These results, and comparison of restriction sites mapped
on the 4.2-kb fragment (Fig. 3) with the published restrictionmaps of DNA fragments containing tetM genes (3, 24, 31,34), allowed us to align the putative location and direction ofthe Tn919 tet gene (Fig. 3).
E. coliJM83
JM109
DH1
MC1061
B. subtilisPSL-1
NonepGV1pCI172
None
NonepGL101pAM554pCI160pCI182pCI186pCI188pKM1
pMLB1034pMG4pMG7
None
S. lactisMG1363 None
pCI172
CH919
IL1403
IL5919
VOL. 54, 1988
on June 5, 2020 by guesthttp://aem
.asm.org/
Dow
nloaded from

APPL. ENVIRON. MICROBIOL.
FIG. 1. Restriction endonuclease site map of pAM554 (21.0 kb) and pCI160 (18.0 kb). Symbols: , region corresponding to Tn919;-, DNA of pAD1 origin; , pGL101 DNA; -----, extent of the in vivo deletion in pAM554. The inner circle in pCI160 depicts the eightHind II fragments (A to H). Restriction endonucleases: V, HindII; E, EcoRI; H, HindlII; P, Pstl; Sa, Sacl.
Subcloning the 4.2-kb Hindll fragment on a pWVO1 origin.The expression of the Tcr determinant (tet) in S. lactis and B.subtilis was examined by subcloning the Hindll B fragmentfrom pCIl60 to a plasmid containing an origin capable ofdirecting replication in those genera. A number of plasmidshave been constructed based on the streptococcal origin ofthe small cryptic plasmid pWVO1 which can replicate inthose hosts in addition to E. coli (19, 35, 37). Attempts toclone the 4.2-kb fragment in one such plasmid, pGKV210(35), proved unsuccessful in E. coli, selecting on tetracyclineat 5 pLg/ml. No transformants were obtained even when thefragment was inserted next to strong promoters previouslycloned in pGKV210 (data not shown). All efforts to clonelarger fragments of both pCI160 and pCI182 also failed.However, pCI160 fragments A and B could be cloned in
pGV1, a vector constructed by Vosman et al. (37) whichcontains the multiple cloning site and the alpha-lacZ regionof M13mplO in addition to a Kanr gene (originally frompJH1) on a pWVO1 origin (37). This plasmid allows for theselection of recombinants in E. coli JM83 on plates contain-ing 5-bromo-4-chloro-3-indolyl-i-D-galactopyranoside (X-Gal). Fragments A and B were extracted from a gel andcloned in the unique SmaI site of pGV1. Blue and white
(insertionally inactivated) colonies arose on plates contain-ing kanamycin, but not on plates containing tetracycline,with or without kanamycin. A total of 20 white colonies werelysed, and 18 contained plasmids with inserts, equally di-vided between fragments A and B. On being restreaked ontoplates containing tetracycline, only those with fragment Bshowed growth. A representative plasmid, pCI172, wasmapped and found to contain the entire 4.2-kb insert (Fig. 2,line d). When pCI172 was retransformed to both E. coliJM83 and DH1, transformants could be obtained only byselecting on kanamycin, not on tetracycline. The Kanrcolonies could again grow on tetracycline on subsequentrestreaking.
Cell-free transcription-translation. Cell-free transcription-translation studies provide an alternative to minicell exper-iments in visualizing proteins produced by DNA inserts. Theproteins produced by some of the plasmids described aboveare shown in Fig. 4. The proteins derived from pGL101 (21)are shown in lane 1. The three heavy bands are products ofthe ampicillin gene and represent pre-3-lactamase, mature3-lactamase, and a specific breakdown product. Other minorbands are probably nonspecific degradation products due toenzymes present in the cell extract. The gene products of
k I I IT IIkb 1 ~~2 A 4 S 6 7 8 9
(a)pCI182 v.
(b)pCI188 T-
(c)pCI186 I
H
H
Sa vScf
Sa Sc P
"qr HIFI II
E
v VEBS H SBE
t -, G onKan
Sa ,S,P E
Kan
(d) H Sa 1HP HEa
pCI172 I Sa , H - _ ESaV
Kan pWVOlFIG. 2. Restriction endonuclease site maps of pCI182 (line a), pCI188 (line b), pCI186 (line c), and pCI172 (line d). Symbols:
Tn919; , pGL101; -, pADI; -, pGV1. Restriction endonucleases: V, Hindll; E, EcoRI; H, HindIll; P, PstI; Sa, Sacl; Sc, ScaI;v ,HindIl-SmaI fusion; ov. HindII- HindIll fusion.
i~i
It 11 i a i w
it-
1232 HILL ET AL.
v
on June 5, 2020 by guesthttp://aem
.asm.org/
Dow
nloaded from
tet FROM Tn919 1233
I I \11a A-- X
I I I 1kb 1 2 3 4
TET
FIG. 3. The 4.2-kb Hindll B fragment of pCI160. Arrow indi-cates putative location and direction of tet.
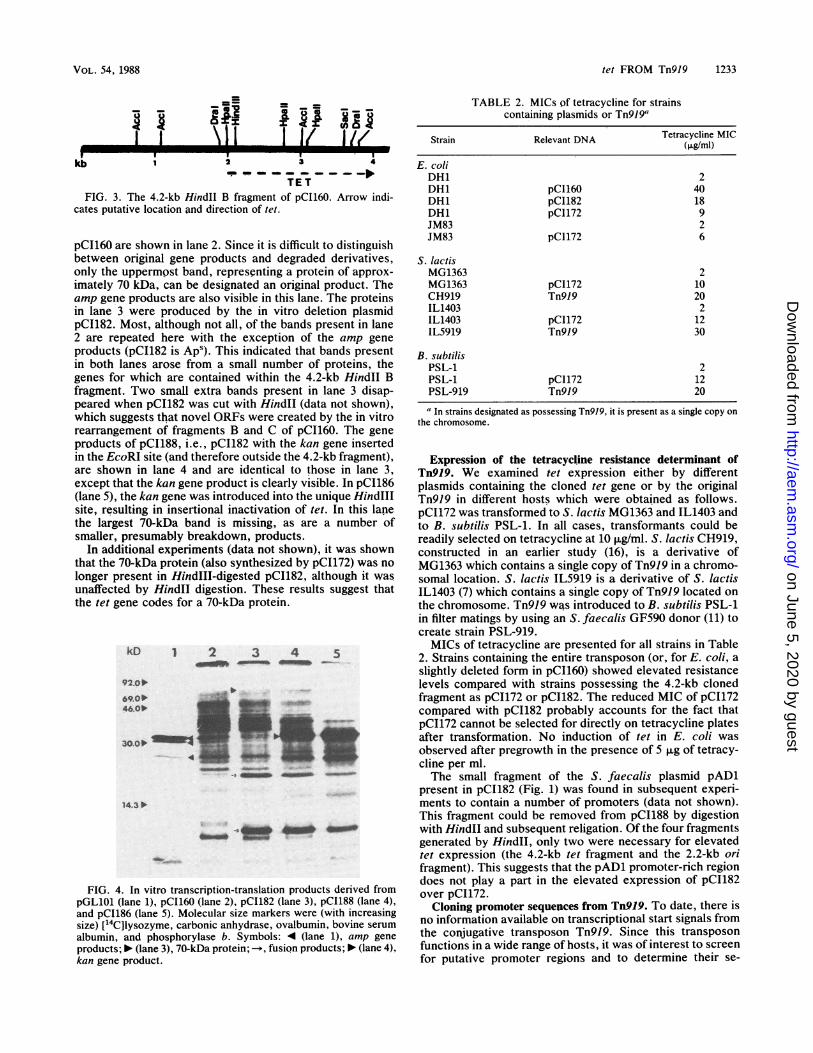
pCI16Q are shown in lane 2. Since it is difficult to distinguishbetween original gene products and degraded derivatives,only the uppermpst band, representing a protein of approx-
imately 70 kDa, can be designated an original product. Theamp gene products are also visible in this lane. The proteinsin lane 3 were produced by the in vitro deletion plasmidpCI182. Most, although not all, of the bands present in lane2 are repeated here with the exception of the amp gene
products (pCI182 is Aps). This indicated that bands presentin both lanes arose from a small number of proteins, thegenes for which are contained within the 4.2-kb HindII Bfragment. Two small extra bands present in lane 3 disap-peared when pCI182 was cut with HindII (data not shown),which suggests that novel ORFs were created by the in vitrorearrangement of fragments B and C of pCI160. The gene
products of pCI188, i.e., pCI182 with the kan gene insertedin the EcoRI site (and therefore outside the 4.2-kb fragment),are shown in lane 4 and are identical to those in lane 3,except that the kan gene product is clearly visible. In pCI186(lane 5), the kan gene was introduced into the unique HindIIIsite, resulting in insertional inactivation of tet. In this lanethe largest 70-kDa band is missing, as are a number ofsmaller, presumably breakdown, products.
In additional experiments (data not shown), it was shownthat the 70-kDa protein (also synthesized by pCI172) was no
longer present in HindIII-digested pCI182, although it wasunaffected by Hindll digestion. These results suggest thatthe tet gene codes for a 70-kDa protein.
kD 1 2 3 4 5
t14m
FIG. 4. In vitro transcription-translation products derived from
pGL1O1 (lane 1), pCI16O (lane 2), pCI182 (lane 3), pCI188 (lane 4),
and pCI186 (lane 5). Molecular size markers were (with increasing
size) [14C]lysozyme, carbonic anhydrase, ovalbumin, bovine serum
albumin, and phosphorylase b. Symbols: 4 (lane 1), amp gene
products; 10 (lane 3), 70-kDa protein; --*, fusion products; 10 (lane 4),
kan gene product.
TABLE 2. MICs of tetracycline for strainscontaining plasmids or Tn919a
Strain Relevant DNA Tetracycline MIC
E. coliDH1 2DH1 pCI160 40DH1 pCI182 18DH1 pCI172 9JM83 2JM83 pCI172 6
S. lactisMG1363 2MG1363 pCI172 10CH919 Tn919 20IL1403 2IL1403 pCI172 12IL5919 Tn919 30
B. subtilisPSL-1 2PSL-1 pCI172 12PSL-919 Tn919 20
a In strains designated as possessing Tn9O9, it is present as a single copy onthe chromosome.
Expression of the tetracycline resistance determinant ofTn919. We examined tet expression either by differentplasmids containing the cloned tet gene or by the originalTn919 in different hosts which were obtained as follows.pCI172 was transformed to S. lactis MG1363 and IL1403 andto B. subtilis PSL-1. In all cases, transformants could bereadily selected on tetracycline at 10 ,ugtml. S. lactis CH919,constructed in an earlier study (16), is a derivative ofMG1363 which contains a single copy of Tn919 in a chromo-somal location. S. lactis IL5919 is a derivative of S. lactisIL1403 (7) which contains a single copy of Tn9J9 located onthe chromosome. Tn919 was introduced to B. subtilis PSL-1in filter matings by using an S. faecalis GF590 donor (11) tocreate strain PSL-919.MICs of tetracycline are presented for all strains in Table
2. Strains containing the entire transposon (or, for E. coli, aslightly deleted form in pCI160) showed elevated resistancelevels compared with strains possessing the 4.2-kb clonedfragment as pCI172 or pCI182. The reduced MIC of pCI172compared with pCI182 probably accounts for the fact thatpCI172 cannot be selected for directly on tetracycline platesafter transformation. No induction of tet in E. coli wasobserved after pregrowth in the presence of 5 ,ug of tetracy-cline per ml.The small fragment of the S. faecalis plasmid pAD1
present in pCI182 (Fig. 1) was found in subsequent experi-ments to contain a number of promoters (data not shown).This fragment could be removed from pCI188 by digestionwith Hindll and subsequent religation. Of the four fragmentsgenerated by Hindll, only two were necessary for elevatedtet expression (the 4.2-kb tet fragment and the 2.2-kb orifragment). This suggests that the pAD1 promoter-rich regiondoes not play a part in the elevated expression of pCI182over pCI172.
Cloning promoter sequences from Tn919. To date, there isno information available on transcriptional start signals fromthe conjugative transposon Tn9J9. Since this transposonfunctions in a wide range of hosts, it was of interest to screenfor putative promoter regions and to determine their se-
. - -I . . . . .No=
VOL. 54, 1988
on June 5, 2020 by guesthttp://aem
.asm.org/
Dow
nloaded from

APPL. ENVIRON. MICROBIOL.
TABLE 3. Tn9J9 promoter sequences and E. coli consensus
Promoter Nucleotide sequence" Chloramphenicol MIC-35 Spacing -10 (,ugml)
144 TTGACA AATATCTCTTAACGTGC (17 ntb) TATTAT 40142 TTGACA TACGCTGAAATTTTG (15 nt) TAAATC 15139 CTGACA AACAGATTCACCAGTAG (17 nt) TAGAAT 15Consensus TTGACA 16 of 18 nt TATAAT
a Nucleotide sequence of three Tn919 putative promoters. The sequences shown were the only ones within each fragment which resembled the consensuspromoter sequence as determined for E. coli and B. subtilis. The consensus sequence is included for comparison.
b nt, Nucleotide.
quence. pGKV210 is a promoter-screening vector whichreplicates in B. subtilis, S. lactis, and E. coli (35). Thisplasmid was used to select for promoterlike regions of Tn9J9by cloning small fragments into the multiple cloning site ofthe vector and selecting for expression of the promoterlesschloramphenicol acetyltransferase (CAT) gene in B. subtilis.Using both HindII and Sau3A to generate fragments ofpCI160, we isolated a large number of promoter-bearingrecombinants. At least seven different promoter-bearingSau3A fragments were found by Southern hybridization tohave originated from the HindII C fragment (Fig. 1), origi-nating in part from pGL101 and in part from pAD1. Becauseof the profusion of promoters in this region, the HindII Cfragment was removed from a gel and the remaining sevenfragments of pCI60 were eluted and subsequently rediges-ted with RsaI. This generated a large number of fragments(>10) which were less than 1.0 kb in size. These wereshotgun cloned in the single SmaI site of pGKV210 andtransformed to B. subtilis PSL-1, selecting for erythromycin(5 ,ug/ml) and chloramphenicol (5 ,ug/ml). Transformantswere divided into different classes on the basis of spot testson plates of increasing chloramphenicol concentration. Rep-resentatives of different classes were lysed, and all werefound to contain inserts. Four randomly chosen recombinantplasmids (pCI139, pCI140, pCI142, and pCI144, containinginserts of 260, 780, 300, and 280 base pairs, respectively)hybridized to the 4.2-kb Hindll B fragment of pCI160, whichcontains only Tn9J9 DNA. The three smaller inserts (frompCI139, pCI142, and pCI144) were cloned in M13mpl8 andM13mpl9 and sequenced by the dideoxy method (the frag-ment contained in pCI144 could not be subcloned inM13mpl9, a problem often encountered with promoter-bearing fragments, and the sequence had to be deduced fromrepeated sequencing in one direction). Only one putativepromoter sequence was identified on each fragment. Thesequences of interest are shown in Table 3, along with the
MICs obtained after growth for 6 h in broth containingvarious levels of chloramphenicol. The putative promotersequences closely resemble the canonical consensus se-quences of E. coli, B. subtilis, and S. cremoris (36). This isto be expected, since fragments were screened in B. subtilisand also since the transposon functions in all three genera.However, we have as yet no evidence that these sequencespromote transcription of Tn919 genes.Only promoter 144 has an ORF following immediately
downstream. This ORF also contains a putative ribosomalbinding site (RBS; free energy, -13.3 kcal [-55.6 kJ]) in asuitable location (Fig. 5). However, the ORF is only 7 aminoacids in length. Although this may be the gene product forwhich the promoter is meant, there is a second possibility.Soon after this ORF there is a second, stronger, RBS (freeenergy -16 kcal [-66.9 kJ]), and within 10 base pairs thereis a possible start codon TTG. It is impossible to determinewhether this represents a second ORF, since the limit of theinserted DNA does not extend beyond this point. It ispossible, however, by conducting fusion experiments with alacZ gene missing its first nine codons and expressionsignals, to determine whether the second RBS and startcodon can act as translational start signals. Consequently,promoter 144 was cut with EcoRI and Hindll and ligated tothe EcoRI-Smal-digested vectors pMLB1034, pMG4, andpMG7, which contain the SmaI site in three different framesrelative to the truncated lacZ gene (Fig. 5). Only fusions withpMLBiO34 showed any expression of lacZ, this being theonly instance in which the TTG of the putative second ORFis in frame. Expression was weak (light-blue colonies onX-Gal) compared with that in a control experiment in whicha fragment containing promoter 144 plus the first codons ofthe CAT gene was ligated in frame to pMLB1034 (intenselyblue colonies; data not shown). These data confirm thatpromoter 144 can direct, if only weakly, transcription and
bp 80 17 14 9-TTGACA----TATTAT----AAAAGGAG----ATG.CGG.CAA.GGT.ATT.CTT.AAA.TAA--
1 -35 -10 RBS stopEcoRl
145 10 -------------pGKV210-*--------------AGGAGG----TTG.TGG.GGA.TCC.AGA.GTC.GAC------* CAT gene
RBS IHind 11
EcoRl Smal
----- ---- CCCGGG.GAT.CCC.GTC--lacZ___...--------------CC.CGG.GCC.GTC--lacZ----__ -------------C.CCG.GGG.ATC.GAT.CCC.GTC--lacZ
pMLB1 034pMG4pMG7
FIG. 5. Relevant base sequences of pCI144, pMLB1034, pMG4, and pMG7. The upper two lines represent the entire insert in pCI144. Thespacings between relevant sequences are shown above the line. The lower three lines indicate the reading frame for lacZ in pMLB1034,pMG4, and pMG7.
1234 HILL ET AL.
on June 5, 2020 by guesthttp://aem
.asm.org/
Dow
nloaded from

tet FROM Tn919 1235
translation of a second ORF, assuming that one exists,starting from the TTG at the end of the sequenced insert.
DISCUSSION
Tn919 and related elements show great potential as genetictools for the analysis of lactic streptococci (9, 17). However,little information is available regarding the characterizationof Tn9J9. In this study we describe the cloning of both theTcr determinant (tet) and promoter-bearing fragments ofTn919. Although more than 90% of Tn919 has been sub-cloned in our laboratory (unpublished data), the work pre-sented here focuses on an internal 4.2-kb HindII fragmentwhich was found by in vitro deletion analysis to contain theresistance gene. Initial attempts to subclone this fragment inE. coli on an origin which can also function in S. lactis andB. subtilis, selecting on tetracycline at 5 ,ug/ml, provedunsuccessful, even though the original in vitro deletionderivative, pCI182, could be readily selected after transfor-mation under similar selection conditions. However, thefragment was cloned by using the vector pGV1, in whichdirect selection for Tcr resistance is not necessary to selectrecombinants in E. coli. The resultant plasmid, pCI172,showed limited Tcr in E. coli JM83, but was still unable totransform this strain with selection for tetracycline. To ruleout any possible strain influence, pCI172 was introduced toE. coli DH1, but again no Tcr transformants were obtained,even though Kanr colonies did display Tcr upon furtherrestreaking. The possibility that the difference betweenpCI172 and pCI182 was due to a strong promoter regionpresent in pCI182 and originating from pAD1 was ruled out,since this promoter-rich region could be removed frompCI182 without preventing transformation and selection ontetracycline at 5 p.g/ml. The most obvious remaining differ-ence between pCI172 and pCI182 was their relative copynumber, which was significantly higher (as determined visu-ally from agarose gels) for the latter plasmid.The tet determinant was expressed in S. lactis and B.
subtilis, and pCI172 could be readily selected in these strainsafter transformation, with tetracycline at 10 ,ug/ml. Thisresult, together with the higher tetracycline MICs for pCI172in gram-positive than gram-negative strains, suggests thatthe gene product either functions, or is expressed, moreefficiently in those genera. It does appear that the tet gene ismore efficiently expressed by the full transposon than whensubcloned on the 4.2-kb fragment. Strains containing a singlechromosomal copy of Tn919 were resistant to higher levelsof tetracycline than were those containing more than onecopy of pCI172. For E. coli also, a higher level of resistancewas observed in strains harboring pCI160 (which containsalmost the entire transposon) than in those containing thedeletion derivative pCI182. This suggests that DNA fromoutside the 4.2-kb fragment has a role in determining theexpression of the tet gene. Tobian et al. (33) also found thata tetM gene cloned from the chromosome of Streptococcusmutans V825 gave higher levels of resistance in E. coli(tetracycline MIC > 20 ,ug/ml) than in Streptococcuis sanguis(tetracycline MIC, ca. 5 ,ug/ml). The levels of resistance in S.sanguis were comparable to those in S. mutans V825.However, the cloned and chromosomal determinants werenot directly compared in an isogenic background.
Insertional inactivation of tet at the HindlIl site of pCI182results in the loss of a 70-kDa protein, suggesting that this isthe tet gene product. Similar results have been reported forthe tetM gene products of Tn1545 (24) and the Campylobac-terjejuni plasmid pUA466 (34). In contrast, the product of a
tetM gene isolated from the chromosome of S. mutans codesfor at least one of two proteins of 33 and 35 kDa (33). At arestriction site level, similarities are also apparent betweendifferent tetM genes (e.g. six of seven enzyme sites mappedwithin the Tn919 gene can be located on the sequence of theTn1545 tetM gene in similar positions). However, the se-quences are not identical, since no ClaI, Sau3A, or KpnI sitecould be identified, although all appear in Tn1545. It may beconcluded that although the tetM determinants encoded bythese elements are similar, there has been some divergenceat a sequence level, resulting in altered restriction patterns.
Since Tn9J9 can function in a number of genera, it was ofinterest to clone and sequence transposon promoter regions.The information generated could be useful in at least tworespects: first, in constructing expression vectors capable offunctioning in different genera, and second, in helping tolocate different ORFs on the transposon. Initial attemptswere unsuccessful, in that promoter-bearing fragments wereisolated at a high frequency from one particular 3.3-kbfragment of pCI160 which contains DNA of pGL101 andpADI origin. When this fragment was removed prior tocloning, a large number of Cmr transformants were isolated.Four recombinant plasmids were chosen and probed againstpCI160. All four showed homology with the 4.2-kb HindIIfragment. The three smaller inserts were cloned in M13 andsequenced. One promoterlike sequence was found in eachinsert, all of which closely resemble the canonical consensussequences reported for E. coli, B. subtilis, and, most re-cently, S. cremoris (36). It was found that the promoter mostclosely resembling the consensus sequence was strongest. Asimilar finding was reported by van der Vossen et al. (36) forpromoters from S. cremoris. The same authors suggested apossible involvement of a TG sequence separated from the-10 region by a single nucleotide in strong promoters. ThisTG sequence was also found in promoter 144.
It is difficult to determine whether the promoters isolatedin this study are actually involved in the expression oftransposon functions, since in only one case was an ORFfound on the same fragment. This probably reflects the smallsizes of the inserts which were sequenced. It is notable inthis respect that no obvious promoter was found for the tetMgene of Tn1545 in the 130 base pairs preceeding the ORF.One promoter in this study, cloned in pCI144, does have anextremely small ORF and a RBS downstream, but this wouldcode for a peptide of only 7 amino acids. A second RBS andstart codon were detected downstream of the ORF, justwithin the limits of the insert. The start codon in thisinstance is TTG, which is less common than ATG, but iscertainly capable of functioning in this role (the promoterlessCAT gene used in this study begins at TTG). Fusion exper-iments demonstrated that this second RBS and start codonallow expression of lacZ in E. coli, although weakly whencompared with the expression of a fused CAT-lacZ underthe same promoter. It has been found that a small ORFpreceding the ermC gene in B. subtilis (1) is most probably a
regulatory device, but the kind of hairpin structures de-scribed in that study were not found in this instance.The combined results of transcription-translation experi-
ments and cloning of the promoter-bearing fragments maysuggest a concentration of expression signals and geneproducts on the 4.2-kb Hindll fragment. An alternativeexplanation may be that only expression signals recognizedby E. coli and B. subtilis lie in this region. It is expected thatthe resistance gene and promoters isolated in this study,which function in three unrelated genera, will find a wide
VOL. 54, 1988
on June 5, 2020 by guesthttp://aem
.asm.org/
Dow
nloaded from

APPL. ENVIRON. MICROBIOL.
application in the design of shuttle vectors, a number ofwhich are currently being constructed in our laboratory.
ACKNOWLEDGMENTS
We thank J. van der Vossen, M. van der Guchte, J. Kiel, and B.Vosman for providing some of the plasmids used in this study.C.H. was the recipient of a Commission of the European Com-
munities Fellowship to work in the laboratory of G.V. This workwas funded partly by contract BAP0008-IRL from the Commissionof the European Communities and partly by the National Board forScience and Technology, Ireland.
LITERATURE CITED1. Bechhofer, D. H., and D. Dubnau. 1987. Induced mRNA stabil-
ity in Bacillus subtilis. Proc. Natl. Acad. Sci. USA 84:498-502.2. Burdett, V. 1986. Streptococcal tetracycline resistance medi-
ated at the level of protein synthesis. J. Bacteriol. 165:564-569.3. Burdett, V., J. Inamine, and S. Rajagoplan. 1982. Heterogeneity
of tetracycline resistance determinants in Streptococcus. J.Bacteriol. 149:995-1004.
4. Carlier, C., and P. Courvalin. 1982. Resistance of streptococcito aminoglycoside-aminocyclitol antibiotics, p. 162-166. In D.Schlessinger (ed.), Microbiology-1982. American Society forMicrobiology, Washington, D.C.
5. Casadaban, M. J., and S. N. Cohen. 1980. Analysis of genecontrol signals by DNA fusion and cloning in Escherichia coli.J. Mol. Biol. 138:179-207.
6. Chang, S., and S. N. Cohen. 1979. High frequency transforma-tion of Bacillus subtilis protoplasts by plasmid DNA. Mol. Gen.Genet. 168:111-115.
7. Chopin, A., M.-C. Chopin, A. Moillo-Batt, and P. Langella.1984. Two plasmid-determined restriction systems in Strepto-coccus lactis. Plasmid 11:260-263.
8. Christie, P. J., R. Z. Korman, S. A. Zahler, J. C. Adsit, andG. M. Dunny. 1987. Two conjugation systems associated withStreptococcus faecalis plasmid pCF10: identification of a con-jugative transposon that transfers between Streptococcus fae-calis and Bacillus subtilis. J. Bacteriol. 169:2529-2536.
9. Clewell, D. B., and C. Gawron-Burke. 1986. Conjugative trans-posons and the dissemination of antibiotic resistance in strep-tococci. Annu. Rev. Microbiol. 40:635-659.
10. Davis, R. W., D. Botstein, and J. R. Roth (ed.). 1980. Advancedbacterial genetics, p. 140-141 and 201. Cold Spring HarborLaboratory, Cold Spring Harbor, N.Y.
11. Fitzgerald, G. F., and D. B. Clewell. 1985. A conjugativetransposon (Tn919) in Streptococcus sanguis. Infect. Immun.47:415-420.
12. Gasson, M. J. 1983. Plasmid complements of Streptococcuslactis NCDO712 and other lactic streptococci after protoplast-induced curing. J. Bacteriol. 154:1-9.
13. Gawron-Burke, C., and D. B. Clewell. 1982. A transposon inStreptococcus faecalis with fertility properties. Nature(London) 300:281-284.
14. Hanahan, D. 1983. Studies on transformation of Escherichia coliwith plasmids. J. Mol. Biol. 166:620-626.
15. Hartley, D. L., K. R. Jones, J. A. Tobian, D. J. LeBlanc, andF. L. Macrina. 1984. Disseminated tetracycline resistance inoral streptococci: implication of a conjugative transposon. In-fect. Immun. 45:13-17.
16. Hill, C., C. Daly, and G. F. Fitzgerald. 1985. Conjugativetransfer of the transposon Tn919 to lactic acid bacteria. FEMSMicrobiol. Lett. 30:115-119.
17. Hill, C., C. Daly, and G. F. Fitzgerald. 1987. Development ofhigh-frequency delivery system for transposon Tn919 in lacticstreptococci: random insertion in Streptococcus lactis subsp.diacetylactis 18-16. Appl. Environ. Microbiol. 53:74-78.
18. Kiel, J. A. K. W., J. P. M. J. Vossen, and G. Venema. 1987. Ageneral method for the construction of Escherichia coli mutantsby homologous recombination and plasmid segregation. Mol.
Gen. Genet. 207:294-301.19. Kok, J., J. M. B. M. van der Vossen, and G. Venema. 1984.
Construction of plasmid cloning vectors for lactic streptococciwhich also replicate in Bacillus subtilis and Escherichia coli.Appl. Environ. Microbiol. 48:726-731.
20. Laemmli, U. K. 1970. Cleavage of structural proteins during theassembly of the head of bacteriophage T4. Nature (London)227:680-685.
21. Lauer, G., R. Pastrana, J. Sherley, and M. Ptashne. 1981.Construction of overproducers of the bacteriophage 434 repres-sor and cro proteins. J. Mol. Appl. Genet. 1:139-147.
22. Mandel, M., and A. Higa. 1970. Calcium-dependent bacterio-phage DNA infection. J. Mol. Biol. 53:159-162.
23. Maniatis, T., E. F. Fritsch, and J. Sambrook. 1982. Molecularcloning: a laboratory manual. Cold Spring Harbor Laboratory,Cold Spring Harbor, N.Y.
24. Martin, P., P. Trieu-Cuot, and P. Courvalin. 1986. Nucleotidesequence of the tetM tetracycline resistance determinant of thestreptococcal conjugative shuttle transposon Tn1545. NucleicAcids Res. 14:7047-7057.
25. Ostroff, G. R., and J. J. Pene. 1983. Molecular cloning withbifunctional plasmid vectors in Bacillus subtilis: isolation of aspontaneous mutant of Bacillus subtilis with enhanced transfor-mability for Escheric hia coli-propagated chimeric plasmidDNA. J. Bacteriol. 156:934-936.
26. Sanger, F., S. Nicklen, and A. R. Coulson. 1977. DNA sequenc-ing with chain-terminating inhibitors. Proc. Natl. Acad. Sci.USA 74:5463-5467.
27. Silhavy, T. J., M. L. Berman, and L. W. Enquist. 1984.Experiments with gene fusions. Cold Spring Harbor Labora-tory, Cold Spring Harbor, N.Y.
28. Skinner, M. K., and M. D. Griswold. 1983. Fluorographicdetection of radioactivity in polyacrylamide gels with 2,5-diphenyloxazole in acetic acid and its comparison with existingprocedures. Biochem. J. 209:281-284.
29. Smith, M. D., S Hazum, and W. R. Guild. 1981. Homologyamong tet determinants in conjugative elements of streptococci.J. Bacteriol. 148:323-340.
30. Tautz, D., and M. Renz. 1983. An optimized freeze-squeezemethod for the recovery of DNA fragments from agarose gels.Anal. Biochem. 132:14-19.
31. Taylor, D. E., K. Hiratsuka, H. Ray, and E. K. Manavathu.1987. Characterization and expression of a cloned tetracyclineresistance determinant from Campylobacter jejuni plasmidpUA466. J. Bacteriol. 169:2984-2989.
32. Terzaghi, B. E., and W. E. Sandine. 1975. Improved medium forlactic streptococci and their bacteriophages. Appl. Microbiol.29:807-813.
33. Tobian, J. A., M. L. Cline, and F. L. Macrina. 1984. Charac-terization and expression of a cloned tetracycline resistancedeterminant from the chromosome of Streptococcus mutans. J.Bacteriol. 160:556-563.
34. Tobian, J. A., and F. L. Macrina. 1982. Helper plasmid cloningin Streptococcus sanguis: cloning of a tetracycline resistancedeterminant from the Streptococcus mutans chromosome. J.Bacteriol. 152:215-222.
35. van der Vossen, J. M. B. M., J. Kok, and G. Venema. 1985.Construction of cloning, promoter-screening and terminator-screening shuttle vectors for Bacillus subtilis and Streptococcuslactis. Appl. Environ. Microbiol. 50:540-542.
36. van der Vossen, J. M. B. M., D. van der Lelie, and G. Venema.1987. Isolation and characterization of Streptococcus cremorisWg2-specific promoters. Appl. Environ. Microbiol. 53:2452-2457.
37. Vosman, B., J. Kooistra, J. Olijve, and G. Venema. 1987.Cloning in Escherichia coli of the gene specifying the DNA-entry nuclease of Bacillus subtilis. Gene 52:175-183.
38. Yanisch-Perron, C., J. Vieira, and J. Messing. 1985. ImprovedM13 cloning vectors and host strains: nucleotide sequences ofthe M13mpl8 and pUC19 vectors. Gene 33:103-119.
1236 HILL ET AL.
on June 5, 2020 by guesthttp://aem
.asm.org/
Dow
nloaded from





























