Embed Size (px)
Citation preview

Bleeding Disorders
Elisabeth M. Battinelli, M.D./Ph.D.
Assistant Professor of Medicine
Harvard Medical School
Associate Physician
Hematology Division
Brigham and Women’s Hospital

Elisabeth M. Battinelli, M.D./Ph.D.
Assistant Professor of Medicine
Associate Physician
Disclosures
Sanofi, Consultant

http://medicinembbs.blogspot.com/2011/02/normal-hemostasis.html

Durkan, CVC 2008
Primary Hemostasis
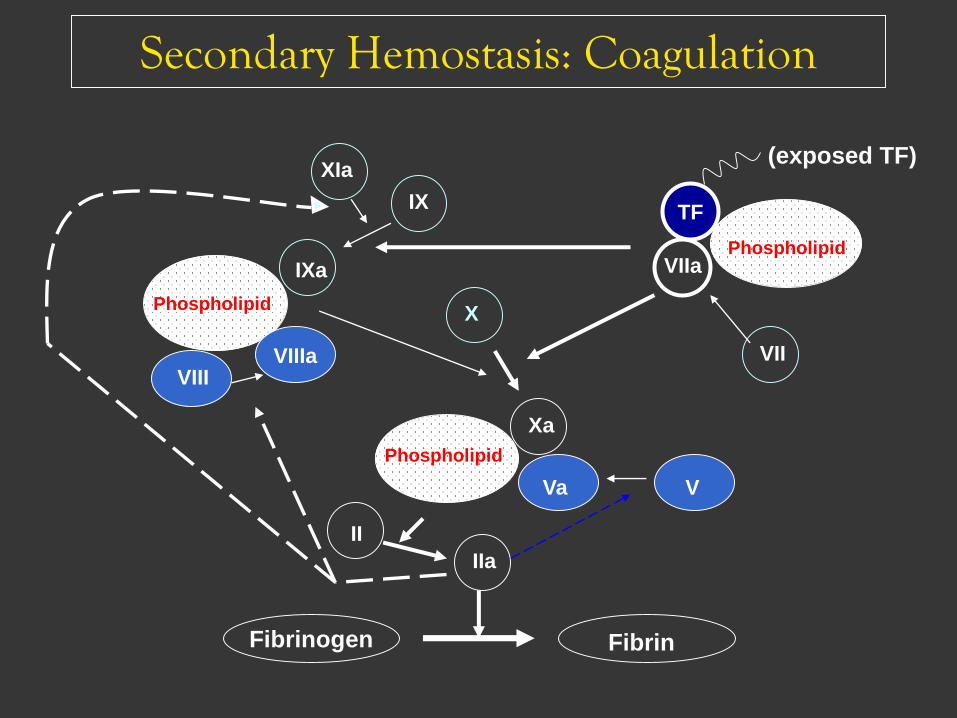
Fibrinogen
Xa
Va
II
IIa
Fibrin
VIIaPhospholipid
TF
X
V
VIIIa
IXa
XIa
IX
VIII
Phospholipid
Phospholipid
Secondary Hemostasis: Coagulation
VII
(exposed TF)

Coagulation Cascade Screening Tests
Fibrin stabilizes platelet plug
Endothelial Injury
Soluble fibrinogen
aPTT PT
TT

Case 1: Pregnancy Panic
• A 32 yo woman from another state, pregnant with
her first child, is visiting friends and unexpectedly
goes into labor.
• She tells the covering obstetrician and OB
anesthetist at your hospital that she has von
Willebrand’s Disease and wants to know if it is
okay for her to have an epidural.
• You are asked to see her and provide advice re the
advisability of an epidural and possible treatment
for the von Willebrand’s disease.

Laboratory results• Chemistries including BUN, creatinine and
glucose are normal
• CBC:– Hb is 12.3 gm/dl
– Hct 35.5%
– WBC 9500
– Platelet count 450,000
– Differential is reported as normal
• Coagulation Parameters:
– PT 11.5 seconds (INR 1.1)
– PTT 28 seconds (nl < 35)

Your consultation
• She tells you that she has had occassional nosebleeds since she was a child and has had menorrhagia, also she bled a lot when she had her wisdom teeth extracted.
• She is usually treated with the intravenous or intra-nasal administration of ddavp for bleeding or before surgeries.
• Her mother and one sister have similar symptoms and respond to the same medication regimen.
• You try but are not able to reach her primary hematologist.


Mannucci, 2004

Coagulation in Pregnancy
Increase
•Fibrinogen •vWF•VIII•VII•X•IX•XII
Decrease
•Protein S•Acquired resistance to activated protein C•Fibrinolysis (inhibition)•Platelet Count

vWF
• Large multimeric protein
– Synthesized in endothelial cells, megakaryocytes
– Stored in Weibel-Palade bodies of endothelial cells, alpha granules of platelets
• 3 main functions
– Bind to collagen on subendothelial matrix
– Bind to platelet GPIb receptor
– Prolong half life of factor VIII from 24 mins to 12 hours

vWD
• Most common inherited hemostatic disorder,
prevalence up to 1%
• Mucocutaneous bleeding • Epistaxis
• Easy bruising
• GI bleeding
• Post-op
• Surgery--immediately
• Dental procedures
• “Women’s health issue• 75% women with vWD have menorrhagia
• often dx with first menstrual period or PPH

vWF: Laboratory Evaluation
• aPTT
• FVIII:C
• vWF:Ag
• vWF:RCo Ristocetin cofactor activity
• Multimer gel electrophoresis

vWD
• Mild vWD can be difficult to diagnose
• Levels vary and are affected by:
– Blood type
– Estrogen
– Inflammation
– Stress
– Smoking

vWD: Classification
– Type I: autosomal dominant, quantitative
decrease in vWF and concordant decrease in
all functions
• 70-80% of vWD cases
– Type III: homozygous recessive, almost no
detectable vWD
• Very Rare

vWD: Classification
Type II: Qualitative Abnormalities
A: decreased large mw multimers
10-15% of VWD
B: gain of function mutations, increased binding to
gpIb
M: loss of function mutations, decreased binding to
gpIb
N: loss of function mutations, decreased FVIII
binding

TYPE 1 vWD
• Quantitative deficiency
• Autosomal dominant trait
• 85% of patients
• Mis-sense mutations detected in some but not all patients

vWD and Pregnancy
• Antigen levels increase with increased estrogen
• Function does not change
• Increased risk of bleeding
– Miscarriage?
– 1st trimester
– Perineal hematomas
• 3/49 vaginal deliveries vs
• 2.2/1000 from 26,187 cohort study

vWD and Pregnancy
• Increased risk of post-partum hemorrhage
– Levels return to baseline over 7-21 days
– 20-25% patients with vWD have delayed PPH
– Mean onset 15.7 + 5.2 days

vWD: Treatment
DDAVP• Release of stored vWF from Weibel-Palade bodies
• Onset of action within 30 mins
• Watch for Tachyphylaxis with repeated dosing
• Side effects include:
– Headache, HTN, flushing,N/V, hyponatremia and seizures, uterine contractions
• Pre-procedure DDAVP challenge test to monitor response

vWD:Treatment
vWF containing concentrates
• plasma derived pathogen-inactivated.
• Dose by FVIII or vWF levels.
– Humate-P
– Alphanate
Cryoprecipitate
• Pooled product from 10 donors*
• Only as last resort

Next steps
• Assume she has von Willebrand’s as
suggested by her history?
• Administer DDAVP to treat her
presumed vWD?
• Tell the patient and her doctors that you
are not certain what she has and how to
treat it and she must not have epidural
anesthesia?

What happens next?
• At 3 am your patient delivers a 7 lb 4 oz baby boy without any bleeding
• You reach primary hematologist and learn that she indeed has Type I vWD.
• Labs prior to pregnancy vWF Ag 30%, activity 35%, VIII activity 45%
• Values two weeks before delivery vWF Ag 105%, activity 120%, VIII activity 150%

And then ….
• 48 hours after delivery you are making
rounds at the hospital and are paged by her
obstetrician because she has developed
uterine hemorrhage.
• What has happened?

VON WILLEBRAND’S DISEASE AND
PREGNANCY
• SELF CORRECTION IN (MOST) TYPE 1 PATIENTS
• POSTPARTUM BLEEDING IN 25% OF PATIENTS
• INCREASED THROMBOCYTOPENIA IN TYPE 2B PATIENTS

vWD: NHLBI Guidelines
• “We suggest that VWF ristocetin cofactor activity and factor VIII levels of at least 50 IU/dL be achieved before delivery and maintained for three to five days afterward (Grade
2C).”
• For patients who do not have this degree and length of response on their own or following the use of DDAVP, we suggest the use of VWF concentrates (Grade 2C).”
NHLBI Guidelines for vWD 2008

vWD: NHLBI Guidelines
“In patients who are known to adequately
respond to DDAVP, we suggest that
DDAVP be given after the beginning of
labor and as close to the time of delivery as
can be estimated (Grade 2C).”
NHLBI Guidelines for vWD 2008

vWD: NHLBI Guidelines
“We suggest that regional anesthesia can be
considered if vWF and FVIII levels can be
maintained above 50 IU/dl (Grade 2C).”
NHLBI Guidelines for vWD 2008

Case 2: Can’t Stop Bleeding
• 23 year old female who presents with bleeding after wisdom tooth
removal. Patient has been referred to ED from dental clinic after the
dentist could not control the bleeding.
• Pt. has no history of epistaxis, gingival bleeding but does report that at
onset of menses her period was very heavy prompting her to regulate
menses with OCPs.
• No history of any other tooth extractions or surgical procedures.
• Family Hx: Mother with von Willebrand disease per report of patient.
No other bleeding history in the Family.

Can’t Stop the Bleeding: Labs
• WBC 8.6
• Hct 38
• Plt 235, normal appearance on smear
• PT 1.1
• PTT 32.5
• Fibrinogen 365
• von Willebrand Panel is normal
• Platelet Aggregation studies: Abnormal

Can’t Stop the Bleeding:
Platelet Aggregation Studies
• Absent platelet aggregation in response
to multiple agonists:
– ADP
– Thrombin
– Collagen
– Arachadonic Acid
• Normal Aggregation to Ristocetin

Signs of Thrombocytopathies
• Prolonged bleeding times
• Defective clot formation
• Bleeding tendency from childhood
• Low prevalence of these disorders but high
percentage of patients with unclassified
platelet dysfunction.

Classify platelet disorders by platelet
functionality
• Adhesion/Aggregation
• Activation/Signal Transduction
• Storage Granule Disorder/Release
• Aggregation

Platelet aggregation studies
• Aggregation
– Light transmission aggregometry using Chrono-Log instrument
– ADP, arachidonic acid, collagen, epinephrine, ristocetin
• ATP secretion
– Luciferin-Luciferase assay
– ADP, arachidonic acid, collagen,
epinephrine, ristocetin, thrombin

Platelet aggregation studies
• Requirements:– Platelet count minimum 100K/uL
– 8 blue-top (citrate) tubes
• Common interferences:– EDTA
– NSAIDs (e.g., aspirin, ibuprofen, indomethacin, COX-2 inhibitors)
– ADP receptor antagonists (e.g., clopidogrel, ticlopidine)
– GPIIb/IIIa antagonists (e.g., abciximab, eptifibatide, tirofiban)
– Various other drugs (see Arch Pathol Lab Med. 2002;126:133–146)

Problems with Platelet Aggregation
Studies
• Numerous variables affect aggregation:• Anticoagulant (sodium citrate best)
• Plt count in PRP
• Plt size distribution
• Time of day
• Temporal relation to meals and physical activity

Aggregometry
•Platelets shift from disc-like to a rounded form with pseudopods transiently decreasing light transmission.
•Then they aggregate into clumps, increasing light transmission.

The Aggregometer Tracings
Secondary wave (Dense granule
release, thromboxane A2
production)
Primary wave (Agonist
interacts with receptor)

http://medicinembbs.blogspot.com/2011/02/normal-hemostasis.html
Disorders in Platelet Aggregation

Aggregation Disorders: Glanzmann’s
Thrombasthenia
• History of muco-cutaneous bleeding
• Autosomal Recessive Inheritance
• Deficiency of GPIIb/GPIIIa complex

Diagnosis• Platelet count and morphology is normal
• Bleeding time prolonged
• The hallmark of the disease is severely reduced or absent platelet aggregation in response to multiple agonists ie ADP, thrombin, or collagen (except Ristocetin)
• Flow cytometry: decreased mAb expression of CD41 (GPIIb) and CD61 (GPIIIa)

Can’t Stop the Bleeding:
Management
• Platelet transfusions
• Supportive care: ddavp and amicar
• She improved after transfusions of platelets.

Case 3: Is it safe to operate?
• 75 y.o. female comes to you for pre-op
clearance for cardiac surgery.
• She is referred to your clinic for a prolonged
PTT.
• She has not seen a physician in 40 years.
• No bleeding history per the patient

Laboratory testing
• Factor XIII screen is normal
• Factor VIII level is 102%
• Factor IX level is 89%
• Factor XI level is 22%

Factor XI Deficiency
• Rare bleeding disorder
• Inherited as Autosomal Recessive
• Prolongs the PTT
• Characterized by variable bleeding history
• Incidence is 1 in 450 in the Ashkenazi
Jewish population and 1 in a million in non-
Jewish population.

Factor XI deficiency
• Usual sights of bleeding:
– Skin and mucousa
– Genitourinary tract
– Gastrointestinal tract


Factor XI deficiency
• Bleeding manifestations do not correlate with factor XI
levels
• Most bleeding episodes in patients with severe deficiency
are injury-related
• Spontaneous bleeding is rare
• May be associated with bruising, epistaxis, menorrhagia,
GI/GU bleeding, umbilical stump bleeding or bleeding
after surgery, trauma, dental procedures, pregnancy or
circumcision
• Up to 33% of patients with severe deficiency develop
inhibitors after replacement therapy

Treatment for Factor XI
Deficiency
• 10-20 ml fresh frozen plasma/kg, then 5-10 ml/kg
every 24 hours as necessary
• Antifibrinolytic therapy has been used in women
with factor XI deficiency and menorrhagia
• Patients with inhibitors have been treated
successfully with plasma, prothrombin complex
concentrates, and recombinant activated factor VII
• Note: factor XI concentrates available in Europe
monitor for thromboembolic complications

CASE 4
25 yo healthy woman
• CC: rash on lower extremities
• PE:• skin: rash
• HEENT: hemorrhagic bullae in mouth
• Otherwise normal
• Labs: • WBC 3.9 with normal diff; Hct 35
with normal MCV; Plts 14K
• PT/PTT normal
• electrolytes, LFTs normal

Thrombocytopenia
DECREASED PRODUCTION
• Marrow Failure
• Drugs
• Myelophthysis
• Nutritional deficiency (megaloblastic anemia)
SEQUESTRATION
• Splenomegaly (rarely <50,000)
INCREASED DESTRUCTION
• ITP
• TTP
• DIC

Evaluation of Thrombocytopenia
History• Intercurrent illnesses
• Medication history
• History of autoimmune disease, LPD
Physical• Bleeding manifestations (purpura, petechiae)
• Splenomegaly
• Adenopathy
Laboratory• Examination of the peripheral smear
• Platelet reticulocyte count
• Antiplatelet antibodies??
• Bone marrow examination??

Laboratory Evaluation for
Thrombocytopenia
• CBC
• Peripheral Blood Smear
• Liver Function tests
• Thyroid Function tests
• Quantitative immunoglobulin levels
• Direct antiglobulin test
• Antiphospholipid antibodies
• ANA
• H pylori testing
• HIV, HCV, HBV
• VWD type IIb testing

ITP Therapy: Initial Treatment
Agents used for initial therapy of ITP:
• Corticosteroids
• High dose methylprednisolone
• Prednisone
• Dexamethasone
• IV Immunoglobulin
• Anti-D

ITP Therapy: Initial Treatment
“Standard of care”
• Prednisone 1mg/kg/day
• Taper—schedules undefined
• Response: 65-90%
• Long term response: 5-30%
• Of note, study that reported 30% assessed at
6 months.
• Most studies with longer follow-up report <10%
long term response to prednisone

ITP Therapy: Initial TreatmentDexamethasone:
• Single course, 40mg/day X 4 (NEJM 2003; 349: 831)
• Initial response: 85%• Relapse: 50%; 2nd course response: 100%• Sustained: 42%• Persistent response after D/C maintenance: 18%
• Multiple course, 40mg/day X 4 (BLOOD 2007;109: 1401)
• Two protocols: 6 cycles q28 days/4 cycles q14 days• Initial response: 89%/86%• RFS at 15 mos: 90% /81%• Long term response:
• median 26mos with 6 cycles: 68%• median 8 mos with 4 cycles: 74%
• Randomized trial Dex vs Prednisone (BLOOD 2016;127: 296)
• Durable responses with less toxicity• Only a single course, so still need a trial of repeated courses of
dexamethasone
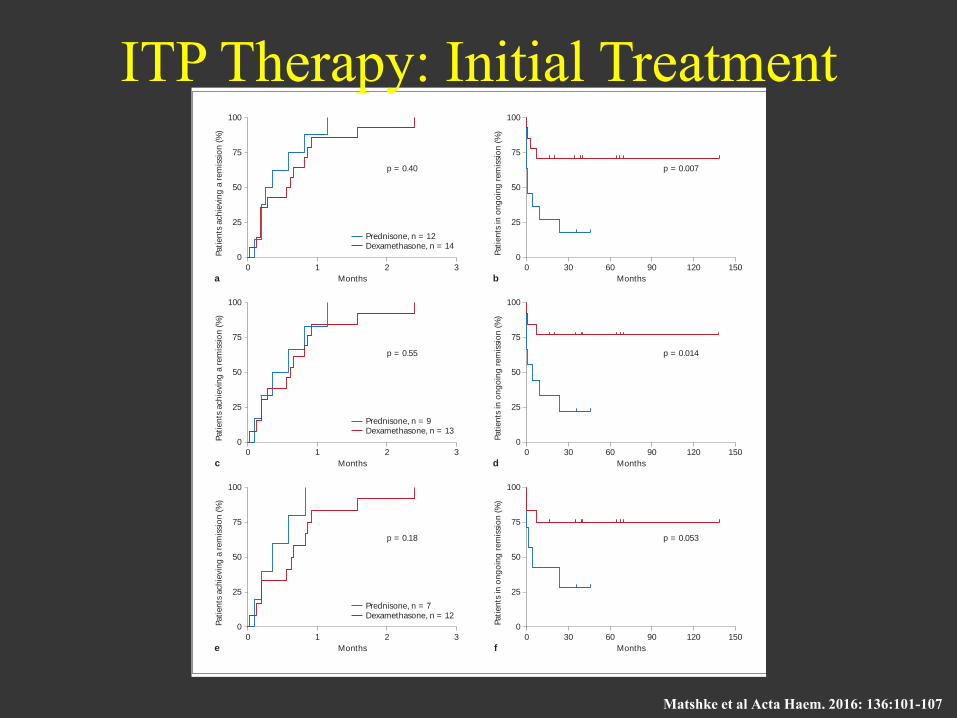
Prednisone versus Pulsed Dexamethasone in Adult Patients with ITP
Acta Haematol 2016;136:101–107DOI: 10.1159/000445420
103
100
75
50
25
0
0 1 2
p = 0.40
Prednisone, n = 12Dexamethasone, n = 14
Months
Pati
en
ts a
ch
ievin
g a
rem
issi
on
(%
)
a
3
100
75
50
25
0
0 30 60 90 120
p = 0.007
Months
Pati
en
ts in
on
go
ing
rem
issi
on
(%
)
b
150
100
75
50
25
0
0 1 2
p = 0.55
Prednisone, n = 9Dexamethasone, n = 13
Months
Pati
en
ts a
ch
ievin
g a
rem
issi
on
(%
)
c
3
100
75
50
25
0
0 30 60 90 120
p = 0.014
Months
Pati
en
ts in
on
go
ing
rem
issi
on
(%
)
d
150
100
75
50
25
0
0 1 2
p = 0.18
Prednisone, n = 7Dexamethasone, n = 12
Months
Pati
en
ts a
chie
vin
g a
rem
issi
on
(%
)
e
3
100
75
50
25
0
0 30 60 90 120
p = 0.053
Months
Pati
en
ts in
on
go
ing
rem
issi
on
(%
)
f
150
Fig. 1. Time to remission (PLT 50 × 10 9 /l; a , c , e ) and remission duration ( b , d , f ) in ITP patients receiving daily prednisone versus pulsed dexamethasone in the intent-to-treat ( a , b ), per-protocol ( c , d ) and primary ITP per-protocol populations ( e , f ). All patients achieved a remission. There was no statistically significant differ-
ence in the interval between treatment initiation and the first doc-umentation of remission. The remission duration was significant-ly longer in the dexamethasone arm as compared to the prednisone arm (Kaplan-Meier analysis, log-rank test).
Co
lor
vers
ion
availab
le o
nlin
e
Do
wnlo
aded
by:
Ha
rvard
Un
ive
rsity
12
8.1
03.1
49
.52 -
4/1
0/2
017 6
:12
:26 P
M
ITP Therapy: Initial Treatment
Matshke et al Acta Haem. 2016: 136:101-107

Therapy: Relapsed or Refractory ITP
TherapyResponse
Rate
Time to
Response ToxicityDuration of
Response
Splenectomy 80% 1-24 days Surgical
complications,
thrombosis
2/3 with no
further Rx
Rituximab 60% response
40% complete
1-8 weeks Allergic rxns;
immune supp.
15-20%
sustained
Can be retreated
TPO Mimetics >80% response
(plts >50K)
2-3 weeks Unknown long-
term toxicity
Increased reticulin
Rapid fall of plts
with D/C of drug
Up to 1.5-4 yrs
with continuous
drug
Adapted from Blood. 2010;115:168-186

TPO Response to Thrombocytopenia
Hematol Oncol Clin North Am.23:1193, 2009.

Thrombopoietin Mimetics
Romiplostim• Fc-peptide fusion
protein (peptibody)
• Binds to and activates the TPO receptor to increase platelet counts
• Approved for treatment of chronic ITP in US, EU, Canada and Australia
Eltrombopag
• Small molecule, oral TPO receptor agonist
• Increases platelet production by increasing megakaryocyte growth
Fc DomainTPOr binding
domain
Both approved for the treatment of chronic ITP

Take Home Messages: Case 4
• The treatment of ITP continues to be somewhat controversial
• First-line therapy is corticosteroids—my recommendation is pulse dexamethasone
• Second line therapy is probably still splenectomy
• However, rituximab is effective in many patients, is probably the third most likely to give a long remission, and can be repeated
• TPO mimetics work but have some known and many possible unknown downsides.

Summary• In bleeding disorders you must think about
primary and secondary hemostasis to
understand etiology.
Primary: Platelets disorder and vWD
Secondary: Coagulation factors and
fibrinolysis
History is Key for diagnosis and management.

Board Review Questions

Question 1• You are asked to see a 37 yo woman
with history of vWD for consultation. She asks: “Is it safe for me to have elective knee surgery?”
• Pt. never had an surgery. She had frequent epistaxis as a child and heavy menses.
• FH: Mother has hx vWD but died young (trauma). 5 uncles, one with bleeding disorder.

Question 1
– F VIII:C 27
– vWF:ag 89
– vWF:RCo 75
– PTT 46 sec
• What type of vWD does this patient have?

Question 1
• A. Types I vWD. Treat with ddavp prior to
surgery.
• B. She does not have vWD because her
VWF:ag and :Rco are normal.
• C. You need more info. She could have type
2N or be a hemophilia A carrier.

2N vs. Hemophilia A carrier
• 2N should be consider in AR inheritance
with disproportionately low FVIII:C levels
compared with VWF levels.
• To prove 2N, a FVIII-VWF binding assay is
required.
• VWF gene mutation screening:R854Q at
amino-terminus of VWF subunit is most
frequent

2N vs. Hemophilia A carrier• Treatment Plan?
• Hemophilia A carrier
– Low production FVIII
– Normal vWF
– Treat with recombinant FVIII
– Maintain levels in postop period
• vWD Type II N
– Normal FVIII production
– Endogenous vWF does not bind FVIII
– Treat with Humate-P
– Monitor levels in postop period

Question 1
• A. Types I vWD. Treat with ddavp at
delivery and postpartum.
• B. She does not have vWD because her
VWFag and Rco are normal.
• C. You need more info. She could have type
2N or be a hemophilia A carrier.

Question 2
• 38 y.o. Female is seen in the ER for
bleeding gums after a dental procedure.
She tells you that she has essential
thrombocytosis and takes daily
hydroxyurea and aspirin.
• A CBC is drawn and her platelet count
is 1600 X109/L million.
The ER team asks why this patient is
bleeding

• A. The patient is on aspirin and
bleeding due to its anti-platelet effects.
• B. The patient has developed an
acquired von willebrand disease.
• C. This is a laboratory error and the
platelet count should be repeated.
Possible Answers

Question 2
• This patient has developed an
acquired von willebrand disease in
the setting of thrombocytosis.
• Diagnosis is confirmed by von
Willebrand panel demonstrates low
ristocetin cofactor activity with normal
antigen levels.
• Aspirin should be used cautiously in
patients with platelets >1000 x 109/L

References
Mannucci, PM. Treatment of von Willebrand’s Disease. N Engl J Med. 2004 Aug 12;351(7):683-94.
Giuseppe L Diego A, Roberto Q, and Gianfranco C. Urgent monitoring of direct oral anticoagulants in
patients with atrial fibrillation: a tentative approach based on routine laboratory tests. Journal of Thrombosi
and Thrombolysis, May 2014 (epub).
Conolly, S. et al. Dabigatran versus Warfarin in Patients with Atrial Fibrillation. N Engl
J Med 2009; 361: 1139-1151.
Duga, S., Salomon O., Congenital Factor XI deficiency: an update. Semin Thromb Hemost. 2013
Sep;39(6):621-31. doi: 10.1055/s-0033-1353420. Epub 2013 Aug 8.
Gernsheimer, T., James, AH, and Stasi, R. How I treat thrombocytopenia in pregnancy. Blood. 2013 Jan
3;121(1):38-47. doi: 10.1182/blood-2012-08-448944. Epub 2012 Nov 13.

Elisabeth M. Battinelli, M.D./Ph.D.
Assistant Professor of Medicine
Associate Physician
No Disclosures





























